Область техники, к которой относится изобретение
Настоящее изобретение относится к новому тиазольному производному (соединению, представленному приведенной ниже формулой (I) (далее в данном описании также называемое соединением (I)) и его фармацевтически приемлемой соли, которую далее в данном описании иногда называют совместно соединением настоящего изобретения). Кроме того, настоящее изобретение относится к ингибитору сосудистого адгезивного белка-1, фармацевтическому агенту для профилактики или лечения заболевания, связанного с сосудистым адгезивным белком-1, и подобных, который содержит соединение настоящего изобретения в качестве активного ингредиента.
Уровень техники
Сосудистый адгезивный белок-1 (далее в данном описании сокращенно обозначаемый VAP-1) представляет собой аминооксидазу (чувствительную к семикарбазиду аминооксидазу, SSAO), в большом количестве присутствующую в плазме человека, экспрессия которой заметно повышается в сосудистом эпителии и гладких мышцах сосудов при воспалительном поражении. Хотя физиологическая роль VAP-1 до недавнего времени не была прояснена, ген VAP-1 был клонирован в 1998, и сообщалось, что VAP-1 является мембранным белком, который в качестве адгезионной молекулы контролирует движение и миграцию лимфоцитов и NK клеток при контроле экспрессии воспалительных цитокинов. Хотя амин, который является субстратом, неизвестен, считают, что он представляет собой метиламин, синтезированный в любой части живого организма. Также известно, что пероксид водорода и альдегид, синтезированные в результате внутримолекулярной аминооксидазной активности, являются важными факторами для адгезивной активности.
Недавние сообщения продемонстрировали, что ферментативная активность VAP-1 в плазме увеличивается у пациентов с диабетом как типа I, так и типа II, и увеличение является особенно заметным у диабетиков, страдающих от осложнений в результате ретинопатии (Diabetologia, 42 (1999) 233-237 (непатентный документ 1), Diabetes Medicine, 16 (1999) 514-521 (непатентный документ 2)).
Кроме того, также сообщалось, что VAP-1 связан со следующими заболеваниями (1)-(6), такими как: (1) цирроз печени, эссенциальная стабилизированная гипертензия, диабет, артериосклероз (см. JP-A-61-239891 (патентный документ 1) и патент США No. 4888283 (патентный документ 2)); (2) эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанная с подагрой и артритом, ретинопатия (у диабетиков) (см. WO 1993/23023 (патентный документ 3)); (3) воспалительное заболевание или симптом (соединительных тканей) (ревматоидный артрит, анкилозирующий спондилит, псориатический артрит и остеоартрит или дегенеративное заболевание сустава, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системная красная волчанка, дискоидная красная волчанка, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическая полимиалгия, васкулит, темпоральный артрит, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит); воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление (стоматит) слизистой мембраны рта и рецидивирующий афтозный стоматит]; воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера, и ишемическое/реперфузионное повреждение, связанное с ишемическим инсультом); легочное воспалительное заболевание или симптом (астма, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания); (хроническое) воспалительное заболевание или симптом кожи (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз); заболевание, связанное с метаболизмом углеводов (диабет и осложнения, вызванные диабетом), включая диабет микрососудов и больших сосудов (артериосклероз, сосудистая ретинопатия, ретинопатия, нефропатия, нефротический синдром и невропатия (множественная невропатия, мононевропатия и вегетативная невропатия), язва стопы, проблема суставов и увеличение риска инфекции); заболевание, связанное с нарушениями в дифференциации или функционировании адипоцитов или функционировании клеток гладких мышц (артериосклероз и ожирение); заболевание сосудов [атеросклероз, неатеросклеротическое заболевание, ишемические заболевания сердца, включая инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)]; хронический артрит; воспалительное заболевание кишечника; заболевание кожи (см. WO 2002/02090 (патентный документ 4), WO 2002/02541 (патентный документ 5) и US 2002/0173521 A (патентный документ 6)); (4) диабет (см. WO 2002/38152 (патентный документ 7)); (5) SSAO-опосредованные осложнения [диабет (инсулинозависимый диабет (IDDM) и инсулиннезависимый диабет (NIDDM)) и сосудистые осложнения (инфаркт, стенокардия, апоплексия мозга, ампутация, медицинская слепота и почечная недостаточность)] (см. WO 2002/38153 (патентный документ 8)); (6) заболевание, связанное со сверхпроницаемостью сосудов [возрастная дегенерация желтого пятна, возрастная дисковидная дегенерация желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическая ретинопатия, хориоретинопатия, неоваскулярная макулопатия, неоваскулярная глаукома, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическая офтальмия, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язва роговицы, язва конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующая разъедающая язва роговицы, воспалительное заболевание глаз, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаз, вызванное физическим повреждением глаз, симптом, вызванный воспалительным заболеванием глаз, включающий зуд, воспаление, отек и язву, эритема, многоформная экссудативная эритема, узловатая эритема, кольцевидная эритема, склередема взрослых, дерматит, ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит] (см. WO 2004/087138 (патентный документ 9)); и подобные.
В WO 2004/067521 (патентный документ 10), WO 2004/087138 (патентный документ 9), WO 2006/011631 (патентный документ 11) и WO 2006/028269 (патентный документ 12) описаны тиазольные производные, имеющие специфическую структуру, и возможность их применения для профилактики или лечения связанного с VAP-1 заболевания, такого как отек желтого пятна, заболевания, связанного со сверхпроницаемостью сосудов и подобного.
Тиазольные производные, имеющие специфические структуры, которые описаны в WO 2004/067521 (патентный документ 10), WO 2004/087138 (патентный документ 9) и WO 2006/028269 (патентный документ 12), также концептуально включают соединение, содержащее гидразиновую группу или гидразинокарбонильную группу на конце молекулы. Однако в них не описано соединение, содержащее специфическую функциональную группу настоящего изобретения (эфирную группу карбазиновой кислоты, тиоэфирную группу карбазиновой кислоты или семикарбазидную группу).
Тогда как в WO 2008/066145 (патентный документ 13) описано тиазольное производное, имеющее конкретную структуру, в нем не описано новое соединение настоящего изобретения.
Патентный документ 1: JP-A-61-239891
Патентный документ 2: патент США No. 4888283
Патентный документ 3: WO 1993/23023
Патентный документ 4: WO 2002/02090
Патентный документ 5: WO 2002/02541
Патентный документ 6: US 2002/0173521 A
Патентный документ 7: WO 2002/38152
Патентный документ 8: WO 2002/38153
Патентный документ 9: WO 2004/087138
Патентный документ 10: WO 2004/067521
Патентный документ 11: WO 2006/011631
Патентный документ 12: WO 2006/028269
Патентный документ 13: WO 2008/066145
Непатентный документ 1: Diabetologia, 42 (1999) 233-237
Непатентный документ 2: Diabetes Medicine, 16 (1999) 514-521
Описание настоящего изобретения
Задачи, которые нужно решить с помощью настоящего изобретения
Цель настоящего изобретения заключается в предложении нового тиазольного производного, пригодного в качестве ингибитора VAP-1, фармацевтического средства для профилактики или лечения связанных с VAP-1 заболеваний и подобных.
Способы решения задач
В результате интенсивных исследований авторы настоящего изобретения обнаружили, что тиазольное производное, содержащее специфическую функциональную группу (эфирная группа карбазиновой кислоты, тиоэфирная группа карбазиновой кислоты или семикарбазидная группа) на конце молекулы, обладает превосходной ингибирующей VAP-1 активностью, является высокоселективным в отношении фермента и может устранять опасные побочные эффекты, и провели дополнительные исследования, которые привели в результате к созданию настоящего изобретения.
Соответственно, настоящее изобретение включает следующее.
(1) Соединение, представленное формулой (I):
R1-NH-X-Y-Z
где
R1 представляет собой ацил;
X представляет собой двухвалентный остаток, полученный из необязательно замещенного тиазола;
Y имеет формулу (III):
J-L-M
где J представляет собой связь, низший алкилен, низший алкенилен, низший алкинилен, -(CH2)n-O-, -(CH2)n-NH-, -(CH2)n-CO- или -(CH2)n-SO2- (где n равно целому числу от 0 до 6);
L представляет собой связь, -O-, -NH-, -CO- или -SO2-;
M представляет собой связь, низший алкилен, низший алкенилен или низший алкинилен, при условии, что, когда J представляет собой -(CH2)n-O-, L не является -O-, -NH- и -SO2-, когда J представляет собой -(CH2)n-NH-, L не является -O- и -NH-, когда J представляет собой -(CH2)n-CO-, L не является -CO-, когда J представляет собой -(CH2)n-SO2-, L не является -O- и -SO2- (где n является таким, как определено выше),
Z имеет формулу (II):
A-B-D-E
где A представляет собой двухвалентный остаток, полученный из необязательно замещенного бензола, или двухвалентный остаток, полученный из необязательно замещенного тиофена;
B представляет собой -(CH2)l-NR2-CO-, где R2 представляет собой водород, низший алкил или ацил, l равно целому числу от 1 до 6, -(CH2)m-O-CO- или -(CH2)m-S-CO- (где m равно целому числу от 1 до 6);
D представляет собой -NR3-, где R3 представляет собой водород, низший алкил, алкоксикарбонил или ацил; и
E представляет собой необязательно замещенный амино;
или его фармацевтически приемлемая соль.
(2) Соединение по п.(1), где соединением, представленным указанной выше формулой (I), является
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат,
2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил-гидразинкарбоксилат,
2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил-гидразинкарбоксилат,
4-{2-[(гидразинокарбонил)окси]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилат,
2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-гидразинкарбоксилат,
3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат,
2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил]фенил)этил-гидразинкарбоксилат,
{5-[2-(2-ацетиламино-1,3-тиазол-4-ил)этил]тиофен-2-ил}метил-гидразинкарбоксилат,
2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил-гидразинкарбоксилат,
3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил-гидразинкарбоксилат,
3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил-гидразинкарбоксилат,
3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил-гидразинкарбоксилат,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
N-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил)гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил)гидразинкарбоксамид,
N-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метил}амино)бензил]гидразинкарбоксамид,
2-(ацетиламино)-N-(4-{[(гидразинокарбонил)амино]метил}фенил)-1,3-тиазол-4-карбоксамид,
N-[2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил]гидразинкарбоксамид,
4-{2-[(гидразинокарбонил)амино]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилат,
N-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
N-[2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамид,
N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарбоксамид,
N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил]гидразинкарбоксамид,
N-[3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил]гидразинкарбоксамид,
N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил]гидразинкарбоксамид,
N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил]гидразинкарбоксамид,
S-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарботиоат,
S-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарботиоат или
S-[(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарботиоат,
или его фармацевтически приемлемая соль.
(3) Соединение по приведенному выше (1), где соединением, представленное указанной выше формулой (I), является 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат, 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат или N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид или его фармацевтически приемлемую соль.
(4) Соединение по любому из приведенных выше п.п.(1)-(3), которое применяют в качестве фармацевтического агента, или его фармацевтически приемлемая соль.
(5) Фармацевтическая композиция, содержащая соединение по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемую соль в качестве активного ингредиента.
(6) Ингибитор VAP-1, содержащий соединение по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемую соль в качестве активного ингредиента.
(7) Фармацевтический агент для профилактики или лечения связанного с VAP-1 заболевания, который содержит соединение по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемую соль в качестве активного ингредиента.
(8) Фармацевтический агент по приведенному выше п.(7), где указанное выше связанное с VAP-1 заболевание представляет собой отек желтого пятна (диабетический и недиабетический отек желтого пятна), возрастную дегенерацию желтого пятна, возрастную дисковидную дегенерацию желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическую ретинопатию, хориоретинопатию, неоваскулярную макулопатию, неоваскулярную глаукому, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаз, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаз, вызванное физическим повреждением глаз, симптом, вызванный воспалительным заболеванием глаз, включая зуд, воспаление, отек и язву, эритему, многоформную экссудативную эритему, узловатую эритему, кольцевидную эритему, склередему взрослых, дерматит (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз), ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит, цирроз печени, эссенциальную стабилизированную гипертензию, диабет, артериосклероз, эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанную с подагрой и артритом, воспалительное заболевание или симптом соединительной ткани (ревматоидный артрит, анкилозирующий спондилит, псориатический артрит и остеоартрит или дегенеративное заболевание суставов, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системную красную волчанку, дискоидную красную волчанку, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическую полимиалгию, васкулит, темпоральный артрит, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит), воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление слизистой мембраны рта (стоматит и рецидивирующий афтозный стоматит)], воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера и ишемическое/реперфузионное повреждение, связанное с ишемическим инсультом), легочное воспалительное заболевание или симптом (астма, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания), заболевание, связанное с метаболизмом углеводов (диабет и осложнения, полученный в результате диабета (диабетическую невропатию, диабетическую нефропатию)), включая заболевание микрососудов и больших сосудов (артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (множественную невропатию, мононевропатию и вегетативную невропатию), язву стопы, проблему суставов и увеличение риска инфекции), заболевание, связанное с нарушениями дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (артериосклероз и ожирение), сосудистое заболевание [атероматозный атеросклероз, неатероматозное атеросклеротическое заболевание, ишемическое заболевание сердца, включая инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)], хронический артрит, воспалительное заболевание кишечника или SSAO-опосредованное осложнения [диабет (инсулин-зависимый диабет (IDDM) и инсулин-независимый диабет (NIDDM)) и сосудистые осложнения (инфаркт, стенокардию, апоплексию мозга, ампутацию, медицинскую слепоту и почечную недостаточность)], заболевание глаз, связанное с гипоксией или ишемией [ретинопатию недоношенных, пролиферативную диабетическую ретинопатию, полипообразную хориоидальную васкулопатию, ретинальную ангиоматозную пролиферацию, окклюзию центральной артерии сетчатки, окклюзию ветви центральной вены сетчатки, болезнь Коутса, семейную экссудативную витреоретинопатию, болезнь отсутствия пульса (болезнь Такаясу), болезнь Илза, антифосфолипидный синдром, лейкемическую ретинопатию, синдром повышенной вязкости крови, макроглобулинемию, связанную с интерфероном ретинопатию, гипертоническую ретинопатию, ретинопатию при облучении, дефицит стволовых клеток роговичного эпителия] или катаракту.
(9) Применение соединения по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемой соли для получения фармацевтического агента в качестве ингибитора VAP-1.
(10) Применение соединения по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемой соли для получения фармацевтического агента для профилактики или лечения связанного с VAP-1 заболевания.
(11) Применение по приведенному выше п.(10), где указанное выше связанное с VAP-1 заболевание представляет собой отек желтого пятна (диабетический и недиабетический отек желтого пятна), возрастную дегенерацию желтого пятна, возрастную дисковидную дегенерацию желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическую ретинопатию, хориоретинопатию, неоваскулярную макулопатию, неоваскулярную глаукому, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаз, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаз, вызванное физическим повреждением глаз, симптом, вызванный воспалительным заболеванием глаз, включая зуд, воспаление, отек и язву, эритему, многоформную экссудативную эритему, узловатую эритему, кольцевидную эритему, склередему взрослых, дерматит (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз), ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит, цирроз печени, эссенциальную стабилизированную гипертензию, диабет, артериосклероз, эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанную с подагрой и артритом, воспалительное заболевание или симптом соединительной ткани (ревматоидный артрит, анкилозирующий спондилит, псориатический артритом и остеоартрит или дегенеративное заболевание суставов, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системную красную волчанку, дискоидную красную волчанку, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическую полимиалгию, васкулит, темпоральный артрит, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит), воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление слизистой мембраны рта (стоматит и рецидивирующий афтозный стоматит)], воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера и ишемическое/реперфузионное повреждение, связанное с ишемическим инсультом), легочное воспалительное заболевание или симптом (астму, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания), заболевание, связанное с метаболизмом углеводов (диабет и осложнения, полученные в результате диабета (диабетическую невропатию, диабетическую нефропатию)), включая заболевание микрососудов и больших сосудов (артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (множественную невропатию, мононевропатию и вегетативную невропатию), язву стопы, проблему суставов и увеличение риска инфекции), заболевание, связанное с нарушением дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (артериосклероз и ожирение), сосудистое заболевание [атероматозный атеросклероз, неатероматозное атеросклеротическое заболевание, ишемическое заболевание сердца, включая инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)], хронический артрит, воспалительное заболевание кишечника или SSAO-опосредованные осложнения [диабет (инсулин-зависимый диабет (IDDM) и инсулин-независимый диабет (NIDDM)) и сосудистые осложнения (инфаркт, стенокардию, апоплексию мозга, ампутацию, медицинскую слепоту и почечную недостаточность)], болезнь глаз, связанную с гипоксией или ишемией [ретинопатию недоношенных, пролиферативную диабетическую ретинопатию, полипообразную хориоидальную васкулопатию, ретинальную ангиоматозную пролиферацию, окклюзию центральной артерии сетчатки, окклюзию ветви центральной вены сетчатки, болезнь Коутса, семейную экссудативную витреоретинопатию, болезнь отсутствия пульса (болезнь Такаясу), болезнь Илза, антифосфолипидный синдром, лейкемическую ретинопатию, синдром повышенной вязкости крови, макроглобулинемию, связанную с интерфероном ретинопатию, гипертоническую ретинопатию, ретинопатию после облучения, дефицит стволовых клеток роговичного эпителия] или катаракту.
(12) Способ ингибирования VAP-1 у субъекта, включающий введение эффективного количества соединения по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемой соли данному субъекту.
(13) Способ профилактики или лечения связанного с VAP-1 заболевания у субъекта, включающий введение эффективного количества соединения по любому из приведенных выше п.п.(1)-(3) или его фармацевтически приемлемой соли данному субъекту.
(14) Способ по приведенному выше п.(13), в котором указанное выше связанное с VAP-1 заболевание представляет собой отек желтого пятна (диабетический и недиабетический отек желтого пятна), возрастную дегенерацию желтого пятна, возрастную дисковидную дегенерацию желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическую ретинопатию, хориоретинопатию, неоваскулярную макулопатию, неоваскулярную глаукому, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаз, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаз, вызванное физическим повреждением глаза, симптом, вызванный воспалительным заболеванием глаз, включая зуд, воспаление, отек и язву, эритему, многоформную экссудативную эритему, узловатую эритему, кольцевидную эритему, склередему взрослых, дерматит (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз), ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит, цирроз печени, эссенциальную стабилизированную гипертензию, диабет, артериосклероз, эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанную с подагрой и артритом, воспалительное заболевание или симптом соединительной ткани (ревматоидный артрит, анкилозирующий спондилит, псориатический артрит и остеоартрит или дегенеративное заболевание суставов, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системную красную волчанку, дискоидную красную волчанку, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическую полимиалгию, васкулит, темпоральный артрит, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит), воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление слизистой мембраны рта (стоматит и рецидивирующий афтозный стоматит)], воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера и ишемическое/реперфузионное, связанное с ишемическим инсультом), легочное воспалительное заболевание или симптом (астма, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания), заболевание, связанное с метаболизмом углеводов (диабет и осложнения, полученные в результате диабета (диабетическую невропатию, диабетическую нефропатию)), включая заболевание микрососудов и больших сосудов (артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (множественную невропатию, мононевропатию и вегетативную невропатию), язву стопы, проблему суставов и увеличение риска инфекции), заболевание, связанное с нарушением дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (артериосклероз и ожирение), сосудистое заболевание [атероматозный атеросклероз, неатероматозное атеросклеротическое заболевание, ишемическое заболевание сердца, включая инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)], хронический артрит, воспалительное заболевание кишечника или SSAO-опосредованное осложнения [диабет (инсулин-зависимый диабет (IDDM) и инсулин-независимый диабет (NIDDM)) и сосудистые осложнения (инфаркт, стенокардию, апоплексию мозга, ампутацию, медицинскую слепоту и почечную недостаточность)], заболевание глаз, связанное с гипоксией или ишемией [ретинопатию недоношенных, пролиферативную диабетическую ретинопатию, полипообразную хориоидальную васкулопатию, ретинальную ангиоматозную пролиферацию, окклюзию центральной артерии сетчатки, окклюзию ветви центральной вены сетчатки, болезнь Коутса, семейную экссудативную витреоретинопатию, болезнь отсутствия пульса (болезнь Такаясу), болезнь Илза, антифосфолипидный синдром, лейкемическую ретинопатию, синдром повышенной вязкости крови, макроглобулинемию, связанную с интерфероном ретинопатию, гипертоническую ретинопатию, ретинопатию при облучении, дефицит стволовых клеток роговичного эпителия] или катаракту.
Действие соединений настоящего изобретения
Соединение настоящего изобретения обладает превосходной ингибирующей VAP-1 активностью и превосходной селективностью в отношении фермента и, следовательно, может устранять побочные эффекты и подобное, которые являются нежелательными, в качестве фармацевтического продукта. Следовательно, соединение является пригодным в качестве ингибитора VAP-1, фармацевтического агента для профилактики или лечения связанного с VAP-1 заболевания и подобного.
Лучший способ осуществления настоящего изобретения
Термины, используемые в настоящем изобретении в приведенном выше и ниже описании настоящего изобретения, объясняются подробно далее.
Термин "низший" используют для обозначения группы, содержащей 1-6 атомов углерода, предпочтительно 1-4, если не указано особо.
Примеры "низшего алкила" включают алкил с нормальной или разветвленной цепью, содержащей 1-6 атомов углерода (например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, трет-пентил и гексил) и подобные. Среди них C1-C4алкил является наиболее предпочтительным.
Примеры "низшего алкилена" включают алкилен с нормальной или разветвленной цепью, содержащей 1-6 атомов углерода (например, метилен, этилен, триметилен, пропилен, этилиден и пропилиден) и подобные. Среди них C1-C4алкилен является наиболее предпочтительным.
Примеры "низшего алкенилена" включают алкенилен с нормальной или разветвленной цепью, содержащей 2-6 атомов углерода (например, винилен, 1-пропенилен, 1-метил-1-пропенилен, 2-метил-1-пропенилен, 2-пропенилен, 2-бутенилен, 1-бутенилен, 3-бутенилен, 2-пентенилен, 1-пентенилен, 3-пентенилен, 4-пентенилен, 1,3-бутадиенилен, 1,3-пентадиенилен, 2-пентен-4-инилен, 2-гексенилен, 1-гексенилен, 5-гексенилен, 3-гексенилен, 4-гексенилен, 3,3-диметил-1-пропенилен, 2-этил-1-пропенилен, 1,3,5-гексатриенилен, 1,3-гексадиенилен, 1,4-гексадиенилен) и подобные. Среди них C2-C4алкенилен является наиболее предпочтительным.
Указанный выше низший алкенилен может быть в E-форме или Z-форме. Когда соединение настоящего изобретения содержит остаток низшего алкенилена, соединение настоящего изобретения включает любой стереоизомер, где остаток низшего алкенилена имеет E-структуру или Z-структуру.
Примеры "низшего алкинилена" включают алкинилен с нормальной или разветвленной цепью, содержащей 2-6 атомов углерода, которая содержит 1-3 тройные связи (например, этинилен, 1-пропинилен, 1-метил-1-пропинилен, 2-метил-1-пропинилен, 2-пропинилен, 2-бутинилен, 1-бутинилен, 3-бутинилен, 2-пентинилен, 1-пентинилен, 3-пентинилен, 4-пентинилен, 2-пентин-4-инилен, 2-гексинилен, 1-гексинилен, 5-гексинилен, 3-гексинилен, 4-гексинилен, 3,3-диэтил-1-пропинилен, 2-этил-1-пропинилен) и подобные. Среди них C2-C4алкинилен является наиболее предпочтительным.
Примеры "арила" включают C6-C10арил (например, фенил и нафтил) и подобные, где "арил" может быть замещен 1-3 заместителями, и положение замещения конкретно не ограничено.
Примеры "аралкила" включают аралкил, где арильная группа содержит 6-10 атомов углерода [т.е. арильная группа представляет собой C6-C10арил указанного выше "арила"], и алкильная группа содержит 1-6 атомов углерода [т.е. алкильная группа представляет собой C1-C6алкил указанного выше "низшего алкила"] (например, бензил, фенетил, 1-нафтилметил, 2-нафтилметил, 3-фенилпропил, 4-фенилбутил и 5-фенилпентил) и подобные.
Примеры "циклического низшего алкила" включают циклоалкил, содержащий 3-6 атомов углерода (например, циклопропил, циклобутил, циклопентил, циклогексил) и подобные.
Примеры "циклического низшего алкоксикарбонила" включают циклоалкоксикарбонил, где циклоалкильная группа содержит 3-6 атомов углерода (например, циклопропилоксикарбонил, циклобутилоксикарбонил, циклопентилоксикарбонил, циклогексилоксикарбонил) и подобные.
Примеры "гетероцикла" включают "ароматический гетероцикл" и "неароматический гетероцикл". Примеры "ароматического гетероцикла" включают 5-10-членный ароматический гетероцикл, содержащий, помимо атомов углерода, 1-3 гетероатома, выбранных из атома азота, кислорода, серы и подобных, например, тиофен, фуран, пиррол, имидазол, пиразол, тиазол, изотиазол, оксазол, изоксазол, пиридин, пиридазин, пиримидин, пиразин и подобные. Примеры "неароматического гетероцикла" включают 5-10-членный неароматический гетероцикл, содержащий, помимо атомов углерода, 1-3 гетероатома, выбранных из атома азота, кислорода, серы и подобных, например, пирролидин, имидазолин, пиразолидин, пиразолин, пиперидин, пиперазин, морфолин, тиоморфолин, диоксолан, оксазолидин, тиазолидин, триазолизин и подобные.
Примеры "ацила" включают алкилкарбонил, арилкарбонил и подобные.
Примеры "алкилкарбонила" включают алкилкарбонил, где алкильная группа содержит 1-6 атомов углерода [т.е. алкильная группа представляет собой C1-C6алкил указанного выше "низшего алкила"] (например, ацетил, пропионил, бутирил, изобутирил, валерил, изовалерил, пивалоил, гексаноил, гептаноил и деканоил) и подобные.
Примеры "арилкарбонила" включают арилкарбонил, где арильная группа содержит 6-10 атомов углерода [т.е. арильная группа представляет собой C6-C10арил указанного выше "арила"] (например, бензоил и нафтоил) и подобные.
Примеры "алкоксикарбонила" включают алкилоксикарбонил, аралкилоксикарбонил и подобные.
Примеры "алкилоксикарбонила" включают алкилоксикарбонил, где алкильная группа содержит 1-10 атомов углерода (например, метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, изобутоксикарбонил, втор-бутоксикарбонил, трет-бутоксикарбонил, пентилоксикарбонил, трет-пентилоксикарбонил, гексилоксикарбонил и децилоксикарбонил, и т.д.) и подобные.
Примеры "аралкилоксикарбонила" включают аралкилоксикарбонил, где арильная группа содержит 6-10 атомов углерода [т.е. арильная группа представляет собой C6-C10арил указанного выше "арила"], и алкильная группа содержит 1-6 атомов углерода [т.е. алкильная группа представляет собой C1-C6алкил указанного выше "низшего алкила"] (например, бензилоксикарбонил, фенетилоксикарбонил, 1-нафтилметилоксикарбонил, 2-нафтилметилоксикарбонил, 3-фенилпропилоксикарбонил, 4-фенилбутилоксикарбонил и 5-фенилпентилоксикарбонил, и т.д.) и подобные.
Примеры "ацила" для R1 в формуле (I) включают группы, определенные выше, и подобные, предпочтительно алкилкарбонил (алкилкарбонил является таким, как определено выше) и подобные, особенно предпочтительно ацетил и подобные.
Примеры "двухвалентного остатка, полученного из необязательно замещенного тиазола", для X в формуле (I) включают
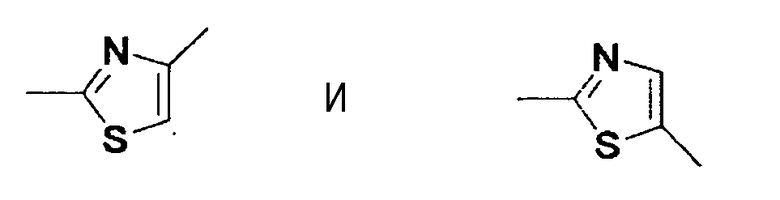
"Тиазол" может иметь заместитель, и положение замещения конкретно не ограничено. Примеры "заместителя" указанного выше "необязательно замещенного тиазола" включают группу, описанную в следующих (1)-(12), и подобные.
(1) галоген (например, фтор, хлор, бром);
(2) алкоксикарбонил, определенный выше (например, этоксикарбонил);
(3) необязательно замещенный арил (арил является таким, как определено выше, и он может быть замещен -SO2-(низшим алкилом), где низший алкил является таким, как определено выше, и подобным, где положение замещения конкретно не ограничено) (например, фенил и 4-(метилсульфонил)фенил);
(4) группу формулы: -CONRaRb, где Ra представляет водород, низший алкил, арил или аралкил, Rb представляет водород, низший алкил, арил или аралкил, где низший алкил, арил и аралкил являются такими, как определено выше (например, N-метиламинокарбонил, N-фениламинокарбонил, N,N-диметиламинокарбонил и N-бензиламинокарбонил);
(5) группу формулы: -CONH-(CH2)k-арил, где k равно целому числу от 0 до 6; арил является таким, как определено выше, необязательно имеет 1-5 заместителей, выбранных из группы, состоящей из -NO2, -SO2-(низшего алкила), где низший алкил является таким, как определено выше, -CF3 и -O-арила, где арил является таким, как определено выше, где положение замещения конкретно не ограничено;
(6) группу формулы: -CONH-(CH2)s-гетероцикл, где s равно целому числу от 0 до 6; и гетероцикл является таким, как определено выше (например, пиридин);
(7) группу формулы: -CO-гетероцикл, где гетероцикл является таким, как определено выше (например, пирролидин, пиперидин, пиперазин, тиоморфолин), и гетероцикл необязательно имеет 1-5 заместителей, выбранных из группы, состоящей из -CO-(низшего алкила), где низший алкил является таким, как определено выше, -CO-O-(низшего алкила), где низший алкил является таким, как определено выше, -SO2-(низшего алкила), где низший алкил является таким, как определено выше, оксо (т.е. =O) и группы формулы: -CONRcRd, где Rc представляет собой водород, низший алкил, арил или аралкил, Rd представляет собой водород, низший алкил, арил или аралкил, и низший алкил, арил и аралкил являются такими, как определено выше, где положение замещения конкретно не ограничено;
(8) группу формулы: -(CH2)t-арил, где t равно целому числу от 1 до 6; арил является таким, как определено выше, и необязательно имеет 1-5 заместителей, выбранных из группы, состоящей из -S-(низшего алкила), где низший алкил является таким, как определено выше, -SO2-(низшего алкила), где низший алкил является таким, как определено выше, -SO2-NRVRW, где Rv представляет собой водород, низший алкил, арил или аралкил, Rw представляет собой водород, низший алкил, арил или аралкил, и где низший алкил, арил и аралкил являются такими, как определено выше, -CO2-(низшего алкила), где низший алкил является таким, как определено выше, -NHCO-O-(низшего алкила), где низший алкил является таким, как определено выше, и группы формулы: -CONReRf, где Re представляет собой водород, низший алкил, арил или аралкил, Rf представляет собой водород, низший алкил, арил или аралкил, и низший алкил, арил и аралкил являются такими, как определено выше, где положение замещения конкретно не ограничено;
(9) группу формулы: -(CH2)O-гетероцикл, где o равно целому числу от 0 до 6; гетероцикл является таким, как определено выше (например, пирролидин, пиперидин, пиперазин, морфолин, тиоморфолин), и необязательно имеет 1-5 заместителей, выбранных из группы, состоящей из оксо (т.е. =O); -CO-(низшего алкила), где низший алкил является таким, как определено выше; -CO-O-(низшего алкила), где низший алкил является таким, как определено выше; -SO2-(низшего алкила), где низший алкил является таким, как определено выше; -CO-(гетероцикла), где гетероцикл является таким, как определено выше (например, пирролидин, пиперазин и морфолин), и необязательно имеет 1-5 заместителей, выбранных из группы, состоящей из низшего алкила (низший алкил является таким, как определено выше) и галогена (например, фтор, хлор, бром), где положение замещения конкретно не ограничено; и группы формулы: -CONRgRh, где Rg представляет собой водород, низший алкил, арил или аралкил, Rh представляет собой водород, низший алкил, арил или аралкил, и низший алкил, арил и аралкил являются такими, как определено выше, где положение замещения конкретно не ограничено;
(10) группу формулы: -(CH2)P-NRiRj, где p равно целому числу от 0 до 6; Ri представляет собой водород, ацил, низший алкил, арил или аралкил, Rj представляет собой водород, ацил, низший алкил, арил или аралкил, и ацил, низший алкил, арил и аралкил являются такими, как определено выше, и низший алкил необязательно имеет 1-5 заместителей, выбранных из группы, состоящей из группы формулы: -CONRkRl, где Rk представляет собой водород, низший алкил, арил или аралкил, Rl представляет собой водород, низший алкил, арил или аралкил, и низший алкил, арил и аралкил являются такими, как определено выше, где положения замещения конкретно не ограничено;
(11) группу формулы: -CON(H или низший алкил)-(CHRm)q-T, где q равно целому числу от 0 до 6; низший алкил является таким, как определено выше; Rm представляет собой водород, аралкил, определенный выше, или алкил, определенный выше (особенно низший алкил), необязательно замещенные 1-3 заместителями, выбранными из группы, состоящей из -OH и -CONH2, где положение замещения конкретно не ограничено; T представляет собой водород; группу формулы: -CONRnRo, где Rn представляет собой водород, низший алкил, арил или аралкил, Ro представляет собой водород, низший алкил, арил или аралкил, и низший алкил, арил и аралкил являются такими, как определено выше; -NH-CO-RP, где Rp представляет собой низший алкил, определенный выше, или аралкил, определенный выше; -NH-SO2-(низший алкил), где низший алкил является таким, как определено выше; -SO2-(низший алкил), где низший алкил является таким, как определено выше; -гетероцикл, где гетероцикл является таким, как определено выше (например, пиридин, пирролидин и морфолин), необязательно имеет 1-3 заместителя (например, оксо (т.е. =O)), где положение замещения конкретно не ограничено; или -CO-(гетероцикл), где гетероцикл является таким, как определено выше (например, пиперидин и морфолин)); и
(12) группу формулы: -(CH2)r-CO-NRtRu, где r равно целому числу от 1 до 6; Rt представляет собой водород, низший алкил, арил или аралкил, Ru представляет собой водород, низший алкил, арил или аралкил, и низший алкил, арил и аралкил являются такими, как определено выше.
Положение замещения на ариле или гетероцикле может быть любым и конкретно не ограничено. Предпочтительный "заместитель" указанного выше "необязательно замещенного тиазола" представляет собой метилсульфонилбензил, сульфамоилбензил (например, 4-сульфамоилбензил) и подобные. Положение замещения метилсульфонильной группой, сульфамоильной группой и подобной конкретно не ограничено.
В качестве "двухвалентного остатка, полученного из тиазольной" группы, "двухвалентного остатка, полученного из необязательно замещенного тиазола", для X в формуле (I), 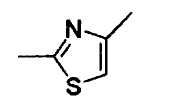 является предпочтительным. В качестве "заместителя" "двухвалентного остатка, полученного из необязательно замещенного тиазола", метилсульфонилбензил, сульфамоилбензил (например, 4-сульфамоилбензил) и подобные являются предпочтительными.
является предпочтительным. В качестве "заместителя" "двухвалентного остатка, полученного из необязательно замещенного тиазола", метилсульфонилбензил, сульфамоилбензил (например, 4-сульфамоилбензил) и подобные являются предпочтительными.
Низший алкилен, низший алкенилен и низший алкинилен для J или M формулы (III): J-L-M для Y в формуле (I) могут быть группами, определенными выше и подобными.
Конкретные примеры формулы (III): J-L-M для Y в формуле (I) включают -(CH2)n-, -(CH2)n-NH-(CH2)n´-, -(CH2)n-O-(CH2)n´-, -(CH2)n-CO-O-(CH2)n´-, -(CH2)n-O-CO-(CH2)n´-, -(CH2)n-CO-NH-(CH2)n´, -(CH2)n-NH-CO-(CH2)n´-, -(CH2)n-SO2-NH-(CH2)n´- и -(CH2)n-NH-SO2-(CH2)n´- (где n и n´, каждый равен целому число от 0 до 6, n предпочтительно равен целому числу от 0 до 3, и n´ предпочтительно равен целому числу от 0 до 3) и подобные. Среди них, -(CH2)n-, -(CH2)n-NH-(CH2)n´-, -(CH2)n-O-(CH2)n´-, -(CH2)n-CO-O-(CH2)n´- и -(CH2)n-CO-NH-(CH2)n´- являются предпочтительными, и -(CH2)n- является особенно предпочтительным. Конкретно можно также указать -(CH2)2-, -CH2-CO-, -CH2-NH-, -CH2-O-, -CO-O-, -CO-NH- и подобные.
Конкретные примеры двухвалентного остатка, полученного из необязательно замещенного бензола, или двухвалентного остатка, полученного из необязательно замещенного тиофена, для A в формуле (II): A-B-D-E для Z в формуле (I) включают
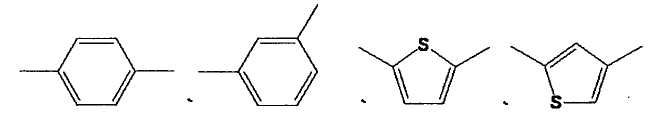
и подобные.
"Бензол" и "тиофен" могут иметь заместитель, и положение замещения конкретно не ограничено. Примеры "заместителей" указанного выше "необязательно замещенного бензола" и "необязательно замещенного тиофена" включают галоген (например, фтор, хлор, бром), низший алкил (например, метил, этил), низший алкокси (например, метокси), ацил (например, ацетил), галогенированный алкил (например, трифторметил) и подобные.
Примеры низшего алкила и ацила для R2 в -(CH2)l-NR2-CO-, представленной B, включают такие, как определено выше, и подобные. l в -(CH2)l-NR2-CO-, представленной B, равно целому числу от 1 до 6 (предпочтительно 1-3). m в -(CH2)m-O-CO- и -(CH2)m-S-CO-, представленной B, равно целому числу от 0 до 6 (предпочтительно 0-3).
Конкретные примеры B включают -O-CO-, -CH2-O-CO-, -(CH2)2-O-CO-, -(CH2)3-O-CO-, -CH2-NH-CO-, -(CH2)2-NH-CO-, -(CH2)3-NH-CO-, -S-CO-, -CH2-S-CO- и -(CH2)2-S-CO-, и подобные.
Примеры низшего алкила, алкоксикарбонила и ацила для R3 в -NR3-, представленного D, включают такие, как определено выше, и подобные. Конкретные примеры D включают -NH-, -N(CH3)- и подобные.
Примеры "необязательно замещенного амино" для E включают незамещенный амино и амино, замещенный 1 или 2 заместителями. "Необязательно замещенный амино" представлен формулой -NR4R5.
Примеры R4 и R5 включают группы низшего алкила, ацила (в частности, низший алкилкарбонил, гидрокси низший алкилкарбонил), алкоксикарбонила, гидроксиалкоксикарбонила, арила, аралкила, низшего циклоалкила, низшего циклоалкоксикарбонила, сульфурила, сульфинила, фосфорила, гетероцикла и подобных, каждый из которых является незамещенным или необязательно замещен гидрокси и т.д., водородом и подобными. Низший алкил, ацил (в частности, низший алкилкарбонил), алкоксикарбонил, арил, аралкил, низший циклоалкил, низший циклоалкоксикарбонил и гетероцикл являются такими, как определено выше.
Конкретные примеры R4 и R5 включают водород, низший алкил (например, метил, этил и подобные), ацетил, бутаноил, деканоил, 3-гидроксипропаноил, 6-гидроксигексаноил, этоксикарбонил, бутоксикарбонил, децилоксикарбонил, 2-гидроксиэтоксикарбонил и подобные.
Аминогруппа "необязательно замещенного амино" для E может быть защищена (т.е. замещенной) согласно способу, описанному в "Protective Groups in Organic Synthesis 3rd Edition" (John Wiley и Sons, 1999), и подобным. R4 и R5 могут быть одинаковыми или различными.
Как -B-D-E часть (окончание молекулы) формулы (II): A-B-D-E, которая показана Z в формуле (I), B представляет собой -O-CO-, -CH2-O-CO-, -(CH2)2-O-CO-, -(CH2)3-O-CO-, -CH2-NH-CO-, -(CH2)2-NH-CO-, -(CH2)3-NH-CO-, -S-CO-, -CH2-S-CO- или -(CH2)2-S-CO-; D представляет собой -NH-; и E представляет собой -NH2 и подобные. Конкретно, -B-D-E часть представляет собой, например, -O-CO-NH-NH2, -CH2-O-CO-NH-NH2, -(CH2)2-O-CO-NH-NH2, -(CH2)3-O-CO-NH-NH2, -CH2-NH-CO-NH-NH2, -(CH2)2-NH-CO-NH-NH2, -(CH2)3-NH-CO-NH-NH2, -CH2-S-CO-NH-NH2, -(CH2)2-S-CO-NH-NH2 и подобные. Предпочтительным является -O-CO-NH-NH2, -CH2-O-CO-NH-NH2, -(CH2)2-O-CO-, -(CH2)3-O-CO-NH-NH2, -CH2-NH-CO-NH-NH2, -(CH2)2-NH-CO- или -CH2-S-CO-. Особенно предпочтительным является -CH2-O-CO-NH-NH2 или -CH2-NH-CO-NH-NH2.
Примеры соединения (I) включают
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат,
2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил-гидразинкарбоксилат,
2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил-гидразинкарбоксилат,
4-{2-[(гидразинокарбонил)окси]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилат,
2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-гидразинкарбоксилат,
3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат,
2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил]фенил)этил-гидразинкарбоксилат,
{5-[2-(2-ацетиламино-1,3-тиазол-4-ил)этил]тиофен-2-ил}метил-гидразинкарбоксилат,
2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил-гидразинкарбоксилат,
3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил-гидразинкарбоксилат,
3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил-гидразинкарбоксилат,
3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил-гидразинкарбоксилат,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
N-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил)гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил)гидразинкарбоксамид,
N-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метил}амино)бензил]гидразинкарбоксамид,
2-(ацетиламино)-N-(4-{[(гидразинокарбонил)амино]метил}фенил)-1,3-тиазол-4-карбоксамид,
N-[2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил]гидразинкарбоксамид,
4-{2-[(гидразинокарбонил)амино]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилат,
N-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
N-[2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамид,
N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарбоксамид,
N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил]гидразинкарбоксамид,
N-[3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил]гидразинкарбоксамид,
N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил]гидразинкарбоксамид,
N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил]гидразинкарбоксамид,
S-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарботиоат,
S-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарботиоат,
S-[(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарботиоат и подобные.
Предпочтительными являются 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат, 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат, N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид и подобные.
Когда соединение (I) содержит асимметрический атом углерода в структуре, настоящее изобретение включает все энантиомеры и диастереомеры.
Соединение (I) можно также преобразовать в фармацевтически приемлемую соль. Фармацевтически приемлемую соль в настоящем изобретении конкретно не ограничивают, при условии, если она является нетоксичной фармацевтически приемлемой обычной солью, и можно указать соль с неорганическим или органическим основанием, кислотно-аддитивную соль и подобные. Примеры соли с неорганическим или органическим основанием включают соли щелочных металлов (например, натриевую соль, калиевую соль и подобную), соль щелочноземельного металла (например, кальциевую соль, магниевую соль и подобную), аммониевую соль и соль амина (например, триэтиламинную соль, N-бензил-N-метиламинную соль и подобные) и подобные. Примеры кислотно-аддитивной соли включают соли, полученные из минеральных кислот (например, хлористоводородной кислоты, бромистоводородной кислоты, йодистоводородной кислоты, фосфорной кислоты, метафосфорной кислоты, азотной кислоты и серной кислоты), и соли, полученные из органических кислот (например, винной кислоты, уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, яблочной кислоты, молочной кислоты, фумаровой кислоты, малеиновой кислоты, бензойной кислоты, гликолевой кислоты, глюконовой кислоты, янтарной кислоты и арилсульфоновой кислоты (например, п-толуолсульфоновой кислоты)) и подобных.
Соединение настоящего изобретения можно применять в качестве пролекарства для указанного выше фармацевтического агента и подобных. Термин "пролекарство" относится к любому соединению, которое может преобразовываться в ингибитор VAP-1 в организме после введения. Пролекарство может представлять собой любое необязательно фармацевтически приемлемое пролекарство соединения настоящего изобретения.
Соединение настоящего изобретения можно применять в качестве активного ингредиента фармацевтического агента, такого как ингибитор VAP-1, фармацевтический агент для профилактики или лечения связанного с VAP-1 заболевания и подобного.
"Связанное с сосудистым адгезивным белком-1 (VAP-1) заболевание" конкретно не ограничено, при условии, что оно представляет собой заболевание, в котором VAP-1 связан с проявлением и/или развитием заболевания, и включает заболевание, выбранное из группы, состоящей из заболевания, связанного со сверхпроницаемостью сосудов [например, отека желтого пятна (например, диабетического и недиабетического отека желтого пятна), возрастной дегенерации желтого пятна, возрастной дисковидной дегенерации желтого пятна, кистозного макулярного отека, пальпебрального отека, отека сетчатки, диабетической ретинопатии, хориоретинопатии, неоваскулярной макулопатии, неоваскулярной глаукомы, увеита, воспаления радужной оболочки глаза, васкулита сетчатки глаза, эндофтальмита, панофтальмита, метастатической офтальмии, хориоидита, ретинального пигментного эпителиита, конъюнктивита, циклита, склерита, эписклерита, неврита зрительного нерва, ретробульбарного неврита зрительного нерва, кератита, блефарита, экссудативного отслоения сетчатки, язвы роговицы, язвы конъюнктивы, хронического монетовидного кератита, кератита Тайджесона, прогрессирующей разъедающей язвы роговицы, воспалительного заболевания глаз, вызванного бактериальной или вирусной инфекцией и операцией на глазах, воспалительного заболевания глаз, вызванного физическим повреждением глаза, симптома, вызванного воспалительным заболеванием глаз, включая зуд, воспаление, отек и язву, эритемы, многоформной экссудативной эритемы, узловатой эритемы, кольцевидной эритемы, склередемы взрослых, дерматита (например, псориаза, аллергического поражения, красного плоского лишая, розового лишая, контактного дерматита, атопического дерматита, красного волосистого питириаза), ангионевротического отека, отека гортани, отека языка, подсвязочного ларингита, бронхита, ринита, фарингита, синусита и ларингита или среднего отита], цирроза печени, эссенциальной стабилизированной гипертензии, диабета, артериосклероза, эндотелиального повреждения (например, при диабете, артериосклерозе и гипертензии), сердечно-сосудистого заболевания, связанного с диабетом или уремией, боли, связанной с подагрой и артритом, воспалительного заболевания или симптома соединительной ткани (например, ревматоидного артрита, анкилозирующего спондилита, псориатического артрита и остеоартрита или дегенеративного заболевания сустава, синдрома Рейтера, синдрома Шегрена, синдрома Бехчета, рецидивирующего полихондрида, системной красной волчанки, дискоидной красной волчанки, системного склероза, эозинофильного фасциита, полимиозита, дерматомиозита, ревматической полимиалгии, васкулита, темпорального артрита, нодозного полиартериита, гранулематоза Вегенера, смешанного заболевания соединительной ткани и ювенильного ревматоидного артрита), воспалительного заболевания или симптома желудочно-кишечного тракта [например, болезни Крона, язвенного колита, синдрома раздраженного кишечника (например, слизистого колита), фиброза печени, воспаления слизистой мембраны рта (например, стоматита и рецидивирующего афтозного стоматита)], воспалительного заболевания или симптома центральной нервной системы (например, множественного склероза, болезни Альцгеймера и травмы ишемии/реперфузии, связанной с ишемическим инсультом), легочного воспалительного заболевания или симптома (например, астмы, синдрома расстройства дыхания у взрослых, хронических облитерирующих легочных заболеваний), заболевания, связанного с метаболизмом углеводов (например, диабета и осложнения, полученного в результате диабета (например, диабетической невропатии, диабетической нефропатии)), включая заболевание микрососудов и больших сосудов (например, артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (например, множественную невропатию, мононевропатию и вегетативную невропатию), язвы стопы, проблемы суставов и увеличения риска инфекции), заболевания, связанного с нарушением дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (например, артериосклероза и ожирения), сосудистого заболевания [например, атероматозного атеросклероза, неатероматозного атеросклеротического заболевания, ишемического заболевания сердца, включая инфаркт миокарда и закупорку периферических артерий, болезни Рейно и феномена Рейно, облитерирующего тромбангиита (болезни Бюргера)], хронического артрита, воспалительного заболевания кишечника, SSAO-опосредованного осложнения [например, диабета (например, инсулин-зависимого диабета (IDDM) и инсулин-независимого диабета (NIDDM)) и сосудистых осложнений (например, инфаркта, стенокардии, апоплексии мозга, ампутации, медицинской слепоты и почечной недостаточности)], заболевания глаз, связанного с гипоксией или ишемией [например, ретинопатии недоношенных, пролиферативной диабетической ретинопатии, полипообразной хориоидальной васкулопатии, ретинальной ангиоматозной пролиферации, окклюзии центральной артерии сетчатки, окклюзии ветви центральной вены сетчатки, болезни Коутса, семейной экссудативной витреоретинопатии, болезни отсутствия пульса (болезни Такаясу), болезни Илза, антифосфолипидного синдрома, лейкемической ретинопатии, синдрома повышенной вязкости крови, макроглобулинемии, связанной с интерфероном ретинопатии, гипертонической ретинопатии, ретинопатии при облучении, дефицита стволовых клеток роговичного эпителия] и катаракты и подобных.
"Профилактика или лечение связанного с сосудистым адгезивным белком (VAP-1) заболевания" относится к введению соединения настоящего изобретения, обладающего ингибирующей VAP-1 активностью (т.е. ингибитора VAP-1), субъекту для лечения (включая профилактику, уменьшение интенсивности симптома, ослабление симптома, предотвращение развития и лечение) указанного выше связанного с VAP-1 заболевания.
Субъектами введения фармацевтического агента, фармацевтической композиции, ингибитора VAP-1, фармацевтического агента для профилактики или лечения связанного с VAP-1 заболевания в настоящем изобретении (далее в данном описании их также совместно называют фармацевтический агент настоящего изобретения) являются различные животные (например, млекопитающие, такие как люди, мыши, крысы, свиньи, собаки, кошки, лошади, коровы и подобные, особенно люди) и подобные.
Фармацевтический агент настоящего изобретения можно вводить любым способом. Способ введения в настоящем изобретении включает общее введение (например, пероральное введение или введение инъекцией), местное введение (например, введение малыми дозами или каплями, внутриглазное введение и трансдермальное введение) и подобные. Способ введения фармацевтического агента настоящего изобретения можно соответствующим образом определить согласно тому, является ли применение, направленное на связанное с VAP-1 заболевание, профилактическим или терапевтическим и подобным.
Фармацевтический агент настоящего изобретения предпочтительно вводят сразу после того, как диагностировано, что субъект введения, такой как млекопитающее, в частности, человек, подвергнут риску связанного с VAP-1 заболевания (профилактическое лечение), или вводят сразу после того, как у подвергаемого введению субъекта возникает связанное с VAP-1 заболевание (терапевтическое лечение). План лечения можно соответствующим образом определять согласно типу активного ингредиента, который будут применять, дозе, способу введения, причине и, при необходимости, степени информированности о связанном с VAP-1 заболевании и подобном.
В качестве способа введения фармацевтического агента настоящего изобретения, можно использовать способ, известный per se для стандартных фармацевтических агентов. Соответствующим образом эффективным может быть один способ введения, и можно использовать один или несколько способов. Соответственно, приведенные выше способы введения являются просто примерами, не вводящими каких-либо ограничений.
Доза фармацевтического агента настоящего изобретения для подвергаемого введению субъекта, такого как млекопитающее, включая человека, особенно человека, представляет собой количество, достаточное для оказания желаемой ответной реакции подвергаемого введению субъекта в течение разумного периода времени. Дозу соответствующим образом определяют согласно различным факторам, включая эффективность активного ингредиента, который будут применять, тип, симптом, стадию заболевания, массу тела и тяжесть заболевания у подвергаемого введению субъекта, способ введения, временной режим и частоту введения и подобные. Дозу можно также соответствующим образом контролировать согласно способу, временному режиму и частоте введения, и подобного. В зависимости от симптома или стадии заболевания может быть необходимо продолжительное лечение, включающее многоразовое введение.
Дозу и режим введения можно определить методикой в диапазоне, известном специалистам в данной области техники. Как правило, лечение или профилактика начинается с дозы, меньшей, чем оптимальная доза соединения. Впоследствии дозу постепенно увеличивают до получения оптимального эффекта в данных обстоятельствах. Фармацевтический агент настоящего изобретения (ингибитор VAP-1 и подобный) можно обычно вводить в дозе, составляющей приблизительно от 0,03 нг/кг массы тела/день до приблизительно 300 мг/кг массы тела/день, предпочтительно приблизительно от 0,003 мкг/кг массы тела/день до приблизительно 10 мг/кг массы тела/день, одним введением или 2-4 порциями в день или замедленным способом.
Фармацевтическая композиция настоящего изобретения предпочтительно содержит "фармацевтически приемлемый носитель" и, в качестве активного ингредиента, соединение настоящего изобретения (ингибитор VAP-1) в количестве, достаточном для профилактического или терапевтического лечения связанного с VAP-1 заболевания. Приемлемый носитель может быть любым, который обычно применяют в качестве фармацевтического агента и конкретно не ограничен, за исключением ограничений на рассматриваемые физико-химические свойства (например, растворимость и отсутствие способности реагировать с соединением) и способ введения.
В то время как количество соединения настоящего изобретения в фармацевтическом агенте настоящего изобретения изменяется в зависимости от состава композиции, оно обычно составляет 0,00001-10,0% масс., предпочтительно 0,001-5% масс., более предпочтительно 0,001-1% масс.
Форма для введения фармацевтического агента настоящего изобретения конкретно не ограничена, и его можно вводить в различных формах для достижения требуемой ингибирующей VAP-1 активности. Фармацевтический агент настоящего изобретения составляют в композицию, используя одно соединение настоящего изобретения или в комбинации с фармацевтически приемлемым носителем или добавкой, такой как разбавитель и подобной, и вводят перорально или парентерально. Характеристики и свойства препарата определяются растворимостью и химическими свойствами активного ингредиента, выбранным способом введения и стандартной фармацевтической практикой. Препарат, который будут применять для перорального введения, может представлять собой твердые дозированные формы (например, капсулы, таблетки, порошки) или жидкую форму (например, раствор или суспензию) и подобные. Препарат, который будут применять для парентерального введения, может представлять собой инъекцию, капельную инфузию и подобные, которые находятся в форме асептического раствора или суспензии. Твердый пероральный препарат может содержать стандартный эксципиент и подобный. Жидкий пероральный препарат может содержать различные ароматизаторы, красители, консерванты, стабилизаторы, солюбилизаторы, суспендирующие агенты и подобные. Парентеральный препарат представляет собой, например, асептический водный или неводный раствор или суспензию, и может содержать конкретные различные консерванты, стабилизаторы, буферы, солюбилизаторы, суспендирующие агенты и подобные. В случае необходимости можно добавлять различные изотонические агенты.
Фармацевтический агент настоящего изобретения может содержать другое фармацевтически активное соединение, при условии, что оно не подавляет действие соединения настоящего изобретения.
Фармацевтический агент настоящего изобретения можно одновременно вводить с другим фармацевтически активным соединением, при условии, что оно не подавляет действие соединения настоящего изобретения. "Одновременное введение" означает введение другого фармацевтически активного соединения перед или одновременно (например, в одном или разных препаратах) или после введения фармацевтического агента настоящего изобретения. Например, можно вводить одновременно кортикостероид, преднизон, метилпреднизон, дексаметазон или триамцинолона ацетонид, или некортикоидное противовоспалительное соединение (например, ибупрофен или флурбипрофен). Аналогично можно вводить одновременно витамин и минерал (например, цинк, антиоксидант (например, каротеноид (например, ксантофилльный каротеноидоподобный зеаксантин или лютеин))) и микроэлементы, и подобные.
Соединение настоящего изобретения является пригодным для получения фармацевтического агента, такого как ингибитор VAP-1, и фармацевтического агента для профилактики или лечения связанного с VAP-1 заболевания.
Соединение (I) может быть получено следующими методиками. Однако данными методиками не ограничиваются. Методики можно изменять согласно общему способу, известному per se.
Соединение (I) также может быть представлено формулой:
R1-NH-X-Y-A-B-D-E
где каждый символ является таким, как определено выше.
Стадии методики получения соединения (I) показаны на следующей схеме 1.
Соединение (I) можно получить химической конденсацией четырех соединений, (1), (2), (3) и эквивалента монооксида углерода (4), как показано на конкретных структурах на схеме 1. Соединения (1), (2), (3) могут быть в форме соли.
Порядок конденсации может представлять собой конденсацию соединений (1) и (2) и затем соединения (3) через эквивалент монооксида углерода (4), или сначала конденсацию соединений (2) и (3) через эквивалент монооксида углерода (4) и затем соединения (1). Соединение (I) может быть получено, используя оба порядка конденсаций. В случае необходимости, может быть осуществлено удаление защитной группы D-E, преобразование в фармацевтически приемлемую соль и подобное. Способ получения соединения (I) не ограничивается приведенным выше, и в нем можно соответствующим образом изменять стадии согласно общему способу, известному per se.
Схема 1
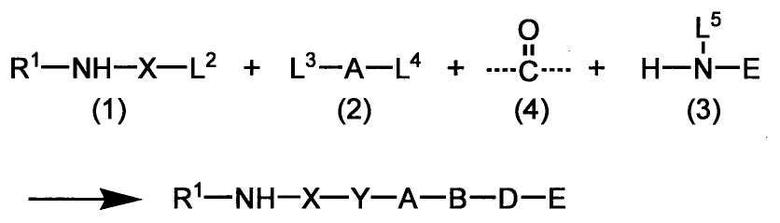
где R1, X, Y, A, B, D и E являются такими, как определено выше. L2 представляет собой реакционноспособную функциональную группу, которая образует химическую связь с L3 соединения (2), образуя Y. L3 представляет собой реакционноспособную функциональную группу, которая образует химическую связь с L2 соединения (1), образуя Y. L4 представляет собой функциональную группу, которая реагирует с соединением (3) через эквивалент монооксида углерода (4), образуя B, посредством чего структура эфира карбазиновой кислоты, структура тиоэфира карбазиновой кислоты и семикарбазидная структура конструируется на конце молекулы соединения (I). L5 представляет собой водород, низший алкил, алкоксикарбонил, ацил или защитную группу.
L2 соединения (1) представляет собой реакционноспособную функциональную группу, которая образует химическую связь с L3 соединения (2), образуя Y. Ее примеры включают, но не ограничиваются ими, -(CH2)U-CHO, -(CH2)u-OH, -(CH2)u-галоген, -(CH2)U-COOH, -(CH2)u-CO-галоген, -(CH2)U-NH2, -(CH2)U-SO3H, -(CH2)u-SO2-галоген, -(CH2)u-O-ацил, полученный из -(CH2)U-OH (например, -(CH2)u-O-ацетил и подобный), эфир -(CH2)u-сульфоновой кислоты (например, -(CH2)U-OSO2CH3 и подобный), реагент Виттига, полученный из -(CH2)u-галогена и подобного, и подобные (где u равно целому числу от 0 до 6, и галоген представляет собой хлор, бром или йод).
Соединение (1) и его соль могут быть коммерчески доступными или их можно также получить согласно способу, известному per se, который описан в WO 2004/067521, и подобных.
L3 соединения (2) представляет собой реакционноспособную функциональную группу, которая образует химическую связь с L2 соединения (1), образуя Y. Ее примеры включают, но не ограничиваются ими, -(CH2)V-CHO, -(CH2)v-OH, -(CH2)v-галоген, -(CH2)V-COOH, -(CH2)v-CO-галоген, -(CH2)v-NH2, -(CH2)V-SO3H, -(CH2)v-SO2-галоген, -(CH2)v-O-ацил, полученный из -(CH2)V-OH (например, -(CH2)v-O-ацетил и подобный), эфир -(CH2)v-сульфоновой кислоты (например, -(CH2)V-OSO2CH3 и подобный), реагент Виттига, полученный из -(CH2)v-галогена и подобного, и подобные (где v равно целому числу от 0 до 6, и галоген представляет собой хлор, бром или йод).
L4 представляет собой функциональную группу, которая реагирует с соединением (3) через эквивалент монооксида углерода (4) или с соединением, полученным предварительной конденсацией эквивалента монооксида углерода (4) с соединением (3), образуя B, посредством чего структура эфира карбазиновой кислоты, структура тиоэфира карбазиновой кислоты и семикарбазидная структура конструируется на конце молекулы соединения (I). Ее примеры включают, но конкретно не ограничены, -(CH2)w-OH, -(CH2)W-SH, -(CH2)t-NHR2, R2-(CH2)w-галоген и подобные (где w равно целому числу от 0 до 6, t равно целому числу от 1 до 6, галоген представляет собой хлор, бром или йод, и R2 является таким, как определено выше.
Соединение (2) и его соль могут быть коммерчески доступными или их можно также получить согласно способу, известному per se, который описан в WO 2004/067521, WO 2006/011631 и подобных.
Соединение (3) представляет собой гидразиновый эквивалент для создания структуры эфира карбазиновой кислоты, структуры тиоэфира карбазиновой кислоты и семикарбазидной структуры на конце молекулы соединения (I), и оно может быть коммерчески доступным или его можно получить согласно способу, известному per se. Защитная группа для L5 представляет собой функциональную группу, вводимую для того, чтобы избежать нежелательных реакций, и удаляемую на подходящей стадии. Ее примеры включают защитные группы (CH3)3C-OCO-, показанные в примерах получения и подобных. Примерами низшего алкила, алкоксикарбонила и ацила для L5 являются такие, которые аналогичны низшему алкилу, алкоксикарбонилу и ацилу для указанной выше R3.
(4) представляет собой синтетический эквивалент (синтон) монооксида углерода, вводящий карбонильную группу в B, и он может быть коммерчески доступным или его можно получить согласно способу, известному per se. Конкретно, в качестве неограничивающих примеров можно использовать 1,1'-карбонилдиимидазол, эфиры хлороформовой кислоты, фосген, бис(трихлорметил)карбонат [трифосген] и подобные.
При получении соединения (I), где Y представляет собой углеродную цепь, соединение (1) или его соль можно химически связать с соединением (2) или его солью (или соединением, полученным конденсацией соединения (2) и (3) заранее через эквивалент монооксида углерода (4)), используя реагент Виттига, реакцию Хорнера-Эванса, реакцию альдольной конденсации, конденсацию Клайзена или аналогичную реакцию образования углерод-углеродной связи для получения Y, содержащего низший алкенилен или низший алкинилен. Подходящие соли соединения (1) и (2) могут быть такими же, как соли приведенные в качестве примеров для соединения (I). Несмотря на то, что можно использовать различные реакции образования углерод-углеродной связи, когда применяют реакцию Виттига или аналогичную реакцию, подходящий пример включает -(CH2)U-CHO для L2 и фосфониевую соль (реагент Виттига), полученную из -(CH2)v-галогена и т.д. для L3, или фосфониевую соль (реагент Виттига), полученную из -(CH2)u-галогена и т.д. для L2, и -(CH2)V-CHO для L3 (где u и v являются такими, как определено выше, и галоген представляет собой хлор, бром или йод). Реакцию обычно проводят в стандартном растворителе, таком как N,N-диметилформамид, диметилсульфоксид, тетрагидрофуран и дихлорметан, или другом органическом растворителе, который не оказывает неблагоприятного влияния на реакцию, или в их смеси, в присутствии стандартного основания, такого как трет-бутоксид калия, гидрид натрия, гидроксид натрия и подобный. Температура реакции в частности не является важной, и реакцию проводят при охлаждении или нагревании. Полученный в результате продукт выделяют или очищают известными методами разделения и очистки, концентрированием, концентрированием при пониженном давлении, экстракцией растворителем, кристаллизацией, перекристаллизацией, фазовым переходом, хроматографией и подобными, или его можно преобразовать в соль, аналогичную солям, приведенным в качестве примеров для соединения (I).
В случае необходимости, низший алкенилен или низший алкинилен гидрируют для преобразования в низший алкилен. Когда Y преобразовывают в алкиленовую связь, реакцию гидрирования проводят в присутствии различных гомогенных катализаторов или гетерогенного катализатора согласно общему способу. В частности, каталитическое гидрирование с использованием гетерогенного катализатора, является предпочтительным, и его проводят в присутствии катализатора, такого как палладий на углероде или никель Ренея.
Когда получают соединение (I), где Y представляет собой эфир, амид или сульфонамид, соединение (1) или его соль конденсируют с соединением (2) или его солью (или соединением, полученным конденсацией соединения (2) и (3) заранее через эквивалент монооксида углерода (4)), с получением эфирной или амидной связи. В данном случае, L2 представляет собой -(CH2)U-OH, -(CH2)U-NH2, -(CH2)u-галоген и подобный, и L3 представляет собой -(CH2)V-COOH, -(CH2)v-CO-галоген, -(CH2)V-SO3H, -(CH2)v-SO2-галоген и подобный, или L2 представляет собой -(CH2)u-COOH, -(CH2)u-CO-галоген, -(CH2)U-SO3H, -(CH2)u-SO2-галоген и подобный, и L3 представляет собой -(CH2)v-OH, -(CH2)V-NH2, -(CH2)v-галоген и подобный, и Y можно получить, исходя из стандартного способа органического синтеза (где u и v являются такими, как определено выше, и галоген представляет собой хлор, бром или йод). Реакцию обычно проводят в стандартном растворителе, таком как дихлорметан, ацетон, тетрагидрофуран, диэтиловый эфир и N,N-диметилформамид, и можно использовать другой органический растворитель, который не оказывает неблагоприятного влияния на реакцию, или в их смеси. В случае необходимости, можно использовать реагент для конденсации, такой как гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида, N,N'-дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол и подобные. Реакцию также проводят в присутствии добавки, такой как N,N-диметил-4-аминопиридин, 1-гидроксибензотриазол, 1-гидроксисукцинимидо и 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазин. Температура реакции не является особенно важной, и реакцию проводят при охлаждении или нагревании.
Когда получают соединение (I), где Y представляет собой группу, содержащую амин, L2 представляет собой -(CH2)U-NH2 или ее соль и подобные, и L3 представляет собой -(CH2)V-CHO, -(CH2)V-галоген и подобные, или L2 представляет собой -(СН2)U-СНO, (СН2)U-галоген и подобные, и L3 представляет собой -(CH2)V-NH2 или ее соль и подобные, и Y может быть получен, исходя из стандартного способа органического синтеза (где u и v являются такими, как определено выше, и галоген представляет собой хлор, бром или йод). Обычно амин и альдегид конденсируют для того, чтобы получить основание Шиффа, которое восстанавливают боргидридом натрия, цианоборгидридом натрия и подобным в стандартном растворителе, таком как тетрагидрофуран, диэтиловый эфир, спирт и подобный, или любом другом органическом растворителе, который не оказывает неблагоприятного влияния на реакцию, или в их смеси в качестве растворителя реакции, создавая, таким образом, структуру вторичного амина. Аналогичную структуру также получают реакцией конденсации амина и галогенового соединения. Когда используют галогеновое соединение, в качестве реагента реакции используют основание, такое как N,N-диизопропиламин, триэтиламин, карбонат калия и подобное, в качестве растворителя реакции используют стандартный растворитель, такой как тетрагидрофуран, ацетонитрил и N,N-диметилформамид, или другой органический растворитель, который не оказывает неблагоприятного влияния на реакцию, или их смесь. Температура реакции не является особенно важной, и реакцию проводят при охлаждении или нагревании. Полученный в результате продукт можно также преобразовать в соль, аналогичную солям, приведенным в качестве примеров для соединения (I).
Когда получают соединение (I), где Y представляет собой группу, содержащую эфирную связь, L2 представляет собой -(CH2)U-OH и подобные, и L3 представляет собой -(CH2)V-OH, -(CH2)v-галоген, эфир -(CH2)v-сульфоновой кислоты и подобные, или L2 представляет собой -(CH2)u-OH, -(CH2)u-галоген, эфир -(CH2)u-сульфоновой кислоты и подобные, и L3 представляет собой -(CH2)V-OH и подобные, и Y может быть получен, исходя из стандартного способа органического синтеза (где u и v являются такими, как определено выше, и галоген представляет собой хлор, бром или йод). Эфирную связь можно образовать способом Уильямсона, способом синтеза эфиров из ароматического галогенида, используя медный катализатор и подобные, реакцией Мицунобу, другими способами получения, известными per se. Данные реакции обычно проводят в стандартном растворителе, таком как ацетонитрил, дихлорметан, ацетон, тетрагидрофуран и N, N-диметилформамид, или любом другом органическом растворителе, который не оказывает неблагоприятного влияния на реакцию, или в их смеси. Температура реакции не является особенно важной, и реакцию проводят при охлаждении или нагревании. Полученный в результате продукт можно также преобразовать в соль, аналогичную солям, приведенным в качестве примеров для соединения (I).
На конце молекулы соединения (I) находится структура эфира карбазиновой кислоты, структура тиоэфира карбазиновой кислоты или семикарбазидная структура.
Один пример способа введения структуры эфира карбазиновой кислоты, структуры тиоэфира карбазиновой кислоты или семикарбазидной структуры в конец молекулы соединения (I) показан на следующей схеме 2.
Схема 2
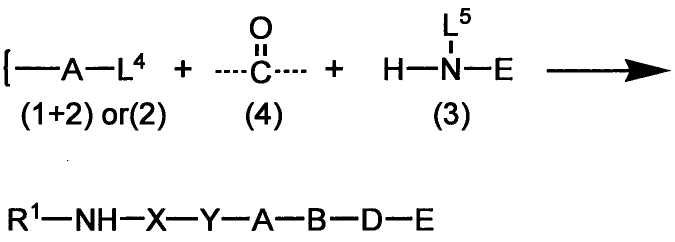
(где R1, X, Y, A, B, D и E являются такими, как определено для соединения (I), и L4 и L5 являются такими, как определено выше).
Когда получают эфир карбазиновой кислоты, т.е. соединение (I), где B представляет собой -(CH2)m-O-CO-, L4 соединения (2) (или соединения, полученного конденсацией соединения (1) и (2)), должно представлять структуру -(CH2)W-OH. Ее можно вводить в виде гидроксигруппы в соединение (2) заранее в качестве исходного соединения, или можно получать, как часть стадии синтеза восстановлением соответствующей карбоновой кислоты, эфира карбоновой кислоты или альдегида, гидролизом галогенида или эфира, гидратированием олефина, гидроборированием и подобным.
L4: -(CH2)W-OH подвергают взаимодействию, например, с 1,1'-карбонилдиимидазолом в качестве синтетического эквивалента монооксида углерода (4), и затем конденсируют с гидразином (или защищенным гидразином), посредством чего создают на конце молекулы соединения (I) структуру эфира карбазиновой кислоты (в формуле (I), B представляет собой -(CH2)W-O-CO-, D представляет собой -NR3-, и E представляет собой необязательно замещенную амино группу), где w является таким, как определено выше. Альтернативно, структуру эфира карбазиновой кислоты можно создать на конце молекулы соединения (I) взаимодействием гидразина (или защищенного гидразина) и 1H-имидазол-1-карбогидразида, синтезированного, например, из 1,1'-карбонилдиимидазола с L4: -(CH2)W-OH, или его алкоголята металла [-(CH2)W-ONa и подобного]. Реакцию обычно проводят в стандартном растворителе, таком как тетрагидрофуран, N,N-диметилформамид, дихлорметан и ацетонитрил, или в любом другом органическом растворителе, который не оказывает неблагоприятного влияния на реакцию, или в их смеси. В случае необходимости, осуществляют удаление защитной группы на подходящей стадии для получения целевого соединения.
Когда получают тиоэфир карбазиновой кислоты, т.е. соединение (I), где B представляет собой -(CH2)m-S-CO-, L4 соединения (2) (или соединения, полученного конденсацией соединения (1) и (2)) должно представлять собой структуру -(CH2)W-SH. Ее можно вводить в виде сульфанильной группы в соединение (2) заранее в качестве исходного соединения, или можно получить, как часть стадии синтеза стандартным способом получения тиола, таким как преобразование функциональной группы соответствующего спирта или галогенового соединения и подобного.
L4: -(CH2)W-SH подвергают взаимодействию, например, с 1,1'-карбонилдиимидазолом в качестве синтетического эквивалента монооксида углерода (4) и затем конденсируют с гидразином (или защищенным гидразином), создавая таким образом структуру тиоэфира карбазиновой кислоты (в формуле (I), B представляет собой -(CH2)W-S-CO-, D представляет собой -NR3- и E представляет собой необязательно замещенную амино группу) на конце молекулы соединения (I), где w является таким, как определено выше. Альтернативно, структуру тиоэфира карбазиновой кислоты можно получить на конце молекулы соединения (I) взаимодействием гидразина (или защищенного гидразина) и 1H-имидазол-1-карбогидразина, синтезированного, например, из 1,1'-карбонилдиимидазола с L4: -(CH2)W-SH или его тиолата металла [-(CH2)w-SNa и подобного]. Реакцию обычно проводят в стандартном растворителе, таком как тетрагидрофуран, N,N-диметилформамид, дихлорметан и ацетонитрил, или в любом другом органическом растворителе, который не оказывает неблагоприятного влияния на реакцию, или в их смеси. В случае необходимости, осуществляют удаление защитной группы на подходящей стадии для получения целевого соединения.
Когда получают семикарбазид, т.е. соединение (I), где B представляет собой -(CH2)l-NR2-CO-, L4 должно представлять собой структуру -(CH2)t-NHR2, где t равно целому числу от 1 до 6. Ее можно вводить в виде аминогруппы или защищенной аминогруппы в соединение (2) заранее в качестве исходного соединения, или можно получать, как часть стадии синтеза восстановлением нитрогруппы, цианогруппы или карбоксамидной группы, преобразованием функциональной группы спирта или галогенида и подобным. Структуру на конце молекулы, где B представляет собой -(CH2)l-NR2-CO-, D представляет собой -NR3-, и E представляет собой необязательно замещенную аминогруппу, можно получить обработкой аминогруппы трет-бутил 2-(1H-имидазол-1-илкарбонил)гидразинкарбоксилатом, полученным, например, из 1,1'-карбонилдиимидазола и трет-бутоксикарбазата, и подобным. Реакцию обычно проводят в стандартном растворителе, таком как дихлорметан, ацетонитрил, тетрагидрофуран и N,N-диметилформамид, или в любом другом органическом растворителе, который не оказывает неблагоприятного влияния на реакцию, или в их смеси. Температура реакции является особенно не важной, и реакцию проводят при нагревании или охлаждении. В случае необходимости, осуществляют удаление защитной группы на подходящей стадии для получения целевого соединения.
Полученное таким образом соединение (I) можно выделить или очистить известными способами выделения и очистки, такими как кристаллизация, перекристаллизация, фазовый переход, хроматография и подобные. Кроме того, его можно преобразовать в фармацевтически приемлемую соль.
Настоящее изобретение будет объяснено более подробно далее со ссылкой на примеры (примеры получения и экспериментальные примеры), которые не расцениваются как ограничивающие.
Примеры
Исходные соединения, использованные в следующих примерах получения, могут быть получены известным способом (WO 2004/067521, WO 2006/011631, WO 2006/028269, WO 2008/066145 и т.д.) или приобретены как коммерчески доступные реагенты.
Пример получения 1
Гидрохлорид 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилата
Стадия 1
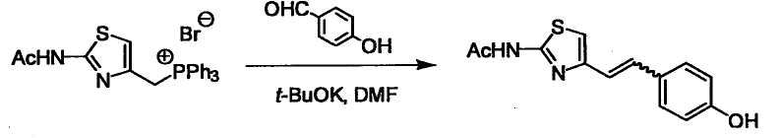
{[2-(Ацетиламино)-1,3-тиазол-4-ил]метил}(трифенил)фосфонийбромид (261,1 мг, 0,525 ммоль) и 4-гидроксибензальдегид (183,2 мг, 1,50 ммоль) растворяли в безводном N,N-диметилформамиде (2 мл) и добавляли при 0°C трет-бутоксид калия (56,1 мг, 0,50 ммоль). После перемешивания при 90°C в течение 12 часа, смесь охлаждали до комнатной температуры. Добавляли воду и этилацетат, смесь перемешивали, оставляли без перемешивания и затем распределяли. Водный слой экстрагировали этилацетатом, объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 25 г, этилацетат:гексан=4:6→5:5), получая N-{4-[2-(4-гидроксифенил)винил]-1,3-тиазол-2-ил}ацетамид (87,9 мг, 0,338 ммоль, выход 67,5%) в виде белого твердого вещества.
Стадия 2
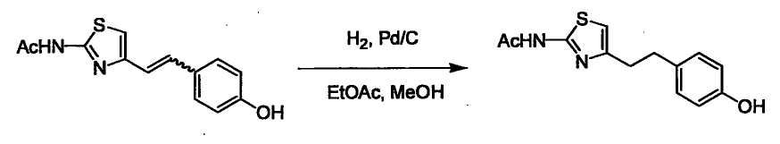
К раствору N-{4-[2-(4-гидроксифенил)винил]-1,3-тиазол-2-ил}ацетамида (932,3 мг, 3,58 ммоль) в этилацетате (50 мл) добавляли 10% палладий на углероде и смесь гидрировали при комнатной температуре и атмосферном давлении. После завершения реакции реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 30 г, дихлорметан:метанол=40:1→20:1), получая N-{4-[2-(4-гидроксифенил)этил]-1,3-тиазол-2-ил}ацетамид (771,3 мг, 2,94 ммоль, выход 82,1%) в виде белого твердого вещества.
Стадия 3
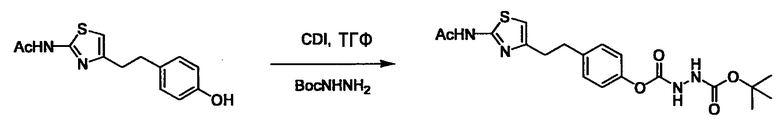
К раствору N-{4-[2-(4-гидроксифенил)этил]-1,3-тиазол-2-ил}ацетамида (655,8 мг, 2,50 ммоль) в безводном тетрагидрофуране (12 мл) добавляли 1,1'-карбонилдиимидазол (608,1 мг, 3,75 ммоль). После перемешивания при 45°C в течение 1 часа смесь охлаждали до комнатной температуры. Добавляли трет-бутилкарбазат (495,6 мг, 3,75 ммоль) и смесь перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 80 г, этилацетат:гексан=1:1→3:2) и колоночной хроматографией на химически модифицированном силикагеле (FUJI SILYSIA CHEMICAL LTD. DM-2035 45 г, дихлорметан:метанол=50:1→20:1), получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-трет-бутил-гидразин-1,2-дикарбоксилат (526,4 мг, 1,252 ммоль, выход 50,0%) в виде белого твердого вещества.
Стадия 4
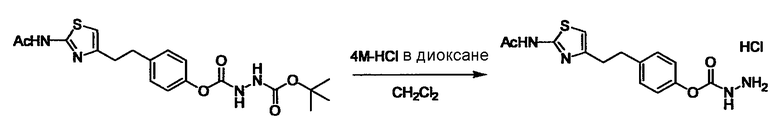
К суспензии 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-трет-бутил-гидразин-1,2-дикарбоксилата (445,2 мг, 1,06 ммоль) в безводном дихлорметане (5,3 мл) добавляли 4M диоксановый раствор хлористого водорода (5,3 мл, 21,3 ммоль). После перемешивания при комнатной температуре в течение 2 часа, реакционную смесь концентрировали при пониженном давлении. Добавляли к концентрированному остатку этилацетат и смесь концентрировали снова при пониженном давлении. Данную операцию повторяли три раза для удаления хлористоводородного газа азеотропно. Остаток суспендировали в этилацетате, твердый остаток отделяли фильтрованием, промывали дважды этилацетатом и сушили при пониженном давлении, получая указанное в заголовке соединение (380,5 мг, количественный) в виде белого твердого вещества, температура плавления: 167-169°C.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,11 (1H, уш.с), 10,97 (1H, уш.с), 7,25 (2H, д, J=8,4 Гц), 7,06 (2H, д, J=8,4 Гц), 6,74 (1H, с), 3,05-2,77 (4H, м), 2,10 (3H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,8, 154,4, 150,1, 148,4, 139,4, 129,6, 121,5, 107,7, 34,0, 32,9, 22,7.
МС(ESI+): 321,1018 [M(свободный)+H]+.
Пример получения 2
Гидрохлорид 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилата
Стадия 1
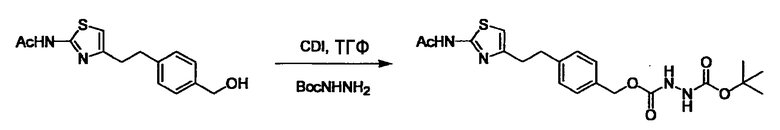
К суспензии N-(4-{2-[4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (161,5 мг, 0,584 ммоль) в безводном тетрагидрофуране (2,4 мл) добавляли 1,1'-карбонилдиимидазол (142,1 мг, 0,876 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли трет-бутилкарбазат (115,9 мг, 0,877 ммоль) и смесь перемешивали в течение 16 часов. Добавляли трет-бутилкарбазат (77,3 мг, 0,584 ммоль) и смесь перемешивали в течение 4 часов. Добавляли снова трет-бутилкарбазат (77,3 мг, 0,584 ммоль) и смесь перемешивали в течение 2 часа. Добавляли снова трет-бутилкарбазат (115,9 мг, 0,877 ммоль) и смесь перемешивали в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 10 г, этилацетат:гексан=5:5→6:4→7:3). Остаток дополнительно очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. DM2035 5 г, этилацетат:гексан=5:5→1:0), получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-трет-бутил-гидразин-1,2-дикарбоксилат (207,0 мг, 0,476 ммоль, выход 81,6%) в виде белого твердого вещества.
Стадия 2
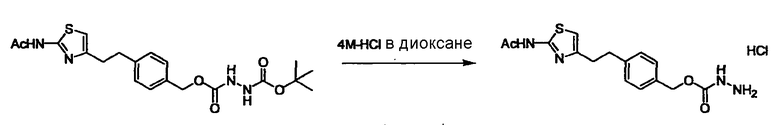
К суспензии 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-трет-бутил-гидразин-1,2-дикарбоксилата (203,0 мг, 0,467 ммоль) в безводном дихлорметане (2,3 мл) добавляли 4M диоксановый раствор хлористого водорода (2,3 мл, 9,2 ммоль) После перемешивания при комнатной температуре в течение 2,5 часов, смесь концентрировали при пониженном давлении. Добавляли к концентрированному остатку этилацетат и смесь концентрировали снова при пониженном давлении. Данную операцию повторяли три раза для удаления хлористоводородного газа азеотропно. Остаток суспендировали в этилацетате и фильтровали. Отфильтрованный продукт промывали этилацетатом и сушили при пониженном давлении, получая указанное в заголовке соединение (179,3 мг, количественный) в виде белого твердого вещества. Температура плавления 162-164°C.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,06 (1H, уш.с), 10,25 (3H, уш.с), 7,29 (2H, д, J=8,2 Гц), 7,20 (2H, д, J=8,2 Гц), 6,71 (1H, с), 5,13 (2H, с), 3,00-2,78 (4H, м), 2,10 (3H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,7, 155,8, 150,2, 141,8, 133,5, 128,6, 128,5, 107,7, 67,2, 34,4, 32,8, 22,7.
MS (ESI+): 357,0965 [M(свободный)+Na]+.
Пример получения 3
Гидрохлорид 2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-гидразинкарбоксилат
Стадия 1
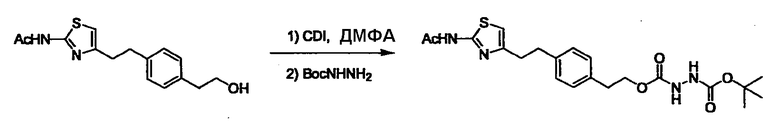
К суспензии N-(4-{2-[4-(2-гидроксиэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (552,5 мг, 1,799 ммоль) в безводном тетрагидрофуране (8 мл) добавляли 1,1'-карбонилдиимидазол (437,8 мг, 2,700 ммоль) и смесь перемешивали при 45°C в течение 0,5 часа. Добавляли трет-бутилкарбазат (356,8 мг, 2,700 ммоль). После перемешивания в течение 1 часа добавляли трет-бутилкарбазат (356,6 мг, 2,698 ммоль). После перемешивания в течение 3 часов добавляли дополнительное количество трет-бутилкарбазата (357,0 мг, 2,701 ммоль). После перемешивания в течение 24 часов смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. DM1025 60 г, этилацетат:гексан=5:5→7:3). Остаток дополнительно очищали колоночной хроматографией на силикагеле (Sep pak-5 г, этилацетат:гексан=7:3), получая 2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-трет-бутил-гидразин-1,2-дикарбоксилат (755,9 мг, 1,685 ммоль, выход 93,6%) в виде белых кристаллов.
Стадия 2
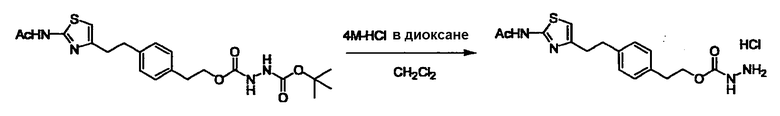
К суспензии 2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-трет-бутил-гидразин-1,2-дикарбоксилата (620,0 мг, 1,382 ммоль) в безводном дихлорметане (6,9 мл) добавляли 4M диоксановый раствор хлористого водорода (6,9 мл, 27,6 ммоль). После перемешивания при комнатной температуре в течение 13 часов, реакционную смесь концентрировали при пониженном давлении. Добавляли к остатку дихлорметан и смесь концентрировали снова при пониженном давлении. Операцию повторяли дважды. Добавляли к остатку дополнительное количество этилацетата и смесь концентрировали при пониженном давлении. Данную операцию повторяли три раза для удаления хлористоводородного газа азеотропно. Остаток сушили при пониженном давлении, получая неочищенный продукт (570,4 мг). Неочищенный продукт растворяли в метаноле (18 мл) и добавляли этилацетат (144 мл) для перекристаллизации неочищенного продукта. Кристаллы отделяли фильтрованием, промывали этилацетатом и сушили при пониженном давлении, получая указанное в заголовке соединение (474,8 мг, 1,234 ммоль, выход 89,3%) в виде белого твердого вещества. Температура плавления 172-174°C.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,09 (1H, уш.с), 11,0-9,6 (3H, уш.с), 7,25-6,95(4H, м), 6,74 (1H, с), 4,27 (2H, т, J=6,7 Гц), 3,01-2,68 (6H, м), 2,11 (3H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,8, 155,9, 150,2, 139,7, 135,3, 129,1, 128,5, 107,6, 66,5, 34,3, 32,9, 22,7.
МС(ESI+): 349,1332 [M(свободный)+H]+, 371,1147 [M(свободный)+Na]+.
Пример получения 4
Гидрохлорид 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-гидразинкарбоксилата
Стадия 1
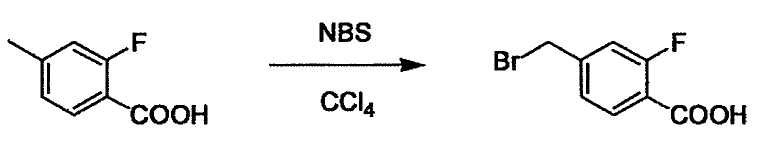
К раствору 2-фтор-4-метилбензойной кислоты (1,029 г, 6,678 ммоль) в тетрахлориде углерода (10 мл) добавляли N-бромсукцинимид (1,189 г, 6,682 ммоль) и 2,2'-азобисизобутиронитрил (43,9 мг, 0,267 ммоль). После перемешивания при 90°C в течение 30 минут и при 100°C в течение 2,5 часов смесь охлаждали до 0°C. Осадок отделяли фильтрованием и промывали гексаном и водой, получая неочищенный продукт. Неочищенный продукт растворяли в этилацетате (5 мл) и добавляли гексан (10 мл). Выпавший осадок отделяли фильтрованием и сушили при пониженном давлении, получая 4-(бромметил)-2-фторбензойную кислоту (838,6 мг, 3,599 ммоль, выход 53,9%) в виде слегка желтого твердого вещества.
Стадия 2
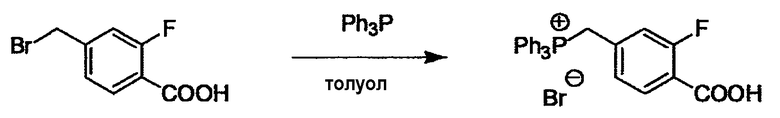
К суспензии 4-(бромметил)-2-фторбензойной кислоты (914,2 мг, 3,923 ммоль) в толуоле (20 мл) добавляли трифенилфосфин (1,029 г, 3,923 ммоль). После кипячения смеси с обратным холодильником в течение 6 часов реакционную смесь охлаждали до комнатной температуры. Осадок отделяли фильтрованием и сушили при пониженном давлении, получая (4-карбокси-3-фторбензил)(трифенил)фосфонийбромид (2,057 г, количественный) в виде белого твердого вещества.
Стадия 3
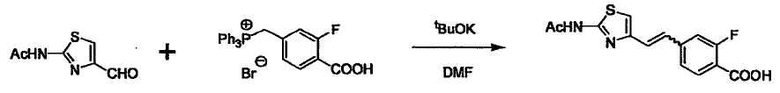
К раствору (4-карбокси-3-фторбензил)(трифенил)фосфонийбромида (2,037 г, 4,112 ммоль) и N-(4-формил-1,3-тиазол-2-ил)ацетамида (599,5 мг, 3,523 ммоль) в безводном N,N-диметилформамиде (15 мл) добавляли трет-бутоксид калия (1,180 г, 10,52 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли к реакционной смеси воду (150 мл) и смесь промывали 3 раза этилацетатом. При перемешивании добавляли к водному слою 1M хлористоводородную кислоту (10,5 мл). Выпавший осадок отделяли фильтрованием и промывали водой. Твердый остаток сушили при пониженном давлении, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}-2-фторбензойную кислоту (753,9 мг, 2,461 ммоль, выход 69,9%) в виде желтого твердого вещества.
Стадия 4
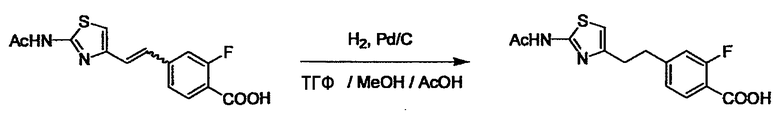
К перемешиваемому раствору 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}-2-фторбензойной кислоты (738,9 мг, 2,412 ммоль) в тетрагидрофуране (105 мл), метаноле (105 мл) и уксусной кислоте (21 мл) добавляли 10% палладий на углероде (593,0 мг, содержащий 50% воды), смесь гидрировали при комнатной температуре и атмосферном давлении. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Полученный в результате твердый остаток отделяли фильтрованием, промывали диизопропиловым эфиром и сушили при пониженном давлении, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензойную кислоту (576,2 мг, 1,869 ммоль, выход 77,5%) в виде белого твердого вещества.
Стадия 5
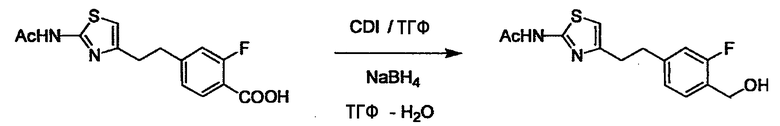
К суспензии 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензойной кислоты (555,0 мг, 1,800 ммоль) в безводном тетрагидрофуране (4 мл) добавляли 1,1'-карбонилдиимидазол (364,8 мг, 2,250 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь добавляли по каплям к смеси боргидрида натрия (1,362 г, 36,0 ммоль), тетрагидрофурана (36 мл) и воды (9 мл), которую перед этим охлаждали до -25°C. После перемешивания при температуре не выше 0°C в течение 1 часа добавляли к реакционной смеси воду и смесь экстрагировали дважды этилацетатом. Объединенные органические слои промывали 1M хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным раствором соли и сушили над безводным сульфатом натрия. Органический слой концентрировали при пониженном давлении и добавляли к остатку смесь метанола (0,5 мл) и диизопропилового эфира (15 мл). Осадок отделяли фильтрованием и сушили при пониженном давлении, получая N-(4-{2-[3-фтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (342,8 мг, 1,165 ммоль, выход 64,7%) в виде белого твердого вещества.
Стадия 6
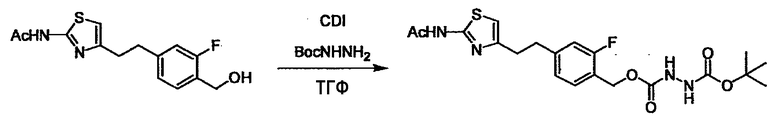
К суспензии N-(4-{2-[3-фтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (118,8 мг, 0,4036 ммоль) в безводном тетрагидрофуране (1,5 мл) добавляли 1,1'-карбонилдиимидазол (98,1 мг, 0,605 ммоль) и смесь перемешивали при комнатной температуре в течение 2,5 часов. Добавляли трет-бутилкарбазат (160,3 мг, 1,213 ммоль) и смесь перемешивали при комнатной температуре в течение 20 часов. Добавляли воду, 1M хлористоводородную кислоту и этилацетат, смесь перемешивали, оставляли без перемешивания и затем распределяли. Органический слой промывали дважды водой и промывали насыщенным раствором соли. После сушки над безводным сульфатом натрия, остаток концентрировали при пониженном давлении. Остаток суспендировали в дихлорметане (15 мл) и фильтровали. После промывки дихлорметаном, остаток сушили при пониженном давлении и очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 12 г, этилацетат:гексан=1:1). Фракции, содержащие целевой продукт, концентрировали, получая твердый остаток, который суспендировали в смеси трет-бутилметилового эфира (5 мл) и гексана (5 мл) и фильтровали. Отфильтрованный продукт сушили при пониженном давлении, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-трет-бутил-гидразин-1,2-дикарбоксилат (153,7 мг, 0,340 ммоль, выход 84,2%) в виде белого твердого вещества.
Стадия 7
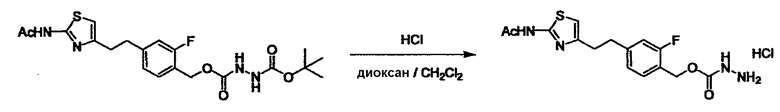
К суспензии 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-трет-бутил-гидразин-1,2-дикарбоксилата (147,0 мг, 0,325 ммоль) в безводном дихлорметане (2 мл) добавляли 4M диоксановый раствор хлористого водорода (2 мл). После перемешивания при комнатной температуре в течение 2 часа смесь концентрировали при пониженном давлении. Добавляли к концентрированному остатку этилацетат и смесь концентрировали снова при пониженном давлении. Данную операцию повторяли три раза для удаления хлористоводородного газа азеотропно. Остаток суспендировали в смеси этанола (2 мл) и этилацетата (8 мл) и фильтровали. Отфильтрованный продукт промывали дважды этилацетатом и сушили при пониженном давлении, получая гидрохлорид 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-гидразинкарбоксилата (129,1 мг, количественный выход) в виде белого твердого вещества, температура плавления 162-165°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,07 (1H, уш.с), 10,5-9,8 (2H, уш.с), 10,28 (1H, уш.с), 7,38 (1H, т, J=7,9 Гц), 7,11 (1H, д, J=11,1 Гц), 7,06 (1H, дд, J=7,9, 1,4 Гц), 6,74(1H, с), 5,19(2H, с), 2,99-2,87 (4H, м), 2,12 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,2, 160,3 (д, J=246,9 Гц), 157,4, 155,4, 149,7, 145,1 (д, J=8,2 Гц), 131,0 (д, J=4,5 Гц), 124,3 (д, J=3,0 Гц), 119,8 (д, J=15,0 Гц), 115,1 (д, J=21,0 Гц), 107,5, 61,2, 33,8, 32,1, 22,4.
19F-ЯМР (376 Гц, ДМСО-d6): δ (м.д.): -120,9.
МС(ESI+): 353,1037 [M(свободный)+H]+, 375,0859 [M(свободный)+Na]+.
Пример получения 5
Гидрохлорид 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил-гидразинкарбоксилата
Стадия 1
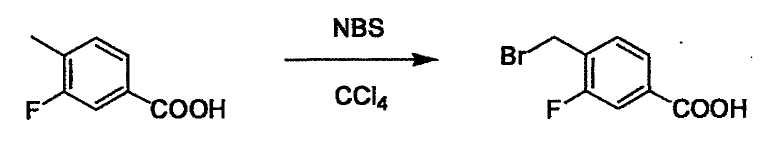
3-Фтор-4-метилбензойную кислоту (2,541 г, 16,49 ммоль) бромировали по аналогичной методике примера получения 4, стадия 1, получая 4-(бромметил)-3-фторбензойную кислоту (2,539 г, 10,90 ммоль, выход 66,1%) в виде белого твердого вещества.
Стадия 2
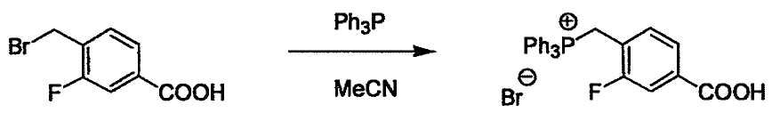
По аналогичной методике примера получения 4, стадия 2, (4-карбокси-3-фторбензил)(трифенил)фосфонийбромид (4,130 г, 8,338 ммоль, выход 76,9%) получали в виде белого твердого вещества из 4-(бромметил)-3-фторбензойной кислоты (2,526 г, 10,84 ммоль).
Стадия 3
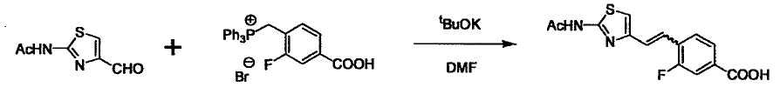
N-(4-Формил-1,3-тиазол-2-ил)ацетамид (941,7 мг, 5,533 ммоль) и (4-карбокси-3-фторбензил)(трифенил)фосфонийбромид (4,111 г, 8,300 ммоль) конденсировали по аналогичной методике примера получения 4, стадия 3, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}-3-фторбензойную кислоту (1,086 г, 3,547 ммоль, выход 64,1%) в виде бледно-желтого твердого вещества.
Стадия 4
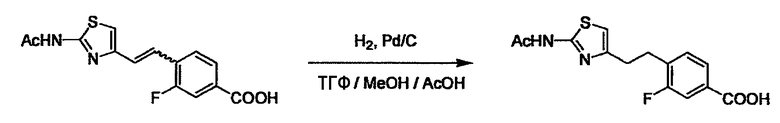
4-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]винил}-3-фторбензойную кислоту (1,000 г, 3,265 ммоль) гидрировали по аналогичной методике примера получения 4, стадия 4, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензойную кислоту (620,0 мг, 2,011 ммоль, выход 61,7%) в виде бледно-желтого твердого вещества.
Стадия 5
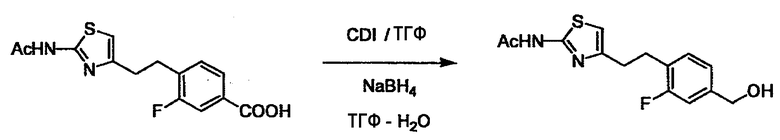
4-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензойную кислоту (593,4 мг, 1,924 ммоль) восстанавливали по аналогичной методике примера получения 4, стадия 5, получая N-(4-{2-[2-фтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (406,9 мг, 1,382 ммоль, выход 71,8%) в виде белого твердого вещества.
Стадия 6
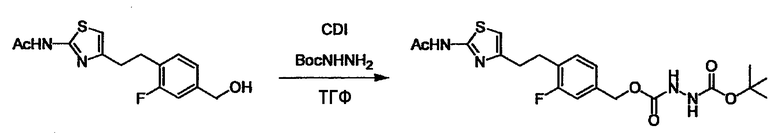
По аналогичной методике примера получения 4, стадия 6, 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил-трет-бутил-гидразин-1,2-дикарбоксилат (196,0 мг, 0,433 ммоль, выход 66,7%) получали в виде бледно-желтого твердого вещества из N-(4-{2-[2-фтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (191,0 мг, 0,649 ммоль).
Стадия 7
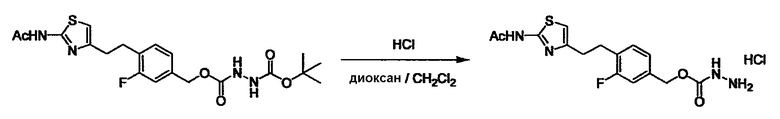
С 4-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил-трет-бутил-гидразин-1,2-дикарбоксилата (154,0 мг, 0,341 ммоль) удаляли защитную группу по аналогичной методике примера получения 4, стадия 7, получая указанное в заголовке соединение (123,0 мг, 0,316 ммоль, выход 92,9%) в виде белого твердого вещества. Температура плавления 204-208°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,09 (1H, уш.с), 10,43 (3H, уш.с), 7,27(1H, т, J=8,0 Гц), 7,17(1H, д, J=10,4 Гц), 7,14(1H, т, J=8,0 Гц), 6,74(1H, с), 5,14(2H, с), 2,97-2,82 (4H, м), 2,10(3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,4, 160,5 (д, J=24,2 Гц), 157,7, 149,9, 136,3 (д, J=7,4 Гц), 131,0, 128,0 (д, J=14,9 Гц), 124,0, 114,8(д, J=14,9 Гц), 107,7, 66,3, 31,5, 27,8, 22,6.
МС(ESI+): 353,1075 [M(свободный)+H]+, 375,0895 [M(свободный)+Na]+.
Пример получения 6
Гидрохлорид 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил-гидразинкарбоксилата
Стадия 1
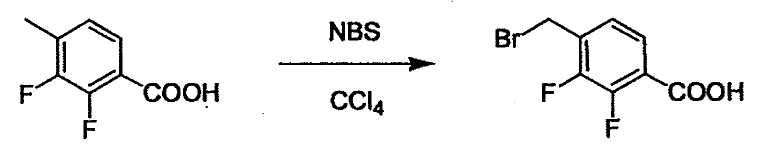
2,3-Дифтор-4-метилбензойную кислоту (4,689 г, 27,24 ммоль) бромировали по аналогичной методике примера получения 4, стадия 1, получая 4-(бромметил)-2,3-дифторбензойную кислоту (1,724 г, 6,869 ммоль, выход 25,2%) в виде слегка желтоватого твердого вещества.
Стадия 2
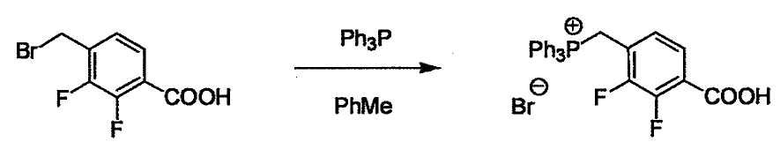
По аналогичной методике примера получения 4, стадия 2, (4-карбокси-2,3-дифторбензил)(трифенил)фосфонийбромид (3,246 г, 6,323 ммоль, выход 95,2%) получали в виде белого твердого вещества из 4-(бромметил)-2,3-дифторбензойной кислоты (1,667 г, 6,640 ммоль).
Стадия 3
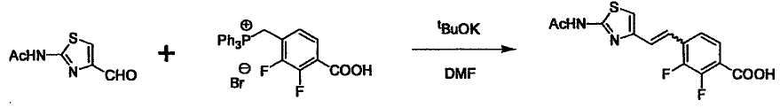
N-(4-Формил-1,3-тиазол-2-ил)ацетамид (1,071 г, 6,292 ммоль) и (4-карбокси-2,3-дифторбензил)(трифенил)фосфонийбромид (3,227 г, 6,287 ммоль) конденсировали по аналогичной методике примера получения 4, стадия 3, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}-2,3-дифторбензойную кислоту (1,550 г, 4,778 ммоль, выход 76,0%) в виде желтого твердого вещества.
Стадия 4
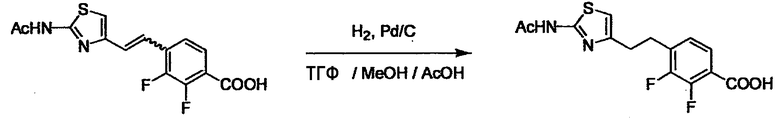
4-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]винил}-2,3-дифторбензойную кислоту (1,533 г, 4,728 ммоль) гидрировали по аналогичной методике примера получения 4, стадия 4, получая 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензойную кислоту (1,325 г, 4,059 ммоль, выход 85,8%) в виде бледно-желтого твердого вещества.
Стадия 5
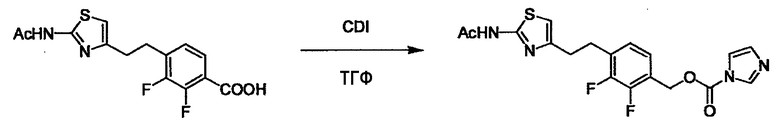
К суспензии 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензойной кислоты (654,0 мг, 2,004 ммоль) в безводном тетрагидрофуране (5 мл) добавляли 1,1'-карбонилдиимидазол (408,2 мг, 2,517 ммоль) и смесь перемешивали при комнатной температуре в течение 2,5 часов. Добавляли 1,1'-карбонилдиимидазол (61,0 мг, 0,376 ммоль) и смесь перемешивали при комнатной температуре в течение 0,5 часа. Реакционную смесь концентрировали при пониженном давлении, осадок суспендировали в этилацетате (10 мл) и фильтровали. Отфильтрованный продукт сушили при пониженном давлении, получая N-(4-{2-[2,3-дифтор-4-(1H-имидазол-1-илкарбонил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (516,0 мг, 1,371 ммоль, выход 68,4%) в виде белого твердого вещества.
Стадия 6
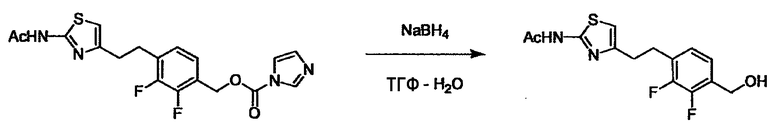
Боргидрид натрия (1,009 г, 26,66 ммоль) суспендировали в смеси тетрагидрофурана (36 мл) и воды (9 мл) и суспензию охлаждали до -20°C. Добавляли по каплям суспензию N-(4-{2-[2,3-дифтор-4-(1H-имидазол-1-илкарбонил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (503,5 мг, 1,338 ммоль) в безводном тетрагидрофуране (4 мл). После перемешивания при температуре, не большей чем 0°C, в течение 2,5 часов, добавляли насыщенный водный аммонийхлорид (50 мл). Смесь экстрагировали 3 раза этилацетатом, объединенные органические слои промывали насыщенным водным аммонийхлоридом и насыщенным раствором соли. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли метанол (0,5 мл) и диизопропиловый эфир (25 мл), смесь перемешивали и фильтровали. Отфильтрованный продукт сушили при пониженном давлении, получая N-(4-{2-[2,3-дифтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (316,8 мг, 1,014 ммоль, 75,8%) в виде белого твердого вещества.
Стадия 7
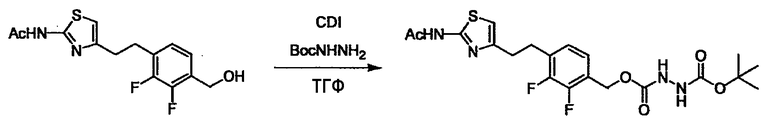
По аналогичной методике примера получения 4, стадия 6, 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил-трет-бутил-гидразин-1,2-дикарбоксилат (173,4 мг, 0,369 ммоль, выход 92,3%) получали в виде белого твердого вещества из N-(4-{2-[2,3-дифтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (125,0 мг, 0,400 ммоль).
Стадия 8
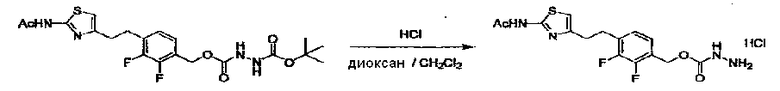
С 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил-трет-бутил-гидразин-1,2-дикарбоксилата (164,8 мг, 0,350 ммоль) удаляли защитную группу по аналогичной методике примера получения 4, стадия 7, получая указанное в заголовке соединение (145,2 мг, количественный выход) в виде белого твердого вещества. Температура плавления 154-160°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,10 (1H, уш.с), 10,41 (4H, уш.с), 7,22 (1H, т, J=7,3 Гц), 7,12 (1H, т, J=7,3 Гц), 6,76 (1H, с), 5,23 (2H, с), 3,03-2,87 (4H, м), 2,12 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,2, 157,5, 155,2, 149,3, 148,1 (дд, J=248,7, 12,6 Гц), 147,9 (дд, J=244,6, 11,9 Гц), 131,0 (д, J=12,8 Гц), 125,2, 125,1, 122,6(д, J=12,0 Гц), 107,7, 60,7, 30,9, 27,5, 22,4.
19F-ЯМР (376 Гц, ДМСО-d6): δ (м.д.): -144,8 (IF, д, JFF=19,1 Гц), -145,9 (IF, д, JFF=19,1 Гц).
МС(ESI+): 371,0950 [M(свободный)+H]+, 393,0768 [M(свободный)+Na]+.
Пример получения 7
2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил-гидразинкарбоксилат
Стадия 1
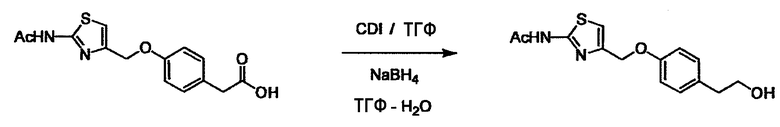
(4-{[2-(Ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)уксусную кислоту (644,0 мг, 2,102 ммоль) восстанавливали по аналогичной методике примера получения 4, стадия 5, получая N-(4-{[4-(2-гидроксиэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамид (577,6 мг, 1,976 ммоль, выход 94,0%) в виде белого твердого вещества.
Стадия 2
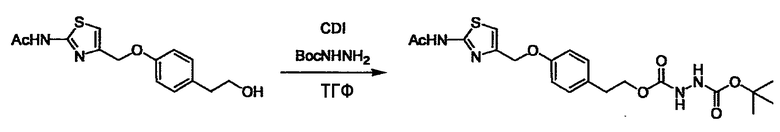
По аналогичной методике примера получения 4, стадия 6, 2-(4{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил-трет-бутил-гидразин-1,2-дикарбоксилат (479,4 мг, количественный выход) получали в виде белого твердого вещества из N-(4-{[4-(2-гидроксиэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамида (250,0 мг, 0,855 ммоль).
Стадия 3
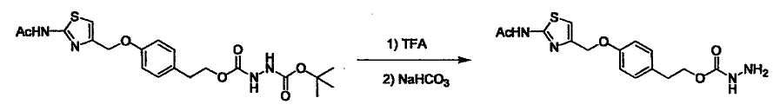
К раствору 2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил-трет-бутил-гидразин-1,2-дикарбоксилата (0,855 ммоль) в дихлорметане (30 мл) добавляли трифторуксусную кислоту (3,18 мл, 42,8 ммоль) при 0°C. После перемешивания при 0°C в течение 30 минут смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали, добавляли к остатку водный раствор гидрокарбоната натрия и смесь экстрагировали 4 раза этилацетатом. Объединенные органические слои промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Добавляли этилацетат (20 мл) для суспендирования остатка. Суспензию фильтровали и промывали один раз этилацетатом и 5 раз диэтиловым эфиром, сушили при пониженном давлении, получая указанное в заголовке соединение (105,9 мг, 0,302 ммоль, выход 35,3%) в виде белого твердого вещества, температура плавления 177-180°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,13 (1H, уш.с), 8,09 (1H, уш.с), 7,16 (1H, с), 7,14 (2H, д, J=8,6 Гц), 6,92 (2H, д, J=8,6 Гц), 5,00 (2H, с), 4,10 (2H, т, J=6,9 Гц), 3,99 (2H, уш.с), 2,77 (2H, т, J=6,9 Гц), 2,12 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,6, 158,2, 156,9, 146,6, 130,4, 130,0, 114,8, 111,4, 65,6, 64,9,34,2, 22,6.
МС(ESI+): 351,1090[M+H]+, 373,0911[M+Na]+.
Пример получения 8
Гидрохлорид 4-{2-[(гидразинокарбонил)окси]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилата
Стадия 1
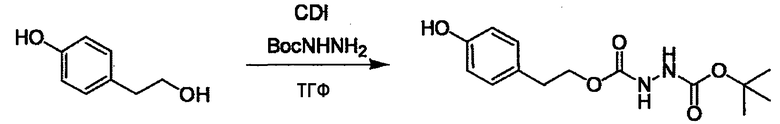
К суспензии 1,1'-карбонилдиимидазола (1,620 г, 9,989 ммоль) в безводном тетрагидрофуране (20 мл) добавляли 2-(4-гидроксифенил)этанол (1,383 г, 10,01 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часа. Добавляли трет-бутилкарбазат (1,323 г, 10,01 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли к реакционной смеси воду (100 мл) и этилацетат (100 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Водный слой экстрагировали этилацетатом, объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 40 г, этилацетат:гексан=4:6→5:5). Фракции, содержащие целевое соединение, концентрировали при пониженном давлении, полученный твердый остаток суспендировали в диизопропиловом эфире (50 мл) и фильтровали. Отфильтрованный продукт промывали 3 раза диизопропиловым эфиром и сушили при пониженном давлении, получая трет-бутил 2-(4-гидроксифенил)этил-гидразин-1,2-дикарбоксилат (616,7 мг, 2,08 ммоль, выход 20,8%) в виде белого твердого вещества.
Стадия 2
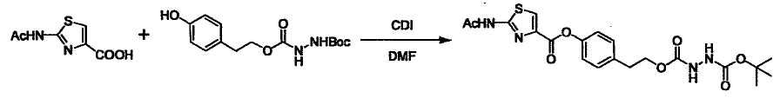
К суспензии 2-(ацетиламино)-1,3-тиазол-4-карбоновой кислоты (466,2 мг, 2,504 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли 1,1'-карбонилдиимидазол (405,7 мг, 2,502 ммоль) и смесь перемешивали при 50°C в течение 2,5 часов. Добавляли трет-бутил 2-(4-гидроксифенил)этил-гидразин-1,2-дикарбоксилат (594,8 мг, 2,001 ммоль) и смесь перемешивали при 50°C в течение 18 часов. Добавляли к реакционной смеси воду (40 мл) и этилацетат (40 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 40 г, гексан:этилацетат=6:4→5:5). Фракции, содержащие целевое соединение, концентрировали при пониженном давлении и полученный твердый остаток суспендировали в смеси гексана (40 мл) и трет-бутилметилового эфира (20 мл). Суспензию фильтровали и сушили при пониженном давлении, получая 2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}окси)фенил]этил-трет-бутил-гидразин-1,2-дикарбоксилат (539,2 мг, 1,161 ммоль, выход 58,0%) в виде белого твердого вещества.
Стадия 3
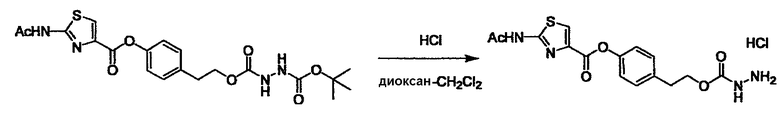
К суспензии 2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}окси)фенил]этил-трет-бутил-гидразин-1,2-дикарбоксилата (371,6 мг, 0,800 ммоль) в безводном дихлорметане (4 мл) добавляли 4M диоксановый раствор хлористого водорода (4 мл). После перемешивания при комнатной температуре в течение 2,5 часов реакционную смесь концентрировали при пониженном давлении. Добавляли к концентрированному остатку этилацетат и смесь концентрировали снова при пониженном давлении. Операцию повторяли дважды для удаления хлористоводородного газа азеотропно. Остаток суспендировали в смеси этанола (5 мл) и этилацетата (30 мл) и суспензию фильтровали. Отфильтрованный продукт промывали дважды этилацетатом и сушили при пониженном давлении, получая указанное в заголовке соединение (317,7 мг, 0,793 ммоль, выход 99,1%) в виде белого твердого вещества, температура плавления 179-184°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,55 (1H, уш.с), 10,09 (4H, уш.с), 8,28 (1H, с), 7,35 (2H, д, J=8,6 Гц), 7,19 (2H, д, J=8,6 Гц), 4,35 (2H, т, J=6,6 Гц), 2,95 (2H, т, J=6,6 Гц), 2,16 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 169,1, 159,4, 158,2, 155,7, 148,9, 139,7, 135,4, 129,9, 124,4, 121,6, 66,0, 33,7, 22,3.
МС(ESI+): 387,0729 [M(свободный)+Na]+, 403,0478 [M(свободный)+K]+.
Пример получения 9
Гидрохлорид 2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-гидразинкарбоксилата
Стадия 1
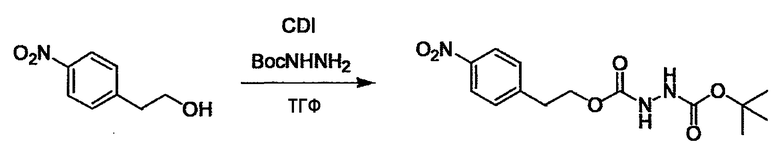
К раствору 2-(4-нитрофенил)этанола (5,015 г, 30,00 ммоль) в безводном тетрагидрофуране (50 мл) добавляли 1,1'-карбонилдиимидазол (5,839 г, 36,01 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли трет-бутилкарбазат (5,956 г, 45,06 ммоль), смесь перемешивали при комнатной температуре в течение 16 часов и дополнительно при 50°C в течение 8 часов. Добавляли к реакционной смеси 0,5M хлористоводородную кислоту (100 мл) и этилацетат (100 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Органический слой промывали 0,5M хлористоводородной кислотой, водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 200 г, этилацетат:гексан=4:6→5:5), получая трет-бутил 2-(4-нитрофенил)этил-гидразин-1,2-дикарбоксилат (9,780 г, количественный) в виде слегка желтоватого твердого вещества.
Стадия 2
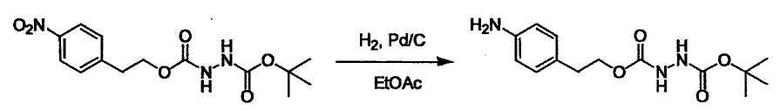
К раствору трет-бутил 2-(4-нитрофенил)этил-гидразин-1,2-дикарбоксилата (9,780 г, 30,00 ммоль) в этилацетате (100 мл) добавляли 10% палладий на углероде (980,0 мг, содержащий 50% воды), смесь гидрировали при комнатной температуре и атмосферном давлении. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток суспендировали в смеси гексана (70 мл) и этилацетата (30 мл), фильтровали и сушили при пониженном давлении, получая 2-(4-аминофенил)этил-трет-бутил-гидразин-1,2-дикарбоксилат (5,207 г, 17,63 ммоль, выход 58,8%) в виде белого твердого вещества.
Стадия 3
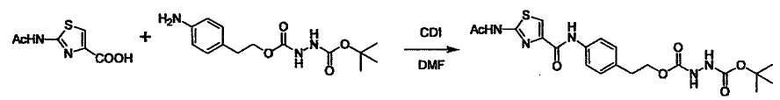
К суспензии 2-(ацетиламино)-1,3-тиазол-4-карбоновой кислоты (557,3 мг, 2,993 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли 1,1'-карбонилдиимидазол (531,7 мг, 3,279 ммоль) и смесь перемешивали при 50°C в течение 2 часа. Добавляли 2-(4-аминофенил)этил-трет-бутил-гидразин-1,2-дикарбоксилат (1,065 г, 3,607 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли к реакционной смеси воду (100 мл) и этилацетат (100 мл), смесь перемешивали, оставляли без перемешивания и распределяли. Органический слой промывали 0,5M хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток суспендировали в трет-бутилметиловом эфире (30 мл), фильтровали, промывали 3 раза трет-бутилметиловым эфиром и сушили при пониженном давлении, получая 2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-трет-бутил-гидразин-1,2-дикарбоксилат (1,098 г, 2,368 ммоль, выход 79,1%) в виде белого твердого вещества.
Стадия 4
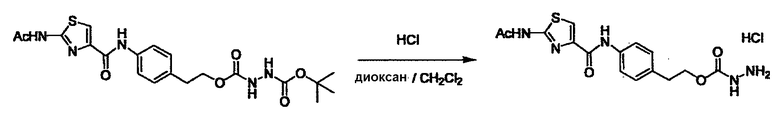
К суспензии 2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-трет-бутил-гидразин-1,2-дикарбоксилата (370,8 мг, 0,800 ммоль) в безводном дихлорметане (4 мл) добавляли 4M диоксановый раствор хлористого водорода (4 мл) и смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении. Добавляли к концентрированному остатку этилацетат и смесь концентрировали снова при пониженном давлении. Операцию повторяли дважды для удаления азеотропно хлористоводородного газа. Остаток суспендировали в этилацетате, фильтровали, промывали 3 раза этилацетатом и 3 раза метанолом, сушили при пониженном давлении, получая гидрохлорид 2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-гидразинкарбоксилата (280,8 мг, 0,702 ммоль, выход 87,8%) в виде белого твердого вещества. Температура плавления 225-233°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,33 (1H, уш.с), 10,18 (3H, уш.с), 9,71 (1H, уш.с), 7,94 (1H, с), 7,67 (2H, д, J=8,5 Гц), 7,24 (2H, д, J=8,4 Гц), 4,30 (2H, т, J=6,7 Гц), 2,89 (2H, т, J=6,6 Гц), 2,18 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 169,2, 159,3, 158,0, 155,9, 144,5, 136,9, 133,2, 129,3, 120,1, 118,3, 66,3, 34,1, 22,6.
МС(ESI+): 364,1066 [M(свободный)+H]+, 386,0889 [M(свободный)+Na]+.
Пример получения 10
Гидрохлорид 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилата
Стадия 1
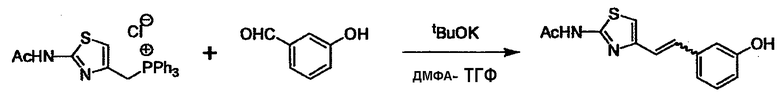
К раствору {[2-(ацетиламино)-1,3-тиазол-4-ил]метил}(трифенил)фосфонийхлорида (3,894 г, 8,598 ммоль) и 3-гидроксибензальдегида (1,000 г, 8,189 ммоль) в безводном N,N-диметилформамиде (42 мл) добавляли по каплям тетрагидрофурановый раствор трет-бутоксида калия (1M, 25,4 мл, 25,4 ммоль) при 0°C. После перемешивания при 0°C в течение 30 минут смесь перемешивали при комнатной температуре в течение 2 часа. Реакционную смесь охлаждали до 0°C и добавляли ледяную воду (100 мл). Смесь промывали дважды этилацетатом и водный слой подкисляли 1M хлористоводородной кислотой (pH 2,5). Смесь экстрагировали 3 раза этилацетатом, объединенные органические слои промывали дважды насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 120 г, этилацетат:гексан=1:1), получая N-{3-[2-(4-гидроксифенил)винил]-1,3-тиазол-2-ил}ацетамид (1,953 г, 7,503 ммоль, выход 91,6%) в виде слегка желтоватого твердого вещества.
Стадия 2
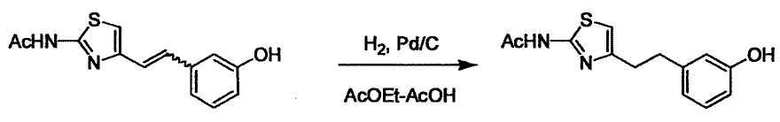
N-{3-[2-(4-Гидроксифенил)винил]-1,3-тиазол-2-ил}ацетамид (1,900 г, 7,299 ммоль) растворяли в смеси этилацетата (300 мл) и уксусной кислоты (50 мл) и добавляли 10% палладий на углероде (760 мг, содержащий 50% воды). Смесь гидрировали при комнатной температуре при 4-5 атм. После завершения реакции реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток растворяли в этилацетате при нагревании, перекристаллизовывали охлаждением, получая N-{3-[2-(4-гидроксифенил)этил]-1,3-тиазол-2-ил}ацетамид (1,834 г, 6,993 ммоль, выход 95,8%) в виде белого твердого вещества.
Стадия 3
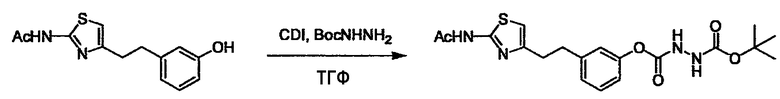
По аналогичной методике примера получения 1, стадия 3, 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-трет-бутил-гидразин-1,2-дикарбоксилат (636,1 мг, 1,513 ммоль, выход 52,9%) получали в виде белого твердого вещества из N-{3-[2-(4-гидроксифенил)этил]-1,3-тиазол-2-ил}ацетамида (750,0 мг, 2,859 ммоль).
Стадия 4
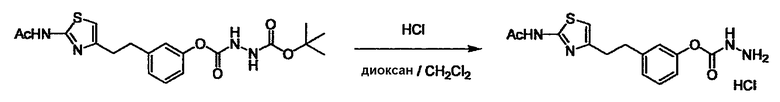
По аналогичной методике примера получения 1, стадия 4, с 4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-трет-бутил-гидразин-1,2-дикарбоксилата (260,0 мг, 0,618 ммоль) удаляли защитную группу, получая указанное в заголовке соединение (219,0 мг, 0,614 ммоль, выход 99,3%) в виде белого твердого вещества. Температура плавления 158-162°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,11 (1H, уш.с), 10,99 (1H, уш.с), 11,2-9,8 (2H, уш.с), 8,8-7,6 (1H, уш.с), 7,32 (1H, т, J=7,7 Гц), 7,11 (1H, д, J=7,4 Гц), 7,02-6,97 (2H, м), 6,74 (1H, с), 2,97-2,86 (4H, м), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,6, 157,9, 154,3, 150,4, 150,3, 143,8, 129,8, 126,4, 121,6, 119,4, 107,9, 34,5, 32,8, 22,9.
МС(ESI+): 321,0972 [M(свободный)+H]+, 343,0793 [M(свободный)+Na]+.
Пример получения 11
Гидрохлорид 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилата
Стадия 1
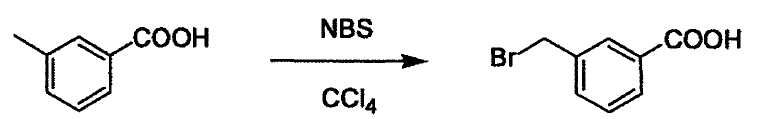
м-Толуиловую кислоту (13,62 г, 100,0 ммоль) бромировали по аналогичной методике примера получения 4, стадия 1, получая 3-(бромметил)бензойную кислоту (16,85 г, 78,36 ммоль, выход 78,4%) в виде бледно-желтого твердого вещества.
Стадия 2
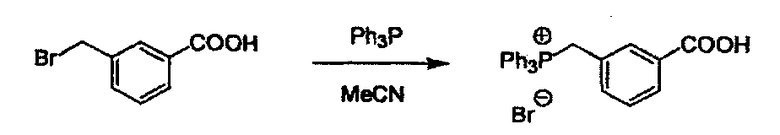
К раствору 3-(бромметил)бензойной кислоты (16,50 г, 76,73 ммоль) в ацетонитриле (76,7 мл) добавляли трифенилфосфин (22,14 г, 84,40 ммоль). После кипячения смеси с обратным холодильником в течение 2 часа, реакционную смесь охлаждали до комнатной температуры. Осадок отделяли фильтрованием и сушили при пониженном давлении, получая (3-карбоксибензил)(трифенил)фосфонийбромид (30,13 г, 63,12 ммоль, выход 82,3%) в виде белого твердого вещества.
Стадия 3
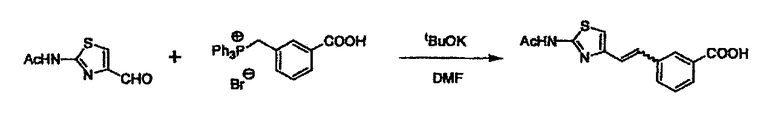
N-(4-Формил-1,3-тиазол-2-ил)ацетамид (1,702 г, 10,00 ммоль) и (3-карбоксибензил)(трифенил)фосфонийбромид (5,251 г, 11,00 ммоль) конденсировали по аналогичной методике примера получения 4, стадия 3, получая 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}бензойную кислоту (2,8 62 г, 9,926 ммоль, выход 99,3%) в виде бледно-желтого твердого вещества.
Стадия 4
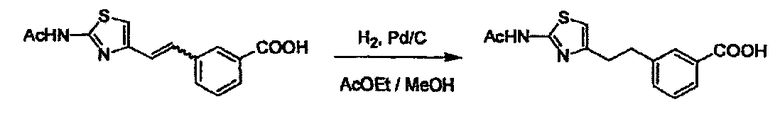
3-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]винил}бензойную кислоту (1,780 г, 6,174 ммоль) гидрировали по аналогичной методике примера получения 4, стадия 4, получая 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензойную кислоту (1,323 г, 4,557 ммоль, выход 73,8%) в виде белого твердого вещества.
Стадия 5
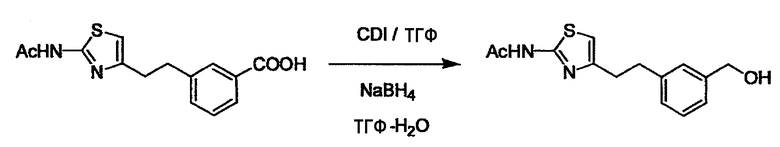
3-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}бензойную кислоту (844,6 мг, 2,909 ммоль) восстанавливали по аналогичной методике примера получения 4, стадия 5, получая N-(4-{2-[3-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (850,0 мг, количественный) в виде не совсем белого твердого вещества.
Стадия 6
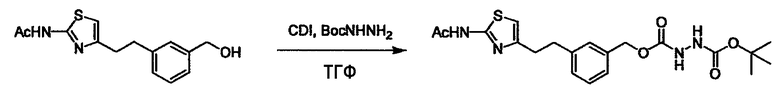
По аналогичной методике примера получения 4, стадия 6, 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-трет-бутил-гидразин-1,2-дикарбоксилат (710,0 мг, 1,634 ммоль, выход 86,2%) получали в виде белого твердого вещества из N-(4-{2-[3-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (524,0 мг, 1,896 ммоль).
Стадия 7
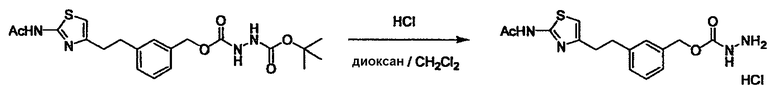
По аналогичной методике примера получения 1, стадия 4, с 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-трет-бутил-гидразин-1,2-дикарбоксилата (406,0 мг, 0,934 ммоль) удаляли защитную группу, получая указанное в заголовке соединение (329,1 ммоль, 0,887 ммоль, выход 95,0%) в виде белого твердого вещества. Температура плавления 113-119°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,07 (1H, уш.с), 10,37 (3H, уш.с), 7,30-7,16 (4H, м), 6,74 (1H, с), 5,14 (2H, с), 3,55-2,86 (4H, м), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,7, 157,9, 156,2, 150,6, 142,2, 136,2, 128,9, 128,7, 128,5, 126,2, 107,9, 67,6, 34,9, 33,2, 22,9.
МС(ESI+): 335,1145 [M(свободный)+H]+, 357,0957 [M(свободный)+Na]+.
Пример получения 12
Гидрохлорид 2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-гидразинкарбоксилата
Стадия 1
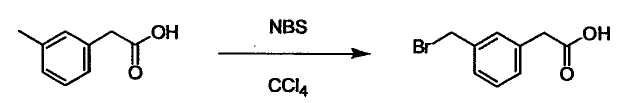
К раствору м-толилуксусной кислоты (25,00 г, 166,5 ммоль) в безводном тетрахлориде углерода (200 мл) добавляли N-бромсукцинимид (30,00 г, 168,6 ммоль) и смесь постепенно нагревали до температуры кипения. После кипячения смеси с обратным холодильником в течение 5,5 часов, реакционную смесь охлаждали до комнатной температуры, нерастворившиеся вещества удаляли фильтрованием и промывали дважды тетрахлоридом углерода (100 мл). Фильтрат концентрировали, добавляли тетрахлорид углерода (60 мл) и остаток растворяли при нагревании приблизительно до 70°C. Раствор охлаждали приблизительно до 40°C и добавляли по каплям гексан (300 мл). После перемешивания при комнатной температуре в течение 30 минут, выпавшие кристаллы отфильтровывали, промывали гексаном и сушили при пониженном давлении, получая (3-бромметилфенил)уксусную кислоту (22,80 г, 99,53 ммоль, выход 59,8%) в виде белого твердого вещества.
Стадия 2
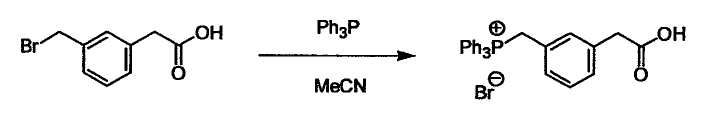
Раствор (3-бромметилфенил)уксусной кислоты (22,00 г, 96,04 ммоль) и трифенилфосфина (30,23 г, 115,2 ммоль) в безводном ацетонитриле (300 мл) кипятили с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры раствор концентрировали при пониженном давлении приблизительно до 100 г. Добавляли диэтиловый эфир (200 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Выпавшие кристаллы отделяли фильтрованием, промывали 3 раза диэтиловым эфиром и сушили при пониженном давлении, получая [(3-карбонилметил)бензил](трифенил)фосфонийбромид (43,60 г, 88,73 ммоль, выход 92,4%) в виде белого твердого вещества.
Стадия 3
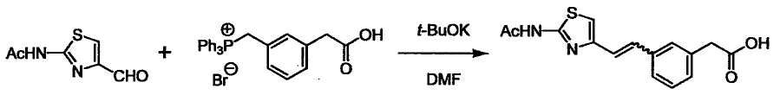
К суспензии [(3-карбонилметил)бензил](трифенил)фосфонийбромида (9,529 г, 19,39 ммоль) в безводном N,N-диметилформамиде (85 мл) добавляли трет-бутоксид калия (5,935 г, 52,89 ммоль) при 0°C небольшими порциями. После перемешивания при комнатной температуре в течение 30 минут добавляли (4-формил-1,3-тиазол-2-ил)ацетамид (3,000 г, 17,63 ммоль) и смесь перемешивали в течение 3 часа. После охлаждения до 0°C добавляли воду (200 мл) и смесь промывали дважды этилацетатом (100 мл). Добавляли по каплям к водному слою при 0°C 6M хлористоводородную кислоту для доведения pH до 3 и смесь перемешивали в течение 30 минут. Осадок отделяли фильтрованием, промывали 3 раза водой и дважды диизопропиловым эфиром, сушили при пониженном давлении, получая 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}фенилуксусную кислоту (4,625 г, 15,30 ммоль, выход 86,8%) в виде белого твердого вещества.
Стадия 4
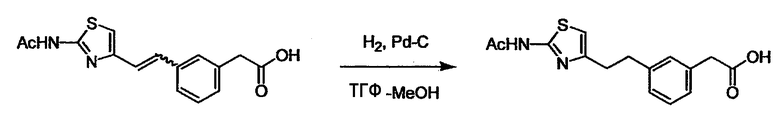
3-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]винил}фенилуксусную кислоту (4,500 г, 14,88 ммоль) растворяли в смеси тетрагидрофурана (225 мл) и метанола (90 мл) и добавляли 20% палладий на углероде (содержащий 50% воды, 1,800 г). Гидрирование проводили при комнатной температуре -30°C, 4 атм. После завершения реакции реакционную смесь фильтровали через целит и фильтрат концентрировали. Добавляли к остатку диэтиловый эфир (100 мл) и осадок отделяли фильтрованием. Отфильтрованный продукт промывали 3 раза диэтиловым эфиром и сушили при пониженном давлении, получая 3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенилуксусную кислоту (4,152 г, 13,64 ммоль, выход 91,7%) в виде белого твердого вещества.
Стадия 5
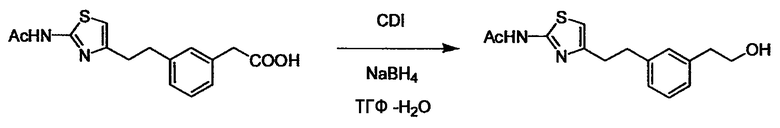
(3-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}фенил)уксусную кислоту (935,0 мг, 3,072 ммоль) восстанавливали по аналогичной методике примера получения 4, стадия 5, получая N-(4-{2-[3-(2-гидроксиэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (670,0 мг, 2,307 ммоль, выход 75,1%) в виде белого твердого вещества.
Стадия 6
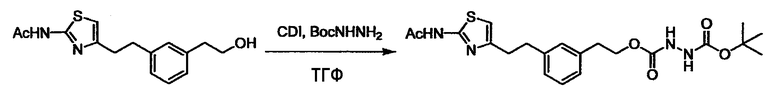
По аналогичной методике примера получения 4, стадия 6, 2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-трет-бутил-гидразин-1,2-дикарбоксилат (534,9 мг, количественный выход) получали в виде белого твердого вещества из N-(4-{2-[3-(2-гидроксиэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (300,0 мг, 1,033 ммоль).
Стадия 7
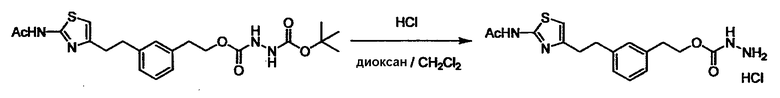
С 2-(3-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-трет-бутил-гидразин-1,2-дикарбоксилата (1,033 ммоль) удаляли защитную группу по аналогичной методике примера получения 4, стадия 7, получая указанное в заголовке соединение (399,5 мг, количественный выход) в виде белого твердого вещества. Температура плавления 138-140°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,06 (1H, уш.с), 10,8-9,8 (4H, уш.с), 7,20 (1H, т, J=7,6 Гц), 7,11-7,04 (3H, м), 6,73 (1H, с), 4,30 (2H, т, J=6,9 Гц), 2,92-2,85 (6H, м), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,7, 157,9, 156,2, 150,8, 142,0, 138,0, 129,3, 128,8, 126,9, 107,8, 66,7, 35,0, 34,9, 33,3, 22,9.
МС(ESI+): 349,1292 [M(свободный)+H]+, 371,1106 [M(свободный)+Na]+
Пример получения 13
Гидрохлорид {5-[2-(2-ацетиламино-1,3-тиазол-4-ил)этил]тиофен-2-ил}метил-гидразинкарбоксилата
Стадия 1
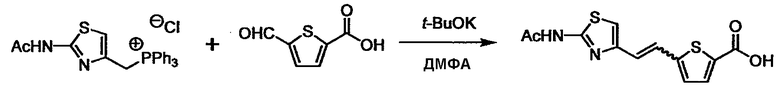
К раствору {[2-(ацетиламино)-1,3-тиазол-4-ил]метил}(трифенил)фосфонийхлорида (3,771 г, 8,325 ммоль) в безводном N,N-диметилформамиде (35 мл) добавляли трет-бутоксид калия (2,515 г, 22,41 ммоль) при 0°C и смесь перемешивали в течение 15 минут. Добавляли небольшими порциями 5-формилтиофен-2-карбоновую кислоту (1,000 г, 6,404 ммоль) и смесь перемешивали при 0°C в течение 30 минут. Смесь нагревали до комнатной температуры, перемешивали в течение 2 часа и выливали в ледяную воду (300 мл). После перемешивания в течение 30 минут смесь промывали дважды этилацетатом. Водный слой охлаждали до 0°C и доводили pH до 3 добавлением по каплям 6M хлористоводородной кислоты. После перемешивания при 0-5°C в течение 2 часа, выпавшие кристаллы отделяли фильтрованием, промывали 3 раза водой и дважды диизопропиловым эфиром, сушили при пониженном давлении, получая 5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}тиофен-2-карбоновую кислоту (1,104 г, 3,751 ммоль, выход 58,6%) в виде желто-охрового твердого вещества.
Стадия 2
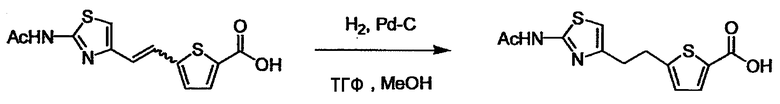
5-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]винил}тиофен-2-карбоновую кислоту (900,0 мг, 3,058 ммоль) растворяли в смеси тетрагидрофурана (250 мл) и метанола (100 мл). Добавляли 20% палладий на углероде и смесь гидрировали при комнатной температуре при 4 атм. После завершения реакции реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Добавляли к концентрированному остатку диэтиловый эфир (100 мл), осадок отделяли фильтрованием, промывали диэтиловым эфиром и сушили при пониженном давлении, получая 5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-карбоновую кислоту (738,1 мг, 2,490 ммоль, выход 81,4%) в виде не совсем белого твердого вещества.
Стадия 3
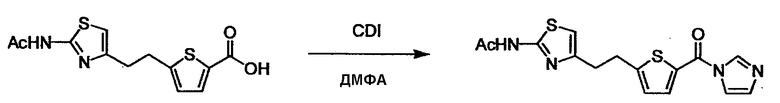
К раствору 5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-карбоновой кислоты (350,0 мг, 1,181 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли 1,1'-карбонилдиимидазол (287,2 мг, 1,771 ммоль) и смесь перемешивали при 50°C в течение 1 часа. После охлаждения до комнатной температуры добавляли по каплям диизопропиловый эфир (15 мл). После перемешивания в течение 5 минут осадок отделяли фильтрованием, промывали дважды диизопропиловым эфиром и 3 раза этилацетатом, сушили при пониженном давлении, получая N-(4-{2-[5-(имидазол-1-карбонил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (346,2 мг, 0,999 ммоль, выход 84,6%) в виде желтого твердого вещества.
Стадия 4
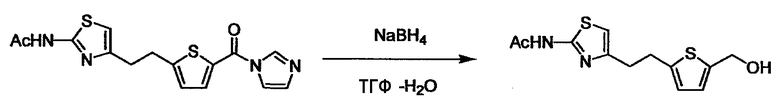
Раствор N-(4-{2-[5-(1H-имидазол-1-илкарбонил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (220,0 мг, 0,635 ммоль) в безводном тетрагидрофуране (17,6 мл) охлаждали до 0°C и добавляли воду (4,4 мл) и боргидрид натрия (240,2 мг, 6,350 ммоль). После перемешивания при 0°C в течение 1,5 часов реакционную смесь концентрировали приблизительно до 2 мл и добавляли по каплям насыщенный водный аммонийхлорид (20 мл). Реакционную смесь экстрагировали 3 раза этилацетатом, объединенные органические слои промывали водным аммонийхлоридом и насыщенным раствором соли. Смесь сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 100 г, этилацетат:гексан=2:1), получая N-(4-{2-[5-(гидроксиметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (157,1 мг, 0,556 ммоль, выход 87,6%) в виде белого твердого вещества.
Стадия 5
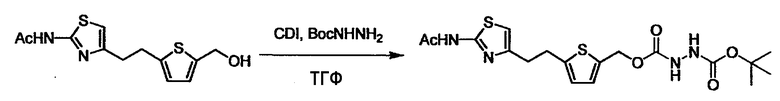
По аналогичной методике примера получения 4, стадия 6, (5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил-трет-бутил-гидразин-1,2-дикарбоксилат (158,2 мг, количественный) получали в виде белого твердого вещества из N-(4-{2-[5-(гидроксиметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (100,0 мг, 0,353 ммоль).
Стадия 6
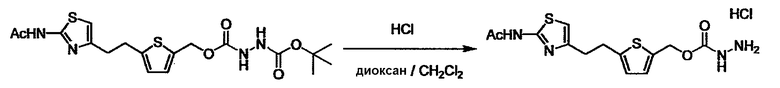
С (5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил-трет-бутил-гидразин-1,2-дикарбоксилата (0,353 ммоль) удаляли защитную группу по аналогичной методике примера получения 4, стадия 7, получая указанное в заголовке соединение (73,0 мг, 0,194 ммоль, выход 54,9%) в виде белого твердого вещества. Температура плавления 137-141°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,06 (1H, уш.с), 7,5-6,7 (4H, уш.с), 6,96 (1H, д, J=3,6 Гц), 6,77 (1H, с), 6,69 (1H, д, J=3,6 Гц), 4,93 (2H, с), 3,12 (2H, т, J=7,5 Гц), 2,90 (2H, т, J=7,5 Гц), 2,10 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,4, 157,7, 149,6, 146,2, 137,9, 128,4, 124,6, 108,1, 41,3, 32,9, 29,1, 22,6.
Пример получения 14
Гидрохлорид 2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил-гидразинкарбоксилата
Стадия 1
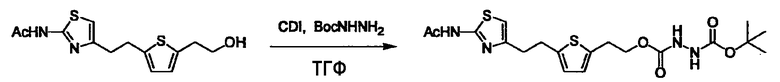
По аналогичной методике примера получения 2, стадия 1, 2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиазол-2-ил)этил-трет-бутил-гидразин-1,2-дикарбоксилат (571,6 мг, количественный выход) получали в виде белого твердого вещества из N-(4-{2-[5-(2-гидроксиэтил)тиазол-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (300,0 мг, 1,012 ммоль).
Стадия 2
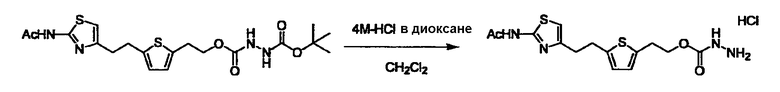
По аналогичной методике примера получения 2, стадия 2, указанное в заголовке соединение (263,4 мг, 0,674 ммоль, выход 66,6%) получали в виде белого твердого вещества из 2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиазол-2-ил)этил-трет-бутил-гидразин-1,2-дикарбоксилата (571,6 мг, соответствует 1,012 ммоль). Температура плавления: (не отчетливая температура плавления, разложение около 240°C).
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,06 (1H, уш.с), 9,06 (1H, уш.с), 7,23 (2H, уш.с), 6,77 (1H, с), 6,69 (2H, д, J=3,3 Гц), 6,65 (2H, д, J=3,3 Гц), 4,17 (2H, т, J=6,5 Гц), 3,15-2,95 (4H, м), 2,95-2,84 (2H, м), 2,10 (3H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,4, 157,7, 157,2, 149,9, 142,4, 137,7, 125,5, 124,5, 107,9, 65,2, 33,2, 29,4, 29,0, 22,7.
MS (ESI+): 355,0896 [M(свободный)+H]+, 377,0724 [M(свободный)+Na]+
Пример получения 15
Гидрохлорид 3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил-гидразинкарбоксилата
К раствору тиофен-3-карбальдегида (25,00 г, 222,9 ммоль) в толуоле (250 мл) добавляли этиленгликоль (69,18 г, 1115 ммоль) и п-толуолсульфокислоту (424,0 мг, 2,229 ммоль). Смесь кипятили с обратным холодильником в течение 4 часов, отделяя образующуюся воду, используя ловушку Дина-Старка. После охлаждения до 0°C добавляли насыщенный водный раствор гидрокарбоната натрия (50 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Водный слой экстрагировали дважды этилацетатом. Объединенные органические слои промывали водой и насыщенным раствором соли и сушили над безводным сульфатом натрия. Остаток концентрировали при пониженном давлении, фильтровали через слой силикагеля (FUJI SILYSIA CHEMICAL LTD. BW-300SP 100 г) и промывали смесью этилацетат/гексан (1:2, 1000 мл). Фильтрат концентрировали при пониженном давлении, получая 2-(тиофен-3-ил)-1,3-диоксолан (33,40 г, 213,8 ммоль, выход 95,9%) в виде желтого масла.
Стадия 2
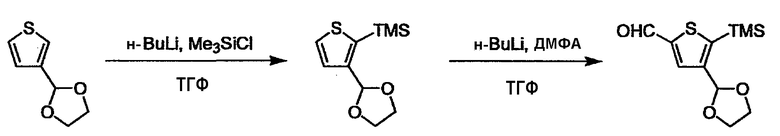
Раствор 2-(тиофен-3-ил)-1,3-диоксолана (10,00 г, 64,02 ммоль) в безводном тетрагидрофуране (50 мл) охлаждали на бане со смесью сухой лед-метанол и добавляли по каплям гексановый раствор н-бутиллития (1,59M, 44,3 мл, 70,4 ммоль), не допуская того, чтобы температура реакционной смеси превышала -55°C. После перемешивания в течение 1 часа добавляли по каплям хлортриметилсилан (8,94 мл, 70,4 ммоль), не допуская того, чтобы температура реакционной смеси превышала -60°C. После перемешивания при (-78)-(-60)°C в течение 30 минут, смесь нагревали до 0°C в течение 30 минут и перемешивали при 0°C в течение 1 часа. Реакционную смесь охлаждали снова на бане со смесью сухой лед-метанол, добавляли по каплям гексановый раствор н-бутиллития (1,59M, 46,3 мл, 73,6 ммоль), не допуская того, чтобы температура реакционной смеси превышала -60°C. После перемешивания при (-78)-(-60)°C в течение 1 часа добавляли по каплям N,N-диметилформамид (12,4 мл, 160 ммоль) и смесь дополнительно перемешивали в течение 1 часа. Смесь нагревали до 0°C в течение 1 часа и добавляли насыщенный водный раствор аммонийхлорида (100 мл). Смесь промывали 3 раза этилацетатом, объединенные органические слои промывали дважды 0,2M хлористоводородной кислотой и один раз насыщенным раствором соли. К органическому слою добавляли безводный сульфат магния и активированный уголь, смесь перемешивали в течение 5 минут и фильтровали. Фильтрат концентрировали при пониженном давлении, получая 4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-карбальдегид (16,09 г, 62,76 ммоль, выход 98,0%) в виде оранжевого масла.
Стадия 3
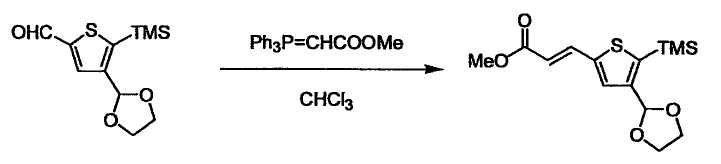
К раствору 4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-карбальдегида (12,82 г, 50,00 ммоль) в хлороформе (128 мл) добавляли небольшими порциями метил(трифенилфосфанилиден)ацетат (17,55 г, 52,50 ммоль) при 0°C. После перемешивания при 0°C в течение 1 часа смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к остатку диэтиловый эфир (200 мл) и смесь перемешивали. Нерастворимое вещество (в основном трифенилфосфиноксид) отфильтровывали и промывали 4 раза диэтиловым эфиром (50 мл). Фильтрат концентрировали при пониженном давлении и концентрированный остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 450 г, этилацетат:гексан=1:6), получая метил (2E)-3-[4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-ил]акрилат (13,76 г, 44,04 ммоль, выход 88,1%) в виде бледно-желтого масла.
Стадия 4
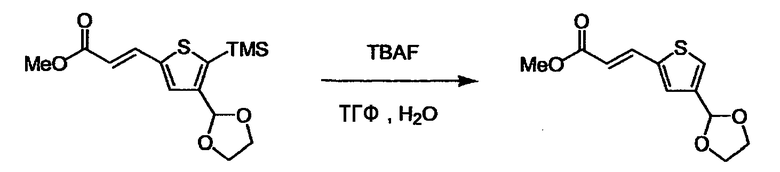
К раствору метил (2E)-3-[4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-ил]акрилата (4,000 г, 12,80 ммоль) в тетрагидрофуране (20 мл) добавляли воду (0,2 мл) и смесь охлаждали до 0°C. Добавляли тетрагидрофурановый раствор тетрабутиламмонийфторида (1M, 13,4 мл, 13,4 ммоль) и смесь перемешивали при 0°C в течение 5 минут и при комнатной температуре в течение 10 минут. Смесь охлаждали до 0°C, добавляли насыщенный водный раствор аммонийхлорида (40 мл) и смесь экстрагировали 3 раза этилацетатом. Объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 200 г, этилацетат:гексан=1:3), получая метил (2E)-3-[4-(1,3- диоксолан-2-ил)тиофен-2-ил]акрилат (3,065 г, 12,76 ммоль, 99,7%) в виде бесцветного масла.
Стадия 5
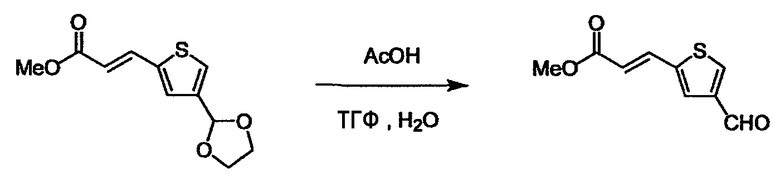
К раствору метил (2E)-3-[4-(1,3-диоксолан-2-ил)тиофен-2-ил]акрилата (3,050 г, 12,69 ммоль) в тетрагидрофуране (10 мл) добавляли уксусную кислоту (30 мл) и воду (10 мл) и смесь перемешивали при 50°C в течение 2 часа. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении и добавляли к остатку воду (30 мл). Смесь экстрагировали 3 раза этилацетатом, объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, концентрировали приблизительно до 50 мл при пониженном давлении и добавляли гексан (200 мл). Смесь концентрировали снова приблизительно до 50 мл при пониженном давлении, осадок отделяли фильтрованием, промывали 3 раза гексаном и сушили при пониженном давлении, получая метил (2E)-3-(4-формилтиофен-2-ил)акрилат (2,213 г, 11,28 ммоль, 88,9%) в виде белого твердого вещества.
Стадия 6
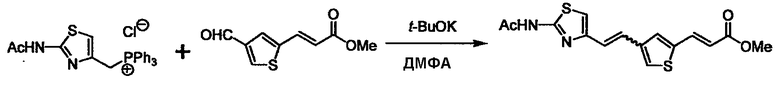
К {[2-(ацетиламино)-1,3-тиазол-4-ил]метил}(трифенил)фосфонийхлориду (6,602 г, 14,58 ммоль) добавляли безводный N,N-диметилформамид (30 мл) и смесь охлаждали до 0°C. Добавляли трет-бутоксид калия (3,146 г, 28,03 ммоль) и смесь перемешивали при 0°C в течение 15 минут. Добавляли по каплям раствор метил (2E)-3-(4-формилтиофен-2-ил)акрилата (2,200 г, 11,21 ммоль) в безводном N,N-диметилформамиде (30 мл) и смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь выливали в ледяную разбавленную хлористоводородную кислоту (600 мл, содержащая 15 ммоль HCl). Добавляли хлорид натрия (10 г) и смесь экстрагировали 3 раза этилацетатом. Объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 500 г, этилацетат:гексан=1:1), получая метил (2E)-3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}тиофен-2-ил)акрилат (3,090 г, 9,241 ммоль, 82,4%) в виде желтовато-белого твердого вещества.
Стадия 7
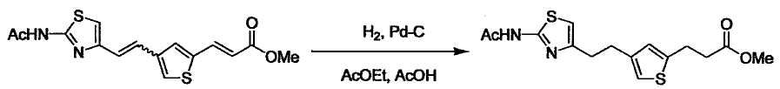
Метил (2E)-3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]винил}тиофен-2-ил)акрилат (3,000 г, 8,971 ммоль) растворяли в этилацетате (400 мл) и уксусной кислоте (100 мл). Добавляли 20% палладий на углероде и смесь гидрировали при комнатной температуре при атмосферном давлении. После завершения реакции катализатор отфильтровывали и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP, 100 г, этилацетат:гексан=2:3), получая метил 3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропионат (2,521 г, 7,449 ммоль, выход 83,0%) в виде бледно-желтого твердого вещества.
Стадия 8

К раствору метил 3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропионата (1,450 г, 4,284 ммоль) в безводном тетрагидрофуране (28 мл) добавляли по каплям 1,5M диизобутилалюмогидрид (10,7 мл, 16,1 ммоль) при -50°C в течение 30 минут. После перемешивания при (-50)-(-40)°C в течение 2 часов добавляли насыщенный водный раствор тартрата калия натрия (60 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 1,5 часов. Смесь экстрагировали 3 раза этилацетатом и промывали 1M хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным раствором соли. После сушки над безводным сульфатом натрия, смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 100 г, этилацетат:гексан=2:1), получая N-(4-{2-[5-(3-гидроксипропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамид (499,9 мг, 1,610 ммоль, выход 37,6%) в виде белого твердого вещества.
Стадия 9
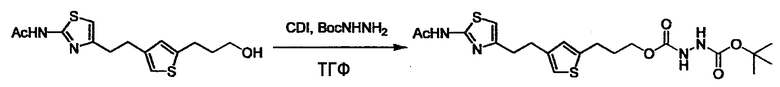
По аналогичной методике примера получения 2, стадия 1, 3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил-трет-бутил-гидразин-1,2-дикарбоксилат (362,9 мг, количественный) получали в виде бесцветного масла из N-(4-{2-[5-(3-гидроксипропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамида (205,0 мг, 0,660 ммоль).
Стадия 10
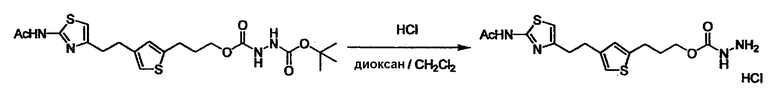
По аналогичной методике примера получения 2, стадия 2, указанное в заголовке соединение (207,9 мг, 0,513 ммоль, выход 77,7%) получали в виде белого твердого вещества из 3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил-трет-бутил-гидразин-1,2-дикарбоксилата (0,660 ммоль). Температура плавления 150-153°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,06 (1H, уш.с), 11,0-9,8 (4H, уш.с), 6,92 (1H, с), 6,73 (1H, с), 6,72 (1H, с), 4,12 (2H, т, J=5,5 Гц), 2,90-2,80 (4H, м), 2,80 (2H, т, J=7,6 Гц), 2,11 (3H, с), 1,90 (2H, тт, J=7,6, 5,5 Гц).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,7, 155,9, 150,3, 143,5, 141,6, 126,2, 118,5, 107,5, 65,0, 31,9, 30,5, 29,5, 25,7, 22,7.
МС(ESI+): 369,1006 [M(свободный)+H]+, 391,0827 [M(свободный)+Na]+.
Пример получения 16
Гидрохлорид 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил-гидразинкарбоксилата
Стадия 1
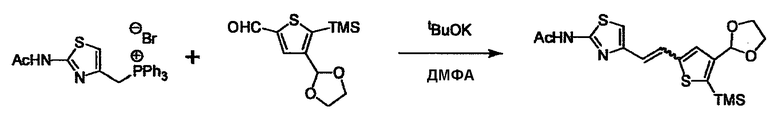
К раствору {[2-(ацетиламино)-1,3-тиазол-4-ил]метил}(трифенил)фосфонийбромида (7,970 г, 16,03 ммоль) в безводном N,N-диметилформамиде (32 мл) добавляли трет-бутоксид калия (3,687 г, 32,86 ммоль) при 0°C и смесь перемешивали в течение 1 часа. Добавляли по каплям раствор 4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-карбальдегида (2,740 г, 10,69 ммоль) в безводном N,N-диметилформамиде (6 мл) и смесь перемешивали в течение 1 часа. Добавляли 1M хлористоводородную кислоту (17,0 мл) и ледяную воду (170 мл) и смесь экстрагировали 3 раза этилацетатом. Объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 200 г, этилацетат:дихлорметан=1:6), получая N-(4-{2-[4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-ил]винил}-1,3-тиазол-2-ил)ацетамид (4,010 г, 10,16 ммоль, выход 95,0%) в виде не совсем белого твердого вещества.
Стадия 2
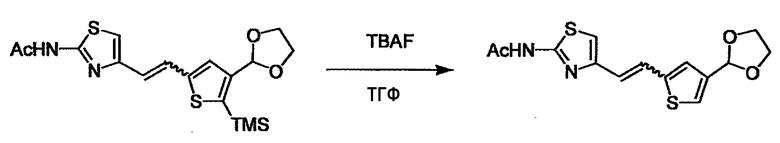
К раствору N-(4-{2-[4-(1,3-диоксолан-2-ил)-5-(триметилсилил)тиофен-2-ил]винил}-1,3-тиазол-2-ил)ацетамида (3,980 г, 10,09 ммоль) в тетрагидрофуране (20 мл) добавляли 1M тетрагидрофурановый раствор тетрабутиламмонийфторида (11,1 мл, 11,1 ммоль) при комнатной температуре и смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (Merck 7734 100 г, этилацетат:дихлорметан=1:4→1:2), получая N-(4-{2-[4-(1,3-диоксолан-2-ил)тиофен-2-ил]винил}-1,3-тиазол-2-ил)ацетамид (3,03 г, 9,40 ммоль, выход 93,2%) в виде не совсем белого твердого вещества.
Стадия 3
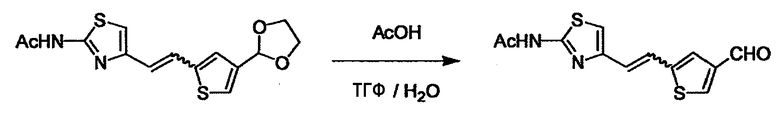
К N-(4-{2-[4-(1,3-диоксолан-2-ил)тиофен-2-ил]винил}-1,3-тиазол-2-ил)ацетамиду (1,418 г, 4,398 ммоль) добавляли уксусную кислоту (16,8 мл), тетрагидрофуран (5,6 мл) и воду (5,6 мл) и смесь перемешивали при 55°C в течение 2,5 часов. Реакционную смесь концентрировали и добавляли диизопропиловый эфир. Выпавший осадок отделяли фильтрованием, промывали диизопропиловым эфиром и сушили при пониженном давлении, получая N-{4-[2-(4-формилтиофен-2-ил)винил]-1,3-тиазол-2-ил}ацетамид (1,136 г, 4,081 ммоль, выход 92,8%) в виде бледно-желтого твердого вещества.
Стадия 4
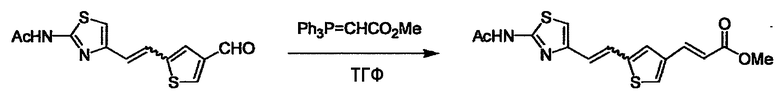
К раствору N-{4-[2-(4-формилтиофен-2-ил)винил]-1,3-тиазол-2-ил}ацетамида (1,133 г, 4,070 ммоль) в безводном тетрагидрофуране (80 мл) добавляли метил(трифенилфосфанилиден)ацетат (1,497 г, 4,477 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. FL60D 60 г, этилацетат:дихлорметан=1:5), получая метил (2E)-3-(5-{2-[2-(ацетиламино)-1,3-тиазол-2-ил]винил}тиофен-3-ил)-2-пропеноат (2,320 г, содержащий трифенилфосфиноксид) в виде бледно-желтого твердого вещества.
Стадия 5
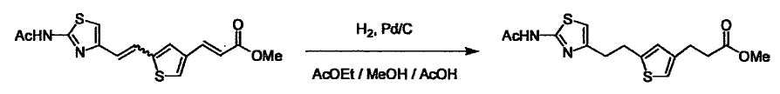
Метил (2E)-3-(5-{2-[2-(ацетиламино)-1,3-тиазол-2-ил]винил}тиофен-3-ил)-2-пропеноат (соответствует 4,070 ммоль) растворяли в смеси этилацетата (120 мл), метанола (40 мл) и уксусной кислоты (40 мл). Добавляли 10% палладий на углероде (700 мг, содержащий 50% воды) и смесь гидрировали при комнатной температуре при атмосферном давлении. После завершения реакции реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 60 г, этилацетат:дихлорметан=1:7), и неочищенные после выделения фракции снова очищали колоночной хроматографией, получая метил 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-2-ил]этил}тиофен-3-ил)пропионат (917,4 мг, 2,711 ммоль, суммарный выход на стадию 4 66,6%) в виде не совсем белого твердого вещества.
Стадия 6

Метил 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-2-ил]этил}тиофен-3-ил)пропионат (845,0 мг, 2,497 ммоль) восстанавливали по аналогичной методике примера получения 15, стадия 8, получая N-(4-{2-[4-(3-гидроксипропил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (555,1 мг, 1,788 ммоль, выход 71,6%) в виде не совсем белого твердого вещества.
Стадия 7
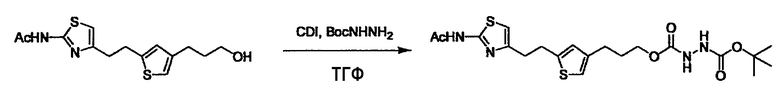
По аналогичной методике примера получения 2, стадия 1, 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил-трет-бутил-гидразин-1,2-дикарбоксилат (277,4 мг, 0,592 ммоль, выход 99,5%) получали в виде бледно-желтого твердого вещества из N-(4-{2-[4-(3-гидроксипропил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (184,6 мг, 0,595 ммоль).
Стадия 8
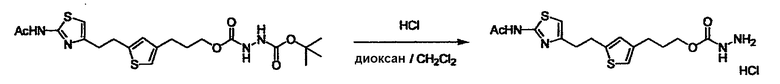
По аналогичной методике примера получения 2, стадия 2, указанное в заголовке соединение (238,4 мг, 0,589 ммоль, выход 99,7%) получали в виде белого твердого вещества из 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил-трет-бутил-гидразин-1,2-дикарбоксилата (277,0 мг, 0,591 ммоль). Температура плавления 98-102°C
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,07 (1H, уш.с), 10,6-9,6 (3H, уш.с), 6,90 (1H, с), 6,76 (1H, с), 6,71 (1H, с), 4,09 (2H, т, J=6,6 Гц), 3,09 (2H, т, J=7,3 Гц), 2,88 (2H, т, J=7,3 Гц), 2,55 (2H, т, J=6,6 Гц), 2,10 (3H, с), 1,85 (2H, кв, J=6,6 Гц).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,4, 158,03, 157,79, 149,8, 144,1, 141,0, 126,0, 118,6, 108,0, 65,4, 33,17, 33,15, 29,1, 26,1, 22,7.
МС(ESI+): 369,1047 [M(свободный)+H]+, 391,0864 [M(свободный)+Na]+.
Пример получения 17
Гидрохлорид 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил-гидразинкарбоксилата
Стадия 1
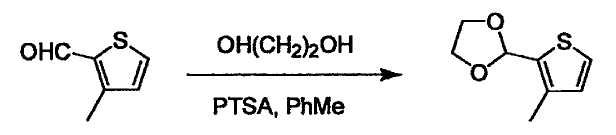
К раствору 3-метилтиофен-2-карбальдегида (3,53 г, 25 28,0 ммоль) в толуоле (75 мл) добавляли этиленгликоль (31,2 мл, 560 ммоль) и каталитическое количество пара-толуолсульфокислоты. Смесь кипятили с обратным холодильником в течение 17 часов, удаляя образующуюся воду, используя ловушку Дина-Старка. Реакционную смесь охлаждали до комнатной температуры, промывали насыщенным водным гидрокарбонатом натрия и насыщенным раствором соли, и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (Merck 7734 80 г, этилацетат:гексан=1:10), получая 2-(3-метилтиофен-2-ил)-1,3-диоксолан (3,93 г, 23,1 ммоль, выход 82,5%) в виде бледно-желтого масла.
Стадия 2
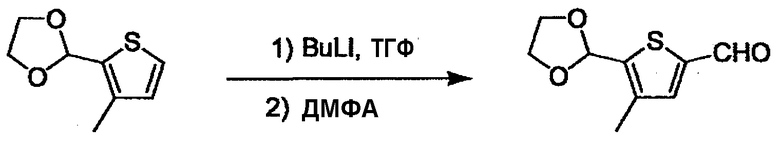
К раствору 2-(3-метилтиофен-2-ил)-1,3-диоксолана (3,198 г, 18,79 ммоль) в безводном тетрагидрофуране (30 мл) добавляли по каплям 1,55M гексановый раствор бутиллития (12,1 мл, 18,8 ммоль) при -78°C. После перемешивания в течение 30 минут добавляли по каплям безводный N,N-диметилформамид (4,36 мл, 56,3 ммоль). После перемешивания при -78°C в течение 1 часа реакционную смесь нагревали, добавляли насыщенный водный аммонийхлорид и смесь экстрагировали дважды этилацетатом. Объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 80 г, этилацетат:гексан=1:5), получая 5-(1,3-диоксолан-2-ил)-4-метилтиофен-2-карбальдегид (3,309 г, 16,69 ммоль, выход 88,8%) в виде коричневого масла.
Стадия 3
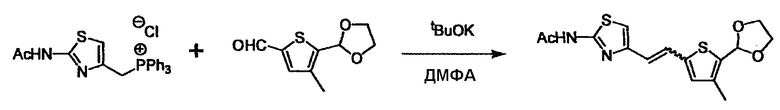
По аналогичной методике примера получения 16, стадия 1, {[2-(ацетиламино)-1,3-тиазол-4-ил]метил}(трифенил)фосфонийхлорид (12,03 г, 26,56 ммоль) и 5-(1,3-диоксолан-2-ил)-4-метилтиофен-2-карбальдегид (3,51 г, 17,7 ммоль) конденсировали, получая N-(4-{2-[5-(1,3-диоксолан-2-ил)-4-метилтиофен-2-ил]винил}-1,3-тиазол-2-ил)ацетамид (4,450 г, 13,23 ммоль, выход 74,7%) в виде желтого твердого вещества.
Стадия 4
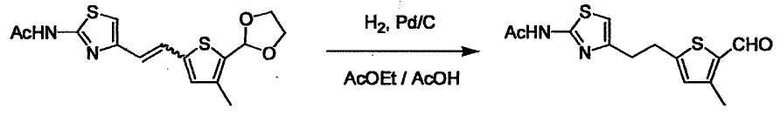
N-(4-{2-[5-(1,3-Диоксолан-2-ил)-4-метилтиофен-2-ил]винил}-1,3-тиазол-2-ил)ацетамид (4,42 г, 13,1 ммоль) растворяли в смеси этилацетата (400 мл) и уксусной кислоты (100 мл) и добавляли 10% палладий на углероде (2,21 г, содержащий 50% воды). Смесь гидрировали при комнатной температуре при атмосферном давлении. Реакционную смесь фильтровали через целит и фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 200 г, этилацетат:дихлорметан=1:6), получая N-{4-[2-(5-формил-4-метилтиофен-2-ил)этил]-1,3-тиазол-2-ил}ацетамид (2,26 г, 7,68 ммоль, выход 58,4%) в виде желтого твердого вещества.
Стадия 5
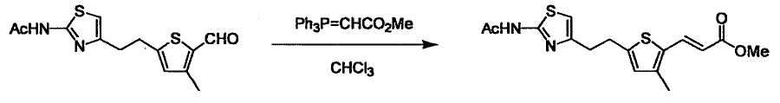
К раствору N-{4-[2-(5-формил-4-метилтиофен-2-ил)этил]-1,3-тиазол-2-ил}ацетамида (468,0 мг, 1,590 ммоль) в хлороформе (4,7 мл) добавляли по каплям раствор метил(трифенилфосфоранилиден)ацетата (797,4 мг, 2,385 ммоль) в хлороформе (2 мл) при 0°C. Смесь нагревали до комнатной температуры и перемешивали в течение 2 часа. Реакционную смесь концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (Merck 9385 60 г, этилацетат:дихлорметан=1:8), получая метил (2E)-3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)-2-пропеноат (591,6 мг, содержащий трифенилфосфиноксид) в виде бледно-желтого твердого вещества.
Стадия 6
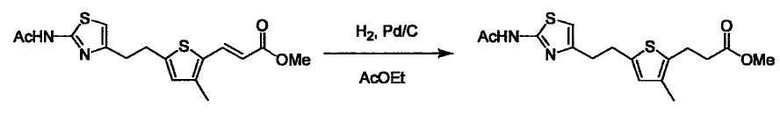
К раствору метил (2E)-3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)-2-пропеноат (соответствует 1,590 ммоль) в этилацетате (30 мл) добавляли 10% палладий на углероде (296 мг, содержащий 50% воды). Смесь гидрировали при комнатной температуре при атмосферном давлении. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении, получая метил 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)-2-пропионат (376,9 мг, 1,069 ммоль, суммарный выход на стадию 5, 67,2%).
Стадия 7
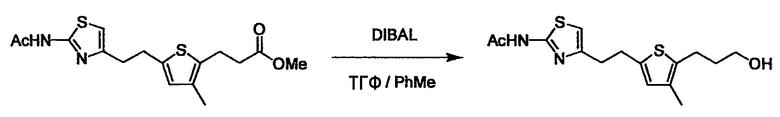
Метил 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)-2-пропионат (214,7 мг, 0,609 ммоль) восстанавливали по аналогичной методике примера получения 15, стадия 8, получая N-(4-{2-[5-(3-гидроксипропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (105,9 мг, 0,326 ммоль, выход 53,6%) в виде белого твердого вещества.
Стадия 8
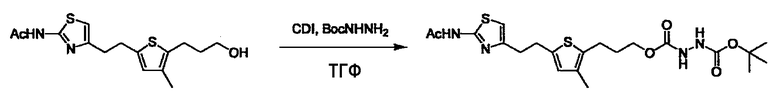
По аналогичной методике примера получения 15, стадия 9, 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил-трет-бутил-гидразин-1,2-дикарбоксилат (155,7 мг, 0,323 ммоль, выход 90,7%) получали в виде белого твердого вещества из N-(4-{2-[5-(3-гидроксипропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (115,6 мг, 0,356 ммоль).
Стадия 9
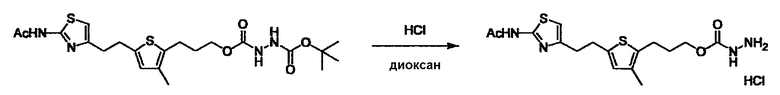
С 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил-трет-бутил-гидразин-1,2-дикарбоксилата (155,7 мг, 0,323 ммоль) удаляли защитную группу по аналогичной методике примера получения 15, стадия 10, получая указанное в заголовке соединение (112,3 мг, 0,258 ммоль, выход 79,9%) в виде бледно-желтого твердого вещества, температура плавления 111-115°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,05 (1H, уш.с), 10,8-9,8 (4H, уш.с), 6,75 (1H, с), 6,53 (1H, с), 4,09 (2H, т, J=6,4 Гц), 3,00 (2H, т, J=7,6 Гц), 2,84 (2H, т, J=7,6 Гц), 2,67 (2H, т, J=7,5 Гц), 2,10 (3H, с), 2,01 (3H, с), 1,80 (2H, тт, J=7,5, 7,6 Гц).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,2, 157,5, 155,8, 149,7, 139,6, 134,0, 132,2, 127,5, 107,7, 64,9, 32,9, 30,2, 28,8, 23,3, 22,5, 13,3.
МС(ESI+): 383,1227 [M(свободный)+H]+, 405,1058 [M(свободный)+Na]+.
Пример получения 18
Гидрохлорид N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамида
Стадия 1
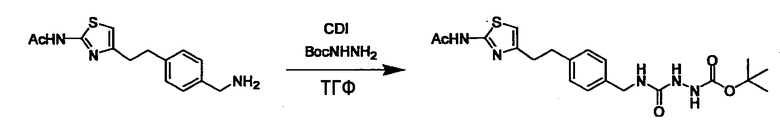
К суспензии 1,1'-карбонилдиимидазола (332,1 мг, 2,048 ммоль) в безводном тетрагидрофуране (1,3 мл) добавляли трет-бутилкарбазат (270,7 мг, 2,048 ммоль). После перемешивания при комнатной температуре в течение 15 минут добавляли N-(4-{2-[4-(аминометил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (282,6 мг, 1,024 ммоль), смесь перемешивали при комнатной температуре в течение 6 часов и концентрировали при пониженном давлении. Добавляли к остатку этилацетат и воду, смесь перемешивали, оставляли без перемешивания и затем распределяли. Водный слой экстрагировали этилацетатом, объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 24 г, дихлорметан:метанол=30:1→20:1). Остаток дополнительно очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. DM-2035 16 г, дихлорметан:метанол=30:1), получая трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)карбамоил]гидразинкарбоксилат (348,3 мг, 0,803 ммоль, выход 78,5%) в виде белого твердого вещества.
Стадия 2
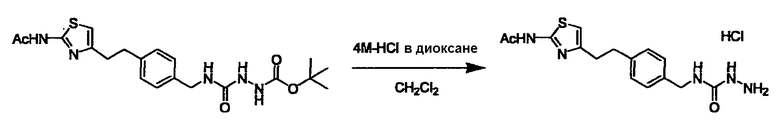
К суспензии трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)карбамоил]гидразинкарбоксилата (281,8 мг, 0,65 ммоль) в безводном дихлорметане (3,25 мл) добавляли 4M диоксановый раствор хлористого водорода (3,25 мл, 13,0 ммоль). После перемешивания при комнатной температуре в течение 1 часа, реакционную смесь концентрировали при пониженном давлении. Добавляли к остатку этилацетат и смесь снова концентрировали при пониженном давлении. Операцию осуществляли 3 раза для удаления хлористоводородного газа азеотропно. Остаток суспендировали в этилацетате и фильтровали. Отфильтрованный продукт промывали этилацетатом, сушили при пониженном давлении, получая указанное в заголовке соединение (245,1 мг, количественный выход) в виде белого твердого вещества. Температура плавления 167-169°C.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,08 (1H, уш.с), 9,92 (3H, уш.с), 8,87 (1H, уш.с), 7,53 (1H, т, J=5,7 Гц), 7,22-7,04 (4H, м), 6,71 (1H, с), 4,22 (2H, д, J=5,7 Гц), 2,98-2,75 (4H, м), 2,10(3H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,7, 157,5, 150,3, 140,2, 137,3, 128,4, 127,3, 107,6, 42,8, 34,3, 33,0, 22,7.
МС(ESI+): 334,1316 [M(свободный)+H]+, 356,1129 [M(свободный)+Na]+.
Пример получения 19
Гидрохлорид N-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамида
Стадия 1
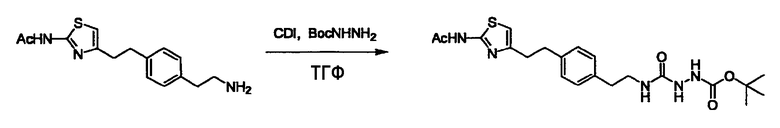
По аналогичной методике примера получения 3, стадия 1, трет-бутил 2-{[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]карбамоил}гидразинкарбоксилат (674,7 мг, 1,508 ммоль, выход 88,7%) получали в виде белого твердого вещества из N-(4-{2-[4-(2-аминоэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (492,0 мг, 1,700 ммоль).
Стадия 2
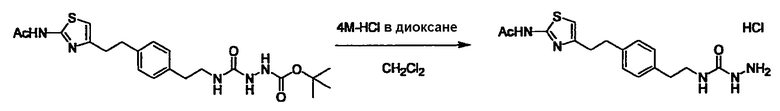
По аналогичной методике примера получения 3, стадия 2, указанное в заголовке соединение (422,1 мг, 1,100 ммоль, выход 95,8%) получали в виде белого твердого вещества из трет-бутил 2-{[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]карбамоил}гидразинкарбоксилата (513,8 мг, 1,148 ммоль). Температура плавления 171-173°C.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,12 (1H, уш.с), 10,00 (3H, уш.с), 8,82 (1H, уш.с), 7,30-6,96 (4H, м), 6,73(1H, с), 3,39-3,18 (2H, м), 2,97-2,77(4H, м), 2,66(2H, т, J=7,2 Гц), 2,10(2H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,7, 157,3, 150,3, 139,4, 136,8, 128,8, 128,5, 107,6, 35,4, 34,4, 32,9, 22,7.
MS (ESI+): 348,1490 [M(свободный)+H]+, 370,1307 [M(свободный)+Na]+.
Пример получения 20
Гидрохлорид N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)гидразинкарбоксамида
Стадия 1
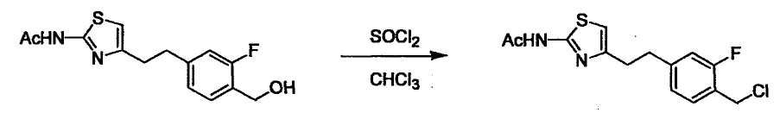
К раствору N-(4-{2-[3-фтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (196,6 мг, 0,668 ммоль) в хлороформе (10 мл) добавляли тионилхлорид (0,144 мл, 2,00 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли тионилхлорид (0,077 мл, 1,00 ммоль), смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали. Добавляли к остатку этилацетат (10 мл) и смесь снова концентрировали при пониженном давлении. Операцию повторяли 3 раза для удаления тионилхлорида азеотропно. Добавляли к остатку смесь этилацетата (1 мл) и диизопропилового эфира (10 мл), осадок отделяли фильтрованием и сушили при пониженном давлении, получая N-(4-{2-[4-(хлорметил)-3-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (205,3 мг, 0,656 ммоль, выход 98,3%) в виде слегка желтоватого твердого вещества.
Стадия 2
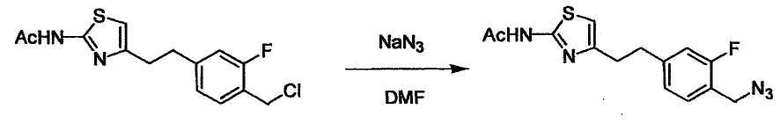
К раствору N-(4-{2-[4-(хлорметил)-3-фторфенил]этил}-1,3-тиазол-2-ил)ацетамида (203,3 мг, 0,650 ммоль) в безводном N,N-диметилформамиде (2 мл) добавляли азид натрия (233,7 мг, 3,595 ммоль) и смесь перемешивали при комнатной температуре в течение 3,5 часов. Добавляли к реакционной смеси воду (20 мл) и этилацетат (20 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Водный слой экстрагировали этилацетатом, объединенные органические слои промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Добавляли к остатку диизопропиловый эфир (10 мл), смесь фильтровали и сушили при пониженном давлении, получая N-(4-{2-[4-(азидометил)-3-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (160,5 мг, 0,503 ммоль, выход 77,3%) в виде белого твердого вещества.
Стадия 3
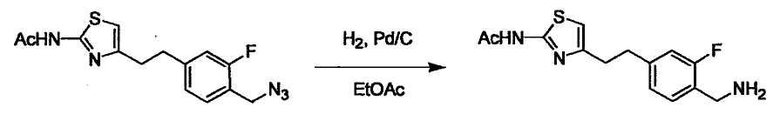
К раствору N-(4-{2-[4-(азидометил)-3-фторфенил]этил}-1,3-тиазол-2-ил)ацетамида (152,3 мг, 0,477 ммоль) в этилацетате (21 мл) добавляли 10% палладий на углероде (30,5 мг, содержащий 50% воды) и смесь гидрировали при комнатной температуре при атмосферном давлении. Добавляли к реакционной смеси этилацетат (5 мл) и метанол (5 мл) и смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении, полученный в результате твердый остаток отделяли фильтрованием, промывали трет-бутилметиловым эфиром (5 мл) и сушили при пониженном давлении, получая N-(4-{2-[4-(аминометил)-3-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (119,3 мг, 0,407 ммоль, выход 85,3%) в виде белого твердого вещества.
Стадия 4
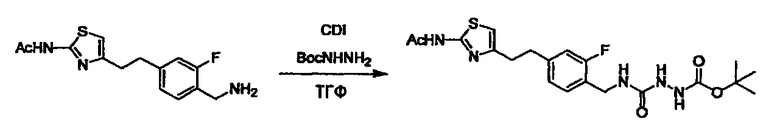
К суспензии 1,1'-карбонилдиимидазола (129,7 мг, 0,800 ммоль) в безводном тетрагидрофуране (0,8 мл) добавляли трет-бутилкарбазат (105,7 мг, 0,800 ммоль) и смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли N-(4-{2-[4-(аминометил)-3-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (114, 2 мг, 0,389 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часа. Добавляли к реакционной смеси этилацетат (15 мл), воду (12 мл) и 1М хлористоводородную кислоту (3 мл) и смесь перемешивали. Смесь оставляли без перемешивания, распределяли и органический слой промывали водой (15 мл) и насыщенным раствором соли (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток суспендировали в смеси этилацетата (5 мл) и гексана (5 мл), суспензию фильтровали и сушили при пониженном давлении, получая трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)карбамоил]гидразинкарбоксилат (150,0 мг, 0,332 ммоль, выход 85,3%) в виде белого твердого вещества. Стадия 5
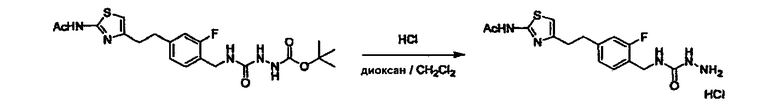
К суспензии трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)карбамоил]гидразинкарбоксилата (141,6 мг, 0,314 ммоль) в безводном дихлорметане (2 мл) добавляли 4М диоксановый раствор хлористого водорода (2 мл). Смесь перемешивали при комнатной температуре в течение 2 часа и концентрировали при пониженном давлении. Добавляли к остатку этилацетат (10 мл) и смесь концентрировали снова при пониженном давлении. Данную операцию повторяли 3 раза для удаления хлористоводородного газа азеотропно. Остаток суспендировали в смеси этанола (2 мл) и этилацетата (4 мл), фильтровали, промывали дважды этилацетатом и сушили при пониженном давлении, получая гидрохлорид N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)гидразинкарбоксамида (123,6 мг, 0,319 ммоль, количественный выход) в виде белого твердого вещества.
Температура плавления 172-175°C.
1H-ЯМР (400 МГц, ДМСО-d6): 5 (м.д.): 12,08 (1Н, уш.с), 9,96 (3H, уш.с), 8,91 (1Н, уш.с), 7,53 (1H, т, J=5,7 Гц), 7,23 (1H, т, J=8,0 Гц), 7,03-6,99 (2H, м), 6,74 (1H, с), 4,27 (2H, д, J=5,7 Гц), 2,96-2,85 (4Н, м), 2,12 (3H, с).
13C-ЯМР (100 МГц, ДMCО-d6): 6 (м.д.): 168, 2, 159, 7 (д, J=243,9 Гц), 157,4, 157,1, 149,8, 142,9 (д, J=7,5 Гц), 129,1 (д, J=4,5 Гц), 124,0 (д, J=3,0 Гц), 123,4 (д, J=14,9 Гц), 114,7 (д, J=20,9 Гц), 107,4,36,6(д, J=3,8 Гц),33,7, 32,2, 22,4.
19F-ЯМР (376 Гц, ДМСО-d6): δ (м.д.): -121,0.
МС(ESI+): 352,1183 [M(свободный)+H]+, 374,1003 [M(свободный)+Na]+.
Пример получения 21
Гидрохлорид N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил)гидразинкарбоксамида
Стадия 1
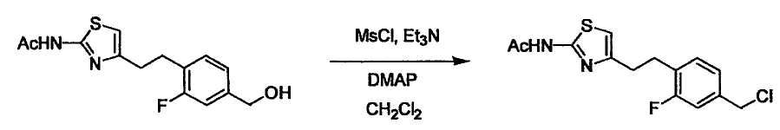
К раствору N-(4-{2-[2-фтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (194,7 мг, 0,665 ммоль) в безводном дихлорметане (2 мл) добавляли триэтиламин (0,17 мл, 1,20 ммоль) и 4-диметиламинопиридин (8,1 мг, 0,066 ммоль) и смесь охлаждали до 0°C. Добавляли по каплям метансульфонилхлорид (77 мкл, 1,0 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь охлаждали до 0°C, добавляли триэтиламин (93 мкл, 0,67 ммоль) и метансульфонилхлорид (51 мкл, 0,67 ммоль) и смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли к реакционной смеси ледяную воду (2 мл), смесь оставляли без перемешивания и распределяли. Водный слой экстрагировали 3 раза дихлорметаном, объединенные органические слои промывали 1M хлористоводородной кислотой и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали колоночной хроматографией на силикагеле (Merck 9385 16 г, этилацетат:гексан=2:3), получая N-(4-{2-[4-(хлорметил)-2-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (59,6 мг, 0,191 ммоль, выход 28,7%) в виде бледно-желтого твердого вещества.
Стадия 2
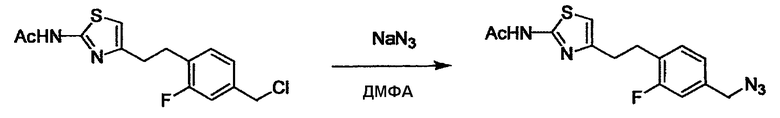
N-(4-{2-[4-(Хлорметил)-2-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (59,1 мг, 0,189 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[4-(азидометил)-2-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (54,8 мг, 0,172 ммоль, выход 91,0%) в виде бледно-желтого твердого вещества.
Стадия 3
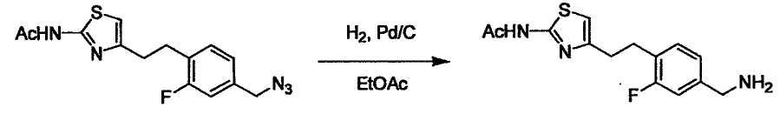
N-(4-{2-[4-(Азидометил)-2-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (54,0 мг, 0,169 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[4-(аминометил)-2-фторфенил]этил}-1,3-тиазол-2-ил)ацетамид (48,8 мг, 0,166 ммоль, выход 98,2%) в виде белого твердого вещества.
Стадия 4
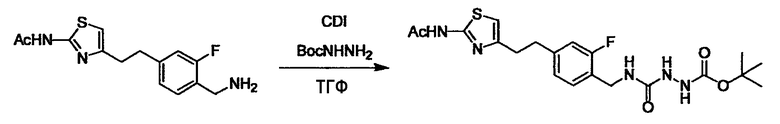
По аналогичной методике примера получения 20, стадия 3, трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил)карбамоил]гидразинкарбоксилат (28,7 мг, 0,064 ммоль, выход 38,3%) получали в виде бледно-желтого твердого вещества из N-(4-{2-[4-(аминометил)-2-фторфенил]этил}-1,3-тиазол-2-ил)ацетамида (53,1 мг, 0,327 ммоль).
Стадия 5
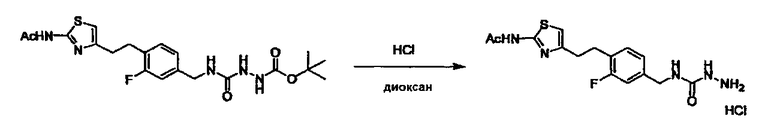
К раствору трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил)карбамоил]гидразинкарбоксилата (12,3 мг, 0,027 ммоль) в диоксане (1,2 мл) добавляли 4М диоксановый раствор хлористого водорода (0,1 мл), смесь перемешивали при комнатной температуре в течение 17 часов и концентрировали при пониженном давлении. Остаток суспендировали в этилацетате (2 мл) и смесь фильтровали. Отфильтрованный продукт промывали 3 раза этилацетатом, и сушили при пониженном давлении, получая указанное в заголовке соединение (10,2 мг, 0,026 ммоль, выход 96,7%) в виде белого твердого вещества. Температура плавления 165-169°C.
1H-ЯМР (400 МГц, ДМСО-d6): 5 (м.д.): 12,07 (1H, уш.с), 10,5-9,0 (3H, уш.с), 8,83 (1Н, уш.с), 7,57(1Н, т, J=2,0 Гц), 7,27-7,14 (1Н, м), 7, 03-6,98 (2Н, м), 6,73 (1Н, с), 4,23 (2Н, д, J=6,0 Гц), 2,92-2,81 (4Н, м), 2,10 (3H, с).
MS (ESI+): 352,1210 [M(свободный)+H]+, 374,1038 [M(свободный)+Na]+.
Пример получения 22
Гидрохлорид N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил)гидразинкарбоксамида
Стадия 1
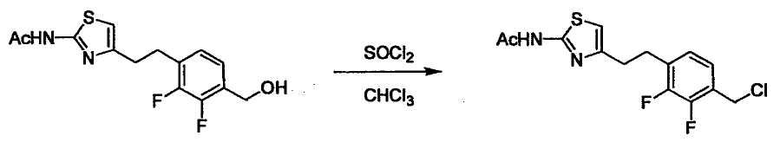
N-(4-{2-[2,3-Дифтор-4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (193,2 мг, 0,619 ммоль) хлорировали по аналогичной методике примера получения 20, стадия 1, получая N-(4-{2-[4-(хлорметил)-2,3-дифторфенил]этил}-1,3-тиазол-2-ил)ацетамид (212,7 мг) в виде белого твердого вещества.
Стадия 2
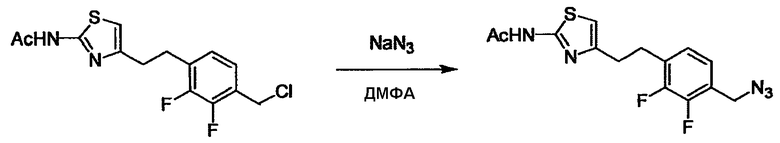
N-(4-{2-[4-(Хлорметил)-2,3-дифторфенил]этил}-1,3-тиазол-2-ил)ацетамид (201,9 мг, 0,610 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[4-(азидометил)-2,3-дифторфенил]этил}-1,3-тиазол-2-ил)ацетамид (180,3 мг, 0,535 ммоль, выход 87,6%) в виде белого твердого вещества.
Стадия 3
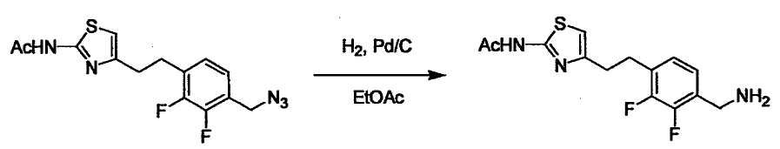
N-(4-{2-[4-(Азидометил)-2,3-дифторфенил]этил}-1,3-тиазол-2-ил)ацетамид (178,0 мг, 0,528 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[4-(аминометил)-2,3-дифторфенил]этил}-1,3-тиазол-2-ил)ацетамид (132,1 мг, 0,424 ммоль, выход 80,4%) в виде белого твердого вещества.
Стадия 4
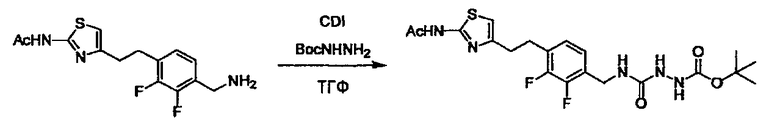
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил)карбамоил]гидразинкарбоксилат (135,2 мг, 0,288 ммоль, выход 7 3,7%) получали в виде белого твердого вещества из N-(4-{2-[4-(аминометил)-2,3-дифторфенил]этил}-1,3-тиазол-2-ил)ацетамида (126,7 мг, 0,781 ммоль).
Стадия 5
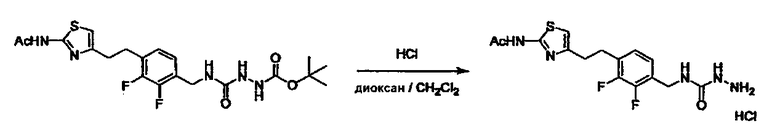
С трет-бутил 2-[(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил)карбамоил]гидразинкарбоксилата (130,2 мг, 0,27 7 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (101,4 мг, 0,250 ммоль, выход 90,1%) в виде белого твердого вещества. Температура плавления 163-166°C.
1H-ЯМР (400 МГц, ДMCО-d6): 5 (м.д.): 12,10 (1H, уш.с), 9,95 (3H, уш.с), 8,96 (1H, уш.с), 7,61 (1H, т, J=6,0 Гц), 7,23 (1H, т, J=8,0 Гц), 7,08-7,03 (2H, м), 6,76 (1H, с), 4,31 (2H, т, J=5,9 Гц), 2,96-2,85 (4H, м), 2,13 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,2, 157,5, 157,1, 149,5, 147,9 (дд, J=243,1, 10,5 Гц), 147,5 (дд, J=240,1, 7,5 Гц), 128,9 (д, J=12,7 Гц), 126,3 (д, J=11,2 Гц), 124,8, 123,6, 107,6, 36,5, 31,1, 27,4, 22,4.
19F-ЯМР (376 Гц, ДМСО-d6): δ (м.д.): -146,2 (IF, д, JFF=19,1 Гц), -146,7 (IF, д, JFF=19,1 Гц).
МС(ESI+): 370,1101 [M(свободный)+H]+, 392,0915 [M(свободный)+Na]+.
Пример получения 23
Дигидрохлорид N-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метил}амино)бензил]гидразинкарбоксамида
Стадия 1
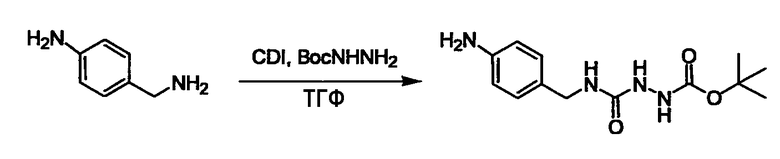
К суспензии 1,1'-карбонилдиимидазола (1,783 г, 11,00 ммоль) в безводном тетрагидрофуране (7 мл) добавляли трет-бутилкарбазат (1,322 г, 10,00 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли раствор 4-(аминометил)анилина (1,228 г, 10,05 ммоль) в безводном тетрагидрофуране (3 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель упаривали при пониженном давлении, добавляли к остатку этилацетат (50 мл) и воду (50 мл). Смесь перемешивали, оставляли без перемешивания и распределяли. Водный слой экстрагировали дважды этилацетатом, объединенные органические слои сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрированный остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 90 г, этилацетат:гексан=7:3→8:2→9:1), получая трет-бутил 2-[(4-аминобензил)карбамоил]гидразинкарбоксилат (2,180 г, 7,778 ммоль, выход 77,8%) в виде белого твердого вещества.
Стадия 2
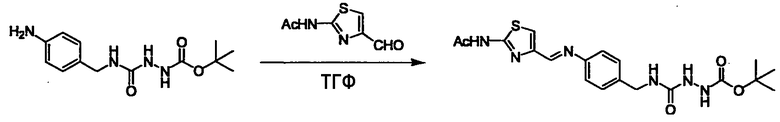
К раствору трет-бутил 2-[(4-аминобензил)карбамоил]гидразинкарбоксилата (700,7 мг, 2,500 ммоль) в безводном тетрагидрофуране (10 мл) добавляли N-(4-формил-1,3-тиазол-2-ил)ацетамид (425,5 мг, 2,500 ммоль). Смесь кипятили с обратным холодильником в течение 1 часа, охлаждали до комнатной температуры и концентрировали при пониженном давлении. Добавляли к остатку тетрагидрофуран (5 мл) и трет-бутилметиловый эфир (10 мл) и смесь перемешивали. Полученный в результате твердый остаток отделяли фильтрованием, промывали дважды трет-бутилметиловым эфиром и сушили при пониженном давлении, получая трет-бутил 2-{[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метилиден}амино)бензоил]карбамоил}гидразинкарбоксилат (1,097 г, количественный) в виде белого твердого вещества.
Стадия 3
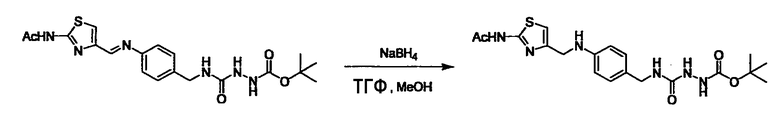
трет-Бутил 2-{[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метилиден}амино)бензоил]карбамоил}гидразинкарбоксилат (778,5 мг, 1,800 ммоль) растворяли в смеси безводного тетрагидрофурана (18 мл) и безводного метанола (9 мл) и смесь охлаждали до 0°C. Добавляли боргидрид натрия (68,1 мг, 1,80 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли уксусную кислоту (0,22 мл, 3,85 ммоль), смесь перемешивали в течение 15 минут и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 40 г, дихлорметан:метанол=20:1→15:1). Чистые фракции концентрировали при пониженном давлении, добавляли к остатку этилацетат (30 мл) и трет-бутилметиловый эфир (10 мл), полученный в результате твердый остаток отделяли фильтрованием и сушили при пониженном давлении, получая трет-бутил 2-{[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метил}амино)бензоил]карбамоил}гидразинкарбоксилат (653,1 мг, 1,503 ммоль, выход 83,5%) в виде белого твердого вещества.
Стадия 4
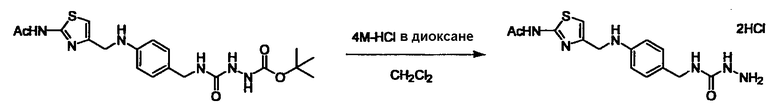
К суспензии трет-бутил 2-{[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метил}амино)бензоил]карбамоил}гидразинкарбоксилата (306,2 мг, 0,705 ммоль) в безводном дихлорметане (7 мл) добавляли 4M диоксановый раствор хлористого водорода (7,0 мл, 28,0 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часа и концентрировали при пониженном давлении. Добавляли к остатку этилацетат и смесь снова концентрировали при пониженном давлении. Данную операцию повторяли 3 раза для удаления хлористого водорода азеотропно. Остаток суспендировали в этилацетате, суспензию фильтровали, промывали дважды этилацетатом и сушили при пониженном давлении. Остаток перекристаллизовывали растворением в метаноле (3,6 мл) и затем добавлением этилацетата (12 мл). Кристаллы отделяли фильтрованием, сушили при пониженном давлении, получая указанное в заголовке соединение (272,8 мг, 0,670 ммоль, выход 95,0%) в виде белого твердого вещества.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,07 (1H, уш.с), 9,87 (3H, уш.с), 8,75 (1H, уш.с), 7,40 (1H, т, J=5,7 Гц), 7,06 (2H, д, J=8,2 Гц), 6,93 (1H, с), 6,74 (2H, д, J=8,2 Гц), 4,27 (2H, с), 4,12 (2H, J=5,7 Гц), 2,11 (3H, с).
13C-ЯМР (50 МГц, D2O): δ (м.д.): 174,6, 162,2, 160,8, 143,1, 142,2, 135,8, 131,2, 125,7, 119,1, 57,1, 53,1, 24,9.
МС(ESI+): 335,1293[M(свободный)+H]+.
Пример получения 24
Гидрохлорид 2-(ацетиламино)-N-(4-{[(гидразинокарбонил)амино]метил}фенил)-1,3-тиазол-4-карбоксамида
Стадия 1
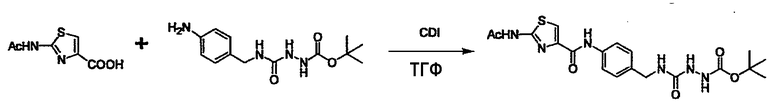
К суспензии 2-(ацетиламино)-1,3-тиазол-4-карбоновой кислоты (558,6 мг, 3,000 ммоль) в безводном тетрагидрофуране (10 мл) добавляли 1,1'-карбонилдиимидазол (535,2 мг, 3,301 ммоль) и безводный тетрагидрофуран (10 мл) и смесь перемешивали при 50°C в течение 2 часа. Добавляли трет-бутил 2-[(4-аминобензил)карбамоил]гидразинкарбоксилат (841,0 мг, 3,000 ммоль) и смесь перемешивали в течение 24 часов. К раствору 2-(ацетиламино)-1,3-тиазол-4-карбоновой кислоты (280,2 мг, 1,505 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли 1,1'-карбонилдиимидазол (243,2 мг, 1,500 ммоль) и смесь перемешивали при 50°C в течение 30 минут. Полученный раствор добавляли к смеси, полученной выше, смесь перемешивали при 50°C в течение 3 часа и при комнатной температуре в течение 65 часов. Добавляли к реакционной смеси воду и этилацетат и смесь перемешивали. Выпавший осадок фильтровали, промывали этилацетатом и сушили при пониженном давлении, получая трет-бутил 2-{[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)бензил]карбамоил}гидразинкарбоксилат (1,223 г, 2,728 ммоль, выход 90,9%) в виде белого твердого вещества.
Стадия 2
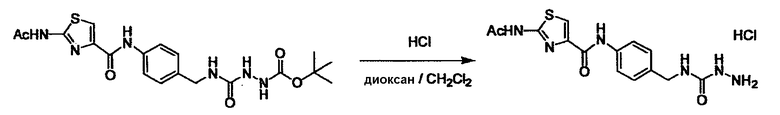
С трет-бутил 2-{[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)бензил]карбамоил}гидразинкарбоксилата (298,0 мг, 0,664 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (257,4 мг, количественный выход) в виде белого твердого вещества. Температура плавления 231-234°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,33 (1H, уш.с), 9,83 (3H, уш.с), 9,72 (1H, уш.с), 8,80 (1H, уш.с), 7,93 (1H, с), 7,68 (2H, д, J=8,6 Гц), 7,52 (1H, т, J=5,9 Гц), 7,25 (2H, д, J=8,6 Гц), 4,24 (2H, д, J=5,9 Гц), 2,18 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 169,2, 159,3, 158,0, 157,4, 144,4, 137,2, 135,1, 127,7, 119,9, 118,3, 42,7, 22,5.
МС(ESI+): 371,0872 [M(свободный)+Na]+, 387,0612 [M(свободный)+K]+.
Пример получения 25
Трифторацетат N-[2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил]гидразинкарбоксамида
Стадия 1
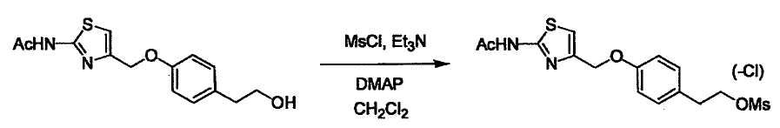
К раствору N-(4-{[4-(2-гидроксиэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамида (310,0 мг, 1,060 ммоль) в безводном дихлорметане (8 мл) добавляли триэтиламин (0,27 мл, 1,91 ммоль) и 4-диметиламинопиридин (13,0 мг, 0,106 ммоль) и смесь охлаждали до 0°C. Добавляли по каплям при 0°C метансульфонилхлорид (0,12 мл, 1,60 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Добавляли к реакционной смеси ледяную воду (20 мл), смесь перемешивали в течение 20 минут и экстрагировали 3 раза этилацетатом. Объединенные органические слои промывали 1M хлористоводородной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая 2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этилметансульфонат (410,8 мг, содержащий N-(4-{[4-(2-хлорэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамид) в
виде оранжевого твердого вещества.
Стадия 2
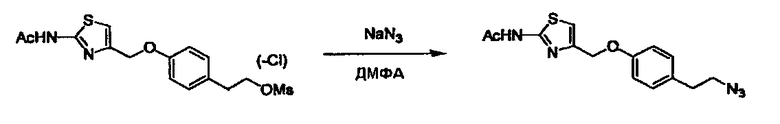
2-(4-{[2-(Ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этилметансульфонат (соответствует 1,060 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{[4-(2-азидоэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамид (240,7 мг, 0758 ммоль, выход 71,5%) в виде белого твердого вещества.
Стадия 3
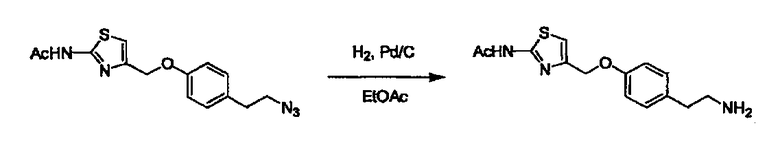
N-(4-{[4-(2-Азидоэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамид (240,0 мг, 0,756 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{[4-(2-аминоэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамид (217,6 мг, 0,7 47 ммоль, выход 98,5%) в виде белого твердого вещества.
Стадия 4
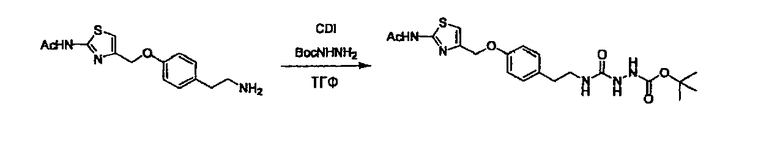
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил}метокси}фенил)этил]карбамоил}гидразинкарбоксилат (309,7 мг, 0,68 9 ммоль, выход 97,9%) получали в виде белого твердого вещества из N-(4-{[4-(2-аминоэтил)фенокси]метил}-1,3-тиазол-2-ил)ацетамида (205,0 мг, 0,704 ммоль).
Стадия 5
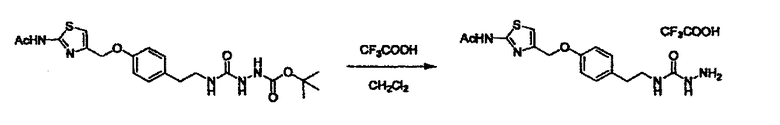
К раствору трет-бутил 2-{[2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил]карбамоил}гидразинкарбоксилата (305,0 мг, 0,667 ммоль) в безводном дихлорметане (20 мл) добавляли трифторуксусную кислоту (2,48 мл, 33,4 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часа и концентрировали при пониженном давлении. Добавляли к остатку дихлорметан (20 мл) и смесь концентрировали снова при пониженном давлении. Данную операцию повторяли 3 раза для удаления трифторуксусной кислоты азеотропно. Добавляли к остатку этилацетат (30 мл) и смесь перемешивали. Осадок отделяли фильтрованием, промывали 5 раза этилацетатом и 5 раз диэтиловым эфиром, сушили при пониженном давлении, получая указанное в заголовке соединение (253,4 мг, 0,547 ммоль, выход 81,9%) в виде белого твердого вещества. Температура плавления 195-197,5°C.
1H-ЯМР (400 МГц, ДМСО-de): 5 (м.д.): 12,14 (1H, уш.с), 10,0-9,0 (2H, уш.с), 8,52 (1H, уш.с), 7,16 (1H, с), 7,03 (1H, уш.с), 7,11 (2H, д, J=8,6 Гц), 6,92 (2H, д, J=8,6 Гц), 5,00 (2H, с), 3,40-3,34 (2H, м), 2,65 (2H, т, J=7,3 Гц), 2,12 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,6, 158,3, 157,5, 156,9, 146,7, 131,5, 129,8, 114,8, 111,4, 65,6, 41,3, 39,1, 34,8, 22,6 (пик трифторуксусной кислоты был слабее по интенсивности и сложно отличим от шума).
19F-ЯМР (376 Гц, ДМСО-d6): δ (м.д.): -75,1.
МС(ESI+): 350,1262 [M(свободный)+H]+, 372,1085 [M(свободный)+Na]+.
Пример получения 26
Гидрохлорид 4-{2-[(гидразинокарбонил)амино]этил}фенил 2-(ацетиламино)-1,3-тиазол-4-карбоксилата
Стадия 1
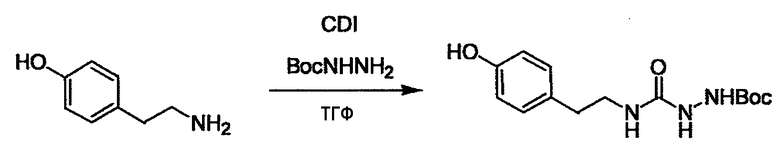
К суспензии 1,1'-карбонилдиимидазола (1,621 г, 9,994 ммоль) в безводном тетрагидрофуране (20 мл) добавляли трет-бутилкарбазат (1,322 г, 10,00 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли 4-(2-аминоэтил)фенол (1,371 г, 9,991 ммоль) и смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли к реакционной смеси воду (40 мл), осадок отделяли фильтрованием, промывали 3 раза водой и дважды этилацетатом и сушили при пониженном давлении, получая трет-бутил 2-{[2-(4-гидроксифенил)этил]карбамоил}гидразинкарбоксилат (2,417 г, 8,184 ммоль, выход 81,9%) в виде белого твердого вещества.
Стадия 2
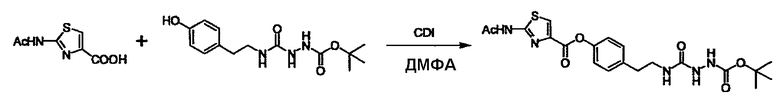
К суспензии 2-(ацетиламино)-1,3-тиазол-4-карбоновой кислоты (559,5 мг, 3,005 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли 1,1'-карбонилдиимидазол (534,2 мг, 3,294 ммоль) и смесь перемешивали при 50°C в течение 2 часа. Добавляли трет-бутил 2-{[2-(4-гидроксифенил)этил]карбамоил}гидразинкарбоксилат (591,6 мг, 2,003 ммоль), смесь перемешивали при 50°C в течение 24 часов и при комнатной температуре в течение 17 часов. Добавляли к реакционной смеси воду (50 мл) и этилацетат (50 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали 0,5M хлористоводородной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Добавляли к остатку метанол (20 мл) и трет-бутилметиловый эфир (40 мл), выпавший осадок отделяли фильтрованием и сушили при пониженном давлении, получая 4-[2-({[2-(трет-бутоксикарбонил)гидразино]карбонил}амино)этил]фенил 2-(ацетиламино)-1,3-тиазол-4-карбоксилат (417,7 мг, 0,890 ммоль, выход 44,5%) в виде белого твердого вещества.
Стадия 3
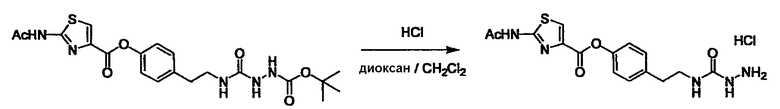
С 4-[2-({[2-(трет-бутоксикарбонил)гидразино]карбонил}амино)этил]фенил 2-(ацетиламино)-1,3-тиазол-4-карбоксилата (375,2 мг, 0,810 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (300,8 мг, 0,752 ммоль, выход 92,9%). Температура плавления 204-207°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,56 (1H, уш.с), 9,87 (3H, уш.с), 8,74 (1H, уш.с), 8,28 (1H, с), 7,30 (2H, д, J=8,6 Гц), 7,17 (2H, д, J=8,4 Гц), 7,07 (1H, т, J=5,5 Гц), 3,35-3,30 (2H, м), 2,76 (2H, т, J=7,1 Гц), 2,16 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 169,1, 159,4, 158,2, 157,1, 148,6, 139,7, 136,9, 129,6, 124,4, 121,5, 40,8, 34,7, 22,3.
МС(ESI+): 386,0897 [M(свободный)+Na]+, 402,0639 [M(свободный)+K]+.
Пример получения 27
Гидрохлорид N-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамида
Стадия 1
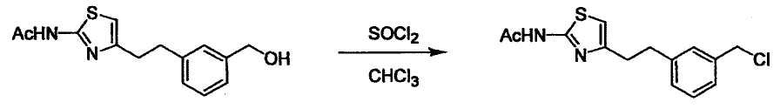
N-(4-{2-[3-(Гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (2,909 ммоль) хлорировали по аналогичной методике примера получения 20, стадия 1, получая N-(4-{2-[3-(хлорметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (718,7 мг, 2,438 ммоль, выход 83,8%) в виде светло-желтоватого твердого вещества.
Стадия 2
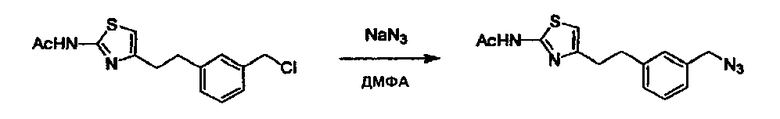
N-(4-{2-[3-(Хлорметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (716,0 мг, 2,42 9 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[3-(азидометил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (555,9 мг, 2,019 ммоль, выход 83,1%) в виде белого твердого вещества.
Стадия 3
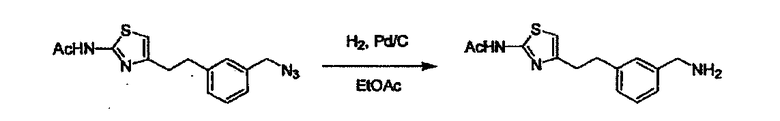
N-(4-{2-[3-(Азидометил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (553,0 мг, 1,835 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[3-(аминометил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (467,5 мг, 1,698 ммоль, выход 92,5%) в виде белого твердого вещества.
Стадия 4
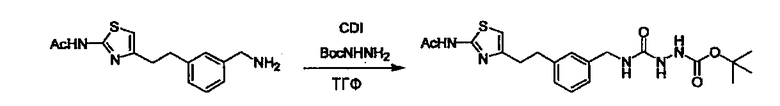
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-[(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)карбамоил]гидразинкарбоксилат (483,2 мг, 1,115 ммоль, выход 69,0%) получали в виде белого твердого вещества из N-(4-{2-[3-(аминометил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (4 44,7 мг, 1,615 ммоль).
Стадия 5
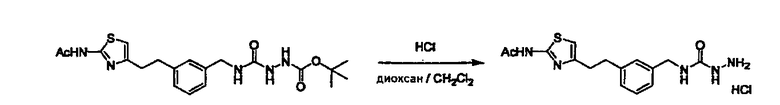
С трет-бутил 2-[(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)карбамоил]гидразинкарбоксилата (412,6 мг, 0,952 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (341,0 мг, 0,922 ммоль, выход 96,8%) в виде белого твердого вещества. Температура плавления 123-127°C.
1H-ЯМР (400 МГц, ДМСО-d6): 5 (м.д.): 12,08 (1H, уш.с), 9,96 (3H, уш.с), 8,93 (1H, уш.с), 7,56 (1H, уш.т, J=5,8 Гц), 7,21 (1H, т, J=7,6 Гц), 7,14 (1H, с), 7,09-7,05 (2H, м), 6,75 (1H, с), 4,24 (2H, д, J=5,8 Гц), 2,91-2,83 (4H, м), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДMCO-d6): 6 (м.д.): 168,4, 157,7, 157,5, 150,3, 141,6, 139,7, 128,4, 127,2, 126,9, 124,9, 107,6, 43,0, 34,8, 32,9, 22,6.
MC(ESI+): 334,1328 [M(свободный)+H]+, 356,1147 [M(свободный)+Na]+.
Пример получения 28 Гидрохлорид N-[2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамида
Стадия 1
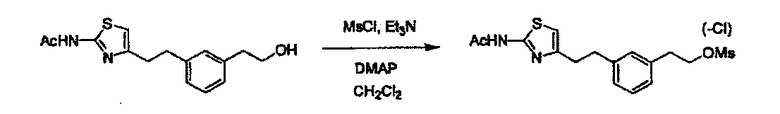
N-(4-{2-[3-(2-Гидроксиэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (360,0 мг, 1,240 ммоль) мезилировали по аналогичной методике примера получения 25, стадия 1, получая 2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этилметансульфонат (454,9 мг, содержащий N-(4-{2-[3-(2-хлорэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид) в виде желтого твердого вещества.
Стадия 2
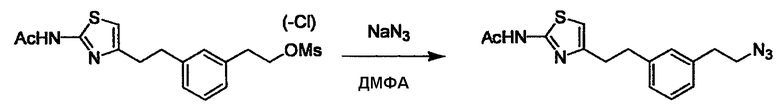
2-(3-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этилметансульфонат (соответствующий 1,240 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[3-(2-азидоэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (304,7 мг, 0,966 ммоль, суммарный выход на стадию 1 77,9%) в виде белого твердого вещества.
Стадия 3
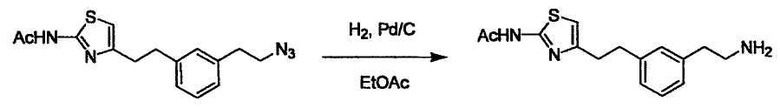
N-(4-{2-[3-(2-Азидоэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (300,0 мг, 0,951 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[3-(2-аминоэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (329,8 мг) в виде белого твердого вещества.
Стадия 4
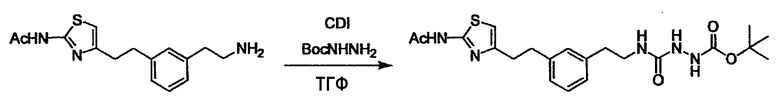
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]карбамоил}гидразинкарбоксилат (238,2 мг, 0,532 ммоль, суммарный выход на стадию 3 55,9%) получали в виде белого твердого вещества из N-(4-{2-[3-(2-аминоэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (соответствующий 0,951 ммоль).
Стадия 5
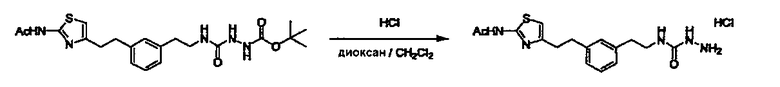
С трет-бутил 2-{[2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]карбамоил}гидразинкарбоксилата (225,0 мг, 0,503 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (191,6 мг, 0,499 ммоль, выход 99,2%) в виде белого твердого вещества.
Температура плавления 176-179°C.
1H-ЯМР (400 МГц, ДMCO-d6): 5 (м.д.): 12,09 (1H, уш.с), 9,95 (3H, уш.с), 8,79 (1H, уш.с), 7,18 (1H, т, J=7,6 Гц), 7,06-7,01 (4H, м), 6,73 (1H, с), 3, 40-3,20 (2H, м), 2,91-2,84 (4H, м), 2,68 (2H, т, J=7,3 Гц), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): 5 (м.д.): 168, 4, 157, 6, 157, 3, 150,4, 141,6, 139,2, 128,8, 128,5, 126,4, 126,3, 107,5, 41,1, 35,7, 34,8,33,0, 22,6.
MC(ESI+): 348,1429 [M(свободный)+H]+, 370,1252 [M(свободный)+Na]+.
Пример получения 29
Гидрохлорид N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарбоксамида
Стадия 1
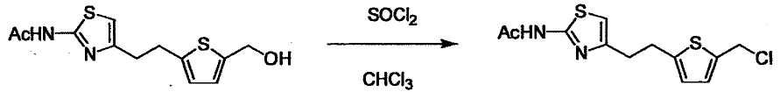
На водяной бане приблизительно при 20°C к суспензии N-(4-{2-[5-(гидроксиметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (500,0 мг, 1,771 ммоль) в безводном хлороформе (2,5 мл) добавляли по каплям тионилхлорид (0,77 мл, 10,6 ммоль). После перемешивания при комнатной температуре в течение 1 часа, реакционную смесь концентрировали при пониженном давлении. Добавляли к остатку безводный хлороформ (5 мл) и смесь концентрировали снова при пониженном давлении. Операцию повторяли 3 раза для удаления тионилхлорида азеотропно. Остаток сушили при пониженном давлении, получая N-(4-{2-[5-(хлорметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (633,2 мг) в виде светло-коричневого твердого вещества.
Стадия 2
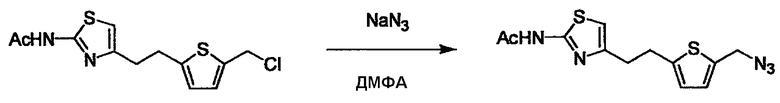
N-(4-{2-[5-(Хлорметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (соответствующий 1,771 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[5-(азидометил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (489,8 мг, 1,593 ммоль, выход 90,0%) в виде слегка желтоватого твердого вещества.
Стадия 3
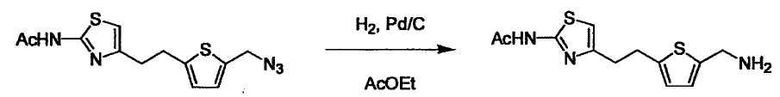
N-(4-{2-[5-(Азидометил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (480,0 мг, 1,562 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[5-(аминометил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (376,1 мг, 1,337 ммоль, выход 85,6%) в виде белого твердого вещества.
Стадия 4
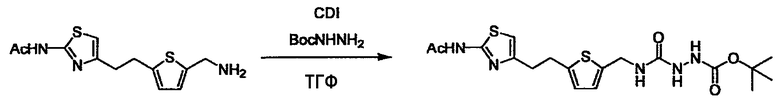
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]карбамоил}гидразинкарбоксилат (307,9 мг, 0,700 ммоль, выход 56,3%) получали в виде белого твердого вещества из N-(4-{2-[5-(аминометил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (350,0 мг, 1,244 ммоль).
Стадия 5
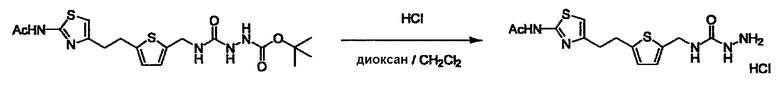
С трет-бутил 2-{[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]карбамоил}гидразинкарбоксилата (250,0 мг, 0,569 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (208,7 мг, 0,555 ммоль, выход 97,6%) в виде белого твердого вещества, температура плавления 158-161°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,08 (1H, уш.с), 9,98 (3H, уш.с), 8,93 (1H, уш.с), 7,54 (1H, уш.т, J=5,6 Гц), 6,77 (1H, с), 6,74 (2H, д, J=3,2 Гц), 6,64 (2H, д, J=3,2 Гц), 4,32 (2H, д, J=5,6 Гц), 3,08 (2H, д, J=7,5 Гц), 2,87 (2H, д, J=7,5 Гц), 2,10 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,5, 157,7, 157,1, 149,8, 143,3, 140,2, 125,3, 124,2, 108,0, 38,4, 33,1, 29,0, 22,7.
МС(ESI+): 340,0855 [M(свободный)+H]+, 362,0670 [M(свободный)+Na]+.
Пример получения 30
Гидрохлорид N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил]гидразинкарбоксамида
Стадия 1
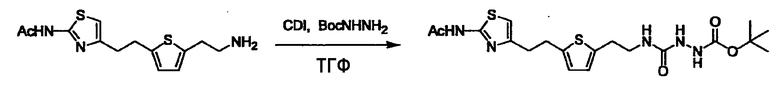
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил]карбамоил}гидразинкарбоксилат (658,6 мг, 1,452 ммоль, выход 90,3%) получали в виде белого твердого вещества из N-(4-{2-[5-(2-аминоэтил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (475,0 мг, 1,608 ммоль).
Стадия 2
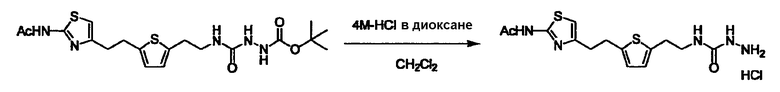
С трет-бутил 2-{[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил]карбамоил}гидразинкарбоксилата (650,0 мг, 1,433 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5. Неочищенный продукт растворяли в воде и перекристаллизовывали добавлением ацетонитрила. Осадок отделяли фильтрованием, промывали 3 раза ацетонитрилом и сушили при пониженном давлении, получая указанное в заголовке соединение (485,1 мг, 1,244 ммоль, выход 86,8%) в виде белого твердого вещества, температура плавления 178-181°C.
1H-ЯМР (200 МГц, ДМСО-d6): δ (м.д.): 12,09 (1H, уш.с), 9,97 (3H, уш.с), 8,85 (1H, уш.с), 7,11 (1H, уш.т, J=5,0 Гц), 6,78 (1H, с), 6,70-6,60 (2H, м), 3,35-5,15 (2H, м), 3,15-3,00 (2H, м), 2,95-2,80 (4H, м), 2,11 (3H, с).
13C-ЯМР (50 МГц, ДМСО-d6): δ (м.д.): 168,7, 158,0, 157,6, 150,8, 142,4, 139,4, 125,3, 124,8, 108,3, 41,5, 38,7, 30,4, 29,3, 22,9.
МС(ESI+): 354,1049 [M(свободный)+H]+, 370,1252 [M(свободный)+Na]+.
Пример получения 31
Гидрохлорид N-[3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил]гидразинкарбоксамида
Стадия 1
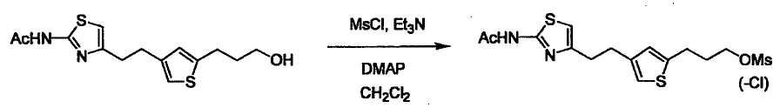
N-(4-{2-[5-(3-Гидроксипропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамид (290,0 мг, 0,934 ммоль) мезилировали по аналогичной методике примера получения 25, стадия 1, получая 3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропилметансульфонат (362,4 мг, содержащий небольшое количество N-(4-{2-[5-(3-хлорпропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамида, 0,933 ммоль в виде сульфоната, выход 99,8%) в виде бледно-оранжевого твердого вещества.
Стадия 2
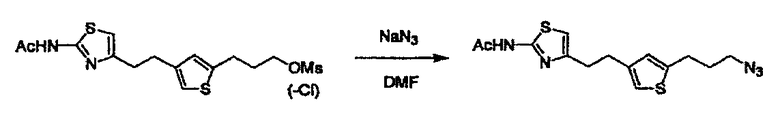
3-(4-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропилметансульфонат (360,0 мг, 0,927 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[5-(3-азидопропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамид (286,4 мг, 0,854 ммоль, выход 92,1%) в виде белого твердого вещества.
Стадия 3
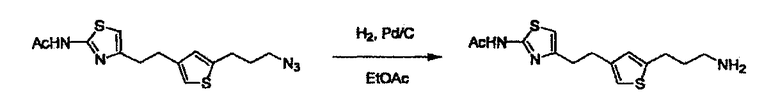
N-(4-{2-[5-(3-Азидопропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамид (280,0 мг, 0,843 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[5-(3-аминопропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамид (247,1 мг, 0,7 99 ммоль, выход 94,7%) в виде белого твердого вещества.
Стадия 4
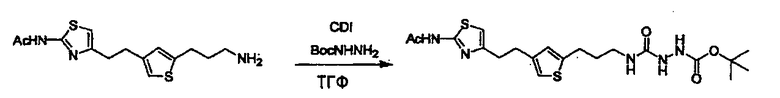
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил]карбамоил}гидразинкарбоксилат (180, 3 мг, 0,38 6 ммоль, выход 70,2%) получали в виде белого твердого вещества из N-(4-{2-[5-(3-аминопропил)тиофен-3-ил]этил}-1,3 -тиазол-2-ил)ацетамида (170,0 мг, 0,549 ммоль).
Стадия 5
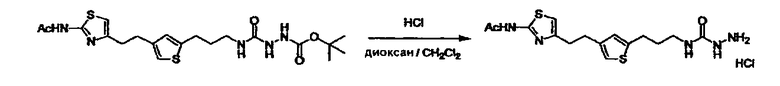
С трет-бутил 2-{[3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил]карбамоил}гидразинкарбоксилата (180,0 мг, 0,385 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (102,7 мг, 0,254 ммоль, выход 66,0%) в виде белого твердого вещества, температура плавления 153-156°C.
1H-ЯМР (400 МГц, ДМСО-d6): 5 (м.д.): 12,04 (1H, уш.с), 9,72 (3H, уш.с), 8,61 (1H, уш.с), 7,10 (1H, уш.т), 6,91 (1H, с), 6,73 (1H, с), 6,71 (1H, с), 3,12-3,06 (2H, м), 2, 90-2,80 (4H, м), 2,73 (2H, т, J=7,6 Гц), 2,10 (3H, с), 1,72 (2H, тт, J=7,6, 6,4 Гц).
13C-ЯМР (100 МГц, ДMCO-d6): 5 (м.д.): 168,4, 157, 6, 150,6, 144,1, 141,6, 126,0, 118,4, 107,4, 32,0, 31,7, 29,5, 26,9, 22,6.
MS (ESI+): 368,1182 [М(свободный)+Н]+, 390,0097 [M(свободный)+Na]+.
Пример получения 32
Гидрохлорид N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил]гидразинкарбоксамида
Стадия 1
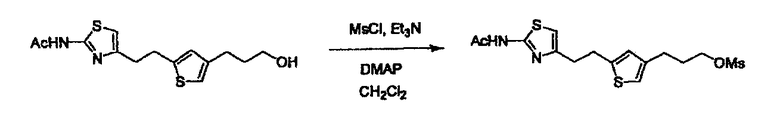
N-(4-{2-[5-(3-Гидроксипропил)тиофен-3-ил]этил}-1,3-тиазол-2-ил)ацетамид (380,0 мг, 1,224 ммоль) мезилировали по аналогичной методике примера получения 25, стадия 1. Неочищенный продукт очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 70 г, дихлорметан:метанол=40:1), получая 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропилметансульфонат (461,3 мг, 1,187 ммоль, выход 97,0%) в виде не совсем белого твердого вещества.
Стадия 2
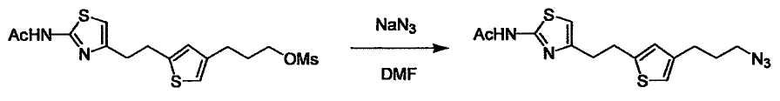
3-(5-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропилметансульфонат (460,0 мг, 1,184 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[4-(3-азидопропил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (322,8 мг, 0,962 ммоль, выход 81,3%) в виде белого твердого вещества.
Стадия 3
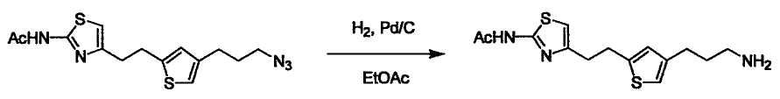
N-(4-{2-[4-(3-Азидопропил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (310,4 мг, 0,925 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[4-(3-аминопропил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (284,5 мг, 0,919 ммоль, выход 99,4%) в виде белого твердого вещества.
Стадия 4
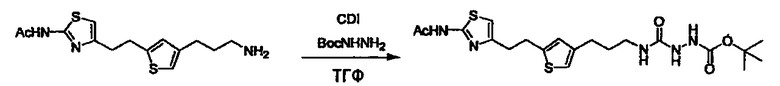
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил]карбамоил}гидразинкарбоксилат (423,0 мг, 0,905 ммоль, выход 100%) получали в виде не совсем белого твердого вещества из N-(4-{2-[4-(3-аминопропил)тиофен-2-ил]этил}-1, 3-тиазол-2-ил)ацетамида (280, 1 мг, 0, 905 ммоль).
Стадия 5

С трет-бутил 2-{[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил]карбамоил}гидразинкарбоксилат (420,1 мг, 0,898 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (333,5 мг, 0,831 ммоль, выход 92,5%) в виде белого твердого вещества. Температура плавления 111-115°C.
1H-ЯМР (200 МГц, ДМСО-d6): 5 (м.д.): 12,08 (1H, уш.с), 9,96 (3H, уш.с), 8,80 (1H, уш.с), 7,15 (1H, уш.с), 6,89 (1H, с), 6,77 (1H, с), 6,70 (1H, с), 3,30-2,95 (6H, м), 2, 95-2,80 (2H, м), 2,11 (3H, с), 1,98-1,64 (2H, м).
13C-ЯМР (50 МГц, MMCО-d6): 5 (м.д.): 168,4, 157,7, 157,4, 149,8, 143,9, 141,5, 126,1, 118,4, 108,0, 33,2, 30,3, 29,1, 27,2, 22,7.
MC(ESI+): 368,1194 [M(свободный)+H]+, 390,1015 [М(свободный)+Na]+.
Пример получения 33
Гидрохлорид N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил]гидразинкарбоксамида
Стадия 1
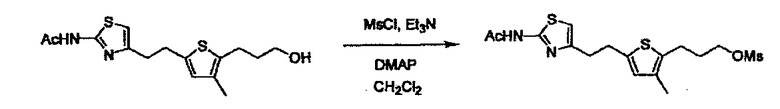
N-(4-{2-[5-(3-Гидроксипропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (102,1 мг, 0,315 ммоль) мезилировали по аналогичной методике примера получения 25, стадия 1. Неочищенный продукт очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 15 г, дихлорметан:метанол=30:1), получая 3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропилметансульфонат (93,0 мг, 0,231 ммоль, выход 73,3%) в виде бледно-желтого твердого вещества.
Стадия 2
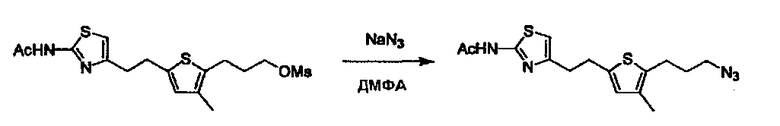
3-(5-{2-[2-(Ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропилметансульфонат (88,0 мг, 0,219 ммоль) азидировали по аналогичной методике примера получения 20, стадия 2, получая N-(4-{2-[5-(3-азидопропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (52,0 мг, 0,149 ммоль, выход 68,0%) в виде бледно-желтого твердого вещества.
Стадия 3
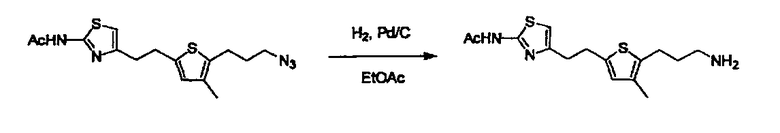
N-(4-{2-[5-(3-Азидопропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (47,2 мг, 0,135 ммоль) гидрировали по аналогичной методике примера получения 20, стадия 3, получая N-(4-{2-[5-(3-аминопропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (42,7 мг, 0,132 ммоль, выход 97,8%) в виде бледно-желтого твердого вещества.
Стадия 4
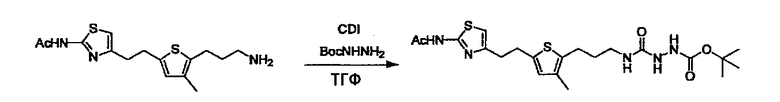
По аналогичной методике примера получения 20, стадия 4, трет-бутил 2-{[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил]карбамоил}гидразинкарбоксилат (60,9 мг, 0,12 6 ммоль, выход 96,2%) получали в виде бледно-желтого твердого вещества из N-(4-{2-[5-(3-аминопропил)-4-метилтиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (42,5 мг, 0,131 ммоль).
Стадия 5
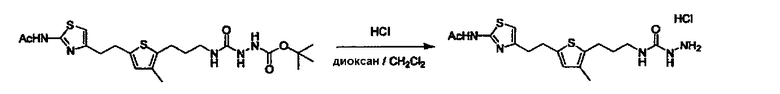
трет-Бутил 2-{[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил]карбамоил}гидразинкарбоксилат (48,4 мг, 0,101 ммоль) удаляли защитную группу по аналогичной методике примера получения 20, стадия 5, получая указанное в заголовке соединение (41,9 мг, 0,100 ммоль, выход 99,0%) в виде белого твердого вещества. Температура плавления 152-157°C.
1H-ЯМР (400 МГц, ДМСО-d6): 5 (м.д.): 12,06 (1H, уш.с), 9,87
(3H, уш.с), 8,72 (1H, уш.с), 7,13 (1H, уш.т), 6,76 (1H, с), 6,51 (1H, с), 3,11-3,05 (2H, м), 3,01 (2H, т, J=7,4 Гц), 2,85 (2H, т, J=7,4 Гц), 2,60 (2H, т, J=7,6 Гц), 2,10 (3H, с), 2,01 (3H, с), 1,63 (2H, кв, J=7,6 Гц).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,3, 157,5, 157,3, 149,8, 139,3, 134,8, 131,9, 127,5, 107,8, 33,0, 31,5, 28,9, 24,6, 22,5, 13,4.
МС(ESI+): 382,1351 [M(свободный)+H]+, 405,1098 [M(свободный)+Na]+.
Пример получения 34
Гидрохлорид S-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарботиоата
Стадия 1
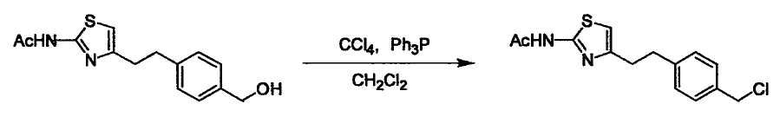
К суспензии N-(4-{2-[4-(гидроксиметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (1,383 г, 5,005 ммоль) в безводном дихлорметане (60 мл) добавляли тетрахлорид углерода (5,8 мл, 60 ммоль) и трифенилфосфин (1,574 г, 6,000 ммоль) и смесь перемешивали при комнатной температуре в течение 22,5 часов. Добавляли трифенилфосфин (786,0 мг, 2,997 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 100 г, этилацетат:гексан=2:3), получая N-(4-{2-[4-(хлорметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (1,224 г, 4,153 ммоль, выход 83,0%) в виде белого твердого вещества.
Стадия 2
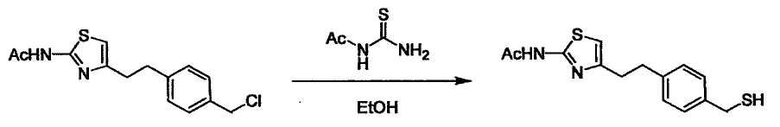
К суспензии N-(4-{2-[4-(хлорметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (589,7 мг, 2,000 ммоль) в этаноле (10 мл) добавляли 1-ацетил-2-тиомочевину (472,6 мг, 4,000 ммоль) и смесь кипятили с обратным холодильником в течение 19 часов. После охлаждения до комнатной температуры, смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 30 г, этилацетат:гексан=2:3), получая N-(4-{2-[4-(сульфанилметил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (507,7 мг, 1,736 ммоль, выход 86,8%) в виде белого твердого вещества.
Стадия 3
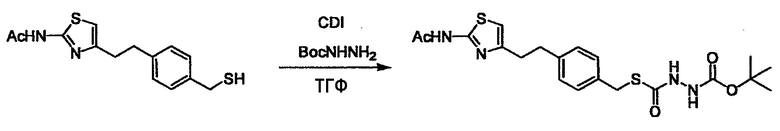
К раствору N-(4-{2-[4-(сульфанилметил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (468,9 мг, 1,604 ммоль) в безводном тетрагидрофуране (5 мл) добавляли 1,1'-карбонилдиимидазол (390,6 мг, 2,409 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часа. Добавляли трет-бутилкарбазат (422,4 мг, 3,196 ммоль) и смесь перемешивали при комнатной температуре в течение 21 часов. Добавляли воду (15 мл), 1M хлористоводородную кислоту (15 мл) и этилацетат (30 мл), смесь перемешивали, оставляли без перемешивания и затем распределяли. Органический слой промывали водой, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток суспендировали в дихлорметане (15 мл), суспензию фильтровали, промывали дихлорметаном и сушили при пониженном давлении, получая трет-бутил 2-({[2-(4-{2-[(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)метил]сульфанил}карбонил)гидразинкарбоксилат (602,2 мг, 1,337 ммоль, выход 83,3%) в виде белого твердого вещества.
Стадия 4
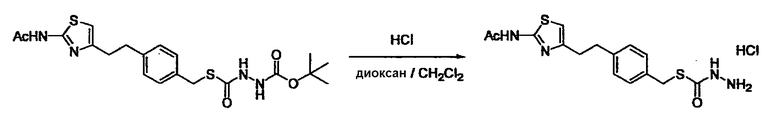
К суспензии трет-бутил 2-({[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)метил]сульфанил}карбонил)гидразинкарбоксилата (450,4 мг, 1,000 ммоль) в безводном дихлорметане (5 мл) добавляли 4M диоксановый раствор хлористого водорода (5 мл) и смесь перемешивали при комнатной температуре в течение 2 часа. Реакционную смесь концентрировали при пониженном давлении. Добавляли к остатку этилацетат (20 мл) и смесь концентрировали снова при пониженном давлении. Данную операцию повторяли 3 раза для удаления хлористого водорода азеотропно. Остаток суспендировали в смеси этанола (5 мл) и этилацетата (15 мл), суспензию фильтровали, промывали дважды этилацетатом и сушили при пониженном давлении, получая указанное в заголовке соединение (375,6 мг, 0,971 ммоль, выход 97,1%) в виде белого твердого вещества. Температура плавления 149-158°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,03 (1H, уш.с), 10,99 (1H, уш.с), 10,8-9,6 (3H, уш.с), 7,22 (2H, д, J=8,2 Гц), 7,13 (2H, д, J=8,1 Гц), 6,72 (1H, с), 4,11 (2H, с), 2,92-2,81 (4H, м), 2,10 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,1, 167,2, 157,4, 150,0, 140,3, 135,3, 128,6, 128,3, 107,3, 34,0, 32,5, 32,4, 22,4.
MS (ESI+): 351,0931 [M(свободный)+H]+, 373,0757 [M(свободный)+Na]+.
Пример получения 35
Гидрохлорид S-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарботиоата
Стадия 1
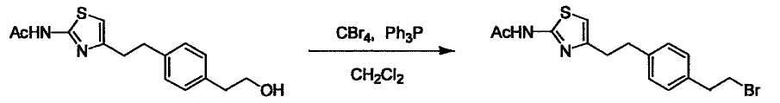
К суспензии N-(4-{2-[4-(2-гидроксиэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (1,452 г, 4,998 ммоль) в безводном дихлорметане (60 мл) добавляли тетрабромид углерода (2,001 г, 6,035 ммоль) и трифенилфосфин (1,571 г, 5,989 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (BW-300SP 10 г, этилацетат:гексан=2:3→1:1), получая N-(4-{2-[4-(2-бромэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (1,553 г, 4,396 ммоль, выход 88,0%) в виде белого твердого вещества.
Стадия 2
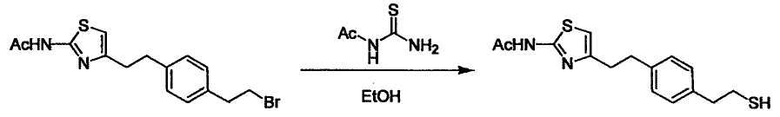
К суспензии N-(4-{2-[4-(2-бромэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (353,3 мг, 1,000 ммоль) в этаноле (4 мл) добавляли 1-ацетил-2-тиомочевину (177,4 мг, 1,501 ммоль) и смесь кипятили с обратным холодильником в течение 7 часов. Добавляли 1-ацетил-2-тиомочевину (176,6 мг, 1,495 ммоль) и смесь кипятили с обратным холодильником в течение 17 часов. Смесь охлаждали до комнатной температуры, добавляли этилацетат (10 мл), и выпавший осадок отфильтровывали. Фильтрат концентрировали при пониженном давлении и очищали дважды колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. DM-2035 21 г, этилацетат:гексан=2:3→1:1), получая N-(4-{2-[4-(2-сульфанилэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамид (210,4 мг, 0,687 ммоль, 68,7%) в виде белого твердого вещества.
Стадия 3
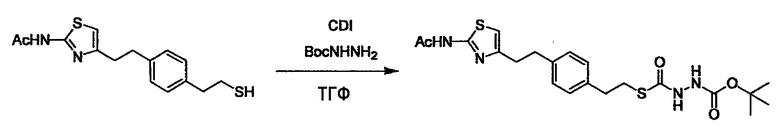
К раствору N-(4-{2-[4-(2-сульфанилэтил)фенил]этил}-1,3-тиазол-2-ил)ацетамида (368,0 мг, 1,201 ммоль) в безводном тетрагидрофуране (30 мл) добавляли 1,1'-карбонилдиимидазол (291,4 мг, 1,797 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли трет-бутилкарбазат (477,9 мг, 3,616 ммоль), безводный тетрагидрофуран (2 мл) и смесь перемешивали при комнатной температуре в течение 19 часов. Добавляли воду (20 мл), 1M хлористоводородную кислоту (10 мл) и этилацетат (30 мл), смесь перемешивали, оставляли без перемешивания и распределяли. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 30 г, этилацетат:гексан=1:1), получая трет-бутил 2-({[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]сульфанил}карбонил)гидразинкарбоксилат (469,9 мг, 1,011 ммоль, выход 84,2%) в виде белого твердого вещества.
Стадия 4
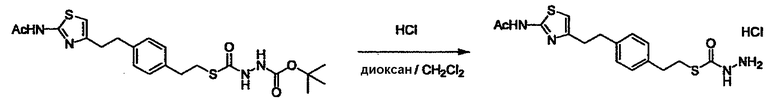
С трет-бутил 2-({[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]сульфанил}карбонил)гидразинкарбоксилата (449,3 мг, 0,967 ммоль) удаляли защитную группу по аналогичной методике примера получения 33, стадия 4, получая указанное в заголовке соединение (308,0 мг, 0,769 ммоль, выход 79,5%) в виде белого твердого вещества. Температура плавления 146-149°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,05 (1H, уш.с), 11,22 (1H, уш.с), 11,0-9,8 (3H, уш.с), 7,13 (4H, с), 6,72 (1H, с), 3,10 (2H, т, J=7,5 Гц), 2,92-2,78 (6H, м), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,1, 167,5, 157,4, 150,1, 139,4, 137,1, 128,3, 128,2, 107,2, 35,2, 34,1, 32,6, 30,1, 22,4.
МС(ESI+): 365,1057 [M(свободный)+H]+, 387,0875 [M(свободный)+Na]+.
Пример получения 36
Гидрохлорид S-[(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарботиоата
Стадия 1
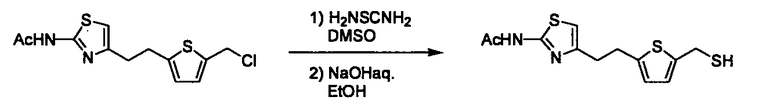
К суспензии N-(4-{2-[5-(хлорметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида в безводном диметилсульфоксида (1,5 мл) добавляли тиомочевину (479,9 мг, 6,304 ммоль) и смесь перемешивали при комнатной температуре в течение 21 часов. Добавляли к реакционной смеси этилацетат (200 мл) и помещали в ультразвуковую ванну при 20-40°C в течение 1 часа. После перемешивания при комнатной температуре в течение 45 минут, осадок отделяли фильтрованием, промывали 5 раза этилацетатом и сушили при пониженном давлении, получая желтовато-белый твердый остаток (1,489 г). Твердый остаток растворяли в смеси этанола (150 мл) и воды (30 мл) и охлаждали до 0°C. Добавляли охлажденный на льду 8M водный гидроксид натрия (30 мл, 240 ммоль) и перемешивали при 0°C в течение 30 минут. Для подкисления реакционной смеси добавляли охлажденную на льду 1M хлористоводородную кислоту (600 мл, 600 ммоль). После перемешивания при 0°C в течение 30 минут смесь концентрировали приблизительно до 350 мл. Смесь экстрагировали 5 раза этилацетатом, объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 120 г, этилацетат:гексан=1:1), получая N-(4-{2-[5-(сульфанилметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамид (493,3 мг, 1,653 ммоль, выход 52,4%) в виде белого твердого вещества.
Стадия 2
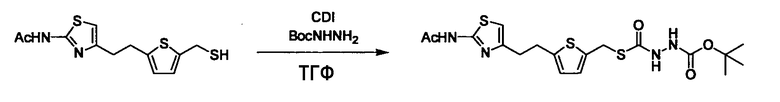
К раствору 1,1'-карбонилдиимидазола (326,0 мг, 2,010 ммоль) в безводном тетрагидрофуране (8 мл) добавляли по каплям при 0°C раствор N-(4-{2-[5-(сульфанилметил)тиофен-2-ил]этил}-1,3-тиазол-2-ил)ацетамида (400,0 мг, 1,340 ммоль) в безводном тетрагидрофуране (10 мл). Смесь перемешивали при 0°C в течение 15 минут и при комнатной температуре в течение 1 часа. Добавляли трет-бутилкарбазат (351,4 мг, 4,021 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. Смесь концентрировали, к остатку добавляли воду (40 мл) и экстрагировали 3 раза этилацетатом. Объединенные органические слои промывали охлажденной на льду 1M хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (FUJI SILYSIA CHEMICAL LTD. BW-300SP 100 г, этилацетат:гексан=1:1), получая трет-бутил 2-({[(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]сульфанил}карбонил)гидразинкарбоксилат (316,7 мг, 0,694 ммоль, выход 51,8%) в виде белого твердого вещества.
Стадия 3
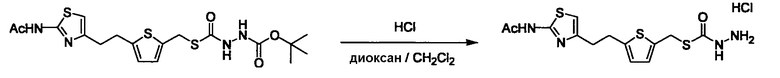
С трет-бутил 2-({[(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]сульфанил}карбонил)гидразинкарбоксилата (315,0 мг, 0,690 ммоль) удаляли защитную группу по аналогичной методике примера получения 1, стадия 4, получая указанное в заголовке соединение (222,8 мг, 0,567 ммоль, выход 82,2%) в виде слегка желто-белого твердого вещества. Температура плавления 147-153°C.
1H-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 12,07 (1H, уш.с), 11,36 (1H, уш.с), 11,0-9,8 (3H, уш.с), 6,80 (1H, д, J=3,3 Гц), 6,77 (1H, с), 6,63 (1H, д, J=3,3 Гц), 4,31 (2H, с), 3,07 (2H, т, J=7,6 Гц), 2,87 (2H, т, J=7,6 Гц), 2,11 (3H, с).
13C-ЯМР (100 МГц, ДМСО-d6): δ (м.д.): 168,5, 167,1, 157,1, 149,7, 144,1, 138,4, 126,8, 124,4, 108,0, 33,0, 29,1, 28,0, 22,7.
МС(ESI+): 357,0485[M(свободный)+H]+.
Экспериментальный пример 1
Ингибирующий эффект по отношению к активности человеческого и крысиного VAP-1 фермента (SSAO)
Соединения настоящего изобретения, полученные в примерах получения, исследовали на ингибирующий эффект по отношению к активности человеческого и крысиного VAP-1 фермента (SSAO) следующим способом.
Активность VAP-1 фермента (SSAO) как у людей, так и у крыс измеряли радиохимическим ферментным анализом, используя 14C-бензиламин в качестве искусственного субстрата. Человеческий или крысиный VAP-1 клонировали из кДНК библиотеки и экспрессировали в клетке. Клеточный экстракт предварительно инкубировали с раствором испытуемого соединения (конечная концентрация 1×10-7-1×10-11 моль/л) при комнатной температуре в течение 20 минут. Затем добавляли 14C-бензиламин (конечная концентрация 1×10-5 моль/л) и смесь инкубировали при конечной концентрации 200 мкл при 37°C в течение 2 часов. Ферментативную реакцию останавливали добавлением 2 моль/л (200 мкл) лимонной кислоты. Продукт окисления экстрагировали 1 мл смеси толуол/этилацетат (1:1), и его радиоактивность измеряли на жидкостном сцинтилляционном счетчике. Результаты показаны в таблице 1.
Как показано в таблице 1, соединение настоящего изобретения заметно ингибировало ферментативную активность человеческого и крысиного SSAO.
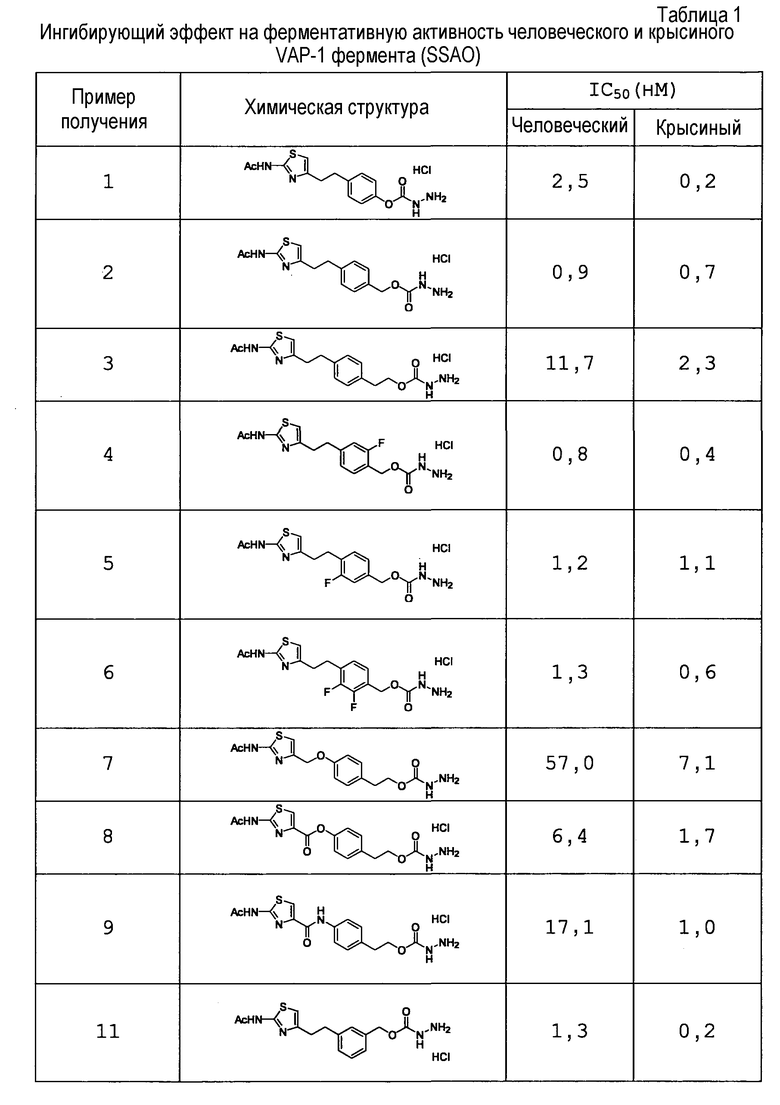
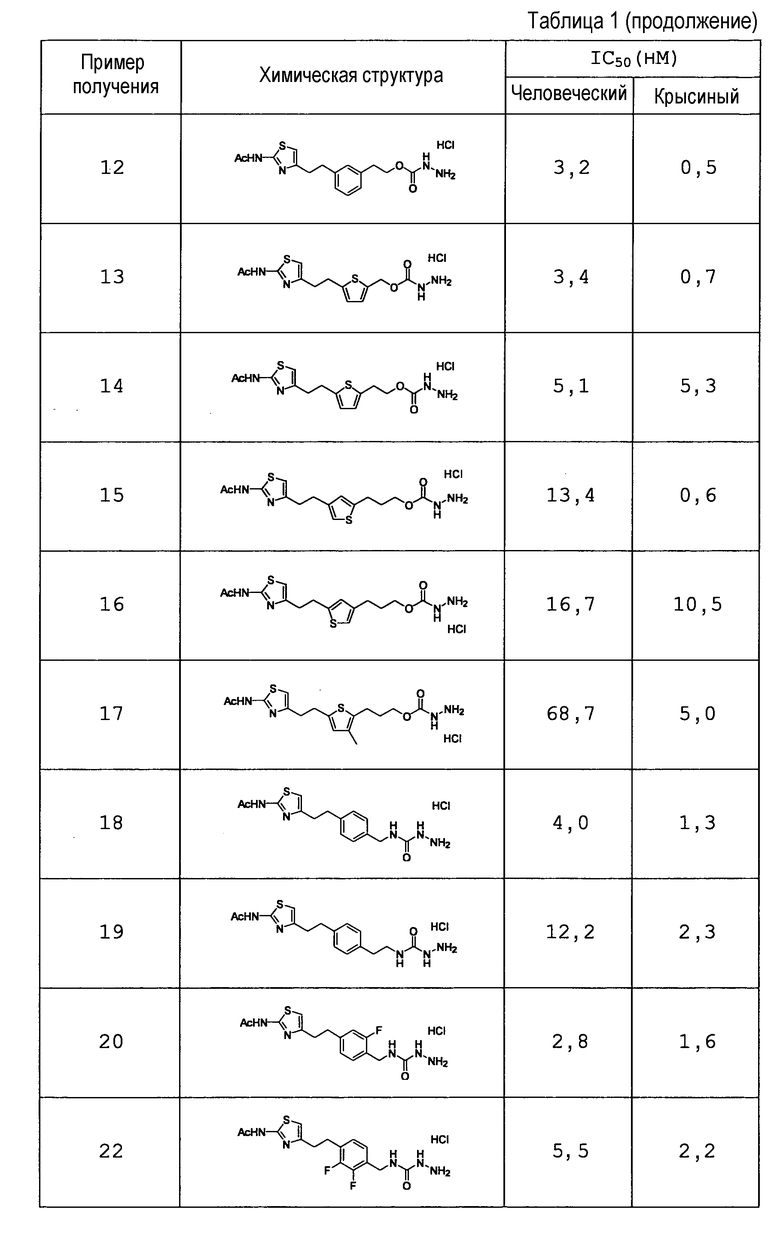
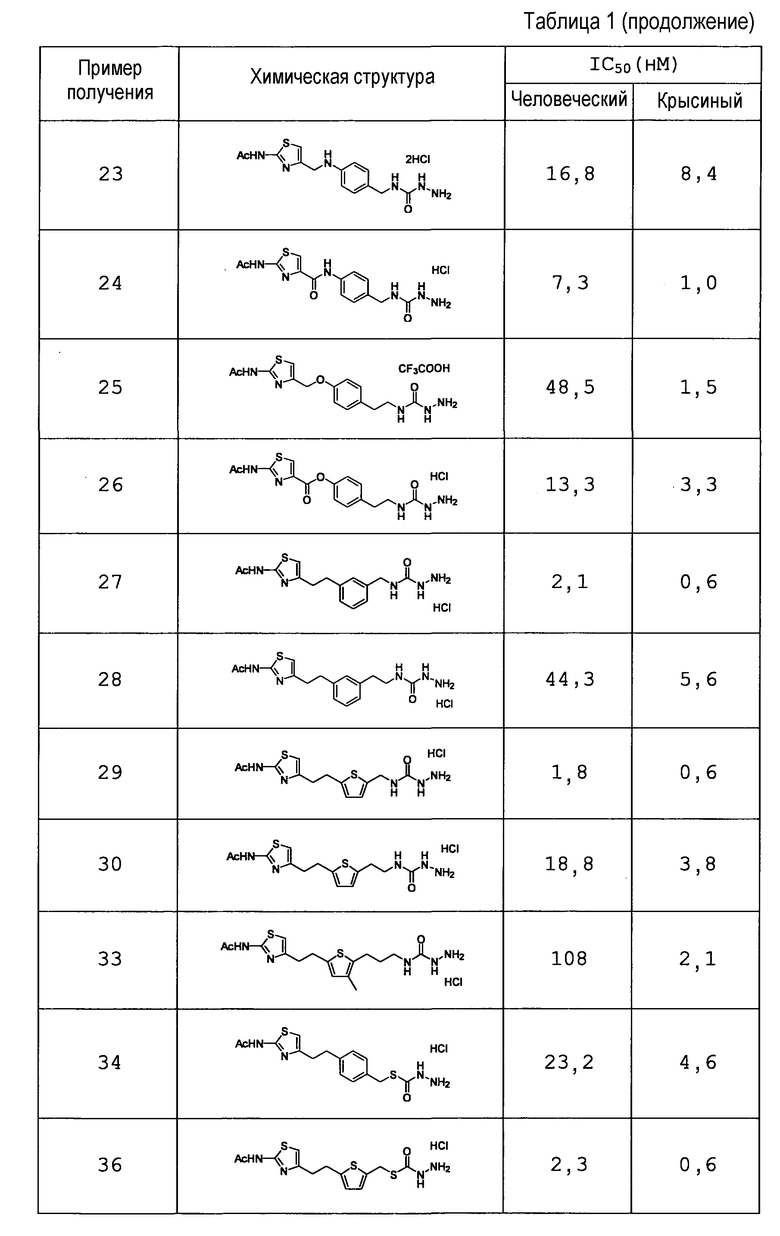
Экспериментальный пример 2
Ингибирующий эффект по отношению к ферментативной активности человеческих моноаминооксидазных ферментов (MAO-A и MAO-B)
Соединения настоящего изобретения, полученные в примерах получения, исследовали на ингибирующий эффект по отношению к ферментативной активности человеческих моноаминооксидазных ферментов (MAO-A и MAO-B) следующим способом.
Рекомбинантные человеческие MAO-A и MAO-B ферменты получали у Sigma Ltd. Активность человеческих MAO-A и MAO-B измеряли, используя набор для обнаружения MAO (Fluoro MAO, Cell Technology Inc.). Анализ проводили, используя 96-луночный планшет. 1× реакционный буфер (40 мкл) добавляли в каждую лунку и дополнительно добавляли 50 мкл MAO-A или MAO-B. Затем, добавляли раствор тестируемого соединения (10 мкл, конечная концентрация 1×10-5-1×10-10 моль/л) и смесь инкубировали при 37°C в течение 20 минут. Добавляли реакционный коктейль (100 мкл) и смесь инкубировали при конечном объеме 200 мкл при 37°C в течение 2 часов. Затем определяли флуоресценцию при 590 нм на мультиспектральном планшет-ридере (Varioskan, Thermo Fisher Scientific K.K.), при волне возбуждения 570 нм. Результаты показаны в таблице 2.
Как показано в таблице 2, соединение настоящего изобретения не обладает заметной ингибирующей активностью по отношению к человеческому MAO-A или MAO-B. Так как соединение практически не обладает ингибирующей активностью по отношению к другим моноаминооксидазам, ясно, что соединение настоящего изобретения обладают селективностью и специфической ингибирующей активностью по отношению к SSAO.
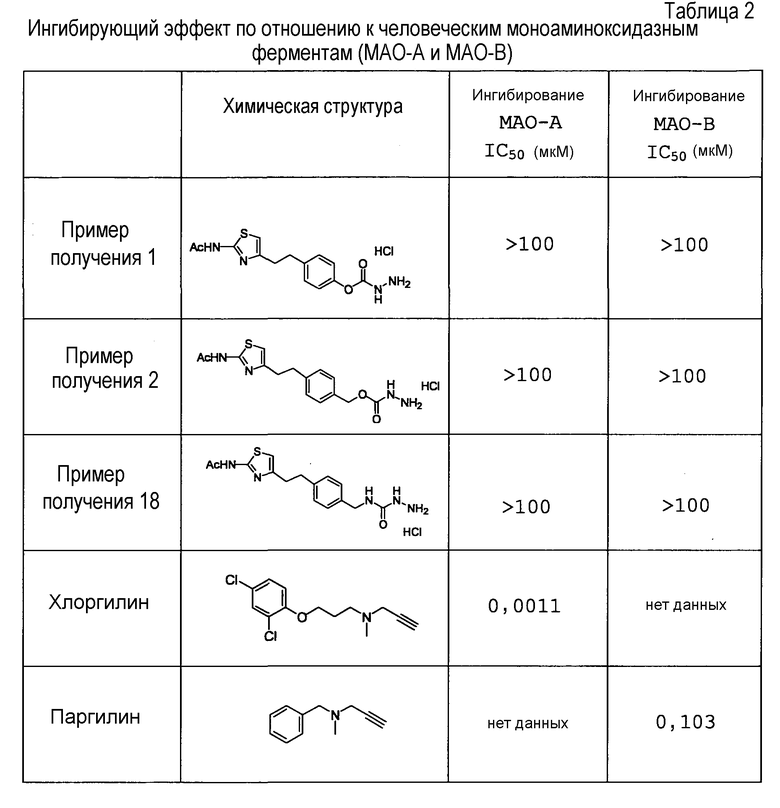
Промышленная применимость
Настоящее изобретение относится к соединению, представленному формулой (I)
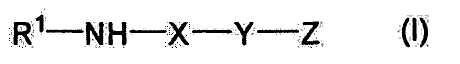
где каждый символ является таким, как определено выше, полезному в качестве ингибитора VAP-1, или его фармацевтически приемлемой соли, фармацевтической композиции, фармацевтическому агенту для профилактики или лечения связанных с VAP-1 заболеваний, таких как отек желтого пятна, заболевание, связанное со сверхпроницаемостью сосудов, глазные заболевания, связанные с гипоксией или ишемией, и катаракта, и подобных.
Данная заявка основана на патентной заявке No.2008-021588, поданной в Японии, содержание которой включено в настоящее описание посредством ссылки во всей его полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ БЕНЗОЛА ИЛИ ТИОФЕНА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА VAP-1 | 2009 |
|
RU2526256C2 |
| АМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КСАНТИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ ФОСФОЕНОЛПИРУВАТКАРБОКСИКИНАЗЫ (ФЕПКК), СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2295525C2 |
| ИНГИБИТОРЫ ТИРОЗИНФОСФАТАЗЫ БЕЛКА ЧЕЛОВЕКА И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2435763C2 |
| НОВОЕ ПРОИЗВОДНОЕ АМИНОПИРИДИНА С ИНГИБИТОРНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ SYK. | 2006 |
|
RU2363699C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ ГЛАЗНОГО ОТЕКА, НЕОВАСКУЛЯРИЗАЦИИ И СВЯЗАННЫХ С НИМИ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2600794C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| 2, 5-ДИОКСОИМИДАЗОЛИДИН-4-ИЛ-МЕТИЛКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2003 |
|
RU2326117C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АКТИВАТОРОВ GK | 2002 |
|
RU2374236C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ МЕТАСТАЗОВ РАКОВЫХ КЛЕТОК | 2010 |
|
RU2519123C2 |
Изобретение относится к соединениям общей формулы (I), где R1 представляет собой низший алкилкарбонил; X представляет собой двухвалентный остаток, полученный из тиазола; Z имеет формулу (II), где А представляет собой двухвалентный остаток, полученный из бензола, необязательно замещенного 1 или 2 атомами галогена, или двухвалентный остаток, полученный из тиофена, необязательно замещенного низшим алкилом; В представляет собой -(CH2)l-NR2-СО-, где R2 представляет собой водород, l равно целому числу от 1 до 6, -(СН2)m-O-CO- или -(CH2)m-S-CO- (где m равно целому числу от 0 до 6); D представляет собой -NR3-, где R3 представляет собой водород; E представляет собой амино; Y имеет формулу (III), где J представляет собой связь, низший алкилен, -(CH2)n-O-, -(СН2)n-CO- (где n равно целому числу от 0 до 6); L представляет собой связь, -О-, -NH-; M представляет собой связь, низший алкилен. Также изобретение относится к фармацевтической композиции, содержащей указанные соединения и обладающей ингибирующей активностью в отношении сосудистого адгезивного белка-1 (VAP-1), ингибитору VAP-1, фармацевтическому агенту, к применению соединений формулы (I) и фармацевтического агента для профилактики или лечения связанного с VAP-1 заболевания, способу ингибирования VAP-1 и к способу профилактики или лечения связанного с VAP-1 заболевания. Технический результат - соединения формулы (I) в качестве ингибиторов VAP-1. 8 н. и 6 з.п. ф-лы, 2 табл., 38 пр.
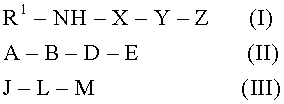
1. Соединение, представленное формулой (I):
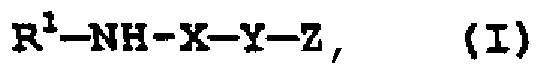
где R1 представляет собой низший алкилкарбонил;
X представляет собой двухвалентный остаток, полученный из тиазола;
Y имеет формулу (III):
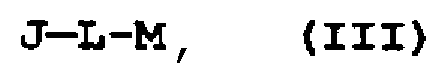
где J представляет собой связь, низший алкилен, -(СН2)n-O, -(СН2)n-СО-
(где n равно целому числу от 0 до 6);
L представляет собой связь, -O-, -NH-;
М представляет собой связь, низший алкилен;
Z имеет формулу (II):

где A представляет собой двухвалентный остаток, полученный из бензола, необязательно замещенного 1 или 2 атомами галогена, или двухвалентный остаток, полученный из тиофена, необязательно замещенного низшим алкилом;
В представляет собой -(CH2)l-NR2-СО-, где R2 представляет собой водород, l равно целому числу от 1 до 6, -(СН2)m-O-CO- или -(CH2)m-S-CO- (где m равно целому числу от 0 до 6);
D представляет собой -NR3-, где R3 представляет собой водород; и
E представляет собой амино;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где соединением, представленным указанной выше формулой (I), является
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат,
2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил-гидразинкарбоксилат,
2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил-гидразинкарбоксилат,
4-{2-[(гидразинокарбонил)окси]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилат,
2-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]карбонил}амино)фенил]этил-гидразинкарбоксилат,
3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат,
2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил-гидразинкарбоксилат,
{5-[2-(2-ацетиламино-1,3-тиазол-4-ил)этил]тиофен-2-ил}метил-гидразинкарбоксилат,
2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил-гидразинкарбоксилат,
3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил-гидразинкарбоксилат,
3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил-гидразинкарбоксилат,
3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил-гидразинкарбоксилат,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
N-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2-фторбензил)гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-фторбензил)гидразинкарбоксамид,
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-2,3-дифторбензил)гидразинкарбоксамид,
N-[4-({[2-(ацетиламино)-1,3-тиазол-4-ил]метил}амино)бензил]гидразинкарбоксамид,
2-(ацетиламино)-N-(4-{[(гидразинокарбонил)амино]метил}фенил)-1,3-тиазол-4-карбоксамид,
N-[2-(4-{[2-(ацетиламино)-1,3-тиазол-4-ил]метокси}фенил)этил]гидразинкарбоксамид,
4-{2-[(гидразинокарбонил)амино]этил}фенил-2-(ацетиламино)-1,3-тиазол-4-карбоксилат,
N-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
N-[2-(3-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарбоксамид,
N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарбоксамид,
N-[2-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)этил]гидразинкарбоксамид,
N-[3-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)пропил]гидразинкарбоксамид,
N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-3-ил)пропил]гидразинкарбоксамид,
N-[3-(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}-3-метилтиофен-2-ил)пропил]гидразинкарбоксамид,
S-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарботиоат,
S-[2-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил)этил]гидразинкарботиоат или
S-[(5-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}тиофен-2-ил)метил]гидразинкарботиоат,
или его фармацевтически приемлемая соль.
3.Соединение по п.1, где соединением, представленным указанной выше формулой (1), является
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}фенил-гидразинкарбоксилат,
4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил-гидразинкарбоксилат или
N-(4-{2-[2-(ацетиламино)-1,3-тиазол-4-ил]этил}бензил)гидразинкарбоксамид,
или его фармацевтически приемлемая соль.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, которое применяют в качестве активного ингредиента фармацевтического агента, обладающего ингибирующей активностью в отношении сосудистого адгезивного белка-1 (VAP-1).
5. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении сосудистого адгезивного белка-1 (VAP-1), содержащая соединение по любому из пп.1-3 или его фармацевтически приемлемую соль в качестве активного ингредиента в количестве, достаточном для профилактического или терапевтического лечения связанного с VAP-1 заболевания, и фармацевтически приемлемый носитель.
6. Ингибитор VAP-1, содержащий соединение по любому из пп.1-3 или его фармацевтически приемлемую соль в качестве активного ингредиента.
7. Фармацевтический агент для профилактики или лечения связанного с VAP-1 заболевания, который содержит соединение по любому из пп.1-3 или его фармацевтически приемлемую соль в качестве активного ингредиента в количестве, достаточном для профилактического или терапевтического лечения связанного с VAP-1 заболевания.
8. Фармацевтический агент по п.7, где указанное выше связанное с VAP-1 заболевание представляет собой отек желтого пятна (диабетический и недиабетический отек желтого пятна), возрастную дегенерацию желтого пятна, возрастную дисковидную дегенерацию желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическую ретинопатию, хориоретинопатию, неоваскулярную макулопатию, неоваскулярную глаукому, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаза, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаза, вызванное физическим повреждением глаза, симптом, вызванный воспалительным заболеванием глаза, включая зуд, воспаление, отек и язву, эритему, многоформную экссудативную эритему, узловатую эритему, кольцевидную эритему, склередему взрослых, дерматит (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз), ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит, цирроз печени, эссенциальную стабилизированную гипертензию, диабет, артериосклероз, эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанную с подагрой и артритом, воспалительное заболевание или симптом соединительной ткани (ревматоидный артрит, анкилозирующий спондилит, псориатический артрит и остеоартрит или дегенеративное заболевание суставов, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системную красную волчанку, дискоидную красную волчанку, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическую полимиалгию, васкулит, темпоральный артрит, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит, воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление слизистой мембраны рта (стоматит и рецидивирующий афтозный стоматит)], воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера и ишемическое/реперфузионное повреждение, связанное с ишемическим инсультом), легочное воспалительное заболевание или симптом (астму, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания), заболевание, связанное с метаболизмом углеводов (диабет и осложнения, полученные в результате диабета (диабетическую невропатию, диабетическую нефропатию)), включая заболевание микрососудов и больших сосудов (артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (множественную невропатию, мононевропатию и вегетативную невропатию), язву стопы, проблему суставов и увеличение риска инфекции), заболевание, связанное с нарушением дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (артериосклероз и ожирение), сосудистое заболевание [артероматозный атеросклероз, неартероматозное атеросклеротическое заболевание, ишемическое заболевание сердца, включая инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)], хронический артрит, воспалительное заболевание кишечника или SSAO-опосредованные осложнения [диабет (инсулин-зависимый диабет (IDDM) и инсулин-независимый диабет (NIDDM)) и сосудистое осложнение (инфаркт, стенокардию, апоплексию мозга, ампутацию, медицинскую слепоту и почечную недостаточность)], глазное заболевание, связанное с гипоксией или ишемией [ретинопатию недоношенных, пролиферативную диабетическую ретинопатию, полипообразную хориоидальную васкулопатию, ретинальную ангиоматозную пролиферацию, окклюзию центральной артерии сетчатки, окклюзию ветви центральной вены сетчатки, болезнь Коутса, семейную экссудативную витреоретинопатию, болезнь отсутствия пульса (болезнь Такаясу), болезнь Илза, антифосфолипидный синдром, лейкемическую ретинопатию, синдром повышенной вязкости крови, макроглобулинемию, связанную с интерфероном ретинопатию, гипертоническую ретинопатию, ретинопатию при облучении, дефицит стволовых клеток роговичного эпителия] или катаракту.
9. Применение соединения по любому из пп.1-3 или его фармацевтически приемлемой соли для получения фармацевтического агента в качестве ингибитора VAP-1.
10. Применение соединения по любому одному из пп.1-3 или его фармацевтически приемлемой соли для получения фармацевтического агента для профилактики или лечения связанного с VAP-1 заболевания.
11. Применение по п.10, в котором указанное выше связанное с VAP-1 заболевание представляет собой отек желтого пятна (диабетический и недиабетический отек желтого пятна), возрастную дегенерацию желтого пятна, возрастную дисковидную дегенерацию желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическую ретинопатию, хориоретинопатию, неоваскулярную макулопатию, неоваскулярную глаукому, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаз, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаз, вызванное физическим повреждением глаз, симптом, вызванный воспалительным заболеванием глаз, включая зуд, воспаление, отек и язву, эритему, многоформную экссудативную эритему, узловатую эритему, кольцевидную эритему, склередему взрослых, дерматит (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз), ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит, цирроз печени, эссенциальную стабилизированную гипертензию, диабет, артериосклероз, эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанную с подагрой и артритом, воспалительное заболевание или симптом соединительной ткани (ревматоидный артрит, анкилозирующий спондилит, псориатический артрит и остеоартрит или дегенеративное заболевание суставов, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системную красную волчанку, дискоидную красную волчанку, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическую полимиалгию, васкулит, темпоральный артрит, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит), воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление слизистой мембраны рта (стоматит и рецидивирующий афтозный стоматит)], воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера и ишемическое/реперфузионное повреждение, связанное с ишемическим инсультом), легочное воспалительное заболевание или симптом (астму, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания), заболевание, связанное с метаболизмом углеводов (диабет и осложнения, полученные в результате диабета (диабетическую невропатию, диабетическую нефропатию)), включая заболевание микрососудов и больших сосудов (артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (множественную невропатию, мононевропатию и вегетативную невропатию), язву стопы, проблему суставов и увеличение риска инфекции), заболевание, связанное с нарушением дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (артериосклероз и ожирение), сосудистое заболевание [артероматозный атеросклероз, неартероматозное атеросклеротическое заболевание, ишемическое заболевание сердца, включающее инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)], хронический артрит, воспалительное заболевание кишечника или SSAO-опосредованное осложнение [диабет (инсулин-зависимый диабет (IDDM) и инсулин-независимый диабет (NIDDM)) и сосудистое осложнение (инфаркт, стенокардию, апоплексию мозга, ампутацию, медицинскую слепоту и почечную недостаточность)], глазное заболевание, связанное с гипоксией или ишемией [ретинопатию недоношенных, пролиферативную диабетическую ретинопатию, полипообразную хориоидальную васкулопатию, ретинальную ангиоматозную пролиферацию, окклюзию центральной артерии сетчатки, окклюзию ветви центральной вены сетчатки, болезнь Коутса, семейную экссудативную витреоретинопатию, болезнь отсутствия пульса (болезнь Такаясу), болезнь Илза, антифосфолипидный синдром, лейкемическую ретинопатию, синдром повышенной вязкости крови, макроглобулинемию, связанную с интерфероном ретинопатию, гипертоническую ретинопатию, ретинопатию при облучении, дефицит стволовых клеток роговичного эпителия] или катаракту.
12. Способ ингибирования VAP-1 у субъекта, включающий введение эффективного количества соединения по любому из пп.1-3 или его фармацевтически приемлемой соли данному субъекту.
13. Способ профилактики или лечения связанного с VAP-1 заболевания у субъекта, включающий введение эффективного количества соединения по любому из пп.1-3 или его фармацевтически приемлемой соли данному субъекту.
14. Способ по п.13, в котором указанное выше связанное с VAP-1 заболевание представляет собой отек желтого пятна (диабетический и недиабетический отек желтого пятна), возрастную дегенерацию желтого пятна, возрастную дисковидную дегенерацию желтого пятна, кистозный макулярный отек, пальпебральный отек, отек сетчатки, диабетическую ретинопатию, хориоретинопатию, неоваскулярную макулопатию, неоваскулярную глаукому, увеит, воспаление радужной оболочки глаза, васкулит сетчатки глаза, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, ретинальный пигментный эпителиит, конъюнктивит, циклит, склерит, эписклерит, неврит зрительного нерва, ретробульбарный неврит зрительного нерва, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюнктивы, хронический монетовидный кератит, кератит Тайджесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаз, вызванное бактериальной или вирусной инфекцией и операцией на глазах, воспалительное заболевание глаз, вызванное физическим повреждением глаза, симптом, вызванный воспалительным заболеванием глаз, включая зуд, воспаление, отек и язву, эритему, многоформную экссудативную эритему, узловатую эритему, кольцевидную эритему, склередему взрослых, дерматит (псориаз, аллергическое поражение, красный плоский лишай, розовый лишай, контактный дерматит, атопический дерматит, красный волосистый питириаз), ангионевротический отек, отек гортани, отек языка, подсвязочный ларингит, бронхит, ринит, фарингит, синусит и ларингит или средний отит, цирроз печени, эссенциальную стабилизированную гипертензию, диабет, артериосклероз, эндотелиальное повреждение (при диабете, артериосклерозе и гипертензии), сердечно-сосудистое заболевание, связанное с диабетом или уремией, боль, связанную с подагрой и артритом, воспалительное заболевание или симптом соединительной ткани (ревматоидный артрит, анкилозирующий спондилит, псориатический артрит и остеоартрит или дегенеративное заболевание суставов, синдром Рейтера, синдром Шегрена, синдром Бехчета, рецидивирующий полихондрид, системную красную волчанка, дискоидную красную волчанку, системный склероз, эозинофильный фасциит, полимиозит, дерматомиозит, ревматическую полимиалгию, васкулит, темпоральный артритом, нодозный полиартериит, гранулематоз Вегенера, смешанное заболевание соединительной ткани и ювенильный ревматоидный артрит), воспалительное заболевание или симптом желудочно-кишечного тракта [болезнь Крона, язвенный колит, синдром раздраженного кишечника (слизистый колит), фиброз печени, воспаление слизистой мембраны (стоматит и рецидивирующий афтозный стоматит)], воспалительное заболевание или симптом центральной нервной системы (рассеянный склероз, болезнь Альцгеймера и ишемическое/реперфузионное повреждение, связанное с ишемическим инсультом), легочное воспалительное заболевание или симптом (астму, синдром расстройства дыхания у взрослых, хронические облитерирующие легочные заболевания), заболевание, связанное с метаболизмом углеводов (диабет и осложнения, полученные в результате диабета (диабетическую невропатию, диабетическую нефропатию)), включая заболевание микрососудов и больших сосудов (артериосклероз, ретинопатию, нефропатию, нефротический синдром и невропатию (множественную невропатию, мононевропатию и вегетативную невропатию), язву стопы, проблему суставов и увеличение риска инфекции), заболевание, связанное с нарушением дифференциации или функционирования адипоцитов или функционирования клеток гладких мышц (артериосклероз и ожирение), сосудистое заболевание [артероматозный атеросклероз, неартероматозное атеросклеротическое заболевание, ишемическое заболевание сердца, включая инфаркт миокарда и закупорку периферических артерий, болезнь Рейно и феномен Рейно, облитерирующий тромбангиит (болезнь Бюргера)], хронический артрит, воспалительное заболевание кишечника, или SSAO-опосредованное осложнения [диабет (инсулин-зависимый диабет (IDDM) и инсулин-независимый диабет (NIDDM)) и сосудистое осложнение (инфаркт, стенокардию, апоплексию мозга, ампутацию, медицинскую слепоту и почечную недостаточность)], глазное заболевание, связанное с гипоксией или ишемией [ретинопатию недоношенных, пролиферативную диабетическую ретинопатию, полипообразную хориоидальную васкулопатию, ретинальную ангиоматозную пролиферацию, окклюзию центральной артерии сетчатки, окклюзию ветви центральной вены сетчатки, болезнь Коутса, семейную экссудативную витреоретинопатию, болезнь отсутствия пульса (болезнь Такаясу), болезнь Илза, антифосфолипидный синдром, лейкемическую ретинопатию, синдром повышенной вязкости крови, макроглобулинемию, связанную с интерфероном ретинопатию, гипертоническую ретинопатию, ретинопатию при облучении, дефицит стволовых клеток роговичного эпителия] или катаракту.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Сифонное устройство для отвода воды из рек и верховых озер и подвода ее к водяному двигателю | 1926 |
|
SU5375A1 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА | 1991 |
|
RU2048468C1 |

