Настоящее изобретение относится к кристаллическим формам макроциклического ингибитора HCV.
Вирус гепатита C (HCV) является главной причиной хронических заболеваний печени во всем мире. После начальной острой инфекции у большинства инфицируемых индивидуумов развивается хронический гепатит, поскольку HCV реплицируется предпочтительно в гепатоцитах, но не оказывает прямого цитопатического действия. Хронический гепатит может прогрессировать до фиброза печени, приводящего к циррозу, конечной стадии заболевания печени, и HCC (гепатоклеточной карциноме), что делает хронический гепатит основной причиной трансплантаций печени. Указанные обстоятельства и большое число поражаемых пациентов сделали HCV центральным вопросом масштабного медицинского исследования. Репликация генома HCV опосредована рядом ферментов, среди которых сериновая протеаза NS3 HCV и ассоциированный с ней кофактор NS4A. Считается, что сериновая протеаза NS3 важна для репликации вируса, и она является привлекательной мишенью для доставки лекарственных средств.
Современные способы терапии, направленной против HCV, основаны на (пэгилированном) интерфероне-альфа (IFN-α) в сочетании с рибавирином. Мало того, что такая терапия имеет в итоге ограниченную эффективность, которая заключается в том, что успешно можно лечить только часть пациентов, но также при этом сталкиваются с существенными побочными эффектами, и такая терапия плохо переносится многими пациентами. Следовательно, существует необходимость в дополнительных ингибиторах HCV, которые позволяют преодолеть недостатки современной терапии HCV, такие как побочные эффекты, ограниченная эффективность, плохая переносимость, появление резистентности и другие недостатки.
Описаны различные средства, которые ингибируют сериновую протеазу NS3 HCV. В WO 05/073195 описаны линейные и макроциклические ингибиторы сериновой протеазы NS3 с центральным замещенным остатком пролина, а в WO 05/073216 - с центральным остатком циклопентила. Среди таких ингибиторов макроциклические производные являются привлекательными благодаря тому, что они позволяют преодолевать недостатки современной терапии, направленной против HCV.
Обнаружено, что соединение формулы (I), имеющее структуру, изображенную ниже, особенно подходит для применения в терапии, направленной против HCV:

Соединение формулы (I) является ингибитором сериновой протеазы вируса гепатита C (HCV) и описано в WO 2007/014926, опубликованной 8 февраля 2007. Указанное соединение позволяет преодолеть несколько недостатков современной анти-HCV-терапии и, в частности, проявляет выраженную активность против HCV, имеет привлекательный фармакокинетический профиль и хорошо переносится. Используя способ синтеза, описанный в примере 5 в WO 2007/014926, получают аморфную твердую форму.
Обнаружено, что соединение формулы (I) может быть превращено в кристаллические формы, которые предпочтительно можно применять в качестве активных ингредиентов при анти-HCV-терапии. С этой целью такие кристаллические формы превращают в фармацевтические препараты.
Аморфная форма представляет собой форму, в которой не существует трехмерного дальнего упорядочения. В аморфной форме положение молекул относительно друг друга по существу является случайным, т.е. отсутствует регулярное расположение молекул в структуре решетки. Аморфные вещества могут обладать интересными свойствами, но при создании и стабилизации такого состояния обычно сталкиваются с трудностями, так как кристаллическое состояние обычно является более стабильным состоянием. Соединения в аморфной форме могут частично или полностью превращаться в кристаллические формы с течением времени или под влиянием внешних факторов, таких как температура, влажность, следовые количества кристаллического вещества в среде и т.д. Обычно кристаллическая форма активного ингредиента является предпочтительной для производства и хранения лекарственных форм.
Кристалл или кристаллическая форма представляет собой форму, в которой положение молекул относительно друг друга организовано в соответствии с трехмерной решетчатой структурой. Кристаллические формы могут включать полиморфы и псевдополиморфы. Полиморфы представляют собой разные кристаллические формы одного и того же соединения, возникающие вследствие разного расположения молекул в твердом состоянии. Полиморфы отличаются друг от друга по своим физико-химическим свойствам, но не отличаются по своему химическому составу. Полиморфизм трудно контролировать, и он может создавать проблемы для разработки лекарственных форм. Термин псевдополиморфы относится к разным кристаллическим формам, возникающим вследствие разного количества или типов растворителя в решетчатой структуре соединения.
Химия твердого тела представляет интерес для фармацевтической промышленности, в особенности с точки зрения того, что касается разработки подходящих лекарственных форм. Твердофазные переходы могут серьезно влиять на стабильность фармацевтических средств (срок хранения). Метастабильная фармацевтическая твердая форма может изменяться по своей кристаллической структуре (например, от аморфной до кристаллической) или подвергаться сольватации/десольватации в ответ на изменения условий окружающей среды, на обработку или с течением времени.
Различные кристаллические формы или аморфная форма данного лекарственного средства могут иметь существенные различия в таких фармацевтически важных свойствах, как скорость растворения, термодинамическая растворимость и биодоступность. Скорость растворения активного ингредиента в желудочном соке пациента может иметь терапевтические последствия, так как она устанавливает верхний предел скорости, с которой перорально вводимый активный ингредиент может достигать кровотока пациента. Соответственно, скорость растворения учитывается при приготовлении твердых и жидких лекарственных форм. Подобным образом разные твердые формы могут иметь разные свойства, влияющие на обработку, такие как гигроскопичность, текучесть, сжимаемость и тому подобное, которые могут влиять на их применимость в качестве фармацевтических средств в случае коммерческого производства.
При клинической разработке фармацевтических средств в том случае, если полиморфная форма не сохраняется постоянной, применяемые или исследуемые дозированные формы из разных партий могут точно не совпадать. Также желательно иметь способы получения соединения в выбранной полиморфной форме с высокой чистотой в том случае, когда соединение используют в клинических исследованиях или коммерческих продуктах, так как присутствующие примеси могут приводить к нежелательным токсикологическим эффектам. Некоторые полиморфные формы могут проявлять повышенную термодинамическую стабильность или их можно легче получить в больших количествах и с высокой чистотой, и, следовательно, они больше подходят для включения в фармацевтические препараты.
Целью настоящего изобретения является получение ингибирующего HCV средства формулы (I) в кристаллической форме, обладающей полезными свойствами, обусловливающими одну или несколько из следующих возможностей: возможность приготовления композиций, хранения и введения так, чтобы эффективно влиять на его противовирусные свойства.
Описание чертежей
На фигуре 1 представлена рентгенограмма порошковой дифракции (XPRD) формы I.
На фигуре 2 представлен спектр инфракрасного излучения (ИК) формы I.
Фигура 3 представляет собой кривую дифференциальной сканирующей калориметрии (ДСК) формы I.
На фигуре 4 представлена картина XPRD формы II.
На фигуре 5 представлен ИК-спектр формы II.
Фигура 6 представляет собой кривую ДСК формы II.
На фигуре 7 представлена XPRD формы III.
На фигуре 8 представлен ИК-спектр формы III.
Фигура 9 представляет собой кривую ДСК формы III.
На фигуре 10 представлена XPRD формы IV.
На фигуре 11 представлен ИК-спектр формы IV.
Фигура 12 представляет собой кривую ДСК формы IV.
На фигуре 13 представлена XPRD формы V.
На фигуре 14 представлена XPRD формы VI.
На фигуре 15 представлена XPRD соединения формулы (I) в аморфной форме.
Описание изобретения
Настоящее изобретение относится к ингибитору HCV, который представляет собой соединение формулы (I) в кристаллической форме. В частности, изобретение относится к кристаллическим формам, названным формой I, формой II, формой III, формой IV, формой V и формой VI. Указанные формы охарактеризованы ниже. Особый интерес представляют форма I и форма II.
В одном варианте осуществления изобретение относится к кристаллической форме соединения формулы (I), которую называют формой I соединения формулы (I) или коротко «формой I». Данная форма имеет рентгенограмму порошковой дифракции и картину ИК, указанные ниже.
Форма I имеет рентгенограмму порошковой дифракции, характеризующуюся пиками 8,5º±0,2º, 10,7º±0,2º и 17,1º±0,2º при угле дифракции два тета. Форма I характеризуется типичными дифракционными пиками в положениях при угле дифракции два тета 8,5º±0,2º, 10,7º±0,2º, 13,7º±0,2º, 14,8º±0,2º и 17,1º±0,2º. Форма I дополнительно характеризуется пиками порошковой дифракции рентгеновских лучей в положениях при угле дифракции два тета 6,51º±0,2º, 8,9º±0,2º, 13,0º±0,2º, 18,6º±0,2º и 21,0º±0,2º. Форма I имеет ИК-картину, характеризующуюся пиками 3405±1 см-1, 3066±1 см-1, 1517±1 см-1, 1427±1 см-1, 1301±1 см-1, 1285±1 см-1, 1149±1 см-1, 1132±1 см-1, 1111±1 см-1, 975±1 см-1, 956±1 см-1 и 800±1 см-1. Или Форма I имеет ИК-картину, характеризующуюся пиками 3405 (сл), 3066 (сл), 1712 (ср), 1665 (ср), 1517 (с), 1427 (с), 1387 (ср), 1351 (ос), 1300 (ср), 1285 (ср), 1132 (с), 1111 (ос), 1082 (ср), 1072 (ср), 1049 (с), 975 (ср), 885 (с), 872 (с), 838 (с), 813 (с), 800 (с), 760 (ср) и 742 (ср), где указанные числа выражены в волновых числах (см-1), и ср означает среднюю интенсивность, с означает сильную интенсивность и ос означает очень сильную интенсивность.
В другом варианте изобретение относится к кристаллической форме соединения формулы (I), которую называют формой II соединения формулы (I) или коротко «формой II». Данная форма имеет рентгенограмму порошковой дифракции и картину ИК, указанные ниже.
Форма II имеет рентгенограмму порошковой дифракции, характеризующуюся пиками 6,5º±0,2º, 10,2º±0,2º, 12,9º±0,2º и 14,4º±0,2 при угле дифракции два тета. Форма II характеризуется типичными пиками дифракции в положениях при угле дифракции два тета 4,6º±0,2º, 6,5º±0,2º, 10,2º±0,2º, 12,9º±0,2º и 14,4º±0,2º. Форма II дополнительно характеризуется пиками порошковой дифракции рентгеновских лучей в положениях при угле дифракции два тета 9,1º±0,2º, 16,5º±0,2º, 18,1º±0,2º, 20,4º±0,2º и 22,8º±0,2º. Форма II имеет ИК-картину, характеризующуюся пиками 1592±1 см-1. Или форма II имеет ИК-картину, характеризующуюся пиками 1711 (ср), 1435 (с), 1349 (с), 1065 (ср), 1038 (ср), 881 (с), 873 (с), 834 (ср) и 746 (ср), где указанные числа выражены в волновых числах (см-1), и ср, с и ос имеют значения, указанные выше.
В другом варианте осуществления изобретение относится к кристаллической форме соединения формулы (I), которую называют формой III соединения формулы (I) или коротко «формой III». Данная форма имеет картину порошковой дифракции рентгеновских лучей и картину ИК, указанные ниже.
Форма III имеет рентгенограмму порошковой дифракции, характеризующуюся пиками 9,8º±0,2º и 17,8º±0,2º при угле дифракции два тета. Форма III характеризуется типичными пиками дифракции в положениях при угле дифракции два тета 6,5º±0,2º, 9,8º±0,2º и 17,8º±0,2º. Форма III дополнительно характеризуется пиками порошковой дифракции рентгеновских лучей в положениях при угле дифракции два тета 8,6º±0,2º, 10,6º±0,2º, 11,7º±0,2º, 12,9º±0,2º, 13,7º±0,2º, 14,8º±0,2º и 19,5º±0,2º. Форма III имеет ИК-картину, характеризующуюся пиками 3120±1 см-1, 2870±1 см-1 и 1063±1 см-1. Или форма III имеет ИК-картину, характеризующуюся пиками 1718 (ср), 1664 (ср), 1434 (с), 1353 (с), 1113 (с), 1076 (ср), 1063 (ср), 1039 (с), 881 (с), 836 (с), 810 (ср), 799 (ср) и 758(ср), где указанные числа выражены в волновых числах (см-1), и ср, с и ос имеют значения, указанные выше.
В другом варианте осуществления изобретение относится к кристаллической форме соединения формулы (I), которую называют формой IV соединения формулы (I) или коротко «формой IV». Данная форма имеет рентгенограмму порошковой дифракции и картину ИК, указанные ниже.
Форма IV имеет картину порошковой дифракции рентгеновских лучей, характеризующуюся пиками 9,6º±0,2º, 11,8º±0,2º и 17,1º±0,2º при угле дифракции два тета. Форма IV характеризуется типичными пиками дифракции в положениях при угле дифракции два тета 5,6º±0,2º, 9,6º±0,2º, 11,8º±0,2º, 15,9º±0,2º и 17,1º±0,2º. Форма IV дополнительно характеризуется пиками порошковой дифракции рентгеновских лучей в положениях при угле дифракции два тета 6,8º±0,2º, 7,8º±0,2º, 11,1º±0,2º, 3,0º±0,2º и 14,4º±0,2º. Форма IV имеет ИК-картину, характеризующуюся пиками 1369±1 см-1 и 846±1 см-1. Или форма IV имеет ИК-картину, характеризующуюся пиками 1713 (ср), 1436 (с), 1348 (с), 1075 (ср), 1038 (с), 883 (с), 872 (с), 801(ср) и 743(ср), где числа выражены в волновых числах (см-1), и ср, с и ос имеют значения, указанные выше.
В другом варианте осуществления изобретение относится к кристаллической форме соединения формулы (I), которую называют формой V соединения формулы (I) или коротко «формой V». Данная форма имеет картину порошковой дифракции рентгеновских лучей и картину ИК, указанные ниже.
Форма V имеет картину порошковой дифракции рентгеновских лучей, характеризующуюся пиками 9,6º±0,2º и 19,0º±0,2º при угле дифракции два тета.
В другом варианте изобретение относится к кристаллической форме соединения формулы (I), которую называют формой VI соединения формулы (I) или коротко «формой VI». Данная форма имеет рентгенограмму порошковой дифракции и картину ИК, указанные ниже.
Форма VI имеет картину порошковой дифракции рентгеновских лучей, характеризующуюся пиками 4,4º±0,2º, 16,5º±0,2º, 9,9º±0,2º, 10,5º±0,2º и 12,9º±0,2º при угле дифракции два тета. Форма VI характеризуется типичными пиками дифракции в положениях при угле дифракции два тета 4,4º±0,2º, 6,5º±0,2º, 9,9º±0,2º, 10,5º±0,2º и 12,9º±0,2º. Форма VI дополнительно характеризуется пиками порошковой дифракции рентгеновских лучей в положениях при угле дифракции два тета 13,9º±0,2º, 15,0º±0,2º, 18,3º±0,2º, 19,1º±0,2º и 19,9º±0,2º.
Могут иметь место изменения интенсивности вследствие процессов, которые влияют на интенсивности, в частности характер обработки образца.
Настоящее изобретение также относится к смесям двух или более кристаллических форм соединения формулы (I) и смесям одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I).
Кроме того, настоящее изобретение относится к способам получения кристаллических форм соединения формулы (I).
В одном варианте осуществления предлагается способ получения формы I, включающий:
a) растворение соединения формулы (I) в C1-4-алканоле, в частности в 1-бутаноле или 2-пропаноле, с нагреванием при температуре образования флегмы растворителя; и
b) обеспечение возможности того, чтобы полученный раствор a) остыл до температуры ниже 60ºC, например до температуры в диапазоне от 60ºC до комнатной температуры, в частности ниже 40ºC, например, в диапазоне от 40ºC до комнатной температуры, более предпочтительно до комнатной температуры.
В одном варианте осуществления предлагается способ получения формы I, включающий:
c) растворение соединения формулы (I) в C1-4-алканоле, в частности в 1-бутаноле или 2-пропаноле, с нагреванием при температуре образования флегмы растворителя; и
d) обеспечение возможности спонтанного охлаждения.
В другом варианте осуществления предлагается способ получения формы I, включающий:
- суспендирование формы II в спиртовом растворителе, выбранном из C1-4-алканола, в частности из 2-пропанола, этанола, 1-бутанола, метанола, смеси спирта (такого как метанол, этанол, пропанол, изопропанол, 1-бутанол или 2-бутанол) и дихлорметана или воды или их смеси, при температуре образования флегмы спиртового растворителя; или
- суспендирование смеси формы I и формы II в растворителе, выбранном из C1-4-алканола, в частности этанола, 2-пропанола, 1-бутанола, метанола или из метилизопропилкетона (MIK), ТГФ, ацетонитрила, ацетона, 1-метоксипропан-2-ола (1-M-2-P), метилэтилкетона (MEK), дихлорметана, смеси спирта, в частности смеси C1-4-алканола (такого как метанол, этанол, пропанол, изопропанол, 1-бутанол или 2-бутанол) и дихлорметана или воды или их смеси, при температуре по меньшей мере примерно 30ºC, в частности по меньшей мере примерно 50ºC, например, в диапазоне от 30ºC до комнатной температуры до 60ºC или в диапазоне от 40ºC до температуры образования флегмы смеси.
В другом варианте осуществления предлагается способ получения формы II, включающий:
a) получение суспензии аморфной формы соединения формулы (I) в C1-4-алканоле, в частности в 2-пропаноле;
b) перемешивание суспензии при комнатной температуре; и
c) затравку суспензии затравочными кристаллами формы II или формы I.
В другом варианте осуществления предлагается альтернативный способ получения формы II, включающий:
a) растворение соединения формулы (I) в C1-4-алканоле, в частности в 2-пропаноле; и
b) выдерживание раствора со стадии a) при комнатной температуре в течение по меньшей мере 1 суток, например в течение периода времени в диапазоне от 1 суток до 4 суток или от 1 суток до 2 суток, или при температуре около 0ºC в течение по меньшей мере 4 ч, например в течение периода времени в диапазоне от 4 ч до 24 ч или от 4 ч до 12 ч, или от 4 ч до 8 ч.
В других вариантах осуществления предлагаются способы получения форм III, IV, V и VI.
Настоящее изобретение также относится к кристаллической форме соединения формулы (I) для применения в качестве лекарственного средства. Настоящее изобретение также относится к кристаллической форме соединения формулы (I) для применения в качестве ингибитора HCV или для применения при лечении связанных с HCV состояний. Изобретение также относится к применению кристаллической формы соединения формулы (I) в производстве лекарственного средства для ингибирования HCV или для лечения связанных с HCV состояний. Кроме того, изобретение относится к способу лечения млекопитающего, страдающего от связанных с HCV состояний, включающему введение эффективного количества кристаллических форм соединения формулы (I), их смесей указанному млекопитающему. Млекопитающее предпочтительно является человеком. В одном варианте осуществления кристаллическая форма в указанных выше применениях и способах выбрана из форм I, II, III, IV, V и VI, включая их смеси.
Кроме того, изобретение относится к фармацевтической композиции, содержащей кристаллическую форму соединения формулы (I) или, в частности, форму, выбранную из форм I, II, III, IV, V и VI, включая их смеси, и фармацевтически приемлемый носитель. Указанная кристаллическая форма соединения формулы (I) предпочтительно присутствует в эффективном количестве, т.е. в количестве, которое является эффективным для профилактики или лечения HCV-инфекции или состояний, ассоциированных с HCV-инфекцией.
Кроме того, предлагаются затравочные кристаллы формы I, формы II или смесь аморфной формы соединения формулы (I) и формы II, которые применимы для получения формы II соединения формулы (I).
В одном варианте осуществления изобретение относится к полиморфным формам, названным форма I, форма II, форма III, форма IV, форма V и форма VI, соединения формулы (I), которые указаны выше, по существу не содержащим примесей. В конкретном варианте осуществления такие формы содержат не более 10% примесей, или не более 5% примесей, или не более 1% примесей, или не более 0,5% примесей, или не более 0,1% примесей. Примеси могут представлять собой другие соединения или могут представлять собой любые другие твердые формы соединения формулы (I), в частности другие полиморфные формы или аморфную форму. Чистоту полиморфов можно проверить с помощью XPRD, при этом для расчета чистоты полиморфов используют площадь под пиками.
Настоящее изобретение, кроме того, относится к смеси двух или более кристаллических форм соединения формулы (I), при этом кристаллические формы выбраны из формы I, формы II, формы III, формы IV, формы V и формы VI. В одном варианте осуществления предлагается смесь, содержащая форму II и форму I соединения формулы (I). В другом варианте осуществления предлагается смесь, содержащая форму III и форму II соединения формулы (I).
Настоящее изобретение, кроме того, относится к смеси одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I), при этом кристаллические формы выбраны из формы I, формы II, формы III, формы IV, формы V и формы VI. В одном варианте осуществления предлагается смесь, содержащая форму II и аморфную форму соединения формулы (I). Указанная смесь формы II и аморфной формы соединения формулы (I), в частности, применима в качестве затравочного вещества для получения формы II.
Положения характерных пиков интенсивности XPRD (в градусах при угле 2 тета) для каждой из форм показаны в следующей далее таблице 1. Наиболее характерные положения пиков интенсивности XPRD каждой формы отмечены жирным шрифтом.
Положения пиков интенсивности XPRD полиморфных форм соединения формулы (I)
Рентгенограмма порошковой дифракции формы I по существу представляет собой картину, изображенную на фигуре 1. Рентгенограмма порошковой дифракции формы II по существу представляет собой картину, изображенную на фигуре 4. Рентгенограмма порошковой дифракции формы III по существу представляет собой картину, изображенную на фигуре 7. Рентгенограмма порошковой дифракции формы IV по существу представляет собой картину, изображенную на фигуре 10. Рентгенограмма порошковой дифракции формы V по существу представляет собой картину, изображенную на фигуре 13. Рентгенограмма порошковой дифракции формы VI по существу представляет собой картину, изображенную на фигуре 14.
Данные XPRD и представления картин для всех форм I-VI могут быть получены с использованием дифрактометра Philips X'PertPRO MPD PW3050/60 с генератором PW3040. Прибор оборудован Cu LFF-рентгеновской трубкой PW3373/00. Анализируемое соединение распределяли на держателе образца с нулевым фоном. Использовали следующие параметры прибора:
- напряжение генератора: 45 кВ;
- сила тока генератора: 40 мА;
- геометрия: Брегга-Брентано;
- платформа: поворотная платформа.
Использовали следующие параметры сканирования для форм I, II, III и IV: диапазон составлял от 3º до 50º при угле 2 тета с непрерывным сканированием со скоростью 0,01675º/шаг, 29,845 с/шаг. Время одного оборота вращателя составляло 1 с, тип излучения CuKα, и длина волны излучения 1,54056 Å.
Для форм V и VI использовали следующие параметры сканирования: диапазон составлял от 3º до 35º при угле 2-тета с непрерывным сканированием со скоростью 0,0502448º/шаг, 90,17 с/шаг. Время одного оборота вращателя составляло 1 с, тип излучения CuKα, и длина волны излучения 1,54056 Å. Использовали следующие параметры пути падающего луча для форм I, II, III, IV, V и VI:
- программируемая щель расходимости: 15 мм;
- щель Соллера: 0,04 рад;
- диафрагма луча: 15 мм;
- антирассеивающая щель: 1º;
- отсечка луча: +.
Параметры пути дифрагированного луча для форм I, II, III, IV, V и VI следующие:
- антирассеивающая диафрагма с длинной щелью: +;
- щель Соллера: 0,04 рад;
- Ni-фильтр: +;
- детектор: X'Celerator.
Положения пиков XPRD, устанавливаемых для форм I, II, III, IV, V и VI, определяют с точностью до 0,2º вследствие экспериментальных различий, таких как различия в приборах, приготовлении образцов и тому подобном.
Положение характеристических пиков ИК-поглощения (в волновых числах в см-1) форм I, II, III и IV приведены в следующей таблице 2. Положение наиболее характерных пиков ИК-поглощения для каждой формы выделены жирным шрифтом.
Положение пиков ИК-поглощения полиморфных форм соединения формулы (I)
Картина ИК-поглощения формы I практически является такой, как она изображена на фигуре 2. Картина ИК-поглощения формы II практически является такой, как она изображена на фигуре 5. Картина ИК-поглощения формы III практически является такой, как она изображена на фигуре 8. Картина ИК-поглощения формы IV практически является такой, как она изображена на фигуре 11.
ИК-данные и картины ИК-поглощения получали используя инфракрасную микроспектрометрию нарушенного полного внутреннего отражения (microATR), на спектрофотометре Nexus FTIR. Вспомогательным оборудованием для microATR являлся Harrick Split Pea с кристаллом Si. Использовали детектор DTGS с окнами KBr. Для форм I, II, III и IV использовали следующие параметры сканирования:
- количество сканирований: 32;
- разрешение: 1 см-1;
- область длин волн: от 4000 до 400 см-1;
- коррекция относительно исходного уровня: да;
- расщепитель пучка: Ge на KBr.
Положения пиков ИК-поглощения, устанавливаемых для форм I, II, III и IV, определяют с точностью до 1 см-1 вследствие экспериментальных различий, таких как различия в приборах, приготовлении образцов и тому подобном.
Положения характеристических эндотермических пиков ДСК и их ширина (в ºC) для форм I, II, III и IV приведены в следующей таблице 3:
Положения или диапазон эндотермических пиков ДСК для полиморфных форм соединения формулы (I)
Кривая ДСК формы I практически является такой, как она изображена на фигуре 3. Кривая ДСК формы II практически является такой, как она изображена на фигуре 6. Кривая ДСК формы III практически является такой, как она изображена на фигуре 9. Кривая ДСК формы IV практически является такой, как она изображена на фигуре 12.
Данные ДСК и изображения кривых ДСК получали с использованием TA-Instruments Q1000 MTДСК, оборудованного блоком охлаждения RCS. Масса образцов составляла примерно 3 мг, и образцы помещали в стандартную алюминиевую кювету для образцов TA-Instrument. Образцы сканировали со скоростью 10ºC/мин, начиная с 25ºC и до конечной температуры 300ºC. Печь постоянно продували газообразным азотом со скоростью потока 50 мл/мин.
Допустимое отклонение кривых ДСК, полученных для форм I и II, определено равным 3ºC вследствие экспериментальных различий, таких как различия в приборах, приготовлении образцов и тому подобном.
Обнаружено, что полиморфная форма I является наиболее стабильной формой. Кроме того, указанная форма является наименее гигроскопичной формой. Вышесказанное делает форму I особенно привлекательной для применения в качестве активного ингредиента в фармацевтических дозированных формах.
Обнаружено, что полиморфная форма II является менее стабильной, но, тем не менее, достаточно стабильной для применения в фармацевтических дозированных формах. Обнаружено, что присущая данной форме растворимость выше, чем в случае формы I. Следовательно, форма II может найти применение в фармацевтических дозированных формах, которые используют в тех случаях, когда требуется более высокая свойственная форме растворимость. Более высокая свойственная форме растворимость может положительно влиять на фармакокинетические свойства активного ингредиента формулы (I), например активный ингредиент может быстрее достигать кровотока или локализации в теле, где он должен проявлять свою противовирусную активность.
На основании данных ДСК можно сделать вывод, что полиморфные формы I и II образуют монотропную систему. В случае монотропной системы графики зависимости свободной энергии различных полиморфов от температуры не пересекаются до того момента, когда все полиморфы подвергаются плавлению, другими словами, любой переход от одного полиморфа к другому будет необратимым. В случае энантиотропной системы на графике зависимости свободной энергии от температуры видна точка пересечения раньше различных точек плавления, и возможно обратимое превращение между двумя полиморфами при нагревании и охлаждении.
Получение кристаллических форм
Соединение формулы (I) может быть получено, как описано в примерах.
Форма I соединения формулы (I) может быть получена способом, включающим:
a) растворение соединения формулы (I) в C1-4-алканоле при температуре, составляющей от 65ºC до температуры кипения раствора;
b) обеспечение возможности охлаждения раствора до комнатной температуры.
В используемом в настоящем описании смысле термин «C1-4-алканол» относится к C1-4-алкиловому спирту, полученному из алкана, имеющего от одного до четырех атомов углерода, такому как метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол, 2-бутанол, 2-метил-1-пропанол, трет-бутанол. Подгруппа среди «C1-4-алканолов» представлена «C3-4-алканолами», которые получают из алкана, имеющего от трех до четырех атомов углерода, такими как 1-пропанол, 2-пропанол, 1-бутанол, 2-бутанол, 2-метил-1-пропанол, трет-бутанол.
Предпочтительными для применения при получении формы I являются 1-пропанол, 2-пропанол, 1-бутанол, 2-бутанол, особенно 1-бутанол или 2-пропанол. На стадии a) указанного выше способа получения формы I соединение формулы (I) в C1-4-алканоле предпочтительно нагревают до температуры образования флегмы смеси. В одном варианте осуществления соединение формулы (I) смешивают с C1-4-алканолом с образованием суспензии, и полученную суспензию нагревают до температуры образования флегмы смеси, после чего смесь титруют дополнительным количеством C1-4-алканола вплоть до получения раствора. Охлаждение до комнатной температуры в указанном выше способе предпочтительно осуществляют медленно, например, в течение периода времени от примерно 12 ч до примерно 48 ч, например в течение периода времени, составляющего примерно 12 ч, или примерно 24 ч, или примерно 48 ч. В одном варианте осуществления раствору дают возможность спонтанно остыть, т.е. не контролируя температуру. В другом варианте осуществления раствору дают возможность остыть, контролируя при этом температуру. Исходное соединение формулы (I) в указанном выше способе может иметь любую форму, например аморфную или любую кристаллическую форму или их смесь, например смесь формы I и формы II.
Количество 1-бутанола или 2-пропанола, которое добавляют на стадии a), может быть в диапазоне от примерно 15 до примерно 25 л/моль или от примерно 17 до примерно 19 л/моль, предпочтительно в количестве 17,85 л/моль или 18,5 л/моль. В одном варианте осуществления указанный выше способ получения формы I дополнительно включает на стадии b) охлаждение раствора до 65ºC или выше. В другом варианте осуществления указанный выше способ получения формы I дополнительно включает на стадии b) частичное выпаривание растворителя, особенно в том случае, когда не происходит преципитация при 65ºC или выше.
В одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы I, включающему:
a) растворение соединения формулы (I) в 1-бутаноле или 2-пропаноле при нагревании до температуры образования флегмы растворителя; и
b) обеспечение возможности спонтанного охлаждения до комнатной температуры.
В одном варианте осуществления указанный в приведенном выше абзаце способ получения формы I включает добавление 1-бутанола в концентрации от 17 до 19 л/моль, предпочтительно в концентрации 17,85 л/моль или 18,5 л/моль. В другом варианте осуществления указанный в последнем варианте способ получения формы I дополнительно включает на стадии b) использование медленного охлаждения раствора. В другом варианте осуществления указанный в последнем абзаце способ получения формы I дополнительно включает на стадии b) охлаждение раствора до 65ºC или выше. В другом варианте осуществления указанный в последнем абзаце способ получения формы I дополнительно включает на стадии b) частичное выпаривание растворителя, особенно в том случае, когда не происходит преципитация при 65ºC или выше.
Настоящее изобретение дополнительно относится к способу суспендирования для получения кристаллической формы I, включающему:
- суспендирование формы II в спиртовом растворителе, в частности в C1-4-алканоле, который может быть выбран из 2-пропанола, этанола, 1-бутанола, метанола, смеси спирта, в частности C1-4-алканола (такого как метанол, этанол, пропанол, изопропанол, 1-бутанол или 2-бутанол) и дихлорметана или воды или их смеси, при температуре образования флегмы спиртового растворителя; или
- суспендирование смеси формы I и формы II в растворителе, выбранном из C1-4-алканола (в частности, 2-пропанола, 1-бутанола, метанола, этанола), метилизопропилкетона (MIK), ТГФ, ацетонитрила, ацетона, 1-метоксипропан-2-ола (1-M-2-P), метилэтилкетона (MEK), дихлорметана, смеси спирта (такого как C1-4-алканол, например, метанол, этанол, пропанол, изопропанол, 1-бутанол или 2-бутанол) и дихлорметана или воды или их смеси, при температуре в диапазоне от примерно 30ºC до температуры образования флегмы смеси, или при температуре в диапазоне от примерно 30ºC до примерно 100ºC, или при температуре в диапазоне от примерно 40ºC до примерно 80ºC, или при температуре, составляющей по меньшей мере примерно 30ºC.
Способы суспендирования для получения формы I могут дополнительно включать перемешивание суспензии формы II при комнатной температуре в спиртовом растворителе, например в C1-4-алканоле, или суспензии смеси формы I и формы II в растворителе, который указан выше.
Способы суспендирования для получения формы I могут дополнительно включать перемешивание в течение периода времени, составляющего от примерно 2 ч до примерно 24 ч или от примерно 2 ч до примерно 12 ч, в одном варианте в течение периода времени, составляющего по меньшей мере 2 ч, суспензии формы II в спиртовом растворителе или суспензии смеси формы I и формы II в растворителе, который указан выше. Суспендирование может быть осуществлено в течение по меньшей мере 4 ч, например в течение по меньшей мере 8 ч.
Способы суспендирования для получения формы I могут дополнительно включать фильтрование преципитатов, полученных после суспендирования формы II в спиртовом растворителе или после суспендирования смеси формы I и формы II в растворителе, который указан выше.
Способы суспендирования для получения формы I могут дополнительно включать после стадии фильтрования, указанной в приведенном выше абзаце, промывку отфильтрованных преципитатов, полученных после суспендирования формы II в спиртовом растворителе или после суспендирования смеси формы I и формы II в растворителе, который указан выше, при этом стадию промывки осуществляют таким же растворителем, который использовали во время стадии суспендирования.
При получении любой из твердых форм согласно настоящему изобретению, которая образуется из прозрачного раствора соединения формулы (I), твердая форма исходного вещества не оказывает влияния на твердую форму конечного продукта, и контроль полученной твердой формы осуществляют в результате контролирования параметров способа.
Изобретение также относится к способу получения формы II, включающему:
a) получение суспензии аморфной формы соединения формулы (I) в C1-4-алканоле, в частности в 2-пропаноле и;
b) перемешивание суспензии при комнатной температуре; и
c) затравку суспензии затравочными кристаллами формы II или формы I.
В том случае, когда способ внесения затравки согласно указанной выше стадии c) осуществляют с использованием затравочных кристаллов формы I, форма II будет получена с минимальным содержанием формы I.
В одном варианте осуществления способ получения формы II дополнительно включает после стадии c) перемешивание суспензии после внесения затравки при комнатной температуре.
Способ получения формы II может дополнительно включать после стадии c) перемешивание суспензии после внесения затравки в течение периода времени от 15 мин до 72 ч. Перемешивание можно осуществлять в течение периода времени от 5 до 60 ч, в частности в течение от 10 до 48 ч.
Способ получения формы II дополнительно может включать фильтрование преципитата, полученного после стадии c). Способ получения формы II дополнительно может включать после стадии фильтрования, указанной в приведенном выше абзаце, промывку отфильтрованного преципитата, полученного после стадии c), изопропанолом.
Настоящее изобретение также относится к альтернативному способу получения формы II, включающему:
a) растворение соединения формулы (I) в C1-4-алканоле, в частности в 2-пропаноле; и
b) выдерживание раствора со стадии a) при комнатной температуре в течение по меньшей мере 1 суток, в частности в течение периода времени в диапазоне от примерно 1 суток до примерно 4 суток, или в диапазоне от примерно 1 суток до примерно 2 суток; или при температуре около 0ºC в течение по меньшей мере 4 ч, в частности в течение периода времени в диапазоне от примерно 4 ч до примерно 12 ч.
В одном варианте осуществления указанный выше альтернативный способ получения формы II включает перед стадией a) растворение соединения формулы (I) в дихлорметане, затем добавление C1-4-алканола, в частности добавление 2-пропанола, как указано для стадии a), и перед стадией b) частичное или полное удаление дихлорметана. Удаление дихлорметана может быть осуществлено выпариванием с использованием, например, роторного испарителя в вакууме.
В другом варианте осуществления указанный выше альтернативный способ получения формы II включает выдерживание раствора со стадии a) при комнатной температуре в течение периода времени, составляющего от примерно 5 ч до примерно 48 ч, в частности в течение периода времени, составляющего от примерно 14 ч до примерно 36 ч. Указанный выше альтернативный способ получения формы II может включать выдерживание раствора со стадии a) при комнатной температуре в течение по меньшей мере 14 ч, 16 ч, 18 ч, 20 ч, 22 ч, 24 ч, 26 ч, 28 ч, 30 ч, 32 ч, 34 ч или 36 ч.
В другом варианте осуществления указанный выше альтернативный способ получения формы II включает выдерживание раствора со стадии a) при температуре около 0ºC в течение периода времени, составляющего от примерно 5 ч до примерно 48 ч, в частности в течение периода времени, составляющего от примерно 5 ч до примерно 36 ч, более предпочтительно в течение периода времени, составляющего от примерно 5 ч до примерно 16 ч. Указанный выше альтернативный способ получения формы II может включать выдерживание раствора со стадии a) при температуре около 0ºC в течение по меньшей мере 5 ч, 6 ч, 7 ч, 8 ч, 9 ч, 10 ч, 11 ч, 12 ч, 13 ч, 14 ч, 15 ч или 16 ч.
Указанный выше альтернативный способ получения формы II также может включать выдерживание раствора со стадии a) при температуре от -10ºC до 10ºC, в частности при температуре от -5ºC до 5ºC, например при температуре -10ºC, -9ºC, -8ºC, -7ºC; -6ºC, -5ºC, -4ºC, -3ºC, -2ºC, -1ºC, 0ºC, 1ºC, 2ºC, 3ºC, 4ºC, 5ºC, 6ºC, 7ºC, 8ºC, 9ºC или 10ºC, в течение по меньшей мере4 ч, в частности в течение периода времени в диапазоне от примерно 4 ч до примерно 12 ч.
В другом варианте указанный выше альтернативный способ получения формы II включает на стадии b) перемешивание раствора при его выдерживании или его хранении при комнатной температуре в течение по меньшей мере 1 суток, в частности в течение периода времени в диапазоне от примерно 1 суток до примерно 4 суток или в диапазоне от примерно 1 суток до примерно 2 суток; или при температуре около 0ºC в течение по меньшей мере 4 ч, в частности в течение периода времени в диапазоне от примерно 4 ч до примерно 12 ч.
Изобретение также относится к способу получения формы III, включающему:
a) получение насыщенного или почти насыщенного раствора соединения формулы (I) в ацетонитриле и насыщенного или почти насыщенного раствора соединения формулы (I) в воде;
b) нагревание двух насыщенных или почти насыщенных растворов со стадии a) по меньшей мере при 40ºC;
c) смешивание двух насыщенных или почти насыщенных растворов со стадии b) в объемном соотношении 50/50.
В одном варианте осуществления способ получения формы III включает на стадии b) нагревание двух насыщенных или почти насыщенных растворов при температуре от примерно 40ºC до примерно 70ºC, предпочтительно примерно от 45ºC до 65ºC, более предпочтительно примерно от 50ºC до 60ºC. Способ получения формы III может дополнительно включать фильтрование двух растворов со стадии b) перед их смешиванием. Способ получения формы III дополнительно может включать перемешивание раствора при комнатной температуре после смешивания насыщенных или почти насыщенных растворов на стадии c). Способ получения формы III дополнительно может включать обеспечение возможности упаривания раствора после осуществления смешивания на стадии c) и предпочтительно после его перемешивания при комнатной температуре.
Изобретение также относится к способу получения формы IV, включающему:
a) получение насыщенного или почти насыщенного раствора соединения формулы (I) в 1-метокси-2-пропаноле;
b) нагревание насыщенного или почти насыщенного раствора при температуре образования флегмы 1-метокси-2-пропанола;
c) смешивание насыщенного или почти насыщенного раствора со стадии b) с водой в процентном соотношении по объему 30%-70% раствор/вода или в объемном соотношении 4/10.
Способ получения формы IV дополнительно может включать перемешивание раствора при комнатной температуре после смешивания с водой на стадии c). Перемешивание раствора при комнатной температуре может быть осуществлено в течение от примерно 4 до примерно 24 ч, или в течение от примерно 6 до примерно 18 ч, или в течение от примерно 8 до примерно 16 ч. Способ получения формы IV дополнительно может включать фильтрование раствора после его смешивания с водой на стадии c) и предпочтительно после его перемешивания при комнатной температуре.
Изобретение также относится к способу получения формы V, включающему:
a) получение насыщенного или почти насыщенного раствора соединения формулы (I) в 2-бутаноле и насыщенного или почти насыщенного раствора соединения формулы (I) в воде;
b) нагревание двух насыщенных или почти насыщенных растворов со стадии a) по меньшей мере при 40ºC;
c) смешивание двух насыщенных или почти насыщенных растворов со стадии b) в объемном соотношении 50/50.
Способ получения формы V может включать на стадии b) нагревание двух насыщенных или почти насыщенных растворов при температуре от примерно 40ºC до примерно 70ºC, предпочтительно от примерно 45ºC до примерно 65ºC, более предпочтительно от примерно 50ºC до примерно 60ºC. Способ получения формы V дополнительно может включать фильтрование двух растворов со стадии b) перед их смешиванием. Способ получения формы V дополнительно может включать перемешивание раствора при комнатной температуре после смешивания на стадии c). Способ получения формы V дополнительно может включать обеспечение возможности для упаривания раствора после смешивания на стадии c) и предпочтительно после его перемешивания при комнатной температуре.
Изобретение также относится к способу получения формы VI, включающему:
a) получение суспензии соединения формулы (I) в воде;
b) нагревание суспензии со стадии a), по меньшей мере, при комнатной температуре в течение по меньшей мере примерно 4 суток.
В одном варианте осуществления способ получения формы VI включает на стадии a) получение раствора, предпочтительно суспензии, соединения формулы (I) в воде, при этом количественное соотношение формы I и формы II составляет примерно 1/99, 5/95, 10/90, 20/80, 40/60, 50/50, 60/40, 80/20, 90/10, 95/5 или 99/1, предпочтительно примерно 1/99, 5/95, 10/90, 20/80, 40/60 или 50/50, более предпочтительно примерно 5/95, 10/90 или 20/80, еще более предпочтительно примерно 10/90.
В другом варианте способ получения формы VI включает на стадии a) получение раствора формы I и формы II в воде, при этом количество воды находится в избытке по отношению к количеству формы I и формы II. Способ получения формы VI может включать на стадии b) нагревание раствора со стадии a) примерно при 30ºC в течение по меньшей мере примерно 4 суток, или примерно при 40ºC в течение по меньшей мере примерно 4 суток, или примерно при 50ºC в течение по меньшей мере примерно 4 суток. В одном варианте указанный период, составляющий по меньшей мере 4 суток на стадии b), представляет собой период от примерно 4 суток до примерно 10 суток, в частности от примерно 4 суток до примерно 6 суток.
Изобретение также относится к способу, в котором полученную кристаллическую форму выделяют фильтрованием или центрифугированием, необязательно в сочетании с промывкой и сушкой. Исходным веществом, используемым в способах согласно настоящему изобретению, может быть любая кристаллическая или аморфная форма соединения формулы (I), включая его гидрат. В случае способов кристаллизации кристаллическая форма исходного вещества обычно не влияет на конечный результат. В случае растирания конечный продукт может варьировать в зависимости от исходного вещества. Специалисту в данной области может быть известен подходящий способ обработки исходного вещества для получения требуемой формы в случае растирания. Настоящее изобретение не ограничено исходной формой, используемой для растирания, если такая форма необходима для получения другой формы.
В одном варианте растворители, используемые для получения кристаллических форм согласно настоящему изобретению, являются фармацевтически приемлемыми или фармацевтически неприемлемыми растворителями, при этом предпочтительны первые. Фармацевтически неприемлемые растворители необходимо будет удалять перед использованием полиморфа в фармацевтическом препарате.
В смесях воды и смешиваемых с водой растворителей количество воды может варьировать от примерно 5% об. до примерно 95% об., предпочтительно от примерно 25% до примерно 75% об., более предпочтительно от примерно 40% до примерно 60% об.
Способы получения кристаллических форм согласно настоящему изобретению обычно включают получение кристаллического твердого вещества из раствора или дисперсии соединения формулы (I) в растворяющей среде или в результате суспендирования соединения формулы (I), которое исходно может быть в аморфной или кристаллической форме.
Условия кристаллизации могут быть модифицированы, для того чтобы улучшить способ кристаллизации или индуцировать преципитацию, не влияя на форму получаемого полиморфа. Такие условия включают доведение раствора, дисперсии или суспензии соединения формулы (I) и растворителя(ей) до требуемой концентрации, охлаждение, следуя определенной кривой охлаждения/температуры, добавление затравочных кристаллов, доведение указанного раствора, дисперсии или суспензии до требуемой температуры, воздействие определенного подходящего давления, удаление и/или отделение любого нежелательного вещества или примесей, сушку образованных кристаллов с получением полиморфов в твердом состоянии, если такое состояние требуется.
Предпочтительным способом индукции преципитации является снижение растворимости соединения формулы (I). Растворимость соединения может быть снижена, например, посредством охлаждения раствора. Растворимость соединения формулы (I) может быть снижена при добавлении антирастворителя.
Доведение раствора, дисперсии или суспензии соединения формулы (I) и растворителей до требуемой концентрации не обязательно предполагает увеличение концентрации соединения формулы (I). В некоторых случаях может быть предпочтительным уменьшение или отсутствие изменения концентрации соединения формулы (I). Методики, используемые для получения требуемой концентрации, включают, например, упаривание в результате атмосферной дистилляции, вакуумную дистилляцию, фракционную дистилляцию, азеотропную дистилляцию, испарение пленки, нагревание, охлаждение, другие методики, хорошо известные в данной области, и их сочетания. Необязательный способ получения требуемой концентрации также может включать насыщение раствора соединения формулы (I) и растворителя, например, добавлением к раствору достаточного объема вещества, не являющегося растворителем, чтобы достичь точки насыщения. Другой подходящий способ насыщения раствора включает в качестве примера введение в раствор дополнительного количества соединения формулы (I) и/или выпаривание части растворителя из раствора. В используемом в настоящем описании смысле насыщенный раствор охватывает растворы в точках их насыщения или превышающие точки их насыщения, т.е. перенасыщенные. Почти насыщенный раствор относится к растворам, которые близки к насыщению, но не достигли своих точек насыщения.
Способ улучшения процесса кристаллизации согласно настоящему изобретению, в частности ускорения кристаллизации, заключается во внесении затравки кристалла продукта или нанесении царапин на внутреннюю поверхность сосуда для кристаллизации стеклянной палочкой. В других случаях кристаллизация может происходить спонтанно без какого-либо стимула. Настоящее изобретение охватывает оба варианта, в которых кристаллизация конкретной формы соединения формулы (I) происходит спонтанно или может быть индуцирована или ускорена, если такая индукция или ускорение не являются критическим фактором получения конкретной формы.
Термин «внесение затравки» относится к добавлению кристаллического вещества для облегчения кристаллизации. Термин «затравочные кристаллы» означает порошок предварительно полученной кристаллической формы соединения формулы (I). Конкретными затравочными кристаллами или затравочным веществом согласно настоящему изобретению, которые применимы для получения формы II, являются следующие затравочные кристаллы:
- затравочные кристаллы смеси формы II и аморфной формы соединения формулы (I);
- затравочные кристаллы формы I; и
- затравочные кристаллы формы II.
Доведение указанного раствора, дисперсии или суспензии до требуемой температуры следует понимать как действия, связанные с нагреванием, охлаждением или оставлением при температуре окружающей среды. Нагревание раствора, дисперсии или суспензии может быть необходимо для полного растворения соединения формулы (I).
Удаление и/или отделение любого нежелательного вещества или примесей можно осуществить, используя способы очистки, фильтрования, промывки, преципитации или подобные способы. Отделение, например, можно осуществить известными способами разделения твердых веществ и жидкости. Фильтрование можно осуществить наряду с другими способами, пропуская раствор, дисперсию или суспензию через бумагу, фильтр из пористого стекла или другой мембранный материал, посредством центрифугирования или с использованием фильтра типа воронки Бюхнера, фильтра или пластин Rosenmund или рамного пресса. Предпочтительно поточное фильтрование или безопасное фильтрование может быть успешно введено в описанные выше способы, чтобы повысить чистоту получаемой полиморфной формы. Кроме того, также можно использовать такие средства для фильтрования, такие как силикагель, Celite®, Arbocel®, дикалит диатомит или тому подобные, чтобы отделить примеси от представляющих интерес кристаллов.
Полученные кристаллы также могут быть высушены, и такой способ сушки необязательно можно использовать на разных этапах кристаллизации, если применяют более одного этапа кристаллизации. Способы сушки включают все методики, известные специалистам в данной области, такие как нагревание, применение вакуума, циркулирующего воздуха или газа, добавление десиканта, сушка вымораживанием, распылительная сушка, упаривание или тому подобные, или любое их сочетание.
Способы кристаллизации полиморфов соединения формулы (I) могут включать различные сочетания методик и их вариантов. Кристаллизация полиморфов соединения формулы (I) может быть осуществлена посредством растворения, диспергирования или суспендирования соединения формулы (I) при подходящей температуре в растворителе, при этом часть указанного растворителя испаряется, увеличивая концентрацию соединения формулы (I) в указанном растворе, дисперсии или суспензии, посредством охлаждения указанной смеси и необязательно промывки и/или фильтрования и сушки полученных кристаллов соединения формулы (I). Необязательно полиморфы соединения формулы (I) могут быть получены растворением, диспергированием или суспендированием соединения формулы (I) в растворяющей среде, охлаждением полученного таким образом раствора, дисперсии или суспензии и последующим фильтрованием и сушкой полученного полиморфа. Другим примером получения кристаллических форм соединения формулы (I) может быть насыщение растворяющей среды соединением формулы (I) и необязательно фильтрование, промывка и сушка полученных кристаллов.
При образовании кристаллов также можно использовать более одного способа кристаллизации. В некоторых случаях предпочтительно могут быть осуществлены одна, две или более дополнительных стадий кристаллизации по разным причинам, например, чтобы повысить качество получаемой кристаллической формы. Например, полиморфы согласно настоящему изобретению также могут быть получены добавлением растворителя к исходному основному веществу соединения формулы (I), перемешиванием раствора при постоянной температуре до тех пор, пока вещества не растворятся полностью, концентрированием раствора посредством вакуумной дистилляции и охлаждением. Может происходить первая кристаллизация, и образованные кристаллы могут быть промыты растворителем с последующим растворением соединения формулы (I) в растворителе с образованием требуемого полиморфа. Может быть осуществлена перекристаллизация реакционной смеси с последующей стадией охлаждения от температуры образования флегмы. Образованный полиморф необязательно можно отфильтровать и высушить.
При растворении, диспергировании или суспендировании соединения формулы (I) в растворителе можно получить разные степени дисперсии, например суспензии, взвеси или смеси; или предпочтительно получить гомогенные однофазные растворы. Термин «суспензия» относится к двухфазной системе, состоящей из тонко диспергированного твердого вещества, т.е. соединения формулы (I), в аморфной, кристаллической форме или смеси таких форм, диспергированного (суспендированного) в жидкой или дисперсионной среде, обычно в растворителе. Термин «суспензия» относится к суспензии, образованной в том случае, когда определенное количество порошка перемешивают в жидкости, в которой твердое вещество только слаборастворимо (или не растворимо). «Суспендирование» относится к получению суспензии.
Необязательно растворяющая среда может содержать добавки, например диспергирующие агенты, поверхностно-активные вещества или другие добавки или их смеси такого типа, который обычно используют для получения суспензий кристаллического вещества. Добавки преимущественно могут быть использованы при модификации формы кристалла посредством увеличения мягкости и уменьшения площади поверхности.
Растворяющую среду, содержащую твердое вещество, необязательно можно перемешивать в течение определенного периода времени или энергично встряхивать с использованием, например, мешалки с большими сдвиговыми усилиями или гомогенизатора или их сочетание, чтобы получить требуемый размер частиц в случае органического соединения.
Дополнительно можно использовать контроль температуры преципитации и внесение затравки, чтобы повысить воспроизводимость процесса кристаллизации, распределения размера частиц и формы продукта. Как таковая кристаллизация может быть осуществлена без внесения затравки в виде кристаллов соединения формулы (I) или предпочтительно в присутствии кристаллов соединения формулы (I), которые вводят в раствор в ходе затравки. Затравку также можно осуществлять несколько раз при разных температурах. Количество затравочного вещества зависит от масштаба эксперимента и без труда может быть определено специалистом в данной области. Обычно количество затравочного вещества составляет от примерно 0,1 до 1% мас. от количества кристаллического вещества, ожидаемого в результате реакции.
Время кристаллизации на каждой стадии кристаллизации будет зависеть от используемых условий, применяемых методик и/или используемых растворителей.
Измельчение крупных частиц или агрегатов частиц может быть дополнительно осуществлено после превращения кристаллов, чтобы получить требуемый и гомогенный размер частиц. Соответственно кристаллы, агрегаты порошка и крупный порошок полиморфных форм соединения формулы (I) необязательно могут быть размолоты и отсортированы по размеру после того, как их подвергли превращению. Размалывание или измельчение относится к физическому делению на более мелкие части крупных частиц или агрегатов частиц с использованием способов и устройств, хорошо известных в данной области для уменьшения размера частиц порошков. Размер полученных частиц может быть в диапазоне от миллиметров до нанометров с получением, например, нанокристаллов, микрокристаллов. Предпочтительным устройством для размалывания или измельчения является струйная мельница или мельница тончайшего помола, благодаря ее способности давать частицы небольшого размера с узким диапазоном распределения по размеру.
Фармацевтическое применение кристаллических форм
Настоящее изобретение, кроме того, относится к кристаллической форме соединения формулы (I), смеси двух или более кристаллических форм соединения формулы (I) или смеси одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I) для применения в качестве лекарственного средства. В одном варианте кристаллическая форма, отдельно или в любой из указанных выше смесей для применения в качестве лекарственного средства, выбрана из форм I, II, III, IV, V и VI.
Настоящее изобретение, кроме того, относится к применению кристаллической формы соединения формулы (I), смеси двух или более кристаллических форм соединения формулы (I) или смеси одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I) в производстве лекарственного средства для лечения связанных с HCV состояний. В одном варианте осуществления кристаллическая форма, отдельно или в любой из указанных выше смесей, используемых в производстве лекарственного средства, выбрана из форм I, II, III, IV, V и VI.
Настоящее изобретение также относится к способу лечения млекопитающего, страдающего от связанных с HCV состояний, включающему введение кристаллической формы соединения формулы (I), смеси двух или более кристаллических форм соединения формулы (I) или смеси одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I) млекопитающему, нуждающемуся в таком введении. В одном варианте осуществления способ лечения включает введение кристаллической формы, отдельно или в любой из указанных выше смесей, выбранной из форм I, II, III, IV, V и VI.
Связанные с HCV состояния включают такие патологические состояния, которые вызваны HCV и другими патогенными флавивирусами, такими как вирусы желтой лихорадки, лихорадки денге (типы 1-4), энцефалита Сент-Луиса, японского энцефалита, энцефалита долины Мюррей, вирус Западного Нила и вирус Кунджин. Заболевания, ассоциированные с HCV, включают прогрессирующий фиброз печени, воспаление и некроз, приводящие к циррозу, заболевание печени в конечной стадии и гепатоклеточную карциному (HCC); и в случае других патогенных флавивирусов заболевания включают желтую лихорадку, лихорадку денге, геморрагическую лихорадку и энцефалит. HCV и другие патогенные флавивирусы включают штаммы HCV как дикого типа, так и мутантные штаммы.
Термин «лечение» относится к любому лечению патологического состояния у млекопитающего, в частности у человека, и включает в себя одно или несколько из следующих действий:
(i) профилактику появления патологического состояния у субъекта, который может быть предрасположен к такому состоянию, но у которого состояние еще не диагностировано, и, соответственно, лечение является профилактическим лечением патологического состояния;
(ii) подавление патологического состояния, т.е. задержка его развития;
(iii) облегчение патологического состояния, т.е. вызывание регрессии патологического состояния; или
(iv) ослабление симптомов, опосредованных патологическим состоянием.
Настоящее изобретение, кроме того, относится к фармацевтической композиции, содержащей кристаллическую форму соединения формулы (I), смесь двух или более кристаллических форм соединения формулы (I) или смесь одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I) и фармацевтически приемлемый эксципиент. В одном варианте осуществления фармацевтическая композиция содержит кристаллическую форму, одну или в любой из указанных выше смесей, выбранную из форм I, II, III, IV, V и VI.
Фармацевтические композиции могут быть получены в виде лекарственных средств для введения перорально, парентерально (включая подкожное, внутримышечное и внутривенное введение), ректально, трансдермально, буккально или назально. Подходящие формы для перорального введения включают порошки, гранулы, агрегаты, таблетки, прессованные или имеющие покрытие пилюли, драже, саше, твердые или желатиновые капсулы, сиропы и суспензии. Подходящие формы для парентерального введения включают водный или неводный раствор или эмульсию, тогда как в случае ректального введения подходящие для введения формы включают суппозитории с гидрофильным или гидрофобным носителем. Для местного введения в изобретении предлагаются системы трансдермальной доставки, известные в данной области, а для назальной доставки предлагаются подходящие аэрозольные системы доставки, известные в данной области. Хотя наиболее подходящее введение в любом конкретном случае будет зависеть от природы и тяжести состояния, подвергаемого лечению, наиболее предпочтительным путем введения согласно настоящему изобретению является пероральный.
Дозы могут быть для удобства представлены в виде стандартной лекарственной формы и приготовлены любым способом, хорошо известным в данной области. Альтернативно дозированные формы могут быть представлены в виде одной, двух, трех или четырех или более поддоз, вводимых с соответствующими интервалами в течение суток. Используемая стандартная доза предпочтительно составляет от примерно 1 мг до примерно 1000 мг соединения формулы (I), эквивалентных форме основания, или от примерно 5 до примерно 800 мг, или от примерно 5 до примерно 400 мг, или от примерно 50 до примерно 600 мг, или от примерно 100 до примерно 400 мг.
Фармацевтические композиции согласно настоящему изобретению содержат указанные выше полиморфные формы соединения формулы (I). Фармацевтическая композиция может содержать только одну форму соединения формулы (I) или смесь различных форм соединения формулы (I) с аморфной формой или без нее. Кроме активного ингредиента (ингредиентов) фармацевтическая композиция содержит один или несколько эксципиентов или адъювантов.
Примерами подходящих эксципиентов являются аравийская камедь, окись магния, карбонат магния, фосфат калия, лактоза, глюкоза или крахмал, в частности кукурузный крахмал. Подходящими масляными эксципиентами или растворителями являются растительные масла или животные жиры, такие как подсолнечное масло или жир печени трески. Подходящими растворителями для водных или спиртовых растворов являются вода, этанол, растворы сахаров или их смеси. Полиэтиленгликоли и полипропиленгликоли также применимы в качестве дополнительных вспомогательных средств для других форм введения.
В случае подкожного или внутривенного введения полиморфы соединения формулы (I), при необходимости с веществами, обычно используемыми для такого введения, такими как солюбилизаторы, эмульгаторы или другие вспомогательные средства готовят в виде суспензии в жидком носителе, таком как, например, вода, физиологический раствор соли или спирты, например, этанол, пропанол, глицерин, дополнительно также растворы сахаров, такие как растворы глюкозы или маннита, или альтернативно смеси различных указанных растворителей.
Подходящими фармацевтическими композициями для введения в форме аэрозолей или спреев являются, например, суспензии полиморфов соединения формулы (I) в фармацевтически приемлемом жидком носителе, таком как этанол или вода или их смесь. При необходимости препарат дополнительно также может содержать другие фармацевтические вспомогательные средства, такие как поверхностно-активные вещества, эмульгаторы и стабилизаторы, а также пропеллент. Такой препарат обычно содержит активное соединение в концентрации примерно от 0,1 до 50%, в частности примерно от 0,3 до 3% мас.
В дополнение к ингредиентам, конкретно упомянутым выше, фармацевтические композиции согласно настоящему изобретению могут содержать другие средства, обычно используемые в данной области, имеющие отношение к данному типу препарата, например средства, подходящие для перорального введения, могут включать корригенты или средства для маскировки вкуса.
В используемом в настоящем описании смысле термин «примерно» имеет свое обычное значение. В конкретных вариантах в отношении числового значения можно интерпретировать, что «примерно» означает числовое значение ±10%, или ±5%, или ±2%, или ±1%, или ±0,5% или ±0,1%. В других подразумевают точное значение, т.е. убирают слово «примерно».
ПРИМЕРЫ
Следующие примеры предназначены для иллюстрации настоящего изобретения, но не для его ограничения.
Пример 1: Получение 17-[2-(4-изопропилтиазол-2-ил)-7-метокси-8-метил-хинолин-4-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (16)
Синтез 4-гидрокси-2-(4-изопропилтиазол-2-ил)-7-метокси-8-метилхинолина (6)
Стадия 1: синтез N-(трет-бутилоксикарбонил)-3-метокси-2-метиланилина (2)
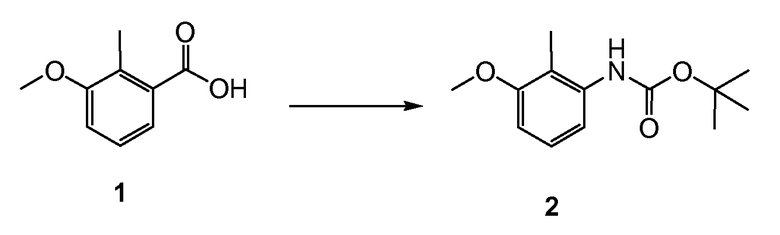
Триэтиламин (42,4 мл, 302 ммоль) добавляли к суспензии 3-метокси-2-метилбензойной кислоты (45,6 г, 274 ммоль) в безводном толуоле (800 мл). Получали прозрачный раствор. Затем медленно добавляли dppa (65,4 мл, 302 ммоль) в толуоле (100 мл). После 1 часа при комнатной температуре реакционную смесь последовательно нагревали при 50ºC в течение 0,5 ч, при 70ºC в течение 0,5 ч, затем при 100ºC в течение 1 ч. К полученному раствору добавляли t-BuOH (30,5 г, 411 ммоль) в толуоле (40 мл) при 100ºC и полученную смесь кипятили с обратным холодильником в течение 7 ч. Раствор охлаждали до комнатной температуры, затем последовательно промывали водой, 0,5 н. HCl, 0,5 н. NaOH и насыщенным раствором соли, сушили (Na2SO4) и упаривали, получая 67 г целевого продукта: m/z = 237 (M)+.
Стадия 2: Синтез 3-метокси-2-метиланилина (3)

ТФУ (40,7 мл, 548 ммоль) добавляли к раствору N-(трет-бутилоксикарбонил)-3-метокси-2-метиланилина в дихлорметане (500 мл). Спустя 2 ч при комнатной температуре добавляли ТФУ (40,7 мл, 548 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Затем летучие вещества выпаривали. Остаток растирали в толуоле (100 мл) и диизопропиловом эфире (250 мл), отфильтровывали и промывали диизопропиловым эфиром (100 мл), получая 56,3 г указанного в заголовке продукта в виде ТФУ-соли: m/z = 138 (M+H)+. ТФУ-соль превращали в свободный анилин обработкой NaHCO3.
Стадия 3: Синтез (2-амино-4-метокси-3-метилфенил)(метил)кетона (4)

Раствор BCl3 (1,0 М, 200 мл, 200 ммоль) в CH2Cl2 медленно добавляли в атмосфере азота к раствору 3-метокси-2-метиланилина (26,0 г, 190 ммоль) в ксилоле (400 мл). Во время добавления контролировали температуру и поддерживали ее ниже 10ºC. Реакционную смесь перемешивали при 5ºC в течение 0,5 ч. Затем добавляли безводный ацетонитрил (13 мл, 246 ммоль) при 5ºC. После 0,5 ч при 5ºC раствор переносили в капельную воронку и медленно добавляли при 5ºC к суспензии AlCl3 (26,7 г, 200 ммоль) в CH2Cl2 (150 мл). После 45 мин при 5ºC реакционную смесь нагревали при 70ºC в потоке азота. После выпаривания CH2Cl2 температура реакционной смеси достигала 65ºC. После выдерживания в течение 12 ч при 65ºC реакционную смесь охлаждали при 0ºC, вливали в лед (300 г) и медленно кипятили с обратным холодильником в течение 7 ч. После выдерживания в течение 2 дней при комнатной температуре добавляли 6 н. NaOH (50 мл). Значение pH полученного раствора составляло 2-3. Слой ксилола декантировали. Органический слой экстрагировали CH2Cl2. Слои ксилола и CH2Cl2 объединяли, последовательно промывали водой, 1 н. NaOH и насыщенным раствором соли, сушили (Na2SO4) и упаривали. Остаток растирали в диизопропиловом эфире при 0ºC, отфильтровывали и промывали диизопропиловым эфиром, получая 13,6 г (40%) указанного в заголовке продукта в виде твердого вещества желтоватого цвета: m/z = 180 (M+H)+.
Стадия 4: Синтез 2'-[[(4-изопропилтиазол-2-ил)(оксо)метил]амино]-4'-метокси-3'-метилацетофенона (5)
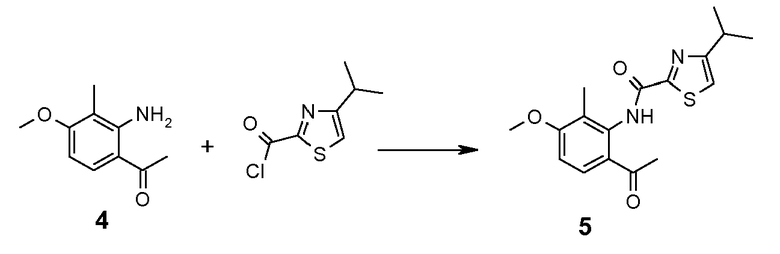
Раствор соединения 4 (18,6 г, 104 ммоль) в диоксане (50 мл) добавляли в атмосфере азота к суспензии 4-изопропилтиазол-2-карбонилхлорида в диоксане (250 мл). После выдерживания в течение 2 ч при комнатной температуре реакционную смесь концентрировали досуха. Затем остаток распределяли между водным раствором NaHCO3 и AcOEt, органический слой промывали насыщенным раствором соли, сушили (Na2SO4) и упаривали. Остаток растирали в диизопропиловом эфире, отфильтровывали и промывали диизопропиловым эфиром, получая 30,8 г (90%) указанного в заголовке продукта 5.
Стадия 5: Синтез 4-гидрокси-2-(4-изопропилтиазол-2-ил)-7-метокси-8-метилхинолина (6)
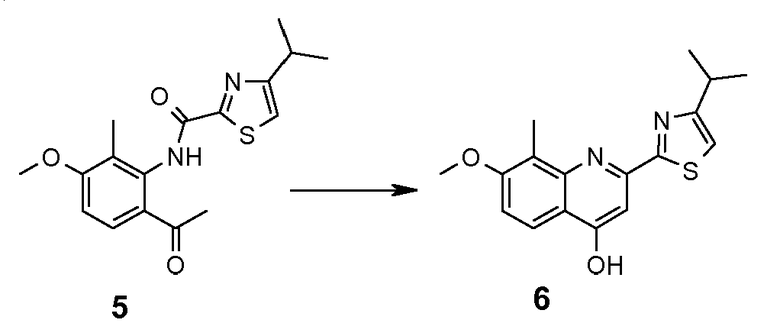
Трет-бутоксид калия (21,8 г, 195 ммоль) добавляли к суспензии соединения 5 (30,8 г, 92,7 ммоль) в трет-бутаноле. Полученную реакционную смесь нагревали при 100ºC в течение ночи. Затем реакционную смесь охлаждали при комнатной температуре и разбавляли эфиром (100 мл). Осадок отфильтровывали и промывали Et2O, получая порошок (фракция A). Маточный раствор концентрировали в вакууме, растирали в эфире, отфильтровывали и промывали эфиром, получая порошок (фракция 2). Фракции 1 и 2 смешивали и вливали в воду (250 мл). Значение pH полученного раствора доводили до 6-7 (контроль с помощью pH-бумаги), используя 1 н. HCl. Осадок отфильтровывали, промывали водой и сушили. Затем твердое вещество растирали в диизопропиловом эфире, отфильтровывали и сушили, получая 26 г (88%) соединения 6 в виде твердого вещества коричневатого цвета: m/z = 315 (M+H)+.
Синтез (гекс-5-енил)(метил)амина (8)
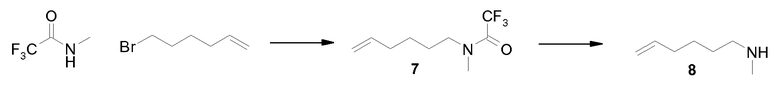
(a) Гидрид натрия (1,05 эквивалента) медленно добавляли при 0ºC к раствору N-метилтрифторацетамида (25 г) в ДМФА (140 мл). Смесь перемешивали в течение 1 ч при комнатной температуре в атмосфере азота. Затем по каплям добавляли раствор бромгексена (32,1 г) в ДМФА (25 мл) и смесь нагревали до 70ºC в течение 12 ч. Реакционную смесь вливали в воду (200 мл) и экстрагировали эфиром (4×50 мл), сушили (MgSO4), фильтровали и упаривали, получая 35 г целевого продукта 7 в виде масла желтоватого цвета, которое использовали без дополнительной очистки на следующей стадии.
(b) Раствор KOH (187,7 г) в воде (130 мл) по каплям добавляли к раствору 7 (35 г) в метаноле (200 мл). Смесь перемешивали при комнатной температуре в течение 12 ч. Затем реакционную смесь вливали в воду (100 мл) и экстрагировали эфиром (4×50 мл), сушили (MgSO4), фильтровали и эфир отгоняли при атмосферном давлении. Полученное масло очищали дистилляцией в вакууме (давление 13 мм Hg, 50ºC), получая 7,4 г (34%) указанного в заголовке продукта 8 в виде бесцветного масла: 1H-ЯМР (CDCl3): δ 5,8 (м, 1H), 5 (ддд, J=17,2 Гц, 3,5 Гц, 1,8 Гц, 1H), 4,95 (м, 1H), 2,5 (т, J=7,0 Гц, 2H), 2,43 (с, 3H), 2,08 (кв., J=7,0 Гц, 2H), 1,4 (м, 4H), 1,3 (ушир.с, 1H).
Получение 17-[2-(4-изопропилтиазол-2-ил)-7-метокси-8-метилхинолин-4-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 lоктадец-7-ен-4-карбоновой кислоты (16)
Стадия A

3-Оксо-2-оксабицикло[2.2.1]гептан-5-карбоновую кислоту 9 (500 мг, 3,2 ммоль) в 4 мл ДМФА добавляли при 0ºC к HATU (1,34 г, 3,52 ммоль) и N-метилгекс-5-ениламину (435 мг, 3,84 ммоль) в ДМФА (3 мл), с последующим добавлением DIPEA. После перемешивания в течение 40 мин при 0ºC смесь перемешивали при комнатной температуре в течение 5 ч. Затем растворитель выпаривали, остаток растворяли в EtOAc (70 мл) и промывали насыщенным раствором NaHCO3 (10 мл). Водный слой экстрагировали EtOAc (2×25 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (20 мл), сушили (Na2SO4) и упаривали. Очистка флэш-хроматографией (EtOAc/петролейный эфир, 2:1) давала 550 мг (68%) целевого продукта 10 в виде бесцветного масла: m/z = 252 (M+H)+.
Стадия B
Раствор LiOH (105 мг в 4 мл воды) добавляли при 0ºC к амиду лактона 10. Через 1 ч превращение завершалось (ВЭЖХ). Смесь подкисляли до pH 2-3, используя 1 н. HCl, экстрагировали AcOEt, сушили (MgSO4), упаривали, несколько раз упаривали совместно с толуолом и сушили в условиях высокого вакуума в течение ночи, получая 520 мг (88%) целевого продукта 11: m/z = 270 (M+H)+.
Стадия C
Гидрохлорид этилового эфира 1-(амино)-2-(винил)циклопропанкарбоновой кислоты 12 (4,92 г, 31,7 ммоль) и HATU (12,6 г, 33,2 ммоль) добавляли к 11 (8,14 г, 30,2 ммоль). Смесь охлаждали на бане со льдом в атмосфере аргона и затем последовательно добавляли ДМФА (100 мл) и DIPEA (12,5 мл, 11,5 ммоль). После выдерживания в течение 30 мин при 0ºC раствор перемешивали при комнатной температуре еще в течение 3 ч. Затем реакционную смесь распределяли между EtOAc и водой, последовательно промывали 0,5 н. HCl (20 мл) и насыщенным раствором NaCl (2×20 мл) и сушили (Na2SO4). Очистка флэш-хроматографией (AcOEt/CH2Cl2/петролейный эфир, 1:1:1) давала 7,41 г (60%) целевого продукта 13 в виде бесцветного масла: m/z = 407 (M+H)+.
Стадия D
DIAD (1,02 мл, 5,17 ммоль) добавляли при -15ºC в атмосфере азота к раствору 13 (1,5 г, 3,69 ммоль), хинолина 6 (1,39 г, 4,43 ммоль) и трифенилфосфина (1,26 г, 4,80 ммоль) в безводном ТГФ (40 мл). После выдерживания в течение 4,5 ч при -15ºC реакционную смесь распределяли между ледяной водой и AcOEt, сушили (Na2SO4) и упаривали. Сырое вещество очищали флэш-хроматографией (градиент петролейный AcOEt/CH2Cl2, от 1:9 до 2:8), получая 1,45 г (56%) целевого продукта 14: m/z = 703 (M+H)+.
Стадия E

Раствор 14 (1,07 г, 1,524 ммоль) и катализатора первого поколения Ховейды-Граббса (33 мг, 0,03 эквивалента) в безводном и дегазированном 1,2-дихлорэтане (900 мл) нагревали при 75ºC в атмосфере азота в течение 12 ч. Затем растворитель выпаривали и остаток очищали хроматографией на силикагеле (25% EtOAc в CH2Cl2). Получали 620 мг (60%) чистого макроцикла 15. m/z = 674 (M+H)+. 1H-ЯМР (CDCl3): 1,18-1,39 (м, 12H), 1,59 (м, 1H), 1,70-2,08 (м, 5H), 2,28 (м, 1H), 2,38 (м, 1H), 2,62 (м, 2H), 2,68 (с, 3H), 2,83 (м, 1H), 3,06 (с, 3H), 3,19 (септ., J=6,7 Гц, 1H), 3,36 (м, 1H), 3,83 (м, 1H), 3,97 (с, 3H), 4,09 (м, 2H), 4,65 (тд, J=4 Гц, 14 Гц, 1H), 5,19 (дд, J=4 Гц, 10 Гц, 1H), 5,31 (м, 1H), 5,65 (тд, J=4 Гц, 8 Гц, 1H), 7,00 (с, 1H), 7,18 (с, 1H), 7,46 (д, J=9 Гц, 1H), 7,48 (с, 1H), 8,03 (д, J=9 Гц, 1H).
Стадия F
Раствор гидроксида лития (1,65 г, 38,53 ммоль) в воде (15 мл) добавляли к перемешиваемому раствору сложного эфира 15 (620 мг, 0,920 ммоль) в ТГФ (30 мл) и MeOH (20 мл). После выдерживания в течение 16 ч при комнатной температуре реакционную смесь гасили насыщенным раствором NH4Cl, концентрировали при пониженном давлении, подкисляли до pH 3, используя 1 н. HCl, и экстрагировали CH2Cl2, сушили (MgSO4) и упаривали, получая 560 мг (88%) карбоновой кислоты 16. m/z = 647 (M+H)+. 1H-ЯМР (CDCl3): 1,11-1,40 (м, 8H), 1,42-1,57 (м, 2H), 1,74 (м, 2H), 1,88-2,00 (м, 2H), 2,13 (м, 1H), 2,28 (м, 1H), 2,40 (м, 1H), 2,59 (м, 2H), 2,67 (с, 3H), 2,81 (м, 1H), 2,97 (с, 3H), 3,19 (м, 1H), 3,31 (м, 1H), 3,71 (м, 1H), 3,96 (с, 3H), 4,56 (дт, J=4 Гц, 12 Гц, 1H), 5,23 (м, 2H), 5,66 (м, 1H), 7,01 (с, 1H), 7,10 (с, 1H), 7,22 (д, J=10 Гц, 1H), 7,45 (с, 1H), 8,00 (д, J=10 Гц, 1H).
Пример 2: Получение N-[17-[2-(4-изопропилтиазол-2-ил)-7-метокси-8-метилхинолин-4-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (17)
Раствор соединения 16 (560 мг, 0,867 ммоль), полученного согласно примеру 4, и карбонилдиимидазола (308 мг, 1,90 ммоль) в безводном ТГФ (10 мл) перемешивали при кипячении с обратным холодильником в атмосфере азота в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли циклопропилсульфонамид (400 мг, 3,301 ммоль) и DBU (286 мг, 1,881 ммоль). Полученный раствор нагревали при 50ºC в течение 15 ч. Затем реакционную смесь охлаждали при комнатной температуре и концентрировали при пониженном давлении. Остаток распределяли между CH2Cl2 и 1 N HCl, органический слой промывали насыщенным раствором соли, сушили (MgSO4) и упаривали. Очистка флэш-хроматографией (градиент EtOAc (0-25%) в CH2Cl2) давала 314 мг твердого вещества не совсем белого цвета, которое затем промывали водой, затем изопропиловым эфиром и сушили в вакуумной печи, получая 282 мг (40%) очищенного указанного в заголовке продукта 17, который представляет собой соединение формулы (I), в виде порошка белого цвета: m/z = 750 (M+H)+. 1H-ЯМР (CDCl3): 0,99-1,52 (м, 14H), 1,64-2,05 (м, 4H), 2,77 (м, 1H), 2,41 (м, 2H), 2,59 (м, 2H), 2,69 (с, 3H), 2,92 (м, 2H), 3,04 (с, 3H), 3,19 (м, 1H), 3,40 (м, 2H), 3,98 (с, 3H), 4,60 (т, J=13 Гц, 1H), 5,04 (т, J=11 Гц, 1H), 5,37 (м, 1H), 5,66 (м, 1H), 6,21 (с, 1H), 7,02 (с, 1H), 7,22 (д, J=10 Гц, 1H), 7,45 (с, 1H), 7,99 (д, J=10 Гц, 1H), 10,82 (ушир.с, 1H).
Пример 3: Получение полиморфа I
2 г смеси полиморфа I и II кипятили с обратным холодильником в небольшом количестве 1-бутанола. К кипящей суспензии небольшими порциями добавляли 1-бутанол вплоть до получения прозрачного раствора. В указанной точке количество добавленного 1-бутанола составляло 17,85 л/моль. Раствор дополнительно перемешивали и охлаждали спонтанно до комнатной температуры в течение двух выходных дней. Твердое вещество извлекали фильтрованием и 2 раза промывали 5 мл 1-бутанола. Анализ XPRD показал, что полученное вещество представляло собой кристаллический полиморф I.
Пример 4: Получение соединения формулы (I) в аморфной форме
1 г смеси полиморфа I и II растворяли в дихлорметане (120 мл). Полученный прозрачный раствор фильтровали через фильтр P4 (с размером пор 10-16 мкм) и упаривали досуха (роторный испаритель; 40ºC; 750-50 мбар), и при этом получали аморфное соединение (I), подтвержденное в результате анализа XPRD (см. фигуру 21).
Пример 5: Получение полиморфа II
3.1. В результате затравки полиморфом II
К 1 г аморфного вещества соединения (I) (которое получено в примере 2) добавляли 25 мл 2-PrOH, и суспензию перемешивали при комнатной температуре в течение примерно 15 мин. Затем добавляли небольшое количество затравочного вещества полиморфа II, и суспензию перемешивали при комнатной температуре. В течение 15 мин в суспензии начинало образовываться вещество белого цвета, и суспензию дополнительно перемешивали в течение двух выходных дней. Белый осадок отфильтровывали, промывали 10 мл 2-prOH и сушили в течение ночи при 60ºC/в вакууме.
Выход по массе составлял 92% мас. и анализ XPRD показал, что полученное вещество представляло собой кристаллический полиморф II вероятно со следовыми количествами полиморфа I согласно ИК-анализу.
3.2. В результате затравки полиморфом I
0,2 г смеси полиморфов I и II растворяли в дихлорметане (10 мл). Полученный прозрачный раствор упаривали досуха (роторный испаритель), и остаток соскребали со стенок колбы. К полученному аморфному веществу добавляли 5 мл 2-PrOH (25 мл/г), и суспензию перемешивали при комнатной температуре в течение примерно 15 мин. Затем в качестве затравочного вещества добавляли полиморф I (который получали согласно любому из примеров 3, 10 или 11), и затем суспензию перемешивали при комнатной температуре. В суспензии начинал образовываться осадок белого цвета, и суспензию дополнительно перемешивали в течение ночи. Осадок отфильтровывали, промывали небольшим количеством 2-prOH и сушили в течение ночи при 60ºC/в вакууме.
Извлекали твердое вещество белого цвета, и анализ XPRD показал, что полученное вещество представляло собой кристаллический полиморф II со следами полиморфа I.
3.3. С использованием способа в масштабе нескольких грамм (масштаб 20 г)
1-я партия
Примерно 20 г смеси полиморфа I и II растворяли в дихлорметане (100 мл) и фильтровали через фильтр P4 (с размером пор 10-16 мкм). Полученный прозрачный раствор упаривали досуха (роторный испаритель; 40ºC; 750-50 мбар). К остатку добавляли 250 мл 2-PrOH (12,5 мл/г), и суспензию перемешивали при комнатной температуре в течение примерно 15 мин. Затем в качестве затравочного вещества добавляли полиморф II (который получен согласно примеру 12), и затем суспензию перемешивали при комнатной температуре. В суспензии начинал образовываться осадок белого цвета, и суспензию дополнительно перемешивали в течение ночи. Осадок отфильтровывали, промывали 10 мл 2-PrOH и сушили в течение ночи при 60ºC/в вакууме. Извлекали 7,8 г твердого вещества белого цвета, и анализ XPRD показал, что полученное вещество представляло собой кристаллический полиморф II.
2-я партия
Маточный раствор вместе с веществом, которое оставалось на стенках реактора, собирали, и растворитель выпаривали. В середине выпаривания брали образец суспензии, фильтровали, сушили, анализировали и обнаружили, что образец главным образом представляет собой аморфное вещество с полиморфами I и II, присутствующими вместе с некоторым количеством неидентифицированного кристаллического вещества (веществ). Остаток суспензии упаривали досуха (масса=11 г).
Остаток растворяли в дихлорметане и фильтровали через фильтр P4. Полученный прозрачный раствор упаривали досуха (роторный испаритель; 40ºC; 750-50 мбар). К полученному аморфному веществу добавляли 275 мл 2-PrOH (25 мл/г), и суспензию перемешивали при комнатной температуре в течение примерно 15 мин. Затем в качестве затравочного вещества добавляли полиморф II (который получен согласно примеру 10), и затем суспензию перемешивали при комнатной температуре. В пределах 15 мин в суспензии начинал образовываться белый осадок, и суспензию дополнительно перемешивали в течение ночи. Осадок отфильтровывали, два раза промывали 10 мл 2-PrOH и сушили в течение ночи при 60ºC в вакууме. Маточный раствор вместе с веществом, которое оставалось на стенках реактора, собирали, и растворитель выпаривали досуха (масса=6,51 г).
Извлекали 4,6 г твердого вещества белого цвета, и анализ XPRD показал, что полученное вещество представляло собой кристаллический полиморф II.
Пример 6: Получение полиморфа III
Готовили два насыщенных раствора полиморфа II в ацетонитриле и в воде при 50ºC. Полученные растворы фильтровали после выдерживания в течение 1,5 ч при 50ºC. По 225 мкл каждого фильтрата вносили в одну и ту же лунку, и смеси давали возможность кристаллизоваться при комнатной температуре, растворитель выпаривали при комнатной температуре досуха. Получали форму III.
Пример 7: Получение полиморфа IV
40 мг полиморфа I и 4 мл 1-метокси-2-пропанола кипятили с обратным холодильником при перемешивании. К раствору добавляли 10 мл воды, и давали возможность раствору кристаллизоваться при комнатной температуре в течение ночи при перемешивании. Осадок отфильтровывали, используя фильтр Millipore, и продукт сушили при комнатной температуре в течение 1 ч. Получали форму IV.
Пример 8: Получение полиморфа V
Готовили два насыщенных раствора полиморфа II в 2-бутаноне и в воде при 50ºC. Полученные растворы фильтровали после выдерживания в течение 1,5 ч при 50ºC. По 225 мкл каждого фильтрата вносили в одну и ту же лунку, и смеси давали возможность кристаллизоваться при комнатной температуре, растворитель выпаривали при комнатной температуре досуха. Получали форму V.
Пример 9: Получение полиморфа VI
Готовили суспензию, взвешивая 15 мг полиморфа II и 1,5 мг полиморфа I во флаконе для ВЭЖХ. Добавляли 100 мкл воды, и закрытый флакон хранили в течение 4 дней при 30ºC и 7 дней при 40ºC. Продукт сушили на бумажном фильтре при комнатной температуре. Получали форму VI.
Пример 10: Превращение смеси полиморфов II и I в полиморф I с использованием суспензионного способа
1 г смеси полиморфов I и II кипятили с обратным холодильником в параллельных экспериментах в фиксированном количестве растворителя (11 л/моль каждого из MeOH, EtOH, EtOH/H2O, 2-PrOH и 1-бутанола). Суспензии кипятили с обратным холодильником в течение примерно 2 ч, и давали возможность спонтанно остыть до комнатной температуры, и перемешивали в течение двух выходных дней. В случае отдельной параллельной реакции в 2-пропаноле осуществляли горячее фильтрование. Твердое вещество извлекали фильтрованием и 2 раза промывали 5 мл соответствующего растворителя.
В таблице 4 для каждого эксперимента показан используемый растворитель, выход, чистота полученных полиморфов или их смесей и тип полиморфизма.
(% мас.)
(объемное соотношение 95/5)
(2) Выделение вещества посредством горячего фильтрования.
Пример 11: Превращение полиморфа II в полиморф I с использованием суспензионного способа, выявляемое с применением технологии анализа процессов (PAT)
В реактор MultiMax объемом 250 мл загружали 3,7 г полиморфа II и добавляли 100 мл 2-пропанола (20,3 л/моль). Реактор устанавливали в MultiMax, и в суспензию вводили зонды комбинационного рассеяния, NIR и FTIR, при этом суспензию перемешивали при комнатной температуре. Реактор защищали от дневного света и начинали измерения. Примерно через 30 мин реакционную смесь нагревали до 80ºC со скоростью примерно 2º/мин. После выдерживания примерно в течение 1 часа при 80ºC наблюдали прозрачный раствор, и поэтому в реактор добавляли дополнительное количество 1,85 г полиморфа II, доводя общее количество полиморфа II до 5,55 г. В указанной точке использовали 18 мл растворителя/г полиморфа II (по сравнению с 15 мл/г в более ранних экспериментах, проводимых с использованием суспензии).
Суспензию перемешивали при 80ºC в течение ночи. Примерно через 20 ч 1,11 г полиморфа II, 20% от исходного количества, добавляли к горячей суспензии, которую перемешивали еще в течение примерно 2 ч. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали.
Спектры комбинационного рассеяния получали каждые 2 мин, используя спектрометр комбинационного рассеяния RXN1/785 Kaiser Optical Systems в сочетании с погружным зондом. Использовали анализ главных компонентов (PCA) (без предварительной обработки данных, диапазон 1200-1400 см-1), чтобы проанализировать изменение во времени. Первые 2 главных компонента были сходными в спектрах полиморфов I и II. Смотри таблицу 5 ниже.
(в см-1)
Временная кривая единиц поглощения показала превращение полиморфа II в полиморф I. В течение первых 4 ч имело место растворение полиморфа II. Спустя 1 ч начинал образовываться полиморф I, и превращение заканчивалось еще через 5 ч. Добавление дополнительного количества полиморфа II (в точке 20 ч) приводило к быстрому превращению полиморфа II в I.
Спектры в ближней инфракрасной области (NIR) получали каждые 2 мин, используя NIR-спектрометр Bruker-Matrix-F (32 сканирования, разрешение 4 см-1, 10000-5000 см-1) и отражательный зонд (Solvias Reflector). Спектры суспензии полиморфа I и II калибровали по значению 1 и 2 соответственно (PLS, 6800-5600 см-1, нормализация векторов и ранг=1). Указанную модель использовали для мониторинга изменений направления в полиморфизме с течением времени. В течение первых 4 ч имело место растворение полиморфа II. Спустя 1 ч начинал образовываться полиморф I, и превращение заканчивалось спустя еще 5 ч.
Анализ XPRD выделенного продукта показал, что полученное вещество представляло собой кристаллический полиморф I. С использованием спектров комбинационного рассеяния и спектров ближнего инфракрасного излучения показали, что превращение полиморфа II в полиморф I начиналось примерно через 5 ч и занимало примерно 3 часа. Добавление дополнительного количества полиморфа II после полного превращения в полиморф I приводило к немедленному началу превращения полиморфа II в полиморф I.
Указанный эксперимент повторяли в смеси 2-пропанол/дихлорметан (97/3) (об./об.), но это приводило к идентичным результатам в отношении периода индукции, времени превращения и полиморфизма конечного продукта.
Пример 12: Получение полиморфа II посредством кристаллизации внесением затравки или без затравки
a. 20 мл раствора соединения формулы (I) в дихлорметане (10 л/моль) вносили в колбу объемом 100 мл. Раствор перемешивали при комнатной температуре и добавляли 20 мл изопропанола. Полученный раствор частично упаривали (используя роторный испаритель) в условиях умеренного вакуума (750 мбар) при комнатной температуре вплоть до удаления большей части дихлорметана, получая прозрачный раствор.
b. К 2 мл раствора, полученного согласно пункту a), добавляли в виде затравочного вещества небольшое количество полиморфа I (который получали согласно любому из примеров 3, 10 или 11) при комнатной температуре. Сразу же образовывался объемный осадок белого цвета, который отфильтровывали, промывали 2 мл 2-пропанола и сушили при 60ºC и атмосферном давлении (фракция 9.1).
c. 2 мл раствора, полученного согласно пункту a), охлаждали до 0ºC и перемешивали в течение 14 ч при указанной температуре. Образовывалось некоторое количество клейкого вещества, которое выделяли посредством декантации, промывали и сушили в течение 72 ч при 60ºC и атмосферном давлении. Получали твердое вещество (фракция 9.2).
d. Раствор соединения формулы (I), полученный согласно пункту a), выдерживали 3 дня при комнатной температуре. Образованный осадок отфильтровывали, и выделенное твердое вещество состояло из полусферических частиц вместе с веществом, похожим на тонкие белые иглы. Обе фракции собирали отдельно:
- фракция 9.3: иглоподобное вещество;
- фракция 9.4: твердое вещество полусферической формы.
Оба образца сушили в течение 14 ч при 60ºC и атмосферном давлении.
Анализ XPRD (см. фигуры 27-30) показал, что полученное вещество было кристаллическим.
Пример 13: Определение растворимости формы I и формы II в разных растворителях
Избыток продукта (форма I или форма II в соответствующих случаях) встряхивали с соответствующим растворителем в течение 24 ч при 20ºC. После фильтрования определяли концентрацию продукта в растворе, используя УФ-спектрометрию. Результаты растворимости формы I и формы II показаны в таблице 6 ниже:
н.о. = не определяли.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ КАРБАМОИЛПИРИДОНОВЫХ ИНГИБИТОРОВ ИНТЕГРАЗЫ ВИЧ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2527451C2 |
| Полиморфная форма гиодезоксихолата натрия (NAHDC) и способ ее получения | 2014 |
|
RU2669785C2 |
| КРИСТАЛЛИЧЕСКИЕ СОЛЬВАТЫ ГИДРОХЛОРИДА 6-(ПИПЕРИДИН-4-ИЛОКСИ)-2Н-ИЗОХИНОЛИН-1-OHA | 2012 |
|
RU2619129C2 |
| КРИСТАЛЛ ПРОИЗВОДНОГО БЕНЗОКСАЗОЛА | 2019 |
|
RU2787767C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МОНОГИДРАТА ГИДРОХЛОРИДА (R)-7-ХЛОР-N-(ХИНУКЛИДИН-3-ИЛ)БЕНЗО[b]ТИОФЕН-2-КАРБОКСАМИДА | 2011 |
|
RU2577334C2 |
| НОВЫЕ ПОЛИМОРФЫ И СОЛИ | 2011 |
|
RU2573384C2 |
| КОМПОЗИЦИЯ РЕБАУДИОЗИДА А И СПОСОБ ОЧИСТКИ РЕБАУДИОЗИДА А | 2007 |
|
RU2438353C2 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТРИАЗОЛО (4,5-d) ПИРИМИДИНА | 2005 |
|
RU2418802C2 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ И АМОРФНАЯ ФОРМА ТРИАЗОЛО[4,5-d]ПИРИМИДИНА | 2001 |
|
RU2325391C2 |
| БЕНЗОЙНОКИСЛАЯ СОЛЬ ОТАМИКСАБАНА | 2012 |
|
RU2597423C2 |
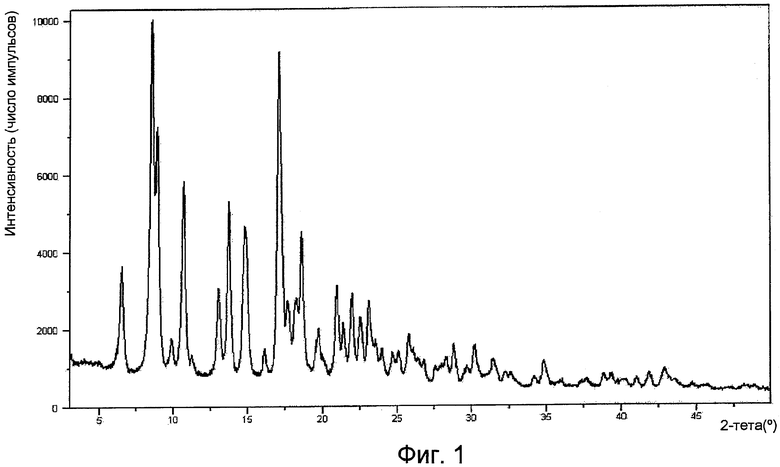
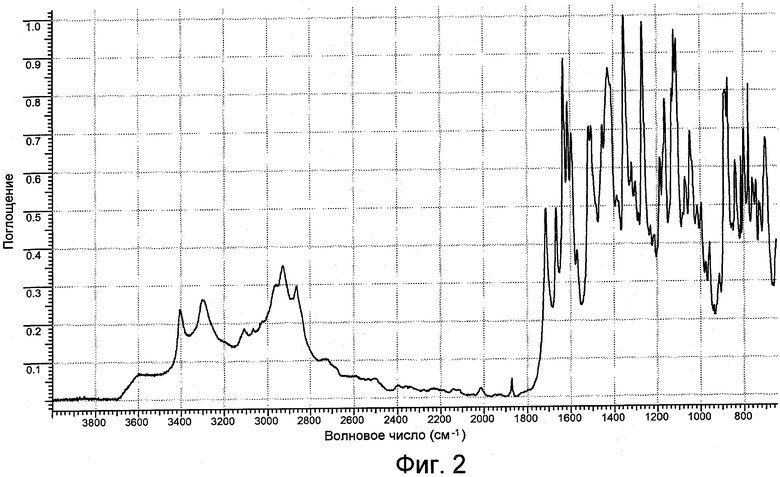

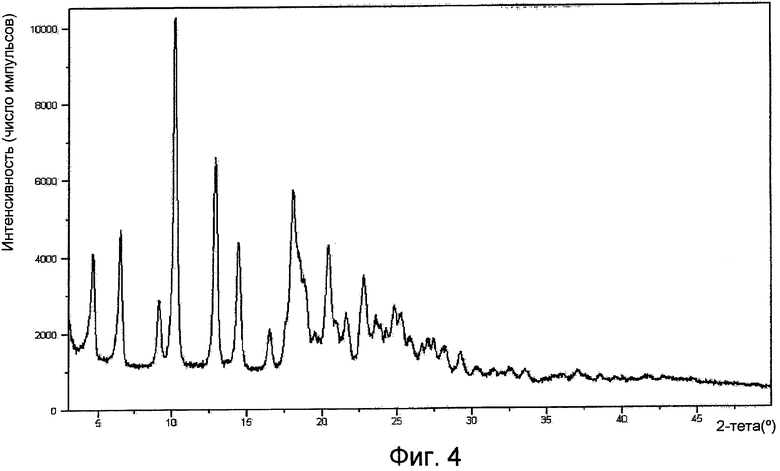
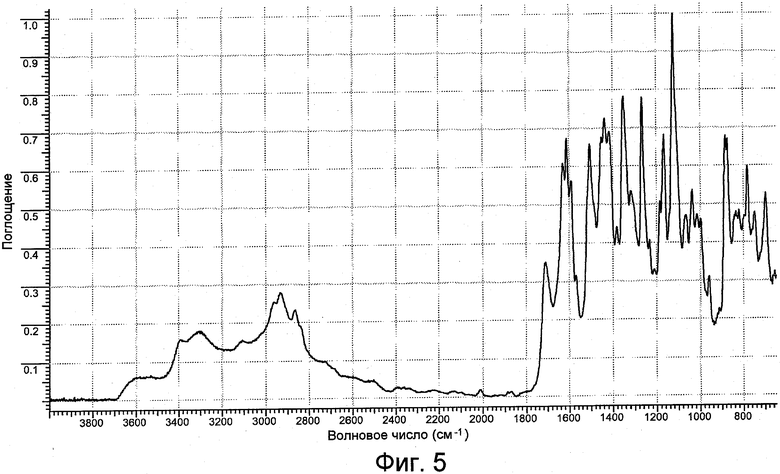
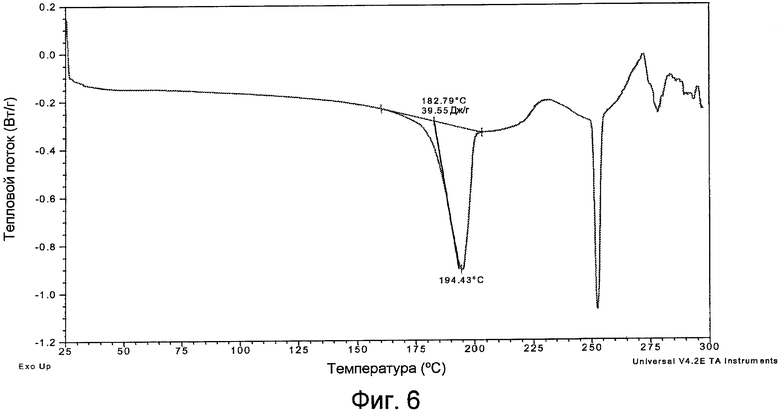
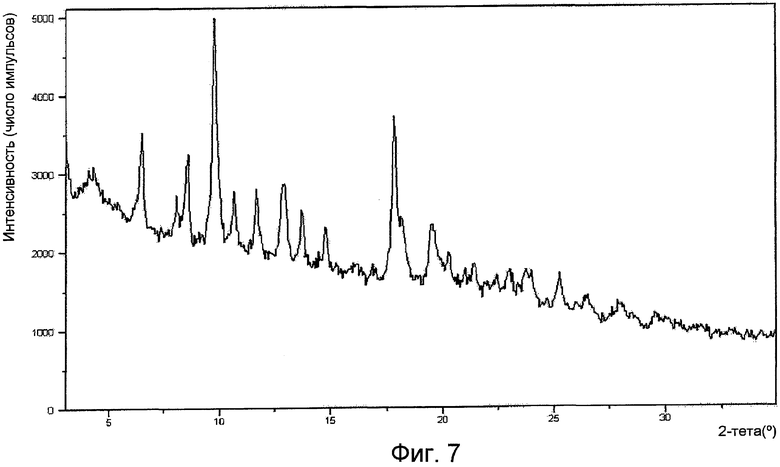
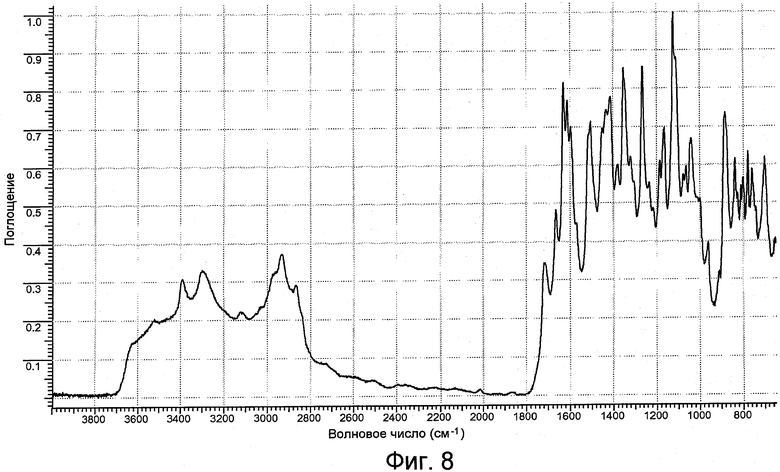

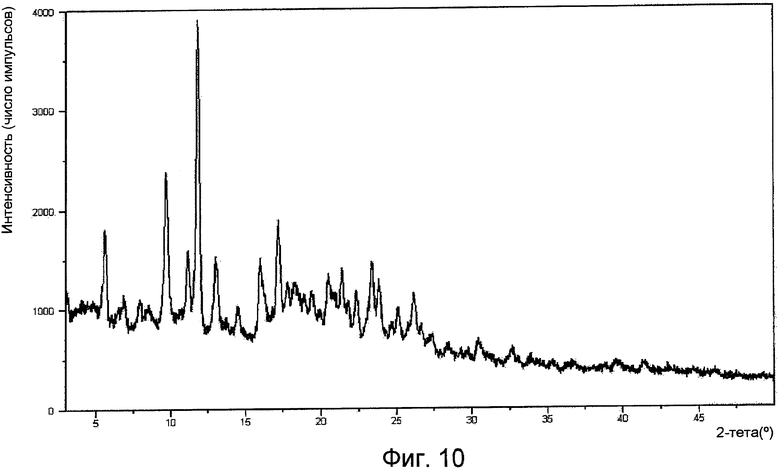
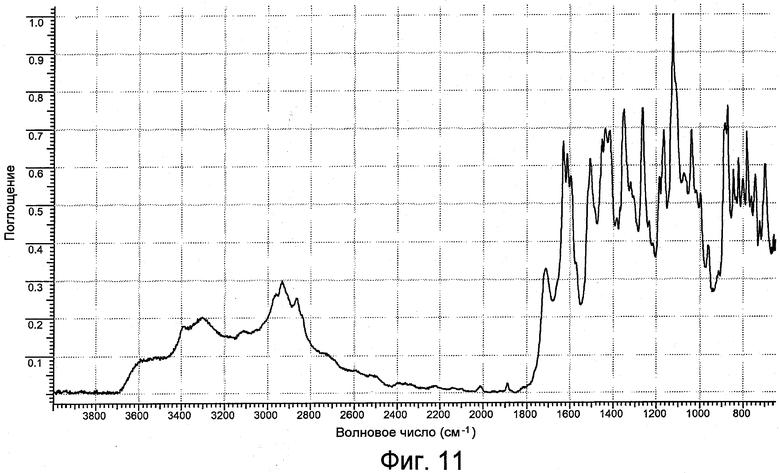
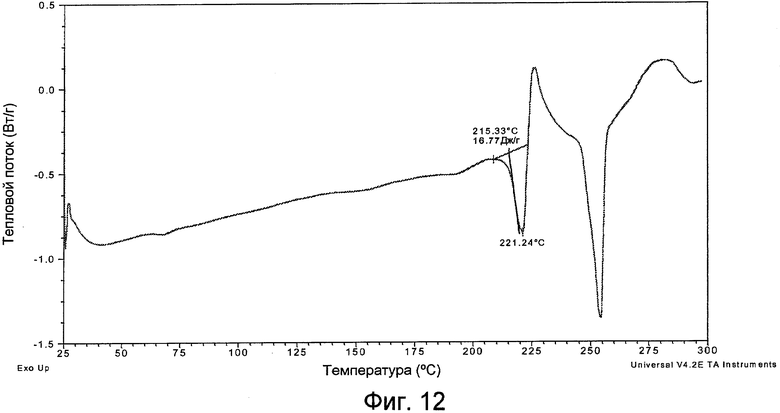
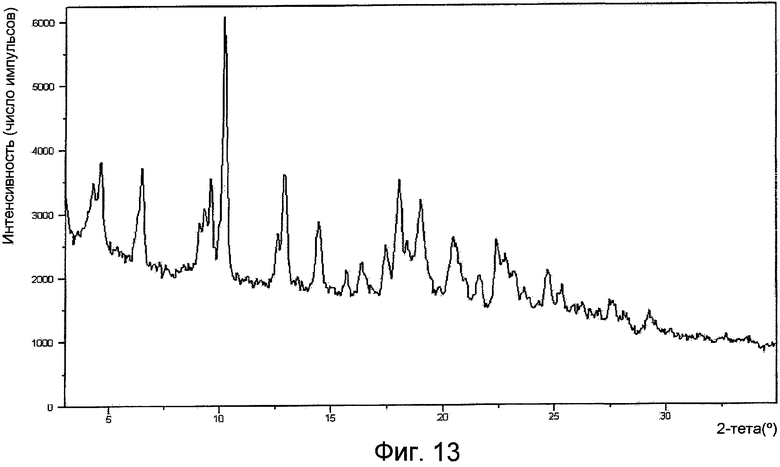
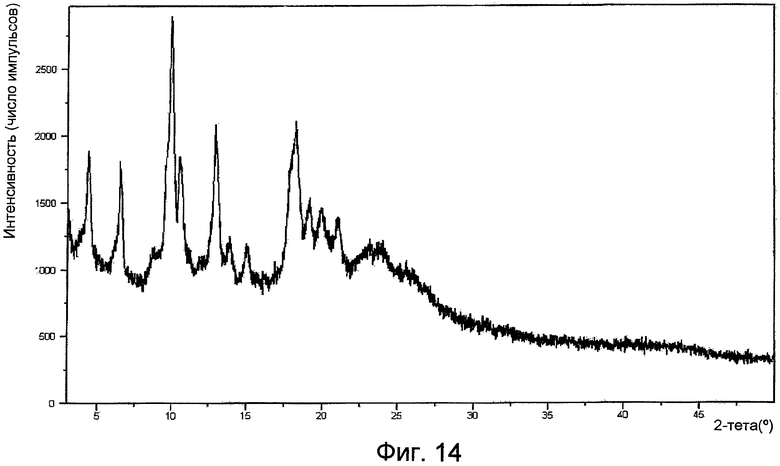
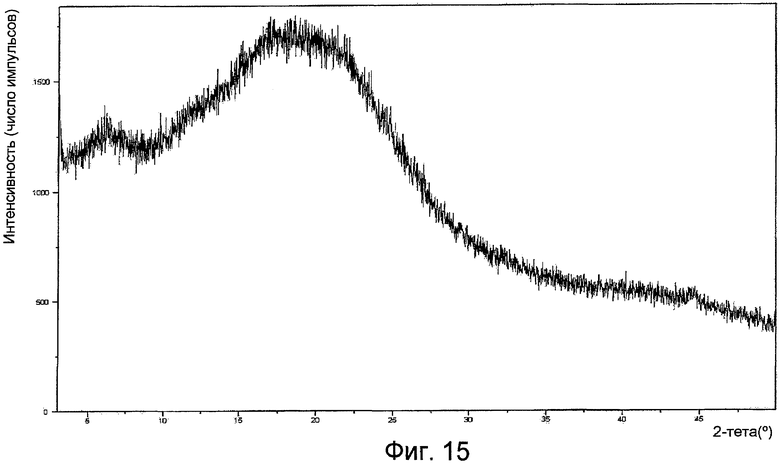
Изобретение относится к кристаллической форме соединения формулы (I) в твердом состоянии, имеющей рентгенограмму порошковой дифракции, характеризующуюся пиками: (i) 8,5°±0,2°, 10,7°±0,2°, 13,7°±0,2°, 14,8°±0,2° и 17,1°±0,2° при угле дифракции два тета (форма I); (ii) 4,6°±0,2°, 6,5°±0,2°, 10,2°±0,2°, 12,9°±0,2° и 14,4°±0,2° при угле дифракции два тета (форма II); (iii) 6,5°±0,2°, 9,8°±0,2° и 17,8°±0,2° при угле дифракции два тета (форма III); (iv) 5,6°±0,2°, 9,6°±0,2°, 11,8°±0,2°, 15,9°±0,2° и 17,1°±0,2° при угле дифракции два тета (форма IV); (v) 9,6°±0,2° и 19,0°±0,2° при угле дифракции два тета (форма V); (vi) 4,4°±0,2°, 6,5°±0,2°, 9,9°±0,2°, 10,5°±0,2° и 12,9°±0,2° при угле дифракции два тета (форма VI). Изобретение также относится к смеси двух или более кристаллических форм I, II, III, IV, V и VI соединения формулы (I) для лечения HCV и к смеси одной или нескольких кристаллических форм I, II, III, IV, V и VI соединения формулы (I) и аморфной формы соединения формулы (I) для лечения HCV. Кристаллическую форму (I) получают: a) кипячением смеси полиморфов I и II в 1-бутаноле или 2-пропаноле с обратным холодильником до получения прозрачного раствора; и b) самопроизвольным охлаждением раствора до комнатной температуры при перемешивании с последующим фильтрованием и извлечением кристаллической формы I. Способ получения кристаллической формы II включает: a) получение суспензии аморфной формы соединения формулы (I) в изопропаноле; b) перемешивание суспензии при комнатной температуре; и c) затравку суспензии затравочными кристаллами формы II или формы I при перемешивании с последующим фильтрованием и сушкой при 60°С. Также предложены варианты способа получения кристаллической формы (I) и (II) и форм (III), (IV) (V) и (VI). Кроме того, изобретение относится к фармацевтической композиции для лечения HCV, содержащей кристаллическую форму соединения формулы (I), смесь двух или более кристаллических форм соединения формулы (I) и фармацевтически приемлемый эксципиент. Технический результат - кристаллическая форма ингибитора сериновой протеазы вируса гепатита С (HCV). 13 н. и 6 з.п. ф-лы, 15 ил., 6 табл., 12 пр.

1. Кристаллическая форма соединения формулы (I):
в твердом состоянии, имеющей рентгенограмму порошковой дифракции, характеризующуюся пиками:
(i) 8,5°±0,2°, 10,7°±0,2°, 13,7°±0,2°, 14,8°±0,2° и 17,1°±0,2° при угле дифракции два тета (форма I);
(ii) 4,6°±0,2°, 6,5°±0,2°, 10,2°±0,2°, 12,9°±0,2° и 14,4°±0,2° при угле дифракции два тета (форма II);
(iii) 6,5°±0,2°, 9,8°±0,2° и 17,8°±0,2° при угле дифракции два тета (форма III);
(iv) 5,6°±0,2°, 9,6°±0,2°, 11,8°±0,2°, 15,9°±0,2° и 17,1°±0,2° при угле дифракции два тета (форма IV);
(v) 9,6°±0,2° и 19,0°±0,2° при угле дифракции два тета (форма V);
(vi) 4,4°±0,2°, 6,5°±0,2°, 9,9°±0,2°, 10,5°±0,2° и 12,9°±0,2° при угле дифракции два тета (форма VI).
2. Соединение по п.1, в котором кристаллическая форма
(i) форма I имеет ИК-картину, характеризующуюся пиками 3405±1 см-1, 3066±1 см-1, 1517±1 см-1, 1427±1 см-1, 1301±1 см-1, 1285±1 см-1, 1149±1 см-1, 1132±1 см-1, 1111±1 см-1, 975±1 см-1, 956±1 см-1 и 800±1 см-1;
(ii) форма II имеет ИК-картину, характеризующуюся пиками 1592±1 см-1;
(iii) форма III имеет ИК-картину, характеризующуюся пиками 3120±1 см-1, 2870±1 см-1 и 1063±1 см-1;
(iv) форма IV имеет ИК-картину, характеризующуюся пиками 1369±1 см-1 и 846±1 см-1.
3. Смесь двух или более кристаллических форм соединения формулы (I) для лечения HCV, в которой кристаллические формы выбраны из формы I, формы II, формы III, формы IV, формы V и формы VI, где формы определены в пп.1 и 2.
4. Смесь по п.3, которая содержит форму II и форму I соединения формулы (I).
5. Смесь по п.3, которая содержит форму III и форму II соединения формулы (I).
6. Смесь одной или нескольких кристаллических форм соединения формулы (I) и аморфной формы соединения формулы (I) для лечения HCV, в которой кристаллические формы выбраны из формы I, формы II, формы III, формы IV, формы V и формы VI, где формы определены в п.1 или 2.
7. Смесь по п.6, которая содержит форму II и аморфную форму соединения формулы (I).
8. Способ получения кристаллической формы (I), определенной по п.1 или 2, включающий:
a) кипячение смеси полиморфов I и II в 1-бутаноле или 2-пропаноле с обратным холодильником до получения прозрачного раствора; и
b) самопроизвольное охлаждение раствора до комнатной температуры при перемешивании с последующим фильтрованием и извлечением кристаллической формы I.
9. Способ получения кристаллической формы (I), определенной по п.1 или 2, включающий:
- суспендирование формы II в спиртовом растворителе, выбранном из 2-пропанола, этанола, 1-бутанола, метанола, смеси спирта (такого как метанол, этанол, пропанол, изопропанол, 1-бутанол или 2-бутанол) при температуре кипения;
перемешивание суспензии при 80°С от 2 до 24 часов;
охлаждение смеси до комнатной температуры и фильтрование; или
- суспендирование смеси формы I и формы II в растворителе, выбранном из 2-пропанола, метилизопропилкетона (MIK), ТГФ, ацетонитрила, этанола, ацетона, 1-метоксипропан-2-ола (1-М-2-Р), метилэтилкетона (МЕК), дихлорметана, 1-бутанола, метанола, смеси спирта (такого как метанол, этанол, пропанол, изопропанол, 1-бутанол или 2-бутанол) и дихлорметана или воды или их смеси, при температуре в диапазоне от примерно 30°С до температуры образования флегмы смеси;
самопроизвольное охлаждение смеси до комнатной температуры при перемешивании, фильтрованием и промыванием твердого вещества.
10. Способ получения кристаллической формы II, определенной по п.1 или 2, включающий:
a) получение суспензии аморфной формы соединения формулы (I) в изопропаноле;
b) перемешивание суспензии при комнатной температуре; и
c) затравку суспензии затравочными кристаллами формы II или формы I при перемешивании с последующим фильтрованием и сушкой при 60°С.
11. Способ получения кристаллической формы II, определенной по п.1 или 2, включающий:
a) растворение соединения формулы (I) в 2-пропаноле; и
b) выдерживание раствора со стадии а) при комнатной температуре в течение по меньшей мере 1 суток или при температуре около 0°С в течение по меньшей мере 4 часов.
12. Способ получения кристаллической формы III, определенной по п.1 или 2, включающий:
a) получение насыщенного или почти насыщенного раствора соединения формулы (I) в ацетонитриле и насыщенного или почти насыщенного раствора соединения формулы (I) в воде при 50°С;
b) выдерживание двух насыщенных или почти насыщенных растворов со стадии а) в течение 1,5 часов при 50°С;
c) смешивание двух насыщенных или почти насыщенных растворов со стадии b) в объемном соотношении 50/50 с последующим выпариванием растворителя.
13. Способ получения кристаллической формы IV, определенной по п.1 или 2, включающий:
a) получение насыщенного или почти насыщенного раствора соединения формулы (I) в 1-метокси-2-пропаноле;
b) кипячение с обратным холодильником насыщенного или почти насыщенного раствора при температуре образования флегмы 1-метокси-2-пропанола;
c) смешивание насыщенного или почти насыщенного раствора со стадии b) с водой в объемном соотношении 4/10;
d) предоставление раствору кристаллизоваться при перемешивании с последующей фильтрацией и сушкой продукта при комнатной температуре.
14. Способ получения кристаллической формы V, определенной по п.1, включающий:
a) получение насыщенного или почти насыщенного раствора соединения формулы (I) в 2-бутаноне и насыщенного или почти насыщенного раствора соединения формулы (I) в воде;
b) нагревание двух насыщенных или почти насыщенных растворов со стадии а) при 50°С в течение 1,5 часов;
c) смешивание двух насыщенных или почти насыщенных растворов со стадии b) в объемном соотношении 50/50;
d) предоставление смеси кристаллизоваться при комнатной температуре с последующим выпариванием растворителя.
15. Способ получения кристаллической формы VI, определенной по п.1, включающий:
a) получение суспензии соединения формулы (I) в воде;
b) нагревание суспензии со стадии а), по меньшей мере, при комнатной температуре в течение по меньшей мере 4 суток.
16. Фармацевтическая композиция для лечения HCV, содержащая кристаллическую форму соединения формулы (I), определенную по п.1 или 2, смесь двух или более кристаллических форм соединения формулы (I), определенную по пп.3-7, и фармацевтически приемлемый эксципиент.
17. Соединение по п.1 или 2 для производства лекарственного средства для лечения HCV.
18. Соединение по п.1 или 2 для лечения вируса гепатита С (HCV).
19. Применение соединения по п.1 или 2 для производства лекарственного средства для лечения HCV.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| EA 200600732 A1, 25.08.2006 | |||
| BERNSTEIN J | |||
| et al: "Concomitant polymorphs", ANGEWANDTE CHEMIE | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| CAIRA M.R.: "CRYSTALLINE POLYMORPHISM OF ORGANIC COMPOUNDS", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, p.163-208 | |||

