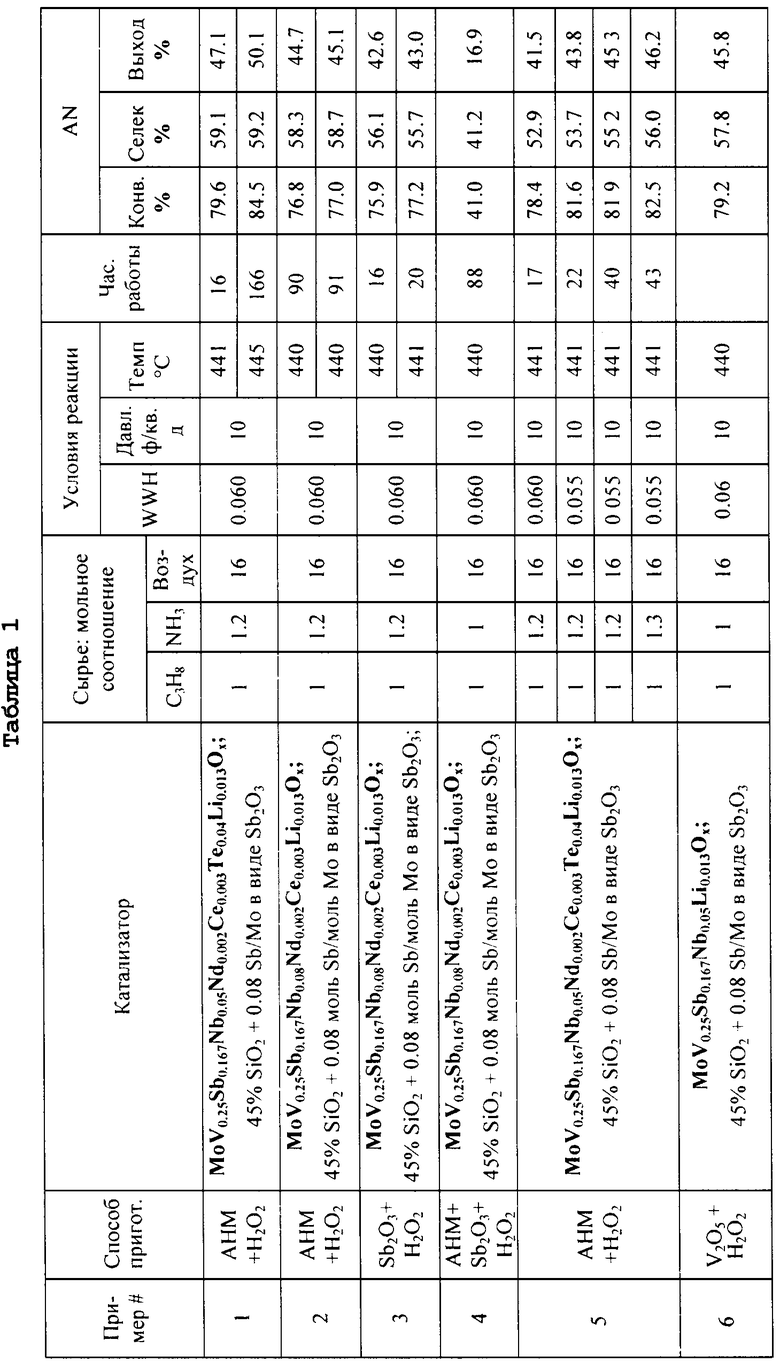
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способу приготовления твердых композиций, содержащих смешанные оксиды металлов, каталитически активные в окислительном аммонолизе или окислении низших алканов, для получения с высоким выходом ненасыщенного мононитрила или органической кислоты. Смешанные металлоксидные катализаторы по данному изобретению включают в качестве компонентов молибден (Мо), ванадий (V), сурьму (Sb), ниобий (Nb), кислород (О).
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Нитрилы типа акрилонитрила и метакрилонитрила давно получают в промышленном масштабе, т.к. они являются важными промежуточными продуктами в производстве синтетических волокон, синтетических смол, синтетических каучуков и т.п.Акрилонитрил в основном применяют в виде волокон. Акрилонитрил-бутадиен-стирольные терполимеры (ABS) представляют собой важные термопластичные структурные пластики. Нитрильные каучуки, которые впервые начали производить в Германии в 1930 г.под маркой Buna-N, являются сополимерами акрилонитрила и диена, обычно бутадиена.
В применяемых в настоящее время промышленных способах получения нитрилов, таких как акрилонитрил и метакрилонитрил, алкен, т.е. пропилен или изобутен, вводят в газофазную реакцию с аммиаком и кислородом в присутствии катализатора при высокой температуре. Обычно применяемые составы катализаторов запатентованы поставщиком катализаторов, но их технологии хорошо известны. Более того, известны способы введения наряду с исходными углеводородами дополнительных исходных веществ, таких как молекулярный кислород и/или водяной пар, газ и инертные вещества типа азота и диоксида углерода.
Ввиду большей доступности и дешевизне низших алканов по сравнению с соответствующими алкенами, например при сравнении пропана с пропиленом или изобутана с изобутиленом, большое внимание привлекли исследования, направленные на разработку усовершенствованных катализаторов получения нитрилов из более доступных низших алканов. Пропан или изобутан используют в качестве сырья в способе окислительного аммонолиза аммиаком и кислородом в газовой фазе в присутствии катализатора.
Было показано, что катализаторы, содержащие молибден, ванадий, сурьму и ниобий, эффективны в превращении пропана в акрилонитрил и изобутана в метакрилонитрил (по реакции окислительного аммонолиза), и способы приготовления указанных катализаторов описаны в многочисленных публикациях, патентах и патентных заявках. См., например, патент США №5750760, Ushikubo и др., патент США №6036880, Komada и др., патент США №6143916, Hinago и др., и патент США №6514902, Inoue и др.
Оксидные катализаторы, содержащие молибден, теллур, ванадий и ниобий, и способы приготовления указанных катализаторов описаны в патентах США №5049692, 5231214, 5281745, 5380933 и 5422328. Катализаторы, содержащие молибден, ванадий, ниобий и сурьму, также описаны, например, в патентах США 4760159, 4797381 и 7087551.
В целом способы приготовления указанных катализаторов можно разделить на две группы - гидротермальные и негидротермальные. В так называемом гидротермальном способе водную смесь ингредиентов обычно обрабатывают при повышенной температуре (например, 150-250°С) и повышенном давлении (например, 200-300 фунт/кв. дюйм), что приводит предположительно к образованию каталитически активных фаз смешанных оксидов. В негидротермальном способе водную смесь ингредиентов обычно обрабатывают при температуре ниже 100°С при атмосферном давлении с последующей сушкой и получают предшественник катализатора. Предшественник катализатора нагревают или прокаливают с образованием каталитически активных фаз. Например, в патентах США 5750760 6514902, 6610629, 7087551, 7109144, патентах ЕР 1632287, ЕР 1806178 и WO 2007/119376 раскрыты способы негидротермального приготовления катализаторов, содержащих молибден, ванадий, сурьму и ниобий. В патенте США №5750760 описан способ приготовления водного раствора предшественника оксидного катализатора эмпирической формулы MoaVbSbcXxOn (где Х представляет собой элемент, который выбирают из группы, состоящей из Mb, Та, W, Ti, Zr, Cr, Mn, Fe, Ru, Co, Rh, Ni, Pd, Pt, B, In, Ce, щелочного металла и щелочноземельного металла), включающий (1) способ добавления и смешения соединения, содержащего Мо, и соединения, содержащего элемент X, с водным раствором, содержащим компонент V и компонент Sb, с образованием водного раствора, или (2) способ добавления и смешения соединения, содержащего V, и соединения, содержащего элемент X, с водным раствором, содержащим компонент Мо и компонент Sb, с образованием водного раствора.
В патенте США 6514902 описан способ приготовления оксидного катализатора, включающего сложный оксид, содержащий Мо, V и Sb в качестве основных компонентов, который включает специфическую окислительную обработку в воде и/или спирте раствора и/или суспензии исходной смеси, содержащей соединение Мо, соединение V и соединение Sb в качестве основного источника, с использованием окисляющего газа и/или окисляющей жидкости до сушки раствора или суспензии и последующим прокаливанием. Получаемые этими способами катализаторы недостаточно селективны и не обеспечивают необходимый выход продуктов для промышленного применения.
Настоящее изобретение предлагает катализаторы селективного окисления и окислительного аммонолиза алканов и способы их приготовления.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В широком аспекте настоящее изобретение относится к способу приготовления смешанных металлоксидных катализаторов, способных ускорять окислительный аммонолиз или окисление насыщенного углеводорода в соответствующий ненасыщенный нитрил или ненасыщенную карбоновую кислоту с высоким выходом, и к способам применения этих катализаторов в экономичном превращении низших алканов. Обычно смешанные металлоксидные катализаторы по данному изобретению включают в качестве компонентов молибден (Мо), ванадий (V), сурьму (Sb) и ниобий (Nb). В одном варианте композиции по данному изобретению включают оксиды молибдена, ванадия, сурьмы, теллура, ниобия и по меньшей мере один элемент, который выбирают из группы, состоящей из лития, цезия, рубидия, титана, олова, германия, циркония, гафния, лантана, празеодима, неодима, самария, европия, гадолиния, диспрозия, гольмия, эрбия, туллия, иттербия и лютеция.
Настоящее изобретение описывает усовершенствованный способ приготовления предшественника смешанного оксидного катализатора получения акрилонитрила или метакрилонитрила из пропана или изобутана путем газофазного окислительного аммонолиза, причем указанный катализатор содержит молибден (Мо), ванадий (V), сурьму (Sb), ниобий (Mb), кислород (О), а указанный способ включает приготовление реакционной смеси из соединения молибдена, соединения ванадия, соединения сурьмы и пероксида водорода, причем предлагаемое усовершенствование включает: контактирование любого одного из соединения сурьмы, соединения молибдена и соединения ванадия с пероксидом водорода перед смешением с остальными ингредиентами, причем количество используемого пероксида водорода таково, что мольное соотношение пероксида водорода и сурьмы в катализаторе находится в интервале 0.1-5, и далее включает сушку полученной смеси с образованием твердого предшественника.
Настоящее изобретение также описывает катализатор, представляющий собой смешанный оксид эмпирической формулы:
MolVaSbbNbcTedMeXfZgOn,
где М может быть одним или несколькими щелочными металлами, которые выбирают из группы, состоящей из Li, Cs и Rb; X может быть одним или несколькими элементами из Y, Ti, Sn, Ge, Zr и Hf; и Z может быть одним или несколькими редкоземельными металлами, которые выбирают из группы, состоящей из Pr, La, Nd, Се и Ей, и где 0.1≤а≤1.0, 0.05≤b≤1.0, 0.001≤с≤1.0, 0≤d≤1.0, 0≤e≤0.1, 0≤f≤0.6, 0≤g≤0.1; и n является числом атомов кислорода, необходимых для насыщения валентности всех остальных элементов твердого предшественника при условии, что один или несколько остальных элементов в твердом предшественнике могут находиться в степени окисления, более низкой по сравнению с высшей степенью окисления, а, b, с, d, e, f и g представляют собой отношение числа молей соответствующего элемента к числу молей Мо, причем указанный катализатор приготовлен из предшественника, полученного усовершенствованным способом, описанным выше.
Для более полного понимания настоящего изобретения будут рассмотрены варианты, описанные ниже более подробно с помощью примеров.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖА
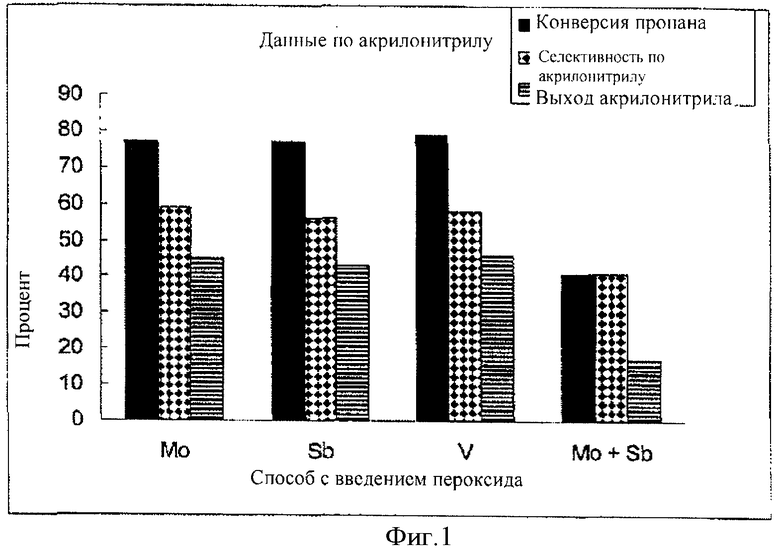
Фигура 1 схематически показывает, что настоящее изобретение предлагает повышенный выход акрилонитрила.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает усовершенствованный способ приготовления твердого предшественника смешанного оксидного катализатора получения акрилонитрила или метакрилонитрила из пропана или изобутана газофазным окислительным аммонолизом, причем указанный катализатор содержит элементы: молибден (Мо), ванадий (V), сурьму (Sb), ниобий (Nb), кислород (О), и способ включает приготовление реакционной смеси, содержащей соединение молибдена, соединение ванадия, соединение сурьмы и пероксид водорода, причем указанную реакционную смесь получают путем контактирования любого одного из соединений сурьмы, молибдена и ванадия с пероксидом водорода до смешения с исходными соединениями остальных элементов, содержащихся в смешанном оксидном катализаторе, и количество используемого пероксида водорода таково, что мольное соотношение пероксида водорода и сурьмы в катализаторе находится в интервале 0.01-20, и затем включает сушку полученной смеси с образованием твердого предшественника.
Использованный термин «исходное соединение» означает любое соединение, содержащее один или несколько элементов, входящих в состав смешанного оксидного катализатора.
В одном варианте настоящего изобретения соединение молибдена вводят в контакт с пероксидом водорода с образованием реакционной смеси Мо-пероксид водорода, и соединение ванадия смешивают с соединением сурьмы с образованием реакционной смеси V-Sb, и реакционную смесь V-Sb вводят в контакт с указанной реакционной смесью Мо-пероксид водорода с образованием объединенной смеси Mo-V-Sb. В одном варианте реакционную смесь V-Sb нагревают с обратным холодильником при температуре примерно от 80°С до примерно температуры кипения смеси в течение примерно 15-45 мин до контактирования с реакционной смесью Мо-пероксид. В другом варианте реакционную смесь V-Sb нагревают при температуре примерно от 80°С до примерно 100°С в течение примерно 15-45 мин до контактирования с реакционной смесью Мо-пероксид. В другом варианте реакционную смесь V-Sb нагревают при температуре примерно 90°С в течение 30 мин до контактирования с реакционной смесью Мо-пероксид. Использованный здесь термин «температура кипения с обратным холодильником» означает температуру, при которой реакционная смесь кипит при атмосферном давлении. Водная реакционная смесь (т.е. водные растворы исходных соединений металлов объединяют с образованием реакционной смеси) будет иметь температуру кипения с обратным холодильником примерно 100°С.
В одном варианте настоящего изобретения соединение ванадия вводят в контакт с пероксидом водорода с образованием реакционной смеси V-пероксид и затем соединение молибдена смешивают с соединением сурьмы с образованием реакционной смеси Mo-Sb, и реакционную смесь Mo-Sb вводят в контакт с указанной реакционной смесью V-пероксид с образованием объединенной смеси Mo-V-Sb. В одном варианте реакционную смесь Mo-Sb нагревают при температуре от примерно 80°С до примерно температуры кипения смеси с обратным холодильником в течение примерно 15-45 мин до контактирования с реакционной смесью V-пероксид. В одном варианте реакционную смесь Mo-Sb нагревают при температуре примерно от 80°С до примерно 100°С в течение примерно 15-45 мин до контактирования с реакционной смесью V-пероксид. В другом варианте реакционную смесь Mo-Sb нагревают при температуре примерно от 90°С в течение примерно 30 мин до контактирования с реакционной смесью V-пероксид.
В одном варианте настоящего изобретения соединение сурьмы вводят в контакт с пероксидом водорода с образованием реакционной смеси Sb-пероксид и соединение молибдена смешивают с соединением ванадия с образованием реакционной смеси Mo-V, и реакционную смесь Mo-V приводят в контакт с указанной реакционной смесью Sb-пероксид с образованием объединенной смеси Mo-V-Sb. В другом варианте реакционную смесь Mo-V нагревают при температуре примерно от 80°С до примерно 80°С в течение примерно 15-45 мин до контактирования с реакционной смесью Sb-пероксид. В одном варианте реакционную смесь Mo-V нагревают при температуре примерно от 80°С до примерно 100°С в течение примерно 15-45 мин до контактирования с реакционной смесью Sb-пероксид. В другом варианте реакционную смесь Mo-V нагревают при температуре примерно 90°С в течение примерно 30 мин до контактирования с реакционной смесью Sb-пероксид.
В другом варианте объединенную реакционную смесь Mo-V-Sb нагревают при 80°С или ниже в течение по меньшей мере примерно часа до контактирования с исходными соединениями остальных элементов, содержащихся в смешанном оксидном катализаторе. Используемое выражение «по меньшей мере в течение одного часа» означает примерно один час или более. В еще одном варианте объединенную реакционную смесь Mo-V-Sb нагревают при 70°С или выше в течение примерно двух часов до контактирования с исходными соединениями остальных элементов, содержащихся в смешанном оксидном катализаторе.
В одном варианте настоящего изобретения мольное соотношение H2O2 и Sb находится в интервале 1-2.
В другом варианте настоящего изобретения мольное соотношение H2O2 и Sb составляет 0.5, 1.0 и 1.5.
В одном варианте настоящего изобретения катализатор представляет собой смешанный оксид эмпирической формулы:
MolVaSbbNbcTedMeXfZgOn,
где М может быть одним или несколькими щелочными металлами, которые выбирают из группы, состоящей из Li, Cs и Rb; X может быть одним или несколькими элементами, которые выбирают из группы, состоящей из Y, Ti, Sn, Ge, Zr, Hf; и Z может быть одним или несколькими редкоземельными металлами, которые выбирают из группы, состоящей из Pr, La, Nd, Се и Eu; и где 0.1≤а≤1.0, 0.05≤b≤1.0, 0.001≤с≤1.0, 0≤d≤1.0 0≤e≤0.1, 0≤f≤0.6, 0≤g≤0.1; и n является числом атомов кислорода, необходимых для насыщения валентности всех остальных элементов твердого предшественника при условии, что один или несколько остальных элементов в твердом предшественнике могут находиться в степени окисления более низкой по сравнению с высшей степенью окисления, а, b, с, d, e, f и g представляют собой отношение числа молей соответствующего элемента к числу молей Мо, причем указанный катализатор приготовлен из предшественника, полученного способом по п.1.
В одном варианте настоящего изобретения компонент катализатора Х представляет собой Li.
В другом варианте настоящего изобретения компонент катализатора Z выбирают из группы, включающей Nd, Се и смесь Nd и Се.
В еще одном варианте настоящего изобретения b+d≥а. В другом варианте 0≤d≤0.06.
Настоящее изобретение включает нагревание твердого предшественника, содержащего соединения молибдена (Мо), ванадия (V), ниобия (Nb), кислорода (О), и контактирование с потоком газа при первой скорости нагрева выше примерно 15°С/мин до тех пор, пока смесь твердого предшественника не достигнет температуры предварительного прокаливания не выше 400°С.
В одном варианте настоящее изобретение включает нагревание твердого предшественника, содержащего соединения молибдена (Мо), ванадия (V), ниобия (Nb), кислорода (О), и контактирование с потоком газа при первой скорости нагревания выше примерно 20°С/мин до тех пор, пока смесь твердого предшественника не достигнет температуры предварительного прокаливания не выше 300 С, и контактирование при второй скорости нагрева выше примерно 1°С/мин до тех пор, пока температура смеси твердого предшественника не достигнет значения между 300°С и 650°С.
В одном варианте предлагается способ окислительного аммонолиза или окисления насыщенного или ненасыщенного углеводорода или смеси насыщенного и ненасыщенного углеводородов с образованием ненасыщенного нитрила или ненасыщенной органической кислоты, причем указанный способ включает: физическое смешение сухого металлоксидного катализатора и модификатора активности с образованием каталитически активной смеси, причем модификатор активности выбирают из группы, состоящей из соединений алюминия, соединений сурьмы, соединений мышьяка, соединений бора, соединений церия, соединений германия, соединений лития, соединений неодима, соединений ниобия, соединений фосфора, соединений селена, соединений тантала, соединений титана, соединений вольфрама, соединений ванадия, соединений циркония и их смесей; и контактирование насыщенного или ненасыщенного углеводорода или смеси насыщенного и ненасыщенного углеводородов с кислородсодержащим газом или кислородсодержащим газом и аммиаком в присутствии каталитической смеси, причем указанный сухой металлоксидный катализатор получают из предшественника по настоящему изобретению.
Модификатор активности по настоящему изобретению может включать оксид сурьмы(III), триоксид сурьмы (Sb2O3), оксалат сурьмы(III), тартрат сурьмы(III), оксид сурьмы(V), тетроксид сурьмы Sb6O13, оксид германия (IV), теллуровую кислоту (H6TeO6), диоксид титана (TiO2), оксид циркония (ZrO2), гидроксид лития (LiOH), оксид церия(IV) или их смесь.
В одном варианте модификатор активности составляет по меньшей мере примерно 0.01 моль на моль Мо в составе смешанного металлоксидного катализатора.
Настоящее изобретение предлагает усовершенствованный способ приготовления предшественника смешанного оксидного катализатора получения акрилонитрила или метакрилонитрила из пропана или изобутана газофазным окислительным аммонолизом, причем указанный катализатор содержит элементы: молибден (Мо), ванадий (V), сурьму (Sb), теллур (Те), ниобий (Nb), кислород (О), и указанный способ включает приготовление реакционной смеси, содержащей соединение молибдена, соединение ванадия, соединение сурьмы и пероксид водорода, причем предлагаемое усовершенствование включает: контактирование любого одного из соединений сурьмы, молибдена и ванадия с пероксидом водорода до смешения с остальными ингредиентами, причем количество используемого пероксида водорода таково, что мольное соотношение пероксида водорода и сурьмы в катализаторе находится в интервале 0.01-20.
В одном варианте настоящего изобретения количество используемого пероксида водорода таково, что мольное соотношение пероксида водорода и сурьмы в катализаторе находится в интервале 0.1-5.
В другом варианте количество используемого пероксида водорода таково, что мольное соотношение пероксида водорода и сурьмы в катализаторе находится в интервале 0.5-3.
В еще одном варианте Nb вводят в виде соединения ниобия, представляющего собой ниобиевую кислоту, кислый оксалат ниобия, аммоний-ниобий оксалат или их смесь.
В одном варианте изобретения предварительно смешивают гептамолибдат аммония (АНМ) с пероксидом водорода (H2O2). Продукт реакции метаванадата аммония (AMV) с триоксидом сурьмы (Sb2O3) добавляют к предварительно приготовленной смеси гептамолибдата аммония (АНМ) с пероксидом водорода (H2O2) и получают водную смесь (А).
Альтернативно, предварительно смешивают метаванадат аммония (AMV) с пероксидом водорода (Н2О2). Продукт реакции гептамолибдата аммония (АНМ) с триоксидом сурьмы (Sb2O3) добавляют к предварительно приготовленной смеси метаванадата аммония (AMV) с пероксидом водорода (H2O2) и получают водную смесь (А).
Альтернативно, предварительно смешивают триоксид сурьмы (Sb2O3) с пероксидом водорода (H2O2). Продукт реакции гептамолибдата аммония (АНМ) с метаванадатом аммония (AMV) добавляют к предварительно приготовленной смеси триоксида сурьмы (Sb2O3) с пероксидом водорода (H2O2) и получают водную смесь (А).
В одном варианте водную смесь (А) нагревают при перемешивании. Эту водную смесь лучше нагревать до температуры в интервале от 30°С до нормальной температуры кипения смеси. Можно нагревать с обратным холодильником с использованием оборудования, снабженного обратным холодильником. При нагревании с обратным холодильником температура кипения обычно находится в интервале примерно 101-102°С. Повышенные температуры поддерживают в течение 0.5 час или дольше. В случае низкой температуры нагрева (например, ниже 50°С) нагревать надо длительное время. При температуре нагрева в интервале 80-100°С, время нагрева составляет 1-5 час.
После прогрева к водной смеси (А) добавляют золь оксида кремния и пероксид водорода. Пероксид водорода берут в таком количестве, чтобы мольное соотношение пероксида водорода и соединения сурьмы (мольное соотношение H2O2/Sb) в расчете на сурьму находилось в интервале 0.01-20, в интервале 0.5-3, в интервале примерно 1-2.5.
После добавления пероксида водорода водную смесь (А) перемешивают при температуре в интервале 30-70°С в течение времени от 30 мин до 2 час.
Водную жидкость (В) готовят добавлением соединения ниобия (например, ниобиевой кислоты) к воде с последующим нагреванием полученной смеси до температуры в интервале от 50°С до почти 100°С. Лучше, чтобы водная жидкость (В) содержала наряду с соединением ниобия дикарбоновую кислоту (например, щавелевую кислоту). Обычно мольное соотношение дикарбоновой кислоты и соединения ниобия в расчете на ниобий находится в интервале 1-4, лучше в интервале 2-4. Т.е. в этом случае ниобиевую кислоту и щавелевую кислоту добавляют в воду, затем нагревают и перемешивают полученную смесь и получают водную жидкость (В).
Способ приготовления указанной водной жидкости (В) включает следующие стадии: (1) смешение воды, дикарбоновой кислоты (например, щавелевой кислоты) и соединения ниобия (например, ниобиевой кислоты) и получение предварительного ниобийсодержащего водного раствора или ниобийсодержащей водной смеси с суспендированной в ней частью соединения ниобия; (2) охлаждение предварительно приготовленного ниобийсодержащего водного раствора или ниобийсодержащей водной смеси и осаждение части дикарбоновой кислоты; и (3) удаление осажденной дикарбоновой кислоты из предварительно приготовленного ниобийсодержащего водного раствора или удаление осажденной дикарбоновой кислоты и суспендированного соединения ниобия из ниобийсодержащей водной смеси с образованием ниобийсодержащей водной жидкости (В). В водных жидкостях (В), полученных указанным способом, мольное соотношение дикарбоновая кислота/ниобий обычно находится в интервале примерно 2-4.
В одном варианте дикарбоновая кислота представляет собой щавелевую кислоту, а соединение ниобия на стадии (1) включает ниобиевую кислоту, кислый оксалат ниобия и аммоний-ниобий оксалат. Эти соединения ниобия можно использовать в виде твердого вещества, смеси или дисперсии в соответствующей среде. Если в качестве соединения ниобия используют кислый оксалат ниобия или аммоний-ниобий оксалат, дикарбоновую кислоту можно не добавлять. В случае, когда в качестве соединения ниобия используют ниобиевую кислоту, для удаления кислотных примесей, которыми была загрязнена ниобиевая кислота в ходе ее получения, ниобиевую кислоту перед использованием можно промыть водным раствором аммиака и/или водой. В одном варианте в качестве соединения ниобия используют свежеприготовленное соединение ниобия. Однако в указанном способе можно использовать соединение ниобия с измененными свойствами (например, дегидратированное) в результате длительного хранения и т.п. На стадии (1) данного способа растворение соединения ниобия можно ускорить, добавляя небольшое количество водного аммиака или нагревая.
Концентрацию соединения ниобия (в расчете на ниобий) в предварительно приготовленном ниобийсодержащем водном растворе или в водной смеси поддерживают в интервале 0.2-0.8 моль/кг раствора или смеси. В одном варианте используют дикарбоновую кислоту в таком количестве, чтобы мольное соотношение дикарбоновой кислоты и соединения ниобия в расчете на ниобий составляло примерно 3-6. При использовании избытка дикарбоновой кислоты в водном растворе дикарбоновой кислоты можно растворить большое количество соединения ниобия; однако недостатком является то, что количество дикарбоновой кислоты, которое осаждается при охлаждении полученного предварительного ниобийсодержащего водного раствора или смеси, становится слишком большим, что уменьшает количество использованной дикарбоновой кислоты. С другой стороны, при использовании недостаточного количества дикарбоновой кислоты может проявиться другой недостаток, обусловленный тем, что большое количество соединения ниобия может остаться не растворенным и суспендированным в водном растворе дикарбоновой кислоты с образованием смеси, причем суспендированное соединение ниобия удаляют из водной смеси, что уменьшает степень его использования.
На стадии (2) можно применять любой способ охлаждения. Например, можно охлаждать просто на ледяной бане.
Осажденную дикарбоновую кислоту (или осажденную дикарбоновую кислоту и диспергированное соединение ниобия) на стадии (3) можно легко отделить традиционными способами, например, декантацией или фильтрацией.
В случае, когда мольное соотношение дикарбоновая кислота/ниобий в полученном ниобийсодержащем водном растворе находится за пределами интервала примерно 2-4, к водной жидкости (В) можно добавить либо соединение ниобия, либо дикарбоновую кислоту, с тем чтобы мольное соотношение дикарбоновая кислота/ниобий в растворе попало в указанный интервал.
Однако обычно такая операция не нужна, т.к. водную жидкость (В) с мольным соотношением дикарбоновая кислота/ниобий в интервале 2-4 можно приготовить, регулируя концентрацию соединения ниобия, соотношение дикарбоновой кислоты и ниобия и температуру охлаждения указанного выше предварительного ниобийсодержащего водного раствора или водной смеси.
Для приготовления водной жидкости (В) можно также использовать другие компоненты. Например, по меньшей мере часть водной жидкости (В), содержащей соединение ниобия или смесь соединения ниобия с дикарбоновой кислотой, используют вместе с пероксидом водорода. В этом случае оптимальное количество пероксида водорода подбирают таким, чтобы мольное соотношение пероксида водорода и соединения ниобия (мольное соотношение H2O2/Nb) в расчете на ниобий находилось в интервале 0.5-20 или 1-20.
В другом примере по меньшей мере часть водной жидкости (В), содержащей соединение ниобия или смесь соединения ниобия и дикарбоновой кислоты или их смесь с пероксидом водорода, содержит также соединение сурьмы (например, триоксид сурьмы), соединение титана (например, диоксид титана, который может быть смесью рутила и анатаза) и/или соединение церия (например, ацетат церия). В этом случае количество пероксида водорода таково, что мольное соотношение пероксида водорода и соединения ниобия (мольное соотношение H2O2/Nb) в расчете на ниобий находится в интервале 0.5-20, от 1 до 20. В другом примере количество соединения сурьмы, смешанного по меньшей мере с частью водной жидкости (В) и пероксида водорода, таково, что мольное соотношение (мольное соотношение Sb/Nb) соединения сурьмы в расчете на сурьму и соединения ниобия в расчете на ниобий составляло не более 5 и находилось в интервале 0.01-2.
Водную смесь (А) и водную жидкость (В) смешивают в нужном соотношении для получения заданного состава катализатора с образованием водной смеси ингредиентов, обычно в виде суспензии. Содержание ингредиентов в водной смеси обычно находится в интервале выше примерно 50 масс.%, от 70 до 95 масс.%, от 75 до 90 масс.%.
Для приготовления нанесенного на оксид кремния катализатора по настоящему изобретению водную смесь исходных веществ готовят так, чтобы она содержала источник оксида кремния (в частности золь оксида кремния или плавленый оксид кремния). Количество источника оксида кремния устанавливают в зависимости от количества носителя - оксида кремния в катализаторе, который надо приготовить. Для получения сухого предшественника катализатора водную смесь высушивают. Сушку можно осуществлять традиционными способами, например, распылительной сушкой или испарением. Особенно пригодна распылительная сушка, т.к. в результате получают мелкие сферические частицы сухого предшественника катализатора. Распылительную сушку можно проводить центрифугированием, продувкой через сопло двухфазного потока или продувкой через сопло при высоком давлении. В качестве источника тепла для сушки в одном варианте можно использовать воздух, нагретый водяным паром, электронагреватель и т.п. В одном из вариантов температура распылительной сушилки на входе в зону сушки составляет 150-300°С.
Далее изобретение включает нагревание твердого предшественника, содержащего соединения молибдена (Мо), ванадия (V), сурьмы (Sb), ниобия (Nb) и кислорода (О), контактирование с потоком газа при первой скорости нагрева выше примерно 15°С/мин до тех пор, пока смесь температура твердого предшественника не станет равной температуре предварительного прокаливания - не выше 400°С. В одном варианте настоящее изобретение включает способ приготовления смешанного оксидного катализатора синтеза акрилонитрила или метакрилонитрила из пропана или изобутана окислительным аммонолизом в газовой фазе, содержащего элементы: молибден (Мо), ванадий (V), сурьму (Sb), теллур (Те), ниобий (Nb) и кислород (О), путем нагревания твердой смеси предшественника, содержащей соединения молибдена (Мо), ванадия (V), сурьмы (Sb), теллура (Те), ниобия (Nb) и кислорода (О), и контактирования с потоком газа при первой скорости нагрева выше примерно 15°С/мин до тех пор, пока температура смеси твердого предшественника не станет равной температуре предварительного прокаливания - не выше 400°С.
Один вариант приготовления смешанного оксидного катализатора включает нагревание твердой смеси предшественника при второй скорости нагрева выше примерно 0.5°С/мин, до тех пор, пока температура твердой смеси предшественника не достигнет значения примерно 590-680°С. В настоящем изобретении возможна вторая скорость нагрева выше примерно 1°С/мин, 2°С/мин или 5°С/мин. Кроме того, нагревание со второй скоростью проводят в атмосфере, практически не содержащей кислорода. Кроме того, твердую смесь предшественника выдерживают при температуре примерно 590-680°С в течение примерно двух (2) часов.
Способ прокаливания по настоящему изобретению включает применение инертного газа. Инертный газ может быть благородным газом. Инертный газ может представлять собой азот. В качестве такого газа можно использовать воздух, водяной пар, перегретый пар, монооксид углерода и диоксид углерода. В том случае, когда для приготовления твердой смеси предшественника предварительно смешивают соединение ванадия и пероксид водорода, предпочтительным способом прокаливания является предварительное прокаливание на воздухе.
Газ можно продувать со скоростью примерно 1.33-1.67 см3/г/мин. Скорость потока газа зависит от размера реактора. В одном варианте первая скорость нагрева составляет более примерно 20°С/мин.
На стадии прокаливания сухой предшественник катализатора превращают в смешанный металлоксидный катализатор. Прокаливание можно проводить в барабанной печи, в печи с кипящим слоем, в реакторе с кипящим слоем, в реакторе с неподвижным слоем и т.п. Условия прокаливания выбирают такими, чтобы полученный катализатор имел удельную поверхность от примерно 5 м2/г до примерно 35 м2/г или от примерно 15 м2/г до примерно 20 м2/г.
Прокаливание включает нагревание сухого предшественника катализатора до конечной температуры в интервале примерно 550-680°С.
В настоящем изобретении способ прокаливания включает непрерывное или импульсное нагревание сухого предшественника катализатора от температуры ниже 200°С до температуры предварительного прокаливания не выше примерно 400°С, не выше примерно 350°С, не выше примерно 300°С, со скоростью выше 15°С/мин. В одном варианте температура предварительного прокаливания равна 300°С. В другом варианте скорость нагрева составляет примерно 20°С/мин. В еще одном варианте скорость нагрева равна 25°С/мин. В следующем варианте скорость нагрева составляет 30°С/мин. В еще одном варианте сухой предшественник катализатора вводят в горячий аппарат прокаливания с температурой примерно 300°С или несколько выше, с тем чтобы температура предшественника быстро достигла величины примерно 300°С.
Скорость нагревания от температуры предварительного прокаливания до конечной температуры может составлять примерно 0.5°С/мин, 1°С/мин, 2°С/мин или 5°С/ мин или может быть любой в интервале 0.5-5°С/мин. В одном варианте скорость нагрева в температурном интервале от примерно 300°С до промежуточной температуры составляет примерно 1°С/мин и от промежуточной температуры до конечной температуры скорость нагрева равна или превышает 15°С/мин или равна 20°С/мин и выше, или равна 25°С/мин и выше, или равна 30°С/мин. В другом варианте твердое вещество можно охладить после достижения промежуточной температуры и затем нагреть до конечной температуры со скоростью выше примерно 15°С/мин и выше или равной 20°С/мин и выше, или равной 25°С/мин и выше, или равной 30°С/мин.
В одном варианте изобретения прокаливание проводят в две стадии: (1) до промежуточной или предварительной температуры прокаливания и (2) от промежуточной или предварительной температуры до конечной температуры. В одном варианте твердое вещество со стадии прокаливания (1), необязательно охлажденное, вводят в горячий аппарат прокаливания с температурой, почти равной конечной температуре, с тем чтобы температура предшественника быстро достигла конечной температуры.
В другом варианте скорость нагрева в температурном интервале от примерно 300°С до примерно 340-350°С, 345°С составляет примерно 0.5°С/мин или 1°С/мин или примерно 2°С/мин или примерно 5°С/мин или может быть любой в интервале 0.5-5°С/мин. В одном варианте твердое вещество выдерживают при температуре в интервале 300-400°С, в интервале 340-350°С, при 345°С в течение примерно 1-4 час. В другом варианте твердое вещество нагревают со скоростью 2.45°С/мин в интервале температур 345-680°С.
По достижении конечной температуры твердое вещество выдерживают при этой температуре в течение примерно 1-3 час, примерно 2 час.
Конечная температура может быть равной 550°С, 560°С, 570°С, 580°С, 590°С, 600°С, 610°С, 620°С, 630°С, 640°С, 650°С, 660°С, 670°С и 680°С или любой величине в интервале 550-680°С. В одном варианте твердое вещество нагревают со скоростью 0.5°С/мин от примерно 600°С до примерно 680°С. В другом варианте твердое вещество нагревают со скоростью 1°С/мин от примерно 600°С до примерно 680°С.
Прокаливать можно на воздухе или в токе воздуха. Однако по меньшей мере часть операции прокаливания проводят в атмосфере газа (например, в токе газа), такого как азот, практически не содержащий кислорода. Настоящее изобретение предполагает инертный газ. Инертный газ может представлять собой благородный газ. Инертный газ может представлять собой азот. Таким газом может быть воздух, водяной пар, перегретый водяной пар, монооксид углерода и диоксид углерода. В одном варианте настоящего изобретения прокаливание можно проводить в токе азота, практически не содержащего кислорода, в обоих температурных интервалах: (1) до примерно 400-450°С и (2) выше примерно 400-450°С. В другом варианте настоящего изобретения прокаливание проводят в токе воздуха в температурном интервале (1) до примерно 400-450°С и в токе азота, практически не содержащего кислорода, в температурном интервале (2) выше примерно 400-450°С. Скорость потока газа является критичной особенно в температурном интервале (1) до примерно 400-450°С, Скорость потока газа может находиться в интервале от примерно 0.67 до примерно 2.5 см3 на г предшественника катализатора в минуту.
В одном варианте смешанный оксидный катализатор включает элементы: молибден (Мо), ванадий (V), ниобий (Nb), сурьму (Sb) и кислород (О). Кроме того, в другом варианте смешанный оксидный катализатор содержит элементы: молибден (Мо), ванадий (V), ниобий (Nb), сурьму (Sb), теллур (Те) и кислород (О).
Кроме того, в одном варианте твердая смесь предшественника по настоящему изобретению включает элементы: молибден (Мо), ванадий (V), ниобий (Nb), сурьму (Sb) и кислород (О). В другом варианте твердая смесь предшественника по настоящему изобретению включает элементы: молибден (Мо), ванадий (V), ниобий (Nb), сурьму (Sb), теллур (Те) и кислород (О).
В одном варианте изобретения смешанный оксидный катализатор окислительного аммонолиза смешивают с модификатором активности в твердом состоянии, который выбирают из группы, состоящей из соединений алюминия, соединений сурьмы, соединений мышьяка, соединений бора, соединений церия, соединений германия, соединений лития, соединений неодима, соединений ниобия, соединений фосфора, соединений селена, соединений тантала, соединений теллура, соединений титана, соединений вольфрама, соединений ванадия, соединений циркония и их смесей.
В одном варианте изобретения смешанный оксидный катализатор смешивают с твердым соединением, который выбирают из группы, состоящей из триоксида сурьмы (Sb2O3), теллуровой кислоты (Н6ТеО6), диоксида титана (TiO2) и оксида циркония (ZrO2).
В одном варианте твердый предшественник отвечает эмпирической формуле:
MolVaSbbTecNbdOn,
где 0.1≤а≤1.0, 0≤b≤1.0, 0≤с≤1.0, 0.001≤d≤0.25; n является числом атомов кислорода, необходимых для насыщения валентности всех других элементов твердого предшественника при условии, что один или несколько других элементов в твердом предшественнике могут находиться в степени окисления более низкой по сравнению с высшей степенью окисления, а, b, с и d представляют собой отношение числа молей соответствующего элемента к числу молей Мо.
В другом варианте твердый предшественник отвечает эмпирической формуле:
MolVaSbbTecNbdOn,
где 0.1≤а≤1.0, 0.05≤b≤3.0, 0.001≤с≤1.0, 0≤d≤1.0, b+с≥a; n является числом атомов кислорода, необходимых для насыщения валентности всех других элементов твердого предшественника при условии, что один или несколько других элементов в твердом предшественнике могут находиться в степени окисления более низкой по сравнению с высшей степенью окисления, а, b, с и d представляют собой отношение числа молей соответствующего элемента к числу молей Мо.
Настоящее изобретение предлагает способ приготовления смешанного оксидного катализатора получения акрилонитрила или метакрилонитрила из пропана или изобутана окислительным аммонолизом в газовой фазе, содержащего элементы: молибден (Мо), ванадий (V), сурьму (Sb), ниобий (Nb) и кислород (О), причем способ включает нагревание твердой смеси предшественника, содержащего соединения молибдена (Мо), ванадия (V), сурьмы (Sb), ниобия (Nb) и кислорода (О), контактирование с потоком газа при первой скорости нагрева выше примерно 15°С/мин до тех пор, пока твердая смесь предшественника не достигнет температуры не выше 400°С, затем контактирование твердой смеси предшественника с горячей зоной при температуре выше примерно 100°С. Настоящее изобретение предлагает вариант, в котором твердая смесь предшественника контактирует с потоком газа при температуре горячей зоны выше примерно 100°С, выше примерно 200°С, выше примерно 300°С или выше примерно 400°С до стадии со второй скоростью нагрева. В одном варианте настоящее изобретение предлагает способ приготовления смешанного оксидного катализатора для получения акрилонитрила или метакрилонитрила из пропана или изобутана окислительным аммонолизом в газовой фазе, содержащего элементы: молибден (Мо), ванадий (V), сурьму (Sb), теллур (Те), ниобий (Nb) и кислород (О), контактирование с потоком газа при первой скорости нагрева выше примерно 15°С/мин до тех пор, пока твердая смесь предшественника не достигнет температуры не выше 400°С, затем контактирование твердой смеси предшественника с горячей зоной при температуре выше примерно 100°С.
В одном варианте твердую смесь предшественника нагревают в интервале температур 100-250°С в течение времени не более 7.5 мин, 10 мин, 15 мин или 30 мин.
Температура предварительного прокаливания в настоящем изобретении составляет не выше 400°С, 350°С или 300°С.
Катализатор по настоящему изобретению можно использовать либо нанесенным, либо ненанесенным (т.е. катализатор может содержать носитель). Подходящие носители включают оксид кремния, оксид алюминия, оксид циркония, оксид титана или их смеси. Однако при использовании в качестве носителей оксидов циркония или титана соотношение молибдена и циркония или титана увеличивается по сравнению с значениями в приведенных формулах, и соотношение Мо и Zr или Ti составляет примерно 1-10. Обычно носитель служит для катализатора связующим, который позволяет получить более твердый катализатор с повышенной прочностью к истиранию. Однако в промышленности соответствующая смесь активной фазы (т.е. комплекса описанных выше каталитически активных оксидов) и носителя должна способствовать получению приемлемой активности и твердости (прочности к истиранию) катализатора. Носитель составляет примерно 10-90 масс.% нанесенного катализатора. Обычно носитель составляет примерно 40-60 масс.% нанесенного катализатора. В одном варианте настоящего изобретения носитель составляет всего примерно 10 масс.% нанесенного катализатора. В другом варианте данного изобретения носитель составляет всего примерно 30 масс.% нанесенного катализатора. В еще одном варианте этого изобретения носитель составляет до примерно 70 масс.% нанесенного катализатора. Промышленные носители содержат один или несколько промоторов, и эти промоторы вводят в катализатор вместе с носителем.
Изобретение предполагает непрерывные способы выделения и очистки ценных органических продуктов из горячих газовых смесей, образующихся при каталитическом окислительном аммонолизе легких алканов. Более конкретно, данное изобретение относится к выделению и очистке ценных азотсодержащих органических соединений, образующихся при каталитическом окислении по меньшей мере одного соединения в исходном сырье, которое выбирают из группы, состоящей из пропана и изобутана, в присутствии аммиака и кислорода, с образованием отходящего газового потока из реактора, содержащего соответствующий ненасыщенный мононитрил.
Пропан превращают в акрилонитрил, а изобутан в метакрилонитрил в присутствии одного или нескольких из указанных выше катализаторов в газофазном проточном реакторе путем контактирования катализатора с пропаном или изобутаном в присутствии кислорода (например, поступившего в зону реакции в потоке сырья, содержащего кислородсодержащий газ, например, воздух) и аммиака в условиях реакции, эффективных для получения акрилонитрила или метакрилонитрила. Поток сырья для этой реакции содержит пропан или изобутан, кислородсодержащий газ, такой как воздух, и аммиак при следующих мольных соотношениях: пропана или изобутана и кислорода в интервале примерно 0.1-10, примерно 0.125-5, примерно 0.25-2.5, и пропана или изобутана и аммиака в интервале примерно 0.2-20, примерно 0.3-2.5, примерно 0.5-2.0. Поток сырья может также содержать один или несколько дополнительных компонентов сырья, включая полученный акрилонитрил или метакрилонитрил (например, из потока рецикла или с предшествующей стадии многостадийной системы реакторов) и водяной пар. Например, это могут примерно 5-30 масс.% относительно всего потока сырья или в молях относительно количества пропана или изобутана в потоке сырья. В одном варианте описанный катализатор используют для окислительного аммонолиза пропана в акрилонитрил в однопроходном способе, т.е. работающем без рецикла выделенного, но непрореагировавшего сырья.
Конкретная конструкция газофазного проточного реактора не является строго критичной. Следовательно, газофазный проточный реактор может быть реактором с неподвижным слоем, реактором с кипящим слоем или реактором другого типа. Реактор может быть одиночным или может быть частью многостадийной системы реакторов. В одном или нескольких вариантах реактор содержит один или несколько входов для подачи потока исходных реагентов в зону реакции реактора, содержащую смешанный металлоксидный катализатор, и выход для выгрузки продуктов реакции и непрореагировавших реагентов.
Условия реакции регулируют таким образом, чтобы обеспечить эффективное превращение пропана в акрилонитрил или изобутана в метакрилонитрил. Обычно реакция протекает в интервале температур примерно 300-550°С, в одном варианте примерно 325-500°С, в некоторых вариантах примерно 350-450°С, а в других вариантах примерно 430-520°С.
Обычно скорость потока сырья, содержащего пропан или изобутан, через зону реакции в газофазном проточном реакторе можно регулировать таким образом, чтобы массовая часовая объемная скорость (WHSV) была в интервале примерно 0.02-5, в некоторых вариантах примерно 0.05-1 и в других вариантах примерно 0.1-0.5, в каждом случае, например, в граммах пропана или изобутана на грамм катализатора.
Давление в зоне реакции можно регулировать в интервале от примерно 0 фунт/кв. дюйм до примерно 200 фунт/кв. дюйм, в одном варианте от примерно 0 фунт/кв. дюйм до примерно 100 фунт/кв. дюйм, от примерно 0 фут/кв. дюйм до примерно 50 фут/кв. дюйм от примерно 0 фунт/кв. дюйм до примерно 20 фунт/кв. дюйм.
Полученные акрилонитрил или метакрилонитрил можно при необходимости отделить от других побочных продуктов и от непрореагировавших реагентов способами, известными специалистам в данной области техники. Полученные акрилонитрил или метакрилонитрил можно при необходимости отделить от других побочных продуктов или от непрореагировавших реагентов способами, известными в данной области.
Описанные здесь катализаторы окислительного аммонолиза пропана за один проход (т.е. без рецикла) способны образовать акрилонитрил наряду с COX (диоксид углерода + монооксид углерода), синильной кислотой (HCN) и ацетонитрилом или метилцианидом (CH3CN). Отходящие из реактора газы могут содержать непрореагировавший углеводород (пропан или изобутан), кислород (О2), аммиак (NH3) и унесенные мелкие частицы катализатора.
Способы выделения и очистки продуктов реакции включают охлаждение отходящего газового потока из реактора с помощью водной гасящей жидкости; образование водного раствора, содержащего соответствующий ненасыщенный мононитрил, синильную кислоту и другие органические дополнительные продукты, и последующие процессы дистилляции и разделения фаз для выделения водной жидкости на рецикл и получения ценных азотсодержащих органических соединений и синильной кислоты.
Смешение пропана, аммиака и кислорода в реакторе и окисление пропилена в присутствии аммиака протекают на поверхности частиц катализатора в кипящем слое. В результате сложных экзотермических реакций образуются следующие продукты: акрилонитрил, синильная кислота, диоксид углерода, монооксид углерода, ацетонитрил, акролеин, акриловая кислота, вода, другие высшие нитриты, альдегиды, кетоны, уксусная кислота и различные неизвестные органические соединения. Конверсия трех исходных реагентов обычно бывает менее 100%, так что в отходящем газе реактора содержатся непрореагировавший пропан, аммиак, кислород и азот. Источник пропана обычно содержит небольшое количество пропилена и некоторых более тяжелых углеводородов, основная часть которых отводится непрореагировавшими. Часть тепла экзотермической реакции отводят с помощью ряда паровых змеевиковых теплообменников, которые генерируют и перегревают паровые стоки при давлении примерно 600 фунт/кв. дюйм для нужд процесса, таких как подача тепла для дистилляции в секции выделения продуктов и очистки. Отходящий из реактора газ проходит через циклоны, в которых из газа удаляются мелкие частицы катализатора. Затем поток отходящего из реактора газа дополнительно охлаждают в холодильнике, включающем оболочку и трубчатый теплообменник, с использованием подпиточной воды в качестве источника холода.
Как хорошо известно в данной области, активность катализаторов окисления является важным фактором, возможно наиболее важным фактором, в экономике данного и других процессов окисления. Активность катализаторов определяется конверсией реагентов, селективностью, т.е. конверсией реагента в целевой продукт, скоростью образования целевого продукта на единицу объема реактора в единицу времени и сроком службы катализатора, т.е. эффективным временем работы до снижения активности или селективности.
Факторы, от которых зависит активность катализатора, включают состав, способы приготовления, носитель и условия прокаливания. Помимо требований к химическому составу, другие определяющие свойства включают величину поверхности, пористость, плотность, распределение пор по размерам, твердость, прочность и устойчивость к механическому истиранию, особенно для катализаторов в кипящем слое.
Обычно окислительный аммонолиз проводят в реакторе с кипящим слоем. Для достижения высокой конверсии алканов время контакта в однопроходной системе должно составлять несколько секунд. Необязательными дополнительными продуктами являются ацетонитрил и синильная кислота, которые можно выделить в промышленных условиях. В кипящий слой частиц катализатора вводят почти стехиометрические количества пропана, аммиака и кислорода. Рабочие условия включают давление в интервале примерно 3-35 фунт/кв. дюйм (20.7-241.4 кПа по манометру), примерно 5-25 фунт/кв. дюйм (34.5-172.4 кПа по манометру). Обычно температура находится в интервале примерно 700-1000°F (371-538°С), в интервале примерно 750-950°F (399-510°C). Для регулирования температуры тепло реакции отводят путем генерирования водяного пара при температурах примерно 300-500°С и повышенном давлении.
Для иллюстрации настоящего изобретения были приготовлены образцы катализатора и затем испытаны в сходных условиях реакции. Приведенные ниже составы являются номинальными в расчете на все металлы, взятые для приготовления катализатора. Поскольку некоторые металлы могут быть потеряны или не могут полностью вступить в реакцию в ходе приготовления катализатора, реальный состав конечного катализатора может слегка отличаться от приведенных ниже номинальных составов.
Испытание катализаторов Катализатор испытывали в лабораторном реакторе с кипящим слоем объемом 40 см3 и диаметром 1 дюйм. В реактор загружали примерно 20-45 г частиц катализатора или каталитической смеси. Пропан подавали в реактор со скоростью 0.04-0.15 WWH (т.е. грамм пропана/грамм катализатора в час). Давление в реакторе поддерживали в интервале примерно 2-15 фунт/кв. дюйм. Температура реакции была в интервале примерно 420-460°С. Обычно аммиак подавали в реактор с такой скоростью, чтобы соотношение аммиака и пропана составляло примерно 1-1.5. Кислород подавали в реактор с такой скоростью, чтобы соотношение кислорода и пропана было равным примерно 3.4. Азот подавали в реактор с такой скоростью, чтобы соотношение азота и пропана составляло примерно 12.6.
Использованный термин «примерно» при указании любого количества относится к изменению количества в общепринятых реальных условиях получения катализатора или предшественников катализатора, например, в лаборатории, на пилотной или на промышленной установке. Например, количество ингредиента в смеси со словом «примерно» включает вариации и степень точности, обычно используемые при работе на промышленных или лабораторных установках получения катализаторов или предшественников катализаторов. Например, количество компонента продукта со словом «примерно» включает вариации между порциями катализатора, полученными на промышленной установке или в лаборатории получения катализаторов или предшественников катализаторов, и вариации, присущие аналитическим методам. Независимо от того, имеют ли количества добавку «примерно», количества включают эквиваленты этих величин. В настоящем изобретении можно использовать любые приведенные количества, снабженные словом «примерно», а также количества без добавки «примерно».
ПРИМЕРЫ
Пример 1 Способ с пероксидом и Мо.
Mo1.0V0.25Sb0.167Nb0.05Nd0.002Се0.003Те0.04Li0.013Ох
В 5-галлонном реакционном контейнере приготовили реакционный раствор А1 путем (i) добавления гептамолибдата аммония (2343 г) к 7537 мл деионизированной воды и затем (ii) добавления по каплям при перемешивании пероксида водорода (30 масс.%, 360 г) в течение 15 мин.
Реакционный раствор А2 приготовили аналогично реакционной смеси А1.
Реакционную смесь В приготовили в 20-галонном реакторе. Сначала загрузили 20159 г деионизированной воды и нагрели ее до 90°С. Затем добавили при перемешивании метаванадат аммония (776 г), поддерживая температуру 90°С. Затем добавили 619 г Sb2O3. В полученной смеси протекала реакция при 90°С в течение часа при перемешивании, в результате чего образовалась реакционная смесь В.
Реакционный раствор С приготовили путем растворения 835 г аммоний-ниобий оксалата в 2170 г деионизированной воды при 50°С, и затем перемешивали раствор в течение 15 мин при 50°С.
Затем реакционные растворы А1 и А2 последовательно добавили к реакционной смеси В при перемешивании. Перемешанную объединенную реакционную смесь выдержали при 90°С еще в течение часа.
Объединенную реакционную смесь охладили до 70°С. Затем к объединенной реакционной смеси добавили золь оксида кремния (Nalco, 32.5 масс.% SiO2, 9206 г). Объединенную реакционную смесь продолжали перемешивать при 70°С в течение еще 30 мин.
Затем объединенную реакционную смесь охладили до 50°С. Далее к объединенной реакционной смеси добавили при перемешивании реакционный раствор С.
К полученной смеси добавили дисперсию 1496 г плавленого оксида кремния в 13464 г деионизированной воды и затем добавили Се(ООССН3)3·1.5H2O (27.4 г), Nd(ООССН3)3·H2O (18.0 г), Те(ОН)6 (243.8 г) и LiOH·H2O (14.5 г).
Полученную реакционную смесь затем высушили в распылительной сушилке фирмы Bowen. Температуры входа и выхода из распылительной сушилки составляли 325 и 125°С соответственно при давлении в сопле 25 фунт/кв. дюйм.
Часть (550 г) высушенного в распылительной сушилке вещества затем прокалили во вращающейся обжиговой печи (стеклянная трубка диаметром 3'') в токе азота (500 см3/мин). Процедура прокаливания включала нагревание со скоростью 20°С/мин до 300°С и затем со скоростью 1°С/мин до 630°С. Температуру 630°С поддерживали 2 час и затем охладили до комнатной температуры.
Результаты окислительного аммонолиза:
Сырье: О2 3.39/С3 1.0/NH3 1.20/N2 12.61, (16 воздух) 10 фунт/кв. дюйм.
Пример 2 Способ с пероксидом и молибденом
Mo1.0V0.25Sb0.167Nb0.08Nd0.002Ce0.003Li0.013Ox
Реакционную смесь А приготовили, добавляя гептамолибдат аммония (189.1 г) к 500 мл деионизированной воды, и затем добавили по каплям пероксид водорода (30 масс.%, 30.4 г) при перемешивании в течение 15 мин.
Реакционную смесь В приготовили путем (i) добавления метаванадата аммония (31.3 г) к 400 см3 воды, (ii) нагревания раствора при перемешивании до 90°С, затем (iii) добавления оксида сурьмы (Sb2O3, 26.1 г) и затем (iv) реакции при перемешивании при 90°С в течение часа.
Реакционную смесь А затем добавили к реакционной смеси В при перемешивании и объединенную смесь нагрели до 90°С при перемешивании. Затем объединенную реакционную смесь нагревали при 90°С при перемешивании еще час.
Объединенную реакционную смесь охладили до 70°С. Затем добавили золь оксида кремния (Nalco, 369 г, 32.5% оксида кремния). Реакционную смесь продолжали перемешивать при 70°С еще в течение 30 мин.
Объединенную реакционную смесь охладили до 50°С. К объединенной реакционной смеси добавили при перемешивании раствор оксалата ниобия (112.0 г, 0.765 моль Nb/кг раствора). Затем добавили смесь 60.0 г плавленого оксида кремния в 900 мл деионизированной воды. Далее добавили Се(ООССН3)3·1.5SH2O (1.11 г), Nd (ООССН3)3·H2O (0.727 г) и LiOH·Н2О (0.584 г). Полученную реакционную суспензию охладили до комнатной температуры при перемешивании.
Затем реакционную смесь высушили в распылительной сушилке Niro. Температуры входа и выхода из сушилки с распылением составляли 325 и 125°С соответственно при давлении в сопле 25 фунт/кв. дюйм.
Затем часть (60 г) вещества, высушенного распылительной сушкой, прокалили в аппарате для прокаливания с кипящим слоем диаметром 1'' в токе азота 100 см3/мин. Процедура прокаливания включала нагревание со скоростью 20°С/мин до 300°С и затем со скоростью 1°С/мин до 630°С. Температуру 630°С поддерживали 2 час и затем охладили до комнатной температуры.
Результаты окислительного аммонолиза:
Конверсия 77%, селективность 59%, выход 45%.
Пример 3 Способ с пероксидом и сурьмой
Mo1.0V0.25Sb0.167Nb0.056Nd0.002Ce0.003Li0.013Ox
Приготовили реакционную смесь А, добавив гептамолибдат аммония (189.0 г) и метаванадат аммония (31.3 г) к 500 мл деионизированной воды.
Реакционную смесь В приготовили, добавляя оксид сурьмы (Sb2O3, 26.1 г) к 400 см3 воды.
Реакционную смесь А нагрели при перемешивании до 90°С и выдержали при этой температуре один час. К концу этого часа к реакционной смеси В добавили при перемешивании пероксид водорода (30 масс.%, 30.4 г). Через две минуты после добавления пероксида водорода реакционную смесь А добавили к реакционной смеси В. Объединенную смесь нагрели до 90°С и затем выдержали при этой температуре в течение трех часов при перемешивании.
Объединенную реакционную смесь затем охладили до 70°С. Добавили золь оксида кремния (Nalco, 369 г, 32.5% оксида кремния). Реакционную смесь перемешивали при 70°С в течение 30 мин.
Объединенную реакционную смесь охладили до 50°С. Добавили при перемешивании аммоний-ниобий оксалат (26.5 г, 21.0 масс.% Nb), растворенный в 100 г деионизированной воды. Далее добавили смесь 60.0 г плавленого оксида кремния с 900 мл деионизированной воды. Затем добавили Се(ООССН3)3·1.5H2O (1.11 г), Nd(ООССН3)3·Н2О (0.727 г) и LiOH·H2O (0.899 г). Затем реакционную суспензию охладили до комнатной температуры при перемешивании.
Затем реакционную смесь высушили в распылительной сушилке Niro. Температуры входа и выхода из распылительной сушилки составляли 325 и 125°С соответственно при давлении в сопле 25 фунт/кв. дюйм.
Часть (60 г) высушенного с распылением вещества затем прокалили в аппарате для прокаливания с кипящим слоем диаметром 1'' в токе азота 100 см3/мин. Процедура прокаливания включала нагревание со скоростью 20°С/мин до 300°С и затем со скоростью 1°С/мин до 630°С. Температуру 630°С поддерживали 2 час и затем охладили до комнатной температуры.
Результаты окислительного аммонолиза:
Конверсия 77%, селективность 56%, выход 43%.
Пример 4 - Сравнительный пример способа с пероксидом водорода, молибденом и сурьмой
Mo1.0V0.25Sb0.167Nb0.08Nd0.002Ce0.003Li0.013Ox
Приготовили реакционную смесь А путем (i) добавления гептамолибдата аммония (189.1 г) к 500 мл деионизированной воды, (ii) нагревания смеси до 90°С при перемешивании, (iii) добавления оксида сурьмы (Sb2O3, 26.1 г), затем (iv) перемешивания смеси в течение 10 мин и затем (v) поддержания температуры 90°С, добавления пероксида водорода (30 масс.%, 30.4 г) по каплям при перемешивании в течение 30 мин.
Реакционную смесь В приготовили путем добавления метаванадата аммония (31.3 г) к 400 см3 воды и нагревания раствора при перемешивании до 90°С.
Затем реакционную смесь В добавили к реакционной смеси А при перемешивании и объединенную смесь выдержали при 90°С, перемешивая, в течение часа.
Объединенную реакционную смесь затем охладили до 70°С. Добавили золь оксида кремния (Nalco, 369 г, 32.5% оксида кремния). Реакционную смесь перемешивали при 70°С в течение 30 мин.
Объединенную реакционную смесь охладили до 50°С. Добавили при перемешивании раствор оксалата ниобия (112.0 г, 0.765 моль Nb/кг раствора). Далее добавили смесь 60.0 г плавленого оксида кремния с 900 мл деионизированной воды. Затем добавили Се(ООССН3)3·1.5H2O (1.11 г), Nd(ООССН3)3·H2O (0.727 г) и LiOH·H2O (0.584 г). Затем реакционную суспензию охладили до комнатной температуры при перемешивании.
Затем реакционную смесь высушили в распылительной сушилке Niro. Температуры входа и выхода из распылительной сушилки составляли 325 и 125°С соответственно при давлении в сопле 25 фунт/кв. дюйм.
Часть (60 г) высушенного в распылительной сушилке вещества затем прокалили в аппарате для прокаливания с кипящим слоем диаметром 1'' в токе азота 100 см3/мин. Процедура прокаливания включала нагревание со скоростью 20°С/мин до 300°С и затем со скоростью 1°С/мин до 630°С. Температуру 630°С поддерживали 2 час и затем охладили до комнатной температуры.
Результаты окислительного аммонолиза: конверсия 41%, селективность 41%, выход 17%
Пример 5 Способ с пероксидом и молибденом при промотировании Са
Mo1.0V0.25Sb0.167Nb0.056Nd0.002Ce0.003Li0.013Te0.04Ca0.02Ox
Приготовили реакционную смесь А путем (i) добавления гептамолибдата аммония (182.2 г) к 500 мл деионизированной воды и затем (ii) добавления по каплям при перемешивании пероксида водорода (30 масс.%, 29.3 г) в течение 15 мин.
Реакционную смесь В приготовили путем (i) добавления метаванадата аммония (30.2 г) к 400 см3 воды, (ii) нагревания полученного раствора при перемешивании до 90°С и затем (iii) добавления оксида сурьмы (Sb2O3, 25.1 г) и взаимодействия смеси при перемешивании при 90°С в течение одного часа.
Затем реакционную смесь А добавили к реакционной смеси В при перемешивании. Затем объединенную смесь нагревали, перемешивая, при 90°С еще в течение часа.
Объединенную реакционную смесь охладили до 70°С. Добавили золь оксида кремния (Nalco, 369 г, 32.5% оксида кремния). Эту смесь перемешивали при 70°С в течение 30 мин.
Полученную смесь охладили до 50°С. Добавили при перемешивании аммоний-ниобий оксалат (25.5 г, 21.0 масс.% Nb), растворенный в 100 г деионизированной воды. Далее добавили смесь 60.0 г плавленого оксида кремния с 900 мл деионизированной воды. Затем добавили Се(ООССН3)3·1.5H2O (1.07 г), Nd(ООССН3)3·H2O (0.700 г), Са(ООССН3)2·1Н2О (3.64 г) и LiOH·H2O (0.563 г). Затем реакционную суспензию охладили до комнатной температуры при перемешивании.
Затем реакционную смесь высушили в распылительной сушилке Niro. Температуры на входе и выходе из распылительной сушилки составляли 325 и 125°С соответственно при давлении в сопле 25 фунт/кв. дюйм.
Часть (60 г) высушенного в распылительной сушилке вещества затем прокалили в аппарате для прокаливания с кипящим слоем диаметром 1'' в токе азота 100 см3/мин. Процедура прокаливания включала нагревание со скоростью 20°С/мин до 300°С и затем со скоростью 1°С/мин до 630°С. Температуру 630°С поддерживали 2 час и затем охладили до комнатной температуры.
Результаты окислительного аммонолиза: конверсия 83%, селективность 56%, выход 46%.
Пример 6 Пероксид и ванадий
MoV0.25Sb0.167Nb0.08Li0.013Ox
25.1 г пентоксида ванадия (V2O5) добавили к 600 мл дистиллированной воды в пробирке при перемешивании и затем добавили три (3) аликвоты 30% пероксида водорода (70 г, 70 г, 35 г) с интервалами примерно 10 мин с образованием бордово-красного раствора пероксида ванадия. В отдельной пробирке растворили 194.6 г гептамолибдата аммония в 600 мл дистиллированной воды в присутствии порошка триоксида сурьмы.
К этой смеси добавили раствор пероксида ванадия и полученную смесь нагревали с обратным холодильником в течение 2.5 час. После охлаждения смеси до 70°С добавили 369 г золя оксида кремния с 30 масс.% оксида кремния и перемешивали 30 мин.
Далее полученную смесь охладили до 50°С и добавили при перемешивании смесь 60 г плавленого оксида кремния и 34.7 г аммоний-ниобий оксалата в 900 мл воды. Затем добавили 0.6 г гидроксида лития.
Эту конечную смесь перемешивали еще 30 мин, затем высушили в распылительной сушилке и получили микросфероидный порошок. Этот порошок нагревали при 350°С на воздухе в муфельной печи в течение 3 час в закрытой пробирке и затем прокалили в токе азота при 630°С в течение 2 час. Примерно 35 г этого вещества поместили в реактор с кипящим слоем объемом 40 см3 и испытали в окислительном аммонолизе пропана. При 440°С, величине WWH, равной 0.06, давлении 10 фунт/кв. дюйм и составе сырья 1 пропан/1 аммиак/3.39 кислород/12.61 азот получили выход акрилонитрила 45.8% при селективности 57.8%. Кроме того, получили 5.3% HCN, 3.0% ацетонитрила и 2.2% акриловой кислоты.
Пример 7 Способ с пероксидом и молибденом
Mo1.0V0.25Sb0.167Nb0.056Nd0.002Ce0.003Li0.013Te0.04
Реакционную смесь А приготовили путем (1) добавления гептамолибдата аммония (183.4 г) к 500 мл деионизированной воды и затем (ii) добавления по каплям при перемешивании пероксида водорода (30 масс.%, 29.5 г) в течение 15 мин.
Реакционную смесь В приготовили путем (i) добавления метаванадата аммония (30.38 г) к 400 см3 воды, (ii) нагревания раствора при перемешивании до 90°С, (iii) введения оксида сурьмы (Sb2O3, 25.3 г) и затем (iv) реакции смеси при перемешивании при 90°С в течение 30 мин.
Затем реакционную смесь А добавили к реакционной смеси В и объединенную реакционную смесь затем нагревали при 70°С, перемешивая, еще в течение часа.
Объединенную реакционную смесь охладили до 70°С. Добавили золь оксида кремния (Nalco, 369 г, 32.5% оксида кремния). Эту смесь перемешивали при 70°С в течение 30 мин. К этой смеси добавили при перемешивании аммоний-ниобий оксалат (36.8 г, 21.0 масс.% Nb), растворенный в 200 г деионизированной воды. Затем добавили 60.0 г плавленого оксида кремния в 800 мл деионизированной воды. Затем добавили Се(ООССН3)3·1.5Н2О (1.073 г), Nd(ООССН3)3·H2O (0.705 г) и LiOH·H2O (0.567 г). Полученную реакционную суспензию затем охладили до комнатной температуры при перемешивании.
Затем полученную реакционную смесь высушили в распылительной сушилке Niro. Температуры на входе и выходе из распылительной сушилки составляли 325 и 125°С соответственно при давлении в сопле 25 фунт/кв. дюйм.
Часть (60 г) высушенного в распылительной сушилке вещества затем прокалили в аппарате для прокаливания с кипящим слоем диаметром 1'' в токе азота 100 см3/мин. Процедура прокаливания включала нагревание со скоростью 20°С/мин до 300°С и затем со скоростью 1°С/мин до 630°С. Температуру 630°С поддерживали 2 час и затем охладили до комнатной температуры.
Результаты окислительного аммонолиза: конверсия 81.98%, селективность 61.16%, выход 50.1%.
Пример 8
Mo1.0V0.25Sb0.167Nb0.056Nd0.002Ce0.003Li0.013Te0.04
Такой же, как пример 7, за исключением того, что объединенную смесь из реакционной смеси А и реакционной смеси В вводили в реакцию в течение 2 час при 70°С.
Результаты окислительного аммонолиза: конверсия 85.9%, селективность 61.0%, выход 52.4%.
Пример 9
Mo1.0V0.25Sb0.167Nb0.056Nd0.002Ce0.003Li0.013Te0.04
Такой же, как пример 7, за исключением того, что объединенную смесь из реакционной смеси А и реакционной смеси В вводили в реакцию при 90°С в течение 1 час.
Результаты окислительного аммонолиза: конверсия 74.1%, селективность 60.7%, выход 45.0%.
Пример 10
Mo1.0V0.25Sb0.167Nb0.056Nd0.002Ce0.003Li0.013Te0.04
Такой же, как пример 7, за исключением того, что реакционную смесь В вводили в реакцию при 70°С в течение 1 час и объединенную смесь из реакционной смеси А и реакционной смеси В вводили в реакцию при 70°С в течение 1 час.
Результаты окислительного аммонолиза: конверсия 59.9%, селективность 60.4%, выход 36.2%.
Пример 11
Mo1.0V0.25Sb0.0167Nb0.056Nd0.002Ce0.003Li0.013Te0.04
Такой же, как пример 7, за исключением того, что объединенную смесь из реакционной смеси А и реакционной смеси В вводили в реакцию при 70°C в течение 20 мин.
Результаты окислительного аммонолиза: конверсия 78.2%, селективность 55.8%, выход 43.6%.
Пример 12
Mo1.0V0.25Sb0.0167Nb0.056Nd0.002Ce0.003Li0.013Te0.04
Такой же, как пример 7, за исключением того, что реакционную смесь В вводили в реакцию при 70°C в течение 30 мин и объединенную смесь из реакционной смеси А и реакционной смеси В вводили в реакцию при 70°C в течение 1 час.
Результаты окислительного аммонолиза: конверсия 60.8%, селективность 59.7%, выход 36.3%.
Изобретение относится к способам приготовления предшественников катализаторов. Описаны способы приготовления твердых предшественников смешанных оксидных катализаторов получения акрилонитрила или метакрилонитрила из пропана или изобутана окислительным аммонолизом в газовой фазе, содержащих молибден (Мо), ванадий (V), сурьму (Sb), ниобий (Nb), кислород (О), включающие приготовление реакционной смеси, включающей указанные выше элементы, причем реакционную смесь готовят путем контактирования только одного из соединений сурьмы, молибдена и ванадия с пероксидом водорода до смешения с исходными соединениями остальных элементов, содержащихся в смешанных оксидных катализаторах, и пероксид водорода берут в таком количестве, чтобы мольное соотношение пероксида водорода и сурьмы в катализаторах находилось в интервале 0.01-20. Технический результат - увеличение активности и селективности катализаторов. 2 н. и 17 з.п. ф-лы, 1 ил., 4 табл., 12 пр.
1. Способ приготовления твердого предшественника смешанного оксидного катализатора получения акрилонитрила или метакрилонитрила из пропана или изобутана окислительным аммонолизом в газовой фазе, причем катализатор содержит элементы: молибден (Мо), ванадий (V), сурьму (Sb), ниобий (Nb), кислород (О), и в котором катализатор представляет собой смешанный оксид эмпирической формулы:
Mo1VaSbbNbcTedMeXfZgOn, где М является одним или несколькими щелочными металлами, которые выбирают из группы, состоящей из Li, Cs и Rb; X может представлять один или несколько элементов из группы, состоящей из Y, Ti, Sn, Ge, Zr, Hf; и Z может быть одним или несколькими редкоземельными металлами, которые выбирают из группы, состоящей из Pr, La, Nd, Се и Eu, где 0.1≤а≤1.0, 0.05≤b≤1.0, 0.001≤с≤1.0, 0≤d≤1.0, 0≤е≤0.1, 0≤f≤0.6, 0≤g≤0.1; и n является числом атомов кислорода, необходимых для насыщения валентности всех остальных элементов твердого предшественника при условии, что один или несколько остальных элементов в твердом предшественнике могут находиться в степени окисления, более низкой по сравнению с высшей степенью окисления, а, b, с, d, e, f и g представляют собой отношение числа молей соответствующего элемента к числу молей Мо, и данный способ включает приготовление реакционной смеси, содержащей соединение молибдена, соединение ванадия, соединение сурьмы и пероксид водорода, причем реакционную смесь готовят путем контактирования только одного из соединений сурьмы, молибдена и ванадия с пероксидом водорода до смешения с исходными соединениями остальных элементов, содержащихся в смешанном оксидном катализаторе, и пероксид водорода берут в таком количестве, чтобы мольное соотношение пероксида водорода и сурьмы в катализаторе находилось в интервале 0.01-20; и сушку полученной смеси с образованием твердого предшественника.
2. Способ по п.1, в котором соединение молибдена приводят в контакт с пероксидом водорода с образованием реакционной смеси Мо-пероксид, а соединение ванадия смешивают с соединением сурьмы с образованием реакционной смеси V-Sb, и реакционную смесь V-Sb вводят в контакт с реакционной смесью Мо-пероксид с образованием объединенной реакционной смеси Mo-V-Sb.
3. Способ по п.2, в котором реакционную смесь V-Sb перед контактированием с реакционной смесью Мо-пероксид нагревают с обратным холодильником при температуре от примерно 80°C до примерно температуры кипения в течение примерно 15-45 мин.
4. Способ по п.2, в котором реакционную смесь V-Sb перед контактированием с реакционной смесью Мо-пероксид нагревают при температуре примерно 90°C в течение примерно 30 мин.
5. Способ по п.2, в котором объединенную реакционную смесь Mo-V-Sb перед контактированием с исходными соединениями остальных элементов, содержащихся в смешанном оксидном катализаторе, нагревают при температуре ниже или равной примерно 80°C по меньшей мере в течение часа.
6. Способ по п.2, в котором объединенную реакционную смесь Mo-V-Sb перед контактированием с исходными соединениями остальных элементов, содержащихся в смешанном оксидном катализаторе, нагревают при температуре примерно 70°C в течение примерно двух часов.
7. Способ по п.1, в котором соединение ванадия вводят в контакт с пероксидом водорода с образованием реакционной смеси V-пероксид, соединение молибдена смешивают с соединением сурьмы с образованием реакционной смеси Mo-Sb и реакционную смесь Mo-Sb вводят в контакт с указанной реакционной смесью V-пероксид.
8. Способ по п.1, в котором соединение сурьмы вводят в контакт с пероксидом водорода с образованием реакционной смеси Sb-пероксид, соединение молибдена смешивают с соединением ванадия с образованием реакционной смеси Mo-V и приводят в контакт реакционную смесь Mo-V с указанной реакционной смесью Sb-пероксид.
9. Способ по п.1, в котором мольное соотношение H2O2 и Sb находится в интервале 1-2.
10. Способ по п.1, в котором X в эмпирической формуле представляет собой Li.
11. Способ по п.1, в котором Z в эмпирической формуле выбирают из группы, включающей Nd, Се и смесь Nd и Се.
12. Способ по п.1, в котором в эмпирической формуле b+d≥a.
13. Способ по п.1, в котором в эмпирической формуле 0≤d≤0.06.
14. Способ по п.1, дополнительно включающий нагревание предшественника путем контактирования с потоком газа при первой скорости нагрева выше примерно 15°C/мин до тех пор, пока смесь предшественника не достигнет температуры предварительного прокаливания не выше 400°C.
15. Способ по п.1, дополнительно включающий нагревание предшественника путем контактирования с потоком газа при первой скорости нагрева выше примерно 20°C/мин до тех пор, пока твердая смесь предшественника не достигнет температуры предварительного прокаливания не выше 300°C, и контактирования при второй скорости нагрева выше примерно 1°C/мин до тех пор, пока твердая смесь предшественника не достигнет температуры в интервале 300-650°C.
16. Способ приготовления предшественника смешанного оксидного катализатора получения акрилонитрила или метакрилонитрила из пропана или изобутана газофазным окислительным аммонолизом, причем катализатор содержит элементы: молибден (Мо), ванадий (V), сурьму (Sb), теллур (Те), ниобий (Nb), кислород (О), в котором катализатор представляет собой смешанный оксид эмпирической формулы: MO1VaSbbNbcTedMcXfZgOn, где М является одним или несколькими щелочными металлами, которые выбирают из группы, состоящей из Li, Cs и Rb; X может представлять один или несколько элементов из группы, состоящей из Y, Ti, Sn, Ge, Zr, Hf; и Z может быть одним или несколькими редкоземельными металлами, которые выбирают из группы, состоящей из Pr, La, Nd, Се и Eu, где 0.1≤а≤1.0, 0.05≤b≤1.0, 0.001≤c≤1.0, 0≤d≤1.0. 0≤е≤0.1, 0≤f≤0.6, 0≤g≤0.1; и n является числом атомов кислорода, необходимых для насыщения валентности всех остальных элементов твердого предшественника при условии, что один или несколько остальных элементов в твердом предшественнике могут находиться в степени окисления, более низкой по сравнению с высшей степенью окисления, a, b, c, d, e, f и g представляют собой отношение числа молей соответствующего элемента к числу молей Мо, и данный способ включает приготовление реакционной смеси, содержащей соединение молибдена, соединение ванадия, соединение сурьмы и пероксид водорода, причем реакционную смесь готовят путем контактирования только одного из соединений сурьмы, молибдена и ванадия с пероксидом водорода до смешения с остальными ингредиентами и пероксид водорода вводят в таком количестве, чтобы мольное соотношение пероксида водорода и сурьмы в катализаторе находилось в интервале 0.01-20.
17. Способ по п.1, в котором пероксид водорода вводят в таком количестве, чтобы мольное соотношение пероксида водорода и сурьмы в катализаторе находилось в интервале 0.1-5.
18. Способ по п.1, в котором пероксид водорода вводят в таком количестве, чтобы мольное соотношение пероксида водорода и сурьмы в катализаторе находилось в интервале 0.5-3.
19. Способ по п.1, в котором Nb вводят в виде соединения ниобия, в том числе ниобиевой кислоты, кислого оксалата ниобия, аммоний-ниобий оксалата или их смеси.
| Стенд для нагружения образцов горных пород | 1981 |
|
SU945432A1 |
| US 2003017944 A1, 23.01.2003 | |||
| US 2005054869 A1, 10.03.2005 | |||
| ЕР 1632287 A1, 08.03.2006 | |||
| US 6514902 B1, 04.02.2003 | |||
| US 20020115879 A1, 22.08.2002 | |||
| СПОСОБ ПАРОФАЗНОГО АММОКСИДИРОВАНИЯ С - С-МОНООЛЕФИНОВ | 1993 |
|
RU2105757C1 |

