Область техники, к которой относится изобретение
Настоящее изобретение относится к стереоспецифичному синтезу (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, его солевых форм и новых полиморфных форм этих солей. Настоящее изобретение также включает фармацевтические композиции этих солевых форм и способы лечения широкого ряда заболеваний и расстройств, включая боль, воспаление, и заболеваний и расстройств, связанных с нарушением функции центральной и автономной нервных систем.
Уровень техники
Соединение (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин представляет собой агонист нейронального никотинового рецептора (NNR) с селективностью к никотиновому подтипу α4β2 по отношению к другим никотиновым подтипам, например, подтипу α7, ганглиозному и мускульному подтипам. (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин создает преимущества в лечении или предотвращении расстройств центральной нервной системы (CNS) и боли.
(R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин имеет следующую структурную формулу:
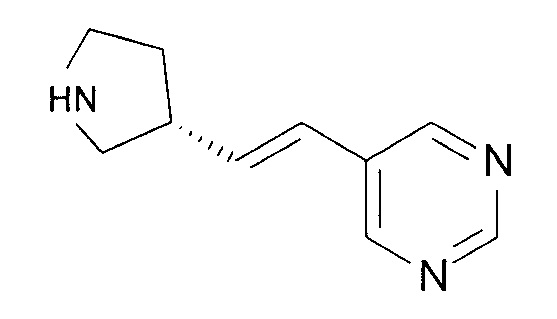
Разработка производства в коммерческих целях потенциального лекарственного средства, такого как (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин, включает много стадий, включающих разработку способа экономически эффективного синтеза, который является адаптируемым к крупномасштабному производственному процессу. Разработка производства в коммерческих целях также включает исследования, касающиеся солевых форм лекарственного вещества, которые обладают подходящей чистотой, химической стабильностью, фармацевтическими свойствами и характеристиками, которые способствуют удобному обращению и обработке. Далее, композиции, содержащие лекарственное вещество, должны иметь адекватную продолжительность хранения. То есть, они не должны проявлять значительных изменений в физико-химических характеристиках, таких как, но не ограничиваясь ими, химический состав, содержание воды, плотность, гигроскопичность и растворимость при хранении в течение существенного периода времени. Вдобавок, воспроизводимые и постоянные профили концентрации лекарственного средства в плазме по введению пациенту также являются важными факторами.
Твердые солевые формы обычно являются предпочтительными для пероральных препаратов, поскольку именно они склонны к проявлению таких свойств; и в случае основных лекарственных средств, таких как (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин, соли присоединения кислоты часто являются предпочтительной солевой формой. Однако различные солевые формы сильно различаются по их способности придавать такие свойства, и такие свойства не могут быть предсказаны с достаточной точностью. Например, некоторые соли представляют собой твердые вещества при температуре окружающей среды, в то же время другие соли представляют собой жидкости, вязкие масла или смолы при температуре окружающей среды. Далее, некоторые солевые формы являются стабильными к воздействию тепла и света в экстремальных условиях, а другие легко разлагаются при гораздо более мягких условиях. Таким образом, разработка подходящей формы соли присоединения кислоты основного лекарственного средства для использования в фармацевтической композиции является крайне непредсказуемым процессом.
Синтез (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина и его полугалактаратной соли, ее разделение при помощи хиральной хроматографии на оптические изомеры и галактаратные соли изомеров раскрыты в опубликованной заявке WO 04/078752 и патенте США № 7098331, каждый из которых включен в данное описание путем ссылки. Однако, являются желательными стереоспецифические синтезы (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, которые являются масштабируемыми для крупномасштабного производства. Далее, так как (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин в форме свободного основания представляет собой вязкое масло с ограниченной растворимостью в воде и стабильностью, существует необходимость в солевых формах, которые проявляют улучшенные свойства, включающие чистоту, стабильность, растворимость и биодоступность. Предпочтительные характеристики этих новых солевых форм включают такие, которые увеличили бы простоту или эффективность изготовления активного ингредиента и его состава до коммерческого продукта. Наконец, существует необходимость в стабильных полиморфных формах этих солей, которая делает возможным увеличение простоты или эффективности изготовления активного ингредиента и его состава до коммерческого продукта.
Сущность изобретения
Одним аспектом изобретения является соль присоединения кислоты (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. В определенных вариантах осуществления кислоту выбирают из хлороводородной, серной, метансульфоновой, малеиновой, фосфорной, 1-гидрокси-2-нафтойной, кетоглутаровой, малоновой, L-винной, фумаровой, лимонной, L-яблочной, гиппуровой, L-молочной, бензойной, янтарной, адипиновой, уксусной, никотиновой, пропионовой, оротовой, 4-гидроксибензойной, ди-(п-толуоил)-D-винной, ди-п-анизоил-D-винной, дибензоил-D-винной, 10-камфорсульфоновой, камфорной или фенцифоса.
Одним аспектом изобретения является соль малеиновой кислоты (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другим аспектом изобретения является соль оротовой кислоты (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Дальнейшим аспектом изобретения является соль лимонной кислоты (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Одним аспектом изобретения является моноцитрат (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другим аспектом изобретения является кристаллическая полиморфная форма моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Одним аспектом изобретения является стереоспецифический синтез (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другие аспекты и варианты осуществления настоящего изобретения будут описаны в данном документе. Объем настоящего изобретения включает комбинации аспектов, вариантов осуществления и предпочтения.
Краткое описание чертежей
Фиг. 1 представляет собой диаграмму XRPD аморфной формы моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг. 2 представляет собой диаграмму XRPD формы I моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг. 3 представляет собой диаграмму XRPD формы II моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг. 4 представляет собой диаграмму XRPD формы III моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг. 5 представляет собой диаграмму XRPD формы IV моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг. 6 представляет собой диаграмму XRPD формы I монооротата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг. 7 представляет собой диаграмму XRPD формы I мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Фиг .8 представляет собой диаграмму XRPD формы II мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Подробное описание
Определения
Следующие определения имеют целью прояснить, но не ограничить определенные термины. Если конкретный термин, используемый в данном документе, не является конкретно определенным, то такой термин не должен считаться неограниченным. Скорее, термины используются в рамках их принятых значений.
Выражение "соединения настоящего изобретения" в том виде, как она используется в данном документе, относится к (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидину или его соли присоединения кислоты. Кислоту выбирают из хлороводородной кислоты, серной кислоты, метансульфоновой кислоты, малеиновой кислоты, фосфорной кислоты, 1-гидрокси-2-нафтойной кислоты, кетоглутаровой кислоты, малоновой кислоты, L-винной кислоты, фумаровой кислоты, лимонной кислоты, L-яблочной кислоты, гиппуровой кислоты, L-молочной кислоты, бензойной кислоты, янтарной кислоты, адипиновой кислоты, уксусной кислоты, никотиновой кислоты, пропионовой кислоты, оротовой кислоты, 4-гидроксибензойной кислоты, ди-(п-толил)-D-винной кислоты, ди-п-анизоил-D-винной кислоты, дибензоил-D-винной кислоты, 10-камфорсульфоновой кислоты, камфорной кислоты или 2-гидрокси-5,5-диметил-4-фенил-1,2,3-диоксафосфоринан-2-она (фенцифоса). Данное понятие охватывает гидратную или сольватную форму.
Далее, в том виде, как он используется в данном документе, термин "соединение" может быть использован для обозначения формы свободного основания или, альтернативно, солевой формы (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, в зависимости от контекста, который будет легко очевидным. Специалисты в данной области техники будут в состоянии легко различить отличие.
В том виде, как он используется в данном документе, термин "фармацевтически приемлемый" относится к носителю(ям), разбавителю(ям), инертному(ым) наполнителю(ям) или солевым формам, которые являются совместимыми с другими компонентами состава и не вредными для получателя фармацевтической композиции.
В том виде, как он используется в данном документе, термин "фармацевтическая композиция" относится к соединению настоящего изобретения, необязательно смешанному с одним или более фармацевтически приемлемыми носителями, разбавителями, инертными наполнителями или адъювантами. Фармацевтические композиции предпочтительно проявляют относительную стабильность по отношению к условиям окружающей среды так, чтобы сделать их пригодными для целей изготовления и промышленного производства.
В том виде, как они используются в данном документе, термины "эффективное количество", "терапевтическое количество" или "эффективная доза" относятся к количеству активного ингредиента, достаточному для вызывания желаемых фармакологических или терапевтических эффектов, таким образом приводя к эффективному предотвращению или лечению расстройства. Предотвращение расстройства может проявляться как в замедлении или предотвращении прогрессирования расстройства, так и в замедлении или предотвращении возникновения симптомов, связанных с расстройством. Лечение расстройства может проявляться в снижении или удалении симптомов, ингибировании или обращении прогрессирования расстройства, а также в любом другом вкладе в хорошее самочувствие пациента.
Эффективная доза может варьироваться, в зависимости от факторов, таких как состояние пациента, серьезность симптомов расстройства, и способа, каким вводится фармацевтическая композиция. Обычно для введения в эффективной дозе необходимо вводить соединения в количестве менее чем 5 мг/кг массы пациента. Часто соединения могут быть введены в количестве от менее, чем примерно 1 мг/кг массы пациента до менее, чем примерно 100 мкг/кг массы пациента, и иногда между примерно 10 мкг/кг и менее, чем 100 мкг/кг массы пациента. Вышеуказанные эффективные дозы обычно представляют такое количество, как введенное в качестве одной дозы, или в качестве одной или более доз, введенных в течение периода в 24 часа. Для пациента-человека эффективные дозы соединений могут потребовать ввода соединения в количестве, по меньшей мере, примерно 1 мг/24 ч/пациент, но не более, чем примерно 1000 мг/24 ч/пациент, и часто не более, чем 500 мг/24 ч/пациент.
В том виде, как она используется в данном документе, выражение "по существу кристаллический" включает более чем на 20%, предпочтительно, более чем на 30%, и более предпочтительно, более чем на 40% (например, более чем любое из 50, 60, 70, 80 или 90%) кристаллический.
Термин "стабильность", в том виде, как он определен в данном документе, включает химическую стабильность и стабильность твердого состояния, где выражение "химическая стабильность" включает возможность хранения солей изобретения в изолированной форме или в форме состава, в которой она присутствует в смеси с фармацевтически приемлемыми носителями, разбавителями, инертными наполнителями или адъювантами, так что в пероральной лекарственной форме, такой, как таблетка, капсула или им подобная при нормальных условиях хранения с незначительной степенью химического распада или разложения, и выражение "стабильность твердого состояния" включает возможность хранения солей изобретения в изолированной форме или в форме состава, в котором она присутствует, в смеси с фармацевтически приемлемыми носителями, разбавителями, инертными наполнителями или адъювантами, так что в пероральной лекарственной форме, такой как таблетка, капсула или им подобная, при нормальных условиях хранения с незначительной степенью трансформации твердого состояния, такой как кристаллизация, перекристаллизация, фазовое превращение в твердом состоянии, гидратация, дегидратация, сольватация или десольватация.
Примеры "нормальных условий хранения" включают одну или более из температур от -80°С до 50°С, предпочтительно, от 0°С до 40°С, и, более предпочтительно, комнатная температура, такая как 15-30°С, давление 0,1-2 бар, предпочтительно атмосферное давление, относительную влажность 5-95%, предпочтительно 10-60%, и воздействие 460 люкс или меньше УФ/видимого света, в течение продолжительных периодов времени, таких как более или равных шести месяцам. При таких условиях можно установить, что соли изобретения являются менее чем на 5%, более предпочтительно, менее чем на 2%, и, особенно, менее чем на 1% химически распавшимися или трансформировавшими твердое состояние, чем это является приемлемым. Специалист в данной области техники в полной мере поймет, что вышеуказанные верхние и нижние пределы для температуры, давления и относительной влажности представляют крайние значения нормальных условий хранения и что определенные комбинации этих крайних значений не будут наблюдаться при нормальном хранении (например, температура в 50°С и давление в 0,1 бар).
Соединения
Один вариант осуществления настоящего изобретения включает (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин (формула I) или его фармацевтически приемлемую соль.
В одном варианте осуществления соединение формулы I или его фармацевтически приемлемая соль являются по существу чистыми. В одном варианте осуществления соединение формулы I или его фармацевтически приемлемая соль являются по существу свободными от (S)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. В одном варианте осуществления соединение формулы I или его фармацевтически приемлемая соль присутствуют в количестве примерно 75% масс. по сравнению с (S)-5-((E)-2-(пирролидин-3-илвинил)пиримидином, предпочтительно, более чем 85% масс., более предпочтительно, более чем 95% масс., более предпочтительно, более чем 98% масс. и, наиболее предпочтительно, 99% масс. или более.
Один вариант осуществления настоящего изобретения включает способ получения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли, содержащих менее чем 25%, предпочтительно, менее чем 15%, более предпочтительно, менее чем 5%, даже более предпочтительно, менее чем 2%, и, наиболее предпочтительно, менее чем 1% масс. (S)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другой вариант осуществления настоящего изобретения включает способ получения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли, содержащих менее чем 25%, предпочтительно, менее чем 15%, более предпочтительно, менее чем 5%, даже более предпочтительно, менее чем 2%, и, наиболее предпочтительно, менее чем 1% масс. (S)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, без использования стадии хирального хроматографического разделения.
Один вариант осуществления настоящего изобретения включает способ получения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли, содержащих менее чем 25%, предпочтительно, менее чем 15%, более предпочтительно, менее чем 5%, даже более предпочтительно, менее чем 2%, и, наиболее предпочтительно, менее чем 1% масс. (S)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другой вариант осуществления настоящего изобретения включает способ получения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли, содержащих менее чем 25%, предпочтительно, менее чем 15%, более предпочтительно, менее чем 5%, даже более предпочтительно, менее чем 2%, и, наиболее предпочтительно, менее чем 1% масс. (S)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, без использования стадии хирального хроматографического разделения. Таким образом, в одном варианте осуществления настоящего изобретения создан способ получения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина повышенной чистоты без зависимости от хроматографического разделения. Один вариант осуществления настоящего изобретения включает способ получения соединения настоящего изобретения в промышленном масштабе, а именно, где способ представляет собой полностью аттестованное по cGMP получение активного фармацевтического ингредиента (API) в промышленном масштабе, со ссылкой на части 210 и 211 CFR, включенные в данный документ путем ссылки.
Один вариант осуществления настоящего изобретения включает использование (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли в изготовлении лекарственного средства.
Один вариант осуществления настоящего изобретения включает способ лечения или предотвращения ряда расстройств и нарушений функций, включающий введение требующему такого лечения млекопитающему терапевтически эффективного количества (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли. Более конкретно, расстройство или нарушение функции можно выбрать из группы, состоящей из расстройств ЦНС, воспаления, воспалительного ответа, связанного с бактериальной и/или вирусной инфекцией, боли, метаболического синдрома, аутоиммунных расстройств или других расстройств, описанных в данном документе более подробно. Другой вариант осуществления настоящего изобретения включает соединения, которые полезны в качестве диагностических средств и в исследованиях связывания с рецептором, как описано в данном документе.
Один вариант осуществления настоящего изобретения включает фармацевтическую композицию, включающую терапевтически эффективное количество (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли, и один или более фармацевтически приемлемых носителей. Один вариант осуществления настоящего изобретения включает применение фармацевтической композиции настоящего изобретения в изготовлении лекарственного средства для лечения расстройств и нарушений функций центральной нервной системы. Другой вариант осуществления настоящего изобретения включает (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин или его фармацевтически приемлемую соль по отношению к любому из примеров. Другой вариант осуществления настоящего изобретения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин или его фармацевтически приемлемую соль для использования в качестве активного терапевтического вещества. Другой вариант осуществления настоящего изобретения включает (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин или его фармацевтически приемлемую соль для использования для модуляции NNR у субъекта, которому она требуется. Другой вариант осуществления настоящего изобретения включает (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин или его фармацевтически приемлемую соль для использования при лечении или предотвращении заболеваний или расстройств, управляемых NNR. Другой вариант осуществления настоящего изобретения включает использование (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли в изготовлении лекарственного средства для использования в модуляции NNR у субъекта, которому она требуется. Другой вариант осуществления настоящего изобретения включает использование (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли для использования в изготовлении лекарственного средства для использования при лечении или предотвращении заболеваний или расстройств, управляемых NNR. Другой вариант осуществления настоящего изобретения включает способ модуляции NNR у субъекта, которому она требуется, при помощи введения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина или его фармацевтически приемлемой соли.
Если не указано иное, структуры, изображенные в данном документе, также имеют целью включить соединения, которые отличаются только присутствием одного или более изотопно обогащенных атомов. Например, соединения, имеющие настоящую структуру, за исключением замены атома водорода на дейтерий или тритий, или замены атома углерода на 13С или 14С, или замены атома азота на 15N, или замены атома кислорода на 17О или 18О, находятся в рамках изобретения. Такие изотопно меченные соединения являются полезными в качестве исследовательских или диагностических средств.
Как отмечено в данном документе, настоящее изобретение включает конкретные репрезентативные соединения, которые подробно идентифицированы в данном документе. Соединения настоящего изобретения можно получить рядом способов, включая широко известные стандартные синтетические способы. Иллюстративные общие синтетические способы приведены ниже, и затем конкретные соединения изобретения получают в рабочих примерах.
Во всех примерах, описанных ниже, при необходимости используют защитные группы для чувствительных или реакционноспособных групп, в соответствии с общими принципами синтетической химии. Защитными группами управляют в соответствии со стандартными способами органического синтеза (T.W. Green и P.G.M. Wuts, Protecting Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, New York (1999)). Эти группы удаляют на удобной стадии синтеза соединения с использованием способов, которые являются вполне очевидными специалистам в данной области техники. Выбор процессов так же, как и условий реакций и порядка их выполнения будет соответствовать получению соединений настоящего изобретения.
В настоящем изобретении также создан способ синтеза соединений, пригодных в качестве промежуточных.
Общие способы синтеза
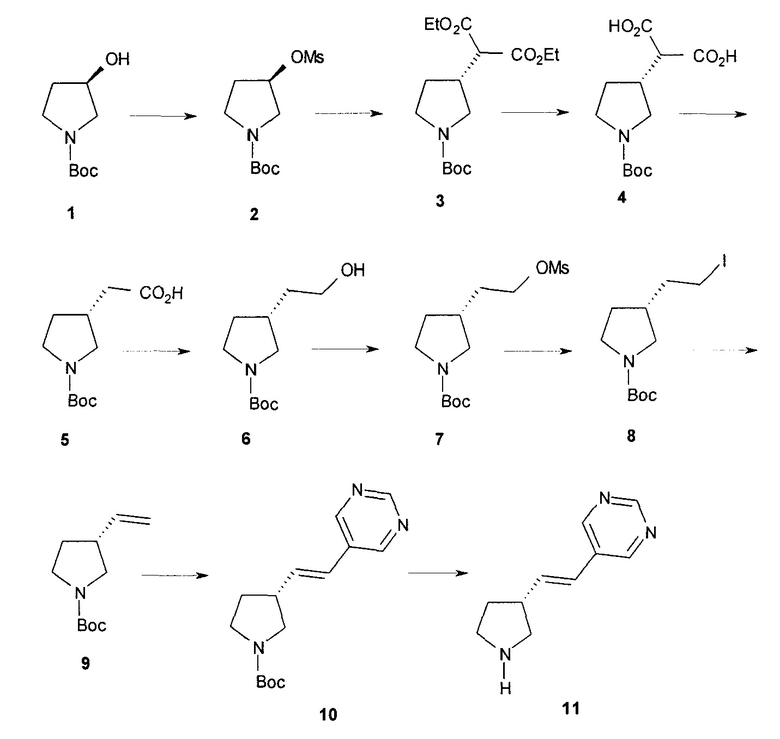
Один аспект настоящего изобретения включает способ стереоспецифического синтеза (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина (11), приведенного на схеме 1. Комерчески доступный трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилат (соединение 1) обрабатывают метансульфонилхлоридом с образованием трет-бутил-(R)-3-(метилсульфонилокси)пирролидин-1-карбоксилата (соединение 2), который затем взаимодействует с диэтилмалонатом и подходящим основанием (например, трет-бутоксидом калия или этоксидом натрия) с образованием диэтил-(R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоната (соединение 3) с обращенной стереохимией у хирального атома углерода.
Подходящие растворители для этих реакций можно выбрать из группы, состоящей из толуола, ксилолов, 1-метил-2-пирролидинона, диметилформамида, диметилацетамида, этанола, трет-бутанола, тетрагидрофурана, 1,2-диметоксиэтана, диоксана и их смесей. В одном варианте осуществления растворитель для образования метансульфонового сложного эфира представляет собой толуол и растворитель для введения малоната представляет собой 1-метил-2-пирролидинон. В другом варианте осуществления растворитель для введения малоната представляет собой этанол. Подходящие основания для этих реакций можно выбрать из группы, состоящей из триэтиламина, диэтилизопропиламина, диизопропилэтиламина, трет-бутоксида калия, металлического натрия, гидрида натрия, этоксида натрия, гидрида калия и гидрида лития. В одном варианте осуществления основание для образования метансульфонового сложного эфира представляет собой триэтиламин, и основание для введения малоната представляет собой трет-бутоксид калия. В другом варианте осуществления основание для введения малоната представляет собой этоксид натрия.
Гидролиз диэфира 3 водным гидроксидом калия приводит к образованию (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (соединение 4), которое декарбоксилируют с получением (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (соединение 5). Подходящие растворители для этих реакций можно выбрать из группы, состоящей из воды, этанола, тетрагидрофурана, диметилформамида, диметилацетамида, 1,2-диметоксиэтана, диоксана, 1-метил-2-пирролидинона, толуола, диметилсульфоксида и их смесей. В одном варианте осуществления растворитель для гидролиза сложного эфира представляет собой водный тетрагидрофуран, и растворитель для декарбоксилирования представляет собой 1-метил-2-пирролидинон. В другом варианте осуществления растворитель для гидролиза сложного эфира представляет собой этанол, и растворитель для декарбоксилирования представляет собой смесь диметилсульфоксида и толуола. Подходящие основания для реакции гидролиза можно выбрать из группы, состоящей из гидроксида калия, гидроксида натрия, карбоната калия, карбоната натрия, гидроксида бария и карбоната цезия. В одном варианте осуществления основание представляет собой гидроксид калия. Восстановление соединения 5 приводит к образованию трет-бутил-(R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилата (соединение 6), которое можно ввести в реакцию с метансульфонилхлоридом и затем иодидом натрия с образованием трет-бутил-(R)-3-(2-метилсульфонилокси)этил)пирролидин-1-карбоксилата (соединение 7) и трет-бутил-(R)-3-(2-йодэтил)пирролидин-1-карбоксилата (соединение 8), соответственно. Подходящие растворители для реакции восстановления можно выбрать из группы, состоящей из тетрагидрофурана, простого эфира, диоксана, 1,2-диметоксиэтана и их смесей. В одном варианте осуществления растворитель представляет собой тетрагидрофуран. Подходящие восстанавливающие агенты можно выбрать из группы, состоящей из борана, диборана, комплекса боран-тетрагидрофуран, комплекса боран-диметиловый эфир и комплекса боран-диметилсульфид. Подходящие растворители для образования метансульфонового эфира можно выбрать из группы, состоящей из толуола, ксилолов, простого эфира, тетрагидрофурана, 1,2-диметоксиэтана, диоксана и их смесей. В одном варианте осуществления растворитель для образования метансульфонового эфира представляет собой толуол. Подходящие основания для образования метансульфонового эфира можно выбрать из группы, состоящей из триэтиламина, диэтилизопропиламина и дизопропилэтиламина. В одном варианте осуществления основание для образования метансульфонового эфира представляет собой триэтиламин. Подходящие растворители для замещения иодида можно выбрать из группы, состоящей из 1-метил-2-пирролидинона, диметилформамида, диметилацетамида, этанола, трет-бутанола, тетрагидрофурана, 1,2-диметоксиэтана, диоксана, диметилсульфоксида и их смесей. В одном варианте осуществления растворитель для замещения иодида представляет собой 1,2-диметоксиэтан.
Наконец, обработка соединения 8 трет-бутоксидом калия дает соединение 9. Подходящие растворители для этой реакции можно выбрать из группы, состоящей из 1,2-диметоксиэтана, 1-метил-2-пирролидинона, диметилформамида, диметилацетамида, этанола, тетрагидрофурана, диоксана и их смесей. В одном варианте осуществления растворитель представляет собой 1,2-диметоксиэтан. Подходящие основания для этой реакции можно выбрать из группы, состоящей из трет-бутоксида калия, этоксида натрия и диазабициклоундекана. В другом варианте осуществления основание представляет собой трет-бутоксид калия.
Катализируемое палладием сочетание соединения 9 с 5-бромпиримидином приводит к образованию (R)-1-(трет-бутоксикарбонил)-5-((E)-2-(пирролидин-3-илвинил)пиримидина (10), с которого снимают защитную группу на конечной стадии для получения (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина (11). Подходящие растворители для катализируемой палладием реакции сочетания можно выбрать из группы, состоящей из 1-метил-2-пирролидинона, диметилформамида, диметилацетамида и ацетонитрила. В одном варианте осуществления растворитель представляет собой диметилацетамид. Подходящие основания для катализируемой палладием реакции сочетания можно выбрать из группы, состоящей из триэтиламина, диэтилизопропиламина, диизопропилэтиламина и ацетата натрия. В одном варианте осуществления основание представляет собой ацетат натрия. Подходящие фосфиновые лиганды для катализируемой палладием реакции сочетания можно выбрать из группы, состоящей из три-н-бутилфосфина, три-трет-бутилфосфина, трициклогексилфосфина, трифенилфосфина, три-о-толилфосфина и 1,1ʹ-бис(дифенилфосфино)ферроцена. В одном варианте осуществления фосфиновый лиганд представляет собой 1,1ʹ-бис(дифенилфосфино)ферроцен. Подходящие палладиевые катализаторы для катализируемой палладием реакции сочетания можно выбрать из группы, состоящей из ацетата палладия, хлорида палладия и дипалладий-трис(дибензилацетона). В одном варианте осуществления палладиевый катализатор представляет собой ацетат палладия. Подходящие растворители для реакции снятия защитной группы можно выбрать из группы, состоящей из воды, дихлорметана, хлороформа и дихлорэтана. В одном варианте осуществления растворитель представляет собой воду. Подходящие кислоты для реакции снятия защитной группы можно выбрать из группы, состоящей из трифторуксусной кислоты, хлороводородной кислоты и серной кислоты. В одном варианте осуществления кислота представляет собой хлороводородную кислоту.
Специалисты в данной области техники - органического синтеза легко поймут, что существуют многочисленные способы получения соединений настоящего изобретения, которые являются меченными радиоактивным изотопом, подходящим для различных диагностических целей. Например, сочетание меченого 11С 5-бромпиримидина с соединением 9 или с последующим удалением защитных групп, как описано, приведет к получению соединения, подходящего для использования в позитронно-эмиссионной томографии.
Схема 1
Солевые формы
Один аспект настоящего изобретения относится к новым солевым формам (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин в форме свободного основания представляет собой вязкое масло с ограниченной растворимостью в воде. Однако свободное основание будет реагировать и с неорганическими, и с органическими кислотами с образованием определенных солей присоединения кислоты, которые имеют физические свойства, которые являются преимущественными для изготовления фармацевтических композиций, такие как кристалличность, растворимость в воде и стабильность по отношению к химическому распаду. Обычно эти солевые формы представляют собой фармацевтически приемлемые соли.
Один аспект настоящего изобретения включает соли присоединения кислоты (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Кислоту выбирают из хлороводородной, серной, метансульфоновой, малеиновой, фосфорной, 1-гидрокси-2-нафтойной, кетоглутаровой, малоновой, L-винной, фумаровой, лимонной, L-яблочной, гиппуровой, L-молочной, бензойной, янтарной, адипиновой, уксусной, никотиновой, пропионовой, оротовой, 4-гидроксибензойной, ди-(п-толуоил)-D-винной, ди-п-анизоил-D-винной, дибензоил-D-винной, 10-камфорсульфоновой, камфорной кислоты и фенцифоса. Настоящее изобретение также включает гидраты и сольваты этих солевых форм.
Стехиометрический состав солей, составляющих настоящее изобретение, может варьироваться. Например, является обычным, что молярное соотношение кислоты и (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина составляет 1:2 или 1:1, но и другие соотношения, такие как 3:1, 1:3, 2:3, 3:2 и 2:1 являются возможными. В зависимости от способа, каким получены соли, описанные в данном документе, соли могут иметь кристаллические структуры, включающие растворители, которые присутствуют во время образования соли. Таким образом, соли могут существовать в виде гидратов и других сольватов с различным стехиометрическим составом растворителя относительно (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
В одном варианте осуществления настоящего изобретения соль имеет стехиометрический состав кислоты по отношению к (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидину 1:2. В другом варианте осуществления соль имеет стехиометрический состав кислоты по отношению к (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидину 1:1.
Другой вариант осуществления настоящего изобретения включает моноцитрат (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, или его гидрат, или сольват. Другой вариант осуществления настоящего изобретения включает монооротат (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, или его гидрат или сольват. Другой вариант осуществления настоящего изобретения включает мономалеат (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, или его гидрат, или сольват.
Дальнейший аспект настоящего изобретения включает способы получения солей. Точные условия, при которых образуются соли, можно установить эмпирически. Соли можно получить путем кристаллизации в контролируемых условиях.
Способ получения солевых форм может варьироваться. Получение солевых форм (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина обычно включает
(i) смешивание свободного основания или раствора свободного основания (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина требуемой степени чистоты в подходящем растворителе, с любой из кислот в чистом виде или в виде раствора любой из кислот в подходящем растворителе, обычно 0,5-1 эквивалентов кислоты,
(ii) (a) охлаждение получившегося раствора соли, если необходимо, для вызывания выпадения осадка, или
(ii) (b) прибавление подходящего антирастворителя для вызывания выпадения осадка, или
(ii) (c) испарение первого растворителя и добавление нового растворителя и повторение либо стадий (ii) (a) или стадии (ii) (b) и
(iii) фильтрование для выделения соли и необязательную перекристаллизацию.
Используемые стехиометрический состав, смесь растворителей, концентрация растворенного вещества и температура могут варьироваться. Репрезентативные растворители, которые можно использовать, чтобы получить или перекристаллизовать солевые формы, включают, без ограничения, этанол, метанол, изопропиловый спирт, изопропилацетат, ацетон, этилацетат, толуол, воду, метилэтилкетон, метилизобутилкетон, трет-бутилметиловый эфир, тетрагидрофуран, дихлорметан, н-гептан и ацетонитрил.
Один вариант осуществления настоящего изобретения включает соли хлороводородной кислоты, серной кислоты, метансульфоновой кислоты, малеиновой кислоты, фосфорной кислоты, 1-гидрокси-2-нафтойной кислоты, кетоглутаровой кислоты, малоновой кислоты, L-винной кислоты, фумаровой кислоты, лимонной кислоты, L-яблочной кислоты, гиппуровой кислоты, L-молочной кислоты, бензойной кислоты, янтарной кислоты, адипиновой кислоты, уксусной кислоты, никотиновой кислоты, пропионовой кислоты, оротовой кислоты, 4-гидроксибензойной кислоты, ди-(п-толуоил)-D-винной кислоты, ди-п-анизоил-D-винной кислоты, дибензоил-D-винной кислоты, 10-камфорсульфоновой кислоты, камфорной кислоты и солей фенцифоса с (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидином в по существу кристаллической форме.
Степень (%) кристалличности может быть определена специалистом в данной области техники с использованием рентгеновской порошковой дифрактометрии (XRPD). Можно также использовать другие методы, такие как спектроскопию твердофазного ЯМР, FT-IR, рамановскую спектроскопию, дифференциальную сканирующую калориметрию (DSC) и микрокалориметрию. Для соединений настоящего изобретения было обнаружено, что является возможным получить соли в формах, которые являются кристаллическими более чем на 80%.
Некоторые из этих кристаллических форм проявляли стабильность, достаточную для установления их перспективности в изготовлении фармацевтических составов. Такую стабильность можно продемонстрировать различными способами. Склонность захватывать и отдавать атмосферную влагу можно оценить при помощи динамической сорбции пара (DVS). Стабильность по отношению к повышенным температурам и влажности можно изучить путем хранения твердых солей при 40°С/75%RH в течение вплоть до восьми дней, и затем заново изучить каждую по массе, виду под микроскопом и XRPD.
Полиморфы
Соединения настоящего изобретения могут кристаллизоваться более чем в одной форме; эта характеристика известна как полиморфизм, и такие полиморфные формы ("полиморфы") подпадают под объем притязаний настоящего изобретения. Полиморфизм, в общем, может возникать в качестве ответа на изменения в температуре, давлении, или и того, и другого. Полиморфизм также может быть вызван изменениями в процессе кристаллизации. Полиморфы можно различить по различным физическим характеристикам, известным из уровня техники, таким как диаграммы XRPD (дифрактограммы), растворимость в различных растворителях и точка плавления.
Настоящее изобретение включает различные полиморфные формы солевых форм (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, включая гидраты и сольваты солей. Такие полиморфные формы характеризуются по их диаграммам рентгеновской порошковой дифрактометрии (XRPD) (дифрактограммам).
Один вариант осуществления настоящего изобретения включает кристаллическую форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другой вариант осуществления настоящего изобретения включает аморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина. Другой вариант осуществления настоящего изобретения включает аморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, чья диаграмма XRPD по существу соответствует приведенной на фиг. 1.
Один вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму I, характеризуемую диаграммой XRPD, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму I, чья диаграмма XRPD по существу соответствует приведенной на фиг. 2.
Один вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму II, характеризуемую диаграммой порошковой рентгеновской дифрактометрии, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму II, чья диаграмма XRPD по существу соответствует приведенной на фиг. 3.
Один вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму III, характеризуемую диаграммой XRPD, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму III, чья диаграмма XRPD по существу соответствует приведенной на фиг. 4.
Один вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму IV, характеризуемую диаграммой XRPD, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму моноцитрата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму IV, чья диаграмма XRPD по существу соответствует приведенной на фиг. 5.
Один вариант осуществления настоящего изобретения включает кристаллическую форму монооротата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Один вариант осуществления настоящего изобретения включает полиморфную форму монооротата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму I, характеризуемую диаграммой XRPD, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму монооротата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму I, чья диаграмма XRPD по существу соответствует приведенной на фиг. 6.
Один вариант осуществления настоящего изобретения включает кристаллическую форму мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина.
Один вариант осуществления настоящего изобретения включает полиморфную форму мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму I, характеризуемую диаграммой XRPD, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму I, чья диаграмма XRPD по существу соответствует приведенной на фиг. 7.
Один вариант осуществления настоящего изобретения включает полиморфную форму мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму II, характеризуемую диаграммой XRPD, включающей, по меньшей мере, один из следующих пиков:
Другой вариант осуществления настоящего изобретения включает полиморфную форму мономалеата (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина, форму II, чья диаграмма XRPD по существу соответствует приведенной на фиг. 8.
Как указано, солевые формы (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина могут существовать в качестве сольватированных, например гидратированных, так же, как и в виде несольватированных форм. Настоящее изобретение охватывает все такие формы.
Настоящее изобретение также включает изотопно меченные соединения, в которых один или более атомов заменены на атом, имеющий атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые можно включить в соединения изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O и 17O. Такие изотопно меченные соединения являются полезными в качестве исследовательских или диагностических средств.
Фармацевтические композиции
Хотя является возможным ввести соединение настоящего изобретения в форме активного химического вещества самого по себе, является предпочтительным вводить соединение в форме фармацевтической композиции или состава. Таким образом, один аспект настоящего изобретения включает фармацевтические композиции, включающие соединение настоящего изобретения и один или более из фармацевтически приемлемых носителей, разбавителей или инертных наполнителей. Другой аспект настоящего изобретения предоставляет процесс получения фармацевтической композиции, включающей смешивание соединения настоящего изобретения с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Способ, которым соединение настоящего изобретения вводят, может варьироваться. Соединение настоящего изобретения, предпочтительно, вводят перорально. Предпочтительные фармацевтические композиции для перорального введения включают таблетки, капсулы, таблетки в форме капсул, сиропы, растворы и суспензии. Фармацевтические композиции настоящего изобретения можно создать в виде лекарственных форм замедленного высвобождения, таких как составы таблеток и капсул с задержкой высвобождения.
Фармацевтические композиции также можно ввести путем инъекции, а именно внутривенно, внутримышечно, подкожно, внутриперитонально, внутриартериально, подоболочечно и интрацеребровентрикулярно. Внутривенное введение является предпочтительным способом инъекции. Подходящие носители для инъекции являются хорошо известными специалистам в данной области техники и включают 5% растворы декстрозы, физиологический раствор и буферизованный фосфатами физиологический раствор.
Составы также можно ввести с использованием других способов, например, ректального введения. Составы, пригодные для ректального введения, такие как суппозитории, являются хорошо известными специалистам в данной области техники. Соединения также можно ввести путем ингаляции, например, в виде аэрозоля; местным образом, таким как в виде лосьона; трансдермально, так как, например, с использованием трансдермального пластыря (например, путем использования технологии, которая является коммерчески доступной от Novartis и Alza Corporation), путем порошковой инъекции, или путем ротовой, подъязычной или внутриносовой абсорбции.
Фармацевтические композиции могут быть составлены в виде дозированной лекарственной формы или в виде многократных или субъединичных форм.
Введение фармацевтических композиций, описанных в данном документе, может быть прерывистым, или с постепенной, непрерывной, постоянной или контролируемой скоростью. Фармацевтические композиции можно ввести теплокровному животному, например, млекопитающему, такому как мышь, крыса, кошка, кролик, собака, свинья, корова или обезьяна; но, преимущественным образом, их вводят человеку. Дополнительно, время суток и число раз в день, когда фармацевтическую композицию вводят, могут варьироваться.
Соединение настоящего изобретения можно использовать в лечении различных расстройств и заболеваний само по себе, можно использовать в комбинации с различными другими пригодными терапевтическими средствами, пригодными в лечении или профилактике этих расстройств или заболеваний. Так, один вариант осуществления настоящего изобретения включает введение соединения настоящего изобретения в комбинации с другими терапевтическими соединениями. Например, соединение настоящего изобретения можно использовать в комбинации с другими лигандами NNR (такими как варениклин), антиоксидантами (такими как средства-ловушки свободных радикалов), антибактериальными средствами (такими как пенициллиновые антибиотики), противовирусными средствами (такими как нуклеозидные аналоги, типа зидовудина и ацикловира), антикоагулянтами (такими как варфарин), противовоспалительными средствами (NSAID, нестероидные противовоспалительные средства), антипиретиками, анальгетиками, анестетиками (такими как используемые в хирургии), ингибиторами ацетилхолинэстеразы (такими как донепезил и галантамин), антипсихотическими средствами (такими как галоперидол, клозапин, оланзапин и кветиапин), иммунодепрессантами (такими как циклоспорин и метотрексат), нейрозащитными средствами, стероидами (такими как стероидные гормоны), кортикостероидами (такими как дексаметазон, предизон и гидрокортизон), витаминами, минералами, нутрицевтиками, антидепрессантами (такими как имипрамин, флуоксетин, пароксетин, эсциталопрам, сертралин, венлафаксин и дулоксетин), анксиолитиками (такими как алпразолам и буспирон), антиконвульсантами (такими как фенитоин и габапентин), вазодилятаторами (такими как празосин и силденафил), стабилизаторами настроения (такими как валпроат и арипиразол), противораковыми лекарственными средствами (такими как антипролиферативные средства), антигипертензивными средствами (такими как атенолол, клонидин, амлопидин, верапамил и олмесартан), слабительными, размягчителями стула, диуретиками (такими как фуросемид), противоспазменными средствами (такими как дицикломин), противодискинетическими и противоязвенными медикаментами (такими как эзомепразол). Такую комбинацию фармацевтически активных средств можно ввести вместе или по отдельности и, при введении по отдельности, введение может происходить одновременно или последовательно, в любом порядке. Количества соединений или средств и относительные периоды времени введения будут выбираться с целью достигнуть желаемого терапевтического эффекта. Введение соединения настоящего изобретения в комбинации с другими лечебными средствами может состоять в комбинации путем введения параллельно в (1) общей фармацевтической композиции, включающей оба соединения, или (2) отдельных фармацевтических композиций, где каждая включает одно из соединений. Альтернативным образом, комбинацию можно ввести по отдельности последовательным способом, в котором одно лечебное средство вводят первым, а другое вторым. Подобное последовательное введение может быть близким по времени или отдаленным во времени.
Другой аспект настоящего изобретения включает комбинированную терапию, включающую введение субъекту терапевтически или профилактически эффективного количества соединения настоящего изобретения и проведение одного или более других видов терапии, включающих химиотерапию, радиационную терапию, генную терапию или иммунотерапию.
Способ лечения
(R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин, его фармацевтически приемлемую соль или содержащую их фармацевтическую композицию, можно использовать для предотвращения или лечения различных заболеваний или расстройств, для которых были предложены или были показаны как пригодные в качестве терапевтически активных другие типы никотиновых соединений, таких как расстройства ЦНС, воспаление, воспалительный ответ, связанный с бактериальной и/или вирусной инфекцией, боль, метаболический синдром, аутоиммунные расстройства или другие расстройства, описанные в данном документе более подробно. Это соединение можно также использовать в качестве диагностического средства в исследованиях по связыванию с рецепторами (in vitro и in vivo). Такие терапевтические и другие идеи описаны, например, в ссылках, ранее перечисленных в данном документе, включая Drug News Perspec. 7(4): 205 (1994), Arneric et al., CNS Drug Rev. 1(1): 1-26 (1995), Arneric et al., Exp. Opin. Invest Drugs 5(1): 79-100 (1996), Bencherif et al., J. Pharmacol. Exp. Ther. 279: 1413 (1996), Lippiello et al., J. Pharmacol. Exp. Ther. 279: 1422 (1996), Damaj et al., J. Pharmacol. Exp. Ther. 291: 390 (1999); Chiari et al., Anesthesiology 91: 1447 (1999), Lavand'homme and Eisenbach, Anesthesiology 91: 1455 (1999), Holladay et al., J. Med. Chem. 40(28): 4169-94 (1997), Bannon et al., Science 279: 77 (1998), PCT WO 94/08992, PCT WO 96/31475, PCT WO 96/40682, и патенты США № 5583140, Bencherif et al., 5597919, Dull et al., 5604231, Smith et al. и 5852041, Cosford et al.
Расстройства ЦНС
(R)-5-((E)-2-(пирролидин-3-илвинил)пиримидин, его фармацевтически приемлемая соль или содержащие их фармацевтические композиции, являются пригодными в лечении или предотвращении различных заболеваний ЦНС, включая нейродегенеративные расстройства, нейропсихиатрические расстройства, нейрологические расстройства и зависимости. Соединения и их фармацевтические композиции можно использовать для лечения или предотвращения когнитивных нарушений и нарушений функций, связанных с возрастом, и других; расстройств внимания и помешательств, включая обусловленные инфекционными средствами или метаболическими нарушениями; для создания нейропротективного действия; для лечения конвульсий и многочисленных церебральных инфарктов; для лечения расстройств настроения, компульсивностей и поведения, связанного с зависимостями; для создания обезболивания; для контроля воспаления, такого как управляемого цитокинами и ядерным фактором каппа-В; для лечения воспалительных расстройств; для создания облегчения боли; и для лечения инфекций, в качестве противоинфекционных средств для лечения бактериальных, грибковых и вирусных инфекций. Среди расстройств, болезней и заболеваний, для лечения или предотвращения которых можно использовать фармацевтические композиции настоящего изобретения, находятся возрастное нарушение памяти (AAMI), умеренные когнитивные нарушения (MCI), возрастное снижение когнитивных способностей (ARCD), предсенильное слабоумие, ранняя стадия болезни Альцгеймера, сенильное слабоумие, слабоумие альцгеймеровского типа, болезнь Альцгеймера, снижение когнитивных способностей без слабоумия (CIND), болезнь диффузных телец Леви, ВИЧ-деменция, комплекс умственных расстройств при СПИД, сосудистая деменция, синдром Дауна, травма головы, травматическое повреждение мозга (TBI), деменция боксеров, болезнь Крейцфельда-Якоба и прионные заболевания, удар, ишемия, расстройство дефицита внимания, синдром гиперактивности с расстройством внимания, дислексия, шизофрения, шизофреноформное расстройство, шизоаффективное расстройство, снижение когнитивных способностей при шизофрении, когнитивные расстройства при шизофрении, паркинсонизм, включая болезнь Паркинсона, постэнцефалитический паркинсонизм, паркинсонизм-деменция Гаума, лобно-височная деменция паркинсоновского типа (FTDP), болезнь Пика, болезнь Ниманна-Пика, болезнь Хантингтона, хорея Хантингтона, поздняя дискинезия, гиперкинезия, прогрессирующий надъядерный паралич, синдром беспокойных ног, болезнь Крейтцфельда-Якоба, множественный склероз, амиотрофический латеральный склероз (ALS), болезни двигательного нейрона (MND), множественная системная атрофия (MSA), кортико-базальная дегенерация, синдром Гийома-Барре (GBS) и хроническая воспалительная демиелинизирующая полиневропатия (CIDP), эпилепсия, аутосомно-доминантная ночная лобная эпилепсия, мания, беспокойство, депрессия, предменструальная дисфория, панические расстройства, булимия, анорексия, нарколепсия, избыточная дневная сонливость, биполярные расстройства, генерализованное тревожное расстройство, обсессивно-компульсивное расстройство, вспышки ярости, оппозиционно-вызывающее расстройство, синдром Туретта, аутизм, наркотическая и алкогольная зависимость, табачная зависимость и расстройства питания.
Когнитивные нарушения или дисфункции могут быть связаны с психиатрическими расстройствами или заболеваниями, такими как шизофрения, и другими психотическими расстройствами, но без ограничения, шизофрениформным расстройством, шизоаффективным расстройством, бредовым расстройством, кратким психотическим расстройством, индуцированным психотическим расстройством и психотическими расстройствами вследствие соматических заболеваний, деменций и других когнитивных расстройств, включающих (но не ограничивающихся ими) умеренные когнитивные нарушения, предсенильное слабоумие, болезнь Альцгеймера, сенильное слабоумие, слабоумие альцгеймеровского типа, возрастное снижение памяти, деменцию телец Леви, сосудистую деменцию, комплекс умственных расстройств при СПИД, дислексию, паркинсонизм, включая болезнь Паркинсона, когнитивные нарушения и слабоумие при болезни Паркинсона, когнитивные нарушения при множественном склерозе, когнитивные нарушения, вызванные травматическим повреждением мозга, деменции, связанные с другими соматическими заболеваниями, тревожные расстройства, включающие (но не ограничивающиеся ими) паническое расстройство без агорафобии, паническое расстройство с агорафобией, агорафобию без истории панического расстройства, специфическую фобию, социальную фобию, обсессивно-компульсивное расстройство, посттравматическое стрессовое расстройство, острое стрессовое расстройство, генерализованное тревожное расстройство и генерализованное тревожное расстройство вследствие соматического заболевания, расстройства настроения, включающие (но не ограничивающиеся ими) большое депрессивное расстройство, дистимическое расстройство, биполярную депрессию, биполярную манию, биполярное расстройство I, депрессию, связанную с манией, депрессивные или смешанные случаи, биполярное расстройство II, циклотимическое расстройство и расстройства настроения вследствие соматических заболеваний, расстройства сна, включающие (но не ограничивающиеся ими) расстройства диссомнии, первичную инсомнию, первичную гиперсомнию, нарколепсию, парасомниальные расстройства, расстройства с ночными кошмарами, связанное со страхом нарушение сна и лунатизм, умственную отсталость, расстройства обучаемости, расстройства двигательных навыков, коммуникативные расстройства, первазивные расстройства развития, расстройство дефицита внимания и расстройство дизруптивного поведения, дефицитарное расстройство внимания, расстройство дефицита внимания с гиперактивностью, расстройства кормления и питания у младенцев, детей или взрослых, тиковые расстройства, расстройства от удаления, связанные с веществами расстройства, включающие (но не ограничивающиеся ими) зависимость от веществ, злоупотребление веществами, отравление веществами, абстиненцию от веществ, связанные с алкоголем расстройства, связанные с амфетамином или амфетаминоподобными веществами расстройства, связанные с кофеином расстройства, связанные с марихуаной расстройства, связанные с кокаином расстройства, связанные с галлюциногенами расстройства, связанные с ингаляциями расстройства, связанные с никотином расстройства, связанные с опиоидами расстройства, связанные с фенциклидином или фенциклидиноподобными веществами расстройства и связанные с седативными, гипнотическими или транквилизирующими веществами расстройства, расстройства личности, включающие (но не ограничивающиеся ими) обсессивно-компульсивное расстройство личности и расстройства побуждений.
Вышеуказанные заболевания и расстройства обсуждаются более подробно, например, в American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision, Washington, DC, American Psychiatric Association, 2000.
Воспаление
Известно, что нервная система, преимущественно через блуждающий нерв регулирует магнитуду врожденного иммунного ответа путем ингибирования выброса макрофагового фактора некроза опухолей (TNF). Физиологический механизм известен как "холинергический противовоспалительный путь" (см., например, Tracey, "The inflammatory reflex", Nature 420: 853-9(2002)). Избыточное воспаление и фактор некроза опухолей вызывают осложнения и даже смертность в ряде заболеваний. Эти заболевания включают (но не ограничиваются ими) эндотоксемию, ревматоидный артрит, остеоартрит, псориаз, астму, атеросклероз, идиопатический пневмосклероз и воспалительную болезнь кишечника.
Воспалительные состояния, которые можно лечить или предотвратить путем введения соединений, описанных в данном документе, включают (но не ограничиваются ими) хроническое и острое воспаление, псориаз, эндотоксемию, подагру, острую псевдоподагру, острый подагрический артрит, артрит, ревматоидный артрит, остеоартрит, отторжение аллотрансплантата, хроническое отторжение трансплантата, астму, атеросклероз, зависимое от моноядерных фагоцитов поражение легких, идиопатический пневмофиброз, атопический дерматит, хроническое обструктивное заболевание легких, респираторный дистресс-синдром у взрослых, острый грудной синдром в заболевании серповидных клеток, воспалительную болезнь кишечника, беспокоящий кишечный синдром, болезнь Крона, язвенный колит, острый холангит, афтозный стоматит, паучит, гломерулонефрит, волчаночный нефрит, тромбоз и реакцию трансплантат-против-хозяина.
Воспалительный ответ, связанный с бактериальной и/или вирусной инфекцией
Многие бактериальные и/или вирусные инфекции связаны с побочными эффектами, привносимыми образованием токсинов, и естественным ответом организма на бактерии или вирус, и/или на токсины. Как обсуждалось выше, ответ организма на инфекцию часто включает генерацию значительного количества TNF и/или других цитокинов. Сверхэкспрессия этих цитокинов может привести к значительному поражению, такому как септический шок (когда бактерии представляют собой сепсис), эндотоксический шок, уросептическую вирусную пневмонию и синдром токсического шока.
Экспрессия цитокинов управляется NNR и может быть ингибирована путем введения агонистов или частичных агонистов этих рецепторов. Соединения, описанные в данном документе, которые являются агонистами или частичными агонистами этих рецепторов, могут быть, таким образом, использованы для минимизации воспалительного ответа, связанного с бактериальной инфекцией так же, как и с вирусными и грибковыми инфекциями. Примеры таких бактериальных инфекций включают сибирскую язву, ботулизм и сепсис. Некоторые из этих соединений могут также обладать противомикробными свойствами.
Эти соединения также можно использовать в качестве вспомогательной терапии в комбинации с существующими терапиями для борьбы с бактериальными, вирусными и грибковыми инфекциями, такими как антибиотики, противовирусные и противогрибковые препараты. Противоядия также можно использовать для связывания с токсинами, производимыми инфекционными агентами, и сделать возможным для связанных токсинов проходить через организм без генерации воспалительного ответа. Примеры противоядий раскрыты, например, в патенте США № 6310043, выданном Bundle et al. Другие средства, эффективные против бактериальных и других токсинов, и их терапевтическое действие могут сопровождаться совместным введением с соединениями, описанными в данном документе.
Боль
Соединения настоящего изобретения и их фармацевтические композиции являются особенно полезными в лечении и предотвращении боли, включая острую, постоянную и хроническую боль. Типы боли и болезненные состояния, которые можно лечить или предотвращать с использованием соединений и их фармацевтических композиций, включают ноцицептивную боль, нейрологическую боль, невропатическую боль, специфичную для женщин боль, воспалительную боль, фибромиалгию, послеоперационную боль, боль, связанную с соматическим заболеванием (таким, как СПИД или другое расстройство) боль, боль при артрите, расстройство височно-челюстного сустава, жгучую боль, боль при ранении, боль в спине, ишиалгию, боль в стопе, головную боль, боль в животе, боль в мышцах и соединительной ткани, боль в суставе, прорывную боль, боль при раке, соматическую боль, висцеральную боль, синдром хронической усталости, психогенную боль и болевые расстройства.
Невропатические болевые синдромы являются последствием ненормальных изменений, происходящих внутри болевых сигнальных систем и периферийной, и центральной нервной системы. Разнообразное происхождение и симптоматика традиционно делали их особенно трудными для лечения при любом типичном течении. Примеры невропатических болевых синдромов включают связанные с тригеминальной или герпетической невралгией, периферийными невропатиями (диабетическая невропатия, вызванная хемотерапией невропатия), постгерпетическую невралгию, невропатии сдавления (синдром запястного канала), радикулопатию, комплексный региональный болевой синдром, каузалгию, боль в нижней части спины, спонтанную боль (боль без внешнего раздражителя) и деафферентационные синдромы, такие как авульсия плечевого сцепления и повреждение спинного мозга. Гипералгезия (сильная боль, связанная со средней силы раздражителем), аллодиния (боль, связанная с нейтральным раздражителем), парестезии (ощущение онемелости или покалывания в отсутствие внешнего раздражителя) и дизестезия (спонтанные или вызванные неприятные ненормальные ощущения) также обычно характеризуются как типы невропатической боли. Соединения настоящего изобретения и их фармацевтические композиции являются особенно полезными в лечении и предотвращении этих типов невропатической боли и связанных заболеваний.
Другие расстройства
Дополнительно, к лечению расстройств ЦНС, воспаления и боли соединения настоящего изобретения также можно использовать для предотвращения или лечения определенных заболеваний другого рода, болезней и расстройств, в которых NNR играют роль. Примеры включают аутоиммунные расстройства, такие как волчанка, расстройства, связанные с выбросом цитокинов, вторичная кахексия после инфекции (например, как происходит при СПИД, комплексе, связанном со СПИД, и неоплазии), ожирение, пузырчатка, недержание мочи, ретинальные болезни, инфекционные болезни, миастения, синдром Итона-Ламберта, гипертензию, остеопороз, вазоконстрикцию, вазодилатацию, сердечные аритмии, диабет I типа, диабет II типа, булимию, анорексию, диарею, запор и язвы, также, как и указания, приведенные в опубликованной заявке PCT WO 98/25619. Соединения настоящего изобретения также можно вводить для лечения конвульсий, таких как те, которые являются симптоматичными для эпилепсии, и для лечения заболеваний, таких как сифилис и болезнь Крейтцфельда-Якоба.
Диагностические применения
Соединения можно использовать в диагностических композициях, таких как зонды, особенно, когда они являются модифицированными, чтобы включать подходящие метки. Зонды можно использовать, например, для определения относительного количества и/или функции определенных рецепторов, в особенности, рецепторного подтипа α4β2. Для этой цели соединения настоящего изобретения наиболее предпочтительно метят радиоактивной изотопной частью, такой как 11С, 18F, 76Br 123I или 125I.
Введенные соединения можно детектировать с использованием известных способов детектирования, подходящих для использованной метки. Примеры способов детектирования включают позитронно-эмиссионную томографию (PET) и однофотонную эмиссионную компьютерную томографию (SPECT). Радиоактивные метки, описанные выше, являются пригодными в визуализациях PET (например, 11С, 18F или 76Br) и SPECT (например, 123I) с периодами полураспада примерно 20,4 минуты для 11С, примерно 109 минут для 18F, примерно 13 часов для 123I и примерно 16 часов для 76Br. Желательна высокая удельная активность для визуализации выбранных рецепторных подтипов при ненасыщающих концентрациях. Введенные дозы обычно находятся ниже диапазона токсичности и позволяют получить высоко контрастные изображения. От соединений ожидается способность поглощения их в нетоксичных количествах. Определение дозы проводят способом, известным специалисту в данной области техники - радиоизотопной визуализации. См., например, патент США № 5969144, выданный London et al.
Соединения можно вводить с использованием известных способов. См., например, патент США № 5969144, выданный London et al. Соединения можно вводить в составах-композициях, которые включают другие ингредиенты, такие как типы ингредиентов, которые являются пригодными для составления диагностической композиции. Соединения, пригодные в соответствии с осуществлением настоящего изобретения, наиболее предпочтительно используют в форме высокой чистоты. См. патент США № 5853696, выданный Elmach et al.
После того, как соединения ввели субъекту (например, человеку), наличие этого соединения внутри субъекта можно визуализировать и оценить количественно при помощи подходящих способов с целью установления наличия, количеств и функциональности выбранных подтипов NNR. Помимо людей, соединения можно также вводить другим животным, таким как мыши, крысы, собаки и обезьяны. Визуализацию SPECT и PET можно проводить с использованием любого подходящего способа и устройства. См. Villemagne et al., In: Arneric et al., (Eds.) Neuronal Nicotinic receptors: Pharmacology and Therapeutic Opportunities, 235-250 (1998) и патент США № 5853696, выданный Elmach et al.
Меченные радиоактивными метками соединения связываются с высоким сродством с селективными подтипами NNR (например, α4β2) и, предпочтительно, проявляют ничтожное неспецифическое связывание с другими подтипами никотиновых холинергических рецепторов (например, таких рецепторных подтипов, которые связаны с мышцами и ганглием). В связи с этим соединения можно использовать в качестве средств для неинвазивной визуализации подтипов никотиновых холинергических рецепторов внутри тела субъекта, в особенности, внутри мозга для диагностирования, связанного с рядом болезней и расстройств ЦНС.
В одном аспекте диагностические композиции можно использовать в способе для диагностирования болезни у субъекта, такого как пациент-человек. Способ включает введение такому пациенту детектированным образом меченого соединения, как описано в данном документе, и детектирование связывания этого соединения с выбранными подтипами NNR (например, с рецепторным подтипом α4β2). Специалисты в области техники использования диагностических инструментов, таких как PET и SPECT, могут использовать меченные радиоизотопами соединения, описанные в данном документе, для диагностирования широкого ряда заболеваний и расстройств, включая заболевания и расстройства, связанные с нарушением функции центральной и автономной нервных систем. Такие расстройства включают широкий ряд болезней и расстройств ЦНС, включая болезнь Альцгеймера, болезнь Паркинсона и шизофрению. Эти и другие репрезентативные болезни и расстройства, которые можно оценить, включают указанные в патенте США № 5952339, Bencheriff et al.
В другом аспекте диагностические композиции можно использовать в способе для мониторинга селективных никотиновых рецепторных подтипов у субъекта, такого как пациент-человек. Способ включает введение такому пациенту детектированным образом меченного соединения, как описано в данном документе, и детектирование связывания этого соединения с выбранными никотиновыми рецепторными подтипами, а именно, рецепторными подтипами α4β2.
Связывание с рецептором
Соединения настоящего изобретения можно использовать в качестве сравнительных лигандов в анализах на связывание для соединений, которые связываются с подтипами NNR, в особенности, с рецепторными подтипами α4β2. Для этой цели соединения настоящего изобретения предпочтительно метят радиоактивной изотопной частью, такой как 3Н или 14С. Примеры таких анализов на связывание подробно описаны ниже.
Примеры
Следующие примеры приведены для того, чтобы проиллюстрировать настоящее изобретение и не должны толковаться как ограничивающие. В этих примерах все части и процентные доли приведены по массе, если не указано иначе.
Пример 1
Инструментальные и экспериментальные последовательности для характеристик солевых форм (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина
Рентгеновская порошковая дифрактометрия (XRPD)
Диаграммы рентгеновской порошковой дифрактометрии получали на дифрактометре Bruker AXS C2 GADDS с использованием излучения CuKα (40 кВ, 40 мА), автоматического XYZ-столика, лазерного видеомикроскопа для автоматического позиционирования образца и 2-мерного детектора площади HiStar. Рентгеновская оптика состоит из одиночного многослойного зеркала Gubel, соединенного с точечным коллиматором в 0,3 мм. Расходимость пучка (т.е. эффективный размер рентгеновского луча на образце) составляла приблизительно 4 мм. Использовали непрерывный режим сканирования θ-θ, с расстоянием образец-детектор в 20 см, что дает эффективный диапазон θ-θ в 3,2°-29,7°. Обычно образец подвергали воздействию рентгеновского луча в течение 120 секунд. Образцы, исследованные при температуре окружающей среды, изготовили в виде образцов - плоских пластин с использованием порошка, в том виде, как он был получен, без растирания. Приблизительно 1-2 мг образца слегка придавили на стеклянной пластинке для получения плоской поверхности. Образцы, исследованные при условиях, отличающихся от условий окружающей среды, смонтировали на кремниевую пластину с теплопроводящим соединением. Образец затем нагревали до подходящей температуры примерно при 10°С/мин и затем выдерживали изотермически в течение 5 мин прежде, чем начинали сбор данных. Положения пиков приведены в виде °2θ с точностью ±0,1°.
XRD монокристалла (SXD)
Данные получали на дифрактометре Bruker AXS 1K SMART CCD, оборудованном охлаждающим устройством Oxford Cryosystems Cryostream. Структуры расшифровали с использованием программ либо SHELXS, либо SHELXD и уточнили с помощью программы SHELXL как части программного пакета Bruker AXS SHELXL. Если не указано иначе, атомы водорода, связанные с углеродом, располагали геометрически и уточняли при помощи скользящего изотропного параметра смещения. Положение атомов водорода, присоединенных к гетероатому, устанавливали в дифференциальном синтезе Фурье и уточнили свободным образом при помощи изотропного параметра смещения.
Спектрометрия ядерного магнитного резонанса (ЯМР)
Спектры ЯМР получали либо на спектрометре Varian Unity 300 МГц, либо на приборе Bruker 400 МГц, оборудованном автодозатором и управляемом консолью DRX400. Автоматические эксперименты осуществляли с использованием ICONNMR v4.0.4 (build 1), работающим вместе с Topspin v 1.3 (patch level 8) с использованием стандартных загруженных шаблонов Bruker. Для нерутинной спектроскопии данные получали с использованием только Topspin.
Температура плавления
Использовали аппарат Фишера-Джонса для определения температуры плавления с горячим столиком, с установками, соответствующими скорости нагрева примерно в 5°С в минуту.
Дифференциальная сканирующая калориметрия (DSC)
Данные получали на TA Instruments Q1000 или Mettler DSC 823e, оборудованных 50-позиционным автодозатором. Инструмент калибровали для калибровки энергии и температуры с использованием сертифицированного индия. Обычно 0,5-1,5 мг каждого образца в алюминиевой чашке с игольчатым отверстием нагревали при 10°С/мин от 25°С до 175-200°С. Над образцом поддерживали продувку азотом в 30 мл/мин.
Термогравиметрический анализ (TGA)
Данные TGA получали на TA Instruments Q500 TGA, оборудованном 16-позиционным автодозатором, либо на Mettler TGA/SDTA 851e, оборудованном 34-позиционным автодозатором. TA Instruments Q500: Прибор калибровали по температуре с использованием сертифицированного алюмеля. Обычно 5-10 мг каждого образца помещали в заранее тарированный платиновый тигель и алюминиевую DSC-чашку, и нагревали при 10°С/мин от комнатной температуры до 350°С. Над образцом поддерживали продувку азотом в 60 мл/мин. Mettler TGA/SDTA 851e: Прибор калибровали по температуре с использованием сертифицированного индия. Обычно 5-10 мг каждого образца помещали в заранее тарированный алюминиевый тигель и нагревали при 10°С/мин от температуры окружающей среды до 350°С. Над образцом поддерживали продувку азотом в 50 мл/мин.
Микроскопия в поляризованном свете (PLM)
Образцы изучали на микроскопе поляризованного света Leica LM/DM с цифровой видеокамерой для захвата изображений. Маленькое количество каждого образца помещали на стеклянный слайд, опускали в иммерсионное масло и покрывали покровным стеклом, при этом индивидуальные частицы разделяли насколько возможно. Образец рассматривали с подходящим увеличением и в частично поляризованном свете, совмещенном с λ-искусственным цветовым фильтром.
Высокотемпературная микроскопия (HSM)
Высокотемпературную микроскопию проводили с использованием микроскопа поляризованного света Leica LM/DM, комбинированного с высокотемпературным столиком Mettler-Toledo MTFP82HT и цифровой видеокамерой для захвата изображений. Маленькое количество каждого образца помещали на стеклянный слайд, при этом индивидуальные частицы разделяли, насколько возможно. Образец рассматривали с подходящим увеличением и в частично поляризованном свете, совмещенном с λ-искусственным цветовым фильтром, при нагревании в то же время от температуры окружающей среды обычно при 10°С/мин.
Динамическая сорбция пара (DVS)
Изотермы сорбции определяли с использованием анализатора сорбции влаги SMS DVS Intrinsic, управляемого программным обеспечением SMS Analysis suite. Температуру образца поддерживали при 25°С при помощи элементов управления прибора. Влажность контролировали путем смешивания потоков сухого и влажного азота, с общей скоростью потока в 200 мл/мин. Относительную влажность измеряли при помощи калиброванного зонда Rotronic (динамический диапазон 1,0-100% RH), расположенного рядом с образцом. Изменение массы (релаксация массы) образца как функцию от %RH постоянно отслеживали при помощи микровесов (точность ±0,005 мг).
Обычно 5-20 мг образца помещали в тарированную сетчатую корзину из нержавеющей стали при условиях окружающей среды. Образец загружали и выгружали при 40% RH и 25°С (обычные условия окружающей среды). Изотерму сорбции влаги построили так как указано ниже (2 скана, дающих 1 полный цикл). Стандартную изотерму построили при 25°С с интервалами в 10% RH в диапазоне 0-90% RH.
Основные параметры метода DVS
Образцы выделили после завершения изотермы и заново проанализировали при помощи XRPD.
Определение воды по Карлу Фишеру (KF)
Содержание воды в каждом образце определяли на кулонометре Mettler Toledo DL39 с использованием реактива Hydranal Coulomat AG и продувки аргоном. Взвешенные твердые образцы вводили в сосуд на платиновой чашке TGA, которая была присоединена к септе во избежание попадания воды. Приблизительно 10 мг образца использовали на одно титрование, и были сделаны дублирующие определения.
Термодинамическая растворимость в воде по ВЭЖХ
Растворимость в воде определяли путем суспендирования достаточного количества соединения в воде, чтобы получить максимальную конечную концентрацию в ≥10 мг/мл исходной свободной формы соединения. Суспензию приводили в равновесие при 25°С в течение 24 ч и затем измеряли рН. Суспензию затем фильтровали через фильтр из стекловолокна класса С в 96-луночный планшет. Фильтрат затем разбавили в 101 раз. Количественное измерение осуществляли по ВЭЖХ по отношению к стандартному раствору приблизительно в 0,1 мг/мл в ДМСО. Вводили различные объемы растворов стандарта, разбавленного и неразбавленного образца. Растворимость рассчитали с использованием областей пиков, установленных путем интегрирования пика, найденного с тем же временем удерживания, что и основной пик во вводе стандарта. Если на фильтровальной пластине находилось достаточно твердого вещества, то получали данные XRPD.
Параметры метода ВЭЖХ для метода термодинамической растворимости в воде
Анализ проводили на системе серии Agilent HP1100, оборудованной диодной матрицей и программным обеспечением ChemStation vB.02.01-SR1.
Химическая чистота по ВЭЖХ
Анализ чистоты проводили на системе серии Agilent HP1100, оборудованной диодной матрицей и программным обеспечением ChemStation vB.02.01-SR1.
Параметры метода ВЭЖХ для определения химической чистоты
Ионная хроматография
Данные получали с использованием Metrohm 761 Advanced Compact IC (для катионов) и Metrohm 861 Advanced Compact IC (для анионов) с использованием программного обеспечения IC NET v2.3. Образцы приготавливали в виде 1000 м.д. (ppm) растворов в ДМСО. Образцы разбавляли до 100 м.д. ДМСО перед исследованием. Количественный анализ осуществляли путем сравнения со стандартными растворами с известной концентрацией анализируемого иона.
Метод ионной хроматографии для анионов
1,0 мМ гидрокарбонат натрия в воде
Метод ионной хроматографии для канатов
0,75 мМ дипиколиновая кислота в воде
Определение и предсказание рКа
Данные получали на приборе Sirius GlpKa с приставкой D-PAS. Измерения проводили при 25°С в водном растворе при помощи УФ и в метанольно-водных смесях при помощи потенциометрии. Титровальная среда была настроена по ионной силе (ISA) при помощи 0,15 М KCl (водного). Значения, найденные в метанольно-водных смесях скорректировали до 0% сорастворителя при помощи экстраполяции по Ясуда-Шидловскому. Данные уточняли с использованием программного обеспечения Refinement Pro v1.0. Предсказание значений рKа осуществляли с использованием программного обеспечения для предсказания рKа ACD v9.
Определение Log P
Данные получали при помощи потенциометрического титрования на приборе Sirius GlpKa с использованием трех соотношений октанол:вода с настроенной ионной силой (ISA) для генерации значений Log P, Log Pion и Log D. Данные уточняли с использованием программного обеспечения Refinement Pro v1.0. Предсказание значений Log P осуществляли с использованием программного обеспечения ACD v9 и Syracuse KOWWIN v1.67.
Пример 2
Синтез трет-бутил-(R)-3-(метилсульфонилокси)пирролидин-1-карбоксилата (2)
Процедура А: К раствору трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилата (200 г, 1,07 моль) и триэтиламина (167 г, 1,63 моль) в толуоле (700 мл) при температуре от -20 до -30°С по каплям прибавляли метансульфонилхлорид (156 г, 1,36 моль), поддерживая при этом температуру -10 до -20°С. Раствор прогрели до температуры окружающей среды и оставили перемешиваться. Из реакционного раствора каждый час отбирали пробу и анализировали при помощи ВЭЖХ для установления завершения реакции. По завершении реакции суспензию отфильтровали для удаления гидрохлорида триэтиламина. Фильтрат промыли ~600 мл разбавленного водного раствора бикарбоната натрия. Органический слой высушили и сконцентрировали при пониженном давлении и получили 2 в виде вязкого масла (260 г, 92%), которое использовали без дальнейшей очистки.
1Н ЯМР (CDCl3, 400 МГц) δ 5,27 (м, 1Н), 3,44-3,76 (м, 4Н), 3,05 (с, 3Н), 2,26 (м, 1Н), 2,15 (м, 1Н), 1,47 (с, 9Н).
Процедура В: В реактор загрузили трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилат (2,00 кг, 10,7 моль), толуол (8,70 кг) и триэтиламин (1,75 кг, 17,3 моль). Реактор продули азотом в течение 15 мин. Смесь перемешивали и охлаждали до 3°С. Метансульфонилхлорид (1,72 кг, моль) медленно прибавляли (в течение периода в 2 ч) при непрерывном охлаждении на ледяной бане (экзотермическая реакция) (после полного прибавления температура составляла 14°С). Смесь, теперь вязкую от выпавшего гидрохлорида триэтиламина перемешивали 12 ч, по мере того как она нагревалась до 20°С. И анализ по ГХ, и по ТСХ (нингидриновая проба) показали, что исходного материала не осталось. Смесь отфильтровали для удаления гидрохлорида триэтиламина и фильтрат вернули в реактор. Фильтрат затем промыли (2×3 кг) 5% водным бикарбонатом натрия, используя 15-минутное перемешивание и 15-минутное время отстаивания для каждой промывки. Получившийся органический слой высушили над безводным сульфатом натрия и отфильтровали. Летучие вещества удалили из фильтрата при пониженном давлении, сначала при 50°С в течение 4 ч, затем при температуре окружающей среды в течение 10 ч. Остаток весил 3,00 кг (106% выход) и являлся идентичным по данным хроматографического и ЯМР-анализа ранее полученным образцам, за исключением того, что он содержал толуол.
Пример 3
Синтез диэтил-(R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоната (3)
Получение А: К раствору трет-бутоксида калия (187 г, 1,62 моль) в 1-метил-2-пирролидиноне (1,19 л) добавляли диэтилмалонат (268 г, 1,67 моль), поддерживая при этом температуру ниже 35°С. Раствор нагрели до 40°С и перемешивали в течение 20-30 мин. Добавили трет-бутил-(R)-3-(метилсульфонилоксил)пирролидин-1-карбоксилат (112 г, 420 ммоль) и раствор нагрели до 65°С, и перемешивали в течение 6 ч. Из реакционного раствора отбирали пробу каждые 2 ч и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции (10-12 ч) смесь охладили приблизительно до 25°С. К раствору добавили деионизованную воду (250 мл) и рН довели до 3-4 путем прибавления 2н хлороводородной кислоты (650 мл). Получившуюся суспензию отфильтровали и добавили воду (1,2 л) и хлороформ (1,4 л). Раствор тщательно перемешали, отделили хлороформенный слой и упарили при пониженном давлении с образованием желтого масла. Масло растворили в гексанах (2,00 л) и промыли деионизованной водой (2×1,00 л). Органический слой сконцентрировали при пониженном давлении при 50-55°С с образованием бледно-желтого масла (252 г), которое, как свидетельствует анализ по 1Н ЯМР, состоит из 49,1% 3 (123,8 г) вместе с 48,5% диэтилмалоната (122 г) и 2% 1-метил-2-пирролидинона (5 г). Материал использовали далее в следующей стадии без дальнейшей очистки.
1Н ЯМР (CDCl3, 400 МГц) δ 4,20 (кв, 4Н), 3,63 (м, 1Н), 3,48 (м, 1Н), 3,30 (м, 1Н), 3,27 (д, J=10 Гц, 1Н), 3,03 (м, 1Н), 2,80 (м, 1Н), 2,08 (м, 1Н), 1,61 (м, 1Н), 1,45 (с, 9Н), 1,27 (т, 6Н).
Получение В: В реактор, поддерживаемый в атмосфере азота, загрузили этанол крепости 200 (5,50 кг) и 21% масс. этоксида натрия в этаноле (7,00 кг, 21,6 моль). Смесь перемешивали и нагрели до 30°С. В течение периода времени в 20 минут добавили диэтилмалонат (3,50 кг, 21,9 моль). Реакционную смесь затем нагревали до 40°С в течение 1,5 ч. Добавили раствор трет-бутил-(R)-3-(метилсульфонилоксил)пирролидин-1-карбоксилата (3,00 кг из продукта примера 2, процедуры В, 10,7 моль) в этаноле крепости 200 (5,50 кг), и получившуюся смесь нагревали при кипении (78°С) в течение 2 ч. Анализ и по ГХ, и по ТСХ (нингидриновая проба) показал, что исходного материала не осталось. Перемешиваемую смесь затем охладили до 25°С, разбавили водой (2,25 кг) и медленно обрабатывали раствором концентрированной хлороводородной кислоты (1,27 кг, 12,9 моль) в воде (5,44 кг). Эту смесь промыли дважды метил-трет-бутиловым эфиром (MTBE) (14,1 кг и 11,4 кг) с использованием 15 мин перемешивания и 15 минут отстаивания для каждой промывки. Объединенные промывки MTBE высушили над безводным сульфатом натрия (1 кг), отфильтровали и сконцентрировали под вакуумом при 50°С в течение 6 ч. Остаток (красное масло) весил 4,45 кг и состоял на 49% из желаемого продукта по данным анализа по ГХ (62% общий выход из трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилата).
Пример 4
Синтез (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (4)
Процедура А: К раствору продукта из примера 3, процедуры А (232 г), содержащему 123,8 г (380 ммоль) 3 и 121,8 г (760 ммоль) диэтилмалоната в тетрагидрофуране (1,2 л) добавили 21% раствор гидроксида калия (450 г в 0,50 л деионизованной воды), поддерживая температуру при этом ниже 25°С. Реакционную смесь нагрели до 45°С и перемешивали в течение 1 ч. Из реакционного раствора каждый час отбирали пробу и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции (2-3 ч) смесь охладили приблизительно до 25°С. Отделили водный слой и охладили до 5°С. рН довели до 2 путем прибавления 4н хлороводородной кислоты (750 мл), и получившуюся суспензию выдерживали при 5-10°С в течение 30 мин. Смесь отфильтровали и осадок на фильтре промыли гексанами (1 л). Водный фильтрат экстрагировали хлороформом (1 л) и хлороформенный слой отставили. Твердые вещества, полученные на стадии фильтрации, заново растворили в хлороформе путем нагревания до 40°С. Раствор отфильтровали для удаления нерастворившихся неорганических твердых веществ. Хлороформенные слои объединили и сконцентрировали при пониженном давлении при 50-55°С с образованием не совсем белого твердого вещества (15 г). Твердые вещества объединили и растворили в этилацетате (350 мл) с образованием суспензии, которую нагревали при 55-60°С в течение 2 ч. Суспензию отфильтровали горячей и получившийся осадок промыли этилацетатом (2×150 мл) и гексанами (2×250 мл) с образованием 83,0 г (80,1%) 4 в виде твердого белого вещества, которое использовали на следующей стадии без дальнейшей очистки.
1Н ЯМР (d4-CH3OH, 400 МГц) δ 3,60 (м, 1Н), 3,46 (м, 1Н), 3,29-3,32 (м, 2Н), 2,72 (м, 1Н), 2,09 (м, 1Н), 1,70 (м, 1Н), 1,45 (с, 9Н).
Процедура В: Раствор продукта из примера 3, процедуры В (4,35 кг), содержащего 2,13 кг (6,47 моль) 3 в тетрагидрофуране (13,9 кг) добавили к перемешиваемому охлажденному раствору гидроксида калия (1,60 кг, 40,0 моль) в деионизованной воде (2,00 кг) в атмосфере азота, поддерживая при этом температуру ниже 35°С. Реакционную смесь нагрели и поддерживали при 40-45°С в течение 24 ч, через это время анализ по ГХ и ТСХ показал, что реакция завершилась. Смесь охладили до 25°С и промыли МТБЕ (34 кг) перемешивали в течение 15 мин и дали время отстояться в течение 15 мин. Водный слой собрали и охладили до 1°С. Затем медленно добавили смесь концентрированной хлороводородной кислоты (2,61 кг, 26,5 моль) в деионизованной воде (2,18 кг), поддерживая температуру смеси <15°С в течение прибавления и в течение 15 мин после. рН раствора довели до 3,7 путем дальнейшего прибавления хлороводородной кислоты. Твердое вещество выделили при помощи фильтрования, промыли водой (16 кг) и высушили в вакууме при температуре окружающей среды в течение 6 суток. Твердое вещество весило 1,04 кг. Фильтрат охладили до <10°С и поддерживали при этой температуре, пока рН не была понижена добавлением еще большего количества хлороводородной кислоты (использовали 1,6 л 6н; 9,6 моль, окончательный рН=2). Твердое вещество выделили при помощи фильтрования, промыли водой (8 л) и высушили в вакууме при 40°С в течение 3 суток. Твердое вещество весило 0,25 кг. Объединенные порции твердого вещества (1,29 кг, выход 73%) являлись хроматографически идентичными ранее полученным образцам.
Пример 5
Синтез (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (5)
Процедура А: Раствор (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (83 г) в 1-метил-2-пирролидиноне (0,42 л) перемешивали в атмосфере азота при 110-112°С в течение 2 ч. Из реакционного раствора каждый час отбирали пробу и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции раствор охладили до 20-25°С. Раствор смешали с деионизованной водой (1,00 л) и добавили MTBE (1,00 л). Фазы разделили и отделили органический слой. Водную фазу экстрагировали MTBE (1,00 л), затем хлороформом (1,00 л). Органические слои объединили и сконцентрировали при пониженном давлении при 50-55°С с образованием масла. Это масло растворили в MTBE (2,00 л) и дважды промыли 0,6н хлороводородной кислотой (2×1,00 л). Органический слой отделили и сконцентрировали при пониженном давлении при 50-55°С с образованием полутвердого вещества. Полутвердое вещество суспендировали в смеси этилацетат/гексаны 1:4 (100 мл), нагрели до 50°С, выдержали в течение 30 мин, охладили до -10°С и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с образованием масла, которое растворили в MTBE (250 мл) и промыли дважды 0,6н хлороводородной кислотой (2×100 мл). Органический слой сконцентрировали при пониженном давлении с образованием полутвердого вещества, которое суспендировали в смеси этилацетат/гексаны 1:4 (50 мл), нагрели до 50°С, выдержали в течение 30 мин, охладили до -10°С и отфильтровали. Твердые вещества объединили, суспендировали в гексанах (200 мл) и выделили при помощи фильтрования с образованием 54,0 г (77,6%) 5.
1Н ЯМР (CDCl3, 400 МГц) δ 11,0 (ушир.с, 1Н), 3,63 (м, 1Н), 3,45 (м, 1Н), 3,30 (м, 1Н), 2,97 (м, 1Н), 2,58 (м, 1Н), 2,44 (м, 2Н), 2,09 (м, 1Н), 1,59 (м, 1Н), 1,46 (с, 9Н).
Процедура В: Раствор (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (1,04 кг, 3,81 моль) в 1-метил-2-пирролидиноне (6,49 кг) перемешивали в атмосфере азота при 110°С в течение 5 ч, за это время анализ по ТСХ и ВЭЖХ показал, что реакция завершилась. Реакционную смесь охладили до 25°С (4 ч) и смешали с водой (12,8 кг) и MTBE (9,44 кг). Смесь тщательно перемешивали в течение 20 мин и фазам дали время разделиться (10 ч). Органическую фазу отделили и водную фазу смешали с MTBE (9,44 кг), перемешивали в течение 15 мин, и дали время отстояться (45 мин). Органическую фазу отделили и водную фазу смешали с MTBE (9,44 кг), перемешивали в течение 15 мин и дали время отстояться (15 мин). Три органические фазы объединили и промыли три раза 1н хлороводородной кислотой (порции по 8,44 кг) и один раз водой (6,39 кг) с использованием 15 мин перемешивания и 15 мин отстаивания для каждой промывки. Получившийся раствор высушили над безводным сульфатом натрия (2,0 кг) и отфильтровали. Фильтрат сконцентрировали при пониженном давлении при 31°С (2 ч) с образованием твердого вещества. Это твердое вещество нагревали в вакууме в течение 4 ч при 39°С, и в течение 16 ч при 25°С, получив 704 г (81%) 5 (чистота 99,7% по ГХ).
Процедура С (направленный синтез 5 с использованием 2 в качестве исходного материала): перемешиваемую смесь этоксида натрия в этаноле (21 массовый процент, 343 г, 1,05 моль), этанола (безводного, 300 мл) и диэтилмалоната (168 г, 1,05 моль) нагревали при 40°С в течение 1,5 ч. К этой смеси добавили раствор (R)-трет-бутил-3-(метилсульфонилокси)пирролидин-1-карбоксилата (138 г, 0,592 моль) в этаноле (100 мл), и реакционную смесь нагревали при 78°С в течение 8 ч. Охлажденную реакционную смесь разбавили водой (2,0 л) и подкислили до рН=3 при помощи 6М HCl (100 мл). Водную смесь с этанолом экстрагировали толуолом (1,0 л) и органическую фазу сконцентрировали под вакуумом с получением 230 г красного масла. Это красное масло добавили при 85°С к водному раствору гидроксида калия с массовым содержанием 22,5 процента (748 г, 3,01 моль). После того как завершили прибавление, температуру реакции медленно повысили до 102°С, в то время как происходила отгонка этанола. Когда температура реакции достигла 102°С и отгонка ослабла, нагревание продолжали в течение дополнительных 90 мин. Реакционную смесь охладили до температуры окружающей среды и промыли толуолом (2×400 мл). К водному слою добавили 600 мл 6М хлороводородной кислоты, поддерживая при этом внутреннюю температуру ниже 20°С. Это привело к образованию осадка, начавшемуся при рН приблизительно в 4-5. Суспензию отфильтровали и осадок на фильтре промыли 300 мл воды. Твердое вещество высушили под вакуумом с образованием 77 г (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты в виде не совсем белого твердого вещества (выход 54% по отношению к (R)-трет-бутил-3-(метилсульфонилокси)пирролидин-1-карбоксилату).
1Н ЯМР (ДМСО-d6, 400 МГц) δ 3,47 (м, 1Н), 3,32 (м, 1Н), 3,24 (м, 1Н), 3,16 (м, 1Н), 3,92 (м, 1Н), 2,86 (м, 1Н), 1,95 (м, 1Н), 1,59 (м, 1Н), 1,39 (с, 9Н).
Суспензию (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)малоновой кислоты (15 г, 55 ммоль) в толуоле (150 мл) и диметилсульфоксиде (2 мл) нагревали при кипении в течение периода времени в 2 ч. Температуре смеси позволили достичь комнатной и разбавили ее MTBE (150 мл). Органический раствор промыли 10% водной лимонной кислотой (2Ч200 мл) и растворитель удалили под вакуумом с получением 11,6 г (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты в виде не совсем белого твердого вещества (выход 92%).
1Н ЯМР (ДМСО-d6, 400 МГц) δ 12,1 (с, 1Н), 3,36-3,48 (м, 1Н), 3,20-3,34 (м, 1Н), 3,05-3,19 (м, 1Н), 2,72-2,84 (м, 1Н), 2,30-2,42 (м, 1Н), 2,22-2,30 (м, 2Н), 1,85-2,00 (м, 1Н), 1,38-1,54 (м, 1Н), 1,35 (2, 9Н).
Пример 6
Синтез трет-бутил-(R)-3-(2-гидроксиэтил)пирролидин-1-карбоксилата (6)
Процедура А: Раствор (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (49,0 г, 214 ммоль) в тетрагидрофуране (ТГФ) (200 мл) охладили до -10°С. 250 мл (250 ммоль) 1М раствора борана в ТГФ медленно прибавляли в колбу, поддерживая при этом температуру ниже, чем 0°С. Раствор нагрели до температуры окружающей среды и перемешивали в течение 1 ч. Из раствора каждый час отбирали пробу и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции раствор охладили до 0°С и по каплям добавили 10% раствор гидроксида натрия (80 мл) в течение периода времени в 30 мин для контроля выделения газа. Раствор экстрагировали 500 мл раствора гексанов/этилацетата 1:1. Органический слой промыли насыщенным раствором хлорида натрия и высушили при помощи 10 г силикагеля. Силикагель удалили фильтрованием и промыли 100 мл гексанов/этилацетата 1:1. Органические слои объединили и сконцентрировали под вакуумом с получением 6 (42 г, 91,3%) в виде светло-оранжевого масла, которое затвердевало по отстаивании.
1Н ЯМР (CDCl3, 400 МГц) δ 3,67 (м, 2Н), 3,38-3,62 (м, 2Н), 3,25 (м, 1Н), 2,90 (м, 1Н), 2,25 (м, 1Н), 1,98-2,05 (м, 1Н), 1,61-1,69 (м, 2Н), 1,48-1,59 (м, 2Н), 1,46 (с, 9Н).
Процедура В: Комплекс боран-ТГФ (3,90 кг или л 1М в ТГФ, моль) медленно добавили к перемешиваемому раствору (R)-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)уксусной кислоты (683 г, 3,03 моль) в ТГФ (2,5 кг), выдержали под газообразным азотом, и с использованием водяной бани для поддержания температуры между 23 и 28°С. Прибавление заняло 1,75 ч. Перемешивание при 25°С продолжали в течение 1 ч, после чего анализ по ГХ показал завершение реакции. Реакционную смесь охладили до <10°С и поддерживали ниже 25°С по мере того, как медленно прибавляли водный 10% гидроксид натрия (1,22 кг). Прибавление заняло 40 мин. Смесь перемешивали в течение 1 ч при 25єС и затем объединили с 1:1 гептаном/этилацетатом (об./об.). Смесь перемешивали в течение 15 мин и дали время разделиться на фазы (1 ч). Органическую фазу удалили и водную фазу объединили со второй порцией гептана/этилацетата 1:1 в 7 л. Это перемешивали в течение 15 мин и дали время разделиться на фазы (20 мин). Органическую фазу снова удалили и объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (4,16 кг) с использованием 15 минут перемешивания и 1 часа времени отстаивания. Органическую фазу объединили с силикагелем (140 г) и перемешивали 1 ч. Добавили безводный сульфат натрия (700 г) и перемешивали смесь в течение 1,5 ч. Смесь отфильтровали и осадок на фильтре промыли 1:1 гептана/этилацетата (2 л). Фильтрат сконцентрировали под вакуумом при <40°С в течение 6 ч. Получившееся масло весило 670 г (выход 103%), и содержит следы гептана, но по всему остальному является идентичным ранее приготовленным образцам 6 по данным анализа ЯМР.
Пример 7
трет-Бутил-(R)-3-(2-(метилсульфонилокси)этил)пирролидин-1-карбоксилат (7)
Процедура А: К раствору трет-бутил-(R)-3-(2-гидроксиметил)пирролидин-1-карбоксилата (41,0 г, 190 ммоль) добавили триэтиламин (40 мл) в толуоле (380 мл) и охладили до -10°С. Медленно добавили метансульфонилхлорид (20,0 мл, 256 ммоль) так, чтобы поддерживать температуру около -5-0°С. Раствор нагрели до комнатной температуры и перемешивали в течение 1 ч. Из раствора каждый час отбирали пробу и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции раствор отфильтровали и фильтрат промыли 5% раствором бикарбоната натрия (250 мл). Органический слой отделили и промыли насыщенным раствором хлорида натрия (250 мл). Органический слой отделили, высушили над силикагелем (10 г) и сконцентрировали в вакууме с получением 7 (53,0 г, 92,8%) в виде светло-желтого вязкого масла.
1Н ЯМР (CDCl3, 400 МГц) δ 4,26 (т, J=6,8 Гц, 2Н), 3,41-3,63 (м, 2Н), 3,27 (м, 1Н), 3,02 (с, 3Н), 2,92 (м, 1Н), 2,28 (м, 1Н), 2,05 (м, 1Н), 1,83 (м, 2Н), 1,50-1,63 (м, 1Н), 1,46 (с, 9Н).
Процедура В: В атмосфере азота раствор триэтиламина (460 г, 4,55 моль) и трет-бутил-(R)-3-(2-гидроксиметил)пирролидин-1-карбоксилата (весь образец из примера 7, процедуры В, 3,03 моль) в толуоле (5,20 кг) перемешивали и охлаждали до 5°С. Медленно, в течение 1,25 ч добавили метансульфонилхлорид (470 г, 4,10 моль), поддерживая температуру ниже 15°С с использованием охлаждения на ледяной бане. Смесь постепенно нагрели (в течение 1,5 ч) до 35°С, и эту температуру поддерживали в течение 1,25 ч, в этот момент анализ по ГХ показал, что реакция завершилась. Смесь охладили до 25°С, и твердые вещества отфильтровали и осадок на фильтре промыли толуолом (1,28 кг). Фильтрат перемешивали в течение 15 мин с 10% водным бикарбонатом натрия (4,0 кг), и фазам дали время разделиться в течение 30 мин. Органическую фазу затем перемешивали с насыщенным водным хлоридом натрия (3,9 кг) в течение 30 мин, и фазам дали время разделиться в течение 20 мин. Органическую фазу объединили с силикагелем (160 г) и перемешивали в течение 1 ч. Добавили безводный сульфат натрия (540 г) и смесь перемешивали в течение дополнительных 40 мин. Смесь затем отфильтровали и осадок на фильтре промыли толуолом (460 г). Фильтрат сконцентрировали под вакуумом при 50°С в течение 5 ч, и получившееся масло держали под вакуумом при 23°С в течение дополнительных 8 ч. После этого осталось 798 г 7, чистота 93% по данным анализа по ГХ.
Пример 8
Синтез трет-бутил-(R)-3-винилпирролидин-1-карбоксилата (9)
Процедура А: Раствор трет-бутил-(R)-3-((метилсульфонилокси)этил)пирролидин-1-карбоксилата (49,0 г, 167 ммоль), иодида натрия (30,0 г, 200 ммоль) и 1,2-диметоксиэтана (450 мл) перемешивали при 50-60°С в течение 4 ч. Из раствора каждый час отбирали пробу и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции раствор охладили до -10°С и добавили трет-бутоксид калия (32,0 г, 288 ммоль) в виде твердого вещества, поддерживая при этом температуру ниже 0°С. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. Из смеси каждый час отбирали пробу и анализировали по ВЭЖХ для установления завершения реакции. По завершении реакции смесь профильтровали через подушку из диатомовой земли (25 г сухой массы). Осадок промыли 1,2-диметоксиэтаном (100 мл). Объединенные фильтраты сконцентрировали под вакуумом с получением оранжевого масла с суспендированным твердым веществом. Масло растворили в гексанах (400 мл), перемешивали в течение 30 мин и отфильтровали для удаления твердых веществ. Органический слой высушили над силикагелем (10 г) и сконцентрировали в вакууме с получением 9 (26,4 г, 82,9%) в виде бесцветного масла.
1Н ЯМР (CDCl3, 400 МГц) δ 5,77 (м, 1Н), 5,10 (дд, J=1,2 Гц, J=16 Гц, 1Н), 5,03 (дд, J=1,2 Гц, J=8,8 Гц, 1Н), 3,41-3,59 (м, 2Н), 3,29 (м, 1Н), 3,05 (м, 1Н), 2,78 (м, 1Н), 2,01 (м, 1Н), 1,62-1,73 (м, 1Н), 1,46 (м, 9Н).
Процедура В: Раствор трет-бутил-(R)-3-((метилсульфонилокси)этил)пирролидин-1-карбоксилата (792 г из продукта из примера 7, процедуры В, ~2,5 моль), иодида натрия (484 г, 3,27 моль) и 1,2-диметоксиэтана (7,2 л) перемешивали при 55°С в атмосфере азота в течение 4,5 ч, через это время анализ по ГХ показал, что реакция завершилась. Раствор охладили до <10°С, и порциями добавили твердый трет-бутоксид калия (484 г, 4,32 моль) (время прибавления 1,25 ч), поддерживая при этом температуру ниже 15°С. Реакционную смесь перемешивали в течение 1 ч при 5°С, медленно нагрели (6 ч) до 20°С, и перемешивали при 20°С в течение 1 ч. Раствор профильтровали через подушку диатомовой земли (400 г сухой массы). Осадок на фильтре промыли 1,2-диметоксиэтаном (1,6 кг). Объединенные фильтраты сконцентрировали под вакуумом и полутвердый осадок перемешивали с гептаном (6,0 л) в течение 2 ч. Твердые вещества удалили фильтрованием (осадок на фильтре промыли 440 мл гептана), и фильтрат сконцентрировали под вакуумом при 20°С с получением 455 г 9 (чистота 90,7%). Образец этого вещества подвергли фракционной перегонке при 20-23 торр с получением 296 г очищенного 9 (Ткип 130-133°С) (чистота >99% по данным анализа по ГХ).
Пример 9
Синтез (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина (11)
Азот пропускали через раствор (R)-трет-бутил-3-винилпирролидин-1-карбоксилата (25 г, 127 ммоль), 5-бромпиримидина (30,3 г, 190 ммоль), 1,1ʹ-бис(дифенилфосфино)ферроцена (2,11 г, 3,8 ммоль) и ацетата натрия (18,8 г, 229 ммоль) в N,N-диметилацетамиде (250 мл) в течение 1 ч, и добавили ацетат палладия (850 мг, 3,8 ммоль). Реакционную смесь нагревали до 150°С со скоростью 40°С/ч и перемешивали в течение 16 ч. Смесь охладили до 10°С и быстро охладили водой (750 мл), поддерживая при этом внутреннюю температуру ниже 20°С. Добавили MTBE (300 мл), затем диатомовую землю (40 г, сухой массы). Суспензию перемешивали в течение 1 ч при температуре окружающей среды, и профильтровали через подушку диатомовой земли. Осадок промыли MTBE (2×100 мл) и фильтрат перенесли в 2-л сосуд, снабженный верхнеприводной мешалкой, и загрузили туда активированный уголь (40 г). Суспензию перемешивали в течение 2 ч при температуре окружающей среды и профильтровали через диатомовую землю. Осадок промыли MTBE (2×100 мл), и фильтрат сконцентрировали в вакууме с получением 28,6 г оранжевого масла. Масло растворили в MTBE (100 мл) и добавили Si-Thiol® (2,0 г, 1,46 ммоль тиола/г, Silicycle Inc.). Суспензию перемешивали в атмосфере азота при комнатной температуре в течение 3 ч, профильтровали через плотный фильтр и хранили в стеклянной емкости.
К раствору 6M HCl (70 мл) добавили фильтрат в течение периода времени в 30 мин, поддерживая при этом внутреннюю температуру между 20°С и 23°С. Смесь тщательно перемешивали в течение 1 ч и удалили органический слой. Оставшийся водный слой подщелочили 45% масс. КОН (50 мл) и получившуюся суспензию экстрагировали один раз хлороформом (300 мл). Упаривание растворителя в вакууме (температура бани 45°С) привело к получению 16,0 г (71,8%) (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания в виде красного масла, которое немедленно растворили в изопропаноле (50 мл) и использовали для образования соли.
Пример 10
Синтез моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
К раствору лимонной кислоты (17,6 г, 91,6 ммоль) в изопропаноле (250 мл) и воде (25 мл) по каплям добавили раствор (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (16,0 г, 91,2 ммоль) в изопропаноле (50 мл) при 55°С. В получившийся раствор внесли затравку моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы II (200 мг) и перемешивали в течение 15 мин. Суспензию нагревали до 65°С и перемешивали в течение 1 ч, после чего суспензию охладили до 20°С со скоростью -10°С/ч и дали постоять при 20°С в течение 12 ч. Суспензию отфильтровали через крупнопористый стеклянный фильтр, и выделенное твердое вещество промыли изопропанолом (64 мл) и метил-трет-бутиловым эфиром (64 мл). Полученное сыпучее твердое вещество желтовато-коричневого цвета высушили в вакууме при 70°С с получением 17,4 г (36%) моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина (смесь форм II и III) в виде желтовато-коричневого твердого вещества.
1Н ЯМР (D2O, 400 МГц) δ: 8,85 (с, 1Н), 8,70 (с, 1Н), 6,50 (д, J=17 Гц, 1Н), 6,35 (дд, J=7 Гц, J=17 Гц, 1Н), 3,43-3,50 (м, 1Н), 3,34-3,43 (м, 1Н), 3,20-3,30 (м, 1Н), 3,08-3,19 (м, 1Н), 3,00-3,08 (м, 1Н), 2,77 (д, J=16 Гц, 2Н), 2,65 (д, J=16 Гц, 2Н), 2,16-2,26 (м, 1Н), 1,80-1,92 (м, 1Н).
Пример 11
Отбор солей присоединения хлороводородной кислоты (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(R)-5-((E)-2-пирролидин-3-илвинил)пиримидиновое свободное основание растворили в любом из изопропилацетата, тетрагидрофурана, метилизобутилкетона, ацетонитрила или изопропилового спирта. Получившийся раствор обработали 1 экв. HCl, приготовленном в одном из следующих видов: 1 М в диэтиловом эфире, 1 М в воде, 5 М в изопропиловом спирте или 4 М в диоксане. Смесь выдержали при 50°С/температуре окружающей среды (циклы по 4 ч) в течение 24 ч. Там, где эксперимент приводил к образованию твердого вещества, материал анализировали по XRPD.
Пример 12
Отбор солей "моно"-присоединения кислоты к (R)-5-((E)-2-пирролидин-3-илвинил)пиримидину
(R)-5-((E)-2-пирролидин-3-илвинил)пиримидиновое свободное основание (10 мг, 0,057 ммоль) растворили либо в изопропилацетате, либо в ацетонитриле. Растворы обработали 1 экв. соответствующей кислоты (см. ниже), прогрели до 50°С и медленно охладили до комнатной температуры в течение ночи. Растворитель затем упарили под вакуумом без нагревания и осадки проанализировали по XRPD. Твердые вещества затем хранили в камере влажности при 40°С и 75% RH в течение недели и заново анализировали по XRPD.
В тех случаях, где эксперимент не приводил к образованию кристаллического твердого вещества, образцам давали созреть в тетрагидрофуране и изопропиловом спирте, и там, где получали твердое вещество, там твердое вещество анализировали по XRPD и хранили в камере влажности в течение недели для установления стабильности.
Перебирали следующие кислоты с использованием вышеуказанных процедур для получения солей "моно"-присоединения кислоты: хлороводородную кислоту, серную кислоту, метансульфоновую кислоту, малеиновую кислоту, фосфорную кислоту, 1-гидрокси-2-нафтойную кислоту, кетоглутаровую кислоту, малоновую кислоту, L-винную кислоту, фумаровую кислоту, лимонную кислоту, L-яблочную кислоту, гиппуровую кислоту, L-молочную кислоту, бензойную кислоту, янтарную кислоту, адипиновую кислоту, уксусную кислоту, никотиновую кислоту, пропионовую кислоту, оротовую кислоту, 4-гидроксибензойную кислоту и ди-п-толил-D-винную кислоту.
Пример 13
Отбор солей "полу"-присоединения кислоты к (R)-5-((E)-2-пирролидин-3-илвинил)пиримидину
(R)-5-((E)-2-пирролидин-3-илвинил)пиримидиновое свободное основание (10 мг, 0,057 ммоль) растворили либо в изопропилацетате, либо в ацетонитриле. Растворы обработали 0,5 экв. соответствующей кислоты (см. ниже), прогрели до 50°С и медленно охладили до температуры окружающей среды в течение ночи. Растворитель затем упарили под вакуумом без нагревания и осадки проанализировали по XRPD. Твердые вещества затем хранили в камере влажности при 40°С и 75% RH в течение недели и заново анализировали по XRPD.
В тех случаях, когда эксперимент не приводил к образованию кристаллического твердого вещества, образцам давали созреть в тетрагидрофуране и изопропиловом спирте, и там, где получали твердое вещество, его анализировали по XRPD и хранили в камере влажности в течение недели для установления стабильности.
Перебирали следующие кислоты с использованием вышеуказанных процедур для получения солей присоединения "полу"-кислоты: серную кислоту, малеиновую кислоту, фосфорную кислоту, кетоглутаровую кислоту, малоновую кислоту, L-винную кислоту, фумаровую кислоту, лимонную кислоту, L-яблочную кислоту, янтарную кислоту, адипиновую кислоту и ди-п-толуоил-D-винную кислоту.
Пример 14
Общая процедура масштабирования для выбранных солей (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
Ряд солей (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина выбрали для масштабирования вплоть до ~200 мг для получения дальнейших характеристик. Эти солевые формы включают цитрат (моно- и полу-), оротат (моно-), 4-гидроксибензоат (моно-), ди-п-толил-D-тартрат (моно- и полу-), малеат (моно- и полу-) и фумарат (моно- и полу-).
(R)-5-((E)-2-пирролидин-3-илвинил)пиримидиновое свободное основание (189 мг, 1,077 ммоль) растворили в ацетонитриле. Раствор затем обработали 1,1 экв. соответствующей кислоты для получения моносоли и 0,5 экв. для получения полусоли. Смесь прогрели до 50°С и медленно охладили до температуры окружающей среды в течение ночи.
Полученное твердое вещество отфильтровали и высушили с отсасыванием прежде, чем анализировать по XRPD и 1Н ЯМР. Провели эксперименты по TGA для определения содержания воды или других растворителей и провели эксперименты по DSC для установления стабильности выделенных форм и возможностей новых форм для каждой соли. Для оценки гигроскопичности солей использовали эксперименты DVS. Также для каждой соли измерили чистоту и термодинамическую растворимость по ВЭЖХ.
Пример 15
Моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма I
Моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форму I получили в соответствии с процедурой отбора для моносоли, из изопропилацетата путем испарения и созревания в тетрагидрофуране. Альтернативно, моноцитрат формы I получили согласно процедуре отбора для моносоли, из ацетонитрила путем выпаривания и созревания в изопропиловом спирте. Дифрактограмма XRPD моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы I приведена на фиг. 2.
Пример 16
Моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма II
Суспензию моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, смеси форм II и III, в метаноле нагревали до 50°С и перемешивали в течение 1 ч. Суспензию впоследствии охладили до 20°С со скоростью -30°С/ч с последующим немедленным нагреванием снова до 50°С со скоростью +30°С/ч. Нагревание прекратили по достижении 50°С, и суспензию охладили, и перемешивали при температуре окружающей среды в течение 16 ч. Суспензию отфильтровали, и какой-либо остаточный материал в колбе сполоснули дополнительно метанолом. Осадок сушили при 70°С в вакууме в течение 16 ч с получением моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы II. Дифрактограмма XRPD моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы II приведена на фиг.3.
Пример 17
Аморфный моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
Аморфный моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина получили путем лиофильной сушки раствора моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы II в воде. Дифрактограмма XRPD аморфного моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина приведена на фиг. 1.
Пример 18
Моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма III
Моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форму III получили путем оставления аморфного моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина при температуре окружающей среды в течение двух часов. Дифрактограмма XRPD моноцитрата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы III приведена на фиг. 4.
Пример 19
Моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма IV
Моноцитрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форму IV получили путем созревания формы II в ацетоне/метилизобутилкетоне. Дифрактограмма формы IV моноцитрата приведена на фиг. 5.
Пример 20
Моно-(R)-(-)-оротатная соль (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
Оротовую кислоту (0,965 г, 6,18 ммоль) добавили в виде твердого вещества к перемешиваемому, горячему раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,084 г, 6,18 ммоль) в 2-пропаноле (10 мл) в круглодонной колбе. Получившуюся смесь твердых веществ нагревали при кипении с обратным холодильником в течение 5 мин, охладили до комнатной температуры и перемешивали в течение ночи. Светло-бежевый порошок отфильтровали, промыли 2-пропанолом (10,8 мл) и высушили в вакуумной печи (с выпуском воздуха) при 50°С в течение 20 ч с получением 1,872 г (77,9%) от не совсем белого до белого цвета комковатого твердого вещества, Тпл 230-233°С.
1Н ЯМР (D2O): δ 8,80 (с, 1Н), 8,60 (с, 2Н), 6,40 (д, 1Н), 6,25 (дд, 1Н), 5,93 (с, 1Н, =СН оротовой кислоты, указывающий на стехиометрический состав моносоли), 3,38 (дд, 1Н), 3,29 (м, 1Н), 3,17 (м, 1Н), 3,04 (м, 1Н), 2,97 (дд, 1Н), 2,13 (м, 1Н, 1,78 (м, 1Н).
Результаты элементного анализа заставляют сделать вывод о наличии избытка оротовой кислоты и стехиометрического состава 1:1,1 основание:оротовая кислота.
Элементный анализ: вычислено для C10H13N3⋅C5H4N2O4: (C, 54,38%, H, 5,17%, N, 21,14%); найдено: (C, 53,49%, 53,44%; H, 5,04%, 5,10%; N, 20,79%, 20,84%).
Пример 21
Монооротат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма I
(R)-5-((E)-2-пирролидин-3-илвинил)пиримидиновое свободное основание (189 мг, 1,077 ммоль, свежеполученное) растворили в ацетонитриле (5 мл). Раствор затем обработали 1,1 экв. раствора оротовой кислоты (1 М в этаноле) при температуре окружающей среды. Смесь прогрели до 50°С и медленно охладили до комнатной температуры в течение ночи. Полученное твердое вещество отфильтровали и высушили с отсасыванием прежде, чем анализировать по XRPD и 1Н ЯМР. Дифрактограмма XRPD монооротата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы I приведена на фиг. 6.
Пример 22
Мономалеат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма I
(R)-5-((E)-2-пирролидин-3-илвинил)пиримидиновое свободное основание (189 мг, 1,077 ммоль, свежеполученное) растворили в ацетонитриле (5 мл). Раствор затем обработали 1,1 экв. раствора малеиновой кислоты (1 М в этаноле) при температуре окружающей среды. Смесь прогрели до 50°С и медленно охладили до температуры окружающей среды в течение ночи. Полученное твердое вещество отфильтровали и высушили с отсасыванием прежде, чем анализировать по XRPD и 1Н ЯМР. Дифрактограмма XRPD мономалеата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы I приведена на фиг. 7.
Пример 23
Мономалеат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, форма II
Мономалеат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина (форму I) суспендировали в этаноле и выдерживали при 50єС/комнатной температуре 4-часовыми циклами в течение 48 ч. Данные анализа твердого вещества по XRPD показали форму II. Дифрактограмма XRPD мономалеата (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина, формы II приведена на фиг. 8.
Пример 24
Монооксалат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
Щавелевую кислоту (0,516 г, 5,73 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина (1,00 г, 5,70 ммоль) в этаноле (10 мл). Соль выпала в осадок при дальнейшем прогревании раствора. Для облегчения перемешивания смесь разбавили этанолом (6 мл) и комки раздавили шпателем. Смесь охладили до комнатной температуры и оставили стоять в течение ночи. Светло-бежевый порошок отфильтровали, промыли этанолом и высушили в вакуумной печи при 50°С в течение 6 ч с получением 1,40 г (92,3%) кремово-белого пушистого порошка, Тпл 149-151°С.
1Н ЯМР (ДМСО-d6): δ 9,03 (с, 1Н), 8,86 (с, 2Н), 6,56 (м, 2Н), 3,40 (дд, 1Н), 3,31 (м, 1Н), 3,18 (м, 1Н), 3,08 (м, 1Н), 3,08 (м, 1Н), 2,96 (дд, 1Н), 2,15 (м, 1Н), 1,80 (м, 1Н).
13С ЯМР (ДМСО-d6): δ 164,90 (С=О щавелевой кислоты), 156,97, 154,17, 133,66, 130,31, 124,20, 48,70, 44,33, 40,98, 30,42.
Элементный анализ: вычислено для C10H13N3⋅C2H2O4 (C, 54,33%; H, 5,70%; N, 15,84%); найдено (C, 54,39%, 54,29%; H, 5,68%, 5,66%; N, 15,68%, 15,66%).
Пример 25
Полу-ди-п-толуоил-D-тартрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
Твердые ди-п-толуоил-D-тартратные соли получали в соответствии с процедурой отбора для “полусолей” из изопропилацетата или ацетонитрила путем упаривания, либо упариванием изопропилацетата с последующим созреванием с тетрагидрофураном, либо путем упаривания ацетонитрила с последующим созреванием с изопропиловым спиртом.
Следующую процедуру использовали для получения большего количества соли. (+)-О,Oʹ-ди-п-толуоил-D-винную кислоту (1,103 г, 2,85 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,007 г, 5,74 ммоль) в этаноле (10 мл). Некоторое количество нерастворимых твердых веществ, не растворившихся при нагревании смеси до кипения, выпало в осадок. Раствор светло-янтарного цвета (с некоторым количеством мелких твердых веществ) перемешивали в течение 4-5 ч и затем оставили стоять при температуре окружающей среды в течение ночи. Выпадение соли в осадок в виде светло-бежевого порошка было медленным. После перемешивания в течение 15 дней твердые вещества отфильтровали, промыли этанолом (5 мл) и высушили в вакуумной печи при 50°С в течение 21 ч с получением 1,50 г (71,5%) порошка от не совсем белого до светло-желтого оттенка, Тпл 178-180°С. 1Н ЯМР (ДМСО-d6) подтверждает стехиометрический состав 1:0,5 основание:кислота.
1Н ЯМР (ДМСО-d6): δ 10,30 (ушир.с, ~1Н), 9,02 (с, 1Н), 8,80 (с, 2Н), 7,87 (д, 2Н, -С6Н4-, указывает на стехиометрический состав полусоли), 7,27 (д, 2Н, -С6Н4-, указывает на стехиометрический состав полусоли), 6,40 (дд, 1Н), 6,34 (д, 1Н), 5,58 (с, 1Н, СН(СО2Н)-О- кислотной части, указывает на стехиометрический состав полусоли), 3,21 (дд, 1Н), 3,14 (м, 1Н), 3,00 (м, 1Н), 2,86 (м, 1Н), 2,75 (дд, 1Н), 2,30 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав полусоли), 1,93 (м, 1Н), 1,61 (м, 1Н).
Пример 26
Полудибензоил-D-тартрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(+)-O-Oʹ-дибензоил-D-винную кислоту (1,025 г, 2,72 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,003 г, 5,72 ммоль) в этаноле (10 мл). Смесь нагрели на плитке почти до кипения, получив светло-янтарный раствор. Получившийся раствор охладили до комнатной температуры и оставили стоять в течение ночи. Так как не было твердых веществ, раствор медленно упарили в вытяжном шкафу, получив светло-рыжевато-коричневые твердые вещества. Добавили изопропилацетат (10 мл) и, при помешивании и растирании шпателем, в осадок выпали светло-бежевые твердые вещества. Смесь перемешивали в течение ночи. Твердые вещества отфильтровали, промыли изопропилацетатом (2×5 мл) и высушили в вакуумной печи при 50°С в течение 24 ч с получением 1,93 г (95,2%) не совсем белого порошка, Тпл 155-160°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав основание:кислота в 1:0,5.
1Н ЯМР (ДМСО-d6): δ 10,25 (ушир.с, ~1H), 9,02 (с, 1Н), 9,80 (с, 2Н), 7,98 (д, 2Н, С6Н5-), 7,57 (м, 1Н, С6Н5-), 7,48 (м, 2Н, С6Н5-), 6,38 (м, 2Н), 5,61 (с, 1Н, -СН(СО2Н)-О- кислотной части, указывает на стехиометрический состав полусоли), 3,22 (дд, 1Н), 3,14 (дт, 1Н), 3,00 (дт, 1Н), 2,88 (м, 1Н), 2,77 (дд, 1Н), 1,92 (м, 1Н), 1,61 (м, 1Н).
Пример 27
Полу-ди-п-анизоил-D-тартрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(+)-Ди-п-анизоил-D-винную кислоту (1,199 г) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (0,999 г) в этаноле (10 мл). Получившийся раствор с небольшим количеством твердых веществ перемешивали и нагревали в попытке растворить все твердые вещества. Раствор превратился в густую массу. После отстаивания при комнатной температуре в течение 4-5 ч добавили дополнительно этанол (10 мл). Смесь, содержащую твердые вещества от светло-бежевого до кремового цвета, перемешивали в течение ночи. Твердые вещества отфильтровали, промыли этанолом (10 мл) и высушили в вакуумной печи при 50°С в течение 21 ч с получением 1,91 г (87,3%) белого порошка, Тпл 173-177°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав основание:кислота в 1:0,5.
1Н ЯМР (ДМСО-d6): δ 10,20 (ушир.с, ~1H), 9,02 (с, 1Н), 8,80 (с, 2Н), 7,93 (д, 2Н, -С6Н4-, указывает на стехиометрический состав полусоли), 7,00 (д, 2Н, -С6Н4-, указывает на стехиометрический состав полусоли), 6,40 (дд, 1Н), 6,34 (д, 1Н), 5,56 (с, 1Н, СН(СО2Н)-О- кислотной части, указывает на стехиометрический состав полусоли), 3,76 (с, 3Н, -ОСН3 кислотной части, указывает на стехиометрический состав полусоли), 3,22 (дд, 1Н), 3,14 (м, 1Н), 3,01 (м, 1Н), 2,85 (м, 1Н), 2,75 (м, 1Н), 1,92 (м, 1Н), 1,61 (м, 1Н).
Пример 28
Моно-ди-п-толуоил-D-тартрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
Твердые ди-п-толуоил-D-тартратные соли получили в соответствии с процедурой отбора для "моносоли" из изопропилацетата или ацетонитрила путем упаривания.
Следующую процедуру использовали для получения большего количества соли. (+)-O,Oʹ-Ди-п-толуоил-D-винную кислоту (2,205 г, 5,71 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,000 г, 5,70 ммоль) в этаноле (21 мл). Выпадение соли в осадок было немедленным. После аккуратного нагревания перемешиваемой смеси на горячей плите почти до кипения получившуюся смесь охладили до температуры окружающей среды. Получившиеся твердые вещества отфильтровали, промыли этанолом (3×5 мл) и высушили в вакуумной печи при 50°С в течение 13 ч с получением 3,081 г (96,1%) светло-бежевого порошка, Тпл 181-184°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав соли в 1:1.
1Н ЯМР (ДМСО-d6): δ 9,60 (ушир.с, ~1H), 9,03 (с, 1Н), 8,82 (с, 2Н), 7,83 (д, 4Н, -С6Н4-, указывает на стехиометрический состав моносоли), 7,27 (д, 4Н, -С6Н4-, указывает на стехиометрический состав моносоли), 6,44 (д, 2Н), 5,62 (с, 2Н, СН(СО2Н)-О- кислотной части, указывает на стехиометрический состав моносоли), 3,30 (дд, 1Н), 3,23 (м, 1Н), 3,09 (м, 1Н), 2,95 (м, 1Н), 2,85 (дд, 1Н), 2,33 (6Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли), 2,02 (м, 1Н), 1,69 (м, 1Н).
Пример 29
Моно-ди-п-бензоил-D-тартрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(+)-O,Oʹ-Дибензоил-D-винную кислоту (2,05 г, 5,72 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (0,999 г, 5,69 ммоль) в этаноле (21 мл) в круглодонной колбе, получив раствор. После перемешивания и дальнейшего нагревания в теплом растворе произошло выпадение соли в осадок. Получившуюся смесь охладили до температуры окружающей среды за два выходных дня. Получившиеся твердые вещества отфильтровали на воронке Бюхнера, промыли этанолом (4×5 мл) и высушили в вакуумной печи (с выпуском воздуха) при 50°С в течение 13 ч с получением 2,832 г (93,0%) порошка от светло-бежевого до не совсем белого цвета, Тпл 165-171°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав соли в 1:1.
1Н ЯМР (ДМСО-d6): δ 9,65 (ушир.с, ~1H), 9,03 (с, 1Н), 9,83 (с, 2Н), 7,94 (д, 4Н, С6Н5-), 7,60 (м, 2Н, С6Н5-), 7,50 (м, 4Н, С6Н5-), 6,45 (м, 2Н), 5,67 (с, 2Н, -СН(СО2Н)-О- кислотной части, указывает на стехиометрический состав моносоли), 3,31 (дд, 1Н), 3,22 (м, 1Н), 3,08 (м, 1Н), 2,96 (м, 1Н), 2,85 (дд, 1Н), 2,01 (м, 1Н), 1,69 (м, 1Н).
Пример 30
10-Камфорсульфонат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(1S)-(+)-10-Камфорсульфоновую кислоту (1,329 г, 5,72 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,00 г) в 2-пропаноле (23 мл, 5,70 ммоль) в круглодонной колбе. При охлаждении до температуры окружающей среды выпадения в осадок твердых веществ не произошло. Раствор оставили стоять в течение ночи. Наблюдали желатиновый материал, содержащий белые твердые вещества. После перемешивания в течение 2 дней смесь разбавили 2-пропанолом (10,5 мл), так как перемешивание этой желеподобной белой массы было затруднительным. После перемешивания в течение ночи получившийся белый порошок отфильтровали на воронке Бюхнера, промыли 2-пропанолом (5 мл) (ПРИМЕЧАНИЕ: похоже, что твердые вещества обладают определенной растворимостью в 2-пропаноле) и высушили в вакуумной печи (с выпуском воздуха) при 50°С в течение 21 ч с получением 0,245 г светло-бежевых игл, Тпл 173-174°С.
1Н ЯМР (ДМСО-d6): δ 9,03 (с, 1H), 8,87 (с, 2Н), 6,57 (м, 2Н), 3,41 (дд, 1Н), 3,33 (м, 1Н, частично перекрывается с Н2О), 3,21 (м, 1Н), 3,10 (м, 1Н), 2,98 (дд, 1Н), 2,89 (д, 1Н, -СН2- кислотной части, указывает на стехиометрический состав моносоли), 2,64 (м, 1Н), 2,41 (д, 1Н, -СН2- кислотной части, указывает на стехиометрический состав моносоли), 2,25 (т, 0,5Н), 2,20 (т, 0,5Н), 2,15 (м, 1Н), 1,93 (т, 1Н), 1,82 (м, 3Н), 1,28 (м, 2Н, -СН2- кислотной части, указывает на стехиометрический состав моносоли), 1,03 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли), 0,73 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли).
Пример 31
(+)-Камфорат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(1R,2S)-(+)-камфорную кислоту (1,149 г, 5,74 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,00 г, 5,70 ммоль) в этаноле (14 мл) в круглодонной колбе. При нагревании все твердые вещества растворились, образовав желтый раствор. После отстаивания при температуре окружающей среды в течение ночи выпадения осадка не происходит. Раствор сконцентрировали при помощи роторного упаривания до янтарно-коричневой пены, которую высушили под вакуумом при 50°С (с выпуском воздуха) в течение 6 ч с получением 2,098 г вязкого янтарного масла. Добавили изопропилацетат (10 мл) и раствору дали постоять при комнатной температуре в течение ночи. Были заметны определенные признаки образования центров кристаллизации в смолистом, красно-янтарном масле. Добавили еще изопропилацетата (10 мл) и 2-пропанола (20 капель), и смесь аккуратно нагревали и перемешивали в течение 48 ч. Получившиеся молочного, кремового цвета твердые вещества с небольшим количеством оранжевых комков раздавили шпателем и смесь (бесцветный сироп) перемешивали в течение ночи. Твердые вещества не совсем белого цвета отфильтровали на воронке Бюхнера, промыли холодным изопропилацетатом (10 мл) и высушили в вакуумной печи (с выпуском воздуха) при 50°С в течение 21 ч с получением 2,034 г (94,9%) порошка от не совсем белого до кремового цвета, Тпл 157-159°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав соли в 1:1.
1Н ЯМР (ДМСО-d6): δ 9,00 (с, 1H), 8,85 (с, 2Н), 6,58 (дд, 1Н), 6,47 (д, 1Н), 3,17 (дд, 1Н), 3,08 (м, 1Н), 2,97 (м, 1Н), 2,92 (дд, 1Н), 2,74 (дд, 1Н), 2,61 (дд, 1Н), 2,30 (секстет, 1Н), 2,00 (м, 2Н), 1,65 (м, 2Н), 1,32 (м, 1Н), 1,15 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли), 1,07 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли), 0,75 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли).
Пример 32
Моно-ди-п-анизоил-D-тартрат (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(+)-Ди-п-анизоил-D-винную кислоту (2,388 г, 5,71 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,008 г, 5,75 ммоль) в этаноле (22 мл) в круглодонной колбе. Выпадение соли в осадок произошло до того, как добавили всю (+)-ди-п-анизоил-D-винную кислоту. Соль не растворилась при нагревании, но внешний вид твердых веществ изменился, с переходом в светлый, пушистый белый порошок. Смесь охладили до температуры окружающей среды и перемешивали в течение 48 ч. Получившиеся твердые вещества отфильтровали на воронке Бюхнера, промыли этанолом (5×5 мл) и высушили в вакуумной печи (с выпуском воздуха) при 50°С в течение 13 ч с получением 3,20 г (94,4%) мучнистого порошка от не совсем белого до белого цвета, Тпл 173-176°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав соли в 1:1.
1Н ЯМР (ДМСО-d6): δ 9,65 (ушир.с, ~1H), 9,03 (с, 1Н), 8,82 (с, 2Н), 7,89 (д, 4Н -С6Н4-, указывает на стехиометрический состав моносоли), 7,01 (д, 4Н, -С6Н4-, указывает на стехиометрический состав моносоли), 6,44 (м, 2Н), 5,60 (с, 2Н, СН(СО2Н)-О- кислотной части, указывает на стехиометрический состав моносоли), 3,79 (с, 6Н, -ОСН3 кислотной части, указывает на стехиометрический состав моносоли), 3,30 (дд, 1Н), 3,22 (м, 1Н), 3,09 (м, 1Н), 2,95 (м, 1Н), 2,84 (м, 1Н), 2,01 (м, 1Н), 1,69 (м, 1Н).
Пример 33
Моно-(R)-(-)-фенцифосная соль (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
(R)-(-)-фенцифос (1,391 г, 5,77 ммоль) добавили в виде твердого вещества к перемешиваемому, теплому раствору (R)-5-((E)-2-пирролидин-3-илвинил)пиримидинового свободного основания (1,006 г, 5,73 ммоль) в этаноле (10 мл) в круглодонной колбе. Большая часть твердых веществ растворилась при перемешивании при комнатной температуре, и все твердые вещества растворились при аккуратном нагревании. Перемешиваемый янтарного цвета раствор нагрели до кипения, охладили до температуры окружающей среды и оставили стоять в течение ночи. Получившиеся белого цвета иглоподобные кристаллы отфильтровали на воронке Бюхнера, промыли холодным этанолом (5 мл) и высушили в вакуумной печи (с выпуском воздуха) при 50°С в течение 18 ч с получением 0,811 г (33,9%) не совсем белых кристаллов, Тпл 197-201°С.
1Н ЯМР (ДМСО-d6) подтвердил стехиометрический состав соли в 1:1.
1Н ЯМР (ДМСО-d6): δ 9,81 (ушир.с, ~1H), 9,02 (с, 1Н), 8,85 (с, 2Н), 7,27 (м, 5Н, С6Н5-), 6,56 (дд, 1Н), 6,48 (д, 1Н), 5,00 (д, 1Н, -О-СН- кислотной части, указывает на стехиометрический состав моносоли), 4,00 (д, 1Н, -О-СН2- кислотной части, указывает на стехиометрический состав моносоли), 3,48 (дд, 1Н, -О-СН2- кислотной части, указывает на стехиометрический состав моносоли), 3,36 (дд, 1Н), 3,30 (м, 1Н), 3,17 (м, 1Н), 3,07 (м, 1Н), 2,93 (дд, 1Н), 2,12 (м, 1Н), 1,78 (м, 1Н), 0,79 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли), 0,60 (с, 3Н, -СН3 кислотной части, указывает на стехиометрический состав моносоли).
Хотя конкретные варианты осуществления настоящего изобретения проиллюстрированы и подробно описаны в данном документе, изобретение ими не ограничивается. Вышеуказанные подробные описания приведены в качестве примерных для настоящего изобретения и не должны истолковываться как составляющие какое-либо ограничение изобретения. Модификации будут очевидными для специалистов в данной области техники, и все модификации, которые не выходят за пределы сущности изобретения, имеют целью быть включенными в объем притязаний прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ И НОВЫЕ СОЛЕВЫЕ ФОРМЫ (R)-5-((E)-2-(ПИРРОЛИДИН-3-ИЛВИНИЛ)ПИРИМИДИНА | 2009 |
|
RU2533819C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МОНОЦИТРАТА 3-{(3R,4R)-4-МЕТИЛ-3-[МЕТИЛ-(7Н-ПИРРОЛО[2,3-d] ПИРИМИДИН-4-ИЛ)АМИНО]ПИПЕРИДИН-1-ИЛ}-3-ОКСО-ПРОПИОНИТРИЛА И ЕЕ ПРИМЕНЕНИЕ | 2002 |
|
RU2315052C2 |
| КРИСТАЛЛЫ СОЛИ | 2013 |
|
RU2652116C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2507197C2 |
| ПРОИЗВОДНЫЕ N-(ИМИДАЗОПИРИМИДИН-7-ИЛ)-ГЕТЕРОАРИЛАМИДОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10A | 2011 |
|
RU2562066C2 |
| НОВОЕ КОНДЕНСИРОВАННОЕ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2014 |
|
RU2666349C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| 3-ФЕНОКСИМЕТИЛПИРРОЛИДИНОВЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2535669C2 |
| МАКРОЦИКЛ, СОДЕРЖАЩИЙ АМИНОПИРАЗОЛ И ПИРИМИДИН, И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2779497C2 |
| Новые модулирующие регуляцию дыхания соединения и способы их получения и применения | 2016 |
|
RU2734506C2 |
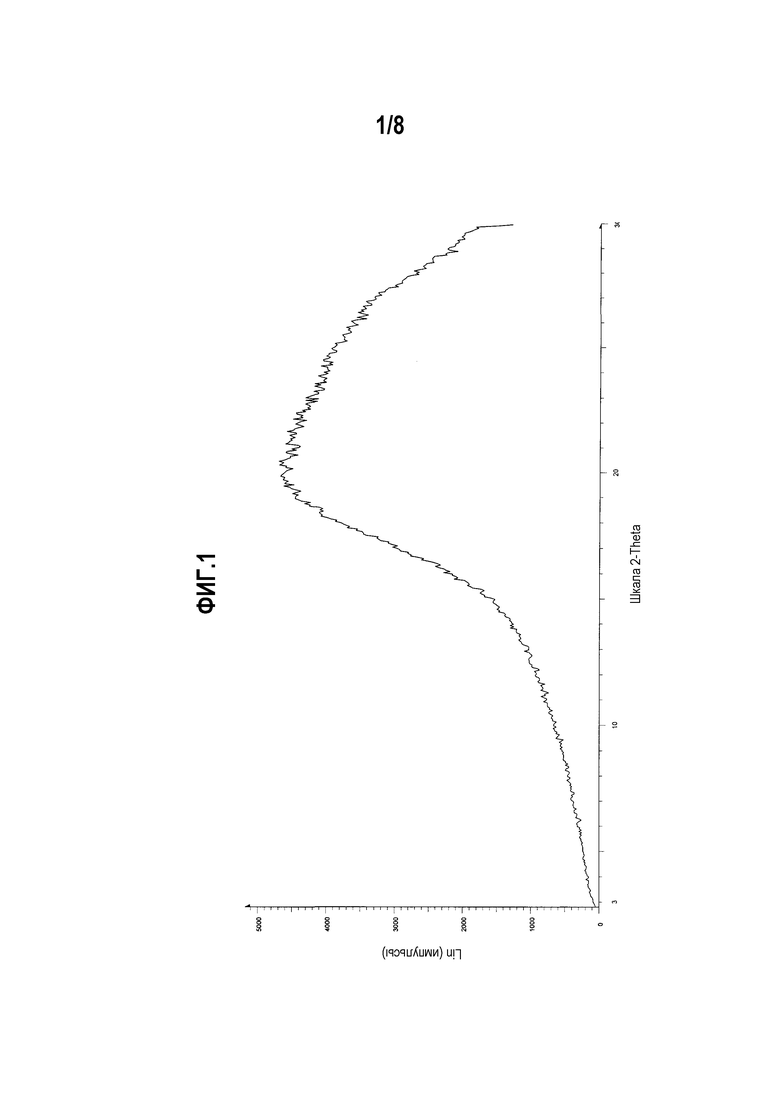
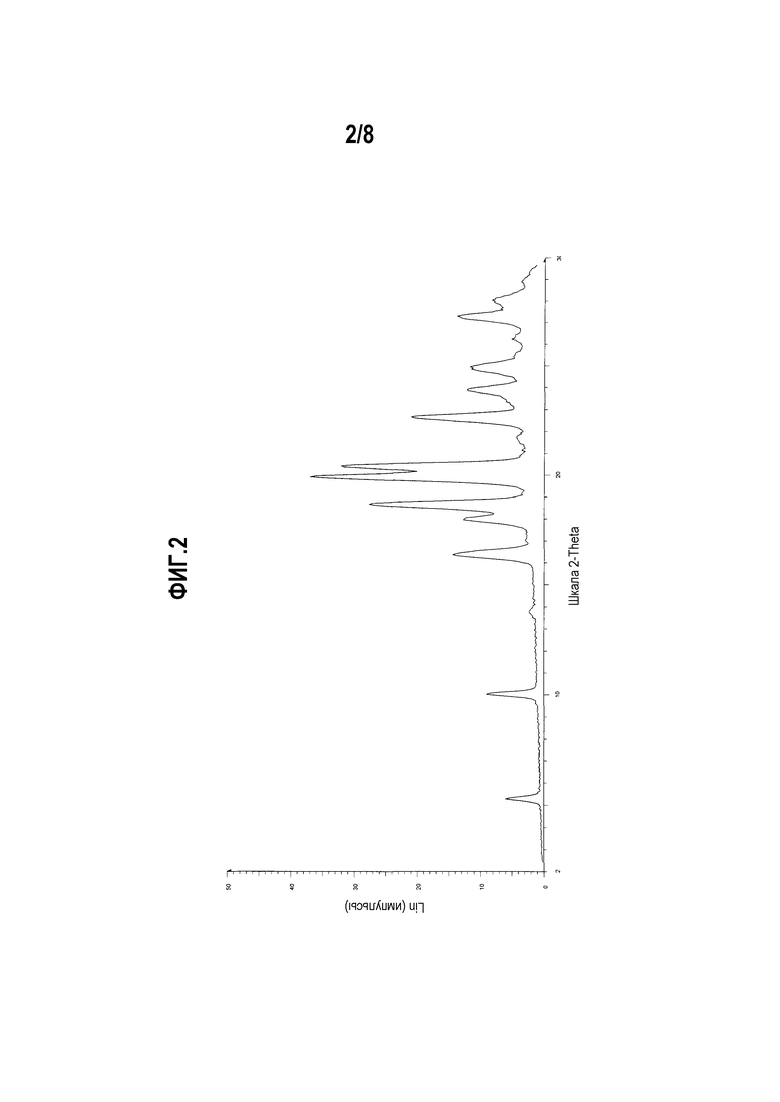
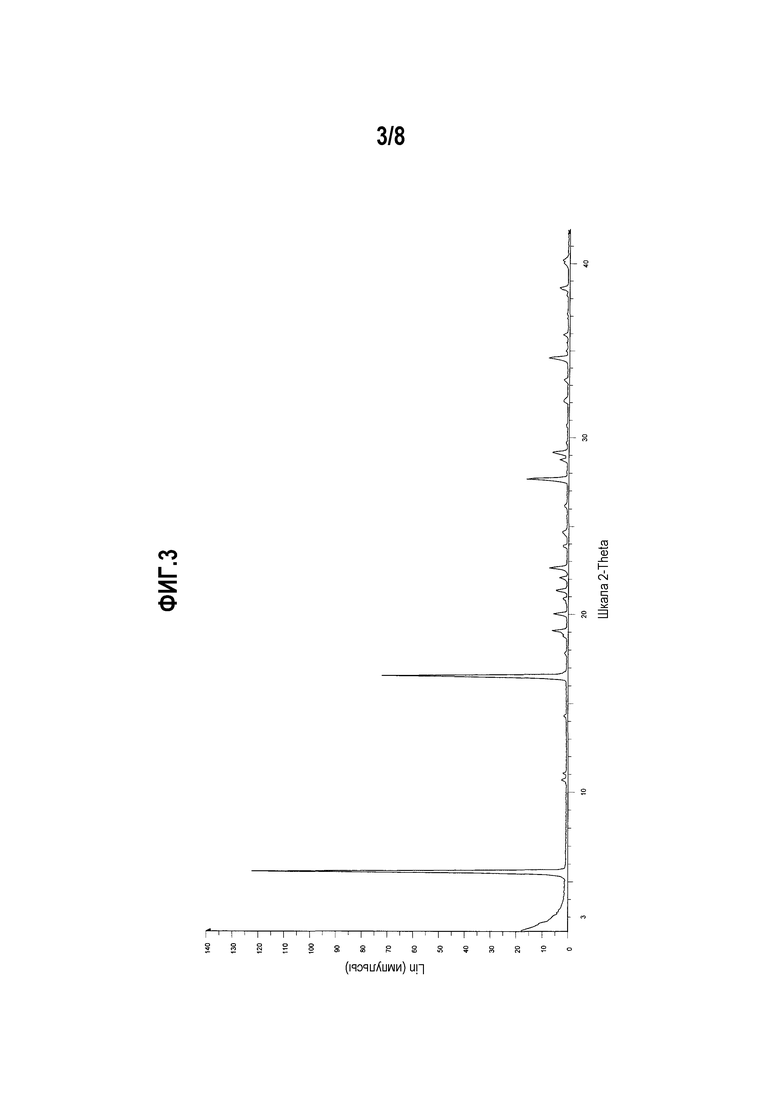
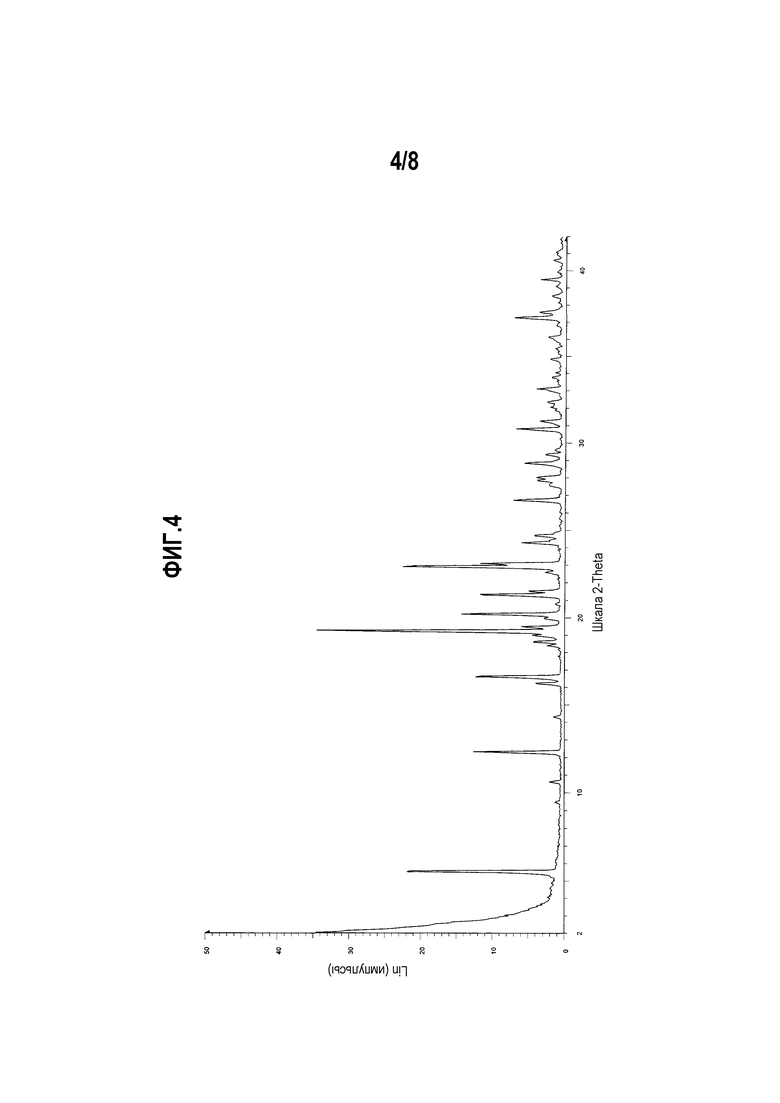
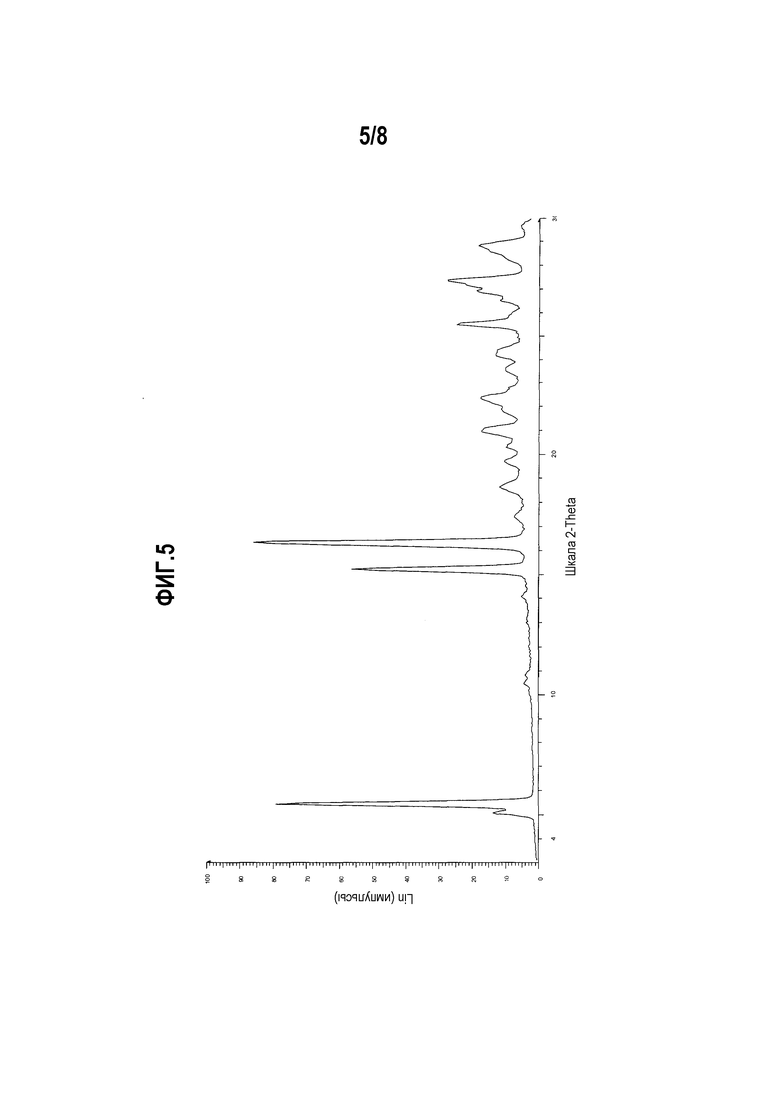
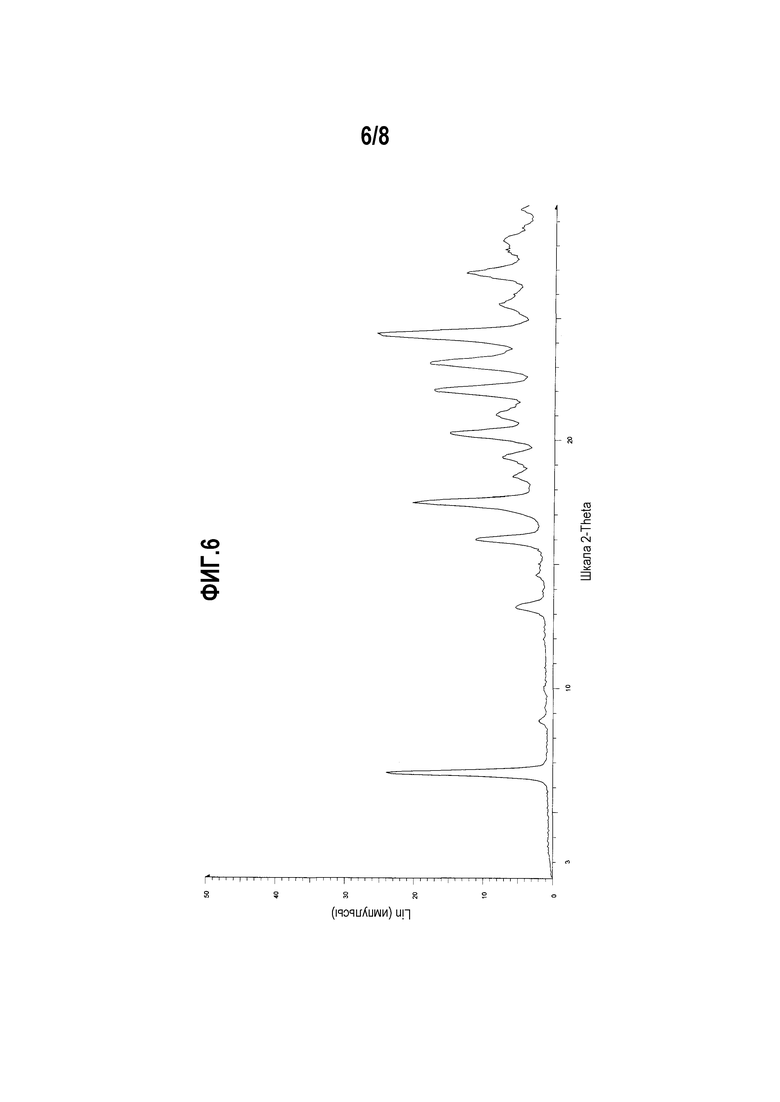
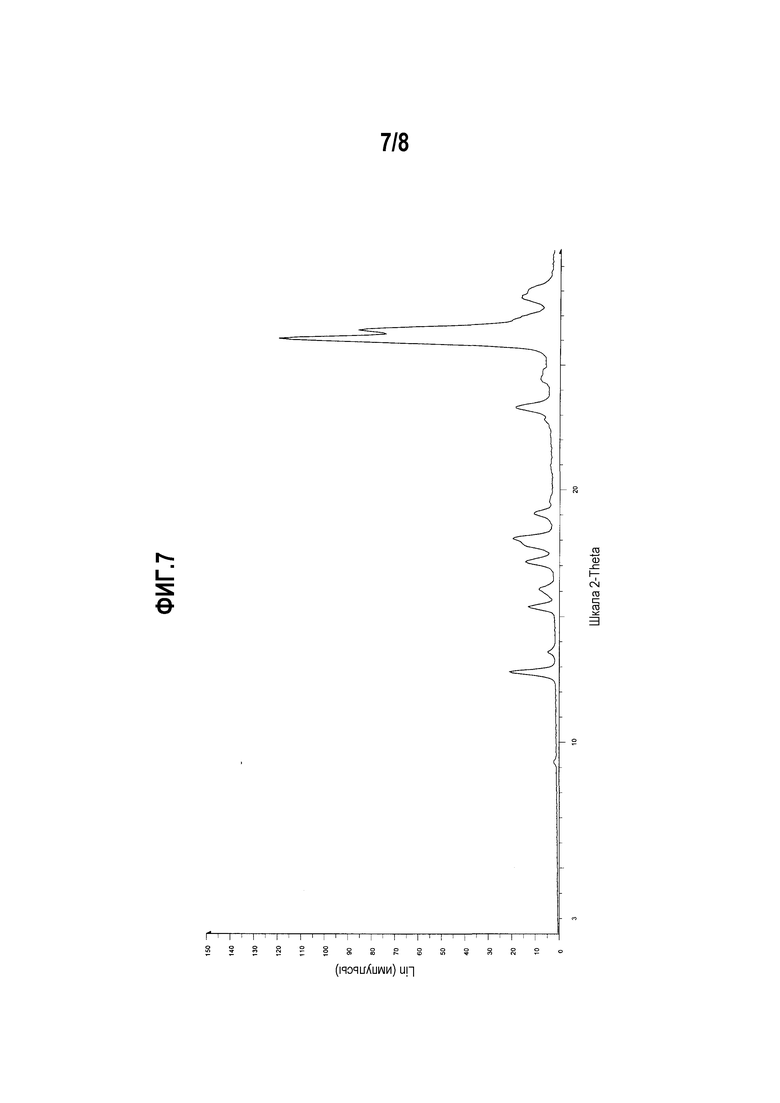
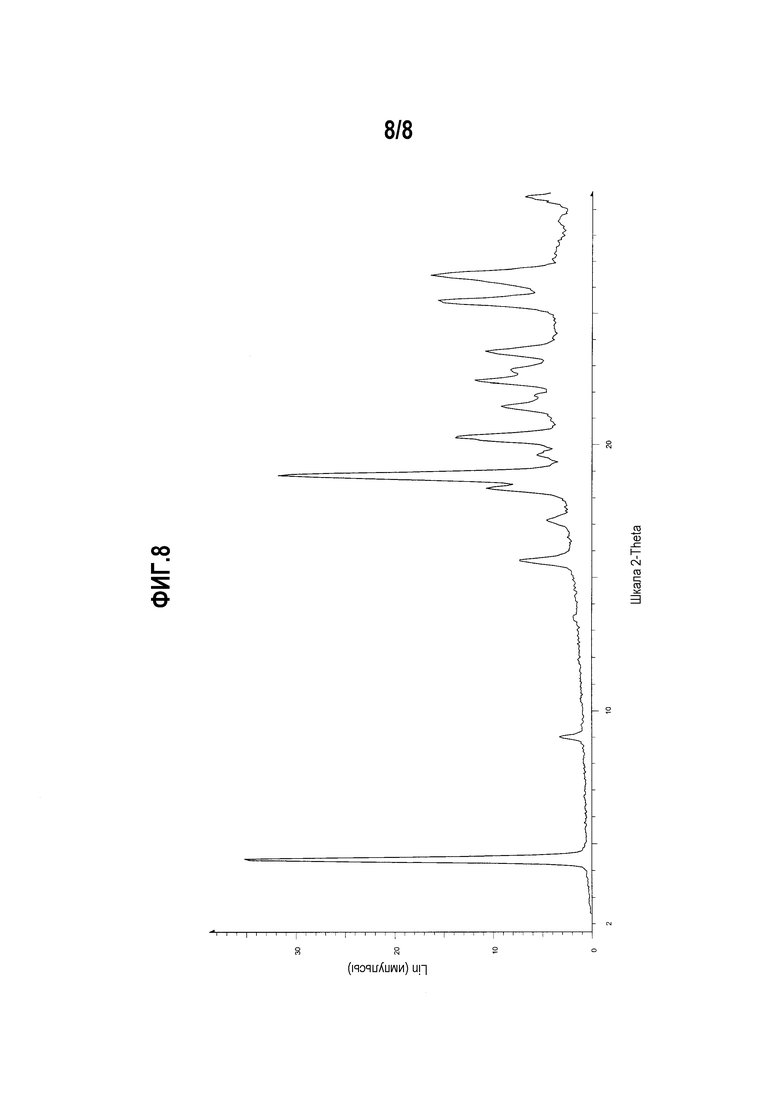
Изобретение относится к стереоспецифичному синтезу (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина и его соли. (R)-5-((E)-2-(Пирролидин-3-илвинил)пиримидин является агонистом нейронального никотинового рецептора (NNR), селективного к никотиновому подтипу α4β2, и может найти применение при лечении и профилактике расстройств ЦНС и боли. Способ получения включает стадии а) обработки соединения 1 метансульфонилхлоридом с получением соединения 2; (b) обработки соединения 2 диэтилмалонатом и подходящим основанием с получением соединения 3; (с) гидролиза соединения 3 с получением соединения 4; (d) декарбоксилирования соединения 4 с получением соединения 5; (е) восстановления соединения 5 с получением соединения 6;
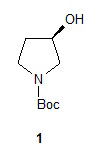
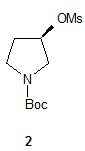
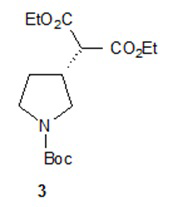
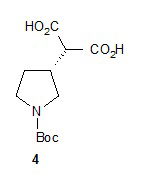
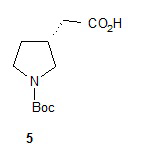
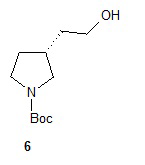
(f) обработки соединения 6 метансульфонилхлоридом с получением соединения 7; (g) взаимодействия соединения 7 с иодидом натрия с получением соединения 8;(h) обработки соединения 8 трет-бутоксидом калия с образованием соединения 9 - трет-бутил-(R)-3-винилпирролидин-1-карбоксилата;(i) взаимодействия соединения 9 с 5-бром-пиридином с получением соединения 10 и (j) снятия защитной группы с соединения 10 с получением соединения 11 - (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина
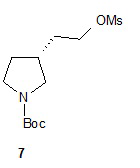
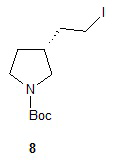
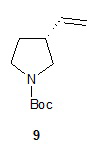
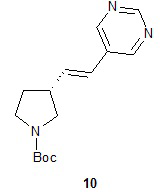
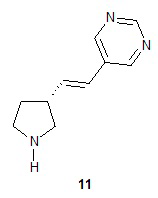
Способ получения соли (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина включает стадии: (i)смешивания свободного основания или раствора свободного основания (R)-5-((E)-2-(пирролидин-3-илвинил)пиримидина в подходящем растворителе, с кислотой в чистом виде или в виде раствора кислоты в подходящем растворителе;(ii)(а) охлаждения полученного раствора соли для того, чтобы вызвать выпадение осадка; или (ii)(b) добавления подходящего анти-растворителя для того, чтобы вызвать выпадение осадка; или (ii)(c) выпаривания первого растворителя и добавления нового растворителя и повторения стадии (ii) (a) или (ii) (b); и (iii) фильтрования для выделения соли (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина. Способ позволяет упростить процесс за счет использования новых промежуточных соединений, не требует использования хроматографии и позволяет получить целевой продукт высокого качества (чистотой 99%). 2 н. и 14 з.п. ф-лы, 8 ил., 33 пр.
1. Способ получения €-5-((Е)-2-пирролидин-3-илвинил)пиримидина, включающий стадии:
(а) обработки соединения 1
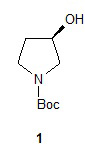
метансульфонилхлоридом с получением соединения 2
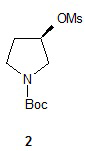 ;
;
(b) обработки соединения 2 диэтилмалонатом и подходящим основанием с получением соединения 3
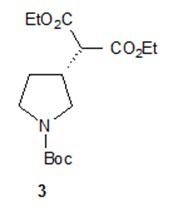 ;
;
(с) гидролиза соединения 3 с получением соединения 4
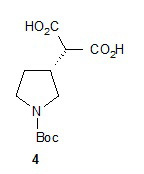 ;
;
(d) декарбоксилирования соединения 4 с получением соединения 5
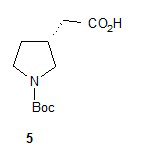 ;
;
(е) восстановления соединения 5 с получением соединения 6
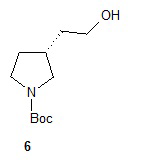 ;
;
(f) обработки соединения 6 метансульфонилхлоридом с получением соединения 7
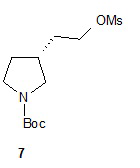 ;
;
(g) взаимодействия соединения 7 с иодидом натрия с получением соединения 8
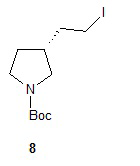 ;
;
(h) обработки соединения 8 трет-бутоксидом калия с образованием соединения 9, трет-бутил-€-3-винилпирролидин-1-карбоксилата
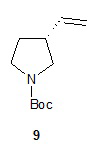 ;
;
(i) взаимодействия соединения 9 с 5-бром-пиридином с получением соединения 10
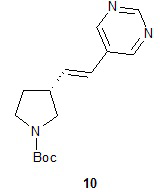 и
и
(j) снятия защитной группы с соединения 10 с получением соединения 11, €-5-(€-2-пирролидин-3-илвинил)пиримидина
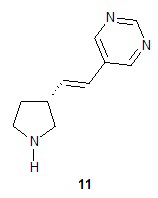 .
.
2. Способ по п.1, где стадия (b) не включает колоночную хроматографию.
3. Способ по п.1, где стадия (е) не включает колоночную хроматографию.
4. Способ по п.1, где стадия (h) не включает колоночную хроматографию.
5. Способ по п.1, где подходящее основание, которое используют для обработки соединения 2, представляет собой трет-бутоксид калия или этоксид натрия.
6. Способ по п.1, где гидролиз соединения 3 проводят путем взаимодействия с гидроксидом калия.
7. Способ по п.1, где декарбоксилирование соединения 4 проводят в подходящем растворителе при нагревании.
8. Способ по п.7, где подходящий растворитель для декарбоксилирования выбирают из воды, этанола, тетрагидрофурана, диметилформамида, диметилацетамида, 1,2-диметоксиэтана, диоксана, 1-метил-2-пирролидинона, диметилсульфоксида, толуола и их смесей.
9. Способ по п.8, где подходящий растворитель для декарбоксилирования выбирают из 1-метил-2-пирролидинона, диметилсульфоксида, толуола и их смесей.
10. Способ по п.1, где восстановление соединения 5 проводят путем взаимодействия с реагентом, выбранным из борана, диборана, комплекса боран-тетрагидрофуран, комплекса боран-диметиловый эфир и комплекса боран-диметилсульфид.
11. Способ по п.10, где восстановление соединения 5 проводят путем взаимодействия с комплексом боран-тетрагидрофуран.
12. Способ по п.1, где реакция стадии (i) катализируется палладием.
13. Способ по п.11, где снятие защитной группы в реакции стадии (j) проводят с помощью реагента, выбранного из трифторуксусной кислоты, хлороводородной кислоты и серной кислоты.
14. Способ получения соли €-5-(€-2-пирролидин-3-илвинил)пиримидина, включающий стадии:
(i) смешивания свободного основания или раствора свободного основания €-5-(€-2-(пирролидин-3-илвинил)пиримидина в подходящем растворителе, с кислотой в чистом виде или в виде раствора кислоты в подходящем растворителе;
(ii) (а) охлаждения полученного раствора соли для того, чтобы вызвать выпадение осадка; или
(i) (b) добавления подходящего анти-растворителя для того, чтобы вызвать выпадение осадка; или
(i) € выпаривания первого растворителя и добавления нового растворителя, и повторения стадии (ii) (a) или (ii) (b); и
(ii) фильтрования для выделения соли €-5-(€-2-пирролидин-3-илвинил)пиримидина.
15. Способ по п.14, где кислоту выбирают из хлороводородной, серной, метансульфоновой, малеиновой, фосфорной, 1-гидрокси-2-нафтойной, кетоглутаровой, малоновой, L-винной, фумаровой, лимонной, L-яблочной, гиппуровой, L-молочной, бензойной, янтарной, адипиновой, уксусной, никотиновой, пропионовой, оротовой, 4-гидроксибензойной, ди-(п-толуоил)-D-винной, ди-п-анизоил-D-винной, дибензоил-D-винной, 10-камфорсульфоновой, камфорной или фенцифоса.
16. Способ по п. 14 или 15, где кислота представляет собой лимонную кислоту и полученная соль (R)-5-((E)-2-пирролидин-3-илвинил)пиримидина представляет собой (R)-5-((E)-2-пирролидин-3-илвинил)пиримидин моноцитрат.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2011127004A, 01,07.2013, приоритет 01.12.2008 & RU2507197, 20.08.2014 | |||
| WO 2006113837 A1, 26 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |

