Данная заявка испрашивает приоритет временной заявки на патент США 61/139875, поданной 22 декабря 2008 г, полностью включенной в настоящее описание путем ссылки.
Настоящее изобретение относится к новым липопептидным соединениям. Изобретение также относится к фармацевтическим композициям указанных соединений и к способам применения данных соединений в качестве противобактериальных средств. Изобретение также относится к способам получения указанных новых липопептидных соединений и промежуточных соединений, используемых при получении данных соединений, а также к композициям с их использованием.
Быстрое возрастание частоты грамположительных инфекций, включая те, которые вызываются устойчивыми бактериями, пробудил возобновленный интерес к разработке новых классов антибиотиков. Класс соединений, которые проявили потенциал в качестве полезных антибиотиков, включает A-21978C липопептиды, описанные, например, в патентах США RE 32333; RE 32455; RE 32311; RE 32310; 4482487; 4537717; 6911525; 7335725; 7408025; 6794490; 7262268; 7335726; и RE 39071. Даптомицин, член данного класса, имеет высокую бактерицидную активность in vitro и in vivo против клинически релевантных грамположительных бактерий, которые вызывают тяжелые и угрожающие жизни заболевания. Данные бактерии включают устойчивые патогенны, такие как устойчивые к ванкомицину энтерококки (VRE), устойчивый к метициллину Staphylococcus aureus (MRSA), восприимчивый к гликопептидным промежуточным соединениям Staphylococcus aureus (GISA), коагулазонегативные стафилококки (CNS) и устойчивый к пенициллину Streptococcus pneumoniae (PRSP), по поводу инфекции которыми имеется мало терапевтических альтернатив. См., например, публикацию Tally et al., 1999, Exp. Qpin. Invest. Drugs 8:1223-1238.
Несмотря на перспективу, которую открывают такие противобактериальные средства, как даптомицин, сохраняется потребность в новых антибиотиках. Многие патогенны повторно подвергались воздействию обычно используемых антибиотиков. Данное воздействие привело к отбору вариантных бактериальных штаммов, устойчивых к широкому спектру антибиотиков. Утрата активности и эффективности антибиотика, вызванная механизмами устойчивости, делает антибиотик неэффективным и, следовательно, может привести к угрожающим жизни инфекциям, которые, в сущности, неизлечимы. По мере поступления на рынок новых антибиотиков у патогенов может развиться устойчивость или промежуточная устойчивость к указанным новым лекарственным средствам, эффективно создавая потребность в потоке новых противобактериальных средств для борьбы с данными появляющимся штаммами. Кроме того, соединения, которые проявляют бактерицидную активность, имели бы преимущества перед имеющимися в настоящее время бактериостатическими соединениями. Таким образом, следовало бы ожидать, что новые синтетические противобактериальные средства могут применяться для лечения не только «естественных» патогенов, но также патогенов, устойчивых к промежуточным лекарственным средствам и к лекарственным средствам, потому что патоген никогда не подвергался воздействию нового противобактериального средства. Кроме того, новые противобактериальные средства могут проявлять дифференциальную эффективность против различных типов патогенов.
Один патоген, вызывающий особую озабоченность, представляет собой Clostridium difficile. C. difficile стал огромной проблемой для общественного здравоохранения и в последние годы стал самой частой причиной внутрибольничной инфекционной диареи. Современные варианты лечения заболеваний, связанных с Clostridium difficile, часто недостаточно эффективны, что приводит к несостоятельности лечения и высокой частоте рецидивов у некоторых пациентов. Кроме того, постоянно развиваются новые, высоковирулентные штаммы C. difficile. Представляется, что один новый эпидемический штамм (PFGE типа BI/NAP1, также называемый Nap1, риботип 027 или NAP1/027) более вирулентен, чем многие другие штаммы. Поскольку частота связанных с Clostridium difficile заболеваний продолжает возрастать и развиваются высоковирулентные штаммы, то требуются новые противобактериальные средства для лечения или профилактики данного заболевания.
Настоящее изобретение направлено на решение данной проблемы предоставлением новых липопептидных соединений, которые обладают противобактериальной активностью против широкого спектра бактерий, включая устойчивые к лекарственным средствам бактерии и C. difficile. Кроме того, соединения по настоящему изобретению проявляют бактерицидную активность.
Настоящее изобретение в одном аспекте относится к антибактериальным соединениям формулы (I):

и к их фармацевтически приемлемым солям, где:
R представляет:
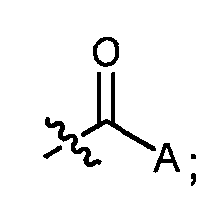 и
и
группа A выбрана из алкила, алкенила, циклоалкила, арила, гетероарила и NHRA,
где группа RA выбрана из алкила и циклоалкила.
В другом варианте осуществления изобретение также относится к фармацевтическим композициям, содержащим соединения формулы I и способам их применения.
В еще одном варианте осуществления изобретение также относится к способам получения соединений формулы I и их фармацевтическим композициям.
В еще одном варианте осуществления изобретение относится к способам применения соединений формулы I для лечения бактериальных инфекций у людей.
Следует понимать, что и предшествующее общее описание, и следующее детальное описание являются лишь иллюстративными и объяснительными, а не ограничивающими изобретение, заявленное в формуле.
Пока нет иных уточнений, молекулярные термины при использовании в настоящей заявке имеют их обычное значение.
Термин «ацил» определяется как карбонильный радикал, присоединенный к алкильной, алкенильной, алкинильной, циклоалкильной, гетероциклильной, арильной или гетероарильной группе, причем примеры включают без ограничения такие радикалы, как ацетил и бензоил.
Термин «амино» обозначает азотный радикал, содержащий два заместителя, независимо выбранные из группы, состоящей из H, алкила, циклоалкила, карбоалкокси, гетероциклила, арила, гетероарила и сульфонила. Поднаборами термина «амино» являются (1) термин «незамещенный амино», который обозначает радикал NH2, (2) термин «монозамещенный амино», которые определяется как азотный радикал, содержащий атом водорода и группу заместителя, выбранную из алкила, циклоалкила, гетероциклила, арила или гетероарила, и (3) термин «дизамещенный амино» определяется как азотный радикал, содержащий две группы заместителей, независимо выбранные из алкила, циклоалкила, гетероциклила, арила и гетероарила. Иллюстративные монозамещенные амино радикалы представляют собой «амино(низшие) монозамещенные» радикалы, посредством чего группа заместителя представляет собой группу низшего алкила. Иллюстративными дизамещенными аминорадикалами являются «амино (низшие) дизамещенные» радикалы, посредством чего группы заместителей представляют низший алкил.
Термин «ацилокси» обозначает кислородный радикал, примыкающий к ацильной группе.
Термин «ациламино» обозначает азотный радикал, примыкающий к ацильной группе.
Термин «карбоалкокси» определяется как карбонильный радикал, примыкающий к алкокси или арилоксигруппе.
Термин «карбоксиамидо» обозначает карбонильный радикал, примыкающий к аминогруппе.
Термин «галоид» определяется как бром-, хлор-, фтор- или йод- радикал.
Термин «тио» обозначает радикал двухвалентной серы, содержащий группу заместителя, независимо выбранную из H, алкила, циклоалкила, гетероциклила, арила и гетероарила. Примеры включают метилтио и фенилтио.
Пока нет иных уточнений, термин «алкил» определяется как линейный или разветвленный, насыщенный радикал, имеющий от одного до примерно двадцати атомов углерода. Иллюстративные алкильные радикалы включают C1-C12, C1-C8, C1-C6 и C4-C6 алкильные группы. Один или более атомов водорода могут быть также замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, оксо, гуанидино, формила и боковой цепи аминокислот. Примеры алкильных групп включают без ограничения метил, трет-бутил, изопропил и метоксиметил. Поднаборами термина «алкил» являются (1) «незамещенный алкил», который определяется как алкильная группа, не несущая группы заместителей; (2) «замещенный алкил», который обозначает алкильный радикал, в котором (a) один или более атомов водорода замещен группой заместителя, выбранной из ацила, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, нитро, тио, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, N-сульфонилкарбоксиамидо, N-ациламиносульфонила или (b) каждый из двух или более атомов водорода замещен группой заместителя, независимо выбранной из гидроксила, карбокси, C1-C3 алкокси, амино, ациламино, оксо или гуанидино; и (3) термин «выбранный замещенный алкил», который обозначает алкильный радикал, в котором (a) одна часть замещена группой, выбранной из гидроксила, карбокси C1-C3 алкокси, незамещенной амино, ациламино или ациламинофенила или (b) от одного до трех протонов замещен галоидным заместителем.
Алкил может быть также «прерван», по меньшей мере, одной «прерывающей функциональной группой», выбранной из арила, циклоалкила, гетероциклоалкила, O, S и N. Используемая в настоящем описании фраза «прерван» означает, что внутренняя метиленовая единица замещена, по меньшей мере, одной из функциональных групп, как определено выше. Примеры алкильных цепей, которые были «прерваны» O, включают -CH2OCH2-, -CH2O(CH2)2-, -CH2O(CH2)S-, -CH2O(CH2)4-, -(CH2)2OCH2, -(CH2)2O(CH2)2-, -(CH2)2O(CH2)3-, - (CH2)3O(CH2)-, -(CH2)3O(CH2)2- и -(CH2)4O(CH2)-. Другие примеры алкильных цепей, которые были «прерваны» функциональными группами, включают -CH2ZCH2-, - CH2Z(CH2)2-, -CH2Z(CH2)3-, -CH2Z(CH2J4-, -(CH2)2ZCH2-, -(CH2)2Z(CH2)2-, -(CH2)2Z(CH2)3-, -(CH2)3Z(CH2)-, -(CH2)3Z(CH2)2- и -(CH2)4Z(CH2)-, где Z представляет одну из «прерывающих функциональных групп», перечисленных выше.
Термин «алкенил» определяется как линейные или разветвленные радикалы, имеющие от двух до примерно двадцати атомов углерода, например, от трех до примерно десяти атомов углерода, и содержащие, по меньшей мере, одну межуглеродную двойную связь. Один или более атомов водорода могут также быть замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, формила, оксо и гуанидино. Часть (части) с двойной связью ненасыщенной углеводородной цепи может быть или в цис, или транс конфигурации. Примеры алкенильных групп включают без ограничения этиленил или фенил этиленил.
Термин «алкинил» обозначает линейные или разветвленные радикалы, имеющие от двух до примерно десяти атомов углерода, и содержащие, по меньшей мере, одну межуглеродную тройную связь. Один или более атомов водорода могут также быть замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, формила, оксо и гуанидино. Пример алкинильной группы включает без ограничения пропинил.
«Алкил», «алкенил» и «алкинил» могут также быть «прерваны», по меньшей мере, одной группой, выбранной из арила, циклоалкила, гетероциклоалкила, O, S и N.
Термин «арил» или «арильное кольцо» обозначает ароматические радикалы в одной или конденсированной карбоциклической кольцевой системой, имеющей от 5 до 14 членов кольца. В одном варианте осуществления, кольцевая система имеет от 6 до 10 членов кольца. Один или более атомов водорода могут также быть замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, азидо, алкилтио, карбоалкокси, корбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сфльфонила и формила. Примеры арильных групп включают без ограничения фенил, нафтил, бифенил, терфенил. Поднаборами термина арил являются (1) термин «фенил», который обозначает соединение формулы:
(2) термин «замещенный фенил», который определяется как фенильный радикал, в котором один или более атомов водорода замещены группой заместителя, выбранной из ацила, амино, ацилокси, азидо, алкилтио, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, N-сульфонилкарбоксиамидо и N-ациламиносульфонила и (3) термин «ациламинофенил» обозначает фенильный радикал, в котором один атом водорода замещен ациламиногруппой. Один или более дополнительных атомов водорода могут быть также замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, азидо, алкилтио, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, N-сульфонилкарбоксиамидо и N-ациламиносульфонила.
«Гетероарил» или «гетероарильное кольцо» обозначает ароматический радикал, который содержит от 1 до 4 гетероатомов или гетерогрупп, выбранных из O, N, S,  или
или  , в одной или конденсированной гетероциклической кольцевой системе, имеющей от 5 до 15 членов кольца. В одном варианте осуществления гетероарильная кольцевая система имеет от 6 до 10 членов кольца. Один или более атомов водорода могут также быть замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, тиокарбонила, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила и формила. Примеры гетероарильных групп включают без ограничения пиридинильную, тиазолильную, тиодиазолильную, изохинолинильную, пиразолильную, оксазолильную, оксадиазолильную, триазолильную и пирролильную группы. Поднаборами термина гетероарил являются (1) термин «пиридинил», который обозначает соединения формулы:
, в одной или конденсированной гетероциклической кольцевой системе, имеющей от 5 до 15 членов кольца. В одном варианте осуществления гетероарильная кольцевая система имеет от 6 до 10 членов кольца. Один или более атомов водорода могут также быть замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, тиокарбонила, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила и формила. Примеры гетероарильных групп включают без ограничения пиридинильную, тиазолильную, тиодиазолильную, изохинолинильную, пиразолильную, оксазолильную, оксадиазолильную, триазолильную и пирролильную группы. Поднаборами термина гетероарил являются (1) термин «пиридинил», который обозначает соединения формулы:
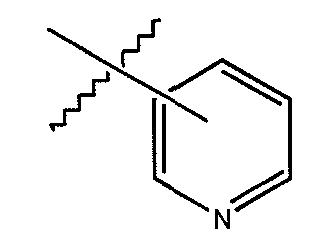
(2) термин «замещенный пиридинил», который определяется как пиридинильный радикал, в котором один или более атомов водорода замещены группой заместителя, выбранной из ацила, амино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, N-сульфонилкарбоксиамидо и N-ациламиносульфонила, и (3) термин «ациламинопиридинил», который обозначает пиридинильный радикал, в котором один атом водорода замещен ациламиногруппой, дополнительно, один или более дополнительных атомов водорода также могут быть замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, тиокарбонила, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила, N-сульфонилкарбоксиамидо и N-ациламиносульфонила.
Термин «циклоалкил» или «циклоалкильное кольцо» определяется как насыщенное или частично ненасыщенное карбоциклическое кольцо в одной или конденсированной карбоциклической кольцевой системе, имеющей от 3 до 12 членов кольца. В одном варианте осуществления циклоалкил представляет собой кольцевую систему, имеющую от 3 до 12 членов кольца. Один или более атомов водорода могут быть также замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила и формила. Примеры циклоалкильной группы включают без ограничения циклопропил, циклобутил, циклогексил и циклогептил.
Термин «гетероциклил», «гетероциклический» или «гетероциклильное кольцо» определяется как насыщенное или частично ненасыщенное кольцо, содержащее от 1 до 4 гетероатомов или гетерогрупп, выбранных из O, N, NH, 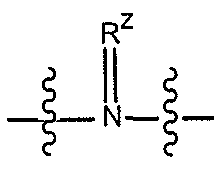 , где радикал RZ представляет, как определено для Rx,
, где радикал RZ представляет, как определено для Rx,  или
или  , в одной или конденсированной гетероциклической кольцевой системе, имеющей от 3 до 12 членов кольца. В одном варианте осуществления гетероциклил представляет собой кольцевую систему, имеющую от 3 до 7 членов кольца. Один или более атомов водорода могут быть также замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, оксо, тиокарбонила, имино, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила и формила. Примеры гетероциклильной группы включают без ограничения морфолинил, пиперидинил и пирролидинил.
, в одной или конденсированной гетероциклической кольцевой системе, имеющей от 3 до 12 членов кольца. В одном варианте осуществления гетероциклил представляет собой кольцевую систему, имеющую от 3 до 7 членов кольца. Один или более атомов водорода могут быть также замещены группой заместителя, выбранной из ацила, амино, ациламино, ацилокси, оксо, тиокарбонила, имино, карбоалкокси, карбокси, карбоксиамидо, циано, галоида, гидроксила, нитро, тио, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, алкокси, арилокси, сульфинила, сульфонила и формила. Примеры гетероциклильной группы включают без ограничения морфолинил, пиперидинил и пирролидинил.
Термин «алкокси» обозначает оксисодержащие радикалы, замещенные алкильной, циклоалкильной или гетероциклильной группой. Примеры включают без ограничения метокси, трет-бутокси, бензилокси и циклогексилокси.
Термин «арилокси» обозначает оксисодержащие радикалы, замещенные арильной или гетероарильной группой. Примеры включают без ограничения фенокси.
Термин «аминокислотная боковая цепь» обозначает любую боковую цепь (R группу) из естественно встречающейся или не встречающейся в естественных условиях аминокислоты.
Термин «сульфинил» определяется как радикал четырехвалентной серы, замещенный оксозаместителем, и вторым заместителем, выбранным из группы, состоящей из алкильной, циклоалкильной, гетероциклильной, арильной или гетероарильной группы.
Термин «сульфонил» определяется как радикал шестивалентной серы, замещенный двумя оксозаместителями, и третьим заместителем, выбранным из алкильной, циклоалкильной, гетероциклильной, арильной или гетероарильной группы.
Термин «карбаматная аминозащитная группа» определяется как признанная аминозащитная группа, которая при связывании с аминогруппой образует карбамат. Примеры карбаматных аминозащитных групп можно найти в публикации «Protective Groups in Organic Synthesis» by Theodora W. Greene, John Wiley and Sons, New York, 1981. Примеры карбаматных аминозащитных групп включают бензилоксикарбонил, т-бутоксикарбонил, т-амилоксикарбонил, изолборнилоксикарлбонил, адамантилоксикарбонил, хлорбензилоксикарбонил, нитробензилоксикарбонил или тому подобные.
Соли соединений по изобретению включают кислотно-аддитивные соли и основно-аддитивные соли. В одном варианте осуществления соль представляет собой фармацевтически приемлемую соль соединения формулы I. Термин «фармацевтически приемлемые соли» включает соли, обычно используемые для образования солей щелочных металлов и для образования аддитивных солей свободных кислот или свободных оснований. Природа соли не имеет решающего значения при условии, что она фармацевтически приемлема. Подходящие фармацевтически приемлемые кислотно-аддитивные соли соединений по изобретению могут быть получены из неорганической кислоты или органической кислоты. Примеры таких неорганических кислот включают без ограничения хлористоводородную, бромистоводородную, йодистоводородную, азотную, карбоновую, серную и фосфорную кислоту. Примеры соответствующих органических кислот могут быть выбраны из алифатических, циклоалифатических, ароматических, арилалифатических, гетероциклических, карбоновых и сульфоновых классов органических кислот, примеры которых включают без ограничения муравьиную, уксусную, пропионовую, янтарную, гликолевую, глюконовую, малеиновую, эмбоновую (памоевую), метансульфоновую, этансульфоновую, 2-гидроксиэтансульфоновую, пантотеновую, бензолсульфоновую, толуолсульфоновую, сульфанильную, месиловую, циклогексиламиносульфоновую, стеариновую, альгеновую, β-гидроксимасляную, малоновую, галактовую и галактуроновую кислоту. Подходящие фармацевтически приемлемые основно-аддитивные соли соединений по изобретению включают без ограничения соли металлов, полученные из алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, полученные из N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, N-метилглюкамина, лизина и прокаина. Все указанные соли могут быть получены обычными средствами из соответствующего соединения по изобретению обработкой, например, соединения по изобретению соответствующей кислотой или основанием.
Соединения по изобретению могут иметь один или более асимметричных атома углерода и, таким образом, способны существовать в форме оптических изомеров, а также в форме их рацемических или нерацемических смесей. Соединения по изобретению могут использоваться в настоящем изобретении в виде одного изомера или в виде смеси стереохимических изомерных форм. Диастереоизомеры, т.е. не налагаемые стереохимические изомеры, могут отделяться обычными средствами, такими как хроматография, отгонка, кристаллизация или сублимация. Оптические изомеры могут быть получены разделением рацемических смесей в соответствии с обычными способами, например, образованием диастереоизомерных солей обработкой оптически активной кислотой или основанием. Примеры соответствующих кислот включают без ограничения винную, диацетилвинную, дибензоилвинную, дитолуолвинную и камфорсульфоновую кислоту. Смесь диастереомеров может быть разделена кристаллизацией с последующим освобождением оптически активных оснований из данных солей. Альтернативный способ разделения оптических изомеров включает использованием хиральной хроматографической колонки, оптимально выбранной для максимизации разделения энантиомеров. Еще один доступный способ включает синтез ковалентных диастереоизомерных молекул взаимодействием соединений по изобретению с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Синтезированные диастереоизомеры могут быть разделены обычными средствами, такими как хроматография, отгонка, кристаллизация или сублимация, а затем гидролизоваться для получения энантимерно чистого соединения. Оптически активные соединения по изобретению могут быть аналогичным образом получены использованием оптически активных исходных материалов. Эти изомеры могут быть представлены в форме свободной кислоты, свободного основания, сложного эфира или соли.
Изобретение также охватывает изолированные соединения. Изолированное соединение относится к соединению, которое представляет, по меньшей мере, 10%, например, по меньшей мере, 20, по меньшей мере, 50% и, кроме того, например, по меньшей мере, 80% соединения, присутствующего в смеси. В одном варианте осуществления соединение, его фармацевтически приемлемая соль или фармацевтическая композиция, содержащая соединение, проявляет выявляемую (т.е. статистически значимую) противомикробную активность при тестировании в обычных биологических анализах, таких как анализы, описанные в настоящей заявке.
В одном варианте осуществления изобретение относится к соединениям, имеющим формулу (I):
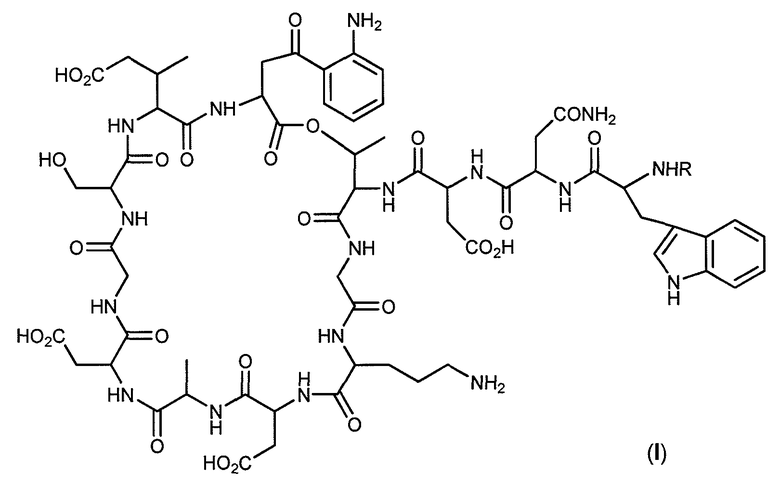
и к их фармацевтически приемлемым солям, где:
R представляет:
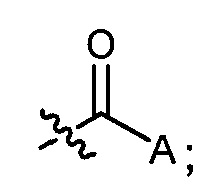 и
и
группа A выбрана из арила, алкенила, циклоалкила, арила, гетероарила и NHRA,
где группа RA выбрана из алкила и циклоалкила.
В одном варианте осуществления группа A выбрана из алкила, такого как C1-14 алкил, такого как C1-6 алкил, и, кроме того, такого как C4-6 алкил. В еще одном варианте осуществления группа A выбрана из алкила, замещенного одним или более циклоалкилом или арилом.
В другом варианте осуществления группа A выбрана из:
a) незамещенного C1-14 алкила;
b) C4-C6 алкила, замещенного одним или более циклоалкилом;
c) C4-C6 алкила, прерванного одним или более арилом;
d) C1-C6 алкила, замещенного одним или более арилом; и
e) C6 алкила, прерванного одним или более циклоалкилом.
В другом варианте осуществления, группа A представляет C1-C6 алкил, замещенный одним или более циклогексилом. В другом варианте осуществления, группа A представляет C1-C6 алкил, прерванный одним или более фенилом. В другом варианте осуществления, группа A представляет C1-C6 алкил, замещенный одним или более фенилом.
В одном варианте осуществления, группа A выбрана из:
a) незамещенного C1-C12 алкенила;
b) 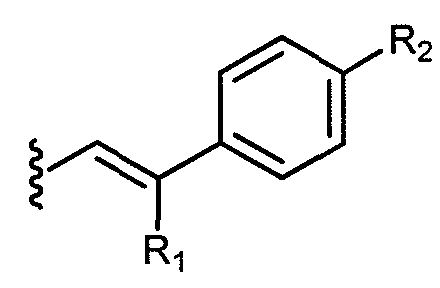 , где
, где
группа R1 выбрана из водорода или метила; и группа R2 выбрана из фенила, незамещенного C3-C7 алкила и OR3, где R3 представляет незамещенный C1-C6 алкил. В другом варианте осуществления R1 представляет метил, а R2 представляет незамещенный C3-C7 алкил.
В одном варианте осуществления группа A выбрана из:
a) C3-C8 циклоалкила, замещенного одним или более незамещенных C1-C-10 алкилом;
b) незамещенного C10-C12 циклоалкила;
c) C3-C8 циклоалкила, замещенного C3-C8 циклоалкилом; и
d) C3-C8 циклоалкила, замещенного фенилом, где указанный фенил необязательно замещен одним или более галогенами. В другом варианте осуществления группа A выбрана из циклогексила и циклопропила, каждый из которых может быть замещен одним или более незамещенным C1-C10 алкилом. В другом варианте осуществления A представляет C3-C8 циклоалкил, замещенный одни или более фенилом, где указанный фенил необязательно замещен одним или более атомами хлора.
В одном варианте осуществления группа A выбрана из:
a) фенила, замещенного незамещенным C1-C8 алкилом;
b) фенила, замещенного OR4, где R4 представляет незамещенный C1-C15 алкил;
c) фенила, замещенного циклоалкилом, где указанный циклоалкил замещен одним или более незамещенным C1-C8 алкилом;
d) фенила, замещенного фенил-OR4, где R4 представляет незамещенный C1-C8 алкил;
e) фенила, замещенного фенилом, необязательно замещенным одним или более атомами галогена; и
f) фенила, замещенного одним или более фенилом.
В другом варианте осуществления, где группа A представляет фенил, замещенный циклогексилом, где указанный циклогексил замещен незамещенным C1-C8 алкилом. В другом варианте осуществления группа A представляет фенил, замещенный фенилом, необязательно замещенным одним или более атомами хлора.
В одном варианте осуществления группа A выбрана из:
a) тиофенила, замещенного незамещенным C1-C8 алкилом; и
b) тиофенила, замещенного фенил-R5, где R5 выбран из водорода, хлор-, фенил-OR6 и SR6, где R6 представляет незамещенный C1-C6 алкил.
В одном варианте осуществления группа A выбрана из NHRA, где группа RA выбрана из незамещенного C1-C12 алкила, циклогексила, замещенного одним или более незамещенным C1-C6 алкилом, или  , где R6* представляет фенил, необязательно замещенный C1-C7 незамещенный алкил.
, где R6* представляет фенил, необязательно замещенный C1-C7 незамещенный алкил.
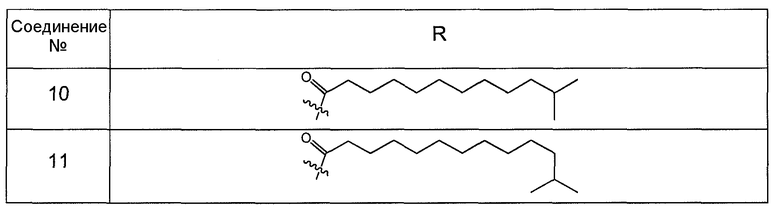
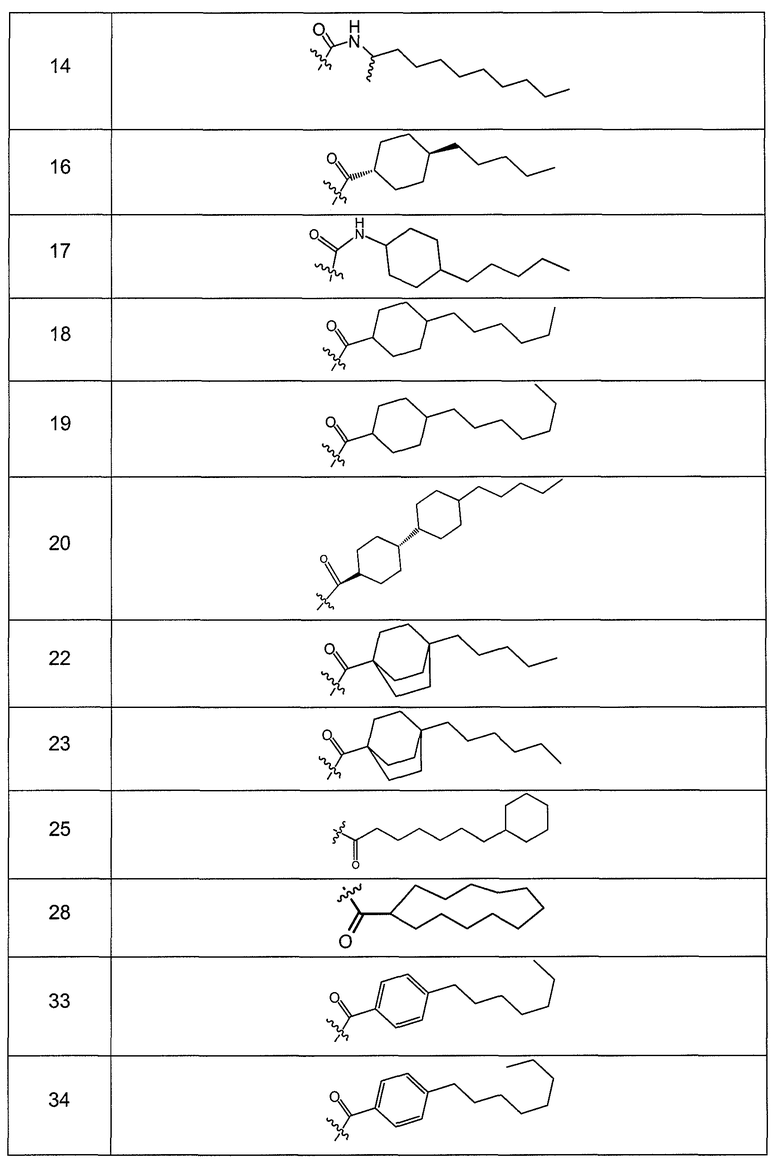
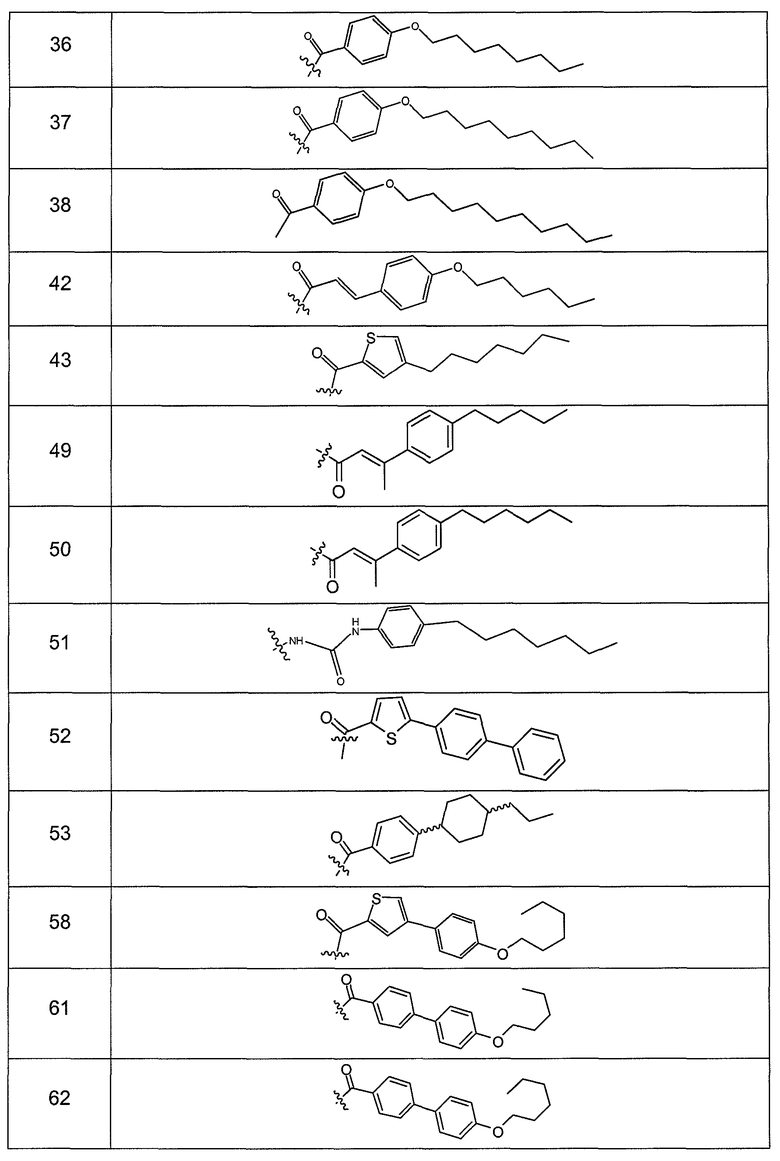
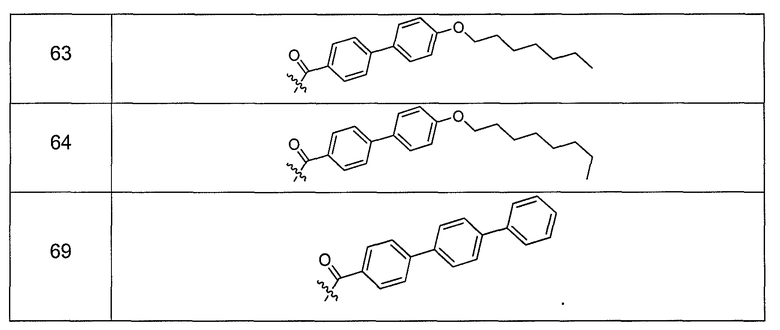
В одном варианте осуществления группа R выбрана из заместителя из таблицы I:

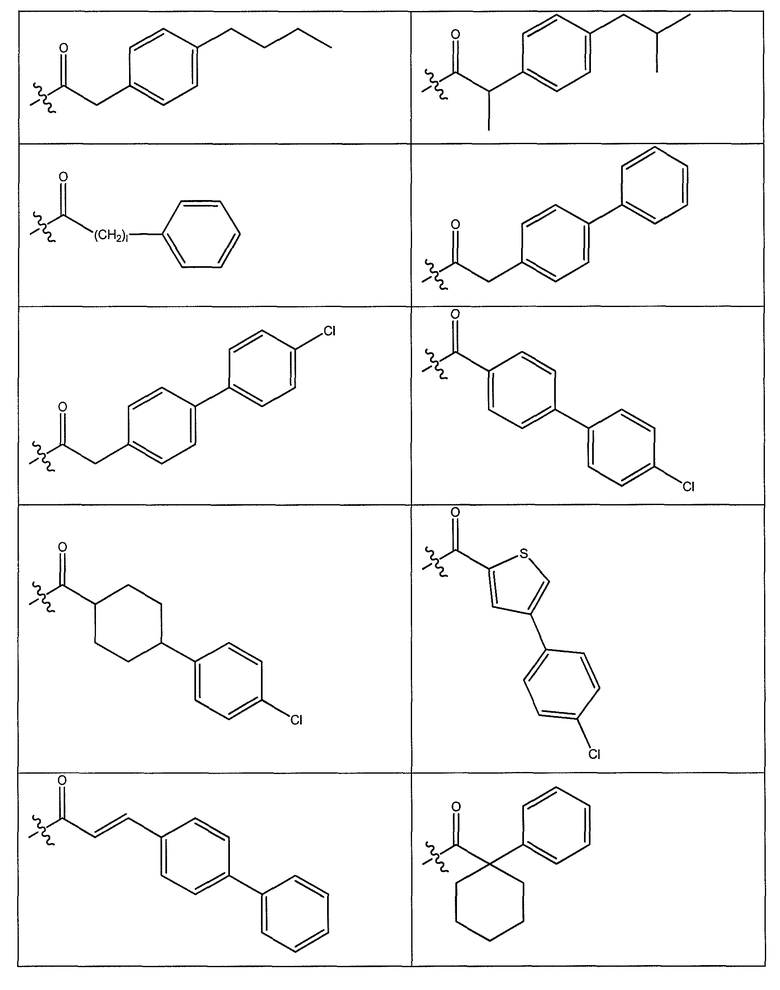
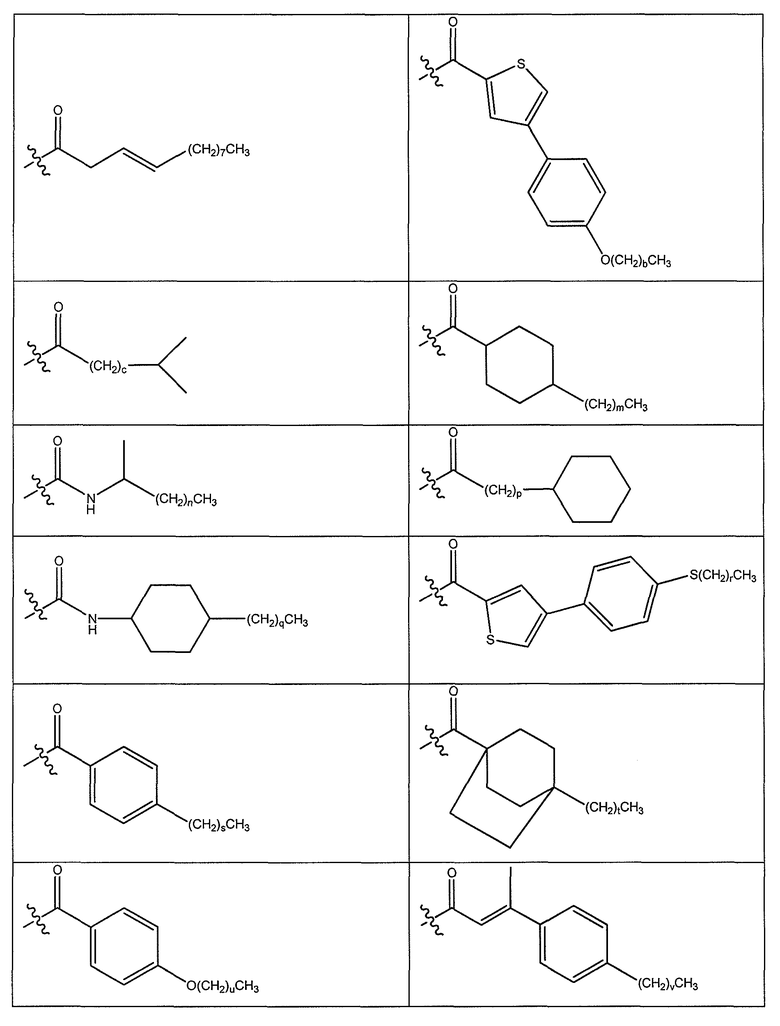
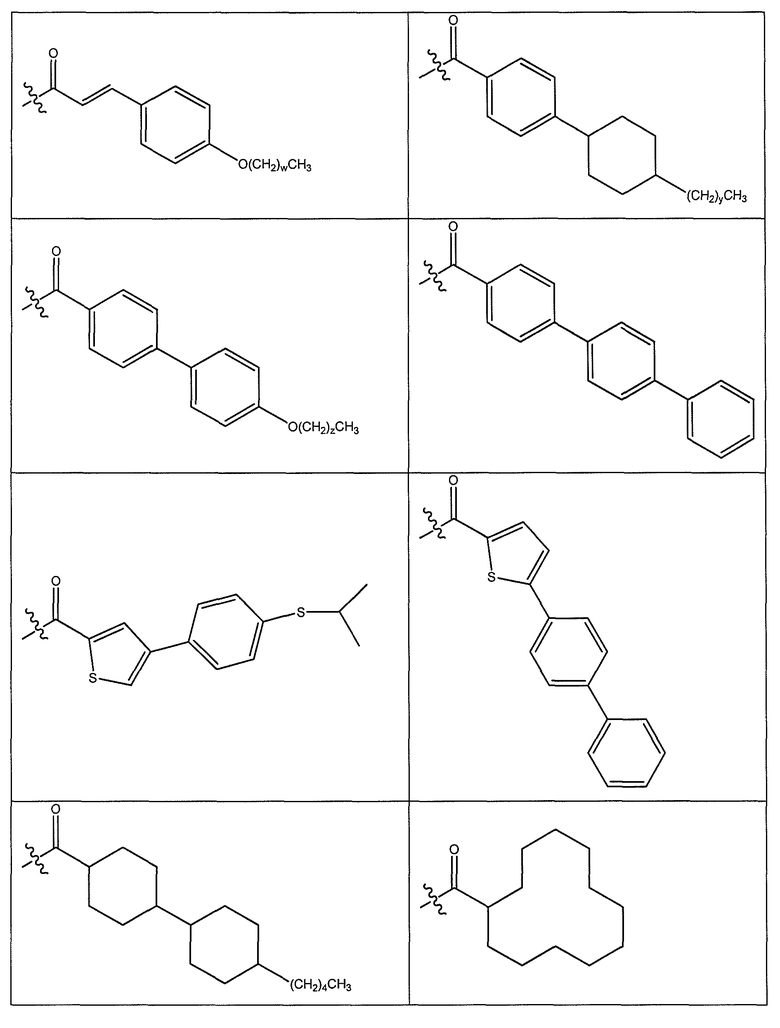

В одном варианте осуществления переменная величина b из таблицы I представляет целое число, выбранное из 0 до 7. В другом варианте осуществления переменная величина b равна 0, 1, 2, 3, 4, 5, 6 или 7. В другом варианте осуществления переменная величина b равна 5. В одном варианте осуществления переменная величина c из таблицы I представляет целое число от 6 до 10. В другом варианте осуществления переменная величина c равна 6, 7, 8, 9 или 10. В другом варианте осуществления переменная величина c равна 6, 9 или 10. В одном варианте осуществления переменная величина d из таблицы I представляет целое число, выбранное из диапазона от 6 до 14. В другом варианте осуществления переменная величина d равна 6, 7, 8, 9, 10, 11, 12, 13, или 14. В другом варианте осуществления переменная величина d равна 7, 9, 10, 11, 12 или 13. В еще одном варианте осуществления переменная величина d равна 10, 11, 12 или 13. В одном варианте осуществления переменная величина e из таблицы I представляет целое число, выбранное из диапазона от 6 до 8. В другом варианте осуществления, переменная величина e равна 6, 7 или 8. В еще одном варианте осуществления переменная величина e равна 7. Каждая из переменных величин f и f* из таблицы I представляет независимо целое число, выбранное из диапазона от 2 до 6. В одном варианте осуществления каждая из переменных величин f и f* равна 2, 3, 4, 5 или 6. В другом варианте осуществления каждая из переменных величин f и f* равна независимо 3, 4 или 5. В еще одном варианте осуществления переменная величина f равна 3, а переменная величина f* равна 5. В одном варианте осуществления переменная величина g в таблице I представляет целое число, выбранное из диапазона от 6 до 10. В другом варианте осуществления переменная величина g равна 6, 7, 8, 9 или 10. В другом варианте осуществления, переменная величина g равна 6. В одном варианте осуществления переменная величина g* представляет целое число, выбранное из диапазона от 6 до 10. В другом варианте осуществления, переменная величина g* равна 6, 7, 8, 9 или 10. В еще одном варианте осуществления переменная величина g* равна 8. В одном варианте осуществления, переменная величина I из таблицы I равна целому числу, выбранному из 5 или 6. В одном варианте осуществления переменная величина m из таблицы I равна целому числу, выбранному из диапазона от 3 до 9. В другом варианте осуществления переменная величина m равна 3, 4, 5, 6, 7, 8 или 9. В еще одном варианте осуществления переменная величина m равна 3, 4, 5 или 6. В одном варианте осуществления переменная величина n из таблицы I представляет целое число, выбранное из диапазона от 8 до 10. В другом варианте осуществления переменная величина n равна 8, 9 или 10. В еще одном варианте осуществления переменная величина n равна 8. В одном варианте осуществления переменная величина p представляет целое число, выбранное из диапазона от 4 до 8. В другом варианте осуществления переменная величина p равна 4, 5, 6, 7 или 8. В еще одном варианте осуществления переменная величина p равна 4, 5 или 6. В одном варианте осуществления переменная величина q из таблицы I представляет целое число, выбранное из диапазона от 4 до 7. В другом варианте осуществления переменная величина q равна 4, 5, 6 или 7. В еще одном варианте осуществления переменная величина q равна 4. В одном варианте осуществления переменная величина r из таблицы I представляет целое число, выбранное из диапазона от 2 до 6. В другом варианте осуществления переменная величина r равна 2, 3, 4, 5 или 6. В другом варианте осуществления переменная величина r равна 2, 3, 4 или 5. В еще одном варианте осуществления переменная величина r равна 2 или 5. В одном варианте осуществления переменная величина s представляет целое число, выбранное из диапазона от 4 до 9. В другом варианте осуществления переменная величина s равна 4, 5, 6, 7, 8 или 9. В другом варианте осуществления переменная величина s равна 4, 5, 6 или 7. В еще одном варианте осуществления, переменная величина s равна 6 или 7. В одном варианте осуществления переменная величина t из таблицы I представляет целое число, выбранное из диапазона от 4 до 9. В другом варианте осуществления переменная величина t равна 4, 5, 6, 7, 8 или 9. В еще одном варианте осуществления переменная величина t равна 4 или 5. В одном варианте осуществления переменная величина u из таблицы I представляет целое число, выбранное из диапазона от 4 до 14. В другом варианте осуществления переменная величина u равна 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14. В другом варианте осуществления переменная величина u равна 5, 7, 8, 9, 11 или 14. В еще одном варианте осуществления переменная величина u равна 5, 7, 8, 9 и 14. В одном варианте осуществления переменная величина v из таблицы I представляет целое число, выбранное из диапазона от 3 до 7. В другом варианте осуществления переменная величина v равна 3, 4, 5, 6 или 7. В другом варианте осуществления переменная величина v равна 3, 4 или 5. В одном варианте осуществления переменная величина w из таблицы I представляет целое число, выбранное из диапазона от 3 до 7. В другом варианте осуществления, переменная величина w равна 3, 4, 5, 6 или 7. В другом варианте осуществления переменная величина w равна 4 или 5. В одном варианте осуществления переменная величина x из таблицы I представляет целое число, выбранное из диапазона от 6 до 10. В другом варианте осуществления переменная величина x равна 6, 7, 8, 9 или 10. В другом варианте осуществления переменная величина x равна 6. В одном варианте осуществления переменная величина y из таблицы I представляет целое число, выбранное из диапазона от 1 до 5. В другом варианте осуществления переменная величина y равна 1, 2, 3, 4 или 5. В другом варианте осуществления переменная величина y равна 2, 3 или 4. В еще одном варианте осуществления переменная величина y равна 2 или 4. В одном варианте осуществления переменная величина z из таблицы I представляет целое число, выбранное из диапазона от 0 до 7. В другом варианте осуществления переменная величина z=0, 1, 2, 3, 4, 5, 6 или 7. В еще одном варианте осуществления переменная величина z равна 2, 3, 4, 5, 6 или 7.
В другом варианте осуществления R выбран из заместителя из таблицы II:
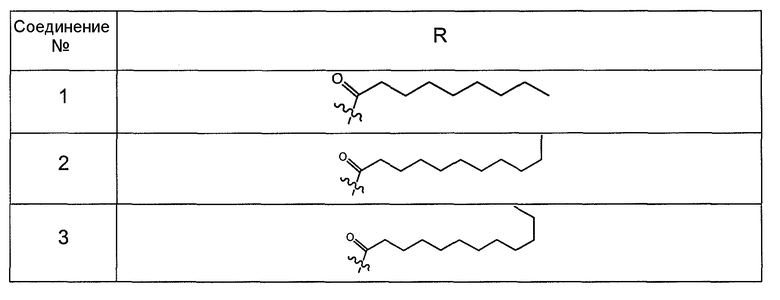
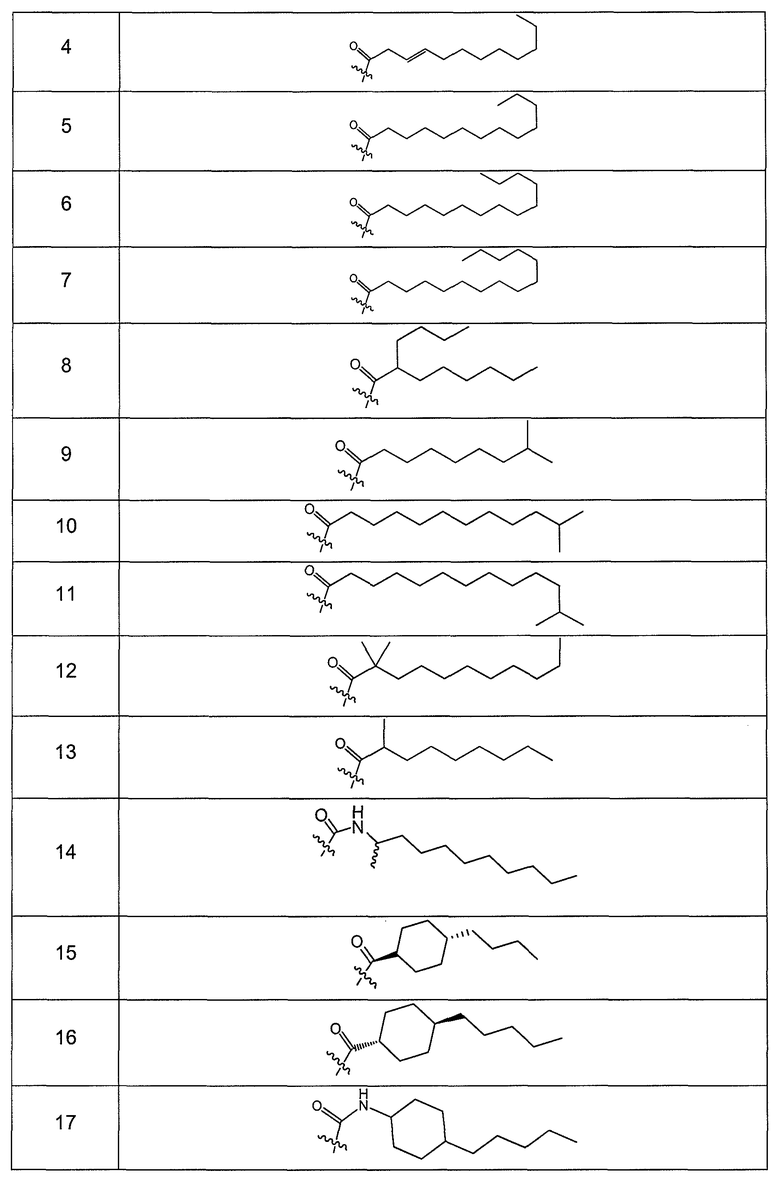
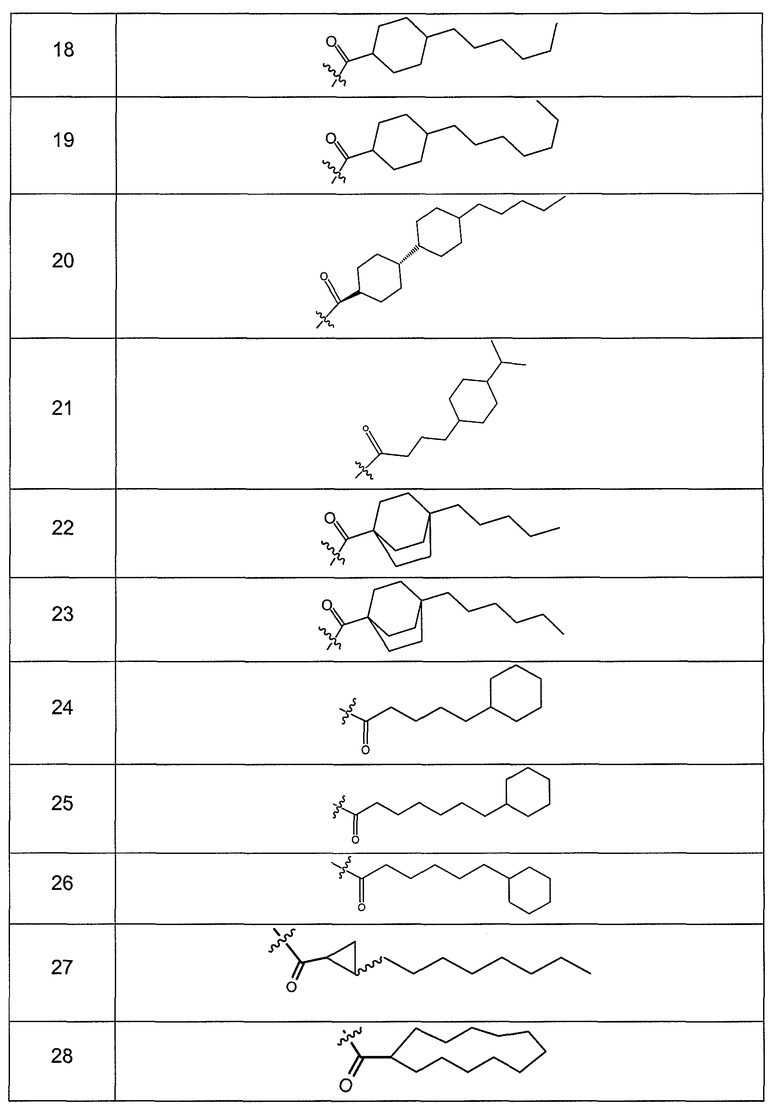
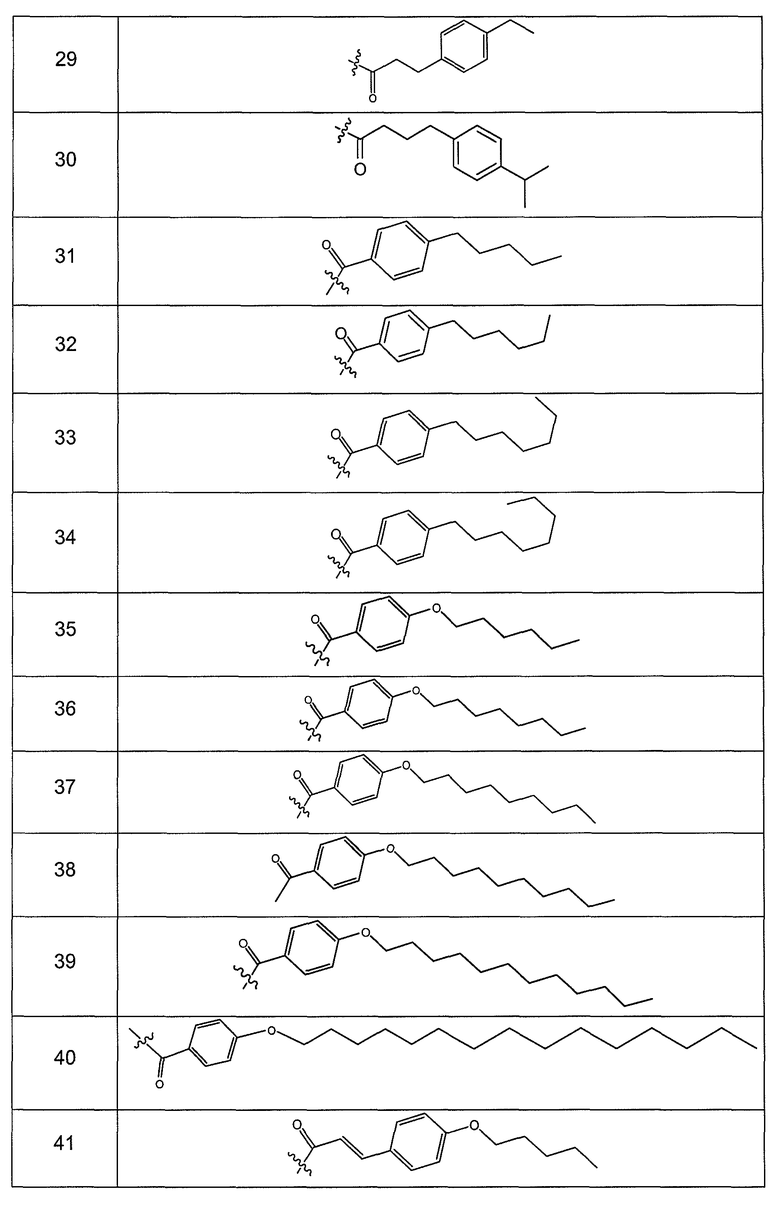
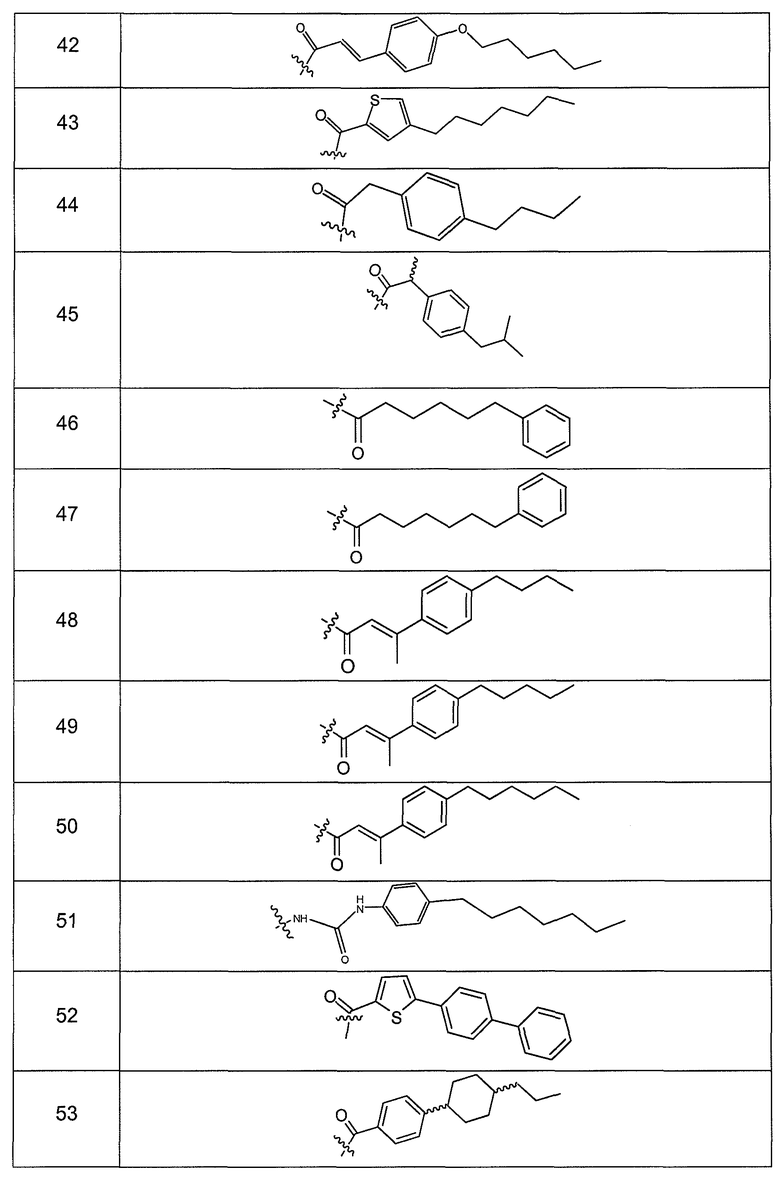
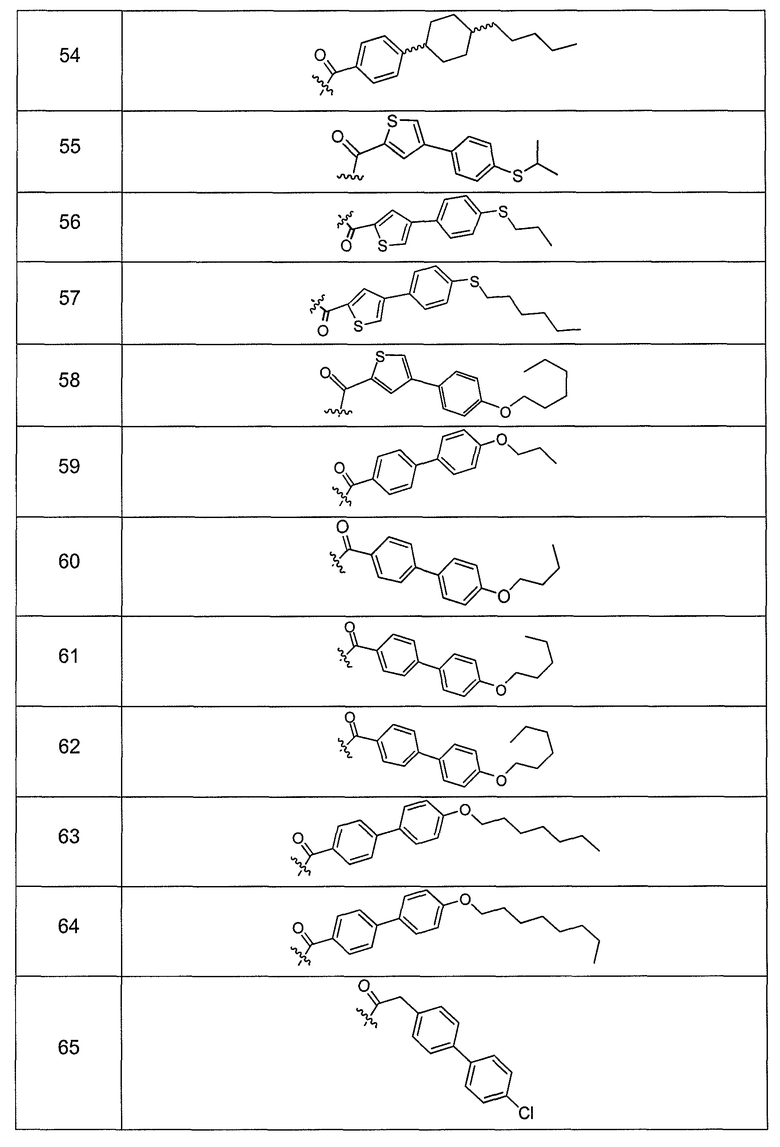
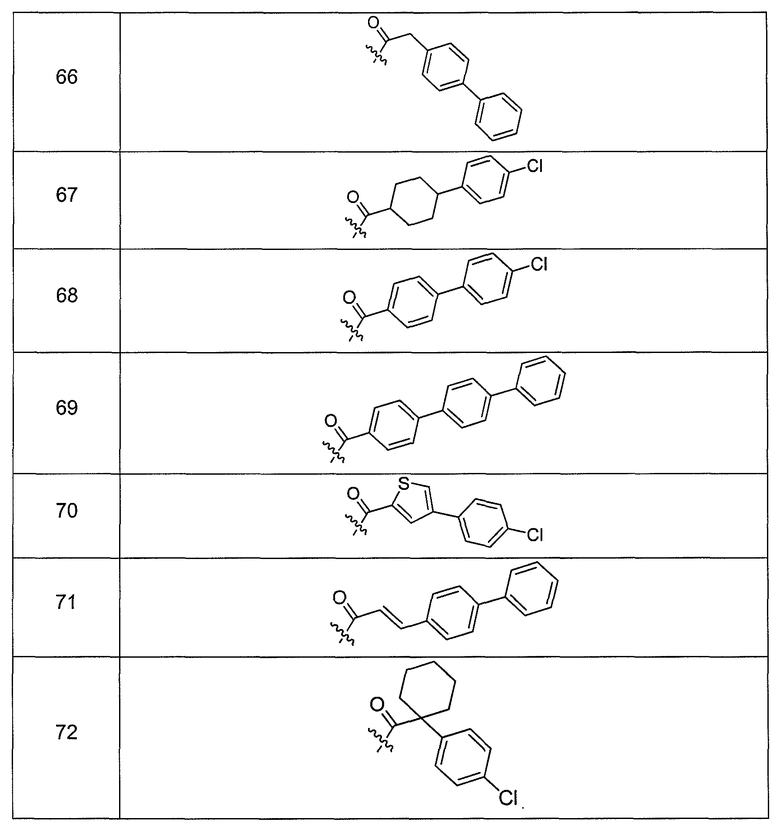
В еще одном варианте осуществления, R выбрана из группы заместителей из таблицы III:
В еще одном варианте осуществления R выбран из группы заместителей из таблицы IV:
Еще одной целью изобретения является фармацевтические композиции или препаративные формы, содержащие соединения, описанные в настоящей заявке, или их соли.
Фармацевтические композиции могут составляться для перорального, внутривенного, внутримышечного, подкожного или парентерального введения для терапевтического или профилактического лечения заболеваний, таких как бактериальные инфекции.
Фармацевтические препараты, описанные в настоящей заявке, могут быть получены в соответствии со стандартными процедурами и вводятся в дозировках, которые выбираются для уменьшения, предотвращения или устранения инфекции (см., например, руководство Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA and Goodman and Gilman's "The Pharmaceutical Basis of Therapeutics," Pergamon Press, New York, NY, содержание которого включено в настоящее описание путем ссылки, где представлено общее описание способов введения различных противомикробных средств для лечения людей).
Фармацевтические композиции могут содержать одно или более соединений, описанных в настоящей заявке, в сочетании с одним или более нетоксичными, фармацевтически приемлемыми носителями и/или разбавителями и/или адъювантами и/или эксципиентами. Используемая в настоящем описании фраза «фармацевтически приемлемый носитель» относится к любому и всем растворителям, дисперсионным средам, покрытиям, противобактериальным и противогрибковым средствам, изотоническим и задерживающим всасывание средствам и тому подобным, которые совместимы с фармацевтическим введением. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области. Не ограничивающие примеры носителей и эксципиентов включают кукурузный крахмал или желатин, лактозу, сахарозу, микрокристаллическую целлюлозу, каолин, маннит, дикальций фосфат, хлорид натрия и альгиновую кислоту. Композиции могут содержать кроскармеллозу натрия, микрокристаллическую целлюлозу, кукурузный крахмал, гликолят крахмал натрия и альгиновую кислоту.
Таблеточные связывающие вещества, которые могут быть включены, представляют собой акацию, метилцеллюлозу, карбоксиметилцеллюлозу натрия, поливинилпирролидон (повидон), гидроксипропилметилцеллюлозу, сахарозу, крахмал и этилцеллюлозу.
Смазывающие вещества, которые могут использоваться, включают стеарат магния или другие стеараты металлов, стеариновую кислоту, силиконовую жидкость, тальк, воски, масла и коллоидные частицы диоксида кремния.
Могут также использоваться ароматизирующие агенты, такие как мята перечная, масло грушанки, вишневая отдушка или тому подобные. Может быть также желательным добавление красящего агента для придания лекарственной форме более эстетичного внешнего вида или содействия идентификации продукта.
Для перорального или парентерального введения, соединения по настоящему изобретению могут смешиваться с обычными фармацевтическим носителями и эксципиентами и применяться в форме таблеток, капсул, эликсиров, суспензий, сиропов, облаток и тому подобных лекарственных форм. Композиции, включающие соединение по настоящему изобретению, могут содержать от примерно 0,1% до примерно 99% масс. активного соединения, например, от примерно 10% до примерно 30%.
Для перорального применения могут использоваться твердые препартивные формы, такие как таблетки и капсулы. Могут быть также получены препараты длительного высвобождения или препараты с энтеросолюбильным покрытием. Для применения в педиатрии и гериатрии один вариант осуществления относится к суспензиям, сиропам и жевательным таблеткам. Для перорального введения фармацевтические композиции представлены в форме, например, таблетки, капсулы, суспензии или жидкости.
Фармацевтические композиции могут быть получены в виде стандартной лекарственной формы, содержащей терапевтически эффективное количество активного ингредиента. Примерами таких лекарственных форм являются таблетки и капсулы. В целях лечения таблетки и капсулы, которые могут содержать, в дополнение к активному ингредиенту, обычные носители, такие как связывающие агенты, например, смолу акации, желатин, поливинилпирролидон, сорбит или трагакант; наполнители, например фосфат кальция, глицин, лактозу, маисовый крахмал, сорбит или сахарозу; смазывающие агенты, например стеарат магния, полиэтиленгликоль, диоксид кремния или тальк; разрыхлители, например картофельный крахмал, ароматизирующие или красящие агенты, или приемлемые смачивающие агенты. Пероральные жидкие препараты в целом представлены в форме водных или масляных растворов, суспензий, эмульсий, сиропов или эликсиров; препараты по изобретению могут содержать обычные добавки, такие как суспендирующие агенты, эмульгирующие агенты, неводные агенты, консерванты, красящие агенты и ароматизаторы. Не ограничивающие примеры добавок для жидких препаратов включают акацию, миндальное масло, этиловый спирт, фракционированное кокосовое масло, желатин, глюкозный сироп, глицерин, гидрированные пищевые жиры, лецитин, метилцеллюлозу, метил или пропил пара-гидробензоат, пропиленгликоль, сорбит или сорбиновую кислоту.
Для внутривенного (IV) применения фармацевтическая композиция может быть растворена или суспендирована в любом из обычно используемых жидкостей для внутривенного введения и вводиться инфузией. Жидкости для внутривенного введения включают без ограничения физиологический солевой раствор или раствор Рингера. Внутривенное введение может осуществляться использованием без ограничения шприца, мини-насоса или внутривенной магистрали.
Фармацевтические композиции по настоящему изобретению для парентеральной инъекции включают фармацевтически приемлемые водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления влагосодержания с превращением в стерильные инъецируемые растворы или дисперсии непосредственно перед применением. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или носителей включают воду, этанол, бензиловый спирт, полиолы (такие как глицерол, поропиленгликоль и полиэтиленгликоль) и их подходящие смеси, растительные масла (такие как кукурузное масло или оливковое масло) и инъецируемые органические сложные эфиры, такие как этилолеат. Должная текучесть может поддерживаться, например, использованием материалов покрытия, таких как лецитин, поддержанием требуемого размера частиц в случае дисперсий и использованием поверхностно-активных веществ (ПАВ). Композиции могут включать различные буферы.
Данные композиции могут также содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгирующие агенты и разрыхлители. Они могут также содержать метящие агенты или другие агенты против подделки, которые хорошо известны в данной области. Предотвращение действия микроорганизмов может быть обеспечено включением различных противобактериальных и противогрибковых средств, например, парабена, хлорбутанола и фенолсорбиновой кислоты. Может быть также желательным включение изотонических агентов, таких как сахара и хлорид натрия. Длительное всасывание инъецируемой фармацевтической формы может достигаться включением агентов, которые задерживают всасывание, таких как моностеарат аммония и желатин.
Инъецируемые депо-формы могут быть получены образованием микроинкапсулирующих капсул лекарственного средства в биологически разлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и природы конкретного используемого полимера можно регулировать скорость высвобождения лекарственного средства. Примеры других биологически разлагаемых полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Депоинъецируемые препаративные формы могут быть также получены включением лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями организма.
Инъецируемые прапаративные формы могут стерилизоваться, например, фильтрацией через удерживающие бактерии фильтры или включением стерилизующих агентов в форме стерильных твердых композиций, которые могут растворяться или диспергироваться в стерильной воде или другой стерильной инъецируемой среде непосредственно перед применением.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. Такие формы могут включать формы, которые быстро растворяются или разрушаются в среде ротовой полости. В таких твердых лекарственных формах активное соединение может быть смешано, по меньшей мере, с одним инертным, фармацевтически приемлемым эксципиентом или носителем. Подходящие эксципиенты включают, например, (a) наполнители или придающие объем агенты, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и силициловая кислота; (b) связывающие агенты, такие как целлюлоза и производные целлюлозы (такие как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза и карбоксиметилцеллюлоза), альгинаты, желатин, поливинилпирролидон, сахароза и акация; (c) увлажнители, такие как глицерол; (d) разрыхлители, такие как гликоляткрахмал натрия, кроскармеллоза, агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, определенные силикаты и карбонат натрия; (e) задерживающие растворение агенты, такие как парафин; (f) ускорители всасывания, такие как соединения четвертичного аммония; (g) смачивающие агенты, такие как цетиловый спирт и глицерол моностеарат, сложные эфиры жирных кислот сорбитана, палоксамеры и полиэтиленгликоли; (h) абсорбенты, такие как каолин и бентонитовая глина; (i) смазывающие агенты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси; и (j) глянцеватели, такие как тальк и диоксид кремния. Другие подходящие эксципиенты включают, например, цитрат натрия или дикальцийфосфат. Лекарственные формы могут также включать буферные агенты.
Твердые лекарственные формы, включая таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытиями и оболочками, такими как функциональные и эстетичные энетросолюбильные покрытия и другие покрытия, хорошо известные в области составления фармацевтических препаративных форм. Возможно содержание в них контрастирующих агентов и красящих агентов. Они могут быть также представлены в форме, способной к контролируемому или длительному высвобождению. Примеры заливочных композиций, которые могут применяться для таких целей, включают полимерные вещества и воски.
Фармацевтические композиции могут доставляться с использованием систем доставки контролируемой (например, капсул) или длительного высвобождения (например, биологически разлагаемых матриц). Иллюстративные системы доставки отсроченного высвобождения для доставки лекарственных средств, которые подходят для введения фармацевтических композиций, описаны в патентах США №№ 4452775 (выданном Kent), 5039660 (выданном Leonard) и 3854480 (выданном Zaffaroni).
В некоторых случаях для продления эффекта лекарственного средства может быть желательным замедление всасывания лекарственного средства после подкожной или внутримышечной инъекции. Это может быть достигнуто использованием жидкой суспензии кристаллического или аморфного материала с низкой растворимостью в воде. При необходимости, аморфный материал может использоваться отдельно или вместе со стабилизаторами. Скорость всасывания лекарственного средства затем зависит от скорости растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы.
Альтернативно, отсроченное всасывание парентерально введенной лекарственной формы средства может быть достигнуто растворением или суспендированием лекарственного средства в масляном носителе.
Для внутримышечных препаратов стерильная препартивная форма соединений в соответствии с формулой I, или ее подходящая растворимая солевая форма, например, гидрохлориды, может быть растворена и введена в фармацевтическом разбавителе, таком как вода для инъекций (WFI), физиологический солевой раствор или 5% глюкоза. Подходящая нерастворимая форма соединения может быть получена и введена в виде суспензии на водной основе или фармацевтически приемлемой масляной основе, например, сложном эфире длинноцепочечной жирной кислоты, такой как этилолеат.
Доза внутривенной, внутримышечной или парентеральной препаративной формы соединения в соответствии с формулой I может вводиться в виде болюса или медленной инфузией. Болюс представляет собой дозу, которая вводится менее чем за 30 мин. В одном варианте осуществления болюс вводится менее чем за 15 мин или менее чем за 10 мин. В другом варианте осуществления болюс вводится менее чем за 5 мин. В еще одном варианте осуществления болюс вводится за одну минуту или менее. Инфузия представляет собой дозу, которая вводится со скоростью 30 мин или более. В одном варианте осуществления, инфузия продолжается один час или больше. В другом варианте осуществления инфузия производится по существу постоянно.
Для местного применения фармацевтические композиции могут быть также получены в формах, подходящих для нанесения на кожу или слизистые оболочки носовой полости и горла, и могут принимать форму кремов, мазей, жидких спреев или ингаляционных форм, пастилок или средств для смазывания горла. Такие местные препаративные формы, кроме того, могут включать химические соединения, такие как диметилсульфоксид (DMSO), для содействия проникновению активного ингредиента через поверхность.
Для подачи в глаза или уши фармацевтическая композиция может быть представлена в жидкой или полужидкой форме, составленной в гидрофобных или гидрофильных основах, таких как мази, кремы, лосьоны, смазки или порошки.
Для ректального введения фармацевтические композиции могут вводиться в форме суппозиторий, смешанных с обычными носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиторий, или другой глицерид, которые являются твердыми при комнатной температуре, но жидкими при температуре тела, и поэтому плавятся в прямой кишке или влагалищной полости и высвобождают активное соединение.
Альтернативно, фармацевтические композиции могут быть представлены в форме порошка для восстановления влагосодержания в соответствующем фармацевтически приемлемом носителе во время доставки. В другом варианте осуществления стандартная лекарственная форма соединений в соответствии с формулой I может представлять собой раствор одного или более соединений или их соли в подходящем растворителе, в стерильных герметически запаянных ампулах или стерильных шприцах. Концентрация соединений в соответствии с формулой I в стандартной лекарственной форме может варьироваться, например, от примерно 1% до примерно 50%, в зависимости от используемого соединения и его растворимости и дозы, желательной для врача. Если композиции содержат стандартные лекарственные формы, то каждая стандартная лекарственная форма может содержать от 1 до 500 мг активного материала. Для лечения взрослого человека используемые дозировки могут находиться в диапазоне от 5 мг до 10 г в день, в зависимости от пути и частоты введения.
Фармацевтические композиции, описанные в настоящей заявке, могут помещаться в фармацевтически приемлемый носитель и доставляются объекту-реципиенту (например, человеку) в соответствии с известными способами доставки лекарственных средств. В целом, в способах доставки фармацевтических композиций in vivo используются принятые в данной области последовательности операций для доставки средства, причем единственная существенная модификация процедуры представляет собой замещение соединениями по настоящему изобретению лекарственных средств в принятых в данной области последовательностях операций. Аналогичным образом, в способах применения заявленных композиций для обработки клеток в культуре, например, для устранения или снижения уровня бактериального заражения клеточной культуры, используются признанные в данной области последовательности операций для обработки клеточных культур антибактериальным средством (средствами), причем единственной существенной процедурной модификацией является замещение соединениями по настоящему изобретению лекарственных средств в принятых в данной области последовательностях операций.
В одном варианте осуществления изобретение относится к способу лечения инфекции у объекта введением терапевтически эффективного количества одного или более соединений формулы I или его композиций. В одном варианте осуществления, способ включает введение нуждающемуся в нем объекту фармацевтической композиции, содержащей, по меньшей мере, одно из соединений, описанных в настоящей заявке. В одном варианте осуществления фармацевтическая композиция может содержать любое из соединений, описанных в настоящей заявке, в качестве одного активного соединения или в комбинации с другим соединением, композицией или биологическим материалом.
Термины «лечение», «способ лечения» и их однокоренные термины относятся и к терапевтическому лечению, и к профилактическим/превентивным мерам. Нуждающихся в лечении могут включать объекты, уже имеющие конкретное медицинское заболевание, а также тех, которые имеют риск развития заболевания (т.е. те, которые вероятно или, в конечном счете, приобретут расстройство). Способ лечения приводит к предотвращению или облегчению симптомов или к иному желательному биологическому исходу и может оцениваться уменьшением клинических признаков, задержкой начала заболевания, сниженными/повышенными уровнями лимфоцитов и/или антител и т.д.
Иллюстративные процедуры доставки антибактериального средства описаны в патентах США №№ 6468967; 6852689 и 5041567, выданных Rogers, и в патентной заявке PCT № EP94/02552 (№ публикации WO 95/05384), описания которых полностью включены в настоящую заявку путем ссылки. В одном варианте осуществления, одно или более соединений формулы I или его фармацевтические композиции вводятся перорально, ректально или путем инъекции (внутривенной, внутримышечной или подкожной). В другом варианте осуществления одно или более соединений формулы I или его фармацевтические композиции вводятся перорально, ректально или путем инъекции (внутривенной, внутримышечной или подкожной) для лечения инфекции, вызванной патогеном C. difficile. В другом варианте осуществления одно или более соединений формулы I или его фармацевтические композиции вводятся перорально для лечения инфекции, вызванной патогеном C. difficile. Используемые в настоящем описании фразы «терапевтически эффективная доза» и «терапевтически эффективное количество» относятся к количеству соединения, которое предотвращает начало, облегчает симптомы, останавливает прогрессирование бактериальной инфекции или приводит к другому желательному биологическому исходу, такому как, например, уменьшение клинических признаков или сниженные/повышенные уровни лимфоцитов и/или антител. Термин «лечение» определяется как введение объекту терапевтически эффективного количества одного или более соединений и для предотвращения возникновения инфекции, и для борьбы или устранения инфекции. Используемый в настоящем описании термин «объект» относится к млекопитающему, растению, низшему животному или клеточной культуре. В одном варианте осуществления объект представляет собой человека или другое животное - пациента, нуждающегося в антибактериальном лечении.
Способы по настоящему изобретению включают введение одного или более соединений формулы I или их фармацевтических композиций нуждающемуся в них объекту в количестве, которое эффективно для уменьшения или устранения бактериальной инфекции. Соединение может вводиться перорально, парентерально, путем ингаляции, местно, ректально, интраназально, буккально, вагинально или имплантированным резервуаром, наружным насосом или катетером. Соединение может быть получено для внутриглазного или аэрозольного применения. Соединения по настоящему изобретению могут вводиться в виде аэрозоля для лечения пневмонии или других легочных инфекций. В одном варианте осуществления носитель аэрозольной доставки представляет собой ингалятор для безводных распыляемых средств или сухих порошков. Одно или более соединений формулы I или их фармацевтические композиции также могут инъецироваться или вводиться непосредственно в абсцесс, желудочек или сустав. Парентеральное введение включает подкожную, внутривенную, внутримышечную, внутрисуставную, интрасиновиальную, цистернальную, подоболочечную, внутрипеченочную, внутриочаговую и внутричерепную инъекцию или инфузию. В одном варианте осуществления, одно или более соединений формулы I вводятся внутривенно, подкожно или перорально. В одном варианте осуществления для введения одного или более соединений в соответствии с формулой I в клеточную культуру данное одно или более соединений могут вводиться в питательной среде.
В одном варианте осуществления одно или более соединений в соответствии с формулой I могут использоваться для лечения объекта, имеющего бактериальную инфекцию, у которого инфекция вызвана или обострена любым типом бактерий, таким как грамположительные бактерии. В одном варианте осуществления одно или более соединений в соответствии с формулой I или их фармацевтических композиций вводятся пациенту в соответствии со способами по настоящему изобретению. В другом варианте осуществления, бактериальная инфекция может быть вызвана или обострена грамположительными бактериями. Указанные грамположительные бактерии включают без ограничения восприимчивые к метициллину и устойчивые к метициллину стафилококки (включая Staphylococcus aureus, S. epidermidis, S. haemolyticus, S. hominis, S. saprophytics, и коагулазонегативные стафилококки), S. aureus с промежуточной восприимчивостью к гликопептидным антибиотикам (GISA), резистентный к ванкомицину Staphylococcus aureus (VRSA), восприимчивые к пенициллину и устойчивые к пенициллину стрептококки (включая Streptococcus pneumoniae, S. pyogenes, S. agalactiae, S. avium, S. bovis, S. lactis, S. sangius и стрептококки группы C, стрептококки группы G и зеленящие стрептококки), энтерококки (включая восприимчивые к ванкомицину и устойчивые к ванкомицину штаммы, такие как Enterococcus faecalis и E. faecium), Clostridium difficile, C. clostridiiforme, C. innocuum, C. perfringens, C. ramosum, Listeria monocytogenes, Corynebacterium jeikeium, Bifidobacterium spp., Eubacterium aerofaciens, E. lentum, Lactobacillus acidophilus, L. casei, L. plantarum, Lactococcus spp., Leuconostoc spp., Pediococcus, Peptostreptococcus anaerobius, P. asaccarolyticus, P. magnus, P. micros, P. prevotii, P. productus, Propionibacterium acnes, Actinomyces spp., Moraxella spp. (включая M. catarrhalis).
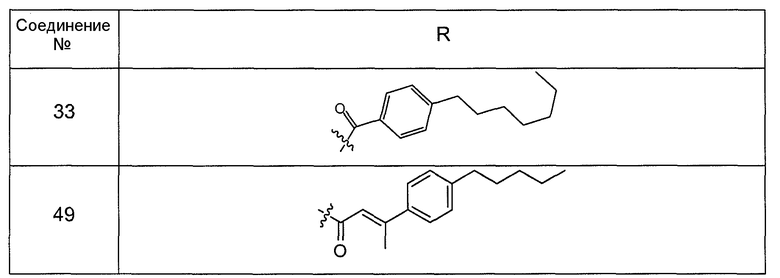
В одном варианте осуществления соединения формулы I так же активны или более активны, чем даптомицин. Соединения по настоящему варианту осуществления включают соединения 3, 4, 5, 6, 7, 9, 10, 11, 14, 16, 17, 18, 19, 20, 21, 22, 23, 25, 26, 27, 28, 32, 33, 34, 35, 36, 37, 38, 39, 42, 43, 48, 49, 50, 51, 52, 53, 54, 55, 57, 58, 61, 62, 63, 64 и 69.
В одном варианте осуществления соединения формулы I неожиданно более активны против бактерий, чем даптомицин. Соединения, которые более эффективны, чем даптомицин, представляют собой, например, соединения 3, 5, 6, 7, 10, 11, 14, 17, 19, 20, 23, 25, 32, 33, 34, 36, 37, 38, 39, 43, 49, 50, 51, 52, 53, 54, 58, 61, 62, 63, 64 и 69.
В одном варианте осуществления бактериальная инфекция может быть вызвана или обострена Clostridium difficile. Соединения, которые могут применяться против бактерий Clostridium difficile, представляют собой, например, соединения 10, 11, 14, 16, 17, 18, 19, 22, 23, 25, 26, 27, 28, 33, 34, 36, 37, 38, 42, 43, 48, 49, 50, 51, 52, 53, 61, 62, 63, 64 и 69.
Соединения, которые особенно полезны против Clostridium difficile, представляют собой, например, соединения 16, 18, 19, 22, 23, 28, 33, 34, 36, 42, 43, 49, 50 и 51.
В одном варианте осуществления соединения формулы I неожиданно активны против бактерий, которые устойчивы к даптомицину, включая устойчивый к даптомицину Staph, aureus (DRSA), устойчивый к даптомицину E. faecium (DREfm) и устойчивый к даптомицину E. faecalis (DREfs). Соединения по изобретению, которые могут применяться против устойчивых к даптомицину штаммов бактерий, представляют собой, например, соединения 3, 4, 5, 6, 7, 10, 11, 14, 17, 18, 19, 20, 21, 22, 23, 25, 26, 27, 32, 33, 34, 36, 37, 38, 39, 41, 42, 43, 48, 49, 50, 51, 52, 53, 54, 55, 57, 58, 61, 62, 63, 64 и 69.
В одном варианте осуществления соединения формулы I неожиданно активны против мутантных штаммов Clostridium difficile, таких как, белок 1 сборки нуклеосомы (NAP-1). Соединения данного варианта осуществления включают, например, соединение 49.
Соединения формулы I, которые неожиданно более активны, чем даптомицин, могут применяться против Clostridium difficile и являются неожиданно активными против бактерий, которые устойчивы к даптомицину, представляют собой соединения 33 и 49. Кроме того, соединения 33 и 49 проявили превосходные результаты и in vitro, и in vivo.
Соединение формулы I, которое неожиданно более активно, чем даптомицин, может применяться против Clostridium difficile, неожиданно активно против бактерий, которые устойчивы к даптомицину, и может применяться против NAP1, представляет собой соединение 49. Кроме того, соединение 49 проявило превосходные результаты и in vitro, и in vivo.
В другом варианте осуществления антибактериальная активность соединений формулы I против классически «устойчивых» штаммов сравнима с активностью против классически «восприимчивых» штаммов в экспериментах in vitro. В одном варианте осуществления одно или более соединений формулы I или их фармацевтические композиции вводятся в соответствии со способами по настоящему изобретению пациенту, у которого проявляется бактериальная инфекция, устойчивая к другим соединениям, включая ванкомицин или даптомицин. Кроме того, в отличие от гликопептидных антибиотиков, липопептидные соединения проявляют быструю, зависимую от концентрации бактерицидную активность против грамположительных организмов. Таким образом, в одном варианте осуществления одно или более соединений формулы I или их фармацевтические композиции вводятся в соответствии со способами по настоящему изобретению пациенту, нуждающемуся в быстродействующей антибиотикотерапии.
Способ по настоящему изобретению может использоваться по поводу любой бактериальной инфекции любого органа или ткани в организме. В одном случае бактериальная инфекция вызвана грамположительными бактериями. Указанные органы или ткани включают без ограничения скелетные мышцы, кожу, кровеносную систему, почки, сердце, легкие и кости. Способ по изобретению может применяться для лечения без ограничения инфекций кожи и мягких тканей, бактериемии и инфекций мочевых путей. Способ по изобретению может применяться для лечения заразных респираторных инфекций, включая без ограничения средний отит, синусит, хронический бронхит и пневмонию, включая пневмонию, вызванную устойчивыми к лекарственным средствам S. pneumoniae или H. influenzae. Способ по изобретению может также применяться для лечения смешанных инфекций, которые включают различные типы грамположительных бактерий или которые включают и грамположительные, и грамотрицательные бактерии. Данные типы инфекций включают внутрибрюшные инфекции и акушерско-гинекологические инфекции. Способ по изобретению может также применяться для лечения инфекции, включая без ограничения эндокардит, нефрит, септический артрит, внутрибрюшной сепсис, инфекции костей и суставов и остеомиелит. Способ по изобретению может также применяться для лечения заболеваний, связанных с Clostridium difficile (CDAD). В одном варианте осуществления способ по изобретению может применяться для лечения CDAD, которое возникает или обостряется NAP-1. В одном варианте осуществления способ по изобретению может применяться для лечения колита, связанного с C. Difficile, и диареи, связанной с C. difficile. В одном варианте осуществления любое из описанных выше заболеваний можно лечить с использованием липопептидных соединений в соответствии с настоящим изобретением или их фармацевтических композиций.
Способ по настоящему изобретению может также осуществляться при одновременном введении одного или более других противомикробных средств, таких как антибактериальные средства (антибиотики) или противогрибковые средства. В одном аспекте способ может осуществляться введением одного или более соединений в соответствии с формулой I. В другом варианте осуществления способ может осуществляться введением одного или более соединений в соответствии с формулой I с другим липопептидным соединением, таким как даптомицин.
Антибактериальные средства и их классы, которые могут совместно вводиться с одним или более соединениями формулы I, включают без ограничения пенициллины карбапенемы, цефалоспорины, аминогликозиды, бацитрацин, грамицидин, мупироцин, хлорамфеникол, тиамфеникол, фузидат натрия, линкомицин, клиндамицин, макролиды, новобиоцин, полимиксины, рифамицины, спектиномицин, тетрациклины, ванкомицин, тейкопланин, стрептограмины, антифолатные средства, триметоприм пириметамин, синтетические антибактериальные средства, нитроимидазолы, хинолоны, фторхинолоны, изониазид, этамбутол, пиразинамид, пара-аминосалициловая кислота (PAS), циклосерин, капреомицин, этионамид, протионамид, тиацетазон, виомицин, эверниномицин, гликопептид, глицилциклин, кетолиды, оксазолидиноны, имипинен, амикацин, нетилмицин, фосфомицин, гентамицин, цефтриаксон, зирацин (56-деацетил-57-деметил-45-O-де(2-метил-1-оксопропил)-12-О-(2,3,6-тридеокси-3-C-метил-4-О-метил-3-нитро-альфа-L-арабьино-гексопиранозил)фламбамицин), LY333328 (оритаванцин), линезолид (N-[[(5S)-3-[3-фтор-4-(4-морфолинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид), синерцид (далфопристин-хинупристин), азтреонам (2-[[(Z)-[1-(2-амино-4-тиазолил)-2-[[(2S,3S)-2-метил-4-оксо-1-сульфо-3-азетидинил]амино]-2-оксоэтилиден]амино]окси]-2-метил-пропановую кислоту), метронидазол (2-метил-5-нитро-1H-имидазол-1-этанол), эпироприм (5-[[3,5-диэтокси-4-(1H-пиррол-1-ил)фенил]метил]-2,4-пиримидиндиамин), OCA-983 (1-[[(2S)-2-амино-3-метил-1-оксобутил]амино]-2,5-ангидро-3-S-[(4R,5S,6S)-2-карбокси-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-3-ил]-1,4-дидеокси-3-тио-D-трео-пентитол), GV-143253 (тринем), санфетринем ((1S,5S,8aS,8bR)-1,2,5,6,7,8,8a,8b-октагидро-1-[(1R)-1-гидроксиэтил]-5-метокси-2-оксо-азето[2,1-a]изоиндол-4-карбоновую кислоту), (сложный 2,2-диметил-1-оксопропокси)метиловый эфир), CS-834 ((4R,5S,6S)-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-3-[[(3R)-5-оксо-3-пирролидинил]тио]-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты, биапенем (6-[[(4R,5S,6S)-2-карбокси-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксио-1-азабицикло[3.2.0]гепт-2-ен-3-ил]тио]-6,7-дигидро-5H-пиразол[1,2-a][1,2,4]триазол-4-ия внутреннюю соль), KA 159 (стипиамид), динемицин A ((1 S,4R,4aR,14S,14aS,18Z)-1,4,7,12,13,14-гексагидро-6,8,11-тригидрокси-3-метокси-1-метил-7,12-диоксо-4a,14a-эпокси-4,14-[3]гексен[1,5]диинонафто[2,3-c]фенантридин-2-карбоновую кислоту), DX8739 ((4R,5S,6S)-3-[[(3S,5S)-5-[[4-[(2S)-5-амино-2-гидрокси-1-оксопентил]-1-пиперазинил]карбонил]-3-пирролидинил]тио]-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), DU 6681 ((4R,5S,6S)-3-[[(6S)-6,7-дигидро-5H-пиррол[1,2-a]имидазол-6-ил]тио]-6-[(1R)-1-гидроксиэтил]-4-метил-7-oxo-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), цефлюпренам ((2E)-N-(2-амино-2-оксоэтил)-3-[(6R,7R)-7-[[(2Z)-(5-амино-1,2,4-тиадиазол-3-ил)[(фторметокси)имино]ацетил]амино]-2-карбокси-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-3-ил]-N-этил-N-метил-2-пропен-1-аминия внутреннюю соль), ER 35786 (моногидрохлорид (4R,5S,6S)-6-[(1R)-1-гидроксиэтил]-3-[[(3S,5S)-5-[(R)-гидрокси(3R)-3-пирролидинилметил]-3-пирролидинил]тио]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты), цефоселис ((6R,7R)-7-[[(2Z)-(2-амино-4-тиазолил)(метоксиимино)ацетил]амино]-3-[[2,3-дигидро-2-(2-гидроксиэтил)-3-имино-1H-пиразол-1-ил]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновую кислоту), сайфетринем целексетил (сложный 1-[(циклогексилокси)карбонил]окси]этиловый эфир (1S,5S,8aS,8bR)-1,2,5,6,7,8,8a,8b-октагидро-1-[(1R)-1-гидроксиэтил]-5-метокси-2-оксо-азето[2,1-a]изоиндол-4-карбоновой кислоты), цефпиром (1-[[(6R,7R)-7-[[(2Z)-(2-амино-4-тиазолил)(метоксиимино)ацетил]амино]-2-карбокси-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-3-ил]метил]-6,7-дигидро-5H-циклопента[b]пиридиния внутреннюю соль), HMR-3647 (3-де[(2,6-дидеокси-3-C-метил-3-O-метил-альфа-L-рибо-гексопиранозил)окси]-11,12-дидеокси-6-O-метил-3-оксо-12,11-[оксикарбонил[[4-[4-(3-пиридинил)-1H-имидазол-1-ил]бутил]имино]]-эритромицин), RU-59863 (C-7 катехолзамещенный циклоспорин), KP 736 (динатриевая соль (6R,7R)-7-[[(2Z)-(2-амино-4-тиазолил)[[(1,4-дигидро-1,5-дигидрокси-4-оксо-2-пиридинил)метокси]имино]ацетил]амино]-8-оксо-3-[(1,2,3-тиадиазол-5-илтио)метил]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты), рифалазил (1',4-дидегидро-1-деокси-1,4-дигидро-3'-гидрокси-5'-[4-(2-метилпропил)-1-пиперазинил]-1-оксо-рифамицин VIII), MEN 10700 ((5R,6S)-3-[[(2-амино-2-оксоэтил)метиламино]метил]-6-[(1R)-1-гидроксиэтил]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), ленапенем ((4R,5S,6S)-6-[(1R)-1-гидроксиэтил]-3-[[(3S,5S)-5-[(1R)-1-гидрокси-3-(метиламино)пропил]-3-пирролидинил]тио]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), BO 2502A ((4R,5S,6S)-3-[(2S,3'S,4S)-[2,3'-бипирролидин]-4-илтио]-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), NE-1530 (3'-сиалиллакто-N-неотетраозу), K130 (5-[[4-[3-[[4-[(4-аминофенил)сульфонил]фенил]амино]пропокси]-3,5-диметоксифенил]метил]-2,4-пиримидиндиамин), PD 138312 ((R)-7-[3-(1-амино-1-метилэтил)-1-пирролидинил]-1-циклопропил-6-фтор-1,4-длигидро-4-оксо-1.8-нафтиридин-3-карбоновую кислоту), PD 140248 (7-[(3R)-3-[(1S)-1-аминоэтил]-1-пирролидинил]-1-(2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3- карбоновую кислоту), CP 111905 (5-деокси-5-[[(2E)-3-[3-гидрокси-4-(2-пропенилокси)фенил]-2-метил-1-оксо-2-пропенил]амино]-1,2-O-метилен-D-нео-инозитол), сулопенем ((5R,6S)-6-[(1R)-1-гидроксиэтил]-7-оксо-3-[[(1R,3S)-тетрагидро-1-оксидо-3-тиенил]тио]-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), ритипенам акоксил ((сложный ацетилокси)метиловый эфир) (5R,6R)-3-[[(аминокарбонил)окси]метил]-6-[(1R)-1-гидроксиэтил]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты, RO-65-5788 (мононатриевая соль (6R,7R)-7-[[(2Z)-(5-амино-1,2,4-тиадиазол-3-ил)(гидроксиимино)ацетил]амино]-3-[(E)-[(3'R)-1'-[[(5-метил-2-оксо-1,3-диоксол-4-ил)метокси]карбонил]-2-оксо[1,3'-бипирролидин]-3-илиден]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты), Sch-40832 (стереоизомер сложного метилового эфира N-[[48-[1-[[2,6-дидеокси-3-O-(2,6-дидеокси-D-арабино-гексапиранозил)-D-арабино-гексапиранозил]окси]этил]-15-этилиден-1,3a,4,5,10,11,12,13,14,15,19,20,21,22,28,29,41,42-октадекагидро-41-гидрокси-12,45-бис(1-гидроксиэтил)-1-(гидроксиметил)-22-(1-гидрокси-1-метилпропил)-36-метил-51,54,57-трис(метилен)-3-(метилтио)-10,13,20,27,38,49,52,55,58-нонаоксо-18H,27H-5a,29-(иминоэтаниминоэтаниминоэтаниминоэтанимино[7,2]
хинолинметаноксиметано)-9,6:19,16:26,23:33,30-тетранитрило-16H,33aH-имидазо[1',5':1,6]пиридо[3,2-m][1,11,17,24,4,7,20,27]тетратиатетраазациклотриаконтин-1-ил]карбонил]-2,3-дидегидроаланил-2,3-дидегидро-аланина), микакоцидин A ((OC-6-26-A)-[(4S)-2-[(2S)-2-[(2R,4R)-2-[(4R)-4,5-дигидро-2-[2-(гидрокси-.каппа.O)-6-пентилфенил]-4-тиазолил-.каппа.N3]-3-метил-4-тиазолидинил-.каппа.N3]-2-(гидрокси-.каппа.O)-1,1-диметилэтил]-4,5-дигидро-4-метил-4-тиазолкарбоксилато(2-)-.каппа.N3,.каппа.O4]-Цинк), SR-15402((1S,5S,8aS,8bR)-1,2,5,6,7,8,8a,8b-октагидро-1-[(1R)-1-гидроксиэтил]-2-оксо-5-[(3S)-3-пирролидинилтио]-азето[2,1-a]изоиндол-4-карбоновую кислоту), TOC 39 (1-(2-амино-2-оксоэтил)-4-[[(1E)-2-[(6R,7R)-7-[[(2Z)-(2-амино-4-тиазолил)(гидроксиимино)ацетил]амино]-2-карбокси-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-3-ил]этенил]тио]-пиридиния внутреннюю соль), карумонам ([[(Z)-[2-[[(2S,3S)-2-[[(аминокарбонил)окси]метил]-4-оксо-1-сульфо-3-азетидинил]амино]-1-(2-амино-4-тиазолил)-2-оксоэтилиден]амино]окси]-уксусную кислоту), Цефозопран (1-[[(6R,7R)-7-[[(2Z)-(5-амино-1,2,4-тиадиазол-3-ил)(метоксиимино)ацетил]амино]-2-карбокси-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-3-ил]метил]-имидазо[1,2-b]пиридазиния внутреннюю соль), Цефетамет пивоксил (сложный (2,2-диметил-1-оксопропокси)метиловый эфир) (6R,7R)-7-[[(2Z)-(2-амино-4-тиазолил)(метоксиимино)ацетил]амино]-3-метил-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты, и T 3811 (дес-F(6)-хинолон).
Противогрибковые средства, которые могут совместно вводиться с одним или более соединений в соответствии с изобретением, включают без ограничения каспофунген, вориконазол, сертаконазол, IB-367, FK-463, LY-303366, Sch-56592, ситафлоксацин, DB-289 полиены, такие как амфотерицин, нистатин, примарицин; азолы, такие как флуконазол, итраконазол и кетоконазол; аллиламины, такие как нафтифин и тербинафин; и антиметаболиты, такие как флуцитозин. Другие противогрибковые средства включают без ограничения те, которые описаны в публикации Fostel, et al., 2000, Drug Discovery Today 5: 25-32, включенной в настоящее описание путем ссылки. В публикации Fostel et al. описаны противогрибковые соединения, включая коринекандин, Mer-WF3010, фузакандины, артрихитин/LL 15G256, сордарины, циспентацин, азоксибациллин, ауреобасидин и хафрефунгин.
Действительные уровни дозировки активных ингредиентов в фармацевтических композициях одного или более соединений в соответствии с формулой I могут варьироваться с тем, чтобы получить терапевтически эффективное количество активного соединения (соединений) для достижения желательной терапевтической реакции для конкретного пациента, композиций и пути введения. Эффективное количество может быть определено, как описано в настоящей заявке. Выбранный уровень дозировки зависит от активности конкретного соединения, пути введения, тяжести подлежащего лечению состояния и состояния и предшествующего медицинского анамнеза получающего лечение пациента. Однако в пределах компетенции специалистов в данной области находится начало лечения дозами соединения на уровне, который ниже, чем тот, который требуется для достижения желательного терапевтического эффекта, и постепенное увеличение дозировки до тех пор пока не будет достигнут желательный эффект. В одном варианте осуществления, данные, полученные по анализам, могут использоваться при составлении диапазона дозировки для применения у людей.
Способ включает введение объекту эффективной дозы одного или более соединений формулы I. Эффективная доза составляет в целом от 125 мг/день до 1000 мг/день. В одном варианте осуществления эффективная доза составляет от примерно 0,1 до примерно 100 мг/кг одного или более соединений формулы I или их фармацевтически приемлемых солей. В одном варианте осуществления доза составляет от примерно 0,1 до примерно 50 мг/кг одного или более соединений формулы I или их фармацевтически приемлемых солей. В другом варианте осуществления доза составляет от примерно 1 до примерно 25 мг/кг одного или более соединений формулы I или их фармацевтически приемлемых солей. В другом варианте осуществления доза составляет от примерно 1 до примерно 12 мг/кг одного или более соединений формулы I. В другом варианте осуществления, доза составляет примерно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 мг/кг одного или более соединений формулы I. Эффективная доза для клеточной культуры составляет обычно от примерно 0,1 до примерно 1000 мкг/мл. В одном варианте осуществления эффективная доза для клеточной культуры составляет от примерно 0,1 до примерно 200 мкг/мл.
В целом, уровни дозировки от примерно 0,1 мкг/кг до примерно 50 мг/кг, например, уровень в диапазоне от примерно 5 до примерно 20 мг активного соединения на 1 кг массы тела в день, могут вводиться местно, перорально или внутривенно пациенту-млекопитающему. Другие уровни дозировки находятся в диапазоне от примерно 1 мкг/кг до примерно 20 мг/кг, от примерно 1 мкг/кг до примерно 10 мг/кг, от примерно 1 мкг/кг до примерно 1 мг/кг, от 10 мкг/кг до 1 мг/кг, от примерно 10 мкг/кг до примерно 100 мкг/кг, от примерно 100 мкг до примерно 1 мг/кг, и от примерно 500 мкг/кг до примерно 5 мг/кг в день. При желании, эффективная суточная доза может быть разделена на множественные дозы в целях введения, например, 2, 3 или 4 отдельных доз в день. В одном варианте осуществления фармацевтическая композиция может вводиться один раз в день.
Одно или более соединений формулы I могут также вводиться в рационе или корме пациента или животного. При введении в виде части общего потребляемого рациона количество используемого соединения может составлять менее чем 1% масс. рациона, например, не более чем 0,5% масс. Рацион для животных может представлять собой обычные корма, к которым может добавляться соединение, или оно может добавляться к премиксу.
Одно или более соединений формулы I могут вводиться в виде одной суточной дозы или множества доз в сутки. В одном варианте осуществления, одно или более соединений формулы I вводятся в виде одной дозы в сутки. В другом варианте осуществления, одно или более соединений формулы I вводятся в виде двух равных доз в день. В другом варианте осуществления, одно или более соединений формулы I вводятся в виде трех равных доз в день. Схема лечения может требовать введения в течение длительных периодов времени, например, в течение нескольких дней или в течение от 2 до 4 недель. Количество на введенную дозу или общее введенное количество зависит от таких факторов, как природа и тяжесть инфекции, возраст и общее состояние здоровья пациента, переносимости пациентом соединения и микроорганизма или микроорганизмов, участвующих в инфекции. Схема лечения для одного типа инфекции могут в значительной степени отличаться от схемы лечения другой инфекции. Например, один тип инфекции может требовать введения посредством внутривенного введения один раз в день, тогда как другая инфекция может требовать схемы лечения из множественного перорального введения. Способ введения пациенту даптомицина, другого члена класса липопетидных соединений, описан в патентах США №№ 6468967 и 6852689.
Одно или более соединений формулы I могут вводиться в соответствии с данным способом до тех пор, пока не будет искоренена или уменьшена бактериальная инфекция. В одном варианте осуществления одно или более соединений формулы I вводятся в течение периода времени от 3 дней до 6 месяцев. В другом варианте осуществления, одно или более соединений формулы I вводятся в течение 7-56 дней. В другом варианте осуществления одно или более соединений формулы I вводятся в течение 7-28 дней. В еще одном варианте осуществления одно или более соединений формулы I вводятся в течение 7-14 дней. Соединения по настоящему изобретению могут вводиться в течение более длительного или более короткого периода времени, если это желательно.
Варианты осуществления, описанные в настоящей заявке, относятся к соединениям формулы I, которые являются новыми и активными агонистами грамположительных бактерий. Другие варианты осуществления, описанные в настоящей заявке, относятся к новым соединениям формулы I, которые проявляют повышенную активность против Clostridium difficile. Другие варианты осуществления, описанные в настоящей заявке, относятся к новым соединениям формулы I, которые проявляют неожиданную активность против бактерий, которые устойчивы к даптомицину.
Другие варианты осуществления изобретения будут очевидны для специалистов в данной области в результате рассмотрения описания и осуществления изобретения, описанного в настоящей заявке. Предполагается, что описание и примеры должны рассматриваться только как иллюстративные, причем действительный объем и сущность изобретения указан следующей формулой изобретения.
ПРИМЕРЫ
Для более полного понимания настоящего изобретения, ниже представлены следующие примеры. Данные примеры представлены только в иллюстративных целях и никоим образом не должны рассматриваться как ограничивающие объем изобретения.
Пока нет других указаний в представленных ниже примерах, следующие аббревиатуры имеют следующие значения. Аббревиатуры, не определенные ниже, имеют их общепринятое значение.
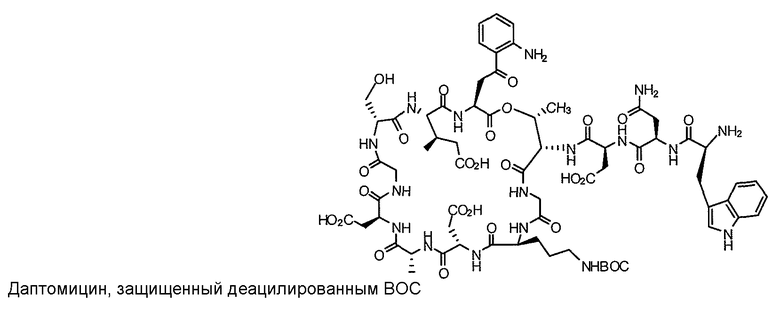
Пока нет других указаний, все величины температуры, приведенные в следующих примерах, представлены в градусах Цельсия (°C). Также, пока не отмечено иначе, реагенты, исходные материалы и растворители были закуплены у промышленных поставщиков (таких как Aldrich, Fluka, Sigma и тому подобные) и использовались без дальнейшей очистки. Даптомицин, защищенный деацилированным BOC, получали на основании способа, описанного в патенте США № 6911525 B2.
Конечные продукты обычно очищали ВЭЖХ в обращенной фазе с использованием колонки C8.
Условия аналитической ВЭЖХ для мониторинга реакций были следующие: ВЭЖХ система Waters Alliance с колонкой SunFire™ C8 (5 мкм, 4,6×150 мм) с использованием возрастающего градиента (например, от 10 до 90% B в A через 20 мин; A=0,01% TFA в воде; B=0,01% TFA в ацетонитриле).
Условия аналитической ВЭЖХ для тестов чистоты были следующие: ВЭЖХ система Waters Alliance с колонкой SunFire™ C8 (5 мкм, 4,6×150 мм) с использованием возрастающего градиента (например, от 30 до 50% B в A через 20 мин; A=O,01% TFA в воде; B=0,01% TFA в ацетонитриле).
Условия препаративной ВЭЖХ: ВЭЖХ система Varian Prep с колонкой SunFireTM prep C8 OBDTM (10 мкм, 19×250 мм) с использованием возрастающего градиента (например, от 25 до 45% B в A через 20 мин; A=0,01% TFA в воде; B=0,01% TFA в ацетонитриле).
Соединения по настоящему изобретению могут быть получены в соответствии с процедурами, описанными в патенте США № 6911525 B2, который полностью включен в настоящее описание путем ссылки, и/или как детально описано ниже:
Схема реакции I
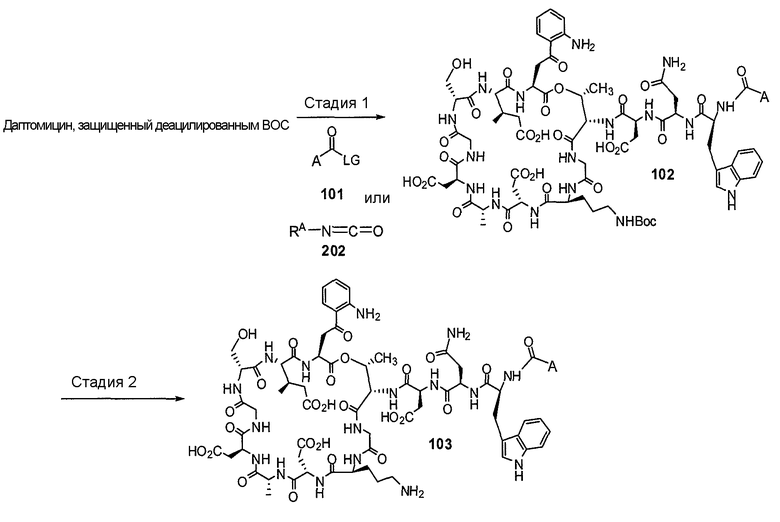
На стадии 1 в схеме реакции 1 Даптомицин, защищенный деацилированным BOC (см. патент США № 6911525 B2), обрабатывается модифицирующим агентом, выбранным из соединений, имеющих формулу 101, где LG представляет соответствующую уходящую группу, или 202. Взаимодействие амина с модифицирующими агентами, как определено в настоящем описании, хорошо известно специалистам в данной области. Например, обработка Даптомицина, защищенного деацилированным BOC, активированным сложным эфиром, лактоном или хлоридом кислоты с последующим удалением защитной группы (стадия 2), дает соединения по настоящему изобретению, где A представляет алкил, алкенил, циклоалкил, арил или гетероарил. Альтернативно, обработка Даптомицина, защищенного деацилированным BOC, изоцианатом, таким как соединение, имеющее формулу 202, с последующим удалением защитной группы (стадия 2), обеспечивает получение соединений формулы I, где A представляет NHRA.
В одном варианте осуществления активированный сложный эфир представляет сложный пентафторфениловый эфир (PFP-эфир). Сложные пентафторфениловые эфиры (PFP) получали обработкой карбоновой кислоты пентафторфенолом и ацилирующим агентом, таким как без ограничения дициклогексилкарбодиимид (DCC) в растворителе, таком как дихлорметан (Synthesis, 2007, 23, 3731-3735).
Ацилхлориды получали обработкой карбоновой кислоты реагентом, таким как без ограничения тионилхлорид или оксалилхлорид, каталитическим DMF, в растворителе, таком как дихлорметан.
Получение соединений формулы (I), где R представляет
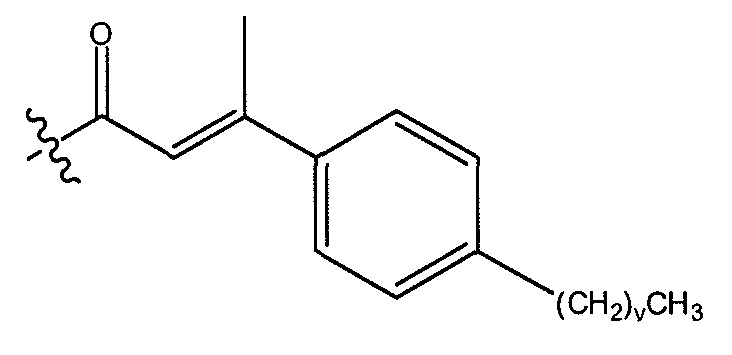
Пример 1
Получение (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (49).
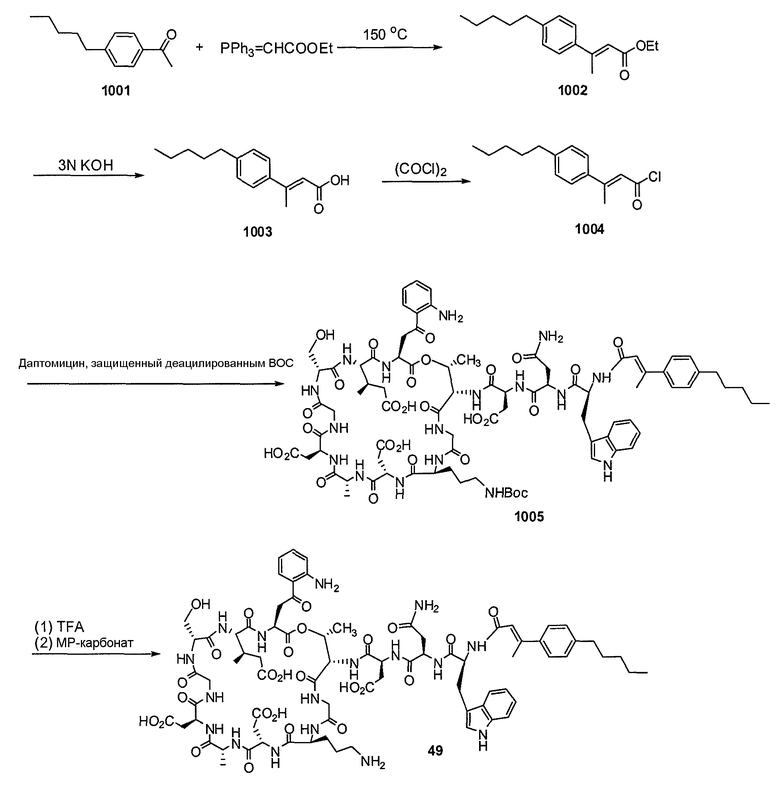
Стадия 1: Получение (E)-этил 3-(4-пентилфенил)бут-2-еноата (1002).
Смесь выпускаемого промышленностью 1-(4-пентилфенил)этанона (5 г, 26,3 ммоль) и (этоксикарбонилметилен)-трифенилфосфорана (18,3 г, 52,5 ммоль) перемешивали при 150°C в течение 48 часов в атмосфере азота. Реакционную смесь охлаждали до окружающей температуры и разбавляли этилацетатом (50 мл) и простым петролейным эфиром (200 мл). Суспензию фильтровали через воронку с пористым фильтром. Концентрированный фильтрат очищали флэш-хроматографией на колонке с силикагелем (простой петролейный эфир:этилацетат=80:1 ) для получения указанного в заголовке соединения (1,6 г), имеющего следующие физические данные: 1H ЯМР (300 МГц, δ, CDCl3) 0,90 (шир., 3H), 1,36 (шир., 7), 1,63 (шир., 2H), 2,58 (с, 3H), 2,63 (шир., 2H), 4,22 (кв., 2H), 6,15 (с, 1H), 7,20 (д, 2H), 7,41 (д, 2H).
Стадия 2: Получение (E)-3-(4-пентилфенил)бут-2-еновой кислоты (1003).
Раствор соединения 1002 (1,5 г, 5,77 ммоль) в этаноле (50 мл) и 3н гидроксида калия (25 мл) перемешивали при 45°C в течение 3 часов. Реакционную смесь концентрировали и полученный остаток разбавляли водой (50 мл). Водный раствор подкисляли до pH 2 1н хлористоводородной кислотой и экстрагировали EtOAc (2×30 мл). Комбинированные органические слои сушили безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на колонке (силикагель, простой петролейный эфир:этилацетат=10:1) для получения указанного в заголовке соединения (0,95 г), имеющего следующие физические данные: 1H ЯМР (300 МГц, δ, CDCl3) 0,90 (шир., 3H), 1,33 (шир., 4H), 1,62 (шир., 2H), 2,60 (шир., 5H), 6,18 (с, 1H), 7,18 (д, 2H), 7,42 (д, 2H).
Стадия 3: Получение (E)-3-(4-пентилфенил)бут-2-аноил хлорид (1004).
Оксалилхлорид (3,2 мл, 36,60 ммоль) и DMF (50 мкл) по каплям добавляли к раствору соединения 1003 (5,0 г, 21,52 ммоль) в дихлорметане (100 мл) при 0°C. Реакционный раствор согревали до комнатной температуры и перемешивали в течение 4 часов. Реакционную смесь концентрировали в вакууме и остаток сушили в высоком вакууме в течение 3 часов. Неочищенный продукт использовали на следующей стадии без дальнейшей очистки.
Стадия 4: Получение (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-[(N-трет-бутоксикарбонил)-орнитил]-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (1005).
Даптомицин, защищенный деацилированным BOC (3,5O г, 2,23 ммоль), и бикарбонат натрия (1,13 г, 61,0 ммоль) растворяли в THF (130 мл) и воде (50 мл). Раствор даптомицина, защищенного деацилированным BOC, и бикарбоната натрия охлаждали до 0°C, и затем вводили раствор соединения 1004 (1,96 г, 7,82 ммоль) в THF (20 мл). Реакционную смесь согревали до комнатной температуры и перемешивали в течение 4 часов. Смесь концентрировали в вакууме для удаления THF. Остающийся водный раствор загружали на колонку C18 для флэш-хроматографии (35 мм×300 мм, смола Bondesil HF C18, закупленная у компании Varian). Данную колонку сначала промывали водой для удаления соли, а затем метанолом для вымывания продукта. Неочищенное соединение 1005 (3,46 г) получали в виде белого твердого вещества после удаления метанола. MS m/z 1780,8 (M+H)+.
Стадии 5-6: Получение (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-дамино-γ-оксобензолбутановой кислоты (49).
TFA (10 мл) добавляли к раствору соединения 1005 (3,46 г) в DCM (50 мл) при комнатной температуре. Реакционную смесь энергично перемешивали в течение 45 минут и медленно добавляли к энергично перемешиваемому простому диэтиловому эфиру (100 мл). Полученный желтый осадок собирали фильтрацией. Неочищенный продукт очищали препаративной ВЭЖХ для получения соли TFA соединения 6 (0,75 г). Карбонатную смолу MP (закупленную у компании Biotage) добавляли к раствору соли TFA соединения 6 (0,70 г, 0,39 ммоль) в безводном метаноле (30,0 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. Смолы удаляли фильтрацией и промывали метанолом. Раствор метанола концентрировали в вакууме для получения продукта в виде не совсем белого твердого вещества (408 мг). MS m/z 1680,7 (M+H)+.
Пример 1b
Альтернативное получение (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (49).
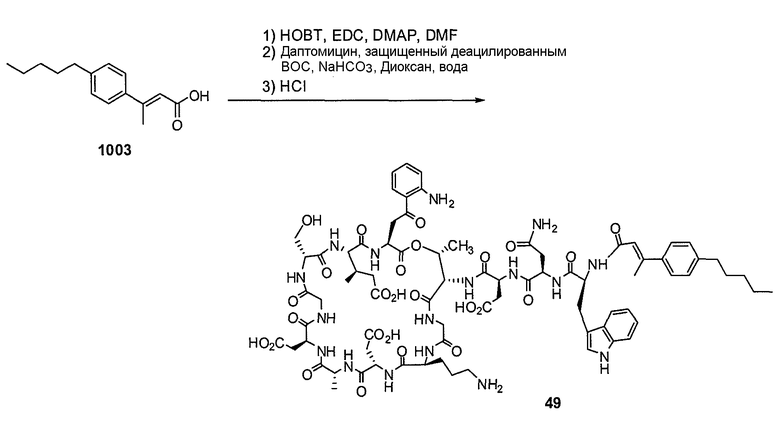
Раствор (E)-3-(4-пентилфенил)бут-2-еновой кислоты (1100 г, 4,73 моль), N-Этил-N'-(3-диметиламинопропил)карбодиимида гидрохлорида (907 г, 4,73 моль), HOBT (640 г, 4,73 моль) и 4-(диметиламино)пиридина (22 г, 0,18 моль) в DMF (11 л) перемешивали при комнатной температуре в течение 4 часов, и в этой точке активация (E)-3-(4-пентилфенил)бут-2-еновой кислоты считалась полной, по данным ВЭЖХ. Данную реакционную смесь добавляли к суспензии даптомицина, защищенного деацилированным BOC (2600 г, 1,66 моль), бикарбоната натрия (804 г, 9,57 моль) в воде (11,25 л) и 1,4-диоксана (33,75 л). Смесь перемешивали при комнатной температуре в течение 2,5 часов, и в это время ВЭЖХ указывала на полное потребление даптомицина, защищенного деацилированным BOC. Реакционную смесь разбавляли водой (22,5 л) и охлаждали ледяной баней. Добавляли концентрированную хлористоводородную кислоту (5,25 л) при поддержании внутренней температуры ниже 30°C. После добавления раствор перемешивали при комнатной температуре в течение 5 дней, и в это время ВЭЖХ указывала на полное потребление защищенного BOC промежуточного соединения.
Реакционную смесь промывали простым метил трет-бутиловым эфиром (90 л, затем приблизительно 60 л, затем приблизительно 45 л, затем приблизительно 45 л) для удаления 1,4-диоксана. pH оставшегося раствора (приблизительно 44 л) доводили до pH 2,69 2н гидроксидом натрия (11,3 л) и водой (53,4 л). Данный материал обрабатывали фильтрацией в тангенциальном потоке (TTF) мембраной 1K до тех пор, пока общий объем не снижался до 54 л. Воду (120 л) добавляли двумя порциями, и раствор концентрировали до 52 л продолжающейся TTF. Водный раствор (30 л из 52 л) очищали хроматографией, используя следующий протокол: Объем водного раствора доводили до трехкратного объема (30 л → 90 л) 20% IPA в водном растворе ацетата аммония (50 мМ). Разбавленный раствор наносили на колонку емкостью 38 л с ионообменной смолой HP20SS со скоростью 1,5 л/мин. Колонку элюировали раствором IPA в водном 50 мМ ацетате аммония (25%→30%→35%, 60 л каждой концентрации). Фракции (приблизительно 11 л) собирали и анализировали ВЭЖХ. Фракции с ВЭЖХ чистотой менее чем 80% объединяли и снова очищали с использованием того же способа. Ключевые фракции от обоих хроматографических разделений (с ВЭЖХ чистотой >80%) объединяли и подкисляли концентрированной HCl до pH 2-3. Полученный раствор обессоливали на ионообменной колонке (смола HP20SS, 16 л), которую элюировали WFI (до проводимости=4,8 мкС) с последующей IPA в WFI (36 л 10% → 40 л 60%). Желтую полосу, которую элюировали 60% IPA (приблизительно 19 л), собирали, pH доводили до pH 2-3 концентрированной HCl и лиофилизировали для выхода 636,5 г Соединения 49 (ВЭЖХ чистота 87,0%). MS m/z 1680,7 (M+H)+.
Пример 1c
Получение (13→4)-лактона N-{1-[(E)-3-(4-гексилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (50).
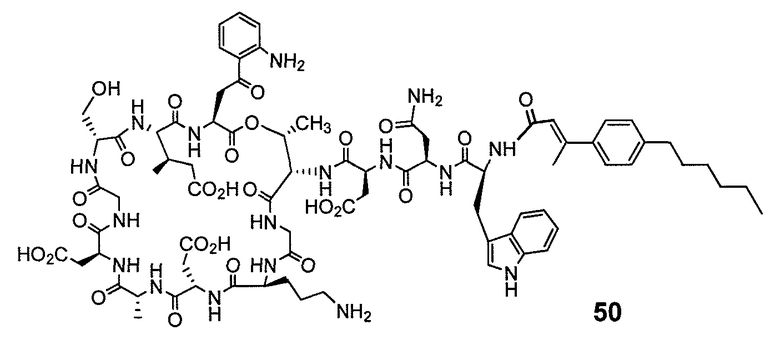
Указанное выше соединение получали таким же образом, как описано на стадиях 1-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(aS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемый промышленностью 1-(4-гексилфенил)этанон использовали вместо 1-(4-пентилфенил)этанона (на стадии 1). MS m/z 1694,8 (M+H)+.
Пример 1d
Получение (13→4)-лактона N-{1-[(E)-3-(4-бутилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксабензолбутановой кислоты (48).
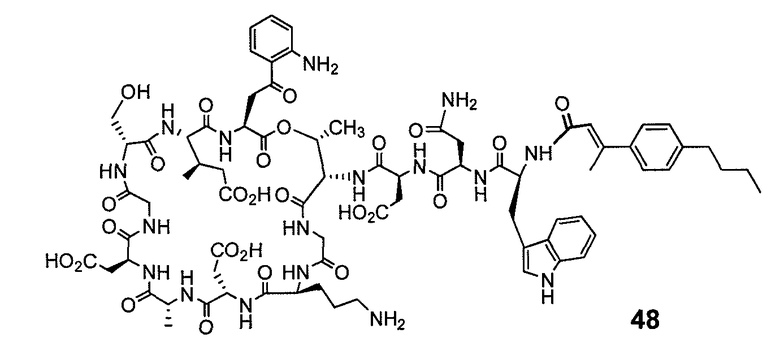
Указанное выше соединение получали таким же образом, как описано в стадиях 1-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемый промышленностью 1-(4-бутилфенил)этанон использовали вместо 1-(4-пентилфенил)этанона (на стадии 1). MS m/z 1666,8 (M+H)+.
Получение соединений формулы (I), где R представляет
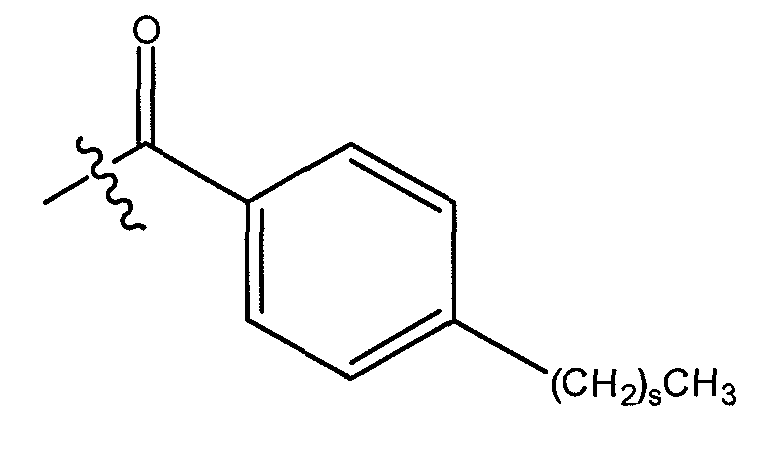
Пример 2
Получение (13→4)-лактона N-(4-гептилбензоил)-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (33).
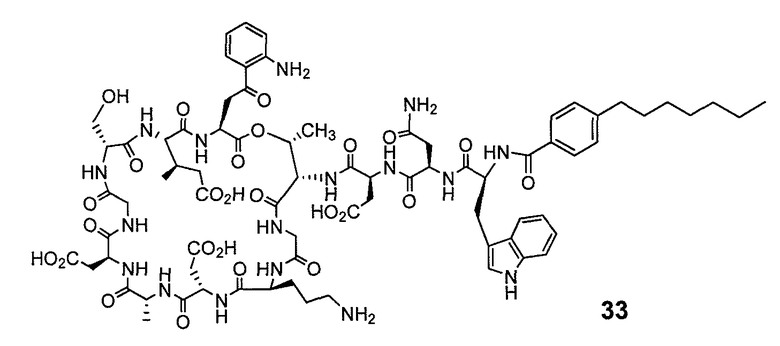
Указанное выше соединение получали таким же образом, как описано в стадиях 4-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-енол]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемый промышленностью 4-гептилбензоил хлорид использовали вместо (E)-3-(4-пентилфенил)бут-2-еноил хлорида. MS m/z 1668,8 (M+H)+.
Пример 2b
Получение (13→4)-лактона N-(4-октилбензоил)-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (34)
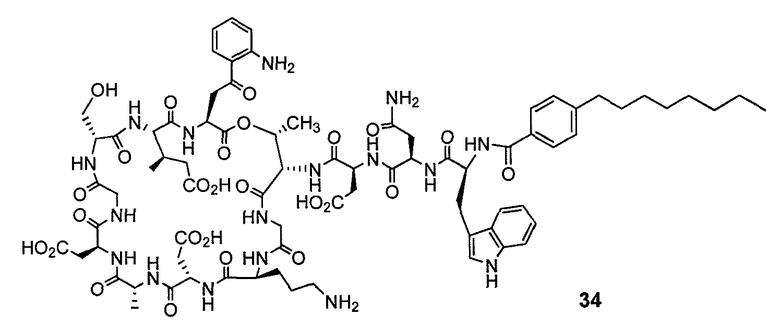
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью 4-октилбензойную кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1682,8 (M+H)+.
Получение соединения формулы (I), где R представляет
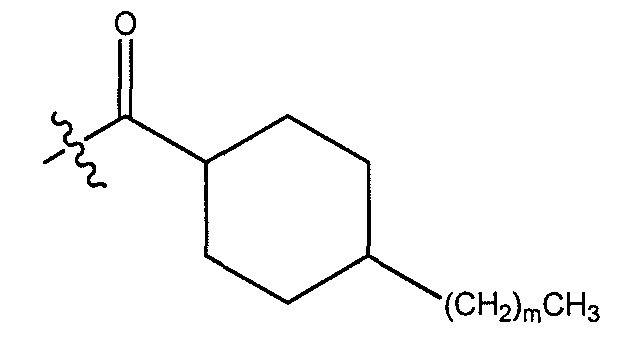
Пример 3
Получение (13→4)-лактона N-(транс-4-пентилциклогексанкарбонил-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (16)
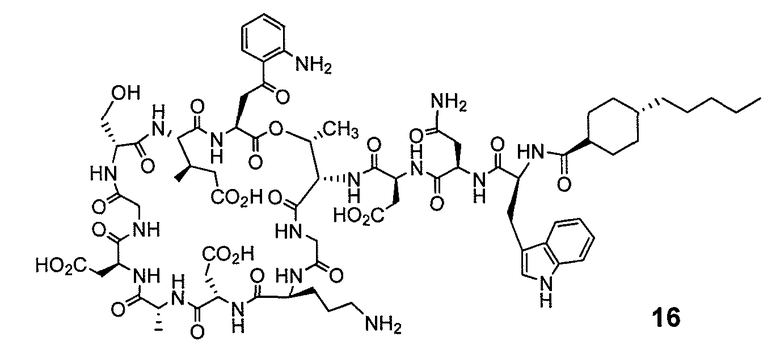
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью Транс-4-пентилциклогексанкарбоновую кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1646,7 (M+H)+.
Получение соединений формулы (I), где R представляет
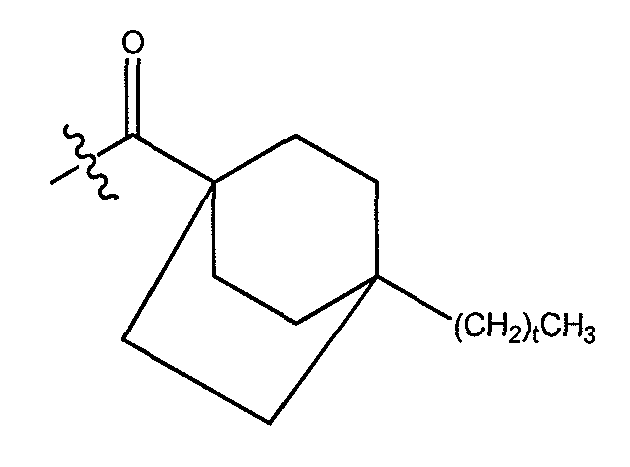
Пример 4
Получение (13→4)-лактона N-(4-пентилбицикло[2.2.2]октан-1-карбонил)-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (22)
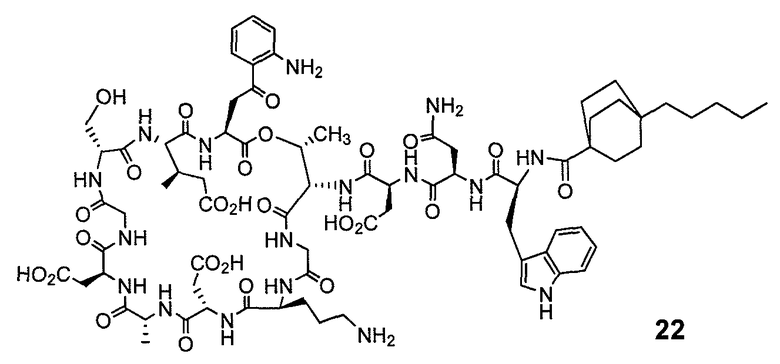
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью 4-пентилбицикло[2.2.2]октан-1-карбоновую кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1672,7 (M+H)+.
Получение соединений формулы (I), где R представляет
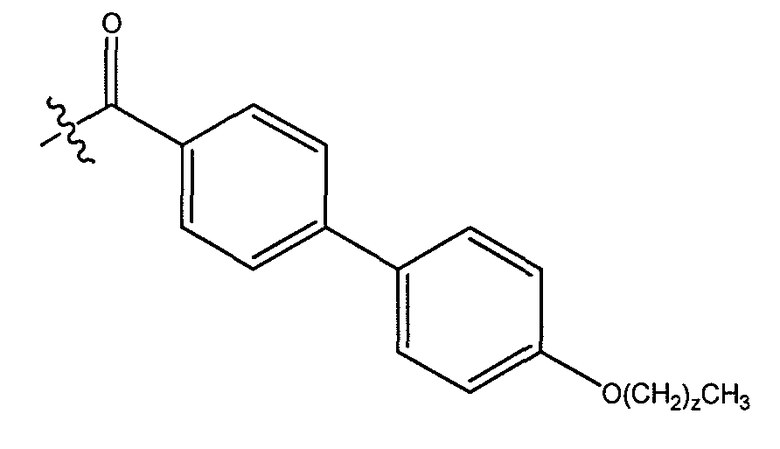
Пример 5
Получение (13→4)-лактона N-[4'-(пентилокси)бифенил-4-карбонил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (61)
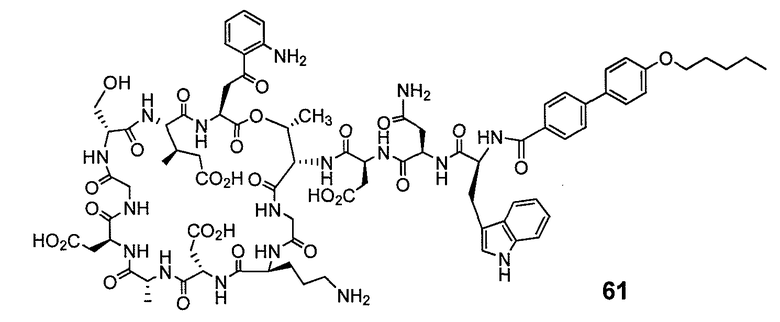
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью 4'-(пентилокси)бифенил-4-карбоновую кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1732,8 (M+H)+.
Получение соединений формулы (I), где R представляет
Пример 6
Получение (13→4)-лактона N-(4-гептилтиофен-2-карбонил)-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (43)
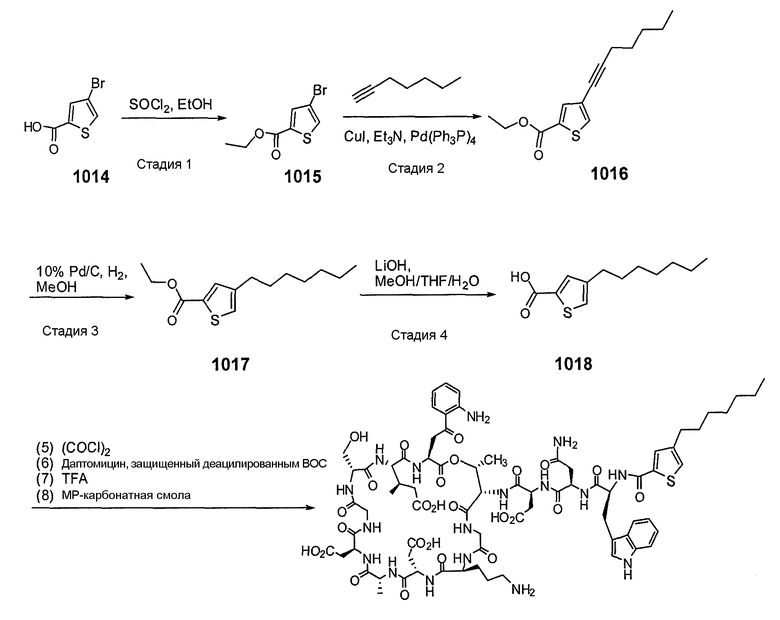
43
Стадия 1: Получение этил 4-бромтиофен-2-карбоксилата (1015)
Тионилхлорид (8,8 мл, 121 ммоль) медленно добавляли к раствору 4-бромтиофен-2-карбоновой кислоты (5,0 г, 24,2 ммоль) в этаноле (30 мл) при комнатной температуре. Реакционную смесь нагревали до 65°C и перемешивали в течение 4 часов. Удаление растворителя и избытка реагента в вакууме давало неочищенный продукт этил 4-бромтиофен-2-карбоксилат (1015), который использовали на следующей стадии без дальнейшей очистки.
Стадия 2: Получение этил 4-(гепт-1-инил)тиофен-2-карбоксилата (1016)
Реакционную смесь соединения 1015 (5,64 г, 24,0 ммоль), DMF (50 мл), йодида меди (I) (1,37, 7,2 ммоль), 1-гептина (9,42 мл, 72,0 ммоль), триэтиламина (6,70 мл, 48,0 ммоль) и тетракиз(трифенилфосфин)палладия(0) (2,77 г, 2,4 ммоль) перемешивали при 65°C в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли простой метил трет-бутиловый эфир (300 мл) и осадок удаляли фильтрацией. Прозрачный органический раствор промывали насыщенным хлоридом натрия, сушили над сульфатом магния и концентрировали в вакууме. Неочищенную коричневую мазь очищали флэш-хроматографией на силикагеле (0%→5% EtOAc в гексане) для получения этил 4-(гепт-1-инил)тиофен-2-карбоксилата (1016, 5,89 г) в виде коричневого масла. 1H ЯМР (300 МГц, δ, CDCl3) 0,89 (т, 3H), 1,28-1,45 (м, 7H), 1,52-1,62 (м, 2H), 2,36 (т, 12,4 Hz, 2H), 4,33 (кв., 2H), 7,48 (с, 1H), 7,72 (с, 1H).
Стадия 3: Получение этил 4-гептилтиофен-2-карбоксилата (1017)
5% Палладий на углероде (2,52 г) добавляли к раствору соединения 1016 (5,94 г, 23,7 ммоль) в метаноле (100 мл). Реакционную смесь перемешивали в атмосфере водорода (1 атм) в течение 12 часов. Реакционную смесь фильтровали через целит и прозрачный раствор концентрировали в вакууме для получения этил 4-гептилтиофен-2-карбоксилата (1017, 4,80 г), которое непосредственно использовали на следующей стадии без дальнейшей очистки. Rf (фактор удерживания)=0,78 (10% EtOAc в гексане).
Стадия 4: Получение 4-гептилтиофен-2-карбоновой кислоты (1018)
Раствор гидрата оксида лития (1,20 г, 28,3 ммоль) в воде (50 мл) добавляли к раствору соединения 1017 (2,4 г, 9,4 ммоль) в THF (50 мл) и метанола (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После удаления органического растворителя, водный раствор подкисляли до pH 4,0 6н раствором хлористоводородной кислоты и экстрагировали EtOAc. Органический слой сушили и концентрировали для получения 4-гептилтиофен-2-карбоновой кислоты (1018, 2,1 г), которую непосредственно использовали на следующей стадии без дальнейшей очистки. Rf=0,03 (10% EtOAc в гексане).
Стадии 5-8: Получение (13→4)-лактона N-(4-гептилтиофен-2-карбонил)-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (43)
Указанное выше соединение получали таким же образом как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что 4-гептилтиофен-2-карбоновую кислоту (соединение 1018) использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1674,7 (M+H)+.
Получение соединений формулы (I), где R представляет
Пример 7
Получение (13→4)-лактона N-[5-(бифенил-4-ил)тиофен-2-карбонил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (52)
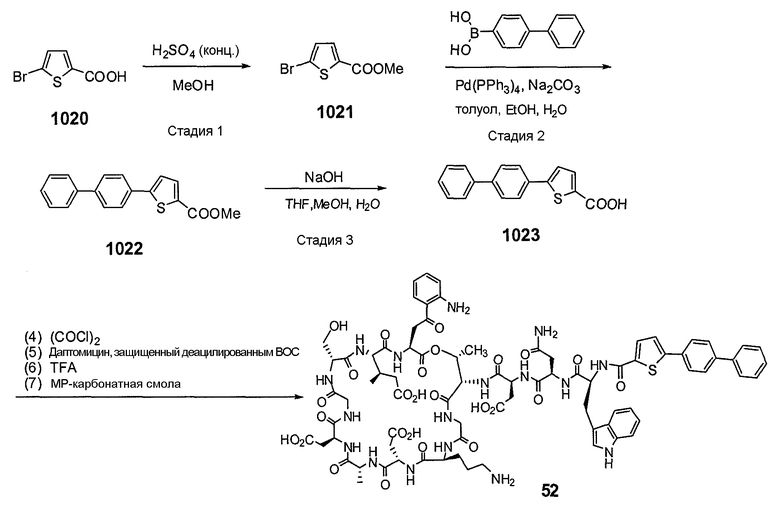
Стадия 1: Получение метил 5-бромтиофен-2-карбоксилата (1021).
Концентрированную H2SO4 (3 мл) добавляли к раствору 5-бромтиофен-2-карбоновой кислоты (5,0 г, 2,42 ммоль) в метаноле (100 мл). Реакционную смесь нагревали с обратным холодильником в течение 16 часов. После охлаждения до окружающей температуры реакционную смесь концентрировали до сухости. Остаток растворяли в EtOAc (60 мл), промывали насыщенным карбонатом натрия (60 мл) и насыщенным хлоридом натрия (40 мл) и сушили над безводным сульфатом натрия. Раствор концентрировали для получения соединения 1021 (4,8 г). 1H ЯМР (CDCl3, 300 МГц) δ 3,90 (с, 3H), 7,09-7,10 (д, 1H), 7,57-7,58 (д, 1H).
Стадия 2: Получение метил 5-(бифенил-4-ил)тиофен-2-карбоксилата (1022)
Смесь соединения 1021 (5,0 г, 22,6 ммоль), бифенил-4-илбороновой кислоты (5,4 г, 27,1 ммоль), карбоната натрия (5,9 г, 56,5 ммоль), тетракиз(трифенилфосфин)палладия(0) (2,65 г, 2,3 ммоль), толуола (200 мл), этанола (100 мл) и воды (50 мл) нагревали с обратным холодильником в течение 12 часов в атмосфере азота. Реакционную смесь охлаждали до окружающей температуры и экстрагировали дихлорметаном (2×250 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией силикагелем (5% EtOAc в гексане) для получения соединения 1022 (5,0 г). 1H ЯМР (CDCl3, 300 МГц) δ 3,92 (с, 3H), 7,32-7,49 (м, 4H), 7,62-7,80 (м, 7H).
Стадия 3: Получение 5-(бифенил-4-ил)тиофен-2-карбоновой кислоты (1023)
Раствор гидроксида натрия в воде (2н, 50 мл) добавляли к суспензии соединения 1022 (3,5 г, 11,9 ммоль) в THF (50 мл) и метанола (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 14 часов и затем концентрировали приблизительно до 1/10 ее первоначального объема (15 мл). pH концентрированного раствора доводили до pH 2-3 с использованием 2н водного раствора хлористоводородной кислоты и экстрагировали смесью DCM и этанола (соотношение 7:3, 200 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали для получения соединения 1023 (2,7 г). 1H ЯМР (CDCl3, 300 МГц) δ 7,39-7,41 (д, 1H), 7,46-7,51 (м, 2H), 7,63-7,64 (д, 1H), 7,71-7,85 (м, 7H).
Стадии 4-7: Получение (13→4)-лактона N-[5-(бифенил-4-ил)тиофен-2-карбонил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (52)
Указанное выше соединение получали таким же образом как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что 5-(бифенил-4-ил)тиофен-2-карбоновая кислота (соединение 1023) использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1728.7 (M+H)+.
Получение соединений формулы (I), где R представляет

Пример 8
Получение (13→4)-лактона N-[4-(4-(гексилокси)фенил)тиофен-2-карбонил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (58)
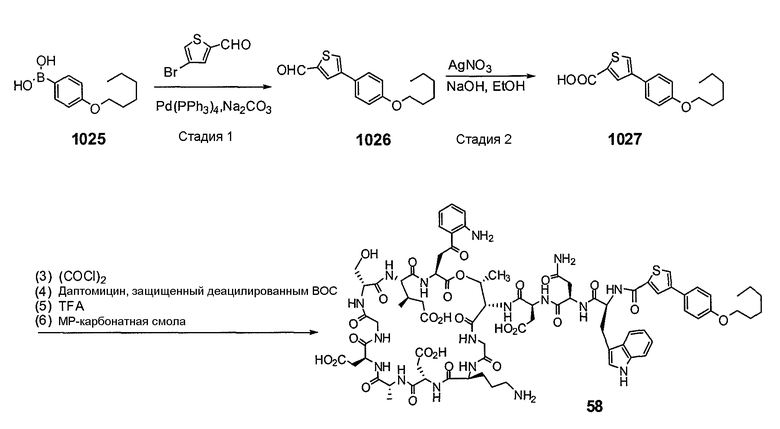
Стадия 1: Получение 4-(4-(гексилокси)фенил)тиофен-2-карбальдегида (1026)
Смесь выпускаемой промышленностью 4-(гексилокси)фенилбороновой кислоты (25, 5,55 г, 25,0 ммоль), 4-бромтиофен-2-карбальдегида (5,25 г, 27,5 ммоль), тетракиз(трифенилфосфин)палладия(0) (0,6 г, 0,13 ммоль), толуола (25 мл), этанола (15 мл) и водного карбоната натрия (2M, 25 мл) перемешивали при кипячении в сосуде с обратным холодильником в течение 18 часов в атмосфере азота. Реакционную смесь охлаждали до окружающей температуры, и водный слой экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным хлоридом натрия (50 мл) и сушили над сульфатом магния. После фильтрации и концентрации неочищенный материал очищали перекристаллизацией из этилацетата/простого петролейного эфира (1/2, об/об) для получения соединения 1026 (3,8 г).
Стадия 2: Получение 4-(4-(гексилокси)фенил)тиофен-2-карбоновой кислоты (1027)
Смесь нитрата серебра (4,07 г, 24 ммоль), соединения 1026 (1,73 г, 6 ммоль), этанола (30 мл) и водного раствора гидроксида натрия (1M, 48 мл) перемешивали при 40°C в течение 3 часов и затем разбавляли водой (150 мл). Водную фазу промывали этилацетатом (2×300 мл) и подкисляли до pH 1 1н водным раствором хлористоводородной кислоты. Водный раствор экстрагировали EtOAc (2×300 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали для выхода соединения 1027 (1,5 г), которое использовали непосредственно на следующей стадии без дальнейшей очистки. 1H ЯМР (300 МГц, DMSO-d6) δ 0,87 (м, 3H), 1,31-1,30 (м, 6H), 1,65 (м, 2H), 3,98 (м, 2H), 6,96 (д, J=8,4 Гц, 2H), 7,66 (д, J=8,7 Гц, 2H), 8,05 (с, 2H), 13,19 (с, 1H). MS m/z =303 [M-H]-.
Стадии 3-6: Получение (13→4)-лактона N-[4-(4-(гексилокси)фенил)тиофен-2-карбонил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (58)
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что 4-(4-(гексилокси)фенил)тиофен-2-карбоновую кислоту (соединение 1027) использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1752,7 (M+H)+.
Получение соединений формулы (I), где R представляет
Пример 9
Получение (13→4)-лактона N-[4-(4-гексилциклогексил)бензоил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (54)
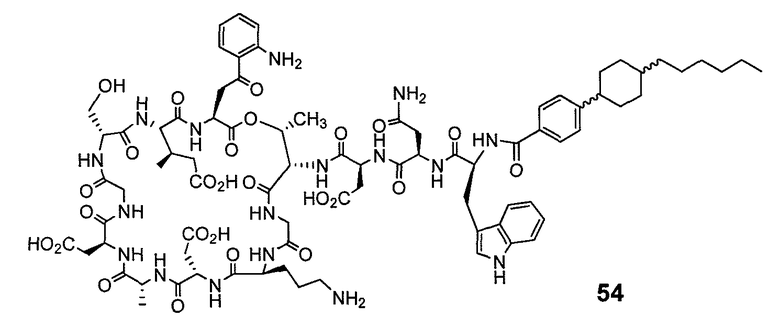
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью 4-(4-гексилциклогексил)бензойную кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1736,8 (M+H)+.
Получение соединений формулы (I), где R представляет
Пример 10
Получение (13→4)-лактона N-циклододеканкарбонил-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диаминол-γ-оксабензолбутановой кислоты (28)
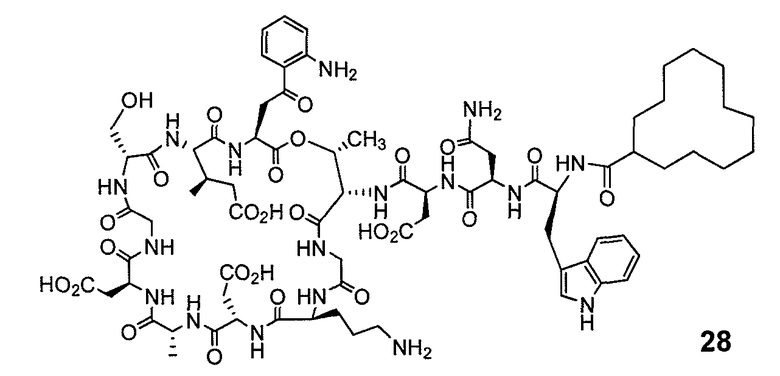
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью циклододеканкарбоновую кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1660,8 (M+H)+.
Получение соединений формулы (I), где R представляет
Пример 11
Получение (13→4)-лактона N-[4-(4-изопропилфенил)бутаноил]-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (30)
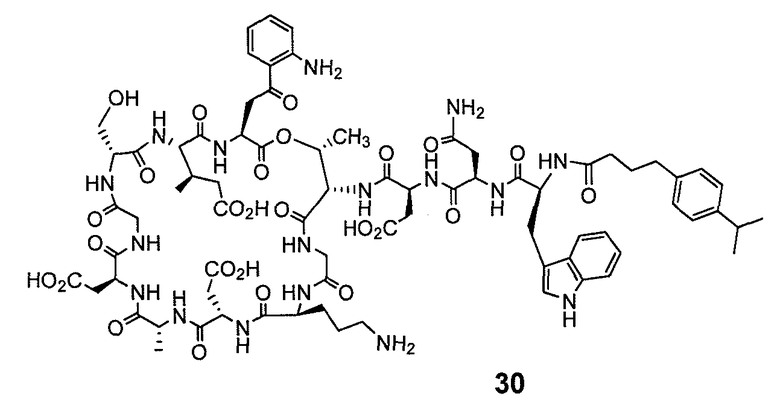
Указанное выше соединение получали таким же образом, как описано в стадиях 3-6 при получении (13→4)-лактона N-{1-[(E)-3-(4-пентилфенил)бут-2-еноил]}-L-триптофил-D-аспарагинил-L-α-аспартил-L-треонилглицил-L-орнитил-L-α-аспартил-D-аланил-L-α-аспартилглицил-D-серил-(3R)-3-метил-L-α-глутамил-(αS)-α,2-диамино-γ-оксобензолбутановой кислоты (см. пример 1), за исключением того, что выпускаемую промышленностью 4-(4-изопропилфенил)бутановую кислоту использовали вместо (E)-3-(4-пентилфенил)бут-2-еновой кислоты. MS m/z 1654,7 (M+H)+.
Дополнительные соединения получали, используя процедуры, аналогичные процедурам, описанным выше в примерах 1-11, из соответствующих выпускаемых промышленностью карбоновых кислот. Карбоновые кислоты превращали или в активированные сложный PFP (пентафторфениловый) эфир или ацилхлорид, как указано в таблице V, при обработке даптомицином, защищенным деацилированным BOC, с последующим удалением защитной группы.
Для получения соединений, где карбоновые кислоты не выпускались промышленностью, карбоновые кислоты получали или процедурами, описанными в литературе, или как описано ниже в примерах 12-17. Затем карбоновые кислоты превращали или в активированный сложный PFP эфир, изоцианат или ацилхлорид, как указано в таблице VI, затем обрабатывали даптомицином, защищенным деацилированным BOC, с последующим удалением защитной группы с использованием процедур, описанных выше в примерах 1-11.
Пример 12: Получение 4-(4-(изопропилтио)фенил)тиофен-2-карбоновой кислоты (73)
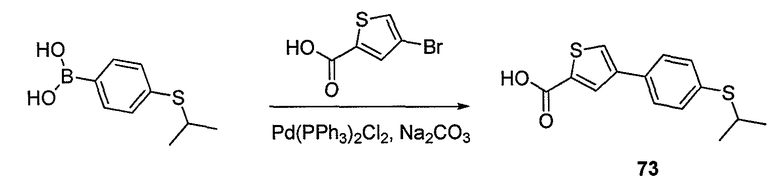
Смесь выпускаемых промышленностью 4-(изопропилтио)фенилбороновой кислоты (735 мг, 3,75 ммоль), 4-бромтиофен-2-карбоновой кислоты (776 мг, 3,75 ммоль), хлорида бис(трифенилфосфин)палладия (II) (131 мг, 0,188 ммоль), ацетонитрила (7,5 мл) и водного карбоната натрия (1M, 7,5 мл) нагревали до 150°C микроволновым облучением (Biotage Intiator™ Sixty, 0-300 W, предварительное перемешивание 2 мин) в течение 5 мин в атмосфере азота. Реакционную смесь охлаждали до окружающей температуры, разбавляли водой (75 мл) и экстрагировали EtOAc (2×50 мл). Затем полученный водный слой подкисляли водной HCl (3,0 M) до pH 2 и экстрагировали EtOAc (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали для получения соединения 73 (990 мг), которое использовали на следующей стадии без дальнейшей очистки.
Пример 13: Получение 4-(4-(гексилтио)фенил)тиофен-2-карбоновой кислоты (74)
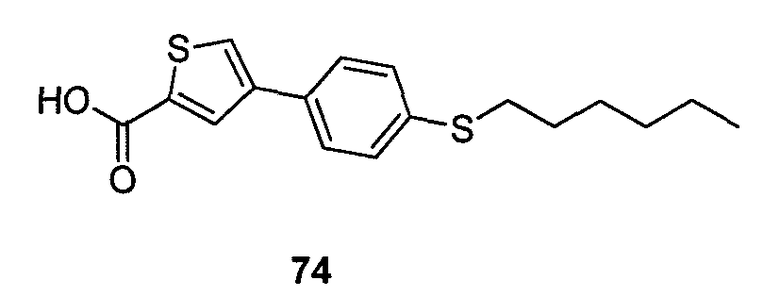
Указанное в заголовке соединение получали таким же образом, как описано при получении 4-(4-(изопропилтио)фенил)тиофен-2-карбоновой кислоты (соединение 73, пример 12), за исключением того, что использовали 4-(гексилтио)фенилбороновую кислоту (Chemical Communications 2003,1, 138-139) вместо 4-(изопропилтио)фенилобороновой кислоты.
Пример 14: Получение 4-(4-(пропилтио)фенил)тиофен-2-карбоновой кислоты (75)
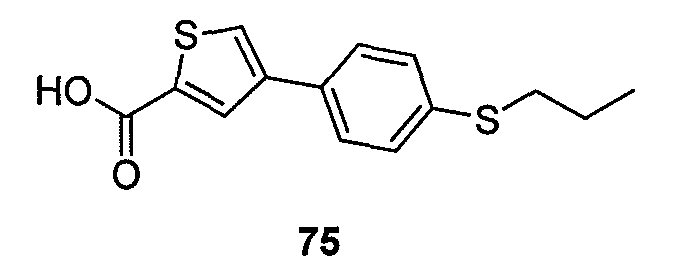
Указанное в заголовке соединение получали таким же образом, как описано при получении 4-(4-(изопропилтио)фенил)тиофен-2-карбоновой кислоты (соединение 73, пример 12), за исключением того, что использовали выпускаемую промышленностью 4-(пропилтио)фенилбороновую кислоту вместо 4-(изопропилтио)фенилобороновой кислоты.
Пример 15: Получение 4-(4-изопропилциклогексил)бутановой кислоты (78)
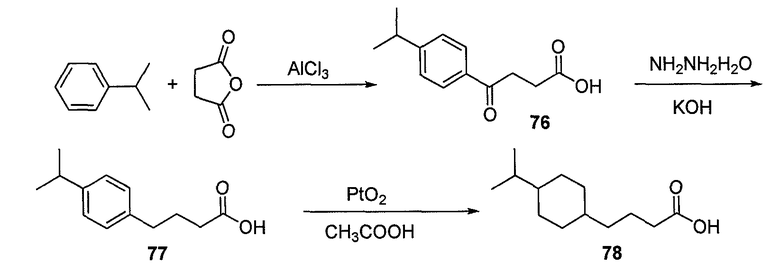
Стадия 1: Получение 4-(4-изопропилфенил)-4-оксобутановой кислоты (76)
К суспензии хлорида алюминия (III) (6,7 г, 50,1 ммоль) и янтарного ангидрида (2 г, 20 ммоль) в 1,2-дихлорэтане добавляли Кумен (2 г, 16,7 ммоль) при 0-5°C. Реакционную смесь перемешивали при окружающей температуре в течение 16 часов, разбавляли 20 мл водного раствора HCl (1M) и экстрагировали этилацетатом (3×80 мл). Объединенные органические фазы промывали насыщенным хлоридом натрия (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле (4×20 см, элюирование простым петролейным эфиром:этилацетатом=8:1) для получения 3,1 г соединения 76. 1H ЯМР (CDCl3, 300 МГц) δ 1,27-1,29 (д, 6H), 2,80-2,84 (т, 2H), 2,95-3,00 (м, 1 H), 3,29-3,33 (т, 2H), 7,32-7,34 (д, 2H), 7,92-7,94 (д, 2H). MS m/z 221 (M+H)+.
Стадия 2: Получение 4-(4-изопропилфенил)бутановой кислоты (77)
Раствор соединения 76 (0,5 г, 2,27 ммоль) и 2 мл 80% гидразин гидрата перемешивали при 45°C в течение 1 часа. К смеси добавляли KOH (1 г, 17,8 ммоль), и реакционную смесь нагревали до 104-150°C в течение 2 часов. После охлаждения до окружающей температуры реакционную смесь разбавляли 10 мл воды и подкисляли до pH 2 водной HCl (1M) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под пониженным давлением. Остаток очищали колоночной флэш-хроматографией на силикагеле (2×15 см, элюирование петролейным эфиром:этилацетатом=8:1) для получения 275 мг соединения 77. MS m/z 207 (M+H)+.
Стадия 3: Получение 4-(4-изопропилциклогексил)бутановой кислоты (78)
Суспензию соединения 77 (1,0 г, 4,8 ммоль) и 0,1 г PtO2 в 50 мл уксусной кислоты перемешивали в атмосфере H2 (50 фунтов/дюйм2 (примерно 3,5 атм)) при окружающей температуре в течение 6 часов. После фильтрации и выпаривания растворителя получали 1,1 г неочищенного продукта. Соединение использовали непосредственно на следующей стадии без дальнейшей очистки. ЖХ-МС m/z 211 (M-H+).
Пример 16: Получение 1-гептил-4-изоцианатобензола (79)
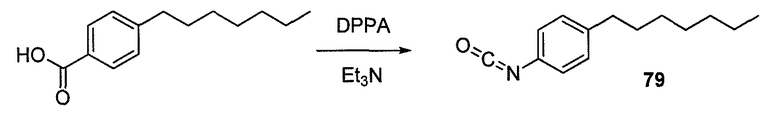
К дегазированному раствору 4-гептилбензойной кислоты (3,0 г, 13,6 ммоль) в безводном толуоле (30 мл) и Et3N (3 мл, 21,5 ммоль) по каплям добавляли дифенилфосфорилазид (2 мл, 9,3 ммоль). После завершения добавления, реакционную смесь нагревали с обратным холодильником и перемешивали в течение 2 часов. Смесь концентрировали до сухости под пониженным давлением для получения продукта, 1-гептил-4-изоцианатбензола (79) в виде масла, который использовали непосредственно в следующей реакции без дальнейшей очистки.
Пример 17: Получение 2-изоцианатундекана (81)
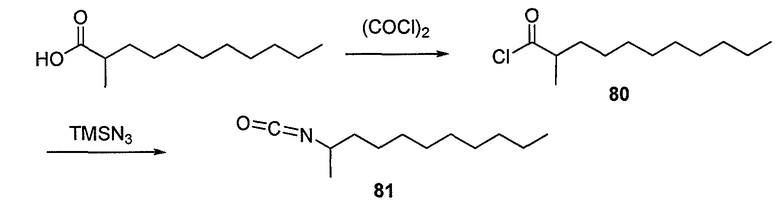
Стадия 1: Синтез 2-метилундеканоилхлорида (80)
К перемешанному раствору 2-метилундекановой кислоты (0,40 г, 2,0 ммоль) в безводном дихлорметане (20 мл) и каталитическому количеству DMF (приблизительно 0,02 мл) добавляли оксалилхлорид (0,4 мл, 4,6 ммоль). Полученную смесь энергично перемешивали в течение 2 часов при комнатной температуре. Органический растворитель выпаривали под пониженным давлением и остаток (80) использовали на следующей стадии.
Стадия 2: Синтез 2-изоцианатундекана (81)
К перемешанному раствору 2-метилундеканоилхлорида (2,0 ммоль) в безводным дихлорметане (10 мл) добавляли азидотриметилсилан (TMSN3) (0,32 моль, 2,4 ммоль). Полученную смесь нагревали с обратным холодильником в течение 5 часов. Избыток азидотриметилсилана удаляли при 50°C в вакууме, и полученный остаток (81) использовали на следующей стадии.
Пример 18: Получение 1-изоцианат-4-пентилциклогексана (82)
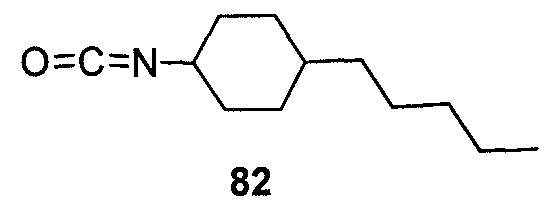
Указанное в заголовке соединение получали таким же образом, как описано при получении 2-изоцианатундекана (соединения 81, пример 17), за исключением того, что выпускаемую промышленностью 4-пентилциклогексанкарбоновую кислоту использовали вместо 2-метилуендекановой кислоты.
Пример 19: Биологическая активность in vitro
Соединения в соответствии с формулой I тестировали на наличие противомикробной активности против набора организмов. Аэробные организмы, Staphylococcus aureus, Enterococcus faecalis и E. faecium тестировали микроразведением бульона в соответствии с документом Института Клинических и Лабораторных Стандартов (CLSI) M7-A7 (Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard - Seventh Edition. CLSI document M7-A7 [ISBN 1-56238-587-9]. Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006) с модификациями, описанными ниже. К бульону Mueller Hinton Broth (MHBc) добавляли 50 мг/л кальция (эквивалентного физиологическим уровням кальция), и аналитические планшеты инкубировали 18-20 часов при 37°C при аэрации (200 об/мин).
Вкратце, соединения растворяли или в смеси 50:50 по объему диметилсульфоксида и стерильной, дистиллированной воды, в зависимости от растворимости соединения, и разбавляли до конечной концентрации (0,03 мкг/мл -32 мкг/мл) в микробной ростовой среде, MHBc. Во всех случаях, конечная концентрация диметилсульфоксида, инкубированного с клетками, составляла менее чем 1% или была равна ей. Для расчетов минимальной ингибиторной концентрации (MIC) двукратные разведения соединений добавляли в лунки титровочных микропланшетов, содержащие 5×105 бактерий в конечном объеме 100 мкл среды. После инкубации (18-20 часов при 37°C с аэрацией при 200 об/мин) рост подтверждали визуально помещением планшетов над визуализирующим прибором (стойку с расположенным ниже зеркалом), и затем оптическую плотность (OD600) измеряли с использованием планшетного ридера SpectraMax 340PC384. Рост определяли как помутнение, которое могло выявляться невооруженным глазом, или достигающее OD600 минимум 0,1. Величины MIC (в мкг/мл) определялись как самая низкая концентрация, не вызывающая видимой мутности.
Для анаэробных Clostridium difficile величины MIC определяли в исследовательской лаборатории R. M. Alden (RMA) Research Laboratory (Culver City, CA) методом разведения на агаре в соответствии с документом CLSI M11-A7 (Clinical and Laboratory Standards Institute. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard - Seventh Edition. CLSI document M11-A7 [ISBN 1-56238-626-3]. Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2007.) Агар, использованный во время тестирования, получали с обеспечением содержания в нем 50 мг/л Ca++ путем приготовления бульонной среды для роста Brucella с концентрацией свободных ионов Ca++, определяемой с использованием кальциевого электрода и добавлением 3,9 мл/л 10 мг/мл раствора CaCl2, и затем 1,5% агара, и в дополнение в среду добавляли витамин K1, гемин и 5% гемолизированной овечьей крови.
Штаммы C. difficile strains извлекали из содержащих токсин фекальных образцов, собранных с 2006 по 2008 гг. Их хранили при -70°C в 20% снятом молоке в коллекции культур RMA. Штаммы удаляли из морозильника и переносили, по меньшей мере, дважды на кровяной агар с добавками для роста Brucella перед тестированием. C. difficile (ATCC (Американская Коллекция Типовых Культур) #700057) и Staphylococcus aureus (ATCC #29213) использовали в качестве контролей.
Серийные двукратные разведения соединений получали в день тестирования и добавляли в расплавленный агар с углублениями, смешивали и выливали в чашки Петри. После отверждения планшеты сушили в инкубаторе в течение 30 минут.
Инокулят получали в анаэробной камере из 48-часовых культур приготовлением суспензий, равных по мутности 0,5 стандарту McFarland в бульоне для роста Brucella и распределяли по лункам головки репликатора Steer. Репликатор удаляли из камеры для инокуляции содержащих лекарственное средство планшетов на лабораторном столе в окружающих условиях. Не содержащие лекарственное средство контрольные планшеты штамповали перед и после каждой серии лекарственных средств для тестирования жизнеспособности бактерий.
Планшеты переносили назад в анаэробную камеру и инкубировали при 37°C в течение 48 часов. Их удаляли из камеры и исследовали для выявления роста бактерий. MIC представляла собой концентрацию лекарственного средства, которая ингибировала рост или заметно уменьшала рост, по сравнению с контрольными планшетами, не содержавшими лекарственное средство, в соответствии с руководством лабораторных стандартов CLSI.
Величины MIC репрезентативных соединений по настоящему изобретению перечислены в таблице VII. Величины C. difficile представлены в виде MIC90, где 90% или 27 из 30 тестированных изолятов имели данную MIC (мкг/мл) или ниже.
Биологическая активность соединений формулы I
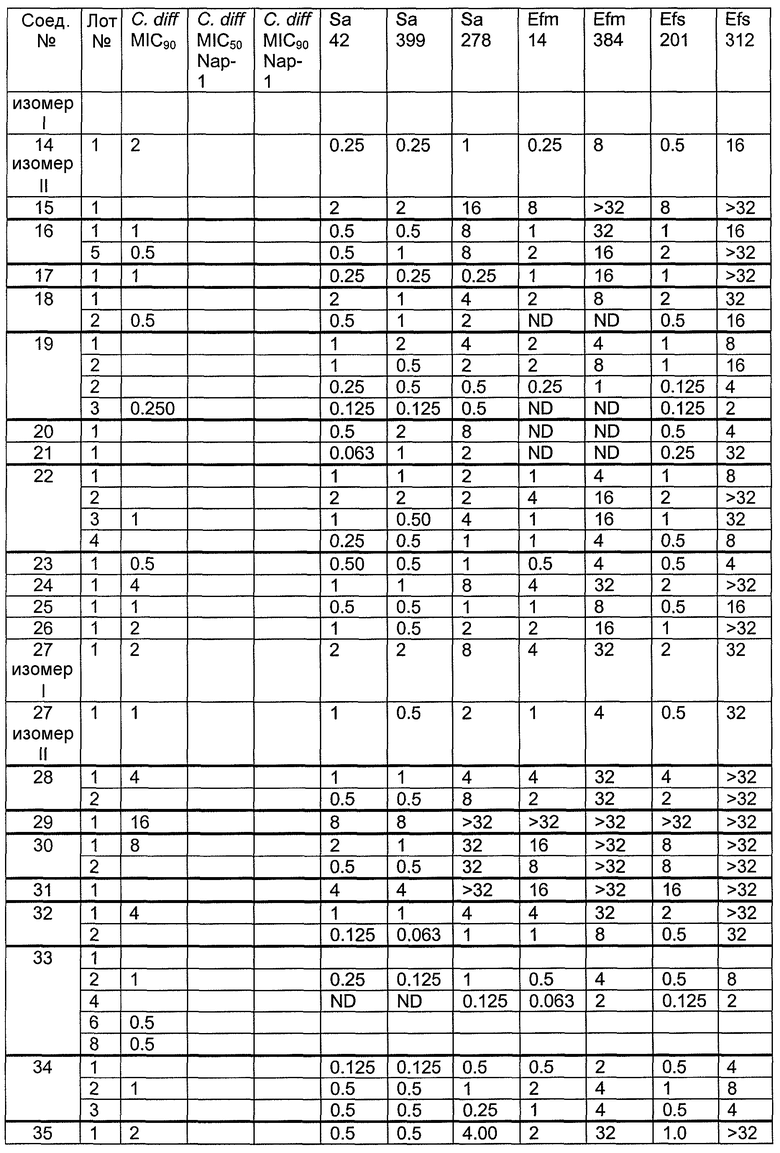

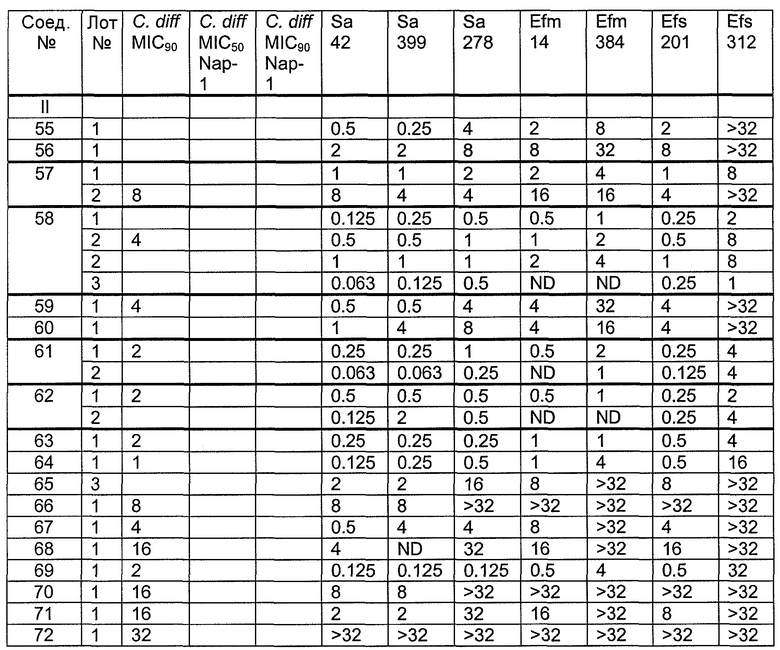
где
Аэробные бактерии:
C.difficile
Анаэробные бактерии, извлеченные из содержащих токсин фекальных образцов:
Штамм Clostridium difficile NAP1, извлеченный из содержащих токсин фекальных образцов № RMA 19139 REA B1,
RMA относится к лабораториям RM Alden Labs, Santa Monica, California.
Пример 20. Биологическая активность in Vivo
Сирийских золотистых хомячков предварительно лечили подкожным введением клиндамицина в дозе 10 мг/г за 24 часа до контрольного бактериального заражения. Инокулят из 20 спор C. difficile (ATCC #43596) в стерильном солевом растворе вводили перорально. Лечение начинали через 4 часа после инокуляции dH2O, етронидазолом (MET), ванкомицином (VAN) или соединением по настоящему изобретению. Схемы перорального введения для VAN и соединения формулы I представляли собой 0,5 мг/кг один раз в день в течение 5 дней. MET вводили в дозе 70 мг/кг 3 раза в день в течение 5 дней. CDAD (заболевание, связанное с Clostridium Difficile) подтверждали как причину смерти наблюдениями при вскрытии, включая влажный хвост и/или макроскопические изменения слепой кишки. Защиту в %, обеспечиваемую каждым тестированным видом лечения, рассчитывали через 1 и 21 день после введения. Средние величины защиты в процентах репрезентативных соединений по настоящему изобретению перечислены в таблице VIII.
Средние величины защиты in vivo
Все контрольные хомячки, инфицированные C. difficile (n=30), которым вводили стерильную воду, умерли в пределах 48 часов после заражения. Лечения MET (n=10) или VAN (n=21 достигли величин защиты в % соответственно 100% или 90% через 1 день после введения и 0% или 62% через 21 день после введения. Соединения по настоящему изобретению обеспечивают величины защиты в %, достигающие 100% и через 1, и через 21 день после введения.
Считается, что соединения со структурой, напоминающей структуры по настоящему изобретению, описаны в Европейском патенте EP No. 0095295('295), и в патентах США №№. 32310, 7335725 ('725) и 6911525 ('525), которые все полностью включены в настоящее описание путем ссылки. См., например, соединение 39 по настоящему изобретению или описанное в патенте '295 (пример 73). Однако указанные ранее известные соединения демонстрируют значительно меньшую активность против грамположительных бактерий, таких как C. Difficile, и другие различные бактериальные штаммы, включая те, которые устойчивы к даптомицину. Например, считается, что соединение 39 патента '295 представляет собой единственное ранее известное соединение, которое структурно напоминает соединения 32 и 33 по настоящему изобретению. Новые соединения 32 и 33 по настоящему изобретению проявляют неожиданные и улучшенные свойства против C. Difficile, о чем свидетельствует таблица VII. Величины MIC50 для соединений 32 и 33 против C. difficile составляют 4 и максимум 1, тогда как MIC90 для соединения 39 оставляет 16. Кроме того, соединения 32 и 33 проявляют улучшенные свойства относительно различных других штаммов грамположительных бактерий, включая те, которые устойчивы к даптомицину.
В еще одном аспекте, как показано в таблице IX, сравнительные анализы данных соединений 48-50 по настоящему изобретению с известными соединениями 111 и 112, как описано в патентах '725 и '525, выявляют, что соединения 48-50 проявляют превосходящую и неожиданную активность против грамположительных бактерий, включая те, которые устойчивы к даптомицину. Например, соединения 49 и 50 имеют величины MIC, максимум 0,3 против Sa42, тогда как величины MIC для соединений 111 и 112 составляют соответственно максимум 1,0 и 0,5. Также, соединение 49 проявляет увеличенную и неожиданную активность в целом против Sa399, Sa278, Efm14, Efm384, Efs201 и Efs312, по сравнению с соединением 111.
Сравнение данных соединений 48-50 с соединениями 111 и 112 патентов '725 и '525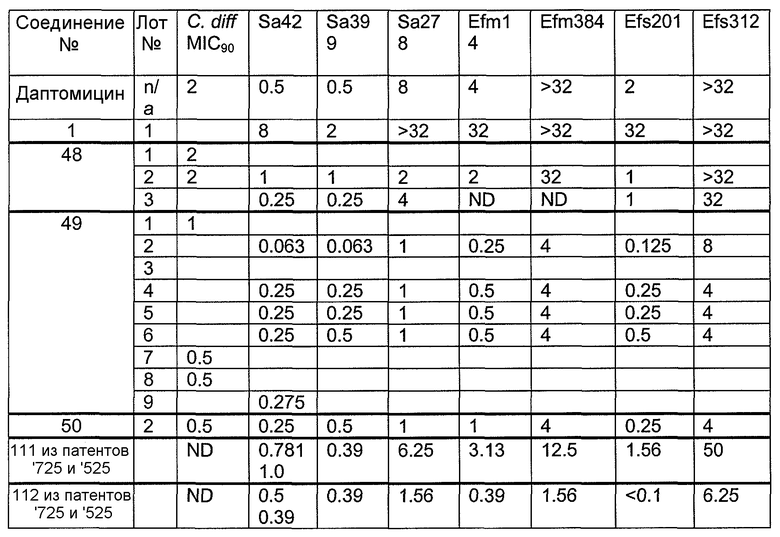
Таким образом, считается, что указанные выше соединения, описанные в предшествующем уровне техники, полностью отличаются от соединений по настоящему изобретению, которые являются новыми и активными против грамположительных бактерий, проявляют увеличенную активность против Clostridium difficile и/или проявляют неожиданную активность против бактерий, которые устойчивы к даптомицину.
Хотя были показаны и описаны некоторые варианты осуществления, в них могут быть внесены различные модификации и замещения без отхода от сущности и объема изобретения. Например, не предполагается, что изложенная ниже формула изобретения должна рассматриваться уже, чем ее буквальное изложение, и не предполагается, что иллюстративные варианты осуществления из описания следует принимать как включенные в формулу изобретения. Соответственно, следует понимать, что настоящее изобретение было описано в настоящей заявке в качестве иллюстрации и что такие описания не ограничивают объем формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения пептидов с последовательностью актг-человека,содержащих в -конечном положении аминооксикислоту | 1973 |
|
SU490284A3 |
| СПОСОБ ОЧИСТКИ ЛИПОПЕПТИДОВ | 2010 |
|
RU2526391C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДОВ КАК ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ ЛАМИНИНА/НИДОГЕНА | 2000 |
|
RU2380371C2 |
| ПРОИЗВОДНЫЕ N-АЦИЛ- α -АМИНОКИСЛОТЫ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОСТЫЕ ИЛИ СЛОЖНЫЕ ЭФИРЫ, АМИДЫ ИЛИ ГИДРАТЫ И КОМПОЗИЦИЯ ИНГИБИРУЮЩАЯ СВЯЗЫВАНИЕ АДГЕЗИВНЫХ ПРОТЕИНОВ С ТРОМБОЦИТАМИ И АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1992 |
|
RU2097378C1 |
| СПОСОБ ЛЕЧЕНИЯ СЕПТИЧЕСКОГО ШОКА И ПРИМЕНЕНИЕ МУРАМИЛОВЫХ СОЕДИНЕНИЙ | 1992 |
|
RU2139086C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ ПРОТИВООПУХОЛЕВЫХ ЦЕЛЕВЫХ ФЕРМЕНТОВ | 2002 |
|
RU2319482C2 |
| КОМБИНАЦИИ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ ДЛЯ ЛЕЧЕНИЯ РАКА | 2007 |
|
RU2449788C2 |
| КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ ИНГИБИТОРЫ Bcr-Abl/c-Kit/PDGF-R TK, ДЛЯ ЛЕЧЕНИЯ РАКА | 2007 |
|
RU2452492C2 |
| ПЕПТИД, ОБЛАДАЮЩИЙ ИММУНОМОДУЛИРУЮЩЕЙ АКТИВНОСТЬЮ | 1995 |
|
RU2091389C1 |
| УСОВЕРШЕНСТВОВАННЫЕ ПЕПТИДНЫЕ СОЕДИНЕНИЯ, ВЫСВОБОЖДАЮЩИЕ ГАСТРИН | 2003 |
|
RU2330859C2 |
Изобретение относится к новым липопептидным соединениям формулы (I):
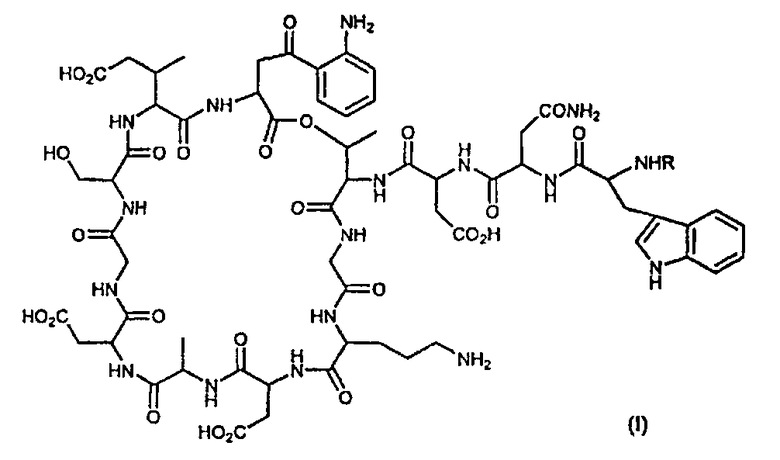
и к его фармацевтически приемлемым солям, где: R представляет: 
и v представляет целое число от 3 до 5. Изобретение также относится к фармацевтической композиции, обладающей противобактериальной активностью, на основе указанного соединения. Технический результат: получено новое соединение формулы (I), которое может найти применение в медицине в качестве антибактериальных средств против Clostridium difficile. 5 н. и 21 з.п. ф-лы, 9 табл., 20 пр.
1. Соединение формулы (I):
и его фармацевтически приемлемые соли, где:
R представляет:
и v представляет целое число от 3 до 5.
2. Соединение формулы (II):
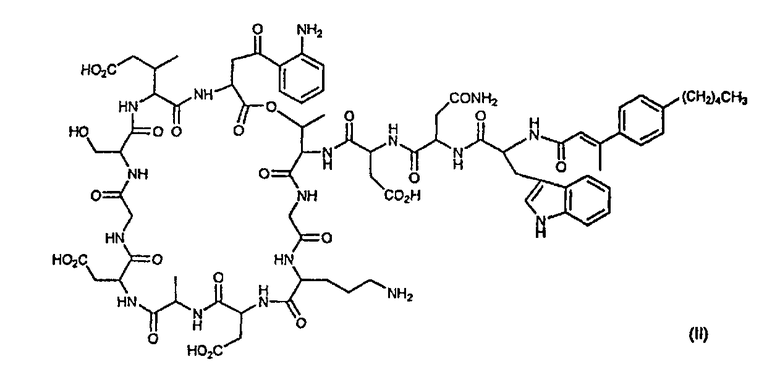
и его фармацевтически приемлемая соль.
3. Фармацевтическая композиция, обладающая противобактериальной активностью, содержащая соединение по п.2 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
4. Способ лечения или профилактики бактериальной инфекции, включающий введение нуждающемуся в нем объекту терапевтически эффективного количества фармацевтической композиции по п.3.
5. Способ по п.4, где объект представляет собой человека, животное или клеточную культуру.
6. Способ по п.5, где объект представляет собой человека.
7. Способ по п.4, где бактериальная инфекция вызвана грамположительными бактериями.
8. Способ по п.7, где бактерия представляет собой штаммом Clostridium difficile.
9. Способ по п.8, где штамм Clostridium difficile представляет собой штамм Nap1 Clostridium difficile.
10. Способ по п.7, где бактерия представляет собой бактерию, устойчивую к антибиотикам.
11. Способ по п.10, где устойчивая к антибиотикам бактерия представляет собой устойчивый к даптомицину Staphylococcus aureus, устойчивый к даптомицину Enterococcus faecium, устойчивый к даптомицину Enterococcus faecalis, устойчивый к метициллину Staphylococcus aureus или смесь бактерий, включающую, по меньшей мере, одну из устойчивых к антибиотикам бактерию.
12. Способ по п.10, где устойчивая к антибиотикам бактерия устойчива к ванкомицину, метициллину, гликопептидным антибиотикам, пенициллину или даптомицину.
13. Способ по п.7, где бактериальная инфекция представляет собой заболевание, связанное с Clostridium Difficile (CDAD).
14. Способ по п.13, где заболевание, связанное с Clostridium Difficile, возникает в результате инфекции и обостряется инфекцией Nap1 Clostridium difficile.
15. Применение соединения по п.2 для получения лекарственного средства для лечения бактериальной инфекции у объекта.
16. Применение по п.15, где объект выбран из человека, животного или клеточной культуры.
17. Применение по п.16, где объект представляет собой человека.
18. Применение по п.15, где бактериальная инфекция вызвана грамположительными бактериями.
19. Применение по п.18, где бактерия представляет собой штамм Clostridium difficile.
20. Применение по п.19, где штамм Clostridium difficile представляет собой штамм Nap1 Clostridium difficile.
21. Применение по п.18, где бактерия представляет собой бактерию, устойчивую к антибиотикам.
22. Применение по п.17, где устойчивая к антибиотикам бактерия представляет собой устойчивый к даптомицину Staphylococcus aureus, устойчивый к даптомицину Enterococcus faecium, устойчивый к даптомицину Enterococcus faecalis, устойчивый к метициллину Staphylococcus aureus или смесь бактерий, включающую, по меньшей мере, одну из устойчивых к антибиотикам бактерию.
23. Применение по п.22, где устойчивая к антибиотикам бактерия устойчива к ванкомицину, метициллину, гликопептидным антибиотикам, пенициллину или даптомицину.
24. Применение по п.18, где бактериальная инфекция представляет собой заболевание, связанное с Clostridium Difficile (CDAD).
25. Применение по п.24, где заболевание, связанное с Clostridium Difficile, возникает в результате инфекции и обостряется инфекцией Nap1 Clostridium difficile.
26. Соединение по п.3, где соединение имеет формулу
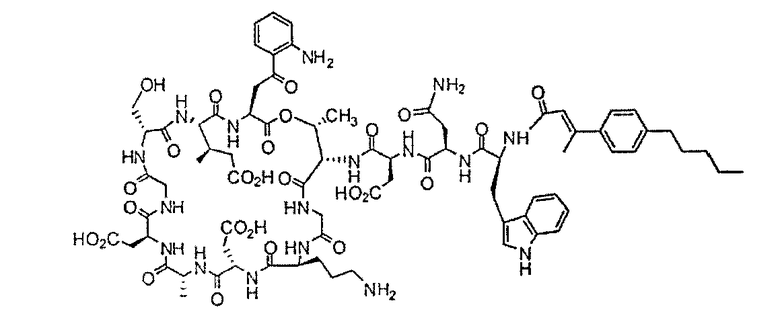
или его фармацевтически приемлемая соль.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Пресс для литниковых и стопорных трубок | 1950 |
|
SU95295A1 |
| RU 2007121705 A, 20.12.2008 | |||

