Область техники, к которой относится изобретение
Настоящее изобретение относится к кристаллам 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, оказывающим ингибирующее воздействие на фосфодиэстеразу (PDE) 4.
Уровень техники
Фармацевтические лекарственные средства в дополнение к эффективности для заболеваний и безопасности должны обладать однородным качеством и стабильностью при хранении. Следовательно, лекарственные вещества фармацевтических лекарственных средств должны обладать превосходной стабильностью по отношению к нагреванию, воздействию влаги и света.
В литературе отсутствуют сообщения о соединении, обладающем превосходным ингибирующим воздействием на PDE4 и превосходной стабильностью по отношению к нагреванию, воздействию влаги и света.
Сущность изобретения
Техническая задача
Объектом настоящего изобретения является получение лекарственного вещества, пригодного для использования в качестве фармацевтического лекарственного средства, с использованием соединения, обладающего превосходным ингибирующим воздействием на PDE4.
Решение задачи
В результате проведения тщательных исследований по решению описанной выше задачи авторы настоящего изобретения смогли успешно провести кристаллизацию соединения, описывающееся формулой (1) и обладающего химической стабильностью, и обнаружили две разные кристаллические формы (кристалл типа I и кристалл типа II).
Кристалл типа I и кристалл типа II соединения, описывающегося формулой (1), обладают достаточной химической стабильностью по данным теста на термическую стабильность, теста на фотостабильность и ускоренного теста стабильности.
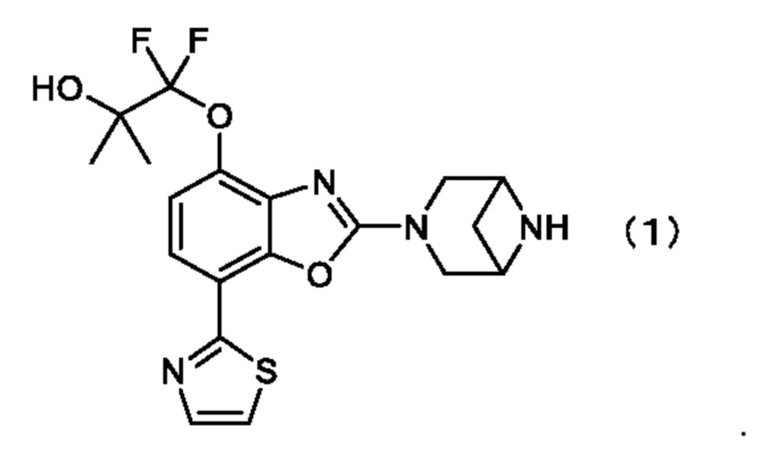
Таким образом, настоящее изобретение относится к кристаллу 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, описывающегося формулой (1) (соединение, описывающееся формулой (1)),
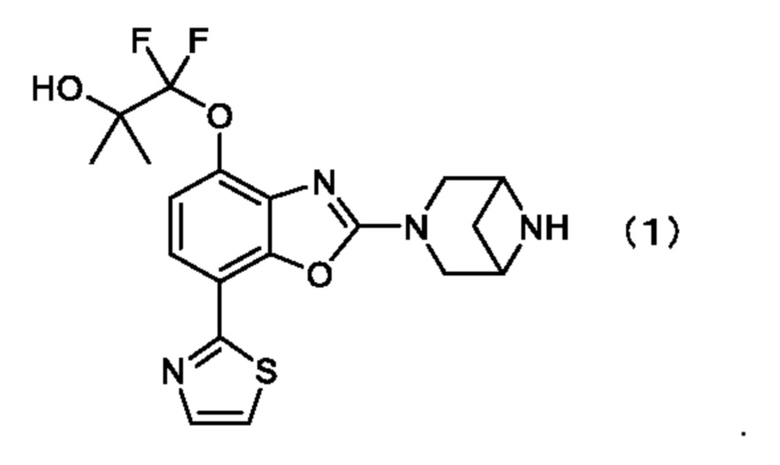 .
.
Полезные эффекты изобретения
Кристаллы, предлагаемые в настоящем изобретении, могут дать лекарственные вещества, пригодные для использования в качестве фармацевтических лекарственных средств.
Кристаллы соединения, описывающегося формулой (1), предлагаемые в настоящем изобретении, применимы для лечения или предупреждения, или для лечения и предупреждения заболеваний, приписываемых влиянию PDE4, или разных заболеваний, связанных с PDE4, поскольку кристаллы обладают превосходным ингибирующим воздействием на PDE4. Примеры заболеваний, приписываемых влиянию PDE4, или заболеваний, связанных с PDE4, включают: разные фиброзные заболевания, такие как астма, COPD, интерстициальную пневмонию, идиопатический фиброз легких, системный склероз и неалкогольный стеатогепатит; воспалительные болезни кишечника, такие как болезнь Крона и язвенный колит; рассеянный склероз; ревматизм; анкилозирующий спондилит; акне; атопический дерматит; гнездную алопецию; аллергический конъюнктивит; сухой кератит; ринит; псориатический артрит; псориаз; саркоидоз; болезнь Бехчета; системную красную волчанку; депрессию; нарушения познавательной способности; болезнь Паркинсона; болезнь Альцгеймера; болезнь Гентингтона; шизофрению; мышечную дистрофию; витилиго; гнойный гидраденит; красный плоский лишай; разные типы рака (например, колоректальный рак, рак легких, рак крови и опухоль головного мозга); и метаболические заболевания (например, диабет и ожирение). Кроме того, кристаллы соединения, описывающегося формулой (1) обладают характеристиками, подходящими для использования в качестве фармацевтических лекарственных средств, такими как растворимость, гигроскопичность и стабильность в растворе.
Краткое описание чертежей
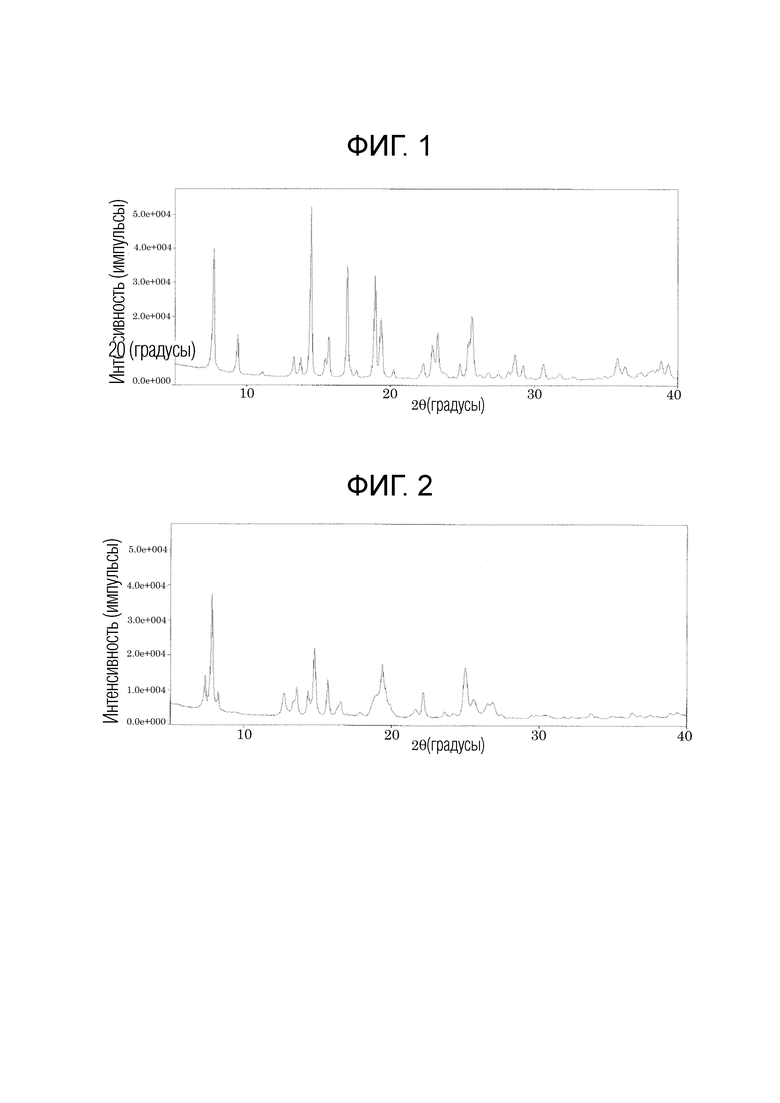
На фиг. 1 приведена порошковая рентгенограмма кристалла типа I соединения, описывающегося формулой (1);
На фиг. 2 приведена порошковая рентгенограмма кристалла типа II соединения, описывающегося формулой (1); и
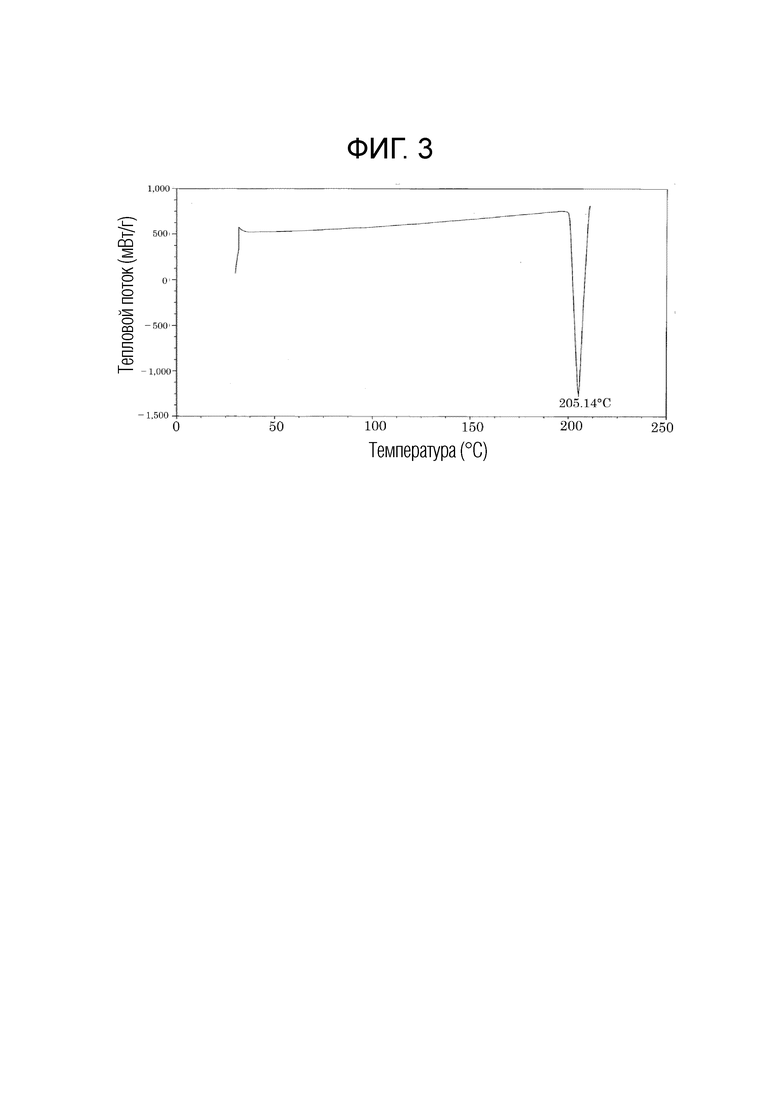
На фиг. 3 приведены данные термического анализа с помощью дифференциальной сканирующей калориметрии (DSC) кристалла типа I соединения, описывающегося формулой (1).
Описание вариантов осуществления
Полиморфизм кристаллов, это явление, при котором образуются два или большее количества типов кристаллов соединения. Обычно известно, что разные полиморфные кристаллические формы кристаллов могут обладать разной стабильностью и физическими характеристиками. Кроме того, если имеются полиморфы кристаллов, обычно может происходить переход кристалл-кристалл. Переход кристалл-кристалл, это явление, часто наблюдающееся, например, при сушке, размоле и хранении в химической промышленности. Кристаллы, для которых не происходит переход кристалл-кристалл в другую кристаллическую форму, являются подходящими для использования в качестве лекарственных веществ фармацевтических лекарственных средств. Следовательно, если получают множество кристаллических форм, важно проверять стабильность каждой.
В настоящем описании эти две разные кристаллические формы называют кристаллом типа I и кристаллом типа II соответственно.
Настоящее изобретение относится к кристаллам 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола (соединение, описывающееся формулой (1)) и содержащим кристаллы фармацевтическим композициям. Соединение, описывающееся формулой (1), можно получить по методике, описанной в Международной публикации № WO 2018/124060 (Международная публикация № PCT/JP2017/046610). Однако методика получения не ограничивается этой методикой.
В настоящем изобретении кристалл типа I на порошковой рентгенограмме обладает характеристическими пиками при углах дифракции (2θ±0,2°), равных 7,7°, 14,5°, 17,0°, 18,9°, 19,4°, 23,3° и 25,7°, и по данным дифференциальной сканирующей калориметрии (DSC) обладает эндотермическим пиком при температуре, равной 205±3°C. Предпочтительно, кристалл типа I обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2θ±0,2°), равных 7,7°, 9,4°, 13,2°, 13,7°, 14,5°, 15,7°, 17,0°, 18,9°, 19,4°, 22,9°, 23,3°, 25,7°, 28,7° и 35,7°, и по данным дифференциальной сканирующей калориметрии (DSC) обладает эндотермическим пиком при температуре, равной 205±3°C.
В настоящем изобретении кристалл типа II обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,8°, 14,7°, 15,7°, 19,3° и 25,0°. Предпочтительно, если кристалл типа II обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,4°, 7,8°, 8,2°, 12,7°, 13,6°, 14,3°, 14,7°, 15,7°, 16,5°, 19,3°, 22,1°, 25,0° и 25,6°.
Кристаллы можно отличить друг от друга, например, с помощью порошковой рентгенографии. Значения углов дифракции на порошковой рентгенограмме могут обладать погрешностями в диапазоне ±0,2° вследствие, например, различий приборов и различия методик анализа. Кроме того, относительные интенсивности пиков на порошковой рентгенограмме могут меняться, например, в зависимости от габитусов кристаллов или условий отбора проб. На порошковой рентгенограмме углы дифракции и общий вид рентгенограммы важны для идентификации кристаллов и они могут немного меняться в зависимости от условий измерений. Кроме того, значения температур для эндотермических пиков, полученные с помощью дифференциальной сканирующей калориметрии (DSC), могут обладать погрешностями в диапазоне ±3°C вследствие, например, различий приборов и различий количеств образцов. Кроме того, при дифференциальной сканирующей калориметрии (DSC), общий вид диаграммы важен для идентификации кристаллов и он может немного меняться в зависимости от условий измерений. Температура для эндотермического пика в дифференциальной сканирующей калориметрии представляет собой температуру для вершины части эндотермического пика.
Кристалл типа I можно получить по разным методикам и можно получить по следующей методике.
Кристалл типа I можно получить путем кристаллизации соединения, описывающегося формулой (1), которое получают по методике, описанной в Международной публикации № WO 2018/124060 (Международная публикация № PCT/JP2017/046610), например, из одного растворителя, выбранного из следующей группы, включающей, или смеси двух или большего количества растворителей, выбранных из группы, включающей спиртовые растворители, сложноэфирные растворители, галогенсодержащие растворители или воду. На количество растворителя не налагают особые ограничения и оно предпочтительно представляет собой, например, количество (об./мас.), которое в 1-200 раз больше массы соединения, описывающегося формулой (1). На температуру не налагают особые ограничения и она предпочтительно равна, например, от 10°C до температуры кипения использующегося растворителя. При кристаллизации раствор соединения, описывающегося формулой (1), в растворителе, описанном выше, обрабатывают с помощью одной или большего количества операций, выбранных из группы, включающей охлаждение, концентрирование, добавление спиртового растворителя, добавление сложноэфирного растворителя, добавление смешанного растворителя, добавление алифатического углеводородного растворителя или добавление воды. На температуру, до которой проводят охлаждение, не налагают особые ограничения и она предпочтительно равна, например, от −10°C до 10°C. На концентрацию не налагают особые ограничения и она предпочтительно представляет собой, например, количество (об./мас.), которое в 1-10 раз больше массы соединения, описывающегося формулой (1). На количество спиртового растворителя, сложноэфирного растворителя, смешанного растворителя, алифатического углеводородного растворителя и воды не налагают особые ограничения и оно предпочтительно представляет собой, например, количество (об./мас.), которое в 0,01-100 раз больше массы соединения, описывающегося формулой (1). Кроме того, после добавления спиртового растворителя или сложноэфирного растворителя, или смешанного растворителя полученный кристалл можно нагреть и дать ему охладиться.
Кристалл типа II можно получить по разным методикам и можно получить по следующей методике.
Кристалл типа II можно получить путем растворения соединения, описывающегося формулой (1), которое получают по методике, описанной в Международной публикации № WO 2018/124060 (Международная публикация № PCT/JP2017/046610), например, в одном из растворителей, выбранном из следующей группы, включающей, или смеси двух или большего количества растворителей, выбранных из группы, включающей спиртовые растворители, сложноэфирные растворители, галогенсодержащие растворители или воду, с последующим охлаждением и кристаллизацией соединения. На количество растворителя не налагают особые ограничения и оно предпочтительно представляет собой, например, количество (об./мас.), которое в 1-200 раз больше массы соединения, описывающегося формулой (1). На температуру для растворения соединения, описывающегося формулой (1), не налагают особые ограничения и она предпочтительно равна, например, от 10°C до температуры кипения использующегося растворителя. На охлаждение для кристаллизации не налагают особые ограничения и его предпочтительно проводят при температуре от −30°C до 10°C и оно предпочтительно является быстрым охлаждением. Затем суспензию, в которой кристаллы осадились путем охлаждения, можно обработать с помощью одной или большего количества операций, выбранных из группы, включающей концентрирование, добавление спиртового растворителя, добавление сложноэфирного растворителя, добавление смешанного растворителя, добавление алифатического углеводородного растворителя или добавление воды. На концентрацию не налагают особые ограничения и она предпочтительно представляет собой, например, количество (об./мас.), которое в 1-10 раз больше массы соединения, описывающегося формулой (1). На количество спиртового растворителя, сложноэфирного растворителя, смешанного растворителя, алифатического углеводородного растворителя и воды не налагают особые ограничения и оно предпочтительно представляет собой, например, количество (об./мас.), которое в 0,01-100 раз больше массы соединения, описывающегося формулой (1).
Спиртовые растворители представляют собой, например, метанол, этанол, 2-пропанол и н-бутанол. Метанол, этанол или 2-пропанол является предпочтительным. Один из этих спиртовых растворителей можно использовать по отдельности или два или большее количество из этих спиртовых растворителей можно использовать в комбинации.
Сложноэфирные растворители представляют собой, например, метилформиат, этилформиат, метилацетат, этилацетат и изопропилацетат. Один из этих сложноэфирных растворителей можно использовать по отдельности или два или большее количество из этих сложноэфирных растворителей можно использовать в комбинации.
Галогенсодержащие растворители представляют собой, например, дихлорметан, хлороформ и 1,2-дихлорэтан. Один из этих галогенсодержащих растворителей можно использовать по отдельности или два или большее количество из этих галогенсодержащих растворителей можно использовать в комбинации.
Алифатические углеводородные растворители представляют собой, например, пентан, гексан, циклогексан и гептан. Гексан или гептан является предпочтительным. Один из этих алифатических углеводородных растворителей можно использовать по отдельности или два или большее количество из этих алифатических углеводородных растворителей можно использовать в комбинации.
Смесь растворители представляет собой, например, смесь одного или большего количества растворителей, выбранных из группы, включающей спиртовые растворители, сложноэфирные растворители, галогенсодержащие растворители, алифатические углеводородные растворители или воду. Смешанный растворитель, включающий метанол и этилацетат, этанол и этилацетат, 2-пропанол и этилацетат, или гексан и этилацетат является предпочтительным. Диапазон отношений количеств растворителей в смеси растворителей составляет от 1/10 до 10/1 (об./об.) и предпочтительно от 1/2 до 2/1 (об./об.).
Примеры
Настоящее изобретение специально описано ниже с помощью примеров. Однако объем настоящего изобретения не следует считать ограниченным этими примерами.
Для порошковой рентгенографии использовали прибор SMARTLAB фирмы Rigaku Corporation (источник излучения: CuKα, длина волны: 1,541862 ангстрем, скорость сканирования: 1,0039°/мин, ширина шага: 0,0100°, выход рентгеновского излучения: 40 кВ, 30 мА и температура, при которой проводят измерения: комнатная температура). Для дифференциальной сканирующей калориметрии (DSC) использовали прибор Q200 фирмы TA Instruments Japan Inc. (скорость повышения температуры: 5°C/мин, скорость потока азота: 50 мл/мин, и чашка: обычная герметизация).
Соединение, описывающееся формулой (1), можно получить по методике, описанной в Международной публикации № WO 2018/124060 (Международная публикация № PCT/JP2017/046610) и приведенной ниже.
Типичный пример синтеза 1
Синтез соединения, описывающегося формулой (1)
(Стадия 1) (((2-Нитро-1,3-фенилен)бис(окси))бис(метилен))дибензол
2-Нитрорезорцин (5 г) растворяли в N, N-диметилформамиде (88 мл), бензилбромид (8,4 мл, 2,2 экв.) и карбонат цезия (25 г, 2,4 экв.) добавляли к полученному продукту и полученный продукт перемешивали при комнатной температуре в течение 12 ч. К реакционной смеси добавляли этилацетат, органический слой промывали 1% водным раствором хлористоводородной кислоты и органический слой повторно промывали дистиллированной водой. Органический слой сушили над безводным сульфатом магния и затем фильтровали. К остатку, полученному концентрированием фильтрата в вакууме, добавляли гексан и осадившееся твердое вещество отфильтровывали и получали искомое соединение (10 г).
(Стадия 2) 3-(Бензилокси)-2-нитрофенол
Соединение (10 г), полученное на стадии 1, растворяли в дихлорметане (270 мл), к полученному продукту при −78°C добавляли 1,0 M раствор трихлорида бора в гептане (45 мл, 1,5 экв.) и полученный продукт перемешивали при −78°C в течение 1 ч. К реакционной смеси добавляли метанол в течение 10 мин, полученный продукт нагревали до комнатной температуры и к полученному продукту добавляли дистиллированную воду. Полученную смесь дважды экстрагировали дихлорметаном и органический слой сушили над безводным сульфатом магния. Полученный продукт фильтровали. Остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (смесью гексан: этилацетат в отношении 9,5:0,5) и получали искомое соединение (4,7 г).
(Стадия 3) 3-(Бензилокси)-6-бром-2-нитрофенол
Ацетонитрил (41 мл), хлортриметилсилан (0,16 мл, 0,1 экв.) и N-бромсукцинимид (2,2 г, 1,0 экв.) добавляли к соединению (3,0 г), полученному на стадии 2, и полученный продукт перемешивали при комнатной температуре в течение 1 ч. К реакционной смеси добавляли воду при 0°C, полученный продукт экстрагировали этилацетатом и затем органический слой сушили над безводным сульфатом магния. Полученный продукт фильтровали, остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (смесью гексан: этилацетат в отношении 7:3) и получали искомое соединение (3,1 г).
(Стадия 4) 2-Амино-3-(бензилокси)-6-бромфенол
Раствор (37 мл) соединения (3,1 г), полученного на стадии 3, в этаноле по каплям добавляли к раствору (63 мл) дитионита натрия (3,1 г, 8 экв.) в воде при 0°C. После того, как температура полученного продукта становилась комнатной, к полученному продукту добавляли воду (30 мл) и этанол (18 мл) и полученный продукт перемешивали в течение 1 ч и 20 мин. Жидкую реакционную смесь фильтровали и промывали этанолом. К остатку, полученному концентрированием фильтрата в вакууме, добавляли воду (67 мл) при 0°C и полученный продукт перемешивали. Твердое вещество отфильтровывали, промывали дистиллированной водой и этилацетатом и сушили в вакууме, и получали искомое соединение (3,3 г).
(Стадия 5) 4-(Бензилокси)-7-бромбензо[d]оксазол-2-тиол
Соединение (3,3 г), полученное на стадии 4, растворяли в этаноле (15 мл), 0,5 M раствор гидроксида калия в этаноле (35 мл) и дисульфид углерода (2,9 мл, 5 экв.) добавляли к полученному продукту и полученный продукт нагревали при 50°C в течение 1 ч и 20 мин. После того, как температура полученного продукта становилась комнатной, к полученному продукту добавляли воду (68 мл) и 5M хлористоводородную кислоту (6 мл). Твердое вещество отфильтровывали и получали искомое соединение (2,3 г).
(Стадия 6) трет-бутил-3-(4-(бензилокси)-7-бромбензо[d]оксазол-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат
м-Ксилол (17 мл) добавляли к соединению (2,3 г), полученному на стадии 5, и трет-бутил-3,8-диазабицикло[3,2,1]октан-8-карбоксилату (1,5 г, 1,1 экв.) и полученный продукт перемешивали при 120°C в течение ночи. К полученному продукту добавляли 1 н. водный раствор гидроксида натрия (20 мл) и полученный продукт экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем полученный продукт фильтровали. Остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (от гексан: этилацетат=50:1 до гексан: этилацетат=7:3) и получали искомое соединение (2,9 г).
(Стадия 7) трет-бутил-3-(4-(бензилокси)-7-(тиазол-2-ил)бензо[d]оксазол-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат
Толуол (19 мл) добавляли к соединению (2,9 г), полученному на стадии 6, к полученному продукту добавляли 0,5 M раствор 2-тиазолилбромидцинка в тетрагидрофуране (29 мл, 2,5 экв.) и комплекс дихлорид 1,1’-бис(дифенилфосфино)ферроценпалладия(II)/дихлорметан (480 мг, 0,1 экв.) и полученный продукт перемешивали в атмосфере аргона при 90°C в течение 6 ч. К жидкой реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия и полученный продукт фильтровали через целит. После экстракции фильтрата этилацетатом органический слой сушили над безводным сульфатом магния. После фильтрования полученного продукта остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (от гексан: этилацетат=9:1 до гексан: этилацетат=1:1) и получали искомое соединение (2,5 г).
(Стадия 8) трет-бутил-3-(4-гидрокси-7-(тиазол-2-ил)бензо[d]оксазол-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат
Соединение (1,5 г), полученное на стадии 7,растворяли в тетрагидрофуране (60 мл), к полученному продукту в атмосфере аргона добавляли 20% гидроксид палладия/уголь (при содержании воды, равном 50%) (2,5 г) и сосуд с продуктом заполняли водородом и перемешивали при 50°C в течение 4,5 ч. Жидкую реакционную смесь фильтровали через целит, остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (от хлороформа до хлороформ: метанол=94:6) и получали искомое соединение (1,0 г).
(Стадия 9) трет-бутил-3-(4-(2-этокси-1,1-дифтор-2-оксоэтокси)-7-(тиазол-2-ил)бензо[d]оксазол-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат
Соединение (1,0 г), полученное на стадии 8, растворяли в ацетонитриле (24 мл), к полученному продукту добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (3,6 мл, 10 экв.) и этил-2-бром-2,2-дифторацетат (3,1 мл, 10 экв.) и полученный продукт перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и полученный продукт трижды экстрагировали этилацетатом. Затем органический слой сушили над безводным сульфатом магния, полученный продукт фильтровали. Остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (от гексан до гексан: этилацетат=5:5) и получали искомое соединение (1,0 г).
(Стадия 10) трет-бутил-3-(4-(1,1-дифтор-2-гидрокси-2-метилпропокси)-7-(тиазол-2-ил)бензо[d]оксазол-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат
Соединение (92 мг), полученное на стадии 9, растворяли в тетрагидрофуране (1,7 мл) и к полученному продукту при 0°C добавляли 0,95 M раствор метилмагнийбромида в тетрагидрофуране (0,85 мл, 5 экв.). Полученный продукт нагревали до комнатной температуры и затем перемешивали в течение 1 ч, к полученному продукту добавляли насыщенный водный раствор хлорида аммония и полученный продукт трижды экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем полученный продукт фильтровали. Остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на силикагеле (от гексана до гексан: этилацетат до этилацетата) и получали искомое соединение (82 мг).
(Стадия 11) Синтез 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола (соединение, описывающееся формулой (1))
Хлороформ (5,9 мл) добавляли к соединению (307 мг), полученному на стадии 10, к полученному продукту при 0°C добавляли трифторуксусную кислоту (1,4 мл) и полученный продукт перемешивали при 0°C в течение 3,5 ч. К жидкой реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия и полученный продукт экстрагировали хлороформом. Затем органический слой сушили над безводным сульфатом магния, полученный продукт фильтровали. Остаток, полученный концентрированием фильтрата в вакууме, очищали с помощью колоночной хроматографии на аминированном силикагеле (от хлороформа до хлороформ: метанол=9:1), и получали соединение, описывающееся формулой (1) в форме белого твердого вещества (223 мг).
ESI-MS (m/z) 423(M+H)+
1H-NMR (хлороформ-d, TMS) δ (част./млн): 1,53 (s, 6H), 1,65 (d, J=9,3 Hz, 1H), 2,82-2,87 (m, 1H), 3,90-4,01 (m, 7H), 7,23-7,25 (m, 1H), 7,44 (d, J=3,3 Hz, 1H), 7,84 (d, J=8,8 Hz, 1H), 7,94 (d, J=3,3 Hz, 1H).
Пример 1
Смешанный растворитель (1:1, об./об.) (21,9 мл), включающий этилацетат и 2-пропанол, добавляли к твердому (219 мг) соединению, описывающемуся формулой (1), полученному в типичном примере синтеза 1, и нагревали при 60°C для растворения соединения. Этот раствор концентрировали досуха и получали остаток, к которому добавляли смешанный растворитель (1:1, об./об.) (4,38 мл), включающий этилацетат и 2-пропанол и полученный продукт перемешивали при 45°C в течение 1 ч. После того, как температура полученного продукта становилась комнатной, к полученному продукту добавляли гексан (21,9 мл) и полученный продукт перемешивали в течение 1 ч. После фильтрования, полученный продукт промывали гексаном и сушили, и получали кристалл типа I (194 мг). Полученный кристалл типа I обладал на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,7°, 9,4°, 13,2°, 13,7°, 14,5°, 15,7°, 17,0°, 18,9°, 19,4°, 22,9°, 23,3°, 25,7°, 28,7° и 35,7°. Порошковая рентгенограмма кристалла типа I приведена на фиг. 1. С помощью анализа полученного кристалла с помощью дифференциальной сканирующей калориметрии (DSC) наблюдали эндотермический пик при 205°C. Данные термического анализа с помощью дифференциальной сканирующей калориметрии (DSC) кристалла типа I приведены на фиг. 3.
Пример 2
Метанол (50 мл) добавляли к кристаллам типа I (584 мг), полученным в примере 1, для растворения кристаллов типа I. Массу полученного гомогенного раствора уменьшали примерно до одной шестой путем концентрирования в вакууме, его охлаждали до 0°C и перемешивали в течение 10 мин для осаждения твердого вещества, затем концентрировали досуха и получали остаток, к которому добавляли смешанный растворитель (2:1, об./об.) (35 мл), включающий гексан и этилацетат. Полученный продукт перемешивали при комнатной температуре в течение 30 мин. После фильтрования полученный продукт промывали смешанным растворителем (2:1, об./об.), включающим гексан и этилацетат, и сушили, и получали кристалл типа II (566 мг). Полученный кристалл типа II обладал на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,4°, 7,8°, 8,2°, 12,7°, 13,6°, 14,3°, 14,7°, 15,7°, 16,5°, 19,3°, 22,1°, 25,0° и 25,6°. Порошковая рентгенограмма кристалла типа II приведена на фиг. 2.
Пример теста 1: Исследование ингибирования PDE4
???Активность ингибирования PDE4 определяли следующим образом с использованием проксимально-сцинтилляционного анализа (SPA). Образец соединения растворяли в диметилсульфоксиде и разводили в 10 раз буферным раствором для проведения реакции, содержащим 50 мМ Tris-HCl (pH=7,4), 8,3 мМ MgCl2, 1,7 мМ EGTA, и к нему добавляли 10 мкл 3 мг/мл бычьего сывороточного альбумина (BSA) и помещали в лунки 96-луночного планшета для анализа. Затем дополнительно добавляли 50 мкл PDE4, разведенного в 375 раз буферным раствором для проведения реакции, затем добавляли 40 мкл триэтиламмониевой соли [2,8-3H]-аденозин-3’,5’-циклического фосфата, разведенной в 1000 раз буферным раствором для проведения реакции, и полученную смесь выдерживали при комнатной температуре в течение 120 мин. Затем добавляли 50 мкл суспензии связывающихся с RNA гранул YSi-SPA, содержащих 200 мМ ZnSO4, и полученную смесь выдерживали при комнатной температуре в течение 15 мин для адсорбции гранулами продукта ферментативной реакции. Затем жидкостным сцинтилляционным счетчиком для 96-луночного планшета измеряли радиоактивность. Степень ингибирования соединением, описывающимся формулой (1), по сравнению с контролем рассчитывали по приведенной ниже формуле, причем холостой раствор представлял собой только буферный раствор для проведения реакции без добавления препарата фермента и контрольный раствор готовили с добавлением препарата фермента и только диметилсульфоксида вместо раствора образца.
Степень ингибирования (%) = {1−(значение, полученное при добавлении образца - значение для холостого раствора)/( значение для контрольного раствора - значение для холостого раствора)}×100
Активность ингибирования PDE4 (концентрация при степени ингибирования, равной 50%) для образца соединения рассчитывали по зависимости для ингибирования, основанной на степенях ингибирования при разных концентрациях.
По данным методики, описанной выше, активность ингибирования PDE4 (концентрация при степени ингибирования, равной 50%) для соединения, описывающегося формулой (1) была меньше 100 нМ.
Пример теста 2: Тест на стабильность
Кристаллы типа I и кристаллы типа II соединения, описывающегося формулой (1), помещали в стеклянные флаконы (примерно по 300 мг в каждый) и хранили при разных условиях. После некоторого периода хранения химическую чистоту отобранных образцов определяли с помощью HPLC. Условия для каждого теста были следующими.
Тест на термическую стабильность: 60°C, герметично закрытая емкость, продолжительность 3 недели
Тест на фотостабильность: 25°C, 1,2 млн люкс/часов (2000 люкс в течение 25 дней)
Ускоренный тест на стабильность (герметично закрытая емкость): 40°C, 75% RH*, продолжительность 1 месяц
Ускоренный тест на стабильность (открытая емкость): 40°C, 75% RH*, продолжительность 1 месяц
*RH: относительная влажность
В результате исследования каждой стабильности количество соединения, описывающегося формулой (1), содержащегося в кристалле, приведено в таблице 1 в виде остаточного отношения (отношение площади по данным HPLC к начальному значению, выраженное в процентах).
Таблица 1
Остаточное отношение для соединения, описывающегося формулой (1), в каждой кристаллической форме было большим и обнаружена высокая стабильность.
Пример теста 3: Тест на гигроскопичность
Проводили исследование изотермической адсорбции для кристалла типа I и кристалла типа II соединения, описывающегося формулой (1), с использованием прибора для измерения адсорбции паров воды (фирмы Surface Measurement Systems Ltd., DVS ADVANTAGE 1) (с использованием образца массой примерно 10 мг, при 25°C, при RH от 0% до 95%).
Определенные с помощью исследования гигроскопичности степени увеличения массы при 95% RH (увеличение массы по сравнению с начальным значением/начальное значение×100, *начальное значение: равновесное значение массы при 0% RH) приведены в таблице 2.
Таблица 2
Объектами настоящего изобретения являются, например, следующие.
1. Кристалл 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, описывающегося формулой (1) (соединение, описывающееся формулой (1)),
.
2. Кристалл по параграфу 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,7°, 14,5°, 17,0°, 18,9°, 19,4°, 23,3° и 25,7°.
3. Кристалл по параграфу 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,7°, 9,4°, 13,2°, 13,7°, 14,5°, 15,7°, 17,0°, 18,9°, 19,4°, 22,9°, 23,3°, 25,7°, 28,7° и 35,7°.
4. Кристалл по параграфу 2 или 3, где кристалл по данным дифференциальной сканирующей калориметрии (DSC) обладает эндотермическим пиком при температуре, равной 205±3°C.
5. Кристалл по параграфу 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,8°, 14,7°, 15,7°, 19,3° и 25,0°.
6. Кристалл по параграфу 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,4°, 7,8°, 8,2°, 12,7°, 13,6°, 14,3°, 14,7°, 15,7°, 16,5°, 19,3°, 22,1°, 25,0° и 25,6°.
7. Фармацевтическая композиция, включающая
кристалл по любому из параграфов 1-6.
Промышленное применение
Кристаллы 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, полученные в настоящем изобретении, применимы в качестве фармацевтических лекарственных средств, поскольку они обладают высокой стабильностью.
1. Кристалл 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, описывающегося формулой (1),
.
2. Кристалл по п. 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2θ±0,2°), равных 7,7°, 14,5°, 17,0°, 18,9°, 19,4°, 23,3° и 25,7°.
3. Кристалл по п. 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2θ±0,2°), равных 7,7°, 9,4°, 13,2°, 13,7°, 14,5°, 15,7°, 17,0°, 18,9°, 19,4°, 22,9°, 23,3°, 25,7°, 28,7° и 35,7°.
4. Кристалл по п. 2, где кристалл по данным дифференциальной сканирующей калориметрии (DSC) обладает эндотермическим пиком при температуре, равной 205±3°C.
5. Кристалл по п. 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2θ±0,2°), равных 7,8°, 14,7°, 15,7°, 19,3° и 25,0°.
6. Кристалл по п. 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2θ±0,2°), равных 7,4°, 7,8°, 8,2°, 12,7°, 13,6°, 14,3°, 14,7°, 15,7°, 16,5°, 19,3°, 22,1°, 25,0° и 25,6°.
7. Фармацевтическая композиция, включающая:
кристалл по любому из п.п. 1-6.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2017 |
|
RU2767878C2 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА СОЕДИНЕНИЯ-БЛОКАТОРА ПРОНИЦАЕМОСТИ СОСУДОВ | 2020 |
|
RU2809162C2 |
| КРИСТАЛЛ СОЕДИНЕНИЯ ПИРИМИДИНА | 2021 |
|
RU2828232C1 |
| КРИСТАЛЛЫ 3,5-ДИЗАМЕЩЕННОГО БЕНЗОЛАЛКИНИЛЬНОГО СОЕДИНЕНИЯ | 2016 |
|
RU2672563C1 |
| НОВЫЕ КРИСТАЛЛЫ 4-{3-[4-(3-{4-[АМИНО(БУТОКСИКАРБОНИЛ-ИМИНО)МЕТИЛ]ФЕНОКСИ}ПРОПИЛ)-1-ПИПЕРИДИНИЛ]ПРОПОКСИ}-N'-(БУТОКСИКАРБОНИЛ)БЕНЗАМИДИНА | 2008 |
|
RU2456272C2 |
| СОЛЬ И КРИСТАЛЛИЧЕСКАЯ ФОРМА СОЕДИНЕНИЯ, ОБЛАДАЮЩЕГО АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К РЕЦЕПТОРУ S1P | 2020 |
|
RU2833357C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТИАЗОЛЬНОГО ПРОИЗВОДНОГО | 2016 |
|
RU2738937C2 |
| КРИСТАЛЛИЧЕСКОЕ ВЕЩЕСТВО ПРОИЗВОДНОГО ГЕТЕРОЦИКЛИДЕНАЦЕТАМИДА | 2018 |
|
RU2779198C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СОЛЬ 3-(3,5-ДИХЛОР-4-ГИДРОКСИБЕНЗОИЛ)- 1,1-ДИОКСО-2,3-ДИГИДРО-1,3-БЕНЗОТИАЗОЛА | 2018 |
|
RU2772043C2 |
| КРИСТАЛЛ ПРОИЗВОДНОГО 1,3,5-ТРИАЗИНА ИЛИ ЕГО СОЛЬВАТА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2837449C1 |
Изобретение относится к кристаллам 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, охарактеризованного формулой (1). Кристалл типа I 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,7°, 14,5°, 17,0°, 18,9°, 19,4°, 23,3° и 25,7°. Кристалл типа II 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,8°, 14,7°, 15,7°, 19,3° и 25,0°. Также изобретение относится к фармацевтической композиции, обладающей ингибирующим воздействием на фосфодиэстеразу (PDE) 4, включающей кристалл 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, охарактеризованного формулой (1) в качестве активного начала. Технический результат – кристаллические формы 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, обладающие свойством стабильности. 3 н. и 3 з.п. ф-лы, 3 ил., 2 табл., 4 пр.
1. Кристалл типа I 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, охарактеризованного формулой (1),
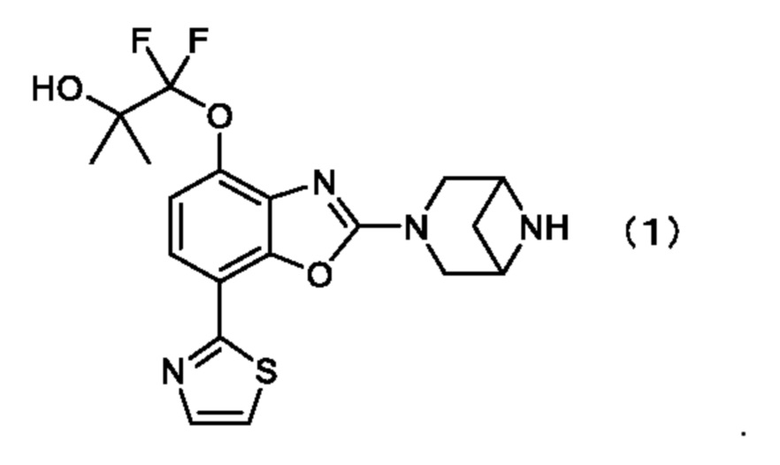 ,
,
где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,7°, 14,5°, 17,0°, 18,9°, 19,4°, 23,3° и 25,7°.
2. Кристалл по п. 1, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,7°, 9,4°, 13,2°, 13,7°, 14,5°, 15,7°, 17,0°, 18,9°, 19,4°, 22,9°, 23,3°, 25,7°, 28,7° и 35,7°.
3. Кристалл по п. 1 или 2, где кристалл по данным дифференциальной сканирующей калориметрии (DSC) обладает эндотермическим пиком при температуре, равной 205±3°C.
4. Кристалл типа II 1-((2-(3,6-диазабицикло[3.1.1]гептан-3-ил)-7-(тиазол-2-ил)бензо[d]оксазол-4-ил)окси)-1,1-дифтор-2-метилпропан-2-ола, охарактеризованного формулой (1),
,
где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,8°, 14,7°, 15,7°, 19,3° и 25,0°.
5. Кристалл по п. 4, где кристалл обладает на порошковой рентгенограмме характеристическими пиками при углах дифракции (2Ɵ±0,2°), равных 7,4°, 7,8°, 8,2°, 12,7°, 13,6°, 14,3°, 14,7°, 15,7°, 16,5°, 19,3°, 22,1°, 25,0° и 25,6°.
6. Фармацевтическая композиция, обладающая ингибирующим воздействием на фосфодиэстеразу (PDE) 4, включающая кристалл по любому из пп. 1-5 в качестве активного начала.
| Токарный резец | 1924 |
|
SU2016A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| MINO R | |||
| CAIRA: "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, pp.163-208 | |||
| Дж | |||
| Бернштейн "Полиморфизм молекулярных кристаллов" Москва, Наука, 2007, гл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

