Изобретение относится к формам химического соединения, в частности кристаллической и аморфной формам, более конкретно, четырем кристаллическим формам и аморфной форме. Изобретение, далее, относится к способам получения таких форм, фармацевтическим композициям, включающим соединение в кристаллической и/или аморфной форме, и к терапевтическому применению таких форм.
При составлении лекарственных композиций важно, чтобы лекарственное соединение находилось в форме, в которой с ним было бы удобно обращаться и обрабатывать. Это важно не только с точки зрения получения коммерчески жизнеспособного производственного процесса, но также с точки зрения последующего производства фармацевтических композиций, включающих активное соединение. Химическая стабильность, стабильность в твердом состоянии и срок хранения активных компонентов также являются очень важными факторами. Лекарственное соединение и композиции, включающие его, должны обладать способностью эффективно храниться в течение значительных периодов времени, не проявляя значительного изменения физико-химических свойств активного компонента (например, его химического состава, плотности, гигроскопичности и растворимости). Кроме того, также является важным представить лекарственный препарат в форме, которая является как можно более чистой. В этом отношении аморфные соединения могут представлять значительные проблемы. Например, с такими соединениями труднее обращаться и включать их в лекарственные формы по сравнению с кристаллическим соединением, за счет сомнительной растворимости, и часто оказывается, что они нестабильны и химически загрязнены. Специалисту в данной области будет понятно, что, если лекарственный препарат можно легко получать в стабильной кристаллической форме, то можно решить вышеуказанные проблемы. Таким образом, при производстве коммерчески жизнеспособных и фармацевтически приемлемых лекарственных композиций, желательно, где это возможно, обеспечить лекарственный препарат в основном в кристаллической и стабильной форме. Следует отметить, однако, что данная цель не всегда является достижимой. Действительно, обычно невозможно предсказать только на основе молекулярной структуры, каким будет поведение соединения при кристаллизации, и обычно это можно установить только эмпирически.
Адгезия и агрегация тромбоцитов являются начальными событиями при тромбозе артерий. Хотя процесс адгезии тромбоцитов к субэндотелиальной поверхности может играть важную роль при восстановлении поврежденных стенок сосудов, агрегация тромбоцитов, которая инициирует этот процесс, может вызвать острую тромботическую окклюзию жизненно важных сосудов, приводя к патологиям с высокой смертностью таким, как инфаркт миокарда и нестабильная стенокардия. Успех вмешательств, используемых для профилактики или облегчения данных состояний, таких, как тромболиз и ангиопластика также находится под угрозой опосредуемой тромбоцитами окклюзии и повторной окклюзии.
Было установлено, что аденозин-5'-дифосфат (AДФ) действует в качестве ключевого медиатора тромбоза. AДФ-индуцируемая агрегация тромбоцитов опосредуется подтипом Р2Т-рецепторов, расположенных на мембране тромбоцитов. Р2Т-рецептор (также известный, как P2YAДФ или P2TAC) в основном участвует в опосредовании агрегации/активации тромбоцитов и является сопряженным с G-белком рецептором, который еще не клонирован. Описаны фармакологические свойства данного рецептора, например, в источниках Humphries et al., Br. J. Pharmacology (1994), 113, 1057-1063 и Fagura et al., Br. J. Pharmacology (1998), 124, 157-164. Недавно было показано, что антагонисты в отношении данного рецептора дают существенные улучшения по отношению к другим антитромботическим средствам (смотри J. Med. Chem. (1999), 42, 213). В заявке на международный патент WO 9905143 раскрывается в общем ряду группа триазоло[4,5-d]пиримидиновых соединений, обладающих активностью в качестве антагонистов Р2Т (P2YAДФ или P2TAC). Соединение формулы (I) (приведено ниже) входит в общий объем заявки на международный патент WO 9905143, но специально там не раскрывается. Данное соединение проявляет высокую эффективность в качестве антагониста P2Т (P2YAДФ или P2TAC). Также оно обладает удивительно высокой метаболической стабильностью и биодоступностью.
Соответственно, настоящее изобретение относится к соединению формулы (I):
(I)
в по существу кристаллической форме.
Соединение формулы (I) обычно называется:
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол.
Соединение формулы (I) может находиться в четырех различных, по существу кристаллических, формах, называемых в последующем полиморф I, полиморф II, полиморф III и полиморф IV. Полиморф представляет собой конкретную кристаллическую форму соединения.
Различные физические свойства полиморфных форм по отношению друг к другу и по отношению к аморфному состоянию могут оказывать заметное влияние на химическую и фармацевтическую обработку соединения, особенно, когда соединение получают или используют в промышленном масштабе.
В одном аспекте изобретения предпочтительной кристаллической формой соединения формулы (I) является форма полиморфа I, полиморфа II, полиморфа III или полиморфа IV.
В альтернативном аспекте изобретения предпочтительной кристаллической формой соединения формулы (I) является полиморф I.
В другом аспекте изобретения предпочтительной кристаллической формой соединения формулы (I) является полиморф II.
В еще одном аспекте изобретения предпочтительной кристаллической формой соединения формулы (I) является полиморф III.
В дополнительном аспекте изобретения предпочтительной кристаллической формой соединения формулы (I) является полиморф IV.
В следующем аспекте изобретения соединение формулы (I) находится в по существу аморфной форме. В аморфной форме трехмерный дальний порядок структуры, который имеется в кристаллической форме (например, в полиморфе) отсутствует, и положения молекул по отношению друг к другу в аморфной форме в основном беспорядочны (смотри B.C.Hancock and G.Zografi, J. Pharm. Sci. (1997) 86 1). Аморфную форму соединения формулы (I) называют форма α.
Заявители выделили соединение формулы (I) в кристаллической и аморфной формах. Данные формы могут существовать в значительной степени или практически свободном от воды виде ("безводные" формы). Следовательно, в одном аспекте изобретения предлагается безводная форма соединения формулы (I) в кристаллической форме или аморфной форме. При использовании термина "в значительной степени чистой и в практически безводной форме" заявители не исключают наличие некоторого количества растворителя, включая воду, в кристаллической решетке или вне кристаллической решетки. Безводная форма имеет менее, чем 0,4 молекулы воды на молекулу соединения (гидратировано менее, чем на 40%). Предпочтительно, безводная форма содержит менее чем 0,1 молекулы воды на молекулу соединения.
Полиморфы I, II, III и IV можно различать по началу плавления, спектрам порошковой дифракции рентгеновских лучей и/или данным рентгеноскопии монокристалла.
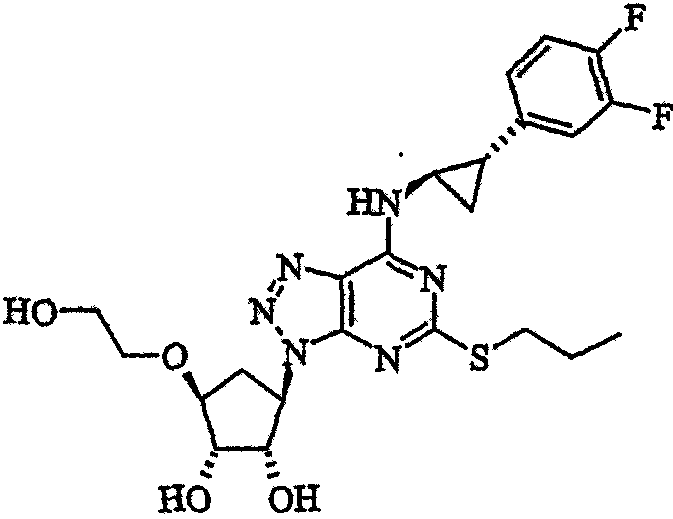
Полиморф I имеет точку начала плавления, которая находится в пределах 146-152°С, например, около 151°С, когда он является в значительной степени чистым и в практически безводной форме.
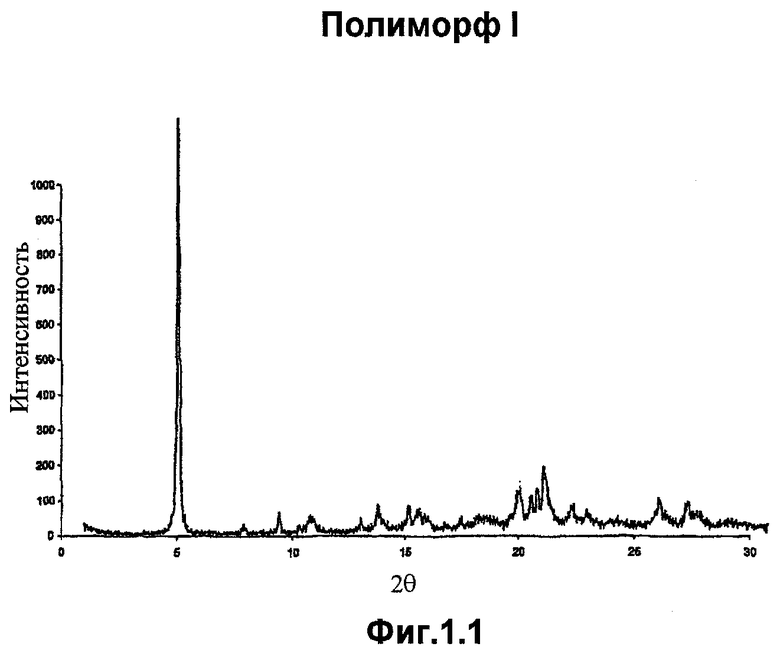
Полиморф II имеет точку начала плавления, которая находится в пределах 136-139°С, например, около 137,5°С, когда он является в значительной степени чистым и в практически безводной форме.
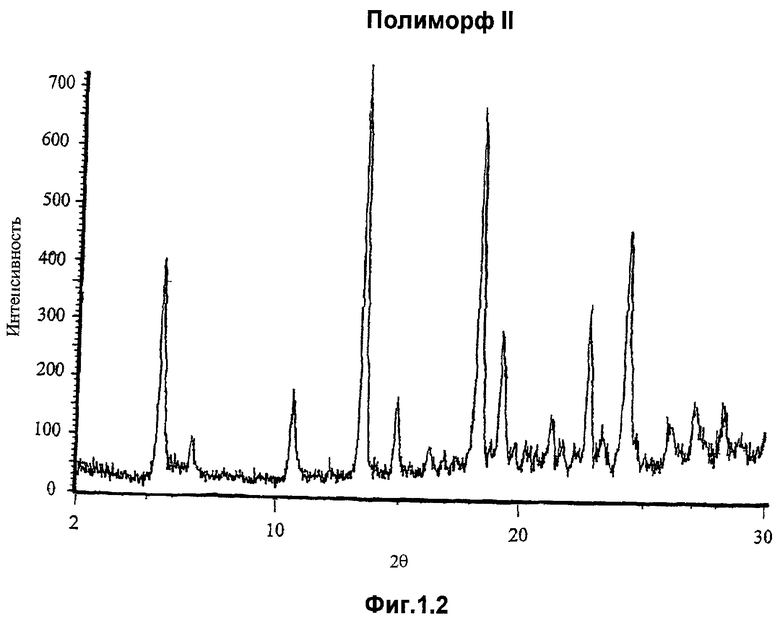
Полиморф III имеет точку начала плавления, которая находится в пределах 127-132°С, например, около 132°С, когда он является в значительной степени чистым и в практически безводной форме.
Полиморф IV имеет точку начала плавления, которая обычно составляет 139°С, когда он является в значительной степени чистым и в практически безводной форме.
Форма α обычно претерпевает стеклование с последующей кристаллизацией в одну из вышеуказанных полиморфных форм, например полиморф II перед плавлением.
Точки плавления определяли с использованием дифференциальной сканирующей калориметрии (ДСК), используя оборудование Perkin Elmer DSC7. Начало плавления определяют как точку, в которой имеет место значительное изменение по сравнению с базовой линией, и его определяют с помощью программного обеспечения Perkin Elmer Pyris. Понятно, что альтернативные данные по точке плавления можно получить с другими типами оборудования или при использовании условий, отличных от описанных здесь. Следовательно, приведенные цифры не следует рассматривать в качестве абсолютных значений. Специалистам в данной области понятно, что на точное значение точки плавления будет влиять чистота соединения, масса пробы, скорость нагревания и размер частиц.
Полиморф I, когда он является в значительной степени чистым и в практически безводной форме, имеет порошковую рентгенограмму, включающую характерные пики высокой интенсивности при 5,3° (±0,1°), 20,1° (±0,1), 20,7° (±0,1°), 21,0° (±0,1°) и 21,3° (±0,1°) 2θ. Более предпочтительно в значительной степени чистый и практически безводный полиморф I имеет порошковую рентгенограмму, включающую характерные пики при 5,3° (±0,1°), 8,0° (±0,1°), 9,6° (±0,1°), 13,9° (±0,1°), 15,3° (±0,1°), 20,1° (±0,1°), 20,7° (±0,1°), 21,0° (±0,1°), 21,3° (±0,1°), 26,2° (±0,1°) и 27,5° (±0,1°) 2θ.
Полиморф II, когда он является в значительной степени чистым и в практически безводной форме, имеет порошковую рентгенограмму, включающую характерные пики высокой интенсивности при 5,5° (±0,1°), 13,5° (±0,1), 18,3° (±0,1°), 22,7° (±0,1°) и 24,3° (±0,1°) 2θ. Более предпочтительно в значительной степени чистый и практически безводный полиморф II имеет порошковую рентгенограмму, включающую характерные пики при 5,5° (±0,1°), 6,8° (±0,1°), 10,6° (±0,1°), 13,5° (±0,1°), 14,9° (±0,1°), 18,3° (±0,1°), 19,2° (±0,1°), 22,7° (±0,1°), 24,3° (±0,1°) и 27,1° (±0,1°) 2θ.
Полиморф III, когда он является в значительной степени чистым и в практически безводной форме, имеет порошковую рентгенограмму, включающую характерные пики высокой интенсивности при 14,0° (±0,1°), 17,4° (±0,1), 18,4° (±0,1°), 21,4° (±0,1°) и 24,1° (±0,1°) 2θ. Более предпочтительно в значительной степени чистый и практически безводный полиморф III имеет порошковую рентгенограмму, включающую характерные пики при 5,6° (±0,1°), 12,5° (±0,1°), 14,0° (±0,1°), 17,4° (±0,1°), 18,4° (±0,1°), 21,4° (±0,1°), 22,2° (±0,1°), 22,9є (±0,1°), 24,1° (±0,1°) и 24,5° (±0,1°) 2θ.
Полиморф IV, когда он является в значительной степени чистым и в практически безводной форме, имеет порошковую рентгенограмму, включающую характерные пики высокой интенсивности при 4,9° (±0,1°), 9,2° (±0,1), 11,6° (±0,1°), 15,6° (±0,1°) и 16,4° (±0,1°) 2θ. Более предпочтительно в значительной степени чистый и практически безводный полиморф IV имеет порошковую рентгенограмму, включающую характерные пики при 4,9° (±0,1°), 6,0° (±0,1°), 9,2° (±0,1°), 11,6° (±0,1°), 12,8° (±0,1°), 15,6° (±0,1°), 16,4° (±0,1°), 17,2° (±0,1°) и 18,1° (±0,1°) 2θ.
Форма α, когда она является в значительной степени чистой и в практически безводной форме, имеет порошковую рентгенограмму, не содержащую острых пиков.
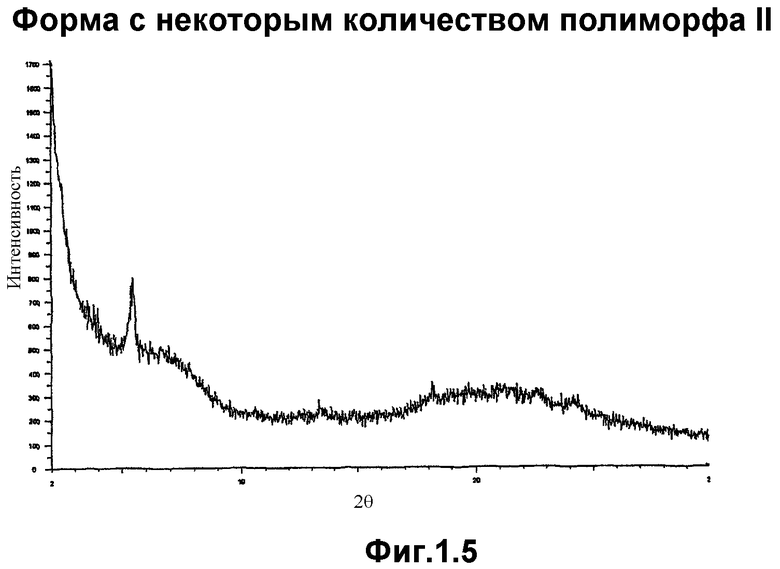
Данные по дифракции рентгеновских лучей для полиморфа II, полиморфа III, полиморфа IV и формы α получали с использованием оборудования Siemens D5000. Данные по дифракции рентгеновских лучей для полиморфа I получали на аппарате Philips X'Pert MPD. Понятно, что другое оборудование и/или условия могут привести к получению несколько других данных. Следовательно, приведенные цифры не следует рассматривать в качестве абсолютных значений.
В альтернативном аспекте изобретения может образоваться сольватированная форма, например, гидратированная форма ("гидрат"). Следовательно, в данном аспекте изобретения предлагается гидрат соединения формулы (I) в кристаллической форме. Гидрат имеет 0,8 или более молекул воды на молекулу соединения (80% и более гидратировано). Гемигидрат имеет от 0,4 до 0,8 молекул воды на молекулу соединения (40-80% гидратировано).
В дополнительном аспекте изобретения предлагается любая смесь кристаллической и/или аморфной форм соединения формулы (I). Предпочтительно смесь представляет полиморф I, полиморф II, полиморф III, полиморф IV и/или форму α. Более предпочтительно изобретение обеспечивает любую смесь полиморфа II и полиморфа III.
В дополнительном аспекте изобретения предлагается способ получения кристаллической формы соединения формулы (I) кристаллизацией соединения формулы (I) из подходящего растворителя. Предпочтительно растворитель выбран из группы: этанол, этилацетат, изопропанол, изооктан, ацетонитрил, вода или их смесь. Более предпочтительно растворитель выбран из группы: этанол, этилацетат, изопропанол, изооктан, вода или их смесь. Преимущественно растворитель выбран из группы: смесь метанол-вода, этанол, этилацетат, смесь этанол-вода, смесь изопропанол-вода, смесь этилацетат-изооктан и ацетонитрил.
Соединение формулы (I) можно получить способами, аналогичными таковым, описанным в WO 9905143.
Для начала кристаллизации может быть необходимым внесение затравочных кристаллов соединения формулы (I). Внесение затравочных кристаллов необходимого полиморфа может быть необходимым для получения выбранного полиморфа. Кристаллизацию соединения формулы (I) из соответствующей системы растворителей можно достичь перенасыщением, например, охлаждением, выпариванием растворителя и/или добавлением антирастворителя (растворителя, в котором соединения формулы (I) слаборастворимы; примеры подходящих антирастворителей включают гептан или изооктан). Температура и время кристаллизации будут варьировать в зависимости от концентрации соединения в растворе, используемой системы растворителей и приемлемого способа кристаллизации.
Соединение формулы (I) в кристаллической форме можно выделить из вышеуказанной реакционной смеси с использованием способов, хорошо известных специалистам в данной области, например декантацией, фильтрованием или центрифугированием. Аналогично соединение формулы (I) в кристаллической форме можно высушить с использованием хорошо известных методов.
Необязательную стадию(и) перекристаллизации можно провести с использованием тех же или других систем растворителей для дальнейшего снижения содержания примесей, таких как аморфное вещество, химические примеси, или превращения кристаллической формы из одного полиморфа в другой полиморф либо в гидратированную или безводную форму. Кроме того, можно провести стадию кондиционирования, подвергнув твердое вещество воздействию высокой влажности для удаления аморфного вещества.
Предпочтительно кристаллизацию проводят непосредственно из реакционного раствора. Альтернативно кристаллизацию проводят из последующего раствора.
В дополнительном аспекте изобретения предлагается способ получения полиморфа I, который включает получение нескольких затравочных кристаллов полиморфа I в результате медленного роста кристаллов полиморфа I из расплава полиморфа II, и использование их для внесения затравочных кристаллов в реакционную смесь, включающую соединение формулы (I) и подходящую смешанную систему растворителей, такую как метанол/вода.
В дополнительном аспекте изобретения предлагается способ получения полиморфа II, который включает кристаллизацию из подходящего растворителя такого, как хлороформ.
В дополнительном аспекте изобретения предлагается способ получения полиморфа III, который включает кристаллизацию из подходящего растворителя, такого как спирт, например, этанол или изопропиловый спирт (ИПС), при внесении затравки конкретно кристаллов полиморфа III или суспендировании соединения формулы (I) в подходящем растворителе, таком как ИПС.
В дополнительном аспекте изобретения предлагается способ получения полиморфа IV, который включает кристаллизацию из подходящего растворителя, такого как ацетонитрил, при внесении затравки конкретно кристаллов полиморфа IV или суспендировании соединения формулы (I) в подходящем растворителе, таком как ацетонитрил.
В дополнительном аспекте изобретения предлагается способ получения полиморфа III, в основном свободного от полиморфа II, который включает, например, суспендирование соединения формулы (I) в системе растворителей C1-6алифатический спирт/вода (предпочтительно ИПС/вода) при температуре в пределах 5-65°С в течение 1-10 суток.
В дополнительном аспекте изобретения предлагается способ получения соединения формулы (I) в основном в аморфной форме, который включает сушку вымораживанием или распылительную сушку раствора соединения формулы (I) с использованием подходящей системы растворителей, например смеси этанол/вода.
Термин "в основном свободный" относится к случаю содержания менее чем 10% другого полиморфа, предпочтительно менее 5%.
В дополнительном аспекте изобретения предлагается соединение, получаемое одним из вышеуказанных способов.
Соединение формулы (I) в кристаллической и/или аморфной форме действует в качестве антагониста рецепторов Р2Т (P2YAДФ или P2TAC). Следовательно, соединение формулы (I) в кристаллической и/или аморфной форме пригодно для лечения, включая комбинированную терапию. В частности, соединение формулы (I) в кристаллической форме показано для применения при лечении или профилактике артериальных тромботических осложнений у пациентов с заболеванием коронарной артерии, цереброваскулярном или периферическом сосудистом заболевании. Артериальные тромботические осложнения могут включать нестабильную стенокардию, первичные артериальные тромботические осложнения при атеросклерозе, такие как тромботический или эмболический инсульт, кратковременные приступы ишемии, периферическое сосудистое заболевание, инфаркт миокарда с тромболизом или без него, артериальные осложнения в результате вмешательств при атеросклерозе, таких как ангиопластика, включая ангиопластику коронарной артерии (РТСА), эндартерэктомию, установку стентов, операцию по пересадке коронарных и других сосудов, тромботические осложнения при хирургическом или механическом повреждении, такие как сохранение тканей после случайной или хирургической травмы, восстановительной хирургии, включая кожные и мышечные лоскуты, состояния с наличием компонента диффузного тромботического/истощения тромбоцитов, такие как распространенная внутрисосудистая коагуляция, тромбогемолитическая тромбоцитопеническая пурпура, гемолитический мочевой синдром, тромботические осложнения при септицемии, дыхательный дистресс - синдром взрослых, антифосфолипидный синдром, индуцированную гепарином тромбоцитопению и преэклампсию/эклампсию или венозный тромбоз, такой как тромбоз глубоких вен, венооклюзивное заболевание, гематологические состояния, такие как миелопролиферативное заболевание, включая тромбоцитемию, серповидную-клеточную болезнь; или при профилактике механически индуцированной активации тромбоцитов in vivo, как, например, экстракорпоральное кровообращение и экстракорпоральное мембранное насыщение кислородом (профилактика микротромбоэмболии), механически индуцированной активации тромбоцитов in vitro, как, например, использование при консервации продуктов крови, например, концентратов тромбоцитов, или окклюзий при шунтировании, таких как при диализе почек и плазмаферезе, тромбоза, вторичного повреждения/воспаления сосудов, такого как васкулит, артериит, гломерулонефрит, воспалительное заболевание кишечника и отторжение при трансплантации органов, таких состояний, как мигрень, синдром Рейно, состояний, при которых тромбоциты вносят свой вклад в основное воспалительное заболевание в стенках сосудов, как, например, образование/прогрессирование атероматозных бляшек, стеноз/рестеноз и при других воспалительных состояниях, таких как астма, при которых тромбоциты и производные от тромбоцитов факторы участвуют в иммунологическом заболевании. Дополнительные показания включают лечение расстройств ЦНС и профилактику роста и распространения опухолей.
Согласно дополнительному аспекту настоящего изобретения предлагается соединение формулы (I) в кристаллической и/или аморфной форме для применения в терапевтическом способе лечения человека или животного.
Согласно дополнительному аспекту настоящего изобретения предлагается соединение формулы (I) в кристаллической и/или аморфной форме для применения в качестве лекарственного препарата. Предпочтительно соединение формулы (I) в кристаллической и/или аморфной форме используется в качестве лекарственного препарата как антагонист рецептора Р2Т (P2YAДФ или P2TAC) у теплокровного животного, такого как человек. Более предпочтительно соединение формулы (I) в кристаллической и/или аморфной форме применяется в качестве лекарственного препарата для лечения или профилактики артериальных тромботических осложнений у пациентов с заболеванием коронарной артерии, цереброваскулярном или периферическом сосудистом заболевании у теплокровного животного, такого как человек.
Согласно изобретению дополнительно предлагается применение соединения формулы (I) в кристаллической и/или аморфной форме в производстве лекарственного препарата для использования в качестве антагониста рецептора Р2Т (P2YAДФ или P2TAC). В частности дополнительно предлагается применение соединения формулы (I) в кристаллической и/или аморфной форме в производстве лекарственного препарата для лечения или профилактики артериальных тромботических осложнений у пациентов с заболеванием коронарной артерии, цереброваскулярном и периферическом сосудистом заболевании.
Изобретение также обеспечивает способ лечения или профилактики артериальных тромботических осложнений у пациентов с заболеванием коронарной артерии, цереброваскулярном или периферическом сосудистом заболевании, который включает введение человеку, страдающему таким заболеванием или расположенному к нему, терапевтически эффективного количества соединения формулы (I) в кристаллической и/или аморфной форме.
Соединение формулы (I) в кристаллической и/или аморфной форме можно вводить местно в легкие и/или дыхательные пути, в виде растворов, суспензий, аэрозолей HFA и сухих порошкообразных композиций; или системно, например, при пероральном введении в виде таблеток, пилюль, капсул, сиропов, порошков или гранул, или парентерально в виде стерильных растворов или суспензий для парентерального введения, подкожно или ректально в виде суппозиториев, или трансдермально.
Соединение формулы (I) в кристаллической и/или аморфной форме можно вводить само по себе или в виде фармацевтической композиции, включающей соединение формулы (I) в кристаллической и/или аморфной форме в комбинации с фармацевтически приемлемым разбавителем, адъювантом и/или носителем. Следовательно, предлагается в качестве дополнительного аспекта изобретения фармацевтическая композиция, включающая соединение формулы (I) в кристаллической и/или аморфной форме в сочетании с фармацевтически приемлемым разбавителем, адъювантом и/или носителем. Особенно предпочтительными являются композиции, не содержащие вещества, способного вызвать побочную реакцию, такую как побочная аллергическая реакция.
Сухие порошкообразные композиции и HFA-аэрозоли под давлением соединения формулы (I) в кристаллической и/или аморфной форме можно вводить перорально или ингаляцией через нос. Для ингаляции соединение формулы (I) в кристаллической и/или аморфной форме желательно мелко измельчить. Также соединение формулы (I) в кристаллической и/или аморфной форме можно вводить с помощью ингалятора для вдыхания сухих порошков. Ингалятор может быть ингалятором с одной или многими дозами и может быть ингалятором для сухих порошков для вдыхания.
Одной возможностью является смешать тонкоизмельченное соединение формулы (I) в кристаллической и/или аморфной форме с веществом-носителем, например, моно-, ди- или полисахаридом, спиртом - производным сахара или другим полиолом. Подходящие носители включают сахара и крахмал. Альтернативно, тонкоизмельченное соединение формулы (I) в кристаллической и/или аморфной форме можно покрыть другим соединением. Порошкообразную смесь можно распределить в твердые желатиновые капсулы, содержащие каждая желаемую дозу активного соединения формулы (I) в кристаллической и/или аморфной форме.
Другой возможностью является превращение тонкоизмельченного порошка в шарики, которые разрушаются при ингаляции. Такой сферонизированный порошок можно поместить в резервуар для лекарственного препарата многодозового ингалятора, например, известного как Turbuhaler®, в котором дозирующее устройство отмеряет желаемую дозу, которая затем вдыхается пациентом. С помощью этой системы активное соединение формулы (I) с веществом-носителем и без него доставляется пациенту. Фармацевтическая композиция, включающая соединение формулы (I) в кристаллической и/или аморфной форме, может представлять такие удобные формы, как таблетки, пилюли, капсулы, сиропы, порошки или гранулы для перорального введения; стерильные растворы для парентерального или подкожного введения; суспензии для парентерального введения или суппозитории для ректального введения.
Для перорального введения соединение формулы (I) в кристаллической и/или аморфной форме можно смешать с адъювантом или носителем, например, лактозой, сахарозой, сорбитом, маннитом, крахмалами, такими как картофельный крахмал, кукурузный крахмал или амилопектин, производными целлюлозы, связующими веществами, такими как желатин или поливинилпирролидон, смазывающими веществами такими, как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и тому подобное, и затем спрессовать в таблетки. Если требуются покрытые оболочкой таблетки, ядра, полученные, как описано выше, можно покрыть концентрированным раствором сахара, который может содержать, например, гуммиарабик, желатин, тальк, диоксид титана и тому подобное. Альтернативно, таблетку можно покрыть подходящим полимером, растворенным либо в легколетучем органическом растворителе, либо в водном растворителе.
Для получения мягких желатиновых капсул соединение формулы (I) в кристаллической и/или аморфной форме можно смешать, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы соединения с использованием либо вышеуказанных наполнителей для таблеток, например, лактозы, сахарозы, сорбита, маннита, крахмалов, производных целлюлозы или желатина. Также жидкие или полужидкие композиции лекарственного препарата можно разлить в твердые желатиновые капсулы.
Жидкие препараты для перорального применения могут быть в виде сиропов или суспензий, например, растворов, содержащих соединение формулы (I) в кристаллической и/или аморфной форме, причем остаток составляет сахар и смесь этанола, воды, глицерина и пропиленгликоля. Необязательно, такие жидкие препараты могут содержать окрашивающие вещества, ароматизаторы, сахарин и карбоксиметилцеллюлозу в качестве загустителя или другие наполнители, известные специалистам в данной области.
Фигура 1.1. представляет рентгенограмму полиморфа I, полученную с использованием прибора Philips X'Pert MPD в θ-θ конфигурации в пределах сканировании от 1° до 40° 2θ при 2 или 5 сек экспозиции на приращение 0,02° 2θ. Рентгеновские лучи получали с помощью медной длиннофокусной трубки, работающей при 40 кB и 50 мА. Длина волны рентгеновских лучей равнялась 1,5406 Å.
Фигура 1.2. представляет рентгенограмму полиморфа II, полученную с использованием прибора Siemens D5000 в θ-θ конфигурации в пределах сканировании от 2° до 30° 2θ при 4 сек экспозиции на приращение 0,02° 2θ. Рентгеновские лучи получали с помощью медной длиннофокусной трубки, работающей при 45 кB и 40 мА. Длина волны рентгеновских лучей равнялась 1,5406 Å. Держатель был сделан из монокристалла кремния, который был срезан вдоль недифрагирующей плоскости и затем отшлифован до оптически плоской отделки. Рентгеновские лучи, падающие на эту поверхность, сводились на нет за счет экстинкции Брэгга.
Фигура 1.3. представляет рентгенограмму полиморфа III, полученную с использованием прибора Siemens D5000, как описано выше.
Фигура 1.4. представляет рентгенограмму полиморфа IV, полученную с использованием прибора Siemens D5000, как описано выше.
Фигура 1.5. представляет рентгенограмму формы α, полученную с использованием прибора Siemens D5000, как описано выше.
Фигура 2 показывает кривые ДСК для полиморфов I, II, III и IV и формы α, полученные с использованием прибора Perkin Elmer DSC 7. Чашечка была алюминиевой с колпаком. Масса пробы составляла 1-3 мг. Процедуру измерения проводили в атмосфере газообразного азота (30 мл/мин), а исследуемый температурный интервал составлял между 30°С и 325°С при постоянной скорости повышения температуры 10°С в мин.
Следует понимать, что анализ проб с зернами размером свыше 30 микрон и неунифицированные соотношения геометрических размеров могут оказывать влияние на относительную интенсивность пиков. Специалисту в данной области будет также понятно, что на положение отражений оказывает влияние точная высота, на которую помещается проба в дифрактометре, и нулевая калибровка дифрактометра. Плоскость поверхности пробы может также иметь небольшой эффект. Следовательно, представленные данные по спектрам дифракции не следует рассматривать в качестве абсолютных значений.
Изобретение можно иллюстрировать следующими неограничивающими примерами.
Пример 1
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол в форме полиморфа I
Часть 1
Соединение формулы (I) в виде полиморфа II (2 мг) нагревали и охлаждали в ДСК следующим образом: от 35 до 143, до 35, до 148, до 35, до 148, до 35°С. Данный процесс отжига приводил к кристаллизации чистого полиморфа I по данным ДСК.
Часть 2
Раствор, содержащий соединение формулы (I), 5 мл/г метанола и 7,3 мл/г воды и небольшое количество затравочных кристаллов полиморфа I, кристаллизовали при 30°С. Данные порошкового РСА и ДСК подтверждали, что образовался в основном чистый полиморф I.
Пример 2
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол в форме полиморфа II
Хлороформ (150 мкл) добавляли к 45 мг соединения формулы (I) и смесь нагревали до растворения на паровой бане. Полученный раствор оставляли для кристаллизации в течение ночи и высушивали в атмосфере азота. Данные порошкового РСА и ДСК подтверждали, что образовался в основном чистый полиморф II.
Пример 3
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол в форме полиморфа III
Этанол (200 мкл) добавляли к 10 мг соединения формулы (I) и смесь нагревали до растворения на паровой бане. Полученный раствор оставляли для кристаллизации в течение ночи. Данные порошкового РСА и ДСК подтверждали, что образовалась смесь полиморфов II и III. Данное вещество использовали для внесения затравки для получения в большем объеме: 191 мг полиморфа II суспендировали в 1 мл 50% водного раствора изопропанола. К данной суспензии добавляли 15 мг затравочных кристаллов смешанного полиморфа II/III. Через 2 суток происходило полное превращение в полиморф III по данным порошкового РСА.
Пример 4
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол в форме полиморфа IV
Ацетонитрил (0,12 мл) добавляли к 10 мг соединения формулы (I) и смесь нагревали до растворения на паровой бане. Теплому раствору давали медленно охладиться в водяной рубашке с горячей водой. Полученные кристаллы высушивали в атмосфере азота. Данные порошкового РСА и ДСК показывали, что это был другой полиморф.
Пример 5
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол в основном в виде формы α
Соединение формулы (I) (218 мг) растворяли в 50% водном растворе этанола (24 мл). К данному раствору по каплям добавляли еще 14,5 мл воды. Затем полученный насыщенный раствор сушили вымораживанием с использованием аппарата Виртиса в следующих условиях (вакуум 2170 мТ, цикл 20,2 ч, температура конденсации -52°С, температура окружающей среды 20,3°С).
Ссылочный пример 1
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол
Раствор {3aR-[3aα,4α,6α(1R*,2S*),6aα]}-2-[6-({7-[2-(3,4-дифторфенил)циклопропил]амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил)окси]этанола (способ А, 0,59 г) в трифторуксусной кислоте (15 мл) и воде (15 мл) перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь осторожно добавляли к раствору бикарбоната натрия (21 г) в воде (150 мл) и перемешивали в течение 30 мин. Смесь экстрагировали этилацетатом, который высушивали и выпаривали. Остаток очищали (SiO2, этилацетат в качестве элюента) с получением указанного в заголовке соединения (0,44 г). МС (АРСI) (химическая ионизация при атмосферном давлении) 523 (М+Н+, 100%); ЯМР: 8,95 (1H, д, J=3,3), 7,39-7,21 (2H, м), 7,10-7,00 (1H, м), 5,12 (1H, д, J=6,4), 5,05 (1H, д, J=3,6), 4,96 (1H, кв, J=9,0), 4,62-4,54 (2H, м), 3,95 (1H, шир. с), 3,79-3,73 (1H, м), 3,55-3,47 (4H, м), 3,20-3,13 (1H, м), 2,98-2,81 (2H, м), 2,63 (1H, дт, J=13,6, 8,5), 2,29-2,21 и 2,16-2,09 (1H, м), 2,07-2,00 (1H, м), 1,73-1,33 (4H, м), 0,99 (3H, т, J=7,4).
Получение исходных соединений
Исходные соединения являются либо промышленно выпускаемыми, либо их легко получить обычными способами из известных соединений. Например, последующие реакции представляют иллюстрации, но не ограничения, получения некоторых исходных соединений, используемых в вышеприведенных реакциях.
Способ А
{3aR-[3aα,4α,6α(1R*,2S*),6aα]}-2-[6-({7-[2-(3,4-дифторфенил)циклопропил]амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил)окси]этанол
DIBAL-H® (1,0 М раствор в гексане, 5,15 мл) добавляли к охлажденному на льду раствору метилового эфира {3aR-[3aα,4α,6α(1R*,2S*),6aα]}-{[6-({7-[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси}уксусной кислоты (способ В, 0,76 г) в ТГФ (1 мл) и раствор перемешивали при данной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате (75 мл). Добавляли насыщенный водный раствор тартрата натрия-калия (75 мл) и смесь энергично перемешивали в течение 16 ч. Органические фракции собирали и водную фракцию повторно экстрагировали этилацетатом (2×50 мл). Объединенные органические фракции высушивали и концентрировали, и остаток очищали (SiO2, смесь изогексан: этилацетат 1:1 в качестве элюента) с получением указанного в заголовке соединения (0,63 г). МС (APCI) 563 (М+Н+, 100%).
Способ В
Метиловый эфир {3aR-[3aα,4α,6α(1R*,2S*),6aα]}-{[6-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидн-3-ил)тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси}уксусной кислоты
К смеси метилового эфира [3aR-(3aα,4α,6α,6aα)]-({6-[7-бром-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол}окси)уксусной кислоты (способ D, 0,80 г) и (1R-транс)-2-(3,4-дифторфенил)циклопропанамина, [R-(R*,R*)]-2,3-дигидроксибутандиоата (1:1) (способ С, 0,61 г) в дихлорметане (25 мл) добавляли N,N-диизопропилэтиламин (0,85 мл). Полученный раствор перемешивали при комнатной температуре в течение 16 ч и затем концентрировали в вакууме. Очистка (SiO2, смесь изогексан:этилацетат 3:1 в качестве элюента) давала указанное в заголовке соединение в виде бесцветной пены (0,77 г). МС (АРСI) 591 (М+Н+, 100%).
Способ С
(1R-транс)-2-(3,4-дифторфенил)циклопропанамин, [R-(R*,R*)]-2,3-дигидроксибутандиоат (1:1)
Указанное в заголовке соединение можно получить по способу, описанному в заявке WO 9905143.
Способ D
Метиловый эфир [3aR-(3aα,4α,6α,6aα)]-({6-[7-бром-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол}окси)уксусной кислоты
Метиловый эфир [3aR-(3aα,4α,6α,6aα)]-({6-[7-амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол}окси)уксусной кислоты (способ Е, 1,1 г) и изоамилнитрит (2,4 мл) в бромоформе (30 мл) нагревали при 80°С в течение 30 мин. Охлажденную реакционную смесь очищали (SiO2, смесь этилацетат:изогексан 1:4 в качестве элюента) с получением указанного в заголовке соединения (0,44 г). МС (АРСI) 502/4 (М+Н+), 504 (100%).
Способ Е
Метиловый эфир [3aR-(3aα,4α,6α,6aα)]-({6-[7-амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол}окси)уксусной кислоты
К раствору [3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола (способ F, 0,50 г) в ТГФ (25 мл) при 0°С добавляли бутиллитий (0,62 мл 2,5 н в гексане). Через 20 мин суспензию обрабатывали раствором метилового эфира трифторметансульфонилоксиуксусной кислоты (0,34 г) (полученого по способу Biton, Tetrahedron, 1995, 51, 10513) в ТГФ (10 мл). Полученному раствору давали подогреться до комнатной температуры, затем концентрировали и очищали (SiO2, этилацетат:гексан 4:6 в качестве элюента) с получением указанного в заголовке соединения (0,25 г). МС (АРСI) 439 (М+Н+, 100%).
Способ F
[3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
[3aR-(3aα,4α,6α,6aα)]-6-[7-хлор-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол (способ G, 13,2 г) в ТГФ (200 мл), содержащем 0,88 аммиак (5 мл), перемешивали в течение 2 ч, затем концентрировали досуха и остаток распределяли в воде и этилацетате. Органические фракции высушивали и затем концентрировали с получением указанного в заголовке соединения (12,5 г). МС (АРСI) 367 (М+Н+, 100%).
Способ G
[3aR-(3aα,4α,6α,6aα)]-6-[7-хлор-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Изоамилнитрит (1,1 мл) добавляли к раствору [3aR-(3aα,4α,6α,6aα)]-6-{[5-амино-6-хлор-2-(пропилтио)пиримидин-4-ил]амино}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола (способ Н, 2,0 г) в ацетонитриле (100 мл) и раствор нагревали при 70°С в течение 1 ч. Охлажденную реакционную смесь концентрировали и очищали (SiO2, смесь этилацетат:изогексан 1:3 в качестве элюента) с получением указанного в заголовке соединения (1,9 г). МС (АРСI) 386 (М+Н+, 100%).
Способ Н
[3aR-(3aα,4α,6α,6aα)]-6-{[5-амино-6-хлор-2-(пропилтио)пиримидин-4-ил]амино}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Порошок железа (3,0 г) добавляли к перемешиваемому раствору [3aR-(3aα,4α,6α,6aα)]-6-{[6-хлор-5-нитро-2-(пропилтио)пиримидин-4-ил]амино}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола (способ I, 2,7 г) в уксусной кислоте (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали до половины объема, разбавляли этилацетатом и промывали водой. Органическую фазу высушивали и концентрировали с получением указанного в заголовке соединения (2,0 г). МС (АРСI) 375 (М+Н+, 100%).
Способ I
[3aR-(3aα,4α,6α,6aα)]-6-{[6-хлор-5-нитро-2-(пропилтио)пиримидин-4-ил]амино}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Раствор [3aR-(3aα,4α,6α,6aα)]-6-аминотетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола гидрохлорида (способ J, 10,0 г) и N,N-диизопропилэтиламина (35 мл) в ТГФ (600 мл) перемешивали в течение 1 ч. Смесь фильтровали и раствор добавляли в течение 1 ч к раствору 4,6-дихлор-5-нитро-2-(пропилтио)пиримидина (WO 9703084, 25,6 г) в ТГФ (1000 мл) и перемешивали еще в течение 2 ч. Объем растворителя уменьшали в вакууме и добавляли этилацетат (1000 мл). Смесь промывали водой и органические слои высушивали и очищали (SiO2, смесь изогексан-этилацетат в качестве элюента) с получением указанного в заголовке соединения (14,2 г). МС (АРСI) 405 (М+Н+, 100%).
Способ J
[3aR-(3aα,4α,6α,6aα)]-6-аминотетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол гидрохлорид
Бис(1,1-диметилэтиловый)эфир [1R-(1α,2β,3β,4α)]-2,3,4-тригидроксициклопентенилимидодикарбоновой кислоты (способ K, 17,4 г) в 6 М НСl (100 мл)/метанола (500 мл) перемешивали в течение 18 ч. Смесь упаривали и затем проводили азеотропную перегонку с толуолом (4×200 мл) с получением бесцветного порошка (8,7 г). Данное твердое вещество суспендировали в ацетоне (250 мл), содержащем 2,2-диметоксипропан (25 мл) и концентрированную НСl (0,2 мл), затем кипятили с обратным холодильником в течение 2 ч. Смесь охлаждали, упаривали и проводили азеотропную перегонку с толуолом (3×200 мл). Остаток растворяли в 20% водной уксусной кислоте и перемешивали в течение 2 ч. Смесь упаривали и проводили азеотропную перегонку с толуолом (4×200 мл) с получением указанного в заголовке соединения (10,1 г). МС (АРСI) 174 (М+Н+, 100%).
Способ K
Бис(1,1-диметилэтиловый)эфир [1R-(1α,2β,3β,4α)]-2,3,4-тригидроксициклопентенилимидодикарбоновой кислоты
К раствору (1R-цис)-бис(1,1-диметилэтил)-4-гидрокси-2-циклопентенилимидодикарбоната (способ L, 17,1 г) в ТГФ (500 мл)/воде (50 мл) добавляли N-метилморфолин-N-оксид (9,4 г), затем четырехокись осмия (10 мл, 2,5% раствор в трет-бутаноле). Смесь перемешивали при комнатной температуре в течение 4 суток, затем обрабатывали гидросульфитом натрия (6,0 г). Суспензию фильтровали через диатомовую землю и продукт очищали (SiO2, смесь этилацетат:гексан 1:1 в качестве элюента) с получением указанного в заголовке соединения (19,1 г). ЯМР: 1,44 (18H, с), 1,46-1,60 (1H, м), 1,97-2,05 (1H, м), 3,55-3,58 (1H, м), 3,66-3,73 (1H, м), 4,11-4,21 (2H, м), 4,54 (1H, д, J=4,8), 4,56 (1H, д, J=5,9), 4,82 (1H, д, J=4,6).
Способ L
(1R-цис)-бис(1,1-диметилэтил)-4-гидрокси-2-циклопентенилимидодикарбонат
К суспензии эфира, промытого гидридом натрия (60% дисперсия в масле; 0,31 г) в ТГФ (30 мл) добавляли бис(1,1-диметилэтиловый)эфир имидодикарбоновой кислоты (1,84 г). Смесь перемешивали при 40°С в течение 1 ч. Затем к смеси при комнатной температуре добавляли (1S-цис)-4-ацетокси-2-циклопентен-1-ол (0,5 г) и тетракис(трифенилфосфин)палладий(0) (0,18 г). Реакционную смесь перемешивали в течение 24 ч, затем очищали (SiO2, смесь этилацетат:гексан 1:9 в качестве элюента) с получением указанного в заголовке соединения в виде бесцветного твердого вещества (0,90 г). ЯМР: 1,43 (18H, с), 1,61 (1H, ддд, J=12,3, 7,7, 6,4), 2,54 (1H, дт, J=12,6, 7,4), 4,51-4,57 (1H, м), 4,86 (1H, ткв, J=8,0, 1,8), 4,91 (1H, д, J=5,4), 5,71-5,77 (2H, м).
Пример 2
Последующее показывает типичные фармацевтические лекарственные формы, содержащие соединение формулы (I) в кристаллической и/или аморфной форме (ниже соединение X), для терапевтического или профилактического применения у людей.
(5% паста мас./об.)
Примечание
Вышеуказанные композиции можно получить обычными способами, хорошо известными в области фармации. Таблетки (а)-(с) можно покрыть энтеросолюбильной оболочкой обычными способами, например, с обеспечением оболочки из ацетатфталатцеллюлозы.
ЯМР-спектры снимали на спектрометре Varian Unity Inova 300 или 400; данные ЯМР даны в форме дельта-значений для основных диагностических протонов, представлены в миллионных долях (ррm) по отношению к тетраметилсилану (TMS) в качестве внутреннего стандарта с использованием пердейтерированного диметилсульфоксида (ДМСО-δ6) в качестве растворителя, если не указано иначе; для примеров, показывающих наличие ротамеров в протонных спектрах ЯМР, представлены только химические сдвиги для основного ротамера; константы взаимодействия (J) представлены в Гц.
Масс-спектры (МС) определяли следующим образом: EI-спектры (электронная ионизация) получали на спектрофотометре VG 70-250S или Finnigan Mat Incos-XL, FAB-спектры (бомбардировка быстрыми атомами) получали на спектрофотометре VG 70-250SEQ, ESI (ионизация электровпрыском) и APCI-спектры химическая ионизация при атмосферном давлении) получали на спектрофотометре Finnigan Mat SSQ7000 или Micromass Platform.
Препаративное разделение ВЭЖХ в основном проводили с использованием колонок Novapak®, Bondapak® или Hypersil®, заполненных обращеннофазовым кремнеземом BDSC-18.
Флэш-хроматографию (указана в примерах, как (SiO2)) проводили с использованием кремнезема Fisher Matrix, 35-70 мкм.
Сокращения
TГФ - тетрагидрофуран
Порошковый РСА - порошковый рентгеноструктурный анализ
ДСК - дифференциальная сканирующая калориметрия.

Изобретение относится к новой кристаллической форме соединения формулы (I), которое обладает свойствами антагониста Р2т и может найти применение для профилактики артериальных тромботических осложнений у пациентов при заболеваниях коронарной артерии, цереброваскулярным и периферическим сосудистым заболеванием.
Кристаллическая форма соединения (I) представляет собой по существу чистый полиморф II, который находится по существу в безводной форме и который характеризуется порошковой рентгенограммой, содержащей характерные основные пики высокой интенсивности при 5,5° (±0,1°), 13,5° (+0,1°), 18,3° (±0,1°), 22,7° (±0,1°) и 24,3° (±0,1°) 2θ, и имеет характерные пики при 5,5° (±0,1°), 6,8° (±0,1°), 10,6° (±0,1°), 13,5° (±0,1°), 14,9° (±0,1°), 18,3° (±0,1°), 19,2° (±0,1°), 22,7° (+0,1°), 24,3° (±0,1°) и 27,1° (±0,1°) 2θ. Кривая дифференциальной сканирующей калориметрии указанного кристаллического полиморфа имеет точку начала плавления, находящуюся в интервале 136-139°С. Изобретение также относится к способу получения предлагаемого полиморфа, согласно которому соединение формулы (I) кристаллизуют из хлороформа. 6 н. и 3 з.п. ф-лы, 6 ил.
1. Соединение формулы (I)
отличающееся тем, что оно представляет собой по существу чистый полиморф II, который находится в, по существу, безводной форме и который характеризуется порошковой рентгенограммой, содержащей характерные пики высокой интенсивности при 5,5° (±0,1°), 13,5° (+0,1°), 18,3° (±0,1°), 22,7° (±0,1°) и 24,3° (±0,1°) 2θ.
2. Соединение формулы (I) по п.1, отличающееся тем, что его порошковая рентгенограмма содержит характерные пики при 5,5° (±0,1°), 6,8° (±0,1°), 10,6° (±0,1°), 13,5° (±0,1°), 14,9° (±0,1°), 18,3° (±0,1°), 19,2° (±0,1°), 22,7° (+0,1°), 24,3° (±0,1°) и 27,1° (±0,1°) 2θ.
3. Соединение формулы (I) по пп.1 или 2, отличающееся тем, что кривая дифференциальной сканирующей калориметрии имеет точку начала плавления, находящуюся в интервале 136-139°С.
4. Способ получения соединения по п.1, в котором соединение формулы (I) кристаллизуют из хлороформа.
5. Соединение по любому из пп.1-3, обладающее свойствами антагониста Р2т, для получения лекарственного препарата.
6. Фармацевтическая композиция, обладающая свойствами антагониста Р2т, включающая терапевтически эффективное количество соединения по любому из пп.1-3 в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
7. Фармацевтическая композиция, включающая соединение по любому из пп.1-3 для применения в целях профилактики артериальных тромботических осложнений у пациентов при заболеваниях коронарной артерии, цереброваскулярным и периферическим сосудистым заболеванием.
8. Применение соединения по любому из пп.1-3 в производстве лекарственного препарата для применения в целях профилактики артериальных тромботических осложнений у пациентов при заболеваниях коронарной артерии, цереброваскулярным и периферическим сосудистым заболеванием.
9. Применение соединения по любому из пп.1-3 в производстве лекарственного препарата для применения в лечении или профилактике артериальных тромботических осложнений у пациентов при заболеваниях коронарной артерии, цереброваскулярным и периферическим сосудистым заболеванием.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| ТРИАЗОЛПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2116308C1 |

