ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым способам получения 1,3-бензодиоксольных гетероциклических соединений и промежуточным соединениям указанных способов. Соединения эффективны в качестве ингибиторов PDE4.
УРОВЕНЬ ТЕХНИКИ
В публикации WO 2011/160632 раскрыты бензодиоксольные и бензодиоксепеновые гетероциклические соединения, эффективные в качестве ингибиторов PDE4, а также подходящие способы их получения.
В публикации WO 2008/104175 раскрыты бензодиоксольные и бензодиоксепеновые гетероциклические соединения, эффективные в качестве ингибиторов PDE4, а также подходящие способы их получения.
В публикации WO 2008/077404 раскрыты замещенные ацетофеноны, эффективные в качестве ингибиторов PDE4, а также подходящие способы их получения.
При разработке новых потенциальных лекарственных средств очень желательно иметь доступ к альтернативным способам получения потенциальных лекарственных средств, так как может оказаться, что некоторые эффективные мелкомасштабные способы синтеза трудно использовать при переходе к получению промышленных количеств. Также, в мелкомасштабных способах синтеза можно применять реагенты и растворители, которые не подходят для применения в промышленных масштабах.
Таким образом, задачей настоящего изобретения является обеспечение альтернативных способов получения 1,3-бензодиоксольных гетероциклических соединений, принадлежащих к типу, раскрытому в WO 2011/160632, так как указанные альтернативные способы обеспечивают преимущества, связанные с одним или более отличительными признаками, такими как количество стадий взаимодействия, чистота, выход, простота очистки, экономичность способа, доступность исходных веществ и реагентов, безопасность, предсказуемость и т.д.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения обнаружили, что альтернативный способ, раскрытый в настоящем описании, обеспечивает преимущества по сравнению с известными способами за счет снижения количества стадий взаимодействия от 10 стадий, требовавшихся ранее, до 4 стадий в настоящем случае, за счет снижения трудоемкости синтеза вещества, улучшенного общего химического и волюметрического выхода и простоты способа получения, так как выделение некоторых промежуточных соединений не проводят.
Таким образом, в настоящем изобретении предложен способ получения 1,3-бензодиоксольных соединений, например, соединения формулы (I).
Также в объем настоящего изобретения входят промежуточные соединения, применяемые в вышеуказанном способе получения соединений формулы (I), и способы получения указанных промежуточных соединений, включающие одну или более вышеуказанных стадий.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно первому аспекту настоящее изобретение относится к способу получения соединения формулы (I)
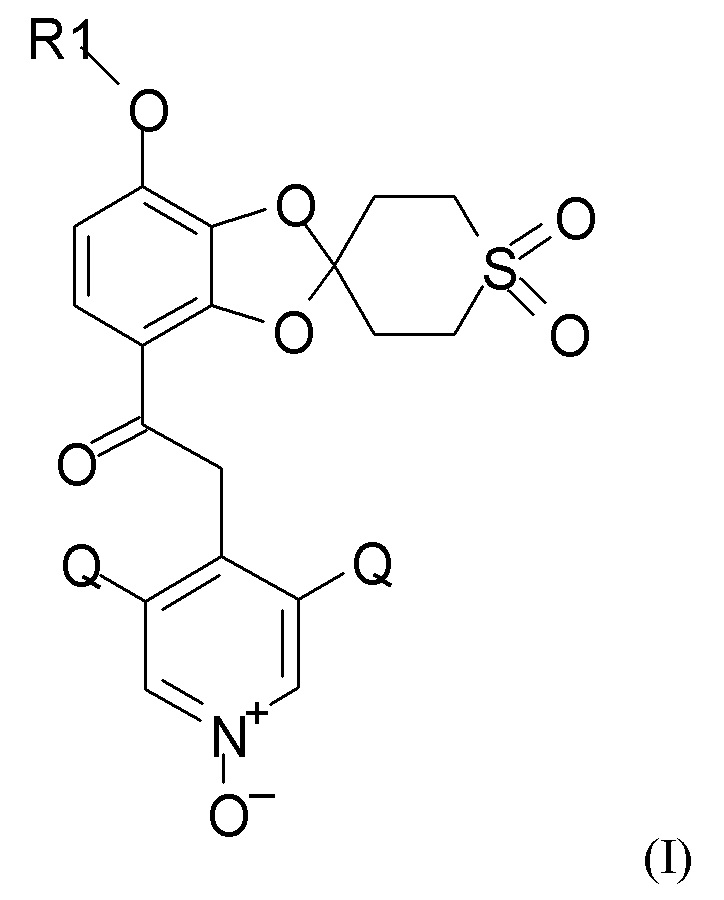
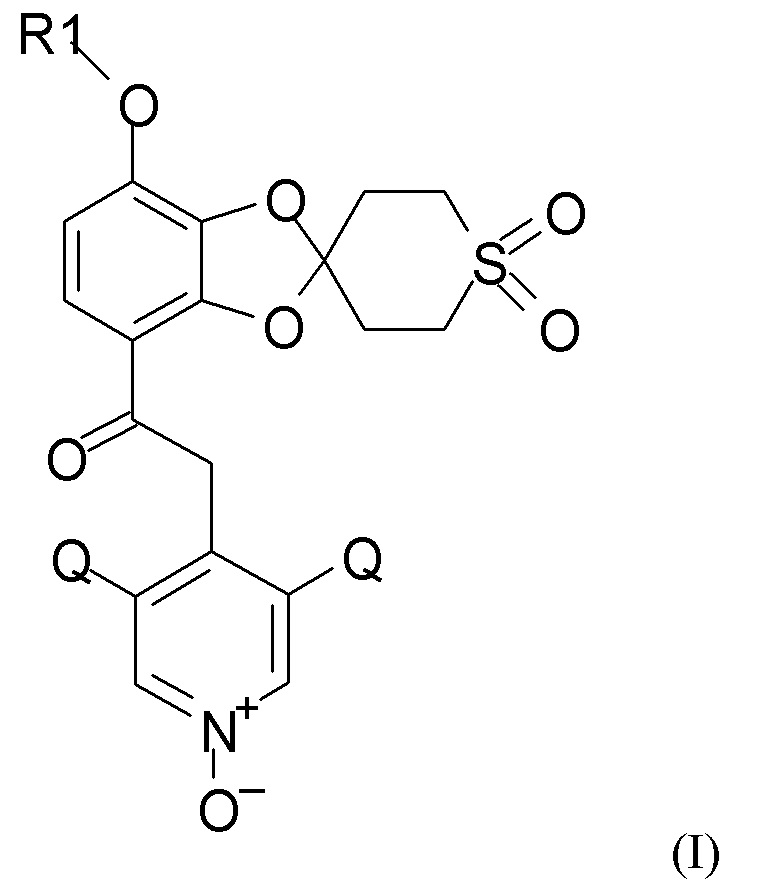
где R1 выбран из CHF2 и CF3, и Q выбран из хлора, брома и фтора.
В соединении формулы (I), R1, как правило, представляет собой CHF2. Q, как правило, выбран из хлора, брома и фтора, предпочтительно представляет собой хлор, при этом Q предпочтительно являются одинаковыми. В одном из вариантов реализации оба Q представляют собой хлор.
Определения
Подразумевается, что термин «C1-6-алкил» обозначает насыщенную линейную или разветвленную углеводородную цепь, содержащую от одного до шести атомов углерода, включая метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил и изогексил. В некоторых вариантах реализации «C1-6-алкил» представляет собой C1-4-алкильную группу, например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Соответственно, «C1 3-алкил» включает метил, этил, пропил и изопропил.
Подразумевается, что термин «галоген» обозначает фтор, хлор, бром и йод. В одном из вариантов реализации термин «галоген» обозначает фтор или хлор. В другом варианте реализации термин «галоген» обозначает хлор.
Подразумевается, что термин «арил» обозначает карбоциклическую ароматическую систему колец, полученную из ароматического углеводорода в результате удаления атома водорода. Кроме того, арил включает би-, три- и полициклические системы колец. Примеры предпочтительных арильных фрагментов включают фенил, нафтил, инденил, инданил, флуоренил и бифенил. Предпочтительными «арилами» являются фенил, нафтил или инданил, в частности фенил, если не указано иное.
Подразумевается, что термин «аралкил» обозначает арильный радикал, такой как определено выше, ковалентно присоединенный к алкильной группе, например, бензил.
Способы получения
Как полагают, способ обеспечивает преимущества по сравнению с известными способами, заключающиеся в применении дешевых исходных веществ и в простоте получения, так как выделение некоторых промежуточных соединений не проводят, что снижает количество стадий взаимодействия. Также общий выход был увеличен в 2,5 раза.
Стадия (1)
Способ получения соединения формулы (I) включает образование соединения формулы (IV), которое получают путем
взаимодействия соединения формулы (II)
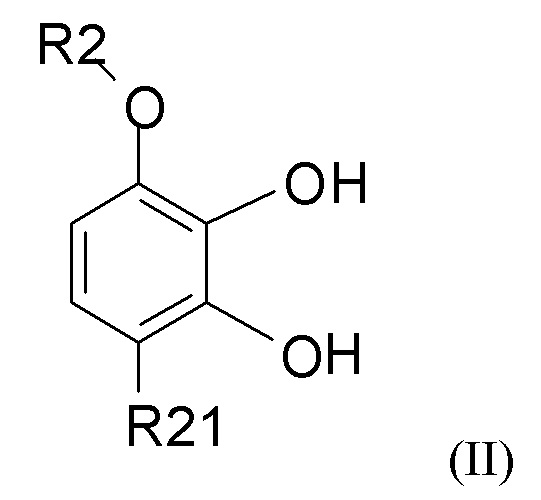
где R2 выбран из водорода, C1-6-алкила и арилалкила, R21 выбран из водорода, C(O)R22 и C(O)OR22, и R22 выбран из водорода и C1-6-алкила; с соединением формулы (III)
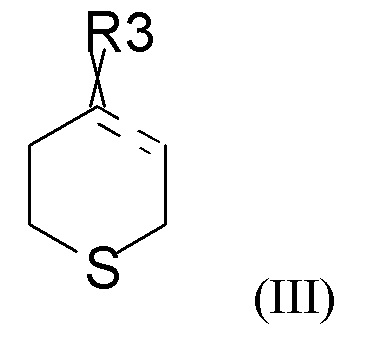
где « » представляет собой простую связь, двойную связь или две простые связи, и если «» представляет собой двойную связь или две простые связи, то «
» представляет собой простую связь, двойную связь или две простые связи, и если «» представляет собой двойную связь или две простые связи, то « » представляет собой простую связь, и если «» представляет собой простую связь, то «» представляет собой двойную связь, R3 представляет собой кислород, если «» представляет собой двойную связь, и R3 представляет собой O-C1-6-алкил, если «» представляет собой простую связь или две простые связи; в присутствии кислотного катализатора для получения соединения формулы (IV)
» представляет собой простую связь, и если «» представляет собой простую связь, то «» представляет собой двойную связь, R3 представляет собой кислород, если «» представляет собой двойную связь, и R3 представляет собой O-C1-6-алкил, если «» представляет собой простую связь или две простые связи; в присутствии кислотного катализатора для получения соединения формулы (IV)
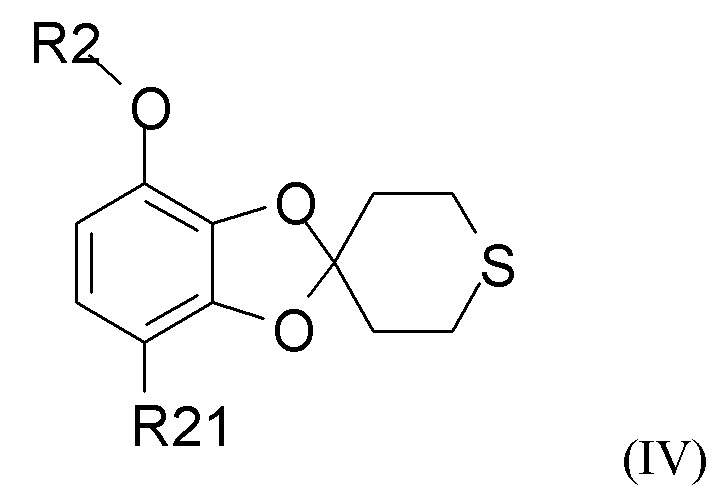
где R2 и R21 такие, как определено выше.
Кислотный катализатор, как правило, имеет форму силикатного минерала. Силикатный минерал, как правило, выбран из монтмориллонита K10, монтмориллонита K30, монтмориллонита KSF, цеолита HSZ-341NHA, цеолита HSZ-331NHA, цеолита HSZ-350HUA и цеолита HSZ-360HUA. В одном из вариантов реализации силикатный минерал выбран из монтмориллонита K10 и цеолита HSZ-360HUA. В другом варианте реализации силикатный минерал представляет собой монтмориллонит K10.
Соединение формулы (III), как правило, выбрано из
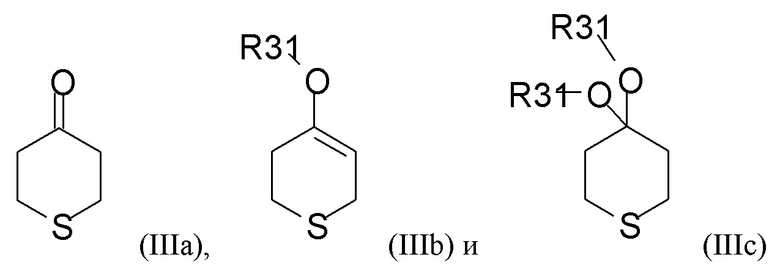
где R31 представляет собой C1-6-алкил. В одном из вариантов реализации соединение формулы (III) выбрано из соединений формулы (IIIa) и формулы (IIIb), где R31 представляет собой метил.
Отношение силикатного минерала к соединению формулы (II) может влиять на продолжительность фильтрования. Таким образом, как правило, предпочтительно количество минерала составляет от 25% (масс./масс.) до 500% (масс./масс.) относительно количества соединения формулы (II). В частности, количество минерала должно составлять от по меньшей мере 50% (масс./масс.) до 200% (масс./масс.).
Взаимодействие, как правило, проводят в толуоле, бензоле, 2-метил-ТГФ (2-метилтетрагидрофуран), EtOAc (этилацетат), ксилолах, гептане, октане, хлорбензоле и дихлорбензоле. В одном из вариантов реализации растворитель представляет собой толуол.
Взаимодействие, как правило, проводят при температуре выше 80°C для ускорения взаимодействия. Таким образом, как правило, температура предпочтительно находится в диапазоне 80-200°C, например, в диапазоне 100-160°C, в частности составляет 110°C. Взаимодействие, как правило, проводят в течение 4-96 часов, например, 24-72 часов.
Полученное соединение формулы (IV) можно выделять при помощи традиционных способов, известных специалистам в данной области техники, например, путем фильтрования.
В одном из вариантов реализации изобретения соединение формулы (II) представляет собой соединение, где R2 выбран из водорода или метила, и R21 выбран из водорода, COCH3 или COOH. В другом варианте реализации соединение формулы (II) представляет собой 2,3-дигидрокси-4-метоксиацетофенон.
В одном из вариантов реализации изобретения соединение формулы (III) представляет собой тетрагидро-4Н-тиопиран-4-он.
В одном из вариантов реализации изобретения соединение формулы (IV) представляет собой соединение, где R2 представляет собой водород, метил, этил, пропил, изопропил, изобутил, втор-бутил, трет-бутил или бензил, и R21 выбран из водорода, COCH3 или COOH. В другом варианте реализации соединение формулы (IV) представляет собой соединение, где R2 представляет собой метил, и R21 представляет собой COCH3.
Стадии (2а) и (2b)
Стадии взаимодействия (2а) и (2b) проводят как реакцию в одном сосуде, это означает, что выделение промежуточного соединения (VI) не проводят.
На стадии (2а) проводят взаимодействие енолятного соединения формулы (IV) с пиридиновым соединением формулы (V)
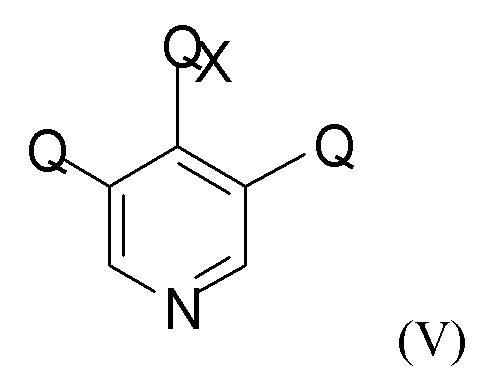
где Q такой, как определено выше, и QX выбран из хлора, брома, фтора и йода, для получения промежуточного соединения формулы (VI)
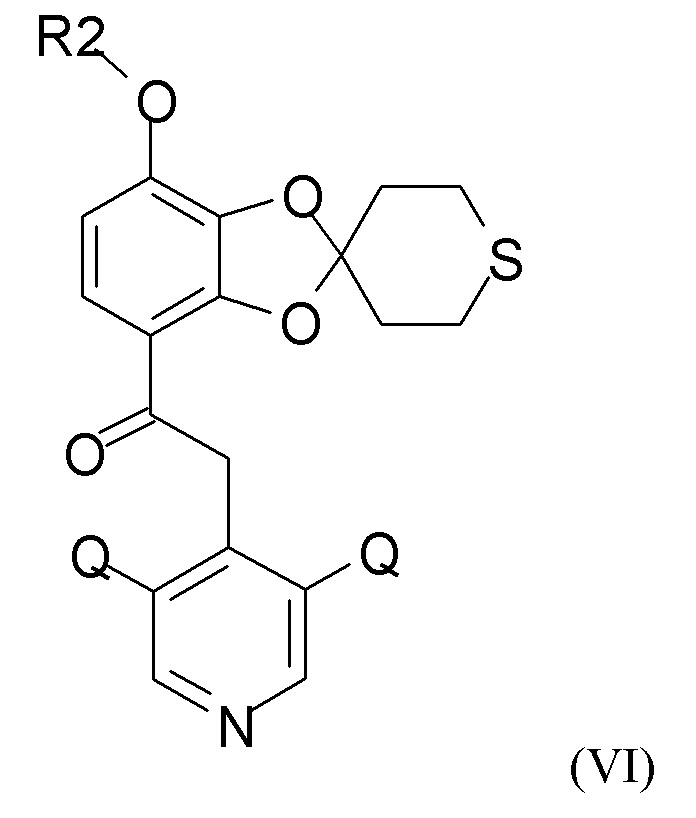
где R2 и Q такие, как определено выше; затем проводят снятие защиты на стадии (2b), где проводят взаимодействие промежуточного соединения формулы (VI) с соединением формулы (VII)
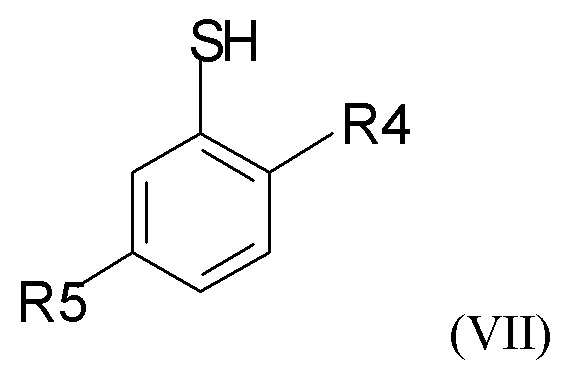
где R4 и R5 независимо представляют собой C1-6-алкил, для получения соединения формулы (VIII)
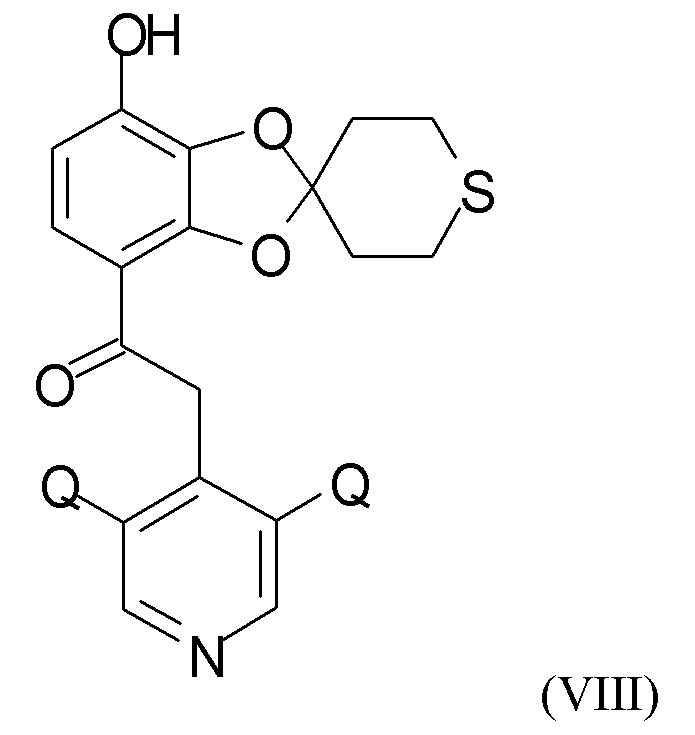
где Q такой, как определено выше.
Сочетание пиридина на стадии (2a), как правило, проводят в апротонном полярном растворителе, например, выбранном из NMP (N-метилпирролидон), ДМФА (N,N-диметилформамид), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), EtOAc (этилацетат), MeCN (ацетонитрил) и ТГФ (тетрагидрофуран) и их смесей, в присутствии основания, например, выбранного из трет-BuONa (трет-бутоксид натрия), трет-BuOK (трет-бутоксид калия), трет-BuOLi (трет-бутоксид лития), K2CO3, Na2CO3, KHCO3, NaHCO3, Et3N (триэтиламин) и DIPEA (N,N-диизопропилэтиламин). В одном из вариантов реализации апротонный растворитель выбран из ДМФА и NMP, в качестве основания присутствует трет-BuONa. В конкретном варианте реализации апротонный растворитель представляет собой NMP, и основание представляет собой трет-BuONa.
Основание, как правило, применяют в примерно стехиометрическом количестве относительно количества соединения формулы (V), например, где эквивалентное отношение (основание)/(формула V) составляет от 1:1 до 3:1, например, от 1,5:1 до 2:1, в частности от 1,7:1 до 1,9:1.
Взаимодействие (2а), как правило, проводят при температуре выше 0°C и ниже 15-20°C, например, в диапазоне 5-10°C.
В одном из вариантов реализации изобретения соединение формулы (V) представляет собой 3,4,5-трихлорпиридин.
Удаление защиты алкильной группы на стадии (2b) можно проводить с применением различных растворителей, например, выбранных из NMP (N-метилпирролидон), ДМСО (диметилсульфоксид), ДМФА (N,N-диметилформамид) и их смесей, в присутствии основания, например, выбранного из K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, ТЭА(триэтаноламин), трет-BuOLi (трет-бутоксид лития) и DIPEA (N,N-диизопропилэтиламин). В одном из вариантов реализации растворитель выбран из NMP, ДМСО и ДМФА, в качестве основания присутствует K2CO3. В другом варианте реализации растворитель представляет собой NMP, и основание представляет собой K2CO3.
Взаимодействие, как правило, проводят при температуре в диапазоне 50-120°С, например, в диапазоне 70-100 °C. Взаимодействие, как правило, проводят в течение 2-36 часов, например, 5-24 часов.
Можно применять различные реагенты формулы (VII). В одном из вариантов реализации изобретения соединение формулы (VII) представляет собой соединение, где R4 и R5 независимо выбраны из метила и трет-бутила. В другом варианте реализации соединение формулы (VII) представляет собой 5-трет-бутил-2-метилтиофенол.
Полученное соединение формулы (VIII) можно выделять при помощи традиционных способов, известных специалистам в данной области техники, например, путем обработки водными растворами и последующей экстракции и, наконец, осаждения и фильтрования.
В одном из вариантов реализации изобретения соединение формулы (VIII) представляет собой соединение, где Q выбран из хлора, брома и фтора. В другом варианте реализации соединение формулы (VIII) представляет собой соединение, где Q представляет собой хлор.
Стадия (2c)
На стадии (2c) проводят взаимодействие соединения формулы (VIII) с водным N(Bu4)+OH- для получения соединения формулы (IX)
N(Bu)4+
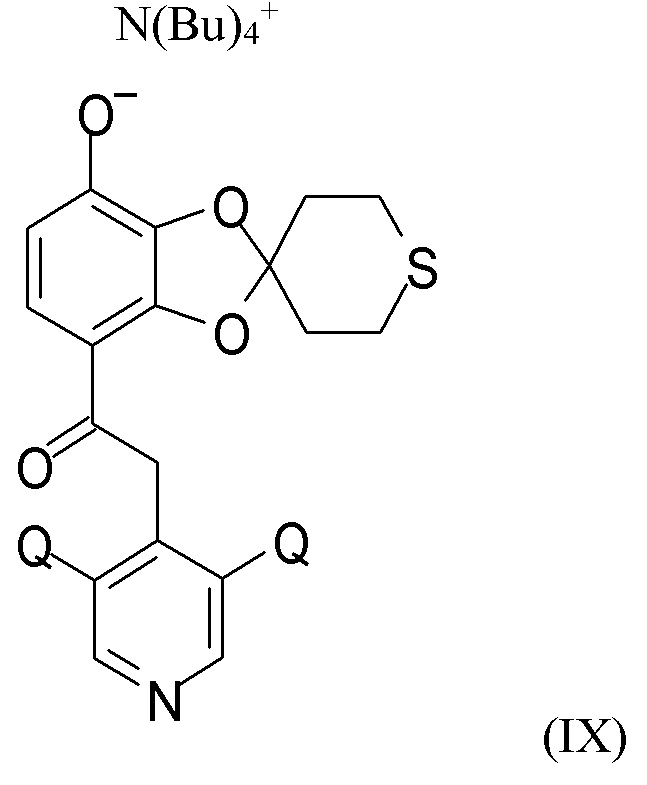
где Q такой, как определено выше.
Неочищенное соединение формулы (VIII) можно растворять, например, в ТГФ, толуоле или EtOAc, после чего добавляют водный N(Bu4)+OH-. В одном из вариантов реализации изобретения неочищенное соединение формулы (VIII) растворяют в ТГФ.
Полученную смесь, как правило, нагревают до температуры в диапазоне 20-60°C, такой как 45°C, и взаимодействие, как правило, проводят в течение 0,5-5 часов, например, 1-2 часов для обеспечения образования соли.
Полученный продукт, как правило, выделяют путем осаждения при помощи первоначального суспендирования неочищенного продукта формулы (IX) в МТБЭ (метил-трет-бутиловый эфир) или гептане, воде и соли (NaCl) в течение 1-2 часов; последующего охлаждения смеси до 0-20°C, например, 5°C, в течение 1-24 часов, например, 1-4 часов, что приводит к осаждению соли TBA (тетрабутиламмоний).
Стадия (3)
Соединение формулы (XI)
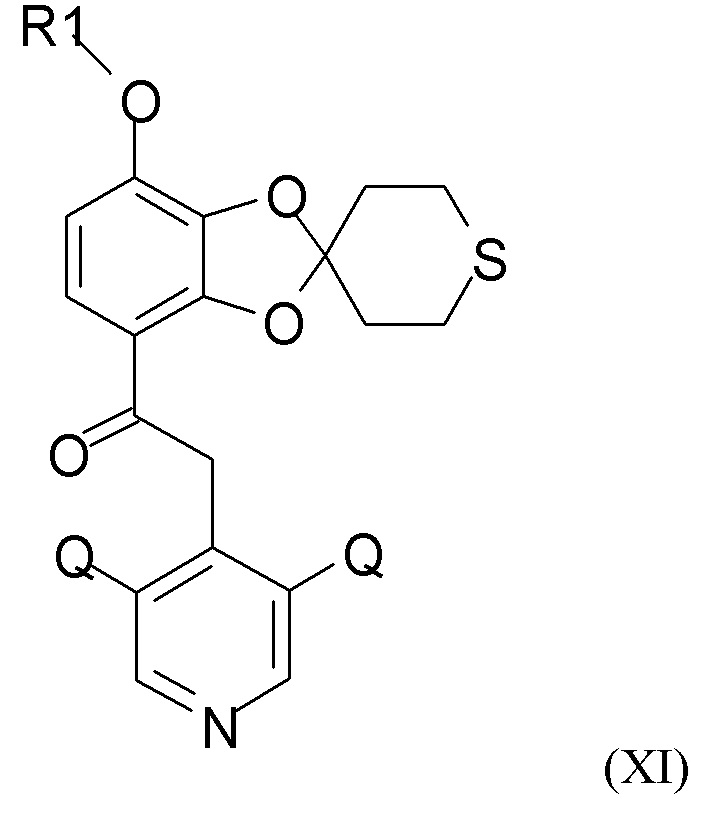
где R1 и Q такие, как определено выше, можно получать путем алкилирования полученного соединения формулы (IX) посредством взаимодействия с хлорфторуглеводородным реагентом, R1-Cl, где R1 такой, как определено выше.
Алкилирование можно проводить с применением одного из различных возможных реагентов, таких как различные хлорфторуглеводородные газы, под давлением. В одном из вариантов реализации реакцию алкилирования проводят с применением хлордифторметана в апротонном полярном растворителе, например, выбранном из ДМФА (N,N-диметилформамид), NMP (N-метилпирролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), EtOAc (этилацетат), MeCN (ацетонитрил) и ТГФ (тетрагидрофуран) и их смесей. В одном из предпочтительных вариантов реализации апротонный растворитель выбран из ДМФА и NMP. В конкретном варианте реализации взаимодействие проводят с применением хлордифторметана в ДМФА.
Взаимодействие, как правило, проводят при температуре в диапазоне 40-120°С, например, в диапазоне 50-70 °C. Взаимодействие, как правило, проводят до завершения.
Полученное соединение формулы (XI) можно выделять при помощи традиционных способов, известных специалистам в данной области техники, например, путем обработки водными растворами и последующей экстракции и, наконец, осаждения и последующего фильтрования.
Стадия (4)
Окисление полученного соединения формулы (XI) проводят для получения соединения формулы (I)
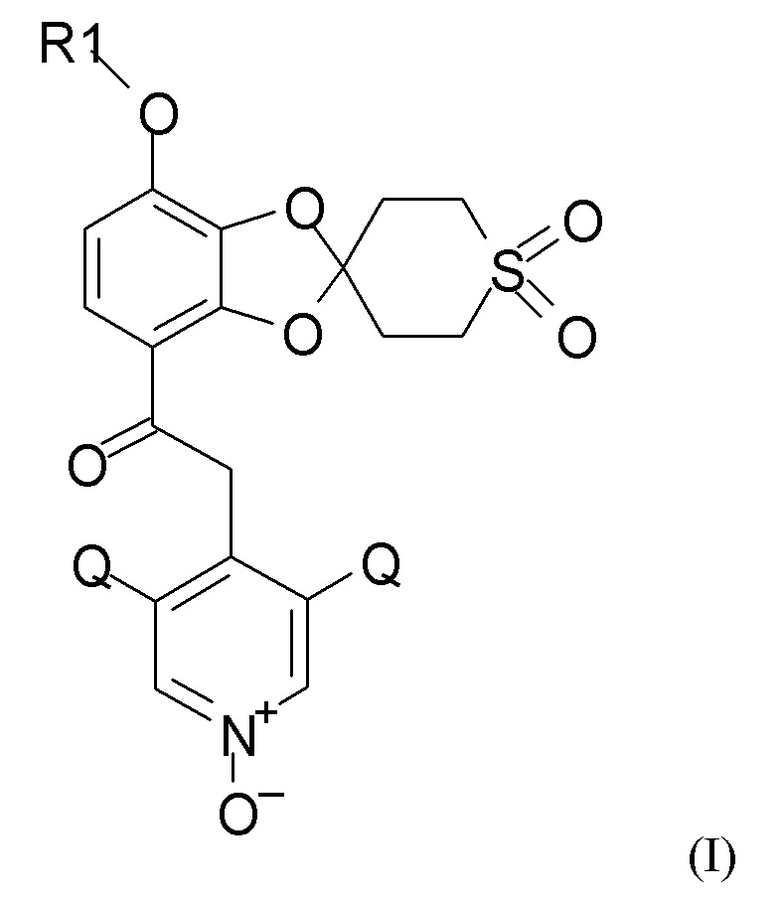
где R1 и Q такие, как определено выше, путем взаимодействия указанного соединения формулы (XI) с окислительным реагентом.
Окислительный реагент, как правило, выбран из надуксусной кислоты (НУК) в AcOH (уксусная кислота), и H2O2 (водн.) в муравьиной кислоте или уксусной кислоте. В одном из предпочтительных вариантов реализации окислительный реагент представляет собой НУК в AcOH. В одном варианте реализации относительное количество НУК по сравнению с (I), как правило, составляет от 3 до 6, в частности 4 экв. Окислительный реагент, как правило, медленно добавляют в течение 1-8 часов, например, 3-5 часов, поддерживая температуру в диапазоне 20-100°С, например, в диапазоне 25-50°C, в частности в диапазоне 25-40°C.
Взаимодействие, как правило, проводят при температуре в диапазоне 30-70°С, таком как 35-45°C, перемешивание проводят в течение 3-24 часов, например, 14-18 часов.
Очистка соединения формулы (I)
Полученный неочищенный продукт формулы (I) можно эффективно очищать путем кристаллизации, осаждения, хроматографии и т.д.
В одном из вариантов реализации полученный неочищенный продукт формулы (I) кристаллизуют из смеси воды и EtOH (этанол) и выделяют путем фильтрования и сушат.
Промежуточные соединения
Согласно другому аспекту настоящее изобретение относится к промежуточным соединениям, которые подходят для получения соединения формулы (I), где R1 выбран из CHF2 и CF3, и Q выбран из хлора, брома и фтора.
В одном из вариантов реализации изобретение относится к промежуточному соединению формулы (IV)
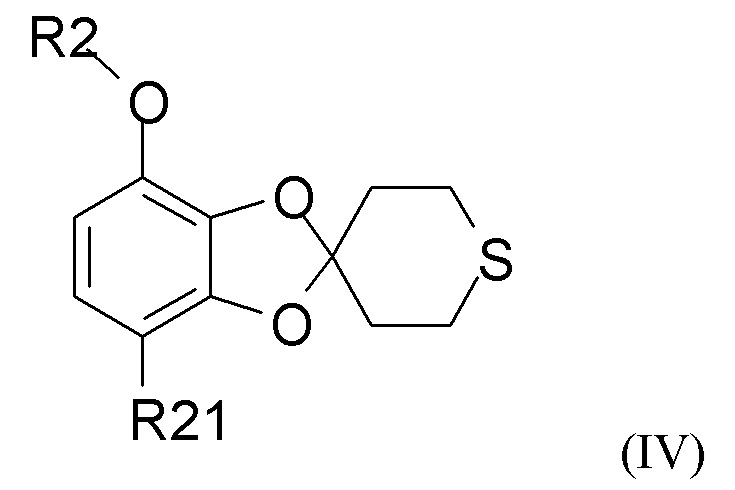
где R2 выбран из водорода, C1-6-алкила и арилалкила, R21 выбран из водорода, C(O)R22 и C(O)OR22, и R22 выбран из водорода и C1-6-алкила. В другом варианте реализации R2 представляет собой водород, метил, этил, пропил, изопропил, изобутил, втор-бутил, трет-бутил или бензил, и R21 выбран из водорода, COCH3 или COOH. В другом варианте реализации промежуточное соединение формулы (IV) представляет собой 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон.
В другом варианте реализации изобретение относится к соединению формулы (IX)
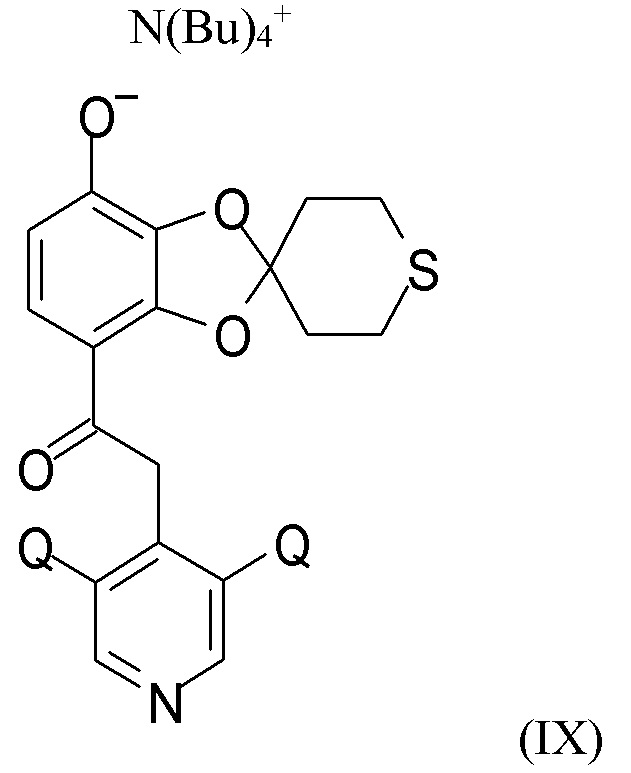
где Q выбран из хлора, брома и фтора, предпочтительно представляет собой хлор, при этом Q предпочтительно являются одинаковыми. В одном из вариантов реализации оба Q представляют собой хлор. В другом варианте реализации промежуточное соединение формулы (IX) представляет собой 2-(3,5-дихлорпиридин-4-ил)-1-(7-тетрабутиламминий-оксидо-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанон.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Способы и реагенты
Все химические агенты и реагенты доступны помимо прочего в Sigma Aldrich Chemicals.
ВЭЖХ:
(мл/мин)
XB-С18
60% H2O, 40% ACN,
0,1% ТФУ
ПРИМЕР 1
Стадия (1): Получение 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона
В реактор помещали 2,3-дигидрокси-4-метоксиацетофенон (1,0 кг, 5,49 моль), тетрагидро-4Н-тиопиран-4-он (0,62 кг, 5,34 моль) и монтмориллонит K10 (0,5 кг), затем добавляли толуол (12 л). Суспензию нагревали до температуры обратной конденсации с применением рубашки реактора, установленной на 150°C, при этом обратный холодильник был снабжен оборудованием Дина-Старка для удаления воды, образующейся в реакции. Температуру обратной конденсации выдерживали в течение 24-72 часов или до достижения конверсии >25%, определенной на основании данных технологического контроля (на основании отношения площадей пиков ВЭЖХ в % 2,3-дигидрокси-4-метоксиацетофенона и титульного соединения). Непрореагировавший 2,3-дигидрокси-4-метоксиацетофенон выделяли путем горячего фильтрования (для удаления K10) реакционной смеси, трехкратной промывки осадка горячим толуолом (по 2 л) и однократной промывки горячим EtOAc (1 л). Объединенные горячие фильтраты охлаждали до 5°C в течение 2-3 часов, что приводило к осаждению непрореагировавшего 2,3-дигидрокси-4-метоксиацетофенона, который собирали путем фильтрования.
Надосадочную жидкость перемешивали с водой (2,67 л) и 27,7% (масс./масс.) NaOH (0,44 кг) в течение 30 минут, оставляли разделяться на 30 минут. Удаляли водную фазу и во второй раз перемешивали органическую фазу в течение 30 минут со свежей водой (2,67 л) и 27,7% (масс./масс.) NaOH (0,44 кг), оставляли разделяться на 30 минут, после чего удаляли водную фазу. Концентрировали органическую фазу до максимально возможного уровня в вакууме, устанавливая температуру рубашки реактора от 65°C до 75°C. Если перегонка происходила медленно, то добавляли EtOH (1,5 л) и концентрировали смесь еще раз до максимально возможного уровня в вакууме, устанавливая температуру рубашки реактора от 65°C до 75°C.
Если перегонка происходила медленно, то в полученную густую суспензию добавляли EtOH (2 л) и нагревали до температуры обратной конденсации, после чего образовывался прозрачный раствор. После медленного добавления воды (1,5 л) со скоростью, которая позволяла поддерживать температуру обратной конденсации, медленно охлаждали до 5°C в течение 10 часов с образованием суспензии титульного соединения. Продукт выделяли путем фильтрования и промывали смесью воды (0,38 л) и EtOH (0,5 л), после чего титульное соединение в виде желтого твердого вещества сушили в вакууме при 40°C, что приводило к получению титульного соединения (0,44 кг, 1,57 моль) с выходом 28%. 1H ЯМР (600 МГц, ДМСО-d6) δ 7,30 (d, J=9,0 Гц, 1H), 6,75 (d, J=9,0 Гц, 1H), 3,88 (s, 3H), 2,90-2,78 (m, 4H), 2,49 (s, 3H), 2,30-2,22 (m, 2H), 2,21-2,14 (m, 2H).
Стадии (2а) и (2b): Получение 2-(3,5-дихлорпиридин-4-ил)-1-(7-гидрокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона
В подходящий реактор помещали 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (1,00 кг, 3,57 моль) и 3,4,5-трихлорпиридин (1,04 кг, 5,70 моль), затем добавляли NMP (2,5 кг). Раствор перемешивали и охлаждали до -5°C. В отдельном сосуде готовили раствор трет-BuONa (1,03 кг, 10,7 моль) в NMP (2,5 кг), который медленно закачивали в реактор, поддерживая температуру при добавлении ниже 15°C.
После завершения добавления поддерживали температуру 15°C и отслеживали прохождение взаимодействия при помощи технологического контроля путем ВЭЖХ. Взаимодействие считали завершенным, если >98% 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона превращалось в 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанон (промежуточное соединение, выделение которого не проводят) согласно отношению площадей пиков ВЭЖХ в % 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона и 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона. После этого при необходимости реакционную смесь можно выдерживать в течение до 2 дней при 5°C.
В реакционную смесь добавляли 5-трет-бутил-2-метилтиофенол (1,03 кг, 5,70 моль) и K2CO3 (0,54 кг, 3,92 моль) и нагревали смесь до 80°C. Взаимодействие считали завершенным, если >85% 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона превращалось в титульное соединение согласно отношению площадей пиков ВЭЖХ в % 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона и титульного соединения.
Реакционную смесь охлаждали до 20°C, добавляли гексан (5 л), 27,7% (масс./масс.) NaOH (0,35 л) и воду (5 л), затем быстро перемешивали в течение 15-30 минут. После прекращения перемешивания разделяли фазы, оставляли водную фазу, при этом органическую фазу отбрасывали. К водной фазе добавляли толуол (0,8 л) и гексан (4,2 л), затем быстро перемешивали в течение 15-30 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Оставляли водную фазу и обрабатывали еще раз толуолом (2 л) и гексаном (3 л) при быстром перемешивании в течение 15-30 минут, затем перемешивание останавливали и оставляли фазы разделяться. Оставляли водную фазу и в третий раз обрабатывали толуолом (2,5 л) и гексаном (2,5 л) при быстром перемешивании в течение 15-30 минут, затем перемешивание останавливали и оставляли фазы разделяться.
Водную фазу возвращали в реактор, добавляли EtOAc (6 л), воду (2 л) и медленно добавляли AcOH (1,03 кг). После завершения добавления AcOH продолжали перемешивать в течение еще 20-30 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Органическую фазу переносили в резервуар для хранения и оставляли, при этом водную фазу возвращали в реактор, добавляли EtOAc (6 л), нагревали до 40°C и перемешивали в течение 20-30 минут, после чего перемешивание останавливали и снова оставляли фазы разделяться. Удаляли водную фазу в отходы, органическую фазу из резервуара для хранения переносили в реактор и объединяли фазы.
К объединенным органическим фазам добавляли воду (4 л) и перемешивали при 40°C в течение 20-30 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Удаляли водную фазу и к органической фазе во второй раз добавляли воду (4 л) и NaCl (нас.) (4 л), затем перемешивали при 40°C в течение 20-30 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Удаляли водную фазу и концентрировали органическую фазу до максимально возможного уровня в вакууме при температуре от 50°C до 60°C. Если перегонка замедлялась, то добавляли EtOAc (2 л), после чего снова концентрировали в вакууме для удаления каких-либо следов сохранившейся воды.
К остатку добавляли ацетон (5,5 л) и смесь нагревали до температуры обратной конденсации для обеспечения полного растворения. Во время кипячения раствора медленно добавляли гексан (12,5 л), при добавлении поддерживали температуру обратной конденсации. После завершения добавления реакционную смесь медленно охлаждали до комнатной температуры в течение 5-8 часов, а затем дополнительно охлаждали до 0°C в течение еще 5-8 часов.
Неочищенный продукт выделяли путем фильтрования, промывали смесью ацетона (1 л) и гексана (2 л), сушили в вакууме при 40°C, что приводило к получению титульного соединения (0,83 кг, 2,01 моль) в виде беловато-желтоватого твердого вещества с выходом 56%. 1H ЯМР (600 МГц, ДМСО-d6) δ 10,76 (s, 1H), 8,65 (s, 2H), 7,26 (d, J=9,0 Гц, 1H), 6,56 (d, J=9,0 Гц, 1H), 4,59 (s, 2H), 2,97-2,89 (m, 2H), 2,86-2,79 (m, 2H), 2,39-2,31 (m, 2H), 2,23-2,15 (m, 2H).
Стадия (2c): Получение 2-(3,5-дихлорпиридин-4-ил)-1-(7-тетрабутиламмоний-оксидо-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона
Неочищенный 2-(3,5-дихлорпиридин-4-ил)-1-(7-гидрокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанон (0,83 кг, 2,01 моль) переносили в реактор, добавляли ТГФ (1,11 л) и перемешивали до растворения, после чего добавляли водный N(Bu4)+OH- (1,83 кг). Реакционную смесь нагревали до 45°C и перемешивали в течение 1-2 часов для обеспечения полноты образования соли. В реактор добавляли МТБЭ (4,15 л), воду (4,15 л) и NaCl (нас.) (1,25 л) при интенсивном перемешивании в течение 1-2 часов, после чего медленно охлаждали до 5°C в течение 1-4 часов, что приводило к осаждению соли TBA 2-(3,5-дихлорпиридин-4-ил)-1-(7-гидрокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона. Останавливали перемешивание и разделяли фазы (три фазы), водную фазу осторожно переносили в отходы, при этом добивались того, чтобы промежуточная фаза титульного соединения сохранялась в реакторе. После завершения разделения добавляли воду (2,08 кг), затем нагревали до 35°C при интенсивном перемешивании в течение 1-2 часов. Реакционную смесь медленно охлаждали до 5°C в течение 1-4 часов, что приводило к повторному осаждению соли TBA 2-(3,5-дихлорпиридин-4-ил)-1-(7-гидрокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона, перемешивание останавливали для обеспечения разделения фаз. Водную фазу, как и ранее, осторожно переносили в отходы, и остаток, содержащий продукт, отфильтровывали и промывали МТБЭ (4,15 л), после чего сушили в вакууме при 40°C. Титульное соединение (1,26 кг, 1,93 моль) выделяли в виде беловатого твердого вещества с общим выходом 55%. 1H ЯМР (600 МГц, ДМСО-d6) δ 8,58 (s, 2H), 6,98 (d, J=9,2 Гц, 1H), 5,76 (d, J=9,2 Гц, 1H), 4,39 (s, 2H), 3,24-3,08 (m, 8H), 2,91-2,82 (m, 2H), 2,82-2,74 (m, 2H), 2,23-2,13 (m, 2H), 2,11-1,99 (m, 2H), 1,67-1,44 (m, 8H), 1,31 (h, J=7,4 Гц, 8H), 0,93 (t, J=7,4 Гц, 12H).
Стадия (3): Получение 2-(3,5-дихлорпиридин-4-ил)-1-(7-дифторметокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона
В реактор, соединенный с газоочистителем, заполненным ДМФА (примерно 5 л) помещали 2-(3,5-дихлорпиридин-4-ил)-1-(7-тетрабутиламмоний-оксидо-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанон (1,0 кг, 1,53 моль), затем добавляли ДМФА (12 л) и перемешивали до полного растворения при комнатной температуре. Закрывали реактор и медленно добавляли хлордифторметан (1,32 кг, 15,3 моль), чтобы давление не повышалось более чем на 0,05 бар. После завершения добавления реактор повторно открывали, что обеспечивало вентиляцию через газоочиститель, и температуру в реакторе повышали до 65°C.
Прохождение взаимодействия отслеживали при помощи технологического контроля и анализировали путем ВЭЖХ каждые два часа. Взаимодействие считали завершенным, если >93% 2-(3,5-дихлорпиридин-4-ил)-1-(7-тетрабутиламмоний-оксидо-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона превращалось в титульное соединение согласно отношению площадей пиков ВЭЖХ в % 2-(3,5-дихлорпиридин-4-ил)-1-(7-тетрабутиламмоний-оксидо-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанона и титульного соединения.
После завершения взаимодействия добавляли воду (1 л), 27,7% (масс./масс.) NaOH (50 мл) и МТБЭ (2 л) в указанном порядке и смесь эффективно перемешивали в течение 30-45 минут. После этого добавляли EtOAc (5 л) и воду (10 л) и смесь перемешивали в течение еще 30-45 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Органическую фазу хранили в резервуаре для хранения и водную фазу возвращали в реактор. В реактор добавляли свежий МТБЭ (2 л), EtOAc (5 л) и смесь эффективно перемешивали в течение 30-45 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Органическую фазу смешивали с полученной ранее органической фазой в резервуаре для хранения и водную фазу возвращали в реактор для третьей экстракции. В реактор добавляли свежий МТБЭ (2 л), EtOAc (5 л) и смесь эффективно перемешивали в течение 30-45 минут, после чего перемешивание останавливали и оставляли фазы разделяться.
Водную фазу отбрасывали в отходы, при этом органические фазы возвращали в реактор, добавляли воду (5 л) и эффективно перемешивали в течение 30-45 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Водную фазу отбрасывали в отходы и добавляли свежую воду (5 л), затем перемешивали в течение 30-45 минут, после чего перемешивание останавливали и оставляли фазы разделяться. Водную фазу отбрасывали в отходы и концентрировали органическую фазу до максимально возможного уровня в вакууме при температуре от 50°C до 60°C. Когда перегонка замедлялась, добавляли 2-PrOH (5 л) и нагревали смесь до температуры обратной конденсации при перемешивании, которое обеспечивало полное растворение, после чего медленно добавляли воду (1,7 л) с такой скоростью, чтобы температура превышала >75°C. После завершения добавления реакционную смесь медленно охлаждали до 5°C в течение 5-12 часов, затем перемешивали в течение еще 3 часов при 5°C. Осажденный продукт выделяли путем фильтрования, промывали смесью воды (2 л) и 2-PrOH (2 л), затем проводили вторую промывку с применением воды (4 л). После сушки в вакууме при 45°C титульное соединение (0,65 кг, 1,40 моль) выделяли в виде беловатого твердого вещества с выходом 92%. 1H ЯМР (600 МГц, ДМСО-d6) δ 8,67 (s, 2H), 7,40 (d, J=9,0 Гц, 1H), 7,39 (t, J=72,9 Гц, 1H), 6,93 (d, J=9,0 Гц, 1H), 4,68 (s, 2H), 2,98-2,89 (m, 2H), 2,88-2,80 (m, 2H), 2,43-2,36 (m, 2H), 2,30-2,18 (m, 2H).
Стадия (4): Получение 2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-дифторметокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран-1ʹ,1ʹ-диоксид]-4-ил)этанона
В реактор помещали уксусную кислоту (3,8 кг), 2-(3,5-дихлорпиридин-4-ил)-1-(7-дифторметокси-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанон (1 кг, 2,2 моль) и недолго перемешивали для обеспечения гомогенной суспензии. Медленно добавляли надуксусную кислоту (40% в уксусной кислоте, 1,65 кг; 8,67 моль; 4 экв.) в течение нескольких часов (3-5 часов), поддерживая температуру 25 40°C для контролирования повышения температуры. Во время добавления суспензия становилась гомогенной.
Реакционную смесь нагревали до 50°С и перемешивали в течение 14-18 часов. Отбирали образец реакционной смеси (IPC, конверсия: >99%=завершение). Реакционную смесь охлаждали до 25°C и медленно добавляли раствор Na2S2O5 (0,42 кг, 2,21 моль) в воде (2,7 кг), поддерживая температуру ниже 35°C. После завершения добавления полученную смесь перемешивали в течение еще 10-20 минут, после чего отбирали пробы для определения остаточных пероксидов, которые были отрицательными. Добавляли IPA (5,0 л) и нагревали до 60°C с образованием гомогенной смеси, которую фильтровали через фильтр-заглушку. К отфильтрованному раствору добавляли воду (15 кг) со скоростью, обеспечивающей поддержание температуры 55-60°C. Реакционную смесь перемешивали при 60°C в течение еще 30-60 минут, после чего охлаждали до 5°C со скоростью 5°C/час. Суспензию перемешивали в течение еще 2 часов при 5°C, после чего фильтровали продукт.
Отфильтровывали кристаллы и промывали водой (1,7 кг). Влажный осадок возвращали в реактор и полностью растворяли в EtOH (20,4 кг) при нагревании до температуры обратной конденсации. Прозрачный раствор охлаждали до 70°C, вводили затравку (10 граммов титульного соединения из предыдущей партии) и затем охлаждали до 5°C со скоростью 5°C/час. Суспензию перемешивали при 5°C в течение не менее чем 2 часов.
Продукт выделяли путем фильтрования, промывали смесью EtOH/вода (2,0 кг EtOH и 0,25 кг воды) и сушили в вакууме (45°C, p< 10 мбар (1 кПа)). Выход титульного соединения составлял 0,8 кг (75%), чистота согласно площадей пиков ВЭЖХ составляла 98,5%. 1H ЯМР (600 МГц, CDCl3) δ 8,24 (s, 2H), 7,52 (d, J=9,0 Гц, 1H), 6,89 (d, J=9,0 Гц, 1H), 6,70 (t, J=72,3 Гц, 1H), 4,49 (s, 2H), 3,47-3,39 (m, 2H), 3,32-3,24 (m, 2H), 2,83-2,76 (m, 2H), 2,75-2,68 (m, 2H).
ПУНКТЫ
С учетом приведенного описания авторы настоящего изобретения предложили, в частности:
Пункт 1. Способ получения соединения формулы (I)
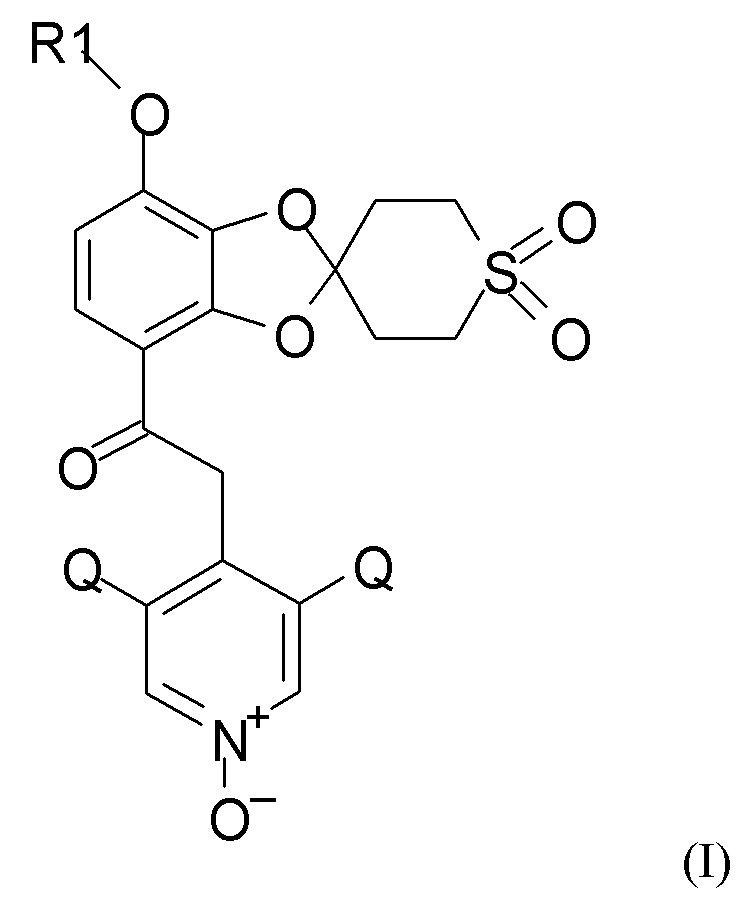
где R1 выбран из CHF2 и CF3, Q выбран из хлора, брома и фтора,
включающий одну или более следующих стадий:
(1) взаимодействие соединения формулы (II)
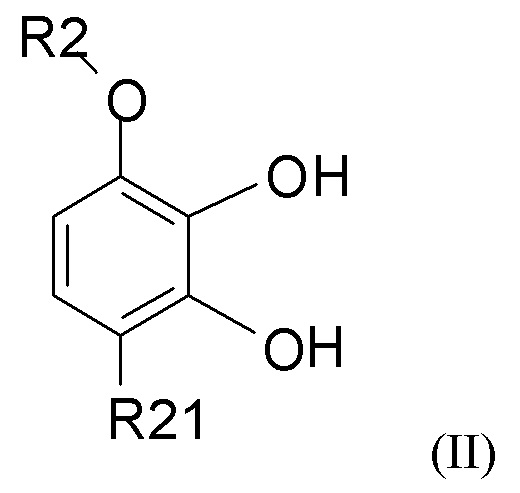
где R2 выбран из водорода, C1-6-алкила и арилалкила, R21 выбран из водорода, C(O)R22 и C(O)OR22, и R22 выбран из водорода и C1-6-алкила; с соединением формулы (III)
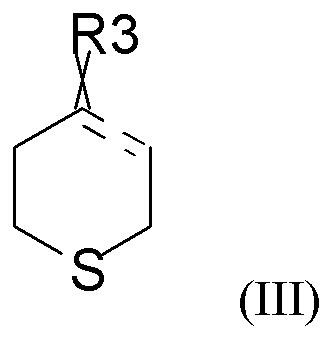
где «» представляет собой простую связь, двойную связь или две простые связи, и если «» представляет собой двойную связь или две простые связи, то «» представляет собой простую связь, и если «» представляет собой простую связь, то «» представляет собой двойную связь, R3 представляет собой кислород, если «» представляет собой двойную связь, и R3 представляет собой O-C1-6-алкил, если «» представляет собой простую связь или две простые связи
в присутствии кислотного катализатора для получения соединения формулы (IV)
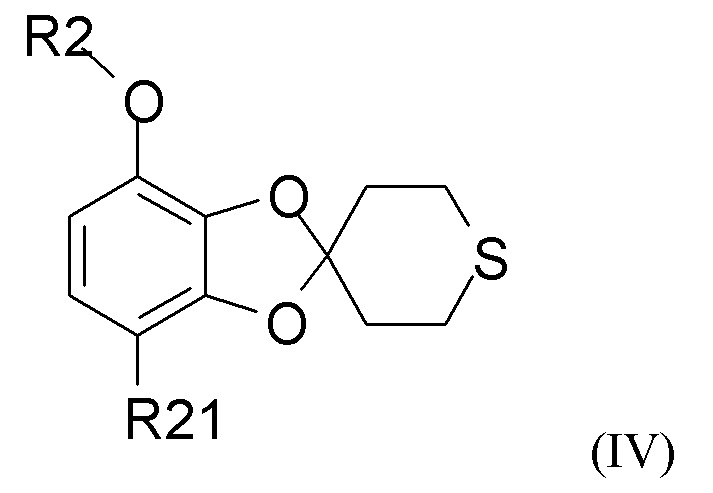
где R2 и R21 такие, как определено выше;
(2а) взаимодействие полученного соединения формулы (IV) с пиридиновым соединением формулы (V)
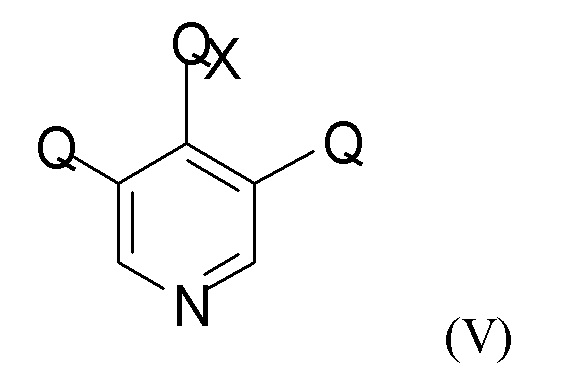
где Q такой, как определено выше, и QX выбран из хлора, брома, фтора и йода, для получения промежуточного соединения формулы (VI)
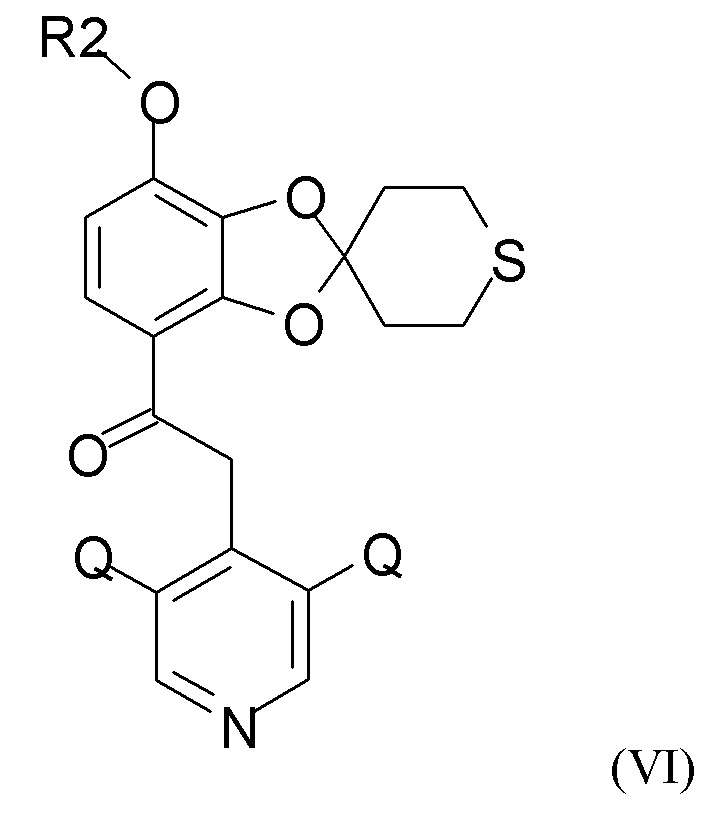
где R2 и Q такие, как определено выше;
(2b) взаимодействие соединения формулы (VI) с соединением формулы (VII)
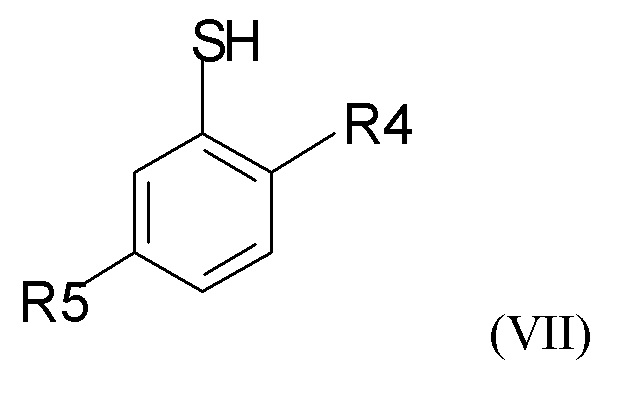
где R4 и R5 представляют собой C1-6-алкил, для получения соединения формулы (VIII)
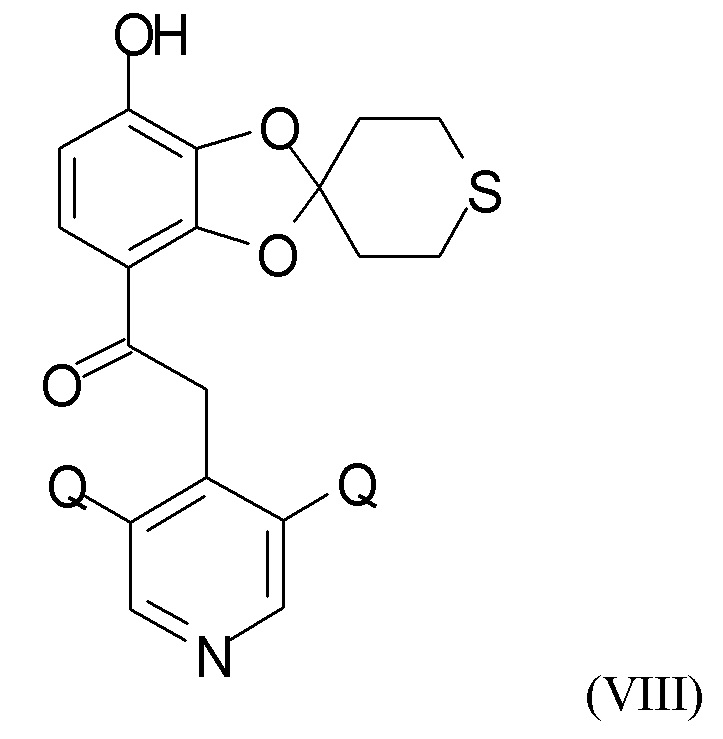
где Q такой, как определено выше;
(2c) взаимодействие соединения формулы (VIII) с водным N(Bu4)+OH- для получения соединения формулы (IX)
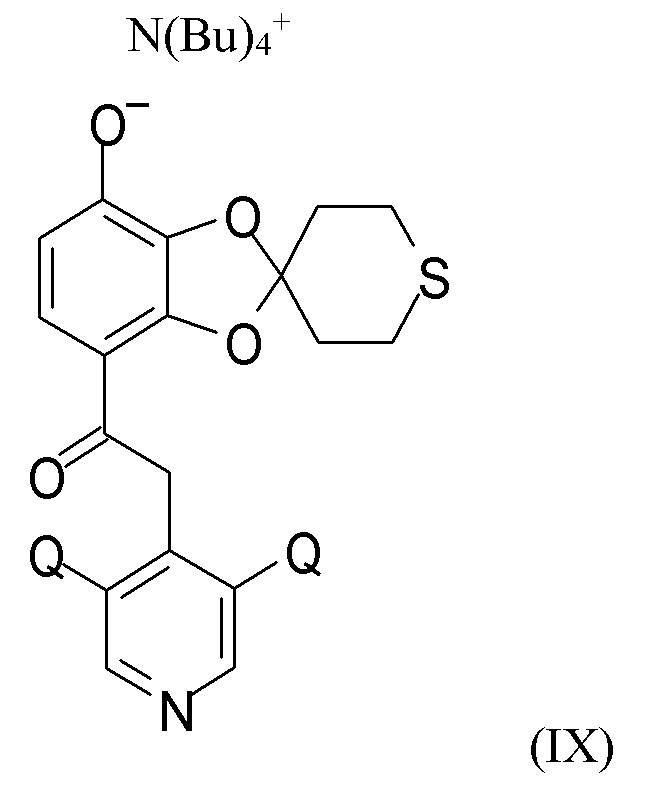
где Q такой, как определено выше; последующее
(3) алкилирование полученного соединения формулы (IX) посредством взаимодействия с хлорфторуглеводородным соединением, R1-Cl, где R1 такой, как определено выше, для получения соединения формулы (XI)
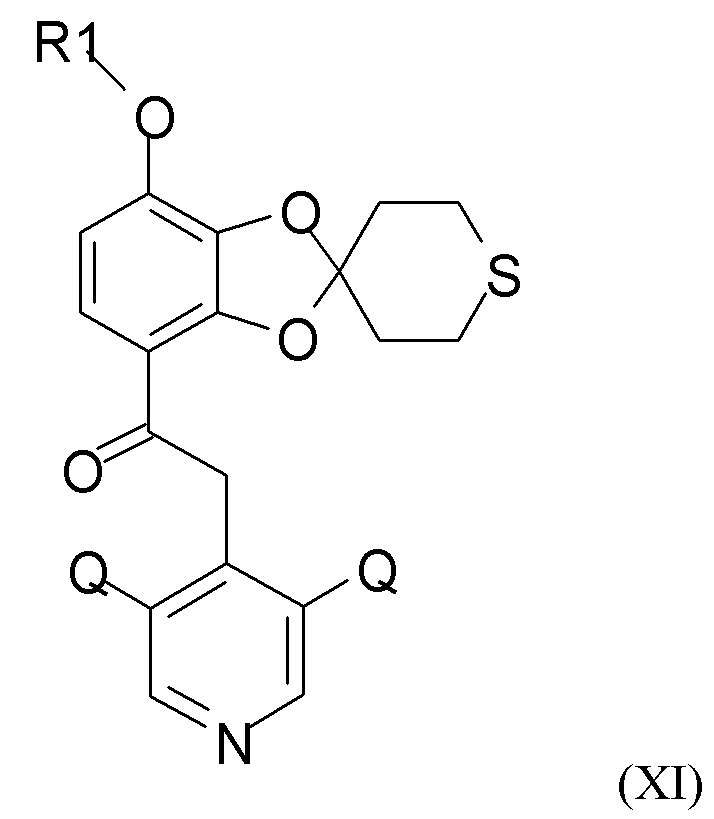
где R1 и Q такие, как определено выше; и
(4) окисление полученного соединения формулы (XI) для получения соединения формулы (I), где R1 и Q такие, как определено выше.
Пункт 2. Способ по п.1, отличающийся тем, что на стадии (1) кислотный катализатор имеет форму силикатного минерала, выбранного из монтмориллонита K10, монтмориллонита K30, цеолита HSZ-350HUA и цеолита HSZ-360HUA.
Пункт 3. Способ по п.2, отличающийся тем, что силикатный минерал представляет собой монтмориллонит K10.
Пункт 4. Способ по любому из предыдущих пунктов, отличающийся тем, что стадию (1) проводят в растворителе, выбранном из толуола, бензола, 2-метил-ТГФ, EtOAc, гептана или дихлорбензола.
Пункт 5. Способ по п.4, отличающийся тем, что растворитель представляет собой толуол.
Пункт 6. Способ по любому из предыдущих пунктов, отличающийся тем, что на стадии (2а) проводят сочетание в апротонном полярном растворителе, например, выбранном из NMP, ДМФА, DMI, ДМСО, EtOAc, MeCN и ТГФ и их смесей, в присутствии основания, например, выбранного из трет-BuONa, трет-BuOK, K2CO3, Na2CO3, KHCO3, NaHCO3, Et3N и DIPEA.
Пункт 7. Способ по п.6, отличающийся тем, что апротонный полярный растворитель представляет собой NMP, и основание представляет собой трет-BuONa.
Пункт 8. Способ по любому из предыдущих пунктов, отличающийся тем, что снятие защиты на стадии (2b) проводят в растворителе, например, выбранном из NMP, ДМСО, ДМФА и их смесей, в присутствии основания, например, выбранного из K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, ТЭА и DIPEA.
Пункт 9. Способ по п.8, отличающийся тем, что растворитель представляет собой NMP, и основание представляет собой K2CO3.
Пункт 10. Способ по любому из предыдущих пунктов, отличающийся тем, что стадию (2с) проводят в присутствии ТГФ, толуола или EtOAc.
Пункт 11. Способ по п.10, отличающийся тем, что растворитель представляет собой ТГФ.
Пункт 12. Способ по любому из предыдущих пунктов, отличающийся тем, что взаимодействие на стадии (3) проводят с применением хлорфторуглеводородного соединения R1-Cl в апротонном полярном растворителе, например, выбранном из ДМФА, NMP, DMI, ДМСО, EtOAc и ТГФ.
Пункт 13. Способ по п.12, отличающийся тем, что взаимодействие проводят с применением хлордифторметана в ДМФА.
Пункт 14. Способ по любому из предыдущих пунктов, отличающийся тем, что взаимодействие на стадии (4) проводят в присутствии надуксусной кислоты в уксусной кислоте или H2O2 (водн.) в муравьиной кислоте или уксусной кислоте.
Пункт 15. Способ по п.14, отличающийся тем, что взаимодействие проводят с применением надуксусной кислоты в уксусной кислоте.
Пункт 16. Способ по любому из предыдущих пунктов, отличающийся тем, что R1 представляет собой CHF2.
Пункт 17. Способ по любому из предыдущих пунктов, отличающийся тем, что все Q и Qx представляют собой хлор.
Пункт 18. Промежуточное соединение формулы (IV)
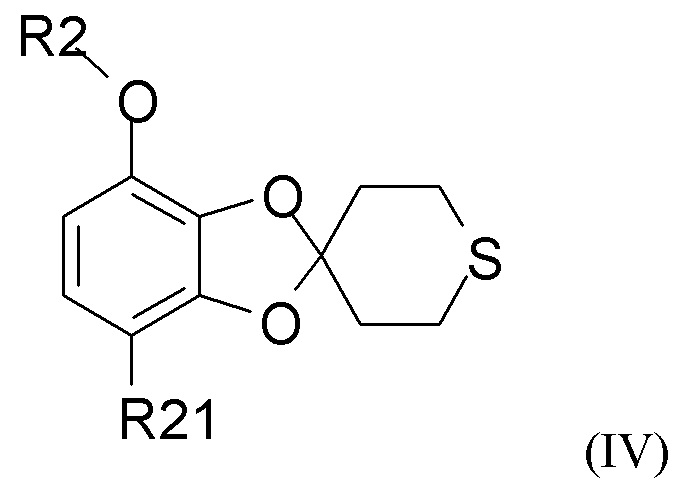
где R2 выбран из водорода, C1-6-алкила и арилалкила, R21 выбран из водорода, C(O)R22 и C(O)OR22, и R22 выбран из водорода и C1-6-алкила.
Пункт 19. Промежуточное соединение по п.18, представляющее собой 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон.
Пункт 20. Промежуточное соединение формулы (IX)
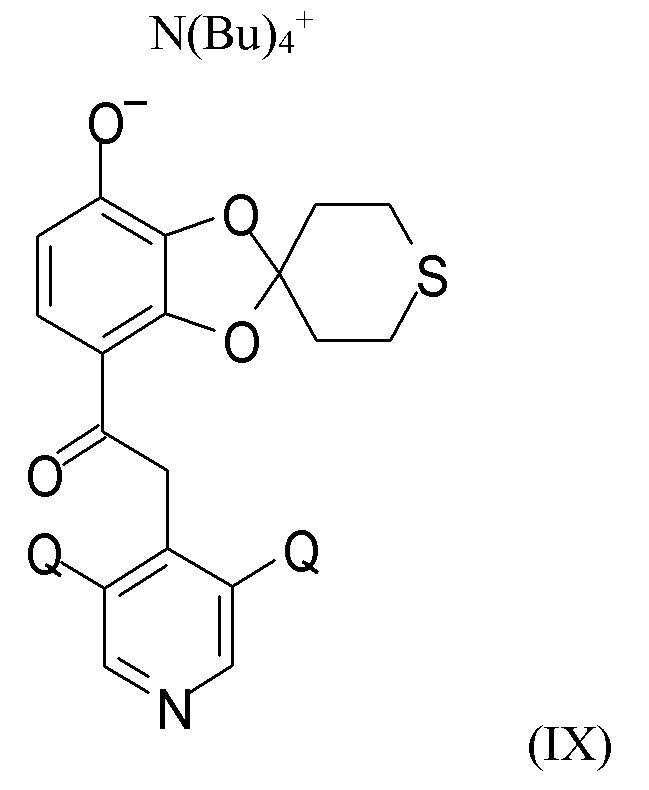
где Q выбран из хлора, брома и фтора.
Пункт 21. Промежуточное соединение по п.20, представляющее собой 2-(3,5-дихлорпиридин-4-ил)-1-(7-тетрабутиламминий-оксидо-2ʹ,3ʹ,5ʹ,6ʹ-тетрагидро-спиро[1,3-бензодиоксол-2,4ʹ-(4H)-тиопиран]-4-ил)этанон.
Пункт 22. Способ получения соединения формулы (IV)
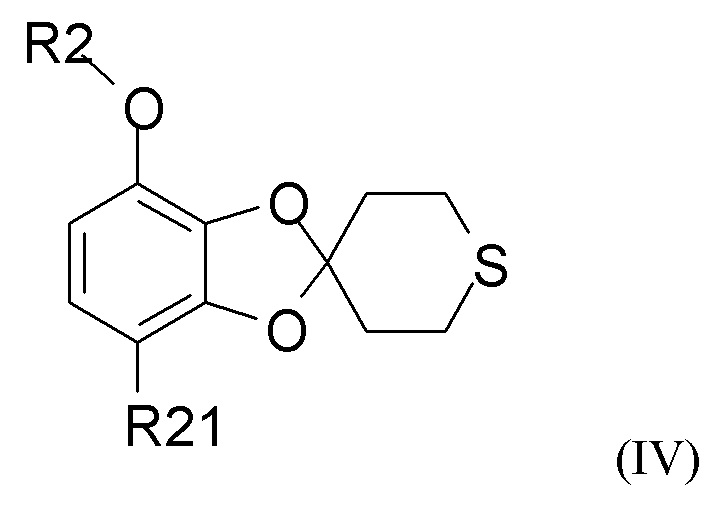
где R2 выбран из водорода, C1-6-алкила и арилалкила, R21 выбран из водорода, C(O)R22 и C(O)OR22, и R22 выбран из водорода и C1-6-алкила; включающий стадию (1), такую как определено по п.1.
Пункт 23. Способ получения соединения формулы (IX)
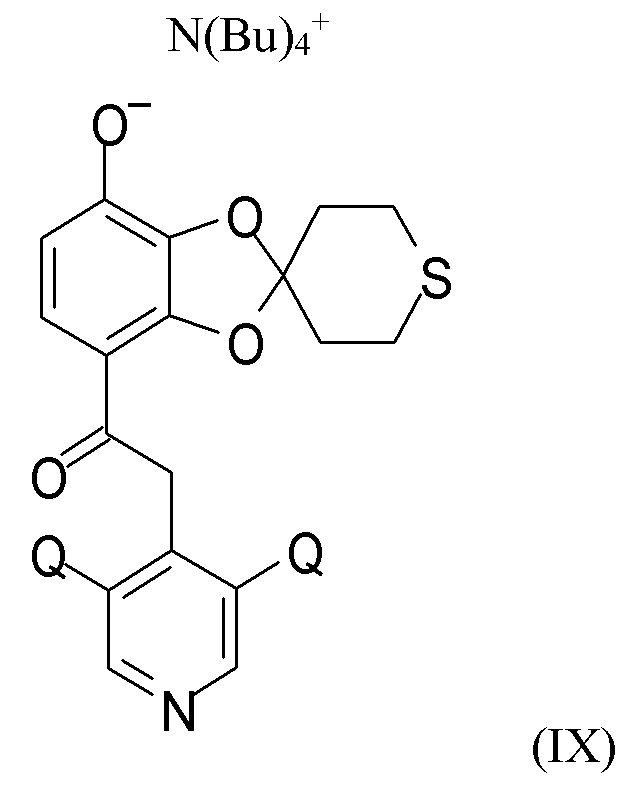
где Q выбран из хлора, брома и фтора, включающий стадию (2а), (2b) и (2с), такую как определено по п.1.
Пункт 24. Способ получения соединения формулы (IX)
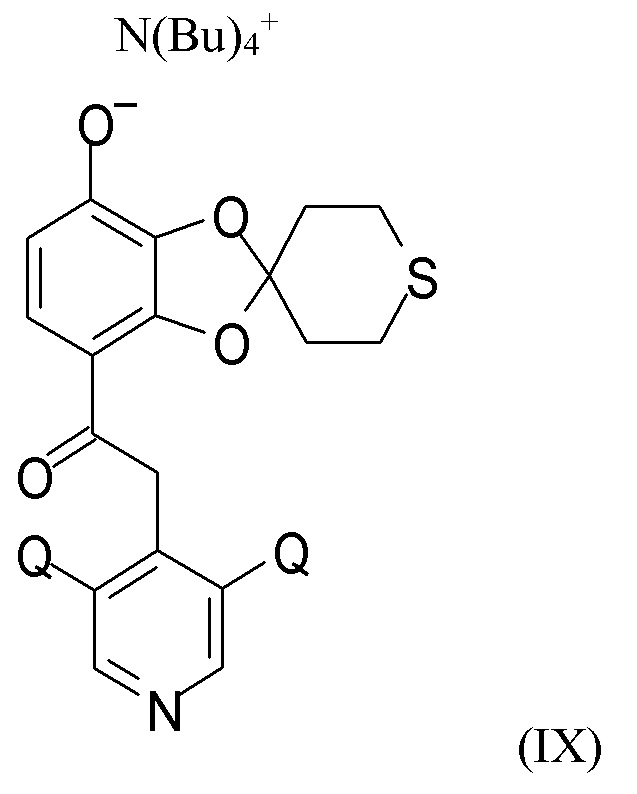
где Q выбран из хлора, брома и фтора, включающий стадию (1), (2а), (2b) и (2с), такую как определено по п.1.
Пункт 25. Способ получения соединения формулы (I)
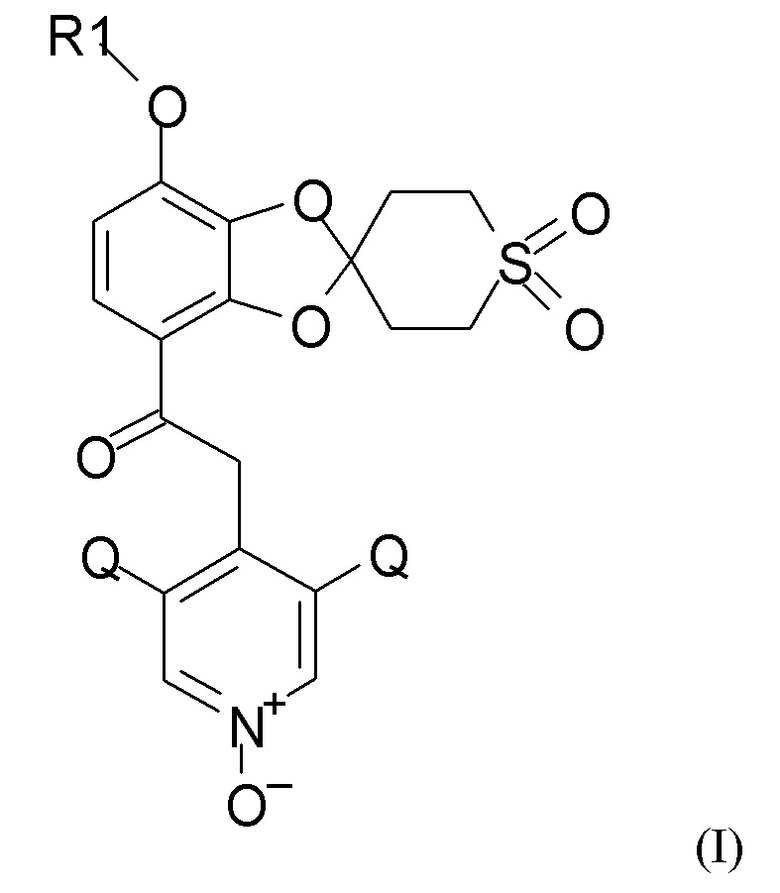
где R1 выбран из CHF2 и CF3, и Q выбран из хлора, брома и фтора, включающий каждую из стадий (1), (2a), (2b) и (2c), таких как определено по п.1, алкилирование и последующее окисление полученного соединения.
Пункт 26. Способ получения соединения формулы (I)
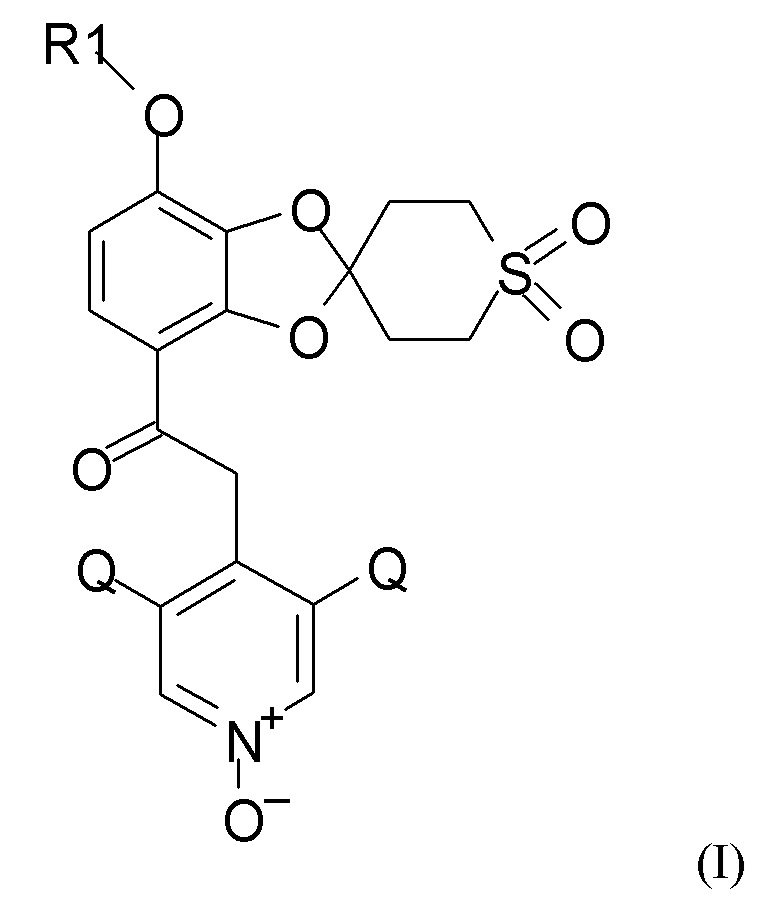
где R1 выбран из CHF2 и CF3, и Q выбран из хлора, брома и фтора, включающий каждую из стадий (1), (2a), (2b) и (2c), (3) и (4), таких как определено по п.1.
Пункт 27. Соединение Формулы (I)
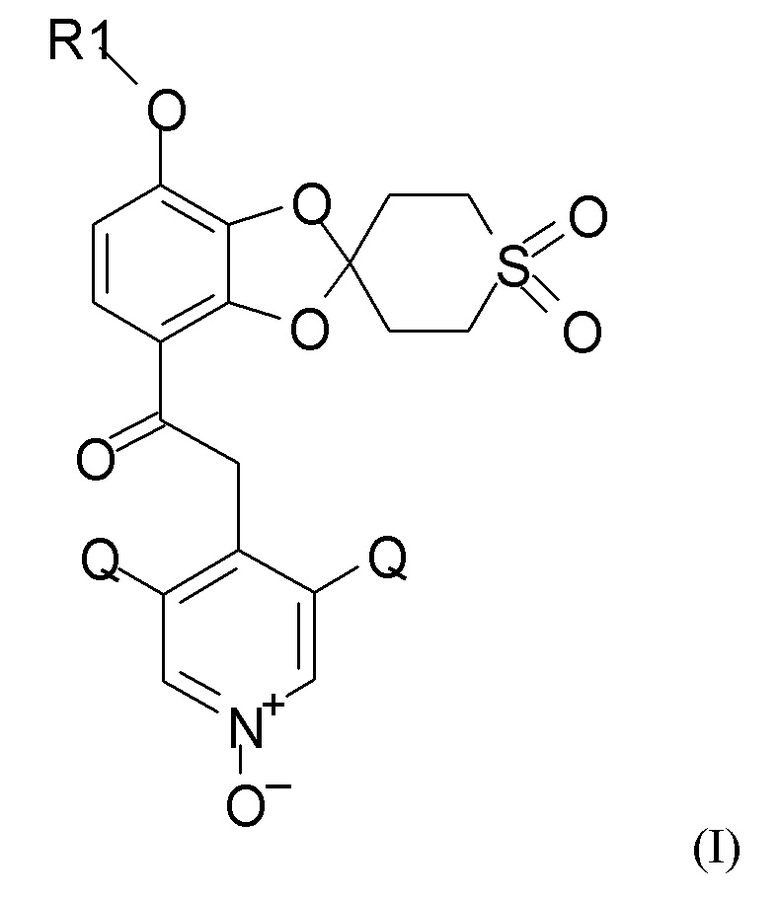
где R1 выбран из CHF2 и CF3, и Q выбран из хлора, брома и фтора, полученное способом по п.1.
Пункт 28. Соединение Формулы (I)
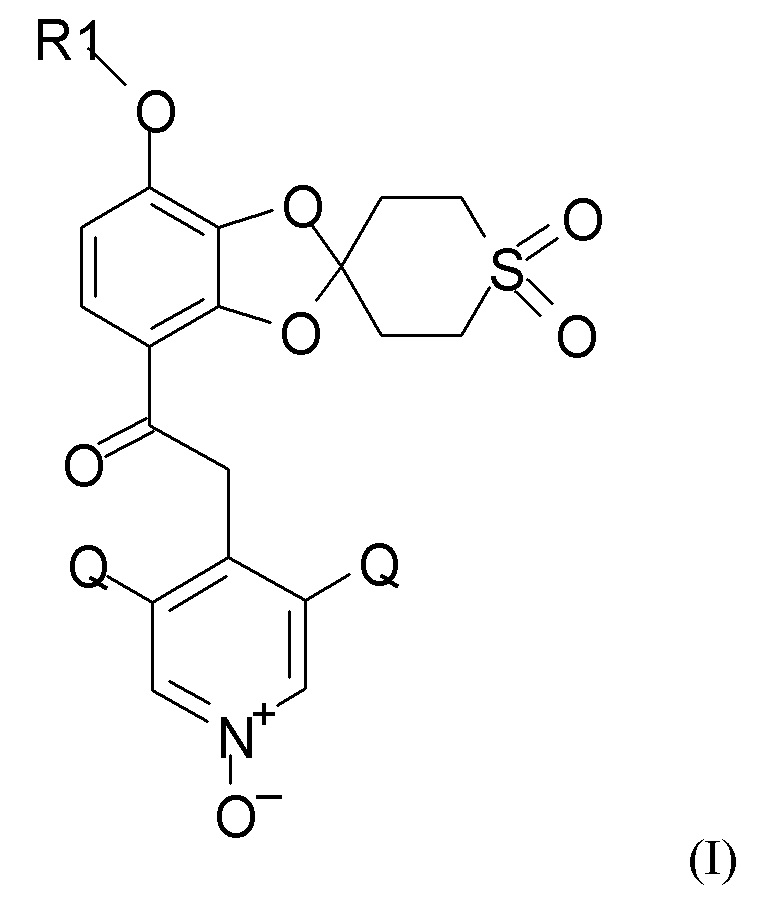
где R1 выбран из CHF2 и CF3, и Q выбран из хлора, брома и фтора, полученное с применением стадий (1), (2a), (2b) и (2c), таких как определено по п.1, алкилирования и последующего окисления полученного соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТАГОНИСТЫ РЕЦЕПТОРА СОМАТОСТАТИНА ПОДТИПА 5 (SSTR5) | 2014 |
|
RU2671958C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОДИОКСОЛА ИЛИ БЕНЗОДИОКСЕПИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2011 |
|
RU2583787C2 |
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ 1, 3-БЕНЗОДИОКСОЛА | 2016 |
|
RU2761337C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1,3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2018 |
|
RU2814175C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛФЕНИЛКЕТОНЫ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2007 |
|
RU2493149C2 |
| КОНЪЮГАТЫ АНТАГОНИСТОВ ИНТЕГРИНА ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ К КЛЕТКАМ, ЭКСПРЕССИРУЮЩИМ LFA-1 | 2013 |
|
RU2624732C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2012 |
|
RU2619184C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ИНГИБИТОР JAK | 2015 |
|
RU2674262C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
Изобретение относится к новым способам получения 1,3-бензодиоксольных гетероциклических соединений формулы (I)
 ,
,
где R1 выбран из CHF2 и CF3, Q выбран из хлора, брома и фтора. Соединения эффективны в качестве ингибиторов PDE4. 3 н. и 8 з.п. ф-лы, 1 пр.
1. Способ получения соединения формулы (I)
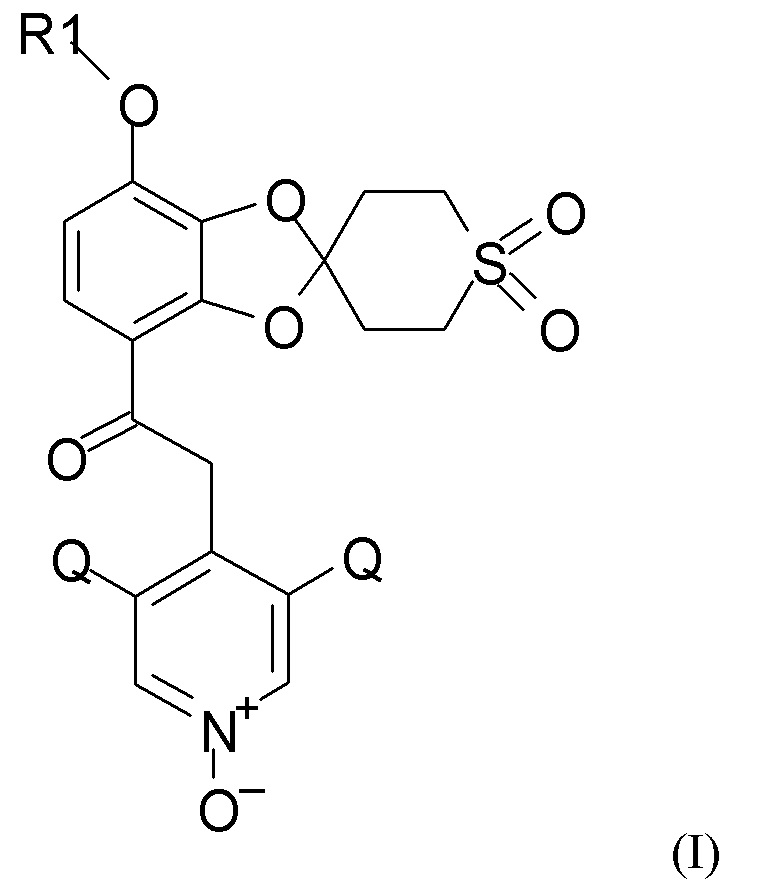
где R1 выбран из CHF2 и CF3, Q выбран из хлора, брома и фтора,
включающий каждую из стадий:
(1) взаимодействие соединения формулы (II)
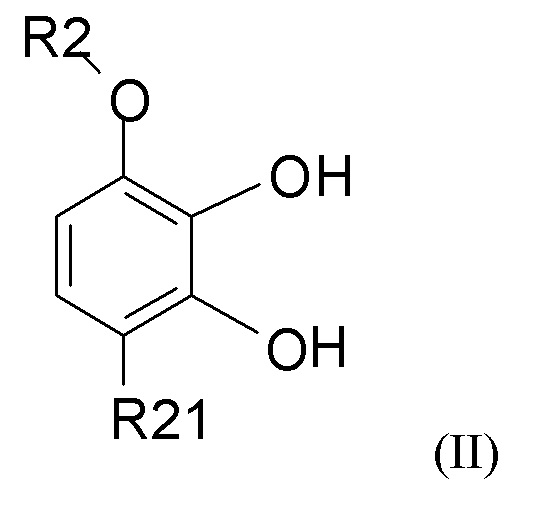
где R2 выбран из водорода, C1-6-алкила и арилалкила, R21 выбран из водорода, C(O)R22 и C(O)OR22 и R22 выбран из водорода и C1-6-алкила; с соединением формулы (III)
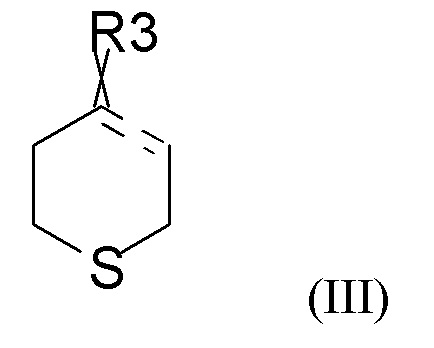
где  представляет собой простую связь, двойную связь или две простые связи, и если представляет собой двойную связь или две простые связи, то
представляет собой простую связь, двойную связь или две простые связи, и если представляет собой двойную связь или две простые связи, то 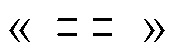 представляет собой простую связь, и если представляет собой простую связь, то представляет собой двойную связь, R3 представляет собой кислород, если представляет собой двойную связь, и R3 представляет собой O-C1-6-алкил, если представляет собой простую связь или две простые связи
представляет собой простую связь, и если представляет собой простую связь, то представляет собой двойную связь, R3 представляет собой кислород, если представляет собой двойную связь, и R3 представляет собой O-C1-6-алкил, если представляет собой простую связь или две простые связи
в присутствии кислотного катализатора с получением соединения формулы (IV)
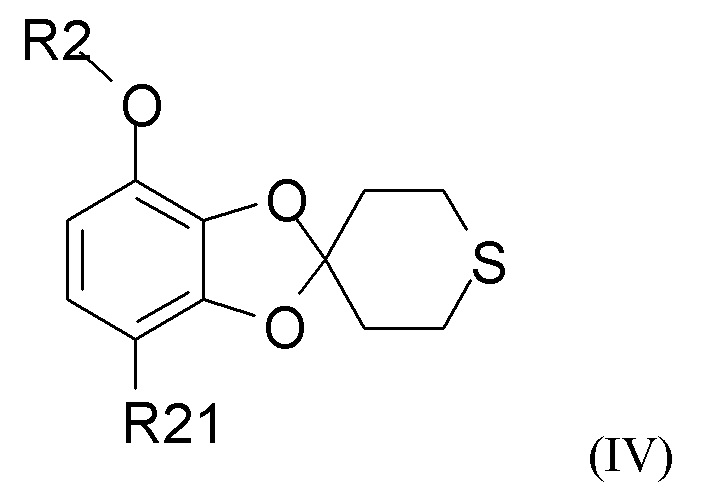
где R2 и R21 такие, как определено выше;
(2а) взаимодействие полученного соединения формулы (IV) с пиридиновым соединением формулы (V)
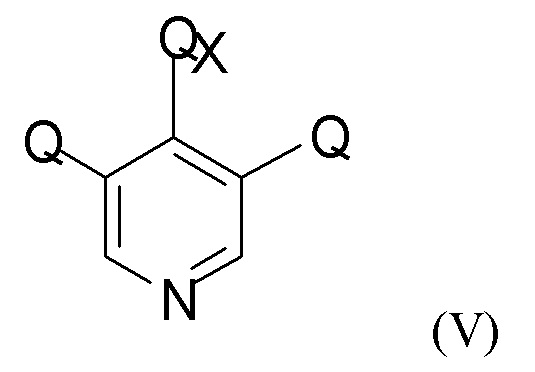
где Q такой, как определено выше, и QX выбран из хлора, брома, фтора и йода, с получением промежуточного соединения формулы (VI)
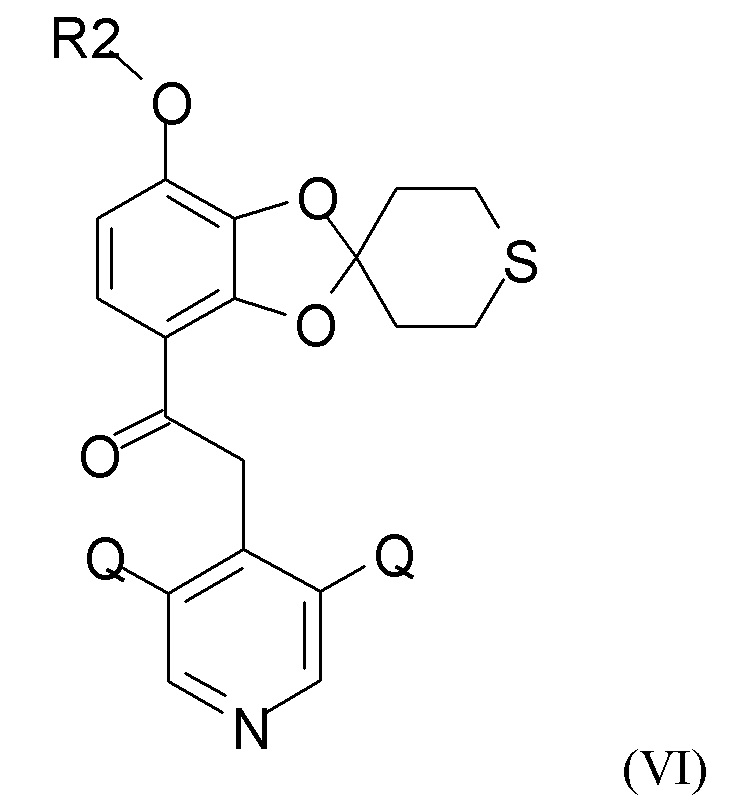
где R2 и Q такие, как определено выше;
(2b) взаимодействие соединения формулы (VI) с соединением формулы (VII)
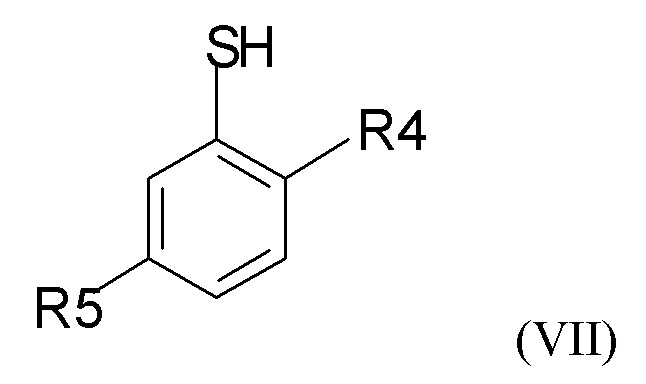
где R4 и R5 представляют собой C1-6-алкил, с получением соединения формулы (VIII)
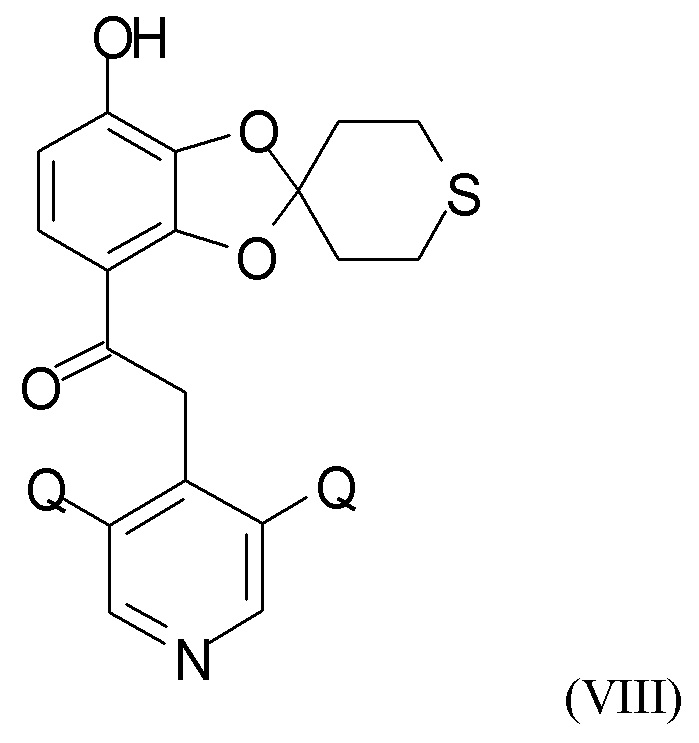
где Q такой, как определено выше;
(2c) взаимодействие соединения формулы (VIII) с водным N(Bu4)+OH- с получением соединения формулы (IX)
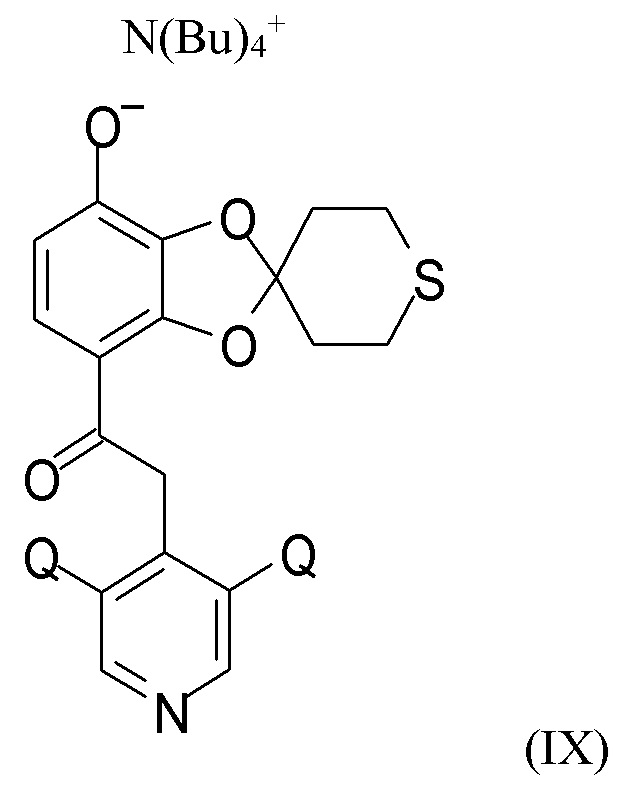
где Q такой, как определено выше; последующее
алкилирование и последующее окисление полученного соединения с получением соединения формулы (I), где R1 и Q такие, как определено выше.
2. Способ по п.1, отличающийся тем, что указанные алкилирование и последующее окисление представляют собой:
(3) алкилирование полученного соединения формулы (IX) посредством взаимодействия с хлорфторуглеводородным соединением, R1-Cl, где R1 такой, как определено выше, с получением соединения формулы (XI)
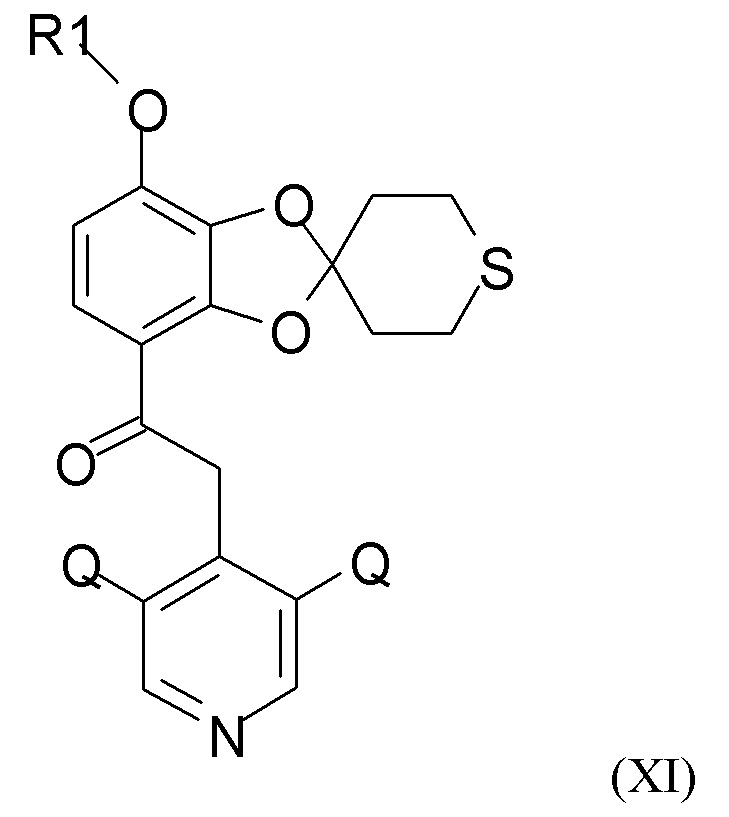
где R1 и Q такие, как определено выше; и
(4) окисление полученного соединения формулы (XI) с получением соединения формулы (I), где R1 и Q такие, как определено выше.
3. Способ по п.1 или 2, отличающийся тем, что на стадии (1) кислотный катализатор имеет форму силикатного минерала монтмориллонита K10.
4. Способ по любому из пп. 1-3, отличающийся тем, что на стадии (2а) проводят сочетание в апротонном полярном растворителе в присутствии основания.
5. Способ по любому из пп. 1-4, отличающийся тем, что снятие защиты на стадии (2b) проводят в растворителе, например, выбранном из NMP, ДМСО, ДМФА и их смесей, в присутствии основания.
6. Способ по любому из пп. 2-5, отличающийся тем, что взаимодействие на стадии (3) проводят с применением хлорфторуглеводорода в присутствии апротонного полярного растворителя.
7. Способ по любому из пп. 2-6, отличающийся тем, что взаимодействие на стадии (4) проводят с применением надуксусной кислоты в уксусной кислоте.
8. Способ по любому из пп. 1-7, отличающийся тем, что R1 представляет собой CHF2.
9. Способ по любому из пп. 1-8, отличающийся тем, что все Q и Qx представляют собой хлор.
10. Способ получения соединения формулы (I)
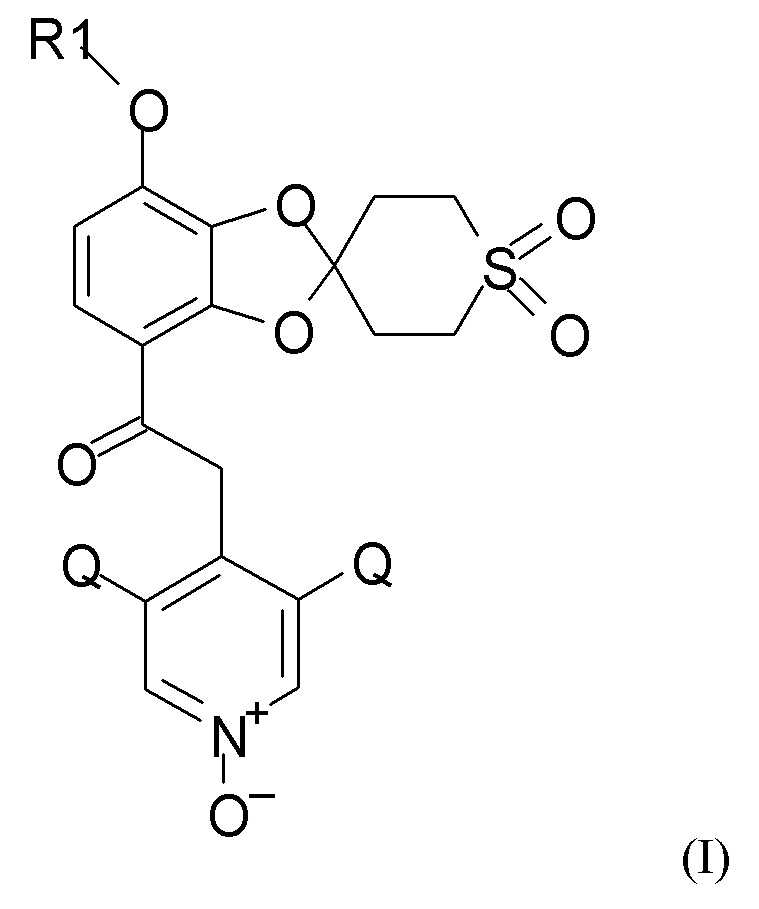
где R1 выбран из CHF2 и CF3 и Q выбран из хлора, брома и фтора, включающий каждую из стадий (1), (2a), (2b) и (2c), таких как определено по п.1, алкилирование и последующее окисление полученного соединения.
11. Способ получения соединения формулы (I)
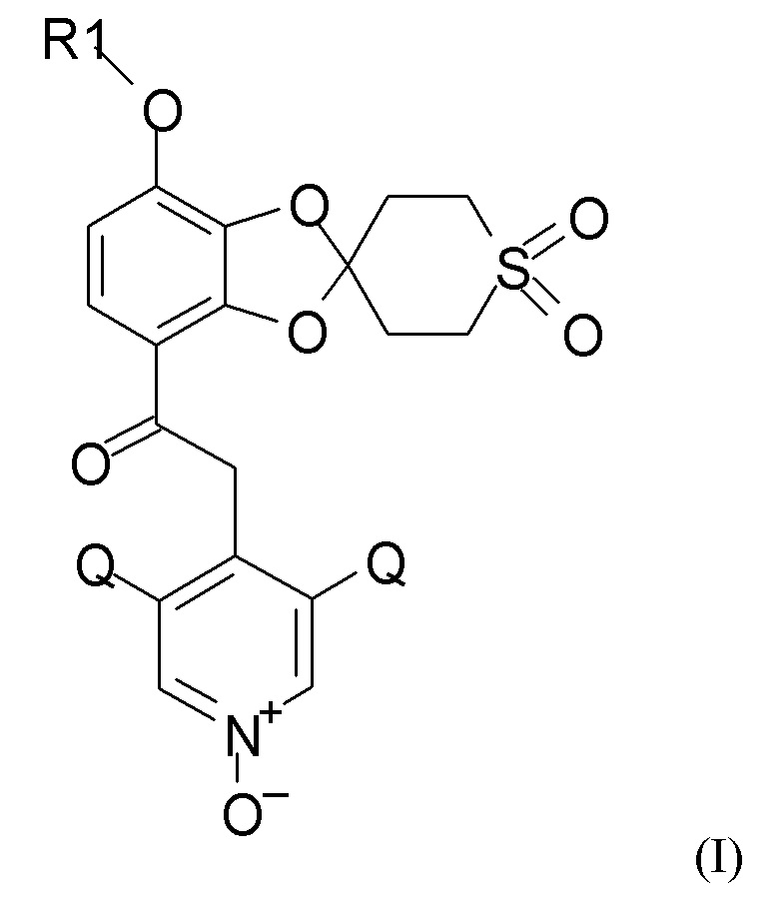
где R1 выбран из CHF2 и CF3 и Q выбран из хлора, брома и фтора, включающий каждую из стадий (1), (2a), (2b) и (2c), (3) и (4), таких как определено по пп.1 и 2.
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| WO 2011160632 A1, 29.12.2011 | |||
| WO 1996036624 A1, 21.11.1996. | |||

