ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к кристаллическим солям ситаглиптина, ингибитора дипептидилпептидазы-IV, полученным прибавлением одноосновных, двухосновных и трехосновных кислот.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Фермент дипептидилпептидаза-IV (ДПП-IV) (dipeptidyl peptidase-IV, DPP-IV) ответственен за деградацию инкретинов, таких как глюкагон-подобный пептид-I (ГПП-I) (glucagon-like peptide-1, GLP-I) и гастроингибиторный полипептид (ГИП) (gastric inhibitory polypeptide, GIP), также известный как глюкозозависимый инсулинотропный пептид (ГИП) (glucose-dependent insulinotropic peptide).
Ингибирование фермента ДПП-IV представляет собой новый подход в лечении сахарного диабета 2 типа, также известного как инсулиннезависимый сахарный диабет (ИНЗСД) (non-insulin dependent diabetes mellitus, NIDDM).
Новый терапевтический подход ранее был описан в научной литературе: C.F. Deacon and J.J. Hoist, "Dipeptidyl peptidase IV inhibition as an approach to the treatment and prevention of Type 2 diabetes: a historical perspective" - [«Ингибирование дипептидилпептидазы-IV как подход для лечения и профилактики диабета 2 типа: историческая перспектива»], Biochem. Biophys. Res. Commun., 294 (2000) 1-4; K. Augustyns, et al., "Dipeptidyl peptidase IV inhibitors as new therapeutic agents for the treatment of Type 2 diabetes" - [«Ингибиторы дипептидилпептидазы-IV как новые терапевтические средства для лечения диабета 2 типа»]. Expert. Opin. Ther. Patents, 13 (2003) 499-510; D.J. Drucker, "Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of Type 2 diabetes", [«Терапевтический потенциал ингибиторов дипептидилпептидазы-IV для лечения диабета 2 типа»], Expert Opin. Investig. Drugs, 12 (2003) 87-100, и М.A. Nauck et al., "Incretins and Their Analogues as New Antidiabetic Drugs" - [«Инкретины и их аналоги в качестве новых антидиабетических лекарственных средств»]. Drug News Perspect., 16 (2003) 413-422.
В EP 0896538 описывается применение ингибитора ДПП-IV для снижения уровня глюкозы в крови у млекопитающих.
В WO 2003/004498 сообщается о бета-амино-тетрагидроимидазо-(1,2а)-пиразине и бета-амино-тетрагидроимидазоло-(4,3а)-пиразине в качестве ингибиторов дипептидилпептидазы-IV для лечения и профилактики диабета.
Одним из этих пиразинов является ситаглиптин (7-[(3R)-3-амино-1-оксо-4-(2,4,5-трифторфенил)бутил]-5,6,7,8-тетрагидро-3-(трифторметил)-1,2,4-триазоло[4,3-а]пиразин) со следующей структурной формулой:
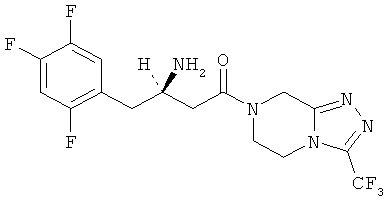
Перечень фармацевтических приемлемых солей большей частью включен в WO 03/004498. Кристаллические соли, приведенные в данном изобретении, в нем не раскрыты.
WO 2005/072530 раскрывает конкретные кристаллические соли ситаглиптина с соляной кислотой, бензолсульфоновой кислотой, п-толуолсульфоновой кислотой, D- и L-винной кислотой и (1S)-(+)- and (1R-(-)-10-камфорсульфоновой кислотой.
Д. Ким с сотр. (D. Kim et al.) упоминает в публикации J. Med. Chem. 48 (2005) 141-151, что ситаглиптин фумарат со стехиометрией 2:1, применялся в исследованиях in-vivo.
В WO 2005/003135 описан ситаглиптин дигидрогенфосфат и его кристаллический моногидрат. Четыре кристаллические полиморфные формы безводного ситаглиптина дигидрогенфосфата раскрыты в WO 2005/020920 и WO 2005/030127. Аморфный ситаглиптин дигидрогенфосфат заявлен в WO 2006/033848.
Поскольку соли ситаглиптина из предшествующего уровня техники имеют ограниченную стабильность, то, следовательно, при создании твердых лекарственных форм может, в определенной степени, наблюдаться разложение. Кроме того, многие соли обладают нежелательной гигроскопичностью.
В фармацевтической промышленности имеется потребность в стабильных солях ситаглиптина, которые обладают хорошей стабильностью и могут подвергаться технологической обработке даже после длительных сроков хранения. В твердых лекарственных формах соли должны быть стабильны, предпочтительно они должны показывать большую стабильность, чем соли, описанные в предшествующем уровне техники, и меньшую гигроскопичность.
Поэтому цель настоящего изобретения состоит в создании ситаглиптина в форме, имеющей хорошую химическую и/или физическую стабильность, низкую гигроскопичность, хорошую растворимость, хорошую биодоступность и/или имеющей хорошую технологичность, как во время получения, так и при приготовлении фармацевтических композиций, содержащих ситаглиптин.
К настоящему времени установлено, что одну или несколько из вышеперечисленных проблем можно решить получением ситаглиптина в форме кристаллической соли одноосновной, двухосновной или трехосновной кислоты. Таким образом, настоящее изобретение относится к кристаллической соли ситаглиптина одноосновной, двухосновной или трехосновной кислоты.
Кристаллическая соль, описанная в данном изобретении, может быть без растворителя или может содержать молекулы растворителя в ее кристаллической структуре. Таким образом, настоящее изобретение также охватывает любую гидратную или безводную или сольватную форму кристаллической соли ситаглиптина.
Кислота для кристаллической соли, описанной в данном изобретении, может быть выбрана, например, из HCl, H2SO4, H3PO4, сульфокислот, таких как метансульфокислота, а также моно-, ди- и трикарбоновых кислот. В одном из вариантов кислота не является H3PO4.
Если кислота является карбоновой кислотой, она может иметь общую структурную формулу R1-COOH, где R1 представляет собой атом водорода, карбоксильную группу, C1-4 алкильную группу или C2-4 алкенильную группу, где C1-4 алкильная группа и C2-4 алкенильная группа возможно могут замещаться 1-2-карбоксильными группами, 1-3 гидроксильными группами, 1-3 аминогруппами, 1-3 фенильными группами и/или 1-3 C1-5 алкильными группами.
Любая из вышеприведенных алкильных и алкенильных групп может быть с прямой цепью или разветвленной цепью, или, если она содержит по меньшей мере три атома углерода, может образовать кольцо. Алкильные группы могут, например, быть метальными, этильными, пропильными, н-бутильными, трет-бутильными и пентильными группами.
Предпочтительными кислотами, отвечающими вышеприведенной общей структурной формуле R1-COOH, являются такие, в которых R1 представляет C2 алкильную группу или С2 алкенильную группу, каждая из которых замещается одной карбоксильной группой и возможно одной гидроксильной группой или аминогруппой. Винная кислота предпочтительно не применяется.
Примерами вышеописанных карбоновых кислот являются фумаровая кислота, малоновая кислота, яблочная кислота, янтарная кислота, молочная кислота, гликолевая кислота, малеиновая кислота, лимонная кислота, аспарагиновая кислота и миндальная кислота.
Примерами дикарбоновых кислот являются янтарная кислота, малоновая кислота и фумаровая кислота, D- и L-винная кислота, D- и L-яблочная кислота и L-аспарагиновая кислота. Одним из примеров трехосновной кислоты является лимонная кислота. Примерами монокарбоновых кислот являются гликолевая кислота и молочная кислота.
Еще другой вариант данного изобретения относится к соли ситаглиптина, в которой соотношение ситаглиптина и кислоты на основе стехиометрии практически составляют 1:1, где двухосновная или трехосновная кислота только однократно депротонирована.
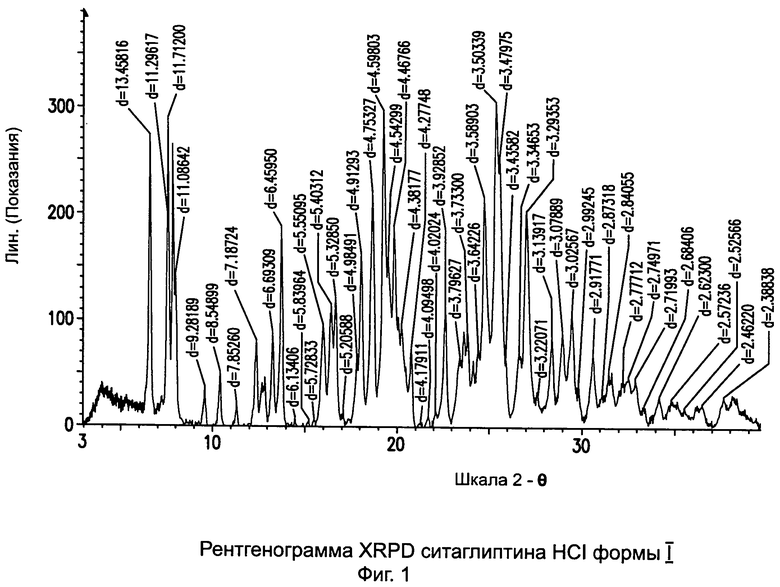
В одном варианте данного изобретения описана новая форма ситаглиптина гидрохлорида. Новая полиморфная форма ситаглиптина гидрохлорида характеризуется дифференциальной сканирующей калориметрией (DSC) (differential scanning calorimetry, DSC) и дифракцией рентгеновских лучей (XRD) (X-ray diffraction, XRD). DSC показывает начало температуры фазового перехода при 195,8°С±2°C и пик точки плавления при 202,3°C±2°C, измеренной с помощью метода DSC. Далее соль ситаглиптина гидрохлорида, описанная в данном изобретении, показывает pH 5,8±0,1 в 2%-ном водном растворе. Кроме того, новая форма соли ситаглиптина гидрохлорида не гигроскопична даже при длительном хранении при 93% относительной влажности. Кроме того, соль ситаглиптина гидрохлорида, описанная в данном изобретении, показывает отличную стабильность при хранении при 60°C в течение 4 недель.
В другом варианте осуществления данного изобретения описан гемисульфат ситаглиптина. Отношение ситаглиптина к серной кислоте составляет 2:1. Ситаглиптин гемисульфат формы II характеризуется DSC и XRD. DSC показывает начало фазового перехода при 169,3°C±2°C и пик точки плавления при 175,9°C±2°C, измеренной с помощью метода DSC. Кроме того, соль ситаглиптина гемисульфата, описанная в данном изобретении, показывает отличную стабильность при хранении при 60°C в течение 4 недель. Сульфатная соль показывает очень высокую растворимость в воде по сравнению с другими известными солями ситаглиптина.
Конкретные полиморфные формы или кристаллические соли ситаглиптина, описанные в данном изобретении, приведены в примерах. Эти полиморфные формы характеризуются данными DSC и/или данными порошковой рентгеновской дифракции XRPD. В одном из вариантов эти полиморфные формы характеризуются тремя характеристическими пиками в рентгенограмме XRPD при углах 2θ (2 тета), приведенными в формуле изобретения для каждой полиморфной формы. В другом варианте эти полиморфные формы характеризуются углами 2 θ пяти пиков в рентгенограмме XRPD, имеющих самые высокие интенсивности. Наиболее предпочтительно, когда полиморфные формы характеризуются рентгенограммой XRPD, как показано на фигурах.
Другим аспектом данного изобретения является фармацевтический состав, содержащий одну или несколько солей, описанных в данном изобретении, и фармацевтически приемлемый носитель или разбавитель.
Другим аспектом данного изобретения является фармацевтический состав, содержащий комбинацию одной или нескольких солей, описанных в данном изобретении, и одного или нескольких активных фармацевтических ингредиентов для одновременного, раздельного или последовательного применения в терапии. Примерами активных фармацевтических ингредиентов являются средства против диабета, например, метформин, пиоглитазон или розиглитазон.
Еще одним вариантом данного изобретения является применение солей ситаглиптина для получения лекарственного препарата для лечения или профилактики неинсулинозависимого сахарного диабета, ожирения, инсулинорезистентности, синдрома X и диабета 2 типа.
Другим объектом данного изобретения является способ приготовления соли ситаглиптина, описанной в данном изобретении, включающий стадии:
I) образование раствора или дисперсии, содержащей ситаглиптин и растворитель, при необходимости нагревание раствора,
II) прибавление кислоты,
III) стимулирование кристаллизации соли ситаглиптина, возможно, прибавлением кристаллов для затравки или анти-растворителя или охлаждением раствора,
IV) выделение кристаллов, и, возможно,
V) сушки кристаллов.
Соли ситаглиптина, описанные в данном изобретении, и их сольваты, гидраты и полиморфные формы обладают улучшенными свойствами по сравнению со свободным основанием. Соли ситаглиптина, описанные в данном изобретении, более стабильны и имеют улучшенное качество по сравнению со свободным основанием, что является преимуществом при хранении сырья, а также для распределения конечного продукта.
Кроме того, соли, описанные в данном изобретении, проявляют более высокую растворимость в водном растворе, в частности, при физиологических условиях, по сравнению со свободным основанием или фосфатной солью. Эти свойства являются полезными, потому что растворение происходит более быстро, и меньшее количество воды требуется для полного растворения. Это может привести к более высокой биодоступности по сравнению со свободным основанием, особенно в случае твердых лекарственных форм.
По сравнению с солями ситаглиптина предшествующего уровня техники новые соли ситаглиптина проявляют более низкую гигроскопичность. Это приводит к улучшенной стабильности в связи с пониженным разложением, вызванным гидролизом. В фармацевтическом секторе существует постоянная потребность в солях ситаглиптина с улучшенной стабильностью, которые могут далее подвергаться технологической обработке в фармацевтические составы. В твердых фармацевтических составах эти соли должны быть по меньшей мере такими же стабильными, как соли из предшествующего уровня техники, и показывать низкую гигроскопичность.
Кроме того, соли ситаглиптина, описанные в данном изобретении, имеют меньшую тенденцию к разложению при гидролизе. Как правило, водный раствор 0,020 кг ситаглиптина в 1 литре воды показывает pH 4-7, предпочтительно 4,5-6,0.
Соли ситаглиптина, описанные в данном изобретении, имеют улучшенную растворимость и преимущественно подходят для прямого прессования. Вследствие их превосходной кристалличности, соли, описанные в данном изобретении, особенно пригодны для приготовления таблеток с хорошей растворимостью.
Соли ситаглиптина, описанные в данном изобретении, имеют очень высокую степень хиральной чистоты, причем степень хиральной чистоты, как правило, составляет от 99% до 100%.
Соли ситаглиптина, описанные в данном изобретении, можно приготовить из свободного основания методами, известными специалисту в данной области.
Как правило, соли можно приготовить по реакции свободного основания с соответствующей кислотой в воде или подходящих органических растворителях или в смеси воды и одного или нескольких подходящих органических растворителей.
Кроме того, соли ситаглиптина, описанные в данном изобретении, можно приготовить путем взаимодействия соли ситаглиптина, соответствующей первой кислоте, со второй кислотой, при условии, что вторая кислота сильнее и вытесняет в свободном виде первую, более слабую кислоту.
Примерами неводных растворителей являются простые эфиры, предпочтительно диэтиловый эфир, сложные эфиры, предпочтительно этилацетат, спирты, предпочтительно этанол и изопропанол, и ацетонитрил. Предпочтительными органическими растворителями являются такие, которые по меньшей мере частично смешиваются с водой. Примерами таких растворителей являются спирты, например, метанол, этанол, н-пропанол, изо-пропанол и бутанол, кетоны, предпочтительно ацетон или метилэтилкетон, ацетонитрил, диметилформамид (ДМФ) и диметилсульфоксид (ДМСО).
В случае смесей воды и органических растворителей доля органических растворителей составляет от 10% до 90%, предпочтительно от 20% до 70%, наиболее предпочтительно от 30% до 50%. В случае высших спиртов, которые являются менее полярными растворителями и лишь частично смешиваются с водой, процент органического растворителя может быть ниже.
В предпочтительном варианте кристаллизация стимулируется прибавлением по меньшей мере одного затравочного кристалла. Для того чтобы достичь количественной кристаллизации раствор можно охладить.
Еще другой вариант изобретения включает применение анти-растворителя. При прибавлении анти-растворителя растворимость соли в определенном растворителе снижается. Примерами анти-растворителей являются C3-7 алкилнитрилы, в частности, ацетонитрил, сложные эфиры, C1-5 алкилэфиры C2-7 алкилуглекислоты, в частности, этиловый эфир уксусной кислоты или изопропиловый эфир уксусной кислоты, ди-(C1-C5-алкиловый)-эфир, например, метил-трет-бутиловый эфир (МТБЭ) и тетрагидрофуран (ТГФ), C5-C8-алканы, в частности, пентан, гексан или гептан. МТБЭ особенно предпочтителен.
Эти соли могут также иметь преимущество в том, что они более эффективны, менее токсичны, обладают более длительным действием, имеют более широкий интервал активности, более мощные, дают меньшие побочные эффекты, более легко усваиваются, чем соли ситаглиптина, известные из предшествующего уровня техники, или имеют другие полезные фармакологические свойства по сравнению с последними. Такие преимущества особенно могут встречаться во время комбинированной терапии с другим активным ингредиентом, например вторым средством против диабета, таким как метформин, пиоглитазон или розиглитазон, или антигипертензивным средством, таким как валсартан или в комбинации со статинами.
Соли, описанные в данном изобретении, могут не содержать растворителей, преимущественно не содержать воды.
В еще другом варианте соли ситаглиптина, описанные в данном изобретении, находятся в форме гидратов, сольватов, например геми-, моно-, ди-, три-, тетра-, пента- или гекса-сольватов или гидратов, соответственно. Растворители, применяемые для процесса кристаллизации, в частности спирты, предпочтительно метанол или этанол, кетоны, сложные эфиры, предпочтительно этилацетат, могут внедряться в кристаллическую структуру. Предпочтительно, когда растворитель является фармацевтически приемлемым.
В соответствии с другим аспектом изобретения предлагается фармацевтическая композиция, содержащая соль ситаглиптина, описанную в данном изобретении, в смеси с фармацевтически приемлемым наполнителем или носителем. Твердые лекарственные формы для перорального введения включают капсулы, таблетки, драже, порошки и гранулы. В таких твердых лекарственных формах активное соединение, как правило, смешивается с по меньшей мере одним инертным, фармацевтически приемлемым наполнителем или носителем, наполнителем и сухим разбавителем, связующим веществом, увлажнителем, разрыхлителем и смазочным веществом. Предпочтительно, если соединения изобретения могут быть перорально активными, иметь быстронаступающую активность и низкую токсичность. Соединение, описанное в изобретении, предпочтительно находится в форме таблетки, предпочтительно таблетки, получаемой прямым прессованием.
На чертежах изображено:
на Фиг.1 - рентгенограмма XRPD ситаглиптина HCI формы I;
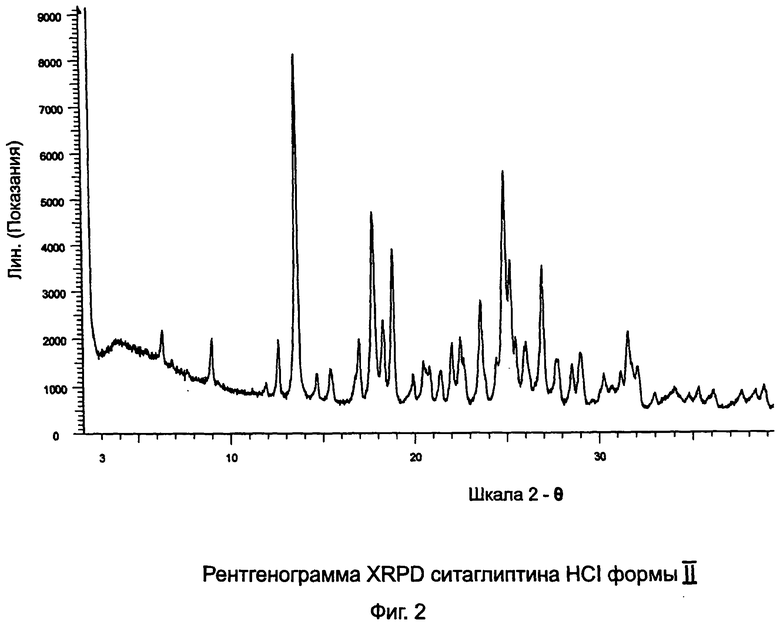
на Фиг.2 - рентгенограмма XRPD ситаглиптина HCI формы II;
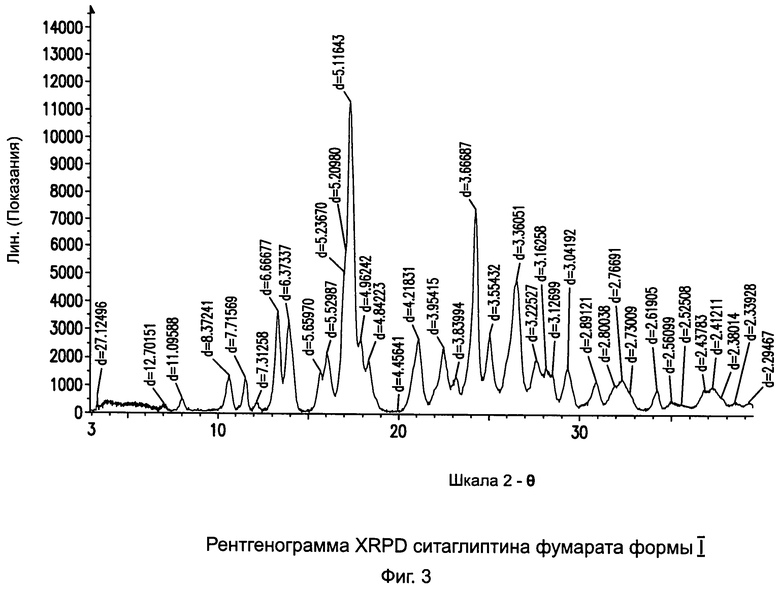
на Фиг.3 - рентгенограмма XRPD ситаглиптина фумарата формы I;
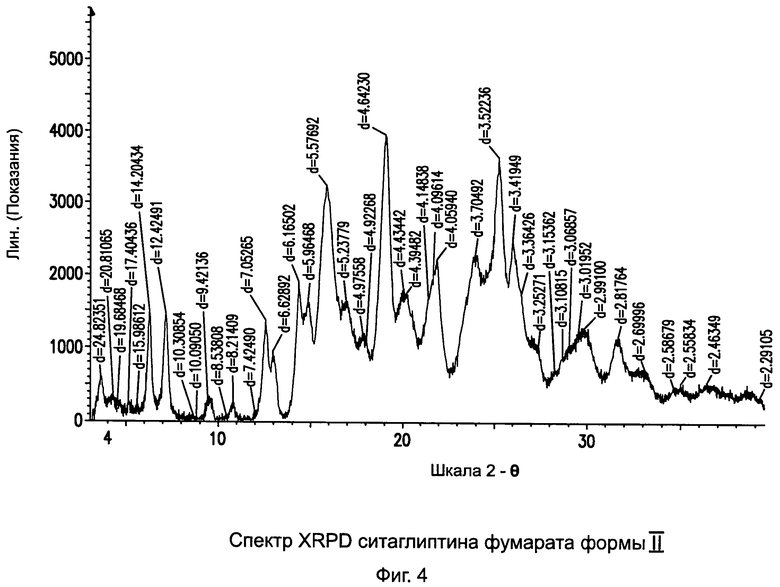
на Фиг.4 - рентгенограмма XRPD ситаглиптина фумарата формы II;
на Фиг.5 - рентгенограмма XRPD ситаглиптина малата;
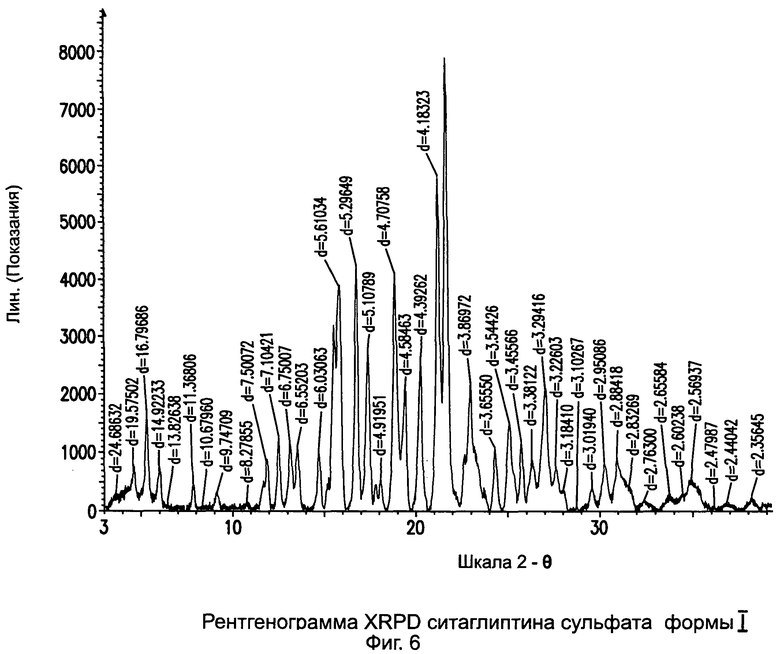
на Фиг.6 - рентгенограмма XRPD ситаглиптина сульфата формы I;
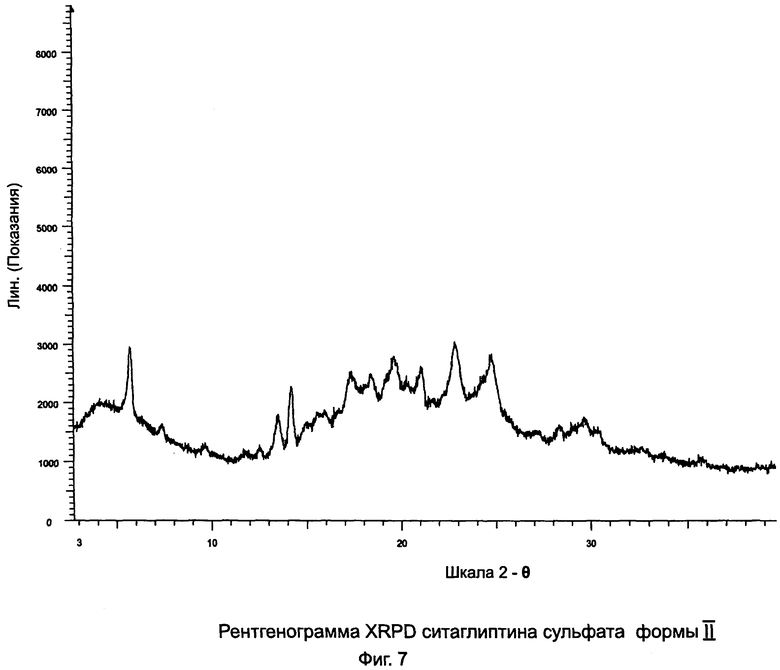
на Фиг.7 - рентгенограмма XRPD ситаглиптина сульфата формы II;
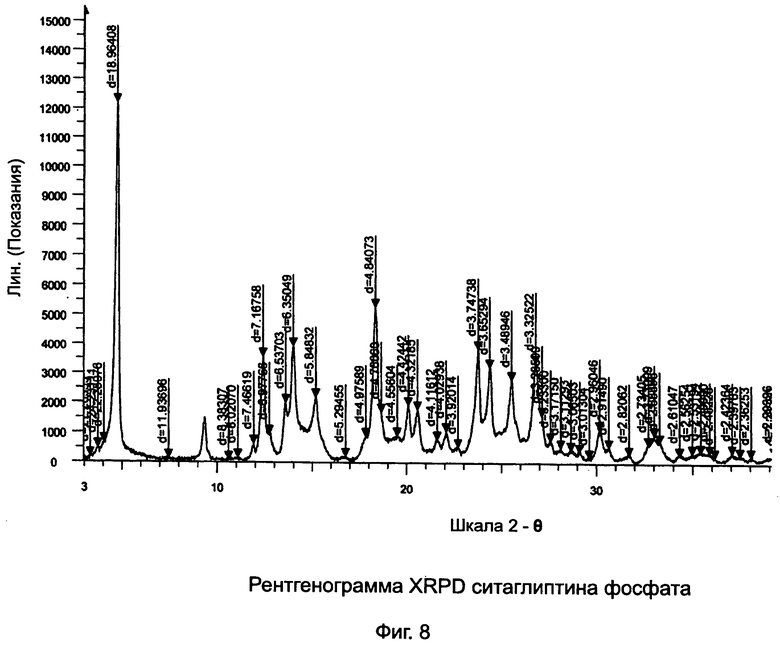
на Фиг.8 - рентгенограмма XRPD ситаглиптина фосфата;
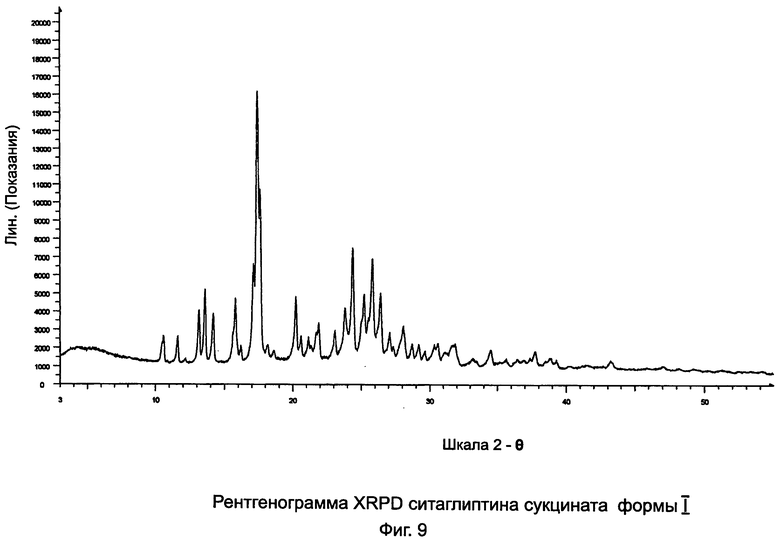
на Фиг.9 - рентгенограмма XRPD ситаглиптина сукцината формы I;
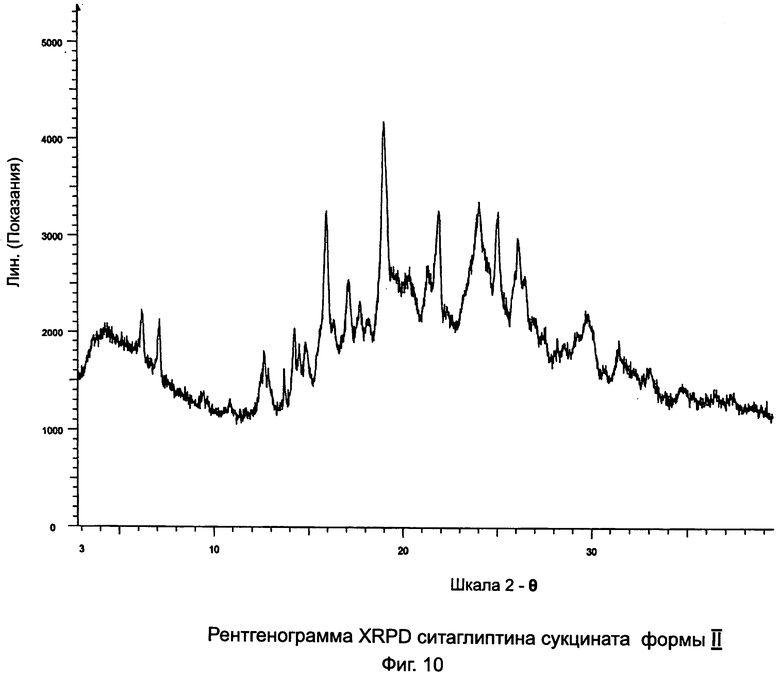
на Фиг.10 - рентгенограмма XRPD ситаглиптина сукцината формы II;
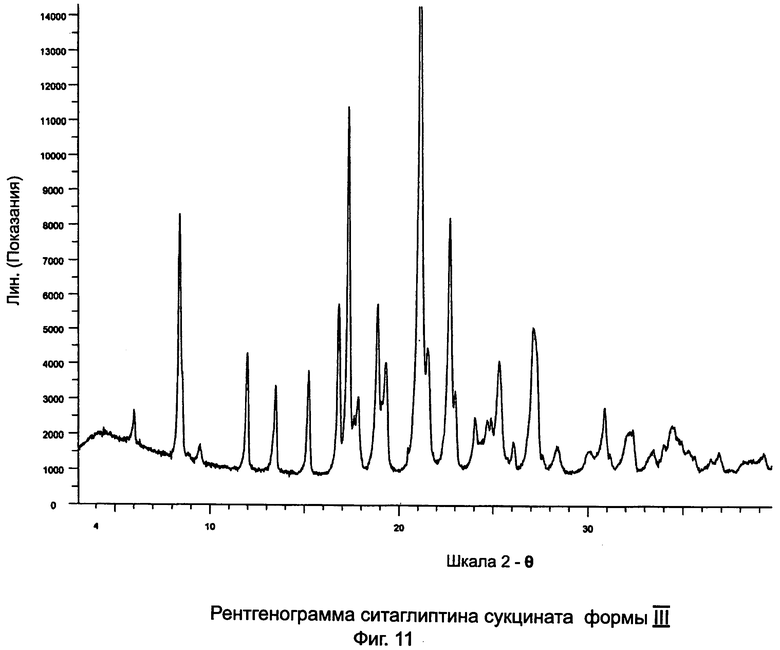
на Фиг.11 - рентгенограмма XRPD ситаглиптина сукцината формы III;
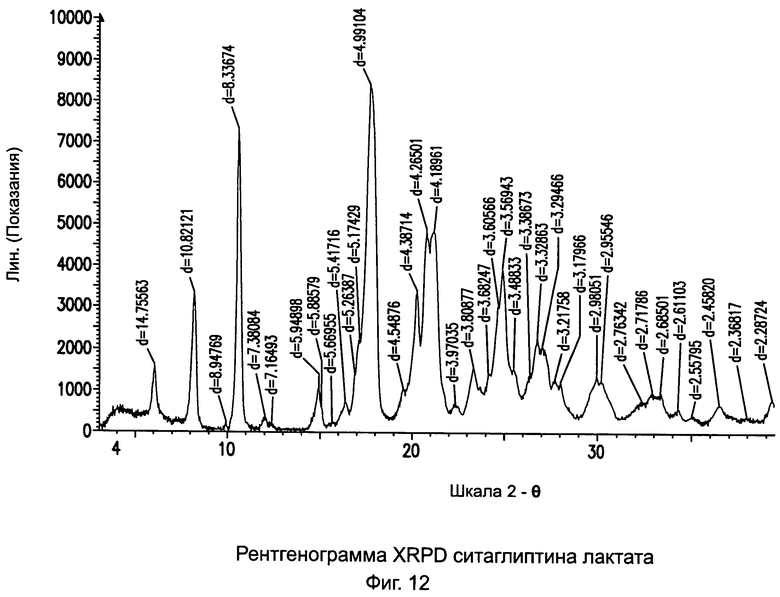
на Фиг.12 - рентгенограмма XRPD ситаглиптина лактата;
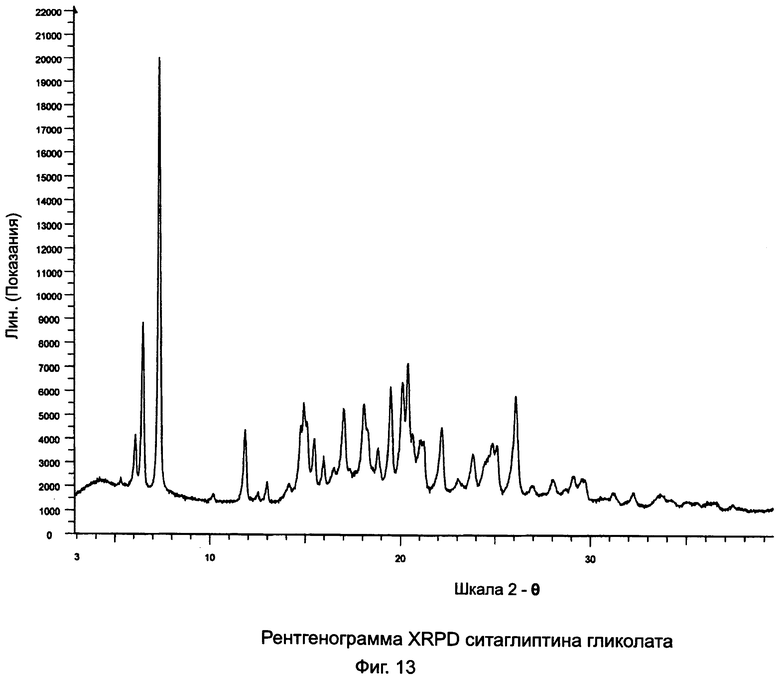
на Фиг.13 - рентгенограмма XRPD ситаглиптина гликолата;
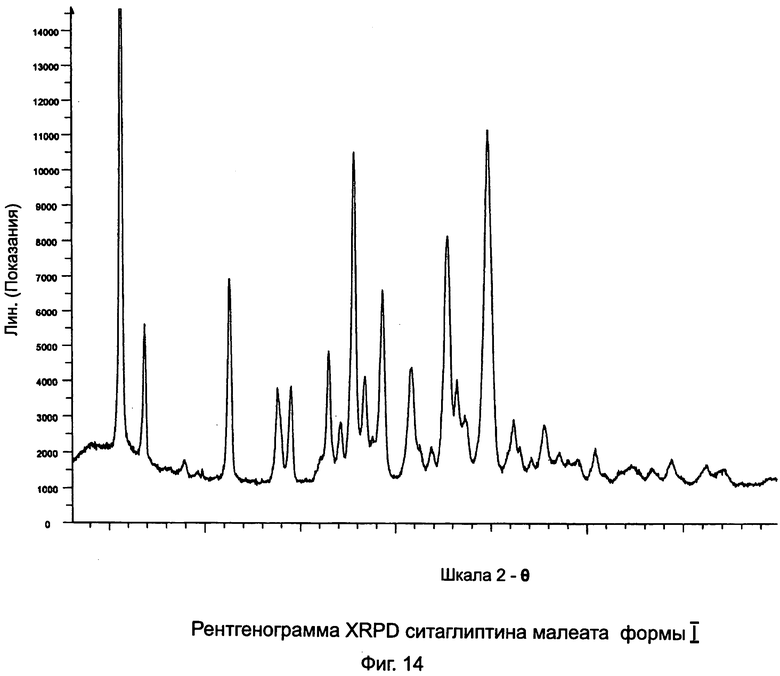
на Фиг.14 - рентгенограмма XRPD ситаглиптина малеата формы I;
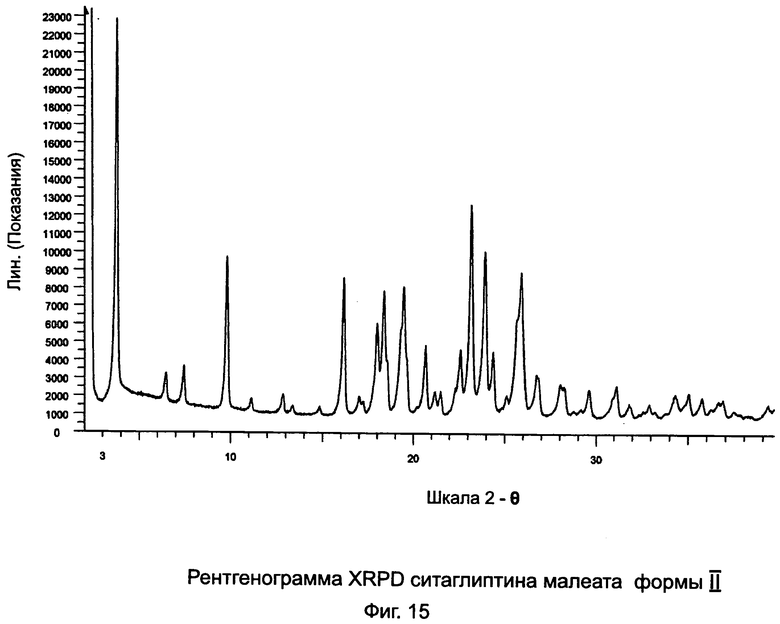
на Фиг.15 - рентгенограмма XRPD ситаглиптина малеата формы II;
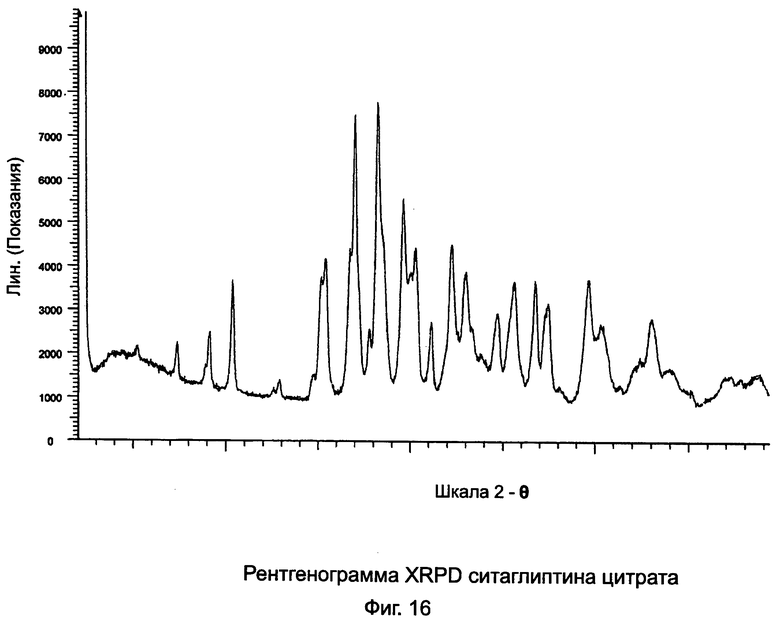
на Фиг.16 - рентгенограмма XRPD ситаглиптина цитрата;
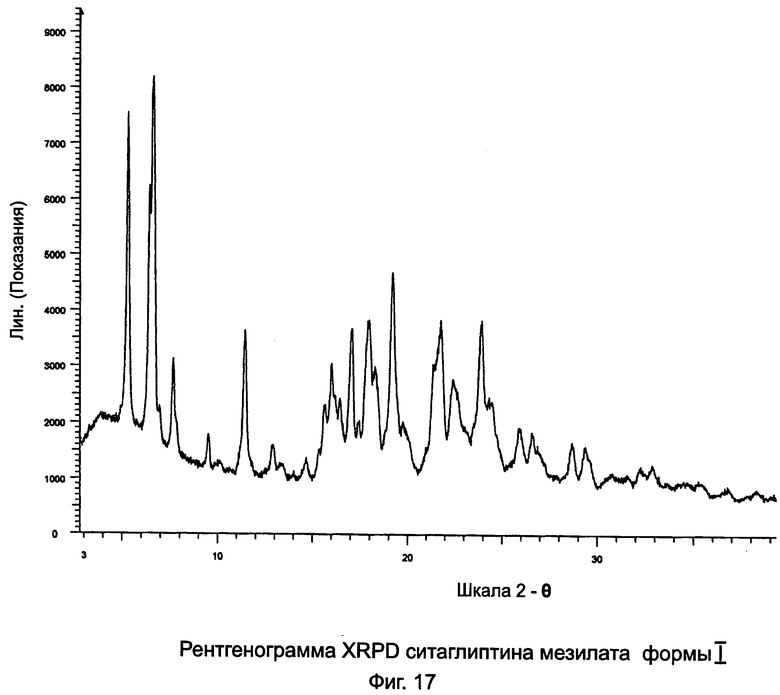
на Фиг.17 - рентгенограмма XRPD ситаглиптина мезилата формы I;
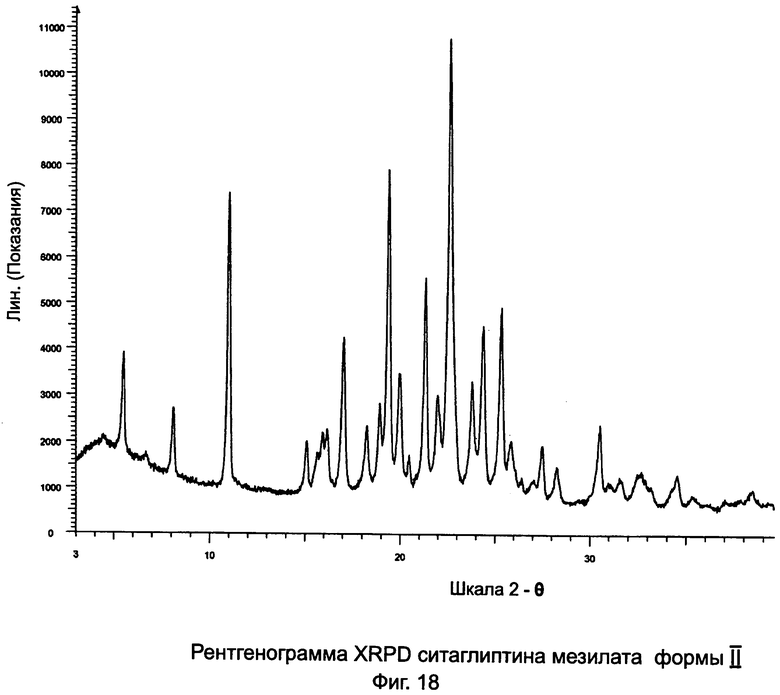
на Фиг.18 - рентгенограмма XRPD ситаглиптина мезилата формы II.
В дальнейшем изобретение будет описано на примерах, которые не следует рассматривать в качестве ограничивающих.
ПРИМЕРЫ
Характеристика образцов
Образцы были охарактеризованы методами рентгеновской порошковой дифракции (XRPD) и дифференциальной сканирующей калориметрии (DSC).
Для характеристики с помощью XRD, образец, в случае необходимости, измельчался в агатовой ступке и должным образом подготавливался (плоская поверхность в zO) на держателе образца, изготовленного из РММА. Затем образцы анализировали на усовершенствованном порошковом рентгеновском дифрактометре Bruker AXS-D8 (Bruker AXS, Карлсруэ, Германия). Держатель образца во время измерения вращался в плоскости zO при 20 об/мин. Условия измерений были следующие: ионизирующее излучение - Cu Ka, источник 40 кВ/40 мА, расходимость щели 0,6 мм, детектор: Vantec-1 антирассеивающая щель 5,59 мм, щель детектора 10,28 мм, начальный угол 2°, конечный угол 55°, шаг 0,016° 2θ. Исходные данные оценивались с помощью программы EVA (Broker AXS, Карлсруэ, Германия).
В таблицах представленных ниже примеров углы 2θ приведены с двумя или более знаками после запятой. Однако, реальная точность измерения меньше, и, поэтому, все углы 2θ должна быть математически округлены до одной десятой. Например, угол 2θ, равный 6,562°, округляется до 6,6°. Кроме того, во всех случаях следует учитывать погрешность ±0,2°. То же самое справедливо для величины d, которая приведена в ангстремах и которая должна округляться до одной десятой и иметь погрешность ±0,2 ангстрем.
Для записи кривых DSC применяли прибор DSC Mettler Toledo 822e. Образец в количестве от 1,5 до 2 мг помещался в ячейку калориметра и нагревался от 30°C до 300°C со скоростью 10°C/мин и в потоке азота - 50 мл/мин. Результаты опытов DSC суммированы в таблицах 25-27.
Пример 1: Ситаглиптин гидрохлорид формы I
В 250 мл двухгорлой круглодонной колбе растворяли свободное основание ситаглиптина (8,0 г, 19,65 ммоль) в 80 мл изопропанола. Реакционную смесь перемешивали на магнитной мешалке с получением прозрачного раствора. В течение 10 минут по каплям прибавляли водный раствор соляной кислоты 2,4 мл (30%) и оставляли перемешиваться при комнатной температуре. В течение следующих 15 минут наблюдали образование густого белого осадка. Для облегчения перемешивания прибавляли дополнительные 40 мл изопропанола в течение еще 20 минут и фильтровали и сушили при температуре 60°C в течение 3 часов при вакууме 5 мбар с получением 8,0 г (выход 92,4%) белого порошка ситаглиптина гидрохлорида. Содержание остаточного растворителя составляло 2,02% изопропанола, что определено с помощью газожидкостной хроматографии (GC).
DSC=109,3±2°C и 171,0±2°C.
Перекристаллизация
Для перекристаллизации часть ситаглиптина гидрохлорида (7,5 г, белый порошок) растворяли в 50 мл этанола при нагревании на водяной бане при 60°C в течение 15-20 минут. Прозрачный раствор выдерживали при комнатной температуре в течение ночи для кристаллизации. В колбе наблюдали образование густого кластера белого осадка и фильтровали через воронку Бюхнера и сушили при температуре 60°C в течение 4 часов под вакуумом 5 мбар на роторном испарителе с получением 6,27 г ситаглиптина гидрохлорида.
DSC: 95,9±2°C, 173,3°С±2°C. Чистота по ВЭЖХ (высокоэффективная жидкостная хроматография) = 99,99%. Удельное оптическое вращение = -24,44°
Маточный раствор вышеописанной реакции выдерживали в течение ночи с получением дополнительного количества кристаллической соли. Ее аналогично фильтровали и сушили и получали еще 0,574 г.
Пример 2: Ситаглиптин гидрохлорид формы II
В двухгорлой колбе растворяли ситаглиптин гидрохлорид (2,0 г, форма I из примера 1) в 125 мл этилацетата и нагревали на масляной бане, которую предварительно нагрели при 125°C в течение приблизительно 30 минут. Раствор становился прозрачным и колбу удаляли из масляной бани и оставляли охлаждаться до комнатной температуры. Белый осадок кристаллизовался через 10 минут. Для облегчения фильтрации прибавляли 10 мл этилацетата и соединение фильтровали и сушили при 70°C в течение 6 часов. Выход = 1,66 г. DSC=202,3°C±2°C.
В случае, если полученный продукт представляет собой смесь двух форм ситаглиптина гидрохлорида, он должен быть перекристаллизован из этилацетата.
Пример 3: Методика приготовления ситаглиптина фумарата формы I
В двухгорлой колбе растворяли свободное основание ситаглиптина (8,0 г, 19,65 ммоль) в 80 мл этилацетата. Реакционную смесь нагревали при 50°C в течение примерно 7 минут на водяной бане до получения прозрачного раствора и перемешивали с помощью тефлоновой лопасти/механической мешалки при 200 об/мин.
В другой колбе растворяли фумаровую кислоту (2,286 г, 19,65 ммоль) в смеси 80,0 мл этилацетата и 60,0 мл изопропанола и нагревали на водяной бане при 50°C в течение 5 минут до получения прозрачного раствора. Раствор фумаровой кислоты переносили в капельную воронку и добавляли по каплям к раствору свободному основания ситаглиптина в течение 6 минут при скорости перемешивания 305 об/мин. Температура реакции повысилась на 1,2°C (с 25,5 до 26,7°C). Прозрачный раствор превратился в мутный во время прибавления и в течение последующих пяти минут выделился густой осадок. Эта смесь перемешивалась в течение 30 минут, фильтровалась и сушилась при температуре 70°C в течение 3 часов при вакууме 5 мбар с получением 9,42 г (91%) ситаглиптина фумарата.
DSC=177,0°C±2°C, общая чистота = 99,98%. Удельное оптическое вращение = -20,77°.
Пример 4: Методика приготовления ситаглиптина фумарата формы II
Ситаглиптин фумарат (1,6 г, форма I, полученная согласно примеру 3) растворяли в 35,0 мл этанола и перемешивали на магнитной мешалке. В течение 10 минут полученный прозрачный раствор становился мутным и начиналась кристаллизация. Смесь перемешивали в течение 3 часов при комнатной температуре, прибавляли 20 мл этанола и фильтровали. Отфильтрованный осадок сушили при 70°C в течение 4 часов. Выход = 1,547 г. DSC=188,36±2°С.
Пример 5: Ситаглиптин малат
В двухгорлой колбе свободное основание ситаглиптина (7,0 г, 17,199 ммоль) растворяли в 70 мл этилацетата и нагревали при 60°C на водяной бане с получением прозрачного раствора при перемешивании на магнитной мешалке. В другой колбе смешивали L-яблочную кислоту (2,306 г, 17,199 ммоль) и 14,0 мл этанола и нагревали на водяной бане при 60°C в течение 2 минут с получением прозрачного раствора. Прозрачный раствор яблочной кислоты переносили в капельную воронку и прибавляли по каплям к раствору свободного основания ситаглиптина в течение 5 минут. В прозрачный раствор вносили затравку образца кристаллического ситаглиптина малата, полученного из предыдущего опыта, и оставляли для кристаллизации. После двух дней выкристаллизованную соль соскребали, фильтровали и сушили при температуре 70°C в течение 3 часов при вакууме 5 мбар с получением 8,7 г ситаглиптина малата.
DSC показывает два пика при 136,6±2°C (меньший пик) и 153,5±2°C (больший пик).
Очистка
В двухгорлой круглодонной колбе растворяли 8,50 г ситаглиптина малата (твердый белый порошок) в 150 мл этилацетата. Содержимое колбы перемешивали с помощью механической мешалки и помещали в масляную баню, которую предварительно нагревали до 80°C, и перемешивали в течение одного часа со скоростью вращения 313 об/мин. Реакционную смесь оставляли охлаждаться до комнатной температуры. Полученное твердое вещество фильтровали через воронку Бюхнера и сушили под вакуумом 5 мбар при 60°C в течение 3 часов, при 70°C в течение 1 часа и при 80°C в течение 1 часа с получением 8,25 г ситаглиптина малата.
DSC=154,4±2°C, указывая на чистую форму ситаглиптина малата.
ВЭЖХ: Площадь в % для яблочной кислоты = 2,44%, площадь в % для ситаглиптина = 97,55%
Общая чистота = 99,99%
Пример 6: Ситаглиптин сульфат формы I
В двухгорлой колбе свободное основание ситаглиптина (8,0 г, 19,65 ммоль) растворяли в 50 мл этанола. Реакционную массу нагревали при 50°C на водяной бане в течение 5 минут с получением прозрачного раствора и помещали на магнитную мешалку. В другой колбе смешивали серную кислоту (98%, 0,9639 г, 9,828 ммоль) и 30,0 мл этанола с получением прозрачного раствора. Прозрачный раствор H2SO4 переносили в капельную воронку и по каплям прибавляли к раствору свободного основания ситаглиптина в течение 6 минут. Реакционную смесь перемешивали в течение 10 минут, начал появляться белый осадок и становиться густым. Смесь оставляли перемешиваться в течение еще 30 минут, фильтровали и сушили при 60°C в течение 1 часа при вакууме 5 мбар, при 70°C в течение 2 часов при вакууме 5 мбар и при 80°C в течение 3 часов при вакууме 5 мбар с получением 8,50 г ситаглиптина сульфата (выход 94,8%).
ВЭЖХ=100%; DSC=148,2°С±2C (больший пик) и 197,4°C±2°C (меньший пик).
Анализ остаточного растворителя показал наличие 3,26% этанола, указывая, что данный ситаглиптин сульфат формы I является этанольным сольватом.
Пример 7: Ситаглиптин сульфат формы II
Ситаглиптин сульфат (0,530 г, полученный в примере 6) растворяли в 0,5 мл воды и раствор выдерживали в морозильнике в течение 2 часов, а затем выдерживали при комнатной температуре в течение 2 дней для кристаллизации. Соединение сушили при 60°C в течение 2 часов и при 40°C в течение ночи.
Выход = 0,470 г. DSC=101,1°C±2°C, 175,9°C±2°C (больший пик), 195,9°C±2°C (меньший пик). Анализ остаточного растворителя по ВЭЖХ показал, что данный продукт не являлся сольватом.
Пример 8: Ситаглиптин фосфат
В двухгорлой колбе свободное основание ситаглиптина (8,0 г, 19,65 ммоль) растворяли в 20 мл этанола и нагревали при 50°C на водяной бане в течение 5 минут с получением прозрачного раствора. Прозрачный раствор перемешивали при комнатной температуре с помощью магнитной мешалки.
В другой колбе смешивали фосфорную кислоту (88%, 1,920 г; 19,591 ммоль) с 5,0 мл этанола с получением прозрачного раствора. Прозрачный раствор H3PO4 переносили в капельную воронку и прибавляли по каплям к раствору свободного основания ситаглиптина в течение 10 минут. Во время прибавления H3PO4 наблюдался густой осадок, поэтому, к реакционной массе прибавляли еще 30 мл этанола для обеспечения надлежащего перемешивания. При прибавлении раствора H3PO4 температура реакционной смеси повышалась на 3,0°C (с 26,0 до 29,0°C). Реакционную смесь перемешивали в течение 30 минут и фильтровали. Полученные кристаллы сушили при 60°C в течение 1 часа и при 70°C в течение 3 часов при вакууме 5 мбар с получением 7,58 г ситаглиптина фосфата (94,8%).
DSC: 213,7°C±2°C, ВЭЖХ: 99,67%.
Пример 9: Ситаглиптин сукцинат формы I и формы II
В двухгорлой колбе свободное основание ситаглиптина (10,0 г, 24,570 ммоль) растворяли в 30 мл этилацетата и нагревали до 70°C на водяной бане с получением прозрачного раствора и перемешивали с помощью магнитной мешалки. В другой колбе растворяли янтарную кислоту (2,9 г, 24,557 ммоль) в 16 мл этанола и нагревали на водяной бане при 50°C в течение 5 минут с получением прозрачного раствора. Данный раствор переносили в капельную воронку и прибавляли по каплям в течение 5 минут к раствору свободного основания ситаглиптина. К прозрачному раствору прибавляли 22,0 мл н-гексана и смесь оставляли для кристаллизации.
Выход первой порции составлял 1,95 г; с пиком DSC при 126,1°C±2°C, что характерно для ситаглиптина сукцината формы I.
Выход второй порции составлял 2,46 г. DSC показал пики при 122,9±2°C (больший) и 135,1±2°C, указывая, что форма I загрязнена следами формы II.
Выход третьей порции составлял 4,90 г с пиками DSC при 124,2±2°C и 136,3±2°C в соотношении 75:25 формы I и формы II, соответственно.
Выход четвертой порции составлял 0,2 г с пиком DSC при 137,0°C±2°C (ситаглиптин сукцинат формы II).
Пример 10: Ситаглиптин сукцинат формы III
В двухгорлой колбе растворяли свободное основание ситаглиптина (4,0 г, 9,828 ммоль) в 12,0 мл этилацетата и нагревали до 60°C на водяной бане с получением прозрачного раствора и перемешивали с помощью магнитной мешалки. В другой колбе растворяли янтарную кислоту (1,16 г, 10,0 ммоль) в 6 мл этанола и нагревали на водяной бане при 50°C в течение 5 минут с получением прозрачного раствора. Данный раствор переносили в капельную воронку и прибавляли по каплям в течение 5 минут к свободному основанию ситаглиптина. К прозрачному раствору прибавляли 8,8 мл н-гексана и в смесь вводили затравку кристаллической соли ситаглиптина сукцината (полученной в примере 9) и оставляли для кристаллизации. Данный осадок фильтровали и сушили при 60°C в течение 3 часов. Получали 4,9 г ситаглиптина сукцината.
Очистка
Соль ситаглиптина сукцината, описанную в данном примере, можно, при желании, дополнительно очистить с удалением следов примесей другой полиморфной формы с пиком DSC при 133,3°C±2°C.
К 4,5 г выделенной соли ситаглиптина сукцината, описанной в данном примере, прибавляли 50 мл этилацетата и перемешивали при комнатной температуре в течение 2 часов, фильтровали и сушили при 60°C в течение 3 часов. Выход составлял 4,2 г ситаглиптина сукцината с пиком DSC при 167,8°C±2°C.
Пример 11: Ситаглиптин лактат
В двухгорлой колбе растворяли свободное основание ситаглиптина (10,0 г, 24,57 ммоль) в 40 мл этилацетата и нагревали до 50°C на водяной бане с получением прозрачного раствора и перемешивали с помощью магнитной мешалки. В другой колбе растворяли L-молочную кислоту (88%, 2,5 г, 24,20 ммоль) в 10,0 мл этилацетата с получением прозрачного раствора. Данный раствор переносили в капельную воронку и прибавляли по каплям в течение 5-7 минут. Прозрачный раствор начинал становиться мутным в первые 10 минут, затем становился густым осадком с повышением температуры реакционной смеси на 2°C. Чтобы отфильтровать этот осадок можно прибавить дополнительные 60,0 мл этилацетата и отфильтровать. Сушку осуществляли при 60°C в течение 1 часа и при 80°C в течение 2 часов при вакууме в 1 мбар.
Выход 7,75 г. DSC пик при 150,3°C±2°C. Удельное оптическое вращение = -25,28°.
Пример 12: Ситаглиптин гликолат
В двухгорлой колбе растворяли свободное основание ситаглиптина (7,0 г, 17,99 ммоль) в 70 мл изопропанола и нагревали до 50°C на водяной бане с получением прозрачного раствора и перемешивали с помощью магнитной мешалки. В другой колбе растворяли гликолевую кислоту (1,307 г, 17,99 ммоль) в 21,0 мл изопропанола и нагревали на водяной бане при 50°C в течение 5 минут с получением прозрачного раствора. Данный раствор переносили в капельную воронку и добавляли по каплям в течение 5-7 минут. Прозрачный раствор становился мутным в первые 10 минут и высаживалась густая масса. Чтобы отфильтровать этот осадок, можно дополнительно прибавить 50,0 мл изопропанола. Смесь фильтровали и сушили при 60°C в течение 1 часа, при 50°C в течение 6 часов и при комнатной температуре в течение ночи под вакуумом 1 мбар. Выход = 7,019 г. Наблюдались пики DSC при 89,1°C±2°C и 101,6°С±2°C. Анализ остаточного растворителя показал наличие 5,93% изопропанола, указывая, что ситаглиптин гликолат был в виде изопропанольного сольвата.
Пример 13: Ситаглиптин малеат формы I
В двухгорлой колбе свободное основание ситаглиптина (7,0 г, 17,19 ммоль) растворяли в 16 мл этанола и нагревали до 50°C на водяной бане с получением прозрачного раствора и перемешивали с помощью магнитной мешалки. В другой колбе растворяли малеиновую кислоту (1,996 г, 17,19 ммоль) в 4,0 мл этанола и нагревали на водяной бане при 50°C в течение 5 минут с получением прозрачного раствора. Данный раствор переносили в капельную воронку и добавляли по каплям в течение 5 минут к раствору ситаглиптина при скорости перемешивания 305 об/мин. Прозрачный раствор выдерживали в течение ночи для кристаллизации. При желании, в раствор можно внести затравку кристаллов соли ситаглиптина малеата, полученной в предыдущем опыте. На следующий день твердое вещество отделяли и фильтровали. Чтобы облегчить фильтрацию, можно использовать 50 мл диизопропилового эфира. Отфильтрованный осадок сушили при 50°C в течение 1 часа и при 60°C в течение 1 часа под вакуумом 5 мбар с получением 9,00 г ситаглиптина малеата.
Очистка
160 мл диизопропилового эфира прибавляли к 9 мг ситаглиптина малеата и суспензию энергично перемешивали в течение 24 часов при комнатной температуре. Прибавляли 100 мл диизопропилового эфира и твердое вещество фильтровали и сушили при 60°C под вакуумом в течение 10 часов.
DSC=92,5°C±2°C.
Анализ на остаточный растворитель показал 5,76% этанола и 1,07% диизопропилового эфира.
Пример 14: Получение ситаглиптина малеата формы II
В двухгорлой колбе ситаглиптин малеат (2,6 г, из примера 13) суспендировали в 50 мл этилацетата и перемешивали на магнитной мешалке в течение 30 минут и оставляли стоять в течение 1 часа. Ситаглиптин малеат осаждали и фильтровали. Для облегчения фильтрации прибавляли 10 мл этилацетата и отфильтрованный осадок сушили при 60°C в течение 3 часов.
Выход составил 2,54 г ситаглиптина малеата с пиком DSC при 159,5°C±2°C.
Пример 15: Кристаллический ситаглиптин цитрат
В двухгорлой колбе свободное основание ситаглиптина (2,0 г, 4,91 ммоль) и лимонную кислоту (0,944 г, 4,91 ммоль) растворяли в 10 мл этанола и нагревали до 80°C на водяной бане в течение 5 минут с получением прозрачного раствора. В раствор вводили затравку кристалла ситаглиптина цитрата и оставляли для кристаллизации. После трех дней первый осадок фильтровали и маточный раствор оставляли для дальнейшей кристаллизации. Поскольку кристаллизация была медленной в качестве анти-растворителя прибавляли 10 мл этилацетата и кристаллизацию продолжали.
Выход первой части осадка составлял 1,0 г ситаглиптина цитрата с пиком DSC при 163,1°C±2°C. Выход второй части осадка составлял 1,6 г. Оба осадка имели одинаковые кривые DSC.
Пример 16: Аморфный ситаглиптин цитрат
В двухгорлой колбе свободное основание ситаглиптина (5,0 г, 12,28 ммоль) растворяли в 20 мл этилацетата и нагревали при 60°C с получением прозрачного раствора. По каплям прибавляли раствор безводной лимонной кислоты (2,36 г, 12,28 ммоль) в 20,0 мл этилацетата и 6 мл этанола в течение 5 минут. Во время прибавления отмечалось образование густого белого осадка. Прибавляли 10 мл 2-пропанола и реакционную массу упаривали досуха с получением 7,39 г ситаглиптина цитрата. С помощью DSC и XRD было подтверждено, что соль является аморфной.
Пример 17: Ситаглиптин мезилат формы I и ситаглиптин мезилат формы II
Свободное основание ситаглиптина (6,0 г, 14,742 ммоль) растворяли в 24 мл этилацетата и нагревали до 65°C на водяной бане с получением прозрачного раствора. По каплям прибавляли раствор метансульфоновой кислоты (1,4167 г, 14,742 ммоль) в 3,0 мл этанола в течение 5 минут. Прозрачный раствор выдерживали в течение ночи для кристаллизации. При желании, в раствор можно внести затравку кристаллической соли ситаглиптина метансульфоната для того, чтобы улучшить кристаллизацию.
Первая порция кристаллизовалась через 2 дня с получением 2,94 г ситаглиптина мезилата формы I с пиком DSC при 125,0°С±2°C. Содержание этанола составляло 2,97%, показывая, что ситаглиптин мезилат формы I является этанольным сольватом.
Вторая порция давала еще 0,8 г.
Третья порция давала 3,16 г ситаглиптина мезилата формы II с пиком DSC при 161,8°С±2°C. Согласно анализу на остаточный растворитель в третьей порции остаточный этанол не присутствовал.
Другие характеристики солей ситаглиптина
Исследования гигроскопичности
Соли, описанные в данном изобретении, и свободное основание исследовали при трех различных условиях влажности (43% относительная влажность, 75% относительная влажность и 93% относительная влажность). Результаты приведены в таблицах 19-20. Свободное основание, фосфат моногидрат, фосфат, фумарат формы I, малат и малеат не показали значительных изменений во время исследований гигроскопичности, что также подтверждено с помощью DSC образцов, выдержанных при 93% относительной влажности в течение 2 недель.
Новый ситаглиптин сульфат (несольватированный; таблица 21) показал увеличение в содержании воды, соответствующее превращению полугидрата в дигидрат. DSC также показал увеличение пика в связи с поглощением воды.
После двух недель хранения при 93% относительной влажности новый аморфный ситаглиптин цитрат (см. таблицы 20 и 26) показал DSC пик при 160,7±2°C, что связано с неожиданным частичным превращением в соответствующую кристаллическую форму. Данный процесс превращения может применяться для кристаллизации аморфного ситаглиптина цитрата или для увеличения кристалличности ситаглиптина цитрата.
Исследования стабильности
Стабильность солей ситаглиптина, описанных в данном изобретении, и свободного основания исследовалась при хранении образцов при 60°C в течение 2 и 4 недель в закрытых флаконах и при хранении при 40°C и 75% относительной влажности (relative humidity, RH) в открытом флаконе в течение 12 недель. Образцы анализировались с помощью ВЭЖХ (HPLC) и содержание примесей, которые являются следствием деградации ситаглиптина, приводится в таблицах 22-24.
После хранения в закрытом флаконе в течение двух недель при температуре 60°C следующие соли были определены как устойчивые соединения: свободное основание и соли: фосфат, фосфат моногидрат, HCI формы I, фумарат формы I, сульфат (сольватированный), малат, сульфат (несольватированный) и малеат (несольватированный). В этих условиях деградация наблюдалась в случае лактата, сукцината, гликолата, цитрата и малеата (сольватированного).
После хранения в закрытом флаконе в течение четырех недель при температуре 60°C следующие соли были определены как устойчивые соединения: свободное основание, фосфат, HCI формы I, фумарат формы I, сульфат (сольватированный) и малат. Лактат, сукцинат формы I, гликолат, цитрат (аморфный) и малеат показали деградацию.
После хранения в закрытом флаконе в течение двенадцати недель при температуре 40°C и 75% RH следующие соли были определены как устойчивые соединения: свободное основание, фосфат, HCI формы I и фумарат формы I. Только лактат показал деградацию.
ПРИМЕРЫ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ:
Кристаллические соли, описанные в данном изобретении, могут входить в состав таблетки, полученной методом прямого прессования. Содержание действующих веществ в 50 мг таблетке составляет: 73,58 мг ситаглиптина цитрата, 176 мг маннита, 5 мг кроскармеллозы натрия и 6 мг стеарата магния.
Сначала смешивают активный ингредиент, микрокристаллическую целлюлозу и кроскармеллозу, и затем вводят смазочное вещество - стеарат магния и прессуют в таблетки.
иссл. гигроскопич. (93% RH, 2 недели), °C
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛЕЗНЫЕ ФАРМАЦЕВТИЧЕСКИЕ СОЛИ 7-[(3R,4R) -3-ГИДРОКСИ-4- ГИДРОКСИМЕТИЛ- ПИРРОЛИДИН -1- ИЛМЕТИЛ ]- 3,5- ДИГИДРО-ПИРРОЛО [ 3,2-D] ПИРИМИДИН-4-ОНА | 2010 |
|
RU2489435C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ, СОЛИ И ПОЛИМОРФЫ ТРАНСНОРСЕРТРАЛИНА И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2578956C2 |
| СОЛИ N-(4-ФТОРБЕНЗИЛ)-N-(1-МЕТИЛПИПЕРИДИН-4-ИЛ)-N`-(2-МЕТИЛПРОПИЛОКСИ)ФЕНИЛМЕТИЛ)КАРБАМИДА И ИХ ПРИГОТОВЛЕНИЕ | 2005 |
|
RU2387643C2 |
| ПОЛИМОРФЫ 2-(4-(2-(1-ИЗОПРОПИЛ-3-МЕТИЛ-1H-1,2,4-ТРИАЗОЛ-5-ИЛ)-5,6-ДИГИДРОБЕНЗО[F]ИМИДАЗО[1,2-D][1,4]ОКСАЗЕПИН-9-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАНАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2658009C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 6-(1Н-ИМИДАЗОЛ-1-ИЛ)-2-ФЕНИЛХИНАЗОЛИНА И ЕГО СОЛЕЙ | 2010 |
|
RU2557547C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ИЗОХИНОЛИНОНОВ И ТВЕРДЫЕ ФОРМЫ ИЗОХИНОЛИНОНОВ | 2012 |
|
RU2626883C2 |
| КРИСТАЛЛИЧЕСКИЕ СОЛИ ИНГИБИТОРА B-RAF-КИНАЗЫ | 2018 |
|
RU2798091C2 |
| ПОЛИМОРФНЫЕ ФОРМЫ БЕНЗОАТА НАТРИЯ И ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2782627C2 |
| ТВЕРДЫЕ ФОРМЫ РОМИДЕПСИНА И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2607634C2 |
| ПОЛИМОРФНЫЕ И АМОРФНЫЕ ФОРМЫ (R)-2-ГИДРОКСИ-2-МЕТИЛ-4-(2,4,5-ТРИМЕТИЛ-3,6-ДИОКСОЦИКЛОГЕКСА-1,4-ДИЕНИЛ)БУТАНАМИДА | 2015 |
|
RU2770091C2 |
Описывается новая кристаллическая соль ситаглиптина - ситаглиптин малат и фармацевтическая композиция, ее содержащая; описываемая соль является ингибитором дипептидилпептидазы-IV и может найти применение в медицине при лечении сахарного диабета 2 типа. 2 н.п. ф-лы, 17 пр., 29 табл., 18 ил.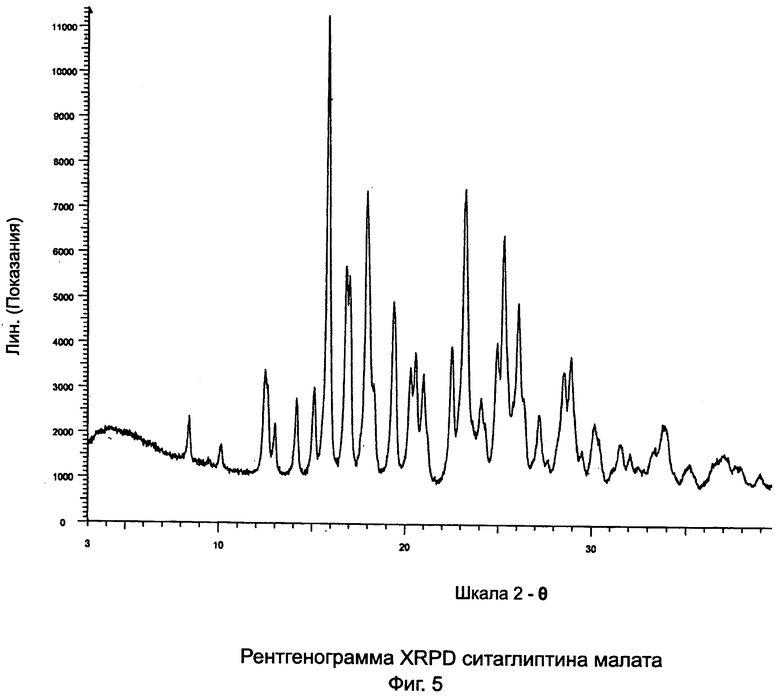
1. Ситаглиптин малат с характеристическими пиками в рентгенограмме XRPD при углах 2 θ - 15,9±0,2°, 18,0±2° и 23,2±0,2°.
2. Фармацевтическая композиция для использования в качестве ингибитора дипептидилпептидазы - IV, содержащая кристаллическую соль ситаглиптина по п.1.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2008115114 A, 27.11.2009 | |||

