Область изобретения
Данное изобретение относится к соединениям, которые связываются с нейронными никотиновыми ацетилхолиновыми рецепторами и модулируют их активность, к новым их солям, к способам получения этих соединений, к фармацевтическим композициям, содержащим эти соединения, и к способам применения этих соединений для лечения большого разнообразия состояний и расстройств, включая те, которые связаны с дисфункцией центральной нервной системы (ЦНС).
Предпосылки к созданию изобретения
Терапевтический потенциал соединений, которые нацелены на нейронные никотиновые ацетилхолиновые рецепторы (NNRs), известные также как никотиновые ацетилхолиновые рецепторы (nAChRs), был темой нескольких обзоров (См., например, Breining et al., Ann. Rep. Med. Chem. 40: 3(2005), Hogg and Bertrand, Curr. Drug Targets: CNS Neurol. Disord. 3: 123 (2004)). К числу показаний для применения лигандов NNR в качестве терапевтических средств относятся указанные ниже расстройства ЦНС. Существует гетерогенное распределение подтипов nAChR как в центральной, так и в периферической нервных системах. Например, подтипами nAChR, которые доминируют в спинном мозге, являются α4β2, α7 и α3β2, тогда как подтипами, которые доминируют при вегетативном ганглии, являются α3β4, и подтипами при нервно-мышечном синапсе являются 1β1γδ и 1β1γε.
Ограничением некоторых никотиновых соединений является то, что они связаны с различными нежелательными побочными эффектами, благодаря неспецифическому связыванию с многочисленными подтипами nAChR. Например, связывание с мышечными и ганглиозными подтипами nAChR и их стимуляция может приводить к побочным эффектам, которые могут ограничивать полезность конкретного никотинового связывающего соединения как терапевтического агента.
Коммерческая разработка лекарственного кандидата вовлекает многие стадии, включая разработку рентабельного метода синтеза, легко приспосабливаемого к способу производства в промышленных масштабах. Коммерческая разработка также предусматривает изыскание солевых форм лекарственной субстанции, которые демонстрируют соответствующие чистоту, химическую стабильность, фармацевтические свойства и характеристики, которые облегчают удобное обращение с ними и их обработку. Кроме того, композиции, содержащие лекарственную субстанцию, должны иметь адекватную сохранность. То есть, они не должны показывать значительных изменений в физико-химических характеристиках, таких как, но без ограничения указанным, химический состав, содержание воды, плотность, гигроскопичность, стабильность и растворимость, при хранении в течение существенного периода времени. К тому же, воспроизводимые и постоянные профили концентрации лекарства в плазме после введения пациенту также являются важными факторами.
Твердые солевые формы, как правило, предпочтительны для пероральных составов, благодаря их тенденции проявлять эти свойства преимущественным образом; и в случае лекарств основного характера аддитивные соли с кислотами часто являются предпочтительными солями. Однако различные солевые формы сильно отличаются по их способности проявлять эти свойства, и такие свойства не могут быть предсказаны с приемлемой точностью. Например, некоторые соли являются твердыми при температурах окружающей среды, тогда как другие соли являются жидкостями, вязкими маслами или камедями при температурах окружающей среды. Более того, некоторые солевые формы являются устойчивыми к теплу и свету в экстремальных условиях, а другие легко разлагаются в значительно более мягких условиях. Соли также сильно различаются по их гигроскопичности, менее гигроскопичные более предпочтительны. Таким образом, разработка подходящей кислотной аддитивной солевой формы лекарства основного характера для применения в фармацевтической композиции является чрезвычайно непредсказуемым способом.
Рацемический 5-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин, его синтез и его гемигалактаратная солевая форма раскрыты в публикации WO 04/078752, которая приобщена ссылкой, и ее дубликатах. Благодаря полезным фармакологическим свойствам единственного энантиомера по сравнению с его рацематом, существует потребность в стереоспецифическом синтезе, предпочтительно в способе, подходящем для крупномасштабного производства. Более того, существует потребность в солевых формах, которые проявляют усовершенствованные свойства, такие как, например, чистота, стабильность, растворимость и биодоступность. Предпочтительные характеристики таких новых солевых форм включают те, которые будут способствовать облегчению и эффективности производства активного ингредиента и его фармацевтической композиции в коммерческий лекарственный продукт, и улучшенной стабильности лекарства в течение продолжительного периода времени.
Сущность изобретения
Один аспект данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин моно-L-малату или его гидрату, или сольвату. Другой аспект относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин гемигалактарату или его гидрату, или сольвату. Другой аспект относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин оксалату или его гидрату, или сольвату. Другой аспект относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин ди-п-толуил-D-тартрату или его гидрату, или сольвату.
Один аспект данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину или его фармацевтически приемлемой соли, по существу не содержащей (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина. В одном варианте осуществления, (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин присутствует в количестве менее чем 25% по массе. В одном варианте осуществления, (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин присутствует в количестве менее чем 15% по массе. В одном варианте осуществления, (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин присутствует в количестве менее чем 5% по массе. В одном варианте осуществления, (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин присутствует в количестве менее чем 2% по массе. В одном варианте осуществления, (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин присутствует в количестве менее чем 1% по массе. В одном варианте осуществления, (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин не содержит значительного количества (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина.
Один аспект данного изобретения относится к фармацевтической композиции, содержащей соединение, которое раскрыто в данном описании, и один или несколько из фармацевтически приемлемых адъюванта, носителя или наполнителя. В одном варианте осуществления, фармацевтическая композиция дополнительно содержит один или несколько дополнительных терапевтических агентов.
Один аспект данного изобретения включает соединение (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин моно-L-малат или гемигалактарат, или оксалат, или ди-п-толуил-D-тартрат для применения в качестве медикамента при лечении NNR-опосредуемого расстройства.
Другой аспект включает способ лечения или профилактики NNR-опосредуемого расстройства, содержащий введение млекопитающему, при необходимости такого лечения, терапевтически эффективного количества (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли.
Другой аспект включает применение соединения (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли при получении медикамента для лечения NNR-опосредуемого расстройства.
В одном варианте осуществления указанного соединения, способа или применения расстройство выбрано из группы, состоящей из расстройств ЦНС, воспаления, воспалительной ответной реакции, связанной с бактериальной и/или вирусной инфекцией, боли, аутоиммунных расстройств с совокупностью метаболических симптомов. В одном варианте осуществления, расстройство ЦНС выбрано из когнитивной дисфункции при шизофрении (CDS), болезни Альцгеймера (AD), расстройства с дефицитом внимания (ADD), пресенильной деменции (раннего начала болезни Альцгеймера), деменции по типу Альцгеймера, легкого ухудшения познавательной способности, возрастного ухудшения памяти и расстройств с дефицитом внимания и гиперактивностью (ADHD).
Другой аспект данного изобретения включает режим введения фармацевтической композиции, содержащий введение (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли в количествах между 7-2200 мкг/кг.
В аспектах и вариантах осуществления, другой вариант осуществления включает то, что (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин предоставляют как его соль моно-L-малат, гемигалактарат, оксалат или ди-п-толуил-D-тартрат.
Другой аспект включает новые промежуточные соединения, включая диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малонат, (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновую кислоту, трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилат и трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилат.
Другой аспект включает способ получения (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина через один или несколько интермедиатов: диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоната, (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты, трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилата и трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилата.
Другой аспект включает способ получения трет-бутил (R)-3-винилпирролидин-1-карбоксилата через один или несколько интермедиатов: диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоната, (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты, трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилата и трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилата.
Другой аспект включает способ очистки (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина, что касается изомерного (R)-3-((Z)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина и (R)-3-(1-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина, превращением в оксалатную соль и регенерацией свободного основания.
Сочетания аспектов и вариантов осуществления образуют дополнительные варианты осуществления данного изобретения.
Краткое описание рисунков
Фигура 1 отображает узнавание нового объекта (NOR) против дозы для (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли, упоминаемых здесь далее как соединение A. Статистически регулярный эффект наблюдали для доз настолько низких, как 0,004 мкМ/кг.
Фигура 2 отображает узнавание нового объекта (NOR) против времени для (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли, соединения A, при дозе 0,004 мкМ/кг. Статистически регулярный эффект наблюдали свыше 8 ч после дозирования.
Фигура 3 отображает результаты исследования в радиальном лучевом лабиринте (RAM), в которых (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль, соединение A, преодолевали индуцированные скополамином дефициты в радиальном лучевом лабиринте.
Подробное описание изобретения
Определения
Следующие определения предназначены для пояснения, но не для ограничения определяемых терминов. Если специфический используемый здесь термин конкретно не определен, такой термин следует понимать как неограниченный. Преимущественно термины использованы в их принятых значениях.
Используемый здесь термин "соединение (соединения)" может быть использован для обозначения формы свободного основания или, в качестве варианта, солевой формы (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина (формула I), в зависимости от контекста, что должно быть очевидно. Специалисты в этой области способны различить эту разницу.
Используемое здесь выражение "фармацевтически приемлемый" относится к носителю (носителям), разбавителю (разбавителям), наполнителю (наполнителям) или солевым формам соединения формулы I, которые совместимы с другими ингредиентами композиции и не опасны для реципиента фармацевтической композиции.
Используемое здесь выражение "фармацевтического качества" относится к соединению или композиции стандарта, подходящего для применения в качестве медицинского средства. Что касается обсуждения здесь, соединения фармацевтического качества по данному изобретению, особенно их солевые формы, проявляют подходящие свойства, включая чистоту, стабильность, растворимость и биодоступность для применения в лекарственном продукте. Предпочтительные характеристики включают те, которые будут способствовать облегчению и эффективности производства активного ингредиента и его композиции в коммерческий лекарственный продукт. Более того, соединения фармацевтического качества по данному изобретению могут быть синтезированы с применением стереоспецифического синтеза, масштаб которого может быть расширен до крупного производства, а именно проявляющего адекватную чистоту и выход.
Используемый здесь термин "фармацевтическая композиция" относится к соединению по данному изобретению, необязательно смешанному с одним или несколькими из фармацевтически приемлемых носителей, разбавителей или наполнителей. Фармацевтические композиции предпочтительно обнаруживают такую степень стойкости к окружающим условиям, которая делает их подходящими для целей производства и коммерциализации.
Используемые здесь термины "эффективное количество", "терапевтическое количество" или "эффективная доза" относятся к количеству соединения по данному изобретению, достаточному, чтобы вызвать желательный фармакологический или терапевтический эффект, имея таким образом результатом эффективная профилактика или лечение расстройства. Профилактика расстройства может быть выражена замедлением или предотвращением прогрессирования расстройства, а также начала симптомов, связанных с расстройством. Лечение расстройства может быть выражено уменьшением или устранением симптомов, ингибированием или обратным развитием прогрессирования расстройства, а также любым другим вкладом в улучшение самочувствия пациента.
Как будет обсуждаться более подробно ниже и со ссылкой на фигуры 1 и 2, статистически регулярный эффект наблюдается для доз соединения формулы I или его фармацевтически приемлемой соли настолько низких, как 0,004 мкМ/кг, включая эффекты, наблюдаемые через 8 ч после дозирования. Эффективная доза может изменяться в зависимости от таких факторов, как состояние пациента, тяжесть симптомов расстройства и пути введения фармацевтической композиции. Так, используемая здесь эффективная доза может быть менее чем 100 мг, в другом варианте осуществления менее чем 50 мг, в другом варианте осуществления менее чем 10 мг или в другом варианте осуществления менее чем 1 мг. Такие эффективные дозы обычно представляют собой количество, вводимое как единственная доза или как одна или несколько доз, вводимых в течение 24 ч периода.
Используемое здесь выражение "существенно" или "достаточно" качественный, чистый или беспримесный, включает более чем 20%, предпочтительно более чем 30% и более предпочтительно, более чем 40% (например, более чем что-либо из 50, 60, 70, 80 или 90%) качественный или чистый.
Термин "стабильность", как определено здесь, включает химическую стабильность и стабильность в твердом состоянии, где выражение "химическая стабильность" включает возможность хранить соли по изобретению в изолированной форме или в форме фармацевтической композиции, в которой они представлены в смеси с фармацевтически приемлемыми носителями, разбавителями, наполнителями, адъювантами, такими как в дозированной форме для перорального введения, такой как таблетка, капсула или тому подобное, в нормальных условиях хранения при незначительной степени химического разложения или деструкции, и выражение "стабильность твердого состояния" включает возможность хранить соли по изобретению в изолированной твердой форме или в форме твердой фармацевтической композиции, в которой они представлены в смеси с фармацевтически приемлемыми носителями, разбавителями, наполнителями или адъювантами, такой как в дозированной форме для перорального введения, такой как таблетка, капсула или тому подобное, в нормальных условиях хранения при незначительной степени трансформации твердого состояния, такой как кристаллизация, перекристаллизация, фазовое превращение твердого состояния, гидратация, дегидратация, сольватация или десольватация.
Примеры "нормальных условий хранения" включают одну или несколько из температур между -80°C и 50°C, предпочтительно между 0°C и 40°C и более предпочтительно, температуры окружающей среды, такие как от 15°C до 30°C, давлений между 0,1 и 2 бар, предпочтительно при атмосферном давлении, относительной влажности между 5 и 95%, предпочтительно 10-60% и воздействии 460 люкс или менее УФ/видимого света в течение продолжительных периодов, таких как более чем или равных шести месяцам. При таких условиях соли по изобретению, менее чем на 5%, более предпочтительно, менее чем на 2% и особенно менее чем на 1%, химически разложившиеся или деградировавшие, или изменившие твердое состояние, могут быть признаны как подходящие. Специалист будет учитывать, что указанные верхний и нижний пределы температуры, давления и относительной влажности представляют крайние пределы нормальных условий хранения, и что конкретные сочетания указанных пределов не будут встречаться во время нормального хранения (например, температура 50°C и давление 0,1 бар).
Используемый здесь термин "расстройство", если не установлено иное, означает какое-либо состояние, дисфункцию или болезнь, связанные с активностью рецептора NNR.
Соединения
Один вариант осуществления данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину (Формула I) или его фармацевтически приемлемой соли.
Формула I
Как будут учитывать специалисты в этой области, различные правила наименования могут называть соединение по-разному. Так, соединение A может быть названо как (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или, в качестве варианта, (R)-3-(2-пирролидин-3-ил)винил)-5-((тетрагидро-2H-пиран-4-ил)окси)пиридин. Такие правила наименования не следует использовать, чтобы вносить двусмысленность в данное описание.
В одном варианте осуществления, соединение формулы I или его фармацевтически приемлемая соль является по существу чистым. В одном варианте осуществления, соединение формулы I или его фармацевтически приемлемая соль по существу не содержат (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина. В одном варианте осуществления, соединение формулы I или его фармацевтически приемлемая соль присутствуют в количестве около 75% по массе по отношению (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину, предпочтительно более чем 85% по массе, более предпочтительно, более чем 95% по массе, более предпочтительно, более чем 98% по массе и наиболее предпочтительно, 99% по массе или более. Один вариант осуществления относится к 100% чистому (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину (Формула I).
Способ
Один вариант осуществления данного изобретения относится к способу получения (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, по существу не содержащих (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина по массе. Другой вариант осуществления данного изобретения относится к способу получения (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, содержащих менее чем 25%, предпочтительно менее чем 15%, более предпочтительно, менее чем 5%, еще более предпочтительно, менее чем 2% и наиболее предпочтительно, менее чем 1% (S)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина по массе, без применения стадии хирального хроматографического разделения. В одном варианте осуществления данного изобретения предложен способ получения по существу чистого (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина без хроматографического разделения.
Общие способы синтеза
Рацемический 3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин может быть синтезирован, как сообщается в публикации PCT WO2004/078752, приобщенной ссылкой, с применением катализируемого палладием связывания трет-бутил 3-винилпирролидин-1-карбоксилата с 3-бром-5-(тетрагидро-2H-пиран-4-илокси)пиридином с последующим удалением трет-бутоксикарбонильной защитной группы. В рацемическом синтезе необходимый трет-бутил 3-винилпирролидин-1-карбоксилат получали обработкой трет-бутил 3-формилпирролидин-1-карбоксилата метилентрифенилфосфораном (реагент Виттига). Несмотря на то, что трет-бутил 3-формилпирролидин-1-карбоксилат может быть получен различными способами, он не был идеальным промежуточным соединением для синтеза единственного энантиомера, ввиду того, что он подвержен рацемизации во время реакции Виттига. Соответственно, был разработан новый путь синтеза, характеризующийся стереохимической точностью.
Соединения могут быть получены согласно следующим способам с использованием коммерчески доступных исходных материалов и реагентов.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин может быть получен путем катализируемого палладием связывания трет-бутил (R)-3-винилпирролидин-1-карбоксилата (соединение 9) и 3-бром-5-(тетрагидро-2Н-пиран-4-илокси)пиридина (соединение 12), как показано в общих чертах на схеме 3.
Получение соединения 9 показано в общих чертах на схеме 1. Коммерчески доступный трет-бутил (R)-3-гидроксипирролидин-1-карбоксилат (соединение 1) обрабатывают метансульфонил хлоридом, чтобы получить трет-бутил (R)-3-(метилсульфонилокси)пирролидин-1-карбоксилат (соединение 2), который затем подвергают реакции с диэтилмалонатом и подходящим основанием (например, трет-бутоксидом калия или этоксидом натрия), чтобы получить диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малонат (соединение 3) с инвертированной стереохимией вокруг хирального атома углерода. Подходящие растворители для указанных реакций могут быть выбраны из группы толуола, ксилолов, 1-метил-2-пирролидинона, диметилформамида, диметилацетамида, этанола, трет-бутанола, тетрагидрофурана, 1,2-диметоксиэтана, диоксана и их смесей. В одном варианте осуществления растворителем для образования метансульфонового сложного эфира служит толуол, и растворителем для вытеснения малоната является 1-метил-2-пирролидинон. В другом варианте осуществления растворителем вытеснения малоната является этанол. Подходящие основания для указанных реакций могут быть выбраны из группы триэтиламина, диэтилизопропиламина, диизопропилэтиламина, трет-бутоксида калия, металлического натрия, гидрида натрия, этоксида натрия, гидрида калия и гидрида лития. В одном варианте осуществления основанием для образования метансульфонового сложного эфира является триэтиламин, и основанием для вытеснения малоната является трет-бутоксид калия. В другом варианте осуществления основанием для вытеснения малоната является этоксид натрия.
Гидролиз сложного диэфира 3 водным гидроксидом калия дает (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновую кислоту (соединение 4), которую декарбоксилируют, чтобы получить (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусную кислоту (соединение 5). Подходящие растворители для указанных реакций могут быть выбраны из группы воды, этанола, тетрагидрофурана, диметилформамида, диметилацетамида, 1,2-диметоксиэтана, диоксана, 1-метил-2-пирролидинона, толуола, диметилсульфоксида и их смесей. В одном варианте осуществления растворителем для гидролиза сложного эфира является водный тетрагидрофуран, и растворителем для декарбоксилирования является 1-метил-2-пирролидинон. В другом варианте осуществления растворителем для гидролиза сложного эфира является этанол, и растворителем для декарбоксилирования является смесь диметилсульфоксида и толуола. Подходящие основания для реакции гидролиза могут быть выбраны из группы гидроксида калия, гидроксида натрия, карбоната калия, карбоната натрия, гидроксида бария и карбоната цезия. В одном варианте осуществления основанием является гидроксид калия.
Восстановление соединения 5 дает трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилат (соединение 6), который может быть подвергнут взаимодействию с метансульфонилхлоридом и затем иодидом натрия для получения трет-бутил (R)-3-(2-(метилсульфонилокси)этил)пирролидин-1-карбоксилата (соединение 7) и трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилата (соединение 8), соответственно. Подходящие растворители для реакции восстановления могут быть выбраны из группы тетрагидрофурана, простого эфира, диоксана, 1,2-диметоксиэтана и их смесей. В одном варианте осуществления растворителем является тетрагидрофуран. Подходящие восстановители могут быть выбраны из группы борана, диборана, комплекса боран-тетрагидрофуран, комплекса боран-диметиловый простой эфир и комплекса боран-диметилсульфид. Подходящие растворители для образования метансульфонового сложного эфира могут быть выбраны из группы толуола, ксилолов, простого эфира, тетрагидрофурана, 1,2-диметоксиэтана, диоксана и их смесей. В одном варианте осуществления растворителем для образования метансульфонового сложного эфира является толуол. Подходящие основания для образования метансульфонового сложного эфира могут быть выбраны из группы триэтиламина, диэтилизопропиламина и диизопропилэтиламина. В одном варианте осуществления основанием для образования метансульфонового сложного эфира является триэтиламин. Подходящие растворители для вытеснения иодида могут быть выбраны из группы 1-метил-2-пирролидинона, диметилформамида, диметилацетамида, этанола, трет-бутанола, тетрагидрофурана, 1,2-диметоксиэтана, диоксана, диметилсульфоксида и их смесей. В одном варианте осуществления растворителем для вытеснения иодида является 1,2-диметоксиэтан.
В конце концов, обработка соединения 8 трет-бутоксидом калия дает соединение 9. Подходящие растворители для указанной реакции могут быть выбраны из группы 1,2-диметоксиэтана, 1-метил-2-пирролидинона, диметилформамида, диметилацетамида, этанола, тетрагидрофурана, диоксана и их смесей. В одном варианте осуществления растворителем является 1,2-диметоксиэтан. Подходящие основания для указанной реакции могут быть выбраны из группы трет-бутоксида калия, этоксида натрия и диазабициклоундекана. В другом варианте осуществления основанием является трет-бутоксид калия.
Один вариант осуществления изобретения относится к способу получения соединения 9 с использованием реакционных стадий, которые представлены в общих чертах на схеме 1 и в обсуждении выше.

Схема 1
Получение 3-бром-5-(тетрагидро-2H-пиран-4-илокси)пиридина (соединение 12) показано в общих чертах на схеме 2. Связывание 3-бром-5-гидроксипиридина (соединение 10) с 4-гидрокситетрагидро-2Н-пираном (соединение 11) дает соединение 12. Подходящие условия для связывания включают такие, в которых фосфин (например, трифенилфосфин) и азосоединение (например, диэтил азодикарбоксилат, известный также как DEAD) используют в инертном растворителе для осуществления связывания (например, в толуоле). В качестве варианта, другие условия, в которых оксо-анион 3-бром-5-гидроксипиридина вытесняет уходящую группу из положения 4 тетрагидропирана, могут быть использованы.
Один вариант осуществления изобретения относится к способу получения соединения 12 с использованием реакционных стадий, как показано в общих чертах на схеме 2 и выше.
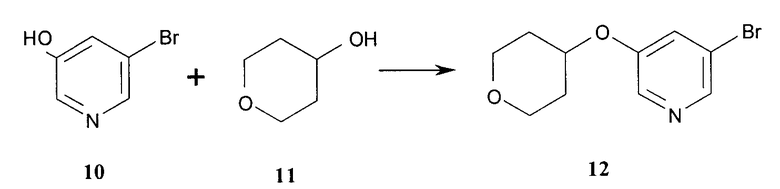
Схема 2
Конечные стадии получении (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина (форма свободного основания) пояснены на схеме 3. Соединения 9 и 12 соединяют путем опосредуемой ацетатом палладия реакции соединения, чтобы получить трет-бутил (R)-(E)-3-(2-(5-(тетрагидро-2H-пиран-4-илокси)пиридин-3-ил)винил)пирролидин-1-карбоксилат (известный также как (R)-5-(1-(трет-бутоксикарбонил)-(E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин, соединение 13), который лишают защитной группы, чтобы получить (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин (соединение 14). Подходящие растворители для катализируемой палладием реакции присоединения могут быть выбраны из группы 1-метил-2-пирролидинона, диметилформамида, диметилацетамида и ацетонитрила. В одном варианте осуществления растворителем является 1-метил-2-пирролидинон. Подходящие основания для катализируемой палладием реакции присоединения могут быть выбраны из группы триэтиламина, диэтилизопропиламина, диизопропилэтиламина. В одном варианте осуществления основанием является диизопропилэтиламин. Подходящие фосфиновые лиганды для катализируемой палладием реакции присоединения могут быть выбраны из группы три-н-бутилфосфина, три-трет-бутилфосфина, трициклогексилфосфина, трифенилфосфина и три-o-толилфосфина. В одном варианте осуществления фосфиновым лигандом является трициклогексилфосфин. Подходящие палладиевые катализаторы для катализируемой палладием реакции присоединения могут быть выбраны из группы ацетата палладия, хлорида палладия и дипалладий трис(дибензилацетона). В одном варианте осуществления палладиевым катализатором является ацетат палладия. Подходящие растворители для реакции удаления защитной группы могут быть выбраны из группы воды, дихлорметана, хлороформа и дихлорэтан. В одном варианте осуществления растворителем является дихлорметан. В другом варианте осуществления растворителем для реакции удаления защитной группы является вода.
Подходящие кислоты для реакции удаления защитной группы могут быть выбраны из группы трифторуксусной кислоты, хлороводородной кислоты и серной кислоты. В одном варианте осуществления кислота является трифторуксусной кислотой.
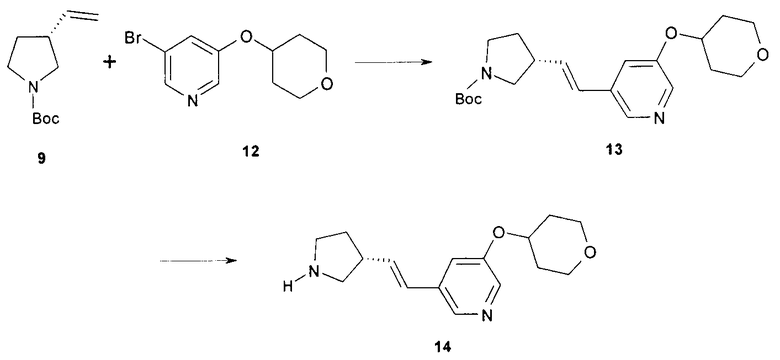
Схема 3
Один вариант осуществления изобретения относится к способу получения соединения 14 с использованием реакционных стадий, которые показаны в общих чертах выше на схемах 1, 2 и 3. Изобретение дополнительно относится к способу получения солевой формы (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин малата, содержащему дополнительную стадию взаимодействия свободного основания с L-яблочной кислотой в смеси 2-пропанола и изопропилацетата или другого подходящего растворителя, как описано ниже.
Другой вариант осуществления изобретения относится к образованию оксалатной соли соединения 14 и применению оксалатной соли в качестве очищенного промежуточного соединения при получении соединения 14. Катализируемое палладием связывание соединений 9 и 12 дает смесь материалов, в которой преобладает соединение 13, обычно составляющее 75-80% продуктов связывания. Остальные продукты связывания включают соответствующий Z изомер, трет-бутил (R)-(Z)-3-(2-(5-(тетрагидро-2H-пиран-4-илокси)пиридин-3-ил)винил)пирролидин-1-карбоксилат, и так называемый "экзо" изомер, трет-бутил (R)-3-(1-(5-(тетрагидро-2H-пиран-4-илокси)пиридин-3-ил)винил)пирролидин-1-карбоксилат, обычно представляющие ~5% и вплоть до 20% продуктов связывания, соответственно. Удаление указанных малых изомеров из главного желательного изомера неожиданно и легко достигалось путем удаления защитных групп смеси изомеров с последующим превращением свободного основания в оксалатную соль. Первоначальное осаждение оксалатной соли в смесях вода/2-пропанол, например, дает соединение 14, в котором изомерные примеси уменьшены до <1%, каждая, или еще лучше. Дополнительно очистка может быть осуществлена перекристаллизацией.
Примерами соединений по данному изобретению, которые метят радиоизотопом, подходящим для различных диагностических применений, являются, например, 11C- или 18F-меченные аналоги соединения 14, которые были бы подходящими для применения в позитронной эмиссионной томографии.
Если не установлено иное, описанные здесь структуры, как подразумевается, включают также соединения, которые различаются только наличием одного или нескольких изотопно обогащенных атомов. Например, соединения, имеющие данную структуру за исключением замещения атома водорода дейтерием или тритием, или замещения атома углерода 13C или 14C, или замещения атома азота 15N, или замещения атома кислорода 17О или 18O, находятся в сфере действия изобретения. Такие изотопно меченные соединения применимы в качестве исследовательских или диагностических инструментов.
Во всех примерах, описанных ниже, защитные группы для чувствительных или реактивных групп используют, где необходимо, в соответствии с общими принципами химического синтеза. Защитными группами манипулируют в соответствии со стандартными способами органического синтеза (T. W. Green and P. G. M. Wuts, Protecting Groups in Organic Synthesis,, 3 rd Edition, John Wiley & Sons, New York (1999)). Такие группы удаляют на удобной стадии синтеза соединения, используя способы, которые очевидны для специалистов. Выбор способов, а также условий реакции и порядка их выполнения должны быть согласующимися с получением соединений по данному изобретению.
Данное изобретение также относится к способу синтеза новых соединений, применимых в качестве промежуточных соединений, таких как диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малонат (соединение 3), (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновая кислота (соединение 4), трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилат (соединение 6) и трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилат (соединение 8).
Солевые формы
Один аспект данного изобретения относится к новым солевым формам (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина. (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин в форме свободного основания является вязким маслом с ограниченной растворимостью в воде. Однако свободное основание может взаимодействовать как с неорганическими, так и с органическими кислотами с образованием солей с присоединенной кислотой, которые имеют физические свойства, полезные для получения фармацевтических композиций, такие как кристалличность, растворимость в воде и стабильность в отношении химического разложения.
Данное изобретение относится к фармацевтически приемлемым солям (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина. Примеры подходящих фармацевтически приемлемых солей включают аддитивные соли неорганических кислот, такие как хлорид, бромид, сульфат, фосфат и нитрат, аддитивные соли органических кислот, такие как ацетат, галактарат, пропионат, сукцинат, лактат, гликолят, малат, тартрат, цитрат, малеат, фумарат, метансульфонат, п-толуолсульфонат и аскорбат, соли с аминокислотами кислотного характера, такие как аспартат и глутамат, соли щелочных металлов, такие как соль натрия и соль калия, соли щелочноземельных металлов, такие как соль магния и соль кальция, соли органических аммониевых оснований, такие как соль триметиламина, соль триэтиламина, соль пиримидина, соль пиколина, соль дициклогексиламина и соль N,N'-дибензилэтилендиамина, и соли с аминокислотами основного характера, такие как соль лизина и соль аргинина. Соли могут быть в некоторых случаях гидратами или сольватами, такими как этанол сольваты.
Один вариант осуществления данного изобретения относится к кислотным аддитивным солям (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина, где кислота выбрана из хлороводородной кислоты, метансульфоновой кислоты, малеиновой кислоты, фосфорной кислоты, 1-гидрокси-2-нафтойной кислоты, малоновой кислоты, L-винной кислоты, фумаровой кислоты, лимонной кислоты, L-яблочной кислоты, R-миндальной кислоты, S-миндальной кислоты, янтарной кислоты, 4-ацетамидобензойной кислоты, адипиновой кислоты, галактаровой кислоты, ди-п-толуил-D-винной кислоты, щавелевой кислоты, D-глюкуроновой кислоты, 4-гидроксибензойной кислоты, 4-метоксибензойной кислоты, (1S)-(+)-10-камфорсульфоновой кислоты, (1R,3S)-(+)-камфорной кислоты и п-толуолсульфоновой кислоты. Данное изобретение также включает гидраты и сольваты указанных солевых форм.
Стехиометрия солей по данному изобретению может изменяться. Например, обычно, что молярное отношение кислоты к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину 1:2 или 1:1, но другие отношения, такие как 3:1, 1:3, 2:3, 3:2 и 2:1, возможны и также включены в сферу действия данного изобретения.
В зависимости от способа, которым описанные здесь соли получены, соли могут иметь кристаллические структуры, которые окклюдируют растворители, которые присутствуют во время солеобразования. Таким образом, соли могут существовать как гидраты и другие сольваты переменной стехиометрии растворителя по отношению к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину.
В одном варианте осуществления данного изобретения, соль имеет стехиометрию кислоты к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину 1:2. В другом варианте осуществления, соль имеет стехиометрию кислоты к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину 1:1.
Другой вариант осуществления данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин моно-L-малату, или его гидрату, или сольвату.
Другой вариант осуществления данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин гемигалактарату или его гидрату, или сольвату.
Другой вариант осуществления данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин оксалату или его гидрату, или сольвату.
Другой вариант осуществления данного изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин ди-п-толуил-D-тартрату или его гидрату, или сольвату.
Один вариант осуществления данного изобретения относится к следующим солям (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина:
Дополнительный аспект данного изобретения относится к способам получения солей. Соли могут быть получены кристаллизацией в контролируемых условиях.
Изобретение также относится к способу получения солевых форм (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина, содержащему следующие стадии:
(i) смешивание свободного основания или раствора свободного основания по существу чистого (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина в подходящем растворителе с какой-либо из указанных выше кислот в чистом виде или в виде раствора какой-либо из кислот в подходящем растворителе, обычно 0,5-1 эквивалент кислоты;
(ii)(a) охлаждение полученного раствора соли, если необходимо вызвать осаждение, или
(ii)(b) добавление подходящего анти-растворителя, чтобы вызвать осаждение, или
(ii)(c) выпаривание растворителя и добавление нового растворителя и повторение или стадии (ii)(a), или стадии (ii)(b), и
(iii) фильтрование и собирание соли.
Используемые стехиометрия, смесь растворителей, концентрация растворенного вещества и температура могут изменяться.
Типичные растворители, которые могут быть использованы для получения или перекристаллизации солевых форм, включают, без ограничения, этанол, метанол, пропанол, 2-пропанол, изопропилацетат, ацетон, этилацетат, толуол, воду, метилэтилкетон, метилизобутилкетон, трет-бутилметиловый простой эфир, тетрагидрофуран, дихлорметан, н-гептан и ацетонитрил.
В одном варианте осуществления растворитель выбран из этанола, пропанола, изопропилацетата, воды, гексана или их смесей, температура, используемая для осаждения, находится между 16°C и 25°C.
В одном варианте осуществления кислотой является L-яблочная кислота и используемым растворителем является 2-пропанол, один или в комбинации с изопропилацетатом. В другом варианте осуществления кислотой является щавелевая кислота, и используемым растворителем является водный 2-пропанол.
В дополнительном варианте осуществления солями являются (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин моно-L-малат или гемигалактарат, или оксалат, или ди-п-толуил-D-тартрат.
Стабильность полученных солей может быть продемонстрирована различными путями. Склонность к приобретению и высвобождению атмосферной влаги может быть определена динамической сорбцией пара (DVS).
Способы лечения
(R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин, или его фармацевтически приемлемые соли, или фармацевтическая композиция, содержащая указанные соединения, могут быть использованы для профилактики или лечения различных состояний или расстройств, для которых другие типы никотиновых соединений были предложены или показаны как применимые в качестве терапевтических средств, таких как расстройства ЦНС, воспаление, воспалительная ответная реакция, связанная с бактериальной и/или вирусной инфекцией, боль, метаболический синдром, аутоиммунные расстройства или другие расстройства, описанные более подробно здесь. Соединения могут быть также использованы в качестве диагностического агента в исследованиях связывания рецептора (in vitro и in vivo). Такие терапевтические и другие доктрины описаны, например, в предварительно перечисленных здесь ссылках, включая Williams et al., Drug News Perspec. 7(4) 205 (1994), Arneric et al., CNS Drug Rev. 1(1) 1-26 (1995), Arneric et al., Exp. Opin. Invest. Drugs 5(1) 79-100 (1996), Bencherif et al., J. Pharmacol. Exp. Ther. 279: 1413 (1996), Lippiello et al., J. Pharmacol. Exp. Ther. 279: 1422 (1996), Damaj et al., J.Pharmacol. Exp. Ther. 291: 390 (1999), Chiari et al., Anesthesiology 91: 1447 (1999), Lavand'homme and Eisenbach, Anesthesiology 91: 1455 (1999), Holladay et al., J. Med. Chem. 40(28) 4169-94 (1997), Bannon et al, Science 279: 77 (1998), PCT WO 94/08992, PCT WO 96/31475, PCT WO 96/40682 и патенты США № 5583140, Bencherif et al., 5597919, Dull et al., 5604231, Smith et al. и 5852041, Cosford et al.
Один вариант осуществления данного изобретения относится к применению (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли в производстве медикамента.
Другой вариант осуществления данного изобретения относится к применению (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин моно-L-малата, или гемигалактарата, или оксалата, или ди-п-толуил-D-тартрата для применения в качестве медикамента.
Один вариант осуществления данного изобретения относится к способу лечения или профилактики расстройств центральной нервной системы (ЦНС), содержащему введение млекопитающему при необходимости такого лечения терапевтически эффективного количества (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, или моно-L-малата, или гемигалактарата, или оксалата, или ди-п-толуил-D-тартрата. Более конкретно, расстройство может быть выбранным из группы, состоящей из расстройств ЦНС, воспаления, воспалительной ответной реакции, связанной с бактериальной и/или вирусной инфекцией, боли, метаболического синдрома, аутоиммунных расстройств или других расстройств, описанных более подробно здесь.
Один вариант осуществления данного изобретения относится к фармацевтической композиции, содержащей терапевтически эффективное количество (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, или его соли моно-L-малата, или гемигалактарата, или оксалата, или ди-п-толуил-D-тартрата, и один или несколько таких ингредиентов, как фармацевтически приемлемые носители, разбавители, наполнители или адъюванты.
Один вариант осуществления данного изобретения относится к применению фармацевтической композиции по данному изобретению в производстве медикамента для лечения расстройств ЦНС.
Другой вариант осуществления данного изобретения относится к применению (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, или его соли моно-L-малата, или гемигалактарата, или оксалата, или ди-п-толуил-D-тартрата в производстве медикамента для лечения или профилактики расстройств, опосредуемых NNR.
Другой вариант осуществления данного изобретения относится к способу модулирования NNR у субъекта, нуждающегося в этом, путем введения (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, или его соли моно-L-малата, или гемигалактарата, или оксалата, или ди-п-толуил-D-тартрата.
Расстройства ЦНС
(R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его фармацевтически приемлемая соль, или его соль моно-L-малат, или гемигалактарат, или оксалат, или ди-п-толуил-D-тартрат, или фармацевтическая композиция, содержащая указанные соединения, применимы для лечения или профилактики разнообразных расстройств ЦНС, включая нейродегенеративные расстройства, нейропсихиатрические расстройства, неврологические расстройства и пагубные привычки. Соединения и их фармацевтические композиции могут быть использованы для лечения или профилактики когнитивных недостаточностей и дисфункций, возрастных и иных; нарушений внимания и деменций, включая вызываемые инфекционными агентами или метаболическими расстройствами; для обеспечения нейрозащиты; для лечения конвульсий и множественных церебральных инфарктов; для лечения психических расстройств, компульсивностей и аддикций; для обеспечения аналгезии; для борьбы с воспалением, таким как опосредуемое цитокинами и ядерным фактором каппа В; для лечения воспалительных расстройств; для облегчения боли и для лечения инфекций в качестве анти-инфекционных агентов для лечения бактериальных, грибковых и вирусных инфекций. К числу расстройств, болезней и состояний, для лечения которых могут быть применены соединения и фармацевтические композиции по данному изобретению, относятся возрастное ухудшение памяти (ААМ), умеренное нарушение познавательной способности (MCI), возрастное угасание познавательной способности (ARCD), пресенильная деменция, преждевременная болезнь Альцгеймера, сенильная деменция, деменция типа Альцгеймера, болезнь Альцгеймера, ухудшение познавательной способности без деменции (CIND), деменция, связанная с тельцами Леви, ВИЧ-деменция, комплекс деменции СПИД, сосудистая деменция, синдром Дауна, травма головы, травматическое повреждение головного мозга (TBI), dementia purgilistica, болезнь Крейцфельда-Якоба и прионовые болезни, удар, ишемия, расстройство с недостаточностью внимания, расстройство с проявлением гиперактивности и недостаточности внимания, дизлексия, шизофрения, шизофреноподобный психоз, шизоаффективный психоз, познавательная дисфункция при шизофрении, недостаточности познавательной способности при шизофрении, паркинсонизм, включая болезнь Паркинсона, постэнцефалитический паркинсонизм, паркинсонизм-деменцию по Гауму, лобно-височная деменция паркинсоновского типа (FTDP), болезнь Пика, болезнь Нейманна-Пика, болезнь Хантингтона, хорея Хантингтона, поздняя дискинезия, гиперкинезия, прогрессивный супрануклеарный паралич, прогрессивный супрануклеарный парез, синдром "усталых ног", болезнь Крейцфельда-Якоба, рассеянный склероз, боковой амиотрофический склероз (ALS), болезни двигательных нейронов (MND), множественная системная атрофия (MSA), кортикобазальная дегенерация, синдром Гийена-Барре (GBS) и хроническая воспалительная димиелинизирующая полиневропатия (CIDP), эпилепсия, автосомальная доминантная ночная фронтально дольная эпилепсия, мания, тревожное состояние, депрессия, пременструальная дисфория, панические расстройства, булимия, анорексия, нарколепсия, чрезмерная дневная сонливость, биполярные расстройства, генерализованное тревожное состояние, навязчивое состояние, вспышки гнева, поведение вызывающего неповиновения, синдром Туретта, аутизм, лекарственная и алкогольная зависимость, табачная зависимость и расстройства питания.
Когнитивные ухудшения или дисфункции могут быть связаны с психиатрическими расстройствами или состояниями, такими как шизофрения и другие психотические расстройства, включая, но без ограничения указанными, психотическое расстройство, шизофреноподобное расстройство, шизоаффективный психоз, бредовое расстройство, кратковременное психотическое расстройство, общее психотическое расстройство и психотические расстройства из-за общего медицинского состояния, деменции и другие когнитивные расстройства, включая, но без ограничения указанными, умеренное ухудшение познавательной способности, пресенильную деменцию, болезнь Альцгеймера, сенильную деменцию, деменцию типа Альцгеймера, возрастное ухудшение памяти, деменцию, связанную с тельцами Леви, сосудистую деменцию, комплекс деменции СПИД, неспособность к чтению, паркинсонизм, включая болезнь Паркинсона, ухудшение познавательной способности и деменцию при болезни Паркинсона, ухудшение познавательной способности при рассеянном склерозе, ухудшение познавательной способности, вызванное травматическим повреждением головного мозга, деменции из-за общего медицинского состояния, тревожные состояния, включая, но без ограничения указанными, паническое расстройство без агарофобии, паническое расстройство с агарофобией, агарофобию без панического расстройства в анамнезе, специфическую фобию, социальную фобию, навязчивое состояние, пост-травматическое стрессовое расстройство, острое стрессовое расстройство, генерализованное тревожное расстройство и генерализованное тревожное расстройство из-за общего медицинского состояния, умственные расстройства, включая, но без ограничения указанными, главное депрессивное расстройство, дистимическое расстройство, биполярную депрессию, биполярную манию, биполярное расстройство типа I, депрессию, связанную с маниакальными, депрессивными или смешанными эпизодами, биполярное расстройство типа II, циклотимическое расстройство и умственные расстройства из-за общего медицинского состояния, нарушения сна, включая, но без ограничения указанными, диссомнию, первичную инсомнию, первичную гиперсомнию, нарколепсию, парасомнию, ночные кошмары, боязнь заснуть и хождение во сне, задержку умственного развития, нарушение обучаемости, нарушение двигательных навыков, нарушение общения, распространяющиеся связанные с развитием расстройства, расстройства с дефицитом внимания и нарушением порядка, расстройство с дефицитом внимания, гиперактивность с дефицитом внимания, нарушения кормления и питания младенцев, детей или взрослых, тиковые расстройства, элиминационные расстройства, относящиеся к веществу расстройства, включая, но без ограничения указанными, зависимость от вещества, злоупотребление веществом, интоксикацию веществом, отказ от вещества, связанные с алкоголем расстройства, связанные с амфетамином или амфетаминоподобным веществом расстройства, связанные с кофеином расстройства, связанные с марихуаной расстройства, связанные с кокаином расстройства, связанные с галюциногеном расстройства, связанные с применяемым при ингаляции средством расстройства, связанные с никотином расстройства, связанные с опиоидом расстройства, связанные с фенциклидином или фенциклидиноподобным веществом расстройства и расстройства, связанные с седативными, снотворными или анксиолитическими средствами, расстройства личности, включая, но без ограничения указанными, расстройство личности с навязчивым состоянием и расстройства импульсивного контроля.
Указанные состояния и расстройства обсуждаются более подробно, например, в American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, Forth Edition, Text Revision, Washington, DC, American Psychiatric Association, 2000. К этому справочнику можно также обратиться по поводу более подробных симптомов и диагностических признаков, связанных с применением веществ, злоупотреблением ими и зависимостью от них.
Один вариант осуществления относится к способу лечения или профилактики расстройств ЦНС у субъекта, при необходимости этого, содержащему введение субъекту (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли или его соли моно-L-малата или геми-галактарата или оксалата или ди-п-толуил-D-тартрата, или фармацевтической композиции, содержащей указанные соединения.
В другом варианте осуществления расстройства ЦНС выбраны из когнитивной дисфункции при шизофрении (CDS), болезни Альцгеймера (AD), расстройства с дефицитом внимания (ADD), пресенильной деменции (раннего начала болезни Альцгеймера), деменции по типу Альцгеймера, умеренного ухудшения познавательной способности, возрастного ухудшения памяти и расстройства с гиперактивностью и дефицитом внимания (ADHD). В одном варианте осуществления расстройства ЦНС выбраны из ухудшения памяти и ухудшения обучаемости.
Воспаление
Известно, что нервная система, главным образом через блуждающий нерв, регулирует величину врожденного иммунного отклика путем ингибирования выделения макрофагового фактора некроза опухоли (TNF). Этот физиологический механизм известен как "холинергический противовоспалительный путь" (см., например, Tracey, "The inflammatory reflex", Natura 420: 853-9 (2002)). Чрезмерное воспаление и синтез фактора некроза опухоли служат причиной заболеваемости и даже смертности при различных болезнях. К таким болезням относятся, но без ограничения указанным, эндотоксемия, ревматоидный артрит, остеоартрит, псориаз, астма, атеросклероз, идиопатический легочный фиброз и воспалительная болезнь кишечника.
Воспалительные состояния, которые можно лечить или предотвратить введением соединений, описанных здесь, включают, но без ограничения указанными, хроническое и острое воспаление, псориаз, эндотоксемию, подагру, острую псевдоподагру, острый подагрический артрит, артрит, ревматоидный артрит, остеоартрит, отторжение аллотрансплантата, хроническое отторжение трансплантата, астму, атеросклероз, зависимое от мононуклеарных фагоцитов поражение легких, идиопатический легочный фиброз, атопический дерматит, хроническую обструктивную легочную болезнь, респираторный дистресс-синдром взрослых, острый синдром грудной клетки при серповидно-клеточной болезни, воспалительную болезнь кишечника, болезнь Крона, язвенный колит, острое воспаление желчных путей, афтозный стоматит, резервуарный илеит, гломерулонефрит, люпус-нефрит, тромбоз и реакцию "трансплантат против хозяина".
Воспалительная реакция, связанная с бактериальной и/или вирусной инфекцией
Многие бактериальные и/или вирусные инфекции связаны с побочными эффектами, вызываемыми образованием токсинов, и естественной ответной реакцией организма на бактерии или вирусы и/или токсины. Как обсуждалось выше, ответная реакция организма на инфекцию часто сопряжена с образованием значительного количества TNF и/или других цитокинов. Сверх-экспрессия этих цитокинов может иметь результатом значительное повреждение, такое как септический шок (когда бактерия вызывает общее заражение крови), эндотоксический шок, уросепсис, вирусный пульмонит и синдром токсического шока.
Экспрессия цитокина опосредуется NNRs и может быть подавлена введением агонистов или частичных агонистов указанных рецепторов. Те описанные здесь соединения, которые являются агонистами или частичными агонистами указанных рецепторов, могут быть, следовательно, применены для минимизации воспалительной ответной реакции, связанной с бактериальной инфекцией, а также с вирусными и грибковыми инфекциями. Примеры таких бактериальных инфекций включают сибирскую язву, ботулизм и сепсис. Некоторые из этих соединений могут иметь также антимикробные свойства.
Соединения по данному изобретению могут быть также использованы в качестве вспомогательного лечебного средства в комбинации с существующими лечебными средствами для борьбы с бактериальными, вирусными и грибковыми инфекциями, такими как антибиотики, антивирусные средства и антигрибковые средства. Антитоксины также могут быть использованы для связывания с токсинами, продуцируемыми инфекционными агентами, и предоставления возможности связанным токсинам проходить через организм, не вызывая воспалительного отклика. Примеры антитоксинов раскрыты, например, в патенте США № 6310043, Bundle и др. Другие агенты эффективные против бактериальных и других токсинов могут быть действенными, и их терапевтический эффект может быть дополнен одновременным введением с соединениями, описанными здесь.
Боль
Соединения могут быть введены для лечения и/или предотвращения боли, включая острую, неврологическую, воспалительную, невропатическую и хроническую боль. Аналгезирующее действие соединений, описанных здесь, может быть продемонстрировано на моделях постоянной воспалительной боли и невропатической боли, осуществленных, как описано в опубликованной патентной заявке США № 20010056084 А1 (Allgeier и др.) (например, механическая гипералгезия на крысиной модели с полным адъювантом Фрейнда воспалительной боли и механическая гипералгезия на модели невропатической боли с частичной лигатурой седалищного нерва мыши).
Аналгезирующий эффект является подходящим для лечения боли различного происхождения или этиологии, в особенности при лечении воспалительной боли и сопутствующей гипералгезии, невропатической боли и сопутствующей гипералгезии, хронической боли (например, тяжелой хронической боли, постоперационной боли и боли, связанной с различными состониями, включая рак, стенокардию, почечные и/или печеночные колики, менструацию, мигрень и подагру). Воспалительная боль может быть иного происхождения, включая артрит и ревматоидную болезнь, тендосиновит и васкулит. Невропатическая боль включает тригеминальную или герпетическую невралгию, невропатии, диабетическую невропатическую боль, казуалгию, поясничную боль и синдромы деафферентации, такие как разрыв плечевого сплетения.
Один вариант осуществления относится к способу лечения боли у субъекта, нуждающегося в этом, содержащему введение указанному субъекту (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли или его соли моно-L-малата, или геми-галактарата, или оксалата, или ди-п-толуил-D-тартрата, или фармацевтической композиции, содержащей указанные соединения.
Другие расстройства
В дополнение к лечению расстройств ЦНС, воспаления и боли соединения по данному изобретению могут быть использованы для профилактики или лечения некоторых других состояний, болезней и расстройств, в которых NNRs играют роль. Примеры включают аутоиммунные расстройства, такие как волчанка, расстройства, связанные с выделением цитокинов, худосочие вторичное по отношению к инфекции (например, как происходит при СПИД, комплексе, относящемся к СПИД и неоплазии), тучность, пузырчатку, недержание мочи, болезни, относящиеся к сетчатке, инфекционные болезни, миастению, синдром Итона-Ламберта, дистонию, гипертонию, остеопороз, ангиоспазм, сосудистую дилатацию, сердечную аритмию, диабет типа I, диабет типа II, язвы, булимию, анорексию, констипацию и диарею, а также показания, установленные в опубликованной заявке РСТ WO 98/25619. Соединения по данному изобретению могут быть также введены для лечения конвульсий, таких как те, которые являются симптоматичными для эпилепсии, и для лечения состояний, таких как сифилис и болезнь Якоба-Крейцфельда.
Диагностические применения
Другой вариант осуществления данного изобретения относится к соединениям, которые находят применение в качестве диагностических агентов и в исследованиях связывания рецептора, как описано здесь.
Соединения могут быть использованы в диагностических композициях, таких как зонды, особенно когда они модифицированы введением соответствующих меток. Зонды могут быть использованы, например, для определения относительного числа и/или функции специфических рецепторов, особенно подтипа рецептора α4β2. Для этой цели соединения по данному изобретению, наиболее предпочтительно, метят радиоактивной изотопной частью молекулы, такой как 11С, 18F, 76Br, 123I или 125I.
Один вариант осуществления изобретения относится к (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридину или его фармацевтически приемлемой соли, или его соли моно-L-малату, или геми-галактарату или оксалату или ди-п-толуил-D-тартрату, где от одного до трех атомов представляют собой обнаруживаемый изотоп, выбранный из 3Н, 19F и 13С, где один из атомов является обнаруживаемым изотопом, выбранным из 19F, 11С и 14С.
Введенные соединения могут быть обнаружены с помощью известных методов обнаружения, подходящих для используемых меток. Примерами методов обнаружения являются сцинтилляционный подсчет, позиционная эмиссионная топография (PET), однофотонная эмиссионная компьютерная томография (SPECT), гамма-визуализация, магниторезонансная визуализация (MRI) или магниторезонансная спектроскопия (MRS). Радиоактивные метки, описанные выше, применимы в визуализации РЕТ (например, 11С, 18F или 76Br) и SPECT (например, 123I) с периодами полураспада около 20,4 мин для 11С, около 109 мин для 18F, около 13 ч для 123I и около 16 ч для 76Br. Высокая специфическая активность желательна для визуализации выбранных подтипов рецептора при ненасыщенных концентрациях. Введенные дозы обычно являются небольшими и обеспечивают высоко контрастные изображения. Определение дозы проводят способом, известным специалистам в области визуализации радиоактивных меток. Соединения могут быть введены в композициях, которые включают другие ингредиенты, такие как те типы ингредиентов, которые применимы в составлении рецептур диагностической композиции. Соединения, применимые в соответствии с осуществлением данного изобретения, наиболее предпочтительно, используют в формах высокой чистоты. После того, как соединения вводят субъекту (например, человеку), присутствие соединения в теле субъекта может быть визуализовано и количественно оценено соответствующими методами, чтобы показать присутствие, количество и функциональность выбранных подтипов NNR. Помимо людей, соединения могут быть также введены животным, таким как мыши, крысы, лошади, собаки и обезьяны. SPECT и РЕТ визуализация может быть проведена с использованием любой подходящей методики и аппарата. Радиоактивно меченные соединения связываются с высоким сродством с селективными подтипами NNR (например, α4β2) и предпочтительно обнаруживают пренебрежимо малое неспецифическое связывание с другими подтипами никотинового холинергического рецептора (например, такими подтипами рецептора, связанными с мышцей и ганглием). Как таковые, соединения могут быть использованы в качестве агентов для неинвазивной визуализации подтипов никотинового холинергического рецептора в теле субъекта, особенно в головном мозге, для диагностики, связанной с разнообразными болезнями и расстройствами ЦНС.
В одном аспекте диагностические композиции могут быть использованы в методе постановки диагноза болезни у субъекта, такого как человек. Метод предусматривает введение этому пациенту обнаружимо меченного соединения, как описано здесь, и обнаружение связывания этого соединения с выбранными подтипами NNR (например, подтипами рецептора α4β2). Специалисты в области применения диагностических инструментов, таких как РЕТ и SPECT, могут применять радиоактивно меченные соединения, описанные здесь, для диагностирования широкого разнообразия состояний и расстройств, включая состояния и расстройства, связанные с дисфункцией центральной и автономной нервных систем. Такие расстройства включают широкое разнообразие болезней и расстройств ЦНС, таких как болезнь Альцгеймера, болезнь Паркинсона и шизофрения или какое-либо расстройство, упомянутое здесь.
Связывание рецептора
Соединения по данному изобретению могут быть использованы в качестве лигандов сравнения в анализах связывания для соединений, которые связываются с подтипами NNR, особенно подтипами рецептора α4β2. Для этой цели соединения по данному изобретению предпочтительно метят радиоактивной изотопной частью молекулы, такой как 3Н или 14С. Примеры таких анализов связывания описаны подробно ниже.
Фармацевтические композиции
В одном аспекте данное изобретение относится к фармацевтическим композициям, содержащим соединение по данному изобретению, и один или несколько из фармацевтически приемлемого носителя, разбавителя или наполнителя. Другой аспект данного изобретения относится к способу получения фармацевтической композиции, содержащему смешивание соединения по данному изобретению с одним или несколькими из фармацевтически приемлемого носителя, разбавителя или наполнителя.
Способ, которым соединение по данному изобретению вводят, может изменяться. Соединение по данному изобретению предпочтительно вводят перорально. Предпочтительные фармацевтические композиции для перорального введения включают таблетки, капсулы, каплетки, сиропы, растворы и суспензии. Фармацевтические композиции по данному изобретению могут быть предоставлены в дозированных формах с модифицированным высвобождением, таких как рецептуры в форме таблеток и капсул с высвобождением во времени.
Фармацевтические композиции могут быть также введены посредством инъекции, а именно внутривенно, внутримышечно, подкожно, интраперитонеально, интраартериально, интратекально и интрацеребровентрикулярно. Носители для инъекций могут включать 5% растворы декстрозы, физиологический раствор и забуференный фосфатом физиологический раствор.
Композиции могут быть также введены с использованием других средств, например, ректальным введением. Композиции могут быть также введены путем ингаляции, например, в виде аэрозоля, местно, например, в виде лосьона, трансдермально, например, с помощью трансдермального пластыря (например, с использованием технологии, которая коммерчески доступна от Novartis and Alza Corporation), путем впрыскивания порошка или путем буккальной, сублингвальной или интраназальной адсорбции.
Фармацевтические композиции могут быть приготовлены в унифицированной дозированной форме или в формах множественных или разделенных доз.
Введение фармацевтических композиций, описанных здесь, может быть периодическим или постепенным, непрерывным при постоянной или регулируемой скорости. Фармацевтические композиции могут быть введены теплокровному животному, например, млекопитающему, такому как мышь, крыса, кошка, морская свинка, кролик, лошади, собака, свинья, корова или обезьяна, но преимущественно их вводят человеку.
Комбинации
Соединение по данному изобретению может быть использовано при лечении разнообразных расстройств и состояний и, как таковое, может быть использовано в комбинации с разнообразными другими терапевтическими агентами, применимыми при лечении или профилактике этих расстройств. Так, один вариант осуществления данного изобретения относится к введению соединения по данному изобретению в комбинации с другими терапевтическими агентами. Например, соединение по данному изобретению может быть использовано в комбинации с другими лигандами NNR (такими как варениклин), антиоксидантами (такими как агенты поглощения свободных радикалов), антибактериальными агентами (такими как пенициллиновые антибиотики), антивирусными агентами (такими как нуклеозидные аналоги, подобные зидовудину и ацикловиру), антикоагулянтами (такими как варфарин), противовоспалительными агентами (такими как NSAIDs), жаропонижающими, аналгетиками, анестетиками (такими как используемые в хирургии), ингибиторами ацетилхолинэстеразы (такими как донепезил и галантамин), антипсихотическими средствами (такими как галоперидол, клозапин, оланзапин и куетиапин), препаратами для подавления иммунитета (такими как циклоспорин и метотрексат), нейрозащитными агентами, стероидами (такими как стероидные гормоны), кортикостеродами (такими как дексаметазон, предизон и гидрокортизон), витаминами, минеральными добавками, препаратами лечебного питания, антидепрессантами (такими как имипрамин, флуоксетин, пароксетин, эсциталопрам, сетралин, венлафаксин и дулоксетин), анксиолитиками (такими как алпразолам и буспирон), противосудорожными агентами (такими как фенитоин и габапентин), сосудорасширяющими средствами (такими как празозин и силденафил), стабилизаторами настроения (такими как валпроат и арипипразол), противораковыми лекарствами (такими как антипролиферативные средства), гипотензивными агентами (такими как атенолол, клонидин, амлопидин, верапамил и олмесартан), слабительными, разжижителями стула, диуретиками (такими как фуросемид), спазмолитическими средствами (такими как дицикломин), антидискинетическими агентами и противоязвенными медикаментами (такими как эзомепразол). Такая комбинация терапевтических агентов может быть введена вместе или раздельно и, когда вводят раздельно, введение может происходить одновременно или последовательно в любом порядке. Количества соединений или агентов и относительное распределение во времени введения следует выбирать так, чтобы достичь желательного терапевтического эффекта. Введение в комбинации соединения по данному изобретению с другими терапевтическими агентами может быть в объединении путем сопутствующего введения: (1) единой фармацевтической композиции, включающей оба соединения; или (2) раздельных фармацевтических композиций, каждая из которых содержит одно из соединений. В качестве варианта, комбинация может быть введена раздельно последовательным образом, когда один лечебный агент вводят первым, а другой вторым. Такое последовательное введение может быть близким по времени или отдаленным по времени.
Другой аспект данного изобретения относится к комбинированной терапии, содержащей введение субъекту терапевтически или профилактически эффективного количества соединения по данному изобретению и одного или нескольких других терапевтических агентов, включая химиотерапевтические средства, радиационные терапевтические агенты, генные терапевтические агенты или агенты, применяемые в иммунотерапии.
Низкая доза
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль является субстратом для мозгового насоса Pgp, который находится в гематоэнцефалическом барьере. Насос Pgp является ответственным за выведение веществ из мозга. Благодаря этому насосу, часто трудно ввести лекарства в мозг в терапевтически эффективных количествах. Результатом этого часто является введение высоких доз лекарств, которые при таких высоких уровнях могут создавать побочные эффекты в других частях тела человека.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин, хотя и является субстратом для насоса PGP, может быть введен в малых дозах, хотя в то же время имеет относительно большую продолжительность действия. Например, по сравнению с ацетилхолином, естественным агонистом NNR, отклик на (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин в два раза больше в анализе in vitro при α4β2.
Один вариант осуществления изобретения относится к введению фармацевтической композиции, содержащей (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его фармацевтически приемлемую соль, в некоторых вариантах осуществления, его соль моно-L-малат, или гемигалактарат, или оксалат, или ди-п-толуил-D-тартрат, в количествах между 1-2200 мкг/день. В другом варианте осуществления, в количестве от 50 до 1500 мкг/день. В дополнительном варианте осуществления, в количестве от 50 до 1000 мкг/день. В одном варианте осуществления, в количестве от 50 до 500 мкг/день. В другом варианте осуществления, в количестве от 75 до 300 мкг/день. В еще одном варианте осуществления, в количестве от 75 до 200 мкг/день. В еще одном дополнительном варианте осуществления, в количестве от 75 до 150 мкг/день.
Доза (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли, в некоторых вариантах осуществления его соли моно-L-малата, или гемигалактарата, или оксалата, или ди-п-толуил-D-тартрата, может быть введена один, два или три раза в день. Один вариант осуществления относится к введению один раз в день. Другой вариант осуществления относится к введению дважды в день.
Другой вариант осуществления изобретения относится к агонисту NNR, который имеет полупериод существования (t1/2) между 5 и 8 часами. В одном варианте осуществления t1/2 между 6 и 7 часами. В другом варианте осуществления t1/2 6,8 часа.
Другой вариант осуществления изобретения относится к агонисту NNR, который имеет продолжительность действия между 5 и 10 часами. В одном варианте осуществления продолжительность действия между 6 и 9 часами. В дополнительном варианте осуществления продолжительность действия 8 часов.
В дополнительном варианте осуществления агонист является α4β2 агонистом.
В еще одном варианте осуществления агонистом является (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин.
Примеры
Следующие примеры приведены для пояснения данного изобретения и не должны быть истолкованы как ограничивающие его. В этих примерах все части и проценты даны по массе, если не указано иное.
Пример 1: Оборудование и экспериментальные протоколы для характеристики (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина и его солевых форм
Спектрометрия ядерного магнитного резонанса (ЯМР)
Спектр ЯМР получали или на приборе Varian Unity 300 МГц или на приборе Bruker 400 МГц, снабженном автоматическим пробоотборником и регулируемой консолью DRX400. Автоматизированные эксперименты осуществляли, используя ICONNMR v4,0.4 (конструкция 1) работающий с Topspin v 1.3 (уровень пятна 8) с использованием стандартных экспериментов на приборе Bruker. Для нерутинной спектроскопии данные получали через использование только Topspin.
Температура плавления
Для определения температуры плавления использовали аппарат Fisher-Johns при настройке, соответствующей скорости нагревания около 5°C в мин.
Дифференциальная сканирующая калориметрия (DSC)
Данные DSC собирали на TA приборах Q1000 или Mettler DSC 823e, снабженных 50-позиционным автоматическим пробоотборником. Калибровку прибора по энергии и температуре осуществляли, используя сертифицированный индий. Обычно 0,5-1,5 мг каждой пробы в перфорированном алюминиевом сосуде нагревали при 10°C/мин от 25°C до 175-200°C. Ток азота при 30 мл/мин поддерживали над пробой.
Динамическая сорбция пара (DVS)
Изотермы сорбции определяли, используя анализатор внутренней сорбции влаги SMS DVS, управляемый комплектом программного обеспечения SMS Analysis suite software. Температуру пробы поддерживали при 25°C регулирующими устройствами. Влажность регулировали смешиванием потоков сухого и влажного азота при общем расходе 200 мл/мин. Относительную влажность измеряли калиброванным Rotronic зондом (динамический диапазон 1,0-100%RH), расположенным вблизи пробы. Изменение массы (облегчение массы) пробы как функцию %RH постоянно отслеживали с помощью микровесов (точность ±0,005 мг).
Обычно пробу 5-20 мг помещали на решето из нержавеющей стали с тарированными отверстиями при окружающих условиях. Пробу нагружали и разгружали при 40% RH и 25°C (обычные окружающие условия). Изотерму сорбции влаги получали, как описано в общих чертах ниже (2 сканирования, дающие 1 полный цикл). Стандартную изотерму получали при 25°C при 10% RH интервалах в диапазоне 0-90% RH.
Общие параметры метода DVS
Химическая чистота путем HPLC
Анализ чистоты проводили на серийной системе Agilent HP1100, снабженной диодным матричным детектором и использующей программное обеспечение ChemStation software vB.02.01-SR1.
Параметры метода HPLC для определения химической чистоты:
150×4,6 мм, 5 мкм
длина волны, полоса (нм):
Ионная хроматография
Данные собирали на Metrohm 761 Advanced Compact 1C (для катионов) и Metrohm 861 Advanced Compact 1C (для анионов), используя программное обеспечение 1C Net software v2.3. Пробы готовили как 1000 м.д. исходного вещества в DMSO. Пробы разбавляли DMSO до 100 м.д. перед испытанием. Количественное определение проводили путем сравнения со стандартными растворами известной концентрации анализируемого иона.
Метод ионной хроматографии для анионов:
1,0 мМ гидрокарбонат натрия в воде
Метод ионной хроматографии для катионов:
0,75 мМ дипиколиновая кислота в воде
Пример 2. Синтез трет-бутил (R)-3-(метилсульфонилокси)пирролидин-1-карбоксилата (2)
Процедура A: К раствору трет-бутил (R)-3-гидроксипирролидин-1-карбоксилата (200 г, 1,07 моль) и триэтиламина (167 г, 1,63 моль) в толуоле (700 мл) при от -20 до -30°C добавляли метансульфонилхлорид (156 г, 1,36 моль) по каплям, поддерживая температуру от -10 до -20°C. Раствор нагревали до окружающей температуры и создавали возможность перемешивания. Пробы реакционного раствора отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции суспензию фильтровали, чтобы удалить триэтиламин гидрохлорид. Фильтрат промывали ~600 мл разбавленного водного раствора бикарбоната натрия. Органический слой сушили и концентрировали при пониженном давлении, получая соединение 2 в виде вязкого масла (260 г, 92%), которое использовали без дополнительной очистки.
1H ЯМР (CDCl3, 400 МГц) δ 5,27 (м, 1Н), 3,44-3,76 (м, 4H), 3,05 (с, 3H), 2,26 (м, 1Н), 2,15 (м, 1Н), 1,47 (с, 9H).
Процедура B: В реактор загружали трет-бутил (R)-3-гидроксипирролидин-1-карбоксилат (2,00 кг, 10,7 моль), толуол (8,70 кг) и триэтиламин (1,75 кг, 17,3 моль). Реактор продували азотом в течение 15 мин. Смесь перемешивали и охлаждали до 3°C. Метансульфонилхлорид (1,72 кг, моль) медленно добавляли (в течение 2 ч периода) с непрерывным охлаждением на бане со льдом (экзотермическая реакция) (после завершения добавления температура была 14°C). Смесь, теперь вязкую, с осажденным триэтиламингидрохлоридом, перемешивали 12 ч, пока она нагревалась до 20°C. Оба анализа и GC, и TLC (окрашивание нингидрином) показали, что никакого исходного материала не осталось. Смесь фильтровали, чтобы удалить триэтиламин гидрохлорид, и фильтрат возвращали в реактор. Фильтрат затем промывали (2×3 кг) 5% водным раствором бикарбоната натрия, используя время перемешивания 15 мин и время осаждения 15 мин для каждого промывания. Полученный органический слой сушили над безводным сульфатом натрия и фильтровали. Летучие вещества удаляли из фильтрата в вакууме, вначале при 50°C в течение 4 ч и затем при окружающей температуре в течение 10 ч. Остаток весил 3,00 кг (106% выход) и был идентичен по хроматографическому и ЯМР анализу предварительно приготовленным пробам за исключением того, что он содержал толуол.
Пример 3. Синтез диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоната (3)
Получение A: К раствору трет-бутоксида калия (187 г, 1,62 моль) в 1-метил-2-пирролидиноне (1,19 л) добавляли диэтилмалонат (268 г, 1,67 моль), поддерживая температуру ниже 35°C. Раствор нагревали до 40°C и перемешивали в течение 20-30 мин, трет-Бутил (R)-3-(метилсульфонилоксил)пирролидин-1-карбоксилат (112 г, 420 ммоль) добавляли и раствор нагревали до 65°C и перемешивали в течение 6 ч. Пробы реакционного раствора отбирали каждые 2 ч и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции (10-12 ч) смесь охлаждали до около 25°C. Деионизированную воду (250 мл) добавляли к раствору и pH доводили до 3-4 добавлением 2 н. хлороводородной кислоты (650 мл). Полученную суспензию фильтровали и добавляли воду (1,2 л) и хлороформ (1,4 л). Раствор тщательно перемешивали и слой хлороформа собирали и выпаривали при пониженном давлении, получая желтое масло. Масло растворяли в гексанах (2,00 л) и промывали деионизированной водой (2×1,00 л). Органический слой концентрировали при пониженном давлении при 50-55°С, получая бледно-желтое масло (252 г), которое, как показал 1H ЯМР анализ, содержало 49,1% соединения 3 (123,8 г) наряду с 48,5% диэтилмалоната (122 г) и 2% 1-метил-2-пирролидинона (5 г). Материал переносили на следующую стадию без дополнительной очистки.
1H ЯМР (CDCl3, 400 МГц) δ 4,20 (кв, 4H), 3,63 (м, 1Н), 3,48 (м, 1Н), 3,30 (м, 1Н), 3,27 (д, J=10 Гц, 1Н), 3,03 (м, 1Н), 2,80 (м, 1Н), 2,08 (м, 1Н), 1,61 (м, 1Н), 1,45 (с, 9H), 1,27 (т, 6H).
Получение B: В реактор, в котором поддерживали атмосферу азота, загружали этанол крепости 200 (5,50 кг) и 21% (по массе) этоксид натрия в этаноле (7,00 кг, 21,6 моль). Смесь перемешивали и нагревали до 30°C. Диэтилмалонат (3,50 кг, 21,9 моль) добавляли в течение 20 мин периода. Реакционную смесь затем нагревали при 40°C в течение 1,5 ч. Раствор трет-бутил (R)-3-(метилсульфонилоксил)пирролидин-1-карбоксилата (3,00 кг продукта из примера 2, процедура B, 10,7 моль) в этаноле крепостью 200 (5,50 кг) добавляли, и полученную смесь нагревали при кипении с возвращением флегмы (78°C) в течение 2 ч. Оба анализа GC и TLC (окрашивание нингидрином) показали, что никакого исходного материала не остается. Перемешиваемую смесь затем охлаждали до 25°C, разбавляли водой (2,25 кг) и медленно обрабатывали раствором концентрированной хлороводородной кислоты (1,27 кг, 12,9 моль) в воде (5,44 кг). Эту смесь промывали дважды метил трет-бутиловым простым эфиром (MTBE) (14,1 кг и 11,4 кг), используя время перемешивания 15 мин и время осаждения 15 мин для каждого промывания. Объединенные смывы MTBE сушили над безводным сульфатом натрия (1 кг), фильтровали и концентрировали в вакууме при 50°C в течение 6 ч. Остаток (красное масло) весил 4,45 кг и был на 49% желательным продуктом по GC анализу (62% общий выход из трет-бутил (R)-3-гидроксипирролидин-1-карбоксилата).
Пример 4. Синтез (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (4)
Процедура A: К раствору продукта из примера 3, процедура A (232 г), содержащему 123,8 г (380 ммоль) соединения 3 и 121,8 г (760 ммоль) диэтилмалоната в тетрагидрофуране (1,2 л) добавляли 21% раствор гидроксида калия (450 г в 0,50 л деионизированной воды), поддерживая температуру ниже 25°C. Реакционную смесь нагревали до 45°C и перемешивали в течение 1 ч. Пробы реакционного раствора отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции (2-3 ч) смесь охлаждали до около 25°C. Водный слой собирали и охлаждали до 5°C. pH доводили до 2 добавлением 4 н. хлороводородной кислоты (750 мл) и полученную суспензию выдерживали при 5-10°C в течение 30 мин. Смесь фильтровали, осадок на фильтре промывали гексанами (1 л). Водный фильтрат экстрагировали хлороформом (1 л) и слой хлороформа оставляли в стороне. Твердые вещества, собранные на стадии фильтрования, повторно растворяли в хлороформе (1 л) путем нагревания до 40°C. Раствор фильтровали, чтобы удалить нерастворенные неорганические твердые вещества. Слои хлороформа объединяли и концентрировали при пониженном давлении при 50-55°C, получая грязно-белое твердое вещество (15 г). Твердые вещества объединяли и растворяли в этилацетате (350 мл), получая суспензию, которую нагревали до 55-60°C в течение 2 ч. Суспензию фильтровали, пока она была горячей, и полученный осадок на фильтре промывали этилацетатом (2×150 мл) и гексанами (2×250 мл), получая 83,0 г (80,1%) соединения 4 в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (d4-CH3OH, 400 МГц) δ 3,60 (м, 1Н), 3,46 (м, 1Н), 3,29-3,32 (м, 2H), 2,72 (м, 1H), 2,09 (м, 1H), 1,70 (м, 1H), 1,45 (с, 9H).
Процедура B: Раствор продукта из примера 3, процедура B, (4,35 кг), содержащий 2,13 кг (6,47 моль) соединения 3 в тетрагидрофуране (13,9 кг), добавляли к перемешиваемому охлажденному раствору гидроксида калия (1,60 кг, 40,0 моль) в деионизированной воде (2,00 кг) в атмосфере азота, поддерживая температуру ниже 35°C. Реакционную смесь нагревали и поддерживали при 40-45°C в течение 24 ч, по истечении этого времени GC и TLC анализы показали, что реакция завершилась. Смесь охлаждали до 25°C и промывали MTBE (34 кг), используя время перемешивания 15 мин и время осаждения 15 мин. Водный слой собирали и охлаждали до 1°C. Смесь концентрированной хлороводородной кислоты (2,61 кг, 26,5 моль) в деионизированной воде (2,18 кг) затем добавляли медленно, поддерживая температуру смеси при <15°C во время и в течение 15 мин после добавления. pH раствора доводили до 3,7 дополнительным добавлением хлороводородной кислоты. Белое твердое вещество собирали фильтрованием, промывали водой (16 кг) и сушили в вакууме при окружающей температуре в течение 6 дней. Сухое твердое вещество весило 1,04 кг. Фильтрат охлаждали до <10°C и выдерживали при этой температуре, когда pH снижали добавлением еще хлороводородной кислоты (использовали 1,6 л 6 н.; 9,6 моль; конечный pH=2). Белое твердое вещество собирали фильтрованием, промывали водой (8 л) и сушили в вакууме при 40°C в течение 3 дней. Сухое твердое вещество весило 0,25 кг. Объединенные твердые вещества (1,29 кг, 73% выход) были хроматографически идентичными предварительно приготовленным пробам.
Пример 5. Синтез (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (5)
Процедура A: Раствор (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (83 г) в 1-метил-2-пирролидиноне (0,42 л) перемешивали в атмосфере азота при 110-112°C в течение 2 ч. Пробы реакционного раствора отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции реакционный раствор охлаждали до 20-25°C. Раствор перемешивали с деионизированной водой (1,00 л) и добавляли MTBE (1,00 л). Фазы разделяли и органический слой собирали. Водную фазу экстрагировали MTBE (1,00 л), затем хлороформом (1,00 л). Органические слои объединяли и концентрировали при пониженном давлении при 50-55°C, получая масло. Это масло растворяли в MTBE (2,00 л) и промывали дважды 0,6 н. хлороводородной кислотой (2×1,00 л). Органический слой собирали и концентрировали при пониженном давлении при 50-55°C, получая полутвердое вещество. Полутвердое вещество суспендировали в смеси 1:4 этилацетат/гексаны (100 мл), нагревали до 50°C, выдерживали в течение 30 мин, охлаждали до -10°C и фильтровали. Фильтрат концентрировали при пониженном давлении, получая масло, которое растворяли в MTBE (250 мл) и промывали дважды 0,6 н. хлороводородной кислотой (2×100 мл). Органический слой концентрировали при пониженном давлении при 50-55°C, получая полутвердое вещество, которое суспендировали в смеси 1:4 этилацетат/гексаны (50 мл), нагревали до 50°C, выдерживали в течение 30 мин, охлаждали до -10°C и фильтровали. Твердые вещества собирали, суспендировали в гексанах (200 мл) и собирали фильтрованием, получая 54,0 г (77,6%) соединения 5.
1H ЯМР (CDCl3, 400 МГц) δ 11 00 (ушир. с, 1Н) 3,63 (м, 1Н), 3,45 (м, 1Н), 3,30 (м, 1Н), 2,97 (м, 1Н), 2,58 (м, 1Н), 2,44 (м, 2H), 2,09 (м, 1Н), 1,59 (М, 1Н), 1,46 (с, 9H).
Процедура B: Раствор (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (1,04 кг, 3,81 моль) в 1-метил-2-пирролидиноне (6,49 кг) перемешивали в атмосфере азота при 110°C в течение 5 ч, по истечении этого времени TLC и HPLC анализы показали, что реакция завершилась. Реакционную смесь охлаждали до 25°C (4 ч) и объединяли с водой (128 кг) и MTBE (9,44 кг). Смесь перемешивали энергично в течение 20 мин и фазам давали возможность разделиться (10 ч). Органическую фазу собирали, и водную фазу объединяли с MTBE (9,44 кг), перемешивали в течение 15 мин и давали возможность отстояться (45 мин). Органическую фазу собирали, и водную фазу объединяли с MTBE (9,44 кг), перемешивали в течение 15 мин, давали возможность отстояться (15 мин). Три органические фазы объединяли и промывали три раза 1 н. хлороводородной кислотой (порции 8,44 кг) и один раз водой (6,39 кг), используя время перемешивания 15 мин и время осаждения 15 мин для каждого промывания. Полученный раствор сушили над безводным сульфатом натрия (2,0 кг) и фильтровали. Фильтрат концентрировали при пониженном давлении при 31°C (2 ч), получая твердое вещество. Это твердое вещество нагревали в вакууме в течение 4 ч, при 39°C в течение 4 ч и в течение 16 ч при 25°C, получая 704 г (81%) соединения 5 (чистота 99,7% по GC).
Процедура C: (модернизированный синтез соединения 5 с использованием соединения 2 в качестве исходного материала) Перемешиваемую смесь этоксида натрия в этаноле (21 массовый процент, 343 г, 1,05 моль), этанола (безводный, 300 мл) и диэтилмалоната (168 г, 1,05 моль) нагревали до 40°C в течение 1,5 ч. К этой смеси добавляли раствор (R)-трет-бутил 3-(метилсульфонилокси)пирролидин-1-карбоксилата (138 г, 0,592 моль) в этаноле (100 мл) и реакционную смесь нагревали до 78°С в течение 8 ч. Охлажденную реакционную смесь разбавляли водой (2,0 л) и подкисляли до pH=3 6M HCl (100 мл). Водную этанольную смесь экстрагировали толуолом (1,0 л) и органическую фазу концентрировали в вакууме, получая 230 г красного масла. Красное масло добавляли при 85°C к 22,5 мас.% водному раствору гидроксида калия (748 г, 3,01 моль). После завершения добавления реакционной температуре давали возможность медленно повышаться до 102°C, в результате чего происходила перегонка этанола. Когда реакционная температура достигала 102°C и перегонка затухала, нагревание продолжали дополнительные 90 мин. Реакционную смесь охлаждали до окружающей температуры и промывали толуолом (2×400 мл). К водному слою добавляли 600 мл 6 M хлороводородной кислоты, поддерживая внутреннюю температуру ниже 20°C. Это имело результатом образование осадка, начинающееся при pH около 4-5. Суспензию фильтровали, и осадок на фильтре промывали 300 мл воды. Твердое вещество сушили в вакууме, получая 77 г (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты в виде грязно-белого твердого вещества (54% выход по отношению к (R)-трет-бутил 3-(метилсульфонилокси)пирролидин-1-карбоксилату).
1H ЯМР (ДМСО-d6, 400 МГц) δ 3,47 (м, 1Н), 3,32 (м, 1Н), 3,24 (м, 1Н), 3,16 (м, 1Н), 3,92 (м, 1Н), 2,86 (м, 1Н), 1,95 (м, 1Н), 1,59 (м, 1Н), 1,39 (с, 9H).
Суспензию (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (15 г, 55 ммоль) в толуоле (150 мл) и диметилсульфоксиде (2 мл) нагревали до кипения с возвращением флегмы в течение периода 2 ч. Смеси позволяли достичь окружающих условий и разбавляли MTBE (150 мл). Органический раствор промывали 10% водной лимонной кислотой (2×200 мл) и растворитель удаляли в вакууме, получая 11,6 г (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты в виде грязно-белого твердого вещества (92% выход).
1H ЯМР (ДМСО-d6, 400 МГц) δ 12,1 (с, 1Н), 3,36-3,48 (м, 1Н), 3,20-3,34 (м, 1Н), 3,05-3,19 (м, 1Н), 2,72-2,84 (м, 1Н), 2,30-2,42 (м, 1Н), 2,22-2,30 (м, 2H), 1,85-2,00 (м, 1Н), 1,38-1,54 (м, 1Н), 1,35 (2, 9H).
Пример 6. Синтез трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилата (6)
Процедура A: Раствор (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (49,0 г, 214 ммоль) в тетрагидрофуране (THF) (200 мл) охлаждали до -10°C. 250 мл (250 ммоль) 1 M раствора борана в THF медленно добавляли в колбу, поддерживая температуру ниже 0°C. Раствор нагревали до окружающей температуры и перемешивали в течение 1 ч. Пробы раствора отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции раствор охлаждали до 0°C и 10% раствор гидроксида натрия (80 мл) добавляли по каплям в течение 30 минутного периода, чтобы контролировать выделение газа. Раствор экстрагировали 500 мл раствора 1:1 гексаны/этилацетат. Органический слой промывали насыщенным раствором хлорида натрия и сушили 10 г силикагеля. Силикагель удаляли фильтрованием и промывали 100 мл смеси 1:1 гексаны/этилацетат. Органические слои объединяли и концентрировали в вакууме, получая соединение 6 (42 г, 91,3%) в виде светло-оранжевого масла, которое затвердевало при отстаивании.
1H ЯМР (CDCl3, 400 МГц) δ 3 67 (м, 2H), 3,38-3,62 (м, 2H), 3,25 (м, 1Н), 2,90 (м, 1Н), 2,25 (м, 1Н), 1,98-2,05 (м, 1Н) 1,61-1,69 (м, 2H), 1,48-1,59 (м, 2H), 1,46 (с, 9H).
Процедура В: Комплекс боран-THF (3,90 кг или л 1M в THF, моль) добавляли медленно к перемешиваемому раствору (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (683 г, 3,03 моль) в THF (2,5 кг), выдерживали в атмосфере азота и использовали водяную баню для поддерживания температуры между 23 и 28°C. Добавление занимало 1,75 ч. Перемешивание при 25°C продолжали в течение 1 ч, по истечении этого времени GC анализ показал завершение реакции. Реакционную смесь охлаждали до <10°C и поддерживали ниже 25°C, когда медленно добавляли 10% водный гидроксид натрия (1,22 кг). Добавление занимало 40 мин. Смесь перемешивали 1 ч при 25°C и затем объединяли со смесью 1:1 (об./об.) гептан/этилацетат (7 л). Смесь перемешивали в течение 15 мин и давали возможность разделиться на фазы (1 ч). Органическую фазу отбрасывали, а водную фазу объединяли со второй 7-л порцией смеси 1:1 гептан/этилацетат. Перемешивали в течение 15 мин и давали возможность разделиться на фазы (20 мин). Органическую фазу снова отбрасывали, и объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (4,16 кг), используя время перемешивания 15 мин и время отстаивания 1 ч. Органическую фазу объединяли с силикагелем (140 г) и перемешивали 1 ч. Безводный сульфат натрия (700 г) добавляли и смесь перемешивали в течение 1,5 ч. Смесь фильтровали, и осадок на фильтре промывали смесью 1:1 гептан/этилацетат (2 л). Фильтрат концентрировали в вакууме при <40°C в течение 6 ч. Полученное масло весило 670 г (103% выход) и содержало следовые количества гептана, но в остальном было идентично предварительно приготовленным пробам соединения 6 по ЯМР анализу.
Пример 7: трет-бутил (R)-3-(2-(метилсульфонилокси)этил)пирролидин-1-карбоксилат (7)
Процедура A: К раствору трет-бутил (R)-3-(2-гидроксиметил)пирролидин-1-карбоксилата (41,0 г, 190 ммоль) добавляли триэтиламин (40 мл) в толуоле (380 мл) и охлаждали до -10°C. Метансульфонилхлорид (20,0 мл, 256 ммоль) добавляли медленно так, чтобы поддерживать температуру около -5 до 0°C. Раствор нагревали до окружающей температуры и перемешивали в течение 1 ч. Пробы раствора отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции раствор фильтровали, и фильтрат промывали 5% раствором бикарбоната натрия (250 мл). Органический слой собирали и промывали насыщенным водным раствором хлорида натрия (250 мл). Органический слой собирали, сушили над силикагелем (10 г) и концентрировали в вакууме, получая соединение 7 (53,0 г, 92,8 %) в виде светло-желтого вязкого масла.
1H ЯМР (CDCl3, 400 МГц) δ 4,26 (т, J=6,8 Гц, 2H), 3,41-3,63 (м, 2H), 3,27 (м, 1Н), 3,02 (с, 3H), 2,92 (м, 1Н), 2,28 (м, 1H), 2,05 (м, 1Н), 1,83 (м, 2H), 1,50-1,63 (м, 1Н), 1,46 (с, 9H).
Процедура B: В атмосфере азота раствор триэтиламина (460 г, 4,55 моль) и трет-бутил (R)-3-(2-гидроксиметил)пирролидин-1-карбоксилата (полная проба из примера 7, процедура B, 3,03 моль) в толуоле (5,20 кг) перемешивали и охлаждали до 5°C. Метансульфонилхлорид (470 г, 4,10 моль) добавляли медленно в течение 1,25 ч, поддерживая температуру ниже 15°C, используя охлаждение на бане со льдом. Смесь постепенно нагревали (в течение 1,5 ч) до 35°C и эту температуру поддерживали в течение 1,25 ч, на этот момент GC анализ показал, что реакция завершилась. Смесь охлаждали до 25°C и твердые вещества отфильтровывали и осадок на фильтре промывали толуолом (1,28 кг). Фильтрат перемешивали с 10% водным бикарбонатом натрия (4,0 кг) в течение 15 мин и фазам давали возможность разделиться в течение 30 мин. Органическую фазу затем перемешивали с насыщенным водным раствором хлорида натрия (3,9 кг) в течение 30 мин и фазам давали возможность разделиться в течение 20 мин. Органическую фазу объединяли с силикагелем (160 г) и перемешивали в течение 1 ч. Безводный сульфат натрия (540 г) добавляли, и смесь перемешивали дополнительно 40 мин. Смесь затем фильтровали и осадок на фильтре промывали толуолом (460 г). Фильтрат концентрировали в вакууме при 50°C в течение 5 ч и полученное масло выдерживали в вакууме при 23°C еще 8 ч. Оставалось 798 г соединения 7, чистота 93% по GC анализу.
Пример 8: Синтез трет-бутил (R)-3-винилпирролидин-1-карбоксилата (9)
Процедура A: Раствор трет-бутил (R)-3-((метилсульфонилокси)этил)пирролидин-1-карбоксилата (49,0 г, 167 ммоль), иодида натрия (30,0 г, 200 ммоль) и 1,2-диметоксиэтана (450 мл) перемешивали при 50-60°C в течение 4 ч. Пробы раствора отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции раствор охлаждали до -10°C и добавляли твердый трет-бутоксид калия (32,0 г, 288 ммоль), поддерживая температуру ниже 0°C. Реакционную смесь нагревали до окружающей температуры и перемешивали в течение 1 ч. Пробы смеси отбирали каждый час и анализировали HPLC, чтобы установить завершение реакции. После завершения реакции смесь фильтровали через прокладку диатомовой земли (25 г на основе сухого вещества). Осадок на фильтре промывали 1,2-диметоксиэтаном (100 мл). Объединенные фильтраты концентрировали в вакууме, получая оранжевое масло с суспендированными твердыми веществами. Масло растворяли в гексанах (400 мл), перемешивали в течение 30 мин и фильтровали, чтобы удалить твердые вещества. Органический слой сушили над силикагелем (10 г) и концентрировали в вакууме, получая соединение 9 (26,4 г, 82,9%) в виде бесцветного масла.
1H ЯМР (CDCl3, 400 МГц) δ 5,77 (м, 1Н), 5,10 (дд, J=1,2 Гц, J=16 Гц, 1Н), 5,03 (дд, J=1,2 Гц, J=8,8 Гц, 1Н), 3,41-3,59 (м, 2H), 3,29 (м, 1Н), 3,05 (м, 1Н), 2,78 (м, 1Н), 2,01 (м, 1Н), 1,62-1,73 (м, 1Н), 1,46 (м, 9H).
Процедура B: Раствор трет-бутил (R)-3-(2-(метилсульфонилокси)этил)пирролидин-1-карбоксилата (792 г продукта из примера 7, процедура B, ~2,5 моль), иодида натрия (484 г, 3,27 моль) и 1,2-диметоксиэтана (7,2 л) перемешивали при 55°C в течение 4,5 ч в атмосфере азота, на этот момент GC анализ показал, что реакция завершилась. Раствор охлаждали до <10°C и твердый трет-бутоксид калия (484 г, 4,32 моль) добавляли порциями (время добавления 1,25 ч), поддерживая температуру ниже 15°C. Реакционную смесь перемешивали 1 ч при 5°C, нагревали медленно (6 ч) до 20°C и перемешивали при 20°C в течение 1 ч. Раствор фильтровали через прокладку из диатомовой земли (400 г на основе сухого вещества). Осадок на фильтре промывали 1,2-диметоксиэтаном (1,6 кг). Объединенные фильтраты концентрировали в вакууме, и полутвердый остаток перемешивали с гептаном (6,0 л) в течение 2 ч. Твердые вещества удаляли фильтрованием (осадок на фильтре промывали 440 мл гептана) и фильтрат концентрировали в вакууме при 20°C, получая 455 г соединения 9 (чистота 90,7%). Пробу этого материала (350 г) подвергали фракционной перегонке при 20-23 торр, получая 296 г очищенного соединения 9 (т. кип. 130-133°C) (чистота >99% по GC анализу).
Пример 9: Синтез 3-бром-5-(тетрагидро-2H-пиран-4-илокси)пиридина (12)
Раствор 5-бромпиридин-3-ола (146 г, 834 ммоль), тетрагидро-2H-пиран-4-ола (128 г, 1250 ммоль) и трифенилфосфина (329 г, 1250 ммоль) в толуоле (2,0 л) нагревали до кипения с возвращением флегмы и 750 мл дистиллята удаляли через ловушку Дина-Старка. Реакционную смесь охлаждали до 60°C и 547 г (1,25 моль) 40% (мас./мас.) раствора DEAD в толуоле добавляли по каплям в течение 1-часового периода. Добавление было экзотермическим с реакционной температурой в конце добавления около 95°C. Реакционную смесь перемешивали при 115°C в течение 18 ч и порцию реакционного раствора отбирали каждый час и анализировали HPLC, чтобы установить, что реакция завершилась. После завершения реакции 500 мл растворителя удаляли перегонкой, и остаток охлаждали до окружающей температуры. Этот органический слой промывали 10% водным раствором гидроксида натрия (2×0,50 л) и концентрировали в вакууме, получая вязкое масло, которое растворяли в 2 н. хлороводородной кислоте (1,0 л). Диатомовую землю (100 г) добавляли с перемешиванием, и полученную суспензию фильтровали. Слой промывали 2 н. хлороводородной кислотой (1,0 л) и фильтраты объединяли и экстрагировали диизопропиловым простым эфиром (500 мл). Слой диизопропилового простого эфира отбрасывали, и водный слой обрабатывали сажей (10 г) и перемешивали при 45-50°C в течение 1 ч. Суспензию фильтровали через слой диатомовой земли (25 г). Фильтрат собирали, охлаждали до 5°C и pH доводили 50% водным раствором гидроксида натрия (250 мл) до pH=13. Раствор экстрагировали дважды хлороформом (1,0 л, 600 мл) и экстракты в хлороформе объединяли и концентрировали в вакууме, получая соединение 12 в виде темно-красного вязкого масла/низкоплавкого твердого вещества (187 г, 87%), которое использовали далее без дополнительной очистки.
1H ЯМР (CDCl3, 400 МГц) δ 8,29 (с, 1Н), 8,24 (с, 1Н), 7,38 (с, 1Н), 4,52 (м, 1Н), 3,98 (м, 2H), 3,60 (м, 2H), 2,05 (м, 2H), 1,81 (м, 2H).
Пример 10: Синтез трет-бутил (R)-(E)-3-(2-(5-(тетрагидро-2H-пиран-4-илокси)пиридин-3-ил)винил)пирролидин-1-карбоксилата (13)
Смесь трет-бутил (R)-3-винилпирролидин-1-карбоксилата 9 (7,00 г, 35,5 ммоль), 3-бром-5-(тетрагидро-2H-пиран-4-илокси)пиридина 12 (10,0 г, 38,8 ммоль), ацетата палладия (0,40 г, 1,8 ммоль), трихлоргексилфосфина (1,0 г, 3,57 ммоль) и диизопропилэтиламина (15 мл) в 1-метил-2-пирролидиноне (130 мл) перемешивали при 130°C в течение 17 ч. Реакционную смесь охлаждали до окружающей температуры, разбавляли водой (800 мл) и экстрагировали этилацетатом (2×200 мл). Объединенные органические экстракты сушили над сульфатом натрия, концентрировали и очищали колоночной хроматографией на силикагеле, используя 60-100% этилацетат в гексанах. Этот продукт дополнительно очищали HPLC с обращенной фазой, используя 0,05% трифторуксусную кислоту в ацетонитриле и 0,05% трифторуксусную кислоту в воде, получая трет-бутил (R)-(E)-3-(2-(5-(тетрагидро-2H-пиран-4-илокси)пиридин-3-ил)винил)пирролидин-1-карбоксилат (11,0 г) в виде камеди.
1H-ЯМР (CDCl3, 400 МГц): δ 8,41 (с, 1Н), 8,37 (д, J=2,3 Гц, 1Н), 7,68 (с, 1Н), 6,48 (д, J=16,1 Гц, 1Н), 6,43 (дд, J=16,0, 6,4 Гц, 1Н), 4,71-4,66 (м, 1Н), 4,02-3,96 (м, 2Н), 3,68-3,52 (м, 4Н), 3,44-3,34 (м, 1Н), 3,28-3,15 (м, 1Н), 3,09-2,98 (м, 1Н), 2,18-2,04 (м, 3Н), 1,90-1,78 (м, 3Н), 1,48 (с, 9Н).
Пример 11: Синтез (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин (14) гемигалактарата
Раствор трет-бутил (R)-(E)-3-(2-(5-(тетрагидро-2H-пиран-4-илокси)пиридин-3-ил)винил)пирролидин-1-карбоксилата (18 г, 48,13 ммоль) в дихлорметане (40 мл) и трифторуксусной кислоте (40 мл) перемешивали при окружающей температуре в течение 2 ч. Реакционную смесь концентрировали на роторном испарителе и остаток распределяли между насыщенным раствором хлорида натрия (50 мл) и хлороформом (100 мл). Смесь подщелачивали до pH 9 10% водным раствором гидроксида натрия. Органический слой отделяли, и водный слой экстрагировали хлороформом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали на роторном испарителе, получая (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидро-2H-пиран-4-илокси)пиридин (8,0 г) в виде камеди. Ее растворяли в метаноле (100 мл) и добавляли галактаровую кислоту (3,0 г, 14,6 ммоль) и смесь нагревали до кипения с возвращением флегмы. Горячий раствор фильтровали и фильтрату давали возможность охладиться до окружающей температуры. Кристаллизованный продукт фильтровали, и твердое вещество суспендировали в 10% воде в этаноле (180 мл). Суспензию нагревали до кипения с возвращением флегмы и горячий раствор фильтровали. Фильтрату давали возможность охладиться до окружающей температуры. Кристаллизованный продукт фильтровали и сушили на высоковакуумном насосе, получая (R)-3-((E)-2-пирролидин-3-илвинил)-5-(тетрагидро-2H-пиран-4-илокси)пиридин гемигалактарат (4,5 г).
1H-ЯМР (CD3OD, 300 МГц): δ 8,04 (с, 1Н), 8,01 (д, J=2,2 Гц, 1Н), 7,36 (с, 1Н), 6,46 (д, J=16,0 Гц, 1Н), 6,21 (дд, J=16,0, 7,5 Гц, 1Н), 4,65-4,4,54 (м, 1Н), 4,12 (с, 1Н), 3,89-3,83 (м, 2Н), 4,80 (с, 1Н), 3,56-3,33 (м, 4Н), 3,27-3,18 (м, 1Н), 3,12-2,96 (м, 2Н), 2,23-2,14 (м, 1Н), 1,98-1,91 (м, 2Н), 1,88-1,78 (м, 1Н), 1,68-1,58 (м, 2Н); MS (m/z): 275 (М+1).
Пример 12: Крупномасштабный синтез (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин (14) моно-L-малата
В атмосфере азота смесь 3-бром-5-(тетрагидро-2H-пиран-4-илокси)пиридина (125 г, чистота 85%, 410 ммоль), (R)-трет-бутил-3-винилпирролидин-1-карбоксилата (67,4 г, 340 ммоль), ацетата палладия (II) (8,1 г, 36 ммоль), три-н-бутилфосфина (15 г, 74 ммоль), карбоната калия (74,0 г, 530 ммоль) и DMAC (0,85 л) перемешивали и нагревали при 130°C, отслеживая завершение реакции по LCMS. После завершения реакции реакционную смесь охлаждали до окружающей температуры и фильтровали через прокладку из диатомовой земли (50 г на основе сухого вещества), промывали прокладку диизопропиловым простым эфиром (0,60 л). Фильтрат объединяли с диизопропиловым простым эфиром (0,60 л) и деионизированной водой (0,50 л) и перемешивали в течение 15 мин. Фазам давали возможность разделиться (15 мин) и органическую фазу собирали. Водную фазу экстрагировали второй порцией диизопропилового простого эфира (0,60 л), используя время перемешивания 15 мин и время отстаивания 15 мин. Объединенные слои диизопропилового простого эфира промывали деионизированной водой (2×0,50 л) и концентрировали при пониженном давлении, получая темно-красное вязкое масло (136 г). Это масло растворяли в диизопропиловом простом эфире (1,40 л) и охлаждали до около 10°C на бане со льдом перед загрузкой 6 н. хлороводородной кислоты (0,40 л) через капельную воронку в течение 15 мин периода, поддерживая температуру ниже 20°C. Двухфазную смесь нагревали до окружающей температуры (при нагревании происходило выделение газа) и перемешивали до тех пор, пока анализ LCMS не показал, что реакция завершилась. После завершения реакции фазам давали возможность разделиться, и органический слой отбрасывали. pH водного слоя доводили до pH 5-6, используя 10% водный раствор гидроксида натрия (0,485 л), и экстрагировали хлороформом (0,25 л). Слой хлороформа отбрасывали. Водный слой затем доводили до pH>13, используя 10% водный раствор гидроксида натрия (0,075 л), и снова экстрагировали хлороформом (0,50 л). Экстракт в хлороформе концентрировали при пониженном давлении, получая красное вязкое масло (55,0 г). Этот материал был смесью желательного (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-тетрагидро-2H-пиран-4-илокси)пиридина (~75%) и соответствующего Z (~5%) и "экзо" (~20%) изомеров по ЯМР анализу. Этот результат был воспроизводимым в течение многих экспериментов.
Z и "экзо" примеси удаляли из желательного (R)-3-((E)2-(пирролидин-3-ил)винил)-5-тетрагидро-2H-пиран-4-илокси)пиридина путем превращения в соль щавелевой кислоты. Раствор щавелевой кислоты (53,2 г, 591 ммоль) в смеси 2-пропанола (0,20 л) и деионизированной воды (0,09 л) готовили перемешиванием и нагреванием при 50-55°C (15 мин). Этот раствор добавляли в течение 5 мин периода к перемешиваемому раствору (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-тетрагидро-2H-пиран-4-илокси)пиридина (82,0 г, чистота 76% по HPLC, 299 ммоль) в 2-пропаноле (1,0 л), поддерживаемом при 70-75°C. Добавление щавелевой кислоты давало экзотерму (4-5°C), которую контролировали, регулируя скорость добавления. Источник нагревания удаляли, и раствор охлаждали медленно до 45-50°C в течение 45 мин. Образование осадка происходило быстро, начинаясь около 65-70°C, и осадок становился более густым при охлаждении полученной суспензии. Твердые вещества собирали фильтрованием при 45-50°C и промывали последовательно 2-пропанолом (2×0,25 л) и гексанами (2×0,20 л). Желтовато-коричневое твердое вещество сушили на воздухе в течение 2 ч, после чего взвешивали (95 г). ЯМР анализ показал, что содержание Z и "экзо" примесей, каждой, было уменьшено до <1%. Этот результат был воспроизводимым в течение многих экспериментов. Материал даже более высокой чистоты получали перекристаллизацией из смеси этанол/вода. Стехиометрия соли была 2,3:1 кислота/основание (см. пример 15).
Раствор (R)-3-((E)-2-(пирролидиний-3-ил)винил)-5-(тетрагидро-2H-пиран-4-илокси)пиридиний оксалата (380 г) в деионизированной воде (2,6 л) перемешивали и охлаждали до около 10°C на бане со льдом. Водный раствор гидроксида натрия (0,40 л, 25%) добавляли в течение периода 15 мин, поддерживая температуру ниже 30°C. Хлороформ (1,6 л) затем добавляли и смесь перемешивали энергично в течение 20 мин и фильтровали, чтобы удалить нерастворимый оксалат натрия. Слоям давали возможность разделиться, и слой хлороформа объединяли с Silicycle Si-Thiol® (21,6 г). Смесь перемешивали и нагревали при 50-55°С в течение 3-4 ч, охлаждали до окружающей температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, получая светло-красное вязкое масло (221 г). Часть этого свободного основания (216 г) растворяли в 2-пропаноле (1,2 л), нагревали до 70-75°C и обрабатывали твердой L-яблочной кислотой (106 г), используя промывание 2-пропанолом (100 мл) для облегчения переноса. Растворение твердого вещества давало экзотерму 5-7°C в течение 3-5 мин. Смесь выдерживали при 75-78°C в течение 10 мин, чтобы гарантировать завершение растворения твердых веществ, и затем охлаждали медленно до окружающей температуры (90 мин). Когда температура достигала 65°C, в раствор вносили затравку из нескольких кристаллов соли, а именно (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидро-2H-пиран-4-илокси)пиридин моно-L-малата. После перемешивания при окружающей температуре в течение 1 ч суспензию фильтровали. Собранные твердые вещества промывали 2-пропанолом (2×0,80 л), сушили на воздухе в течение 30 минут и сушили в вакууме при 78°C в течение 8 ч. Полученный грязно-белый материал весил 297 г и имел чистоту >99% по HPLC.
Пример 13: Процедура отбора солевых форм (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина
Пробирки (4 мл), снабженные магнитными стержневыми мешалками, загружали эквимиллимолярными количествами основания (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина и представляющей интерес кислоты (неразбавленные) и растворяли в 500 мкл или 2-пропанола, или ацетонитрила с помощью нагревания. Если осаждения не происходило при охлаждении, добавляли изопропилацетат (100 мкл) в качестве анти-растворителя.
Если никакого осаждения не происходило, растворитель выпаривали в токе азота с умеренным нагреванием и пробовали альтернативный растворитель. Альтернативные растворители включали ацетон, этилацетат, изопропилацетат, абсолютный этанол, ацетонитрил, гексан, трет-бутанол, трет-бутилацетат и их смеси.
Применения спиртов избегали в случае сульфоновых кислот. Изопропилацетат использовали с осторожностью, тогда как от применения ацетона и этилацетата отказались из-за продемонстрированной реактивности с функциональной вторичной аниногруппой (R)-3-((E)-2-пирролидин-3-илвинил)-5-(тетрагидро-2H-пиран-4-илокси)пиридина с указанными растворителями.
Результаты этих экспериментов суммированы в таблице 1.
Кислоты, использованные в предварительном отборе солей
Как показано в таблице 1, поиск твердых солевых форм (R)-3-((E)-2-пирролидин-3-илвинил)-5-(тетрагидро-2H-пиран-4-илокси)пиридина был требующим усилий. Ниже даны примеры твердых солей и их синтезов.
Пример 14: Получение (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин моно-L-малата
К перемешиваемому раствору (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина (900 мг, 3,28 ммоль) в 2-пропаноле (5 мл), нагреваемому почти до кипения, добавляли L-яблочную кислоту (439,8 мг, 3,28 ммоль, неразбавленная) тремя порциями. Раствор перемешивали вблизи кипения в течение 10 мин. Добавляли изопропилацетат (1 мл), прекращали нагревание и в раствор вносили затравку, пока он еще был горячим. Раствору давали возможность охладиться до окружающей температуры (22°C) с перемешиванием, при этом соль осаждалась в виде белого гранулированного твердого вещества. Соль повторно растворяли при нагревании, повторно вносили затравку, пока раствор был горячим, охлаждали и давали отстояться при окружающей температуре без перемешивания в течение 24 ч. Полученные пластинчатые кристаллы собирали вакуумным фильтрованием, промывали изопропилацетатом (5 мл) и сушили в атмосфере азота в течение 10 мин. Дополнительная сушка в вакуумной печи при 70°C в течение 1,5 ч давала 1,267 г (94,6%) светло-желтых кристаллов (т. пл.=118-119°C). 1H-ЯМР (D2O или d6-DMSO) согласуется со стехиометрией 1:1 кислота:основание. DSC показывает единственную эндотерму с максимумом при 119,62°C. DVS показывает минимальное поглощение воды вплоть до 80% R.H.
1H-ЯМР (D2O, 400 МГц): δ 8,15 (с, 1Н), 8,10 (с, 1Н), 7,58 (с, 1Н), 6,52 (д, 1Н), 6,28 (дд, 1Н), 4,63 (м, 1Н, частично маскируется резонансом остаточной Н2О), 4,22 (дд, 1Н), 3,88 (м, 2Н), 3,55 (м, 2Н), 3,46 (дд, 1Н), 3,38 (м, 1Н), 3,25 (м, 1Н), 3,11 (м, 1Н), 3,02 (м, 1Н), 2,65 (дд, 1Н), 2,42 (дд, 1Н), 2,20 (м, 1Н), 1,96 (м, 2Н), 1,85 (м, 1Н), 1,68 (м, 2Н).
1H-ЯМР (d6-ДМСО, 400 МГц): δ 8,20 (с, 1Н), 8,18 (с, 1Н), 7,52 (с, 1Н), 6,55 (д, 1Н), 6,46 (дд, 1Н), 4,68 (м, 1Н), 3,87 (м, 3Н), 3,49 (м, 2Н), 3,40 (дд, 1Н), 3,32 (м, 1Н), 3,18 (м, 1Н), 3,05 (м, 1Н), 2,93 (м, 1Н), 2,51 (дд, 1Н, частично маскируется остаточным ДМСО), 2,31 (дд, 1Н), 2,14 (м, 1Н), 1,98 (м, 2Н), 1,80 (м, 1Н), 1,58 (м, 2Н).
Пример 15: Получение (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин оксалата
Теплый раствор (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина (1,137 г 85%, 3,52 ммоль, корректированный для чистоты) в 2-пропаноле (8,5 мл) и воды (0,4 мл) обрабатывали щавелевой кислотой (0,373 г, 4,14 ммоль), как твердым веществом, одной порцией. Полученную смесь перемешивали и нагревали до температуры, близкой к кипению с возвращением флегмы. Немного твердого вещества начинало осаждаться из горячего раствора. Смеси давали возможность охладиться до окружающей температуры. Грязно-белые твердые вещества отфильтровали (Бюхнер), промывали 2-пропанолом (10 мл, 8 мл) и сушили в вакууме (с током воздуха) при 50°C в течение 3 ч, получая 0,861 г (50,8% выход на основе 2,3 оксалатной стехиометрии, корректированной для чистоты исходного материала) грязно-белого порошка. Пробу 0,765 г указанного материала перекристаллизовывали из смеси 2-пропанола (8 мл) и воды (1,3 мл), нагревали до кипения с возвращением флегмы. При охлаждении до окружающей температуры полученные твердые вещества отфильтровывали (Бюхнер), промывали 2-пропанолом (10 мл), сушили в вакууме (с продуванием воздухом) при 50°C в течение 4 ч и затем дополнительно сушили в вакууме с продуванием воздухом при 70°C в течение 24 ч, получая 0,441 г (57,6% извлечение) от грязно-белого до белого твердого вещества, т.пл. 180-181°C.
1H-ЯМР (D2O, 400 МГц) δ 8,23 (с, 1Н), 8,20 (с, 1Н), 7,96 (с, 1Н), 6,54 (д, 1Н), 6,40 (дд, 1Н), 4,73 (м, 1Н), 3,84 (м, 2Н), 3,54 (м, 2Н), 3,45 (дд, 1Н), 3,35 (м, 1Н), 3,23 (м, 1Н), 3,12 (м, 1Н), 3,02 (м, 1Н), 2,16 (м, 1Н), 1,96 (м, 2Н), 1,83 (м, 1Н), 1,68 (м, 2Н).
Пример 16: Получение (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин ди-п-толуил-D-тартрата
К перемешиваемому раствору (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина (4,0 г, 15 ммоль) в растворе этанола (12,5 мл), нагретому до 60°C, добавляли твердую ди-п-толуил-D-винную кислоту (5,3 г, 14 ммоль). Раствор выдерживали при 60°C в течение 2-3 мин, чтобы гарантировать завершение растворения твердых веществ, затем источник тепла удаляли и раствор охлаждали до 25-30°C в течение 60 мин. Полученную суспензию выдерживали при 25-30°C в течение 30 мин и затем фильтровали, чтобы собрать твердые вещества. Твердые вещества промывали этанолом (2×20 мл), сушили на воздухе в течение 30 мин, затем сушили в вакуумной печи при пониженном давлении при 50°C до прекращения изменения веса, получая (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин ди-п-толуил-D-тартрат в виде грязно-белого твердого вещества (6,7 г, 72%). ЯМР анализ показал стехиометрию 1:1 соли.
1H-ЯМР (ДМСО-d6, 400 МГц) δ 8,18 (с, 1Н), 8,15 (с, 1Н), 7,86 (д, 4Н), 7,47 (с, 1Н), 7,32 (д, 4Н), 6,43 (д, 1Н), 6,36 (м, 1Н), 5,67 (с, 2Н), 4,69 (м, 1Н), 3,85 (м, 2Н), 3,49 (м, 2Н), 3,25 (м, 2Н), 3,10 (м, 1Н), 2,88 (м, 2Н), 2,39 (с, 6Н), 1,98 (м, 3Н), 1,60 (м, 3Н).
Биологические анализы
Пример 17: Связывание радиолиганда при ЦНС nAChRs: α4β2 NNR подтип
Получение мембран из коры головного мозга крысы: Крыс (самки, Sprague-Dawley) с массой тела 150-250 г содержали при 12 ч цикле свет/темнота и обеспечивали свободный доступ к воде и пище, поставляемой PMI Nutrition International, Inc. Животных анестезировали 70% CO2 и затем обезглавливали. Головы удаляли и помещали на охлаждаемую льдом платформу. Головной мозг удаляли и помещали в 20 объемов (масса:объем) охлаждаемого льдом препаративного буфера (137 мМ NaCl, 10,7 мМ KCl, 5,8 мМ KH2PO4, 8 мМ Na2HPO4, 20 мМ HEPES (свободная кислота), 5 мМ иодацетамид, 1,6 мМ EDTA, pH 7,4), добавляли PMSF, растворенный в метаноле до конечной концентрации 100 мкМ, и суспензию гомогенизировали Polytron. Гомогенат центрифугировали при 18000 x g в течение 20 мин при 4°C и полученный осадок повторно суспендировали в 20 объемах охлаждаемой льдом воды. После 60 мин инкубирования на льду новый осадок собирали центрифугированием при 18000 x g в течение 20 мин при 4°C. Конечный осадок повторно суспендировали в 10 объемах буфера и хранили при -20°C.
Получение мембран из SH-EP1/α4β2 клональных клеток человека: Клеточные осадки из 40 150-мм культуральных чашек объединяли и гомогенизировали с применением Polytron (Kinematica GmbH, Switzerland) в 20 миллилитрах охлаждаемого льдом препаративного буфера. Гомогенат центрифугировали при 48000 g в течение 20 мин при 4°C. Полученный осадок повторно суспендировали в 20 мл охлаждаемого льдом препаративного буфера и хранили при -20°C.
Анализ В: день анализа замороженные мембраны оттаивали и вращали при 48000 x g в течение 20 мин. Супернатант декантировали и отбрасывали. Осадок повторно суспендировали в забуференном фосфатом солевом растворе Дульбекко (PBS, Life Technologies) pH 7,4 и гомогенизировали с применением Polytron в течение 6 секунд. Концентрации белка определяли, используя набор для анализа белка Pierce BCA с бычьим сывороточным альбумином в качестве стандарта (Pierce Chemical Company, Rockford, IL).
Препараты мембран (приблизительно 50 мкг для свойственного человеку и 200-300 мкг белка для крысиного α4β2) инкубировали в PBS (50 мкл и 100 мкл, соответственно) в присутствии конкурентного соединения (0,01 нМ до 100 мкМ) и 5 нМ [3H]никотина в течение 2-3 ч на льду. Инкубирование прекращали быстрым фильтрованием на харвестере мульти-многообразной ткани (Brandel, Gaithersburg, MD), используя фильтры GF/B, предварительно замоченные в 0,33% полиэтиленамине (мас./об.), чтобы уменьшить неспецифическое связывание. Ткань промывали 3 раза в PBS, pH 7,4. Сцинтилляционную жидкость добавляли к фильтрам, содержащим промытую ткань, и давали возможность уравновеситься. Фильтры затем подсчитывали, чтобы определить радиоактивность, связанную с мембранами, путем жидкостного сцинтилляционного подсчета (2200CA Tri-Carb LSC, Packard Instruments, 50% эффективность, или Wallac Trilux 1450 MicroBeta, 40% эффективность, Perkin Elmer).
Данные выражали как расщепления в минуту (DPMs). В каждом анализе каждая точка имела 2-3 повтора. Повторы для каждой точки усредняли и наносили на график против log концентрации лекарства. IC50, то есть концентрацию соединения, которая дает 50% ингибирование связывания, определяли нелинейной регрессией наименьших квадратов. Величины Ki рассчитывали, используя уравнение Cheng-Prussof (1973):
Ki=IC50/(1+N/Kd)
где N означает концентрацию [3H]никотина, Kd означает сродство никотина (3 нМ, определенное в отдельном эксперименте).
Пример 18: Связывание радиолиганда при ЦНС nAChRs: подтип α7 NNR
Крыс (самки, Sprague-Dawley) с массой тела 150-250 г содержали при 12 ч цикле свет/темнота и обеспечивали свободный доступ к воде и пище, поставляемой PMI Nutrition International, Inc. Животных анестезировали 70% CO2 и затем обезглавливали. Головы удаляли и помещали на охлаждаемую льдом платформу. Гиппокампы удаляли и помещали в 10 объемов (масса:объем) охлаждаемого льдом препаративного буфера (137 мМ NaCl, 10,7 мМ KCl, 5,8 мМ KH2PO4, 8 мМ Na2HPO4, 20 мМ HEPES (свободная кислота), 5 мМ иодацетамид, 1,6 мМ EDTA, pH 7,4), добавляли PMSF, растворенный в метаноле до конечной концентрации 100 мкМ, и суспензию ткани гомогенизировали с применением Polytron. Гомогенат центрифугировали при 18000 x g в течение 20 мин при 4°C и полученный осадок повторно суспендировали в 10 объемах охлаждаемой льдом воды. После 60 мин инкубирования на льду новый осадок собирали центрифугированием при 18000 x g в течение 20 мин при 4°C. Конечный осадок повторно суспендировали в 10 объемах буфера и хранили при -20°C. В день анализа ткань оттаивали, центрифугировали при 18000 x g в течение 20 мин и затем повторно суспендировали в охлажденном льдом PBS (забуференный фосфатом солевой раствор Дульбекко 138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4) до конечной концентраци приблизительно 2 мг белка/мл. Белок определяли по методу Lowry et al., J. Biol. Chem. 193: 265 (1951), используя бычий сывороточный альбумин в качестве стандарта.
Связывание [3H]MLA измеряли, используя модификацию методов Davies et al., Neuropharmacol. 38: 679 (1999), что приобщено ссылкой в отношении такого метода. [3H]MLA (удельная активность = 25-35 Ки/ммоль) получали от Tocris. Связывание [3H]MLA определяли, используя 2 ч инкубирование при 21°C. Инкубирования проводили в 48-луночных микротитрационных планшетах, и они содержали около 200 мкг белка на лунку в конечном инкубационном объеме 300 мкл. Буфером для инкубирования был PBS и конечная концентрация [3H]MLA была 5 нМ. Реакцию связывания прекращали фильтрованием белка, содержащего связанный лиганд, на стекловолоконных фильтрах (GF/B, Brandel), используя харвестер ткани Brandel при окружающей температуре. Фильтры замачивали в деионизированной воде, содержащей 0,33% полиэтиленамин, чтобы уменьшить неспецифическое связывание. Каждый фильтр промывали PBS (3×1 мл) при окружающей температуре. Неспецифическое связывание определяли добавлением 50 мкМ нерадиоактивного MLA в выбранные лунки.
Ингибирование связывания [3H]MLA испытуемыми соединениями определяли введением семи различных концентраций испытуемого соединения в выбранные лунки. Каждую концентрацию повторяли трижды. Величины IC50 оценивали как концентрацию соединения, которая ингибирует 50 процентов специфического связывания [3H]MLA. Постоянные ингибирования (величины Ki), выраженные в нМ, рассчитаны из величин IC50 по методу Cheng et al., Biochem. Pharmacol. 22: 3099-3108 (1973).
Селективность против периферических nAChRs
Пример 19: Взаимодействие при мышечном подтипе nAChR челокека
Активацию nAChRs мышечного типа устанавливали на клональной линии человека TE671/RD, которую получали из эмбриональной рабдомиосаркомы (Stratton et al., Carcinogen 10: 899 (1989)). Эти клетки экспрессируют рецепторы, которые имеют фармакологический (Lukas, J Pharmacol. Exp. Ther. 251: 175 (1989)), электрофизиологический (Oswald et al., Neurosci. Lett. 96: 207 (1989)) и молекулярный биологический профили (Luther et al., J. Neurosci. 9: 1082 (1989)), подобные nAChR мышечного типа.
Клетки TE671/RD поддерживали в пролиферативной ростовой фазе согласно традиционным протоколам (Bencherif et al., Mol. Cell. Neurosci. 2: 52 (1991) и Bencherif et al., J. Pharmacol. Exp. Ther. 257: 946 (1991)). Клетки культивировали в модифицированной по Дульбекко среде Игла (Gibco/BRL) с 10% лошадиной сывороткой (Gibco/BRL), 5% телячьей сывороткой (HyClone, Logan UT), 1 мМ пируватом натрия, 4 мМ L-глутамином и 50000 единицами пенициллина-стрептомицина (Irvine Scientific). Когда клетки были на 80% слившимися, их помешали на 12-луночные полистироловые планшеты (Costar). Эксперименты проводили, когда клетки достигали 100% слияния.
Функцию никотинового ацетилхолинового рецептора (nAChR) анализировали, используя эманацию 86Rb+ согласно методу, описанному Lukas et al., Anal. Biochem. 175: 212 (1988). В день эксперимента ростовую среду осторожно удаляли из лунки и ростовую среду, содержащую хлорид 86рубидия (106 мкКи/мл) добавляли в каждую лунку. Клетки инкубировали при 37°C в течение минимум 3 ч. После периода насыщения избыток 86Rb+ удаляли и клетки промывали дважды не содержащим метки забуференным фосфатом солевым раствором Дульбекко (138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4), соблюдая предосторожность, чтобы не разрушить клетки. Затем клетки подвергали воздействию 100 мкМ испытуемого соединения, 100 мкМ L-никотина (Acros Organics) или одного буфера в течение 4 мин. После периода воздействия супернатант, содержащий освобожденный 86Rb+, удаляли и переносили в сцинтилляционные сосуды. Сцинтилляционную жидкость добавляли и освобожденную радиоактивность измеряли жидкостным сцинтилляционным подсчетом.
В каждом анализе каждая точка имела 2 повтора, которые усредняли. Количество освобожденного 86Rb+ сравнивали как с положительным контролем (100 мкМ L-никотин), так и с отрицательным контролем (один буфер), чтобы определить процентное освобождение по отношению к таковому для L-никотина.
Когда свойственно, определяли кривые отклика на дозу испытуемого соединения. Максимальную активацию для отдельных соединений (Emax) определяли как процентную долю максимальной активации, индуцируемой L-никотином. Концентрацию соединения, имеющую результатом половину максимальной активации (EC50) специфического ионного потока также определяли.
Пример 20: Взаимодействие при ганглиозном подтипе nAChR человека
Клеточная линия SH-SY5Y представляет собой непрерывную линию, полученную последовательным субклонированием родительской клеточной линии, SK-N-SH, которая первоначально была получена из периферической нейробластомы человека. Клетки SH-SY5Y экспрессируют подобный ганглиону nAChR (Lukas et al., Mol. Cell, Neurosci. 4: 1 (1993)).
Клетки SH-SY5Y человека поддерживали в пролиферативной ростовой фазе согласно традиционным протоколам (Bencherif et al., Mol. Cell. Neurosci. 2: 52 (1991) и Bencherif et al., J. Pharmacol. Exp. Ther. 257: 946 (1991)). Клетки культивировали в модифицированной по Дульбекко среде Игла (Gibco/BRL) с 10% лошадиной сывороткой (Gibco/BRL), 5% телячьей сывороткой (HyClone, Logan UT), 1 мМ пируватом натрия, 4 мМ L-глутамином и 50000 единицами пенициллина-стрептомицина (Irvine Scientific). Когда клетки были на 80% слившимися, их помешали на 12-луночные полистироловые планшеты (Costar). Эксперименты проводили, когда клетки достигали 100% слияния.
Функцию никотинового ацетилхолинового рецептора (nAChR) анализировали, используя эманацию 86Rb+ согласно методу, описанному Lukas et al., Anal. Biochem. 175: 212 (1988). В день эксперимента ростовую среду осторожно удаляли из лунки и ростовую среду, содержащую хлорид 86рубидия (106 мкКи/мл), добавляли в каждую лунку. Клетки инкубировали при 37°C в течение минимум 3 ч. После периода насыщения избыток 86Rb+ удаляли и клетки промывали дважды не содержащим метки забуференным фосфатом солевым раствором Дульбекко (138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4), соблюдая предосторожность, чтобы не разрушить клетки. Затем клетки подвергали воздействию 100 мкМ испытуемого соединения, 100 мкМ L-никотина или одного буфера в течение 4 мин. После периода воздействия супернатант, содержащий освобожденный 86Rb+, удаляли и переносили в сцинтилляционные сосуды. Сцинтилляционную жидкость добавляли и освобожденную радиоактивность измеряли жидкостным сцинтилляционным подсчетом.
В каждом анализе каждая точка имела 2 повтора, которые усредняли. Количество освобожденного 86Rb+ сравнивали как с положительным контролем (100 мкМ L-никотин), так и с отрицательным контролем (один буфер), чтобы определить процентное освобождение по отношению к таковому для L-никотина.
Когда свойственно, определяли кривые отклика на дозу испытуемого соединения. Максимальную активацию для отдельных соединений (Emax) определяли как процентную долю максимальной активации, индуцируемой L-никотином. Концентрацию соединения, имеющую результатом половину максимальной активации (EC50) специфического ионного потока, также определяли.
Пример 21: Узнавание нового объекта
Память оценивали, используя трехэтапный тест узнавания нового объекта (Luine et al., Pharm. Biochem. Behav. 74, 213-220 (2002)). В первый день (ознакомительный опыт) крысам давали возможность исследовать открытую арену (44,5×44,5×30,5 см) в течение 6 мин. На второй день (опыт приобретения) крысам давали возможность исследовать ту же арену в присутствии двух идентичных объектов (оба объект A) в течение 3 мин. На третий день (опыт удерживания или вспоминания) проявление оценивали, давая возможность тому же животному повторно исследовать арену в течение 3 мин в присутствии двух различных объектов привычного объекта A и нового объекта B. Интервал между опытами 24 ч устанавливали между тремя NOR опытами. Память узнавания оценивали путем сравнения времени, затраченного на исследование нового объекта (объект В) против привычного объекта (объект А) во время опыта воспоминания. Показатель узнавания определяли для каждого животного и выражали как отношение ((время В/время А+В)×100).
Пример 22: Лабиринт с радиальными лучами
Рабочую память определяли по задаче лабиринта с радиальными лучами (RAM). Задачу RAM проводили, используя автоматизированный восьми-лучевой лабиринт (Med Associates, Inc.). Лабиринт размещали на круглом столе приблизительно на высоте 88 см над полом с верхним освещением в специализированных испытуемых окружающих и больших высококонтрастных геометрических формах на стенке. Кроме того, дополнительные визуальные знаки размещали на центральном входе в каждый луч, над каждой кормушкой и на потолке. Центральная платформа была 30,5 см в диаметре с восьмью лучами (9 см W × 45,7 см L × 16,8 см Н), радиально отходящими от нее. Автоматические гильотинные дверцы размещали на входе в каждый путь с емкостью для лепешки на дистальном конце каждого луча. Белый шум будет слышимым во время всех тренировочных и испытательных процедур. Активность в лабиринте контролировали отслеживанием количественной активности (генерируемой прерываниями инфракрасного пучка) на компьютерном интерфейсе и экране монитора.
После установления базисной линии и сеансового критерия повторное приобретение животными оценивали по их чувствительности к химически индуцируемому когнитивному ухудшению, используя мускариновый антагонист скополамин (0,2-0,4 мг/кг, s.c.). Дозу скополамина определяли для каждого животного на основе минимальной дозы, которая производила заметное и достоверное когнитивное ухудшение. Скополамин вводили за 0,5 ч до испытания фазы приобретения, тогда как (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин гемигалактарат (0,03 мг/кг; p.o.) вводили за 0,5 ч до начала испытания фазы воспоминания (или испытания). В опыте приобретения один случайно выбранный луч блокировали плексиглассовым барьером, расположенным внутри луча позади центральной дверцы. Животное помещали в центр лабиринта с опущенными дверцами. Приблизительно через 10 с дверцы в 7 доступных лучей поднимали. Первое вхождение в каждый открытый луч поощряли сахарозной кормовой лепешкой. Сеанс заканчивался после посещения всех 7 доступных лучей или по истечении 5 мин. Порядок посещения лучей, полученные поощрения, ошибки (повторные вхождения), время для завершения задачи, число вхождений и время, потребовавшееся для прохождения 7 доступных лучей, и потребления кормового поощрения регистрировали. В опыте вспоминания все 8 лучей были доступными, однако, поощряли только первый визит в прежде блокированный луч (т.е. луч, который был блокирован во время опыта приобретения). Сеанс заканчивался сразу после посещения прежде блокированного луча и потребления поощрения или по истечении 5 мин. В опыте вспоминания регистрировали ошибки повторного вхождения, число (неправильных) лучей, пройденных перед выбором луча, который был блокированным во время опыта приобретения, и время, затраченное до завершения опыта. Задержка между исследованиями фазы приобретения и испытания была 24 ч.
Пример 23: Исследования ингибирования CYP
Ингибирование каталитической активности CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4 (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридином или его солью определяли с помощью флуорогенного анализа CYP. Пробные субстраты, которые флуоресцируют при катализируемом CYP окислении, использовали для оценки степени ингибирования испытуемого субстрата. Единственную концентрацию каждого пробного субстрата (при приблизительно очевидной величине Km) и две различные концентрации (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин гемигалактарата (2 и 20 мкМ) испытывали в двух экземплярах. Интенсивность флуоресценции при выбранных длинах волн использовали как меру активности фермента. Пониженная флуоресценция в присутствии (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли была показателем ингибирования. Положительные контроли (известные ингибиторы) испытывали параллельно, чтобы продемонстрировать контроль метода и активность CYP. Двойные пробы использовали параллельно с положительными и отрицательными контролями. Инкубирование испытуемых проб осуществляли при 37°С. Экспериментальные параметры приведены в общих чертах в таблице 2.
Экспериментальные условия для флуоресцентных анализов ингибирования CYP
(пмоль/лунка)
1
2
2
3
1
сравнительного
ингибитора
MFC = 7-метокси-4-трифторметилкумарин
MAMC = 7-метокси-4-(аминометил)кумарин
BFC = 7-бензилокси-4-трифторметилкумарин
Сводка биологических данных
Фармакология in vitro
Сводка первичных фармакологических данных in vitro для (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли представлена в таблице 3 и обсуждается подробно ниже.
Первичная фармакология и селективность: Способность (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли связываться с рецепторами α4β2 определяли анализами ингибирования связывания рецептора с использованием рекомбинантных рецепторов α4β2 человека, экспрессированных в SH-EP1 клеточных мембранах, и крысиные нативные рецепторы α4β2 в крысиных кортикальных мембранах.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль ингибировали связывание [3H]-никотина с рекомбинантными никотиновыми рецепторами α4β2 с Ki 2 нМ и [3H]эпибатидина с крысиными нативными рецепторами α4β2 с Ki 4 нМ.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль ингибировали связывание [3H]метилликаконитина (MLA) c крысиными нативными рецепторами α7 в крысиных мембранах гиппокампа с Ki>10000 нМ. (R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль также проявляли пониженное сродство к нативным ганглиозным никотиновым рецепторам (вероятно α3β4), ингибируя связывание [3H]эпибатидина с рецепторами в SH-SY5Y мембранах с Ki 3400 нМ, и пониженное сродство к нативным мышечным никотиновым рецепторам (вероятно α1β1γδ), ингибируя связывание [3H]эпибатидина с рецепторами в TE-671 мембранах с Ki 25000 нМ.
Сведения о фармакологии in vitro (R)-3-(2-(пирролидин-3-ил)винил)-5-((тетрагидро-2H-пиран-4-ил)окси)пиридина или его соли
Клеточная эффективность : Целью этих исследований было определение функциональной активности (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли при рекомбинантных рецепторах α4β2 человека. (R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль являются α4β2 никотиновым агонистом, активирующим рецептор с EC50 0,1 мкМ и Emax 76% по отношению к 10 мкМ никотину в анализе кальциевого потока с клетками SH-EP1/человеческими α4β2 с последующим 24-ч инкубированием при 29°C.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль испытывали в анализах ионного потока ганглиозного или мышечного никотинового рецептора, чтобы исследовать функциональную селективность. В анализах оттока Ca++ (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль имеют EC50 11 мкМ и Emax 13% при человеческих нативных ганглиозных рецепторах в клетках SH-SY5Y, EC50 13 мкМ и Emax 37% при человеческих нативных мышечных рецепторах в клетках TE-671.
In vitro вторичная фармакология: Множественный скрининг-анализ рецепторов
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль испытывали на селективность против панели из 65 рецепторов. При единственной концентрации 10 мкМ (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль ингибировали связывание меченого лиганда только с нейронными никотиновыми рецепторами (α-BnTx нечувствительными) с 99% ингибированием.
Ингибирование hERG
IC50 для ингибирования hERG (клетки HEK-239 человека) (R)-3-((E)-2-пирролидин-3-илвинил)-5-(тетрагидро-2H-пиран-4-илокси)пиридином или его солью определили как 84 мкМ.
In vivo фармакология
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль улучшали долговременную визуальную эпизодическую/декларативную память, которую оценивали по задаче узнавания нового объекта (NOR) после перорального дозирования на обычных крысах. Результаты этих исследований представлены на фигуре 1. Показатель узнавания в группе, получавшей носитель, спустя 24 ч после опыта приобретения, был 50±0,5%, демонстрируя неспособность этой группы узнавать знакомый объект после этой отсрочки (левая панель). Напротив, животные, получавшие (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль обнаружили показатели узнавания 71±2% при уровне дозы 0,04 мкмоль/кг и 61±3% при уровне дозы 1,1 мкмоль/кг (левая панель). В дополнительном NOR исследовании (экспериментальные процедуры были идентичны процедурам первого NOR исследования) уровень минимальной эффективной дозы для 5(R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли был определен как 0,004 мкмоль/кг (правая панель), подтверждая, что крысы способны узнавать знакомый объект при всех испытанных уровнях доз. В двух сессиях "только вспоминание" подгруппе животных перорально давали с водой в день 1 (т.е. исследовательская сессия) и день 2 (т.е. сессия приобретения) и затем перорально давали или 1,1 мкмоль/кг (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли (левая панель) или 0,04 мкмоль/кг (правая панель) на день 3 (т.е. сессия вспоминания). Даже после единственного перорального введения (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль продемонстрировали прокогнитивные эффекты при указанных двух уровнях доз. При обоих уровнях доз (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль проявили показатели узнавания значительно выше контрольных, что свидетельствует об узнавании знакомого объекта после высокого дозирования. На фигуре пунктирная лития при 65% означает субъективный критерий биологической когнитивной усовершенствованной активности. *P<0,05.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль оценивали по продолжительности их действия в задаче NOR на обычных крысах. Результаты этих исследований представлены на фигуре 2. Показатель узнавания в группе, получавшей носитель, через 0,5 ч после дозирования в опыте вспоминания был 52±0,8%, демонстрируя неспособность этой группы узнавать знакомый объект после такой отсрочки. Напротив, животные, получавшие (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль (0,004 мкмоль/кг, перорально), обнаружили показатели узнавания 72±2% при 0,5 ч, 70±3% при 6 ч и 70±4% при 8 ч, подтверждая, что крысы способны узнавать знакомый объект вплоть до 8 ч после дозирования. На фигуре пунктирная лития при 65% означает субъективный критерий биологической когнитивной усовершенствованной активности (*P<0,05).
На основе этих исследований обнадеживающий фармакологический эффект возможен при дозировании (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли в широких пределах, включая относительно низкие уровни доз. Один вариант осуществления данного изобретения относится к дозированию (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его фармацевтически приемлемой соли в пероральных дозах настолько низких как около 0,004 мкмоль/кг. Один вариант осуществления данного изобретения относится к оральной дозе менее чем 100 мг, предпочтительно менее чем 50 мг, более предпочтительно, менее чем 10 мг и наиболее предпочтительно, менее чем 1 мг. Такие эффективные дозы обычно представляют собой количество, вводимое как единственная доза или как одна или несколько доз, вводимых в течение 24 ч периода.
Исследования в радиальнолучевом лабиринте (RAM).
Во втором когнитивном анализе (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль смягчали когнитивные дефициты, индуцируемые скополамином, на животной модели рабочей памяти. Результаты этих экспериментов поясняются на фигуре 3. Во время опыта приобретения крысам давали возможность доступа в 7 из восьми лучей, тогда как в опыте испытания все 8 лучей были доступны, однако, только первый визит в ранее блокированный луч (т.е. луч, который был блокирован во время опыта приобретения) стимулировали. Скополамин (0,3±0,1 мг/кг; s.c.) вводили за 0,5 ч до опыта приобретения и (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль (0,03 мг/кг или 0,1 мкмоль/кг; p.o.) вводили за 0,5 ч до опыта испытания. (R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль были способны давать обратное развитие индуцированным скополамином когнитивным дефицитам (*P<0,05).
Ингибирование, индуцирование, перенос человеческого цитохрома P450 (CYP) и возможность межлекарственного взаимодействия
Анализ ингибирования CYP450 с использованием флуоресцентных субстратов и рекомбинантных ферментов не показал явного ингибирования (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридином или его солью 5 главных CYPs (IC60>20 мкМ, таблица 4). К тому же не наблюдалось никакого явного зависимого от времени ингибирования CYP3A4, CYP2D6, CYP2B6, CYP2C9 или CYP1A2. Никакой активации PXR (прегнановый X рецептор) (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридином не наблюдалось вплоть 10 мкМ, таким образом риск, относящийся к индуцированию P450s, вероятно, незначителен.
(R)-3-((E)-2-(Пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин или его соль обнаруживают низкую скорость гепатического оборота в микросомах или гепатоцитах человека. Предварительные данные фенотипирования подтвердили, что и CYP2D6, и FMO3 вносили свой вклад в метаболизм (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли. В дополнение, почечный клиренс, как предполагалось, является главным путем выведения, составляя более чем 50% всего клиренса у человека. Поэтому какое-либо изменение метаболизма (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридина или его соли из-за полиморфизма CYP предположительно должно быть менее чем 2-кратным, благодаря значительному почечному клиренсу и низкому печеночному клиренсу.
Испытуемые соединения для описанных здесь экспериментов использовали в свободной или солевой форме и, если не установлено иное, испытуемым соединением является (R)-3-((E)-2-(пирролидин-3-ил)винил)-5-(тетрагидропиран-4-илокси)пиридин гемигалактарат.
Наблюдавшиеся конкретные фармакологические отклики могут изменяться в зависимости от выбранного конкретного активного соединения или от присутствия фармацевтических носителей, а также типа фармацевтической композиции и используемого пути введения, и такие ожидаемые изменения или различия в результатах рассматриваются в соответствии с практикой данного изобретения.
Хотя конкретные варианты осуществления данного изобретения поясняются здесь и описаны подробно, изобретение не ограничивается ими, и они не должны рассматриваться как устанавливающие какое-либо ограничение. Модификации будут очевидны специалистам, и все модификации, не выходящие за сферу действия изобретения, предназначаются для включения в объем прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ И НОВЫЕ СОЛЕВЫЕ ФОРМЫ (R)-5-((Е)-2-ПИРРОЛИДИН-3-ИЛВИНИЛ)ПИРИМИДИНА | 2014 |
|
RU2700796C2 |
| СИНТЕЗ И НОВЫЕ СОЛЕВЫЕ ФОРМЫ (R)-5-((E)-2-(ПИРРОЛИДИН-3-ИЛВИНИЛ)ПИРИМИДИНА | 2009 |
|
RU2533819C2 |
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗЫ PIM | 2012 |
|
RU2598846C2 |
| ПРОИЗВОДНЫЕ N-ФОРМИЛГИДРОКСИЛАМИНА | 2006 |
|
RU2412939C2 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| АЗОЛЬНОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2573634C2 |
| ТВЕРДЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ СРЕДСТВА, ВЫЗЫВАЮЩИЕ АПОПТОЗ | 2011 |
|
RU2598345C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| СОЕДИНЕНИЯ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА BRM, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2797832C2 |
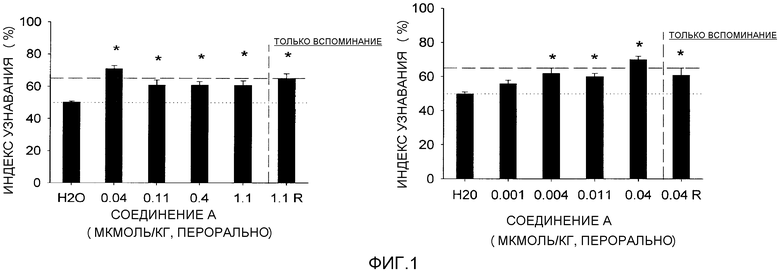
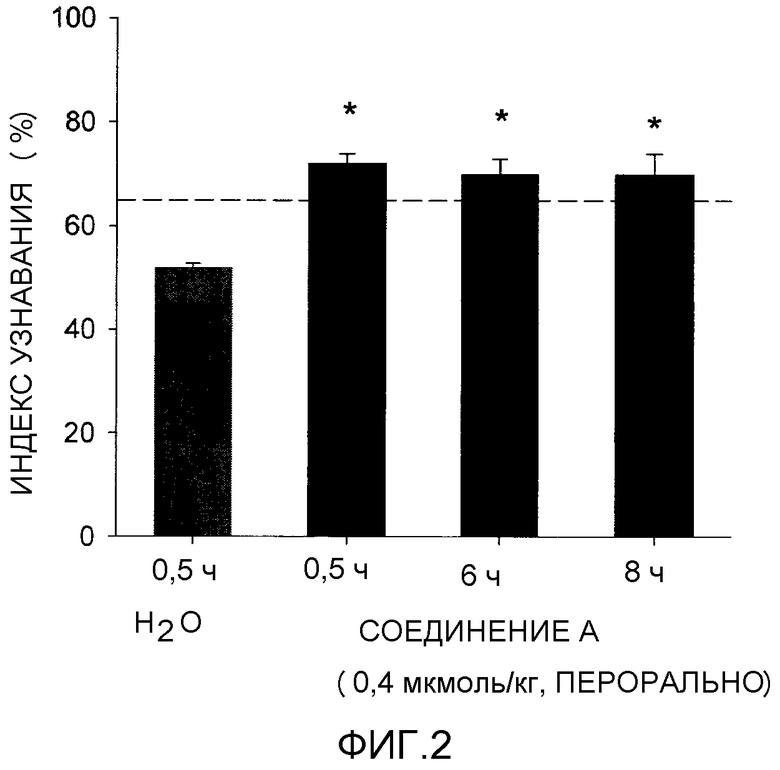
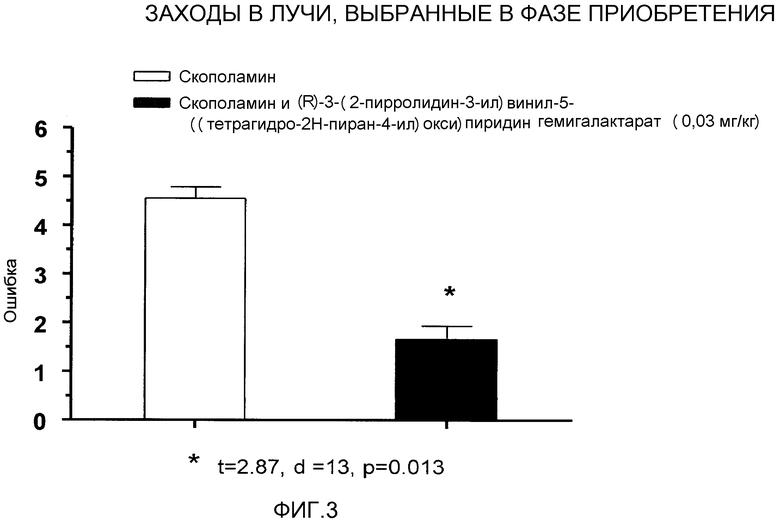
Изобретение относится к новым промежуточным соединениям, как: диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малонат, (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновая кислота, трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилат, трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилат, которые используют в способе получения трет-бутил (R)-3-винилпирролидин-1-карбоксилата. 5 н.п. ф-лы, 3 ил., 4 табл., 23 пр.
1. Соединение диэтил (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малонат.
2. Соединение (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил) малоновая кислота.
3. Соединение трет-бутил (R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилат.
4. Соединение трет-бутил (R)-3-(2-иодэтил)пирролидин-1-карбоксилат.
5. Способ получения трет-бутил (R)-3-винилпирролидин-1-карбоксилата, включающий
a)обработку трет-бутил (R)-3-гидроксипирролидин-1-карбоксилата метансульфонилхлоридом;
b) взаимодействие продукта, полученного на стадии (а), с диэтилмалонатом и основанием;
c) гидролиз продукта, полученного на стадии (b), с водным гидроксидом калия;
d) декарбоксилирование продукта, полученного на стадии (с), подходящим основанием;
e) восстановление продукта, полученного на стадии (d);
f) взаимодействие продукта, полученного на стадии (е) с метансульфонилхлоридом и йодидом натрия;
g) обработку продукта, полученного на стадии (f) трет-бутоксидом калия.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНОПИРРОЛИДИНЫ | 1997 |
|
RU2180901C2 |

