Область техники
Настоящее изобретение относится к новым способам получения 1,3-бензодиоксольных гетероциклических соединений. Данные соединения пригодны в качестве ингибиторов PDE4.
уровень техникиВ WO 2011/160632 раскрыты бензодиоксольные и бензодиоксепеновые гетероциклические соединения, пригодные в качестве ингибиторов PDE4, а также пригодные способы их получения.
В WO 2008/104175 раскрыты бензодиоксольные и бензодиоксепеновые гетероциклические соединения, пригодные в качестве ингибиторов PDE4, а также пригодные способы их получения.
В WO 2008/077404 раскрыты замещенные ацетофеноны, пригодные в качестве ингибиторов PDE4, а также пригодные способы их получения.
В WO 2015/197534 раскрыты способы получения 1,3-бензодиоксольных гетероциклических соединений.
В WO 2017/103058 раскрыты дополнительные способы получения 1,3-бензодиоксольных гетероциклических соединений.
В Zafrani et al. Tetrahedron 65, 2009, pp 5278-5283 описан способ дифторметилирования фенолов и тиофенолов.
В Sperry et al Org. Process Res. Dev. 15, 2011, pp 721-725 также описано дифторметилирование фенолов.
Frey et al. В Tetrahedron 2003, 59, pp. 6363-6373 также описано деметилирование и дифторметилирование фенолов.
В Zhang et al. J. Org. Chem. 2006, 71, 9845-9848 также описано дифторметилирование фенолов.
В Zheng et al. Chem. Commun. 2007, 5149-5151 также описано дифторметилирование фенолов.
При разработке новых препаратов-кандидатов крайне желательно иметь доступ к альтернативным способам получения препаратов-кандидатов, поскольку некоторые эффективные мелкомасштабные синтезы могут оказаться трудными для масштабирования до количеств промышленного масштаба. Кроме того, мелкомасштабные синтезы могут включать реагенты и растворители, которые невозможно использовать на уровне промышленного масштаба.
Следовательно, целью настоящего изобретения является создание альтернативных способов получения 1,3-бензодиоксольных гетероциклических соединений типа, раскрытого в WO 2011/160632, WO 2015/197534 и WO 2017/103058, поскольку такие альтернативные способы обеспечивают преимущества в отношении одного или более признаков, таких как количество стадий реакций, чистота, выход, простота очистки, экономичность процесса, доступность исходных материалов и реагентов, безопасность, предсказуемость и т. д.
По сравнению со стадией (2a) в WO 2017/103058 на стадии (2a) по настоящему изобретению, использовали более дешевые реагенты и более благоприятные для окружающей среды растворители, а раствор нагревали до кипения с обратным холодильником и перемешивали до превращения ≥98%.
На стадии (3), при проведении реакции, как описано в Примере 3 в WO 2017/103058 (Пример 5 по настоящему изобретению) углекислый газ высвобождается в количествах эквимолярных к добавленному количеству хлордифторацетата натрия в реакцию. При увеличении выделения газа и возможном повышении давления в используемом оборудовании процедура может превратиться в потенциальную проблему для безопасности. Поэтому была разработана альтернативная процедура, позволяющая контролировать выделение углекислого газа с течением времени.
Идентификация побочного продукта на данной стадии (3) привела к новым условиям реакции, поскольку указанный побочный продукт не удаляется во время обычной обработки и выделения соединения. Применение ТФУ (трифторуксусная кислота) или МСК (метансульфоновая кислота) в полярном растворителе при повышенной температуре и последующее их удалении путем обработки водным основанием, таким как NaOH или KOH, во время кристаллизации приводит к чистоте данного соединения > 94%.
Идентификация примесей на данной стадии (4) привела к новым условиям реакции. Применение растворителя ДМФА/tBuOH и основания трет-BuOK подавляло образование примесей до минимума, что позволило получить практический выход 70-73%. Это превосходит выход 57%, полученный способом, описанным в WO 2017/103058.
Улучшенный выход, исключение потенциальных проблем для безопасности, применение других условий реакции и применение более дешевых реагентов, оптимизирующих экономичность процесса способа по настоящему изобретению, по сравнению со способом, описанным в WO 2017/103058, WO 2011/160632 и WO 2015/197534, являются довольно неожиданными.
Сущность изобретения
Авторы настоящего изобретения обнаружили, что альтернативные стадии и применение альтернативных реагентов, раскрытых в данном документе, обеспечивают преимущества по сравнению с известными способами благодаря улучшенному общему химическому и объемному выходу, предотвращению потенциальных проблем безопасности и снижению стоимости производства.
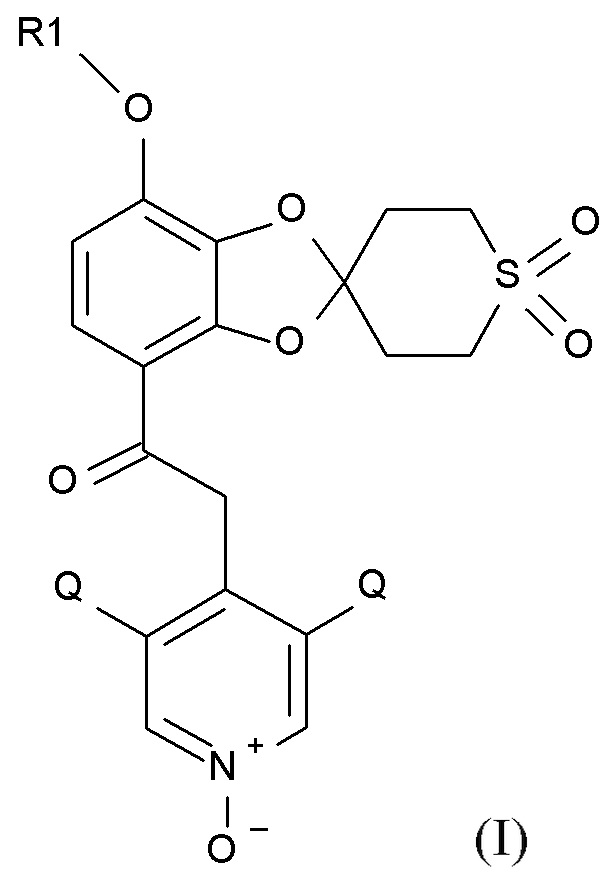
Следовательно, настоящее изобретение обеспечивает способ приготовления 1,3-бензодиоксольных соединений, например соединения формулы (I).
Также в объем изобретения входят промежуточные продукты, применяемые в вышеупомянутом способе получения соединений формулы (I).
Подробное описание изобретения
В первом аспекте настоящее изобретение относится к способу получения соединения формулы (I)
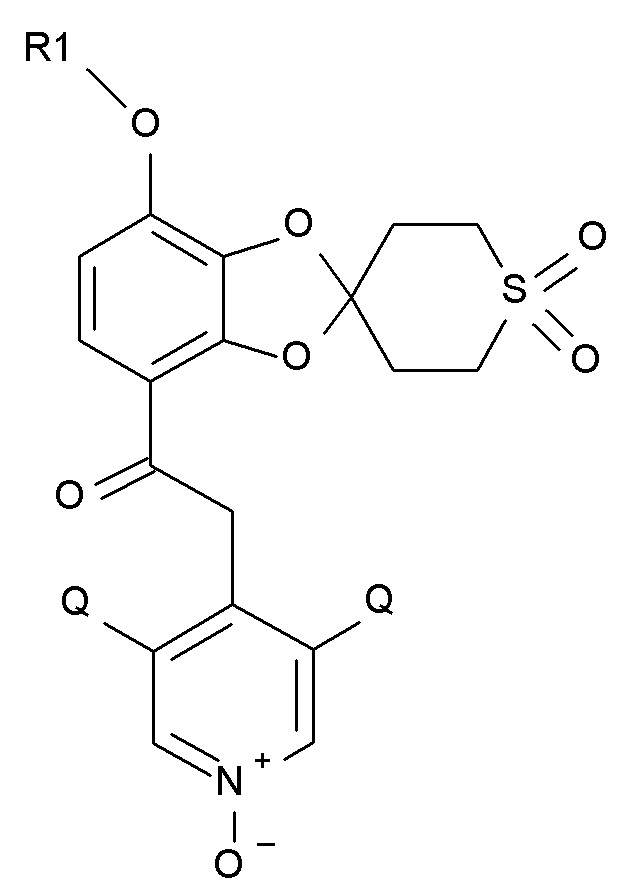 (I),
(I),
где R1 выбирают из CHF2 и CF3, и Q выбирают из хлора, брома и фтора.
В соединении формулы (I) R1 обычно представляет собой CHF2. Q обычно выбирают из хлора, брома и фтора, предпочтительно хлора, где Q предпочтительно одинаковы. В одном варианте осуществления изобретения оба Q представляют собой хлор.
Определения
Термин «C1-6-алкил» означает насыщенную, неразветвленную или разветвленную углеводородную цепь, имеющую от одного до шести атомов углерода, включая метил, этил, пропил, изопропил, бутил, изобутил, вторичный бутил, третичный бутил, пентил, изопентил, неопентил, третичный пентил, гексил и изогексил. В некоторых вариантах осуществления изобретения «C1-6-алкил» представляет собой C1-4-алкильную группу, например метил, этил, пропил, изопропил, бутил, изобутил, вторичный бутил и третичный бутил. Соответственно, «C1-3-алкил» включает метил, этил, пропил, изопропил.
Термин «галоген» предназначен для обозначения одного из фтора, хлора, брома и йода. В одном варианте осуществления изобретения термин «галоген» обозначает фтор или хлор. В другом варианте осуществления изобретения термин «галоген» обозначает хлор.
Термин «арил» предназначен для обозначения карбоциклической ароматической кольцевой системы, полученной из ароматического углеводорода путем удаления атома водорода. Арил дополнительно включает би-, три- и полициклические кольцевые системы. Примеры предпочтительных арильных групп включают фенил, нафтил, инденил, инданил, флуоренил и бифенил. Предпочтительный «арил» представляет собой фенил, нафтил или инданил, в частности фенил, если не указано иное.
Термин «арилалкил» предназначен для обозначения арильного радикала, как определено выше, ковалентно присоединенного к алкильной группе, например бензилу.
Способы получения
Очевидно, что данный способ обеспечивает преимущества по сравнению с известными способами, основываясь на дешевых исходных материалах, простоте способа производства и увеличении выхода в реакциях.
Стадия (1)
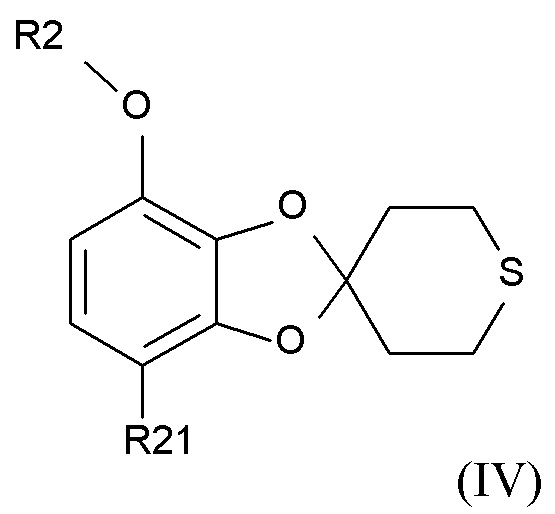
Способ получения соединения формулы (I) включает в себя образование соединения формулы (IV), которое получают путем
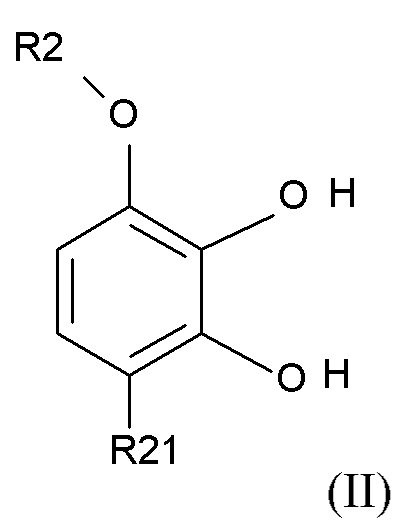
введения в реакцию соединения формулы (II)
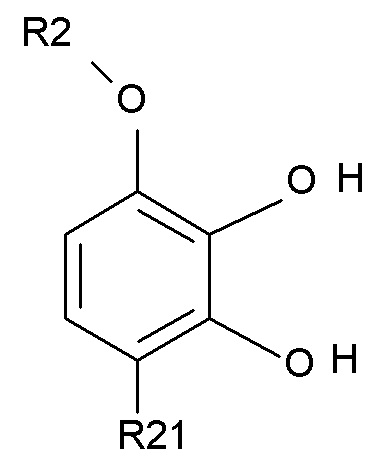 (II),
(II),
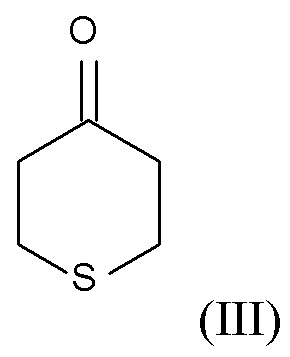
где R2 выбирают из водорода, C1-6-алкила и арилалкила, R21 выбирают из водорода и C(O)R22, и R22 выбирают из водорода и C1-6-алкила; с соединением формулы (III)
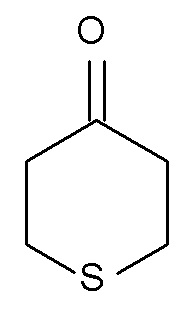 (III),
(III),
в присутствии кислотного катализатора с образованием соединения формулы (IV)
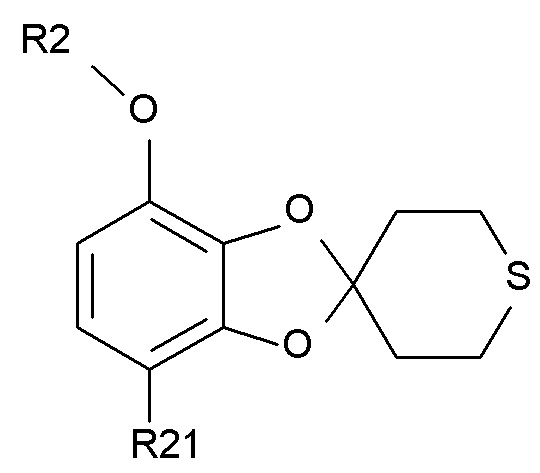 (IV),
(IV),
где R2 и R21 соответствуют определениям выше.
Кислотный катализатор обычно находится в форме глины или цеолита. Цеолит обычно выбирают из CBV 720, CBV 760, CBV 780, HSZ-390HUA. Глину обычно выбирают из монтмориллонита K10, Taiko Classic, Taiko Omega, Actol-10, Actol-20, Actol-20X, Tonsil Supreme 116 FF или Tonsil Supreme 115 FF. В одном варианте осуществления изобретения глину выбирают из монтмориллонита K10, Tonsil Supreme 116 FF или Tonsil Supreme 115 FF. В другом варианте осуществления изобретения глина представляет собой монтмориллонит K10.
Соотношение между цеолитом или глиной и соединением формулы (II) может влиять на время превращения и фильтрации. Следовательно, обычно предпочтительно иметь количество цеолита или глины от 10% мас./мас. до 500% мас./мас. по отношению к соединению формулы (II). В частности, количество минерального вещества должно составлять от 25% мас./мас. до 75% мас./мас., предпочтительно в диапазоне от 45% мас./мас. до 55% мас./мас.
Данную реакцию обычно проводят в толуоле, бензоле, 2-метил-ТГФ (2-метил-тетрагидрофуран), EtOAc (этилацетат), ксилолах, гептане, октане, хлорбензоле и дихлорбензоле. В одном варианте осуществления изобретения растворитель представляет собой толуол или ксилолы. В другом варианте осуществления изобретения растворитель представляет собой толуол.
Реакцию обычно проводят при температуре выше 80°C для того, чтобы способствовать прохождению реакции. Следовательно, обычно предпочтительно, чтобы температура находилась в диапазоне 80-200 °C, например в диапазоне 100-160 °C, особенно при 105-115°C или 135-145 °C. В одном варианте осуществления изобретения реакцию проводят при кипячении реакционной смеси с обратным холодильником. Реакции обычно позволяют протекать в течение 4-96 часов, например 24-84 часов, особенно 48-84 часов.
Полученное соединение формулы (IV) может быть выделено обычными способами, известными специалистам в данной области техники, например водной обработкой с последующей экстракцией и, в конце, осаждением и фильтрацией.
В одном варианте осуществления изобретения соединение формулы (II) представляет собой такое, где R2 выбирают из водорода или метила, и R21 выбирают из водорода, COCH3 или COOH. В другом варианте осуществления изобретения соединение формулы (II) представляет собой 1-(2,3-дигидрокси-4-метоксифенил)этанон.
В одном варианте осуществления изобретения соединение формулы (III) представляет собой тетрагидротиопиран-4-он.
В одном варианте осуществления изобретения соединение формулы (IV) представляет собой такое, где R2 представляет собой водород, метил, этил, пропил, изопропил, изобутил, вторичный бутил, третичный бутил или бензил, и R21 выбирают из водорода, COCH3 или COOH. В другом варианте осуществления изобретения соединение формулы (IV) представляет собой такое, где R2 представляет собой метил и R21 представляет собой COCH3.
Стадия (2a)
Соединение формулы (IV)
(IV),
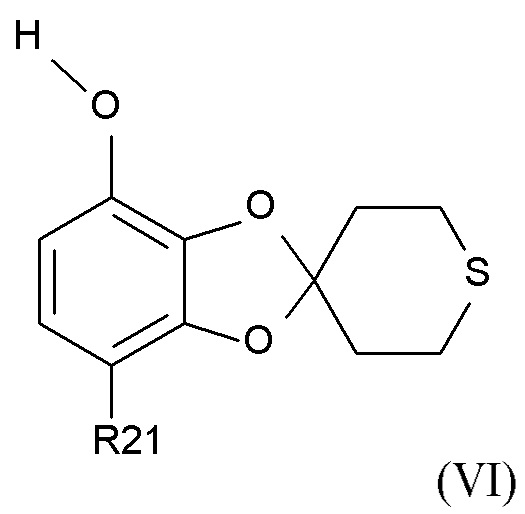
где R2 и R21 соответствуют определениям выше, превращают в соединение формулы (VI)
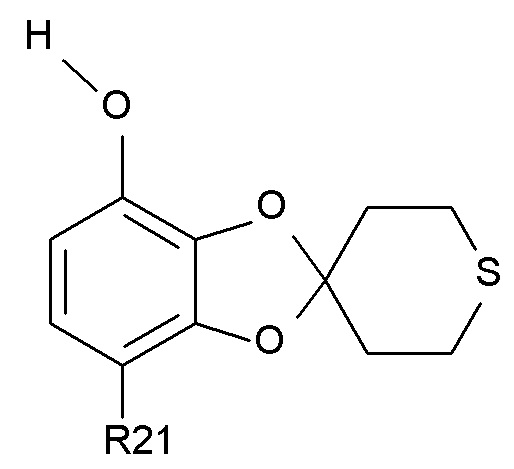 (VI),
(VI),
где R21 соответствует определениям выше, путем снятия защиты с фенольного фрагмента.
Это может быть осуществлено путем введения в реакцию соединения формулы (IV) с ароматическим или алифатическим тиолом в комбинации с основанием.
Ароматический тиол может представлять собой, например, но не ограничиваясь ими, бензолтиол, 4-метилбензолтиол, 3,5-диметилбензолтиол, 2,5-диметилбензолтиол, 4-изопропилбензолтиол или 5-трет-бутил-2-метил-бензолтиол. В одном варианте осуществления изобретения ароматический тиол представляет собой 5-трет-бутил-2-метил-бензолтиол.
Алифатический тиол может представлять собой, например, но не ограничиваясь ими, 1-додекантиол, 1-тетрадекантиол, 1-гексадекантиол или трет-додекантиол. В одном варианте осуществления изобретения алифатический тиол представляет собой 1-додекантиол.
Снятие защиты с фенольной группы на стадии (2а) можно проводить с использованием различных растворителей, например, выбранных из ДМФА (N, N-диметилформамид), NMP (N-метилпирролидон), ДМСО (диметилсульфоксид), метанола, этанола, 1-пропанола, 2-пропанола и их смеси. В одном варианте осуществления изобретения растворитель представляет собой ДМФА. В другом варианте осуществления изобретения растворитель представляет собой смесь ДМФА и метанола. В другом варианте осуществления изобретения растворитель представляет собой этанол. В еще одном варианте осуществления изобретения растворитель представляет собой 1-пропанол.
Снятие защиты с фенольной группы осуществляют в присутствии основания, например, выбранного из K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, TEA (триэтиламин), гидроксида металла, например, выбранного из NaOH, KOH и LiOH; и трет-бутоксида калия, трет-BuOLi (трет-бутоксид лития), метоксида натрия, этоксида натрия и DIPEA (N, N-диизопропилэтиламин). В одном варианте осуществления изобретения основание представляет собой K2CO3. В другом варианте осуществления изобретения основание представляет собой метоксид натрия. В другом варианте осуществления изобретения основание представляет собой гидроксид металла, в другом варианте осуществления изобретения основание представляет собой NaOH.
Реакцию обычно проводят при температуре в диапазоне 50-120 °C, например в диапазоне 70-100 °C. Реакции обычно дают протекать в течение 2-36 часов, например 3-24 часа. Реакции обычно позволяют протекать до тех пор, пока превращение не составит ≥98%.
В конкретном варианте осуществления настоящего изобретения смесь соединения формулы (IV), гидроксида металла, 1-додекантиола и спирта нагревают до кипения с обратным холодильником и перемешивают. В другом конкретном варианте осуществления настоящего изобретения гидроксид металла представляет собой NaOH и спирт представляет собой EtOH. В другом конкретном варианте осуществления настоящего изобретения гидроксид металла представляет собой NaOH и спирт представляет собой 1-пропанол.
Полученное соединение формулы (VI) может быть извлечено обычными способами, известными специалистам в данной области техники, например водной обработкой с последующей экстракцией и, наконец, осаждением и фильтрацией.
В одном варианте осуществления изобретения соединение формулы (VI) является таким, где R21 представляет собой C(O)R22, и R22 выбирают из водорода и C1-6-алкила. В другом варианте осуществления изобретения соединение формулы (VI) представляет собой 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон.
Стадия (2b)
На стадии (2b) соединение формулы (VI) вводят в реакцию с водным N(Bu)4+OH- с образованием соединения формулы (VII)
N(Bu)4+
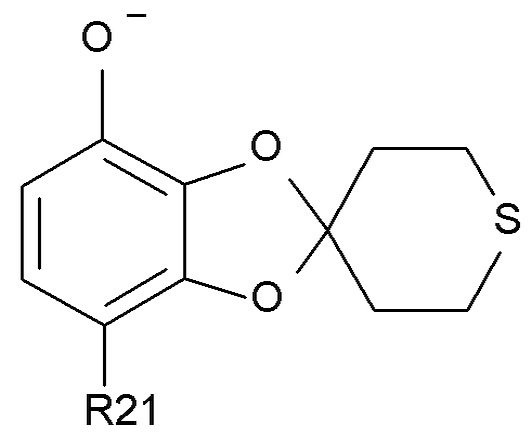 (VII),
(VII),
где R21 соответствует определениям выше.
Смесь обычно нагревают до температуры в диапазоне 20-80 °C, например 55-65 °C, до тех пор, пока все не растворится.
Полученный раствор обычно промывают раствором хлорида натрия в воде при перемешивании при температуре в диапазоне 20-80 °C, например 55-65 °C, в течение > 20 мин. Последующее добавление смеси воды и хлорида натрия с последующим охлаждением смеси от ≥35°C до 0-20 °C, например до 5°C в течение периода, равного 1-24 часам, например 1-4 часам, вызывает осаждение соли TBA (тетрабутиламмония). Соль TBA выделяют, например фильтрацией и сушкой.
Стадия (3)
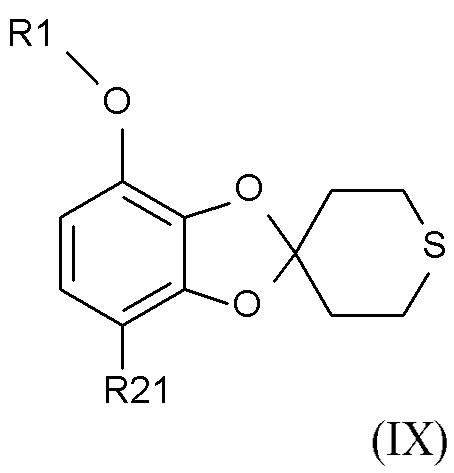
Соединение формулы (IX)
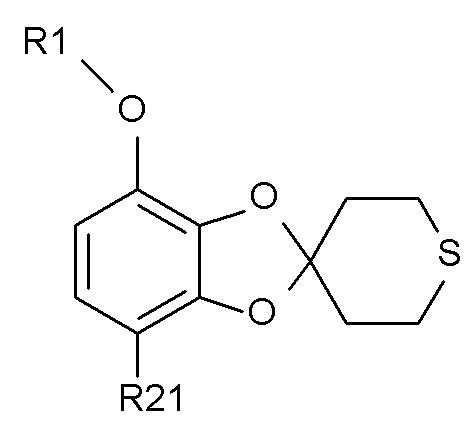 (IX),
(IX),
где R1 и R21 соответствуют определениям выше, может быть получено путем алкилирования полученного соединения формулы (VII)
N(Bu)4+
(VII),
где R21 соответствует определениям выше, путем введения в реакцию с гидрохлорфторуглеродным реагентом,
R1-Cl (VIII),
где R1 соответствует определениям выше.
Алкилирование могут проводить с применением одного из различных возможных реагентов, таких как различные гидрохлорфторуглеродные газы. В одном варианте осуществления изобретения реакцию алкилирования проводят, используя хлордифторметан в апротонном полярном растворителе, например выбранном из ДМФА (N, N-диметилформамид), NMP (N-метилпиролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), EtOAc (этилацетат), MeCN (ацетонитрил) и ТГФ (тетрагидрофуран), и их смеси. В одном предпочтительном варианте осуществления изобретения апротонный растворитель выбирают из ДМФА и NMP. В конкретном варианте осуществления изобретения реакцию проводят, используя хлордифторметан в ДМФА.
Реакцию обычно проводят при температуре в диапазоне 40-120 °C, например в диапазоне 50-70 °C. Реакции обычно позволяют протекать до тех пор, пока в реакционной смеси не останется ≤4% фенола.
Полученное соединение формулы (IX) может быть выделено обычными способами, известными специалистам в данной области техники, например водной обработкой с последующим осаждением и последующей фильтрацией.
В одном варианте осуществления изобретения соединение формулы (IX) представляет собой 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
Альтернативная стадия (2b+3)
Альтернативно, соединение формулы (IX),
(IX),
где R1 и R21 соответствуют определениям выше, может быть получено из соединения формулы (VI),
(VI),
где R21 соответствует определениям выше, без образования промежуточной соли формулы (VII), используя дифторметилатный реагент в полярном растворителе в присутствии основания.
Дифторметилирующий реагент выбирают из например, но не ограничиваясь ими, хлордифторацетата натрия, бромдифторацетата натрия, диэтилбромдифторметилфосфоната, хлордифторметилфенилсульфона и 2-хлор-2,2-дифторацетофенона. Специалист в данной области может легко выбрать другой подходящий аналог упомянутого дифторметилирующего реагента. В одном варианте осуществления изобретения дифторметилирующий реагент представляет собой хлордифторацетат натрия. В другом варианте осуществления изобретения дифторметилирующий реагент представляет собой диэтилбромдифторметилфосфонат.
Реакцию проводят в растворителе, выбранном из, например, ДМФА (N, N-диметилформамид), NMP (N-метилпирролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), EtOAc (этилацетат), MeCN (ацетонитрил), ТГФ (тетрагидрофуран), этанола, метанола, воды, и их смесей. В одном варианте осуществления изобретения растворитель представляет собой смесь воды и ДМФА. В другом варианте осуществления изобретения растворитель представляет собой смесь воды и ацетонитрила.
Реакцию проводят в присутствии основания, выбранного, например, из K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, TEA (триэтиламин), трет-BuOLi (трет-бутоксид лития), метоксида натрия, этоксида натрия, DIPEA (N, N-диизопропилэтиламин), KOH, NaOH, LiOH. В одном варианте осуществления изобретения основание представляет собой K2CO3. В другом варианте осуществления изобретения основание представляет собой NaOH.
Реакцию обычно проводят при температуре в диапазоне 0-120 °C, например 6-115 °C. В одном варианте осуществления изобретения реакцию проводят при 6-20 °C, используя диэтилбромдифторметилфосфонат в качестве дифторметилирующего реагента. В другом варианте осуществления изобретения реакцию проводят при температуре окружающей среды 111 °C, применяя хлордифторацетат натрия в качестве дифторметилирующего реагента.
При выполнении вышеуказанной реакции, как описано в настоящем Примере 5, диоксид углерода выделяется в эквимолярных количествах к добавленному в реакцию количеству хлордифторацетата натрия. При увеличении выделения газа и возможном повышении давления в использованном оборудовании данный способ может превратиться в потенциальную проблему для безопасности.
Поэтому был разработан альтернативный способ, чтобы иметь возможность контролировать выделение диоксида углерода длительное время от начала реакции.
Альтернативная стадия (2b+3’)
Альтернативно, соединение формулы (IX),
(IX),
где R1 и R21 соответствуют определениям выше, может быть получено из соединения формулы (VI),
(VI),
где R21 соответствует определениям выше, путем добавления раствора соединения формулы (VI) и хлордифторацетата натрия в ДМФА к предварительно нагретой смеси ДМФА, воды и карбоната калия в течение длительного периода времени; как описано в настоящем Примере 8.
В одном варианте осуществления изобретения соединение формулы (IX) представляет собой 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
Полученное соединение формулы (IX), где R1 и R21 соответствуют определениям выше, может быть получено обычными способами, известными специалистам в данной области, например водной обработкой с последующим осаждением и последующей фильтрацией.
Во время реакции образования соединения формулы (IX) в значительных количествах образуется побочный продукт формулы (IXb).
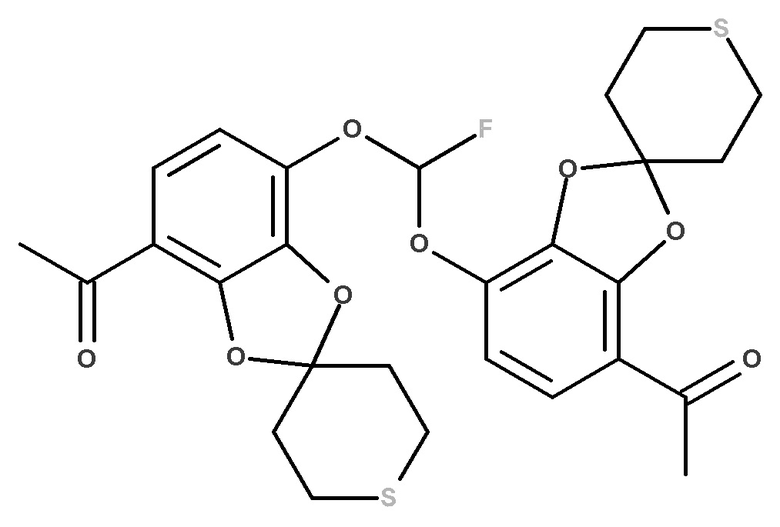 (IXb)
(IXb)
Побочный продукт гидролизуется до соответствующего фенола, соединение которого затем удаляют из продукта обработкой ТФУ или МСК в полярном растворителе, таком как ДМФА, при повышенной температуре и последующим удалением путем обработки водным основанием, таким как NaOH или КОН, при кристаллизации соединения формулы (IX).
Стадия (4)
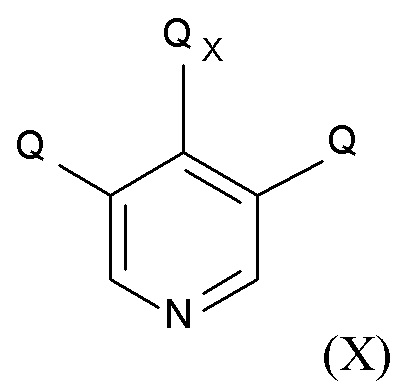
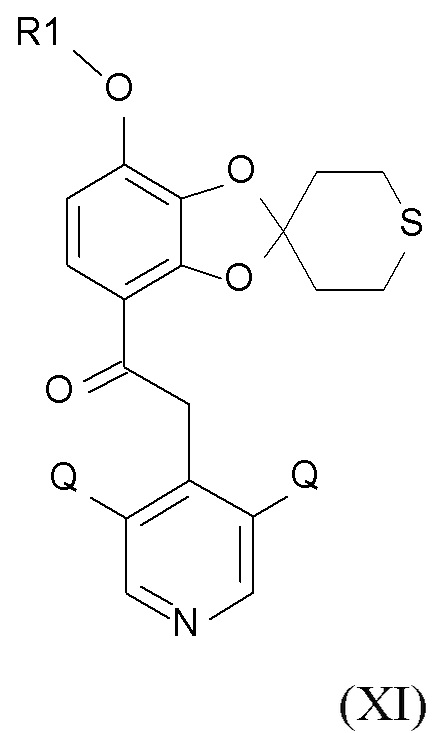
На стадии (4), соединение формулы (IX) вводят в реакцию с соединением пиридина формулы (X)
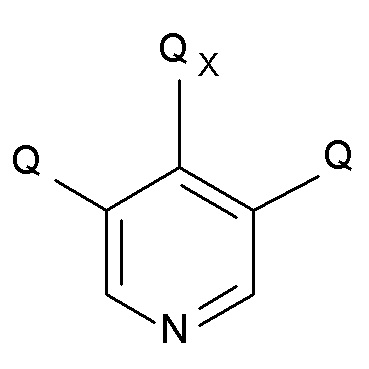 (X),
(X),
где Q соответствует определениям выше и QX выбирают из хлора, брома, фтора и йода с образованием соединения формулы (XI)
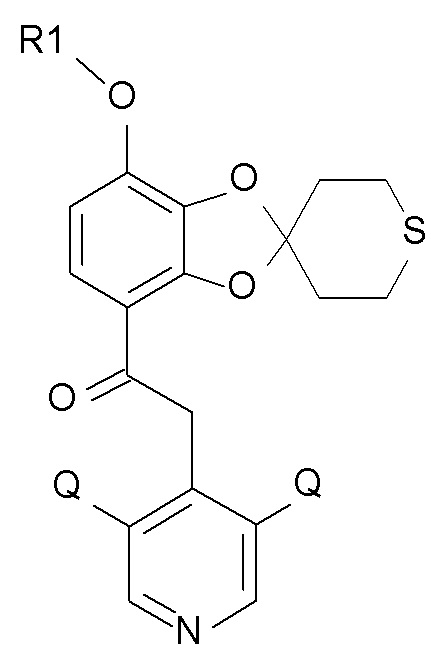 (XI),
(XI),
где R1 и Q соответствуют определениям выше.
Пиридиновое кросс-сочетание на стадии (4) обычно проводят в полярном растворителе, например, выбранном из ДМФА (N, N-диметилформамид), NMP (N-метилпирролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), MeCN (ацетонитрил), ТГФ (тетрагидрофуран), tBuOH (трет-бутиловый спирт), и их смесях, в присутствии основания, например выбранного из трет-BuOK (трет-бутоксид калия), трет-BuOLi (трет-бутоксид лития), трет-BuONa (трет-бутоксид натрия), метоксид натрия или калия, этоксида натрия или калия, K2CO3, Na2CO3, KHCO3, NaHCO3, Et3N (триэтиламин) и DIPEA (N, N-диизопропилэтиламин). В одном варианте осуществления изобретения растворитель представляет собой ДМФА и основание представляет собой трет-BuOK.
Обычно используется два или более эквивалентов основания относительно соединения формулы (IX), например, где молярное соотношение (основание)/(формула IX) составляет от 5:1 до 2:1, например от 3:1 до 2:1, особенно от 2,4:1 до 2,7:1.
Реакцию на стадии (4) обычно проводят при температуре равной 0-40 °C, например 5-25 °C.
В одном варианте осуществления изобретения соединение формулы (X) представляет собой 3,4,5-трихлорпиридин.
В одном варианте осуществления изобретения соединение формулы (XI) представляет собой 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
Во время реакции в значительном количестве образуется примесь формулы (XII).
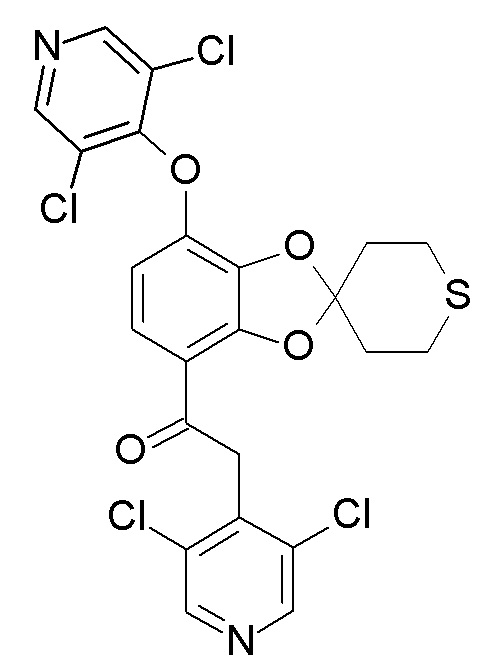 (XII)
(XII)
Продукт очищают от данной примеси путем кристаллизации этого продукта из растворителя, выбранного, например, из диметилформамида (ДМФА), этанола, метанола, этилацетата, гексана, гептана, и их смесей. В одном варианте осуществления изобретения растворитель представляет собой смесь этилацетата и этанола.
Во время детального изучения реакции в значительном количестве была выделена примесь формулы (XIIb).
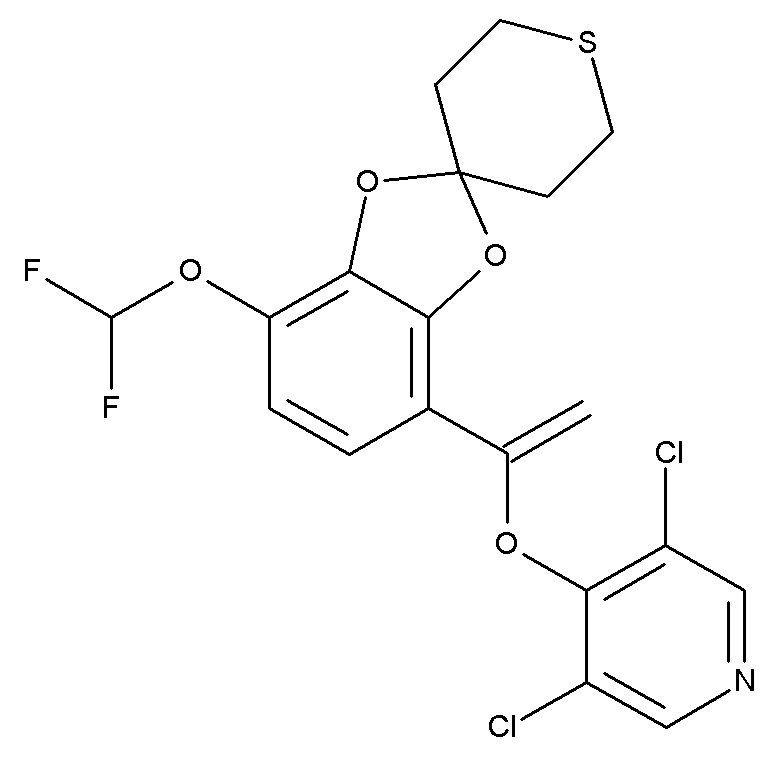 (XIIb)
(XIIb)
Идентификация примеси (XIIb) привела к новым условиям реакции, как описано ниже.
На стадии (4) использованный растворитель представлял собой смесь ДМФА/tBuOH 30/70 об./об. и основание представляло собой трет-BuOK. В этих условиях образование примеси (XIIb) было подавлено до минимума, тогда как продолжительность реакции обычно составляла 3-24 часа. Диапазон температур составлял 20-30 °C. Новый способ позволил достичь практического выхода 70-73%, превысив выход 57%, полученный способом, описанным в WO 2017/103058.
Стадия (5)
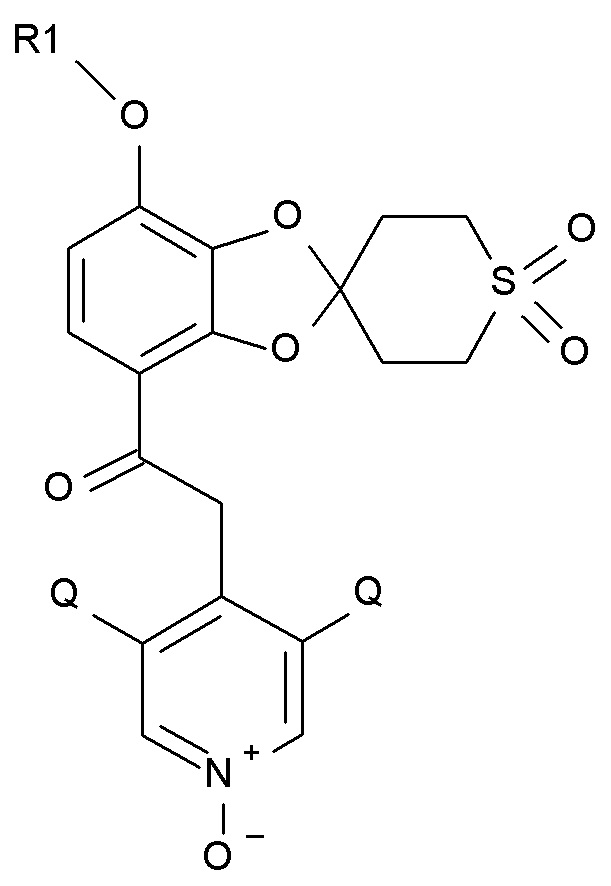
Окисление полученного соединения формулы (XI) проводят с образованием соединения формулы (I)
(I),
где R1 и Q соответствуют определениям выше, путем введения в реакцию указанного соединения формулы (XI) с окисляющим реагентом.
Окисляющий реагент обычно выбирают из PAA (перуксусная кислота) в АсОН (уксусная кислота) и H2O2 (водн.) в муравьиной кислоте или уксусной кислоте. В одном предпочтительном варианте осуществления изобретения окисляющий реагент представляет собой PAA в AcOH. В одном варианте осуществления изобретения количество использованного PAA относительно (XI) (молярное соотношение) обычно составляет от 3 до 6, например от 3,8 до 4,2. Окисляющий реагент обычно медленно добавляют в течение 1-8 часов, например 3-5 часов, поддерживая температуру в диапазоне 15-100 °C, например в диапазоне равном 15-50 °C, особенно в диапазоне равном 15-40 °C.
Реакцию обычно проводят при температуре в диапазоне 30-70 °C, например 40-60 °C, особенно 48-55 °C, и перемешивают в течение 3-48 часов, например 16-24 часов.
Очистка соединения формулы (I)
Полученный сырой продукт формулы (I) может быть преимущественно очищен путем кристаллизации, осаждения, хроматографии или тому подобного.
В одном варианте осуществления изобретения полученный сырой продукт формулы (I) кристаллизуют из смеси воды и EtOH (этанол), выделяют фильтрацией и сушат.
В другом варианте осуществления изобретения первую кристаллизацию из воды пропускают, а соединение формулы (I) кристаллизуют (форма Е) непосредственно из концентрированной реакционной смеси.
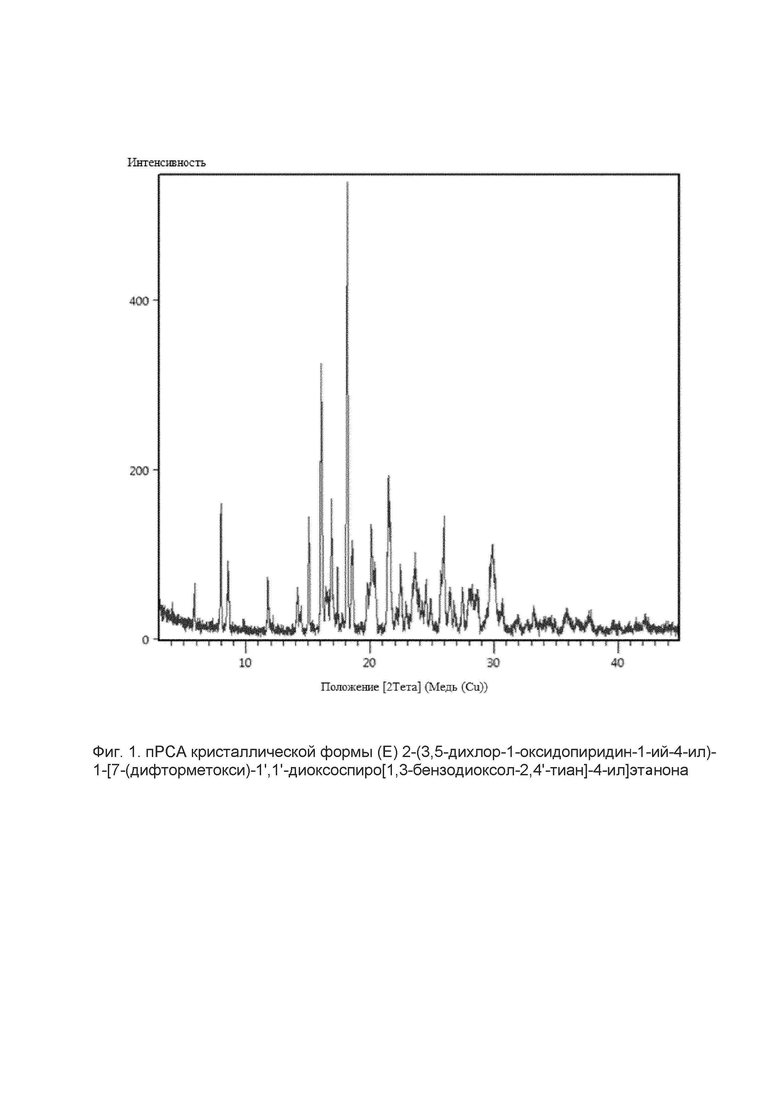
Кристаллическая форма E 2-(3,5-дихлор-1-оксидо-пиридин-1-ий-4-ил)-1-[7-(дифторметокси)-1',1'-диоксо-спиро[1,3-бензодиоксол-2,4'-тиан]-4-ил]этанона имеет порошковую рентгеновскую дифрактограмму, как изображено на Фиг. 1.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы и реагенты
Все использованные химикаты и реагенты были доступны из коммерческих источников.
Спектры ядерного магнитного резонанса (ЯМР) 1H были записаны при указанном магнитном поле, а значения химических сдвигов (δ, в миллионных долях) приведены в указанном растворителе относительно тетраметилсилана (δ=0,00).
ВЭЖХ: Колонка: Aeris Peptide 3,6 мкм XB-C18, 100×4,6 мм, элюент представлял собой градиент A: 10% MeCN; 90% H2O; 0,1% ТФУ и B: 90% MeCN; 10% H2O; 0,1% ТФУ, температура колонки: 35 °C, УФ-детектирование при 220 нм, скорость потока: 1,5 мл/мин. Использовали следующие градиенты элюентов:
Градиенты стадий 2a, 2b, 3 и 5
Градиент стадии 4
ПРИМЕР 1
Стадия (1): Получение 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона
В реактор загружали 1-(2,3-дигидрокси-4-метокси-фенил)этанон (60,0 кг, 329 моль), тетрагидротиопиран-4-он (37,2 кг, 320 моль), монтмориллонит K 10 (30,0 кг) и толуол (720,0 л). Смесь перемешивали при нагревании до кипения с обратным холодильником, применяя температуру в рубашке 140-150°C в течение 84 часов. Смесь охлаждали до 86-90°C и фильтровали через слой фильтрующего средства. Реактор промывали горячим (86-90 °C) толуолом (120 л), а горячий толуол затем использовали для промывания слоя фильтрующего средства. Ополаскивание реактора и последующее промывание слоя фильтрующего средства повторяли два раза горячим толуолом (120 л) и один раз горячим (70 °С) этилацетатом (60 л). Все фильтраты толуола и этилацетата объединяли и охлаждали до 2-6 °С в течение приблизительно 6 часов. Смесь перемешивали при 2-6°C в течение примерно получаса.
Непревращенный исходный материал собирали фильтрацией и сушили в вакууме при 43-47 °C. Выход 32,0 кг.
Фильтрат после выделения непревращенного исходного материала охлаждали до 10-16°C при перемешивании, и прибавляли смесь гидроксида натрия (26,40 кг) и воды (162,0 л) при 10-16 °C. Затем реакционную смесь перемешивали в течение приблизительно получаса при 10-16 °С, затем перемешивание прекращали и фазам давали разделиться. Нижнюю водную фазу отбрасывали, а затем прибавляли смесь гидроксида натрия (26,40 кг) и воды (162 л) при перемешивании при 10-16 °С. Смесь перемешивали в течение приблизительно одного часа, затем перемешивание прекращали, а фазам давали разделиться. Нижнюю водную фазу отбрасывали, а органическую фазу переносили в контейнер. Реактор ополаскивали толуолом, а затем органическую фазу переносили обратно в реактор через картриджный фильтр.
Раствор максимально концентрировали в вакууме, применяя температуру ≤70 °C. Прибавляли этанол (90,0 л), и смесь нагревали до 47-53°C и перемешивали при данной температуре в течение 10-15 минут. Затем смесь максимально концентрировали в вакууме при температуре ≤55 °C. В реактор прибавляли этанол (120,0 л), смесь нагревали до кипения с обратным холодильником при перемешивании, при нагревании прибавляли воду (90,0 л), поддерживая смесь при кипячении с обратным холодильником. Смесь охлаждали до 2-8°C в течение примерно 10 часов и перемешивали при данной температуре в течение примерно получаса.
Продукт выделяли фильтрацией, промывали смесью этанола (30,0 л) и воды (22,8 л) и сушили в вакууме при 43-47 °C. Выход 21,80 кг (24%, но 51% с поправкой на выделенный исходный материал). 1H ЯМР (600 МГц, ДМСО-d6) δ 7,30 (д, J=9,0 Гц, 1H), 6,75 (д, J=9,0 Гц, 1H), 3,88 (с, 3H), 2,91-2,84 (м, 2H), 2,84-2,77 (м, 2H), 2,49 (с, 3H), 2,30-2,22 (м, 2H), 2,22-2,12 (м, 2H).
При необходимости стадию (1) повторяли для получения необходимого количества 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона.
В данном способе использовали другие катализаторы; таблица ниже обобщает выход реакции и количество выделенного исходного материала, когда вышеуказанный способ проводили с серией глин и цеолитов.
%
%
Стадия (2a): Получение 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона
В реактор загружали 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (26,0 кг, 92,7 моль), карбонат калия (14 кг, 101 моль), диметилформамид (104 л) и 5-трет-бутил-2-метил-бензолтиол (26,8 кг, 149 моль). Смесь нагревали при перемешивании до 85-92°C до тех пор, пока не достигали превращения ≥98%, согласно ВЭЖХ. Затем смесь охлаждали до 25 °C, добавляли воду (104 л) и гидроксид натрия (28% в воде, 21,4 кг), и перемешивали в течение ≥10 минут. Если pH смеси был ниже 12, добавляли больше гидроксида натрия (28% в воде). Затем прибавляли толуол (65 л), и перемешивание продолжали в течение ≥15 минут. Перемешивание прекращали, а фазам давали разделиться. Фазы разделяли и органическую фазу отбрасывали. Две нижние водные фазы перемешивали с толуолом (65 л), и смесь перемешивали в течение ≥15 минут. Перемешивание прекращали, что позволяя фазам разделиться. Фазы разделяли и органическую фазу отбрасывали. Две водные фазы возвращали в реактор и при перемешивании медленно прибавляли соляную кислоту (18% в воде, 67,6 кг), чтобы контролировать выделение газа. Полученную смесь перемешивали в течение ≥10 минут. Прибавляли больше соляной кислоты (18% в воде, 10,2 кг) для достижения pH ≤6.
Температуру смеси доводили до 35-45 °С и поддерживали на этом уровне во время следующих экстракций. Прибавляли этилацетат (156 л), и смесь перемешивали в течение ≥30 минут. Перемешивание прекращали, и фазам давали разделиться. Фазы отделяли. Водную фазу перемешивали с этилацетатом (78 л) в течение ≥30 минут. Перемешивание прекращали, а фазам давали разделиться. Водные фазы отбрасывали. Две этилацетатные фазы объединяли в реакторе и перемешивали с водой (78 л) в течение ≥15 минут. Перемешивание прекращали, и фазам позволяли разделиться. Водную фазу отбрасывали.
Органические фазы концентрировали в максимально возможной степени при температуре рубашки 50-60 °С и применении вакуума. Затем прибавляли гептан (39 л), и полученную смесь охлаждали до ≤5°C со скоростью ≤10 °C/ч и поддерживали при данной температуре в течение ≥3 часов. Указанное в заголовке соединение выделяли фильтрацией, промывали холодной (≤5 ° C) смесью этилацетата (10 л) и гептана (10 л) и сушили в вакууме при 40-50 °C. Выход 19,75 кг (80%). 1H ЯМР (600 МГц, ДМСО-d6) δ 10,51 (с, 1H), 7,18 (д, J=9,0 Гц, 1H), 6,50 (д, J=9,0 Гц, 1H), 2,93-2,85 (м, 2H), 2,84-2,78 (м, 2H), 2,46 (с, 3H), 2,31-2,23 (м, 2H), 2,20-2,11 (м, 2H).
Стадия (2b): 7-ацетилспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-олат тетрабутиламмония
1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (19,75 кг, 74,16 моль) загружали в подходящий реактор, с последующим добавлением гидроксида тетрабутиламмония (40% раствор в воде, 53,0 кг, 81,7 моль). Температуру рубашки устанавливали на 60 °C, и смесь перемешивали до тех пор, пока все растворится. Прибавляли насыщенный раствор хлорида натрия в воде (59,2 кг), и перемешивание продолжали при температуре рубашки равной 60°C в течение ≥20 минут. Перемешивание прекращали, позволяя фазам разделиться. Нижнюю водную фазу отбрасывали. Смесь в реакторе снова перемешивали с температурой рубашки равной 60 °C. Прибавляли насыщенный раствор хлорида натрия в воде (29,6 кг), а затем снова воду (25 л). Смесь перемешивали в течение ≥15 минут при температуре в смеси ≥35 °C. Смесь охлаждали до 0-5°C со скоростью приблизительно 20 °C/ч, в смесь вносили затравки при 40°C и снова при 35 °C. Смесь перемешивали при 0-5°C в течение ≥2 часов, а затем указанное в заголовке соединение выделяли фильтрацией и сушили в вакууме при 40-50 °С. Выход 32,9 кг (87%). 1H ЯМР (600 МГц, ДМСО-d6) δ 6,94 (д, J=9,1 Гц, 1H), 5,74 (д, J=9,1 Гц, 1H), 3,23-3,07 (м, 8H), 2,87-2,72 (м, 4H), 2,25 (с, 3H), 2,16-2,07 (м, 2H), 2,06-1,96 (м, 2H), 1,62-1,51 (м, 8H), 1,30 (г, J=7,4 Гц, 8H), 0,93 (т, J=7,4 Гц, 12H).
Стадия (3): 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
7-Ацетилспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-олат тетрабутиламмония (32,93 кг, 64,85 моль) и диметилформамид (198 л) добавляли в реактор. Смесь перемешивали до полного растворения. К раствору прибавляли хлордифторметан (39,5 кг, 457 моль) через погруженную трубку в реакторе. Реакционную смесь нагревали до 50-55°C и перемешивали до тех пор, пока не осталось ≤4% исходного материала, согласно ВЭЖХ. Реакционную смесь охлаждали до 20-25°C и переносили в контейнер через фильтр. Реактор и твердое вещество на фильтре промывали диметилформамидом (10 л), который также прибавляли в контейнер.
Воду (198 л) и гидроксид натрия (28% в воде, 11,0 кг) загружали в реактор и нагревали до 45-55 °C. Реакционную смесь в емкости медленно прибавляли в реактор при перемешивании, поддерживая температуру на уровне 45-55 °C. Смесь затем охлаждали до 5-10 °С и перемешивали при этой температуре в течение ≥2 часов. Продукт выделяли фильтрацией, промывали водой (82 л) и сушили в вакууме при 45-55 °С с отведением азота. Выход 19,08 кг (94%). 1H ЯМР (600 МГц, ДМСО-d6) δ 7,34 (т, J=73,1 Гц, 1H), 7,32 (д, J=9,1 Гц, 1H), 6,86 (д, J=9,1 Гц, 1H), 2,92-2,80 (м, 4H), 2,54 (с, 3H), 2,34-2,27 (м, 2H), 2,27-2,19 (м, 2H).
Стадия (4) 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Диметилформамид (96 л) загружали в подходящий реактор, с последующим добавлением трет-бутоксида калия (17,60 кг, 156,8 моль). Перенос трет-бутоксида калия обеспечивали ополаскиванием диметилформамидом (3 л), и смесь перемешивали до растворения трет-бутоксида калия. Раствор переносили из реактора в контейнер, реактор ополаскивали диметилформамидом (6 л), который также переносили в контейнер.
В реактор загружали 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (19,08 кг, 60,32 моль), 3,4,5-трихлорпиридин (14,30 кг, 78,38 моль) и диметилформамид (96 л). Смесь перемешивали и охлаждали до 10-15 °C, а затем медленно прибавляли раствор трет-бутоксида калия в диметилформамиде, поддерживая температуру реакционной смеси на уровне 5-25 °C. Перенос раствора трет-бутоксида калия обеспечивали ополаскиванием диметилформамидом (6 л). Смесь нагревали до 20-25°C и перемешивали до тех пор, пока превращение не достигало ≥98%, согласно ВЭЖХ.
К реакционной смеси медленно прибавляли воду (96 л) при охлаждении, поддерживая температуру в пределах 20-30 °С. За этим следовало добавление насыщенного хлорида натрия в воде (115,2 кг) и этилацетата (134 л). Смесь перемешивали в течение 20-60 минут и затем перемешивание прекращали, позволяя фазам разделиться. Фазы отделяли, и водную фазу возвращали в реактор. Прибавляли этилацетат (96 л), и смесь перемешивали в течение 20-60 минут. Перемешивание прекращали, что позволяя фазам разделиться. Фазы отделяли. Органические фазы объединяли в реакторе и перемешивали с водой (48 л) и насыщенным хлоридом натрия в воде (57,8 кг) в течение ≥20 минут. Смешивание прекращали, позволяя фазам разделиться. Нижнюю водную фазу отбрасывали и добавляли воду (48 л) и насыщенный хлорид натрия (57,6 кг). Смесь перемешивали в течение 20-60 минут, и затем перемешивание прекращали, позволяя фазам разделиться. Нижнюю водную фазу отбрасывали и добавляли воду (84 л) и гидроксид натрия (28% в воде, 14,0 кг). Смесь перемешивали в течение 20-60 минут и затем перемешивание прекращали, позволяя фазам разделиться. Нижнюю водную фазу отбрасывали.
Органическую фазу в реакторе концентрировали с использованием вакуума и нагревания с температурой рубашки 50-65 °С до остаточного объема приблизительно 40 л. В реактор загружали этанол (57 л), и смесь нагревали до кипения с обратным холодильником до получения прозрачного раствора. Смесь охлаждали до 5°C в течение ≥5 часов и перемешивали при этой температуре в течение ≥3 часов. Продукт выделяли фильтрацией, перенос обеспечивали ополаскиванием этанолом (10 л). Продукт промывали холодным (≤5°) этанолом (48 л) и сушили в вакууме при 45-55 °C. Выход 15,57 кг (56%). 1H ЯМР (600 МГц, хлороформ-d) δ 8,52 (с, 2H), 7,46 (д, J=8,9 Гц, 1H), 6,80 (д, J=8,9 Гц, 1H), 6,73 (т, J=73,3 Гц, 1H), 4,59 (с, 2H), 3,01-2,85 (м, 4H), 2,47-2,30 (м, 4H). ВЭЖХ: Чистота: 97,8%.
Стадия (5): 2-(3,5-дихлор-1-оксидо-пиридин-1-ий-4-ил)-1-[7-(дифторметокси)-1',1'-диоксо-спиро[1,3-бензодиоксол-2,4'-тиан]-4-ил]этанон
В реактор загружали 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (15,6 кг, 33,7 моль) и ледяную уксусную кислоту (78,0 кг), и смесь охлаждали до 13-20 °C. Медленно прибавляли перуксусную кислоту (36-40% в уксусной кислоте, 6,52 кг, 32,6 моль), поддерживая температуру ниже 40 °C. Смесь нагревали до 40-50°C и перемешивали в течение 10-25 минут. Смесь охлаждали до 13-20°C и медленно прибавляли вторую порцию перуксусной кислоты (36-40% в уксусной кислоте, 6,51 кг, 32,5 моль), поддерживая температуру ниже 40 °C. Смесь нагревали до 40-50°C и перемешивали в течение 10-25 минут. Смесь охлаждали до 20-30°C и медленно прибавляли третью порцию перуксусной кислоты (36-40% в уксусной кислоте, 14,3 кг, 71,5 моль). Смесь нагревали до 48-55°C и перемешивали до тех пор, пока превращение не составило ≥98,5%. Смесь охлаждали до 20-25°C и медленно прибавляли смесь метабисульфита натрия (7,21 кг, 37,9 моль) и воды (46 л), поддерживая температуру ниже 35°C.
Прибавляли 2-пропанол (78 л), и смесь нагревали до 60-65°C и фильтровали горячей. Реактор очищали и фильтрованную реакционную смесь возвращали в реактор. Смесь нагревали до 60-65°C и медленно прибавляли воду (234 л), поддерживая температуру выше 55 °C. Смесь перемешивали в течение 30-60 минут при 60-65 °C, медленно охлаждали до 5°C в течение 12 часов, и перемешивали при 0-10°C в течение ≥2 часов. Неочищенный продукт выделяли фильтрацией, промывали водой (27 л) и сушили в вакууме в течение приблизительно двух часов.
Твердое вещество возвращали в реактор и нагревали до кипения с обратным холодильником с этанолом (390 л). Затем смесь охлаждали до 68-72°C и вносили затравки. Смесь охлаждали до 5°C в течение 13 часов и перемешивали при 0-10°C в течение ≥2 часов. Продукт выделяли фильтрацией, промывали холодной (0-10 °C) смесью воды (4 л) и этанола (39 л) и сушили в вакууме при 45-55°C с отведением азота. Выход 14,6 кг (85%). 1H ЯМР (600 МГц, хлороформ-d) δ 8,23 (с, 2H), 7,52 (д, J=9,1 Гц, 1H), 6,90 (д, J=9,1 Гц, 1H), 6,71 (т, J=72,3 Гц, 1H), 4,49 (с, 2H), 3,47-3,38 (м, 2H), 3,33-3,24 (м, 2H), 2,83-2,75 (м, 2H), 2,75-2,68 (м 2H). ВЭЖХ: чистота 98,6%.
ПРИМЕР 2
1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон
Метоксид натрия в метаноле (30%, 64,2 мл, 0,34 моль) прибавляли к раствору 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона (50,0 г, 0,178 моль) в диметилформамиде (250 мл) при 25-30 °C. Затем прибавляли 1-додекантиол (64,88 мл, 0,271 моль) при 25-30 °C, и смесь нагревали до 95-100°C в течение трех часов. Реакционную смесь охлаждали до 25-30°C и добавляли гидроксид натрия (28% в воде, 50 мл) и воду (250 мл). Полученную смесь перемешивали в течение получаса и затем смесь трижды экстрагировали толуолом (250 мл). Водный раствор подкисляли соляной кислотой (6 М) приблизительно до рН 6 и экстрагировали этилацетатом (250 мл) четыре раза. Этилацетатные экстракты объединяли, промывали раствором хлорида натрия (250 мл) четыре раза и концентрировали до приблизительно 50 мл с использованием роторного испарителя. Прибавляли гептан (300 мл), и смесь перемешивали в течение одного часа при температуре окружающей среды. Продукт выделяли фильтрацией, промывали гептаном (100 мл) и сушили. Выход 44,3 г (93%). ЯМР соответствует ЯМР продукта со стадии (2a) в Примере 1.
ПРИМЕР 3
1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон
К раствору 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона (15,0 г, 53,5 ммоль) в EtOH (500 мл) прибавляли гидроксид натрия (31,4 г, 0,785 моль) при 25-30 °C. Затем прибавляли 1-додекантиол (197 мл, 0,87 моль) при 25-30 °C, и смесь нагревали до кипения с обратным холодильником и перемешивали в течение двадцати четырех часов. Из реакционной смеси затем удаляли 300 мл растворителя в вакууме. К оставшейся суспензии затем добавляли воду (500 мл). Потом полученный раствор экстрагировали толуолом (500 мл). Органическую фазу затем отбрасывали, а оставшуюся водную фазу подкисляли соляной кислотой (1 М) приблизительно до рН 3-5. Продукт выделяли фильтрацией, промывали водой (2х100 мл) и сушили в вакууме при 60 °С. Выход 93,0 г (98%). ЯМР соответствует ЯМР продукта со стадии (2a) в Примере 1.
ПРИМЕР 4
1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон
К раствору 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона (100,0 г, 0,357 моль) в 1-пропаноле (75 мл) прибавляли гидроксид натрия (4,7 г, 117,7 ммоль) при 25-30 °C. Затем прибавляли 1-додекантиол (29,5 мл, 123,1 ммоль) при 25-30 °C, и смесь нагревали до кипения с обратным холодильником и перемешивали в течение шести часов. Реакционную смесь охлаждали до 25 °C. Затем к реакционной смеси добавляли воду (75 мл) и потом дважды экстрагировали толуолом (2×75 мл). Органические фазы затем отбрасывали, а оставшуюся водную фазу подкисляли соляной кислотой (1 М) до приблизительно рН 3-5. Продукт выделяли фильтрацией, промывали водой (2х50 мл) и сушили в вакууме при 60 °С. Выход 11,3 г (79%). ЯМР соответствует ЯМР продукта со стадии (2a) в Примере 1.
ПРИМЕР 5
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Смесь 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона (221,6 г, 0,8322 моль), карбоната калия (161,3 г, 1,167 моль), хлордифторацетата натрия (292,0 г, 1,915 моль), диметилформамида (1,50 л) и воды (500 мл) перемешивали в 5 литровой реакционной колбе и медленно нагревали до 106-111 °C, наблюдали выделение газа при приблизительно 78 °C. Реакционную смесь перемешивали при 106-111°C до прекращения выделения газа, приблизительно два часа. Смесь охлаждали на ледяной бане и медленно прибавляли воду (1,00 л) при 30-32 °C. Полученную суспензию дополнительно охлаждали до 6 °С при перемешивании. Сырой продукт выделяли фильтрацией и промывали водой.
Влажный неочищенный продукт перемешивали с этилацетатом (1,66 л) и гидроксидом натрия (1 М, 560 мл) в течение приблизительно 20 минут, а затем фазы разделяли в делительной воронке. Нижнюю водную фазу отбрасывали и органическую фазу дважды промывали водой (два раза по 560 мл). Органическую фазу концентрировали с использованием роторного испарителя (в вакууме при 60°C на водяной бане) до приблизительно 450 мл. Прибавляли этилацетат (1,56 л), а смесь снова концентрировали с использованием роторного испарителя, как описано выше, до приблизительно 450 мл. Прибавляли этилацетат (1,44 л), и непрозрачный раствор фильтровали, переносили и промывали свежей порцией этилацетата (100 мл). Объединенные фильтраты фильтровали через слой активированного угля (6,0 г), переносили и промывали этилацетатом (200 мл). Объединенные фильтраты концентрировали на роторном испарителе, как описано выше, до объема приблизительно 450 мл. Полученный горячий раствор (приблизительно 60 °C) перемешивали при температуре окружающей среды, одновременно медленно добавляли гептан (2,00 л) в течение приблизительно получаса. Суспензию перемешивали при комнатной температуре в течение 14 часов.
Смесь перемешивали на ледяной бане в течение приблизительно 2,5 часов, после чего температура смеси составляла 4 °С. Продукт выделяли фильтрацией, промывали ледяной смесью гептана и этилацетата (10:1, 200 мл) и сушили в вакууме при 50 °С с отведением воздуха. Выход 201 г (76%). ЯМР соответствует ЯМР продукта со стадии 3 в Примере 1.
ПРИМЕР 6
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Гидроксид натрия (6,16 г, 154 ммоль) растворяли в воде (40 мл) и раствор перемешивали при охлаждении на ледяной бане. Прибавляли 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (2,00 г, 7,51 ммоль) и ацетонитрил (20 мл) и продолжали перемешивание с охлаждением. Одной порцией прибавляли диэтилбромдифторметилфосфонат (2,67 мл, 15,0 ммоль) при 6 °C, и продолжали перемешивание при охлаждении в течение приблизительно 20 минут. Убрали охлаждающую баню, и смесь перемешивали в течение приблизительно 21 часа при температуре окружающей среды.
Фазы разделяли с использованием делительной воронки, и водную фазу экстрагировали этилацетатом (20 мл). Объединенные органические фазы промывали водой (20 мл), а затем раствором хлорида натрия (20 мл). Органическую фазу концентрировали досуха с использованием роторного испарителя. К остатку прибавляли этилацетат (20 мл), и смесь снова концентрировали досуха, используя роторный испаритель.
Остаток растворяли в этилацетате (30 мл) и фильтровали, переносили и промывали этилацетатом (20 мл). Объединенные фильтраты концентрировали досуха с использованием роторного испарителя, как указано выше, получая указанное в заголовке соединение в виде желтоватого твердого вещества. Выход 2,14 г (90%). ЯМР соответствует ЯМР продукта со стадии 3 в Примере 1.
ПРИМЕР 7
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Гидроксид натрия (301 г, 7,52 моль) перемешивали с водой (2,0 л), и полученный раствор охлаждали на ледяной бане. Прибавляли 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (100,1 г, 0,3757 моль) и ацетонитрил (1,0 л). Медленно добавляли диэтилбромдифторфосфонат (150,5 г, 0,5637 моль) в течение приблизительно 40 минут при температуре в реакционной смеси 15-20 °С. Перемешивание продолжали еще приблизительно два часа при 15-20 °С. Фазы отделяли.
К органической фазе при перемешивании медленно прибавляли воду (920 мл), и полученную суспензию перемешивали при температуре окружающей среды в течение приблизительно 18 часов. Продукт выделяли фильтрацией, промывали смесью ацетонитрила и воды 1:1 (120 мл) и сушили в вакууме при 50°C с отведением воздуха. Выход 108 г (91%). ЯМР соответствует ЯМР продукта со стадии 3 в Примере 1.
ПРИМЕР 8
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
К смеси карбоната калия (1,45 г, 10,5 ммоль) в ДМФА (8,2 мл) и воды (3,6 мл) при температуре реакционной смеси 110°C медленно прибавляли раствор 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона (2,0 г, 7,51 ммоль) и хлордифторацетата натрия (2,86 г, 18,8 ммоль) в ДМФА (6,2 мл) в течение периода 2-4 ч. После завершения добавления реакционную смесь перемешивали в течение еще 60 мин. Затем температуру реакционной смеси снижали до 70 °С, причем к реакционной смеси добавляли водный 0,5 М раствор NaOH (10 мл).
Полученную реакционную суспензию затем медленно охлаждали до 10-20 °С. Продукт выделяли фильтрацией, промывали водой (40 мл) и сушили в вакууме при 60 °С. Выход 1,73 г (73%). ЯМР соответствует ЯМР продукта со стадии (3) в Примере 1.
ПРИМЕР 9
2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Диметилформамид (128 мл) и трет-бутанол (298 мл) загружали в подходящий реактор с последующим добавлением трет-бутоксида калия (81,4 г, 726 ммоль). Смесь перемешивали до растворения трет-бутоксида калия.
Во второй реактор загружали 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (85,0 г, 269 ммоль), 3,4,5-трихлорпиридин (58,8 г, 322 ммоль), диметилформамид (76,5 мл) и трет-бутанол (179 мл). Смесь перемешивали при 22-25 °C, медленно добавляя раствор трет-бутоксида калия в диметилформамиде и трет-бутаноле, поддерживая температуру реакционной смеси <30 °С. Реакционную смесь перемешивали при 22-25 °С до тех пор, пока превращение не достигло ≥98%, согласно ВЭЖХ.
К реакционной смеси медленно прибавляли воду (340 мл), поддерживая температуру < 30 °C. Реакционную смесь переносили в делительную воронку и разбавляли этилацетатом (850 мл). Водную нижнюю фазу отбрасывали. Органическую фазу промывали водным раствором гидроксида натрия (2 М, 500 мл), и нижнюю водную фазу отбрасывали. Органическую фазу промывали водой (340 мл), и нижнюю водную фазу отбрасывали. Органическую фазу переносили в подходящий реактор и концентрировали при пониженном давлении (100 мбар) при 50 °С (температура рубашки), пока температура пара не достигла 39 °С. Затем прибавляли этанол (255 мл), и суспензию нагревали до кипения с обратным холодильником (90 °С) и перемешивали в течение одного часа. Потом раствор медленно охлаждали до 5 °С. Продукт выделяли фильтрацией, перенос обеспечивали ополаскиванием этанолом (20 мл). Продукт промывали холодным (≤5 °) этанолом (150 мл) и сушили в вакууме при 45-55 °C. Выход 91,0 г (73,2%). 1H ЯМР (600 МГц, хлороформ-d) δ 8,52 (с, 2H), 7,46 (д, J=8,9 Гц, 1H), 6,80 (д, J=8,9 Гц, 1H), 6,73 (т, J=73,3 Гц, 1H), 4,59 (с, 2H), 3,01-2,85 (м, 4H), 2,47-2,30 (м, 4H). ВЭЖХ: Чистота: 97%.
ПРИМЕР 10
Получение 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона
Подходящий реакционный сосуд продували азотом. Поток азота временно останавливали и загружали в реакционный сосуд 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (1,00 кг, 3,57 моль), с последующим добавлением этанола (2,5 л), гидроксида натрия (314 г, 7,85 моль, 2,2 экв.) и 1-додекантиола (1,66 кг, 8,2 моль, 2,3 экв.). Реакционную смесь нагревали до кипения с обратным холодильником при перемешивании, и данную температуру поддерживали в течение 22-24 часов.
ИПХ (ионно-парная хроматография) проводили в этаноле для анализа ВЭЖХ (220 нм). Если исходный материал присутствовал менее чем на 2% площади, реакционную смесь охлаждали до 20-25 °С.
Далее в реакционную смесь при перемешивании загружали воду (5 л). Впоследствии прибавляли толуол (1,5 л), и смесь перемешивали в течение по меньшей мере 15 минут.
Перемешивание останавливали и давали фазам разделиться.
Водную фазу дважды промывали толуолом (2 х 1,5 л).
Прибавляли воду (1 л) с последующим добавлением водного раствора соляной кислоты (18%, 1,5-1,6 кг, 2,1-2,2 экв.).
Осадок отфильтровали и дважды промывали водой (2 × 2 л) и гептаном (2 л). Влажное твердое вещество переносили в реакционный сосуд и прибавляли гептан (5 л). Суспензию перемешивали и нагревали до кипения с обратным холодильником, и удаляли воду азеотропной перегонкой.
Смесь охлаждали до 20-25°C, и твердое вещество отфильтровали и сушили в вакууме.
Выход: 80-90%. ЯМР соответствует ЯМР продукта со стадии (2a) в Примере 1.
Получение 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанона
Карбонат калия (3,12 кг, 22,6 моль, 1,4 экв.) и ДМФА (15,8 кг) прибавляли в подходящий реакционный сосуд (№ 1). Суспензию перемешивали при 20-25 °С, и сосуд и смесь продували азотом в течение по меньшей мере 1 часа при 20-25 °С.
В отдельный реакционный сосуд (№ 2), снабженный трубой для продувания азота, помещали 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (4,3 кг, 16,2 моль, 1 экв.), хлордифторацетат натрия (6,16 кг, 40,4 моль, 2,5 экв.) и ДМФА (12 кг). Суспензию перемешивали при 20-25 °С, и сосуд и смесь продували азотом в течение по меньшей мере 1 часа при 20-25 °С.
Суспензию в сосуде (№ 1) нагревали до 115 °C, и содержимое сосуда (№ 2) переносили в сосуд (№ 1) в течение 7-8 часов.
ИПХ проводили в ацетонитриле для анализа ВЭЖХ (220 нм), с критерием для дальнейшего протекания: < 5% площади 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]4-ил)этанона.
Реакционную смесь охлаждали до 20-25°C, и к реакционной смеси прибавляли водный раствор КОН (0,75 М, 18,8 кг).
Осадок отфильтровали и промывали раствором КОН (0,75 М, 9 кг), а затем водой (86 кг).
Влажный материал промывали в ДМФА (16 кг) и нагревали до 40-45 °C. Прибавляли MСК (метансульфоновая кислота) (3,44 кг, 35,8 моль, 2,2 экв.), поддерживая температуру между 45-50 °C.
ИПХ проводили в ацетонитриле для анализа ВЭЖХ (272 нм), с критерием для дальнейшего протекания: < 4% площади химического побочного продукта формулы (IXb).
Реакционную смесь охлаждали до 35-40°C, и к смеси прибавляли водный раствор KOH (3,25M, 18,7 кг). После завершения добавления смесь охлаждали до 20-25 °C. Критерием дальнейшего протекания было значение рН раствора 10-12.
Твердое вещество отфильтровали и промывали раствором КОН (0,75 М, 9 кг), а затем водой (86 кг). Твердое вещество сушили в вакууме при температуре 60 °C.
Выход: > 85% (чистота > 93%). ЯМР соответствует ЯМР продукта со стадии (3) в Примере 1.
Альтерантивно, вместо MСК для обработки может быть использована ТФУ. Влажный материал после первого выделения промывали в ДМФА (100 мл). Прибавляли ТФУ (трифторуксусная кислота) (13,4 г, 116,4 ммоль, 1,5-3 экв.) вместе с водой (21 мл), реакцию проводили при температуре 60-70 °C.
Реакционную смесь охлаждали до 44-50 °C, и к смеси прибавляли водный раствор NaOH (1,25 M, 140 мл). После завершения добавления смесь перемешивали в течение 1 ч.
Твердое вещество выделяли фильтрацией и промывали 0,5 М NaOH, а затем водой (480 мл). Твердое вещество сушили в вакууме при температуре 50 °C. Выход: > 90% (чистота > 94%). ЯМР соответствует ЯМР продукта со стадии (3) в Примере 1.
Получение 3,4,5-трихлорпиридина
Стадия 1: Перемешиваемый раствор 4-пиридинола (1,0 экв.) в ацетонитриле (15,0 об.) и воде (0,1 об.) нагревали до 40°C и затем порциями добавляли N-хлор сукцинамид (2,2 экв.) при 40-55 °C. Реакционную смесь перемешивали в течение 6-8 ч при 45-55 °C, протекание реакции контролировали с помощью ВЭЖХ. После завершения реакции реакционную смесь охлаждали и перемешивали в течение 3-4 ч. Твердое вещество фильтровали и промывали ацетонитрилом (1х2,0 об.) и водой (5,0 об. + 2,0 об.). Продукт высушивали в печи до получения постоянной массы.
Стадия 2: К перемешиваемой суспензии 3,5-дихлор-4-пиридинола (1,0 экв.) в ацетонитриле (5,0 об.) прибавляли POCl3 (2,0 экв.). Реакционную смесь нагрели до 50-55°C и перемешивали в течение 24 часов. За протеканием реакции наблюдали с помощью ВЭЖХ. После завершения реакции смесь охлаждали. Затем реакционную смесь медленно выливали в воду (5,0 об.) при 2-10 °C. Смесь перемешивали в течение 20-30 минут и pH довели до 9-10 с помощью 50% NaOH(водн.). Температуру повышали до 25-30°C и смесь экстрагировали н-гептаном (1х18,0 об., 2х10,0 об.). Объединенные органические слои промывали водой (1×5,0 об.), а затем добавляли древесный уголь (15% мас./мас.) и перемешивали в течение 1-2 часов. Органическую фазу фильтровали через слой hyflo и осадок на фильтре промывали гептаном (2,0 об.). Собранную органическую фазу концентрировали в вакууме при температуре 40 °С или ниже до тех пор, пока не было достигнуто 1,0 об. Смесь охлаждали до 25-30°C и прибавляли воду (2,0 об.). Смесь снова концентрировали в вакууме, чтобы удалить больше гептана. Концентрирование прекращали, когда было достигнуто менее чем 2,0 об. Прибавляли воду (5,0 об.) и смесь перемешивали в течение 2-3 ч. Твердое вещество выделяли фильтрацией и промывали водой (2,0 об.). Сушили в вакуумной печи при 40-45°C до достижения постоянной массы. 1H ЯМР (600 МГц, хлороформ-d) δ 8,52 (с, 2H)
Получение 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанона
В реакционный сосуд прибавляли ДМФА (5 л) и впоследствии порциями прибавляли трет-бутоксид калия (0,92 л, 0,8 моль, 2,6 экв.) при эффективном перемешивании. Смесь перемешивали при 20-25°C в инертной атмосфере в течение ночи.
В реакционный сосуд (№ 2) добавляли 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (1 кг, 3,16 моль, 1 экв.) и 3,4,5-трихлорпиридин (0,73 кг, 4,1 моль, 1,3 экв.) с последующим добавлением ДМФА (4,5 л). Перемешивание начинали со скоростью, обеспечивающей хорошее перемешивание реагентов.
В сосуд (№2) прибавляли раствор трет-бутоксида калия.
Когда 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон составлял < 2%, реакционную смесь охлаждали и добавляли воду (1 л) и этилацетат (10 л).
Добавляли воду (19 л), и смесь перемешивали. Смешивание прекращали, и фазам позволяли разделиться. Температуру поддерживали при 35-45 °C.
Нижнюю водную фазу отбрасывали. Добавляли этилацетат (5 л), с последующим добавлением воды (20 л). Смесь перемешивали, а после перемешивания фазам позволяли разделиться.
Нижнюю водную фазу отбрасывали. Органическую фазу нагревали до кипения при перемешивании. При кипячении с обратным холодильником в смесь загружали этанол (15 л) со скоростью, позволяющей поддерживать условия кипения. Азеотропную смесь растворителей отгоняли до достижения температуры пара 74-76 °C.
Смесь охлаждали и продукт отфильтровывали и дважды промывали холодным этанолом (2 × 2,5 л).
Продукт сушили в вакууме при 40-50°C. Выход: 45-80%. ЯМР соответствует ЯМР продукта со стадии (4) в Примере 1.
Получение 2-(3,5-дихлор-1-оксидо-пиридин-1-ий-4-ил)-1-[7-(дифторметокси)-1',1'-диоксо-спиро[1,3-бензодиоксол-2,4'-тиан]-4-ил]этанона
В реакционный сосуд помещали 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (16,1 кг, 34,8 моль, 1 экв.) и ледяную уксусную кислоту (33,8 кг). Суспензию охлаждали до 15-20 °C. К смеси порциями добавляли перуксусную кислоту (36-40%, 20,5 кг, 104,7 моль, 3,01 экв.). Смесь нагревали до 60°C и перемешивали в течение 12-24 часов.
ИПХ проводили в ацетонитриле для анализа ВЭЖХ (220 нм). Реакцию завершали, когда площадь 2-(3,5-дихлор-1-оксидо-пиридин-1-ий-4-ил)-1-[7-(дифторметокси)-1',1'-диоксо-спиро[1,3-бензодиоксол-2,4'-тиан]-4-ил]этанона была > 98,5%.
К реакционной смеси добавляли уксусную кислоту (36 кг) и метабисульфит натрия (2,415 кг, 12,7, 0,36 экв.). Смесь нагревали до 40-45°C в течение по меньшей мере 3 часов.
Уксусную кислоту удаляли выпариванием в вакууме, и затем при перемешивании добавляли этанол (386 л), и после - воду (16 л).
К смеси добавляли указанное в заголовке соединение (141 г, форма E), и смесь затем нагревали до кипения с обратным холодильником.
Твердое вещество отфильтровывали, промывали этанолом (39 л) и сушили в вакууме при 50 °С.
Выход: 80-90%). ЯМР соответствует ЯМР продукта со стадии (5) в Примере 1. Порошковая рентгеновская дифрактограмма.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ 1, 3-БЕНЗОДИОКСОЛА | 2016 |
|
RU2761337C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1, 3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2015 |
|
RU2697251C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОДИОКСОЛА ИЛИ БЕНЗОДИОКСЕПИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2011 |
|
RU2583787C2 |
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ ПРОИЗВОДНЫЕ 2,4-ДИАМИНО-1,3,5-ТРИАЗИНА ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЧЕЛОВЕКА И ЖИВОТНЫХ | 2012 |
|
RU2509770C2 |
| СОЛИ ТРИАЗОЛИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PAR1, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2009 |
|
RU2494100C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТРИАЗОЛА | 1991 |
|
RU2036194C1 |
| ТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1991 |
|
RU2114838C1 |
| ПРОИЗВОДНЫЕ ИЗОКСАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ БОРЬБЫ С СОРНЯКАМИ | 1994 |
|
RU2126394C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
Изобретение относится к способу получения соединения формулы (I), где R1 представляет собой CHF2, и Q представляет собой хлор, включающему следующие стадии: (1) введение в реакцию соединения формулы (II), где R2 выбирают из водорода, C1–6-алкила и арилалкила, и R21 представляет собой COСН3; с соединением формулы (III) в присутствии кислотного катализатора в форме глины или цеолита, с образованием соединения формулы (IV); (2a) введение в реакцию соединения формулы (IV) с алифатическим или ароматическим тиолом в присутствии гидроксида металла в подходящем растворителе с получением соединения формулы (VI); (3’) введение в реакцию соединения формулы (VI) путем добавления раствора соединения формулы (VI) и хлордифторацетата натрия в ДМФА к предварительно нагретой смеси ДМФА, воды и карбоната калия в течение продолжительного периода времени с получением соединения формулы (IX); (4) введение в реакцию соединения формулы (IX) с соединением пиридина формулы (X), где QX выбирают из хлора, брома, фтора и йода, с образованием соединения формулы (XI); и (5) окисление полученного соединения формулы (XI) с получением соединения формулы (I). Технический результат - разработан новый способ получения соединения формулы (I) с высоким выходом и чистотой, которое находит свое применение в медицине в качестве ингибиторов PDE4. 3 н. и 13 з.п. ф-лы, 1 ил., 10 пр.
1. Способ получения соединения формулы (I)
 (I),
(I),
где R1 представляет собой CHF2, и Q представляет собой хлор,
включающий каждую из стадий:
(1) введение в реакцию соединения формулы (II)
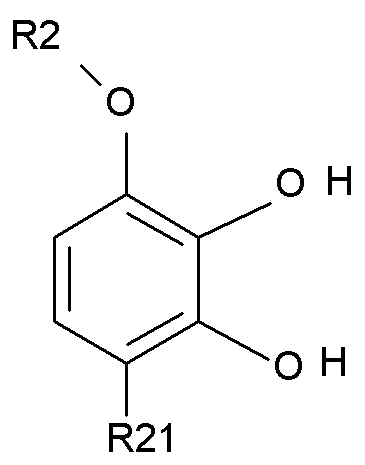 (II),
(II),
где R2 выбирают из водорода, C1–6–алкила и арилалкила, и R21 представляет собой COСН3; с соединением формулы (III)
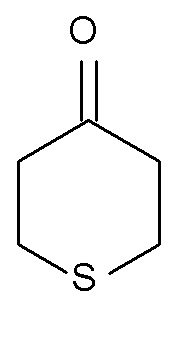 (III)
(III)
в присутствии кислотного катализатора в форме глины или цеолита, с образованием соединения формулы (IV)
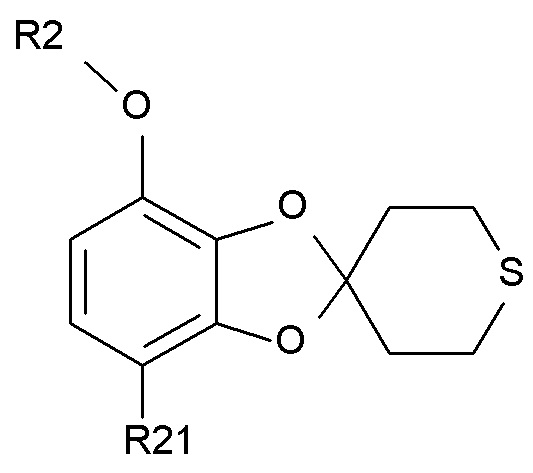 (IV),
(IV),
где R2 и R21 соответствуют определениям выше;
(2a) введение в реакцию соединения формулы (IV)
(IV),
где R2 и R21 соответствуют определениям выше, с алифатическим или ароматическим тиолом в присутствии гидроксида металла в подходящем растворителе с получением соединения формулы (VI)
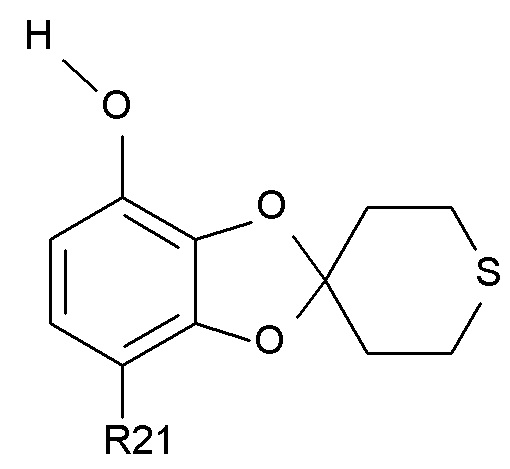 (VI),
(VI),
где R21 соответствует определению выше;
(3’) введение в реакцию соединения формулы (VI),
(VI),
где R21 соответствует определению выше, путем добавления раствора соединения формулы (VI) и хлордифторацетата натрия в ДМФА к предварительно нагретой смеси ДМФА, воды и карбоната калия в течение продолжительного периода времени с получением соединения формулы (IX),
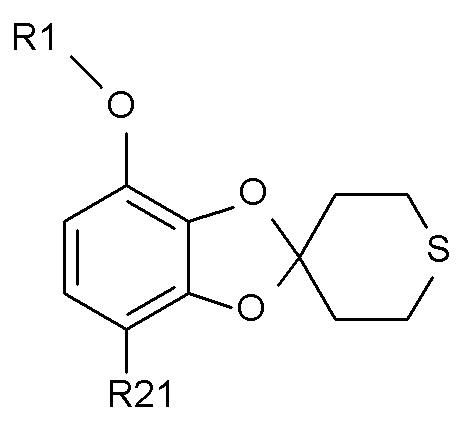 (IX),
(IX),
где R1 и R21 соответствуют определениям выше
(4) введение в реакцию соединения формулы (IX) с соединением пиридина формулы (X)
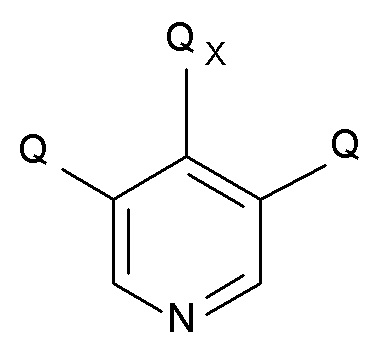 (X),
(X),
где Q соответствует определению выше, и QX выбирают из хлора, брома, фтора и йода, с образованием соединения формулы (XI);
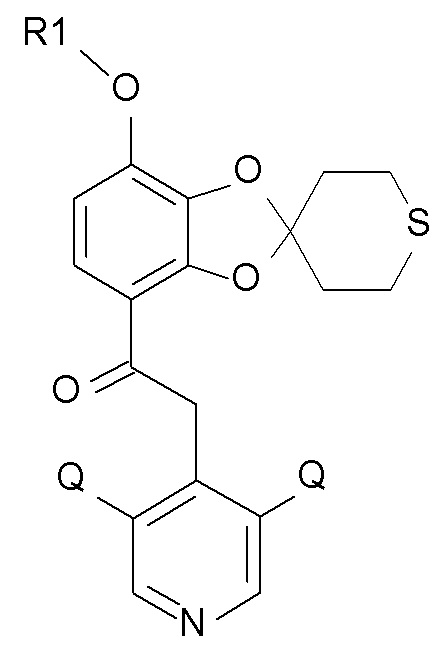 (XI),
(XI),
где R1 и Q соответствуют определениям выше; и
(5) окисление полученного соединения формулы (XI) с получением соединения формулы (I)
(I),
где R1 и Q соответствуют определениям выше.
2. Способ по п. 1, в котором гидроксид металла на стадии (2a) представляет собой NaOH.
3. Способ по п. 1 или 2, в котором реакцию на стадии (2a) проводят, используя NaOH, 1–додекантиол и этанол.
4. Способ по п. 1 или 2, в котором реакцию на стадии (2a) проводят, используя NaOH, 1–додекантиол и 1–пропанол.
5. Способ по любому одному из предшествующих пунктов, в котором на стадии (3’) соединение формулы (IX) обрабатывают ТФУ или МСК в ДМФА при повышенной температуре, с последующим удалением обработкой водным NaOH или КОН во время кристаллизации соединения формулы (IX).
6. Способ по любому одному из предшествующих пунктов, в котором реакцию на стадии (4) проводят в растворителе ДМФА/tBuOH, применяя в качестве основания трет–BuOК.
7. Способ по п. 6, в котором растворитель ДМФА/tBuOH представляет собой смесь 30/70 об./об.
8. Способ по любому одному из предшествующих пунктов, в котором на стадии (5) соединение формулы (I) кристаллизуют непосредственно из концентрированной реакционной смеси.
9. Способ по любому одному из предшествующих пунктов, в котором каждый Q и каждый QХ представляют собой хлор.
10. Способ по любому одному из предшествующих пунктов, в котором на стадии (1) кислотный катализатор представляет собой силикатный материал монтмориллонит K10.
11. Способ получения соединения формулы (I)
(I),
где R1 представляет собой CHF2, и Q представляет собой хлор, включающий каждую из стадий: (2a), (3’) и (4), и последующее окисление полученного соединения, где
стадия (2a) включает введение в реакцию соединения формулы (IV)
(IV),
с алифатическим или ароматическим тиолом в присутствии гидроксида металла в подходящем растворителе с получением соединения формулы (VI)
(VI),
где R2 выбирают из водорода, C1-6-алкила и арилалкила, и R21 представляет собой COCH3;
стадия (3’) включает введение в реакцию соединения формулы (VI)
(VI),
где R21 соответствует определению выше, путем добавления раствора соединения формулы (VI) и хлордифторацетата натрия в ДМФА к предварительно нагретой смеси ДМФА, воды и карбоната калия в течение продолжительного периода времени с получением соединения формулы (IX)
(IX),
где R1 и R21 соответствуют определениям выше;
стадия (4) включает введение в реакцию соединения формулы (IX) с соединением пиридина формулы (X)
(X),
где Q соответствует определению выше, и QX выбирают из хлора, брома, фтора и йода, с образованием соединения формулы (XI)
(XI),
где R1 и Q соответствуют определениям выше.
12. Способ получения соединения формулы (I)
(I),
где R1 представляет собой CHF2, и Q представляет собой хлор, включающий стадии (3’), (4) и (5):
(3’) введение в реакцию соединения формулы (VI)
(VI),
где R21 представляет собой COCH3, путем добавления раствора соединения формулы (VI) и хлордифторацетата натрия в ДМФА к предварительно нагретой смеси ДМФА, воды и карбоната калия в течение продолжительного периода времени с получением соединения формулы (IX)
(IX),
где R1 и R21 соответствуют определениям выше;
(4) введение в реакцию соединения формулы (IX) с соединением пиридина формулы (X)
(X),
где Q представляет собой хлор, и QX выбирают из хлора, брома, фтора и йода, с образованием соединения формулы (XI)
(XI),
где R1 и Q соответствуют определениям выше; и
(5) окисление полученного соединения формулы (XI) с получением соединения формулы (I).
13. Способ по п. 11 или 12, где реакцию на стадии (4) проводят в растворителе ДМФА/tBuOH, применяя в качестве основания трет–BuOК.
14. Способ по п. 13, в котором растворитель ДМФА/tBuOH представляет собой смесь 30/70 об./об.
15. Способ по п. 11 или 12, где соединение формулы (IX) на стадии (3’) обрабатывают ТФУ или МСК в ДМФА при повышенной температуре, с последующим ее удалением обработкой водным КОН или NaOH во время кристаллизации соединения формулы (IX).
16. Способ по любому из пп. 1-15, где способ дополнительно включает получение соединения формулы (Х)
(X),
где каждый Q и QХ представляет собой хлор,
включающий каждую из стадий:
(i) хлорирование 4-пиридинола с образованием 3,5-дихлор-4-пиридинола; и
(ii) хлорирование 3,5-дихлор-4-пиридинола с образованием соединения формулы (X).
| WO 2015197534 A2, 30.12.2015 | |||
| WO 2011160632 A1, 29.12.2011 | |||
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛФЕНИЛКЕТОНЫ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2007 |
|
RU2493149C2 |
