Изобретение относится к способам получения высокочистых порошков наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита (далее Сад-КГА), которые могут быть использованы для производства медицинских материалов, стимулирующих восстановление дефектов костной ткани, в том числе в стоматологии, и в качестве сорбентов для адсорбции ионов тяжелых металлов.
В настоящее время синтетический гидроксиапатит Ca10(PO4)6OH2 (далее ГА) и материалы на его основе находят широкое применение в стоматологии, реконструктивно-восстановительной костной хирургии, ортопедии в качестве заменяющего материала для поврежденных зубов и костей [Suchanek W., Yoshimura M. Processing and properties of HA-based biomaterials for use as hard tissue replacement implants //J. Mater. Res. Soc. 1998. V.13. №1. P.94-103]. Многочисленные исследования подтверждают биологическую совместимость ГА с организмом, а также его близкие показатели физических и химических свойств [Bauer I W, Li S. P., Han Y. C. and Yin L. M. Internalization of hydroxyapatite nanoparticles in liver cancer cells //J Mater Sci: Mater Med. 2008. V.19. P.1091-1095.; Abdel-Gawad E. I. and Awwad S. Biocompatibility of Intravenous Nano Hydroxyapatite in Male Rats //Nat. and Sci.2010.V.8. P.60-68].
Благодаря высокой адсорбционной способности по отношению к ионам тяжелых металлов в настоящее время материалы на основе ГА представляют большой интерес к использованию в качестве сорбентов для решения таких экологических проблем как очистка сточных вод, утилизация промышленных отходов, восстановление загрязненных почв [Janga, S. H., Jeonga,Y.G., Mina, B.G., Lyoob, W.S. and Leea, S.L. Preparation and lead ion removal property of hydroxyapatite polyacrylamide composite hydrogels //J. Hazardous Materials. 2008. V.159. P.294-3002]. Кроме того, гидроксиапатиты могут успешно применяться для извлечения ионов тяжелых металлов из крови и восстановления функциональности печени и почки [Removal of Lead Nitrate Toxicity //Journal of American Science. 2011. Vol.7. №.1. P. 105-119 Abdel-Gawad E.I., Awwad S.A. In-vivo and in-vitro prediction of the efficiency of Nano-Synthesized Material in].
Было установлено, что ГА в живых организмах не находится в чистом виде, а всегда содержит в своей структуре примеси, преимущественно карбонат-ионы, благодаря их способности адаптироваться к постоянно меняющимся условиям внутренней среды. Карбонатные группы, содержащиеся в кости (около 4-8% по массе), находятся в нестабильном положении, замещая либо ОН--группы (А-тип замещения), либо (РО4)3--группы (В-тип замещения). Для костной ткани характерен смешанный АВ-тип замещения [Баринов С.М., Комлев В.С. Биокерамика на основе фосфатов кальция. М.: Наука 2005. 204c]. По результатам исследований ряда работ выяснено, что в костях и зубах ГА не является чистым, стехиометрическим Ca10(PO4)6(OH)2 с молярным соотношением Ca/P=10/6=1,67 [Elliott, J. C. Structure and chemistry of the apatites and other calcium orthophosphates //Elsevier: Amsterdam, Holland. 1994. 404p; Dorozhkin, S. V. Calcium orthophosphates //J. Mater. Sci. 2007. V.42. P.1061-1095; Dorozhkin, S. V. Calcium orthophosphates in nature, biology and medicine //Materials. 2009. V.2. P.399-498].
В основном можно считать, что апатит биологического происхождения является кальций-дефицитным карбонатсодержащим с молярным соотношением Ca/(P+CO3)<1,67.
В связи с вышеизложенным синтез высокобиосовместимого наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита для медицинских целей, а также в качестве высокоэффективного сорбента ионов тяжелых металлов для детоксикации организма человека и восстановления окружающей среды является весьма актуальным.
Известен способ получения карбонатсодержащего гидроксиапатита (далее КГА) по патенту EP 0722772 A1, опубликованному 24.07.1996 г., путем нагрева стехиометрического ГА в среде углекислого газа из атмосферного воздуха при примерно 900°С в течение несколько дней. Это трудно контролируемый процесс, который, с одной стороны, приводит к получению КГА с низким коэффициентом замещения, с другой стороны, не всегда получают однофазные образцы. При этом карбонат-анионы замещаются в неоптимальные позиции т.е. в А-позиции, а не в В-позиции, что соответствует получению биомиметического материала подобного биоапатиту.
Известен также способ получения КГА методом осаждения из водных растворов, описанный в источнике J. C. Merry, I. R. Gibson, S. M. Best, W. Bonfield. Synthesis and characterization of carbonate hydroxyapatite (Journal of materials science: materials in medicine. 1998. V.9. P.779-783.), где в качестве источника ионов кальция и фосфат-ионов использовали четырехводный нитрат кальция - Са(NO3)2·4H2O и гидрофосфат аммония - (NH4)2HPO4. Реакцию проводили при температуре 20°С, а рН поддерживали на уровне ≥ 11 за счет добавления раствора гидроксида аммония. Источником карбонат-ионов являлся гидрокарбонат натрия NaHCO3, который добавлялся к раствору гидрофосфата аммония до синтеза. Синтез осуществляли добавлением по каплям смеси растворов гидрофосфата аммония и гидрокарбоната натрия к раствору нитрата кальция. Полученную суспензию ставили для старения на 24 часа. Затем осадок тщательно промывали дистиллированной водой, отфильтровали под вакуумом с последующим высушиванием и измельчением. Недостатками данного способа являются низкая термостабильность продукта при обжиге/спекании, т.е. потеря больших количеств ионов карбоната при нагревании, и трудность за контролем степени замещения карбоната. Кроме того, полученные частицы КГА имеют большой размер и, следовательно, низкую удельную поверхность, а также происходит негативное дополнительное замещение ионов Na+или NH4 +в структуре гидроксиапатита.
В патенте EP 0722773 A1 (опубликован 24.07.1996 г.) описан метод получения КГА А-типа, у которого карбонат-анионы замещались только по позиции ОН-групп в структуре ГА. К недостатку данного метода относится несходство по структуре полученных образцов к биоапатиту природной кости.
В патенте EP 0625490 A1 (опубликован 23.11.1994 г.) предложен метод получения КГА, который имеет молярное соотношение Са/Р близко к 1,66. В качестве источника ионов СО3 2- использовали Na2CO3. Недостаток данного метода состоит в том, что вместе с карбонат-ионами в кристаллической решетке ГА идет нежелательное примесное замещение катионов Na+.
В патенте WO1994008458 A1 (опубликован 28.04.1994 г.) описан способ получения КГА, где исходные реагенты смешивали так, чтобы сформировать гидроксиапатитный цемент при комнатной или физиологической температуре. Источником карбонат-ионов является твердый карбонат кальция СаСО3. Полученный материал представляет собой низкокристаллический или аморфный апатит, который содержит нежелательные ионы натрия.
Патент EP 0342932 A1 (опубликован 23.11.1989 г.) описывает способ получения КГА путем добавления Ca(OH)2 и СаСО3 к суспензии CaHPO4. Ионы CO3 2-, которые вводятся в реакционную смесь, находятся в виде нерастворимого CaCO3, а не в виде раствора. Молярное соотношение Са/Р согласно этому патенту всегда меньше 1,67. После спекания при температуре от 1000°С до 1100°С содержание карбоната полученного материала уменьшается менее 0,1%. Недостаток данного метода заключается в том, что полученный продукт содержит в своем составе примесь CaCO3 и высокая температура спекания.
В патенте WO 2001083367 A2 (опубликован 08.11.2001 г.) получали КГА путем выдерживания кальция фосфата в сверхкритическом или конденсированном диоксиде углерода. Недостатком данного способа являются техническая сложность и крайне высокая стоимость, которые не позволяют организовать массовое производство продукта.
Наиболее близким по технической сущности является способ, описанный в патенте US 6582672B1 (опубликован 24.06.2003 г. ). Указанный способ получения монофазного КГА включает синтез КГА методом осаждения при рН 10,5-11 путем смешения водных растворов реагентов, содержащих ионы СО3 2-, РО4 3- и Са2+. Причем реагенты вводятся в реакционную смесь при условии обеспечения молярного соотношения Са/Р выше 1,67, содержание СО3 2-, замещенного в Б-позиции или в А и Б позициях, не должно быть выше 5%. Углекислый газ барботируют в деионизированную воду в течение 30 минут, затем к этому раствору прибавляют ортофосфорную кислоту. В полученную смесь при постоянном перемешивании добавляют по каплям суспензию Са(ОН)2 со скоростью 5,5 мл/мин, рН реакционной смеси поддерживают в интервале 10,5-11 путем добавления концентрированного аммиака. Полученную суспензию после синтеза продолжают перемешивать в течение 2 часов и затем оставляют для старения около 12 часов. Вся реакция проходит при комнатной температуре. Осадок фильтруют, промывают дистиллированной водой для удаления избытка аммиака с последующим высушиванием при 80°С в течение 12 часов. После чего высушенный фильтр-корж извлекают, измельчают в мелкий порошок. Частицы порошка имеют средний размер ниже 100 мкм. Это способ позволяет получать монофазовый карбонатсодержащий гидроксиапатит, не содержащий ионы Na+, NH4 +. Основными недостатками данного способа являются низкое содержание карбонат-ионов, трудность точного контроля степени замещения СО3 2-, а кристаллохимическая формула полученного карбонатсодержащего гидроксиапатита не соответствует формуле кальций-дефицитного гидроксиапатита. Кроме того, частицы КГА, полученные данным методом, имеют большой средний размер около 100 мкм.
Задачей данного изобретения является разработка способа получения монофазного наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита наиболее близкого по химическому составу и структуре к природной кости с высокими биоактивностью и удельной поверхностью при молярном соотношении Са/(Р+СО3 2-)<1,67.
Технический результат:
- получение монофазного продукта Сад-КГА наиболее близкого по химическому составу и структуре к природной кости формулы Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+; х - коэффициент или степень замещения карбоната в интервале от 0,76 до 1,21, а массовое содержание карбонат-ионов от 5% до 8%;
- в полученном Сад-КГА молярное соотношение Са/(Р+СО3 2-)<1,67, средний размер кристаллов от 8 нм до 70 нм, удельная поверхность 90-200 м2/г, что обеспечивает повышенную биоактивность и адсорбционную способность готового продукта.
Для решения поставленной задачи получения монофазного наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита предложен способ получения Сад-КГА, включающий синтез путем осаждения из водного раствора реагентов, содержащих ионы СО3 2-, РО4 3- и Са2+, отстаивание при комнатной температуре для завершения процесса фазообразования, выделение осадка и высушивание до постоянной массы с последующем измельчением.
Способ включает следующие новые признаки:
- готовят водный раствор композиции гидроксид кальция/карбонат аммония с использованием 0,08-0,16%-ного водного раствора гидроксида кальция и расчетного количества карбоната аммония для получения готового продукта с массовым содержанием карбонат-ионов от 5 до 8 мас.% и молярным соотношением Са/(Р+СО3 2-) ниже 1,67;
- приливают раствор ортофосфорной кислоты со скоростью 0,5-5 мл/мин на литр водного раствора к композиции гидроксид кальция/карбонат аммония;
- рН реакционной смеси поддерживают выше 11.
В качестве поставщика ионов СО3 2- использовали карбонат аммония, который позволяет более точно контролировать содержание СО3 2- в полученном продукте и вносит вклад в поддержание значения рН реакционной смеси.
Количество карбонат-ионов в продукте составляет от 5 до 8 мас.%, что соответствует оптимальному содержанию карбонат-ионов в природной кости. При этом не требуется проведения контроля содержания карбонат-ионов, что обусловливается точной навеской (NH4)2CO3.
Скорость приливания 10-20%-ного раствора ортофосфорной кислоты менее 0,5 мл/мин на литр водного раствора композиции гидроксида кальция и карбоната аммония нецелесообразна из-за крайне большой длительности процесса синтеза.
Скорость приливания 10-20%-ного раствора ортофосфорной кислоты более 5 мл/мин на литр водного раствора композиции гидроксида кальция и карбоната аммония приводит к возможности получения продукта с большой дефектностью в структуре, т.к. при быстром приливании кислоты карбонат-ионы не успевают встраиваться в структуру гидроксиапатита, большая часть их адсорбируется на поверхности образовавшихся кристаллов.
Молярное соотношение Са/(Р+СО3 2-) выше или равно 1,67, нецелесообразно из-за того, что не дает возможность получить кальций-дефицитный карбонатсодержащий гидроксиапатит и может приводить к появлению побочных продуктов, например СаСО3, Са3(РО4)2.
Если молярное соотношение Са/(Р+СО3 2-) меньше 1,67 вызывается не из-за дефицита Са, а из-за увеличения содержания замещенных карбонат-ионов в дополнительных позициях, то структура полученного гидроксиапатита не соответствует структуре природной кости.
Значение рН реакционной смеси, поддерживающееся выше 11 благодаря использованию насыщенного раствора гидроксида кальция и карбоната аммония, обеспечивает большое количество гидроксильных групп, что позволяет получить продукт с более завершенной структурой.
Теоретическое обоснование предложенного изобретения заключается в следующем.
Карбонатсодержащие кальций-дефицитные гидроксиапатиты могут быть представлены формулой:
Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+; х - коэффициент или степень замещения СО3 2-.
При этом наличие 5-8 мас.% карбоната в составе костной ткани является принципиально важным для формирования полноценной естественной кости с требуемыми строением и функционалом. Для обеспечения необходимого содержания карбоната и кальция в синтетическом карбонатсодержащем гидроксиапатите степень замещения СО3 2- в синтезируемом продукте должна быть в пределах х=0,76-1,21, а молярное соотношение Са/(Р+CO3 2-) ниже 1,67.
В предлагаемом способе источником карбонат-ионов является (NH4)2CO3, который добавляют в раствор гидроксида кальция. В результате диссоциации в реакционную смесь поступают свободные карбонат-ионы по реакции:
(NH4)2CO3 →2NH4 ++CO3 2- (1)
Далее свободные ионы CO3 2- сразу вступают в реакцию с избытком ионов Са2+с образованием малорастворимого карбонат кальция по реакции:
Са2++СО3 2- → СaCO3↓ (2)
и тем самым уходят из реакционного объема.
Затем в реакционную смесь начинают с заданной скоростью дозировать раствор ортофосфорной кислоты. В этот период происходит формирование промежуточного продукта - аморфного фосфата кальция
3Са2++2PО4 3- → Сa3(PO4)2↓ (3)
Аморфный фосфат кальция затем трансформируется в термодинамически наиболее устойчивое и самое малорастворимое соединение в системе:
гидроксиапатит СаО-Р2O5-Н2O.
Поскольку CaCO3 является наиболее растворимым соединением в этом ряду, то по мере формирования новой фазы Са3(РO4)2 и далее ГА карбонат кальция начинает растворяться с высвобождением в объем раствора ионов CO3 2-. Новообразующаяся фаза фосфата кальция, адсорбируя карбонат-ионы и гидроксильные ионы, образует карбонатсодержащий гидроксиапатит.
СaCO3↓ → Са2++СО3 2- (4)
(3-x/2)Сa3(PO4)2+(1-d+3x/2)Са2++xСО3 2-+(2+x-2d)OH- → Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d (5)
Предложенный способ характеризуют следующие фигуры.
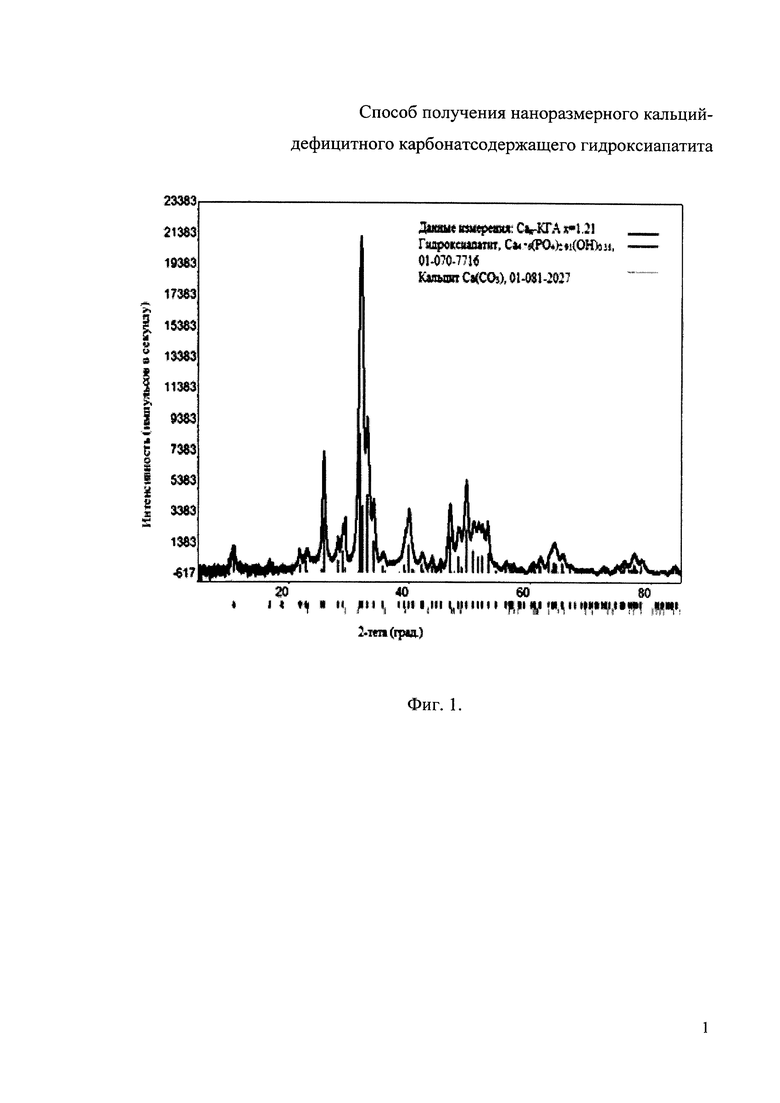
Фиг. 1. Дифрактограмма РФА образца Сад-КГА с молярным соотношением Са/(Р+СО3 2-)=1,50, полученного при степени замещения карбоната х=0,76 и содержании СО3 2- равном 5 мас.%;
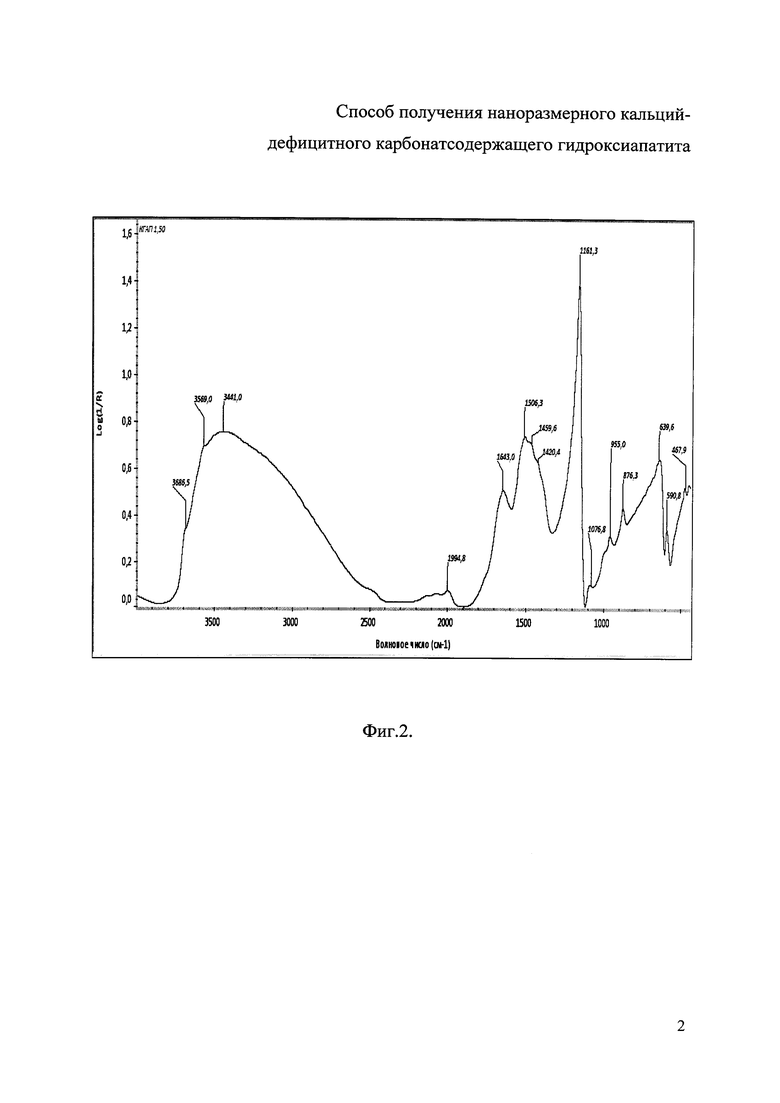
Фиг. 2. Ик-спектр образца Сад-КГА с молярным соотношением Са/(Р+СО3 2-)=1,50, полученного при степени замещения карбоната х=0,76 и содержании СО3 2- равном 5 мас.%;
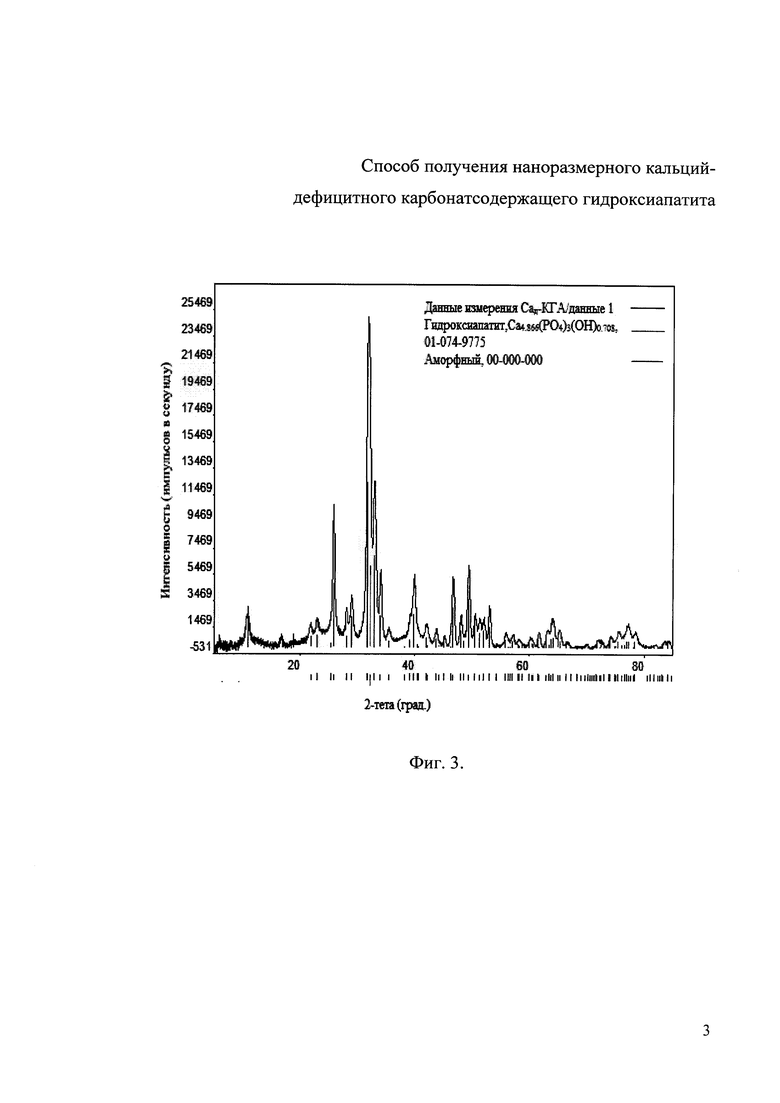
Фиг. 3. Дифрактограмма РФА образца Сад-КГА с молярным соотношением Са/(Р+СО3 2-)=1,67, полученного при степени замещения карбоната х=1,21 и содержании СО3 2- равном 8 мас.%.
Фиг. 1 и 3 были получены на дифрактометре Rigaku Ultima IV (Япония) с детектором D/teX Ultra. Съемку проводили в кварцевых кюветах в режиме на отражение (геометрия Брегга-Брентано) с использованием Cu Kα-излучения (длина волны λ=1.54178 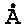 ). Параметры работы генератора: ускоряющее напряжение 40 кВ, ток трубки 250 мА. Параметры съемки интервал углов 2θ=5-85°, шаг по 2θ=0.02°, скорость регистрации спектров 3°/мин. Качественный анализ полученных рентгенограмм и профильный анализ спектров, определение значений параметров решетки проводили с помощью программы PDXL Qualitative Analysis при использовании баз данных ICDD (PDF 2008).
). Параметры работы генератора: ускоряющее напряжение 40 кВ, ток трубки 250 мА. Параметры съемки интервал углов 2θ=5-85°, шаг по 2θ=0.02°, скорость регистрации спектров 3°/мин. Качественный анализ полученных рентгенограмм и профильный анализ спектров, определение значений параметров решетки проводили с помощью программы PDXL Qualitative Analysis при использовании баз данных ICDD (PDF 2008).
Фиг. 2 была снята на ИК-Фурье спектрометре Nicolet 6700 (Thermo Electron Corporation, США) с детектором МСТ-А (50 мкм). ИК-спектры поглощения образцов регистрировали в диапазоне 400-4000 см1 со следующими параметрами: число сканов пробы 32; число сканов 32; разрешение 4,000; усиление 8,0; скорость зеркала 0,6329; диафрагма 100,00. Анализ полученных ИК-спектров, определение значений волновых чисел проводили с помощью программного комплекса OMNIC (версия 7.3) при использовании автофильтра, базовой коррекции.
Описание способа поясняется примерами.
Пример 1
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9(PO4)5,24(CO3)0,76(ОН)0,76 с молярным соотношением Са/(Р+CO3 2-)-1,50, степенью дефицитности Са2+ d=1 и степенью замещения карбоната x=0,76, что соответствует около 5 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,1753 г (NH4)2CO3.
Затем 11,69 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 0,5 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
Дифрактограмма РФА полученного образца Сад-КГА с молярным соотношением Са/(Р+CO3 2-)=1,50, полученного при степени замещения карбоната х=0,76, что соответствует 5 мас.% CO3 2-, представлена на фиг. 1, ИК-спектр образца Сад-КГА с молярным соотношением Са/(Р+CO3 2-)=1,50 и х=0,76 представлен на фиг. 2.
Пример 2
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9(PO4)5(CO3)1(ОН)1 с молярным соотношением Са/(Р+CO3 2-) - 1,50, степенью дефицитности Са2+ d=1 и степенью замещения карбоната х=1,0, что соответствует около 6,5 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 20 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2306 г (NH4)2CO3.
Затем 5,54 мл 20%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 1 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
Пример 3
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9(PO4)4,79(CO3)1,21(ОН)1,21 с молярным отношением Са/(Р+CO3 2-) - 1,50, степенью дефицитности Са2+ d=1 и степенью замещения карбоната х=1,21, что соответствует около 8,0 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,08 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
Навеску гидроксида кальция массой 0,8 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,08%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,1394 г (NH4)2CO3.
Затем 5,35 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/(NH4)2CO3, со скоростью 3,5 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
Пример 4
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9,36(PO4)5,24(CO3)0,76(ОН)1,48 с молярным соотношением Са/(Р+CO3 2-) - 1,56, степенью дефицитности Са2+ d=0,64 и степенью замещения карбоната х=0,76, что соответствует около 5,0 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 15 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,1685 г (NH4)2CO3.
Затем 7,31 мл 15%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 1 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
Пример 5
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9,36(PO4)5(CO3)1(ОН)1,72 с молярным соотношением Са/(Р+CO3 2-) - 1,56, степенью дефицитности Са2+ d=0,64 и степенью замещения карбоната x=1,0, что соответствует около 6,5 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2218 г (NH4)2CO3.
Затем 10,75 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 5 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм
Пример 6
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9,36(PO4)4,79(CO3)1,21(ОН)1,93 с молярным соотношением Са/(Р+CO3 2-) - 1,56, степенью дефицитности Са2+ d=0,64 и степенью замещения карбоната х=1,21, что соответствует около 8,0 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2683 г (NH4)2CO3.
Затем 10,29 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 2 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
Пример 7
Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы с молярным соотношением Са/(Р+CO3 2-) - 1,67 и степенью замещения карбоната х=1,21, что соответствует около 8,0 мас.% CO3 2-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2511 г (NH4)2CO3.
Затем 9,64 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 1 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
Дифрактограмма РФА образца Сад-КГА с молярным соотношением Са/(Р+CO3 2-)=1,67, полученного при степени замещения карбоната х=1,21, что соответствует 8 мас.% CO3 2-, представлена на фиг. 3.
Физико-химические характеристики образцов, полученных по примерам 1-7, приведены в табл.1.
Физико-химические характеристики образцов, полученных по примерам 1-7
Текстурные характеристики образцов, полученных по примерам 1-7,
приведены в табл.2.
Текстурные характеристики образцов, полученных по примерам 1-7
Определение удельной поверхности, объема и среднего размера пор материалов по методу БЭТ проводили на автоматизированной сорбционной установке TriStar II 3020 производства Micromeritics (США) с использованием объемного варианта сорбционного метода. Удельная поверхность была рассчитана по изотерме низкотемпературной сорбции паров азота по одноточечному методу БЭТ в точке P/Po=0,3189. Образцы были выдержаны в инертном газе азота и гелия с одновременным обеспечением нагрева образцов при температуре 95°С.
Известно, что поверхностный заряд наноразмерных частиц, наряду с их размером и формой, являются важными характеристиками ГА, определяющими процессы клеточной адсорбции. В этом аспекте частицы гидроксиапатита, обладающие отрицательно заряженной поверхностью, имеют заметное преимущество [Yingchao Han et al. Nanosize and Surface Charge Effects of Hydroxyapatite Nanoparticles on Red Blood Cell Suspensions //ACS Appl. Mater. Interfaces. 2012. V.4. №9. P. 4616-4622]. Измерение величины и знака дзета-потенциала частиц КГА после созревания осуществляли методом электрофореза на анализаторе Zetasizer Nano ZS фирмы Malvern Instruments.
Значения дзета-потенциала частиц КГА в виде гидрогеля после 24 часов выдерживания в маточном растворе (время созревания) представлены в табл.3
Дзета-потенциал образцов КГА
Проблема загрязнения окружающей среды ионами тяжелых металлов в настоящее время особенно значима. Соединения тяжелых металлов являются распространенными компонентами выбросов транспорта и многих предприятий различных отраслей промышленности. Среди ионов тяжелых металлов наиболее опасными загрязнителями считаются ионы свинца, так как его техногенное накопление в окружающей среде идет высокими темпами. Путем миграции по пищевым цепям ионы Pb2+попадают в организм человека, вызывая единовременные или хронические отравления, и приводят к серьезным нарушениям процессов обмена веществ и жизненно важных функций организма [Herbert L. Needleman, Christine McFarland, Roberta B. Ness, Stephen E. Fienberg, Michael J. Tobin. Bone lead levels in adjudicated delinquents A case control study //J. Neurotoxicology and Teratology. 2002. V.24. P. 711-717; Sharma A., Sharma V., Kansal L. Amelioration of lead-induced hepatotoxicity by Allium sativum extracts in Swiss albino mice //Libyan J. of Med. 2010. V.5. P. 4621-4630]. Одним из наиболее опасных последствий действия ионов свинца считается его способность замещать кальций в костях и быть постоянным источником отравления в течение длительного времени [Silbergeld E.K. Facilitative mechanisms of lead as a carcinogen //Mutation Research: Fundamental and Molecular Mechanisms of Mutagenesis. 2003. V.533. P. 121-133].
Сорбцию ионов свинца на полученных образцах изучали при комнатной температуре при 25±2°С с использованием ацетатного буферного раствора при рН=5,5. В качестве поставщика ионов Pb2+был выбран Pb(CH3COO)2·3H2O. В химические стаканы емкостью 100 мл, содержащие навески 0,1±0,0010 г исследуемых образцов, помещали по 50 мл модельных растворов с разными концентрациями ионов Pb2+в диапазоне концентраций Pb2+от 40 до 4800 мг/л. Определение равновесных концентраций ионов свинца и ионов кальция в модельных растворах было проведено после 48 часов экспозиции.
Концентрацию ионов свинца определяли на вольтамперометрическом анализаторе АКВ-07МК с твердотельным электродом (компания Аквилон, Россия). Для определения концентрации ионов свинца была использована методика с пределом обнаружения 0,2 мг/дм3; диапазон тока - 5000 мА; время накопления - 10 с.
Данные о сорбционной емкости полученных образцов, представленные в табл.4, определили графическим путем, строя график зависимости отношения равновесной концентрации ионов свинца С к величине сорбции Г от величины сорбции Г, используя традиционное
уравнение Лэнгмюра 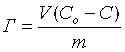
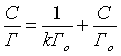
где Г- величина сорбции, мг/г; V - объем раствора ионов свинца, мл; C - равновесная концентрация ионов свинца, мг/л; Cо - исходная концентрация ионов свинца, мг/л; Го - сорбционная емкость, мг/г; k - константа адсорбции, характеризующая степень сродства данного иона к сорбции по Ленгмюру.
Сорбционная емкость образцов, полученных по примерам 1-6, по отношению к ионам тяжелых металлов (при рН=5,5)
Из табл.4 следует, что в зависимости от степени дефицитности и содержания карбоната сорбционная емкость синтетических образцов Сад-КГА по отношению к ионам Pb2+составляет до 1720 мг/г, что превышает почти в 5 раз сорбционную емкость немодифицированного гидроксиапатита [Bailliez S., Nzihou А., Beche А., Flamant G. Removal of lead (Pb) by hydroxyapatite sorbent //Trans IChemE, Part B, Process Safety and Environmental Protection. 2004.V. 82. №2.Р. 175-180].
Таким образом, приведенные примеры подтверждают возможность осуществления предложенного способа с достижением заявленного технического результата:
- получение монофазного продукта Сад-КГА наиболее близкого по химическому составу и структуре к природной кости формулы Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+; х - коэффициент или степень замещения карбоната в интервале от 0,76 до 1,21, а массовое содержание карбонат-ионов от 5% до 8%;
- повышенную биоактивность и адсорбционную способность в полученном Сад-КГА с удельной поверхность 90-200 м2/г, что обеспечивается за счет молярного соотношения Са/(Р+СО3 2-)<1,67 и среднего размера кристаллов от 8 нм до 70 нм.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения наноразмерного гидроксиапатита | 2020 |
|
RU2736048C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО КРЕМНИЙЗАМЕЩЕННОГО ГИДРОКСИАПАТИТА | 2012 |
|
RU2500840C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОРАЗМЕРНОГО КАЛЬЦИЙДЕФИЦИТНОГО СИЛИКАТ- И ФТОРЗАМЕЩЕННОГО ГИДРОКСИАПАТИТА | 2024 |
|
RU2835237C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИОННОГО МАТЕРИАЛА ДЛЯ ЗАМЕЩЕНИЯ КОСТНЫХ ДЕФЕКТОВ С ИСПОЛЬЗОВАНИЕМ ГИДРОЛИТИЧЕСКОЙ КОНВЕРСИИ | 2015 |
|
RU2599022C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКОВОГО МАТЕРИАЛА НА ОСНОВЕ КАРБОНАТГИДРОКСИАПАТИТА И БРУШИТА | 2014 |
|
RU2546539C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО СИЛИКАТЗАМЕЩЕННОГО КАРБОНАТГИДРОКСИАПАТИТА | 2014 |
|
RU2555337C1 |
| СПОСОБ ПОЛУЧЕНИЯ КЕРАМИЧЕСКИХ МАТЕРИАЛОВ НА ОСНОВЕ ФОСФАТОВ КАЛЬЦИЯ | 2006 |
|
RU2321428C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТОГО МАТЕРИАЛА НА ОСНОВЕ ФОСФАТА КАЛЬЦИЯ | 2008 |
|
RU2392007C2 |
| АМОРФНЫЙ, КАРБОНИРОВАННЫЙ И ФТОРИРОВАННЫЙ ГИДРОКСИАПАТИТ ДЛЯ ЗУБНЫХ ПАСТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1999 |
|
RU2179437C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИОСОВМЕСТИМОЙ ПОРИСТОЙ КЕРАМИКИ НА ОСНОВЕ ДИОКСИДА ЦИРКОНИЯ ДЛЯ ЭНДОПРОТЕЗИРОВАНИЯ | 2020 |
|
RU2741918C1 |
Изобретение может быть использовано в производстве медицинских материалов, стимулирующих восстановление дефектов костной ткани, в том числе в стоматологии, и в качестве сорбентов для адсорбции ионов тяжелых металлов. Для получения монофазного нанокристаллического кальций-дефицитного карбонатсодержащего гидроксиапатита Са10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+, а х - степень замещения CO3 2-, величина которой не ниже 0,76, но не выше 1,21, готовят водный раствор композиции гидроксид кальция/карбонат аммония с использованием 0,08-0,16% водного раствора гидроксида кальция. К указанному раствору после перемешивания в течение 10-15 минут и отстаивания до полного растворения гидроксида кальция добавляют расчетное количество карбоната аммония для получения готового продукта с массовым содержанием карбонат-ионов от 5 до 8 мас.% и молярным соотношением Са/(Р+CO3 2-) ниже 1,67. Затем к композиции гидроксид кальция/карбонат аммония приливают 10-20% раствор ортофосфорной кислоты со скоростью 0,5-5 мл/мин на литр водного раствора композиции гидроксид кальция/карбонат аммония при условии поддержания рН реакционной смеси выше 11. Осуществляют отстаивание для завершения процесса фазообразования, выделение и высушивание осадка. После сушки продукта до постоянной массы его подвергают измельчению. Изобретение позволяет получать наноразмерный продукт с высокой биоактивностью, со средним размером кристаллов от 8 нм до 70 нм, удельной поверхностью 90-200 м2/г и сорбционной емкостью к ионам тяжелых металлов до 1720 мг/г. 3 ил., 4 табл., 7 пр.
Способ получения монофазного наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+, а х - степень замещения CO3 2-, величина которой не ниже 0,76, но не выше 1,21, включающий синтез путем осаждения из водного раствора реагентов, содержащих ионы CO3 2-, PO4 3- и Са2+, для чего сначала готовят водный раствор композиции гидроксид кальция/карбонат аммония с использованием 0,08-0,16% водного раствора гидроксида кальция, к которому после перемешивания в течение 10-15 минут и отстаивания до полного растворения гидроксида кальция добавляют расчетное количество карбоната аммония для получения готового продукта с массовым содержанием карбонат-ионов от 5 до 8 мас.% и молярным соотношением Са/(Р+CO3 2-) ниже 1,67, затем к композиции гидроксид кальция/карбонат аммония приливают 10-20% раствор ортофосфорной кислоты со скоростью 0,5-5 мл/мин на литр водного раствора композиции гидроксид кальция/карбонат аммония при условии поддержания рН реакционной смеси выше 11, проводят отстаивание при комнатной температуре для завершения процесса фазообразования, выделение осадка и высушивание до постоянной массы с последующим измельчением.
| US 6582672 B1, 24.06.2003 | |||
| АМОРФНЫЙ, КАРБОНИРОВАННЫЙ И ФТОРИРОВАННЫЙ ГИДРОКСИАПАТИТ ДЛЯ ЗУБНЫХ ПАСТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1999 |
|
RU2179437C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО КРЕМНИЙЗАМЕЩЕННОГО ГИДРОКСИАПАТИТА | 2012 |
|
RU2500840C1 |
| EP 1042222 B1, 27.03.2002 | |||
| WO 2001083367 A2, 08.11.2001. | |||

