Область техники
[0001]
Настоящее изобретение относится к оксидному катализатору и способу получения ненасыщенного нитрила.
Уровень техники
[0002]
В прошлом в качестве катализатора, применяемого для получения ненасыщенной карбоновой кислоты или ненасыщенного нитрила посредством реакции газофазного каталитического окисления или реакции газофазного каталитического аммоксидирования пропилена или изобутилена, использовали сложный оксид, содержащий множество металлов, таких как молибден и ванадий. Сложный оксид, содержащий множество металлов, таких как молибден и ванадий, также использовали в производстве соответствующего ненасыщенного нитрила с применением пропана или изобутана вместо олефина в качестве исходного соединения.
[0003]
Непатентный документ 1 описывает оксидный катализатор, содержащий молибден (Mo), ванадий (V), сурьму (Sb) и ниобий (Nb). Непатентный документ 1 поясняет механизм окислительно-восстановительного действия оксидного катализатора и, в частности, описывает катализатор Mo/V/Sb/Nb=1/0,33/0,15/0,11 в качестве оксидного катализатора.
[0004]
Патентный документ 1 описывает каталитическую композицию для осуществления аммоксидирования пропана в газовой фазе, где каталитическая композиция содержит одну или несколько кристаллических фаз, где, по меньшей мере, одна из кристаллических фаз представляет собой первую фазу, содержащую кристаллическую структуру M1 и содержащую смешанный оксид металла, содержащий молибден (Мо), ванадий (V), сурьму (Sb) и ниобий (Nb); и первая фаза имеет объем единичной решетки в диапазоне от 2250 A3 до 2350 A3, первый размер кристалла и второй размер, который пересекает первый размер кристалла, при условии, что отношение первого размера ко второму размеру находится в диапазоне от 2,5 до 0,7. Описано, что каталитическая композиция проявляет свойства, которые делают аммоксидирование насыщенного углеводорода до соответствующего ненасыщенного нитрила легким с высоким выходом.
Список цитируемой литературы
Патентные документы
[0005]
[Патентный документ 1]: японский патент No. 5547057
Непатентные документы
[0006]
[Непатентный документ 1]: Safonova, O., et. al., J. Phys. Chem. B 2006, 110, 23962 to 23967
[Непатентный документ 2]: Millet, J.M., et. al., Appl. Catal., A 2002, 232, 77
[Непатентный документ 3]: Millet, J.M., et. al., Appl. Catal., A 2003, 244, 359
[Непатентный документ 4]: Baca, M., et. al., Top. Catal. 2003, 23, 39
[Непатентный документ 5]: Shannon et al., Acta A 32 (1976) 751
Сущность настоящего изобретения
Техническая проблема
[0007]
При газофазном каталитическом аммоксидировании пропана или изобутана улучшение выхода продукта и увеличение производительности являются проблемами. Непатентный документ 1 описывает только, как упомянуто выше, механизм окислительно-восстановительного действия оксидного катализатора, содержащего молибден (Mo), ванадий (V), сурьму (Sb) и ниобий (Nb) и имеющего ромбическую кристаллическую структуру, но исследования по повышению выхода ненасыщенного нитрила не проводились.
[0008]
Патентный документ 1 описывает, что выход ненасыщенного нитрила можно увеличить, контролируя соотношение сторон данного оксидного катализатора, но исследования по контролю соотношения элементов, которые образуют кристаллическую структуру оксидного катализатора, не проводились.
[0009]
Настоящее изобретение было выполнено с учетом данных проблем, и цель настоящего изобретения состоит в том, чтобы предложить оксидный катализатор, способный повысить выход ненасыщенного нитрила, который является продуктом реакции газофазного каталитического аммоксидирования пропана или изобутана, и способ получения ненасыщенного нитрила с применением данного оксидного катализатора.
Решение проблемы
[0010]
Авторы настоящего изобретения провели тщательные исследования, чтобы решить проблемы, и обнаружили, что оксидный катализатор, содержащий сложный оксид, имеющий заданное соотношение элементов, в частности сложный оксид, в котором доля содержания Nb увеличена, и доля содержания V относительно снижена, может улучшить выход ненасыщенного нитрила, в результате чего было выполнено настоящее изобретение.
[0011]
То есть, настоящее изобретение включает следующие аспекты.
[1]
Оксидный катализатор для применения в газофазной каталитической реакции аммоксидирования пропана или изобутана, причем оксидный катализатор содержит сложный оксид, в котором
сложный оксид содержит каталитически активные частицы, которые выделяют из сложного оксида с применением раствора пероксида водорода, и
данные каталитически активные частицы имеют средний состав, определенный измерениями с помощью STEM-EDX, представленный следующей формулой (1):
Mo1VaSbbNbcWdXeOn (1)
где X представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Te, Ce, Ti и Ta; a, b, c, и d удовлетворяют соотношениям, представленным формулами 0,050≤a≤0,200, 0,050≤b≤0,200, 0,100≤c≤0,300, 0≤d≤0,100, 0≤e≤0,100, и a≤c; и n представляет собой число, определяемое валентностями других элементов.
[2]
Оксидный катализатор по пункту [1], где формула (1) удовлетворяет соотношению, представленному формулой 1,1×a ≤ c.
[3]
Оксидный катализатор по пункту [1], где формула (1) удовлетворяет соотношению, представленному формулой 1,3×a ≤ c.
[4]
Оксидный катализатор по любому из [1]-[3], где формула (1) удовлетворяет соотношению, представленному формулой 8,00 ≤ 100×b/(1+a) ≤ 10,00.
[5]
Оксидный катализатор по любому из [1]-[4], где
оксидный катализатор содержит диоксид кремния в качестве носителя, на который нанесен сложный оксид, и
массовая доля диоксида кремния составляет от 30 до 70% по массе в пересчете на SiO2 относительно общего количества оксидного катализатора.
[6]
Оксидный катализатор для применения в газофазной каталитической реакции аммоксидирования пропана или изобутана, причем оксидный катализатор содержит сложный оксид, в котором
сложный оксид содержит каталитически активные частицы, содержащие молибден, ванадий, сурьму и ниобий, и
массовая доля каталитически активных частиц составляет 45% по массе или более от общего количества сложного оксида.
[7]
Способ получения ненасыщенного нитрила, причем способ включает получение ненасыщенного нитрила посредством реакции газофазного каталитического аммоксидирования пропана или изобутана в присутствии оксидного катализатора по любому из [1]-[6].
Полезные эффекты настоящего изобретения
[0012]
Согласно настоящему изобретению, обеспечивают оксидный катализатор, способный повысить выход ненасыщенного нитрила, который является продуктом газофазной каталитической реакции аммоксидирования пропана или изобутана, и способ получения ненасыщенного нитрила, применяя данный оксидный катализатор.
Описание вариантов осуществления
[0013]
Далее будет подробно описан вариант осуществления настоящего изобретения (в дальнейшем именуемый «настоящий вариант осуществления»). Следует отметить, что настоящее изобретение не ограничивается следующим настоящим вариантом осуществления и может быть реализовано модификацией настоящего варианта осуществления различными способами в пределах объема настоящего изобретения. В случае, когда числовые значения или значения физических свойств выражены до и после «-», таким образом, что между ними в настоящем описании вставлено «-», значения применяют при условии, что значения до и после «-» являются включенными. Например, обозначение диапазона числовых значений от «1 до 100» включает в себя как верхнее предельное значение «100», так и нижнее предельное значение «1». То же самое относится к обозначениям других диапазонов числовых значений.
[0014]
[Оксидный катализатор]
Оксидный катализатор настоящего варианта осуществления применяют для реакции газофазного каталитического аммоксидирования пропана и изобутана, и он представляет собой оксидный катализатор, содержащий сложный оксид. Далее, реакцию получения ненасыщенного нитрила из пропана или изобутана, источника кислорода, такого как кислород, и источника азота, такого как аммиак, называют просто "реакция газофазного каталитического аммоксидирования".
[0015]
Оксидный катализатор настоящего варианта осуществления содержит предварительно определенный составной оксид и, если необходимо, может содержать носитель, на который нанесен сложный оксид. Далее будет подробно описан каждый компонент.
[0016]
(Сложный оксид)
Сложный оксид содержит каталитически активные частицы, которые выделяют из сложного оксида, применяя раствор пероксида водорода, и каталитически активные частицы имеют средний состав, определенный измерениями по методике STEM-EDX, представленный следующей формулой (1).
Формула:
Mo1VaSbbNbcWdXeOn (1)
где X представляет собой, по меньшей мере, то, что выбрано из группы, состоящей из Te, Ce, Ti и Ta; a, b, c, и d удовлетворяют соотношениям, представленным формулами 0,050≤a≤0,200, 0,050≤b≤0,200, 0,100≤c≤0,300, 0≤d≤0,100, 0≤e≤0,100, и a≤c; и n представляет собой число, определяемое валентностями других элементов.
[0017]
Каталитически активные частицы в настоящем варианте осуществления можно выделять из оксидного катализатора, более конкретно из сложного оксида, применяя раствор пероксида водорода, и таким образом выделенные вещества называют каталитически активными частицами. В этом случае, в случае, когда оксидный катализатор содержит носитель, каталитические частицы можно выделять в состоянии, в котором смешивают носитель, но каждый из носителя и каталитически активных частиц представляет собой разные компоненты. Средний состав выделенных каталитически активных частиц измеряют с помощью STEM-EDX. STEM-EDX в настоящем изобретение представляет собой сканирующий просвечивающий электронный микроскоп, оснащенный рентгеновским спектрометром с энергодисперсионными характеристиками.
[0018]
Каталитически активные частицы в настоящем изобретении представляют собой кристаллическую фазу, содержащую, по меньшей мере, молибден (Mo), ванадий (V), сурьму (Sb) и ниобий (Nb), и сложный оксид металлов, обладающий каталитической активностью для аммоксидирования пропана и/или изобутана. Следует отметить, что при необходимости, по меньшей мере, один дополнительный металл (X), выбранный из группы, состоящей из вольфрама (W), теллура (Te), церия (Ce), титана (Ti) и тантала (Ta) может дополнительно содержаться в каталитически активных частицах.
[0019]
Ранее приведенные каталитически активные частицы описаны, например, в качестве первой фазы, имеющей M1 кристаллическую структуру в патентной литературе 1 (японский патент No. 5547057), и аналогичное объяснение приведено в непатентной литературе 1 (Safonova, O., et. al., J. Phys. Chem. B 2006, 110, 23962 to 23967), непатентной литературе 2 (Millet, J.M., et. al., Appl. Catal., A 2002, 232, 77), непатентной литературе 3 (Millet, J.M., et. al., Appl. Catal., A 2003, 244, 359), непатентной литературе 4 (Baca, M., et. al., Top. Catal. 2003, 23, 39) и подобных.
[0020]
В результате тщательных исследований, проведенных авторами настоящего изобретения, стало ясно, что применяя оксидный катализатор, в котором относительное содержание Nb в каталитически активных частицах увеличено, и относительное содержание V относительно снижено, достигают повышения выхода ненасыщенного нитрила в газофазной каталитической реакции аммоксидирования. То есть, когда формула (1) каталитически активных частиц имеет вышеописанный средний состав, тем самым повышают выход ненасыщенного нитрила, получаемого посредством реакции газофазного каталитического аммоксидирования.
[0021]
Как будет упомянуто ниже в способе получения оксидного катализатора, было обнаружено, что при применении конкретной Nb-содержащей жидкости в качестве исходного материала или/и дополнительном осуществлении конкретной стадии получения катализатора относительное можно повысить содержание Nb в каталитически активных частицах и можно относительно уменьшить относительное количество V. Согласно непатентной литературе 5 (Shannon et al., Acta A 32 (1976) 751), ионные радиусы Nb и V (в случае пяти валентностей и шестикоординированности в оксиде) составляют 0,64 и 0,54 Å соответственно, и между ними есть большая разница до 18,5%, и поэтому обычно считается, что твердый раствор замещения вряд ли образуется по правилам Юма-Розери. Приведенные в настоящем изобретении правила Юма-Розери представляют собой правила, показывающие, что в твердых растворах замещения твердый раствор образуется почти во всем соотношении компонентов, если разница в ионном радиусе соответствующих компонентов составляет примерно до 10%, но, с другой стороны, в случае, когда ионные радиусы отличаются на 15% и более, твердый раствор практически не образуется.
Предполагается, что специфические явления замещения, такие как те, в которых сайт, который занимает Mo, имеющий ионный радиус 0,59 Å, причем ионный радиус таков, что разница в ионном радиусе относительно мала для обоих Nb и V (Nb: 8,5%), замещается Nb, и что дестабилизированный соседний сайт, который занимает V, замещается Мо, являются результатом того, что настоящие каталитически активные частицы имеют чрезвычайно сложную кристаллическую структуру, хотя подробная структура замещения не совсем понятна.
[0022]
Фактор, который может улучшить выход ненасыщенного нитрила увеличением относительного содержания Nb в каталитических активных частицах и относительным уменьшением относительного содержания V, рассматривается следующим образом. Прежде всего, Nb имеет высокую температуру плавления, и когда относительное содержание Nb, имеющего высокую температуру плавления, увеличивают, стабильность каталитически активных частиц в реакционной атмосфере увеличивается. Считают, что таким образом можно улучшить выход ненасыщенного нитрила.
[0023]
В настоящем варианте осуществления, предпочтительно относительно увеличивать относительное содержание Sb относительно относительного содержания Mo-O-V в каталитически активных частицах по сравнению с содержанием в общепринятых катализаторах. Посредством этого, относительное содержание Mo-O-V, которое, как считают, вызывает активность по неселективному разложению, хотя Mo-OV является активным участком реакции дегидрирования пропана или изобутана, можно относительно снизить, и относительное содержание Sb, которая является активным участком газофазной каталитической реакции аммоксидирования, можно относительно увеличить. Считают, что поэтому можно увеличить выход ненасыщенного нитрила.
[0024]
Ненасыщенный нитрил означает акрилонитрил в качестве предпочтительного аспекта настоящего варианта осуществления.
[0025]
Сложный оксид в настоящем варианте осуществления представляет собой оксид металла, который составляет оксидный катализатор настоящего варианта осуществления, по меньшей мере, молибден (Mo), ванадий (V), сурьма (Sb), и ниобий (Nb) включены в качестве металлов оксида металла, и при необходимости, можно включать дополнительный металл.
[0026]
Каталитически активные частицы в настоящем варианте осуществления обозначает металлические компоненты, которые получают обработкой перекисью водорода, как показано в вышеописанном способе, в частности, в способе измерения (физическое свойство 1), который будет упомянут ниже. Обычно, сложный оксид содержит две кристаллические фазы и аморфный компонент во многих случаях. Данные две кристаллические фазы состоят из фазы, обладающей высокой стойкостью к окислению, и фазы, имеющей слабую стойкость к окислению, и известно, что в настоящей каталитической системе фаза, обладающая высокой стойкостью к окислению, соответствует каталитически активным частицам, как описано в непатентной литературе 1. Когда проводят предварительную обработку, применяя раствор пероксида водорода, сложного оксида, фаза, имеющая слабую стойкость к окислению, растворяется, но каталитически активные частицы, которые представляют собой фазу, обладающую высокой стойкостью к окислению, не растворяются, так что может сохраняться состояние до окислительной обработки окислением. Следовательно, проведя окислительную обработку, фаза, имеющая слабую стойкость к окислению, растворяется, так что каталитически активные частицы можно отделить от сложного оксида, и состав каталитически активных частиц можно измерить с высокой точностью.
[0027]
Как показано в способе измерения (физическое свойство 4), который будет упомянут ниже, массовую долю каталитически активных частиц (G1), предполагая, что масса сложного оксида составляет 100%, можно рассчитать, применяя следующее уравнение, из массовой доли носителя, у которого почти не наблюдается увеличения или уменьшения веса до и после обработки. Массовая доля каталитически активных частиц предпочтительно составляет 43% по массе или более, более предпочтительно 45% по массе или более, и еще более предпочтительно 48% по массе или более по массовой доле.
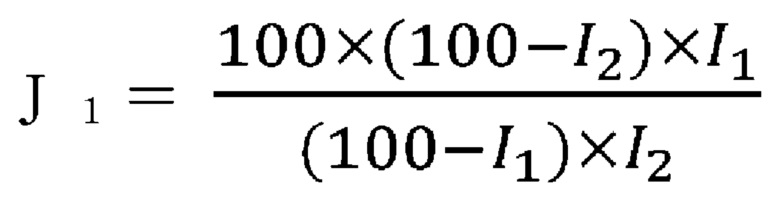
I1: массовая доля носителя в оксидном катализаторе
I2: массовая доля носителя в остатке оксидного катализатора, полученном окислительной обработкой раствором пероксида водорода
[0028]
Каталитически активные частицы могут дополнительно содержать или необязательно содержать дополнительный металлический элемент (X). Примеры дополнительного металлического элемента (X) включают, но конкретно не ограничены, Te, Ce, Ti и Ta. В случае, когда каталитически активные частицы содержит дополнительный металлический элемент (X), дополнительный металлический элемент (X) может быть одним металлическим элементом или может быть множеством металлических элементов. Следует отметить, что в случае, когда каталитически активные частицы не содержат дополнительный металлический элемент (X), формула (1) представлена следующей формулой (1a).
Mo1VaSbbNbcWdOn (1a)
где a, b, c, d, и n имеют те же значения, как переменные в формуле (1).
[0029]
Что касается измерения среднего состава каталитически активных частиц, каталитические активные частицы отделяют от оксидного катализатора, применяя раствор пероксида водорода, и средний состав выделенных каталитически активных частиц можно измерить STEM-EDX, как упомянуто выше, и в частности, измерение среднего состава каталитических частиц можно выполнить способом (физическое свойство 1), описанным в примерах.
[0030]
В формуле (1), a удовлетворяет 0,050≤a≤0,200, предпочтительно удовлетворяет 0,050≤a≤0,150, более предпочтительно удовлетворяет 0,080≤a≤0,150, еще более предпочтительно удовлетворяет 0,100≤a≤0,150 и даже еще более предпочтительно удовлетворяет 0,100≤a≤0,130.
[0031]
В формуле (1), b удовлетворяет 0,050≤b≤0,200, предпочтительно удовлетворяет 0,050≤b≤0,150, более предпочтительно удовлетворяет 0,070≤b≤0,150 и еще более предпочтительно удовлетворяет 0,080≤b≤0,150.
[0032]
В формуле (1), c удовлетворяет 0,100≤c≤0,300, предпочтительно удовлетворяет 0,100≤c≤0,250, более предпочтительно удовлетворяет 0,130≤c≤0,250 и еще более предпочтительно удовлетворяет 0,140≤c≤0,200.
[0033]
В формуле (1), d удовлетворяет 0≤d≤0,100, предпочтительно удовлетворяет 0<d≤0,100, более предпочтительно удовлетворяет 0,010≤d≤0,100 и еще более предпочтительно удовлетворяет 0,020≤d≤0,100.
[0034]
В формуле (1), e удовлетворяет 0≤e≤0,100, предпочтительно удовлетворяет 0≤e≤0,050, более предпочтительно удовлетворяет 0≤e≤0,010 и еще более предпочтительно удовлетворяет e=0. Следует отметить, что e представляет собой относительное содержание, по меньшей мере, одного металла X, выбранного из группы, состоящей из Te, Ce, Ti, и Ta, на один атом молибдена, и в случае, когда содержатся два или более металлов X, e представляет собой суммарное относительное содержание металлов X.
[0035]
Есть тенденция, что когда каждый из a, b, c, d и e удовлетворяет описанному выше диапазону, за счет этого выход ненасыщенного нитрила улучшается. Средний состав каталитически активных частиц можно контролировать регулированием количества исходных компонентов, Соблюдение соотношения, представленного формулой a≤c, в частности, можно контролировать, применяя конкретную Nb-содержащую жидкость в качестве исходного материала.
[0036]
Формула (1) предпочтительно удовлетворяет 0,050≤a≤0,150 и 0,100≤c≤0,250, и более предпочтительно удовлетворяет 0,050≤a≤0,150, 0,080≤b≤0,150, 0,100≤c≤0,250, и 0≤d≤0,100. Кроме того, формула (1) предпочтительно удовлетворяет 0,050≤a≤0,150, 0,100≤c≤0,250, и e=0, и более предпочтительно удовлетворяет 0,050≤a≤0,150, 0,08≤b≤0,150, 0,100≤c≤0,250, 0≤d≤0,100 и e=0. Есть тенденция, что когда формула (1) удовлетворяет описанному выше составу, за счет этого выход ненасыщенного нитрила улучшается.
[0037]
Формула (1) удовлетворяет соотношению, представленному формулой a≤c, и с точки зрения дальнейшего повышения выхода ненасыщенного нитрила, предпочтительно удовлетворяет соотношению, представленному формулой 1,1 × a≤c, более предпочтительно удовлетворяет соотношению, представленному формулой 1,3 × a≤c, и еще более предпочтительно удовлетворяет соотношению, представленному формулой 1,5 × a≤c. Кроме того, формула (1) предпочтительно удовлетворяет соотношению, представленному формулой c≤a × 3,0, более предпочтительно удовлетворяет соотношению, представленному формулой c≤a × 2,5, и еще более предпочтительно удовлетворяет соотношению, представленному формулой c≤a × 2,0.
[0038]
Sb, содержащуюся в каталитически активных частиц, считают активной частью реакции газофазного каталитического аммоксидирования, как приведено выше, и следовательно, она предпочтительно содержится в определенном количестве. Конкретно, формула (1) предпочтительно удовлетворяет соотношению, представленному формулой 0,100≤b/(a+b+c)≤0,400, более предпочтительно удовлетворяет соотношению, представленному формулой 0,150≤b/(a+b+c)≤0,300, и еще более предпочтительно удовлетворяет соотношению, представленному формулой 0,200≤b/(a+b+c)≤0,300. Есть тенденция, что когда формула (1) удовлетворяет описанному выше соотношению, за счет этого выход ненасыщенного нитрила улучшается.
[0039]
Каталитически активные частицы предпочтительно содержит определенное количество Sb, формула (1) предпочтительно удовлетворяет соотношению, представленному формулой 7,00≤100 × b/(1+a)≤11,00, более предпочтительно удовлетворяет соотношению, представленному формулой 8,00≤100 × b/(1+a)≤10,00, и еще более предпочтительно удовлетворяет соотношению, представленному формулой 8,50≤100 × b/(1+a)≤10,00. Выход ненасыщенного нитрила имеет тенденцию к большему увеличению, когда формула (1) удовлетворяет описанному выше соотношению.
[0040]
(Носитель)
Оксидный катализатор настоящего варианта осуществления может содержать носитель в добавление к сложному оксиду. То есть, оксидный катализатор настоящего варианта осуществления может иметь форму такую, что оксидный катализатор переносится носителем. В качестве носителя применяют оксид, такой как диоксид кремния, оксид алюминия, диоксид титана или диоксид циркония, но диоксид кремния подходит с той точки зрения, что снижение селективности для объекта невелико, и износостойкость и прочность сформированных частиц катализатора становятся удовлетворительными.
[0041]
Количество носителя на основе диоксида кремния составляет обычно от 20% по массе до 80% по массе, предпочтительно от 30% по массе до 70% по массе, и более предпочтительно от 40% по массе до 60% по массе, исходя из общей массы носителя на основе диоксида кремния и сложного оксида, а именно общего количества оксидного катализатора.
[0042]
Один из предпочтительных аспектов оксидного катализатора настоящего варианта осуществления представляет собой оксидный катализатор, содержащий диоксид кремния, в качестве носителя, несущего сложный оксид, и массовая доля диоксида кремния составляет от 30% по массе до 70% по массе в пересчете на SiO2, исходя из суммарного количества оксидного катализатора.
[0043]
Примеры исходного материала для носителя на основе диоксида кремния включают, но конкретно не ограничены, золь диоксида кремния (также называемый коллоидным диоксидом кремния) и порошкообразный диоксид кремния. В качестве исходного материала для носителя на основе диоксида кремния, золь диоксида кремния является предпочтительным с точки зрения простоты обращения. Средний диаметр первичных частиц диоксида кремния, содержащегося в золе диоксида кремния, специально не ограничен. В качестве носителя на основе диоксида кремния для применения можно добавлять различные типы золя диоксида кремния, каждый из которых имеет различный диаметр первичных частиц.
[0044]
Форма и размер частиц оксидного катализатора настоящего варианта осуществления конкретно не ограничены, но в случае, когда оксидный катализатор настоящего варианта осуществления применяют в качестве катализатора псевдоожиженного слоя, оксидный катализатор предпочтительно является сферическим и предпочтительно имеет диаметр частиц 10-150 мкм с точки зрения текучести.
[0045]
[Способ получения оксидного катализатора]
Оксидный катализатор настоящего варианта осуществления можно получить способом получения, включающим: стадию смешения исходных материалов для соответствующего смешения исходных материалов с получением, таким образом, суспензии предшественника; стадию сушки суспензии предшественника с получением высушенной частицы; и стадию прокаливания, на которой прокаливают высушенная частица с получением оксидного катализатора. При необходимости способ получения оксидного катализатора согласно настоящему варианту осуществления может включать стадию удаления выступов полученного оксидного катализатора.
[0046]
Следует отметить, что здесь и далее водная смешанная жидкость A соответствует смешанной жидкости (B), не содержащей ниобий в основных применениях (японская патентная заявка № 2019-106015 и японская патентная заявка № 2019-106018). Жидкость B с ниобиевым исходным веществом соответствует составу для получения катализатора, содержащего ниобий, составу (A) для получения катализатора или смешанной жидкости в основных применениях (японская патентная заявка № 2019-106015 и японская патентная заявка № 2019- 106018). Суспензия C предшественников соответствует исходной суспензии или жидкости со смесью исходных веществ в основных применениях (японская патентная заявка № 2019-106015 и японская патентная заявка № 2019-106018).
[0047]
(Стадия смешения исходных веществ)
Стадия смешения исходных веществ конкретно не ограничена, и например, включает: стадию получения A смешения молибденового исходного вещества, ванадиевого исходного вещества, исходного вещества, содержащего сурьму, и воды, посредством этого получая водную смешанную жидкость A; стадию получения B смешения ниобиевого исходного вещества и органической кислоты, посредством этого получая жидкость B ниобиевого исходного вещества; и стадию смешения C водной смешанной жидкости A и жидкости B с ниобиевым исходным веществом, посредством этого получая суспензию C предшественников.
[0048]
Оксидный катализатор настоящего варианта осуществления можно получить приспособлением предпочтительных условий получения, которые будут упомянуты ниже, на стадии получения B, или приспособлением предпочтительных условий смешения, которые будут упомянуты ниже, на этапе смешения C.
[0049]
Затем, жидкость B с ниобиевым исходным веществом, содержащую органическую кислоту и ниобиевое исходное вещество, и водную смешанную жидкость A, которую получили на стадии получения B заранее, смешивают так, чтобы соответствовать предполагаемой композиции, посредством этого получая жидкость со смесью исходных веществ в качестве суспензии предшественников (стадия смешения C). Например, в случае, когда катализатор содержит W или Ce, жидкость со смесью исходных веществ получают подходящим смешением соединения, содержащего W. Соединение, содержащее W или Ce, можно добавлять в водную смешанную жидкость A, или можно добавлять одновременно при смешении жидкости B с ниобиевым исходным веществом и водной смешанной жидкости A.
[0050]
В случае, когда оксидный катализатор содержит носитель на основе диоксида кремния, а именно в случае, когда оксидный катализатор переносится носителем на основе диоксида кремния, жидкость со смесью исходных веществ можно получить таким способом, чтобы она содержала золь диоксида кремния, и в данном случае золь диоксида кремния можно добавлять походящим способом.
[0051]
(Стадия получения A)
Стадия получения A представляет собой стадию смешения молибденового исходного вещества, ванадиевого исходного вещества, исходного вещества, содержащего сурьму, и воды, посредством этого получая водную смешанную жидкость A. Более конкретно, молибденовое исходное вещество, ванадиевое исходное вещество и исходное вещество, содержащее сурьму, добавляют к воде и нагревают при заранее определенной температуре или выше при перемешивании, и посредством этого можно получить водную смешанную жидкость A. В данном случае, когда катализатор содержит W или Ce, можно дополнительно добавлять вольфрамовое исходное вещество или цериевое исходное вещество.
[0052]
Компоненты, являющиеся исходными веществами, которые будут применять для водной смешанной жидкости A, конкретно не ограничены, и например, можно применять следующие компоненты.
[0053]
Примеры молибденового исходного вещества включают, но конкретно не ограничены, оксид молибдена, димолибдат аммония, гептамолибдат аммония, фосфорномолибденовую кислоту и кремнемолибденовую кислоту, и среди прочего можно удобно применять гептамолибдат аммония.
[0054]
Примеры ванадиевого исходного вещества включают, но конкретно не ограничены, пентоксид ванадия, метаванадат аммония и ванадилсульфат и, среди прочего, можно удобно применять метаванадат аммония.
[0055]
В качестве исходного вещества, содержащего сурьму, оксид сурьмы можно удобно применять. Вольфрамовое исходное вещество конкретно не ограничено, и например, удобно применять метавольфрамат аммония. Цериевое исходное вещество конкретно не ограничено, и например, гексагидрат нитрата церия можно удобно применять.
[0056]
Нижний предел температуры при перемешивании обычно составляет 80°C или выше, и предпочтительно 90°C или выше. Есть тенденция, что когда температура при перемешивании устанавливают на 80°C или выше, более легко протекает реакция окисления/восстановления между оксидом сурьма, который, как правило, плохо растворяется в воде, и другим исходным оксидом (например, ванадиевым исходным веществом). С другой стороны, предпочтительно, чтобы верхний предел температуры при перемешивании обычно составлял 100°C или ниже, чтобы избежать бурления.
[0057]
(Стадия получения B)
Стадия получения B представляет собой стадию смешения ниобиевого исходного вещества и органической кислоты, посредством этого получая жидкость B с ниобиевым исходным веществом. На стадии получения B, можно дополнительно добавлять воду. Ниобиевое исходное вещество является, в общем, плохо растворимым в воде и, следовательно, растворяется в воде за счет наличия органической кислоты. В данном случае, предпочтительно, чтобы содержание Nb в жидкости B с ниобиевым исходным веществом было высоким, и диспергируемость Nb была удовлетворительной при получении оксидного катализатора настоящего варианта осуществления.
[0058]
С этой точки зрения предпочтительно смешивать ниобиевое исходное вещество и органическую кислоту при перемешивании и нагревании на стадии получения B, регулируя молярное соотношение органической кислоты к Nb и мутность. Более конкретно, установив молярное соотношение органической кислоты к Nb (органическая кислота/Nb) на 2,40 или меньше, концентрацию Nb можно повышать в достаточной степени, и количество применяемой органической кислоты, которая действует как восстанавливающий агент, можно в достаточной степени уменьшить. Кроме того, установив мутность жидкости B с ниобиевым исходным веществом на 500 НЕМ или меньше, дисперсность Nb можно сохранять на удовлетворительном уровне, даже при наличии достаточно высокой концентрации Nb. Благодаря жидкости B с ниобиевым исходным веществом, которую получают данным способом, относительное содержание Nb в сложном оксиде в оксидном катализаторе настоящего варианта осуществления увеличивается, и относительное содержание V, который, как считают, вызывает разложение алкана, относительно снижается, и таким образом может быть увеличен выход ненасыщенного нитрила.
[0059]
Ниобиевое исходное вещество конкретно не ограничено при условии, что оно представляет собой соединение, содержащее Nb, но любое из данных соединений является труднорастворимым и, поэтому растворяется при наличии органической кислоты. Конкретные примеры ниобиевого исходного вещества включают, но не ограничиваются, гидрооксалат ниобия, оксалат аммония ниобия, NbCl3, NbCl5, Nb2(C2O4)5, Nb2O5, ниобиевую кислоту, Nb(OC2H5)5, галогенид ниобия, галогенированную аммониевую соль ниобия и их комбинации. Среди них, в случае, когда к жидкости B с ниобиевым исходным веществом добавляют дополнительный металл, ниобиевая кислота и гидрооксалат ниобий являются предпочтительными с точки зрения уменьшения влияния на дополнительный металл. Следует отметить, что ниобиевая кислота содержит гидроксид ниобия и оксид ниобия. Ниобиевое исходное вещество меняет свое качество в некоторых случаях из-за длительного хранения или в процессе дегидратации, и, следовательно, предпочтительно применять ниобиевое исходное вещество сразу после получения для получения жидкости B с ниобиевым исходным веществом, но можно применять соединение, которое в некоторой степени претерпело изменение качества.
[0060]
Ниобиевое исходное вещество может быть твердым или быть в виде суспензии при получении жидкости B с ниобиевым исходным веществом. В случае применения ниобиевой кислоты диаметр частиц предпочтительно является небольшим с точки зрения легкости растворения. Перед применением ниобиновую кислоту можно промывать аммиачной водой и/или водой.
[0061]
Примеры органической кислоты включают, но конкретно не ограничены, дикарбоновые кислоты. Конкретные примеры дикарбоновой кислоты включают щавелевую кислоту, малоновую кислоту, янтарную кислоту и глутаровую кислоту, и с точки зрения подавления чрезмерного восстановления оксида металла на стадии кальцинирования во время получения катализатора щавелевая кислота является предпочтительной, более конкретно ангидрид щавелевый кислоты и дигидрат щавелевой кислоты являются предпочтительными. Можно добавить только одну дикарбоновую кислоту или можно добавлять несколько дикарбоновых кислот.
[0062]
Как приведено выше, молярное соотношение органической кислоты к Nb (органическая кислота/Nb) является предпочтительно низким. Когда величина молярного соотношения (органическая кислота/Nb) является большой, концентрация Nb является низкой, и есть тенденция того, что дисперсность и стабильность Nb в жидкости B с ниобиевым исходным веществом являются удовлетворительными, но когда величина молярного соотношения (органическая кислота/Nb) является избыточно большой, есть риск того, что образующийся катализатор чрезмерно восстанавливается под действием органической кислоты, которая функционирует как восстанавливающий агент. Кроме того, количество Nb является недостаточным, в результате чего каталитически активные частицы не образуются в достаточной степени, и эффект подавления разложение продукта не получают в достаточной степени. С другой стороны, когда величина молярного соотношения (органическая кислота/Nb) является небольшой, концентрация Nb является высокой, получают жидкость B с ниобиевым исходным веществом, содержащую достаточное количество Nb, но дисперсность Nb в жидкости B с ниобиевым исходным веществом снижается, и как результат, становится сложно получить равномерное введение Nb в каталитически активные частицы. С данной точки зрения, молярное соотношение (органическая кислота/Nb) устанавливают равным 2,40 или меньше. То есть, установив молярное соотношение на 2,40 или меньше, может быть введено достаточное количество Nb, не делая вводимое количество органической кислоты, которая функционирует как восстанавливающий агент, чрезмерным, и, следовательно, каталитические активные частицы образуются в достаточной степени, и эффект подавления разложения продукта можно достигнуть в достаточной степени, и как результат, характеристики полученного катализатора становятся удовлетворительными. С точки зрения, аналогичной точке зрения, описанной выше, молярное соотношение (органическая кислота/Nb) предпочтительно составляет 2,20 или меньше, более предпочтительно 1,90 или больше и 2,18 или меньше, и еще более предпочтительно 1,95 или больше и 2,15 или меньше.
[0063]
Как приведено ранее, если дисперсность Nb в жидкости B с ниобиевым исходным веществом является неудовлетворительной, наблюдают осаждение Nb, и существует тенденция, что изменение концентрации Nb со временем является заметным. Следовательно, молярное соотношение в настоящем варианте осуществления повышают, исходя из концентрации после того, как жидкость B с ниобиевым исходным веществом остается стоять при нормальной температуре в течение одного дня сразу после получения. Следует отметить, что "нормальная температура" в настоящем изобретении обозначает температуру в районе 15-25°C. Конкретно, молярное соотношение можно измерить способом, описанным в примерах, которые приводятся ниже. Молярное соотношение (органическая кислота/Nb) можно регулировать в описанном выше диапазоне соотношением исходных веществ, которые будут применять.
[0064]
Что касается мутности жидкости B с ниобиевым исходным веществом, большая величина предполагает, что дисперсность Nb является плохой, и меньшая величина предполагает, что дисперсность Nb является более удовлетворительной. С данной точки зрения, мутность жидкости B с ниобиевым исходным веществом составляет 500 НЕМ или меньше. В настоящем варианте осуществления, мутность является в достаточной степени низкой, даже если молярное соотношение (органическая кислота/Nb) снижают к минимуму 2,40 или меньше, как приведено ранее, и, следовательно, каталитически активные частицы, которые являются активными центрами, образуются в достаточной степени, и эффект подавления разложения продукта можно получить в достаточной степени. В добавление к этому, равномерное введение Nb в каталитически активные частицы становится возможным из-за удовлетворительной дисперсности Nb, и как результат, характеристики полученного оксидного катализатора становятся особенно удовлетворительными. С точки зрения, аналогичной точки зрения, описанной выше, мутность составляет предпочтительно 0,5 НЕМ или больше и 400 НЕМ или меньше, и более предпочтительно 1,0 НЕМ или больше и 200 НЕМ или меньше.
[0065]
Как приведено ранее, если дисперсность Nb в жидкости B с ниобиевым исходным веществом не является удовлетворительной, наблюдают осаждение Nb, и существует тенденция, что изменение величины мутности со временем становится заметным. Следовательно, мутность в настоящем варианте осуществления также повышают, исходя из мутности после того, как жидкость B с ниобиевым исходным веществом остается стоять при нормальной температуре в течение одного дня непосредственно перед получением. Конкретно, мутность можно измерить способом, описанным в примерах, которые будут приведены ниже. Мутность можно регулировать в описанном выше диапазоне, приспосабливая предпочтительный способ получения, который будет приведен ниже.
[0066]
Температура смешения на стадии получения B является предпочтительно большей чем 60°C и меньшей чем 80°C, более предпочтительно 63°C или выше и 78°C или ниже, и еще более предпочтительно 65°C или выше и 75°C или ниже. Установив температуру смешения таким способом, чтобы она была больше чем 60°C, существует тенденция, что облегчается достаточное суспендирование Nb. Установив температуру смешения таким образом, чтобы она была ниже 80°C, комплекс органической кислоты и Nb, который образуется в жидкости B с ниобиевым исходным веществом, стабилизирован, так что существует тенденция, что обеспечивают достаточную дисперсность даже при высокой концентрации Nb. Кроме того, перемешивание предпочтительно осуществляют одновременно с нагреванием на стадии получения B. Посредством этого, молярное соотношение (органическая кислота/Nb) контролируют в описанном выше диапазоне, и мутность можно сохранять на низком уровне.
[0067]
Что касается продолжительности смешения на стадии получения B, желательно обеспечить достаточно длительную продолжительность времени реакции с точки зрения обеспечения протекания реакции между органической кислотой и ниобиевым исходным веществом в достаточной степени, но если время реакции будет слишком велико, есть опасения, что стабильность комплекса органической кислоты и Nb снижается, потому что смешанная жидкость находится в нагретом состоянии в течение чрезмерно длительного времени. С точки зрения обеспечения достаточной дисперсности, даже несмотря на то, что концентрация Nb является большой, стадию растворения предпочтительно проводят в течение 1 часа или дольше и 8 часов или меньше, предпочтительно 3 часов или дольше и 7 часов или меньше, и еще более предпочтительно 4 часов или дольше и 6 часов или меньше.
[0068]
В случае, когда органическая кислота и ниобиевое исходное вещество нагревают при перемешивании, температуру смеси органической кислоты и ниобиевого исходного вещества снижают до 5°C или ниже, и затем твердый остаток фильтруют и отделяют фильтрованием с отсасыванием, получая жидкость B с ниобиевым исходным веществом.
[0069]
Из того, что описано выше, предпочтительно устанавливать молярное соотношение органической кислоты к Nb на 2,40 или меньше в качестве органическая кислота/Nb и смешивать органическую кислоту и ниобиевое исходное вещество при 80°C или ниже при перемешивании с точки зрения более эффективного получения оксидного катализатора настоящего варианта осуществления. Условие, которое удовлетворяет им, называют условием получения b.
[0070]
(Стадия смешения C)
Стадия смешения C представляет собой стадию смешения водной смешанной жидкости A и жидкость B с ниобиевым исходным веществом, посредством этого получая суспензию предшественников C. На стадии смешения C, при необходимости можно дополнительно добавлять пероксид водорода, основный водный раствор, дополнительное металлическое исходное вещество, такое как вольфрамовое исходное вещество или цериевое исходное вещество, и/или исходный носитель. Суспензия предшественников C, которую получают данным способом, представляет собой равномерно смешанную жидкость в некоторых случаях, но обычно представляет собой суспензию.
[0071]
Условия смешения на стадии смешения C конкретно не ограничено, но, например, температура смешения предпочтительно составляет 50°C или выше и 80°C или ниже, и более предпочтительно 60°C или выше и 80°C или ниже. Продолжительность перемешивания предпочтительно составляет 1 час или больше и 5 часов или меньше.
[0072]
Примеры способа добавления пероксида водорода на стадии смешения C включают: способ, в котором водную смешанную жидкость A и пероксид водорода смешивают и затем дополнительно добавляют жидкость B с ниобиевым исходным веществом; способ, в котором жидкость B с ниобиевым исходным веществом и пероксид водорода смешивают и затем дополнительно добавляют водную смешанную жидкость A; способ, в котором водную смешанную жидкость A, жидкость B с ниобиевым исходным веществом и пероксид водорода смешивают одновременно; и способ, в котором водную смешанную жидкость A и жидкость B с ниобиевым исходным веществом смешивают, и затем дополнительно добавляют пероксид водорода. Среди них, способ, в котором водную смешанную жидкость A и пероксид водорода смешивают, и затем дополнительно добавляют ниобиевое исходное вещество B, является предпочтительным.
[0073]
В случае, когда пероксид водорода добавляют на стадии смешения C, молярное соотношение пероксида водорода к сурьме (Sb) (H2O2/Sb) в водной смешанной жидкости A составляет предпочтительно 0,01 или больше и 5,0 или меньше, более предпочтительно 2,0 или больше и 4,0 или меньше, и еще более предпочтительно 2,5 или больше и 3,5 или меньше. В случае, когда смешивают водную смешанную жидкость A и пероксид водорода, предпочтительно добавлять пероксид водорода при перемешивании водной смешанной жидкости A в условиях нагревания. В данном случае, температура обычно составляет 30°C-70°C, предпочтительно 60°C или выше. Продолжительность перемешивания предпочтительно составляет 30 минут-2 часа.
[0074]
Примеры способа добавление основного водного раствора на стадии смешения C включают: способ, в котором водную смешанную жидкость A и основный водный раствор смешивают и затем дополнительно добавляют жидкость B с ниобиевым исходным веществом; способ, в котором жидкость B с ниобиевым исходным веществом и основный водный раствор смешивают и затем дополнительно добавляют водную смешанную жидкость A; способ, в котором водную смешанную жидкость A, жидкость B с ниобиевым исходным вещество, и основный водный раствор смешивают одновременно; и способ, в котором смешивают водную смешанную жидкость A и жидкость B с ниобиевым исходным веществом, и затем дополнительно добавляют основный водный раствор. Среди них, способ, в котором водную смешанную жидкость A и жидкость B с ниобиевым исходным веществом смешивают и затем дополнительно добавляют основный водный раствор, является предпочтительным.
[0075]
Примеры основного водного раствора, который будут добавлять на стадии смешения C, включают, но конкретно не ограничены, аммиачную воду, амин, и щелочной водный раствор. Среди них, основный водный раствор является предпочтительно аммиачной водой, поскольку аммиачная вода испаряется по большей части на стадии сушки и не влияет на стадии после стадии сушки.
[0076]
В случае, когда основный водный раствор добавляют на стадии смешения C, молярное соотношение NH3 к ниобию (Nb) в жидкости B с ниобиевым исходным веществом (NH3/Nb) предпочтительно составляет 0,01 или больше и 5 или меньше, более предпочтительно 2,0 или больше и 4 или меньше, и еще более предпочтительно 2,5 или больше и 3,5 или меньше.
[0077]
Среди описанного выше, предпочтительно смешивать пероксид водорода таким способом, чтобы молярное соотношение пероксида водорода к сурьме (Sb) (H2O2/Sb) составляло 2,5 или больше и добавлять аммиак таким способом, чтобы молярное соотношение аммиака к ниобию (Nb) (NH3/Nb) составляло 2,0 или больше на стадии смешения C, с точки зрения более эффективного получения оксидного катализатора настоящего варианта осуществления. Кроме того, предпочтительно устанавливать pH на стадии смешения C равной 5 или больше и получать суспензию предшественников проведением перемешивания в условиях нагревания 60°C или выше. Условия, которые удовлетворяют этому, называют условиями смешения c. Следует отметить, что pH можно регулировать, применяя приведенный выше основный водный раствор.
[0078]
Примеры способа добавления дополнительного металлического исходного вещества, такого как вольфрамовое исходное вещество или цериевое исходное вещество, на стадии смешения C включает: способ, в котором водную смешанную жидкость A и дополнительное металлическое исходное вещество смешивают и затем дополнительно добавляют жидкость B с ниобиевым исходным веществом; способ, в котором жидкость B с ниобиевым исходным веществом и дополнительное металлическое исходное вещество смешивают и затем дополнительно добавляют водную смешанную жидкость A; способ, в котором водную смешанную жидкость A, жидкость B с ниобиевым исходным веществом и дополнительное металлическое исходное вещество смешивают одновременно; и способ, в котором водную смешанную жидкость A и жидкость B с ниобиевым исходным веществом смешивают и затем дополнительно добавляют дополнительное металлическое исходное вещество.
[0079]
Примеры способа добавления исходного вещества, являющегося носителем, на стадии смешения C включают: способ, в котором водную смешанную жидкость A и исходное вещество, являющееся носителем, смешивают и затем дополнительно добавляют жидкость B с ниобиевым исходным веществом; способ, в котором жидкость B с ниобиевым исходным веществом и исходное вещество, являющееся носителем, смешивают и затем дополнительно добавляют водную смешанную жидкость A; способ, в котором водную смешанную жидкость A, жидкость B с ниобиевым исходным веществом и исходное вещество, являющееся носителем, смешивают одновременно; и способ, в котором водную смешанную жидкость A и жидкость B с ниобиевым исходным веществом смешивают и затем дополнительно добавляют исходное вещество, являющееся носителем.
[0080]
В случае, когда получают оксидный катализатор, так как сложный оксид находится на носителе на основе диоксида кремния, золь диоксида кремния можно применять в качестве исходного вещества, являющегося носителем.
[0081]
(Другие условия)
Регулируя оба описанных выше условия получения b и условия смешения c, получают оксидный катализатор, удовлетворяющий указанным выше отношениям, представленным формулами. В частности, регулирование обоих описанных выше условий получения b и условий смешения является предпочтительным, поскольку можно получить оксидный катализатор, удовлетворяющий соотношению, представленному формулой 1,1×a≤c или 1,5×a≤c, или соотношению, представленному формулой 8,00≤100×b/(1+a) ≤10,00.
[0082]
Предпочтительно, чтобы количество ванадиевого исходного вещества, содержащееся в водной смешанной жидкости A, было относительно низким, исходя из количества ниобиевого исходного вещества, содержащегося в жидкости B с ниобиевым исходным веществом, в добавление к приспособлению описанных выше условия получения b или условий смешения c. Конкретно, атомное соотношение V/Nb атома ванадия, содержащегося в ванадиевом исходном веществе, к атому ниобия, содержащемуся в ниобиевом исходном веществе, предпочтительно составляет 2,7 или меньше, более предпочтительно 2,0 или меньше, и еще более предпочтительно 1,7 или меньше. Атомное соотношение V/Nb можно регулировать для каждой концентрации ванадиевого исходного вещества в водной смешанной жидкости A и ниобиевого исходного вещества в жидкости B с ниобиевым исходным веществом или массового соотношения водной смешанной жидкости A к жидкости B с ниобиевым исходным веществом.
[0083]
(Стадия сушки)
Стадия сушки представляет собой стадию сушки суспензии предшественников C, полученной на приведенной выше стадии, посредством этого получая высушенный порошок. Сушку можно осуществлять известным способом, и можно осуществлять, например, сушкой распылением или упариванием досуха. Среди них, мелкодисперсные сферические высушенные частицы предпочтительно получают сушкой распылением. Распыление в способе сушки распылением можно осуществлять центробежной системой, пневмораспылительной системой или распылительной системой высокого давления. В качестве источника тепла для сушки можно применять пар или воздух, нагретый электронагревателем или подобными. Температура на входе сушилки в устройстве для распылительной сушки составляет предпочтительно от 150 до 300°C, и температура на выходе из сушилки составляет предпочтительно от 100 до 160°C.
[0084]
(Стадия прокаливания)
Стадия прокаливания представляет собой стадию прокаливания высушенного порошка, полученного на стадии сушки, посредством этого получая оксидный катализатор. В качестве устройства для прокаливания, можно применять a вращающуюся печь. Форма кальцинатора конкретно не ограничена, но предпочтительно он имеет трубчатую форму, поскольку можно осуществлять непрерывное прокаливание. Форма кальцинационной трубы конкретно не ограничена, но является предпочтительно цилиндрической. В качестве системы нагрева предпочтительным является тип внешнего обогрева, и целесообразно применять электрическую печь.
[0085]
Соответствующий размер, материал и подобные трубы для прокаливания можно выбрать в зависимости от условий прокаливания или получаемого объема, но внутренний диаметр трубы для прокаливания предпочтительно составляет от 70 до 2000 мм, более того от 100 до 1200 мм, и длина трубы для кальцинации предпочтительно составляет от 200 до 10000 мм, и более предпочтительно от 800 до 8000 мм. В случае, когда кальцинатор подвергают ударной нагрузке, толщина кальцинатор предпочтительно составляет 2 мм или больше, и более предпочтительно 4 мм или больше с точки зрения наличия толщины, достаточной для того, чтобы кальцинатор не ломался при ударной нагрузке, и предпочтительно составляет 100 мм или меньше, и более предпочтительно 50 мм или меньше с точки зрения достаточной степени передачи ударной нагрузки внутрь кальцинатора. Материал кальцинатора конкретно не ограничено, за исключением того, что материал обладает такими термостойкостью и прочностью, что материал не разрушается при ударной нагрузке, и можно удобно применять SUS.
[0086]
Труба для прокаливания можно разделить на две или больше зон установкой в трубу для прокаливания отбивной решетки вертикально потоку порошка, отбивной решетки, содержащей отверстие для прохождения порошка в центральную часть. Установка отбивной решетки позволяет легко контролировать время удерживания в трубе для прокаливания. Количество отбивных решеток может составлять одну или несколько. Материал отбивной решетки предпочтительно представляет собой металл, и можно удобно применять тот же металл, что и для трубы для прокаливания. Высоту отбивной решетки можно регулировать в зависимости продолжительности удержания, которое необходимо обеспечить. Например, в случае, когда порошок подают при 250 г/ч во вращающуюся печь, содержащую SUS трубу для прокаливания с внутренним диаметром 150 мм и длинной 1150 мм, отбивная решетка предпочтительно имеет высоту 5-50 мм, более предпочтительно 10-40 мм, и еще более предпочтительно 13-35 мм. Толщина отбивной решетки конкретно не ограничена и предпочтительно устанавливают в зависимости от размера трубы для прокаливания. Например, в случае вращающейся печи, содержащей SUS труба для прокаливания с внутренним диаметром 150 мм и длиной 1150 мм, толщина трубы для прокаливания предпочтительно составляет 0,3 мм или больше и 30 мм или меньше, и более предпочтительно 0,5 мм или больше и 15 мм или меньше.
[0087]
Труба для прокаливания предпочтительно вращается для того, чтобы предотвратить образования трещин, растрескивание и подобных на высушенном порошке и обеспечить равномерное прокаливание. Скорость вращения трубы для прокаливания предпочтительно составляет 0,1-30 об/мин, более предпочтительно 0,5-20 об/мин, и еще более предпочтительно 1-10 об/мин.
[0088]
Когда прокаливают высушенный порошок, предпочтительно, чтобы повышение температуры для нагревания высушенного порошка начиналось с температуры ниже чем 400°C, и температура повышалась непрерывно или периодически вплоть до температуры в пределах диапазона 550-800°C.
[0089]
Атмосфера прокаливания может находиться в атмосфере воздуха или под потоком воздуха, но предпочтительно осуществляет, по меньшей мере, часть прокаливания, позволяя протекать инертному газу, такому как азот, который по существу не содержит кислорода. Подаваемое количество инертного газа составляет 50 N литров или больше, предпочтительно 50-5000 N литров, и еще более предпочтительно 50-3000 N литров на кг высушенного порошка (N литров обозначает литры, измеренные в условиях стандартной температуры/давления, а именно литры, измеренные при 0°C и 1 атм). В данном случае, нет проблем, когда инертный газ и высушенный порошок приводят в контакт в параллельном потоке или противотоке, но контакт противотоком является предпочтительным, учитывая компоненты газа, образующиеся из высушенного порошка, и воздух, который смешивается в небольшом количестве вместе с высушенным порошком.
[0090]
Стадию прокаливания можно осуществлять в одну стадию, но предпочтительно, чтобы прокаливание включало предварительное прокаливание и финальное прокаливание, причем предварительное прокаливание проводят в диапазоне температур 250-400°C, и финальное прокаливание проводят в диапазоне температур 550-800°C. Предварительное прокаливание и финальное прокаливание можно осуществлять непрерывно, или после завершения предварительного прокаливания, финальное прокаливание можно проводить заново. Кроме того, каждое из предварительного прокаливания и финального прокаливания можно разделить на несколько стадий.
[0091]
Предварительное прокаливание проводят при температуре нагревания в диапазоне 250°C-400°C, и предпочтительно 300°C-400°C, предпочтительно в потоке инертного газа. Температуру предпочтительно поддерживают при постоянной температуре в диапазоне температур 250°C-400°C, но не имеет значения, изменяется ли температура, или плавно повышается или понижается в диапазоне от 250°C до 400°C. Время выдержки температуры нагрева предпочтительно составляет 30 минут или дольше, и более предпочтительно 3-12 часов.
[0092]
Относительно схемы повышения температуры до момента времени, когда температура достигает температуры предварительного прокаливания, температуру можно повышать линейно, или температуру можно повышать таким способом, чтобы нарисовать выпуклую дугу, направленную вверх или вниз.
[0093]
Средняя скорость повышения температуры при повышении температуры до того, как температура достигает температуры предварительного прокаливания, конкретно не ограничена, но обычно составляет приблизительно 0,1-15°C/мин, предпочтительно 0,5-5°C/мин и более предпочтительно 1-2°C/мин.
[0094]
Финальное прокаливание осуществляют при 550-800°C, предпочтительно 580-750°C, более предпочтительно 600-720°C и еще более предпочтительно 620-700°C, предпочтительно в атмосфере инертного газа. Температуру предпочтительно поддерживают при постоянной температуре в диапазоне температур 620-700°C, но не имеет значения, изменяется ли температура, или плавно повышается или понижается в диапазоне 620-700°C. Время финального прокаливания составляет 0,5-20 часов и предпочтительно 1-15 часов.
[0095]
В случае, когда труба для прокаливания разделена отбивной решеткой или тарелками, высушенный порошок и/или составной оксидный катализатор последовательно проходит через, по меньшей мере, две, предпочтительно 2-20, и более предпочтительно 4-15 зон. Контроль температуры можно осуществлять, применяя одно или более контрольных устройств, но температуру предпочтительно контролируют установкой нагревателя и контрольного устройства для каждой зоны, разделенной отбивными решетками для того, чтобы получить требуемую схему прокаливания. Например, в случае, когда семь отбивных решеток устанавливают таким способом, чтобы равномерно разделить длину части, который входит в нагревательную печь, трубы для прокаливания на восемь, и применяют трубу для прокаливания, разделенную на восемь зон, установленные температуры восьми зон предпочтительно контролируются нагревателем и контрольным устройством, установленным для соответствующих зон таким образом, что температура высушенного порошка и/или сложного оксидного катализатора показывает требуемую схему прокаливания. Следует отметить, что не имеет значения, добавляет ли окислительный компонент (например, кислород) или восстановительный компонент (например, аммиак), при необходимости, к атмосфере прокаливания в потоке инертного газа.
[0096]
Что касается схемы повышения температуры до момента времени, когда температура достигает температуры финального прокаливания, температура можно повышать линейно, или температуру можно повышать таким способом, чтобы нарисовать выпуклую дугу вверх или вниз.
[0097]
Средняя скорость повышения температуры в процессе повышения температуры до того, как температура достигает температуры финального прокаливания, конкретно не ограничена, но обычно составляет 0,1-15°C/мин, предпочтительно 0,5-10°C/мин и более предпочтительно 1-8°C/мин.
[0098]
Средняя скорость понижения температуры после завершения финального прокаливания предпочтительно составляет 0,05-100°C/мин, и более предпочтительно 0,1-50°C/мин. Временное выдерживание температуры равной температуре, меньше чем температура финального прокаливания, является также предпочтительным. Температура выдержки представляет собой температуру, ниже чем температура финального прокаливания на 10°C, предпочтительно на 50°C и более предпочтительно на 100°C. Продолжительность выдержки составляет 0,5 часа или более, предпочтительно 1 час или более, более предпочтительно 3 часа или более, и еще более предпочтительно 10 часов или более.
[0099]
В случае, когда после завершения предварительного прокаливания заново осуществляют финальное прокаливание, низкотемпературную обработку предпочтительно проводят на финальном прокаливании.
[0100]
Время, требуемое для низкотемпературной обработки, то есть время, необходимое для повышения температуры высушенного порошка и/или сложного оксидного катализатора до температуры прокаливания после понижения температуры, может быть соответствующим образом отрегулировано размером, толщиной и материалом кальцинатора, получаемым количеством катализатора, рядом периодов непрерывного прокаливания высушенного порошка и/или сложного оксидного катализатора, скоростью закрепления/степенью фиксации и подобными. Например, в случае, когда применяют SUS трубу для прокаливания с внутренним диаметром 500 мм, длинной 4500 мм и толщиной 20 мм, время, требуемое для низкотемпературной обработки, предпочтительно находится в пределах 30 дней, более предпочтительно в пределах 15 дней, еще более предпочтительно в пределах 3 дней, и особенно предпочтительно в пределах 2 дней в процессе серии периодов непрерывного прокаливания катализатора.
[0101]
Например, в случае, когда высушенный порошок подают при скорости 35 кг/ч, при этом высушенный порошок вращают при 6 об/мин во вращающейся печи, содержащей SUS трубу для прокаливания с внутренним диаметром 500 мм, длинной 4500 мм и толщиной 20 мм, и температуру финального прокаливания устанавливают на 645°C, стадию понижения температуры до 400°C, и затем повышение температуры до 645°C можно проводить приблизительно за один день. В случае, когда прокаливание проводят в течение одного года непрерывно, проводя данную низкотемпературную обработку с периодичностью один раз в месяц, прокаливание можно проводить, пока температура оксидного слоя поддерживается стабильной.
[0102]
[Способ получения ненасыщенного нитрила]
Способ получения ненасыщенного нитрила настоящего варианта осуществления дает ненасыщенный нитрил при осуществлении реакции газофазного каталитического аммоксидирования на пропане или изобутане в присутствии оксидного катализатора настоящего варианта осуществления.
[0103]
(Исходное вещество)
Пропан или изобутан, и аммиак, которые являются исходными веществами, не обязательно должны быть высокой чистоты, и можно применять газы промышленного класса, такие как пропан, содержащие 3 об.% или меньше примесей, таких как этан, этилен, н-бутан, аммиак, содержащий 3 об.% или меньше примесей, таких как вода. Кислородсодержащий газ конкретно не ограничен, например, воздух, воздух, обогащенный кислородом, чистый кислород, газ, полученный разбавлением любого из них инертным газом, таким как гелий, аргон, диоксид углерода или азот, или пар также можно обеспечивать для реакции. Среди них, в случае, когда кислородсодержащий газ применяют в промышленных масштабах, воздух предпочтительно применяется с точки зрения простоты.
[0104]
(Условия реакции)
Реакцию газофазного каталитического аммоксидирования пропана или изобутана можно проводить, например, но они конкретно не ограничены, в следующих условиях. Молярное соотношение кислорода, который будут подавать для реакции, к пропану или изобутану, предпочтительно составляет 0,1-6, и более предпочтительно 0,5-4. Молярное соотношение аммония, который будут подавать для реакции, к пропану или изобутану предпочтительно составляет 0,3-1,5, и более предпочтительно 0,7-1,2.
[0105]
Температура реакции предпочтительно составляет 350-500°C, и более предпочтительно 380-470°C. Давление реакции предпочтительно составляет 5 × 104-5 × 105 Па, и более предпочтительно 1 × 105-3 × 105 Pa. Продолжительность контакта предпочтительно составляет 0,1-10 сек∙г/см3, и более предпочтительно 0,5-5 сек∙г/см3. Устанавливая условия для реакции газофазного каталитического аммоксидирования в вышеописанных диапазонах, существует тенденция, что подавляется получение побочных продуктов, и может быть дополнительно увеличен выход ненасыщенного нитрила.
[0106]
В настоящем варианте осуществления, продолжительность контакта определяю следующим уравнением.
Продолжительность контакта (сек г/см3) = (W/F) × 273/(273+T)
где W, F, и T определяют следующим образом.
W=количество (г) заполненного катализатора
F=скорость потока (N см3/сек) смешанного газа исходного вещества в стандартном состоянии (0°C, 1,013 × 105 Па)
T=температура реакции (°C)
[0107]
В качестве реакционной системы в реакции газофазного каталитического аммоксидирования можно приспособить стандартную систему, такую как неподвижный слой, псевдоожиженный слой и подвижный слой. Среди них предпочтительным является реактор с псевдоожиженным слоем, с помощью которого легко отводится тепло реакции. Реакция газофазного каталитического аммоксидирования может быть однопоточной или рециркуляционной.
Примеры
[0108]
Далее настоящий вариант осуществления будет описан более подробно с указанием конкретных примеров и сравнительных примеров, но настоящий вариант осуществления вообще не ограничен следующими примерами и сравнительными примерами в пределах, не выходящих за рамки сущности настоящего варианта осуществления. Соответствующие физические свойства и оценки, выполненные в примерах и сравнительных примерах, которые будут упомянуты ниже, измеряли следующими способами.
[0109]
[Пример 1]
<Стадия получения A; получение водной смешанной жидкости (A1)>
К 2572 г воды добавляли 436,4 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O] и 54,6 г метаванадата аммония [NH4VO3], 91,4 г триоксида дисурьмы [Sb2O3] и 5,4 г нитрата церия [Ce(NO3)3∙6H2O], и полученную в результате смесь нагревали при 98°C в течение 2 часа при перемешивании, получая водную смешанную жидкость (A1).
[0110]
<Стадия получения B; получение жидкости ниобиевого исходного вещества (B1)>
Жидкость ниобиевого исходного вещества получали следующим способом. В смеситель добавляли 57,9 кг воды, и затем воду нагревали до 45°C. Затем загружали 72,2 кг дигидрата щавелевой кислоты [H2C2O4∙2H2O] при перемешивании, и затем загружали 19,9 кг ниобиевой кислоты, содержащей 76,0% по массе Nb2O5, для смешения обеих в воде. Данную жидкость перемешивали при нагревании при 70°C в течение 8 часа, и посредством этого получали водную смешанную жидкость. Данную водную смешанную жидкость оставляли стоять и охлаждали на льду, и затем твердый остаток фильтровали и отделяли фильтрованием с отсасыванием, получая однородную жидкость ниобиевого исходного вещества (B1). Анализом, описанным ниже, определяли, что молярное соотношение щавелевая кислота/ниобий в данной жидкости ниобиевого исходного вещества составляет 2,11. Полученную жидкость ниобиевого исходного вещества применяли в качестве жидкости ниобиевого исходного вещества (B1) в получении оксидных катализаторов примеров 2-5, описанных ниже.
[0111]
Молярное соотношение щавелевая кислота/ниобий в жидкости ниобиевого исходного вещества (B1) рассчитывали, как описано ниже. После тепловой обработки такой, что жидкость ниобиевого исходного вещества (B1) перемешивали при 40°C в течение 20 минут по истечении одних суток после осуществления получения, и жидкость ниобиевого вещества (B1) оставляли стоять при 20°C в течение 7 дней после тепловой обработки, измеряли молярное соотношение щавелевая кислота/ниобий и мутность.
[0112]
Концентрацию Nb в водной смешанной жидкости рассчитывали из веса твердого Nb2O5, полученного таким способом, что 10 г жидкости ниобиевого исходного вещества (B1) точно взвешивали в тигле, высушенном при 120°C в течение 2 часов, и затем подвергали тепловой обработки при 600°C в течение 2 часов, определяя, что Концентрация Nb составляет 1,072 моль/кг.
[0113]
В 300-мл аналитическом стакане, точно взвешивали 3 г жидкости ниобиевого исходного вещества (B1), добавляли 20 мл горячей воды приблизительно 80°C, и затем добавляли 10 мл 1:1 серной кислоты. Полученную таким образом смешанную жидкость титровали при перемешивании, применяя 1/4N KMnO4, при этом температуру жидкости поддерживали равной 70°C на водяной бане. Точка, в которой слабый бледно-розовый цвет из-за KMnO4 сохранялся приблизительно 30 секунд или более, определяли как конечную точку. Концентрацию щавелевая кислота рассчитывали из оттитрованного количества согласно следующей формуле, находя, что концентрация щавелевой кислоты составляет 2,26 моль/кг.
2KMnO4+3H2SO4+5H2C2O4 → K2SO4+2MnSO4 +10CO2+8H2O
[0114]
Мутность измеряли, применяя 2100AN турбидиметр, изготовленный HACH после того, как жидкость ниобиевого исходного вещества (B1) оставляли стоять еще в течение одного дня после получения. В измерительную ячейку помещали 30 мл раствора для проведения измерения на основе US EPA способа 180.1, находя, что мутность жидкости ниобиевого исходного вещества (B1) составляла 69 НЕМ.
[0115]
<Стадия смешения; получение суспензии предшественников (C1)>
После охлаждения полученной водной смешанной жидкости (A1) до 70°C, 593,8 г золя диоксида кремния, содержащего 34,1% по массе SiO2, добавляли к водной смешанной жидкости (A1), и затем в момент времени, когда температура жидкости становилась равной 55°C, добавляли 181 г раствора пероксида водорода, содержащего 30% по массе H2O2, получая водную смешанную жидкость (A1'). Сразу после этого, 309,1 г жидкости ниобиевого исходного вещества (B1), 34,2 г (чистота 50%) водного раствора метавольфрамата аммония, и дисперсную жидкость, полученную диспергированием 247,5 г порошкообразного диоксида кремния в 2228 г воды, добавляли последовательно к водной смешанной жидкости (A1), затем добавляли 51,7 г 25% аммиачной воды, и полученную в результате смесь подвергали выдерживания при перемешивании при 65°C в течение 2,5 часов, получая суспензию предшественников (C1).
[0116]
<Стадия сушки; получение высушенного порошка (E1)>
Полученную суспензию предшественников (C1) подавали в центробежную распылительную сушилку и сушили, получая микросферический высушенный порошок (D1). Воздух применяли как источник тепла для сушки. Следует отметить, что один и тот же источник тепла для сушки применяли в центробежной распылительной сушилке в следующих примерах и сравнительных примерах. Температура на входе в осушитель составляла 210°С, и температуры на выходе составляла 120°С. Полученный высушенный порошок (D1) сортировали, применяя сито с отверстием 32 мкм, получая высушенный порошок (E1), который представляет собой сортированный продукт.
[0117]
<Стадия прокаливания; получение оксидного катализатора (F1)>
В PYREX трубу для прокаливания (PYREX представляет собой зарегистрированную торговую марку), имеющую диаметр 3 дюйма, заполняли 100 г полученного в результате высушенного порошка (E1) и прокаливали при 685°C в течение 2 часов в потоке азота 5,0 Nл/мин при вращении трубы, и посредством этого получали оксидный катализатор (F1).
[0118]
<Стадия удаления>
В вертикальную трубу (внутренний диаметр 41,6 мм, длина 70 см), снабженную диском с тремя отверстиями диаметром 1/64 дюйма в нижней части и бумажным фильтром, установленным в верхней части, загружали 50 г оксидный катализатор (F1). Затем воздуху позволяли течь снизу вверх по вертикальной трубе через соответствующие отверстия при комнатной температуре для облегчения контакта между прокаленными частицами. Длина воздушного потока в данном случае в направлении, в котором он проходил, составляла 56 мм, а средняя линейная скорость воздушного потока составляла 332 м/с. Через 24 часа в полученном оксидном катализаторе (F1) не было выступов.
[0119]
<Физическое свойство 1; анализ состава каталитически активных частиц (G1) STEM-EDX
В 500-мл аналитический стакан помещали 200 г раствора пероксида водорода, содержащего 10% по массе H2O2, и температуру воды устанавливали равной 27°C. К данному раствору пероксида водорода добавляли 15 г оксидного катализатора (F1) после удаления выступов и перемешивали, применяя магнитную мешалку при 500 об/мин в течение 5 часов, и затем нерастворимый компонент получали фильтрованием с отсасыванием. Полученный нерастворимый компонент сушили при 50°C в течение 12 часов, собирая остаток (H1). Собранный остаток (H1) был таким, что другие кристаллы, отличные от каталитически активных частиц (G1) и носителя (например, диоксида кремния), были удалены окислительной обработкой раствором пероксида водорода, и он содержит каталитически активные частицы (G1) и носитель.
[0120]
Полученный остаток (H1) растирали в ступке в течение 30 секунд, получая образец порошка. В сосуд с внутренним объемом 10 мл помещали 0,1 г полученного образца порошка и 6 мл этанола. Сосуд помещали в настольную ультразвуковую стиральную машину (YAMATO BRANSON, производимую Yamato Scientific Co., Ltd.) и подвергали ультразвуковой вибрации в течение 60 секунд, диспергируя образец порошка остатка (H1) в этаноле.
[0121]
После этого данную этанольную жидкую дисперсию образца порошка по каплям наносили на сетку из Cu с углеродной поддерживающей мембраной (эластичная углеродная поддерживающая мембрана; шаг сетки 100 мкм; производство Okenshoji Co., Ltd.), и затем каплю жидкости удерживали в течение приблизительно одной минуты и затем удалялся фильтровальной бумагой и подобными. Таким образом, получали образец такой, что частица катализатора в жидкой дисперсии была помещена на углеродную поддерживающую мембрану из Cu-сетки. Подготовленный таким образом образец наблюдали, применяя STEM-EDX.
[0122]
Сканирующий трансмиссионный электронный микроскоп, оснащенный рентгеновским спектрометром с энергодисперсионными характеристиками (STEM-EDX), применяли для количественного определения каталитически активных частиц (G1) в образце порошка. HD2300A (производства Hitachi Hi-Tech Corporation) применяется в качестве сканирующего трансмиссионного электронного микроскопа (STEM), и Apollo XLT2 SUTW (производства AMETEK, Inc) применяли в качестве рентгеновского спектрометра с энергодисперсионными характеристиками (EDX). Genesis (Ver 6.53, производство AMETEK, Inc.) применяли в качестве программного обеспечения для измерения и анализа EDX.
[0123]
Способ калибровки энергии для EDX был следующим. Применяли образец, в котором присутствуют Al и Cu, и образец облучали электронным пучком, ускоренным до 200 кВ, для регистрации характеристического рентгеновского излучения. В спектре характеристического рентгеновского излучения, детектируемого в данном случае, выбирали место измерения, где пиковая интенсивность излучения Al-Kα была приблизительно такой же или несколько большей, чем пиковая интенсивность излучения Cu-Kα, и число импульсов (CPS) становилось равным 1000-5000 имп./с. Калибровку значений энергии проводили, применяя функцию калибровки в программном обеспечении, устанавливая количество измерений на 5, устанавливая пик 1 на значение энергии луча Al-Kα ((1486 эВ) и устанавливая пик 2 на энергию значение Cu-Kα луча (8,04 эВ).
[0124]
Условия измерения характеристического рентгеновского излучения были следующими. Ускоряющее напряжение составляло 200 кВ, рабочий режим; нормальный, апертура объектива была № 2 (диаметр отверстия 60 µmɸ, угол облучения около 15 мрад), угол наклона спектрометра был 26°, апертура EDX была вставлена для измерения спектра анализом площади таким способом, чтобы включать периферию центральной части частицы катализатора. Измерение проводили в условиях детекции EDX: постоянная времени 7,68 мкс и время интегрирования от 70 до 80 с.
[0125]
Способ коррекции в тонкой пленке (Thin Apex) для STEM применяли в качестве способа количественного определения. Количественное определение проводили, применяя L-лучи для Nb, Mo, Sb и W, и K-лучи для V. В качестве K-факторов для применения в количественном определении применяли значения, зарегистрированные в программном обеспечении, Nb-L: 3,52, Mo-L; 3,62, Sb-L; 5,59, V-К; 1,34 и W-L; 7,68 (K-факторы, которые являются рекомендованными значениями производителя устройства; AMETEK EDAX). Плотность образца для количественного определения устанавливали на 7,8 г/см3 (средняя плотность Nb, Mo, Sb и V), и толщину пленки устанавливали на 50 нм. Количественное определение осуществляли, устанавливая устранение фона до автоматическую настройку.
[0126]
Измерение осуществляли в 15 точках, и состав каталитически активных частиц (G1) рассчитывали, беря среднее из них. В случае, когда носитель (например, диоксид кремния) рассчитывали как композицию, диоксид кремния исключали из результатов измерения. Частица, присутствующая в виде кристалла в образце, была только каталитически активными частицами (G1) благодаря приведенной выше окислительной обработке раствором пероксида водорода. Соответственно, наблюдая за частицей, в которой среди наблюдаемых частиц наблюдаются поперечные полосы, отражающие периодичность кристаллов, в поле зрения можно эффективно обнаружить каталитически активные частицы (G1). Состав каталитически активных частиц (G1) представлен в таблице 1.
[0127]
<Физическое свойство 2; оценка реакционноспособности в реакции аммоксидирования пропана>
Пропан подавали в реакцию газофазного каталитического аммоксидирования следующим способом, применяя оксидный катализатор (F1), полученный выше. В реакционную трубу с псевдоожиженным слоем из стекла VYCOR с внутренним диаметром 25 мм заполняли 40 г оксидного катализатора и подавали смешанный газ, имеющий молярное соотношение пропан:аммиак:кислород:гелий=1:1,1:2,9:11,6, при продолжительности контакта 3,0 (сек∙г/см3) при температуре реакции 445°C и давлении реакции 40 кПа.
[0128]
Выход акрилонитрил определяли следующим образом. После получения калибровочной кривой заранее анализом газа акрилонитрила с известной концентрацией с помощью газовой хроматографии (ГХ: название продукта «GC 2014» производства SHIMADZU CORPORATION), газ, полученный реакцией аммоксидирования, количественно вводили в ГХ, и измеряли количество молей акрилонитрила. Выход акрилонитрила определяли по количеству молей акрилонитрила, измеренному в соответствии со следующим уравнением.
Выход акрилонитрила (%)=(количество молей полученного акрилонитрила)/(количество молей поданного пропана)×100
Реакцию непрерывно проводили с данным катализатором (F1), и выход акрилонитрила (AN), измеренный через 10 дней после начала реакции, показан в таблице 1.
[0129]
<Физическое свойство 3; измерение количества катализатора>
Количество катализатора определяли как количество (% по массе) SiO2 носителя, исходя из суммарного количества (100% по массе) оксидного катализатора (F1), и полученный оксидный катализатор (F1) измельчали/перемешивали в течение двух часов, применяя автоматическую агатовую ступку одноосного типа (производимого NITTO KAGAKU CO., LTD.) и формовали прессованием на одноосном прессе в кольцо из винилхлорида (производимое Rigaku Corporation). Измерение с помощью полуколичественного анализа осуществляли на полученном осадке с помощью способа фундаментальных параметров (FP), в котором относительное содержание определяют из библиотеки чувствительности, зарегистрированной в программном обеспечении заранее, применяя рентгенофлуоресцентный анализ с дисперсией по длине волны (торговое название «RIX 1000», производство Rigaku Corporation, трубка из хрома, напряжение трубки 50 кВ, ток трубки 50 мА). Массовая доля (I1) (%) носителя (например, диоксида кремния), определенная данным измерением, показана в таблице 1.
[0130]
<Физическое свойство 4; измерение состава и количества каталитически активных частиц рентгеновским флуоресцентным анализом>
Анализ состава рентгеновской флуоресценцией осуществляли на грануле, полученной измельчением/смешиванием в ступке остатка (H1), полученного при измерении физического свойства 1, для выполнения пресс-формования таким же способом, как и для физического свойства 3. Массовую долю (J1) (%) каталитически активных частиц (G1), предполагая, что масса сложного оксида составляла 100%, можно рассчитать из определенной массовой доли (I2) (%) носителя и массовой доли (I1) (%) носителя в оксидном катализаторе (F1), измеренной для физического свойства 3, применяя следующее уравнение. Полученная массовая доля (J1) (%) каталитически активных частиц представлена в таблице 1.
[0131]
[Пример 2]
Оксидный катализатор получали тем же способом, как в примере 1, за исключением того, что добавляемое количество воды заменяли на 2564 г, добавляемое количество гептамолибдата аммония заменяли на 440,9 г, добавляемое количество метаванадата аммония заменяли на 52,2 г, добавляемое количество триоксида дисурьмы заменяли на 90,5 г, добавляемое количество нитрата церия заменяли на 5,49 г, жидкость ниобиевого исходного вещества (B1), которую добавляли, заменяли на 300,7 г в получении водной смешанной жидкости (A1), и добавляемое количество раствора пероксида водорода заменяли на 179,2 г, добавляемое количество золя диоксида кремния заменяли на 593,8 г, добавляемое количество водного раствора метавольфрамата аммония заменяли на 34,6 г, добавляемое количество порошка диоксида кремния заменяли на 247,5 г, и добавляемое количество воды заменяли на 2228 г на стадии смешения для суспензии предшественников (C1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0132]
[Пример 3]
Оксидный катализатор получали тем же способом, как в примере 1, за исключением того, что добавляемое количество воды заменяли на 2732 г, добавляемое количество гептамолибдата аммония заменяли на 421,1 г, добавляемое количество метаванадата аммония заменяли на 63,8 г, добавляемое количество триоксида дисурьмы заменяли на 76,1 г, добавляемое количество нитрата церия заменяли на 5,25 г, жидкость ниобиевого исходного вещества (B1), которую добавляли, заменяли на 176,8 г при получении водной смешанной жидкости (A1), и добавляемое количество раствора пероксида водорода заменяли на 192,3 г, добавляемое количество золя диоксида кремния заменяли на 862,2 г, добавляемое количество водного раствора метавольфрамата аммония заменяли на 33,0 г, добавляемое количество порошка диоксида кремния заменяли на 196,0 г, и добавляемое количество воды заменяли на 1764 г на стадии смешения для суспензии предшественников (C1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0133]
[Пример 4]
Оксидный катализатор получали тем же способом, как в примере 1, за исключением того, что добавляемое количество воды заменяли на 2909 г, добавляемое количество гептамолибдата аммония заменяли на 448,3 г, добавляемое количество метаванадата аммония заменяли на 67,9 г, добавляемое количество триоксида дисурьмы заменяли на 84,7 г, добавляемое количество нитрата церия заменяли на 5,6 г, жидкость ниобиевого исходного вещества (B1), которую добавляли, заменяли на 211,7 г при получении водной смешанной жидкости (A1), и добавляемое количество раствора пероксида водорода заменяли на 166,7 г, добавляемое количество золя диоксида кремния заменяли на 791,8 г, добавляемое количество водного раствора метавольфрамата аммония заменяли на 35,2 г, добавляемое количество порошка диоксида кремния заменяли на 180,0 г, и добавляемое количество воды заменяли на 1620 г на стадии смешения для суспензии предшественников (C1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0134]
[Пример 5]
Оксидный катализатор получали тем же способом, как в примере 1, за исключением того, что водный раствор метавольфрамата аммония не добавляли на стадии смешения для суспензии предшественников (C1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0135]
[Пример 6]
Водную смешанную жидкость (A1) получали тем же способом, как в примере 1, за исключением того, что воду заменяли на 25 кг, гептамолибдат аммония [(NH4)6Mo7O24∙4H2O] заменяли на 4,088 кг, метаванадат аммония [NH4VO3] заменяли на 0,646 кг, триоксид дисурьмы [Sb2O3] заменяли на 0,907 кг, и нитрат церия заменяли на 0,051 кг, и полученную в результате смесь нагревали при 95°C в течение 1 часа при перемешивании при получении водной смешанной жидкости (A1).
[0136]
Жидкость ниобиевого исходного вещества (B1) получали тем же способом, как в примере 1, за исключением того, что водную смешанную жидкость, полученную заменой воды на 77,3 кг, дигидрата щавелевой кислоты [H2C2O4∙2H2O] на 57,0 кг, и ниобиевой кислоты на 15,7 кг, и перемешиванием полученной в результате смеси при нагревании при 73°C в течение 6 часов, охлаждали до 40°C естественным охлаждением при перемешивании при получении жидкости ниобиевого исходного вещества (B1).
[0137]
Молярное соотношение щавелевая кислота/ниобий и мутность жидкости ниобиевого исходного вещества (B2) измеряли тем же способом, как в примере 1. Как результат, было обнаружено, что концентрация Nb составляла 0,756 моль/кг, концентрация щавелевой кислоты составляла 1,74 моль/кг, и мутность составляла 32 НЕМ.
[0138]
После охлаждения полученной водной смешанной жидкости (A1) до 70°C, добавляли 7,038 кг золя диоксида кремния, содержащего 34,0% по массе SiO2, 1,06 кг раствора пероксида водорода, содержащего 30% по массе H2O2, дополнительно добавляли к водной смешанной жидкости (A1), и перемешивание продолжали при 55°C в течение 30 минут. После дополнительного последовательного добавления дисперсной жидкости, полученной диспергированием 3,850 кг жидкости ниобиевого исходного вещества (B1) и 2,4 кг порошка диоксида кремния (торговое название "AEROSIL 200", изготовитель NIPPON AEROSIL Co., Ltd.) в 197,6 кг воды, и 0,319 кг жидкости метавольфрамата аммония, содержащей 50,2% по весу оксида вольфрама, к жидкости, полученную в результате смесь перемешивали при 50°C в течение 2,5 часов, получая суспензию предшественников (C1).
[0139]
Оксидный катализатор получали тем же способом, как в примере 1, исключая описанное выше, и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0140]
[Пример 7]
Оксидный катализатор получали тем же способом, как в примере 6, за исключением того, что воду заменяли на 83,2 кг, дигидрат щавелевой кислоты [H2C2O4∙2H2O] заменяли на 52,3 кг, и ниобиевую кислота заменяли на 14,5 кг, и полученную в результате смесь перемешивали при нагревании при 78°C в течение 5 часов при получении жидкости ниобиевого исходного вещества (B1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0141]
Молярное соотношение щавелевая кислота/ниобий и мутность полученной жидкости ниобиевого исходного вещества (B1) измеряли тем же способом, как в примере 1. Как результат, было обнаружено, что концентрация ниобия составляла 0,753 моль/кг, концентрация щавелевой кислоты составляла 1,77 моль/кг, и мутность составляла 41 НЕМ.
[0142]
[Сравнительный пример 1]
В смеситель добавляли 88,7 кг воды, и затем воду нагревали до 50°C. Затем, 48,1 кг дигидрата щавелевой кислоты [H2C2O4∙2H2O] загружали при перемешивании, и затем загружали 13,2 кг ниобиевой кислоты, содержащей 76,3% по массе Nb2O5, смешивая оба в воде. Водную смешанную жидкость, полученную перемешиванием данной жидкости при нагревании при 95°C в течение 3 часов, охлаждали до 40°C естественным охлаждением при перемешивании. Затем водную смешанную жидкость охлаждали до 2°C при -10°C/ч и оставляли стоять в течение 1 часа. Затем, смесь осажденного твердого вещества и смешанной жидкости выливали на фильтр, и смешанную жидкость получали фильтрацией осажденного твердого вещества.
[0143]
Оксидный катализатор получали тем же способом, как в примере 1, за исключением того, что полученную в результате смешанную жидкость применяли в качестве жидкости ниобиевого исходного вещества (B1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1. Анализ полученной жидкости ниобиевого исходного вещества (B1) осуществляли тем же способом, как в примере 1, находя, что концентрация Nb данной жидкости ниобиевого исходного вещества (B1) составляет 0,578 моль/кг, молярное соотношение щавелевая кислота/ниобий составляет 2,77, и мутность составляет 201 НЕМ.
[0144]
[Сравнительный пример 2]
Оксидный катализатор получали тем же способом, как в примере 3, за исключением того, что получение осуществляли, применяя жидкость ниобиевого исходного вещества (B1), полученную тем же способом, как в сравнительном примере 1, и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0145]
[Сравнительный пример 3]
Оксидный катализатор получали тем же способом, как в примере 5, за исключением того, что получение осуществляли, применяя жидкость ниобиевого исходного вещества (B1), полученную тем же способом, как в сравнительном примере 1, и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0146]
[Пример 8]
Оксидный катализатор получали тем же способом, как в примере 1, за исключением того, что количество загружаемой воды заменяли на 67,5 кг, дигидрат щавелевой кислоты [H2C2O4∙2H2O] заменяли на 64,6 кг, ниобиевую кислоту заменяли на 17,9 кг, и полученную в результате смесь перемешивали при нагревании при 95°C в течение 4 часов при получении жидкости ниобиевого исходного вещества (B1), и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0147]
[Сравнительный пример 4]
Оксидный катализатор получали тем же способом, как в примере 6, за исключением того, что получение жидкости ниобиевого исходного вещества (B1) осуществляли тем же способом, как в примере 8, и оценку физических свойств 1-3 проводили тем же способом, как в примере 1.
[0148]
[Таблица 1]
Изобретения относятся катализаторам и их использованию для получения ненасышенного нитрила. Описан оксидный катализатор для применения в газофазной каталитической реакции аммоксидирования пропана или изобутана, причем оксидный катализатор содержит сложный оксид, в котором сложный оксид содержит каталитически активные частицы, которые выделяют из сложного оксида с применением раствора пероксида водорода, и данные каталитически активные частицы имеют средний состав, определенный измерениями с помощью STEM-EDX, представленный следующей формулой: Mo1VaSbbNbcWdOn, где a, b, c и d удовлетворяют соотношениям, представленным формулами 0,050≤a≤0,200, 0,050≤b≤0,200, 0,100≤c≤0,300, 0≤d≤0,100, и a≤c; и n представляет собой число, определяемое валентностями других элементов. Описан способ получения ненасыщенного нитрила реакцией газофазного каталитического аммоксидирования пропана или изобутана в присутствии описанного выше оксидного катализатора. Технический результат - получение ненсыщенного нитрила. 2 н. и 5 з.п. ф-лы, 1 табл., 12 пр.
1. Оксидный катализатор для применения в газофазной каталитической реакции аммоксидирования пропана или изобутана, причем оксидный катализатор содержит сложный оксид, в котором
сложный оксид содержит каталитически активные частицы, которые выделяют из сложного оксида с применением раствора пероксида водорода, и
данные каталитически активные частицы имеют средний состав, определенный измерениями с помощью STEM-EDX, представленный следующей формулой (1):
Mo1VaSbbNbcWdOn (1),
где a, b, c и d удовлетворяют соотношениям, представленным формулами 0,050≤a≤0,200, 0,050≤b≤0,200, 0,100≤c≤0,300, 0≤d≤0,100, и a≤c; и n представляет собой число, определяемое валентностями других элементов.
2. Оксидный катализатор по п. 1, где формула (1) удовлетворяет соотношению, представленному формулой 1,1×a≤c.
3. Оксидный катализатор по п. 1, где формула (1) удовлетворяет соотношению, представленному формулой 1,3×a≤c.
4. Оксидный катализатор по любому из пп. 1-3, где формула (1) удовлетворяет соотношению, представленному формулой 8,00≤100×b/(1+a)≤10,00.
5. Оксидный катализатор по любому из пп. 1-4, где оксидный катализатор содержит диоксид кремния в качестве носителя, на который нанесен сложный оксид, и массовая доля диоксида кремния составляет от 30 до 70% по массе в пересчете на SiO2 относительно общего количества оксидного катализатора.
6. Оксидный катализатор по любому из пп. 1-5, в котором сложный оксид содержит каталитически активные частицы, содержащие молибден, ванадий, сурьму и ниобий, и массовая доля каталитически активных частиц составляет 45% по массе или более от общего количества сложного оксида.
7. Способ получения ненасыщенного нитрила, причем способ включает получение ненасыщенного нитрила реакцией газофазного каталитического аммоксидирования пропана или изобутана в присутствии оксидного катализатора по любому из пп. 1-6.
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| CN 104661745 A, 27.05.2015 | |||
| СМЕШАННЫЕ МЕТАЛЛООКСИДНЫЕ КАТАЛИЗАТОРЫ И СПОСОБ КАТАЛИТИЧЕСКОЙ КОНВЕРСИИ НИЗШИХ АЛИФАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2008 |
|
RU2476265C2 |
| US 2013225862 A1, 29.08.2013 | |||
| УЛУЧШЕННЫЕ СЕЛЕКТИВНЫЕ КАТАЛИЗАТОРЫ АММОКСИДИРОВАНИЯ | 2014 |
|
RU2690512C2 |

