Изобретение относится к технологии получения неорганических материалов, а именно к способу получения нанокристаллического кремнийзамещенного гидроксиапатита, который может быть использован для производства медицинских материалов, стимулирующих восстановление дефектов костной ткани, в том числе в стоматологии.
В последние годы в медицинской практике с целью замены, восстановления или реставрации поврежденных костей и суставов в качестве биосовместимых имплантатов весьма эффективно используются кальций-фосфатные материалы.
Ограниченные источники аутогенных материалов, а также риски, связанные с использованием аллогенных или ксеногенных материалов, сделали актуальным широкое применение в стоматологической и ортопедической практике синтетических костнопластических материалов. Решающим фактором при выборе подобных синтетических материалов является то, что они должны быть максимально биосовместимыми и биологически активными материалами, способными к полноценной интеграции в естественный процесс ремоделирования костной ткани.
Одним из наиболее востребованных костнопластических синтетических материалов кальций-фосфатного состава, среди разработанных в течение последних 30-40 лет, является гидроксиапатит (далее ГАП), который соответствует химической формуле Са10(РО4)6(ОН)2.
Гидроксиапатит можно считать кристаллохимическим аналогом минеральной составляющей тканей скелета животных и людей и в связи с этим он успешно служит базовым компонентом синтетических материалов для ортопедии и стоматологии. Поэтому синтетический ГАП является наиболее биосовместимым ортофосфатом кальция, который, как сообщается, часто готовят по керамической технологии и в качестве костнозамещающего материала применяют в поризованной, гранулированной, плазменно-напыленной и компактированной формах [1]. Однако гидроксиапатитовая керамика не очень хорошо растворяется в организме и не резорбирует в течение разумного периода времени [2]. Предлагаемое время для идеально полной резорбции этих материалов составляет от 1 месяца до 3 лет, в течение которых они будут замещены на новые кости. Поэтому повышение скорости интеграции с костной тканью и биоактивности материалов на основе гидроксилапатита является важной задачей.
Одним из путей решения этой проблемы является синтез наноразмерного гидроксиапатита с последующей термической обработкой ниже температуры спекания. Поэтому для получения наноразмерного ГАП сегодня наибольшее распространение получили методы, основанные на осаждении гидроксиапатита из щелочных водных растворов реагентов, содержащих в стехиометрическом соотношении ионы кальция и фосфат-ионы. Подобный метод, в частности, изложен в патенте РФ №2342938 [3].
Вместе с тем, последние результаты клинической апробации препаратов медицинского назначения, приготовленных с использованием ГАП, отвечающего формуле Са10(РO4)6(ОН)2, показывают, что такой материал, наряду с явными преимуществами обладает рядом недостатков: недостаточной скоростью биорезорбции in vivo и остеоиндукцией, т.е. слабым стимулирующим воздействием на рост новой костной ткани.
Перспективным направлением повышения резорбируемости и остеоиндукции кальций-фосфатных материалов является химическое модифицирование гидроксиапатита, прежде всего, путем получения кремний- или силикатзамещенного гидроксиапатита, далее Si-ГАП. Установлено, что гидроксиапатит, модифицированный силикат-анионами, способствует улучшенной пролиферации остеобластов и росту внеклеточного матрикса, ускоренной минерализации костной ткани [4-5]. Кремнийзамещенные гидроксиапатиты чаще всего описываются формулой:
Ca10(PO4)6-x(SiO4)x(OH)2-x, где х - коэффициент или степень замещения кремния.
Известен способ получения кремнийзамещенного гидроксиапатита путем осаждения из водного раствора реагентов [6], где в качестве источника ионов кальция и фосфат-ионов использовали гидроксид кальция - Са(ОН)2 и ортофосфорную кислоту - Н3РО4. Реакцию между ними проводили при комнатной температуре, а рН поддерживали на уровне 10,5 за счет добавления гидроксида аммония. Кремний в состав ГАП вводили с использованием тетраацетата кремния - Si (СН3СОО)4. Материал был приготовлен по стехиометрии ГАП при условии, что кремний замещает позиции фосфора в кристаллической решетке, а молярное отношение Са/(Р+Si) равно 1,67. Осадок ГАП термообрабатывали при 1200°С в течение 2 часов в атмосфере воздуха. Недостатком этого способа, с одной стороны, является невысокая степень замещения кремния x≤1, что соответствует содержанию кремния в материале 0,8-1,5 мас.%, а с другой - появление примесных фаз, например оксида кальция или трикальцийфосфата после термообработки при использовании более высоких степеней замещения, например, при х>1.
Известен также способ получения кремнийзамещенного гидроксиапатита, согласно которому готовили порошки методом осаждения из водных растворов, используя соотношение Са/(Р+Si), равное 1,67 [7]. Порошки содержали карбонат. После прокаливания были получены апатиты свободные от карбонатов. Используя химическую формулу
Ca10(PO4)6-x(SiO4)x(OH)2-x, где 0≤х≤2
для прокаленных декальцинированных продуктов, авторы способа сообщают, что поведение порошков кремнийсодержащего гидроксиапатита с х>1 очень отличается от композиций с х≤1. Если при коэффициенте замещения на силикат-ионы х≤1 после прокаливания был получен Si-ГАП, свободный от карбонатов, то у порошков с коэффициентом замещения х>1 при нагреве выше 700°С одновременно появилось две фазы: гидроксиапатит и α-трикальцийфосфат, что, как и в вышеуказанном способе [6], является существенным недостатком.
Наиболее близким по технической сущности и достигаемому результату является способ, описанный в патенте US 20100173009 A1 [8].
Указанный способ включает синтез Si-ГАП методом осаждения при рН не менее 9 из водного раствора реагентов, содержащих ортофосфорную кислоту, гидроксид кальция и тетраэтилортосиликат (далее ТЭОС). Причем реагенты вводятся в реакционную смесь при условии обеспечения молярного отношения Са/Р в диапазоне от 2,05 до 2,55, содержания атомов кремния в диапазоне от 2,9 до 6 мас.% и молярного отношения Са/(Р+Si) в диапазоне от 1,50 до 1,66. Раствор ортофосфорной кислоты помещают в капельную воронку и добавляют по каплям в композицию гидроксид кальция/ТЭОС, в течение примерно 60-120 минут. Реакционную смесь перемешивают в течение 2 часов, а затем оставляют для старения около 24 часов. Вся реакция проходит при комнатной температуре. Затем суспензию отфильтровывают с помощью воронки Бюхнера с использованием фильтровальной бумаги и вакуумного насоса. После удаления фильтрата мокрый фильтр-корж помещают в сушильный шкаф и сушат при 90°С в течение 2 дней. После чего высушенный фильтр-корж извлекают, измельчают в мелкий порошок с использованием ступки и пестика, а затем помещают в камерную печь и нагревают в атмосфере воздуха до максимальных температур от 400°С до 900°С в течение одного часа. Скорость нагрева 2,5°С/мин, скорость охлаждения 10°С/мин.
Этот способ позволяет получать поликристаллический чистый или монофазовый кремнийзамещенный гидроксиапатит, состоящий из частиц размером 0,05-5 мкм (или 50-5000 нм соответственно), обладающий относительно высокой скоростью растворения и способностью извлекать достаточные количества кремния в раствор.
Указывается, что преимуществом кремнийзамещенного гидроксиапатита этого изобретения является более высокая растворимость по сравнению с керамическим кремнийзамещенным гидроксиапатитом и обычным немодифицированным гидроксиапатитом, а также более высокий выход кремния при контакте с раствором. Также отмечается, что при выдерживании кремнийзамещенного гидроксиапатита в физиологическом растворе концентрация ионов кальция остается неизменной или увеличивается. Это выгодно отличает его от гидроксиапатитов с низким уровнем замещения кремния, для которых содержание кальция в физиологическом растворе снижается в тот же период времени.
Сообщается, что удельная поверхность порошка кремнийзамещенного гидроксиапатита, получаемого с помощью данного способа составляет от 10 до 90 м2/г, что существенно превышает удельную поверхность порошка обычного гидроксиапатита после нагревания образцов при той же температуре. Например, после нагрева при 900°С удельная поверхность порошка кремнийзамещенного гидроксиапатита, полученного с помощью данного способа, составляет 27 м2/г, а удельная поверхность соответствующего порошка незамещенного гидроксиапатита составляет 13 м2/г.
В то же время в описании указывается, что использование молярного отношения Са/(Р+Si)=1,667 и выше может после нагрева при 900°С приводить к появлению двух фаз: фазы Si - ГАП и 2-й нежелательной фазы - СаО. Кроме того, отмечается, что такой кремнийзамещенный гидроксиапатит может быть представлен формулой:
Ca10-δ(PO4)6-x(SiO4)x(OH)2-x, где δ представляет дефицит кальция, поскольку молярное отношение Са/(Р+Si) имеет значение меньше чем 1,667.
Таким образом, основными недостатками данного технического решения является недостаточная биоактивность полученного продукта, т.е. недостаточно высокая скорость растворения в физиологических средах, невозможность получения стабильного монофазового кремнийсодержащего гидроксиапатита с коэффициентом замещения х в диапазоне от 1 до 2 при молярном отношении реагентов Са/(Р+Si)=1,67, и, как следствие, нестехиометричность такого продукта из-за дефицита катионов кальция. А, как известно, недостаток кальция в костных тканях человека и животных может приводить к более серьезным проблемам, чем дефицит кремния. Также следует иметь в виду, что у кремнийсодержащих гидроксиапатитов, отвечающих формуле Ca10(PO4)6-x(SiO4)x(OH)2-x, нарушается принцип электронейтральности. Это, в свою очередь, является основной причиной нестабильности подобных соединений и объясняет их склонность к деструкции с выделением побочных примесных фаз, особенно при нагревании.
Задачей изобретения является создание способа получения монофазового нанокристаллического кремнийзамещенного гидроксиапатита с повышенной биоактивностью и со степенью замещения кремния х=1-2, при молярном отношении Са/(Р+Si), близком к 1,67.
Технический результат:
- получение стехиометричного монофазового Si-ГАП со средним размером кристаллов 9,95-12,53 нм, удельной поверхностью 108,97-132,58 м2/г, что обеспечивает повышенную биоактивность готового продукта;
- получение стабильного монофазового продукта формулы Ca10(PO4)6-x(SiO4)x(OH)2-x, где 1≤х≤2, при нагревании которого не образуются побочные фазы.
Для решения поставленной задачи получения стабильного монофазового нанокристаллического кремнийзамещенного гидроксиапатита со степенью замещения кремния х=1-2, что соответствует содержанию кремния до 6 мас.%, при молярном отношении Са/(Р+Si), близком к 1,67, предложен способ получения Si-ГАП, включающий синтез путем осаждения из водного раствора реагентов, содержащих ортофосфорную кислоту, гидроксид кальция и тетраэтилортосиликат при рН не менее 9 и молярном отношении Са/Р в диапазоне от 2,0 до 2,5, отстаивание для завершения процесса фазообразования, выделение осадка, высушивание и термообработку осадка, причем синтез ведут путем приливания 10-20%-ного раствора ортофосфорной кислоты со скоростью 0,2-0,8 мл/мин на литр водного раствора композиции гидроксид кальция/тетраэтилортосиликат, приготовленной с использованием 0,08-0,16%-ного водного раствора гидроксида кальция и расчетного количества тетраэтилортосиликата для получения готового продукта со степенью замещения кремнием х=1-2 и молярным отношением Са/(Р+Si), равным 1,67, а термообработку ведут при температуре не ниже 300°С, но не более 400°С.
Способ включает следующие новые признаки:
- скорость приливания 10-20%-ного раствора ортофосфорной кислоты составляет 0,2;
- 0,8 мл/мин на литр водного раствора композиции, приготовленной с использованием 0,08;
- 0,16%-ного водного раствора гидроксида кальция и расчетного количества тетраэтилортосиликата для получения готового продукта;
- термообработку осадка ведут при температуре не ниже 300°С, но не более 400°С, т.к. термообработка ниже 300°С приводит к снижению биоактивности готового продукта, а выше 400°С - нецелесообразно из-за дополнительных энергозатрат.
Использование водного раствора гидроксида кальция с концентрацией менее 0,08 мас.% нецелесообразно по экономическим причинам из-за крайне низкого выхода готового продукта.
Получение и использование истинного водного раствора гидроксида кальция с концентрацией более 0,16 мас.% нецелесообразно из-за низкой растворимости гидроксида кальция.
Скорость приливания 10-20%-ного раствора ортофосфорной кислоты менее 0,2 л/мин на литр водного раствора композиции гидроксида кальция и тетраэтилортосиликата нецелесообразно из-за крайне низкого выхода готового продукта.
Скорость приливания 10-20%-ного раствора ортофосфорной кислоты более 0,8 л/мин на литр водного раствора композиции гидроксида кальция и тетраэтилортосиликата приводит к возможности получения нестабильного нестехиометричного кремнийзамещенного гидроксиапатита Ca10-δ(PO4)6-x(SiO4)x(OH)2-x с нарушенным принципом электронейтральности, который далее при нагревании может разлагаться с образованием трехкальциевого фосфата - Са3(РO4)2, гидроксида кальция - Са(ОН)2 или оксида кальция - СаО.
Теоретическое обоснование предложенного изобретения заключается в следующем. Кремнийзамещенные гидроксиапатиты могут быть представлены формулой - Са10(РO4)6-x(SiO4)x(OH)2-x, где х - коэффициент или степень замещения кремния. При этом наличие до 5-6 мас.% кремния в составе костной ткани является принципиально важным для формирования полноценной естественной кости с требуемыми строением и функционалом. Для обеспечения необходимого содержания кремния и кальция в синтетическом кремнийзамещенном гидроксиапатите коэффициент замещения кремнием в синтезируемом продукте должен быть в пределах х=1-2, а молярное отношение Са/(Р+Si) близким к 1,67.
Однако, если природный гидроксиапатит, отвечающий этим характеристикам, успешно формируется в костях человека и животных, то получение подобного монофазового и стабильного синтетического Si-ГАП, как следует из данных зарубежных и отечественных авторов, до сих пор остается нерешенной технической проблемой. Формально для синтеза кремнийзамещенного ГАП данного состава в щелочной раствор с рН более 9 (как источник групп ОН-) должны быть внесены или присутствовать в требуемом соотношении свободные ионы кальция, фосфат-ионы и силикат-ионы. Однако силикат-ионы, в отличие от ионов Са2+ и РО4 3-, имеют особое химическое поведение в водных растворах, а именно высокую склонность к полимеризации с образованием олигомеров и далее частиц коллоидного кремнезема. Причем скорость полимеризации при прочих равных параметрах возрастает по мере увеличения концентрации ионов SiO4 4- и снижения щелочности среды.
В предлагаемом способе, как и в прототипе, источником силикат-ионов является тетраэтилортосиликат (ТЭОС), который добавляют в раствор гидроксида кальция. В результате щелочного гидролиза ТЭОС в реакционную смесь поступают свободные силикат-ионы по реакции:
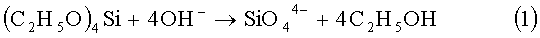
Далее свободные ионы SiO4 4- сразу вступают в реакцию с избытком ионов Са2+ с образованием малорастворимого силиката кальция по реакции:
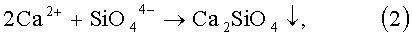
и тем самым уходят из реакционного объема.
Затем в реакционную смесь начинают с заданной скоростью дозировать раствор ортофосфорной кислоты. В этот период происходит формирование промежуточного продукта - аморфного фосфата кальция Са3(РO4)2, который затем трансформируется в термодинамически наиболее устойчивое и самое малорастворимое соединение в системе СаО-Р2O5-Н2O - гидроксиапатит (ГАП). Поскольку Ca2SiO4 является наиболее растворимым соединением в этом ряду, то по мере формирования новой фазы Са3(РO4)2 и далее ГАП, силикат кальция начинает растворяться с высвобождением в объем раствора ионов SiO4 4-. Эти силикат ионы встраиваются в кристаллическую решетку новообразующейся фазы с образованием кремнийзамещенного гидроксиапатита.
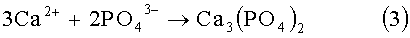
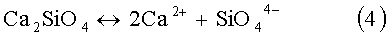
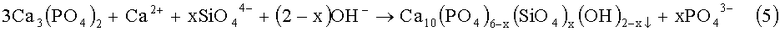
При получении стехиометричного монофазового Si-ГАП со степенью замещения х=1-2 (до 6 мас.% Si) с учетом вышерассмотренного химизма принципиально важно вести процесс синтеза на всех этапах таким образом, чтобы максимально синхронизировать скорости протекания сопряженных процессов (3)-(5). Если часть ионов SiO4 4- после деструкции Ca2SiO4 не успевает встраиваться в структуру ГАП, то происходит быстрая их полимеризация с образованием олигомеров и наночастиц коллоидного кремнезема. Эта часть заполимеризованного кремния уже не может участвовать в изоморфном замещении фосфора, а осаждается на твердых частицах суспензии гидроксида кальция или на поверхности уже образовавшихся кристаллитов незамещенного ГАП. В результате наряду с фазой кремнийзамещенного гидроксиапатита возникают побочные продукты, например нестабильный нестехиометричный гидроксиапатит Ca10-δ(PO4)6-x(SiO4)x(OH)2-x, с нарушенным принципом электронейтральности, который далее может разлагаться с образованием трехкальциевого фосфата - Са3(РO4)2, гидроксида кальция - Са(ОН)2 или оксида кальция - СаО.
Предложенный способ характеризуют следующие фигуры:
Фиг.1. Дифрактограмма РФА образца Si-ГАП при степени замещения кремнием х=2;
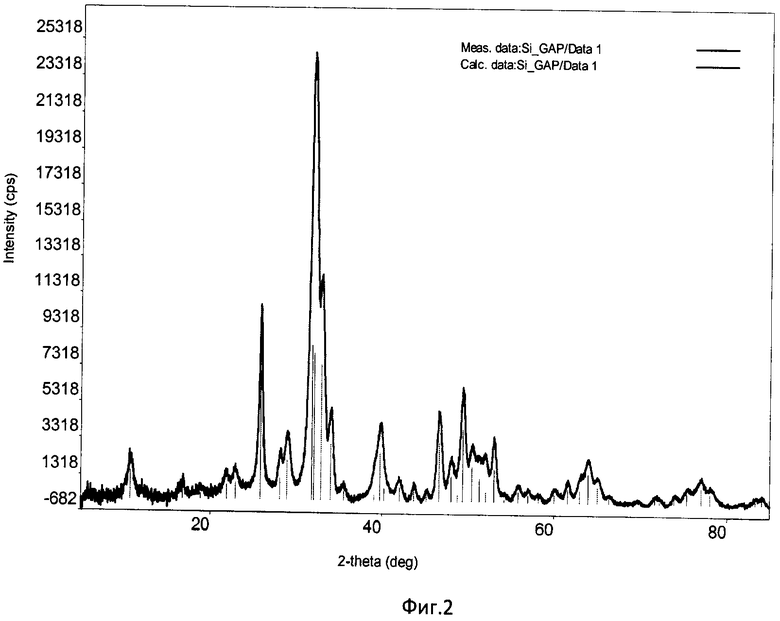
Фиг.2. Дифрактограмма РФА образца Si-ГАП при степени замещения кремнием х=1;
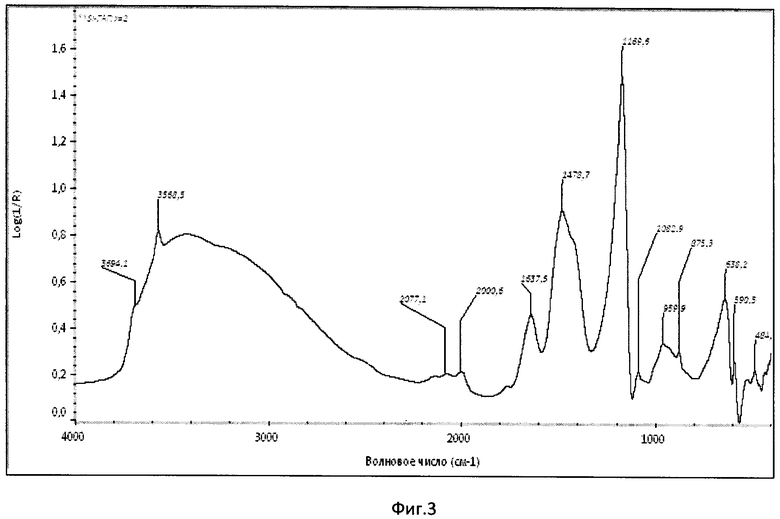
Фиг.3. ИК-спектр образца Si-ГАП при х=2;
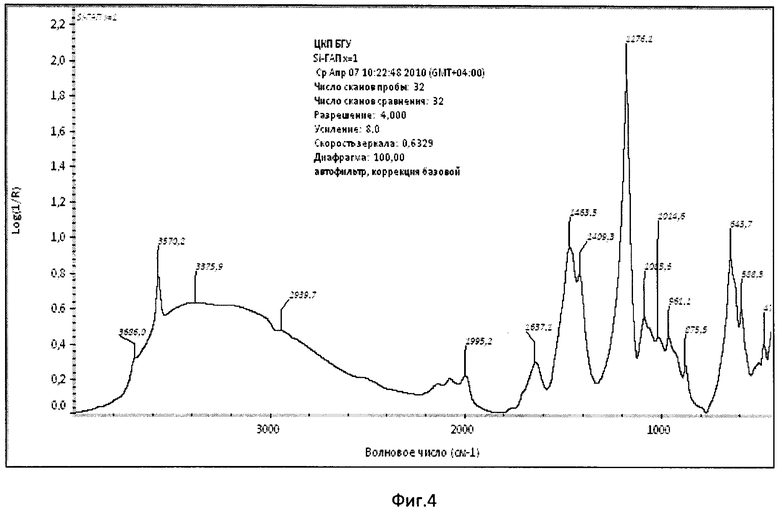
Фиг.4. ИК-спектр образца Si-ГАП при х=1.
Рентгенографические исследования проводили на дифрактометре Rigaku Ultima IV (Япония) с детектором D/teX Ultra. Съемку проводили в режиме на отражение (геометрия Брегга-Брентано) с использованием Cu Kа-излучения (длина волны Х=1.54178 Ǻ). Параметры работы генератора: ускоряющее напряжение 40 кВ, ток трубки 250 мА. Съемку проводили в кварцевых кюветах. Для закрепления порошковых образцов не использовали растворители. Параметры съемки интервал углов 2θ=5-85°, шаг по 20θ 0.02°, скорость регистрации спектров 37 мин. Качественный анализ полученных рентгенограмм и профильный анализ спектров, определение значений параметров решетки проводили с помощью программы PDXL Qualitative Analysis при использовании баз данных ICDD (PDF 2008).
Качественный анализ Si-ГАП на наличие карбонат-ионов в тех или иных позициях кристаллической структуры был проведен при помощи ИК-спектроскопии на ИК-Фурье спектрометре Nicolet 6700 (Thermo Electron Corporation, США) с детектором МСТ-А∗ (50 мкм).
ИК-спектры поглощения образцов регистрировали в диапазоне 400-4000 см-1 со следующими параметрами: число сканов пробы 32; число сканов 32; разрешение 4,000; усиление 8,0; скорость зеркала 0,6329; диафрагма 100,00. Анализ полученных ИК-спектров, определение значений волновых чисел проводили с помощью программного комплекса OMNIC (версия 7.3) при использовании автофильтра, базовой коррекции.
Описание способа поясняется примерами.
Пример 1
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=2 (около 5,8 мас.% Si или 19 мас.% силикат-иона) в случае, когда концентрация водного раствора гидроксида кальция - 0,08 мас.%, а концентрация водного раствора ортофосфорной кислоты - 20 мас.%.
Навеску гидроксида кальция массой 0,8 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,08%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,44 мл ТЭОС (0,42 г, ω=99%) и раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
1,72 мл ортофосфорной кислоты (1,96 г 20% конц. Н3РO4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 0,6 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 300°С в течение 2 часов, скорость нагрева 10°С/мин. Дифрактограмма РФА образца Si-ГАП, полученного при степени замещения кремнием х=2, представлена на фиг.1, ИК-спектр образца Si-ГАП при х=2 представлен на фиг.3.
Пример 2
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=2 (около 5,8 мас.% Si или 19 мас.% силикат-ионов) в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,88 мл ТЭОС (0,84 г, ω=99%) и водный раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
7,4 мл ортофосфорной кислоты (7,84 г 10% конц. Н3РО4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 0,6 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 350°С в течение 2 часов, скорость нагрева 10°С/ мин.
Пример 3
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=2 (около 5,8 мас.% Si или 19 мас.% силикат-ионов) в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а скорость подачи 10%-ного водного раствора ортофосфорной кислоты в реакционную смесь - 0,8 мл/мин.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,88 мл ТЭОС (0,84 г, ω=99%) и водный раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
7,4 мл ортофосфорной кислоты (7,84 г 10% конц. Н3РО4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 0,8 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 300°С в течение 2 часов, скорость нагрева 10°С/мин.
Пример 4
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=2 (около 5,8 мас.% Si или 19 мас.% силикат-ионов) в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а скорость подачи 10%-ного водного раствора ортофосфорной кислоты в реакционную смесь - 0,2 мл/мин.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавили 0,88 мл ТЭОС (0,84 г, ω=99%) и водный раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
Затем 7,4 мл ортофосфорной кислоты (7,84 г 10% конц. Н3РО4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 0,2 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 370°С в течение 2 часов, скорость нагрева 10°С/мин.
Пример 5
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=2 (около 5,8 мас.% Si или 19 мас.% силикат-ионов) в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а скорость подачи 10%-ного водного раствора ортофосфорной кислоты - 1,0 мл/мин.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавили 0,88 мл ТЭОС (0,84 г, ω=99%) и водный раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
Затем 7,4 мл ортофосфорной кислоты (7,84 г 10% конц. Н3РО4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 1,0 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 300°С.
Из таблицы 1 видно, что повышение скорости добавления ортофосфорной кислоты приводит к появлению второй фазы.
Пример 6
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=1 (около 2,85 мас.% Si или 9,35 мас.% силикат-ионов) при концентрации водного раствора гидроксида кальция - 0,16 мас.% и скорости подачи 10%-ного водного раствора ортофосфорной кислоты в реакционную смесь - 0,6 мл/мин.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавили 0,44 мл ТЭОС (0,42 г, ω=99%) и водный раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
Затем 9,3 мл ортофосфорной кислоты (9,80 г 10% конц. Н3РО4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 0,6 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 300°С в течение 2 часов, скорость нагрева 10°С/мин.
Дифрактограмма РФА образца Si-ГАП, полученного при степени замещения кремнием х=1 представлена на фиг.2, а ИК-спектр образца Si-ГАП при х=1 на фиг.4.
Пример 7
Синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/(Р+Si) - 1,67 и степенью замещения на кремний х=2 (около 5,8 мас.% Si или 19 мас.% силикат-иона) в случае, когда концентрация водного раствора гидроксида кальция - 0,08 мас.%, а концентрация водного раствора ортофосфорной кислоты - 20 мас.%.
Навеску гидроксида кальция массой 0,8 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,08%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,44 мл ТЭОС (0,42 г, ω=99%) и раствор интенсивно перемешивали с помощью электрической мешалки еще в течение 5-10 минут.
1,72 мл ортофосфорной кислоты (1,96 г 20% конц. Н3РО4) помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/ТЭОС, со скоростью 0,6 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10, в связи с высоким молярным соотношением Са/Р используемых реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре.
Образовавшийся коллоидный раствор отфильтровывали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 90°С в сушильном шкафу до постоянной массы. После этого Si-ГАП измельчали в мелкий порошок с использованием ступки и пестика и помещали в муфельную печь, где его термообрабатывали при 250°С в течение 2 часов, скорость нагрева 10°С/мин.
Из таблицы 2 видно, что выход кремния после выдерживания в SBF-растворе, несмотря на высокое содержание кремния в образце, равное 5,8 мас.%, примерно соответствует выходу кремния для образца по примеру 6, где содержание кремния в два раза ниже. А по сравнению с оптимальными составами по примерам 1-4, с таким же содержанием кремния в образце, выход кремния в SBF-раствор в полтора раза ниже, что является подтверждением снижения биоактивности в результате термоотбработки при температуре ниже 300°С.
Пример 8
Синтез немодифицированного гидроксиапатита по патенту РФ №2342938.
В связи с тем, что ряд характеристик немодифицированного наногидроксиапатита не был указан в описании данного изобретения их определяли опытным путем. Нанокристаллический гидроксилапатит был получен с помощью «мокрого синтеза» добавлением 20%-ного водного раствора ортофосфорной кислоты (Н3РО4) в насыщенный раствор гидроксида кальция Са(ОН)2 со скоростью 1 мл/мин. Уравнение реакции можно представить следующим образом:
10Са(ОН)2+6Н3РО4=Са10(РO4)6(ОН)2↓+18Н2O
Конечное значение рН доводилось до величины 10,2. Реакционная смесь отстаивалась в течение 24 часа для завершения протекающих процессов кристаллизации. Синтезированная 2%-ная водная суспензия высушивалась в сушильном шкафу при 95°С, затем полученные порошки термообрабатывали в муфельной печи при 350°С в течение 2 часов для дегидратации кристаллов ГАП и его кристаллизации.
Пример 9 (по прототипу)
Синтез кремнийзамещенного гидроксиапатита по патенту US 20100173009 A1.
В связи с тем, что ряд характеристик подобного кремнийзамещенного гидроксиапатита не был указан в описании данного изобретения, их определяли опытным путем. Для этого, в соответствии со способом, приведенным в этом изобретении, проводили синтез кремнийзамещенного гидроксиапатита с молярным отношением Са/Р - 2,5, молярным отношением Са/(Р+Si) - 1,67 и содержанием кремния около 5,8 мас.% (19 мас.% силикат-иона).
0,495 моль гидроксида кальция (36,679 г) добавляли к 1000 мл дистиллированной воды и водную суспензию перемешивали с помощью магнитной мешалки в течение 10-15 минут. Затем 0,099 моль тетраэтилортосиликата (ТЭОС) (20,800 г, ω=99%) добавили непосредственно при перемешивании в суспензию гидроксида кальция. Эту смесь перемешивали в течение 5-10 минут. Приготовление 2%-ного раствора ортофосфорной кислоты - 0,198 моль ортофосфорной кислоты (22,828 г 85% конц. Н3РO4) добавляли в 1000 мл дистиллированной воды и перемешивали с помощью магнитной мешалки в течение 5-10 минут. Этот раствор ортофосфорной кислоты помещали в капельную воронку и добавляли по каплям в реакционную композицию гидроксид кальция/ТЭОС, в течение 60 минут (скорость подачи 2%-ного раствора Н3РО4 - 16 мл/мин). После добавления раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 10. Реакционную смесь перемешивали в течение 2 часов, а затем оставляли для старения около 24 часов. Вся реакция проходила при комнатной температуре. Затем суспензию отфильтровали с помощью воронки Бюхнера, с использованием фильтровальной бумаги и вакуумного насоса. После этого фильтрат был удален и был получен мокрый фильтр-корж, его помещали в сушильный шкаф и сушили при 90°С в течение 2 дней. После этого высушенный фильтр-корж извлекали и измельчали в порошок с использованием ступки и пестика. Этот порошок далее помещали в муфельную лабораторную печь, где его нагревали в атмосфере воздуха до 900°С с выдержкой в течение одного часа. Скорость нагрева составляла 2,5°С/мин, скорость охлаждения - 10°С/мин.
Полученные образцы по примерам 1-9 были подвергнуты исследованиям на определение удельной поверхности, объема и среднего размера пор, а также биоактивности.
Определение удельной поверхности, объема и среднего размера пор материалов осуществлялось по методу БЭТ на автоматизированной сорбционной установке TriStar II 3020 производства Micromeritics (США). Использовался объемный вариант сорбционного метода. Удельная поверхность рассчитывалась по изотерме низкотемпературной сорбции паров азота по одноточечному методу БЭТ в точке Р/Ро=0.318937093. Образцы были выдержаны в инертном газе азота и гелия с одновременным обеспечением нагрева образцов при температуре 350°С.
Определение биоактивности осуществляли следующим образом: точную навеску 0,3 г образца помещали в 50 мл SBF-раствора, моделирующего межтканевую жидкость организма (рН=7,4; катионы: Na+ - mM, К+ - 5 mМ, Са2+ - 2,5 mМ, Mg2+ - 1,5 mМ; анионы: Сl- - 125 mМ, НСО3 - - 27 mМ, НРO4 2- - 1 mМ, SO4 2- - 0,5 mМ). Время экспозиции - 1 час.
Содержание катионов кальция в растворе Са2+ определяли комплексонометрическим методом с помощью раствора 0,005Н Трилона Б, индикатора Эриохром черного Т. Объем аликвоты = 10 мл.
Содержание кремния в растворе определяли рентгенофлюоресцентным методом с использованием анализатора «Спектроскан».
В таблице 1 представлены физико-химические характеристики образцов, полученных по примерам 1-9.
∗SBF-раствор (англ. synthetic body fluid) - среда, моделирующая межтканевую жидкость организма.
Из таблиц 1 и 2 видно, что поставленная задача по созданию способа получения нанокристаллического кремнийзамещенного гидроксиапатита со степенью замещения кремния х=1-2, что соответствует содержанию кремния до 6 мас.%, при молярном отношении Са/(Р+Si), близком к 1,67, и с последующей термической обработкой ниже температуры спекания, решена, и при реализации предложенного способа достигнут заявленный технический результат, заключающийся в том, что получен стехиометричный монофазовый Si-ГАП формулы Ca10(PO4)6-x(SiO4)x(OH)2-x, где 1≤х≤2, со средним размером кристаллов 9,95-12,53 нм, удельной поверхностью 108,97-132,58 м2/г и повышенной биоактивностью, при нагревании которого не образуются побочные фазы.
Литература.
1. Баринов С.М., Комлев B.C. Биокерамика на основе фосфатов кальция. - М.: Наука, 2005. - 187 с.
2. С.Р.А.Т Klein et al., J. Biomed. Mater. Res., 1983, 17, 769.
3. Патент РФ №2342938, МПК (2006.01): A61K 33/42, A61K 33/06, A61K 9/10, A61K 6/033, A61L 27/12, B82B 3/00, C01B 25/32, опубликован 10.01.2009 г.
4. N.Patel, S.М.Best, W.Bonfield et al. A comparative study on the in vivo behavior of hydroxyapatite and silicon substituted hydroxyapatite granules. Journal of materials science: Materials in medicine 13 (2002), p.1199-1206.
5. N.Patel et al. In vivo assessment of hydroxyapatite and silicate-substituted hydroxyapatite granules using an ovine defect model. Journal of materials science: Materials in medicine 16 (2005), p.429-440.
6. I.R.Gibson, S.M.Best, and W.Bonfield. Chemical characterization of silicon-substituted hydroxyapatite. Journal of Biomedical Materials Research, 44:422-428, 1999.
7. Palard et al. Journal of Solid State Chemistry, 181 (2008), p.1950-1960.
8. Патент US 20100173009 A1. Silicate-substituted hydroxyapatite. I.R.Gibson et al.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МОНОФАЗНОГО КРИСТАЛЛИЧЕСКОГО КРЕМНИЙ-ЗАМЕЩЕННОГО ГИДРОКСИЛАПАТИТА | 2014 |
|
RU2580728C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОРАЗМЕРНОГО КАЛЬЦИЙ-ДЕФИЦИТНОГО КАРБОНАТСОДЕРЖАЩЕГО ГИДРОКСИАПАТИТА | 2014 |
|
RU2588525C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОРАЗМЕРНОГО КАЛЬЦИЙДЕФИЦИТНОГО СИЛИКАТ- И ФТОРЗАМЕЩЕННОГО ГИДРОКСИАПАТИТА | 2024 |
|
RU2835237C1 |
| Энтеросорбент на основе кремнийзамещенного биогенного гидроксиапатита кальция | 2023 |
|
RU2811486C1 |
| Способ получения наноразмерного гидроксиапатита | 2020 |
|
RU2736048C1 |
| Способы получения кремнийзамещенного гидроксиапатита и биоактивного покрытия на его основе | 2016 |
|
RU2635189C1 |
| Биологически активное вещество с эффектом разрушения биоплёнок бактерий | 2021 |
|
RU2759294C1 |
| БИОРЕЗОРБИРУЕМЫЙ МАТЕРИАЛ НА ОСНОВЕ АМОРФНОГО ГИДРОКСИАПАТИТА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2510740C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИОННОГО СОРБЕНТА | 2018 |
|
RU2675866C1 |
| ИМПЛАНТАТЫ ДЛЯ ЗАМЕНЫ "НЕСУЩЕЙ НАГРУЗКУ" КОСТИ, ИМЕЮЩИЕ ИЕРАРХИЧЕСКИ ОРГАНИЗОВАННУЮ АРХИТЕКТУРУ, ПОЛУЧЕННЫЕ ПОСРЕДСТВОМ ПРЕВРАЩЕНИЯ РАСТИТЕЛЬНЫХ СТРУКТУР | 2011 |
|
RU2585958C2 |
Изобретение относится к технологии получения неорганических материалов, которые могут быть использованы для производства медицинских материалов, стимулирующих восстановление дефектов костной ткани, в том числе в стоматологии. Способ получения монофазового нанокристаллического кремнийзамещенного гидроксиапатита включает синтез кремнийсодержащего гидроксиапатита методом осаждения из водного раствора реагентов, содержащих ортофосфорную кислоту, гидроксид кальция и тетраэтилортосиликат при рН не менее 9 и молярном отношении Са/Р в диапазоне от 2,0 до 2,5, отстаивание для завершения процесса фазообразования, выделение осадка, высушивание и термообработку осадка, при этом синтез ведут путем приливания 10-20%-ного раствора ортофосфорной кислоты со скоростью 0,2-0,8 мл/мин на литр водного раствора композиции гидроксид кальция /тетраэтилортосиликат, приготовленной с использованием 0,08-0,16%-ного водного раствора гидроксида кальция и расчетного количества тетраэтилортосиликата для получения готового продукта со степенью замещения кремнием х, равным 1-2, и молярным отношением Са/(Р+Si), близким к 1,67, а термообработку ведут при температуре не ниже 300°С, но не более 400°С. Изобретение позволяет получать стехиометрический монофазный продукт фаз со средним размером кристаллов 9,95-12,53 нм, удельной поверхностью 108,97-132,58 м2/г и повышенной биоактивностью, при нагревании которого не образуются побочные фазы. 4 ил., 2 табл., 8 пр. 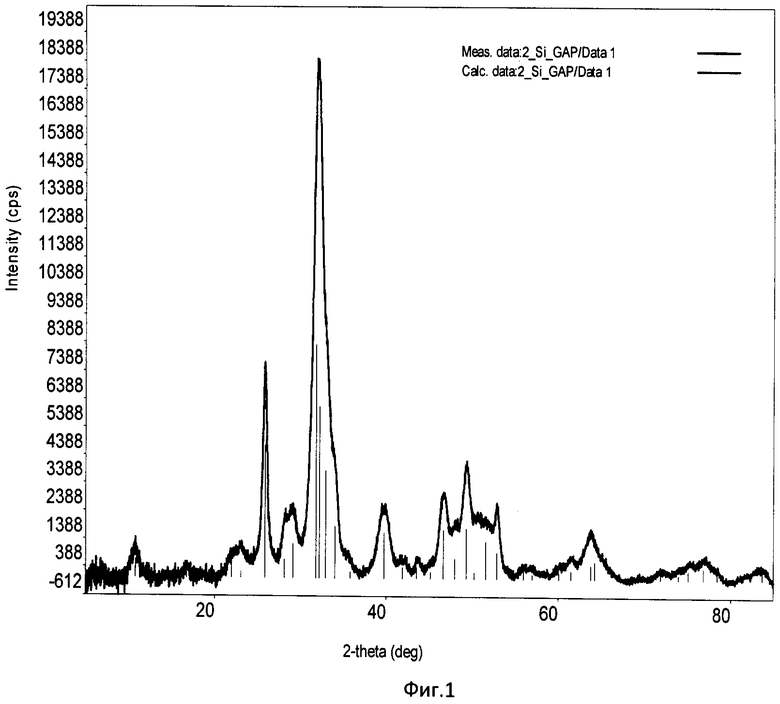
Способ получения монофазового нанокристаллического кремнийзамещенного гидроксиапатита, включающий синтез кремнийсодержащего гидроксиапатита методом осаждения из водного раствора реагентов, содержащих ортофосфорную кислоту, гидроксид кальция и тетраэтилортосиликат при рН не менее 9 и молярном отношении Са/Р в диапазоне от 2,0 до 2,5, отстаивание для завершения процесса фазообразования, выделение осадка, высушивание и термообработку осадка, отличающийся тем, что синтез ведут путем приливания 10-20%-ного раствора ортофосфорной кислоты со скоростью 0,2-0,8 мл/мин на литр водного раствора композиции гидроксид кальция/тетраэтилортосиликат, приготовленной с использованием 0,08-0,16%-ного водного раствора гидроксида кальция и расчетного количества тетраэтилортосиликата для получения готового продукта со степенью замещения кремнием х, равным 1-2, и молярным отношением Са/(Р+Si), близким к 1,67, а термообработку ведут при температуре не ниже 300°С, но не более 400°С.
| MUNIR G | |||
| et al., Novel Patterning of Silicon-Substituted Hydroxyapatite, “Bioceramics Development and Applications”, 2011, vol.1, p.p.1-4 | |||
| БИОЛОГИЧЕСКИ АКТИВНЫЕ НАНОЧАСТИЦЫ ЗАМЕЩЕННОГО КАРБОНАТОМ ГИДРОКСИАПАТИТА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ВКЛЮЧАЮЩИЕ ИХ КОМПОЗИЦИИ | 2006 |
|
RU2426690C2 |
| HUANG J et al, Novel deposition of nano-size silicon substituted hydroxyapatite by electrostatic spraying, “Journal of Materials Science: Materials in | |||

