Настоящее изобретение относится к новым производным N-формилгидроксиламина, к применениям этих соединений в различных медицинских приложениях, включая лечение расстройств, поддающихся лечению ингибиторами пептидилдеформилазы, как то лечение бактериальных инфекций, а также к фармацевтическим композициям, содержащим эти соединения.
Лечение микробных инфекций в организме-хозяине требует эффективных способов уничтожения микроорганизмов, наносящих при этом как можно меньше вреда организму-хозяину. Соответственно, для лечения желательны средства, мишенью которых являются характерные атрибуты, уникальные для обуславливающего патологию микроорганизма.
Пептиддеформилаза является металлопептидазой, присутствующей в прокариотических организмах, таких как бактерии. Синтез белков в прокариотических организмах начинается с N-формилметионина (fMet). После инициирования синтеза белка формильная группа удаляется ферментом пептиддеформилазой (ПДФ). Данная активность является для созревания белков основополагающей.
Металлопротеиназы играют критическую роль во многих аспектах нормального метаболизма. Расстройства, в которых участвуют металлопротеиназы, задействованы в ряде заболеваний, таких как рак, артрит и аутоиммунные заболевания. Вследствие важности матричных металлопротеиназ (ММП) для нормальных физиологических процессов было бы предпочтительно разработать такие средства, которые ингибировали бы ПДФ, избегая существенного ингибирования ММП. Альтернативно те ингибиторы ПДФ, которые также ингибируют ММП, могут быть применимы в таких случаях, когда терапевтические преимущества от ингибирования ПДФ перевешивают риск побочных эффектов ингибирования ММП.
Исследования ингибиторов ПДФ гораздо менее обширны, чем исследования ингибиторов ММП. Производные N-формилгидроксиламина описаны в международных заявках на изобретение WO 99/39704 и WO 02/102790. Ввиду важности нахождения новых антибиотиков для лечения бактерий, резистентных к существующим антибиотикам, было бы желательно разработать новые ингибиторы ПДФ для оценки и применения в качестве антибактериальных и противомикробных средств. Настоящее изобретение направлено на удовлетворение данной потребности.
В частности, объектом настоящего изобретения являются производные N-формилгидроксиламина, именуемые в контексте в своей совокупности "соединениями по настоящему изобретению", их соль или пролекарство, например, соединение формулы (I)
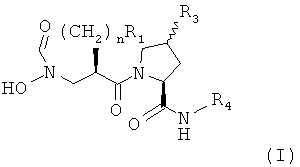 ,
,
в котором R1 является водородом, алкилом, гетероалкилом, гетероциклоалкилом, арилом или гетероарилом,
R3 является водородом, галоидом или алкоксигруппой и
R4 является арилом или гетероарилом,
n принимает значения от 0 до 3,
а также его соль или его пролекарство.
В одном из вариантов осуществления настоящего изобретения R4 является гетероарилом формулы (II)
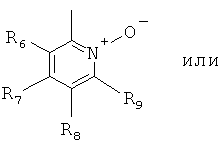
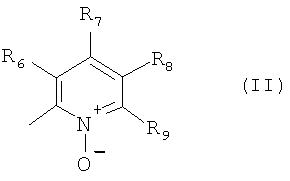
где каждый из заместителей R6, R7, R8 и R9 независимо от остальных может являться водородом, алкилом, замещенным алкилом, фенилом, галоидом. гидроксигруппой или алкоксигруппой, например:
а) R6 и R8 являются водородом, R9 является водородом или алкилом, a R7 является алкилом, замещенным алкилом или фенилом,
б) R6, R7 и R9 являются водородом, a R8 является галоидом, алкилом или замещенным алкилом,
в) R7, R8 и R9 являются водородом, a R6 является гидроксилом.
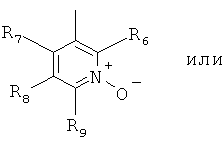
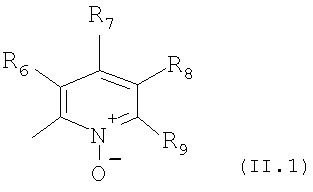
В особенно полезном варианте осуществления настоящего изобретения гетероарил отвечает формуле (II.1)
 ,
,
в которой R6, R7 и R9 отвечают определениям, приведенным выше для формулы (II), a R8 является галоидом, например фтором.
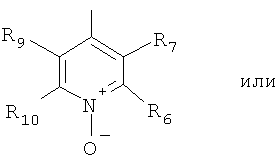
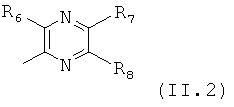
Еще в одном варианте осуществления настоящего изобретения R4 отвечает формуле (11.2)
 ,
,
в которой R6, R7 и R8 отвечают определениям, приведенным выше для формулы (II).
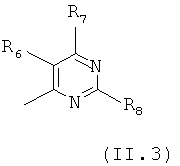
Еще в одном варианте осуществления настоящего изобретения R4 отвечает формуле (II.3)
 ,
,
в которой R6, R7 и R8 отвечают определениям, приведенным выше для формулы (II).
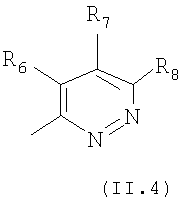
Еще в одном варианте осуществления настоящего изобретения R4 отвечает формуле (II.4)
 ,
,
в которой R6, R7 и R8 отвечают определениям, приведенным выше для формулы (II).
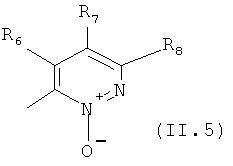
Еще в одном варианте осуществления настоящего изобретения R4 отвечает формуле (II.5)
 ,
,
в которой R6, R7 и R8 отвечают определениям, приведенным выше для формулы (II).
Если иное не оговорено специально, нижеследующие термины, как они употреблены в описании изобретения, имеют следующие значения.
Группа, отвечающая термину "циклоалкан" или "циклоалкил" содержит от 3 до 7 кольцевых атомов углерода и предпочтительно является циклопропилом, циклобутилом, циклопентилом и циклогексилом.
Термин "алифатическая группа" относится к насыщенным или ненасыщенным алифатическим группам, таким как алкил, алкенил или алкинил, циклоалкил или замещенный алкил, включая неразветвленные, разветвленные и циклические группы, содержащие от 1 до 10 атомов углерода. Термин "алкил" или "алк", где бы он ни встречался, обозначает неразветвленную или разветвленную алифатическую группу, содержащую от 1 до 10 атомов углерода или циклоалкил, содержащий от 3 до 10 атомов углерода. Более предпочтительно алкильные группы являются (С1-С7)алкилом, в частности (С1-С4)алкилом. Примеры "алкила" или "алк" включают, не ограничиваясь, однако, перечисленными, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, неопентил, н-гексил или н-гептил, циклопропил и, в особенности, н-бутил.
Термин "замещенный алкил" относится к алкильной группе, которая замещена одним или более заместителями, которые могут включать, не ограничиваясь, однако, перечисленными, такие заместители, как галоид, низшая алкоксигруппа гидроксигруппа, меркаптогруппа, карбоксильная группа, циклоалкил, арил, гетероарил и им подобные. Примеры замещенных алкильных групп включают, не ограничиваясь, однако, перечисленными, -CF3, -CF2-CF3, гидроксиметил, 1- или 2-гидроксиэтил, метоксиметил, 1- или 2-этоксиэтил, карбоксиметил, 1- или 2-карбоксиэтил и им подобные.
Термин "арил" или "Ar" относится к ароматической карбоциклической группе, содержащей от 6 до 14 атомов углерода и имеющей один цикл (включая такие группы, как фенил, но не ограничиваясь ими) или несколько конденсированных циклов (включая такие группы, как нафтил или антраценил, но не ограничиваясь ими), предпочтительно же являющейся фенилом.
Термин "гетероарил" или "HetAr" относится к 4-7-членному моноциклическому ароматическому гетероциклу или к бициклической системе. состоящей из 4-7-членного моноциклического ароматического гетероцикла, конденсированного с бензольным циклом. Гетероарил содержит в цикле хотя бы один гетероатом, предпочтительно хотя бы два гетероатома, включая, но, однако, не ограничиваясь ими, такие гетероатомы, как азот, кислород или сера. Предпочтительный гетероарильный остаток является шестичленным моноциклическим гетероциклом, содержащим в цикле 2, 3 или 4 гетероатома азота. Примерами гетероарильных групп являются пиридинил, пиримидинил, пиразинил, пиридазинил-N-оксид или бенздиоксоланил, триазин или тетразины.
Арил или гетероарил могут быть незамещенными или замещенными одним или более заместителями, которые могут включать, не ограничиваясь, однако, перечисленными, (С1-С7)алкил, в частности (С1-С4)алкил, такой как метил, гидроксигруппу, алкоксигрупу, ацил, ацилоксигруппу, SCN, цианогруппу, нитрогруппу, тиоалкоксигруппу, фенил, гетероалкиларил, алкилсульфонильную группу, галоид и формил.
Термин "гетероалкил" относится к насыщенному или ненасыщенному (C1-C8)алкилу, как он определен выше, и предпочтительно относится к (С1-С4)гетероалкилу, который содержит один или более гетероатомов в качестве составляющей главных, разветвленных или циклических цепочек группы. Гетероатомы могут быть независимо выбраны из группы, включающей -NR-, где R является водородом или алкилом, -S-, -О- и -Р-, предпочтительно -NR-, где R является водородом или алкилом и/или -O-. Гетероалкильные группы могут быть присоединены к остальной части молекулы или по гетероатому (если наличествует свободная валентность), или по атому углерода. Примеры гетероалкильных групп включают, не ограничиваясь, однако, перечисленными, такие группы, как -O-СН3, -СН2-О-СН3, -СН2-СН2-O-СН3, -S-CH2-СН2-СН3, -СН2-СН(СН3)-S-СН3 и -CH2-CH2-NH-CH2-CH2-.
Гетероалкильная группа может быть незамещенной или замещенной одним или более заместителями, предпочтительно одним, двумя или тремя заместителями, включая, не ограничиваясь, однако, перечисленными, алкил, галоид, алкоксигруппу, гидроксил, меркаптогруппу, карбоксильную группу и, в особенности, фенил. В ходящие в группу гетероатом(ы), равно как и атомы углерода, могут быть замещены. Гетероатом(ы) может также находиться в окисленной форме.
Термин "алкоксигруппа", как он употребляется в контексте, относится к (С1-С10)алкилу, присоединенному к атому кислорода, предпочтительно к (С1-С7)алкоксигруппе, более предпочтительно к (С1-С4)алкоксигруппе. Примеры алкоксигрупп включают, не ограничиваясь, однако, перечисленными, такие группы, как метоксигруппа, этоксигруппа, н-бутоксигрупа, трет-бутоксигруппа и аллилоксигруппа.
Термин "галоген" или "галоид", как он употребляется в контексте, относится к хлору, брому, фтору и иоду, предпочтительно к фтору.
Термин "защитная группа" относится к химической группе, проявляющей следующие свойства: 1) селективное взаимодействие с желаемой функциональной группой с хорошим выходом, приводящее к образованию вещества в защищенной форме, устойчивого к предполагаемым взаимодействиям, для которых требуется защита, 2) селективное снятие с вещества в защищенной форме, приводящее к образованию требуемой функциональной группы, и 3) возможность снятия с хорошим выходом под действием реагентов, совместимых с другой функциональной группой(группами), уже присутствовавшей или образованной в ходе упомянутых предполагаемых взаимодействий. Примеры подходящих защитных групп могут быть найдены в Greene и др., "Protective Groups in Organic Synthesis", 2-е издание, John Wiley & Sons, Inc., New York (1991). Предпочтительные защитные группы для аминогруппы включают, не ограничиваясь, однако, перечисленными, бензилоксикарбонильную группу (CBz), трет-бутилоксикарбонильную группу (Boc), трет-бутилдиметилсилильную группу (ТБДМС), 9-флуоренилметилоксикарбонпильную группу (Fmoc) или подходящие фотолабильные защитные группы, такие как 6-нитровератрилоксикарбонильная группа (Nvoc), нитропиперонил, пиренилметоксиарбонильная группа, нитробензил, диметилдиметоксибензил, 5-бром-7-нитроиндолинил и им подобные. Предпочтительные защитные группы для гидроксигруппы включают Fmoc, ТБДМС, фотолабильные защитные группы (такие как нитровератрилоксиметиловый эфир (Nvom)), Mom (метоксиметиловый эфир), и Mem (метоксиэтоксиметиловый эфир). Особо предпочтительные защитные группы включают НФЭОК (4-нитрофенилэтилоксикарбонильную группу) и НФЭОМ (4-нитрофенилэтилоксиметилоксикарбонильную группу).
Следует иметь в виду, что соединения формулы (I) могут существовать в форме оптических изомеров, рацематов или диастереоизомеров. Примером является соединение формулы (I), в котором R3 может находиться в R- или S-конфигурации. Следует понимать, что в объем настоящего изобретения входят все энантиомеры и их смеси. Аналогичные соображения действуют и по отношению к исходным веществам, содержащим асимметрические атомы углерода, как о том упоминается.
Соединения по настоящему изобретению могут существовать в форме твердых кристаллических солей. Предпочтительно кристаллические соли являются солями металлов, предпочтительно двухвалентных металлов, хотя для некоторых соединений возможно образование кристаллических твердых веществ с участием одновалентных противоинов, таких как Na. Предпочтительно противоион является Mg, Ca или Zn.
Соединения по настоящему изобретению могут, как правило, находиться в форме гидрата или смешанного сольвата/гидрата. Обычно кристаллическая соль по настоящему изобретению содержит приблизительно от 2 до 8 гидратных молекул воды, более обычно - приблизительно от 2 до 6 гидратных молекул воды, а наиболее обычно - приблизительно от 2 до 4 гидратных молекул воды. Таким образом, кристаллическая соль по настоящему изобретению обычно содержит более 2% воды, более обычно - от приблизительно 4 до приблизительно 12% воды, а наиболее обычно - от приблизительно 8 до приблизительно 9% воды. Сольваты могут быть с одним или более органическими растворителями, такими как низшие спирты, такие как метанол, этанол, изопропанол, бутанол или их смеси.
Соединения по настоящему изобретению, например соединения формулы (I), могут существовать в свободной форме или в форме соли, например в форме фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" соединения означает физиологически и фармацевтически приемлемую соль, которая обладает требуемой фармакологической активностью исходного соединения и не порождает нежелательных токсикологических эффектов. Подобные соли включают:
(1) кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и им подобные, или образованные с органическими кислотами, такими как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота; гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфокислота, этансульфокислота, 1,2-этандисульфокислота, 2-гидроксиэтансульфокислота, бензолсульфокислота, 4-хлорбензолсульфокислота, 2-нафталинсульфокислота, 4-толуолсульфокислота, камфарсульфокислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и им подобные, или
(2) соли, образованные в результате того, что присутствующий в исходном соединении кислый протон либо заменен на ион металла, например ион щелочного металла, ион щелочноземельного металла или ион алюминия, либо координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и им подобные.
Соединение по настоящему изобретению, например соединение формулы (I), может действовать в качестве пролекарства. Термин "пролекарство" означает любое соединение, которое высвобождает активное исходное соединение согласно формуле (I) in vivo, когда таковое пролекарство введено пациенту-млекопитающему. Пролекарства соединений формулы (I) получают посредством модифицирования функциональных групп, присутствующих в соединении формулы (I), таким образом, что возможно расщепление модифицированных групп in vivo с высвобождением исходного соединения. Пролекарства включают те соединения формулы (I), в которых гидроксигруппа, аминогруппа или сульфгидрильная группа связана с любой группой, которая может быть расщеплена in vivo с целью восстановления соответственно свободной гидроксигруппы, аминогруппы или сульфгидрильной группы. Примеры пролекарств включают, не ограничиваясь, однако, перечисленными, сложные эфиры (например, производные уксусной кислоты, муравьиной кислоты и бензойной кислоты), карбаматы (например, N,N-диметиламинокарбонил) гидроксильных функциональных групп в соединениях формулы (I) и им подобные.
Нижеследующие определения в соединениях формулы (I) являются предпочтительными как по отдельности, так и в любом частичном сочетании.
1. R4 является гетероарилом формулы (II.1), в котором R6, R7 и R9 являются водородом, a R8 является фтором, R6, R7 и R9 являются водородом, a R8 является метилом или трифторметилом, или R6, R7 и R8 являются водородом, а R9 является фтором, или R6, R8 и R9 являются водородом, a R7 является этилом или метоксигруппой, или R7, R8 и R9 являются водородом, a R6 является гидроксигруппой, или R7 и R8 являются водородом, R6 является метоксигруппой, а R9 является метилом, или R4 является гетероарилом формулы (II.2), в котором R6, R7 и R8 являются водородом, или R4 является гетероарилом формулы (II.3), в котором R6, R7 и R8 являются водородом, или R4 является гетероарилом формулы (II.4), в котором R6, R7 и R8 являются водородом, или R4 является гетероарилом формулы (II.5), в которой R6, R7 и R8 являются водородом.
2. R1 является алкилом, предпочтительно н-бутилом, или циклоалкилом, предпочтительно (С3-С7)циклоалкилом, таким как циклогексил, циклопропил или циклопентил.
4. R3 является галоидом, предпочтительно фтором.
Полезность
Соединения по настоящему изобретению могут применяться для лечения или профилактики инфекционных расстройств, вызываемых разнообразными бактериальными или прокариотическими организмами. Примеры включают, не ограничиваясь, однако, перечисленными, грамположительные и грамотрицательные аэробные и анаэробные бактерии, включая Staphylococci (стафилококки), например S. aureus (золотистый стафилококк) и S. epidermidis (эпидермальный стафилококк); Enterococci (энтерококки), например Е. faecalis и Е. faecium; Streptococci (стрептококки), например S. pneumoniae (пневмококк), Haemophilus (гемоглобинофильные бактерии), например Н. influenza (гемофильная инфекция), Moraxella (диплококк, продуцирующий р-лактамазу), например М. catarrhalis, и Escherichia, например Е. coli (кишечная палочка). Прочие примеры включают Mycobacteria (микобактерии), например М tuberculosis (туберкулез), межклеточные микробы, например Chlamydia (хламидии) и Rickettsiae (риккетсии), а также Mycoplasma (микоплазма), например М. pneumoniae (микоплазменная пневмония), и Pseudomonas (псевдомонады), например Р. aeruginosa, H. pylori (спиралевидная грамотрицательная бактерия) и parasites (паразитов), например Plasmodium falciparum (малярийный плазмодий).
Соединения по настоящему изобретению предпочтительно представляют существенное улучшение микробиологической эффективности в отношении либо грамположительных, либо грамотрицательных бактерий. Более конкретно, соединения по настоящему изобретению представляют значительное улучшение в своем спектре микробиологической активности вследствие улучшенного ингибирования грамотрицательных и/или грамположительных бактерий, таких как H. influenza и S. pneumonia. Например, в одном из примеров средний сравнительный индекс (ССИ) превосходит три ступени разбавления для усиления ингибирования H. influenza и, вдобавок, отмечен ССИ, составляющий 0,4 ступени разбавления, для усиления ингибирования S. pneumonia.В другом примере получен ССИ, составляющий 3 ступени разбавления, для усиления ингибирования S. pneumonia и, вдобавок, отмечен ССИ, составляющий 1,2 ступени разбавления, для усиления ингибирования H. influenza.
Соединения по настоящему изобретению также предпочтительно проявляют улучшенные свойства с точки зрения безопасности, токсичности и фармакокинетики, например, понижение или устранение потенциальных неблагоприятных эффектов у человека по сравнению с соединениями, относящимися к известному уровню техники.
В одном из вариантов осуществления настоящего изобретения его объектом являются композиции для лечения или профилактики инфекционных расстройств, включающие соединение по настоящему изобретению, его фармацевтически приемлемую соль или его пролекарство, как они раскрываются в контексте, в сочетании с фармацевтически приемлемым носителем. В другом варианте осуществления настоящего изобретения подобные композиции дополнительно включают другое терапевтическое средство.
В другом варианте осуществления настоящего изобретения его объектом является дозировка соединения по настоящему изобретению, его фармацевтически приемлемой соли или его пролекарства, как они раскрываются в контексте, в количестве, эффективном для лечения, профилактики или облегчения расстройства, такого как инфекционное расстройство. Данные соединения или производные могут быть подвергнуты скринингу на предмет выявления активности по отношению к различным микробным агентам, причем с помощью известных в соответствующей области способов могут быть определены подходящие дозировки.
Соединения по настоящему изобретению могут быть применены для обработки субъекта с целью лечения, профилактики или снижения остроты инфекции. Субъекты включают животных, растения, препараты крови, культуры и поверхности, такие как поверхности медицинского и исследовательского оборудования, такого как стекло, иглы, хирургическое оборудование и трубки, а также объекты, предназначенные для временной или постоянной имплантации в организм. Предпочтительные животные включают млекопитающих, например мышей, крыс, кошек, собак, коров, овец, свиней, лошадей, поросят, приматов, таких как макаки-резус, шимпанзе, гориллы, предпочтительнее же всего - людей. Лечение субъекта включает, не ограничиваясь, однако, указанным, профилактику, снижение или устранение клинических симптомов, вызванных заражением объекта микроорганизмом, профилактику, снижение или устранение заражения объекта микроорганизмом или профилактику, снижение или устранение загрязнения объекта микроорганизмом. Таковой микроорганизм предпочтительно является прокариотом, более предпочтительно - бактерией.
В одном из вариантов осуществления настоящего изобретения его объектом являются способы лечения или профилактики инфекционного расстройства, чувствительного к ингибированию пептидилдеформилазы, у субъекта, такого как человек или иной субъект-животное, посредством введения субъекту эффективно ингибирующего пептидилдеформилазу количества соединения по настоящему изобретению, его фармацевтически приемлемой соли или его пролекарства. В одном из вариантов осуществления настоящего изобретения соединение или его производное вводят в фармацевтически приемлемой форме, необязательно в фармацевтически приемлемом носителе. Соединение по настоящему изобретению, его фармацевтически приемлемая соль или его пролекарство может быть введено отдельно или в сочетании с другим терапевтическим средством. Примеры подобных терапевтических средств включают, не ограничиваясь, однако, перечисленными, β-лактам, хинолон, макролид, гликопептид и оксазолидинон. Термин "инфекционное расстройство", как он употребляется в контексте, означает любое расстройство, характеризуемое присутствием микробной инфекции, как, например, присутствием бактерий. Подобные инфекционные расстройства включают, например, инфекции центральной нервной системы, инфекции наружного уха, инфекции среднего уха, такие как острый средний отит, инфекции церебрального венозного синуса, глазные инфекции, инфекции полости рта, такие как инфекции зубов, десен и слизистой оболочки, инфекции верхних дыхательных путей, инфекции нижних дыхательных путей, инфекции мочеполовой системы, инфекции желудочно-кишечного тракта, гинекологические инфекции, септицемию, костные и суставные инфекции, инфекции кожи и кожных структур, бактериальный эндокардит, ожоги, антибактериальная профилактика при хирургических операциях, антибактериальная профилактика у пациентов с подавленным иммунным ответом, таких как пациенты, подвергаемые химиотерапии рака, или пациенты с трансплантацией органов, а также хронические заболевания, вызванные инфекционными организмами, например артериосклероз. Соединения и композиции, включающие соединения, могут быть введены такими путями, как топический, локальный или системный. Системное введение включает любой способ введения соединения в ткани организма, например интратекальное, эпидуральное, внутримышечное, чрескожное, внутривенное, внутрибрюшинное, подкожное, подъязычное, интраназальное, влагалищное, перректальное и пероральное введение. Конкретная дозировка вводимого противомикробного средства, равно как и продолжительность лечения, может быть приспособлена сообразно необходимости.
В другом варианте осуществления настоящего изобретения его объектом являются способы ингибирования пептидилдеформилазы. В одном из вариантов осуществления настоящего изобретения способ включает введение нуждающемуся в том пациенту эффективно ингибирующего пептидилдеформилазу количества соединения формулы (I), его фармацевтически приемлемой соли или его пролекарства. Термины "субъект" и "эффективно ингибирующее пептидилдеформилазу количество" отвечают вышеприведенным определениям.
Еще в одном варианте осуществления настоящего изобретения его объектом также является применение соединения формулы (I), как оно определено выше, его фармацевтически приемлемой соли или его пролекарства для изготовления лекарственного средства для применения при лечении заболеваний, опосредованных пептидилдеформилазой.
Введение и фармацевтическая композиция
Объектом настоящего изобретения также являются фармацевтические композиции, включающие биологически активные производное N-формилгидроксиламина, его фармацевтически приемлемую соль или пролекарство, а также фармацевтически приемлемый носитель. Композиции по настоящему изобретению включают композиции в форме, приспособленной для перорального, локального или парэнтерального применения. Они могут применяться для лечения бактериальной инфекции у пациента, такого как животные, предпочтительно млекопитающие, наиболее предпочтительно люди. Фармацевтические композиции могут также включать другие терапевтические средства, как это описано ниже.
Антибиотические соединения согласно настоящему изобретению, также именуемые в контексте противомикробными соединениями, могут входить в лекарственные формы для введения любым удобным образом для применения в медицине человека или ветеринарной медицине, по аналогии с другими антибиотиками. Соответствующие способы известны в соответствующей области (см., например. Remington's Pharmaceutical Sciences, Easton, PA: Mack Publishing Co.) и не описываются здесь в подробностях.
Соединение может входить в лекарственные формы для введения любым из известных в соответствующей области путей, таких как подкожный, ингаляционный, пероральный, локальный или парэнтеральный. Композиции могут находиться в любой форме, известной в соответствующей области, включая, но не ограничиваясь, однако, перечисленными, таблетки, капсулы, облатки, быстрорастворимые формы (без облаток), порошки, гранулы, пастилки, кремы или жидкие препараты, такие как растворы или суспензии для перорального введения или стерильные растворы или суспензии для парэнтерального введения. Соединения могут также быть введены в виде таких лекарственных форм, как липосомные, мицеллярные или микроэмульсии. Соединения также могут быть введены в виде пролекарств, причем вводимое пролекарство претерпевает в подвергаемом лечению млекопитающем биологическое преобразование, приводящее к биологически активной форме.
Лекарственные формы по настоящему изобретению для локального применения могут представлять собой, например, мази, кремы или лосьоны, растворы, бальзамы, эмульсии, пластыри, глазные мази и глазные или ушные капли, повязки с пропиткой, пластыри для трансдермальной доставки, спреи и аэрозоли. Они могут содержать подходящие общепринятые добавки, такие как консерванты, растворители, способствующие проникновению лекарства и смягчающие средства в мазях и кремах.
Лекарственные формы могут также содержать общепринятые совместимые носители, такие как основы кремов и мазей и этанол или олеиловый спирт для лосьонов. Подобные носители могут составлять, например, от приблизительно 1% до приблизительно 99% лекарственной формы. Например, они могут составлять до приблизительно 80% лекарственной формы.
Таблетки и капсулы для перорального введения могут быть представлены в форме стандартной дозировки и могут содержать общепринятые наполнители, такие как связующие вещества, например сироп, гуммиарабик, желатин, сорбитол, трагакант или поливинилпирролидон, заполнители, например, лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбитол или глицин, скользящие вещества для изготовления таблеток, например, стеарат магния, тальк, полиэтиленгликоль или окись кремния, вещества, способствующие распадению, например картофельный крахмал, или приемлемые смачивающие вещества, такие как лаурилсульфат натрия. Таблетки могут быть снабжены покрытием согласно способам, хорошо известным в обычной фармацевтической практике.
Жидкие препараты для перорального введения могут находиться в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или же могут находиться в форме сухого вещества для переведения перед применением в воду или в другой подходящий жидкий носитель. Подобные жидкие препараты могут содержать общепринятые добавки, такие как суспендирующие средства, например сорбитол, метилцеллюлозу, сироп глюкозы, желатин гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или съедобные гидрогенизированные жиры, эмульгаторы, например, лецитин, сорбитан моноолеат или гуммиарабик, неводные носители (которые могут включать съедобные масла), например миндальное масло, маслянистые сложные эфиры, такие как глицерин, пропиленгликоль или этиловый спирт, консерванты, например, метиловый или пропиловый эфир пара-гидроксибензойной кислоты или сорбиновую кислоту, а также, если то требуется, общепринятые ароматизаторы и красители.
Для парэнтерального введения жидкие формы стандартной дозировки изготавливают из вещества и стерильного носителя, причем предпочтительной является вода. Соединение может быть либо суспендировано, либо растворено в носителе или в ином подходящем растворителе в зависимости от носителя и используемой концентрации. При приготовлении растворов соединение может быть растворено в воде для инъекции и стерилизовано при фильтровании прежде помещения в подходящий пузырек или ампулу и закупоривания. Может быть благоприятным растворение в носителе таких средств, как местные обезболивающие, консерванты и буферные вещества. Для улучшения устойчивости композиция может быть заморожена после помещения в пузырек, а вода может быть отогнана под вакуумом. После этого сухой лиофилизованный порошок закупоривают в пузырьке, который может быть снабжен сопутствующим пузырьком воды для инъекции с целью переведения в жидкую форму перед применением. Парэнтеральные суспензии изготавливают преимущественно таким же образом, за исключением того, что соединение суспендируют в носителе, а не растворяют, а стерилизацию невозможно осуществить посредством фильтрования. Соединение может быть стерилизовано посредством воздействия окиси этилена, а затем суспендировано в стерильном носителе. Может быть благоприятным включение в композицию поверхностно-активного вещества или смачивающего вещества с целью обеспечения равномерного распределения соединения.
Композиции могут содержать, например, от приблизительно 0,1% по массе до приблизительно 99% по массе, например, от приблизительно 10-60% по массе, активного вещества, в зависимости от способа введения. Если композиции содержат стандартные дозировки, каждая дозировка будет содержать, например, от приблизительно 1-1000 мг активного ингредиента. Дозировка, применяемая для лечения взрослого человека, будет находиться, например, в пределах от приблизительно 1-3000 мг в сутки, например 1500 мг в сутки, в зависимости от пути и частоты введения. Подобная дозировка соответствует приблизительно 0,015-50 мг/кг массы тела в сутки. Подходящая дозировка составляет, например, от приблизительно 5-20 мг/кг массы тела в сутки.
Характерные фармацевтические формы, содержащие соединение формулы (I), описаны ниже.
Объектом настоящего изобретения также является способ получения соединения по настоящему изобретению, например соединения формулы (I), включающий взаимодействие соединения формулы (V)
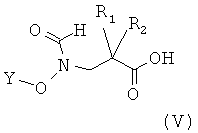 ,
,
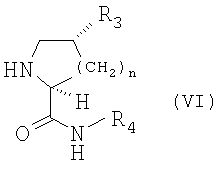
в котором R1 и R2 принимают указанные выше значения, a Y является защитной группой для гидроксигруппы, или же его функционального производного с соединением формулы (VI)
 ,
,
в котором R3 и R4 принимают указанные выше значения, а n равно 1, а также включающий, когда это необходимо, преобразование получаемых в результате соединений в свободной форме в форму солей или наоборот.
Функциональные производные соединений формулы (V) включают, например, хлорангидрид кислоты, ангидрид кислоты или сложный эфир в активированной форме.
Вышеприведенные реакции могут быть осуществлены согласно известным в соответствующей области способам или таким образом, как это раскрывается в нижеприведенных примерах. Реакцию можно легко осуществить в присутствии основания, после чего можно провести гидрирование, предпочтительно в присутствии катализатора гидрирования. Подходящие основания включают, например, основание Хюнига (т.е. диизопропилэтиламин) и неорганические основания, такие как гидрокарбонат натрия. Катализатор гидрирования, предпочтительно палладий на угле или палладиевая чернь, может быть затем добавлен к получаемому в результате продукту, например, после концентрирования, после чего может осуществляться перемешивание в атмосфере водорода в течение, например, от приблизительно 16 до приблизительно 24 часов. Палладиевый катализатор может быть добавлен предпочтительно в количестве от приблизительно 5 мольных процентов до приблизительно 10 мольных процентов от концентрированного продукта.
Соединения формулы (V), применяемые в качестве исходных веществ, могут быть получены, например, посредством взаимодействия соединения формулы (VII)
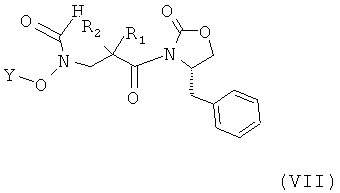
в котором R1, R2 и Y принимают указанные выше значения в, например, мягких основных условиях, например, как это известно в соответствующей области. Как правило, данная реакция может быть осуществлена посредством растворения соединения формулы (VII) в, например, смеси инертного растворителя, такого как тетрагидрофуран (ТГФ), N,N-диметилформамид (ДМФ), толуол, диоксан или CH2Cl2, и воды и добавления перекиси водорода, а затем водного раствора основания к охлаждаемой смеси. Примеры основания включают, например, гидрокарбонат натрия, гидроксид лития, гидроксид натрия и им подобные. Основание может быть задействовано предпочтительно в количестве от приблизительно 1,1 до приблизительно 1,5 эквивалентов по отношению к соединению формулы (VII).
Соединения формулы (VII) могут быть получены, например, посредством взаимодействия соединения формулы (VIII), в котором R1, R2 и Y принимают указанные выше значения, с муравьиной кислотой, как это известно в соответствующей области. Как правило, реакция может быть осуществлена при температуре, например, 0°С посредством добавления раствора уксусного ангидрида в муравьиной кислоте к раствору соединения формулы (VIII) в муравьиной кислоте.
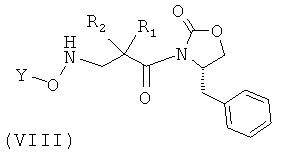
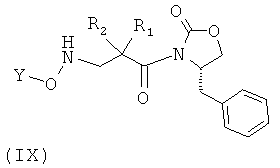
Соединения формулы (VIII) могут быть получены посредством, например, взаимодействия соединения формулы (IX), в котором R1, R2 и Y принимают указанные выше значения, с раствором пара-толуолсульфокислоты в инертном органическом растворителе и с раствором Na2CO3, например, 1 М раствором, как это известно в соответствующей области.
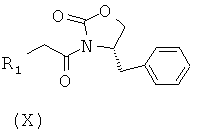
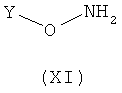
Соединения формулы (IX) могут быть получены посредством, например, взаимодействия соединения формулы (X), в котором R1 принимает указанные выше значения, с защищенным по гидроксильной группе соединением формулы (XI), в котором Y является арилом, алкилом, арилалкилом или силилом, как это известно в соответствующей области.
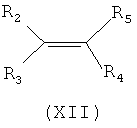
Соединение формулы (X) может быть получено посредством, например, взаимодействия соединения формулы (XII), в котором R4 принимает указанные выше значения, с хлорангидридом триметилуксусной кислоты, как это известно в соответствующей области.
Если получение исходных веществ не описано специально, соединения являются известными или могут быть получены аналогично способам, известным в соответствующей области, или же согласно тому, как это раскрывается в нижеследующих примерах.
Все патенты, заявки на патенты и публикации, цитируемые в данной заявке, включены сюда посредством ссылки во всей их полноте, для всех целей и в такой же степени, как если бы это было указано по отдельности в отношении каждого отдельного патента, заявки на патент или публикации.
В тексте используются следующие сокращения:
АсОН, НОАс = уксусная кислота
Ас2О = уксусный ангидрид
ВОС, Boc = трет-бутоксикарбонил
ДХМ = хлористый метилен
ДИЭА = диизопропилэтиламин
ДМФ = диметилформамид
ДМСО = диметилсульфоксид
Et = этил
EtOAc = этилацетат
Fmoc, ФМОК = 9-флуоренилметилоксикарбонил
ГАТУ = гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-
тетраметилурония
МХПБК = мета-хлорпероксибензойная кислота
Me = метил
МеОН = метанол
ММП = матричная металлопротеиназа
НВОМ = нитровератрилоксиметиловый простой эфир
ПТСК = пара-толуолсульфокислота
КТ = комнатная температура
ТФУК = трифторуксусная кислота
tBu = трет-бутил
ТГФ = тетрагидрофуран
ТГП = 2-тетрагидропиранил
TsOH или ПТСК = толуолсульфокислота
Общая схема синтеза
Соединения по настоящему изобретению могут быть получены с помощью способов, изображенных на нижеприведенных схемах реакций.
Исходные вещества и реагенты, применяемые при получении этих соединений, либо являются доступными от коммерческих производителей, таких как Aldrich Chemical Co., (Милуоки, Висконсин, США), Bachem (Торрэнс, Калифорния, США), Emka-Chemie или Sigma (Сент-Луис, Миссури, США), либо могут быть получены с помощью способов, известных специалистам в соответствующей области, следуя методикам, изложенным в ссылках, таких как Fieser и Fieser, "Reagents for Organic Synthesis", тт.1-15 (John Wiley and Sons, 1991), Rodd, "Chemistry of Carbon Compounds", тт.1-5 и приложения (Elsevier Science Publishers, 1989), "Organic Reactions", тт.1-40 (John Wiley and Sons, 1991), March, "Advanced Organic Chemistry" (John Wiley and Sons, 4-е издание) и Larock, "Comprehensive Organic Transformations" (VCH Publishers Inc., 1989). Данные схемы являются лишь иллюстрациями некоторых из способов, с помощью которых могут быть синтезированы соединения по настоящему изобретению. Могут быть осуществлены различные модификации данных схем, которые легко придут на ум специалисту в соответствующей области, обратившемуся к данному описанию.
Исходные вещества и промежуточные соединения взаимодействия могут быть выделены и очищены с помощью общепринятых способов, включающих, не ограничиваясь, однако, перечисленными, фильтрование, перегонку, кристаллизацию, хроматографию и им подобные. Подобные вещества могут быть охарактеризованы с помощью общепринятых средств, включая физические константы и спектральные данные.
Получение соединений формулы (I)
Соединения формулы (I) могут быть получены с помощью способов, хорошо известных в органической химии. Характерные синтетические методики для получения соединений по настоящему изобретению проиллюстрированы и подробно описаны ниже. Например, соединения формулы (I) могут быть получены таким образом, как это описано на нижеприведенных схемах А-Б.
Общая методика А: синтез амида 1-{2(R)-[(формилгидроксиламино)метил]алканоил}пирролидин-2(S)-карбоновой кислоты
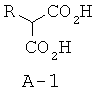
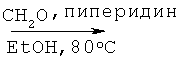
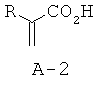
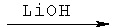
Стадия 1: 2-н-бутилакриловая кислота (А-2)
К раствору алкилмалоновой кислоты А-1 (R=н-бутил, 107,4 ммоля) в этаноле (200 мл) добавляют пиперидин (12,7 мл, 128,8 ммоля, 1,2 экв.) и 37% водного раствора формальдегида (40,0 мл, 536,9 ммоля, 5 экв.). Раствор нагревают до температуры 80°С, при этом выпадает осадок, который затем снова растворяется в течение 1 часа. Перемешивают реакционную смесь при температуре 80°С в течение ночи, а затем охлаждают до комнатной температуры (КТ). Растворители отгоняют при пониженном давлении, остаток растворяют в этилацетате, промывают последовательно 1 М HCl и соляным раствором, высушивают над безводным Na2SO4 и фильтруют. Фильтрат концентрируют, что приводит к указанному в заглавии соединению А-2 в виде прозрачного маслянистого вещества.
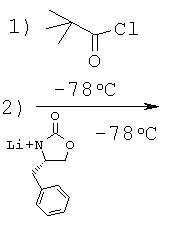
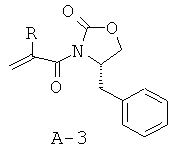
Стадия 2: 4-бензил-3-(2-бутилакрилоил)оксазолидин-2-он (А-3)
2-н-Бутилакриловую кислоту (9,90 г, 77,2 ммоля, 1 экв.) растворяют в безводном ТГФ (260 мл) и охлаждают до температуры -78°С под азотной подушкой. Добавляют основание Хюнига (17,5 мл, 100,4 ммоля, 1,3 экв.) и хлорангидрид триметилуксусной кислоты (9,5 мл, 77,2 ммоля, 1 экв.) с такой скоростью, чтобы температура удерживалась ниже -60°С. Смесь перемешивают при температуре -78°С в течение 30 минут, нагревают до КТ в течение 2 часов и, наконец, охлаждают обратно до температуры -78°С.
В отдельной колбе (S)-(-)-4-бензил-2-оксазолидинон (13,49 г, 77,24 ммоля) растворяют в безводном ТГФ (150 мл) и охлаждают до температуры -78°С под азотной подушкой. Медленно добавляют при температуре -78°С н-бутиллитий (2,5 М раствор в гексанах, 30,9 мл, 77,2 ммоля, 1 экв.) и перемешивают смесь в течение 30 минут при КТ. Получаемый таким образом анион медленно переносят с помощью канюли в исходный реакционный сосуд. Смеси дают нагреться до КТ и перемешивают в течение ночи при КТ. Остатки реагентов разлагают с помощью 1 М КНСО3 и отгоняют растворители при пониженном давлении. Остаток распределяют между фазами этилацетата и воды. Органический слой промывают соляным раствором, высушивают над безводным Na2SО4, фильтруют и концентрируют, что приводит к маслянистому веществу желтого цвета, которое подвергают очистке с помощью флэш-хроматографии (гексан : этилацетат = 4:1), что приводит к указанному в заглавии соединению А-3 в виде твердого вещества белого цвета (15,0 г, 52,2 ммоля, 68%).
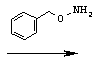
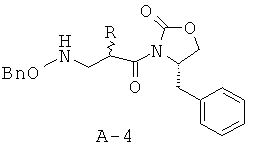
Стадия 3: 4-бензил-3-[2-(бензилоксиаминометил)гексаноил]оксазолидин-2-он (соль пара-толуолсульфокислоты)
Соединение А-3 (8,25 г, 28,7 ммоля) смешивают с О-бензилгидроксиламином (7,07 г, 57,4 ммоля, 2 экв.) и перемешивают в течение 40 часов при КТ в атмосфере азота. Смесь растворяют в этилацетате и добавляют пара-толуолсульфокислоту (21,84 г, 114,8 ммоля, 4 экв.) с целью осаждения избытка О-бензилгидроксиламина в виде твердого вещества белого цвета. Твердое вещество белого цвета отфильтровывают, а фильтрат концентрируют, что приводит к неочищенному маслянистому веществу желтого цвета (анализ с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) показывает присутствие небольших следовых количеств исходного вещества). Обработка неочищенного маслянистого вещества желтого цвета избытком диэтилового эфира и охлаждение до температуры 0°С в течение 30 минут приводят к твердому веществу, которое собирают посредством фильтрования и высушивают под вакуумом, что приводит к указанному в заглавии соединению в виде твердого кристаллического вещества белого цвета (единственный диастереомер).
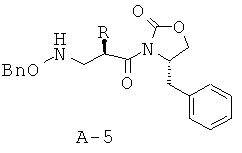
Стадия 4: 4-бензил-3-[2-(бензилоксиаминометил)гексаноил]оксазолидин-2-он (А-5)
К раствору соли ПТСК (22,9 г, 39,3 ммоля), растворенной в этилацетате (400 мл), добавляют 1 М Na2CO3 (200 мл, 5 экв.) и перемешивают при КТ в течение 30 минут. Слои разделяют и экстрагируют водный слой этилацетатом. Объединенные органические слои высушивают над безводным Na2SO4, фильтруют и концентрируют, что приводит к указанному в заглавии соединению в виде бледного непрозрачного маслянистого вещества (15,8 г, 38,6 ммоля, 98%).
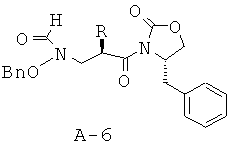
Стадия 5: N-[2-(4-бензил-2-оксооксазолидин-3-карбонил)гексил]-N-бензилоксиформамид (А-6)
Раствор соединения А-5 (5,38 г, 13,1 ммоля, 1 экв.) в муравьиной кислоте (7,4 мл, 196,6 ммоля, 15 экв.) охлаждают до температуры 0°С под азотной подушкой. В отдельной колбе охлаждают муравьиную кислоту (7,4 мл, 196,6 ммоля, 15 экв.) до температуры 0°С под азотной подушкой и добавляют по каплям уксусный ангидрид (2,47 мл, 26,2 ммоля, 2 экв.). Раствор перемешивают при температуре 0°С в течение 15 минут. Получаемый таким образом смешанный ангидрид медленно перемещают с помощью шприца в исходный реакционный сосуд. Смесь перемешивают при температуре 0°С в течение 1 часа, а затем при КТ в течение 3 часов. Смесь концентрируют, переводят в хлористый метилен и промывают последовательно насыщенным раствором NaHCO3 и соляным раствором. Органический слой высушивают над безводным Na2SO4, фильтруют и концентрируют, что приводит к непрозрачному маслянистому веществу, которое подвергают очистке с помощью флэш-хроматографии (гексан : этилацетат = 2:1, а затем хлористый метилен : ацетон = 9:1), что приводит к указанному в заглавии соединению в виде бесцветного маслянистого вещества.
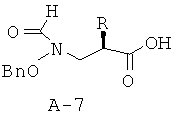
Стадия 6: 2-[(бензилоксиформиламино)метил]гексановая кислота (А-7)
Соединение А-6 (0,163 г, 0,372 ммоля, 1 экв.) растворяют в ТГФ (4,5 мл) и воде (1,5 мл) и охлаждают до температуры 0°С. Добавляют по каплям перекись водорода (30% в воде, 228 мкл, 2,23 ммоля, 6 экв.), а затем медленно добавляют раствор гидроксида лития (0,019 г, 0,446 ммоля, 1,2 экв.) в воде (350 мкл). Получаемую таким образом смесь перемешивают при температуре 0°С в течение 1,5 часов. Реакционную смесь с основной реакцией нейтрализуют с помощью смолы Амберлайт IR-120 (Н+) до pH 4-5 при температуре 0°С. Смолу отфильтровывают и промывают этилацетатом. Смесь концентрируют с целью удаления ТГФ, а затем переводят в этилацетат. Водный слой отделяют, органический слой высушивают над безводным Na2SO4, фильтруют и концентрируют, что приводит к непрозрачному маслянистому веществу, которое подвергают очистке с помощью флэш-хроматографии (хлористый метилен : ацетон = 4:1, а затем ацетон : метанол = 99:1), что приводит к указанному в заглавии соединению А-7 в виде бесцветного маслянистого вещества.
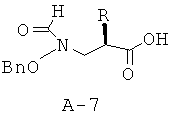
Стадия 7: амид 1-{2-[(бензилоксиформиламино)метил]гексаноил}пирролидин-2-карбоновой кислоты
К раствору соединения А-7 (0,190 г, 0,680 ммоля, 1 экв.) в безводном диоксане (4 мл) добавляют последовательно при КТ в атмосфере азота основание Хюнига (391 мкл, 2,24 ммоля, 3,3 экв.), амин А-8 (0,748 ммоля, 1,1 экв.) и ГАТУ (0,284 г, 0,748 ммоля, 1,1 экв.). Получаемую таким образом смесь перемешивают при КТ в течение 22 часов. Смесь распределяют между фазами этилацетата и 10% лимонной кислоты. Органический слой промывают соляным раствором и насыщенным раствором NaHCO3, высушивают над безводным Na2SO4, фильтруют и концентрируют. Остаток подвергают очистке с помощью флэш-хроматографии (хлористый метилен : ацетон = 3:1), что приводит к указанному в заглавии соединению в виде бесцветного маслянистого вещества.
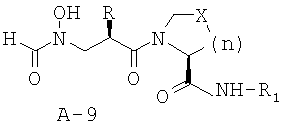
Стадия 8: амид 1-{2-[(формилгидроксиламино)метил]гексаноил}пирролидин-2-карбоновой кислоты (А-9)
Палладий на угле (0,059 г, 0,1 экв.) добавляют к раствору описанного выше соединения (0,550 ммоля, 1 экв.) в растворе этилацетат/этанол 1:1 (12 мл) под азотной подушкой. Смесь перемешивают в атмосфере водорода в течение 36 часов. Катализатор удаляют посредством фильтрования через слой целита. Фильтрат концентрируют и подвергают остаток очистке с помощью препаративной тонкослойной хроматографии (ТСХ) (хлористый метилен : ацетон = 2:1), что приводит к указанному в заглавии соединению в виде аморфного твердого вещества (0,121 г, 0,334 ммоля, 61%).
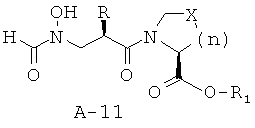
Общая методика Б: синтез 1-{2(RS)-(формилгидроксиламино)метил]алканоил}-пирролидин-2(S)-карбоксилатных сложных эфиров
Х=СН2, -S-, -СН(ОН)-, -CH(OR)-, -CF2- или -CH(F)-, n=0-3.
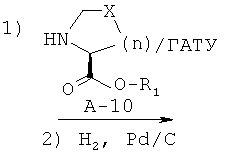
Стадия 1: сложный эфир 1-{2-[(бензилоксиформиламино)метил]гексаноил}пирролидин-2-карбоновой кислоты
К раствору соединения А-7 (0,680 ммоля, 1 экв.) в безводном диоксане (4 мл) при КТ в атмосфере азота добавляют последовательно основание Хюнига (391 мкл, 2,24 ммоля, 3,3 экв.), амин А-10 (0,748 ммоля, 1,1 экв.) и ГАТУ (0,284 г, 0,748 ммоля, 1,1 экв.). Стандартные обработка и очистка приводят к указанному в заглавии соединению.
Стадия 2: сложный эфир 1{2-[(формилгидроксиламино)метил]гексаноил}-пирролидин-2-карбоновой кислоты (А-11)
Палладий на угле (0,059 г, 0,1 экв.) добавляют к раствору описанного выше соединения (0,550 ммоля) в растворе этилацетат/этанол 1:1 (12 мл) под азотной подушкой. Смесь перемешивают в атмосфере водорода в течение 36 часов.
Катализатор удаляют посредством фильтрования через слой целита. Фильтрат концентрируют и подвергают остаток очистке с помощью препаративной ТСХ (хлористый метилен : ацетон = 2:1), что приводит к указанному в заглавии соединению.
Общая методика В: получение пиридин-2-иламида пирролидин-2-S-карбоновой кислоты (А-8) (X=CH2, n=1, R1=2-пиридил)
Стадия 1: бензиловый эфир 2-5-(пиридин-2-илкарбамоил)пирролидин-1-карбоновой кислоты
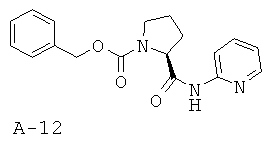
Раствор хлорангидрида N-бензилоксикарбонилпролина (5,0 г, 18,7 ммоля, 1 экв.) в пиридине (40 мл) охлаждают до температуры 0°С под азотной подушкой. Добавляют по каплям 2-аминопиридин (5,27 г, 56,0 ммоля, 3 экв.) в пиридине (10 мл). Получаемую таким образом смесь перемешивают при КТ в течение 4 часов, а затем концентрируют. Маслянистый остаток растворяют в этилацетате и промывают последовательно водой, 10% лимонной кислотой, насыщенным раствором NaHCO3 и соляным раствором. Органический слой высушивают над безводным Na2SO4, фильтруют, и концентрируют, что приводит к указанному в заглавии соединению (4,21 г, 13,0 ммоля, 69%) в виде непрозрачного твердого вещества.
Стадия 2: пиридин-2-иламид пирролидин-2-8-карбоновой кислоты, соль с бромистоводородной кислотой
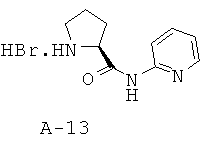
Раствор описанного выше соединения (4,21 г, 13,0 ммоля, 1 экв.) в уксусной кислоте (65 мл) обрабатывают при КТ с помощью HBr (5,7 М, 33% раствор в уксусной кислоте, 110 мл, 649 ммоля, 50 экв.) и перемешивают смесь при КТ в течение 2 часов. Обработка реакционной смеси избытком диэтилового эфира и охлаждение до температуры 0°С в течение 30 минут приводит к твердому веществу, которое собирают с помощью фильтрования и высушивают под вакуумом, что приводит к указанному в заглавии соединению в виде порошка коричневатого цвета.
Общая методика Г: 5-метилпиридин-2-иламид 4-R-гидроксипирролидин-2-S-карбоновой кислоты
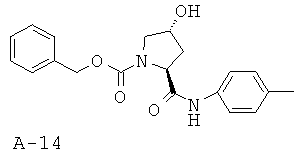
Сочетание производного пролина с О-трет-бутильной защитой (1 ммоль), с 5-пиколином (1,5 ммоля) в ДМФ (5 мл) в присутствии ГАТУ (1,3 ммоля) и N,N-диизопропилэтиламина (5 ммолей) и последующее удаление трет-бутильной группы с атома кислорода с помощью смеси ТФУК-дихлорэтан (1:1) приводит к указанному в заглавии соединению с 85% выходом.
Общая методика Д: 5-метилпиридин-2-иламид 4-S-фторпирролидин-2-S-карбоновой кислоты
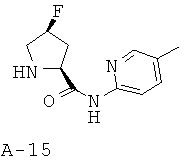
Описанное выше гидроксипроизводное (2 ммоля) в хлористом метилене (20 мл) обрабатывают трифторидом N,N-диэтиламиносеры (ДАСТ, 4 ммоля) при температуре -70°С, после чего реакционную смесь оставляют перемешиваться при КТ в течение 16 часов, промывают ее холодным водным раствором гидрокарбоната натрия, высушивают и концентрируют при пониженном давлении. Очистка с помощью колоночной хроматографии на силикагеле приводит к защищенному по атому азота производному, обработка которого смесью HBr и уксусной кислоты приводит к аминопроизводному.
Общая методика Е: 1-трет-бутиловый эфир 2-метиловый эфир 4-S-гидроксипирролидин-1,2-дикарбоновой кислоты
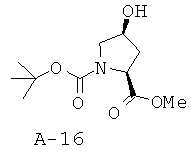
К раствору транс-4-гидроксипроизводного (1 ммоль), трифенилфосфина (1,5 ммоля) и бензойной кислоты (1,5 ммоля) в ТГФ (10 мл) добавляют по каплям при температуре 0°С N,N-диизопропилазодикарбоксилат (1,5 ммоля) в ТГФ (5 мл). Смесь оставляют перемешиваться при КТ в течение 16 часов. Растворитель отгоняют при пониженном давлении и растворяют остаток в диэтиловом эфире. Раствор охлаждают льдом до осаждения окиси фосфина, которую удаляют посредством фильтрования, а фильтрат концентрируют при пониженном давлении. Неочищенное вещество обрабатывают раствором метилата натрия в метаноле в течение 2 часов при температуре 0°С, что приводит к указанному в заглавии цис-гидроксипроизводному.
Общая методика Ж: 5-метилпиридин-2-иламид 4-R-фторпирролидин-2-S-карбоновой кислоты
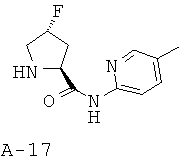
Фторирование описанного выше цис-гидроксипроизводного приводит к транс-4-фторпроизводному, омыление которого приводит к соответствующей кислоте. Амин получают исходя из трет-бутилового эфира 4-R-фторпирролидин-1-карбоновой кислоты и 5-метилпиридин-2-иламина в присутствии ГАТУ, взаимодействие которых приводит к амидопроизводному пролина, обработка которого 4 М HCl в диоксане приводит к искомому амину.
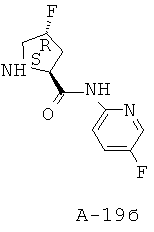
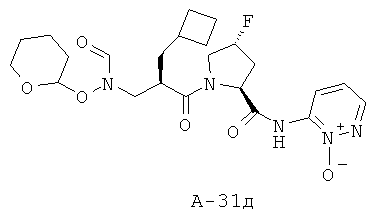
Пример 1: 5-фтор-1-оксипиридин-2-иламид 1-[2-циклопентилметил-3-(формилгидроксиламино)пропионил]пирролидин-2-карбоновой кислоты
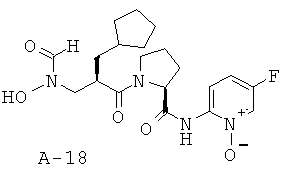
Указанное в заглавии соединение получают согласно общей методике А исходя из 3-бензилоксиформиламино-2-циклопентилметилпропионовой кислоты А-7 (R = циклопентилметил) и пиридин-2-иламида пирролидин-2-карбоновой кислоты А-8 (X=CH2, n=1, R1=5-фтор-2-пиридил).
2-Циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовая кислота
Данное соединение получают исходя из циклопентилметилмалоновой кислоты, как это описано ниже.
Бромметилциклопентан
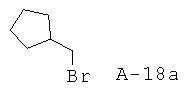
Раствор циклопентилметанола (48,5 г, 484 ммоля), Et3H (88,0 мл, 631 ммоль) и безводного ТГФ (1 л) охлаждают до температуры 4°С и перемешивают в атмосфере азота. Медленно добавляют к перемешиваемому раствору хлорангидрид метансульфокислоты (45,0 мл, 581 ммоль), поддерживая при этом температуру 10°С. Смесь перемешивают в течение еще одного часа при температуре 10°С и медленно добавляют LiBr (300,0 г, 3454 ммоля) (реакция идет с выделением тепла). После этого перемешивают реакционную смесь в течение еще 16 часов при комнатной температуре. Добавляют воду с целью растворения соли и экстрагируют смесь Et2O. Слои Et2O объединяют, высушивают над Na2SO4 и аккуратно концентрируют (25°С, 100 торр). Неочищенный продукт очищают с помощью вакуумной перегонки (35°С, 1 торр, искомое соединение находится в первой из собираемых фракций). Таким образом получают бромметилциклопентан (31,4 г, выход 40%) в виде бесцветного маслянистого вещества.
2-Циклопентилметилмалоновая кислота
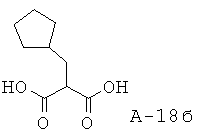
Раствор диэтилового эфира малоновой кислоты (36,91 г, 230,4 ммоля), безводного метанола (400 мл) и NaOMe (25% в метаноле, 49,79 г, 230,4 ммоля) перемешивают при кипячении с обратным холодильником в течение одного часа в атмосфере азота. К смеси добавляют бромметилциклопентан (31,31 г, 192,0 ммоля) и перемешивают в течение еще 3 часов. Затем добавляют раствор NaOH (23,04 г, 576,0 ммоля) в воде (400 мл) и перемешивают смесь в течение еще 1 часа при кипячении с обратным холодильником. Смесь охлаждают, разбавляют водой и экстрагируют диэтиловым эфиром. Эфирный слой удаляют, а водный слой подкисляют 1 н. HCl до рН=1. Водный слой экстрагируют этилацетатом. Этилацетатные слои объединяют, высушивают над Na2SO4 и концентрируют. Таким образом получают 2-циклопентилметилмалоновую кислоту (21,0 г, выход 59%) в виде твердого вещества белого цвета.
2-Циклопентилметилакриловая кислота
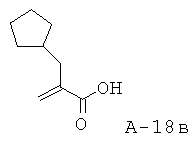
Смесь 2-циклопентилметилмалоновой кислоты (24,90 г, 133,7 ммоля), пиперидина (15,9 мл, 160,8 ммоля), 37% водного раствора формальдегида (51,0 мл, 647,2 ммоля) и EtOH (250 мл) перемешивают при кипячении с обратным холодильником в течение 16 часов. Реакцию останавливают с помощью 1 н. НС1, доводя pH до 1, и экстрагируют смесь этилацетатом. Этилацетатные слои объединяют, высушивают над Na2SO4 и концентрируют. Неочищенный продукт подвергают очистке с помощью флэш-хроматографии (SiO2, 10% ацетон в ДХМ), что приводит к 2-циклопентилметилакриловой кислоте (17,65 г, выход 86%) в виде маслянистого вещества.
4-Бензил-3-(2-циклопентилметилакрилоил)оксазолидин-2-он
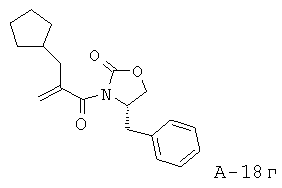
2-Циклопентилметилакриловую кислоту (17,65 г, 114,5 ммоля) растворяют в безводном ТГФ (200 мл) и охлаждают до температуры -78°С в атмосфере азота. Последовательно добавляют N,N-диизопропилэтиламин (25,9 мл, 148,7 ммоля) и хлорид триметилуксусной кислоты (14,1 мл, 114,5 ммоля) с такой скоростью, чтобы температура удерживалась ниже -60°С, а выделение газа происходило контролируемо. Смесь перемешивают при температуре -78°С в течение 30 минут, затем перемешивают при комнатной температуре в течение 2 часов и охлаждают обратно до температуры -78°С.
В отдельной колбе растворяют (S)-(-)-4-бензил-2-оксазолидинон (20,30 г, 114,6 ммоля) в безводном ТГФ (400 мл) и охлаждают до температуры -78°С в атмосфере азота. Медленно добавляют при температуре -78°С BuLi (2,5 M, 45,8 мл, 114,5 ммоля) и перемешивают смесь в течение 30 минут при комнатной температуре. Получаемый таким образом анион медленно перемещают с помощью канюли в исходный реакционный сосуд. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи при комнатной температуре (16 часов). Затем остатки реагентов разлагают с помощью 1 М KHCO3 и экстрагируют реакционную смесь этилацетатом. Органические слои объединяют, промывают соляным раствором, высушивают над Na2SO4 и концентрируют, что приводит к маслянистому веществу желтого цвета. Неочищенный продукт подвергают очистке с помощью флэш-хроматографии (SiO2, 20% этилацетат в гексане), что приводит к 4-бензил-3-(2-циклопентилметилакрилоил)оксазолидин-2-ону (22,9 г, выход 64%) в виде маслянистого вещества.
4-Бензил-3-[2-циклопентилметил-3-(тетрагидропиран-2-илоксиамино)пропионил]оксазолидин-2-он
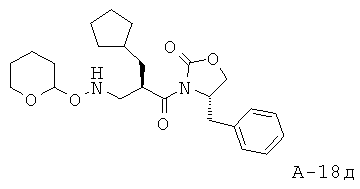
4-Бензил-3-(2-циклопентилметилакрилоил)оксазолидин-2-он (22,90 г, 73,1 ммоля) и O-(тетрагидро-2H-пиран-2-ил)гидроксиламин (34,24 г, 292,3 ммоля) смешивают и перемешивают при температуре 45°С в течение 48 часов в атмосфере азота. Неочищенный продукт подвергают очистке с помощью флэш-хроматографии (SiO2, 0→30% этилацетат в гексане), что приводит к 4-бензил-3-[2-циклопентилметил-3-(тетрагидропиран-2-илоксиамино)пропионил]оксазолидин-2-ону (21,65 г, выход 69%) в виде маслянистого вещества.
N-[3-(4-Бензил-2-оксооксазолидин-3-ил)-2-циклопентилметил-3-оксопропил]-N-(тетрагидропиран-2-илокси)формамид
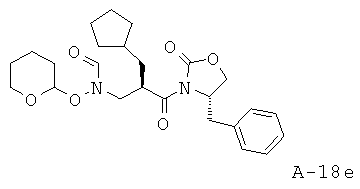
Смесь муравьиной кислоты (45,0 мл, 1193 ммоля) и уксусного ангидрида (90,0 мл, 952 ммоля) перемешивают при температуре 50°С в течение одного часа в атмосфере азота. Во вторую колбу помещают 4-бензил-3-[2-циклопентилметил-3-(тетрагидропиран-2-илоксиамино)пропионил]оксазолидин-2-он (21,62 г, 50,2 ммоля), Et3N (170,0 мл, 1220 ммоля) и безводный ДХМ (450 мл). Данную вторую смесь охлаждают до температуры 4°С в атмосфере азота и медленно добавляют во вторую колбу раствор смешанных кислот, поддерживая температуру 10°С. Объединенную смесь перемешивают в течение 30 минут при температуре 10°С, разлагают остатки реагентов насыщенным раствором, промывают водным раствором NaHCO3 и экстрагируют ДХМ. Слои ДХМ объединяют, высушивают над Na2SO4 и концентрируют. Неочищенный продукт подвергают очистке с помощью флэш-хроматографии (SiO2, 50% этилацетат в гексане), что приводит к N-[3-(4-бензил-2-оксооксазолидин-3-ил)-2-циклопентилметил-3-оксопропил]-N-(тетрагидропиран-2-илокси)формамиду (20,10 г, выход 87%) в виде маслянистого вещества.
2-Циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовая кислота
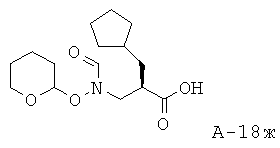
N-[3-(4-Бензил-2-оксооксазолидин-3-ил)-2-циклопентилметил-3-оксопропил]-N-(тетрагидропиран-2-илоксиформамид (3,65 г, 7,96 ммоля), ТГФ (125 мл) и воду (40 мл) охлаждают до температуры 4°С. К данной смеси добавляют 30% H2O2 (5,2 мл, 50,90 ммоля) и моногидрат LiOH (0,40 г, 9,53 ммоля) соответственно. Перемешивают реакционную смесь в течение 1,5 часов. Затем медленно разлагают остатки реагентов 0,5 М Na2SО3, поддерживая при этом температуру ниже 15°С с помощью ледяной бани. После разложения остатков реагентов перемешивают еще в течение 30 минут, концентрируют под вакуумом до отгонки растворителя (ТГФ) и промывают этилацетатом. Смесь с основной реакцией подкисляют с помощью смолы Амберлайт IR-120 (H+) до рН=4,5. К кислому раствору добавляют соляной раствор и экстрагируют объединенную смесь этилацетатом. Органические слои от промывания кислого раствора объединяют, высушивают над Na2SO4 и концентрируют под вакуумом. Таким образом получают 2-циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовую кислоту (1,20 г, выход 50%) в виде маслянистого вещества.
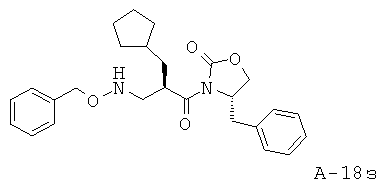
3-(Бензилоксиформиламино)-2-циклопентилметилпропионовая кислота
Данное соединение получают исходя из 2-циклопентилметилакриловой кислоты и O-бензилгидроксиламина, как это описано для синтеза соответствующего полупродукта для построения конечного соединения, в котором атом кислорода защищен тетрагидропиранильной защитой.
4-Бензил-3-(3-бензилоксиамино-2-циклопентилметилпропионил)оксазолидин-2-он
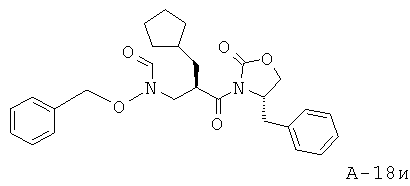
N-[3-(4-Бензил-2-оксооксазолидин-3-ил)-2-циклопентилметил-3-оксопропил]-N-бензилоксиформамид (соединение A-G, в котором R1 циклопентилметил, PG1=бензил)
3-(Бензилоксиформиламино)-2-диклопентилметилпропионовая кислота
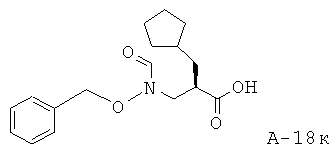
(5-Фтор-1-оксипиридин-2-ил)амид 1-[3-бензилоксиформиламино)-2-циклопентилметилпропионил]пирролидин-2-карбоновой кислоты
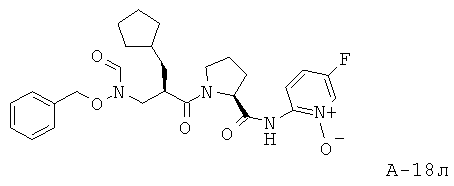
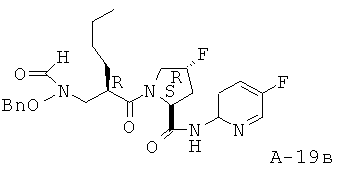
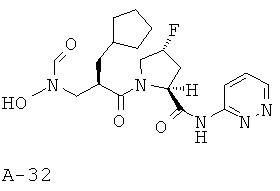
Пример 2: (5-фтор-1-оксипиридин-2-ил)амид 4-фтор-1-{2-[(формилгидроксиламино)метил]гексаноил)}пирролидин-2-карбоновой кислоты
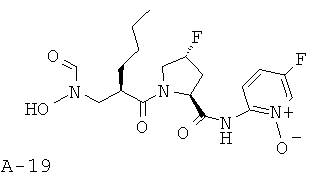
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-[(бензилоксиформиламино)метил]гексановой кислоты А-7 (R=н-бутил) и пиридин-2-иламида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=5-фтор-2-пиридил).
[2-Амино-5-фторпиридин-2-ил]амид 4-транс-фторпирролидин-2-карбоновой кислоты
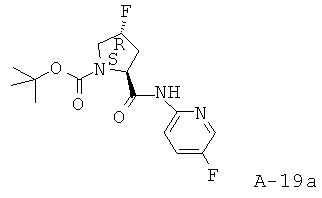
К раствору в ДМФ (15 мл) Boc-L-Pro-4-F-OH (Pro - пролин) (2,5 г, 10,73 ммоля, 1 экв.) добавляют основание Хюнига (диизопропилэтиламин, сокр. ДИЭА) (6,73 мл, 38,61 ммоля, 3,6 экв.) и охлаждают смесь до температуры 0°С. После этого при температуре 0°С добавляют 2-амино-5-фторпиридин (1,44 г, 12,87 ммоля, 1,2 экв.) и ГАТУ (4,89 г, 12,87 ммоля, 1,2 экв.). Получаемую таким образом смесь перемешивают при комнатной температуре в течение 16 часов. Затем смесь распределяют между фазами взятого в избытке этилацетата и 10% лимонной кислоты. Органический слой промывают соляным раствором и насыщенным раствором NaHCO3, высушивают над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают с помощью хроматографии на силикагеле (гексаны: этилацетат = 1:0-7:3), что приводит к указанному в заглавии соединению в виде бесцветного сиропообразного вещества (2,5 г, выход 71%).
[2-Амино-5-фторпиридин-2-ил]амид 4-транс-фторпирролидин-2-карбоновой кислоты (соль с соляной кислотой)
5-Фторпиридин-2-иламид 4-фторпролин, снабженный трет-бутоксикарбонильной защитой (1 г, 3,06 ммоля, 1 экв.), обрабатывают 4 н. HCl в диоксане (30 мл, 120 ммоля, 40 экв.) при комнатной температуре и оставляют перемешиваться в течение 16 часов. Смесь концентрируют и совместно выпаривают остаток с двукратным количеством толуола, после чего опять концентрируют, что приводит к багрянисто-розовому твердому веществу (1 г).
(2-Амино-5-фторпиридин-2-ил)амид 1-{2-[(бензилоксиформиламино)метил]-гексаноил}-4-транс-фторпирролидин-2-карбоновой кислоты
К раствору в ДМФ (10 мл) соли с HCl 5-фторпиридин-2-иламида 4-транс-фторпролина (644 мг, 2,15 ммоля, 1,2 экв.) последовательно добавляют при температуре 0°С основание Хюнига (2 мл, 10,8 ммоля, 5 экв.), Versiacid VRI 172 (500 мг, 1,79 ммоля, 1 экв.) и ГАТУ (818 мг, 2,15 ммоля, 1,2 экв.). Получаемую таким образом смесь перемешивают при комнатной температуре в течение 16 часов. Затем смесь распределяют между фазами взятого в избытке этилацетата и 10% лимонной кислоты. Органический слой промывают соляным раствором и насыщенным раствором NaHCO3, высушивают над безводным Na2SO4, фильтруют и концентрируют. Соединение подвергают очистке с помощью хроматографии на силикагеле в смеси ДХМ : ацетон (1:0-86:14), что приводит к указанному в заглавии соединению в виде порошка белого цвета (630 мг, выход 72%). Масс-спектрометрия с ионизацией распылением в электрическом поле (МС-ИРЭП): рассчитано для C25H30F2N4O5 504,53, найдено 505,4 [М+Н].
(2-Амино-5-фторпиридин-N-оксид-2-ил)амид 1-{2-[(бензилоксиформиламино)метил]гексаноил}-4-транс-фторпирролидин-2-карбоновой кислоты
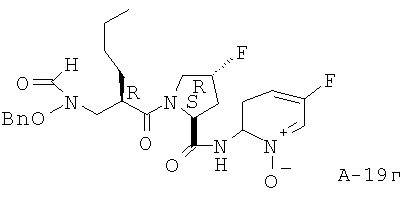
К раствору соединения в ДХМ (1,25 г, 2,56 ммоля, 1 экв.) последовательно добавляют при температуре 0°С МХПБК (1,32 г, 7,68 ммоля, 3 экв.) и перемешивают реакционную смесь в течение 16 часов. Затем реакционную смесь распределяют между фазами NaHCO3 и ДХМ. Органический слой высушивают над Na2SO4 и концентрируют. Остаток подвергают очистке с помощью хроматографии на силикагеле, используя в качестве элюента смесь ДХМ : ацетон (1:0 - 9:1), что приводит к указанному в заглавии соединению (1,2 г).
Пример 3: пиразин-2-иламид 1-{2-[(формилгидроксиламино)метил]гексаноил}-пирролидин-2-карбоновой кислоты
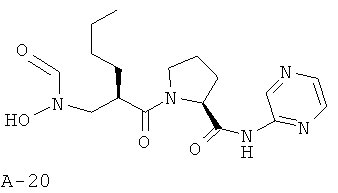
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-[(бензилоксиформиламино)метил]гексановой кислоты А-7 (R=н-бутил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CH2, n=1, R1=2-пиразинил).
Пример 4: пиридазин-3-иламид 1-{2-[(формилгидроксиламино)метил]гексаноил}пирролидин-2-карбоновой кислоты
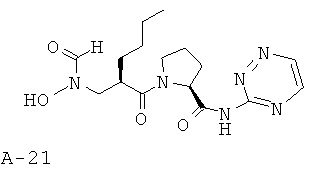
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-{[формил(тетрагидропиран-2-илокси)амино]метил}гексановой кислоты А-7 (R=н-бутил) и пиридазин-3-амида пирролидин-2-карбоновой кислоты А-8 (X=СН2, n=1, R1=3-пиридазинил).
Стадия 1: пиридазин-3-иламин
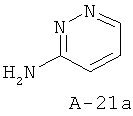
К раствору 6-хлор-2-аминопиридазина (4 г) и NaOH (порошкообразный, 1,4 г) в этаноле (150 мл) добавляют 10% Pd/C (0,6 г). Перемешивают реакционную смесь в атмосфере водорода в течение 16 часов. Затем ее фильтруют через целит и концентрируют растворитель. Получаемый таким образом остаток растирают с диэтиловым эфиром, что приводит к известному аминосоединению.
Стадия 2: трет-бутиловый эфир 2-(пиридазин-3-илкарбамоил)пирролидин-1-карбоновой кислоты
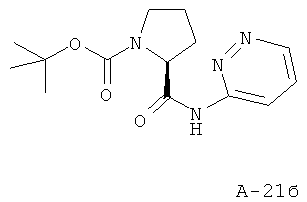
К раствору Вос-Pro-ОН (1 экв.) в ДХМ при температуре 0°С добавляют реактив Ghosez (1-хлор-N,N,2-триметилпропениламин) (1,1 экв.) и перемешивают реакционную смесь при температуре 0°С в течение 1 часа. Затем добавляют к ней амин (1,1 экв.) в пиридине и перемешивают реакционную смесь при комнатной температуре в течение 16 часов. После этого ее концентрируют с целью удаления всех летучих веществ и заново растворяют в избытке ДХМ. Органический слой промывают 10% лимонной кислотой, соляным раствором и NaHCO3, высушивают над Na2SO4 и концентрируют. Получаемый таким образом остаток подвергают очистке с помощью флэш-хроматографии, используя 10-40% этилацетат в гексанах в качестве элюента, что приводит к указанному в заглавии соединению. ВЭЖХ: колонка YMC-Pak Pro С18, S-3 мкм, 120А, 50×4,6 мм (внутренний диаметр); градиент: элюент 0% - 90% MeCN в течение 8,5 минут, 1,5 мл/мин; время удерживания = 4,14 мин.
МС-ИРЭП: рассчитано для C14H20N4O3 292, найдено 293 [М+Н].
Стадия 2: пиридазин-3-иламид пирролидин-2-карбоновой кислоты
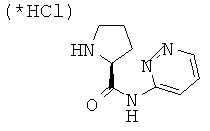
ВЭЖХ: колонка YMC-Pak Pro С18, S-3 мкм, 120А, 50×4,6 мм (внутренний диаметр); градиент: элюент 0%-90% MeCN в течение 8,5 минут, 1,5 мл/мин; время удерживания = 2,398 мин.
МС-ИРЭП: рассчитано для C9H12N4O 192,1, найдено 193,2 [М+Н].
Стадия 3: пиридазин-3-иламид 1-(2-{[формил(тетрагидропиран-2-илокси)амино]метил}гексаноил)пирролидин-2-карбоновой кислоты
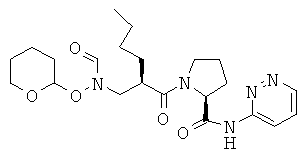
Указанное в заглавии соединение получают в присутствии ГАТУ, как это описано в общей методике А.
ВЭЖХ: колонка YMC-Pak Pro С 18, S-3 мкм, 120А, 50×4,6 мм (внутренний диаметр); градиент: элюент 20% - 90% MeCN в течение 8,5 минут, 1,5 мл/мин; время удерживания = 3,655 мин.
МС-ИРЭП: рассчитано для C22H33H5O5 447, найдено 448 [М+Н].
Пример 5: (5-фтор-1-оксипиридин-2-ил)амид 1-[2-диклопентилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
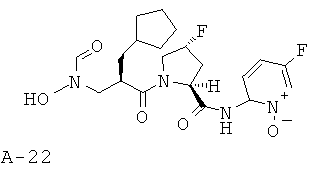
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты и пиридин-2-иламида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=5-фтор-2-пиридил).
(5-Фтор-1-оксипиридин-2-ил)амид 1-{2-циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионил}-4-фторпирролидин-2-карбоновой кислоты
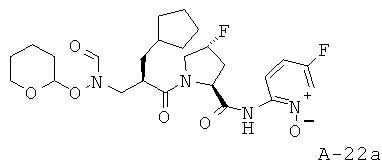
Пример 6: пиридазин-3-иламид 1-[2-циклобутилметил-3-(формилгидроксиламино)пропионил]пирролидин-2-карбоновой кислоты
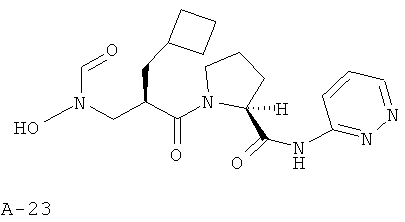
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклобутилметил) и пиридазин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=СН2, n=1, R1=3-пиридазинил).
Пример 7: пиразин-2-иламид 1-[2-циклобутилметил-3-(формилгидроксиламино)-пропионил]пирролидин-2-карбоновой кислоты
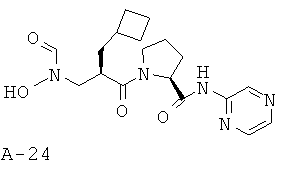
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклобутилметил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=СН2, n=1, R1=2-пиразинил).
Пример 8: 1-[2-циклопентилметил-3-(формилгидроксиламино)пропионил]пирролидин-2-карбоновой кислоты пиразин-2-иламид
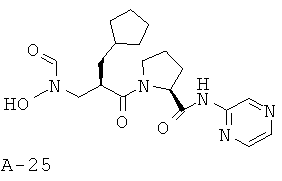
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклопентилметил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CH2, n=1, R1=2-пиразинил).
Пример 9: пиразин-2-иламид 4-фтор-1-{2-[(формилгидроксиламино)метил]-гексаноил}пирролидин-2-карбоновой кислоты
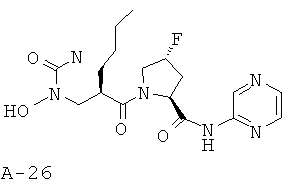
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-[(бензилоксиформиламино)метил]гексановой кислоты А-7 (R=н-бутил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=2-пиразинил).
Пример 10: пиразин-2-иламид 1-[2-циклопентилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
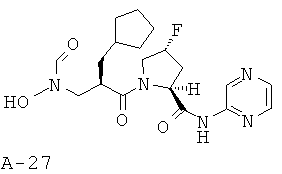
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклопентилметил-3-(формилгидроксиламино)пропионовой кислоты А-7 (R=циклопентилметил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=2-пиразинил).
Пример 11: пиразин-2-иламид 1-[2-циклобутилметил-3-(формилгидроксиламино)-пропионил]-4-фторпирролидин-2-карбоновой кислоты
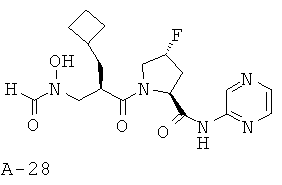
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклобутилметил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=2-пиразинил).
2-Циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовую кислоту получают исходя из 2-циклобутилметилмалоновой кислоты, как это описано для синтеза соответствующего циклопентилметильного производного в примере 1.
2-Циклобутилметилмалоновая кислота
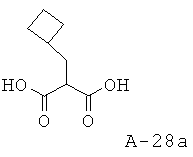
Указанное в заглавии соединение получают исходя из бромметилциклобутана
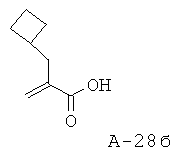
2-Циклобутилметилакриловая кислота
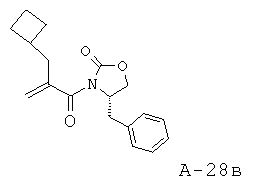
4-Бензил-3-(2-циклобутилметилакрилоил)оксазолидин-2-он
4-Бензил-3-[2-циклобутилметил-3-(тетрагидропиран-2-илоксиамино)пропионил]оксазолидин-2-он
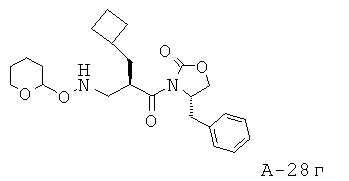
N-[3-(4-Бензил-2-оксооксазолидин-3-ил)-2-циклобутилметил-3-оксопропил]-N-(тетрагидропиран-2-илокси)формамид
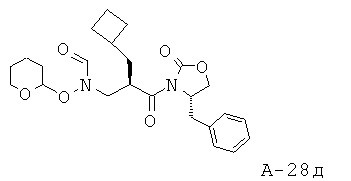
2-Циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовая кислота
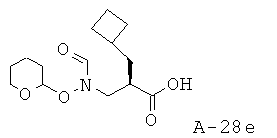
Пиразин-2-иламид 1-{2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионил}-4-фторпирролидин-2-карбоновой кислоты
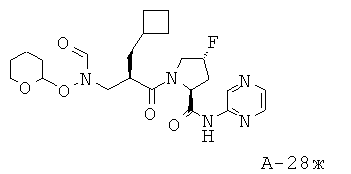
Пример 12: пиримидин-4-иламид 1-[2-циклобутилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
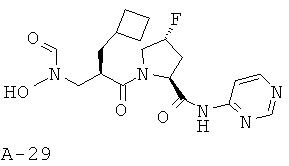
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклобутилметил) и пиримидин-4-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=2-пиримидинил).
Пример 13: пиридазин-3-иламид 1-[2-циклобутилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
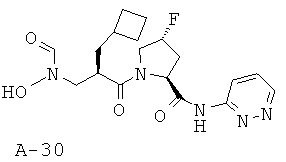
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклобутилметил) и пиридазин-3-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=3-пиридазинил).
(2S,4R)-трет-Бутил-4-фтор-2-(пиридазин-3-илкарбамоил)пирролидин-1-карбоксилат
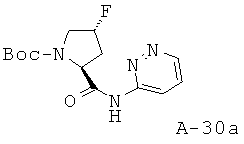
К раствору Boc-Pro(F)-OH (5 г, 21,46 ммоля, 1 экв.) в ДХМ добавляют при температуре 0°С реактив Ghosez (3,1 мл, 23,61 ммоля, 1,1 экв.) и перемешивают реакционную смесь при температуре 0°С в течение 1 часа. Затем к ней добавляют при температуре 0°С амин (2,65 г, 27,9 ммоля, 1,3 экв.) в пиридине и перемешивают реакционную смесь при комнатной температуре в течение 16 часов. После этого ее концентрируют с целью удаления всех летучих веществ и заново растворяют в избытке ДХМ. Органический слой промывают 10% лимонной кислотой, NaCl (насыщ.) и NaHCO3 (насыщ.), высушивают над Na2SO4 и концентрируют. Получаемый таким образом остаток подвергают очистке с помощью флэш-хроматографии, используя 10-15% ацетона в хлористом метилене, что приводит к указанному в заглавии соединению.
(2S,4R)-4-фтор-N-(пиридазин-3-ил)пирролидин-2-карбоксамид
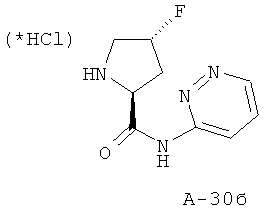
Снабженный трет-бутоксикарбонильной защитной амид переводят в 4 М HCl в диоксане и перемешивают реакционную смесь при комнатной температуре в течение 5 часов. Растворитель отгоняют при пониженном давлении, а остаток растирают с диэтиловым эфиром, что приводит к указанным в заглавии соединениям.
(2S,4R)-1-((2R)-3-Циклобутил-2-((N-(тетрагидро-2Н-пиран-2-илокси)формамидо)-метил)пропаноил)-4-фтор-N-(пиридазин-3-ил)пирролидин-2-карбоксамид
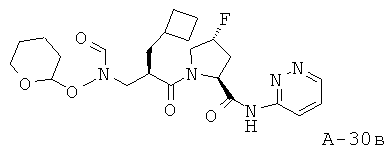
К холодному раствору версатиевой кислоты (500 мг, 1,77 ммоля, 1 экв.) в ДМФ (15 мл) добавляют ДИЭА (1,7 мл, 9,72 ммоля, 5,5 экв.), соль амина с HCl (550 мг, 1,943 ммоля, 1,1 экв.) и ГАТУ (739 мг, 1,943 ммоля, 1,2 экв.). Получаемую таким образом реакционную смесь перемешивают в течение 16 часов при комнатной температуре. Затем смесь распределяют между фазами взятого в избытке этилацетата и 10% лимонной кислоты. Органический слой промывают насыщенным раствором NaCl и насыщенным раствором НаНСО3, высушивают над безводным Na2SO4, фильтруют и концентрируют. Остаток подвергают очистке с помощью хроматографии на силикагеле, используя 10-25% ацетона в ДХМ в качестве элюента, что приводит к указанному в заглавии соединению (53%).
Пример 14: (2-оксипиридазин-3-ил)амид 1-[2-циклобутилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
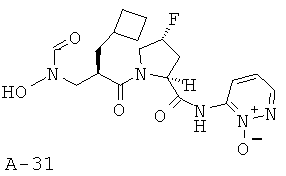
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклобутилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклобутилметил) и пиридазин-1-оксо-3-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=3-пиридазинил-N-оксид).
6-((2S,4R)-1-(трет-Бутоксикарбонил)-4-фторпирролидин-2-карбоксамидо)пиридазин-1-оксид
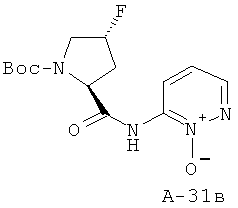
К раствору Boc-Pro(F)-OH (2,047 г, 8,154 ммоля, 1 экв.) в ДХМ добавляют при температуре 0°С реактив Ghosez (1,2 мл, 8,97 ммоля, 1,1 экв.) и перемешивают реакционную смесь при температуре 0°С в течение 1 часа. Затем к ней добавляют при температуре 0°С амин (1,27 г, 11,42 ммоля, 1,4 экв.) в пиридине и перемешивают реакционную смесь при комнатной температуре в течение 16 часов. После этого ее концентрируют с целью удаления всех летучих веществ, а остаток растворяют в избытке ДХМ. Органический слой промывают 10% лимонной кислотой, NaCl (насыщ.) и NaHCO3 (насыщ.), высушивают над Na2SO4 и концентрируют. Получаемый таким образом остаток подвергают очистке с помощью флэш-хроматографии, используя 2-15% ацетона в хлористом метилене в качестве элюента, что приводит к указанному в заглавии соединению (61%).
6-((2S,4R)-4-Фторпирролидин-2-карбоксамидо)пиридазин-1-оксид
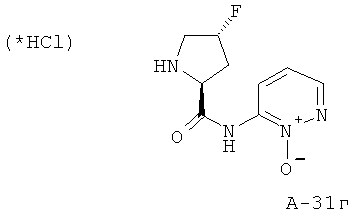
Снабженный трет-бутоксикарбонильной защитой амид переводят в 4 М HCl в диоксане и перемешивают реакционную смесь при комнатной температуре в течение 5 часов. Все летучие вещества отгоняют, а остаток растирают с диэтиловым эфиром, что приводит к указанным в заглавии соединениям.
6-((2S,4R)-1-((2R)-3-Циклобутил-2-((N-тетрагидро-2Н-пиран-2-илокси)-формамидо)метил)пропаноил)-4-фторпирролидин-2-карбосамидо)пиридазин)пиридазин-1-оксид
К холодному раствору Versiacid (571 мг, 2 ммоля, 1 экв.) в ДМФ (20 мл) добавляют ДИЭА (2,51 мл, 14,4 ммоля, 6 экв.), соль амина с НС1 (718 мг, 2,4 ммоля, 1,2 экв.) и ГАТУ (913 мг, 2,4 ммоля, 1,2 экв.). Получаемую таким образом реакционную смесь перемешивают в течение 16 часов при комнатной температуре. Затем смесь распределяют между фазами взятого в избытке этилацетата и 10% лимонной кислоты. Органический слой промывают насыщенным раствором NaCl и насыщенным раствором NaHCO3, высушивают над безводным Na2SO4, фильтруют и концентрируют. Остаток подвергают очистке с помощью хроматографии на силикагеле, используя 10-20% ацетона в ДХМ, а затем 2-8% метанола в ДХМ в качестве элюента, что приводит к указанному в заглавии соединению (44%).
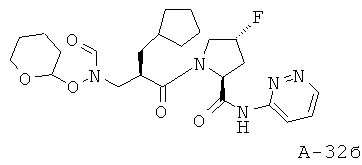
Пример 15: пиридазин-3-иламид 1-[2-циклопентилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклопентилметил) и пиридазин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=3-пиридазинил).
(2S,4R)-1-((2R)-3-Циклопентил-2-((N-(тетрагидро-2Н-пиран-2-илокси)формамидо)метил)пропаноил)-4-фтор-N-(пиридазин-3-ил)пирролидин-2-карбоксамид
Пример 16: (2-оксипиридазин-3-ил)амид 1-[2-циклопентилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
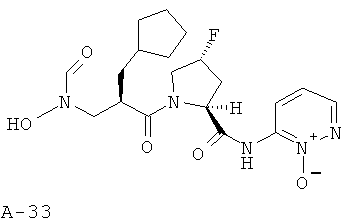
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклопентилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклопентилметил) и пиридазин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=3-пиридазинил-N-оксид).
6-Аминопиридазин-1-оксид
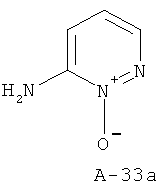
К раствору 6-аминопиридазина в ацетоне добавляют в один прием раствор МХПБК (1 экв.) в ацетоне. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 1 часа. Затем растворитель отгоняют и добавляют к остатку диэтиловый эфир. Твердое вещество отфильтровывают и высушивают, что приводит к указанному в заглавии соединению. Последнее используют как таковое на следующей стадии.
6-((2S,4R)-1-(трет-Бутоксикарбонил)-4-фторпирролидин-2-карбоксамидо)пиридазин-1-оксид
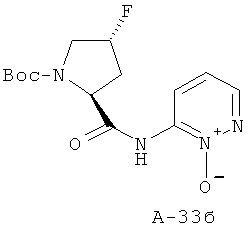
К раствору Boc-Pro(F)-OH (2,047 г, 8,154 ммоля, 1 экв.) в ДХМ добавляют при температуре 0°С реактив Ghosez (1,2 мл, 8,97 ммоля, 1,1 экв.) и перемешивают реакционную смесь при температуре 0°С в течение 1 часа. Затем к ней добавляют при температуре 0°С амин (1,27 г, 11,42 ммоля, 1,4 экв.) в пиридине и перемешивают реакционную смесь при комнатной температуре в течение 16 часов. После этого ее концентрируют с целью удаления всех летучих веществ, а остаток растворяют в избытке ДХМ. Органический слой промывают 10% лимонной кислотой, NaCl (насыщ.) и NaHCO3 (насыщ.), высушивают над Na2SO4 и концентрируют. Получаемый таким образом остаток подвергают очистке с помощью флэш-хроматографии, используя 2-15% ацетона в хлористом метилене в качестве элюента, что приводит к указанному в заглавии соединению (61%).
6-((2S,4R)-4-Фторпирролидин-2-карбоксамидо)пиридазин-1-оксид
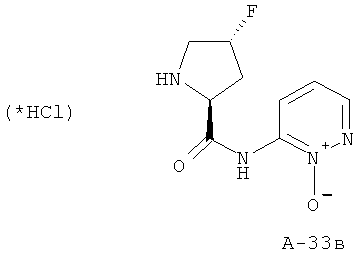
Снабженный трет-бутоксикарбонильной защитой амид переводят в 4 М HCl в диоксане и перемешивают реакционную смесь при комнатной температуре в течение 5 часов. Все летучие вещества отгоняют, а остаток растирают с диэтиловым эфиром, что приводит к указанным в заглавии соединениям.
6-((2S,4R)-1-((2R)-3-циклопентил-2-((N-(тетрагидро-2Н-пиран-2-илокси)формамидо)метил)пропаноил)-4-фторпирролидин-2-карбоксамидо)пиридазин-1-оксид
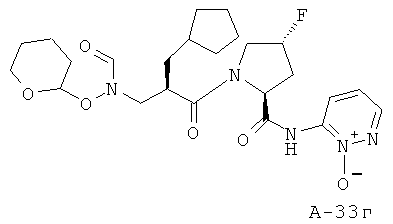
Пример 17: пиридазин-3-иламид 1-[2-циклогексилметил-3-(формилгидроксиламино)пропионил]-4-фторпирролидин-2-карбоновой кислоты
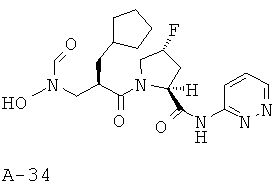
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклогексилметил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклогексилметил) и пиридазин-3-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=3-пиридазинил).
2-Циклогексилметил-3-(формилгидроксиламино)пропионовую кислоту, служащую полупродуктом для сборки конечного соединения, получают исходя из 2-циклогексилметилмалоновой кислоты, как это описано для синтеза соответствующей циклопентилметилмалоновой кислоты в примере 1.
2-Циклогексилметилмалоновая кислота
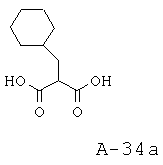
Указанное в заглавии соединение получают исходя из бромметилциклогексана.
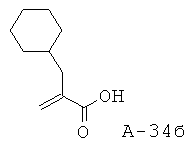
2-Циклогексилметилакриловая кислота
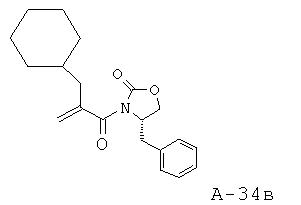
4-Бензил-3-(2-циклогексилметилакрилоил)оксазолидин-2-он
4-Бензил-3-(2-циклогексилметил-3-тетрагидропиран-2-илоксиамино)пропионил]оксазолидин-2-он
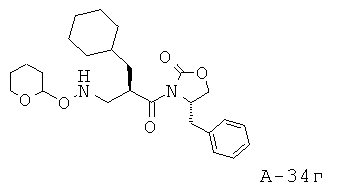
N-[3-(4-Бензил-2-оксооксазолидин-3-ил)-2-циклогексилметил-3-оксопропил]-N-(тетрагидропиран-2-илокси)формамид 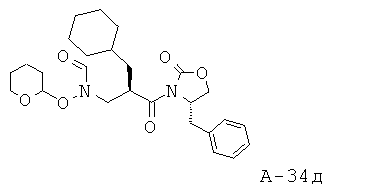
Пример 18: пиразин-2-иламид 1-{4-циклопропил-2-[(формилгидроксиламино)метил]бутирил}-4-фторпирролидин-2-карбоновой кислоты
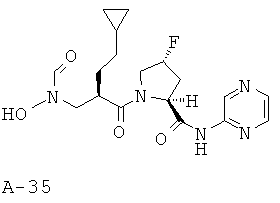
Указанное в заглавии соединение получают согласно общей методике А исходя из 2-циклопропилэтил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовой кислоты А-7 (R=циклопропилэтил) и пиразин-2-амида пирролидин-2-карбоновой кислоты А-8 (X=CHF, n=1, R1=2-пиразинил).
2-Циклопропилэтил-3-(формилгидроксиламино)пропионовую кислоту, служащую полупродуктом для сборки конечного соединения, получают исходя из 2-циклопропилэтилмалоновой кислоты, как это описано для синтеза соответствующей циклопентилметилмалоновой кислоты в примере 1.
Бромэтилциклопропан
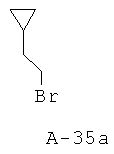
Указанное в заглавии соединение получают исходя из 2-циклопропилэтанола.
2-Циклопропилэтилмалоновая кислота
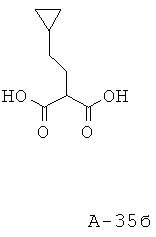
2-Циклопропилэтилакриловая кислота
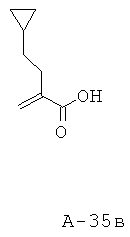
4-Бензил-3-(2-циклопропилэтилакрилоил)оксазолидин-2-он
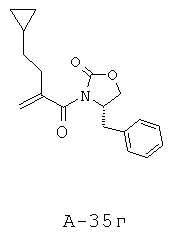
4-Бензил-3-[2-циклопропилэтил-3-(тетрагидропиран-2-илоксиамино)пропионил]оксазолидин-2-он
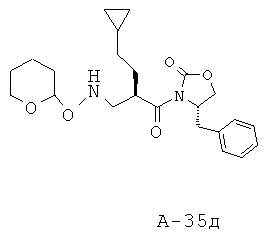
N-[3-(4-Бензил-2-оксооксазолидин-3-ил)-2-циклопропилэтил-3-оксопропил]-N-(тетрагидропиран-2-илокси)формамид
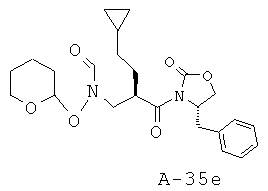
2-Циклопропилэтил-3-[формил(тетрагидропиран-2-илокси)амино]пропионовая кислота
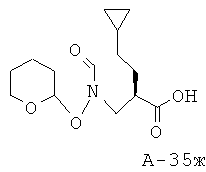
Пиразин-2-иламид 1-{2-циклопропилэтил-3-[формил(тетрагидропиран-2-илокси)амино]пропионил}-4-фторпирролидин-2-карбоновой кислоты
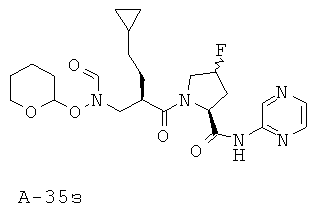
Пример 19: ингибирование активности пептиддеформилазы
Применяют комплексный анализ ПДФ/ФДГ (пептиддеформилаза/фосфатдегидрогеназа) (Lazennec и др., Anal. Biochem., т.224, стр.180-182 (1997)). В данном комплексном анализе формиат, высвобождаемый ПДФ из ее субстрата N-формилметионин-аланин-серина (fMAS), окисляется присоединяющим ферментом FDH, восстанавливая одну молекулу NAD+ (никотинамид-аденин динуклеотид) до NADH, что приводит к увеличению интенсивности поглощения при длине волны 340 нм. Все анализы осуществляют при комнатной температуре в буфере, содержащем 50 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфокислоты (HEPES), pH 7,2, 10 мМ NaCl, 0,2 мг/мл бычьего сывороточного альбумина (BSA), в 96-ячеечных планшетах для микротитрования половинной площади (Corning). Взаимодействие инициируют посредством добавления смеси 0,5 ед./мл ФДГ, 1 мМ NAD+ и fMAS в желаемой концентрации. Для определения величин IC50 (концентрации, обеспечивающей 50% ингибирование активности фермента) ПДФ предварительно инкубируют в течение 10 минут с варьируемыми концентрациями ингибитора, в реакцию деформилирования инициируют посредством добавления реакционной смеси, содержащей 4 мМ fMAS. Начальную скорость реакции, у, измеряют как начальную скорость увеличения интенсивности поглощения при длине волны 340 нМ с помощью детектора для планшетов SpectraMax (Molecular Devices, Sunnyvale, CA). Концентрация ингибитора [In], при которой ингибируется 50% активности фермента, IC50, рассчитывают с помощью следующей формулы:
y=y0/(1+[In]/IC50),
в которой уо обозначает начальную скорость реакции при отсутствии ингибитора. Разрешение данного уравнения относительно IC50 при том значении [In], при котором у=уо/2, приводит к величине IC50. Значение IC50 рассчитывают на основе аппроксимации посредством нелинейной регрессии по методу наименьших квадратов с использованием коммерческого программного обеспечения (Deltapoint, Inc., Chicago, IL.).
С помощью данного анализа определяют величины IC50 различных соединений. Величины IC50 для различных соединений определяют в отношении фермента деформилазы, содержащего в качестве иона металла никель и цинк. Значения IC50 предпочтительных соединений формулы (I), определенные для цинксодержащей деформилазы, находятся в пределах от приблизительно 0,001 мкМ до приблизительно 0,2 мкМ. Значения IC50 предпочтительных соединений формулы (I), определенные для никельсодержащей деформилазы, находятся в пределах от приблизительно 0,005 мкМ до приблизительно 3 мкМ.
Пример 20: анализ для исследования противомикробной активности
Минимальные ингибирующие концентрации (МИК) определяют с помощью метода микроразбавления в 96-ячеечных форматных планшетах. Соединения суспендируют в ДМСО с концентрацией 5 или 10 мг/мл и хранят при температуре 4°С до использования. Их растворяют в бульоне Мюллера-Хинтона (МНВ) или триптиказо-соевом бульоне (TSB) и используют для определения МИК. Диапазон испытуемых концентраций составляет, для конечных концентраций, 64-0,0625 мкг/мл при использовании системы двукратных разбавлений.
Посевной материал изготавливают из клеток, выращенных на триптиказо-соевом агаре (TSA) и инкубированных в течение ночи при температуре 35°С. Для посева в бульоне МНВ или TSB используют 5-10 колоний и инкубируют культуру в течение ночи при температуре 35°С. Инкубированную в течение ночи культуру разбавляют 1:10, инкубируют в течение 1 часа при температуре 35°С, разбавляют до подходящей концентрации посевного материала и помещают в ячейки, содержащие бульон и испытуемое соединение. Концентрации посевного материала составляют 2×104 колониеобразующих единиц (КОЕ)/мл.
Планшеты инкубируют при температуре 35°С в течение 48 часов и определяют МИК после 18 часов инкубирования в присутствии бактерий. МИК определяют как наименьшую концентрацию соединения, при которой не произошло видимого роста после инкубирования.
Минимальная ингибирующая концентрация для различных предпочтительных соединений формулы (I) составляет от приблизительно 0,25 мкг/мл до приблизительно 32 мкг/мл против Н. influenza (четыре штамма), от приблизительно 0,001 мкг/мл до свыше 8 мкг/мл против S. aureus (четыре штамма), от приблизительно 0,016 мкг/мл до приблизительно 16 мкг/мл против S. pneumonia (четыре штамма) и от приблизительно 0,008 мкг/мл до приблизительно 16 мкг/мл против М. catarrhalis. Фермент деформилазу получают из Е. coli.
Ниже представлены характерные фармацевтические лекарственные формы, содержащие соединение формулы (I).
Пример 21: состав таблетки
Нижеследующие ингредиенты тесно смешивают и запрессовывают в таблетки с одной насечкой:
Пример 22: состав капсулы
Нижеследующие ингредиенты тесно смешивают и помещают в желатиновую капсулу с твердой оболочкой:
Пример 23: состав суспензии
Нижеследующие ингредиенты смешивают с целью образования суспензии для перорального введения:
(Vanderbilt Co.)
Пример 24: состав для инъекций
Нижеследующие ингредиенты смешивают с целью образования состава для инъекций:
Пример 25: состав суппозитория
Суппозиторий общей массой 2,5 г получают посредством смешения соединения по настоящему изобретению с Витепсол® Н-15 (триглицериды насыщенной растительной жирной кислоты; Riches-Nelson, Inc., New York). Он имеет следующий состав: соединение по настоящему 500 мг изобретению, Витепсол® Н-15, основа.
| название | год | авторы | номер документа |
|---|---|---|---|
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОБЕНЗО[b][1,4]ДИАЗЕПИН-2-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ MGLUR2 II | 2002 |
|
RU2263112C2 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ОПОСРЕДУЕМОГО ЦИТОКИНАМИ | 2000 |
|
RU2298008C2 |
| НОВЫЕ ЭФИРНЫЕ ПРОИЗВОДНЫЕ ИЗОКСАЗОЛА В КАЧЕСТВЕ ПОЗИТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ РЕЦЕПТОРА ГАМК А АЛЬФА 5 | 2017 |
|
RU2772862C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2000 |
|
RU2242474C2 |
| ИМИДАЗОПИРРОЛОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗ Jak1, Jak3 ИЛИ Syk | 2010 |
|
RU2570416C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
Изобретение относится к соединениям формулы (I)
где R1 означает С1-10алкил, С3-10циклоалкил; R3 означает Н, галоид; R4 означает 6-членный моноциклический гетероарил, содержащий 1-2 атома азота, один из которых может быть окислен, необязательно замещенный галоидом; n равно от 0 до 3, при условии, что R1 означает С3-10циклоалкил и/или R4 означает необязательно замещенный галоидом 6-членный моноциклический гетероарил, содержащий 2 кольцевых гетероатома N, один из которых может быть окислен, или его фармацевтически приемлемая соль. Соединения ингибируют пептидилдеформилазу (ПДФ). 14 з.п. ф-лы.
1. Соединение формулы (I)
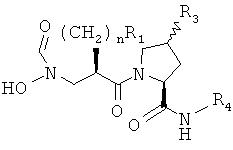
в которой
R1 обозначает С1-10алкил, С3-10циклоалкил;
R3 обозначает водород, галоид; и
R4 обозначает 6-членный моноциклический гетероарил, содержащий 1-2 атома азота, один из которых может быть окислен, необязательно замещенный галоидом; n принимает значения от 0 до 3;
при условии, что R1 обозначает С3-10циклоалкил и/или R4 обозначает необязательно замещенный галоидом 6-членный моноциклический гетероарил, содержащий 2 кольцевых гетероатома азота, один из которых может быть окислен, или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R3 выбран из фтора, хлора или иода.
3. Соединение по п.1, где R1 обозначает С3-10циклоалкил.
4. Соединение по п.1, где R4 обозначает гетероарил формулы (II)
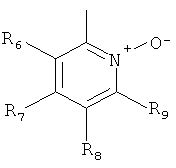 или
или 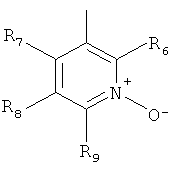 или
или 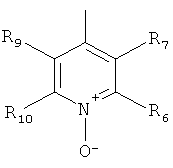 или
или 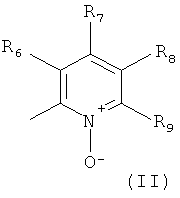
в которой каждый из заместителей R6, R7, R8 и R9 независимо от остальных обозначает водород или галоид, или его фармацевтически приемлемая соль.
5. Соединение по п.1, где R4 обозначает гетероарил формулы (II.1)
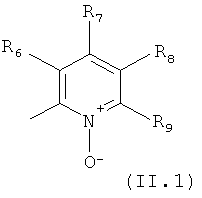
в которой R6, R7 и R9 обозначают водород, a R8 обозначает фтор, или его фармацевтически приемлемая соль.
6. Соединение по п.1, где R4 обозначает гетероарил формулы (II.2)
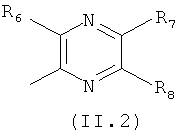
в которой R6, R7 и R8 обозначают водород, или его фармацевтически приемлемая соль.
7. Соединение по п.1, где R4 обозначает гетероарил формулы (II.3)
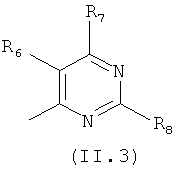
в которой R6, R7 и R8 обозначают водород, или его фармацевтически приемлемая соль.
8. Соединение по п.1, где R4 обозначает гетероарил формулы (II.4)
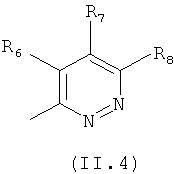
в которой R6, R7 и R8 обозначают водород, или его фармацевтически приемлемая соль.
9. Соединение по п.1, где R1 выбран из циклогексила, циклопентила, циклобутила или циклопропила.
10. Соединение по п.1, где R1 обозначает н-бутилом.
11. Соединение по п.9, где R4 выбран из 5-фторпиридин-N-оксида, 2-пиразина, 3-пиридазина, 3-пиримидина, 4-пиримидина или 3-пиридазин-N-оксида.
12. Соединение по п.10, где R4 выбран из 5-фторпиридин-N-оксида, 2-пиразина, 3-пиридазина, 3-пиримидина, 4-пиримидина или 3-пиридазин-N-оксида.
13. Соединение по п.9, где R3 обозначает фтор.
14. Соединение по п.10, где R3 обозначает фтор.
15. Соединение по п.1, где R4 обозначает гетероарил формулы (II.5)
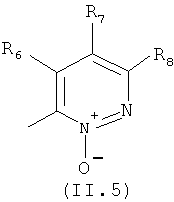
в которой R6, R7 и R8 обозначают водород, или его фармацевтически приемлемая соль.
| American Society for Microbiology, 49, 4, april, 2005, 1468-1476 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 1999 |
|
RU2246941C2 |

