Настоящее изобретение относится к новым оксазолидиниловым антибиотическим соединениям, к фармацевтической антибактериальной композиции, содержащей их, и к применению этих соединений при изготовлении лекарственного средства для лечения инфекций (например, бактериальных инфекций). Эти соединения используются в качестве антимикробных агентов, эффективных по отношению к целому ряду болезнетворных организмов человека и животных, включая, среди прочих, грамположительные и грамотрицательные аэробные и анаэробные бактерии и микобактерии.
Интенсивное применение антибиотиков оказывает селективное эволюционное воздействие на микроорганизмы, способствуя продуцированию в них генетически сформированных механизмов резистентности. Современное медицинское и социо-экономическое поведение обостряет проблему развития резистентности, создавая условия постепенного роста патогенных микроорганизмов, например, в искусственных суставах, и в условиях долговременной поддержки резервов хозяина, например, у иммунологически «скомпрометированных» пациентов.
В больничных условиях увеличивающееся число штаммов Staphylococcus aureus. Streptococcus pneumonia, Enterococcus spp.и Pseudomonas aeruginosa, i.
главных источников инфекций, делает их резистентными в отношении множества лекарственных средств и вызывает трудности при лечении, вплоть до невозможности лечения вообще:
- S. aureus является резистентным по отношению к р-лактаму, хинолонам и теперь даже к ванкомицину;
- S. pneumoniae становится резистентным по отношению к пенициллиновым или хинолоновым антибиотикам и даже к новым макролидам;
- Enteroccocci являются резистентными по отношению к хинолону и ванкомицину, а β-лактамные антибиотики не оказывают никакого эффективного воздействия на эти штаммы;
- Enterobacteriacea являются резистентными по отношению к цефалоспорину и хинолону;
- Р. aeruginosa являются резистентными по отношению к Р-лактаму и хинолону.
Кроме того, скорость распространения мультилекарственно-резистентных грамм-отрицательных штаммов таких, как Enterobacteriacea и Pseudomonas aeruginosa, равномерно увеличивается, вследствие чего вновь появляющиеся организмы, подобные таким организмам, как Acinetobacter spp.или Clostridium difficile, которые были выделены в процессе терапии с использованием применяемых в настоящее время антибиотиков, становятся реальной проблемой в больничных условиях. Поэтому в медицине существует большая потребность в новых антибактериальных агентах, способных преодолевать мультилекарственную резистентность грамм-отрицательных бактерий таких, как A. baumannii, ESBL-продуцирующие Е. coli и Klebsiella штаммы и Pseudomonas aeruginosa (Clinical Infectious Diseases (2006), 42, 657-68).
Помимо этого, микроорганизмы, вызывающие устойчивые инфекции, все более признаются в качестве являющихся причиной или сопутствующих факторов некоторых хронических болезней, подобных пептическим язвам или сердечным болезням.
Нафтиридин-2-оновые и хинолин-2-оновые антибиотические соединения уже были описаны в следующих публикациях: WO 2006/134378, WO 2006/137485, WO 2007/138974, WO 2008/006648, WO 2008/009700, WO 2008/071961, WO 2008/071964 и WO 2008/071981.
Хинолин-, нафтиридин- или хиноксалин-спирооксазолидиноновые антибиотические соединения были, кроме того описаны в WO 2008/026172.
Авторами заявки предложен в настоящее время новый класс оксазолидиниловых антибиотиков, соответствующих формуле (I), приведенной ниже.
Различные варианты осуществления настоящего изобретения представлены далее:
i) Изобретение, прежде всего, относится к соединениям формулы (I)
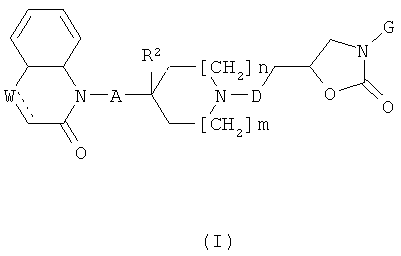
где
R1 представляет собой алкоксигруппу (предпочтительно, метоксигруппу) или галоген (предпочтительно, фтор);
U и V каждый независимо друг от друга представляет собой СИ или N;
 обозначает связь или отсутствует;
обозначает связь или отсутствует;
W представляет собой СН или N или, когда  отсутствует, W представляет собой СН2 или NH,
отсутствует, W представляет собой СН2 или NH,
при условии, что U, V и W не все представляют собой N;
А представляет собой связь или СН2;
R2 представляет собой И или, при условии, что А представляет собой CH2, может также представлять собой ОН;
m и n каждое независимо друг от друга равно 0 или 1;
D представляет собой СН2 или связь;
G представляет собой фенильную группу, которая является однократно или дважды замещенной в м- и/или п-положении(ях) заместителями, выбранными из алкила, С1-3алкоксигруппы и галогена (предпочтительно, фтора), при этом С1-3алкоксизаместитель, предпочтительно, представляет собой линейную С1-3алкоксигруппу и находится в п-положении, или G представляет собой одну из групп G1 и G2
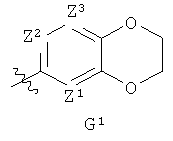
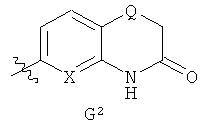
где
Z1, Z2 и Z3 каждый представляет собой СH, или Z1 и Z2 каждый представляет собой СН и Z3 представляет собой N, или Z1 представляет собой СН, Z2 представляет собой N и Z3 представляет собой СН или N, или Z1 представляет собой N и Z2 и Z3 каждый представляет собой СН; и
Х представляет собой N или СН и Q представляет собой О или S;
при этом следует иметь в виду, что если тип каждое равно 0, тогда А представляет собой СН2;
и к солям (в частности, фармацевтически приемлемым солям) соединений формулы (I).
В следующих параграфах представлены определения различных химических фрагментов для соединений согласно изобретению. Упомянутые определения предназначены для единообразного применения во всем описании и в формуле изобретения, если не указано иначе, и если определения не подлежат более широкому или, наоборот, более узкому толкованию.
Термин "алкил", используемый самостоятельно или в комбинации, относится к насыщенной линейной или разветвленной алкильной группе, содержащей от одного до четырех атомов углерода. Типичные примеры алкильных группп включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутнл и трет-бутил. Термин "С1-халкил" (х представляет собой целое число) относится к линейной или разветвленной алкильной группе, содержащей от одного до х атомов углерода. Предпочтительными алкильными группами являются метил и этил. Наиболее предпочтительной алкильной группой является метил.
Термин "алкоксигруппа", используемый самостоятельно или в комбинации, относится к линейной или разветвленной алкильной группе, содержащей от одного до четырех атомов углерода. Термин "Сх-уалкоксигруппа" (при этом, х и у каждый является целым числом) относится к алкоксигруппе по определению, данному выше, содержащей от х до у атомов углерода. Например, C1-3алкоксигруппа содержит от одного до трех атомов углерода. Типичные примеры алкоксигрупп включают метоксигруппу, этоксигруппу, н-пропоксигруппу и изопропоксигруппу. Предпочтительными примерами являются метоксигруппа и этоксигруппа.
Термин «галоген» относится к фтору, хлору, брому или йоду, предпочтительно к фтору или хлору.
В этой патентной публикации связь, прерванная волнистой линией, указывает на точку присоединения радикала к остатку молекулы. Например, радикал, изображенный ниже
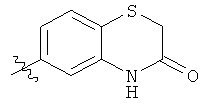
представляет собой 3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил.
Настоящее изобретение включает также меченные изотопом соединения, в частности, меченные с помощью Н (дейтерия) соединения формулы (I), при этом меченные соединения идентичны соединения формулы (I) и отличаются лишь тем, что в них один или более атомов замещены атомом, имеющим такой же атомный номер, но отличающимся по атомной массе от атомов, встречающихся в природе. Меченные изотопом соединения, в частности, меченные 2Н (дейтерием) соединения формулы (I), и их соли включены в объем настоящего изобретения. Замещение водорода более тяжелым изотопом 2Н (дейтерием) приводит к более значительной метаболической стабильности, что, например, в условиях in vivo увеличивает период полураспада или уменьшает требуемое дозирование, или может приводить к уменьшенному ингибированию Р450 ферментов, что улучшает профиль безопасности. В одном варианте осуществления изобретения соединения формулы (I) не являются меченными изотопом, или они мечены только одним или более атомами дейтерия. В подварианте соединения формулы (I) совсем не мечены изотопом. Изотопически меченные соединения формулы (I) могут быть получены аналогично методам описанным далее, но с использованием подходящего изотопного варианта соответствующих реагентов или исходных веществ.
Термин "фармацевтически приемлемые соли" относится к нетоксичным, аддитивным солям неорганической или органической кислоты и/или основания. Более подробные сведения могут быть найдены в публикации "Salt selection for basic drugs". Int. J. Pharm., (1986), 33, 201-217.
Кроме того, термин "комнатная температура", используемый в данном описании, относится к температуре 25°С.
Когда речь идет не о температурах, термин "приблизительно", расположенный перед численной величиной "X", относится в обычном применении к интервалу, составляющему от Х минус 10% Х до Х плюс 10% X, и предпочтительно, к интервалу, составляющему от Х минус 5% Х до Х плюс 5% X. В особом случае, касающемся температур, термин "приблизительно", расположенный перед температурой "Y" относится в обычном применении к температурному интервалу, составляющему от Y минус 10 С до Y плюс 10°С, и предпочтительно, к интервалу, составляющему от Y минус 5 С до Y плюс 5 С.
ii) В частности, изобретение относится к соединениям формулы (I), которые также являются соединениями формулы (ICE)
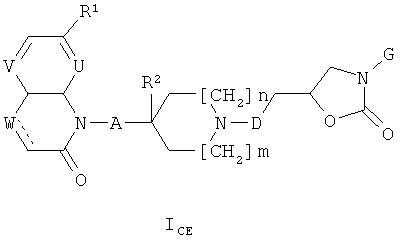
где
R1 представляет собой алкоксигруппу (предпочтительно, метоксигруппу);
V представляет собой СH;
U и W каждый представляет собой СН и обозначает связь, или U представляет собой СН, W представляет собой N и обозначает связь, или U и W каждый представляет собой N и обозначает связь, или U представляет собой СН, W представляет собой СН2 и отсутствует;
А представляет собой связь или СН2;
R2 представляет собой Н или, при условии, что А представляет собой СН2, может также представлять собой ОН;
m и n каждое независимо друг от друга равно 0 или 1;
D представляет собой СН2 или связь;
G представляет собой фенильную группу, которая однократно замещена в м-положении и однократно замещена в п-положении заместителями, выбранными из алкила (предпочтительно, метила) и галогена (предпочтительно, фтора), или G представляет собой одну из групп G1 и G2,
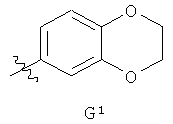 ,
, 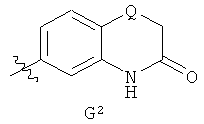
где Q представляет собой О или S;
при этом следует иметь в виду, что, если тип каждое равно 0, тогда А представляет собой СН2;
и к солям (в частности, фармацевтически приемлемым солям) соединений формулы (ICE).
iii) Согласно предпочтительному варианту осуществления настоящего изобретения, соединения формулы (I), представленные выше в вариантах i) или ii), должны быть такими, в которых R1 представляет собой алкоксигруппу или фтор (и предпочтительно, C1-3алкоксигруппу, в частности, метоксигруппу или этоксигруппу, наиболее предпочтительно метоксигруппу).
iv) Другой вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в вариантах i), ii) или iii), где обозначает связь.
v) Согласно одному под-варианту варианта осуществления изобретения iv), соединения формулы (I), представленные выше в варианте iv), должны быть такими, в которых W представляет собой СH.
vi) Согласно другому под-варианту варианта осуществления изобретения iv), соединения формулы (I), представленные выше в варианте iv), должны быть такими, в которых W представляет собой N.
vii) Еще один вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в вариантах i), ii) или iii), где отсутствует.
viii) Согласно одному под-варианту варианта осуществления изобретения vii), соединения формулы (I), представленные выше в варианте vii), должны быть такими, в которых W представляет собой CHz.
ix) Согласно другому под-варианту варианта осуществления изобретения vii), соединения формулы (I), представленные выше в варианте vii), должны быть такими, в которых W представляет собой NH.
х) Один главный вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в вариантах осуществления изобретения i)-ix), в которых V представляет собой СН.
xi) Согласно одному под-варианту главного варианта осуществления настоящего изобретения х), соединения формулы (I), представленные выше в главном варианте осуществления изобретения х), должны быть такими, в которых U представляет собой СН.
xii) Согласно другому под-варианту главного варианта осуществления изобретения х), соединения формулы (I), представленные выше в главном варианте осуществления изобретения х), должны быть такими, в которых U представляет собой N.
xiii) Другой главный вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным в одном из вариантов осуществления изобретения i)-ix), в которых V представляет собой N.
xiv) Следующий вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в варианте осуществления изобретения i)-xiii), в которых А представляет собой связь.
xv) Предпочтительно, соединения формулы (I), представленные выше в варианте осуществления изобретения xiv), должны быть такими, в которых тип каждый представляет собой 1.
xvi) Еще один вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в варианте осуществления изобретения i)-xiii), в которых А представляет собой СН;.
xvii) Согласно одному под-варианту варианта осуществления изобретения xvi), соединения формулы (I), представленные выше в варианте осуществления изобретения xvi), должны быть такими, в которых тип каждый представляет собой 0.
xviii) Согласно другому под-варианту варианта осуществления изобретения xvi), соединения формулы (I), представленные выше в варианте осуществления изобретения xvi), должны быть такими, в которых одно из m и п равно 0, а другое равно 1.
xix) Согласно еще одному под-варианту варианта осуществления изобретения xvi), соединения формулы (I), представленные выше в варианте осуществления изобретения xvi), должны быть такими, в которых тип каждое равно 1.
хх) Один главный вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения xvi)-xix), в которых R представляет собой Н.
xxi) Другой главный вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения xvi)-xix), в которых R представляет собой ОН.
xxii) Еще один другой вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения i)-xxi), в которых D представляет собой СН3.
xxiii) Еще один другой вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения i)-xxi), в которых D представляет собой связь.
xxiv) Один главный вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения i)-xxiii), в которых G представляет собой группу формулы G, или в варианте, относящемся к варианту осуществления изобретения ii), группу G1.
xxv) Предпочтительно, соединения формулы (I), представленные выше в варианте осуществления изобретения xxiv), должны быть такими, в которых каждый из Z, Z hz, если присутствует, представляет собой СН (то есть, G представляет собой 2,3-дигидробензо[1,4]диоксин-6-ил).
xxvi) Другой главный вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения i)-xxiii), в которых G представляет собой группу формулы G2, или в варианте, относящемся к варианту осуществления изобретения ii), группу G2.
xxvii) Предпочтительно, соединения формулы (I), представленные выше в варианте осуществления изобретения xxvi), должны быть такими, в которых X, если присутствует, представляет собой СН (т.е., G представляет собой 3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил или 3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-6-ил).
xxviii) Еще один вариант осуществления настоящего изобретения относится к соединениям формулы (I), представленным выше в одном из вариантов осуществления изобретения i)-xxiii), или в которых G представляет собой фенильную группу, которая однократно или дважды замещена в м- и/или п-положении(ях) заместителями, выбранными независимо друг от друга из С1-4алкила, С1-3алкоксигруппы и галогена (предпочтительно, фтора), при этом С1-3алкоксизаместитель, если присутствует, предпочтительно представляет собой линейную С1-3алкоксигруппу, находящуюся в п-положении, или в вариантах, относящихся к варианту осуществления изобретения ii), где G представляет собой фенильную группу, которая однократно замещена в м-положении и однократно замещена в п-положении заместителями, выбранными независимо друг от друга из С1-4алкила и галогена (предпочтительно, фтора).
xxix) Предпочтительно, соединения формулы (I), представленные выше в варианте осуществления изобретения xxviii), должны быть такими, в которых G представляет собой фенильную группу, которая однократно или дважды замещена в м- и/или п-положении(ях) заместителями, выбранными независимо друг от друга из метила, этила, метоксигруппы, этоксигруппы и галогена (предпочтительно, фтора), при этом метокси- или этоксизаместитель, если присутствует, находится в п-положении, или в варианте осуществления изобретения, относящемся к варианту осуществления изобретения ii), такими, в которых G представляет собой фенильную группу, которая однократно замещена в м-положении и однократно замещена в п-положении заместителями, выбранными независимо друг от друга из метила и фтора.
ххх) В частности, соединения формулы (I), представленные выше в варианте осуществления изобретения xxviii), должны быть такими, в которых G представляет собой 3-фтор-4-метилфенил.
xxxi) Согласно одному особому варианту осуществления настоящего изобретения, соединения формулы (I), приведенные выше в одном из вариантов осуществления изобретения i)-xxx), должны иметь стереохимию, представленную ниже.
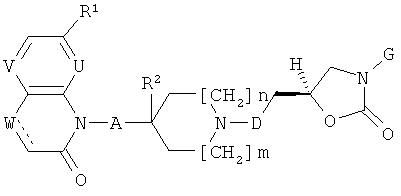
xxxii) Согласно другому особому варианту осуществления настоящего изобретения, соединения формулы (I), приведенные выше в одном из вариантов осуществления изобретения i)-xxx), должны иметь стереохимию, представленную ниже.
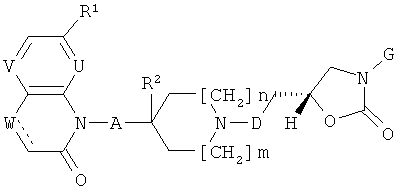
xxxiii) Особенно предпочтительными являются следующие соединения формулы (I), представленные выше в варианте осуществления изобретения i) или ii):
6-{(R)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1H-хинолин-2-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[4-(7-метокси-2-оксо-2H-хинолин-1-илметил)пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-илметил}-7-метокси-1H-хинолин-2-он;
1-{1-[(R)-3-(3-фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]пиперидин-4-илметил}-7-метокси-1H-хинолин-2-он;
6-{(R)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[3-(7-мeтoкcи-2-oкco-3,4-дигидpo-2H-xинoлин-l-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-3,4-Дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1Я-хиноксалин-2-он;
6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4H-пиридо[2,3-6]пиразин-3-он;
6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4H-пиридо[2,3-6]пиразин-3-он;
6-{(R)-5-[(R)-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-((S)-5-{2-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((S)-5-{2-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
4-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-6]пиразин-3-он;
6-метокси-4-(1-{2-[(5)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-6]пиразин-3-он;
4-{1-[(R)-3-(3-фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-6]пиразин-3-он;
6-метокси-4-(1-{2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-6]пиразин-3-он;
а также их соли (в частности, фармацевтически приемлемые соли).
xxxiv) Далее объект настоящего изобретения относится к следующим соединениям формулы (I), представленным выше в вариантах осуществления изобретения i) или ii):
6-{(R)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1Я-хинолин-2-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[4-(7-метокси-2-оксо-2H-хинолин-1-илметил)пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-илметил}-7-метокси-1Я-хинолин-2-он;
1-{1-[(R)-3-(3-фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]пиперидин-4-илметил}-7-метокси-1Я-хинолин-2-он;
6-{(R)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1 -илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1Я-хиноксалин-2-он;
6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4H-пиридо[2,3-b]пиразин-3-он;
6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4H-пиридо[2,3-b]пиразин-3-он;
6-{(R)-5-[(S)-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[(R)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-((S)-5-{2-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[(S)-3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
4-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4Д-пиридо[2,3-6]пиразин-3-он;
6-метокси-4-(1-{2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-6]пиразин-3-он;
4-{1-[(R)-3-(3-фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-6]пиразин-3-он;
6-метокси-4-(1-{2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-6]пиразин-3-он;
а также к их солям (в частности, фармацевтически приемлемым солям).
xxxv) Изобретение, в частности, относится к группам соединений формулы (I), выбранных из соединений, указанных в варианте осуществления изобретения xxxiii), эти группы соединений, кроме того, соответствуют одному из вариантов осуществления изобретения iii)-xxxii), а также к солям (в частности, фармацевтически приемлемым солям) таких соединений.
xxxvi) Изобретение также относится к группам соединений формулы (I), выбранных из соединений, указанных в варианте осуществления изобретения xxxiv), эти группы соединений, кроме того, соответствуют одному из вариантов осуществления изобретения ih)-xxxii), а также к солям (в частности, фармацевтически приемлемым солям) таких соединений.
Соединения формулы (I) согласно изобретению, то есть согласно одному из вариантов i)-xxxvi), являются подходящими для применения в качестве химиотерапевтических активных соединений в медицине и ветеринарии, а также в качестве веществ для сохранения неорганических и органических материалов, в частности, всех типов органических материалов, например, полимеров, смазок, лакокрасочных материалов, волокон, кожи, бумаги и шерсти.
Соединения согласно настоящему изобретению особенно активны против бактерий и бактериально-подобных организмов. Они поэтому являются особенно подходящими для человека, а также животных при профилактике и химиотерапии локальных и системных инфекций, вызываемых этими патогенными организмами, а также заболеваний, связанных с бактериальными инфекциями, включая пневмонию, воспаление среднего уха, синусит, бронхит, тонзиллит и мастоидит, связанные с инфекцией, вызываемой Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus, Enterococcusfaecalis, E.faecium, E. casseliflavus, S. epidermidis, S. haemolyticis или Peptostreptococcus spp.; фарингит, ревматическую лихорадку и гломерулонефрит, связанные с инфекцией, вызываемой Streptococcus pyogenes, группами С и G streptococci, Corynebacterium diphtheriae или Actinobacillus haemolyticum; инфекции верхних дыхательных путей, связанные с инфекцией, вызываемой Mycoplasma pneumoniae, Legionella pneumophila, Streptococcus pneumoniae, Haemophilus influenzae или Chlamydia pneumoniae; инфекции крови и тканей, включая эндокардит и остеомиелит, вызываемые S. aureus, S. haemolyticus, E. faecalis, E. faecium, E. durans, включая наследственную резистентность к известным антибактериальным средствам таким, как, не лимитируя, р-лактамы, ванкомицин, аминогликозиды, хинолоны, хлорамфеникол, тетрациклины и макролиды; неосложненные инфекции кожи и мягких тканей, абсцессы и послеродовой сепсис, связанные с инфекцией, вызываемой Staphylococcus aureus, некоагулируемыми staphylococci (то-есть, S. epidermidis, S. haemolyticus и тому подобные), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcal групп C-F (мельчайшая колония streptococci), viridans streptococci, Corynebacterium MUHUtissimum, Clostridium spp.или Bartonella henselae; неосложненные острые инфекции мочевого тракта, связанные с инфекцией, вызываемой Staphylococcus aureus, некоагулируемыми видами стафилоккоков или Enterococcus spp.; уретрит и цервицит; болезни, передаваемые половым путем, связанные с инфекцией, вызываемой Chlamydia trachomatis, Haemophilus ducreyi, Treponema pallidum, Ureaplasma urealyticum или Neiserria gonorrhoeae; токсикозы, связанные с инфекцией, вызываемой S. aureus (пищевое отравление и токсический шок), или группами А, В и С streptococci; язвы, связанные с инфекцией, вызываемой Helicobacter pylori; системный лихорадочный синдром, связанный с инфекцией, вызываемой Borrelia recurrentis; болезнь Лайма, связанная с инфекцией, вызываемой Borrelia burgdorferi; конъюктивит, кератит и дакриоцистит, связанные с инфекцией, вызываемой Chlamydia trachomatis, Neisseria gonorrhoeae, S. aureus, S. pneumoniae, S. pyogenes, H. Influenzae или Listeria spp.; диссеминированный Mycobacterium avium комплекс (MAC), связанный с инфекцией, вызываемой Mycobacterium avium или Mycobacterium intracellulare; инфекции, вызываемые Mycobacterium tuberculosis, M. leprae, M. paratuberculosis, M. Kansasii или М. chelonei; гастроэнтерит, связанный с инфекцией, вызываемой Campylobacter jejuni; кишечные протозойные инфекции, связанные с инфекцией, вызываемой Cryptosporidium spp.; стоматологическую инфекцию, связанную с инфекцией, вызываемой viridans streptococci; стойкий кашель, связанный с инфекцией, вызываемой Bordetella pertussis; газовую гангрену, связанную с инфекцией, вызываемой Clostridium perfringens или Bacteroides spp.; и атеросклероз или сердечно-сосудистую болезнь, связаннуюе с инфекцией, вызываемой Helicobacter pylori или Chlamydia pneumoniae.
Соединения формулы (I) согласно настоящему изобретению применяются, кроме того, для изготовления лекарственного средства для лечения инфекций, вызываемых такими бактериями, как Е. coli, Klebsiella pneumoniae и другими энтеробактериями, Acinetobacter spp., Stenothrophomonas maltophilia, Neisseria meningitidis. Bacillus cereus. Bacillus anthracis, Corynebacterium spp., Propionibacterium acnes и бактероидами spp.
Соединения формулы (I) согласно одному из вариантов i)-xxxvi) применяются, кроме того, при лечениее протозоальных инфекций, вызываемых Plasmodium malaria, Plasmodiumfalciparum, Toxoplasma gondii, Pneumocystis carimi, Trypanosoma brucei и Leishmania spp.
Представленный перечень патогенных микроорганизмов должен рассматриваться только в качестве примеров и никоим образом в качестве лимитирующего перечня.
Соединения формулы (I) согласно одному из вариантов i)-xxxvi) или их фармацевтически приемлемые соли могут быть использованы для приготовления лекарственного средства, и вследствие этого являются подходящими для профилактики или лечения бактериальной инфекции.
Один аспект настоящего изобретения относится к применению соединения формулы (I) согласно одному из вариантов i)-xxxvi) или его фармацевтически приемлемой соли, для изготовления лекарственного средства для профилактики или лечения (особенно для лечения) бактериальной инфекции. Другой аспект настоящего изобретения относится к соединению формулы (I) согласно одному из вариантов i)-xxxvi), или его фармацевтически приемлемой соли, для профилактики или лечения (особенно для лечения) бактериальной инфекции.
Таким образом, соединения формулы (I) согласно одному из вариантов i)-xcv) или их фармацевтически приемлемые соли могут быть использованы для изготовления лекарственного средства и являются подходящими для профилактики или лечения бактериальной инфекции, выбранной из группы, включающей инфекции дыхательных путей, отит среднего уха, менингит, инфекции кожи и глубоких тканей (осложненные или неосложненные), пневмонию (включая приобретенную госпитальную пневмонию), бактериемию, эндокардит, интраабдоминальные инфекции, желудочно-кишечные инфекции, Clostridium difficile инфекции, инфекции мочевого тракта, инфекции, передаваемые половым путем, инфекции, вызываемые инородными телами, остеомиелит, болезнь Лайма, топические инфекции, офтальмологические инфекции, туберкулез и тропические заболевания (например, малярию), и особенно, для профилактики или лечения бактериальной инфекции, выбранной из группы, включающей инфекции дыхательных путей, отит среднего уха, менингит, инфекции кожи и глубоких тканей (осложненные или неосложненные), пневмонию (включая приобретенную госпитальную пневмонию), бактериемию.
Так же, как и в медицине, бактериальные инфекции подлежат лечению с применением соединений формулы (I) (или их фармацевтически приемлемых солей) в ветеринарии для лечения таких видов животных как, например, свиньи, жвачные животные, лошади, собаки, кошки и домашняя птица.
Настоящее изобретение относится также к фармакологически приемлемым солям и к композициям и рецептурам на основе соединений формулы (I).
Любая ссылка на соединение формулы (I) в этом описании (и особенно в вариантах осуществления изобретения, представленных выше) подразумевает ссылку на соли (в частности, фармацевтически приемлемые соли) таких соединений, как уместную и целесообразную.
Фармацевтическая композиция согласно настоящему изобретению содержит, по крайней мере, одно соединение формулы (I) (или его фармацевтически приемлемую соль) в качестве активного агента и необязательно носители и/или разбавители и/или добавки, и может также содержать дополнительно известные антибиотики.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в качестве лекарственных средств, например, в форме фармацевтических композиций для энтерального и парентерального введения.
Изготовление фармацевтических композиций осуществляется методом, известным любому специалисту в области техники (см., например, ReMHHgton, The Science and Practice of Pharmacy, 21st Edition (2005), Part 5, "Pharmaceutical Manufacturing" [published by Lippincott Williams & Wilkins]) путем введения описанных соединений формулы (I) или их фармацевтически приемлемых солей, необязательно в комбинации с другими фармацевтически приемлемыми веществами, в лекарственную форму вместе с подходящими, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими носителями и, если необходимо, обычными фармацевтическими наполнителями.
Другой аспект изобретения относится к способу профилактики и лечения бактериальной инфекции у пациента, заключающемуся во введении указанному пациенту фармацевтически активного количества соединения формулы (I) согласно одному из вариантов i)-xxxvi) или его фармацевтически приемлемой соли.
Кроме того, любые ссылки и (под-)варианты осуществления изобретения, указанные для соединений формулы (I) (будь то сами соединения, их соли, композиции, содержащие соединения или их соли, применения соединений или их солей и т.п.) применимы также к соединениям формулы (ICE)-
Кроме того, соединения формулы (I) могут быть также использованы для дезинфекции, то есть для удаления патогенных микробов и бактерий с поверхности инструментов или асептической очистки воздушного пространства и самих помещений. Для этих целей соединения формулы (I) могут применяться в виде раствора или спрея.
Соединения формулы (I) могут быть получены в соответствии с настящим изобретением с использованием методов, описанных далее.
Получение соединений формулы (I)
Аббревиатуры:
В описании и примерах используются следующие аббревиатуры:
Ас - ацетил, АсОН - уксусная кислота, AD-смесь α - 1,4-бис(дигидрохинолин)фталазин, К3Fе(СN)6, К2СО3 и К2OsO4 2Н2O, AD-смесь β -1,4-бис(дигидрохинолин)фталазин, К3Fе(СN)6, К2СО3 и K2OsO4 2Н2О, Бок -трет-бутоксикарбонил, Bs - 4-бромфенилсульфонил, Кбз -бензилоксикарбонил, КК - колоночная хроматография на силикагеле, КДИ -1,1 -карбонилдиимидазол, ДХМ - дихлорметан, ДИПЭА - N,N-диизопропилэтиламин, ДМАП - 4-диметиламинопиридин, ДМФ - N,N-диметилформамид, ДМСО - диметилсульфоксид, ЭА - этилацетат, ЭСИ -ионизация электрораспылением, экв. - эквивалент, эфир - диэтиловый эфир, Et -этил, EtOH - этанол, Фмок - 9-флюоренилметоксикарбонил, гекс -гексан, гепт -гептан, ч - час(ы), КНМДС - гексаметилдисилазид калия, ЖХ - жидкостная хроматография, м-ХПБК - м-хлорпербензойная кислота, Me - метил, MeCN -ацетонитрил, МеОН - метанол, мин - минута, Ms - метансульфонил (мезил), MS - масс-спектроскопия, n-BuLi - н-бутиллитий, Nf- нонафторбутансульфонил, NMO - N-оксид N-метилморфолина, Ns - 3-нитрофенилсульфонил, Pd/C -палладий на угле, Pd(OH)2/C - дигидроксид палладия на угле, Ph - фенил, Руr - пиридин, рац - рацемический, КТ - комнатная температура, ТБАФ -тетрабутиламмонийфторид, ТБДМС - трет-бутилдиметилсилил, ТБДФС - трет-бутилдидифенилсилил, ТБМЭ - трет-бутилметиловый эфир, ТЭА -триэтиламин, Tf - трифторметансульфонил (трифлил), ТФК - трифторуксусная кислота, ТТФ - тетрагидрофуран, ТСХ - тонкослойная хроматография, Ts - п-толуолсульфонил.
Общие синтетические методы:
Общий синтетический метод 1 (алкилирование амина):
Соответствующие аминопроизводные вводят в реакцию с соответствующими производными, содержащими группу Y1, Y2 или Y3, где Y1, Y2 и Y3 каждая независимо друг от друга представляет собой OMs, OTf, OTs, Cl, Вr или I, в присутствии неорганического основания такого, как К2СО3, СsСО3, или органического основания такого, как ТЭА, в растворителе таком, как ТГФ, ДМФ или ДМСО, при температуре от 0°С до +80°С. Детали могут быть найдены в публикации: Comprehensive Organic Transformations. A guide to Functionnal Groups Preparations; 2nd Edition, R. C. Larock, Wiley-VC; New York, Chichester, Weinheim, Brisbane, Singapore, Toronto, 1999. Section Amines, p.779.
Общий синтетический метод 2 (удаление аминозащитных групп):
Бензилкарбаматные защитные группы удаляют посредством гидрирования над катализатором из благородного металла (например, Pd/C или Pd(OH)2/C). Бок-группу удаляют в кислых условиях таких, как НСl в органическом растворителе, таком, как МеОН или диоксан, или неразбавленная или разбавленная ТФК в растворителе, таком, как ДХМ. Кроме того, общие методы удаления аминозащитных групп описаны в публикации: T.W. Greene, P.G.M. Wuts, Protecting Groups in Organic Synthesis, 3rd Ed (1999), 494-653 (Publisher: John Wiley and Sons, Inc., New York, N.Y.).
Общий синтетический метод 3 (удаление гидроксильных защитных групп):
Силильные сложноэфирные группы удаляют, используя либо источники фтор-аниона такие, как ТБАФ в ТГФ, в температурном интервале от 0°С до +40°С, или HF в MeCN в температурном интервале от 0°С до +40°С, либо используя кислые условия такие, как АсОН в смеси ТГФ/МеОН или НСl в МеОН. Другие методы, применяемые для удаления ТБДМС и ТБДФС групп, представлены в T.W. Greene, P.G.M. Wuts, Protecting Groups in Organic Synthesis, 3rd Ed (1999), 133-139 и 142-143, соответственно (Publisher: John Wiley and Sons, Inc., New York, N.Y.). Кроме того, общие методы удаления защитных спиртовых групп описаны в T.W. Greene, P.G.M. Wuts, Protecting Groups in Organic Synthesis, 3rd Ed (1999), 23-147 (Publisher: John Wiley and Sons, Inc., New York, N.Y.).
В особом случае алкилкарбоксильной защитной группы свободный спирт может быть получен под действием неорганического основания такого, как К2СО3, в растворителе таком, как МеОН.
Общий синтетический метод 4 (активация спирта):
Спирт вводят в реакцию с BsCI, MsCI, NfCl, NsCl, TfCl или TsCl в присутствии органического основания такого, как ТЭА, ДИПЭА или пиридин, в сухом апротонном растворителе таком, как ДХМ, ТГФ или пиридин, в температурном интервале от -10°С до 25°С. Альтернативно, спирт может быть введен в реакцию с Ms2O или Tf2O. Активированное промежуточное соединение может быть далее превращено в его соответствующее йод- или бромпроизводное посредством реакции активированного спирта с Nal или NaBr в растворителе таком, как ацетон.
Общий синтетический метод 5 (окисление спиртов):
Спирты могут быть превращены в соответствующие альдегиды или кетоны посредством окисления по Шверну (см.: D. Swern et al., J. Org. Chem. (1978), 43, 2480-2482) или Дессу-Мартину (см.: D.B. Dess и J.C. Martin, J. Org. Chem. (1983), 48, 4155), соответственно.
Общий синтетический метод 6 (цис-дигидроксилирование):
Диол получают дигидроксилированием соответствующего этиленового производного, используя каталитические количества тетроксида осмия в присутствии со-оксиданта такого, как N-MO, в водном растворителе таком, как смесь ацетона с водой или ДХМ с водой (см.: Cha, J. К. Chem. Rev. (1995), 95, 1761-1795). Хиральные цис-диолы получают с использованием AD-смеси а или AD-смеси Р в присутствии метансульфонамида в смеси воды с 2-метил-2-пропанолом, как описано в Chem. Rev. (1994), 94, 2483. Ориентация введения зависит от хирального лиганда, содержащегося в AD-смеси: или лиганда, основанного на дигидрохинине, в AD-смеси а, или лиганда, основанного на дигидрохинидине, в AD-смеси р.
Общий синтетический метод 7 (защита спиртов):
Спирты защищают, превращая их в сложные силильные эфиры (обычно ТБДМС или ТБДФС сложные эфиры). Спирт вводят в реакцию с требуемым силилхлоридом (ТБДМСС1 или ТБДФСС1) в присутствии основания такого, как имидазол или ТЭА, в растворителе таком, как ДХМ или ДМФ, в температурном интервале от +10°С до +40°С. Методы, включающие другие спиртовые защитные группы, описаны в публикации: T.W. Greene, P.G.M. Wuts, Protecting Groups in Organic Synthesis, 3rd Ed (1999), 23-147 (Publisher: John Wiley and Sons, Inc., New York, N.Y.).
Общий синтетический метод 8 (гидрирование двойной связи):
Ненасыщенные производные, растворенные в растворителе таком, как МеОН, ЭА или ТГФ, гидрируют над катализатором из благородного металла таким, как Pd/C или Pd(OH)2/C, или над Ni Ренея. В конце реакции катализатор отфильтровывают, а фильтрат выпаривают при пониженном давлении. Альтернативно, восстановление осуществляют путем каталитической обменной гидрогенизации с использованием Pd/C и формиата аммония в качестве источника водорода.
Общие препаративные методы:
Получение соединений формулы (I):
Соединения формулы (I) могут быть получены с помощью методов, приведенных ниже, методов, приведенных в примерах, или аналогичными методами. Оптимальные реакционные условия могут варьироваться в зависимости от особенностей используемых реагентов или растворителей, но такие условия могут быть изменены специалистом в области техники путем обычно принятой оптимизации методик.
В разделах a)-f), представленных далее, описываются общие методы получения соединений формулы (I). В этих разделах, если не указано иначе, общие группы или целые числа m, n, R1, R2, А, В, D, U, V, W, G и Q и необязательно связь имеют значения согласно формуле (I). Другие используемые аббревиатуры приведены в экспериментальной части. В некоторых примерах общие группы U, V, W, R2 и G могут отличаться от изображенных в примерах и в представленных ниже схемах, так как требуют использования защитных групп.Применение защитных групп хорошо известно из области техники (см., например, "Protective Groups in Organic Synthesis", T.W. Greene, P.G.M. Wuts, Wiley-Interscience, 1999).
а) Соединения формулы (I) могут быть получены посредством реакции соединения формулы (II)
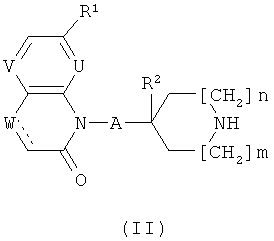
с соединением формулы (III)
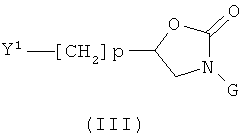 ,
,
где Y1 обозначает галоген такой, как бром или йод, или группу OSO2R3, где Ra обозначает алкил, СF3 или толил и р равно 1 или 2, с использованием общего реакционного метода 1.
b) Соединения формулы (I) могут быть получены посредством реакции соединения формулы (IV) с соединением формулы (V)
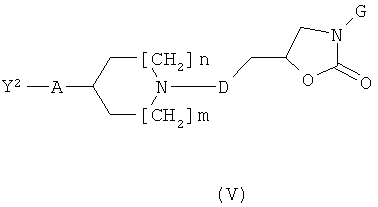
где Y2 обозначает галоген такой, как бром или йод, или группу OSO2Ra, где Ra обозначает алкил, СF3 или толил, с использованием общего реакционного метода 1.
с) Соединения формулы (I), где А обозначает СН2 и R2 обозначает ОН, могут быть получены посредством реакции соединения формулы (IV), представленного выше в разделе b), с соединением формулы (VI)
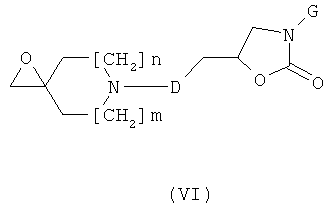
в присутствии Сs2СО3 или NaH в полярном растворителе таком, как ДМФ, в температурном интервале от 60°С до 140°С.
d) Соединения формулы (I), где W обозначает N и "----" обозначает связь, могут быть получены посредством реакции соединения формулы (VII)
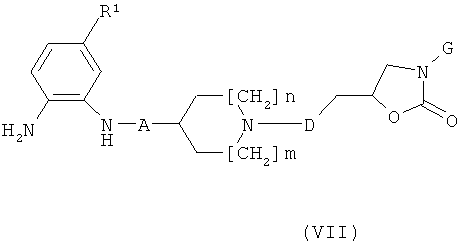
(VII) с соединением формулы CHOCOORb, где Rb обозначает алкил, или с алкилбромацетатом в присутствии основания такого, как К2СО3, с последующей циклизацией в кислой среде такой, как АсОН в горячем толуоле, и ароматизацией путем обработки MnO2 или Н2О2.
е) Соединения формулы (I), кроме того, могут быть получены посредством реакции соединения формулы (VIII)
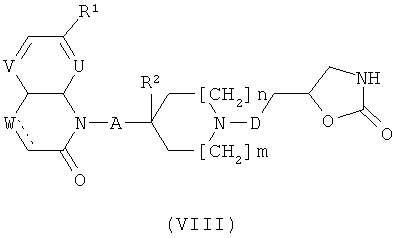
с соединением формулы (IX)
 ,
,
где Xa представляет собой OTf или галоген такой, как хлор, бром или йод. Эту реакцию проводят в условиях, описанных для катализируемого металлами N-арилирования 2-оксазолидинонов или амидов, в частности, с использованием Cul и 1,1,1-трис(гидроксиметил)этана в присутствии Сз2СО3 (Org. Lett. (2006), 8, 5609-5612), или Pd(OAc)2 и DPEphos в присутствии К3РO4, с последующим, при необходимости, удалением защитной группы PG0 согласно общему реакционному методу 2.
f) Соединения формулы (I), где W обозначает СН2 или NH и отсутствует, могут быть получены посредством реакции гидрирования соответствующих соединений формулы (I), где обозначает связь, согласно общему реакционному методу 8 или путем восстановления соответствующих соединений формулы (I) с помощью NaBH4 в растворителе таком, как EtOH.
Соединения формулы (I), полученные таким образом, могут быть, при небходимости, превращены в их соли, предпочтительно, в их фармацевтически приемлемые соли.
Кроме того, всякий раз, когда соединения формулы (I) получают в виде смесей энантиомеров, энантиомеры могут быть разделены с помощью методов, известных любому специалисту в области техники, например, путем получения и разделения диастереомерных солей или с помощью ВЭЖХ на хиральной стационарной фазе такой, как Regis Whelk-01(R,R) (10 мкм) колонка, Daicel ChiralCel OD-H (5-10 мкм) колонка, или Daicel ChiralPak IA (10 мкм) или AD-H (5 мкм) колонка. Типичные условия проведения хиральной ВЭЖХ включают изократную смесь элюента A (EtOH, в присутствии или отсутствии амина такого, как триэтиламин, диэтиламин) и элюента В (гексан), при скорости истечения от 0,8 до 150 мл/мин. Соединения формулы (I), полученные в виде смесей диастереомеров, могут быть разделены путем комбинации хроматографии на силикагеле, ВЭЖХ и методов кристаллизации.
Получение соединений формул (II)-(IX):
Соединения формулы (II) могут быть получены, как описано на приведенной ниже схеме 1.
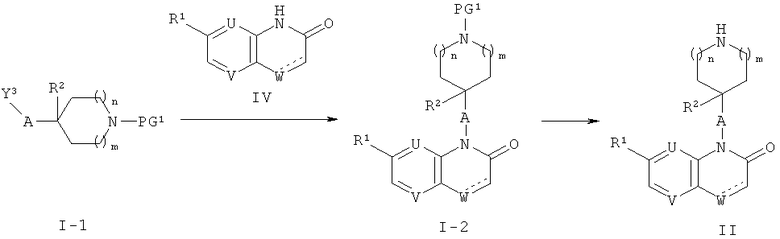
На схеме 1, Y3 обозначает галоген такой, как бром или йод, или группу OSO2Ra где Ra обозначает алкил, СF3 или толил, и PG1 обозначает аминозащитную группу такую, как Кбз, Бок или Фмок.
Соединения формулы (I-1), где А обозначает СН; или связь и R2 обозначает Н, или, при условии, что А обозначает СН2, R2 может также обозначать ОН, вводят в реакцию с соединениями формулы (IV) (общий реакционный метод 1). Аминозащитная группа в промежуточном соединении (1-2) может быть затем удалена (общий реакционный метод 2) с получением соединений формулы (II). Соединения формул (1-2) и (II), где отсутствует, могут быть получены посредством гидрирования соединений формул (1-2) и (II), где обозначает связь, или путем восстановления их с использованием NaBH4 в растворителе таком, как ЕtOН.
Соединения формулы (III) могут быть получены, как описано на приведенной ниже схеме 2.
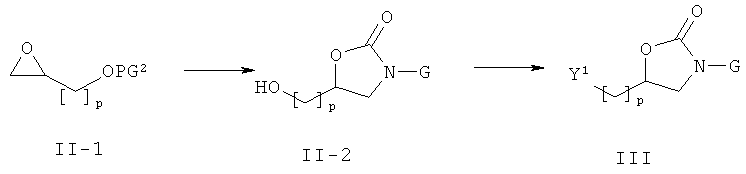
Схема 2
На схеме 2, PG2 представляет собой спиртовую защитную группу такую, как C(O)Rb, где Rb обозначает алкил, или ТБДМС или ТБДФС.
Соединения формулы (III) могут быть получены из соответствующих спиртов формулы (11-2) после активации спиртовой функции (общий реакционный метод 4). Спирты формулы (11-2) могут быть получены с помощью реакции эпоксидов формулы (11-1) с анионами карбаматов формулы GNHCOOR, где R представляет собой алкил или бензил, в присутствии основания такого, как К.НДМС или трет-бутилат лития, с последующим удалением защитной спиртовой группы (общий реакционный метод 3). Альтернативно, эпоксиды формулы (11-1) вводят в реакцию с аминами формулы GNHs в присутствии LiClO4, образовавшиеся аминоспиртовые производные вводят в реакцию с КДИ, удаляют защитную спиртовую группу, используя общий реакционный метод 3 и получая при этом промежуточные соединения формулы (II-2).
Соединения формулы (IV), где обозначает связь, или являются коммерческими продуктами, (U=V=W=СН; R1=МеО или F) или могут быть получены согласно известным методам (например, U=N,V=N, W=СH; U=СН, V=N, W=СН; U=N, V=СН, W=N; U=N, V=СН, W=СН; U=СН, V=СН, W=N и R1=ОМе; см. WO 2008/009700, WO 2006/134378 и J. Heterocycl. Chem. (1986), 23(2), 501-504). Соединения формулы (IV), где U=V=W=СН и отсутствует, могут быть получены согласно WO 2006/134378 или аналогично описанию в WO 2006/090272.
Соединения формулы (V) могут быть получены, как описано на приведенной ниже схеме 3.
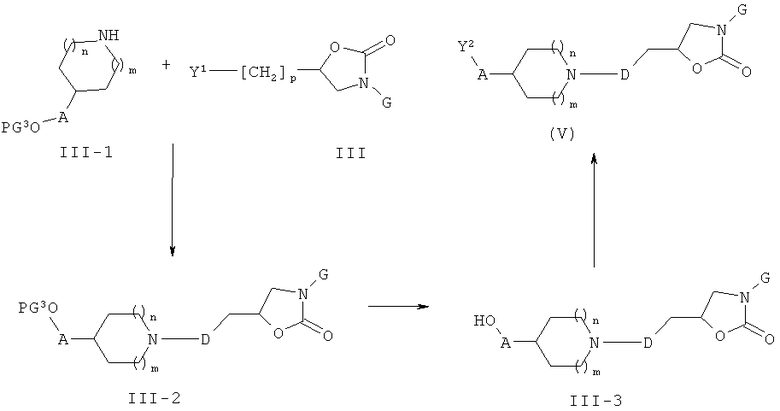
Схема 3
На схеме 3, PG представляет собой защитную спиртовую группу такую, как ТБДМС или ТБДФС.
Соединения формулы (III-1) могут вступать в реакцию с соединениями формулы (III) (общий реакционный метод 1). Защитная спиртовая группа в промежуточных соединениях формулы (III-2) может быть удалена (общий реакционный метод 3). Полученные спиртовые производные формулы (III-3) могут быть превращены в их соответствующие активированные промежуточные соединения формулы (V) (общий реакционный метод 4).
Соединения формулы (VI) могут быть получены, как описано на приведенной ниже схеме 4.
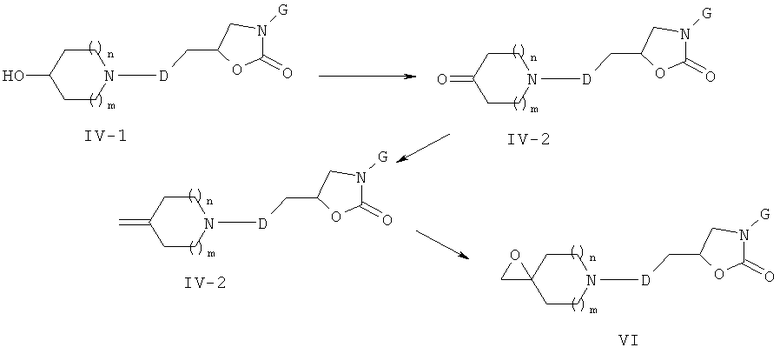
Схема 4
Спирты формулы (IV-1) (то есть соединения формулы (III-3) схемы 3, где А обозначает связь) могут быть окислены в соответствующих кетонные аналоги формулы (IV-2) (общий реакционный метод 5). Эти кетоны могут быть затем превращены в соответствующие эпоксидные производные формулы (VI) либо непосредственным эпоксидированием с помощью йодида триметилсульфония, либо путем последовательно проведенных реакции Виттига с метилентрифенилфосфораном с последующим эпоксидированием промежуточных соединений формулы (IV-3) с м-ХПБК.
Соединения формулы (VII) могут быть получены, как описано на приведенной ниже схеме 5.
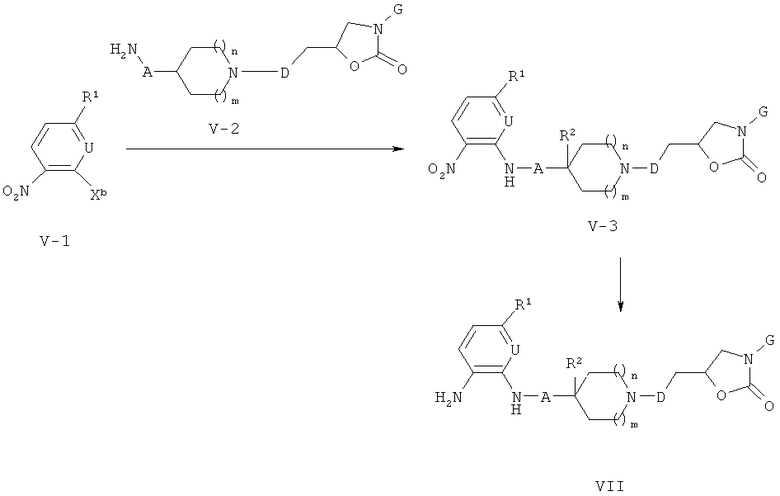
Схема 5
На схеме 5, X представляет собой галоген такой, как бром или хлор. Нитропроизводные формулы (V-1) могут вступать в реакцию с промежуточными соединениями формулы (V-2). Полученные промежуточные соединения формулы (V-3) могут быть затем восстановлены в соответствующие аминопроизводные формулы (VII) путем гидрирования над катализатором из благородного металла таким, как Pd/C, или никелем Ренея. Другие методы восстановления нитросоединений приведены в Comprehensive Organic Transformations. A guide to Functional Group Preparations; 2nd Edition, R. C. Larock, Wiley-VC; New York, Chichester, Weinheim, Brisbane, Singapore, Toronto, (1999). Section Amines; p.821.
Соединения формулы (VIII) могут быть получены, как описано на приведенной ниже схеме 6.
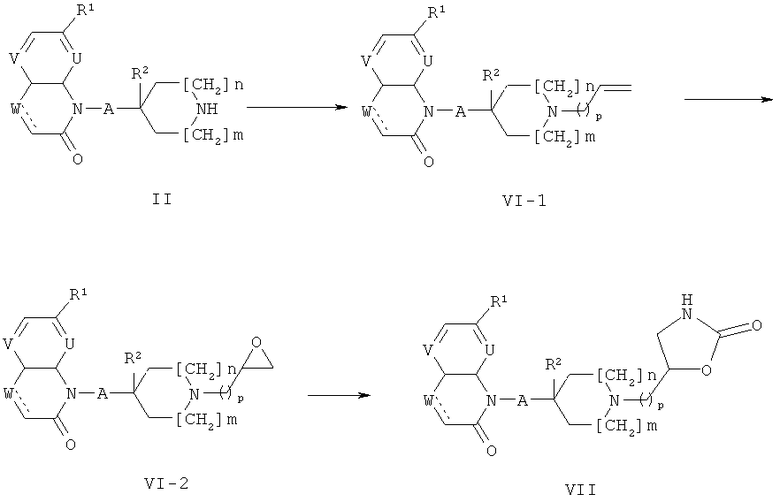
Схема 6
На схеме 6, р представляет собой 1 или 2.
Соответственно, промежуточные соединения формулы (II) могут вступать в реакцию с аллил- или гомоаллилбромидом согласно общему реакционному методу 1. Промежуточные соединения формулы (VI-1) могут быть последовательно дигидроксилированы согласно общему реакционному методу 6, активированы превращением в мономезилаты согласно общему реакционному методу 4 и зациклизованы в присутствии основания такого, как К2СО3, в растворителе таком, как МеОН или ТЭА. Полученные эпоксиды формулы (VI-2) могут быть введены в реакцию с азидом натрия с последующим гидрированием над катализатором из благородного металла таким, как Pd/C, или реакцией с РРЬз в присутствии воды. Полученные промежуточные амины реакцией с бензил- или алкилхлорформиатом могут быть превращены в соответствующие карбаматы, после обработки которых с помощью NaH могут быть получены оксазолидиноны формулы (VIII).
Некоторые соединения формулы (IX) коммерчески доступны (например, соединения, где G=G2, Q=О и Z-N: CAS 337463-99-7; G=G2,Q=S и Z=СН: CAS 6376-70-1; G=G2, Q=О и Z=CH: CAS 7652-29-1).
Соединение формулы (IX), где G обозначает G, Z обозначает N, Q обозначает S и Xa обозначает Сl, могут быть получены, как представлено ниже на схеме 7.
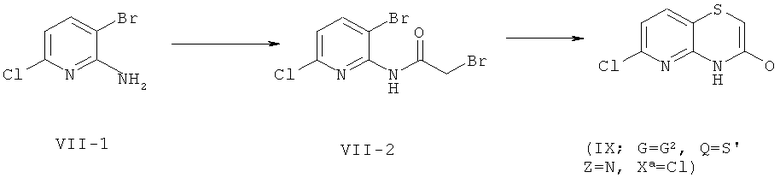
Схема 7
Соответственно, бромпроизводное формулы (VII-1), полученное согласно WO 2008/065198, может быть введено в реакцию с бромацетилбромидом, после чего полученное производное формулы (VII-2) вводят в реакцию с тиоацетатом натрия в присутствии NaOMe, получая при этом соединение формулы (IX), где G обозначает G2, Z обозначает N, Q обозначает S и Хa обозначает Сl.
Соединения формулы (IX), где G обозначает G, Х обозначает СН, Q обозначает О или S и Y4 обозначает OTf, и соединения, где G обозначает G1, каждый из Z1, Z2 и Z3 обозначает СН и Y4 обозначает OTf, могут быть получены из соответствующих спиртовых исходных соединений (Хa=ОН) и Tf2O согласно общему реакционному методу 4. Последние из соединений являются коммерческими продуктами (CAS 53412-38-7; CAS 10288-72-9) или могут быть получены, как описано в ЕР 106 816.
Получение некоторых промежуточных соединений:
Соединения формулы (1-1) могут быть получены, как представлено ниже на схеме 8.
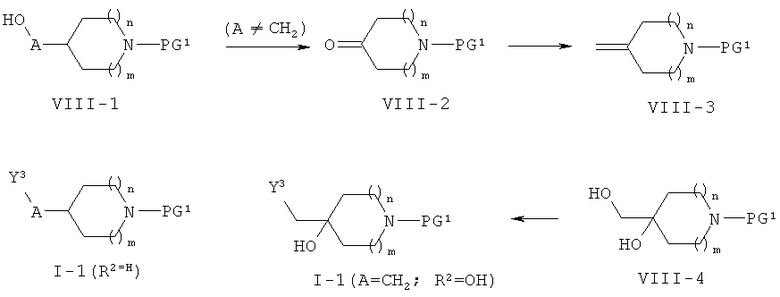
Схема 8
Соединения формулы (1-1) могут быть получены из соответствующих спиртов формулы (VIII-1) или (VIII-4) с использованием общего реакционного метода 4 (см. схему 8). Исходные спирты формулы (VIII-1) коммерчески доступны. Исходные спирты формулы (VIII-4) могут быть получены из спиртов формулы (VIII-1) в результате окисления (общий реакционный метод 5), реакции Виттига с метилентрифенилфосфораном с последующим цис-дигидроксилированием (общий реакционный метод 6).
Соединения формулы (II-1), где р равно 1 и PG2 обозначает C(O)Rb, Rb при этом обозначает алкил, коммерчески доступны. Соединение формулы (11-1), где р равно 2 и PG2 обозначает ТБДМС, могут быть получены согласно WO 2007/144423 или ЕР 518672.
Соединения формулы (III-1) могут быть получены путем защиты спиртовой функции соединений формулы (VIII-1) (общий реакционный метод 7) и удаления аминозащитной группы (общий реакционный метод 2).
Соединения формулы (IV-1) соответствуют соединениям формулы (III-3), где А обозначает связь.
Соединения формулы (V-1), где Хb обозначает бром, R1 обозначает метоксигруппу или фтор и U обозначает N или СН, являются коммерчески доступными.
Соединения формулы (V-2) могут быть получены из соединений формулы (V), где Y2 обозначает галоген такой, как йод, или OSO2Ra, где Ra обозначает алкил, СF3 или толил, путем реакции с NaN3 с последующим восстановлением с помощью РРh3 в присутствии воды.
Особые варианты осуществления изобретения описаны в следующих примерах, которые служат для более детальной иллюстрации изобретения, ни в коей мере не ограничивая его объема.
Примеры
Все температуры приведены в °С. Соединения охарактеризованы с помощью 1Н-ЯМР (300 МГц) (Varian Oxford); или 1Н-ЯМР (400 МГц) (Bruker Advance 400); химические сдвиги приведены в м. д. относительно используемого растворителя; мультиплеты: s=синглет, d=дублет, t=триплет, q=квадраплет, р=пентаплет, гекс=гекстет, гепт=гептет, m=мультиплет, br=расширенный, константы взаимодействия приведены в Гц. Альтернативно соединения охарактеризованы с помощью ЖХ-МС (Sciex API 2000 с Agilent 1100 Binary Pump с DAD и ELSD или Agilent quadrupole MS 6140 с Agilent 1200 Binary Pump, DAD и ELSD); с помощью ТСХ (ТСХ-пластины от фирмы Merck, силикагель 60 F254); яли с помощью температуры плавления. Соединения очищают с помощью хроматографии на силикагеле 60А. NH2OH, используемый для колоночной хроматографии, представляет собой 25%-ный водный раствор.
ВЭЖХ проводят на стационарной фазе такой, как колонка быстрого разрешения Zorbax SB С 18 (1,8 мкм), или колонка быстрого разрешения Zorbax Eclipse Plus С 18 (1,8 мкм). Типичные условия проведения ВЭЖХ включают градиент элюента А (вода/ацетонитрил в соотношении 95:5 с 0,1% муравьиной кислоты, с добавлением или без 5 ммолей/л формиата аммония) и элюента Б (ацетонитрил/вода в соотношении 95:5 с 0,1% муравьиной кислоты, с добавлением или без 5 ммолей/л формиата аммония), со скоростью истечения от 0,8 до 5 мл/мин. Рацематы могут быть разделены на энантиомеры, как описано ранее. Предпочтительные условия для хиральной ВЭЖХ следующие: колонка ChiralPak AD (4,6 х 250 мм, 5 мкм), использование изократной смеси (например, в соотношении 10:90) элюента A (EtOH, в присутствии диэтиламина в количестве, например, 0,1%) и элюента Б (гексан), при комнатной температуре, со скоростью истечения, например, 0,8 мл/мин.
Общие методы:
Метод А (удаление Бок-защитной группы):
Бок-защищенный амин (1 ммоль) растворяют в ДХМ (5 мл) и обрабатывают Et3SiH (необязательно; 0,2 мл, 1,1 экв.) и ТФК (2 мл). Смесь перемешивают при комнатной температуре в течение 1 ч, концентрируют в вакууме и переносят в смесь ДХМ/водный раствор NH4OH. Органический слой промывают водой, высушивают над MgSO4 и концентрируют при пониженном давлении.
Метод В: алкилирование аминов йодидами:
Раствор амина (1 ммоль), йодида (1 ммоль) и ДИПЭА (1,1 ммолей) в сухом ДМСО нагревают до температуры 70°С до полного завершения реакции (1-3 дня). После охлаждения добавляют воду и этилацетат и фазы разделяют.Водный слой более двух раз экстрагируют этилацетатом, объединенные органические слои промывают водой (трижды) и рассолом, высушивают над MgSO4 и концентрируют при пониженном давлении, после чего остаток очищают с помощью колоночной хроматографии.
Метод С: алкилирование аминов мезилатами
Раствор амина (1,0-2,3 ммолей), мезилата (1 ммоль) и ДИПЭА (1,1 ммолей) в сухом ДМСО нагревают до температуры 70°С до полного завершения реакции (2-5 дней). После охлаждения добавляют воду и этилацетат и фазы разделяют.Водный слой более двух раз экстрагируют этилацетатом, объединенные органические слои промывают водой (трижды) и рассолом, высушивают над MgSO4 и концентрируют при пониженном давлении, после чего остаток очищают с помощью колоночной хроматографии.
Эксперимент А: 6-((5')-5-Иодметил-2-оксооксазолидин-3-ил)-4H-бензо[1.4]тиазин-3-он
A.i. 6-((8)-3-Хлор-2-гидроксипропиламино)-4Н-бензо[1Л1 тиазин-3-он
Суспензию 6-амино-4H-бензо[1,4]тиазин-3-она (18,0 г, 100 ммолей; коммерческий продукт) и Ca(OTf)2 (0,5 экв.) в MeCN (800 мл) нагревают при температуре 50° в течение 1 ч. Затем добавляют (5')-эпихлоргидрин (18,5 г, 200 ммолей) и реакционную смесь перемешивают при комнатной температуре в течение 72 ч и при температуре 45°С в течение 24 ч. Летучие фракции удаляют при пониженном давлении. После обработки водой и экстракции этилацетатом названное в заголовке промежуточное соединение кристаллизуют из этилацетата, получая бежевое твердое вещество (17,38 г, 64%-ный выход).
МС (ЭСИ, m/z): 273,2 [М+Н+].
A.ii. 6-((8)-5-Хлорметил-2-оксооксазолидин-3-ил)-4Н-бензо[1,4]тиазин-3-он
Раствор промежуточного соединения (A.i) (39,3 г, 144 ммолей) и КДИ (28,0 г, 1,2 экв.) в ТГФ (1 л) нагревают при температуре 50°С в течение ночи. Реакционную смесь затем концентрируют при пониженном давлении и распределяют между этилацетатом и водой. Водный слой экстрагируют несколько раз этилацетатом, объединенные органические слои высушивают над MgSO4 и концентрируют.Остаток очищают с помощью колоночной хроматографии (ЭА/гептан в соотношении 2:1, ЭА), получая названное в заголовке промежуточное соединение в виде бежевого твердого вещества (34,2 г, 79%-ный выход).
МС (ЭСИ, m/z): 299,1 [М+Н+].
A.iii. 6-((8')-5-Йодметил-2-оксооксазолидин-3-ил)-4Н-бензо[1,4]тиазин-3-он
Смесь промежуточного соединения (A.ii) (14,0 г, 46,9 ммолей) и Nal (3 экв.) в 2-бутаноне (150 мл) нагревают при температуре 85°С в течение 2 дней. После охлаждения до комнатной температуры реакционную смесь разбавляют 10%-ным водным раствором Na2S2O3 (300 мл) и смесью эфир/ЭА (150 мл), энергично перемешивают в течение 10 мин и фильтруют.Твердый остаток промывают водой и эфиром и высушивают в высоком вакууме, получая бледно-бежевое твердое вещество. Фазы объединенных фильтратов разделяют, после чего органическую фазу промывают рассолом, высушивают над MgSO4 и концентрируют, получая бледно-бежевое твердое вещество. Твердые вещества обоих процессов объединяют, получая названное в заголовке соединение в виде бледно-бежевого твердого вещества (15,0 г, 82%-ный выход).
МС (ЭСИ, m/z): 391,4 [М+Н+].
Эксперимент В: (S)-3-(2.3-Дигидробензо[1,4]диоксин-6-ил)-5-йодметилоксазолидин-2-он
B.i. (S)-3-(2.3-Дигидробензо[1,4]диоксин-6-ил)-5-гидроксиметилоксазолидин-2-он
К раствору бензилового эфира (2,3-дигидробензо[1,4]диоксин-6-ил)карбаминовой кислоты (13,0 г, 45,6 ммолей) в ТГФ (220 мл), охлажденному до температуры -78°С, добавляют по каплям н-BuLi (29,5 мл 2,36-молярный раствор в гексане, 1,1 экв.). Реакционную смесь перемешивают при температуре -78°С в течение 1 ч, нагревают до температуры -15°С и при этой температуре добавляют по каплям (S)-глицидилбутират (7,37 г, 1,1 экв.), после чего перемешивают при комнатной температуре в течение ночи. Затем добавляют Сs2СО3 (с помощью шпателя), реакционную смесь нагревают при температуре 40°С до полного завершения реакции, разбавляют этилацетатом и промывают насыщенным водным раствором NH4Cl и водой. Органический слой высушивают над MgSO4 и концентрируют.Остаток очищают с помощью колоночной хроматографии (гексан/ЭА в соотношении 2:1, затем 1:1), получая названное в заголовке промежуточное соединение в виде серого твердого вещества (7,04 г, 62%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 7,13 (d, J=2,5 Гц, 1Н), 6,96 (dd, J=2,5, 8,9 Гц, 1Н), 6,86 (d, J - 8,9 Гц, 1Н), 5,16 (t. J=5,8 Гц, 1Н), 4,70-4,50 (m, 1Н), 4,30-4,10 (m, 4H), 4,10-3,90 (m, 1Н), 4,80-4,70 (m, 1Н), 4,70-4,60 (m, 1Н), 4,60-4,50 (m, 1Н).
B.ii. (8)-3-(2,3-Дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметиловый эфир метансульфоновой кислоты
Раствор промежуточного соединения (B.i) (7,0 г, 27,9 ммолей) в ДХМ (140 мл) охлаждают до температуры 0°С, добавляют ДИПЭА (5,70 мл, 1,2 экв.) и MsCI (2,40 мл, 1,1 экв.), после чего реакционную смесь перемешивают в течение 1 ч при этой температуре. Затем реакционную смесь разбавляют ДХМ и промывают водой. Органическую фазу высушивают над MgSO4 и концентрируют, получая названное в заголовке промежуточное соединение в виде бесцветного твердого вещества (9,0 г, 98%-ный выход).
МС (ЭСИ, m/z): 330,3 [M+H+].
B.iii. (S)-3-(2,3-Дигидробензо[1,4]диоксин-6-ил)-5-йодметилоксазолидин-2-он
Смесь промежуточного соединения (B.ii) (9,0 г, 27,3 ммолей) и Nal (16,4 г, 4 экв.) в ацетоне (150 мл) нагревают с обратным холодильником в течение 20 ч. Растворитель выпаривают, а остаток экстрагируют смесью воды с ДХМ, Органический слой промывают рассолом, высушивают над MgSO4 и концентрируют при пониженном давлении. Остаток растирают со смесью эфир/ЭА, получая названное в заголовке соединение в виде грязно-белого твердого вещества (6,91 г, 70%-ный выход).
1Н ЯМР (CDCl3) δ: 7.07 (d, J=2,6 Гц. 1Н), 6,98 (dd, J=9,1, 2,6 Гц, 1Н), 6,85 (d, J=8,9 Гц, 1Н), 4,68 (m, 1Н), 4,24 (s, 4H), 4,10 (t, J=9,1 Гц, 1Н), 3,72 (dd, J=9,1, 5,9 Гц, 1Н), 3,46 (m, 1Н), 3,33 (m, 1Н).
MC (ЭСИ, m/z): 362,2 [M+H+].
Эксперимент С: 6-[(7?)-5-(2-Йодэтил)-2-оксооксазолидин-3-ил1-4Д-бензо[1,4]тиазин-3-он
C.i. (2R)-трет-Бутилдиметил-(2-оксиранилэтокси)силан и (2S)-4-(трет-бутилдиметилсиланилокси)бутан-1,2-диол
Названные в заголовке промежуточные соединения получены аналогтчно методу, описанному Kishi et al., Org. Lett. (2005), 7, 3997, (промежуточное соединение S2-3) посредством гидролитического кинетического разложения (RS)-трет-бутилдиметил-(2-оксиранилэтокси)силана (получен согласно J. Org. Chem. (2008), 73, 1093). Два соединения были выделены с помощью колоночной хроматографии (гептан/ЭА в соотношении 2:1).
Первое элюируемое соединение: (2R)-трет-бутилдиметил-(2-оксиранилэтокси)силан (бесцветное масло; 25,3 г, 48%-ный выход).
1Н ЯМР (СОСl3) δ: 3,77 (t, J=6,4 Гц, 2Н), 3,04 (m, 1Н), 2,78 (m, 1Н), 2,51 (dd, J=5,0, 2,9 Гц, 1Н), 1,74 (m, 2Н), 0,90 (d, J=0,6 Гц, 9H), 0,06 (s, 6H).
Второе элюируемое соединение: (2S)-4-(трет-бутилдиметилсиланилокси)бутан-1,2-диол (бесцветное масло; 24,9 г, 43%-ный выход).
1Н ЯМР (CDCl3) δ: 3,89 (m, 3H), 3,62 (s, 1Н), 3,53 (m, 1Н), 3,42 (расширенный, s, 1Н), 2,29 (m, 1Н), 1,70 (m, 2Н), 0,90 (s, 9H), 0,09 (s, 6H).
C.ii. 6-[(R)-4-('трет-Бутилдиметилсиланилокси)-2-гидроксибутиламино]-4Н-бензо[1,4]тиазин-3-он
Раствор 6-амино-4H-бензо[1,4]тиазин-3-она (10,68 г, 59,3 ммолей; коммерческий продукт) и (2R)-трет-бутилдиметил-(2-оксиранилэтокси)силана (первое элюируемое соединение стадии C.i, 12,0 г, 59,3 ммолей) в смеси ЕtOН/Н2О (в соотношении 9:1, 320 мл) нагревают при температуре 80°С в течении 2 дней, после чего смесь концентрируют при пониженном давлении. Остаточный исходный анилин может быть удален с помощью смеси Et2O/MeOH с последующим фильтрованием. Фильтрат, содержащий продукт, концентрируют при пониженном давлении, получая названное в заголовке промежуточное соединение в виде коричневого масла (18,8 г, 83%-ный выход), которое используют на следующей стадии без очистки.
МС (ЭСИ. m/z): 383,2 [М+H+].
C.iii. 6-{(R)-5-[2-(трет-Бутилдиметилсиланилокси)этил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
Раствор промежуточного соединения (C.ii) (23,5 г, 49,1 ммолей) и КДИ (9,57 г, 1,2 экв.) в ТГФ (250 мл) нагревают при температуре 50°С в течение ночи, после чего смесь концентрируют при пониженном давлении и распределяют между этилацетатом и водой. Водный слой экстрагируют еще раз этилацетатом, и объединенные органические слои высушивают над MgSO4 и концентрируют.Остаток очищают с помощью колоночной хроматографии (ДХМ/МеОН/NH4ОН в соотношении 1000:50:4), получая названное в заголовке промежуточное соединение в виде бесцветного твердого вещества (8,4 г, 42%-ный выход).
МС (ЭСИ, m/z): 409.3 [М+Н+].
C.iv. 6-[(R)-5-(2-Гидроксиэтил)-2-оксооксазолидин-3-ил]-4Н-бензо[1.4]тиазин-3-он
Раствор промежуточного соединения (C.iii) (8,4 г, 20,6 ммолей) в ТГФ (50 мл) обрабатывают тетрабутиламмонийфторидом (1-молярный раствор в ТГФ, 24,7 мл, 1,2 экв.) при температуре 0°С, после чего раствор перемешивают при этой же температуре в течение 6 ч. Затем реакционную смесь распределяют между водой и этилацетатом и водную фазу экстрагируют этилацетатом (трижды). Объединенные органические слои промывают водой и рассолом, высушивают над MgSO4 и концентрируют.Остаток растирают со смесью Et2О/ЭА, получая названное в заголовке промежуточное соединение в виде грязно-белого твердого вещества (4,79 г, 79%-ный выход).
МС (ЭСИ, m/z): 295,5 [М+Н+].
С.v. 2-[(R)-2-Оксо-3-(3-оксо-3,4-дигидро-2Н-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этиловый эфир метансульфоновой кислоты
Раствор промежуточного соединения (C.iv) (4,7 г, 16,0 ммолей) и ДИПЭА (7,54 мл, 2,9 экв.) в безводном ДХМ (80 мл) охлаждают до температуры 0°С и прибавляют по каплям MsCl (1,50 мл, 1,2 экв.). Образовавшуюся смесь перемешивают при температуре 0°С в течение 1 ч, после чего добавляют воду и ДХМ и фазы разделяют. Органический слой высушивают над MgSO4 и концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии (ДХМ/МеОН/NH4ОН в соотношении 1000:50:4), получая названное в заголовке промежуточное соединение в виде грязно-белого твердого вещества (5,80 г, 98%-ный выход).
МС (ЭСИ, m/z): 373,4 [M+H+].
C.vi. 6-[(R)-5-(2-Йодэтил)-2-оксооксазолидин-3-ил]-4Н-бензо[1,4]тиазин-3-он
Суспензию промежуточного соединения (C.v) (3,5 г, 9,4 ммолей) и Nal (4,23 г, 3 экв.) в 2-бутаноне (35 мл) нагревают при температуре 85°С в течение ночи. После охлаждения реакционную смесь разбавляют смесью эфир/ЭА (20 мл) и обрабатывают 10%-ным водным раствором Na2S2O3 (60 мл). После перемешивания в течение 10 мин фазы разделяют, и водный слой промывают этилацетатом. Объединенные органические слои промывают водой (дважды), высушивают над MgSO4 и концентрируют при пониженном давлении. Остаток растирают со смесью Et2О/ЭА, получая названное в заголовке соединение в виде грязно-белого твердого вещества (3,52 г, 93%-ный выход).
МС (ЭСИ, m/z): 405,0 [М+Н+].
Эксперимент D: (S)-3-(3-Фтop-4-мeтилфeнил)-5-йoдмeтилoкcaзoлидин-2-oн
D.i (S)-3-(3-Фтор-4-метилфенил)-5-гидроксиметилоксазолидин-2-он
Смесь 3-фтор-4-метиланилина (коммерческий продукт; 1,25 г, 10 ммолей), насыщенный водный раствор МаНСОз (10 мл) и ацетон (10 мл) обрабатывают, прибавляя по каплям бензилхлорформиат (1,70 г, 1,41 мл, 1 экв.). После прекращения выделения СО2 реакционную смесь распределяют между этилацетатом и насыщенным водным раствором NаНСО3, органический слой высушивают над MgSO4 и концентрируют при пониженном давлении. Полученный бензилкарбамат растворяют в ТГФ (50 мл), охлаждают в токе аргона при температуре -78°С, после чего прикалывают н-BuLi (2,5-молярный раствор в смеси гексанов, 6,45 мл, 1,1 экв.) и полученный раствор перемешивают в течение 1 ч при этой же температуре. Реакционную смесь оставляют затем самопроизвольно нагреваться до температуры -15°С, при которой добавляют по каплям (5)-глицидилбутират (1,69 мл, 1,1 экв.). Смесь перемешивают при комнатной температуре в течение ночи, добавляют с помощью шпателя Сs2СО3, перемешивают при комнатной температуре в течение 3 ч, а затем добавляют NH4Cl и этилацетат и фазы разделяют.Водную фазу экстрагируют еще раз этилацетатом, объединенные органические экстракты промывают несколько раз водным раствором NH4Cl, затем рассолом, высушивают над Na2SO4 и концентрируют.Полученное оранжевое твердое вещество растирают с этилацетатом, получая названное в заголовке промежуточное соединение в виде бледно-желтого твердого вещества (1,18 г, 53%-ный выход).
МС (ЭСИ, m/z): 226,3 [M+H+].
D.ii. (S)-3-(3-Фтор-4-метилфенил')-2-оксооксазолидин-5-илметиловый эфир метансульфоновой кислоты
Раствор промежуточного соединения (D.i) (4,70 г, 20,9 ммолей) в ДХМ (200 мл) охлаждают до температуры 0°С, добавляют ДИПЭА (9,9 мл, 2,9 экв.) и MsCl (2,0 мл, 1,2 экв.), после чего реакционную смесь перемешивают в течение 1 ч при температуре 0°С, затем разбавляют ДХМ и промывают водой. Органическую фазу высушивают над MgSO4 и концентрируют.Остаток растирают с эфиром, получая названное в заголовке промежуточное соединение в виде желтого твердого вещества (6,37 г, 100%-ный выход).
1Н ЯМР (СDСl3) δ: 7,36 (dd, J=11,7, 2,3 Гц, 1Н). 7,13 (m, 2H), 4,91 (m, 1H), 4,46 (m, 2H), 4,13 (t, J=9,1 Гц, 1H), 3,92 (dd, J=9,1, 6,2 Гц, 1H), 3,10 (s, 3H), 2,25 (d,J=1,8 Гц, 3Н).
МС (ЭСИ, m/z): 330,3 [M+H+].
D.iii. (S)-3-(3-Фтор-4-метилфенил)-5-йодметилоксазолидин-2-он
Смесь промежуточного соединения (D.ii) (6,30 г, 20,8 ммолей) и Nal (12,5 г, 4 экв.) в ацетоне (100 мл) нагревают с обратным холодильником в течение 3 ч. Растворитель выпаривают, а остаток экстрагируют смесью вода/ДХМ. Органический слой промывают рассолом, высушивают над MgSO4 и концентрируют при пониженном давлении. Остаток растирают со смесью эфир/ЭА, получая названное в заголовке соединение в виде розоватого твердого вещества (6,3 г, 91%-ный выход).
МС (ЭСИ, m/z): 335,8 [М+Н+].
Эксперимент Е: (5)-2-Оксо-3-(3-оксо-3.4-дигидро-2Н-бензо[1.4]оксазин-6-ил)оксазолидин-5-илметиловый эфир метансульфоновой кислоты
E.i. 6-[(S)-3-(трет-Бутилдиметилсиланилокси)-2-гидроксипропиламино]-4Н-бензо[1.4]оксазин-3-он
К раствору трет-бутилдиметил-((S)-1-оксиранилметокси)силана (коммерческий продукт; 4,25 г, 22,6 ммолей) в MeCN (70 мл) добавляют LiClO4 (7,20 г, 3 экв.), а затем 6-амино-4H-бензо[1,4]оксазин-3-он (коммерческий продукт; 3,70 г, 1 экв.), после чего реакционную смесь перемешивают при температуре 50°С в течение 6 ч. Затем растворитель удаляют при пониженном давлении, а остаток очищают с помощью колоночной хроматографии (ДХМ/МеОН/NH4ОН в соотношении 1000/25/2), получая названное в заголовке промежуточное соединение в виде бледно-коричневой пены (5,25 г, 66%-ный выход).
МС (ЭСИ, m/z): 353,3 [М+Н+].
E.ii. 6-[(S)-5-(трет-Бутилдиметилсиланилоксиметил)-2-оксооксазолидин-3-ил]-4Н-бензо[1,41оксазин-3-он
Раствор промежуточного соединения (E.i) (10,24 г, 29 ммолей) и КДИ (9,71 г, 2 экв.) в ТГФ (140 мл) нагревают при температуре 50°С в течение 2 ч; после чего реакционную смесь концентрируют при пониженном давлении и распределяют между этилацетатом и водой. Органический слой промывают водой и рассолом, высушивают над MgSO4, концентрируют и растирают с Et2O, получая названное в заголовке промежуточное соединение в виде бледно-желтого твердого вещества (6,30 г, 57%-ный выход).
МС (ЭСИ, m/z): 379,2 [M+H+].
E.iii. 6-((S)-5-Гидроксиметил-2-оксооксазолидин-3-ил)-4Н-бензо[1,4]оксазин-3-он
Суспензию промежуточного соединения (E.ii) (6,30 г, 16,6 ммолей) в ТГФ (15 мл) обрабатывают ТБАФ (1-молярный раствор в ТГФ, 16,6 мл) при температуре 0°С, после чего раствор перемешивают при этой же температуре в течение 3 ч, а затем распределяют между водой и этилацетатом. Водную фазу экстрагируют этилацетатом (трижды). Объединенные органические слои промывают рассолом, высушивают над MgSO4 и концентрируют.Остаток растирают с этилацетатом, получая названное в заголовке промежуточное соединение в виде бесцветного твердого вещества (3,49 г, 79%-ный выход).
МС (ЭСИ, m/z): 265,5 [М+Н+].
E.iv. (S)-2-оксо-3-(3-Оксо-3,4-дигидро-2Н-бензо[1,4]оксазин-6-ил)оксазолидин-5-илметиловый эфир метансульфоновой кислоты
Суспензию промежуточного соединения (E.iii) (4,93 г, 18,7 ммолей) в безводном ДХМ (110 мл) обрабатывают ДИПЭА (12,0 мл, 3,75 экв.), после чего реакционную смесь охлаждают до температуры 0°С и порциями прибавляют к ней Ms2O (4,88 г, 1,5 экв.). Образовавшуюся смесь перемешивают при температуре 0°С в течение 15 мин, затем добавляют воду и перемешивание продолжают в течение 15 мин при комнатной температуре. Выпавший в осадок продукт фильтруют, промывают водой и ДХМ. Полученное твердое вещество растирают со смесью ДХМ/МеОН/NH4ОН (в соотношении 1000:25:2), получая названное в заголовке соединение в виде бесцветного твердого вещества (3,785 г, 60%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,72 (s, 1H), 7,29 (dd, J=2,1, 0,6 Гц, 1Н), 6,94 (m, 2H), 4,95 (m, 1H), 4,52 (s, 2H), 4,49 (m, 2H), 4,11 (t, J=9,1 Гц, 1H), 3,73 (m, 2H), 3,23 (s, 3H).
МС (ЭСИ, m/z): 343,3 [М+Н+].
Эксперимент F: 6-((R)-5-Йодметил-2-оксооксазолидин-3-ил)-4Н-бензо[1,4]тиазин-3-он
F.i. 6-((R)-3-Хлор-2-гидроксипропиламино)-4Н-бензо[1,4]тиазин-3-он Раствор 6-амино-4H-бензо[1,4]тиазин-3-она (18,39 г, 102 ммоля;
коммерческий продукт) и (R)-эпихлоргидрина (8,0 мл, 1 экв.) в смеси ЕtOН/Н2О в соотношении 9:1 (450 мл) нагревают при температуре 80°С в течение ночи. Смесь затем концентрируют при пониженном давлении. Остаточный исходный анилин удаляют путем добавления смеси Еt2О/ЭА с последующим фильтрованием. Фильтрат, содержащий продукт, концентрируют при пониженном давлении, получая названное в заголовке промежуточное соединение в виде бежевого твердого вещества (22,52 г, 81%-ный выход), которое используют на следующей стадии без очистки.
МС (ЭСИ, m/z): 273,2 [М+Н+].
F.ii. 6-((R)-5-Хлорметил-2-оксооксазолидин-3-ил)-4Н-бензо[1.4]тиазин-3-он
Раствор промежуточного соединения (F.i) (22,0 г, 81,0 ммолей) и КДИ (15,7 г, 1,2 экв.) в ТГФ (500 мл) нагревают при температуре 50°С в течение ночи. Реакционную смесь затем концентрируют при пониженном давлении и распределяют между ДХМ и водой. Водный слой экстрагируют еще раз ДХМ, объединенные органические слои промывают 0,5-молярным раствором НС1 (дважды) и водой, высушивают над MgSO4 и концентрируют.Остаток растирают со смесью ДХМ/МеОН, получая названное в заголовке промежуточное соединение в виде желтого твердого вещества (8,79 г, 36%-ный выход). МС (ЭСИ, m/z): 299,1 [М+Н+].
F.iii. 6-((R)-5-Йодметил-2-оксооксазолидин-3-ил)-4Н-бензо[1.4]тиазин-3-он
Смесь промежуточного соединения (F.ii) (8,75 г, 29 ммолей) и Nal (13,17 г, 3 экв.) в 2-бутаноне (75 мл) нагревают при температуре 85°С в течение 4 дней. После охлаждения до комнатной температуры реакционную смесь разбавляют 10%-ным водным раствором Na2S2O3 (150 мл) и смесью эфир/ЭА (75 мл). Затем смесь энергично перемешивают в течение 10 мин и фильтруют. Твердый остаток промывают водой и эфиром и высушивают в высоком вакууме, получая грязно-белое твердое вещество (9,27 г, 81%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,56 (s, 1H), 7,31 (m, 2H), 7,12 (dd, J-8,5, 2,3 Гц, 1Н), 4,71 (m, 1H), 4,14 (t, J=9,1 Гц, 1H), 3,59 (m, ЗН), 3,31 (s, 2H).
МС (ЭСИ, m/z): 390,9 [М+Н+].
Эксперимент G: 6-[(8')-5-(2-Йодэтил)-2-оксооксазолидин-3-ил]-4Н-бензо[1,4]тиазин-3-он
G.i. (S)-4-(трет-Бутилдиметилсиланилокси)-2-гидроксибутиловый эфир толуол-4-сульфоновой кислоты
К раствору (2S)-4-(трет-бутилдиметилсиланилокси)бутан-1,2-диола (23,9 г, 108 ммолей; второе элюируемое соединение в эксперименте С, стадия C.i) и ДМАП (2,65 г, 0,2 экв.) в ДХМ (80 мл), охлажденному до температуры 0°С, добавляют ТЭА (43,8 мл, 2,9 экв.) и раствор TsCl (20,7 г, 1,1 экв.) в ДХМ (15 мл). Затем реакционную месь перемешивают при комнатной температуре в течение 5 ч, переносят в насыщенный водный раствор NaHCOs и экстрагируют ДХМ. Органический слой высушивают над MgSO4 и концентрируют. Остаток очищают с помощью колоночной хроматографии (гептан/ЭА в соотношении 2:1), получая названное в заголовке промежуточное соединение в виде бесцветного масла (31,3 г, 77%-ный выход).
1Н ЯМР (CDCl3) δ: 7,80 (d, J=7,6 Гц, 2H), 7,34 (d, J=7,6 Гц, 2H), 4,02 (m, ЗН), 3,80 (m, 2H), 2,45 (s, ЗН), 1,70 (m, 2H), 1,27 (m, 1H), 0,87 (s, 9H), 0,05 (s, 6H).
G.ii. (2S)-трет-Бутилдиметил-(2-оксиранилэтокси')силан
К раствору промежуточного соединения (G.i) (31,1 г, 83,1 ммолей) в ТГФ (350 мл) добавляют 2-молярный раствор NaOH (35 мл), и реакционную смесь энергично перемешивают при комнатной температуре в течение 3 ч, после чего переносят в 1-молярный раствор NaOH (200 мл) и экстрагируют ТБМЭ (дважды). Объединенные органические слои промывают водой и рассолом, высушивают над MgSO4 и концентрируют.Образовавшееся масло очищают с помощью перегонки через насадку Кюгеля (около 70°С при давлении 0,1 мбар), получая названное в заголовке промежуточное соединение в виде бесцветного масла (14,7 г, 87%-ный выход).
1Н ЯМР (CDCl3) δ: 3,77 (t, J=6,4 Гц, 2H), 3,04 (m, 1H), 2,78 (m, 1H), 2,51 (dd, J=5,0. 2,9 Гц, 1H), 1,74 (m, 2H), 0,90 (d, J=0,6 Гц, 9H), 0,06 (s, 6H).
G.iii. 6-[(8)-4-(трет-Бутилдиметилсиланилокси)-2-гидроксибутиламино]-4Н-бензо[1,4]тиазин-3-он
Раствор 6-амино-4H-бензо[1,4]тиазин-3-OHa (8,0 г, 44,5 ммолей;
коммерческий продукт) и промежуточного соединения (G.ii) (9,0 г, 1 экв.) в смесь EtOH/H2O в соотношении 9:1 (250 мл) нагревают при температуре 80°С в течение 2 дней, после чего реакционную смесь концентрируют при пониженном давлении. Остаточный исходный анилин удаляют путем добавления смеси Et2О/МеОН с последующим фильтрованием. Фильтрат, содержащий продукт, концентрируют при пониженном давлении, получая названное в заголовке промежуточное соединение в виде коричневого масла (14,58 г, 86%-ный выход), которое используют на следующей стадии без очистки.
МС (ЭСИ, m/z): 383,2 [M+H+].
G.iv. 6-{(8)-5-[2-(трет-Бутилдиметилсиланилокси)этил1-2-оксооксазолидин-3-ил}-4Н-бензо[1.4]тиазин-3-он
Раствор промежуточного соединения (G.iii) (14,5 г, 37,9 ммолей) и КДИ (8,60 г, 1,4 экв.) в ТГФ (180 мл) нагревают при температуре 50°С в течение ночи, после чего реакционную смесь концентрируют при пониженном давлении и распределяют между этилацетатом и водой. Водный слой экстрагируют еще раз этилацетатом, объединенные органические слои высушивают над MgSO4 и концентрируют.Остаток очищают с помощью колоночной хроматографии (ДХМ/МеОН/МРЦОН в соотношении 1000:50:4), получая названное в заголовке промежуточное соединение в виде бесцветного твердого вещества (5,56 г, 36%-ный выход).
МС (ЭСИ, m/z): 409.3 [М+Н+].
G.v. 6-[(8)-5-(2-Гидроксиэтил)-2-оксооксазолидин-3-ил]-4Н-бензо[1,4]тиазин-3-он
Раствор промежуточного соединения (G.iv) (5,50 г, 13,6 ммолей) в ТГФ (30 мл) обрабатывают ТБАФ (1-молярный раствор в ТГФ, 16,3 мл, 1,2 экв.) при температуре 0°С. Затем раствор перемешивают при этой же температуре в течение 6 ч, после чего реакционную смесь распределяют между водой и этилацетатом и водную фазу экстрагируют этилацетатом (трижды). Объединенные органические слои промывают водой (трижды) и рассолом, высушивают над MgSO4 и концентрируют.Остаток растирают со смесью Et2O/ЭА, получая названное в заголовке промежуточное соединение в виде грязно-белого твердого вещества (3,08 г, 77%-ный выход).
МС (ЭСИ, m/z): 295,5 [М+Н+].
G.vi. 2-[(8')-2-Оксо-3-(3-оксо-3,4-дигидро-2Н-бензо[1.4]тиазин-6-ил)оксазолидин-5-ил]этиловый эфир метансульфоновой кислоты
Раствор промежуточного соединения (G.v) (3,0 г, 10,2 ммолей) и ДИПЭА (4,8 мл, 2,9 экв.) в безводном ДХМ (50 мл) охлаждают до температуры 0°С и обрабатывают, прибавляя по каплям, MsCl (0,96 мл, 1,2 экв.). Образовавшуюся реакционную смесь перемешивают при температуре 0°С в течение 1 ч, добавляют воду и ДХМ и фазы разделяют.Органический слой высушивают над MgSO4 и концентрируют при пониженном давлении. Остаток растирают с эфиром, получая названное в заголовке промежуточное соединение в виде грязно-белого твердого вещества (3,64 г, 96%-ный выход).
МС (ЭСИ, m/z): 373,4 [М+Н+].
G.vii. 6-[(8)-5-(2-Йодэтил)-2-оксооксазолидин-3-ил]-4Н-бензо[1.4]тиазин-3-он
Суспензию промежуточного соединения (G.vi) (2,5 г, 6,7 ммолей) и Nal (3,02 г, 3 экв.) в 2-бутаноне (25 мл) нагревают при температуре 85°С в течение ночи. После охлаждения реакционную смесь разбавляют смесью эфир/ЭА (20 мл) и обрабатывают 10%-ным водным раствором МазЗзОз (60 мл). После перемешивания в течение 10 мин фазы разделяют, и водный слой экстрагируют этилацетатом. Объединенные органические слои промывают водой (дважды), высушивают над MgSO4 и концентрируют при пониженном давлении. Остаток растирают со смесью EtaO/DA, получая названное в заголовке промежуточное соединение в виде светло-оранжевого твердого вещества (2,11 г, 78%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,55 (s, 1H), 7,30 (m, 2H), 7,04 (dd, J=8,5, 2,3 Гц, 1Н), 4,68 (m, 1H), 4,10 (t, J=8,8 Гц, 1H), 3,70 (dd, J=8,8, 6,7 Гц, 1H), 3,41 (s, 2H), 3,29 (m, 2H), 2,23 (m, 2H).
МС (ЭСИ, m/z): 405,1 [M+H+].
Пример 1: 6-{(R)-5-[3-Гидрокси-3-(7-метокси-2-оксо-2Н-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1Л]тиазин-3-он
l.i. mpew-Бутиловый эфир 3-гидрокси-3-(7-метокси-2-оксо-2Н-хинолин-1-илметил)азетидин-1-карбоновой кислоты
Раствор wpew-бутилового эфира окса-5-азаспиро[2.3]гексан-5-карбоновой кислоты (69 мг; получен согласно WO 2007/044515) в ДМФ (1 мл) добавляют к суспензии 7-метокси-2(1Я)-хинолинона (60 мг; коммерческий продукт) и СздСОз (223 мг, 2 экв.) в ДМФ (2 мл), после чего реакционную смесь перемешивают при температуре 80°С в течение 10 дней. Затем растворитель удаляют при пониженном давлении, а остаток распределяют между водой и этилацетатом. Водный слой экстрагируют этилацетатом и объединенные органические слои высушивают над Na2SO4, фильтруют, выпаривают и очищают с помощью колоночной хроматографии (ДХМ/МеОН в соотношении от 19:1 до 9:1), получая названное в заголовке промежуточное соединение в виде желтого масла (12 мг, 10%-ный выход).
МС (ЭСИ, m/z): 361,3 [М+Н+].
l.ii. 1-(3-Гидроксиазетидин-3-илметил)-7-метокси-1Н-хинолин-2-он
Исходя из промежуточного соединения (l.i) (12 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде желтого масла (8 мг, 92%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 7.61 (d, J=9,4 Гц, 1Н), 7,39 (m, 2H), 6,81 (dd, J=8,5, 1,5 Гц, 1Н), 6,48 (d, J=9,4 Гц. 1Н), 4,65 (s, 2H), 3,86 (s, 4H).
l.iii. 6-{(R)-5-[3-Гидрокси-3-(7-метокси-2-оксо-2Н-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (l.ii) (8 мг) и соединения эксперимента А, и используя метод В, названное в заголовке соединение получают в виде бесцветного твердого вещества (4 мг, 22%-ный выход).
1Н ЯМР (CDCl3) δ: 7,86 (s, 1Н), 7,71 (d, J=9,4 Гц, 1Н), 7,53 (d, J=8,5 Гц, 1Н), 7,42 (m, 2H), 7,29 (d, J=8,5 Гц, 1Н), 6,97 (dd, J=8,8, 2,3 Гц, 1Н), 6,89 (dd, J=8,5, 1,8 Гц, 1Н), 6,60 (d, J=9,4 Гц, 1Н), 6,31 (s, 1Н), 4,65 (m, 3Н), 4,08 (t, J=8,8 Гц, 1Н), 3,87 (m, 4H), 3,58 (m, 3H), 3,41 (s, 2H), 3,08 (m, 2H), 2,87 (dd, J=5,9, 3,5 Гц, 2Н).
МС (ЭСИ, m/z): 522,9 [М+Н+].
Пример 2: 6-{(R)-5-[3-(7-метокси-2-оксо-2Н-хинолин-1-илметил')азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1.4]тиазин-3-он
2.i. трет-Бутиловый эфир 3-(7-метокси-2-оксо-2Н-хинолин-1-илметил)азетидин-1 -карбоновой кислоты
Суспензию 7-метокси-2(1H)-хинолинона (405 мг; коммерческий продукт) в ДМФ (10 мл) обрабатывают NaH (111 мг; 50%-ная дисперсия в масле). Смесь перемешивают при комнатной температуре в течение 30 мин, после чего добавляют раствор трет-бутилового эфира 3-[[(метилсульфонил)окси]метил]-1-азетидинкарбоновой кислоты (674 мг; получен согласно WO 02/066470) в ДМФ (2 мл). Реакционную смесь нагревают при температуре 100°С в течение ночи, а затем распределяют между этилацетатом и водой. Органическую фазу высушивают над MgSO4, концентрируют при пониженном давлении и очищают с помощью колоночной хроматографии (гептан/ЭА в соотношении от 2:1 до 0:1). Второе элюируемое соединение выделяют в виде бесцветной пены (350 мг, 44%-ный выход).
1Н ЯМР (СОСl3) δ: 7,59 (d, J=9,4 Гц, 1Н), 7,48 (d, J=8,5 Гц, 1Н), 6,83 (dd, J=8,5, 2,3 Гц, 1Н), 6,76 (d, J=2,3 Гц, 1Н), 6,52 (d, J=9,4 Гц, 1Н), 4,5 (расширенный, 2H), 3,95 (m, 4H), 3,91 (s, 3H), 3,04 (m, 1Н), 1,42 (s, 9H).
МС (ЭСИ, m/z): 345,2 [М+Н+].
2.ii. 3-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)азетидин
Исходя из промежуточного соединения (2.i) (350 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде желтоватой пены (200 мг, 80%-ный выход).
1Н ЯМР (CDCl3) S: 7,58 (d, J=9,4 Гц, 1Н), 7,45 (d, J=9,1 Гц, 1Н), 6,81 (m, 2H), 6,52 (d, J=9,4 Гц, 1Н), 4,51 (d, J=7,0 Гц, 2H), 3,91 (s, 3H), 3,60 (m, 4H), 2,27 (m, 1Н).
2.iii. 6-{(R)-5-[3-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (2.ii) (65 мг) и соединения эксперимента А, и используя метод В, названное в заголовке соединение получают в виде бежевой пены (28 мг, 21%-ный выход).
МС (ЭСИ, m/z): 507,0 [М+Н+].
Пример 3: 1-{1-[(R)-3-(2,3-Дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1H-хинолин-2-он
Исходя из промежуточного соединения (2.ii) (65 мг) и соединения эксперимента В (106 мг), и используя метод В, названное в заголовке соединение получают в виде бежевой пены (30 мг, 23%-ный выход).
1Н ЯМР (СDСl3) δ: 7,59 (d, J=9,4 Гц, 1Н), 7,46 (d, J=9,1 Гц, 1Н), 7,08 (d, J=2,6 Гц, 1Н), 7,00 (m, 1Н), 6,83 (m, 3Н), 6,52 (d, J=9,4 Гц, 1Н), 4,47 (dd, J=6,7, 1,8 Гц, 2Н), 4,24 (s, 4Н), 3,92 (m, ЗН), 3,81 (m, 1Н), 3,25 (m, 2H), 2,77 (s, 2H).
МС (ЭСИ, m/z): 478,0 [М+Н+].
Пример 4: 6-{(R)-5-[3-(7-Метокси-2-оксо-2Н-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1.4]тиазин-3-он
4.1. трет-Бутиловый эфир 3-(7-метокси-2-оксо-2Н-хиноксалин-1-илметил)азетидин-1-карбоновой кислоты
Используя метод примера 2, стадия 2.i, но исходя из 7-метокси-2(1Я)-хиноксалинона (1,00 г; получен согласно WO 2006/134378) и трет-бутиловото эфира 3-[[(метилсульфонил)окси]метил]-1-азетидинкарбоновой кислоты (1,65 г;
получен согласно WO 02/066470), второе элюируемое соединение было выделено в виде желтого масла (700 мг, 35%-ный выход).
МС (ЭСИ. m/z): 346,2 [М+Н+].
4.ii. 3-(7-Метокси-2-оксо-2Н-хиноксалин-1-илметил)азетидин
Исходя из промежуточного соединения (4.i) (700 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде оранжевой пены (400 мг, 80%-ный выход).
МС (ЭСИ, m/z): 246,4 [М+Н+].
4.iii. 6-{(R)-5-[3-(7-Метокси-2-оксо-2Н-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (4.ii) (90 мг) и соединения эксперимента А (157 мг) и используя метод В, названное в заголовке соединение получают в виде коричневого твердого вещества (20 мг, 11%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,54 (d, J=0,6 Гц, 1Н), 8,03 (s, 1H), 7,73 (d, J=9,7 Гц, 1H), 7,30 (m, 2H), 6,98 (m, 2H), 4,41 (d, J=6,4 Гц, 2Н), 3,99 (d, J - 0,6 Гц, 1H), 3,89 (s, 3Н), 3,68 (m, 1H), 3,42 (s, 2H), 3,06 (m, 2H), 2,68 (m, 2H).
МС (ЭСИ, m/z): 508,2 [M+H+].
Пример 5: 6-((R)-5-{2-[3-(7-Метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил')-4H-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (2.ii) (65 мг) и соединения эксперимента С (118 мг), и используя метод В, названное в заголовке соединение получают в виде бежевой пены (47 мг, 34%-ный выход).
МС (ЭСИ, m/z): 521,4 [М+Н+].
Пример 6: 6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1.4]тиазин-3-он
Исходя из промежуточного соединения (4.ii) (90 мг) и соединения эксперимента С (148 мг) и используя метод В, названное в заголовке соединение получают в виде бежевой пены (54 мг, 28%-ный выход).
МС (ЭСИ, m/z): 522,3 [М+Н+].
Пример 7: 6-{(R)-5-[4-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1.4]тиазин-3-он
7.i. трет-Бутиловый эфир 4-(7-метокси-2-оксо-2Н-хинолин-1-илметил)пиперидин-1-карбоновой кислоты
Используя метод примера 2, стадия 2.i, но исходя из 7-метокси-2(1H)-хинолинона (350 мг; коммерческий продукт) и трет-бутилового эфира 4-[[(метилсульфонил)окси]метил]-1-пиперидинкарбоновой кислоты (645 мг; коммерческий продукт), второе элюируемое соединение выделяют в виде бесцветной пены (227 мг; 30%-ный выход).
МС (ЭСИ, m/z): 373,3 [M+H+].
7.ii. 4-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)пиперидин
Исходя из промежуточного соединения (7.i) (327 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде бесцветной пены (210 мг, 88%-ный выход).
1Н ЯМР (СDСl3) δ: 7,59 (d. J=9,4 Гц, 1Н), 7,47 (m, 1H), 6,82 (m, 2H), 6,53 (d, J=9,4 Гц, 1H), 4,18 (m, 2H), 3,91 (s, 3Н), 3,10 (m, 2H), 2,55 (td, J=12,3, 2,6 Гц, 2H), 2,06 (s, 2H), 1,67 (m, 2H), 1,42 (m, 2H).
7.iii. 6-{(R)-5-[4-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (7.ii) (70 мг) и соединения эксперимента А (110 мг), используя метод В, названное в заголовке соединение получают в виде желтоватой пены (53 мг, 38%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,53 (s, 1H), 7,80 (d, J=9,4 Гц, 1H), 7,63 (d, J=8,8 Гц, 1H), 7,30 (m, 2H), 7,12 (m, 1H), 6,90 (m, 2H), 6,40 (d, J=9,4 Гц, 1H), 4,15 (m, 2H), 4,02 (m, 1H), 3,87 (s, 3Н), 3,43 (s, 2H), 2,60 (m, 2H), 1,99 (m, 2H), 1,50 (m, 2H), l,35(m,2H).
МС (ЭСИ, m/z): 535,5 [M+H+].
Пример 8: 1-{1-[(R)-3-(2,3-Дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-илметил}-7-метокси-1H-хинолин-2-он
Исходя из промежуточного соединения (7.ii) (70 мг) и соединения эксперимента В (102 мг), и используя метод В, названное в заголовке соединение получают в виде желтоватой пены (48 мг, 37%-ный выход).
1Н ЯМР (ДMCO-d6) δ: 7,79 (d, J=9,4 Гц, 1H), 7,63 (d, J=8,5 Гц, 1H), 7,10 (d, J=2,6 Гц, 1H), 6,88 (m, 4H), 6,40 (d, J=9,4 Гц, 1H), 4,21 (m, 6H), 4,03 (d, J=1,8 Гц, 2H), 3,87 (s. ЗН), 2,60 (m, 2H), 1,98 (s, ЗН), 1,50 (m, 2H), 1,35 (m, 2H).
МС (ЭСИ, m/z): 506,3 [M+H+].
Пример 9: 1-{1-[(7?)-3-(3-Фтор-4-метилфенил)-2-оксооксазолидин-5-илметил1пиперидин-4-илметил}-7-метокси-1Я-хинолин-2-он
Исходя из промежуточного соединения (7.ii) (70 мг) и соединения эксперимента D (94 мг), и используя метод В, названное в заголовке соединение получают в виде желтоватой пены (30 мг, 24%-ный выход).
МС (ЭСИ, m/z): 480,3 [М+Н+].
Пример 10: 6-{(R)-5-[3-(7-Метокси-2-оксо-3,4-дигидро-2Н-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1Л]тиазин-3-он
10. i. трет-Бутиловый эфир 3-(7-метокси-2-оксо-3,4-дигидро-2Н-хинолин-1-илметил)азетидин-1-карбоновой кислоты
Используя метод примера 2, стадия 2.i, но исходя из 3,4-дигидро-7-метокси-2(1H)-хинолинона (получен согласно WO 2006/134378; 886 мг) и трет-бутилового эфира 3-[[(метилсульфонил)окси]метил]-1-азетидинкарбоновой кислоты (1459 мг; получен согласно WO 02/066470), названное в заголовке соединение получают в виде бесцветного масла (1223 мг; 71%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 7,07 (dd, J=7,9, 0,6 Гц, 1Н), 6,54 (m, 2H), 4,18 (d, J --7,3 Гц, 2H), 3.94 (t, J=8,5 Гц, 2H), 3,77 (m, 5H), 2,81 (m, 2H), 2,62 (m, 2H), 1,43 (s, 9H).
МС (ЭСИ, m/z): 347,2 [М+Н+].
10. ii. 3-(7-Метокси-2-оксо-3,4-дигидро-2Н-хинолин-1-илметил)азетидин
Исходя из промежуточного соединения (10.i) (1223 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде бесцветного масла (565 мг, 65%-ный выход).
МС (ЭСИ, m/z): 247,5 [М+Н+].
IQ.iii. 6-{(R)-5-[3-(7-Метокси-2-оксо-3.4-дигидро-2Н-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1.4]тиазин-3-он
Исходя из промежуточного соединения (10.ii) (110 мг) и соединения эксперимента А (191 мг), и используя метод В, названное в заголовке соединение получают в виде желтоватой пены (74 мг, 32%-ный выход).
МС (ЭСИ, m/z): 509,1 [М+Н+].
Пример 11: 6-{(S)-5-[3-(7-Метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (10. ii) (110 мг) и соединения эксперимента F (191 мг) и используя метод В, названное в заголовке соединение получают в виде желтоватой пены (75 мг, 33%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,53 (s, 1Н), 7,30 (m, 2H), 7,09 (m, 2H), 6,62 (d, J=2,3 Гц, 1Н), 6,56 (dd, J=8,2, 2,3 Гц, 1Н), 4,61 (s, 1Н), 4,02 (m, ЗН), 3,72 (m. 3H), 3,42 (s, 2H), 3,29 (m, 7H), 2,95 (m, 2H), 2,70 (m, 5H).
МС (ЭСИ, m/z): 509,1 [М+Н+].
Пример 12: 6-{(R)-5-[3-(7-Метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4Д-бензо[1,4]оксазин-3-он
Исходя из промежуточного соединения (10.ii) (110 мг) и соединения эксперимента Е (160 мг), и используя метод С, названное в заголовке соединение получают в виде желтоватой пены (50 мг, 23%-ный выход).
МС (ЭСИ, m/z): 493,1 [М+Н+].
Пример 13: 6-{(R)-5-[3-(7-Метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он
Исходя из промежуточного соединения (4.ii) (90 мг) и соединения эксперимента Е (125 мг) и используя метод С, названное в заголовке соединение получают в виде коричневого твердого вещества (20 мг, 11%-ный выход).
МС (ЭСИ, m/z): 492,2 [М+Н+].
Пример 14: 1-{1-[(R)-3-(2,3-Дигидробензо[1.4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1H-хиноксалин-2-он
Исходя из промежуточного соединения (4.ii) (90 мг) и соединения эксперимента В (135 мг) и используя метод В, названное в заголовке соединение получают в виде коричневой пены (22 мг, 12%-ный выход).
МС (ЭСИ, m/z): 479,3 [М+Н+].
Пример 15: 6-Метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2Н-бензо[1.4]тиазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4Н-пиридо[2.3-b]пиразин-3-он
15. i трет-Бутиловый эфир 4-(6-метокси-3-нитропиридин-2-иламино)пиперидин-1-карбоновой кислоты
Смесь 4-амино-1-Бок-пиперидина (коммерческий продукт; 6,7 г, 35 ммолей), 2-хлор-6-метокси-3-нитропиридина (1 экв.) и К2СО3 (1 экв.) в MeCN (100 мл) и ДМФ (30 мл) нагревают при температуре 60°С в течение 3 ч, после чего реакционную смесь фильтруют и концентрируют в вакууме. Остаток переносят в смесь эфир/вода в соотношении 1:1, органическую фазу высушивают над MgSO4 и концентрируют. Остаток растирают с этилацетатом и фильтруют, получая 4,5 г чистого продукта. Фильтрат концентрируют и очищают с помощью колоночной хроматографии (гептан/ЭА в соотношении 9:1, 4:1, 2:1), получая еще одну порцию продукта. В целом, получено 8,5 г (70%-ный выход) желтого твердого вещества.
1Н ЯМР (CDCl3) δ: 8,62 (m, 1H), 8,31 (d, J=9,4 Гц, 1Н), 6,07 (d, J=9,1 Гц, 1H), 4,32 (m, 1H), 4,05 (m, 2H), 3,95 (s, 3H), 3,02 (s, 2H), 2,10 (m, 2H), 1,60 (s, 3H), l,47(m,9H).
15.ii. трет-Бутиловый эфир 4-(3-амино-6-метоксипиридин-2-иламино)пиперидин-1-карбоновой кислоты
Раствор промежуточного соединения (15.i) (8,45 г) в смеси EtOH/ЭА (в соотношении 1:1; 300 мл) гидрируют над 10%-ным Ро/С в течение 4 ч, после чего катализатор отфильтровывают, фильтрат выпаривают при пониженном давлении, а остаток очищают с помощью колоночной хроматографии (гептан/ЭА в соотношении 1:2), получая красное твердое вещество (5,4 г, 70%-ный выход).
МС (ЭСИ, m/z): 323,3 [М+Н+].
15.iii. трет-Бутиловыи эфир 4-[3-(этоксикарбонилметиламино)-6-метоксипиридин-2-иламино]пиперидин-1-карбоновой кислоты
Суспензию промежуточного соединения (15.ii) (5,38 г), этилбромацетата (2,9 г), К2СО3 (4,6 г) в смеси ДМФ/MeCN (в соотношении 1:2, 120 мл) перемешивают при комнатной температуре в течение ночи. Реакционную смесь фильтруют, и фильтрат выпаривают при пониженном давлении. Остаток растворяют в смеси ЭА/МеОН (в соотношении 19:1, 200 мл) и промывают водой. Органическую фазу высушивают над MgSO4, фильтруют, концентрируют при пониженном давлении и очищают с помощью колоночной хроматографии (гептан/ЭА в соотношении 2:1, затем 1:1), получая темное масло (5,29 г; 77%-ный выход).
МС (ЭСИ, m/z): 409,4 [М+Н+].
15.iv. трет-Бутиловый эфир 4-(6-метокси-3-оксо-3Н-пиридо[2,3-Ь]пиразин-4-ил)пиперидин-1-карбоновой кислоты
Раствор промежуточного соединения (15.iii) (5,26 г) в толуоле (240 мл) обрабатывают АсОН (1 мл) и смесь нагревают с обратным холодильником в течение ночи в атмосфере N2. Реакционную смесь концентрируют при пониженном давлении, остаток растворяют в ДХМ (200 мл) и обрабатывают MnO2 (21,2 г) при комнатной температуре в течение 6 ч. Затем реакционную смесь фильтруют, фильтрат выпаривают при пониженном давлении и очищают с помощью колоночной хроматографии (гептан/ЭА в соотношении 2:1, затем 1:1), получая бежевое твердое вещество (2,3 г, 49%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 8,11 (d, J=8,5 Гц, 1H), 8,07 (s, 1H), 6,83 (d, J=8,5 Гц, 1H), 5,42 (m, 1H), 4,11 (m, 2H), 3,93 (s, ЗН), 2,77 (m, 4H), 1,63 (m, 2H), 1,40 (s,9H).
МС (ЭСИ, m/z): 361,4 [М+Н+].
15. v. 6-Метокси-4-пиперидин-4-ил-4Н-пиридо[2.3-b]пиразин-3-он
Исходя из промежуточного соединения (15.iv) (2,30 г) и используя метод А, названное в заголовке промежуточное соединение получают (223 мг, 13%-ный выход) после очистки с помощью колоночной хроматографии (ДХМ/МеОН в соотношении 19:1, затем 9:1+1% NH40H) в виде желтого твердого вещества.
МС (ЭСИ, m/z): 261,2 [М+Н+].
15.vi. 6-Метокси-4-{1-[(R)-2-оксо-3-('3-оксо-3,4-дигидро-2Н-бензо[1.4]тиазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4Н-пиридо[2,3-b]пиразин-3-он
Исходя из промежуточного соединения (15.v) (101 мг) и соединения эксперимента А (167 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (71 мг, 35%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,57 (d, J - 0,6 Гц, 1Н), 8,11 (d, J=8,5 Гц, 1Н), 8,05 (s, 1Н), 7,33 (m, 2H), 7,14 (m, Н), 6,83 (d, J=8,5 Гц, 1Н), 4,85 (m, 1Н), 4,03 (s, 1Н), 3,91 (s, ЗН), 3,76 (m, 1Н), 3,44 (s, 3H), 3,08 (m, 2H), 2,93 (m, 2H), 2,73 (m, 2H), 2,27 (m, 2H), l,16(m,2H).
MC (ЭСИ, m/z): 523,1 [M+H+].
Пример 16: 6-Метокси-4-{1-[(7?)-2-оксо-3-(3-оксо-3.4-дигидро-2H-бензо[1.4]оксазин-6-ил')оксазолидин-5-илметил]пиперидин-4-ил}-4h-пиридо[2.3-d]пиразин-3-он
Исходя из промежуточного соединения (15.v) (100 мг) и соединения эксперимента Е (145 мг) и используя метод С, названное в заголовке соединение получают в виде бежевого твердого вещества (64 мг, 32%-ный выход).
1Н ЯМР (ДМСО-d6) δ: 10,72 (s, 1 Н), 8,11 (d, J=8,5 Гц, 1Н), 8,05 (s, 1Н), 7,33 (t, J=1,5 Гц, 1Н), 6,97 (d, J=1,5 Гц, 2H), 6,83 (d, J=8,5 Гц, 1Н), 5,25 (m, 1Н), 4,83 (m, 1Н), 4,54 (m, 2H), 4,05 (m, 1Н), 3,92 (s, 3Н), 3,75 (m, 1Н), 3,07 (m, 2H), 2,92 (m, 2H), 2,72 (d, J=5,6 Гц, 2H), 2,27 (m, 2H), 1,59 (m, 2H).
MC (ЭСИ, m/z): 507,2 [М+Н+].
Пример 17: 6-{(R)-5-[(R)-3-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
17.1. трет-Бутиловый эфир (S)-3-(7-метокси-2-оксо-2Н-хинолин-1-илметил-пирролидин-1-карбоновой кислоты
Используя метод примера 2, стадия 1 л, но исходя из 7-метокси-2(1Я)-хинолинона (получен согласно WO 2006/134378; 500 мг) и трет-бутиловото эфира 3-[[(метилсульфонил)окси]метил]-1-пирролидинкарбоновой кислоты (797 мг; получен согласно J. Med. Chem. (1999), 42, 677-690), второе элюируемое соединение получают в виде бесцветного масла (240 мг; 23%-ный выход).
1Н ЯМР (СDСl3) δ: 7,60 (d, J=9,4 Гц, 1Н), 7,48 (d, J=8,8 Гц, 1Н), 6,81 (m, 2H), 6,53 (d, J=9,4 Гц, 1Н), 3,91 (m, 3Н), 3,52 (m, 2H), 3,35 (m, 2H), 2,76 (m, 1Н), 1,89(m,4H), 1,45 (m,9H).
17. ii. (S)-7-Метокси-1-пирролидин-3-илметил-1Н-хинолин-2-он
Исходя из промежуточного соединения (17. i) (220 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде бесцветного масла (120 мг, 75%-ный выход).
1Н ЯМР (CDCl3) δ; 7,62 (d, J=9,4 Гц, 1Н), 7,49 (d, J=8,8 Гц, 1Н), 6,84 (m, 2H), 6,53 (d, J=9,4 Гц. 1Н), 4,41 (m, 1Н), 4,19 (m, 1Н), 3,92 (s, 3Н), 3,48 (s, 1Н), 3,28 (m, 1Н), 3,02 (m, 3Н), 2,80 (m, 1Н), 2,01 (m, 1Н), 1,78 (m, 1Н).
17.iii. 6-{(R)-5-[(R)-3-(7-Метокси-2-оксо-2Н-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1.4]тиазин-3-он
Исходя из промежуточного соединения (17.ii) (100 мг) и соединения эксперимента А (151 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (8 мг, 4%-ный выход).
МС (ЭСИ, т/г): 521,2 [М+Н+].
Пример 18: 6-{(R)-5-[(RS)-3-Гидрокси-3-(7-метокси-2-оксо-2Н-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
18.i. трет-Бутиловый эфир рац-3-гидрокси-3-(7-метокси-2-оксо-2Н-хинолин-1 -илметил)пирролидин-1-карбоновой кислоты
Раствор 7-метокси-1H-хинолин-2-она (650 мг, 3,7 ммолей) и трет-бутилового эфира 1-окса-5-азаспиро[2.4]гептан-5-карбоновой кислоты (1 экв., коммерческий продукт) в ДМФ (10 мл) обрабатывают СзгСОз (1 экв.), после чего нагревают при температуре 70°С в течение ночи. Смесь затем распределяют между этилацетатом и водой, органическую фазу промывают водой и рассолом, высушивают над MgSO4 и концентрируют.Продукт очищают с помощью колоночной хроматографии (ЭА/гептан в соотношении 1:1, ЭА), получая требуемое промежуточное соединение в виде желтоватого масла (650 мг, 47%-ный выход).
1Н ЯМР (CDCl3) δ: 7,70 (d, J=9,4 Гц, 1Н), 7,52 (d, J=8,5 Гц, 1Н), 6,87 (m, 2H), 6,60 (d, J=9,4 Гц, 1Н), 4,59 (m, 1Н), 4,45 (m, 1Н), 3,89 (s, 3Н), 3,49 (m, 6H), 1,40 (расширенный s, 9H).
18.ii. рац-1-(3-Гидроксипирролидин-3-илметил)-7-метокси-1Н-хинолин-2-он
Исходя из промежуточного соединения (18.i) (600 мг) и используя метод А, названное в заголовке промежуточное соединение получают в виде бесцветного масла (440 мг, 100%-ный выход).
1Н ЯМР (СОСl3) δ: 7,67 (d, J=9,4 Гц, 1Н), 7,47 (d, J=8,8 Гц, 1Н), 7,25 (m, 1H), 6,85 (dd, J=8,5, 2,1 Гц, 1Н), 6,55 (d, J=9,4 Гц, 1Н), 4,56 (m, 2H), 3,88 (s, 3Н), 3,21 (m, 5H), 2,01 (m, 2H).
18.iii. 6-{[R]-5-[(RS)-3-Гидрокси-3-(7-метокси-2-оксо-2Н-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4Н-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (18.ii) (100 мг) и соединения эксперимента А (142 мг) и используя метод В, названное в заголовке промежуточное соединение получают в виде бежевого твердого вещества (41 мг, 21%-ный выход).
МС (ЭСИ, m/z): 537,2 [M+H+].
Пример 19: 6-{(R)-5-[(RS)-3-Гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-47^-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (18.ii) (100 мг) и соединения эксперимента F (142 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (31 мг, 16%-ный выход).
МС (ЭСИ, m/z): 537,2 [М+Н+].
Пример 20: 6-((S)-5-{2-[(RS)-3-Гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (18.ii) (100 мг) и соединения эксперимента G (147 мг) и используя метод В, названное в заголовке соединение получают в виде серого твердого вещества (10 мг, 5%-ный выход).
МС (ЭСИ, m/z): 551,2 [М+Н+].
Пример 21: 6-((R)-5-{2-[(RS)-3-Гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он
Исходя из промежуточного соединения (18.ii) (100 мг) и соединения эксперимента С (147 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (47 мг, 23%-ный выход).
МС (ЭСИ, m/z): 551,2 [М+Н+].
Пример 22: 4-{1-[(R)-3-(2,3-Дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-6]пиразин-3-он
Исходя из промежуточного соединения (15.v) (143 мг) и соединения эксперимента В (218 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (82 мг, 30%-ный выход).
МС (ЭСИ, m/z): 494,2 [M+H+].
Пример 23: 6-Метокси-4-(1-{2-[(S)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-6]пиразин-3-он
Исходя из промежуточного соединения (15.v) (130 мг) и соединения эксперимента G (222 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (201 мг, 75%-ный выход).
МС (ЭСИ, m/z): 537,2 [М+Н+].
Пример 24: 4-{1-[(R)-3-(3-Фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-6]пиразин-3-он
Исходя из промежуточного соединения (15.v) (143 мг) и соединения эксперимента D (202 мг) и используя метод В, названное в заголовке соединение получают в виде бежевого твердого вещества (41 мг, 16%-ный выход).
МС (ЭСИ, m/z): 468,2 [М+Н+].
Пример 25: 6-Метокси-4-(1-{2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-6]пиразин-3-он
Исходя из промежуточного соединения (15.v) (130 мг) и промежуточного соединения (C.v) (204 мг) и используя метод С, названное в заголовке соединение получают в виде бежевого твердого вещества (137 мг, 51%-ный выход).
МС (ЭСИ, т/г): 537,2 [М+Н+].
Фармакологические свойства соединений по изобретению
Анализы в условиях in vitro
Экспериментальные методы
Минимальные концентрации ингибирования (MICs; в мг/л) определялись в катион-регулируемой питательной среде Mueller-Hinton Broth с использованием метода микроразбавления, описанного в "Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically", Approved standard, 7 ed., Clinical and Laboratory Standards Institute (CLSI) Document M7-A7, Wayne, PA, USA, 2006.
Результаты
Соединения всех примеров протестированы относительно воздействия на некоторые грамположительные и грамотрицательные бактерии такие, как S. aureus, E.faecalis, S. pneumoniae, M. catarrhalis, A. baumanii, E.coli или Р. aeruginosa. Результаты антибактериальных тестов представлены в нижеприведенной таблице (MIC в мг/л).
ДОПОЛНИТЕЛЬНЫЕ ЭКСПЕРЕМЕНТАЛЬНЫЕ ДАННЫЕ
Пример 26: 6-метокси-4-(1-{2-[(S)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]-тиазин-6-ил)оксазолидин-5-ил]этил)пиперидин-4-ил)-1,4-дигидро-2H-пиридо[2,3-b]пиразин-3-он
К охлажденному до 0°C раствору соединения по примеру 23 (80,5 мг) в ДХМ/метаноле 3:1 (4,8 мл) добавляли NaBH4 (11,3 мг). Реакционную смесь перемешивали при комнатной температуре в течение 8 ч. Смесь разделяли между ДХМ/метанолом в соотношении 9:1 и 1% водным раствором NH4OH. Органическую фазу промывали рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали помощью хроматогорафии на колонке (ДХМ/метанол+NH4OH 1%), получая после перемешивания остатка в эфире, указанное в заголовке соединение в форме твердого вещества белого цвета (53 мг; выход 66%).
МС (ЭСИ, m/z): 539,2 [M+H+].
Пример 21: 6-метокси-4-(1-{2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]-тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-1,4-дигидро-2H-пиридо[2,3-b]пиразин-3-он
Взяв соединение по примеру 25 (53,7 мг) и используя ту же методику, как для приготовления соединения по примеру 26, получали указанное в заголовке соединение в форме твердого вещества белого цвета (30 мг; выход 56%).
МС (ЭСИ, m/z): 539,2 [M+H+].
Пример 28: 6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил)-1,4-дигидро-2H-пиридо[2,3-b]пиразин-3-он
Взяв соединение по примеру 15 (56 мг) и используя ту же методику, как для приготовления соединения по примеру 26, получали указанное в заголовке соединение в виде твердого вещества грязно-белого цвета (29 мг; выход 51%).
МС (ЭСИ, m/z): 525,2 [M+H+].
Пример 29: 6-{(S)-5-[4-(6-метокси-3-оксо-3H-пиридо[2,3-b]пиразин-4-ил)-пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-пиридо[3,2-b][1,4]тиазин-3-он
Взяв промежуточное соединение 15.v (78 мг) и 6-[(5R)-5-[[(метилсульфонил)окси]-метил]-2-оксо-3-оксазолидинил]-2H-пиридо[3,2-6]-1,4-тиазин-3(4H)-он (108 мг; приготовленн согласно документу WO 2010/041194) и используя процедуру С, получили указанное в заголовке соединение в виде бесцветного твердого вещества (18 мг, выход 11%).
МС (ЭСИ, m/z): 524,0 [M+H+].
Пример 30: 6-{(S)-5-[4-(6-метокси-3-оксо-3H-пиридо[2,3-b]пиразин-4-ил)-пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-пиридо[3,2-b][1,4]оксазин-3-он
Взяв промежуточное соединение 15.v (78 мг) и 6-[(5R)-5-[[(метилсульфонил)окси]-метил]-2-оксо-3-оксазолидинио]-2Н-пиридо[3,2-b]-1,4-оксазин-3(4Н)-он (103 мг; приготовлен согласно документу WO 2013/021363) и используя процедуру С, получали указанное в заголовке соединение в в виде бесцветного твердого вещества (25 мг, выход 16%).
1Н ЯМР (ДМСО-d6) δ: 8,09 (m, 1H); 8,03 (m, 1H); 7,58 (m, 1H); 7,42 (m, 1H); 6,82 (m, 1H); 5,25 (m, 1H); 4,82 (m, 1H); 4,59 (m, 2H); 3,97 (m, 3H); 3,85 (m, 1H); 3,36 (m, 1H); 3,04 (m, 2H); 2,87 (m, 2H); 2,73 (m, 2H); 2,26 (m, 2H); 1,58 (m, 2H).
МС (ЭСИ, m/z): 508,1 [M+H+].
Пример 31: 6-(((R)-5-{2-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]-этил}-2-оксооксазолидин-3-ил)-4H-пиридо[3,2-b][1,4]оксазин-3-он
31.i. Этиловый эфир (6-{(R)-5-[2-(трет-бутилдиметилсиланилокси)этил]-2-оксо-оксазолидин-3-ил}-2-нитропиридин-3-илокси)уксусной кислоты:
Суспензия K2CO3 (11,26 г), CuI (388 мг), (R)-5-[2-(трет-бутилдиметилсиланилокси)-этил]оксазолидин-2-она (10,0 г; приготовлен согласно документу WO 2009/104159), этилового эфира (6-бромо-2-нитропиридин-3-илокси)уксусной кислоты (12,43 г; приготовлен согласно документу WO 2004/002992) и N,N-диметилэтилендиамина (0,92 мл) в диоксане (305 мл) дегазировали путем барботирования аргоном и дефлегмировали при 100°C в течение ночи. Полученную темно-коричневую смесь фильтровали через диатомит (Celite), фильтрат испаряли при пониженном давлении и остаток очищали с помощью хроматографии на колонке (Hept/EA 2:1 до 0:1), получая твердое вещество бежевого цвета (16,0 г; выход 84%).
МС (ЭСИ, m/z): 470,3 [M+H+].
31.ii. 6-[(R)-5-(2-гидроксиэтил)-2-оксооксазолидин-3-ил]-4Н-пиридо[3,2-b][1,4]оксазин-3-он:
Суспензию хлорида аммония (13,67 г) и железа (8,56 г) в смеси MeOH/вода (204 мл; 1:1) нагревали до 50°C, затем к суспензии по каплям добавляли раствор промежуточного соединения 31 л (16,0 г) в MeOH (360 мл) и дополнительно перемешивали при 68°C в течении 2,5 ч. Горячую суспензию фильтровали через подложку из диатомита (Celite). Фильтрат разбавляли уксусной кислотой (112 мл) и перемешивали при 95°C в течении 2 ч. Реакционной смеси давали остыть до комнатной температуры и концентрировали при пониженном давлении. Суспензию охлаждали до 0°C, фильтровали и собирали твердую фракцию путем фильтрации, получая твердое вещество бежевого цвета (9,54 г; выход 100%).
МС (ЭСИ, m/z): 280,1 [M+H+].
31.iii. 2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-6-ил)-оксазолидин-5-ил]этиловый эфир метансульфоновой кислоты:
Суспензию промежуточного соединения 31.ii (4,2 г) в ДХМ (65 мл) охлаждали до -40°C и обрабатывали метансульфонилхлоридом (1,6 мл) в течение 1 ч.
Реакционную смесь разбавляли насыщенным водным раствором NaHCO3 и водную фазу экстрагировали ДХМ. Комбинированные органические слои высушивали над MgSO4, концентрировали при пониженном давлении и осадок растирали в ТБМЭ/ДХМ/метаноле, получая твердое вещество розовато-оранжевого цвета (732 мг; выход 17%).
МС (ЭСИ, m/z): 358,2 [M+H+].
31.iv. 6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]-этил}-2-оксооксазолидин-3-ил)-4H-пиридо[3,2-b][1,4]оксазин-3-он:
Взяв промежуточное вещество 2.ii (64 мг) и промежуточное вещество 31.iii (50 мг) и используя процедуру C, получали указанное в заголовке соединение в виде твердого желтоватого вещества (13 мг, выход 18%).
МС (ЭСИ, m/z): 506,0 [M+H+].
Пример 32: 6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]-этил)-2-оксооксазолидин-3-ил)-4H-пиридо[3,2-b)[1,4]тиазин-3-он
Взяв промежуточное вещество 2.ii (61 мг) и 6-[(5R)-5-[2-[(метилсульфонил)окси]-этил]-2-оксо-3-оксазолидинил]-2H-пиридо[3,2-b]-1,4-тиазин-3(4H)-он (50 мг; приготовленный согласно документу WO 2010/041194) и используя процедуру С, получали указанное в заголовке соединение в в виде твердого вещества желтого цвета (4 мг, выход 5%).
МС (ЭСИ, m/z): 522,0 [M+H+].
Пример 33: 6-{(R)-5-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил метил]-2-оксооксазолидин-3-ил}-4H-пиридо[3,2-b][1,4]тиазин-3-он
Взяв промежуточное вещество 2.ii (50 мг) и 6-[(5R)-5-[[(метилсульфонил)окси]-метил]-2-оксо-3-оксазолидинил]-2H-пиридо[3,2-6]-1,4-тиазин-3(4H)-он (64 мг; приготовленный согласно документу WO 2010/041194) и используя процедуру С, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (4 мг, выход 6%).
МС (ЭСИ; m/z): 507,9 [M+H+].
Фармакологические свойства соединений по изобретению Методика эксперимента;
Минимальные ингибирующие концентрации (MIC; мг/л) определяли в катион-регулируемой среде Мюллера-Хинтона (Mueller-Hinton Broth), способом микроразбавления согласно руководству «Методики тестов антимикробной чувствительности с разбавлением для бактерий, растущих в аэробных условиях» ("Procedures for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically"), Утвержденный стандарт, 7-ое изд., Институт клинических и лабораторных стандартов (CLSI) документ М7-А7, Wayne, Пенсильвания, США, 2006.
Результаты:
Протестирована и активность всех соединений против нескольких грамотрицательных и грамположительных видов бактерий, например S.aureus, Е.faecalis, S.pneumoniae, М.catarrhalis, А.baumanii, Е.coli или Р.aeruginosa. Результаты исследования антибактериальной активности против Moraxella catarrhalis А894, Staphylococcus aureus АТСС 29213 и Streptococcus pneumoniae АТСС 49619 приведены в Таблице ниже (MIC в мг/л).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| ТРИЦИКЛИЧЕСКИЕ ОКСАЗОЛИДИНОНОВЫЕ АНТИБИОТИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2530884C2 |
| АНТИБИОТИЧЕСКИЕ ПРОИЗВОДНЫЕ 2-ОКСО-ОКСАЗОЛИДИН-3, 5-ДИИЛА | 2012 |
|
RU2616609C2 |
| ПОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА | 2009 |
|
RU2501799C2 |
| ПРОИЗВОДНЫЕ [4-(1-АМИНОЭТИЛ)ЦИКЛОГЕКСИЛ]МЕТИЛАМИНА И [6-(1-АМИНОЭТИЛ)ТЕТРАГИДРОПИРАН-3-ИЛ]МЕТИЛАМИНА | 2009 |
|
RU2515906C2 |
| ОКСАЗОЛИДИНОВЫЕ АНТИБИОТИКИ | 2008 |
|
RU2470022C2 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОВЫХ АНТИБИОТИКОВ | 2008 |
|
RU2506263C2 |
| ПРОИЗВОДНЫЕ 5-АМИНО-2-(1-ГИДРОКСИЭТИЛ)ТЕТРАГИДРОПИРАНА | 2009 |
|
RU2525541C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МУСКАРИНОВЫЕ АГОНИСТЫ И КОМПОЗИЦИИ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2002 |
|
RU2292346C2 |
| ПРОИЗВОДНЫЕ 2-ГИДРОКСИЭТИЛ-1Н-ХИНОЛИН-2-ОНА И ИХ АЗАИЗОСТЕРИЧЕСКИЕ АНАЛОГИ С АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2540862C2 |
Изобретение относится к соединениям формулы (I), где R1 представляет собой алкоксигруппу или галоген; U и V каждый независимо друг от друга представляет собой СН или N; "----" обозначает связь или отсутствует; W представляет собой СН или N, или если "----" отсутствует, тo W представляет собой СН2 или NH, при условии, что U, V и W не все представляют собой N;
А представляет собой связь или СН2; R2 представляет собой Н, или если А обозначает СН2, то также может представлять собой ОН; m и n каждый независимо друг от друга равен 0 или 1; D представляет собой СН2 или связь; G представляет собой фенильную группу, которая является однократно или дважды замещенной в мета- и/или пара-положении(ях) заместителями, выбранными из алкила, С1-3алкоксигруппы и галогена, или G представляет собой одну из групп G1 и G2: где Z1, Z2 и Z3 каждый представляет собой СН; и Х представляет собой N или СН и Q представляет собой О или S; при этом следует иметь в виду, что если m и n каждый равен 0, тогда А представляет собой СН2; или фармацевтически приемлемой соли такого соединения. Изобретение также относится к фармацевтической композиции для лечения бактериальной инфекции, содержащей в качестве активного компонента соединение формулы (I) или его фармацевтически приемлемую соль, и по крайней мере одно терапевтически инертное вспомогательное вещество. Технический результат - оксазолидиновые соединения, предназначенные для изготовления лекарственного средства для лечения или профилактики бактериальных инфекций. 3 н. и 11 з.п.ф-лы, 8 сх., 2 табл., 33 пр.
R1
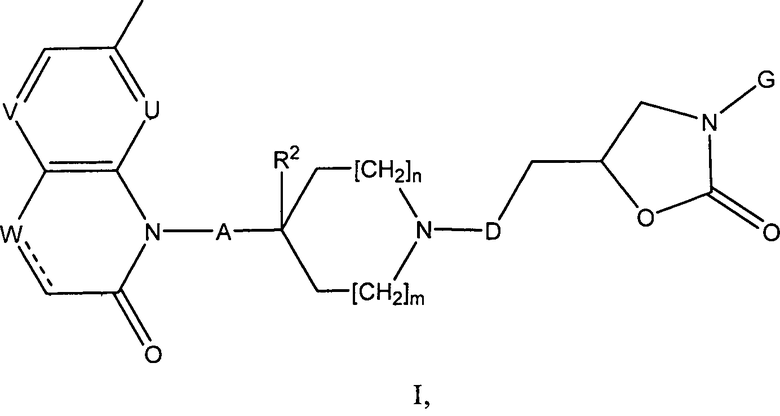
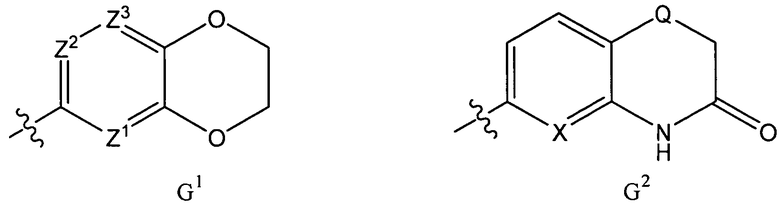
1. Соединение формулы (I)
R1
где
R1 представляет собой алкоксигруппу или галоген;
U и V каждый независимо друг от друга представляет собой СН или N;
"----" обозначает связь или отсутствует;
W представляет собой СН или N, или если "----" отсутствует, тo W представляет собой СН2 или NH,
при условии, что U, V и W не все представляют собой N;
А представляет собой связь или СН2;
R2 представляет собой Н, или если А обозначает СН2, то также может представлять собой ОН;
m и n каждый независимо друг от друга равен 0 или 1;
D представляет собой СН2 или связь;
G представляет собой фенильную группу, которая является однократно или дважды замещенной в мета- и/или пара-положении(ях) заместителями, выбранными из алкила, С1-3алкоксигруппы и галогена, или G представляет собой одну из групп G1 и G2:
где Z1, Z2 и Z3 каждый представляет собой СН; и
Х представляет собой N или СН и Q представляет собой О или S;
при этом следует иметь в виду, что если тип каждый равен 0, тогда А представляет собой СН2;
или фармацевтически приемлемая соль такого соединения.
2. Соединение формулы (I) по п.1, которое также является соединением формулы (ICE)
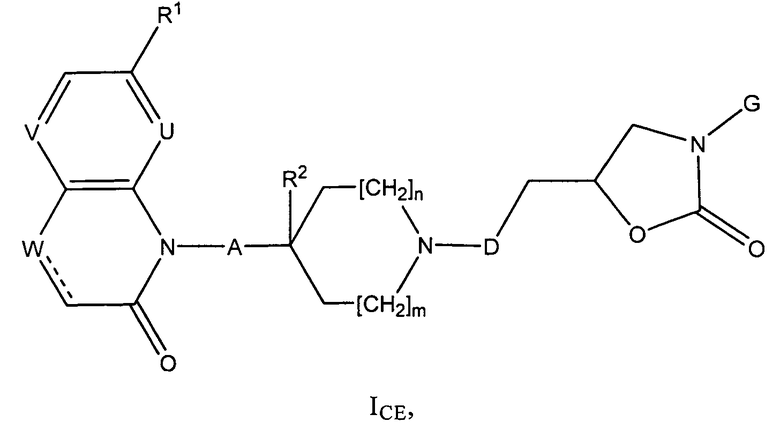
где
R1 представляет собой алкоксигруппу;
V представляет собой СН;
U и W каждый представляет собой СН и "----" обозначает связь, или U представляет собой СН, W представляет собой N и "----" обозначает связь, или U и W каждый представляет собой N и "----" обозначает связь, или U представляет собой СН, W представляет собой СН2 и "----" отсутствует;
А представляет собой связь или СН2;
R2 представляет собой Н, или если А обозначает СН2, то также может представлять собой ОН;
m и n каждый независимо друг от друга равен 0 или 1;
D представляет собой СН2 или связь;
G представляет собой фенильную группу, которая однократно замещена в мета-положении и однократно замещена в пара-положении заместителями, выбранными из алкила и галогена, или G представляет собой одну из групп G1 и G2,:
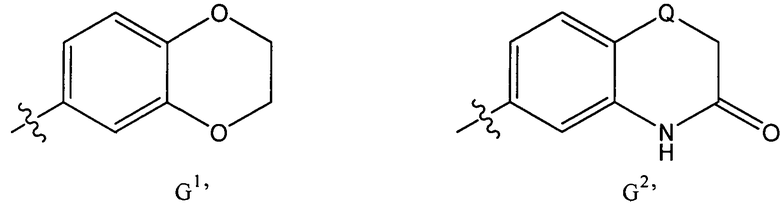
где Q представляет собой О или S;
при этом следует иметь в виду, что если тип каждый равен 0, тогда А представляет собой СН2;
или фармацевтически приемлемая соль такого соединения.
3. Соединение формулы (I) по п.1, где R1 представляет собой метоксигруппу; или фармацевтически приемлемая соль такого соединения.
4. Соединение формулы (I) по п.3, где "----" обозначает связь; или фармацевтически приемлемая соль такого соединения.
5. Соединение формулы (I) по п.3, где "----" отсутствует; или фармацевтически приемлемая соль такого соединения.
6. Соединение формулы (I) по п.4, где А представляет собой связь; или фармацевтически приемлемая соль такого соединения.
7. Соединение формулы (I) по п.4, где А представляет собой СН2; или фармацевтически приемлемая соль такого соединения.
8. Соединение формулы (I) по п.7, где R2 представляет собой ОН;
или фармацевтически приемлемая соль такого соединения.
9. Соединение формулы (I) по одному из п.п.1-8, где G представляет собой группу формулы
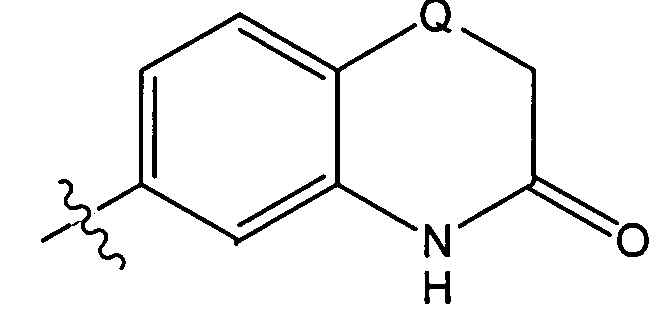
где Q представляет собой О или S,
или фармацевтически приемлемая соль такого соединения.
10. Соединение формулы (I) по п.1, которое выбрано из следующих соединений:
6-{(R)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1H-хинолин-2-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хинолин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-(7-метокси-2-оксо-2H-хиноксалин-1-илметил)азетидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[4-(7-метокси-2-оксо-2H-хинолин-1-илметил)пиперидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-илметил}-7-метокси-1H-хинолин-2-он;
1-{1-[(R)-3-(3-фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]-пиперидин-4-илметил}-7-метокси-1H-хинолин-2-он;
6-{(R)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)-азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)-азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-3,4-дигидро-2H-хинолин-1-илметил)-азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он;
6-{(R)-5-[3-(7-метокси-2-оксо-2H-хиноксалин-l-илметил)азетидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]оксазин-3-он;
1-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]азетидин-3-илметил}-7-метокси-1H-хиноксалин-2-он;
6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4H-пиридо[2,3-b]пиразин-3-он;
6-метокси-4-{1-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-6-ил)оксазолидин-5-илметил]пиперидин-4-ил}-4H-пиридо[2,3-b]пиразин-3-он;
6-{(R)-5-[(R)-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(R)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)-пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-{(S)-5-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)-пирролидин-1-илметил]-2-оксооксазолидин-3-ил}-4H-бензо[1,4]тиазин-3-он;
6-((S)-5-{2-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)-пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
6-((R)-5-{2-[3-гидрокси-3-(7-метокси-2-оксо-2H-хинолин-1-илметил)-пирролидин-1-ил]этил}-2-оксооксазолидин-3-ил)-4H-бензо[1,4]тиазин-3-он;
4-{1-[(R)-3-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-оксооксазолидин-5-илметил]пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-b]пиразин-3-он;
6-метокси-4-(1-{2-[(S)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-b]пиразин-3-он;
4-{1-[(R)-3-(3-фтор-4-метилфенил)-2-оксооксазолидин-5-илметил]-пиперидин-4-ил}-6-метокси-4H-пиридо[2,3-b]пиразин-3-он;
6-метокси-4-(1-{2-[(R)-2-оксо-3-(3-оксо-3,4-дигидро-2H-бензо[1,4]тиазин-6-ил)оксазолидин-5-ил]этил}пиперидин-4-ил)-4H-пиридо[2,3-b]пиразин-3-он;
или фармацевтически приемлемая соль такого соединения.
11. Соединение формулы (I) по одному из п.п.1-10 или его фармацевтически приемлемая соль в качестве лекарственного средства для лечения бактериальной инфекции.
12. Фармацевтическая композиция для лечения бактериальной инфекции, содержащая в качестве активного компонента соединение формулы (I) по одному из п.п.1-10 или его фармацевтически приемлемую соль, и по крайней мере одно терапевтически инертное вспомогательное вещество.
13. Применение соединения формулы (I) по одному из п.п.1-10 или его фармацевтически приемлемой соли для изготовления лекарственного средства для профилактики или лечения бактериальной инфекции.
14. Соединение формулы (I) по одному из п.п.1-10 или его фармацевтически приемлемая соль, предназначенное для профилактики или лечения бактериальной инфекции.
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ 5-HT, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2196140C2 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

