Изобретение относится к химии терпеновых соединений, а именно к арилзамещенным терпенам, в частности способа получения 1-(8-метокси-4,8-диметилнонил)-4-(1-метилэтил) бензола (Pro-Drone, MV-678) формулы 1, из мирцена, выделяемого из растительного возобновляемого сырья.

Соединение 1 наиболее эффективно может быть использовано в сельском хозяйстве, так как обладает рострегулирующей активностью в отношении различных видов насекомых-вредителей и является экологически безопасным инсектицидом (ювеноидом), применяемым для защиты растений.
Ювеноиды представляют собой вещества, которые имитируют или подавляют действие ювенильного гормона, отвечающего в организме насекомого за процессы линьки и метаморфоза. В результате образуются особи, не способные к размножению. Тем самым достигается регуляция численности насекомых, а не полное уничтожение вида. Ювеноиды относятся к экологически безопасным инсектицидам, потому что, как правило, не оказывают действия на других участников биоценоза. (L.M. El-Gazzar, P.G. Koehler, R.S. Patterson, J. Milio, J Med. Entomology, 1986, Vol.23, no.6, pp.651-4; A. Eisa, M.A. El-Fatah, A. El-Nabawi and A.A. El-Dash Phytoparasitica 1991, Vol.19, no., pp.49-55; J.N. Mkhize, Tropical Pest Management, 1986, Vol.32, no.4, pp.324-6; A. Eisa and I.M.A. Ammar, Phytoparasitica", 1992, Vol.20, no.1, pp.7-13; L.S. Mian, M.S. Mulla, J. Economic Entomology, 1982, Vol.75, no.1, pp.80-5; J.N. Mkhize, Insect Science and Its Application 1993, Vol.14, no.3, pp.351-3; N. Sukhapanth, C. Ketavan, J Sci. Soc. of Thailand, 1995, Vol.21, No.3, pp.147-60).
Известен способ получения соединения 1 (патент США №4002769), который состоит из пяти следующих стадий (схема 1):
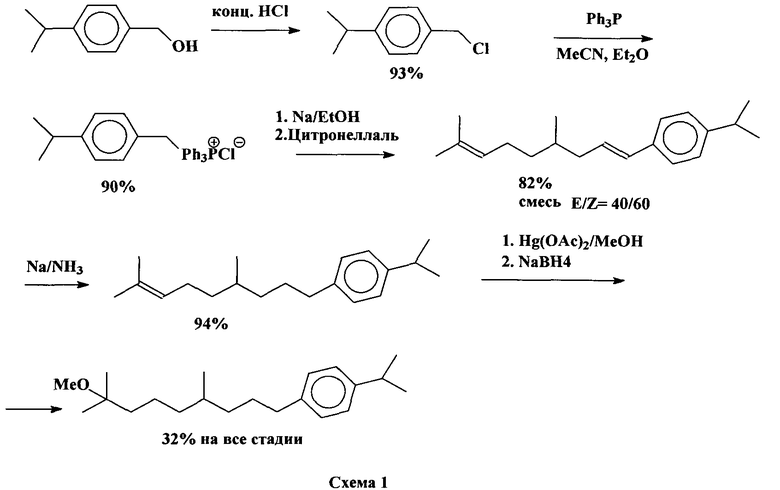
(1) получение p-изопропил-бензилхлорид из p-изопропилбензилового спирта действием концентрированной HCl; с экстракцией продукта гексаном;
(2) взаимодействие p-изопропилбензилхлорида с трифенилфосфином с образованием соответствующей фосфониевой соли с 90%-ным выходом с использованием диэтилового эфира;
(3) реакция Виттига полученной фосфониевой соли с цитронеллалем с образованием 4,8-диметил-1-(n-изопропилфенил)нона-1,7-диена в виде смеси цис- и трансизомеров (40-60). При взаимодействии с натрием в абс. этаноле фосфониевую соль переводят в фосфоран, к которому прибавляют цитронеллаль, отфильтровывают образовавшийся NaCl, после обычной обработки получают 4,8-диметил-1-(n-изопропилфенил)-нона-1,7-диен);
(4) восстановление внутренней двойной связи полученного диена натрием в жидком аммиаке с образованием 4,8-диметил-1-(n-изопропилфенил)-нон-1-ена;
(5) алкоксимеркурирование-демеркурирование 4,8-диметил-1-(p-изопропилфенил)-нон-1-ена (ацетатом ртути в растворе метанола с последующим действием NaBH4 в присутствии NaOH), которое дает целевой продукт 1, с общим выходом 32%.
К недостаткам способа относятся использование большого количества концентрированной соляной кислоты на 1-й стадии, использование пожароопасного серного эфира на 2-й стадии, взрывоопасного металлического натрия на 3-й и 4-й стадиях, высокотоксичного жидкого аммиака на 4-й стадии и токсичных комплексов ртути на 5-й стадии.
Известен способ получения соединения 1 (патенты США №465943 и №4482751 и заявка EP 0116370 A), состоящий из трех стадий:
(1) получение трифенилфосфониевой соли p-изопропилбензилхлорида в растворе метанола и использование метанольного раствора на второй стадии;
(2) взаимодействие полученной фосфониевой соли в растворе метанола с метоксицитронеллалем с образованием 8-метокси-4,8-диметил-1-(4-изопропилфенил)-нон-1-ена (метанольный раствор фосфониевой соли смешивают с раствором метоксида натрия и затем при 10°C перемешивают с метоксицитронеллалем при комнатной температуре, кипятят 1 час, после охлаждения добавляют 30%-ный раствор H2O2 и после обычной обработки получают 8-метокси-4,8-диметил-1-(4-изопропилфенил)-нон-1-ен);
(3) гидрирование двойной связи 8-метокси-4,8-диметил-1-(4-изопропилфенил) - нон-1-ена в растворе гексана в присутствии каталитических количеств палладия на угле при давлении водорода 3-4 атм.
Недостатками способа являются необходимость использования большого количества токсичного метанола на 1-й и 2-й стадиях, легко разлагающегося метоксида натрия и взрывоопасного 30%-ного раствора перекиси водорода, при повышенном давлении водорода на 3-й стадии, а также коммерчески недоступного метоксицитронеллаля на 2-й стадии.
Общим недостатком представленных аналогов является использование эквивалентных количеств трифенилфосфина, который в виде окиси трифенилфосфина выделяется в отходы. Эти вещества являются токсичными. Кроме того, использование фосфорорганических соединений в промышленном производстве не желательно ввиду содержания «биогенного элемента» - фосфора.
Известен способ получения соединения 1 (WO 84/02337), в котором основным промежуточным соединением является дигидромирцен (3,7-диметилокта-1,6-диен), получаемый из альфа- или бета-пинена (схема 2). Этот способ по типу химической реакции наиболее близок к заявляемому и принят за прототип.
Способ-прототип включает следующие стадии:
(1) гидрирование альфа или бета-пинена водородом с образованием пинана (при давлении водорода 350 атм и температуре до 250°C в присутствии скелетного никелевого катализатора);
(2) пиролиз пинана при 600-620°C с образованием дигидромирцена - 3,7-диметилокта-1,6-диена (выход составляет 22%);
(3) метоксилирование дигидромирцена в растворе метанола в присутствии кислотных катализаторов с образованием 7-метокси-3,7-диметилокт-1-ена (в присутствии ионообменной смолы Amberlyst при 50°C, выход составляет 58%);
(4) металлирование полученного 7-метокси-3,7-диметилокт-1-ена пропилмагний хлоридом в присутствии Cp2TiCl2 при 75°C, которое дает 7-метокси-3,7-диметилоктилмагний хлорид (через 19 часов конверсия составляет до 50%);
(5) кросс-сочетание полученного алкилмагнийхлорида с куминил хлоридом в присутствии CuCl или Li2CuCl4 (при 0÷-10°C и перемешивании при комнатной
Схема способа по прототипу:

температуре). В результате целевой продукт 1 получается в виде смеси с p,p'-диизопропилдибензилом. Примесь дибензильного производного может снижать биологическую активность препарата.
Недостатками прототипа являются: необходимость использования высоких температур на стадиях получения пинана и дигидромирцена; низкие и умеренные выходы промежуточных продуктов (так, выход дигидромирцена составляет 22%), также невысокий выход целевого соединения 1 (30-38%) на 5-й стадии; и образование примеси p,p'-диизопропилдибензила. Общий выход по всем стадиям составляет до 3%. Использование пропилмагнийхлорида в качестве металлирующего агента также является недостатком прототипа, поскольку после использования он целиком поступает в отходы, кроме того, он является некоммерческим продуктом и это осложняет процесс.
Задачей настоящего изобретения является разработка технологичного способа получения соединения 1 с высоким выходом из коммерчески доступного сырья.
Техническим результатом изобретения является повышение чистоты целевого продукта 1-(8-метокси-4,8-диметилнонил)-4-(1-метилэтил)бензола и увеличение его выхода.
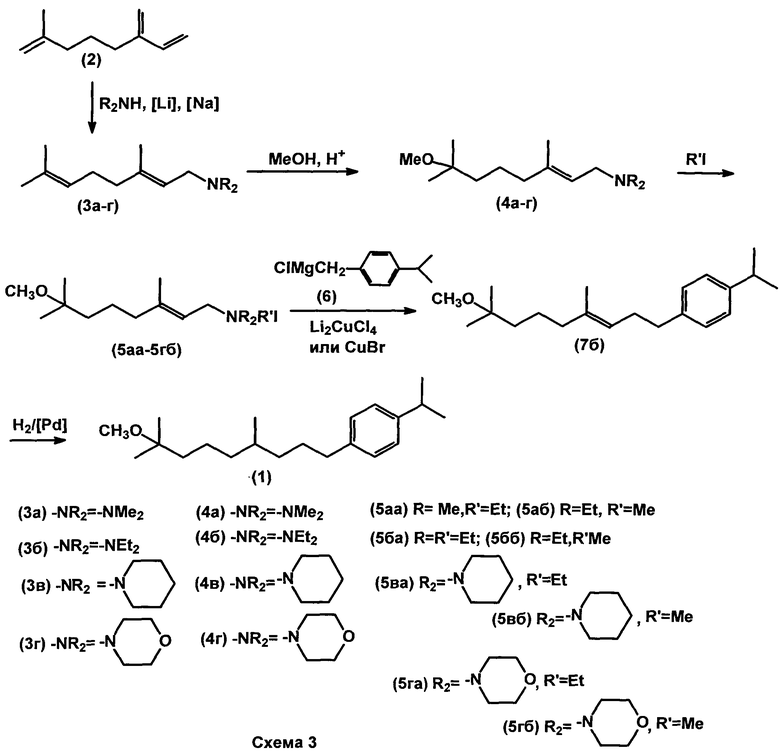
Задача решается предложенными двумя вариантами способа получения соединения 1, один из которых (способ A) заключается в том, что на первой стадии (1) мирцен подвергают взаимодействию с диалкиламином, выбранным из группы: диметиламин, диэтиламин, пиперидин, морфолин, в присутствии катализатора, такого как натрий или литий, при температуре 50-60°C; на стадии (2) полученный геранилдиалкиламин метоксилируют метанолом в присутствии 1-1,2 эквивалентов H2SO4 или HClO4 при 40-50°C с образованием соответствующего N-(7-метокси-3,7-диметил-2E-октенил)диалкиламина; на стадии (3) полученный N-(7-метокси-3,7-диметил-2E-октенил)диалкиламин кватернизируют этил- или метилйодидом; затем на стадии (4) полученный N-(7-метокси-3,7-диметил-2E-октенил)триалкиламмоний йодид подвергают кросс-сочетанию с магнийорганическим соединением, таким как p-изопропилбензилмагнийхлорид, в присутствии медного катализатора, в качестве которого используют CuBr или Li2CuCl4 при температуре (-5)-(-10°C); на стадии (5) полученный 1-(8-метокси-4,8-диметилнонен-3-ил-1)-4-(1-метилэтил)бензол гидрируют в присутствии катализатора Pd/BaSO4 и AcOH в растворе этанола при атмосферном давлении и температуре 20-25°C. (Схема 3).
Второй вариант способа (способ B) заключается в том, что на первой стадии (1) мирцен подвергают взаимодействию с диалкиламином, выбранным из группы: диметиламин, диэтиламин, пиперидин, морфолин, в присутствии катализатора, такого как натрий или литий, при температуре 50-60°C; на стадии (2) полученный геранилдиалкиламин метоксилируют метанолом в присутствии 1-1,2 эквивалентов N2SO4 или HClO4 при 40-50°C с образованием соответствующего N-(7-метокси-3,7-диметил-2E-октенил)диалкиламина; затем на стадии (3) полученный N-(7-метокси-3,7-диметил-2E-октенил)диалкиламин хлорируют фосгеном или алкил(арил)хлоругольным эфиром, выбранным из группы: метилхлоругольный эфир, этилхлоругольный эфир, фенилхлоругольный эфир, с образованием (E)-1-хлор-7-метокси-3,7-диметилокт-2-ена; который на стадии (4) подвергают взаимодействию с p-изопропилбензилмагнийхлоридом в присутствии Li2CuCl4 при температуре (-20)-(-3)°C, с образованием 1-(8-метокси-4,8-диметилнонен-3-ен-1-ил)-4-(1-метилэтил)бензола; который на стадии (5) гидрируют в присутствии катализатора Pd/BaSO4 и AcOH в растворе этанола при атмосферном давлении и температуре 20-25°C. (Схема 4).

Предлагаемый способ получения вещества 1 превосходит прототип по следующим причинам:
Известно, что каталитическое кросс-сочетание, включающее алкилмагниевые компоненты, такие как в прототипе, осложняется большим количеством продуктов гомосочетания (до 50%). Заявляемый продукт отличается высокой чистотой вследствие кросс-сочетания, используемого в способе A, отличного от реакции кросс-сочетания, по прототипу, и заключается в использовании бензилмагниевой компоненты, а не алкильной, как в прототипе, что обусловливает либо отсутствие продукта гомосочетания 8 либо образование его незначительного количества в способе B.
Побочный продукт 8 и изомеры целевого продукта, которые могут образовываться при реакции кросс-сочетания, представлены ниже:

В качестве 2-го компонента реакции кросс-сочетания используют четвертичные аммониевые соли, содержащие аллильный фрагмент. В этом случае продукт гомосочетания 8 не образуется. Состав аммониевой уходящей группы практически не влияет на выход продукта (68-75%). Изомерный состав продуктов варьируется в следующих пределах: 7б: 80-95%, 7а: 10-15%, 7в: 2-10%. 7в является скелетным изомером 7б, а 7а - цис-изомер соединения 7б при гидрировании дает целевой продукт.
Используемые катализаторы CuBr или Li2CuCl4 не оказывают существенного влияния на изомерный состав продукта.
Терпеновое производное реакции кросс-сочетания в заявляемом способе получают в мягких условиях, щелочные металлы используют в каталитических количествах, не превышающих 1,8 мол.%, что не требует специального оборудования. Тогда как в прототипе температуры реакций получения дигидромирцена составляют 250-620°C, причем гидрирование газообразным водородом проводят при высоких температурах и давлениях (250°C и 350 атм).
В заявляемых способах реакция присоединения аминов к мирцену (нетоксичный исходный, выделяемый из растительного сырья) происходит со 100%-ной экономией атомов и не попадает в отходы ни один составляющий элемент реакции. Кроме того, реакция проходит в отсутствие растворителя. Для создания металлоорганической компоненты кросс-сочетания используют дешевый и коммерчески доступный куминилхлорид и металлируют его металлическим магнием, тогда как в прототипе в качестве металлирующего агента используют пропилмагнийхлорид, для получения которого требуется дополнительная стадия получения.
Соединение 1 показало хорошую ювенильную активность на вредителях тропической зоны, таких как термиты или комары.
Заявляемый продукт 1 был исследован в биологических испытаниях на большом мучном хрущаке Tenebrio Molitor L. - основном вредители хлебных запасов в Российской Федерации.
Соединение 1 было протестировано на активность ювенильных гормонов на куколках большого мучного хрущака (Tenebrio molitor Z.) лабораторной разводки Всероссийского научно-исследовательского института химических средств защиты растений (ВНИИХСЗР). Показано, что соединение обладает высокой активностью (оценка 7,6 балла, при максимально возможной оценке 9 баллов).
Газожидкостная хроматография (ГЖХ) выполнена на хроматографе ЛХМ-8МД (5) со стальной колонкой 2000×3 мм с 15% СКТФТ-50 на хроматоне N-AW, газ-носитель - гелий. Спектры ЯМР 1H регистрировали на приборах "Bruker Avance-300 и Avance-400" (CDCl3, внутренний стандарт TMC).
Заявляемые способы иллюстрируются следующими примерами:
Получение соединения 1 способом A (вариант 1)
Пример 1
1.1. Получение N-геранилдиэтиламина 3б. Мелконарезанный металлический Na (0,142 г, 6,0 ммоль, 1,8 мол.%) добавляют в диэтиламин (35,5 мл, 0,344 моль) при комнатной температуре при перемешивании, через 50 мин к полученной смеси прибавляют мирцен (39,1 г, 0,287 моль) и нагревают при температуре 53°C при перемешивании в атмосфере аргона в течение 18 часов. Реакционную смесь выливают в воду, экстрагируют бензолом, затем гексаном. Сушат Na2SO4. Растворители удаляют на роторном испарителе. Фракционная перегонка остатка в вакууме дает 36,7 г (61%) амина 3б с т.кип. 118-124°C/10 мм рт.ст.
1.2. Получение N-(7-метокси-3,7-диметил-2E-октенил)диэтиламина 4б.
40 г (0,18 моль) N-геранилдиэтиламин 3б (40 г, 0, 18 моль) растворяют в 150 мл MeOH и при 0°C прибавляют 24,5 мл (0,36 моль) 57%-ной HClO4. Кипятят 7 дней, выливают в водный раствор KOH (pH 8-8,5), экстрагируют эфиром, сушат Na2SO4. Перегонкой в вакууме получают 25,1 г (52%) N-(7-метокси-3,7-диметил-2E-октенил)диэтиламина 4б, с т.кип. 135-140°C / 10 мм рт.ст., 
1.3.1 N-(7-метокси-3,7-диметил-2E-октенил)триэтиламмоний йодид 5ба.
Смешивают 7 г (28,9 ммоль) амина 4б с 10 мл бензола и прибавляют 9 г (57,8 ммоль) этилйодида. Оставляют в темноте при комнатной температуре. Через 3 дня белый осадок отфильтровают, промывают пентаном. Получают 10,4 г (93%) соли 5ба. Найдено, %: C 51.43, H 8.90, N 3.62. C17H36NOI. Вычислено, %: C 51.38, H 9.13, N 3.52.
1.4. Получение 1-(8-метокси-4,8-диметилнонен-3-ил-1)-4-(1-метилэтил)бензола 7б. Из 2,8 г (16,3 ммоль) p-изопропилбензилхлорида и 0,5 г (20 ммоль) магния получают реагент Гриньяра в 20 мл диэтилового эфира. Полученный реагент прибавляют при -20°C к суспензии 4,0 г (10,9 ммоль) соли (5а) и 0,1 мл (0,697 ммоль, 8 мол.%) Li3CuCl4 в 30 мл ТГФ, перемешивают при -7÷-4°C в течение 5 ч и при комнатной температуре 12 ч. Реакционную смесь выливают в насыщенный раствор NH4Cl, продукт экстрагируют эфиром, сушат Na2SO4. После перегонки в вакууме получают 2,8 г (95%) соединения 7б с т.кип. 123-130°C (2 мм рт.ст.) в виде смеси изомеров: 95% - соединения 7б, 3% его цис-изомера и 2% продукта аллильной перегруппировки 7в. Продукта гомосочетания 8 не обнаружено. Найдено, %: C 83.92, H 11.22. C21H34O. Вычислено, %: C 83.38; H 11.33.
1.5. Получение 1-(8-метокси-4,8-диметилнонанил-1)-4-(1-метилэтил)бензола 1 Соединение 7б (0,8 г, 2,6 ммоль) в 20 мл EtOH гидрируют газообразным водородом при атмосферном давлении и комнатной температуре в присутствии 0,1 г Pd/BaSO4 и 1 мл AcOH. После обычной обработки получают целевой продукт с выходом 97%. 1H ЯМР спектр, δ м.д., J, Гц: 0.90 д (3H, CH3, J=6.7), 1.09 с, с (6H, CH3, CH3) 1.22 д (6H, CH3, CH3, J=6.5), 1.32 м (6H, CH, 5CH2), 2.60 т (2H, CH2-Ph, J=6.7), 2.90 м (1H, CH-Ph), 3.16 с (3H, CH3O), 7.10 м (4H, Ph). Найдено, %: C 82.90, H 11.82. C21H36O. Вычислено, %: C 82.83, H 11.92.
1.3.2. N-(7-метокси-3,7-диметил-2-октен-1-ил)диэтилметиламмоний йодид 5бб. К 3,1 г (12,9 ммоль) амина 4б прибавляют 0,96 мл метилйодида (15,48 ммоль) в атмосфере аргона. Смесь выдерживают при 25°C в темноте 5 дней. Из раствора выпадает коричневое вязкое масло. Масло промывают гексаном методом декантации для удаления непрореагировавшего амина и избытка MeI. Получают: 4,7 г (95%) соли 5бб в виде вязкого масла. Спектр ПМР (CDCl3), м.д., J, Гц: 1,10, 1,11 (с, с, 6H, 8CH3, 7aCH3), 1,34-1,46 (м, 10H, NCH3, 5CH2, 6CH2), 1,85 (с, 3H, 3aCH3), 2,11 (м, 2H, 4CH2), 3,13 (с, 3H, OCH3), 3,14 (с, 3H, NCH3), 3,43-3,63 (м, 4H, NCH2), 4,07 (д, 2H, 1CH2, J=8,33), 5,26 (т, 1H, 2CH, J=8,33).
Далее - аналогично примерам 1.4 и 1.5.
Пример 2
2.1. Получение N-геранилдиметиламина 3а проводят аналогично получению соединения 3б (Пример 1, п.1.1.), используя 15,5 г (0,344 моль) диметиламина, конденсированного из раствора этанола. Получают 30,7 г (59%) амина 3а с т.кип. 95-102°C / 10 мм рт.ст.
2.2. N-(7-метокси-3,7-диметил-2E-октенил)диметиламин 4а получают аналогично получению соединения 4б (Пример 1, п.1.2.), используя 32,6 г (0,18 моль) N-геранилдиметиламина 3а. Получают 21,5 г (56%) продукт 4а с т.кип. 112-117°C / 10 мм рт.ст. Найдено, %: C 72.99, H 12.67, N 6.95. C13H27NO. Вычислено, %: C 73.18, H 12.76, N 6.56.
2.3. Продукт 5аб получают аналогично примеру 1 (п.1.3.2.).
2.4. 1-(8-метокси-4,8-диметилнонен-3-ил-1)-4-(1-метилэтил)бензол 7б получают из продукта 5аб, аналогично примеру 1 (п.1.4).
Получение соединения 1 из соединения 7б описано в примере 1 (п.1.5).
Пример 3.
3.1. N-геранилпиперидин 3в получают аналогично соединению 3б (пример 1, п.1.1), используя 29,3 г (0,344 моль) пиперидина. Получают 45,7 г (72%) амина 3в с т.кип. 112-116°C / 1 мм рт.ст.
3.2. N-(7-метокси-3,7-диметил-2-октенил)пиперидин 4в получают аналогично соединению 4б (пример 1, п.1.2), используя 39,8 г (0,18 моль) N-геранилпиперидина 3в. Получают 23,7 г (52%) амина 4в с т.кип. 124-129°C / 10 мм рт.ст. Найдено, %: C 75.93, H 12.20. C16H31NO. Вычислено, %: C 75.83, H 12.33.
3.3. Соединение 5вб получают аналогично примеру 1, п.1.3.2.
3.4. 1-(8-метокси-4,8-диметилнонен-3-ил-1)-4-(1-метилэтил)бензол 7б получают из продукта 5вб, как описано в примере 1 (п.1.4).
Получение целевого продукта 1 из соединения 7б описано выше (пример 1, п.1.5.).
Пример 4
4.1. Получение N-геранилморфолина 3г проводят аналогично описанному в примере 1 (п.1.1.), используя 30,0 г (0,344 моль) морфолина. Получают 48 г (75%) амина 3 г с т.кип. 118-123°C / 1 мм рт.ст.
4.2 Получение N-(7-метокси-3,7-диметил-2E-октенил)морфолина 4 г проводят аналогично описанному в примере 1 (п.1.2.), используя 40,2 г (0,18 моль) N-геранилморфолина 3г. Получают 27,1 г (59%) амина 4 г с т.кип. 141-147°C / 1 мм рт.ст. Найдено, %: C 70.56; H 11.29; N 5.97. C15H29NO2. Вычислено, %: C 70.54; H 11.45; N 5.48.
Целевое соединение 1 получают из амина 4г (стадии 3-5) аналогично примеру 1 (п.п.1.3.2, 1.4 и 1.5).
Получение соединения 1 способом Б (вариант 2)
Пример 5
Получение соединений 3б и 4б проводят так же, как на стадиях 1 и 2 способа A, описанных в примере 1 (п.п.1.1 и 1.2)
5.3.1. Получение (E)-1-хлор-7-метокси-3,7-диметил-2-октена 5д. В раствор 6,6 г (0,0275 моля) N-(7-метокси-3,7-диметил-2E-октенил)диэтиламина 4б в 20 мл абс. бензола при 0-5°C пропускают фосген до тех пор, пока не поглотилось 6 г (0.06 моля). Бензол упаривают, к остатку добавляют пентан, промывают разб. HCl, сушат Na2SO4. После перегонки получают 3,7 г (65.8%) (E)-7-метокси-3,7-диметил-2-октенил хдорида 5д с т.кип 90-92°C (3 мм рт.ст.). ИК, см-1 (пленка) 2960, 1660, 1445, 1155, 1030, 980, 835, 670; 1H ЯМР (300 MHz, CDCl3), δ: 1,12 с (3H, 7aCH3), 1, 14 с (3H, 8CH3), 1,75 (с, 3H, 3aCH3), 2,11 м (4H,, 4CH2, 5CH2), 3,18 (с, 3H, OCH3), 4,04 д (2H, 1CH2 J=8.5 Hz), 5,42 т (1H, J=8.5 Hz, 2CH).
5.3.2. (E)-1-хлор-7-метокси-3,7-диметил-2-октен 5д получают из 45,1 ммоля N-(7-метокси-3,7-диметил-2E-октен-1-ил)-диэтиламина 4б и 45,1 ммоля фенилхлоругольного эфира (прибавляют при 7°C, затем 3 час при 30°C), выделяют хлорид 5д с т.кип. 60-63°C (1 мм рт.ст.) и выходом 67.8%. ИК, см-1 (пленка) 2960, 1660, 1445, 1155, 1030, 980, 835; 1H ЯМР (300 MHz, CDCl3), δ: 1,12 с (3H, 7aCH3), 1, 14 с (3H, 8CH3), 1,75 (с, 3H, 3aCH3), 2,11 м (4H,, 4CH2, 5CH2), 3,18 (с, 3H, OCH3), 4,09 д (2H, 1CH2, J=8.5 Hz), 5,52 т (1H, J=8.5 Hz, 2CH).
5.4. Из 3,7 г (22 ммоль) p-изопропилбензилхлорида и 0,8 г (30 ммоль) магния получают реагент Гриньяра (6) в 20 мл диэтилового эфира. В течение 20 мин реагент Гриньяра прибавляют к раствору 3,5 г (17 ммоль) хлорида 5д и 1 мл (1.39 ммоль, 0.43 мол.%) Li2CuCl4 в 20 мл ТГФ при -20°C, перемешивают при -5÷-7°C в течение 5 час, выливают в насыщенный раствор NH4Cl, продукт экстрагируют эфиром, сушат Na2SO4. После перегонки в вакууме получают 3,9 г (80%) соединения 7б с т.кип. 123-130°C (2 мм рт.ст.) в виде смеси изомеров 8% - соединения 7а, 75% - соединения 7б, 12% - продукта аллильной перегруппировки 7в и 5% - продукта гомосочетания 8 - пара, пара'-диизопропилдибензил.
5.5. Получение целевого продукта 1 гидрированием соединения 7б описано аналогично примеру 1, п.1.5.
Получение соединений 4а, 4в и 4г проводят так же, как на стадиях 1 и 2 способа A, описанных в примере 1 (п.п.1.1 и 1.2)
(E)-1-хлор-7-метокси-3,7-диметил-2-октен 5д можно получать из соединений 4а, 4в и 4г аналогично получению соединения 5д, описанному в примере 5 (п.п.5.3.1 или 5.3.2).
Получение целевого продукта 1 гидрированием соединения 7б аналогично примеру 1, п.1.5.)
Пример 6
Инсектицидную активность определяли при нанесении на последний сегмента абдомена куколки через 0-6 час после отрождения 4.4 мкг ацетонового раствора, содержащего 10000 мг·литр-1 соединения 1. Соединение было испытано в двух сериях по 17 особей в каждой. Эффект оценивали при отрождении имаго по 9-балльной шкале Шмиалека (Schmialek P. // Z. Naturforsch 1963. Bd. 18В. N 7. Р.516). Оценку 0 приписывали куколке, дающей нормальный имаго, оценку 9 приписывали куколке не дающей имаго и не отличающейся от исходной куколки. Промежуточные оценки соответствовали степени морфогенетических нарушений в превращении куколок в имаго.
После изучения морфологических изменений каждой особи средняя оценка в баллах (N) была определена по формуле:

Абсолютную ошибку (ΔN) определяли по формуле:

Здесь, 1, 2, 3, … n - оценка в баллах от 0 до 9;
x1, x2, x3, … xn - число куколок, получивших соответствующую оценку;
A - общее число куколок.
В результате биологических испытаний на активность ювенильного гормона соединение (V) в растворе с концентрацией 10 г/литр получило оценку N=7.6 баллов (из 9 возможных), абсолютная ошибка измерений ΔN=0.38, относительная ошибка ΔN/N=5,1%.
Таким образом, заявляемый способ основан на использовании мирцена в качестве исходного вещества, который является природным нетоксическим недорогим и коммерчески доступным соединением, выделяемым из растительного сырья; в заявляемом способе более высокий выход целевого продукта (38%) по сравнению с прототипом и аналогами; соединение получают более чистое (до 95% чистоты), причем примесью (2-5%) является скелетный изомер продукта, который не может существенно снижать его биологическую активность, тогда как примесь дибензильного производного в прототипе, отрицательно сказывается на биологической активности и делает препарат токсичным;
условия реакций более получения мягкие с использованием нетоксичных реагентов, опасные щелочные металлы применяют лишь в количествах, не превышающих 1,8 мол.%, и не требует применения взрывоопасного оборудования; в способе используют реакцию со 100%-ной экономией атомов в отсутствие растворителей.
Таким образом, предлагаемый способ является более технологичным, экономичным и экологически безопасным по сравнению с прототипом.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1-(8-МЕТОКСИ-4,8-ДИМЕТИЛНОНИЛ)-4-(1-МЕТИЛЭТИЛ)БЕНЗОЛА ИЗ ИЗОПРЕНА (ВАРИАНТЫ) | 2014 |
|
RU2561272C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(7-МЕТОКСИ-3,7-ДИМЕТИЛ-2Е-ОКТЕН-1-ИЛ)-2-МЕТИЛБЕНЗИМИДАЗОЛА | 2015 |
|
RU2601564C1 |
| 2,4,7-ТРИМЕТИЛОКТ-6-ЕН-1-ОЛ В КАЧЕСТВЕ ДУШИСТОГО ВЕЩЕСТВА | 2016 |
|
RU2712172C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(2,7-ДИМЕТИЛ-2,7-ОКТАДИЕН-1-ИЛ)БЕНЗИМИДАЗОЛА | 2019 |
|
RU2713952C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ГЕРАНИЛБЕНЗИМИДАЗОЛА | 2021 |
|
RU2781210C1 |
| Способ получения полиеновых соединений или их солей | 1974 |
|
SU613718A3 |
| 1-N-АЛКИЛ-N-АРИЛПИРИМИДИНАМИНЫ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2153494C2 |
| Способ получения полиеновых соединений или их солей | 1975 |
|
SU623515A3 |
| УЛУЧШЕННЫЙ СИНТЕЗ ГОНОКИОЛА | 2016 |
|
RU2727202C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
Изобретение относится к вариантам способа получения 1-(8-метокси-4,8-диметилнонил)-4-(1-метилэтил) бензола, обладающего рострегулирующей активностью в отношении различных видов насекомых-вредителей и являющегося безопасным инсектицидом. Первый вариант способа заключается в том, что на первой стадии (1) мирцен подвергают взаимодействию с диалкиламином, выбранным из группы: диметиламин, диэтиламин, пиперидин, морфолин, в присутствии катализатора, такого как натрий или литий, при температуре 50-60°C; на стадии (2) полученный геранилдиалкиламин метоксилируют метанолом в присутствии 1-1,2 эквивалентов H2SO4 или HClO4 при 40-50°C с образованием соответствующего N-(7-метокси-3,7-диметил-2Е-октенил)диалкиламина; на стадии (3) полученный N-(7-метокси-3,7-диметил-2E-октен-1-ил)диалкиламин кватернизируют этил- или метилйодидом; затем на стадии (4) полученный N-(7-метокси-3,7-диметил-2Е-октенил)триалкиламмоний йодид подвергают кросс-сочетанию с магнийорганическим соединением, таким как p-изопропилбензилмагнийхлорид, в присутствии медного катализатора, в качестве которого используют CuBr или Li2CuCl4 при температуре (-5)-(-10°C); на стадии (5) полученный 1-(8-метокси-4,8-диметилнонен-3-ил-1)-4-(1-метилэтил)бензол гидрируют в присутствии катализатора Pd/BaSO4 и AcOH в растворе этанола при атмосферном давлении и температуре 20-25°C. Предлагаемые варианты способа позволяют получить целевой продукт с высоким выходом. 2 н.п. ф-лы, 6 пр.
1. Способ получения 1-(8-метокси-4,8-диметилнонил)-4-(1-метилэтил)бензола, включающий кросс-сочетание магнийорганического соединения с нуклеофилом, катализируемое Li2CuCl4, отличающийся тем, что на первой стадии (1) мирцен подвергают взаимодействию с диалкиламином, выбранным из группы: диметиламин, диэтиламин, пиперидин, морфолин, в присутствии катализатора, такого как натрий или литий, при температуре 50-60°C; на стадии (2) полученный геранилдиалкиламин метоксилируют метанолом в присутствии 1-1,2 эквивалентов H2SO4 или HClO4 при 40-50°C с образованием соответствующего N-(7-метокси-3,7-диметил-2Е-октенил)диалкиламина; на стадии (3) полученный N-(7-метокси-3,7-диметил-2E-октен-1-ил)диалкиламин кватернизируют этил- или метилйодидом; затем на стадии (4) полученный N-(7-метокси-3,7-диметил-2Е-октенил)триалкиламмоний йодид подвергают кросс-сочетанию с магнийорганическим соединением, таким как p-изопропилбензилмагнийхлорид, в присутствии медного катализатора, в качестве которого используют CuBr или Li2CuCl4 при температуре (-5)-(-10°C); на стадии (5) полученный 1-(8-метокси-4,8-диметилнонен-3-ил-1)-4-(1-метилэтил)бензол гидрируют в присутствии катализатора Pd/BaSO4 и AcOH в растворе этанола при атмосферном давлении и температуре 20-25°C.
2. Способ получения 1-(8-метокси-4,8-диметилнонил)-4-(1-метилэтил)бензола, заключающийся в том, что на первой стадии (1) мирцен подвергают взаимодействию с диалкиламином, выбранным из группы: диметиламин, диэтиламин, пиперидин, морфолин, в присутствии катализатора, такого как натрий или литий, при температуре 50-60°C; на стадии (2) полученный геранилдиалкиламин метоксилируют метанолом в присутствии 1-1,2 эквивалентов H2SO4 или HClO4 при 40-50°C с образованием соответствующего N-(7-метокси-3,7-диметил-2Е-октенил)диалкиламина; затем на стадии (3) полученный N-(7-метокси-3,7-диметил-2E-октенил)диалкиламин хлорируют фосгеном или алкил(арил)хлоругольным эфиром, выбранным из группы: метилхлоругольный эфир, этилхлоругольный эфир, фенилхлоругольный эфир, с образованием (E)-1-хлор-7-метокси-3,7-диметил-2-октена; который на стадии (4) подвергают взаимодействию с p-изопропилбензилмагнийхлоридом в присутствии Li2CuCl4 при температуре (-20)-(-3)°C, с образованием 1-(8-метокси-4,8-диметилнонен-3-ен-1-ил)-4-(1-метилэтил)бензола; который на стадии (5) гидрируют в присутствии катализатора Pd/BaSO4 и AcOH в растворе этанола при атмосферном давлении и температуре 20-25°C.
| WO 1984002337A1, 21.06.1984 | |||
| Устройство для использования качки на волнении для передвижения судна | 1935 |
|
SU47562A1 |
| Способ получения 8-метокси-4,8-диметил-1-(4-изопропилфенил)-нонана (его варианты) | 1984 |
|
SU1407396A3 |
| О.С | |||
| Куковинец и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЖОрХ, 2012, 46(6), 808-815 | |||

