Родственные заявки
Данная заявка испрашивает преимущество приоритета нашей заявки на патент Индии IN 201941004693, поданной 6 февраля 2019 года, которая включена в настоящую заявку посредством ссылки.
Область техники
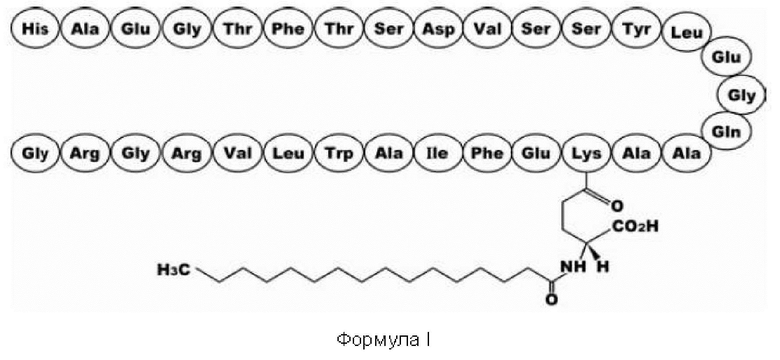
Настоящее изобретение относится к способу очистки неочищенного аналога глюкагоноподобного пептида-1 (GLP-1, от англ. glucagon-like peptide-1), в частности, лираглутида, представленного Формулой I.
Предшествующий уровень техники
Лираглутид (VICTOZA®) представляет собой агонист рецептора глюкагоноподобного пептида-1 (GLP-1), назначаемый в качестве дополнения к рациону питания и физическим упражнениям для улучшения гликемического контроля у взрослых, страдающих сахарным диабетом 2 типа.
Лираглутид является аналогом пролонгированного действия природного человеческого глюкагоноподобного пептида-1 (GLP-1 (7-37)), в котором лизин в положении 34 заменен на аргинин, а к лизину в положении 26 присоединена пальмитоильная группа через глутамоиловый спейсер.
Лираглутид (VICTOZA®), разработанный компанией Novo Nordisk, получил первоначальное одобрение в Соединенных Штатах Америки в 2010 в виде подкожной инъекции.
Благодаря своей длинной пептидной цепи и высокой гидрофобности, обусловленной пальмитоильной группой, лираглутид очень трудно поддается очистке.
Ранее сообщалось о нескольких попытках очистки аналогов GLP-1, включая лираглутид.
В работе, опубликованной в Journal of Medicinal Chemistry 43, 1664-1669, 2000, раскрыт способ очистки лираглутида при помощи обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФ-ВЭЖХ, англ. RP HPLC) с использованием цианопропильной колонки (Zorbax 300SB-CN) и стандартной системы ацетонитрил/TFA (англ. Trifluoroacetic acid - трифторуксусная кислота).
В результате описанного выше способа получают низкий выход очистки, составляющий 35%.
В патентном документе WO 2013117135 раскрыт способ очистки лираглутида методом ОФ-ВЭЖХ с использованием системы изопропиловый спирт/TFA.
Раскрытый способ включает в себя несколько стадий очистки, предполагающих 3 операции ОФ-ВЭЖХ, что является трудоемким процессом.
Пептиды GLP-1, полученные синтетическим или рекомбинантным способом, зачастую содержат близкородственные примеси, которые трудно отделить при помощи ОФ-ВЭЖХ. Эти примеси представляют собой примеси изомеров либо примеси, образующиеся в результате делеций/присоединений, и имеют характеристики, аналогичные исходной молекуле. Такие близкородственные примеси создают сложности при очистке.
Хорошо известно, что использование ОФ-ВЭЖХ ограничено разделением и идентификацией сложных смесей, содержащих компоненты с большим разбросом значений рКа. Вследствие этого разделение при хроматографической очистке близко элюирующихся примесей всегда представляло собой сложную задачу. При разделении органических ионов стандартными методами ВЭЖХ в случаях, когда не работают обычные средства, такие как изменение соотношений элюентов или изменение неподвижной фазы, использование ион-парных реагентов может улучшить форму пиков и время удерживания. Такой способ иногда называют ион-парной хроматографией (англ. IPC, от англ. ion pair chromatography).
IPC представляет собой разновидность ОФ-ВЭЖХ, при которой в подвижную фазу добавляют ион-парные реагенты, способствующие образованию ионных пар с заряженными анализируемыми компонентами, что позволяет использовать колонку с обращенной фазой для разделения ионных молекул с ионной связью. Удерживание/разделение происходит за счет динамического сочетания механизмов обращенно-фазового и ионно-парного-ионного обмена.
Ион-парные реагенты состоят из длинной линейной алкильной цепи (от С3 до С16) и ионной группы, способной обратимо адсорбироваться на алкильных цепях (С8 или С18) RP-фазы (англ. RP, reversed phase - обращенная фаза), образуя динамичный ионообменник, с помощью которого могут быть разделены ионные соединения. Существует два основных типа ион-парных реагентов: анионные алкилсульфонаты для основных соединений и катионные четвертичные амины для кислотных соединений. Обычно при IPC используют алкилсульфонаты, представляющие собой натриевую соль гексансульфоновой кислоты, натриевую соль гептансульфоновой кислоты, натриевую соль 1-октансульфоновой кислоты и т.п. Селективность системы в значительной степени зависит от выбора и количества образователя ионной пары в подвижной фазе. Реагенты с длинной цепью будут значительно лучше адсорбироваться на RP-фазе, что положительно влияет на удерживание.
Отделение лираглутида от таких близкородственных примесей было изучено с помощью ОФ-ВЭЖХ в отсутствие и в присутствии агентов, образующих пары ионов, а именно натриевых солей 1-октансульфоновой кислоты, гексансульфоновой кислоты. Было отмечено, что разделение близкородственных примесей является более эффективным в присутствии агента, образующего пары ионов, приводя к общей более высокой чистоте по сравнению с циклом очистки, в котором агент для образования пары ионов не использовался.
В настоящем изобретении предложен способ очистки лираглутида от таких близкородственных примесей, исследованный на примере ОФ-ВЭЖХ в отсутствие и в присутствии агентов, образующих пары ионов, а именно натриевых солей пропансульфоновой кислоты, бутансульфоновой кислоты, пентансульфоновой кислоты, гексансульфоновой кислоты, гептансульфоновой кислоты, октансульфоновой кислоты, нонансульфоновой кислоты, декансульфоновой кислоты, ундекансульфоновой кислоты, додекансульфоновой кислоты, тридекансульфоновой кислоты.
Было отмечено, что разделение близкородственных примесей является более эффективным в присутствии агента, образующего пары ионов, приводя к общей более высокой чистоте по сравнению с очисткой методом ОФ-ВЭЖХ, где агент для образования пары ионов не использовался
Краткое описание сущности изобретения
Аспекты настоящей заявки предусматривают способы очистки лираглутида.
Один из аспектов настоящего изобретения раскрывает способ очистки неочищенного лираглутида, при этом способ включает в себя:
a. получение раствора лираглутида путем растворения неочищенного лираглутида в смеси, содержащей водный раствор кислоты и ацетонитрил;
b. подвергание раствора неочищенного лираглутида первой очистке методом ВЭЖХ с использованием водного раствора кислоты и агента для образования пары ионов в качестве подвижной фазы А и ацетонитрила, содержащего спирт, в качестве подвижной фазы В;
c. подвергание лираглутида после первой очистки методом ВЭЖХ второй очистке методом ВЭЖХ; и
d. выделение очищенного лираглутида.
Другой аспект настоящего изобретения раскрывает способ очистки неочищенного лираглутида, где агент для образования пары ионов выбирают из соли алкансульфоновой кислоты.
Другой аспект настоящего изобретения раскрывает способ очистки неочищенного лираглутида, где соль алкансульфоновой кислоты выбирают из группы, состоящей из натриевой соли 1-октансульфоновой кислоты или натриевой соли 1-гептансульфоновой кислоты.
Другой аспект настоящего изобретения раскрывает способ очистки неочищенного лираглутида, где соль алкансульфоновой кислоты представляет собой натриевую соль 1-гексансульфоновой кислоты.
Другой аспект настоящего изобретения раскрывает способ очистки неочищенного лираглутида, где водный раствор кислоты выбирают из лимонной кислоты, уксусной кислоты, трифторуксусной кислоты или муравьиной кислоты.
Другой аспект настоящего изобретения раскрывает способ очистки неочищенного лираглутида методом ВЭЖХ с использованием агента для образования пары ионов.
Параметры оборудования:
Параметры оборудования для ВЭЖХ:
Колонка: С8, 150×4,6 мм, 2,7 мкм
Температура колонки: 60°С
Детектирование: УФ
Длина волны: 215 нм
Преимущества настоящего изиобретения
Порошкообразный неочищенный лираглутид (содержание основного вещества 20-25%; чистота 30-50%) подвергают двум последовательным стадиям очистки методом ОФ-ВЭЖХ в разных условиях с последующей лиофилизацией с получением чистого лираглутида. Настоящее изобретение включает в себя использование на одной из стадий ОФ-ВЭЖХ селективных агентов для образования пары ионов для очистки неочищенного лираглутида от близкородственных примесей. При отсутствии во время процесса агентов для образования пары ионов разделения родственных примесей не происходит либо оно является недостаточным, что приводит к появлению примесей в конечном активном фармацевтическом ингредиенте (API, от англ. - active pharmaceutical ingredient).
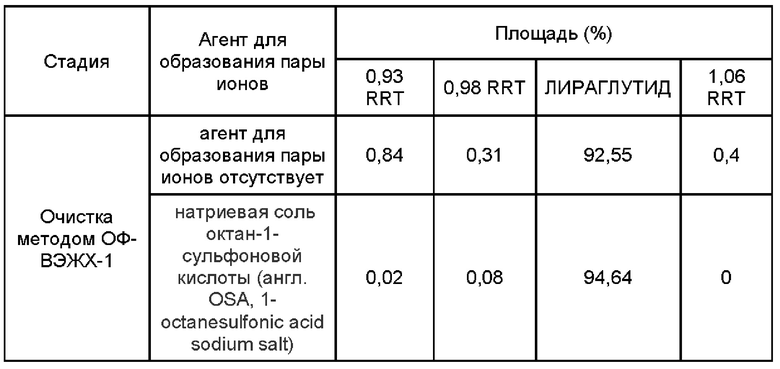
Сравнение профиля чистоты лираглутида без использования и с использованием октан-1-сульфоновой кислоты в качестве агента, образующего пары ионов
В приведенной ниже таблице представлено сравнение профиля чистоты лираглутида в отношении близкородственных примесей, присутствующих при относительном времени удерживания (RRT, от англ. relative retention time) 0,93, 0,98 и 1,06 без использования и с использованием октан-1-сульфоновой кислоты в качестве агента для образования пары ионов.
Краткое описание графических материалов
Для лучшего понимания и применения на практике настоящего раскрытия далее будут представлены примерные варианты осуществления, проиллюстрированные прилагаемыми графическими материалами. Графические материалы вместе с представленным ниже подробным описанием включены в описание, являются его частью и служат для дополнительной иллюстрации вариантов осуществления изобретения и объяснения различных принципов и преимуществ в соответствии с настоящим раскрытием, где:
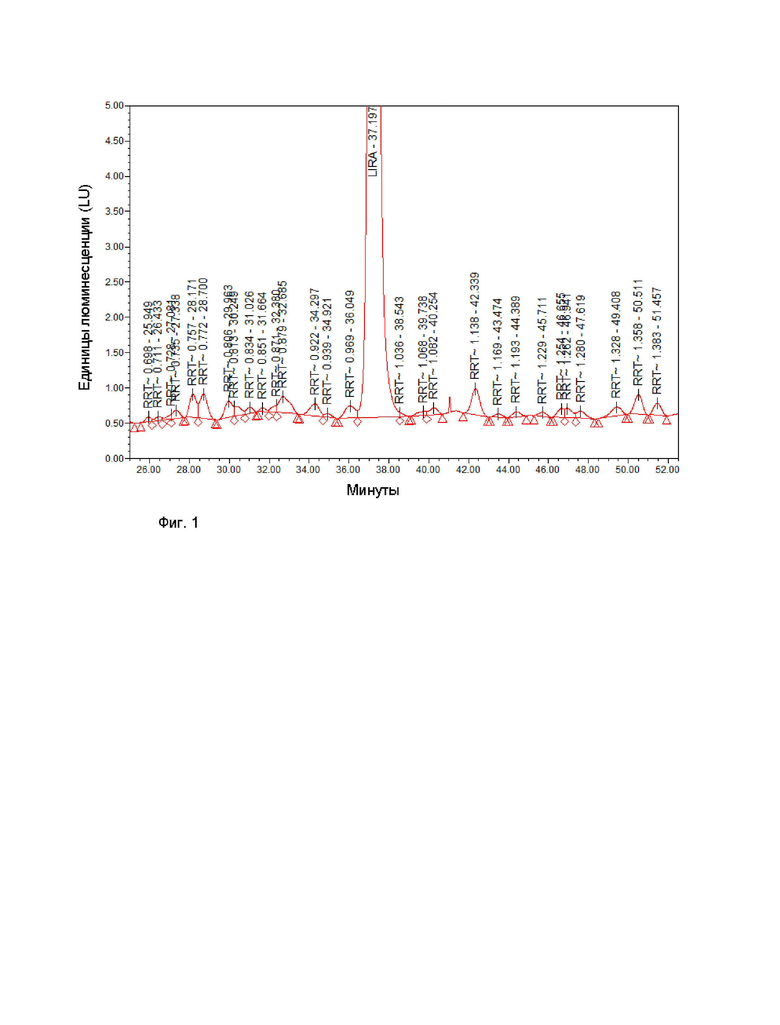
На Фиг. 1 изображена хроматограмма ВЭЖХ неочищенного лираглутида Формулы I.
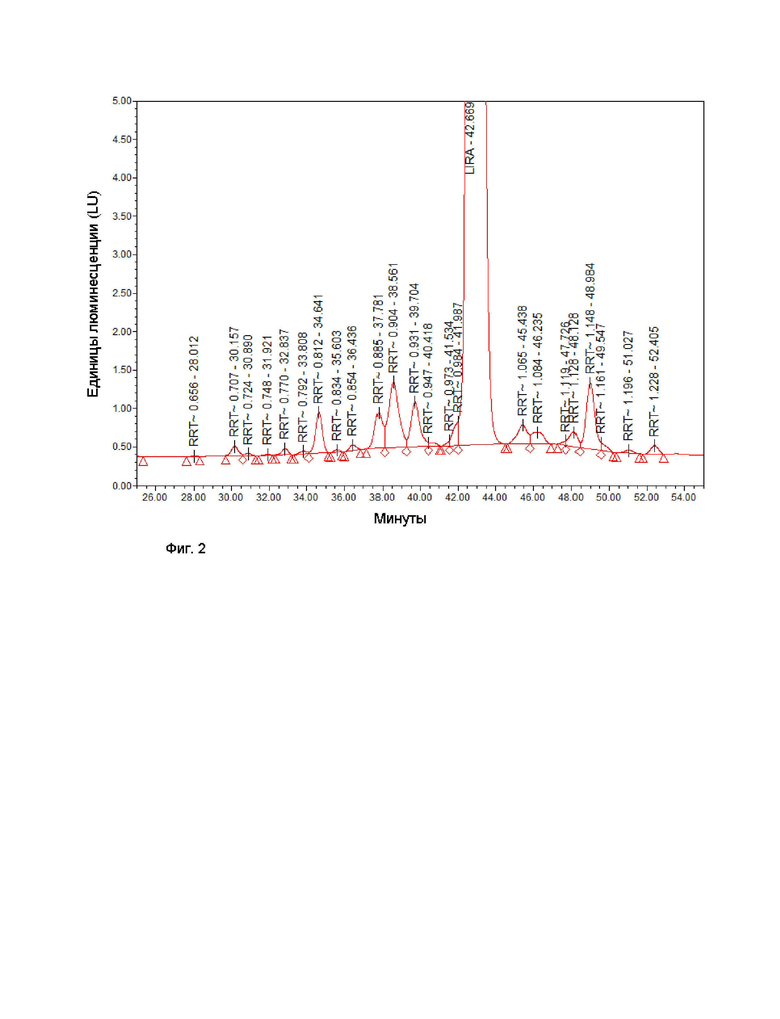
На Фиг. 2 изображена хроматограмма ВЭЖХ лираглутида Формулы I после очистки без использования натриевой соли октан-1-сульфоновой кислоты (OSA).
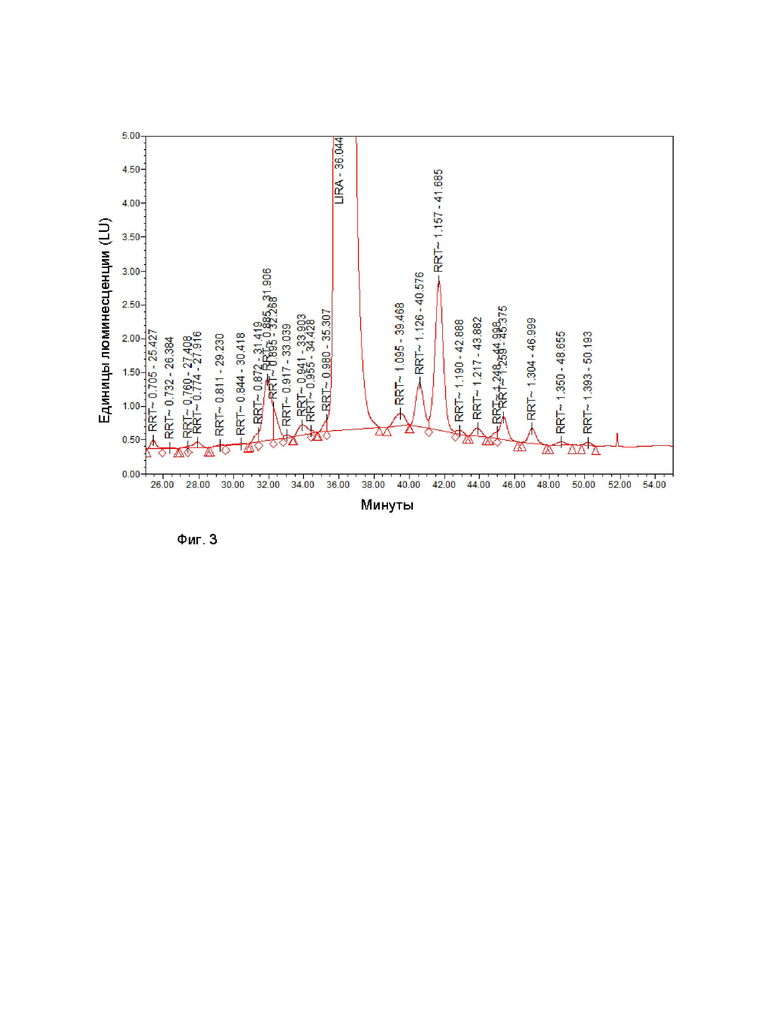
На Фиг. 3 изображена хроматограмма ВЭЖХ лираглутида Формулы I после очистки с использованием OSA.
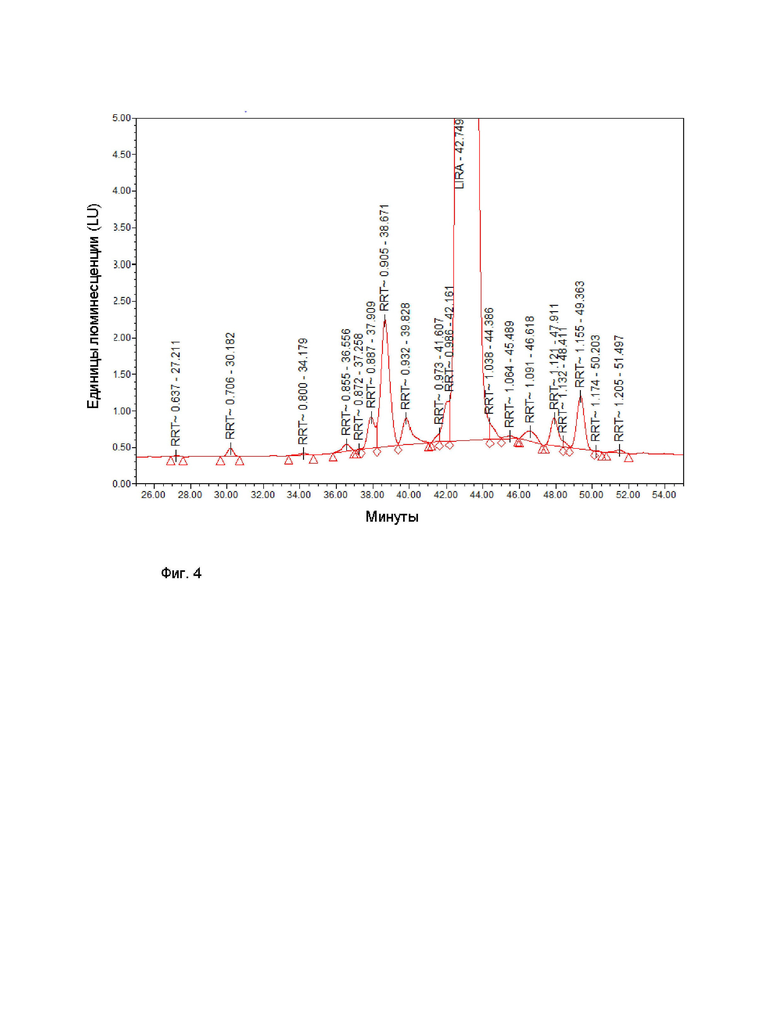
На Фиг. 4 изображена хроматограмма ВЭЖХ лираглутида Формулы I после очистки с использованием HSA (англ. 1-hexane sulfonic acid sodium salt - натриевая соль 1-гексансульфоновой кислоты).
Подробное описание изобретения
Далее описаны варианты осуществления настоящего изобретения с помощью конкретных примеров, приведенных ниже. Примеры представлены для лучшего понимания некоторых вариантов осуществления изобретения, а никоим образом не для ограничения его объема. Возможные модификации и эквиваленты, очевидные для специалистов в данной области техники, использующих идеи настоящего описания и общие знания в области изобретения, также должны составлять часть данного описания и должны быть включены в объем изобретения.
Описание примеров осуществления изобретения
Пример 1
275,5 мг неочищенного лираглутида, полученного твердофазным синтезом, растворяли в 250 мМ растворе моногидрата лимонной кислоты, содержащем 10% ацетонитрила (об./об.), фильтровали и подвергали двухстадийной очистке методом ОФ-ВЭЖХ.
ОФ-ВЭЖХ-1
Раствор неочищенного лираглутида наносили на колонку емкостью 20 мл, заполненную С8 модифицированным силикагелем (размер частиц 10-13 мкм), уравновешенную приблизительно 60 мл 100 мМ раствора лимонной кислоты, содержащего 0,05 мас./об. % натриевой соли октан-1-сульфоновой кислоты (подвижная фаза А), 25% ацетонитрил : изопропанол (7:3) (подвижная фаза В), рН 2,0. После нанесения колонку промывали смесью буфер А: буфер В, 8:2. Продукт элюировали, используя градиент до 60% В. Длину волны детектирования устанавливали на уровне 215 нм. Температуру хроматографирования поддерживали на уровне 25°С. Объединяли фракции с чистотой более 91%, при этом средняя чистота объединенной фракции при RP-1 составляла более 95% с содержанием близкородственных примесей менее 0,50%.
Продукт лираглутид после ОФ-ВЭЖХ-1 использовали далее для ОФ-ВЭЖХ-2
Пример 2
275,5 мг неочищенного лираглутида, полученного твердофазным синтезом, растворяли в 250 мМ растворе моногидрата лимонной кислоты, содержащем 10% ацетонитрила (об./об.), фильтровали и подвергали двухстадийной очистке методом ОФ-ВЭЖХ.
ОФ-ВЭЖХ-1
Раствор неочищенного лираглутида наносили на колонку емкостью 20 мл, заполненную С8 модифицированным силикагелем (размер частиц 10-13 мкм), уравновешенную приблизительно 60 мл 100 мМ раствора лимонной кислоты, содержащего 0,05% натриевой соли 1-гексансульфоновой кислоты (подвижная фаза А), 25% ацетонитрил : изопропанол (7:3) (подвижная фаза В), рН 2,0. После нанесения колонку промывали смесью буфер А: буфер В, 8:2. Продукт элюировали, используя градиент до 60%. Длину волны детектирования устанавливали на уровне 215 нм. Температуру хроматографирования поддерживали на уровне 25°С. Собирали фракции и анализировали их чистоту. Объединяли фракции с чистотой более 91%.
Чистота лираглутида, очищенного с помощью ОФ-ВЭЖХ-1, по хроматограмме ВЭЖХ показана на Фиг. 4.
Чистота по ВЭЖХ лираглутида, очищенного с помощью ОФ-ВЭЖХ, составляла более 95% с содержанием близкородственных примесей менее 2,0%.
рН данной объединенной фракции доводили до 7,8 и упаривали при температуре 35°С для удаления органического растворителя. Осаждение проводили при рН 4,9 и выделяли лираглутид, очищенный с помощью ОФ-ВЭЖХ-1.
Продукт лираглутид после ОФ-ВЭЖХ-1 использовали далее для ОФ-ВЭЖХ-2.
Пример 3
275,5 мг неочищенного лираглутида, полученного твердофазным синтезом, растворяли в 250 мМ растворе моногидрата лимонной кислоты, содержащем 10% ацетонитрила (об./об.), фильтровали и подвергали двухстадийной очистке методом ОФ-ВЭЖХ.
ОФ-ВЭЖХ-1
Раствор неочищенного лираглутида наносили на колонку емкостью 20 мл, заполненную С8 модифицированным силикагелем (размер частиц 10-13 мкм), уравновешенную приблизительно 60 мл 100 мМ раствора лимонной кислоты (подвижная фаза А), 25% ацетонитрил : изопропанол (7:3) (подвижная фаза В), рН 2,0. После нанесения колонку промывали смесью буфер А: буфер В (8:2). Продукт элюировали, используя градиент до 60% В. Длину волны детектирования устанавливали на уровне 215 нм. Температуру хроматографирования поддерживали на уровне 25°С. Собирали фракции и анализировали их чистоту. Объединяли фракции с чистотой более 91%.
Чистота по ВЭЖХ лираглутида, очищенного с помощью ОФ-ВЭЖХ, составляла более 95% с содержанием близкородственных примесей менее 2,0%.
Чистота лираглутида, очищенного с помощью ОФ-ВЭЖХ-1, по хроматограмме ВЭЖХ показана на Фиг. 2.
Продукт лираглутид после ОФ-ВЭЖХ-1 использовали далее для ОФ-ВЭЖХ-2. Пример 4
32,6 г неочищенного лираглутида, полученного твердофазным синтезом, растворяли в 250 мМ растворе моногидрата лимонной кислоты, содержащем 10% ацетонитрила (об./об.), фильтровали и подвергали двухстадийной очистке методом ОФ-ВЭЖХ.
ОФ-ВЭЖХ-1
Раствор неочищенного лираглутида наносили на колонку емкостью 2,4 л, заполненную С8 модифицированным силикагелем (размер частиц 10-13 мкм), уравновешенную приблизительно 7,2 л 100 мМ раствора лимонной кислоты, содержащего 0,05% натриевой соли октан-1-сульфоновой кислоты (подвижная фаза А), 25% ацетонитрил : изопропанол (7:3) (подвижная фаза В), рН 2,0. После нанесения колонку промывали смесью буфер А: буфер В, 8:2. Продукт элюировали, используя градиент до 60% В. Длину волны детектирования устанавливали на уровне 215 нм. Температуру хроматографирования поддерживали на уровне 25°С. Собирали фракции и анализировали их чистоту. Объединяли фракции с чистотой более 91%. Чистота лираглутида, очищенного с помощью ОФ-ВЭЖХ-1, по хроматограмме ВЭЖХ показана на Фиг. 3.
Чистота по ВЭЖХ лираглутида, очищенного с помощью ОФ-ВЭЖХ, составляла более 94% с содержанием близкородственных примесей менее 0,50%.
рН данной объединенной фракции доводили до 7,8 и упаривали при температуре 35°С для удаления органического растворителя. Осаждение проводили при рН 4,9 и выделяли лираглутид, очищенный с помощью ОФ-ВЭЖХ-1.
Продукт лираглутид после ОФ-ВЭЖХ-1 использовали далее для ОФ-ВЭЖХ-2.
ОФ-ВЭЖХ-2
3,1 л лираглутида, очищенного с помощью ОФ-ВЭЖХ-1, растворенного в 50 мМ растворе гидрофосфата натрия, содержащем 25% метанола, в концентрации 3 мг/мл, наносили на колонку емкостью 2,4 л, заполненную С8 модифицированным силикагелем (размер частиц 10-13 мкм), уравновешенную приблизительно 7,2 л 50 мМ натрий-фосфатного буфера с величиной рН 7,5, содержащего 5% ацетонитрила. Продукт элюировали, используя градиент до 41% В. Длину волны детектирования устанавливали на уровне 215 нм. Температуру хроматографирования поддерживали на уровне 25°С. Собирали отдельные фракции и анализировали их чистоту. Фракции с чистотой более 98,0% объединяли и упаривали при температуре 35°С для удаления органического растворителя. Осаждение проводили при величине рН 4,9. Очищенный лираглутид лиофилизировали. Чистота по ВЭЖХ лиофилизированного порошкообразного продукта составляла более 99% с содержанием примесей не более 0,20%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХРОМАТОГРАФИЧЕСКИЕ СПОСОБЫ И СОЕДИНЕНИЯ, ОЧИЩЕННЫЕ ЭТИМИ СПОСОБАМИ | 2010 |
|
RU2508294C2 |
| СПОСОБ ОЧИСТКИ ЦИКЛИЧЕСКОГО ИЛИ НЕЦИКЛИЧЕСКОГО ПЕПТИДА | 2008 |
|
RU2461564C2 |
| ПРЕПАРАТИВНЫЙ ХРОМАТОГРАФИЧЕСКИЙ СПОСОБ НА ОСНОВЕ НЕЛИНЕЙНОГО ГРАДИЕНТА И ПРОДУКТЫ, ОЧИЩЕННЫЕ ЭТИМ СПОСОБОМ | 2010 |
|
RU2489441C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАЛЬКОБУТРОЛА | 2019 |
|
RU2779668C1 |
| ОПТИМИЗИРОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ДИМЕРНОГО ПЕПТИД-ФОСФОЛИПИДНОГО КОНЪЮГАТА | 2019 |
|
RU2784866C1 |
| СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ИНСУЛИНА ГЛАРГИНА | 2008 |
|
RU2495131C2 |
| СИНТЕЗ ЛИРАГЛУТИДА | 2017 |
|
RU2766331C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ИНДИВИДУАЛЬНЫХ СТРЕПТОТРИЦИНОВ | 2006 |
|
RU2322673C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКООЧИЩЕННОГО КРИСТАЛЛИЧЕСКОГО ИНСУЛИНА ЛЮБОГО ПРОИСХОЖДЕНИЯ | 2011 |
|
RU2453331C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТИОФОСФАТНЫХ ОЛИГОНУКЛЕОТИДОВ ДЛЯ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ | 2024 |
|
RU2836132C1 |
В настоящем изобретении предложен способ очистки лираглутида методом обращенно-фазовой высокоэффективной жидкостной хроматографии с использованием селективных агентов для образования пары ионов, позволяющий очищать неочищенный лираглутид от близкородственных примесей. 2 н. и 4 з.п. ф-лы, 4 ил., 1 табл., 3 пр.
1. Способ очистки неочищенного лираглутида, при этом способ включает в себя:
a. получение раствора лираглутида путем растворения неочищенного лираглутида в смеси, содержащей водный раствор кислоты и ацетонитрил;
b. подвергание указанного раствора неочищенного лираглутида первой очистке методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием водного раствора кислоты и агента для образования пары ионов в качестве подвижной фазы А и ацетонитрила, содержащего спирт, в качестве подвижной фазы В;
c. подвергание лираглутида после первой очистки методом ВЭЖХ второй очистке методом ВЭЖХ; и
d. выделение очищенного лираглутида.
2. Способ по п. 1, где агент для образования пары ионов выбирают из соли алкансульфоновой кислоты.
3. Способ по п. 2, где соль алкансульфоновой кислоты выбирают из группы, состоящей из натриевой соли 1-октансульфоновой кислоты, или натриевой соли 1-гептансульфоновой кислоты, или натриевой соли 1-гексансульфоновой кислоты.
4. Способ по п. 2, где соль алкансульфоновой кислоты представляет собой натриевую соль 1-октансульфоновой кислоты.
5. Способ по п. 1, где водный раствор кислоты выбирают из лимонной кислоты, уксусной кислоты, трифторуксусной кислоты или муравьиной кислоты.
6. Способ очистки лираглутида методом ВЭЖХ с использованием агента для образования пары ионов.
| УЗЕЛ КРЕПЛЕНИЯ ПЛАСТИКОВЫХ ТРУБ В ОПОРНЫХ КОНСТРУКЦИЯХ МЕБЕЛИ | 2022 |
|
RU2813514C1 |
| Moldoveanu, S | |||
| C et al "Retention Mechanisms in Different HPLC Types | |||
| Essentials in Modern HPLC Separations, 2013, 145-190, doi:10.1016/b978-0-12-385013-3.00 | |||
| WO 2016059609 A1, 21.04.2016 | |||
| WO2018104922 A1, 14.06.2018 | |||
| Е.Л | |||
| СТЫСКИН, Л.Б | |||
| ИЦИКСОН, Е.В | |||
| БРАУДЕ, Практическая Высокоэффективная Жидкостная ХРОМАТОГРАФИЯ, М., | |||
