Уровень техники изобретения
Настоящее изобретение относится к композициям веществ, каталитическим композициям, способам получения таких композиций веществ и таких каталитических композиций и способам применения таких композиций веществ и таких каталитических композиций. Предпочтительно, чтобы в каждом случае такие композици и такие катализаторы были активны в газофазной конверсии пропана в акриловую кислоту и изобутана в метакриловую кислоту (путем окисления) или пропана в акрилонитрил и изобутилена в метакрилонитрил (путем окислительного аммонолиза) и наиболее предпочтительно с выходом по меньшей мере примерно 50%.
Изобретение в предпочтительном варианте, в частности, относится к композициям веществ и каталитическим композициям, способам приготовления таких композиций веществ и таких каталитических композиций и способам применения таких композиций веществ и таких каталитических композиций, когда в каждом случае они содержат молибден, ванадий, ниобий и сурьму или молибден, ванадий, тантал и сурьму, а в некоторых вариантах еще и германий. Предпочтительные варианты приготовления таких композиций веществ и таких каталитических композиций включают реакции в растворе в закрытых реакционных сосудах при температурах выше 100°С и давлениях выше атмосферного. Особенно предпочтителен гидротермальный синтез с использованием водных растворов.
Вообще область изобретения относится к молибденсодержащим и ванадийсодержащим катализаторам, активным в конверсии пропана в акриловую кислоту (путем окисления) и/или конверсии пропана в акрилонитрил (путем окислительного аммонолиза). Уровень техники в этой области включает множество патентов и патентных заявок, включая, например, патент US №6043185, Cirjak et al., патент US №6514902, Inoue et al., патент US №614391, Hinago et al., патент US №6383978, Bogan, Jr., патентная заявка US №2002/0115879 Al, Hinago et al., патентная заявка US №2003/0004379, Gaffney et al., японская патентная заявка JP №1999/114426 A, Asahi Chemical Co., японская патентная заявка JP №2002/191974 A, Asahi Chemical Co., PCT патентная заявка №WO 01/98246 Al, BASF A. G., а также множество публикаций, включая, например, Watanabe et al.,"Новый путь синтеза смешанных оксидных катализаторов Mo-V-Nb-Te окислительного аммонолиза пропана", Applied Catalysis A: General, 194-195, pp.479-485 (2000), and Ueda et al.,"Селективное окисление низших алканов в присутствии оксидных катализаторов Mo-V-M-O (M=A1, Ga, Bi, Sb и Те), полученных гидротермальным синтезом". Applied Catalysis A: General, 200, pp.135-145.
Несмотря на то, что в области молибденсодержащих и ванадийсодержащих катализаторов достигнут значительный прогресс, предназначенные для промышленности катализаторы, активные в конверсии пропана в акриловую кислоту и изобутана в метакриловую кислоту (путем окисления) и/или конверсии пропана в акрилонитрил и изобутана в метакрилонитрил (путем окислительного аммонолиза), нуждаются в дальнейшем улучшении. Известные каталитические системы для таких реакций отличаются низкими выходами целевого продукта. Кроме того, известные методы приготовления таких каталитических систем трудно воспроизводить, чтобы получать непротиворечивые данные по каталитической активности.
Сущность изобретения
Настоящее изобретение ставит задачу устранить указанный недостаток известных катализатических.
Еще одной задачей изобретения является приготовление катализаторов, характеризующихся повышенными выходами в газофазном окислении и/или окислительном аммонолизе пропана с образованием акриловой кислоты и/или акрилонитрила соответственно и в газофазном окислении и/или окислительном аммонолизе изобутана с образованием метакриловой кислоты и/или метакрилонитрила соответственно.
Еще одной задачей изобретения является разработка способов приготовления катализаторов с воспроизводимой каталитической активностью.
Композиции веществ, композиции катализаторов, способы получения катализаторов, катализаторы, приготовленные этими способами, способы применения таких катализаторов в соответствии с настоящим изобретением имеют преимущества по сравнению с известными системами. Такие катализаторы применимы в лабораторном масштабе (R&D), на пилотных установках и в промышленных реакторах для конверсии пропана в акриловую кислоту путем окисления или акрилонитрил путем окислительного аммонолиза.
Катализатор можно также использовать в тех же масштабах и в таких же системах для конверсии изобутана в метакриловую кислоту и/или метакрилонитрил.
Другие особенности преимущества настоящего изобретения будут частично очевидны специалистам и частично описаны ниже.
Краткое описание чертежей
Фигуры 1А и 1В схематически представляют примеры реакций окисления пропана и изобутана (фиг.1А) и примеры окислительного аммонолиза пропана и изобутана (фиг.1В).
Подробное описание изобретения
Композиции веществ и композиции катализаторов
В одном аспекте настоящее изобретение относится композициям, содержащим молибден, ванадий, ниобий, сурьму, германий и кислород или молибден, ванадий, тантал, сурьму, германий и кислород.
Во втором аспекте изобретение относится к композициям, которые являются катализаторами, содержащими смешанную металлоксидную систему, эффективную в парофазной конверсии пропана в акриловую кислоту и/или акрилонитрил и/или изобутана в метакриловую кислоту и/или метакрилонитрил. Смешанная металлоксидная система имеет состав, в который входят молибден, ванадий, ниобий, сурьма, германий и кислород или молибден, ванадий, тантал, сурьма, германий и кислород. Предпочтительно, чтобы смешанная металлоксидная система имела эмпирическую формулу Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой
а находится в интервале от примерно 0,1 до примерно 0,6, предпочтительно от примерно 0,15 до примерно 0,5 и наиболее предпочтительно от примерно 0,2 до примерно 0,4 и особенно предпочтительно около примерно 0,3,
b находится в интервале от примерно 0,02 до примерно 0,12, предпочтительно от примерно 0,03 до примерно 0,1 и наиболее предпочтительно от примерно 0,04 до примерно 0,08 и особенно предпочтительно около примерно 0,06,
с находится в интервале от примерно 0,1 до примерно 0,5, предпочтительно от примерно 0,15 до примерно 0,35, более предпочтительно от примерно 0,15 до примерно 0,3 и наиболее предпочтительно от примерно 0,2 до примерно 0,3 и особенно предпочтительно около примерно 0,2,
d находится в интервале от примерно 0,01 до примерно 1, в одном варианте нижнее значение интервала d составляет примерно 0,05, в другом варианте нижнее значение интервала d составляет примерно 0,1, еще в одном варианте нижнее значение интервала d больше 0,1, а в другом нижнее значение интервала d равно примерно 0,15, еще в одном варианте нижнее значение интервала d составляет примерно 0,2, еще в одном варианте нижнее значение интервала d составляет примерно больше 0,2; в одном варианте верхнее значение интервала d составляет примерно 0,7, в другом варианте верхнее значение интервала d составляет примерно 0,5, в еще одном варианте d находится в интервале от примерно 0,2 до примерно 0,4 и особенно предпочтительно около примерно 0,3
и х зависит от степени окисления других элементов, входящих в состав смешанной металлоксидной системы.
В третьем аспекте изобретение относится к первому или второму аспектам,как описано выше, и, кроме того, предполагает отсутствие одного или более элементов - теллура, церия и/или галлия - в различных вариациях и комбинациях. Что касается отсутствия теллура, то было обнаружено, что катализаторы, содержащие молибден, ванадий, ниобий и комбинацию сурьмы и германия, более активны в конверсии пропана в акрилонитрил, чем катализаторы, содержащие молибден, ванадий, ниобий и комбинацию теллура и германия.
В еще одном, четвертом, аспекте изобретение относится к композиции веществ или катализатору, содержащему смешанную металлоксидную систему, как в первом или втором аспектах, описанных выше, где композиция веществ или катализатор представляет собой смешанную металлоксидную систему, в каждом случае состоящую из молибдена, ванадия, ниобия, сурьмы, германия и кислорода или молибдена, ванадия, тантала, сурьмы, германия и кислорода.
В любом из приведенных выше аспектов изобретения композиция веществ может иметь стехиометрические соотношения необходимых элементов относительно друг друга. Стехиометрические соотношения могут выражать относительные атомные соотношения или мольные соотношения в веществе (например, в среднем) или альтернативно по меньшей мере части вещества (например, в одной фазе двухфазной системы). Например, соотношение молибден: ванадий находится в интервале от примерно 1:0,1 до примерно 1:0,6, предпочтительно от примерно 1:0,15 до примерно 1:0,5 и наиболее предпочтительно от примерно 1:0,2 до примерно 1:0,4. Соотношение молибден: ниобий или молибден: тантал находится в интервале от примерно 1:0,02 до примерно 1:0,12, предпочтительно от примерно 1:0,03 до примерно 1:0,1 и наиболее предпочтительно от примерно 1:0,04 до примерно 1:0,06. Сооношение молибден: сурьма находится в интервале от примерно 1:0,1 до примерно 1:0,5, предпочтительно от примерно 1:0,15 до примерно 1:0,35, более предпочтительно от примерно 1:0,15 до примерно 1:0,3 и наиболее предпочтительно от примерно 1:0,2 до примерно 1:0,3. Сооношение молибден: германий находится в интервале от примерно 1:0,01 до примерно 1:1, предпочтительно от примерно 1:0,05 до примерно 1:1, еще предпочтительно от примерно 1:0,1 до примерно 1:1, более предпочтительно от примерно 1:0,1 до примерно 1:0,7, даже более предпочтительно от примерно 1:0,1 до примерно 1:0,5 и наиболее предпочтительно от примерно 1:0,2 до примерно 1:0,4. В другом варианте соотношение молибден: германий находится в интервале от 1:>0,1 до примерно 1:1. В еще одном варианте соотношение молибден: германий находится в интервале от 1:0,15 до примерно 1:1. В другом варианте соотношение молибден: германий находится в интервале от 1:>0,2 до примерно 1:1. Заслуживает внимания, что каждый из предпочтительных интервалов для каждой пары компонентов можно реализовать в различных вариациях и комбинациях.
Стехиометрические соотношения компонентов, выраженные, как во втором аспекте, можно определить в виде эмпирической формулы, причем смешанная металлоксидная система имеет эмпирическую формулу Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которых а, b, с, d и х имеют предпочтительные интервалы, как описано выше в связи со вторым аспектом изобретения.
Следовательно, первая предпочтительная композиция катализатора представляет собой смешанную металлоксидную систему Mo1VaNbbSbcGedOx или Мо1VаTabSbсGedОх, в которой а находится в интервале от примерно 0,1 до примерно 0,6, b находится в интервале от примерно 0,02 до примерно 0,12, с находится в интервале от примерно 0,1 до примерно 0,5, d находится в интервале от примерно 0,01 до примерно 1, в другом варианте d находится в интервале от больше, чем 0,1, до примерно 1, в еще одном варианте d находится в интервале от больше, чем 0,2, до примерно 1 и х зависит от степени окисления других элементов, входящих в состав смешанной металлоксидной системы.
Вторая предпочтительная композиция катализатора представляет собой смешанную металлоксидную систему Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой а находится в интервале от примерно 0,15 до примерно 0,5, b находится в интервале от примерно 0,03 до примерно 0,1, с находится в интервале от примерно 0,15 до примерно 0,35, d находится в интервале от примерно 0,05 до примерно 1, в других вариантах d находится в интервале от больше 0,1 до примерно 1, в еще одном варианте d находится в интервале от больше 0,2 до примерно 1 и х зависит от степени окисления других элементов в составе смешанной металлоксидной системы.
Третья предпочтительная композиция катализаторов содержит смешанную металлоксидную систему Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой а находится в интервале от примерно 0,2 до примерно 0,4, b находится в интервале от примерно 0,04 до примерно 0,08, с находится в интервале от примерно 0,15 до примерно 0,3, d находится в интервале от примерно 0,1 до примерно 0,7, предпочтительно больше 0,1 до примерно 0,7, в другом варианте d находится в интервале от примерно 0,2 до примерно 1, предпочтительно больше 0,2 до примерно 0,7 и х зависит от степени окисления других элементов в составе смешанной металлоксидной системы.
Приготовление каталитических композиций
Композиции и катализаторы, приведенные выше, можно приготовить описанными здесь способами гидротермального синтеза. Однако такие способы сами по себе можно эффективно применить для приготовления других композиций и катализаторов, включая более подробно охарактеризованные композиции и катализаторы.
Следовательно, пятый аспект изобретения относится к способу гидротермального синтеза для приготовления композиции смешанных металлоксидных систем и в предпочтительном аспекте катализатора на основе смешанной металлоксидной системы, содержащего молибден, ванадий, ниобий и сурьму или молибден, ванадий, сурьму, германий и кислород, рассмотренному ниже.
Способы гидротермального синтеза раскрыты в патентной заявке US №2003/0004379, Gaffney et al., работах Watanabe et al., «Новый способ синтеза катализаторов на основе смешанных оксидов Mo-V-Nb-Te для окислительного аммонолиза пропана». Applied Catalysis A: General, 194-195, pp.479-485 (2000), и Ueda et al., «Селективное окисление низших алканов в присутствии оксидных катализаторов Mo-V-M-O (M=A1, Ga, Bi, Sb and Те), полученных гидротермальным синтезом», Applied Catalysis A: General, 200, pp.135-145. Соответственно изобретение включает усовершенствованный гидротермальный синтез, в котором предшественники смешанной металлоксидной системы вводят в водный раствор для образования реакционной среды и они взаимодействуют в реакционной среде при повышенном давлении и повышенной температуре в запаянном реакционном сосуде в течение времени, достаточного для образования смешанной металлоксидной системы. Усовершенствование способа заключается в перемешивании реакционной среды во время реакции. Перемешивание реакционной среды, как показано ниже, можно осуществить многими способами, такими как перемешивание внутри реакционного сосуда или, например, переворачивание, встряхивание или вибрационное перемешивание реакционного сосуда. Перемешивание реакционной смеси во время реакции дает много преимуществ. Это усовершенствование приводит к более однородному перемешиванию в ходе реакции, особенно в случае малорастворимых реагентов. Это приводит к более эффективному поглощению исходных веществ и более равномерно перемешанному металлоксидному продукту. Перемешивание реакционной среды в ходе реакции приводит также к образованию смешанной металлоксидной системы в растворе, а не на стенках реакционного сосуда. Это облегчает выделение и отделение смешанной металлоксидной системы такими приемами, как центрифугирование, декантация или фильтрация вместо отделения большей части продукта от стенок реакционного сосуда. (См. заявку US 2003/0004379 А1, в которой продукт гидротермального синтеза образовывался на стенках реакционного сосуда.) Еще одним преимуществом является то, что в растворе частицы смешанного оксида металла могут расти по всем граням, а не на ограниченном числе поверхностей, как это было бы в случае роста на стенке реактора.
Этот пятый аспект изобретения в более широком смысле может также относиться, например, к приготовлению катализатора на основе смешанной металлоксидной системы, включающей по меньшей мере два элемента из молибдена, ванадия, сурьмы и теллура и предпочтительно по меньшей мере молибдена и ванадия, или по меньшей мере молибдена и сурьмы, или по меньшей мере ванадия и сурьмы.
Необязательно, в каждом из таких случаев этого пятого аспекта изобретения способ может относиться к приготовлению катализатора на основе смешанной металлоксидной системы, содержащей один или более элементов из ниобия, тантала, германия и/или других элементов, которые были известны на предыдущем уровне техники в связи с такими системами.
Согласно пятому аспекту изобретение относится к способу приготовления смешанной металлоксидной системы, содержащей молибден, ванадий, ниобий и сурьму или молибден, ванадий, тантал, сурьму, германий и кислород. Способ включает:
введение в реакционный сосуд предшественников Мо, V, Nb или Та и Sb в водном растворителе для образования реакционной среды с начальным рН 4 или меньше;
необязательно, добавление дополнительного водного растворителя в реакционный сосуд;
герметизацию реакционного сосуда;
реакцию в реакционной смеси при температуре больше 100°С и давлении выше атмосферного в течение времени, достаточного для образования смешанной металлоксидной системы;
необязательно, охлаждение реакционной смеси и
выделение смешанной металлоксидной системы из реакционной смеси.
Другой способ согласно пятому аспекту изобретения относится к приготовлению смешанной металлоксидной системы, содержащей молибден, ванадий, ниобий и сурьму или молибден, ванадий, тантал, сурьму и кислород, путем:
введения в реакционный сосуд соединений - предшественников Мо, V, Nb или Та и Sb в водном растворителе с образованием реакционной смеси;
необязательно, добавления дополнительного водного растворителя в реакционный сосуд;
герметизации реакционного сосуда;
реакции в реакционной смеси при температуре выше 100°С и давлении выше атмосферного при перемешивании реакционной смеси в течение времени, достаточного для образования смешанной металлоксидной системы;
необязательно, охлаждения реакционной смеси
и выделения смешанной металлоксидной системы из реакционной смеси.
Когда смешанная металлоксидная система содержит германий, стадия введения компонентов включает введение и соединения Ge.
Шестой аспект изобретения относится к приготовлению катализатора, представляющего собой смешанную металлоксидную систему с эмпирической формулой Mo1VaNbbSbcOx или Мо1VаТаbSbсОх, в которой компонент а находится в пределах от примерно 0,1 до примерно 0,6, предпочтительно от примерно 0,15 до примерно 0,5 и наиболее предпочтительно от примерно 0,2 до примерно 0,4, компонент b находится в пределах от примерно 0,02 до примерно 0,12, предпочтительно от примерно 0,03 до примерно 0,1 и наиболее предпочтительно от примерно 0,04 до примерно 0,08 и в которой компонент с находится в пределах от примерно 0,1 до примерно 0,5, предпочтительно от примерно 0,15 до примерно 0,35, более предпочтительно от примерно 0,15 до примерно 0,3 и наиболее предпочтительно от примерно 0,2 до примерно 0,3. Этот шестой аспект изобретения в широком смысле относится также к приготовлению катализатора, представляющего собой смешанную металлоксидную систему с эмпирической формулой Mo1VaXbYcOx, где Х необязательно, но предпочтительно выбирают из ниобия или тантала, Y необязательно, но предпочтительно выбирают из сурьмы и теллура и компонент а находится в пределах от примерно 0,1 до примерно 0,6, предпочтительно от примерно 0,15 до примерно 0,5 и наиболее предпочтительно от примерно 0,2 до примерно 0,4, в которой компонент b находится в пределах от примерно 0 до примерно 0,12, предпочтительно от примерно 0,02 до примерно 0,12, более предпочтительно от примерно 0,03 до примерно 0,1 и наиболее предпочтительно от примерно 0,04 до примерно 0,08 и в которой компонент с находится в пределах от примерно 0 до примерно 0,5, предпочтительно от примерно 0,1 до примерно 0,5, более предпочтительно от примерно 0,15 до примерно 0,35, более предпочтительно от примерно 0,15 до примерно 0,3 и наиболее предпочтительно от примерно 0,2 до примерно 0,3 и х зависит от степени окисления других элементов в смешанной металлоксидной системе.
Седьмой аспект изобретения относится к приготовлению катализатора, представляющего собой смешанную металлоксидную систему, как описано в пятом и шестом аспектах изобретения, и еще содержит германий. Более конкретно, что касается эмпирической формулы, катализатор может представлять собой смешанную металлоксидную систему с эмпирической формулой Mo1VaNbbSbcGedOx или
Mo1VaTabSbcGedOx, в которой a, b, c и d имеют значения, описанные выше в связи со вторым аспектом изобретения, включая интервалы предпочтительных композиций в пределах приведенных интервалов, и х зависит от степени окисления других элементов в смешанной металлоксидной системе.
В любом из пятого, шестого или седьмого аспектов изобретения способ гидротермального синтеза может включать несколько стадий, как описано выше в общем смысле и конкретно в последующем изложении.
Эти стадии включают стадию формирования водной жидкой реакционной среды (например, в виде раствора, однородной и неоднородной дисперсии типа суспензии или комбинации раствора и дисперсии), которая содержит требуемые компоненты в реакционном сосуде, образуя жидкую реакционную среду (например, раствор и/или суспензию), содержащую компоненты Мо, V, Nb или Та и Sb (а также Ge в седьмом аспекте изобретения) в реакционном сосуде. Предпочтительно в каждом случае жидкую реакционную среду формировать по методике, которая включает комбинирование компонентов в реакционном сосуде в таких относительных мольных количествах, чтобы соблюдалась указанная выше стехиометрия. Также предпочтительно в каждом случае формировать жидкую реакционную среду по методике, включающей перемешивание при соединении по меньшей мере двух компонентов в реакционном сосуде и предпочтительно перемешивание при введении каждого компонента в реакционный сосуд. Предпочтительно, чтобы жидкие реакционные среды представлли собой водный раствор и/или твердые частицы, диспергированные в среде водного носителя. Некоторые компоненты, такие как Мо-содержащие соединения, V-содержащие соединения и Nb- или Та-содержащие соединения можно вводить в реакционный сосуд в виде водных растворов солей металлов Мо, V, Nb или Та и Sb. Некоторые из этих компонентов, а также другие компоненты, такие как Мо-содержащие, V-содержащие, Sb-содержащие и Ge-содержащие соединения, можно вводить в реакционные сосуды в виде твердых веществ или суспензий, содержащих твердые частицы, диспергированные в среде водных носителей.
Предпочтительными предшествененниками для синтеза катализаторов, как описано выше, являются следующие соединения. Предпочтительные источники молибдена включают оксид молибдена(VI), гептамолибдат аммония и молибденовую кислоту. Предпочтительные источники ванадия включают ванадилсульфат, метаванадат аммония и оксид ванадия(V). Предпочтительные источники сурьмы включают оксид сурьмы(III), ацетат сурьмы(III), оксалат сурьмы(III), оксид сурьмы(V), сульфат сурьмы(III) и тартрат сурьмы(III). Предпочтительными источниками ниобия являются оксалат ниобия, оксалат ниобия-аммония и этоксид ниобия. Предпочтительными источниками тантала являются оксалат тантала, оксалат тантала-аммония и этоксид тантала. Предпочтительным источником германия является оксид германия(IV).
Растворители, которые можно использовать для приготовления смешанных металлоксидных систем настоящего изобретения, включают, но не ограничиваются ими, воду; спирты, такие как метанол, этанол, пропанол; диолы (например, этиленгликоль, пропиленгликоль и т.д.), а также другие полярные растворители, известные на уровне техники. Предпочтительно, чтобы предшественники металлов растворялись в растворителе по меньшей мере при температуре и давлении реакции. Вообще предпочтительным растворителем является вода. Можно использовать любую воду квалификации «для химического синтеза». Вода может быть, но не обязательно, дистиллированной и/или деионизированной.
Количество водного растворителя в реакционной среде может варьироваться в зависимости от растворимости предшественников, которые вводят для образования смешанной металлоксидной системы. Количество водного растворителя должно быть по меньшей мере достаточным для образования суспензии реагентов. В гидротермальном синтезе смешанных металлоксидных систем принято оставлять свободное пространство в реакционном сосуде.
В некоторых способах гидротермального синтеза в реакционную среду можно добавлять окислитель для окисления одного или более предшественников металлов до стадии реакции. Например, при гидротермальном синтезе оксидов металлов MoVNbSb или MoVTaSb настоящего изобретения часть V и Sb можно окислить с помощью окислителя до стадии реакции. В этом случае в реакционную среду добавляют окислитель, например, пероксид (Hala). Предпочтительно сделать это до введения предшественников Nb или Та - оксалатов ниобия и тантала, чтобы избежать нежелательной реакции Н2О2 со щавелевой кислотой, входящей в состав раствора оксалата ниобия или тантала. При добавлении окислителя в реакционную среду порядок введения можно выбрать таким образом, чтобы достичь необходимого окисления и/или избежать нежелательных реакций. Предпочтительным окислителем является не содержащий металла оксид типа Н202. Можно использовать металлсодержащие или неорганические оксиданты, когда желательно вводить в смешанную металлоксидную систему конкретные металлы или элементы окислителя.
Способ приготовления включает также стадию герметизации реакционного сосуда, предпочтительно после введения компонентов реакции. Как показано выше, желательно оставлять свободное пространство в реакционном сосуде. Объем свободного пространства может зависеть от конструкции сосуда или способа перемешивания реакционной смеси. При верхнем расположении мешалки объем свободного пространства может, например, составлять 50%. Обычно свободное пространство заполняют воздухом, который вносит в реакцию некоторое количество кислорода. Однако свободное пространство, как известно, можно заполнять и другими газами, в том числе реагентами типа O2 или даже инертным газом типа Ar или N2, и объем свободного пространства и количество газа в нем зависят от проводимой реакции, известной на соответствующем уровне техники.
На следующей стадии предпочтительного способа гидротермального синтеза, как описано здесь, компоненты вводят в реакцию в герметизированном реакционном сосуде при температуре выше 100°С и давлении выше атмосферного для получения предшественника смешанной металлоксидной системы. Предпочтительно, чтобы компоненты вводили в реакцию в герметизированном реакционном сосуде при температуре по меньшей мере примерно 125°С и давлении по меньшей мере примерно 25 фунт/кв.дюйм, более предпочтительно при температуре по меньшей мере примерно 150°С и давлении по меньшей мере примерно 50 фунт/кв.дюйм и в некоторых вариантах при температуре по меньшей мере примерно 175°С и давлении по меньшей мере примерно 100 фунт/кв. дюйм.
В любом случае компоненты предпочтительно вводить в реакцию по методике, по которой компоненты смешивают в герметичном реакционном сосуде на стадии реакции. Конкретный способ смешения не является строго критичным и может включать, например, смешение (например, перемешивание или размешивание) компонентов в реакционном сосуде в ходе реакции любым эффективным способом. Такие способы ключают, например, перемешивание содержимого реакционного сосуда, в частности, путем встряхивания, переворачивания или вибрации реакционного сосуда с компонентами реакции. Такие способы также включают, например, перемешивание с использованием перемешивающего устройства, помещенного по меньшей мере частично внутри реакционного сосуда, и движущей силы, соединенной с перемешивающим устройством или с реакционным сосудом для обеспечения движения перемешивающего устройства относительно реакционного сосуда. Перемешивающим устройством может быть мешалка с приводным валом или без него. Движущая сила может быть напрямую или косвенно (например, через магнитное взаимодействие) связана с перемешивающим устройством. Предпочтительно, чтобы перемешивание было достаточно эффективным для осуществления реакции между компонентами реакционной среды с образованием более гомогенной реакционной среды (например, для образования более однородного предшественника смешанной металлоксидной системы) по сравнению с реакцией без перемешивания. Не прибегая к теории, которая не отражена четко в формуле изобретения, можно утверждать, что хорошо смешанная (например, после хорошего перемешивания) реакционная среда может в некоторых случаях привести к образованию или предшественника смешанной металлоксидной системы, или, после дальнейшей обработки, катализатора на основе смешанной металлоксидной системы и к такой ситуации, когда по меньшей мере часть предшественника или катализатора будет представлять собой существенно однородную смесь необходимых элементов, как показано выше (например, в виде одной фазы), или, например, твердый раствор, или когда по меньшей мере его часть будет иметь кристаллическую структуру, необходимую для активного и селективного окисления и/или окислительного аммонолиза пропана.
Также предпочтительно, чтобы компоненты могли вступать в реакцию в герметичном реакционном сосуде при начальном рН не выше примерно 4. В течение всего гидротермального синтеза рН реакционной смеси может изменяться таким образом, что конечный рН реакционной смеси может оказаться выше или ниже начального рН. Предпочтительно, чтобы компоненты вступали в реакцию в герметичном реакционном сосуде при рН на выше 3,5. В некоторых вариантах компоненты можно вводить в реакцию в герметичном реакционном сосуде при рН не выше примерно 3,0, не выше примерно 2,5, не выше примерно 2,0, не выше примерно 1,5 или не выше примерно 1,0, не выше примерно 0,5 или не выше примерно 0. Предпочтительный интервал значений рН включает рН в интервале от примерно - 0,5 до примерно 4, предпочтительно от примерно 0 до примерно 4, более предпочтительно от примерно 0,5 до примерно 3,5. В некоторых вариантах рН может находиться в интервале от примерно 0,7 до примерно 3,3 или от примерно 1 до примерно 3. Величину рН можно скорректировать путем добавления кислоты или основания к реакционной смеси.
Компоненты можно вводить в реакцию в герметичных реакционных сосудах в указанных выше условиях реакции (включающих, например, температуры реакции, давления реакции, рН, перемешивание и т.д., как описано выше) в течение времени, достаточного для образования смешанной металлоксидной системы, предпочтительно смешанной металлоксидной системы в виде твердого раствора необходимых элементов, как рассмотрено выше, и предпочтительно, чтобы по меньшей мере его часть приобрела кристаллическую структуру, необходимую для катализаторов активного и селективного окисления и/или окислительного аммонолиза пропана или изобутана, как будет показано ниже. Точное время реакции не является строго критичным и может составлять, например, по меньшей мере примерно шесть часов, по меньшей мере примерно двенадцать часов, по меньшей мере примерно восемнадцать часов, по меньшей мере примерно двадцать четыре часа, по меньшей мере примерно тридцать часов, по меньшей мере примерно тридцать шесть часов, по меньшей мере примерно сорок два часа, по меньшей мере примерно сорок восемь часов, по меньшей мере примерно пятьдесят четыре часа, по меньшей мере примерно шестьдесят часов, по меньшей мере примерно шестьдесят шесть часов или по меньшей мере примерно семьдесят два часа. Время реакции может быть даже больше трех дней, составляя, например, по меньшей мере примерно четыре дня, по меньшей мере примерно пять дней, по меньшей мере примерно шесть дней, по меньшей мере примерно семь дней, по меньшей мере примерно две недели, или по меньшей мере примерно три недели, или по меньшей мере примерно один месяц.
Последующие стадии предпочтительных способов приготовления катализаторов можут включать стадии обработки, в том числе, например, охлаждение реакционной среды, содержащей смешанную металлоксидную систему (например, до примерно обычной температуры), отделение твердых частиц смешанной металлоксидной системы от жидкости (например, центрифугированием и/или декантацией верхнего слоя или же альтернативно фильтрованием), промывку отделенных твердых частиц (например, с использованием дистиллированной или деионизированной воды), повторение стадий отделения и промывки один или более раз и проведение конечной стадии отделения.
После стадий обработки промытую и отделенную смешанную металлоксидную систему можно высушить. Смешанную металлоксидную систему можно сушить при обычных условиях (например, при температуре примерно 25°С и атмосферном давлении) и/или в печи, например, при температуре в интервале от 40°С до примерно 150°С и предпочтительно примерно 120°С в течение времени от примерно пяти до примерно пятнадцати часов и предпочтительно примерно двенадцати часов. Сушку можно проводить в регулируемой или нерегулируемой атмосфере, которая может представлять собой инертный газ, газ-окислитель, газ-восстановитель или воздух и обычно и предпочтительно воздух.
На следующей стадии приготовления высушенную смешанную металлоксидную систему можно обработать для формирования смешанного металлоксидного катализатора. Такая обработка может включать, например, прокаливание (например, нагревание в окислительных или восстановительных условиях) в различной атмосфере. Обработанную смешанную металлоксидную систему можно раздробить или измельчить до такой операции и/или делать это периодически во время обработки. Предпочтительно, например, высушенную смешанную металлоксидную систему сначала раздробить и затем прокалить с образованием смешанного металлоксидного катализатора. Прокаливание предпочтительно проводить в инертной атмосфере, например, под азотом. Предпочтительные условия прокаливания включают температуры в интервале от примерно 400°С до примерно 700°С, более предпочтительно от примерно 500°С до примерно 650°С и в некоторых вариантах прокаливают при примерно 600°С.
Обработанная (например, прокаленная) смешанная металлоксидная система может быть далее механически обработана, включая, например, дробление, просеивание и прессование смешанной металлоксидной системы. Предпочтительно просеивать катализатор для получения частиц с таким распределением по размерам, при котором средний размер частиц находится в интервале от примерно 100 мкм до примерно 400 мкм, предпочтительно от примерно 120 мкм до примерно 380 мкм и предпочтительно от примерно 140 мкм до примерно 360 мкм.
Композиции катализаторов, приготовленных приведенными выше способами
В следующем восьмом аспекте изобретение относится к композициям катализаторов, приготовленным по общей методике, описанной выше, включая предпочтительно пятый, шестой и седьмой аспекты изобретения, приведенные выше.
Степень окисления/кристаллические структуры
Степень окисления компонентов катализатора может изменяться, как описано выше, и каждый компонент может иметь более чем одну степень окисления. Смешанный металлоксидный катализатор предпочтительно содержит одну или более фаз с кристаллической структурой, которая активна и селективна в окислении и/или окислительном аммонолизе пропана с образованием акриловой кислоты и/или акрилонитрила соответственно или изобутана с образованием метакриловой кислоты и/или метакрилонитрила соответственно.
Конверсия пропана и изобутана при окислении или окислительном аммонолизе
Композиции и катализаторы на основе смешанной металлоксидной системы, описанные в приведенных выше аспектах изобретения, можно использовать в следующем девятом аспекте для конверсии пропана в акриловую кислоту путем окисления или изобутана в метакриловую кислоту и/или в следующем десятом аспекте изобретения для конверсии пропана в акрилонитрил или изобутана в метакрилонитрил путем окислительного аммонолиза. На фигуре 1А показана общая схема реакций окисления пропана в акриловую кислоту и изобутана в метакриловую кислоту, а на фигуре 1В приведена общая схема реакций окислительного аммонолиза пропана в акрилонитрил и изобутана в метакрилонитрил.
Предпочтительно превращать пропан в акриловую кислоту и изобутан в метакриловую кислоту в присутствии одного или более упомянутых выше катализаторов в газофазном проточном реакторе путем контактирования катализатора с пропаном в присутствии кислорода (например, подаваемого в зону реакции в исходном потоке, представляющем собой кислородсодержащий газ, обычно воздух) в условиях реакции, эффективных для образования акриловой кислоты. Предпочтительно, чтобы поток сырья для этой реакции содержал пропан и кислородсодержащий газ, например, воздух, с мольным соотношением пропана или изобутана к кислороду в интервале от примерно 0,15 до примерно 5 и предпочтительно от примерно 0,25 до примерно 2. Исходный поток может также содержать один или более дополнительный компонент, включая продукт - акриловую кислоту или метакриловую кислоту (например, из потока рецикла или с более ранней стадии многостадийного реактора) и/или водяной пар. Например, исходный поток может содержать примерно от 5 до 30 мас. или мольн.% пропана или изобутана от общей массы исходного потока.
Предпочтительно превращать пропан в акрилонитрил и изобутан в метакрилонитрил в присутствии одного или более из упомянутых выше катализаторов в газофазном проточном реакторе путем контактирования катализатора с пропаном или изобутаном в присутствии кислорода (например, подаваемого в зону реакции в потоке сырья, представляющем собой кислородсодержащий газ, обычно воздух) и аммиака в условиях реакции, эффективных для образования акрилонитрила или метакрилонитрила. Для этой реакции предпочтительно, чтобы поток сырья содержал пропан или изобутан, кислородсодержащий газ, например воздух, и аммиак в следующих мольных соотношениях: пропана или изобутана к кислороду от примерно 0,125 до примерно 5 и предпочтительно от примерно 0,25 до примерно 2,5 и пропана или изобутана к аммиаку в интервале от примерно 0,3 до примерно 2,5 и предпочтительно от примерно 0,5 до примерно 1,5. Исходный поток может содержать один или более дополнительных компонентов сырья, включая продукты - акрилонитрил или метакрилонитрил (например, из потока рецикла или более ранней стадии многостадийного реактора) и/или водяной пар. Например, исходный поток может содержать примерно 5-30 мас. или мольн.% пропана или изобутана от общей массы потока сырья.
Для любой из указанных выше реакций девятого и десятого аспектов изобретения каталитически активную композицию на основе смешанной металлоксидной системы можно поместить в реактор в виде нанесенного катализатора или ненанесенного объемного катализатора. Носители или связующие для использования в нанесенных катализаторах включают силикагель, оксид алюминия, оксид титана, оксид циркония и т.д. Такие нанесенные катализаторы можно приготовить, добавляя эти носители (например, в количестве 20-50 мас.%) в реакционную среду на стадии реакции указанных выше способов. При использовании нанесенных катализаторов предпочтительно, чтобы содержание катализатора находилось в интервале примерно 50-80%.
Конкретная конструкция газофазного проточного реактора не является строго критичной. Так, проточный газофазный реактор может быть реактором с неподвижным слоем, реактором с кипящим слоем или реактором другого типа. Реактор может быть единичным, а может входить в многостадийную систему реакторов. Предпочтительно, чтобы реактор имел один или более входов для подачи исходного потока реагентов в зону реакции, в которую помещен катализатор на основе смешанной металлоксидной системы, и выход для отвода продуктов реакции и непрореагировавших реагентов.
Условия реакции регулируют для достижения максимального эффекта в конверсии пропана в акриловую кислоту или акрилонитрил соответственно или изобутана в метакриловую кислоту или метакрилонитрил соответственно. Условия реакции включают интервал температур от примерно 300°С до примерно 550°С, предпочтительно от примерно 350°С до примерно 450°С и в других вариантах от примерно 430°С до примерно 520°С. Скорость потока пропан- или изобутансодержащего исходного газа через зону реакции газофазного проточного реактора можно регулировать таким образом, чтобы массовая часовая объемная скорость (WHSV) находилась в интервале от примерно 0,02 до примерно 5, предпочтительно от примерно 0,05 до примерно 1 и в некоторых вариантах от примерно 0,1 до примерно 0,5, в каждом случае в граммах пропана или изобутана на грамм катализатора. Давление в зоне реакции можно регулировать в интервале от примерно 0 до примерно 200 фунт/кв.дюйм, предпочтительно от примерно 0 до примерно 100 фунт/кв.дюйм и в некоторых вариантах от примерно 0 до примерно 50 фунт/кв.дюйм.
Кроме того, регулируют условия реакции по теплопереносу и/или температуре. Например, выбирают конфигурацию зоны реакции таким образом, чтобы регулировать теплоперенос в зоне реакции и/или температуру в зоне реакции. Например, окисление пропана и изобутана и окислительный аммонолиз пропана являются экзотермическими реакциями, и поэтому зону реакции надо охлаждать одним или более способами, известными на данном уровне техники.
Для получения целевого продукта реакции (например, акриловой кислоты и/или акрилонитрила или метакриловой кислоты и/или метакрилонитрила) предпочтительно регулировать один или более параметров, включая композицию на основе смешанной металлоксидной системы, состав исходного газа и условия реакции, чтобы выход составлял по меньшей мере примерно 50%, предпочтительно по меньшей мере примерно 53% или больше и наиболее предпочтительно по меньшей мере примерно 55% или больше. Здесь выход в окислении и/или окислительном аммонолизе пропана рассчитан, как показано в примере 5.
Полученный продукт - акриловую кислоту и/или акрилонитрил или метакриловую кислоту и/или метакрилонитрил, - можно отделить, если нужно, от других побочных продуктов и/или непрореагировавших реагентов способами, известными на данном уровне техники.
Полученный продукт - акриловую кислоту и/или акрилонитрил или метакриловую кислоту и/или метакрилонитрил - можно использовать как источник реагентов для многих других целей (например, по ходу потока) в соответствии со способами данного уровня техники.
Следующие примеры иллюстрируют принципы и преимущества изобретения.
ПРИМЕРЫ
Пример 1. Получали катализатор с атомным соотношением Mo/V/Sb/Nb, равным 1/0, 37/0, 13/0, 1 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл помещали 2 мл дистиллированной воды, МоО3 (0,50 г), VOSO4 (1,27 мл 1,0 М раствора) и Sb2O3 (0,0675 г). К суспензии добавляли при перемешивании Н2O2 (0,017 мл 30% раствора). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Отношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,412 М. Добавляли часть раствора оксалата ниобия (0,841 мл 0,413 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Начальный рН реакционной смеси составлял 1,2. Сосуд герметизировали и нагревали до 175°С в течение 48 ч без перемешивания. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 2. Получали катализатор с атомным соотношением Mo/V/Sb/Nb/Ge, равным 1/0, 5/0, 15/0, 1/0, 083 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл помещали 2 мл дистиллированной воды, МоО3 (0,50 г), VOSO4 (1,74 мл 1,0 М раствора), GeO2 (0,030 г) и Sb2О3 (0,076 г). К суспензии добавляли Н2О2 (0,059 мл 30% раствора) при перемешивании. Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Отношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,413 М. Добавляли часть раствора оксалата ниобия (0,841 мл 0,413 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Начальный рН реакционной смеси составлял 1,2. Сосуд герметизировали и нагревали до 175°С в течение 48 ч без перемешивания. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 3. Получали катализатор с атомным соотношением Mo/V/Sb/Nb, равным 1/0, 4/0, 3/0, 06 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл добавляли 2 мл дистиллированной воды. Воду перемешивали магнитной мешалкой при добавлении МоО3 (0,50 г), VOSO4 (1,39 мл 1,0 М раствора) и Sb2О3 (0,152 г). К суспензии добавляли по каплям H2O2 (0,106 мл 30% раствора) и перемешивание продолжали в течение 15 мин. Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Отношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,412 М. Добавляли часть раствора оксалата ниобия (0,506 мл 0,412 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Начальный рН реакционной смеси составлял 1,2. Сосуд герметизировали и нагревали до 175°С в течение 48 час. Во время нагревания сосуд переворачивали для перемешивания реакционной смеси. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 4. Получали катализатор с атомным соотношением Mo/V/Sb/Nb/Ge, равным 1/0, 3/0, 3/0, 06/0,8 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл добавляли 2 мл дистиллированной воды. Воду перемешивали магнитной мешалкой при добавлении МоО3 (0,50 г), VOSO4 (1,04 мл 1,0 М раствора), GeO2 (0,291 г) и Sb2O3 (0,152 г). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Отношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,412 М. Добавляли часть раствора оксалата ниобия (0,506 мл 0,412 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час. Во время нагревания сосуд переворачивали для перемешивания реакционной смеси. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 5. Катализаторы, приготовленные, как описано в примерах 1-4, были испытаны в окислительном аммонолизе пропана до акрилонитрила в реакторе с неподвижным слоем. 150 мг образца катализатора смешивали с трехкратным по объему количеством карбида кремния. Смесь помещали в футерованную стеклом стальную трубку с внутренним диаметром 4 мм. Условия реакции: атмосферное давление, 420 или 430°С, WHSV=0,148 ч-1, соотношение в исходной смеси С3Н8/NH3/O2/Не=1/1,2/3/12. Выходящий поток из реактора анализировали методом газовой хроматографии (колонки с Plot-Q и молекулярными ситами, детекторы пламенно-ионизационный и по теплопроводности соответственно). Конверсию, селективность и выход определяли следующим образом: конверсия = (моль С3Н8 поглощенный/моль С3Н8 введенный) × 100, селективность = (моль продукта/моль С3Н8 поглощенный) × (# С атомов в продукте/3) × 100, выход = (моль продукта/моль
С3Н8 введенный) × (# С атомов в продукте/3) × 100.
Результаты приведены в таблице 1.
Конверсия
Селективность
Пример 6. Получали катализатор с атомным соотношением Mo/V/Sb/Nb/H2O2, равным 1/0, 4/0, 3/0, 06/0, 3 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл добавляли 2 мл дистиллированной воды, МоО3 (0,50 г), VOSO4 (1,39 мл 1,0 М раствора) и Sb2O3 (0,152 г). К суспензии добавляли при перемешивании Н2О2 (0,106 мл 30% раствора). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Сотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,42 М. Добавляли часть раствора оксалата ниобия (0,496 мл 0,42 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час. Во время нагревания сосуд переворачивали для перемешивания реакционной смеси. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Примеры 7. Катализатор готовили таким же способом, как в примере 6, за исключением того, что в смесь для синтеза после добавления Н2О2 добавляли при перемешивании H2SO4 (0,0191 мл 18,2 М раствора).
Пример 8. (1216912) Катализатор готовили таким же способом, как в примере 6, за исключением того, что в смесь для синтеза после добавления Н2О2 добавляли при перемешивании Н2SO4 (0,0954 мл 18,2 М раствора).
Пример 9. Катализатор готовили таким же способом, как в примере 6, за исключением того, что в смесь для синтеза после добавления Н2O2 добавляли при перемешивании Н2SO4 (0,191 мл 18,2 М раствора).
Пример 10. Катализатор готовили таким же способом, как в примере 6, за исключением того, что в смесь для синтеза после добавления Н2О2 добавляли при перемешивании NH4OH (0,233 мл 7,45 М раствора).
Пример 11. Катализатор готовили таким же способом, как в примере 6, за исключением того, что в смесь для синтеза после добавления Н2O2 добавляли при перемешивании NH4OH (0,350 мл 7,45 М раствора).
Пример 12. Получали катализатор с атомным соотношением Mo/V/Sb/Nb/H2O2, равным 1/0, 4/0, 3/0, 06/0, 3 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл добавляли 2 мл дистиллированной воды, МоО3 (0,50 г), NH4VO3 (0,163 г) и Sb2О3 (0,152 г). К суспензии добавляли Н2О2 (0,106 мл 30% раствора) при перемешивании. Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,42 М. Добавляли часть раствора оксалата ниобия (0,496 мл 0,42 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час. Во время нагревания сосуд переворачивали для перемешивания реакционной смеси. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 ч, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 13. Катализатор готовили таким же способом, как в примере 12, за исключением того, что в смесь для синтеза после добавления Н2O2 добавляли при перемешивании H2SO4 (0,0382 мл 18,2 М раствора).
Пример 14. (1216-9-34) Катализатор готовили таким же способом, как в примере 12, за исключением того, что в смесь для синтеза после добавления Н2О2 добавляли при перемешивании Н2SO4 (0,0573 мл 18,2 м раствора).
Пример 15. Катализатор готовили таким же способом, как в примере 12, за исключением того, что в смесь для синтеза после добавления Н2О2 добавляли при перемешивании Н2SO4 (0,0763 мл 18,2 М раствора).
Пример 16. Катализатор готовили таким же способом, как в примере 12, за исключением того, что в смесь для синтеза после добавления Н2О2 добавляли при перемешивании Н2SO4 (0,0954 мл 18,2 М раствора).
Пример 17. Получали катализатор с атомным соотношением Mo/V/Sb/Nb/H2O2, равным 1/0, 4/0, 3/0, 06/0, 3 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл добавляли 2 мл дистиллированной воды, гептамолибдат аммония (0,50 г), NH4VO3 (0,133 г) и Sb2O3 (0,124 г). К суспензии добавляли Н2О2 (0,0868 мл 30% раствора) при перемешивании. Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Отношение оксалат/Nb в этом растворе было равно 3 и концентрация Mb составляла 0,42 М. Добавляли часть раствора оксалата ниобия (0,405 мл 0,42 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час. Во время нагревания сосуд переворачивали для перемешивания реакционной смеси. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в Na при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 18. Получали катализатор с атомным соотношением Mo/V/Sb/Nb/H2O2, равным 1/0, 4/0, 3/0, 06/0, 3 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл добавляли 2 мл дистиллированной воды, гептамолибдат аммония (0,50 г), VOSO4 (1,133 мл 1,0 М раствора) и Sb2О3 (0,124 г). К суспензии добавляли H2O2 (0,0868 мл 30% раствора) при перемешивании. Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,42 М. Добавляли часть раствора оксалата ниобия (0,405 мл 0,42 М раствора). В реакционный сосуд добавляли дистиллированную воду до 75% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час. Во время нагревания сосуд переворачивали для перемешивания реакционной смеси. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 19. Во время синтеза образцов в примерах 6-18 рН реакционной среды измеряли непосредственно перед запаиванием сосуда под давлением для гидротермального синтеза и после того, как сосуд вскрывали после гидротермального синтеза. Электропроводность верхнего слоя жидкости в реакционной среде определяли после гидротермальной обработки. Электропроводность выражали в миллисименсах. Результаты приведены в таблице 2.
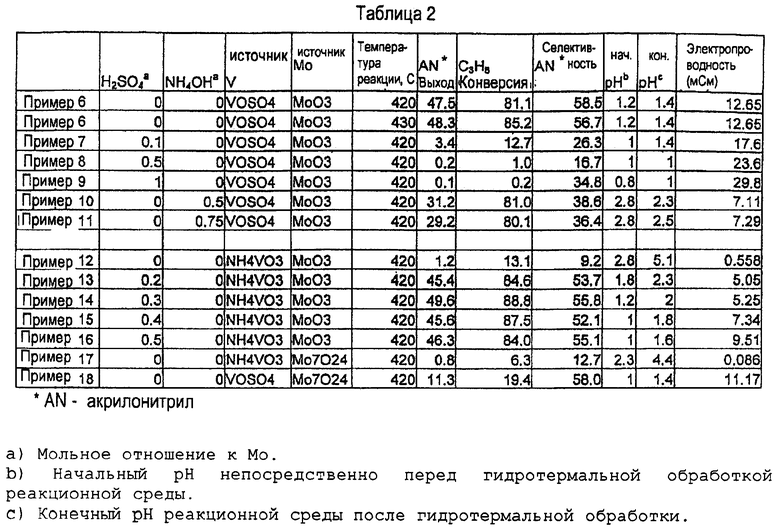
a) Мольное отношение к Mo.
b) Начальный рН непосредственно перед гидротермальной обработкой реакционной среды.
c) Конечный рН реакционной среды после гидротермальной обработки.
Сравнительные примеры 20-24 иллюстрируют катализатор MoVTeNbOx, приготовленный выпариванием растворителя (SE) в присутствии щавелевой кислоты и без нее и прокаленный в различных условиях. Как показано ниже в таблице 3, при добавлении щавелевой кислоты к смеси синтеза и после прокаливания при 600°С в N2 катализатор малоактивен. После прокаливания вещества с добавленной щавелевой кислотой на воздухе при 280°С и затем в N2 при 600°С активность катализатора близка к активности катализатора, приготовленного без щавелевой кислоты. Поэтому в остальных примерах с добавленной щавелевой кислотой или оксалатом Ge вещества прокаливали на воздухе при 280°С и затем в N2 при 600°С.
Сравнительный пример 20. Получали катализатор с атомным соотношением Mo/V/Te/Nb, равным 1/0, 32/0, 2/0, 1 в смеси для синтеза. В 100 мл колбу добавляли 25 мл дистиллированной воды, (NH4)6Mo7O24 (1,412 г) и NH4VO3 (0,299 г). Смесь нагревали до 70°С до растворения твердых веществ. Раствор охлаждали до комнатной температуры, добавляли Те(ОН)6 (0,367 г) и растворяли его. Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,458 М. Добавляли часть раствора оксалата ниобия (1,747 мл 0,458 М раствора). Растворитель удаляли из смеси при пониженном давлении при 50°С. Твердое вещество сушили на воздухе при 120°С в течение 12 час, размалывали и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 21. Катализатор готовили способом, близким к примеру 1, в котором атомное отношение Mo/V/Te/Nb составляло 1/0, 32/0, 2/0, 1 в смеси для синтеза. Перед введением раствора оксалата ниобия к смеси MoVTe добавляли раствор щавелевой кислоты (9,6 мл 0,5 М раствора). Растворитель удаляли из смеси при пониженном давлении при 50°С. Твердое вещество сушили на воздухе при 120°С в течение 12 час, размалывали и прокаливали в атмосфере N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 22. (103791А5) Часть вещества из примера 1, высушенного на воздухе при 120°С, еще нагревали на воздухе при 280°С в течение 2 час. Затем твердое вещество прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 23. (103791А6) Часть вещества из примера 2, высушенного на воздухе при 120°С, далее нагревали на воздухе при 280°С в течение 2 час. Затем вещество прокаливали в N2 при 600°С в течение 2 час. Полученное вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 24. Катализаторы, приготовленные, как описано в примерах 1-4, испытывали в окислительном аммонолизе пропана в акрилонитрил в реакторе с неподвижным слоем. 150 мг образца катализатора смешивали с трехкратным по объему количеством карбида кремния. Смесь загружали в стальную трубку, футерованную стеклом, с внутренним диаметром 4 мм. Условия реакции: атмосферное давление, 420°С, WHSV=0,15 ч-1, соотношение в исходной смеси
С3Н8/NH3/О2/Не=1/1, 2/3/12. Смесь на выходе из реактора анализировали методом газовой хроматографии (колонки с Plot-Q и молекулярными ситами, детекторы пламенно-ионизационный и по теплопроводности соответственно). Конверсию, селективность и выход определяли следующим образом: конверсия = (моль С3Н8 поглощенный/моль C3H8 введенный) × 100, селективность = (моль продукта/моль С3Н8 поглощенные) × (#С атомов в продукте/3) × 100, выход = (моль продукта/моль C3H8 введенный) × (#С атомов в продукте/3) × 100. Результаты приведены в таблице 3.
Конверсию
Селективность
Сравнительные примеры 25-29 иллюстрируют катализаторы MoVTeNbOx+Ge (Ge добавлен в виде оксалата Ge) и MoVTeNbOx + щавелевая кислота, приготовленные выпариванием растворителя. Как показано в таблице 4, добавка Ge понижает активность катализатора, но добавка щавелевой кислоты не вызывает резкого уменьшения активности катализатора. Таким образом, Ge, а не оксалат из предшественника Ge ответственен за уменьшение активности.
Сравнительный пример 25. Получали катализатор с атомным соотношением Mo/V/Te/Nb, равным 1/0, 32/0, 23/0, 1 в смеси для синтеза. В 50 мл колбу добавляли 12 мл дистиллированной воды, (NH4)6MO7O24 (0,500 г) и NH4VO3 (0,106 г). Смесь нагревали до 70°С до растворения твердых веществ. Раствор охлаждали до комнатной температуры и добавляли Те(ОН)6 (1,303 мл 0,5 М раствора). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,458 М. Добавляли часть раствора оксалата ниобия (0,618 мл 0,458 М раствора). Растворитель удаляли из смеси при пониженном давлении при 50°С. Твердое вещество сушили на воздухе при 120°С в течение 12 час, затем нагревали до 280°С на воздухе в течение 2 час, размалывали и прокаливали в N2 при 600°С в течение 2 ч. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 26. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge, равным 1/0, 32/0, 23/0, 1/0, 1 в смеси для синтеза. В 50 мл колбу добавляли 12 мл дистиллированной воды, (NH4)6Mo7O24 (0,500 г) и NH4VO3 (0,106 г). Смесь нагревали до 70°С до растворения твердых веществ. Раствор охлаждали до комнатной температуры и добавляли Те(ОН)6 (1,303 мл 0,5 М раствора). Раствор оксалата германия готовили растворением аморфного оксида германия в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Ge в этом растворе было равно 3 и концентрация Ge составляла 0,5 М. Добавляли часть раствора оксалата германия (0,566 мл 0.5 М раствора). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,458 М. Добавляли часть раствора оксалата ниобия (0,618 мл 0,458 М раствора). Растворитель удаляли из смеси при пониженном давлении при 50°С. Твердое вещество сушили на воздухе при 120°С в течение 12 час, затем нагревали до 280°С на воздухе в течение 2 час, размалывали и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 27. Катализатор готовили, как в сравнительном примере 26, при атомном соотношении Mo/V/Te/Nb/Ge в смеси для синтеза, равном 1/0, 32/0, 23/0, 1/0, 3. Использовали 1,700 мл 0,5 М раствора оксалата германия.
Сравнительный пример 28. Катализатор готовили, как в сравнительном примере 26, при атомном соотношении Mo/V/Te/Nb в смеси для синтеза, равном 1/0, 32/0, 23/0, 1. Перед введением раствора оксалата ниобия к смеси MoVTe добавляли раствор щавелевой кислоты (1,700 мл 0,5 М раствора).
Сравнительный пример 29. Катализатор готовили, как в сравнительном примере 26, при атомном соотношении Mo/V/Te/Nb в смеси для синтеза, равном 1/0, 32/0, 23/0, 1. Перед введением раствора оксалата ниобия к смеси MoVTe добавляли раствор щавелевой кислоты (5,098 мл 0,5 М раствора).
Сравнительные примеры 30-33 иллюстрируют катализатор MoVTeNbOx+Ge, приготовленный гидротермальным (HS) с использованием V2O5 в качестве источника V. Активность таких катализаторов вообще выше, чем активность катализаторов, приготовленных из VOSO4 как источника V. Как показано в таблице 5, при всех концентрациях V, Nb и Те катализатор, не содержащий Ge, всегда активнее, чем образцы, содержащие Ge0.2.
Выход
Конверсия
Селективность
Сравнительный пример 30. Получали катализатор с атомным соотношением Mo/V/Te/Mb, равным 1/0, 36/0, 2/0, 06 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл помещали 2 мл дистиллированной воды, МоО3 (0,50 г), V2O5 (0,1137 г) и TeO2 (0,111 г). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,458 М. Добавляли часть раствора оксалата ниобия (0,455 мл 0,458 М раствора). В реакционный сосуд добавляли дистиллированную воду до 80% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 ч при перемешивании. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 31. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 2. Методика использована такая же, как в сравнительном примере 30, за исключением того, что GeO2 (0,0727 г) добавляли в суспензию синтеза после введения TeO2.
Сравнительный пример 32. Получали катализатор с атомным соотношением Mo/V/Te/Nb в смеси синтеза, равным 1/0, 36/0, 23/0, 06. Была использована методика, описанная в сравнительном примере 30.
Сравнительный пример 33. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 23/0, 06/0, 2. Методика была использована такая же, как в сравнительном примере 30, за исключением того, что GeO2 (0,0727 г) добавляли в суспензию синтеза после введения TeO2 в количестве 0,1275 г.
Выход
Конверсия
Селективность
Сравнительные примеры 34-40 иллюстрируют катализаторы MoVTeNbOx+Ge (6 концентраций), приготовленные гидротермальным синтезом (HS) с использованием V2O5 в качестве источника V. Как показано в таблице 6, при добавлении Ge наблюдается тенденция к понижению активности и увеличению селективности. Общим результатом являются близкие выходы при всех концентрациях Ge, если образцы сравнивают в одинаковых условиях реакции.
Сравнительный пример 34. Получали катализатор с атомным соотношением Mo/V/Te/Nb, равным 1/0, 36/0, 2/0, 06 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл помещали 2 мл дистиллированной воды, МоО3 (0,50 г), V2O5 (0,114 г) и TeO2 (0,111 г). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,399 М. Добавляли часть раствора оксалата ниобия (0,522 мл 0,399 М раствора). В реакционный сосуд добавляли дистиллированную воду до 80% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час при перемешивании. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 ч, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Сравнительный пример 35. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 05. Методика была использована такая же, как в сравнительном примере 34, за исключением того, что GeO2 (0,0182 г) добавляли в суспензию синтеза после введения ТеО2.
Сравнительный пример 36. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 1. Методика была использована такая же, как в сравнительном примере 34, за исключением того, что GeO2 (0,0363 г) добавляли в суспензию синтеза после введения ТеО2.
Сравнительный пример 37. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 15. Методика была использована такая же, как в сравнительном примере 34, за исключением того, что GeO2 (0,0545 г) добавляли в суспензию синтеза после введения TeO2.
Сравнительный пример 38. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 2. Методика была использована такая же, как в сравнительном примере 34, за исключением того, что GeO2 (0,0727 г) добавляли в суспензию синтеза после введения TeO2.
Сравнительный пример 39. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 3. Методика была использована такая же, как в сравнительном примере 34, за исключением того, что GeO2 (0,109 г) добавляли в суспензию синтеза после введения TeO2.
Сравнительный пример 40. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 36/0, 2/0, 06/0, 4. Методика была использована такая же, как в сравнительном примере 34, за исключением того, что GeO2 (0,145 г) добавляли в суспензию синтеза после введения TeO2.
Выход
Конверсия
Селективность
Примеры 41-46 иллюстрируют катализаторы MoVSbNbOx+Ge (6 концентраций), приготовленные гидротермальным (HS) синтезом с использованием VOSO4 в качестве источника V. Данные таблицы 6 показывают, что (i) Ge-содержащие катализаторы более активны, чем катализатор, не содержащий Ge, и (ii) повышение концентрации Ge в катализаторе влияет на активность катализаторов MoVSbNbOx+Ge. Результаты приведены в таблице 7.
Пример 41. Получали катализатор с атомным соотношением Mo/V/Sb/Nb, равным 1/0, 32/0, 2/0, 06 в смеси для синтеза. В футерованный тефлоном реакционный сосуд объемом 7,0 мл помещали 2 мл дистиллированной воды, МоО3 (0,50 г), VOSO4 (1,112 мл 1,0 М раствора) и Sb2O3 (0,1013 г). Раствор оксалата ниобия готовили растворением ниобиевой кислоты в растворе щавелевой кислоты при 60°С. Соотношение оксалат/Nb в этом растворе было равно 3 и концентрация Nb составляла 0,458 М. Добавляли часть раствора оксалата ниобия (0,455 мл 0,458 М раствора) при перемешивании. В реакционный сосуд добавляли дистиллированную воду до 80% объема. Сосуд герметизировали и нагревали до 175°С в течение 48 час при перемешивании. Затем реактору давали остыть до комнатной температуры. Твердые продукты реакции отделяли от жидкости и трижды промывали дистиллированной водой. Затем твердый остаток сушили на воздухе при 120°С в течение 12 час, дробили и прокаливали в N2 при 600°С в течение 2 час. Вещество измельчали в мелкий порошок в шаровой мельнице, прессовали в таблетки, размалывали и просеивали, получая частицы размером 145-355 мкм.
Пример 42. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 32/0, 2/0, 06/0, 05. Методика была использована такая же, как в сравнительном примере 41, за исключением того, что GeO2 (0,0182 г) добавляли в суспензию синтеза после введения Sb2О3.
Пример 43. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 32/0, 2/0, 06/0, 1. Методика была использована такая же, как в сравнительном примере 41, за исключением того, что GeO2 (0,0363 г) добавляли в суспензию синтеза после введения Sb2O3.
Пример 44. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 32/0, 2/0, 06/0, 15. Методика была использована такая же, как в сравнительном примере 41, за исключением того, что GeO2 (0,0545 г) добавляли в суспензию синтеза после введения Sb2O3.
Пример 45. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 32/0, 2/0, 06/0, 2. Методика была использована такая же, как в сравнительном примере 41, за исключением того, что GeO2 (0,0727 г) добавляли в суспензию синтеза после введения Sb2О3.
Пример 46. Получали катализатор с атомным соотношением Mo/V/Te/Nb/Ge в смеси синтеза, составлявшим 1/0, 32/0, 2/0, 06/0, 4. Методика была использована такая же, как в сравнительном примере 41, за исключением того, что GeO2 (0,145 г) добавляли в суспензию синтеза после введения Sb2О3.
Выход
Конверсия
Селективность
Сравнительный пример 47 и примеры 48-50 иллюстрируют конверсию пропана в акрилонитрил в присутствии катализатора MoVSbNbOx+Ge, приготовленного гидротермальным синтезом (HS) различными порциями (23 мл, 450 мл и 1 галлон).
Катализатор с номинальным составом MolV0.3Nb0.06Sb0.20Ge0.30 готовили гидротермальным синтезом следующим образом. Сначала готовили отдельно два раствора. Первый раствор содержал 0,9 г VOSO4, 0,2 г МоО3, 0,41 г Sb2О3 и 0,44 г аморфного GeO2. Второй раствор содержал 0,32 г дигидрата щавелевой кислоты и 0,14 г ниобиевой кислоты, нагретой до 60°С. Второй раствор добавляли к первому раствору и полученную смесь помещали в 23 мл автоклав, облицованный тефлоном. Автоклав герметизировали и нагревали до 175°С в темение 48 час при вращении. Через 48 час реактор охлаждали до комнатной температуры, открывали и отфильтровывали твердые вещества, промывали, сушили на воздухе при 90°С, дробили и прокаливали в азоте при 600°С в течение двух часов. Прокаленное вещество измельчали в тонкий порошок, прессовали в таблетки, дробили и просеивали до нужного размера частиц. Эту процедуру повторяли в автоклавах емкостью 450 мл (пример 49) и 1 галлон (пример 48). Эту же процедуру также повторяли с катализатором, не содержащим Ge (сравнительный пример 47).
Обычно 0,5 г катализатора и 2,5 г инертных кварцевых чешуек помещали в небольшой реактор для испытания. Состав подаваемого газа был следующий: 1,0 С3/1,2 NH3/3 O2/12 N2. Температура в реакторе была 410°С. Результаты тестирования этих катализаторов в окислительном аммонолизе пропана приведены в таблице 8.
Приведенные объяснения и примеры предназначены для того, чтобы ознакомить специалистов с изобретением, его принципами и практическим применением. Специалисты могут адаптировать и применить изобретение в его многочисленных формах, наилучшим образом отвечающих конкретному применению. Соответственно примеры и варианты настоящего изобретения, изложенные здесь, не претендуют на исчерпывающую полноту и не ограничивают изобретение.
Изобретение относится к смешанным металлоксидным катализаторам окисления и окислительного аммонилиза пропана и изобутана, способам их получения и применения. Описана смешанная металлоксидная система, содержащая молибден, ванадий, ниобий, сурьму, германий и кислород или молибден, ванадий, тантал, сурьму, германий и кислород, в которой стехиометрические соотношения элементов включают соотношение молибдена к сурьме в интервале от примерно 1:0,1 до примерно 1:0,5 и соотношение молибдена к германию в интервале от примерно 1:>0,2 до примерно 1:1. Описан катализатор, представляющий собой смешанную металлоксидную систему, эффективную в парофазной конверсии пропана в акриловую кислоту или акрилонитрил или изобутана в метакриловую кислоту или метакрилонитрил, причем смешанная металлоксидная система имеет эмпирическую формулу
Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой
а находится в интервале от примерно 0,1 до примерно 0,6,
b находится в интервале от примерно 0,02 до примерно 0,12,
с находится в интервале от примерно 0,1 до примерно 0,5,
d находится в интервале от более 0,2 до примерно 1
и х зависит от степени окисления других элементов в составе смешанной металлоксидной системы. Описан способ получения указанной выше системы, включающий стадии: введение в реакционный сосуд предшественников Мо, V, Nb или Та, Ge и Sb в водном растворителе для образования реакционной среды с начальным рН 4 или меньше, необязательно, добавление дополнительного водного растворителя в реакционный сосуд; герметизацию реакционного сосуда; реакцию в реакционной смеси при температуре выше 100°С и давлении выше атмосферного в течение времени, достаточного для образования смешанной металлоксидной системы; необязательно, охлаждение реакционной смеси; и выделение смешанной металлоксидной системы из реакционной смеси. Описаны способы превращение пропана в акрилонитрил и изобутана в метакрилонитрил с использованием описанного выше катализатора. Технический эффект - упрощение технологии приготовления катализатора, повышение активности катализатора и выхода целевого продукта в реакциях окислительного аммонолиза пропана и изобутана. 6 н. и 21 з.п. ф-лы, 8 табл.,2 ил.
1. Смешанная металлоксидная система, содержащая молибден, ванадий, ниобий, сурьму, германий и кислород или молибден, ванадий, тантал, сурьму, германий и кислород, в которой стехиометрические соотношения элементов включают соотношение молибдена к сурьме в интервале от примерно 1:0,1 до примерно 1:0,5 и соотношение молибдена к германию в интервале от примерно 1:>0,2 до примерно 1:1.
2. Смешанная металлоксидная система по п.1, в которой стехиометрические соотношения элементов включают соотношение молибдена к ванадию в интервале от примерно 1:0,1 до примерно 1:0,6, соотношение молибдена к ниобию или танталу в интервале от примерно 1:0,02 до примерно 1:0,12, соотношение молибдена к сурьме в интервале от примерно 1:0,1 до примерно 1:0,5 и соотношение молибдена к германию в интервале от примерно 1:>0,2 до примерно 1:1.
3. Катализатор, представляющий собой смешанную металлоксидную систему, эффективную в парофазной конверсии пропана в акриловую кислоту или акрилонитрил или изобутана в метакриловую кислоту или метакрилонитрил, причем смешанная металлоксидная система содержит молибден, ванадий, ниобий, сурьму, германий и кислород или молибден, ванадий, тантал, сурьму, германий и кислород, в которой стехиометрические соотношения элементов включают соотношение молибдена к сурьме в интервале от примерно 1:0,1 до примерно 1:0,5 и соотношение молибдена к германию в интервале от примерно 1:>0,2 до примерно 1:1.
4. Катализатор по п.3, в котором стехиометрические соотношения элементов в смешанной металлоксидной системе включают соотношение молибдена к ванадию в интервале от примерно 1:0,1 до примерно 1:0,6, соотношение молибдена к ниобию или танталу в интервале от примерно 1:0,02 до примерно 1:0,12, соотношение молибдена к сурьме в интервале от примерно 1:0,1 до примерно 1:0,5 и соотношение молибдена к германию в интервале от примерно 1:>0,2 до примерно 1:1.
5. Катализатор, представляющий собой смешанную металлоксидную систему, эффективную в парофазной конверсии пропана в акриловую кислоту или акрилонитрил или изобутана в метакриловую кислоту или метакрилонитрил, причем смешанная металлоксидная система имеет эмпирическую формулу
Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой
а находится в интервале от примерно 0,1 до примерно 0,6,
b находится в интервале от примерно 0,02 до примерно 0,12,
с находится в интервале от примерно 0,1 до примерно 0,5,
d находится в интервале от более 0,2 до примерно 1,
и х зависит от степени окисления других элементов в составе смешанной металлоксидной системы.
6. Катализатор по п.5, в котором смешанная металлоксидная система не содержит теллура.
7. Катализатор по п.5, в котором смешанная металлоксидная система не содержит церия.
8. Катализатор по п.5, в котором смешанная металлоксидная система не содержит галлия.
9. Катализатор по п.5, в котором смешанная металлоксидная система не содержит теллура, церия и галлия.
10. Катализатор по п.5, в котором смешанная металлоксидная система состоит в основном из молибдена, ванадия, ниобия, сурьмы, германия и кислорода или молибдена, ванадия, тантала, сурьмы, германия и кислорода.
11. Катализатор по п.5, в котором смешанная металлоксидная система содержит один или более дополнительных элементов.
12. Катализатор по п.5, в котором смешанная металлоксидная система содержит один или более дополнительных элементов, которые выбирают из группы, состоящей из щелочных металлов, щелочноземельных металлов, редкоземельных металлов, лантанидов и металлов основных групп.
13. Катализатор по п.5, в котором смешанная металлоксидная система представляет собой нанесенную смешанную металлоксидную систему.
14. Катализатор по п.5, в котором смешанная металлоксидная система содержит один или более связующих.
15. Способ получения смешанной металлоксидной системы, содержащей молибден, ванадий, ниобий или тантал, сурьму, германий и кислород, в которой стехиометрические соотношения элементов включают соотношение молибдена к сурьме в интервале от примерно 1:0,1 до примерно 1:0,5 и соотношение молибдена к германию в интервале от примерно 1:>0,2 до примерно 1:1, включающий стадии:
введение в реакционный сосуд предшественников Мо, V, Nb или Та, Ge и Sb в водном растворителе для образования реакционной среды с начальным рН 4 или меньше;
необязательно, добавление дополнительного водного растворителя в реакционный сосуд;
герметизацию реакционного сосуда;
реакцию в реакционной смеси при температуре выше 100°С и давлении выше атмосферного в течение времени, достаточного для образования смешанной металлоксидной системы;
необязательно, охлаждение реакционной смеси;
и выделение смешанной металлоксидной системы из реакционной смеси.
16. Способ по п.15, в котором стадия введения реагентов протекает с перемешиванием.
17. Способ по п.16, в котором стадия введения включает следующие стадии:
введение предшественников Мо, V, Sb и Ge;
добавление окислителя для окисления по меньшей мере части V и Sb;
и после того, как окисление V и Sb существенно заканчивается, добавление водного раствора оксалата ниобия в качестве источника Nb и Та.
18. Способ по п.17, в котором окислителем является Н2О2.
19. Способ по п.15, включающий дополнительно после стадии выделения следующие стадии;
необязательно, промывку выделенной смешанной металлоксидной системы;
сушку выделенной смешанной металлоксидной системы;
и прокаливание выделенной смешанной металлоксидной системы.
20. Способ по п.15, в котором реакционная среда имеет рН не выше примерно 1,5.
21. Способ по п.15, в котором смешанная металлоксидная система имеет эмпирическую формулу Mo1VaNbbSbcGedOx и на стадии введения соединения Мо, V, Nb, Sb и Ge добавляют в таких относительных мольных количествах, чтобы а находился в интервале от примерно 0,1 до примерно 0,6, b находился в интервале от примерно 0,02 до примерно 0,12, с находился в интервале от примерно 0,1 до примерно 0,5, d находился в интервале от примерно 0,01 до примерно 1, а х зависит от степени окисления других элементов в смешанной металлоксидной системе, или эмпирическую формулу Mo1VaTabSbcGedOx и на стадии введения соединения Мо, V, Та, Sb и Ge добавляют в таких относительных мольных количествах, чтобы а находился в интервале от примерно 0,1 до примерно 0,6, b находился в интервале от примерно 0,02 до примерно 0,12, с находился в интервале от примерно 0,1 до примерно 0,5, d находился в интервале от больше 0,2 до примерно 1, а х зависит от степени окисления других элементов в конечной смешанной металлоксидной системе.
22. Способ превращения пропана в акрилонитрил, который включает:
контактирование катализатора с пропаном в газофазном реакторе в присутствии кислорода и аммиака в условиях реакции с образованием акрилонитрила, причем катализатор имеет эмпирическую формулу:
Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой
а находится в интервале от примерно 0,1 до примерно 0,6,
b находится в интервале от примерно 0,02 до примерно 0,12,
с находится в интервале от примерно 0,1 до примерно 0,5,
d находится в интервале от более 0,2 до примерно 1,
и х зависит от степени окисления других элементов в составе катализатора.
23. Способ по п.22, в котором катализатор приводят в контакт с пропаном в реакторе в присутствии кислорода и аммиака в условиях реакции при температуре в интервале от примерно 300°С до примерно 550°С и при давлении в интервале от примерно 0 до примерно 200 фунт/кв.дюйм (14 кг/см2).
24. Способ по п.22, в котором катализатор приводят в контакт с пропаном в реакторе в присутствии кислорода и аммиака в условиях реакции при массовой часовой объемной скорости в интервале от примерно 0,02 до примерно 5.
25. Способ превращения изобутана в метакрилонитрил, который включает:
контактирование катализатора с изобутаном в газофазном реакторе в присутствии кислорода и аммиака в условиях реакции с образованием метакрилонитрила, причем катализатор имеет эмпирическую формулу:
Mo1VaNbbSbcGedOx или Mo1VaTabSbcGedOx, в которой
а находится в интервале от примерно 0,1 до примерно 0,6,
b находится в интервале от примерно 0,02 до примерно 0,12,
с находится в интервале от примерно 0,1 до примерно 0,5,
d находится в интервале от более 0,2 до примерно 1,
и х зависит от степени окисления других элементов в составе катализатора.
26. Способ по п.25, в котором катализатор приводят в контакт с изобутаном в реакторе в присутствии кислорода и аммиака в условиях реакции при температуре в интервале от примерно 300°С до примерно 550°С и давлении в интервале от примерно 0 до примерно 200 фунт/кв.дюйм (14 кг/см2).
27. Способ по п.25, в котором катализатор приводят в контакт с изобутаном в реакторе в присутствии кислорода и аммиака в условиях реакции при массовой часовой объемной скорости в интервале от примерно 0,02 до примерно 5.
| JP 10128112 А, 19.05.1998 | |||
| JP 2000143244 А, 23.05.2000 | |||
| СПОСОБ ПОЛУЧЕНИЯ СОДЕРЖАЩИХ СУРЬМУ КАТАЛИЗАТОРОВ ДЛЯ (АММ) ОКСИДИРОВАНИЯ АЛКАНОВ И АЛКЕНОВ | 1998 |
|
RU2200061C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРОВ НА ОСНОВЕ АНТИМОНАТА ВАНАДИЯ С ПРИМЕНЕНИЕМ SNO•xHO | 1998 |
|
RU2195999C2 |
| JP 11114426 А, 27.04.1999 | |||
| JP 2002219362 А, 06.08.2002 | |||
| WATANABE H | |||
| et al | |||
| "New synthesis route for Mo-V-Nb-Te mixed oxides catalyst for propane ammoxidation", APPLITED CATALYSIS A: GENERAL, ELSEVIER SCIENCE, AMSTERDAM, NL, vol.194-195, 13.03.2000. | |||

