Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения 2,3,3,3-тетрафторпропилена.
Уровень техники
2,3,3,3-тетрафторпропилен, также известен как HFO-1234yf, HFC-1234yf или просто как 1234yf. В дальнейшем, если нет иных указаний, 2,3,3,3-тетрафторпропилен будет обозначаться как 1234yf. Известные способы получения 1234yf обычно отличаются такими недостатками, как низкие выходы и/или использование токсичных и/или дорогих реагентов, и/или использование экстремальных условий, и/или получение токсичных побочных продуктов.
Способы получения 1234yf описаны, например, в Journal Fluorine Chemistry (82), 1997, 171-174. В этой статье 1234yf получают взаимодействием тетрафторида серы с трифторацетилацетоном. Однако этот способ представляет только академический интерес из-за трудностей, связанных с использованием реагентов и их стоимостью.
Другой способ получения 1234yf описан в US-2931840. В этом случае речь идет о пиролизе С1-хлорфторуглеводородов с получением или без получения тетрафторэтилена, приводящим к образованию 1234yf. Однако описанные выходы являются очень низкими и необходимы усилия для работы с вредными химическими реагентами в экстремальных условиях. Кроме того, можно ожидать, что такой способ приведет к получению различных токсичных побочных продуктов.
Помимо указания известных способов, желательно представить новый способ получения 1234yf, использующий только легко доступное сырье.
Нет необходимости перечислять или обсуждать ранее опубликованные документы в этом описании патента в качестве подтверждения, что этот документ частично отражает современный уровень техники или распространенные общие знания.
Раскрытие изобретения
Заявленное изобретение рассматривает недостатки известных способов получения 1234yf, предоставляя способ получения 1234yf, состоящий (а) во взаимодействии 1,1,2,3,3,3-гексафторпропилена (обозначаемого в дальнейшем как 1216 или HFP) с водородом в присутствии катализатора гидрирования, приводящему к получению 1,1,2,3,3,3-гексафторпропана (обозначаемого в дальнейшем как 236еа); (b) в дегидрофторировании 236еа, приводящему к получению 1,2,3,3,3-пентафторпропилена (обозначаемому в дальнейшем как 1225уе); (с) во взаимодействии 1225уе с водородом в присутствии катализатора гидрирования, приводящем к получению 1,2,3,3,3-пентафторпропана (обозначаемого в дальнейшем как 245eb); и (d) в дегидрофторировании 245eb, приводящем к получению 1234yf. Если нет иных указаний, в дальнейшем он будет обозначаться как способ настоящего изобретения.
1225уе существует в виде геометрических изомеров E-1225уе и Z-1225ye. Если нет иных указаний, как используется здесь, 1225уе относится к смеси геометрических изомеров.
Каждая из стадий от (а) до (d) может быть проведена в виде периодического или непрерывного процесса (предпочтительно непрерывного), при использовании любой подходящей аппаратуры, например, статической мешалки, трубчатого реактора, перемешиваемого корпусного реактора или перемешиваемого газо-жидкостного разъединительного реактора. Предпочтительно аппарат изготовляют из одного или нескольких коррозионно-стойких материалов, например, Hastelloy® или Inconel®.
Согласно любому из аспектов способа настоящего изобретения, описанного здесь, продукты со стадий (а), (b), (с) и/или (d) могут быть подвергнуты стадии очистки. Например, очистка может быть произведена, отделением желаемого продукта (продуктов) или реагентов одной или несколькими перегонками, стадиями конденсации или фазового разделения и/или очисткой водой или водным основанием.
Способ настоящего изобретения может быть осуществлен с использованием различных подходящих реакторных топологических схем. Например, способ может быть осуществлен непрерывно с последовательным проведением стадий (а), (b), (с) и (d), в указанном порядке при использовании отдельного реактора на каждой стадии.
В качестве альтернативы способ может быть реализован полунепрерывным путем при использовании одного реактора гидрирования и одного реактора дегидрофторирования, и при последовательном проведении стадий (а), (b), (с) и (d) в указанном порядке. В таком полунепрерывном способе HFP превращается в 236еа в реакторе гидрирования и 236еа превращается в 1225уе реакторе дегидрофторирования. Обе эти реакции, стадии (а) и (b), проходят в течение определенного времени, обычно приблизительно от 1 приблизительно до 1000 часов, например, приблизительно от 10 приблизительно до 500 часов, например, приблизительно от 20 приблизительно до 200 часов. Полученный 1225уе выдерживают в промежуточном резервуаре до тех пор, пока не будут использованы те же самые реакторы гидрирования и дегидрофторирования для превращения 1225уе в 245eb и 245eb в 1234yf, соответственно. В свою очередь, эти реакции, стадии (с) и (d), проходят в течение определенного времени, обычно приблизительно от 1 приблизительно до 1000 часов, например, приблизительно от 10 приблизительно до 500 часов, например, приблизительно от 20 приблизительно до 200 часов.
В следующей предпочтительной реакционной топологии, стадии (а) и (с) способа настоящего изобретения могут быть проведены одновременно в одном и том же реакторе. Известно, что гидрирование фторолефинов, например, HFP и 1225уе представляет собой высоко экзотермический процесс. Считают, что при объединении реакций гидрирования экзотермическую природу реакций можно контролировать, используя преимущества различных значений теплот реакции и теплоемкости продуктов. Это и определяет преимущества более низких капитальных вложений и повышенной эффективности способа настоящего изобретения.
Продукт реакции, при которой стадии (а) и (с) проводят одновременно в одном и том же реакторе, содержит как 236еа, так и 245eb. Они могут быть отделены друг от друга (например, перегонкой) перед загрузкой в отдельные реакторы дегидрофторирования для проведения стадий (b) и (d).
В качестве альтернативы после любых произвольных стадий очистки для удаления нежелательных побочных продуктов (например, CF3CFHCH3 (254eb) и/или Н2), объединенный поток 236еа и 245eb может быть загружен в единственный реактор. Таким образом, стадии дегидрофторирования (b) и (d) могут быть проведены одновременно в одном и том же реакторе. Полагают, что это дает преимущества более низких капитальных вложений и повышенной эффективности способа настоящего изобретения. Конечно, можно провести раздельные загрузки 236еа и 245eb, обусловленные отдельными реакторами гидрирования, в единственный реактор дегидрофторирования, в котором стадии (о) и (d) проходят одновременно.
В дополнительном варианте осуществления экзотермические реакции гидрирования со стадий (а) и/или (с), особенно со стадии (а), можно контролировать использованием потока газа-разбавителя. Чтобы исключить неправильное толкование, поток газа-разбавителя может быть использован только на одной стадии (а), на одной стадии (с) или на объединенных стадиях (а) и (с). В качестве потока газа-разбавителя может выступать такой газ, как азот или 1,1,1,2-тетрафторэтан (134а), избыток одного или нескольких видов сырья (например, HFP и/или 1225уе), или один или два вида продуктов со стадий (а) и (с), 245eb или 236еа.
Следующее описание предпочтительных условий, катализаторов и т.п. для стадий (а) и (с) применимо для всех топологий реакторов (например, описанных выше), которые могут быть использованы для выполнения способа настоящего изобретения.
Реакции гидрирования на стадиях (а) и (с) могут быть проведены в жидкой или паровой фазе, предпочтительно в паровой фазе. На стадиях (а) и (с) могут быть использованы температуры в диапазоне приблизительно от -50 приблизительно до 275°С.Предпочтительные температуры для жидкофазного гидрирования составляют приблизительно от -50 приблизительно до 50°C, например, приблизительно от 15 приблизительно до 40°C. Предпочтительные температуры для гидрирования в паровой фазе составляют приблизительно от 0 приблизительно до 250°C, например, приблизительно от 20 приблизительно до 200°C, например, приблизительно от 50 приблизительно до 150°C.
Стадии (а) и (с) могут быть проведены в присутствии фторированного полярного апротонного растворителя, особенно при проведении в жидкой фазе. Подходящие растворители включают в себя HFC (например, 134а) и PFC (например, перфтордекалин).
Реакции гидрирования на стадии (а) и (с) могут быть проведены при атмосферном давлении, ниже атмосферного давления и выше атмосферного давления, предпочтительно выше атмосферного давления. Например, гидрирование можно проводить при давлении, составляющем приблизительно от 0 приблизительно до 40 бар, например, приблизительно от 1 приблизительно до 30 бар, например, приблизительно от 5 приблизительно до 20 бар.
Соотношение водород: 1216 на стадии (а) и водород: 1225уе на стадии (с) равно соответственно предпочтительно от 0,1:1 приблизительно до 40:1, например, приблизительно от 1:1 приблизительно до 20:1, предпочтительно, от 1,1:1 приблизительно до 10:1, например, приблизительно от 1,5:1 приблизительно до 5:1.
На стадиях (а) и (с) могут быть использованы любые подходящие катализаторы гидрирования, в том числе катализаторы, содержащие переходный металл. Предпочтительные катализаторы гидрирования, содержащие переходный металл, включают в себя катализаторы, содержащие Ni, Pd, Pt, Re, Rh, Ru и их смеси. Такие катализаторы могут быть нанесенными (например, на алюминий, титан, кремний, цирконий (или соответствующие фториды), фторид кальция, углерод или сульфат бария) или ненанесенными (например, Ni Ренея или губчатый палладий). В настоящее время палладий на угле (Pd/C) является предпочтительным катализатором гидрирования для стадий (а) и (с).
Обычно катализатор гидрирования используют в количестве приблизительно от 0,01 приблизительно до 30 масс.% в расчете на общую массу компонентов, которые участвуют на стадиях (а) и (с), например, приблизительно от 0,1 приблизительно до 10%.
Когда в качестве катализатора используют Pd/C, Pd находится в количестве приблизительно от 0,01 приблизительно до 10% от массы катализатора, например, приблизительно от 0,1 приблизительно до 5%.
Время контакта водорода и катализатора для 1216 и 1225уе на стадиях (а) и (с) соответственно составляет приблизительно от 1 приблизительно до 200 секунд, например, приблизительно от 2 приблизительно до 150 секунд.
Соответствующее описание предпочтительных условий, реагентов, катализаторов и т.д. для стадий (b) и (d) применимо для всех топологий реакторов (например, для тех, которые описаны выше), которые могут быть использованы для осуществления способа настоящего изобретения.
Стадии (b) и (d) способа настоящего изобретения могут быть проведены в любых подходящих условиях реакции, эффективных для проведения дегидрофторирования 236еа, приводящего к образованию 1225уе и/или 245eb для получения 1234yf. Дегидрофторирование может быть проведено в паровой или жидкой фазе и при температуре приблизительно от -70 до приблизительно 1000°C (например, приблизительно от 0 приблизительно до 400°C). Способ может быть реализован при атмосферном давлении, ниже атмосферного давления и выше атмосферного давления, предпочтительно приблизительно от 0 приблизительно до 30 бар.
Дегидрофторирование может быть индуцировано термическим путем, может быть опосредовано основанием и/или может быть катализировано любым подходящим катализатором. Подходящие катализаторы включают в себя катализаторы на основе металлов и углерода, например, такие, которые содержат активированный уголь, металлы основной группы (например, катализаторы на основе алюминия) и переходные металлы, например, катализаторы на основе хрома (например, цинк/хромовые) или катализаторы на основе никеля (например, никелевая сетка).
Один из предпочтительных способов осуществления дегидрофторирования на стадиях (b) и (d) состоит во взаимодействии 236еа и 245eb с катализатором на основе хрома, например, с описанным в ЕР-А-0502605, ЕР-А-0773061, ЕР-А-957074, WO 98/10862 и WO 2006/106353 (например, цинк/хромовым катализатором).
Под термином «цинк/хромовый катализатор» мы подразумеваем любой катализатор, содержащий хром и цинк или соединение хрома и цинка или соединение цинка. Обычно хром или соединение хрома присутствующие в цинк/хромововом катализаторе настоящего изобретения, представляют собой оксид, оксифторид или фторид хрома, например, оксид хрома.
Общее количество цинка или соединения цинка, находящегося в цинк/хромововом катализаторе настоящего изобретения, обычно составляет приблизительно от 0,01% приблизительно до 25%, предпочтительно от 0,1% приблизительно до 25%, обычно от 0,01% до 6% цинка, и в некоторых вариантах осуществления предпочтительно от 0,5 масс % приблизительно до 25 масс.% катализатора, предпочтительно приблизительно от 1 до 10 масс.% катализатора, более предпочтительно приблизительно от 2 до 8 масс.% катализатора, например, приблизительно от 4 до 6 масс.% катализатора. В других вариантах осуществления, катализатор обычно содержит от 0,01% до 1%, более предпочтительно от 0,05% до 0,5% цинка.
Предпочтительное количество зависит от ряда факторов, например, от природы хрома или соединения хрома и/или цинка или соединения цинка и/или способа приготовления катализатора. Эти факторы будут описаны более подробно ниже.
Следует понимать, что количество цинка или соединения цинка, цитированного здесь, относится к количеству элементарного цинка, присутствующего в виде элементарного цинка, или соединения цинка.
Цинк/хромововые катализаторы, используемые в настоящем изобретении, могут включать в себя дополнительный металл или его соединение. Обычно дополнительный металл является двухвалентным или трехвалентным металлом, предпочтительно выбранным из никеля, магния, алюминия и их смесей. Обычно дополнительный металл находится в количестве, равном приблизительно от 0,01 масс.% приблизительно до 25 масс.% от массы катализатора, предпочтительно приблизительно от 0,01 масс.% приблизительно до 10 масс.% от массы катализатора. В других вариантах осуществления, катализатор содержит, по меньшей мере, приблизительно 0,5 масс.% или, по меньшей мере, приблизительно 1 масс.% дополнительного металла.
Цинк/хромововые катализаторы, используемые в настоящем изобретении, могут быть аморфными. Это означает, что катализатор не должен проявлять существенных свойств кристаллического вещества, например, дифракции рентгеновских лучей.
В качестве альтернативы катализаторы могут быть частично кристаллическими. Это означает, что от 0,1 до 50 масс.% катализатора находится в форме одного или нескольких кристаллических соединений хрома и/или одного или нескольких кристаллических соединений цинка. Если используют частично кристаллический катализатор, он предпочтительно содержит от 0,2 до 25 масс.%, более предпочтительно от 0,3 до 10 масс.%, еще более предпочтительно от 0,4 до 5 масс.% катализатора в форме одного или нескольких кристаллических соединений хрома и/или одного или нескольких кристаллических соединений цинка.
При использовании реакции дегидрофторирования степень кристалличности может меняться. Таким образом, возможно, что катализатор настоящего изобретения имеет степень кристалличности, как определено выше перед использованием реакции дегидрофторирования, и имеет степень кристалличности, выходящую за эти пределы, в процессе или после использования реакции дегидрофторирования.
Процентное содержание кристаллического материала в катализаторах настоящего изобретения может быть определено любым подходящим способом, известным в данной области техники. Подходящие способы включают в себя дифракцию рентгеновских лучей. (XRD). Когда используют дифракцию рентгеновских лучей, количество кристаллического оксида хрома может быть определено, исходя из известного количества графита, находящегося в катализаторе (например, графита, используемого для получения гранул катализатора) или более предпочтительно сравнением интенсивности XRD образцов материала с эталонами, полученными из подходящих международно-признанных стандартов, например, эталонами NIST (National Institute of Standards and Technology).
Цинк/хромовые катализаторы настоящего изобретения обычно имеют площадь поверхности, равную, по меньшей мере, 50 м2/г и предпочтительно от 70 до 250 м2/г и наиболее предпочтительно от 100 до 200 м2/г, перед тем, как он будет подвергнут предварительной обработке фторсодержащими соединениями, например, фтористым водородом или фторированными углеводородами. В процессе такой предварительной обработки, которая будет описана подробно ниже, по меньшей мере, несколько атомов кислорода в катализаторе замещают атомами фтора.
Цинк/хромовые катализаторы настоящего изобретения обычно характеризуются оптимальным балансом уровней активности и селективности. Предпочтительно они также они также обладают степенью химической устойчивости, что означает, что они имеют относительно большую продолжительность жизни. Катализаторы настоящего изобретения также предпочтительно обладают механической прочностью, что обеспечивает относительно легкий способ эксплуатации, например, их можно загружать в реакторы или выгружать из реакторов, используя известную технику.
Цинк/хромовые катализаторы настоящего изобретения могут быть предоставлены в любой подходящей форме, известной в данной области техники. Например, они могут быть предоставлены в виде таблеток или гранул подходящего размера с целью использования в стационарном слое или псевдоожиженном слое. Катализаторы могут быть нанесенными или ненанесенными. Если катализатор является нанесенным, подходящие подложки включают в себя AIF3, фторированный алюминий или активированный уголь.
Цинк/хромовые катализаторы настоящего изобретения включают в себя промотированные формы таких катализаторов, в том числе содержащие такие, которые имеют повышенную кислотность и/или основность по Льюису или Бренстеду.
Аморфные катализаторы, которые могут быть использованы в настоящем изобретении, могут быть получены любым способом, известным в данной области техники, для получения аморфных катализаторов на основе хрома. Подходящие способы включают в себя соосаждение из растворов нитратов цинка и хрома при добавлении гидроксида аммония. В качестве альтернативы может быть использовано поверхностное импрегнирование цинка или его соединения на аморфный хромовый катализатор.
Дополнительные способы получения аморфных цинк/хромовых катализаторов включают в себя, например, восстановление соединения хрома (VI), например, хромата, бихромата, в особенности, бихромата аммония, до хрома (III), металлическим цинком с последующим соосаждением и промыванием; или смешиванием в виде твердых веществ соединения хрома (VI) и соединения цинка, например, ацетата цинка или оксалата цинка, и нагреванием смеси до высокой температуры, чтобы провести восстановление соединения хрома (VI) в оксид хрома (III) и окислить соединение цинка в до оксида цинка.
Цинк может быть введен внутрь и/или на поверхность аморфного хромового катализатора в виде соединения, например, галогенида, оксигалогенида, оксида или гидроксида в зависимости, по меньшей мере, до некоторой степени от техники приготовления катализатора. В случае, когда получение аморфного катализатора происходит импрегнированием хрома, галогенированного хрома или оксигалогенида хрома, соединение предпочтительно представляет собой водорастворимую соль, например, галогенид, нитрат или карбонат и используется в виде водного раствора или суспензии. В качестве альтернативы гидроксиды цинка и хрома можно соосадить (например, при использовании основания, например, гидроксида натрия или гидроксида аммония), а затем превратить в оксиды для получения аморфного катализатора. Смешение и механическое измельчение нерастворимого соединения цинка с основным хромовым катализатором обеспечивает дополнительный способ получения предшественника аморфного катализатора. Способ получения аморфного катализатора на основе оксигалогенида хрома включает в себя добавление цинка к гидратированному галогениду хрома.
Количество цинка или соединения цинка, введенного в предшественник аморфного катализатора, зависит от использованного способа получения. Считают, что работающий катализатор содержит находящиеся на поверхности катионы цинка, расположенные в решетке, содержащей хром, например, в решетке оксида хрома, оксигалогенида или галогенида. Таким образом, требуемое количество цинка или соединения цинка обычно меньше для катализаторов, полученных импрегнированием, чем для катализаторов, полученных другими способами, например, соосаждением, которые также содержат цинк или соединения цинка, расположенные вне поверхности. Любые из вышеупомянутых способов или другие способы могут быть использованы для получения аморфных катализаторов, которые могут быть использованы в способе настоящего изобретения.
Цинк/хромовые катализаторы, описанные здесь, обычно стабилизируют тепловой обработкой перед использованием, таким образом, чтобы они были стабильными в условиях окружающей среды, действию которых они подвержены при эксплуатации. Такая стабилизация часто является двухстадийным процессом. На первой стадии катализатор стабилизируют тепловой обработкой в атмосфере азота или смеси азот/воздух. В данной области техники эту стадию часто называют "кальцинированием". Катализаторы фторирования затем обычно стабилизируют фтористым водородом тепловой обработкой в атмосфере фтористого водорода. Эту стадию часто называют "предфторированием".
Путем тщательного контроля за условиями, при которых проводятся две эти стадии тепловой обработки в катализаторе может быть индуцирована определенная степень кристалличности.
Например, аморфный катализатор может быть подвергнут тепловой обработке при температуре приблизительно от 300 приблизительно до 600°C, предпочтительно приблизительно от 400 приблизительно до 600°C, более предпочтительно приблизительно от 500 приблизительно до 590°C, например, при 520, 540, 560 или 580°C в течение периода приблизительно от 1 приблизительно до 12 часов, предпочтительно приблизительно от 2 приблизительно до 8 часов, например, приблизительно в течение 4 часов в подходящей газовой среде. Подходящие газовые среды, в которых может быть проведена тепловая обработка, включают в себя атмосферу азота или атмосферу, содержащую уровень кислорода приблизительно от ОД приблизительно до 10% об./об. азота. Альтернативно могут быть использованы другие окислительные среды. Например, среды, содержащие окислительные агенты, включают в себя, но не ограничены теми, которые содержат источник нитратов, С2О3 или О2 (например, воздух). Такая стадия тепловой обработки может быть проведена дополнительно или вместо стадии кальцинирования, которую обычно используют при известном уровне техники для получения аморфных катализаторов.
Условия стадии предфторирования могут быть выбраны так, чтобы индуцировать изменение в кристалличности катализатора или так, чтобы не индуцировать такое изменение. Авторы настоящего изобретения показали, что тепловая обработка предшественника катализатора при температуре приблизительно от 250 приблизительно до 500°C, предпочтительно приблизительно от 300 приблизительно до 400°C при атмосферном или при повышенном давлении в течение времени приблизительно от 1 приблизительно до 16 часов в присутствии фтористого водорода, необязательно в присутствии другого газа, например, воздуха, может привести к получению катализатора, в котором кристалличность является таковой, как определена выше, например, от 0,1 до 8,0 масс.% по катализатору (обычно от 0,1 до менее, чем 8,0 масс.% по катализатору) находящемуся в форме одного или нескольких кристаллических соединений хрома и/или одного или нескольких кристаллических соединений, по меньшей мере, одного дополнительного металла.
Специалисты в данной области техники поймут, что степень кристалличности катализатора можно изменять варьируя условия, описанные выше, например, варьируя температуру и/или время и/или газовую среду, в которой проводят тепловую обработку. Обычно, например, катализаторы с более высокими степенями кристалличности (например, от 8 до 50 масс.% по катализатору) могут быть получены при повышении температуры и/или при увеличении времени кальцинирования и/или увеличении окислительной природы газовой среды, в которой проводят предварительную обработку катализатора.
Варьирование кристалличности катализатора в зависимости от температуры кальцинирования, времени и газовой среды иллюстрируется в следующей таблице, представляющей ряд экспериментов, в которых 8 г образцов 6% цинк/хромового катализатора подвергают кальцинированию в диапазоне условий и уровня индуцируемой кристалличности, определенной дифракцией рентгеновских лучей.
время (t, часы)
Предфторирующая обработка обычно приводит к снижению площади поверхности катализатора. После стадии предфторирования катализаторы настоящего изобретения обычно имеют площадь поверхности от 20 до 200 м2/г, например, от 50 до 150 м2/г, например, менее, чем приблизительно 100 м2/г.
При использовании, цинк/хромовый катализатор может быть периодически регенерирован или реактивирован нагреванием на воздухе при температуре приблизительно от 300°C приблизительно до 500°С. Воздух может быть использован в виде смеси с инертным газом или с фтористым водородом, который выделяет тепло при протекании процесса каталитической обработки и может быть непосредственно применен в процессе фторирования, использующем реактивированный катализатор.
Любой катализатор, используемый на стадиях (b) и (d) может быть использован в количестве приблизительно от 0,01 приблизительно до 50 масс.%, например, приблизительно от 0,1 приблизительно до 30 масс.%, например, приблизительно от 0,5 приблизительно до 20 масс.%, в расчете на массу органических веществ (например, 236еа и/или 245eb).
Катализируемое (металлом или углеродом) дегидрофторирование 236еа и 245eb обычно проводят при температуре приблизительно от 0 приблизительно до 400°C. Например, при проведении стадий (b) и (d) в присутствии катализатора на основе хрома (например, цинк/хромового катализатора), стадии предпочтительно проводят при температуре приблизительно от 200 приблизительно до 360°C, например, приблизительно от 240 приблизительно до 340°C.
Стадии (b) и (d) предпочтительно проводят под давлением приблизительно от 0,01 приблизительно до 25 бар или приблизительно от 0,1 приблизительно до 20 бар, например, приблизительно от 1 до приблизительно 10 бар (например, от 1 до 5 бар).
Время контакта 236еа и/или 245eb с катализатором в реакции каталитического дегидрофторирования, на стадиях (b) и (d), соответственно, равно приблизительно от 1 приблизительно до 500 секунд, например, приблизительно от 5 приблизительно до 400 секунд.
Стадии дегидрофторирования (b) и (d) настоящего изобретения могут проходить в присутствии фтористого водорода (HF). Например, может присутствовать HF, образующийся при дегидрофторировании 23беа и/или 245eb и/или HF из отдельного исходного продукта. В некоторых вариантах осуществления желательно использовать некоторое количество HF для того, чтобы предотвратить и/или задержать избыточное разложение органического исходного продукта и/или спекание катализатора на стадиях (b) и (d). Альтернативно, стадии (b) и (d) могут быть проведены в отсутствие HF, и/или HF может быть удален из реактора для облегчения протекания реакции(й) дегидрофторирования.
Когда HF присутствует на стадиях (b) и (d), молярное соотношение HF:органические вещества (например, 236еа и/или 245eb) предпочтительно находится в диапазоне приблизительно от 0,01:1 приблизительно до 50:1, например, в диапазоне приблизительно от 0,1:1 приблизительно до 40:1, например, в диапазоне приблизительно от 0,5:1 приблизительно до 30:1, или приблизительно от 2:1 приблизительно до 15:1 (например, в диапазоне приблизительно от 5:1 приблизительно до 10:1).
Другой предпочтительный способ проведения дегидрофторирования на стадиях (b) и (d) состоит во взаимодействии 236еа и/или 245eb с основанием (дегидрофторирование, опосредованное основанием). Предпочтительно, основанием является гидроксид или амид металла (предпочтительно гидроксид или амид основного металла, например, гидроксид или амид щелочного или щелочноземельного металла).
Если нет иных указаний, кроме использованных здесь, под термином "гидроксид щелочного металла" мы подразумеваем соединение или смесь соединений, выбранных из гидроксида лития, гидроксида натрия, гидроксида калия, гидроксида рубидия и гидроксида цезия. Аналогично, под термином "амид щелочного металла", мы подразумеваем соединение или смесь соединений, выбранных из амида лития, амида натрия, амида калия, амида рубидия и амида цезия.
Если нет иных указаний, кроме использованных здесь, под термином "гидроксид щелочноземельного металла", мы подразумеваем соединение или смесь соединений, выбранных из гидроксида бериллия, гидроксида магния, гидроксида кальция, гидроксида стронция и гидроксида бария. Аналогично, под термином "амид щелочноземельного металла", мы подразумеваем соединение или смесь соединений, выбранных из амида бериллия, амида магния, амида кальция, амида стронция и амида бария.
Обычно, процесс дегидрофторирования, опосредованного основанием, на стадиях (b) и (d) проводят при температурах в диапазоне приблизительно от -50 приблизительно до 300°С.Предпочтительно процесс проводят при температуре в диапазоне приблизительно от 20 приблизительно до 250°С, например, в диапазоне приблизительно от 50 приблизительно до 200°С. Дегидрофторирование, опосредованное основанием, проводят под давлением в диапазоне приблизительно от 0 приблизительно до 30 бар.
Время проведения реакции для процесса дегидрофторирования, опосредованного основанием, можно варьировать в широком диапазоне. Однако время реакции обычно находится в диапазоне приблизительно от 0,01 приблизительно до 50 часов, например, находится в диапазоне приблизительно от 0,1 приблизительно до 30 часов, например, в диапазоне приблизительно от 1 приблизительно до 20 часов.
Конечно, специалисты в данной области техники понимают, что предпочтительные условия (например, температура, давление и время реакции) для проведения дегидрофторирования, опосредованного основанием, может варьировать в зависимости от ряда факторов, например, природы используемого основания и/или присутствия катализатор и т.д.
Стадии (b) и (d) процесса дегидрофторирования, опосредованного основанием, могут быть проведены в присутствии или отсутствии растворителя. Если не используют растворитель, 23беа и/или 245eb можно пропустить внутрь или над расплавленным основанием или горячим основанием, например, в трубчатом реакторе. При использовании растворителя в некоторых вариантах осуществления предпочтительным растворителем является вода, хотя могут быть использованы и многие другие растворители. В некоторых вариантах осуществления могут быть предпочтительными такие растворители, как спирты (например, пропан- 1-ол), диолы (например, этиленгликоль) и полиолы, например, полиэтиленгликоль (например, ПЭГ200 или ПЭГ300). Эти растворители могут быть использованы индивидуально или в сочетании. В дополнительных вариантах осуществления могут быть предпочтительными растворители, принадлежащие к классу, известному как полярные апротонные растворители. Примеры таких полярных апротонных растворителей включают в себя диглим, сульфолан, диметилформамид (ДМФА), диоксан, ацетонитрил, гексаметиленфосфорамид (ГМФА), диметилсульфоксид (ДМСО) и N-метилпирролидон (НМП). Температура кипения растворителя предпочтительно должна быть таковой, чтобы не создавалось избыточного давления в условиях реакции.
Предпочтительным основанием является гидроксид щелочного металла, выбранный из группы, состоящей из гидроксида лития, гидроксида натрия и гидроксида калия, более предпочтительно, гидроксида натрия и гидроксида калия и наиболее предпочтительно, гидроксида калия.
Другим предпочтительным основанием является гидроксид щелочноземельного металла, выбранный из группы, состоящей из гидроксида магния и гидроксида кальция, более предпочтительно, гидроксида кальция.
Основание обычно находится в количестве в диапазоне приблизительно от 1 до 50 масс.% в расчете на общую массу компонентов, участвующих в стадиях (b) и (d).
Предпочтительно количеств основания находится в диапазоне приблизительно от 5 до 30 масс.%.
Как упоминалось ранее, дегидрофторирование, опосредованное основанием, может предпочтительно использовать воду в качестве растворителя. Поэтому реакция дегидрофторирования может предпочтительно использовать водный раствор, по меньшей мере, одного основания, например, гидроксида щелочного (или щелочноземельного) металла без участия сорастворителя или разбавителя. Однако сорастворитель или разбавитель может быть использован, например, для модификации вязкости системы, для создания предпочтительной фазы для побочных продуктов реакции или для увеличения теплоемкости. Эффективные сорастворители или разбавители включают в себя те, которые не являются реакционноспособными или отрицательно влияющими на равновесие или кинетику процесса и включают в себя спирты, например, метанол и этанол; диолы, например, этиленгликоль; простые эфиры, например, диэтиловый эфир, дибутиловый эфир; сложные эфиры, например, метилацетат, этилацетат и т.п., линейные, разветвленные и циклические алканы, например, циклогексан, метилциклогексан; фторированные разбавители, например, гексафторизопропанол, перфтортетрагидрофуран и перфтордекалин.
Дегидрофторирование, опосредованное основанием, на стадиях (b) и (d) предпочтительно проводят в присутствии катализатора. Катализатор предпочтительно является катализатором фазового переноса, который способствует переносу ионных соединений в органическую фазу, например, из водной фазы. Если в качестве растворителя используют воду, водная или неорганическая фаза присутствует вследствие наличия гидроксида щелочного металла, а органическая фаза присутствует в результате наличия фторуглерода. Катализатор фазового переноса способствует протеканию реакции между этими разнородными компонентами. В то время, как различные катализаторы фазового переноса могут действовать различными способами, механизм их действия не является определяющим для использования в настоящем изобретении, если они способствуют протеканию реакции дегидрофторирования. Катализатор фазового переноса может быть ионным или нейтральным и его обычно выбирают из группы, состоящей из краунэфиров, ониевых солей, криптандов и полиалкиленгликолей и их производных (например, их фторированных производных).
Следует использовать эффективное количество катализатора фазового переноса, чтобы провести данную реакцию, повлиять на селективность в отношении желаемых продуктов или повысить выход; такое количество может быть определено небольшим количеством экспериментов, если выбраны реагенты, условия реакции и катализатор фазового переноса. Обычно количество присутствующего используемого катализатора относительно количества органических соединений на стадиях (b) и (d) находится в диапазоне от 0,001 до 20 моль. %, например, от 0,01 до 10 моль. %, например, от 0,05 до 5 моль. %.
Краунэфиры представляют собой циклические молекулы, в которых эфирные группы связаны диметиленовыми связями. Краунэфиры образуют молекулярную структуру, которая, как считают, способна притягивать или удерживать ион щелочного металла гидроксида и таким образом способствовать протеканию реакции. Особенно эффективные краунэфиры включают в себя 18-краун-6 (особенно в сочетании с гидроксидом калия), 15-краун-5 (особенно в сочетании с гидроксидом натрия) и 12-краун-4 (особенно в сочетании с гидроксидом лития)
Также эффективны производные вышеописанных краунэфиров, например, дибензил-18-краун-6, дициклогексил-18-краун-6, дибензил-24-краун-8 и дибензил-12-краун-4. Другие соединения, аналогичные краунэфирам и используемые в этих же целях, представляют собой соединения, которые отличаются замещением одного или нескольких атомов кислорода другими типами донорных атомов, в частности, N или S. Могут быть также использованы фторированные производные всех вышеописанных соединений.
Криптанды представляют собой другой класс соединений, используемых в дегидрофторировании, опосредованном основанием, в качестве катализаторов фазового переноса. Они представляют собой трехмерные полимакроциклические хелатирующие агенты, образующиеся путем соединения мостиковых структур с цепями, содержащими соответствующим образом расположенные донорные атомы. Все донорные атомы мостиков могут содержать О, N, или S, или соединения могут представлять собой смешанные донорные макроциклы, в которых мостиковая цепь содержит комбинацию таких донорных атомов. Подходящие криптанды включают в себя бициклические молекулы, образующиеся путем присоединения азотных мостиков к цепям, состоящим из (-ОСН2СН2-) групп, например, как в [2.2.2]криптанд (4,7,13,16,21,24-гексаокса-1,10-диазабицикло [8.8.8]гексакозане, доступного под торговыми названиями Криптанд 222 и Криптофикс 222).
Ониевые соли, которые могут быть использованы в качестве катализаторов в процессе, опосредованном основанием, стадии (iii), включают в себя четвертичные фосфониевые соли и четвертичные аммониевые соли, которые могут быть представлены формулами R1R2R3R4P+Z и R1R2R3R4N+Z-, соответственно. В этих формулах каждый из R1, R2, R3 и R4 обычно представляют собой независимо, С1-10 алкильную группу, арильную группу (например, фенил, нафтил или пиридинил) или ариалкильную группу (например, бензил или С1-10 алкилзамещенный фенил), и Z- представляет собой галогенид или другой подходящий противоион (например, гидросульфат).
Отдельные примеры таких фосфониевых солей или четвертичных аммониевых солей включают в себя тетраметиламмоний хлорид, тетраметиламмоний бромид, бензилтриэтиламмоний хлорид, метилтриоктиламмоний хлорид (коммерчески доступный под брендами Aliquat 336 и Adogen 464), тетра-н-бутиламмоний хлорид, тетра-н-бутиламмоний бромид, тетра-н-бутиламмоний гидросульфат, тетра-н-бутилфосфоний хлорид, тетрафенилфосфоний бромид, тетрафенилфосфоний хлорид, трифенилметилфосфоний бромид и трифенилметилфосфоний хлорид. Бензилтриэтиламмоний хлорид предпочтителен для использования в сильно щелочных условиях.
Другие эффективные ониевые соли включают в себя те, которые проявляют высокую термостойкость (например, приблизительно до 200°C), например, 4-диалкиламинопиридиниевые соли, тетрафениларсоний хлорид, бис[трис(диметиламино)фосфин]иминий хлорид и тетракис[трис(диметиламино)фосфинимино]фосфоний хлорид. Описано, что два последних соединения также стабильны при нагревании в присутствии концентрированного гидроксида натрия и, поэтому могут быть особенно эффективными.
Соединения полиалкиленгликоля, эффективные в качестве катализаторов фазового переноса могут быть представлены формулой R6O(R5O)mR7, где R5 представляет собой C1-10 алкиленовую группу, каждый из R6 и R7 представляют собой независимо Н, C1-10 алкильную группу, арильную группу (например, фенил, нафтил или пиридинил) или ариалкильную группу (например, бензил или C1-10 алкилзамещенный фенил), и m представляет собой целое число, равное, по меньшей мере 2. Предпочтительно, что как R6, так и R7 являются одинаковыми, например, оба радикала могут быть Н.
Такие полиалкиленгликоли включают в себя диэтиленгликоль, триэтиленгликоль, тетраэтиленгликоль, пентаэтиленгликоль, гексаэтиленгликоль, диизопропиленгликоль, дипропиленгликоль, трипропиленгликоль, тетрапропиленгликоль и тетраметиленгликоль, простые эфиры моноалкилгликоля, например, монометиловый, моноэтиловьш, монопропиловый и монобутиловый эфиры таких гликолей, диалкиловые эфиры, например, диметиловый эфир тетраэтиленгликоля и диметиловый эфир пентаэтиленгликоля, фениловые эфиры, бензиловые эфиры таких гликолей и полиалкиленгликоли, например, полиэтиленгликоль (средняя молекулярная масса приблизительно 300) и полиэтиленгликоль (средняя молекулярная масса приблизительно 400) и диалкиловые эфиры (например, диметиловый, дипропиловый, дибутиловый) таких полиэтиленгликолей.
Сочетания катализаторов фазового переноса, в которые входят по одному из групп, описанных выше, может быть также эффективно, так же, как и комбинации или смеси, состоящие более, чем из одной группы. В настоящее время краунэфиры и четвертичные аммониевые соли являются предпочтительными группами катализаторов, например, 18-краун-б и его фторированные производные и бензилтриэтиламмоний хлорид. Когда стадии от (а) до (d) проводят отдельно и последовательно, продукт каждой стадии может непосредственно реагировать на следующей стадии без очистки. Например, продукт со стадии (а), содержащий 236еа, может быть загружен непосредственно в отдельный реактор для стадии дегидрофторирования (b). Стадию (b) можно даже проводить в том же реакторе, что и стадию (а), особенно если используют один и тот же катализатор для стадий (а) и (b).
Однако предпочтительно, чтобы продукт с каждой стадии очищали перед проведением реакции на следующей стадии. Очистка может быть проведена отделением нужного продукта от любых других продуктов или реагентов на каждой стадии при помощи одной или нескольких перегонок, стадий конденсации или фазового разделения и/или обработкой водой или водным основанием. Например, 236еа, образующийся на стадии (а), может быть отогнан от водорода и любого оставшегося 1216 перед загрузкой в реактор стадии дегидрофторирования (b).
Когда стадии от (а) и (с) проводят в одном и том же реакторе, продукт из этого реактора можно загрузить в один или несколько реакторов для того, чтобы осуществить стадии (b) и (d) без очистки продуктов со стадий (а) и (с). Однако, предпочтительно, чтобы образующиеся 236еа и 245eb, были отделены (например, перегонкой или другим подходящим способом) от водорода и любых остающихся 1216 и 1225уе, образующихся на стадиях (а) и (с) и загружены в реактор для стадий дегидрофторирования (b) и (d). Если стадии (b) и (d) не объединены, 236еа и 245eb могут быть далее отделены друг от друга перед пропусканием в реакторы для проведения стадий (b) и (d) по отдельности.
Краткое описание чертежей
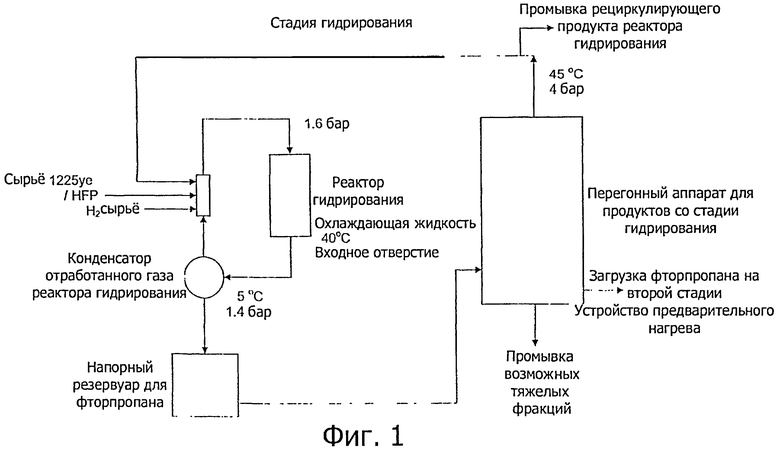
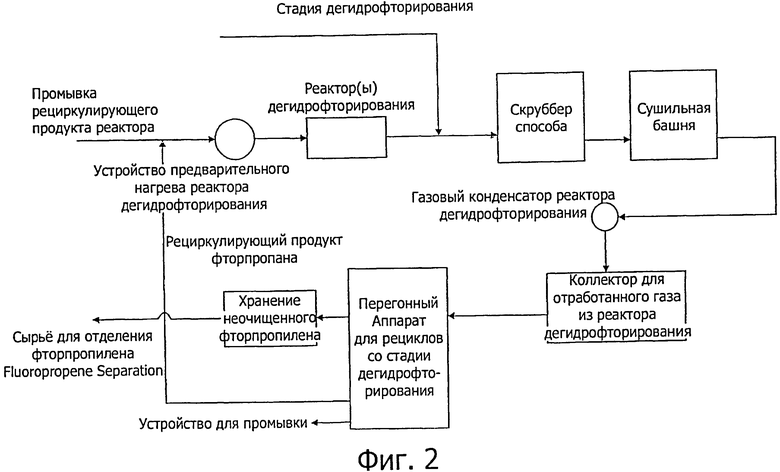
Фиг.1-3 показывают предпочтительное осуществление способа настоящего изобретения, когда стадии гидрирования (а) и (с) проводят непрерывно на одном и том же оборудовании (см. фиг.1), и стадии дегидрофторирования (b) и (d) проводят непрерывно на одном и том же оборудовании (см. фиг.2).
Как показано на фиг.1, HFP и R1225ye могут быть загружены вместе с водородом в реактор гидрирования, обычно вместе с рециклом, который служит для уменьшения динамического воздействия экзотермического эффекта, возникающего при проведении реакции гидрирования. Для уменьшения динамического воздействия экзотермического эффекта может быть использована также охлаждающая жидкость.
Отработанные реакторные газы можно частично конденсировать, причем пар рециркулируют при помощи любого подходящего устройства (например, вентилятора, компрессора или эжектора) на входное отверстие реактора гидрирования. Конденсированную жидкость можно затем перекачать, необязательно через фторпропановый гидроаккумулирующий бак в дистилляционную колонну (обозначенный на фиг.1 как перегонный аппарат для продукта со стадии гидрирования).
Верхнюю фракцию (дистиллят), содержащую более летучие фторпропилены, обычно рециклизуют в реактор гидрирования. Необязательно, чтобы функционирование перегонного аппарата для продукта со стадии гидрирования могло быть приспособлено для рециклизации части фторпропанов, которые служат для снижения экзотермического эффекта реактора гидрирования. Может быть также проведена продувка рециклизованного продукта реактора гидрирования и совмещена с очисткой реактора дегидрофторирования (см. фиг.2). Менее летучие фторпропановые продукты гидрирования (236еа и 245eb) могут быть извлечены из колонны в виде нижней фракции, либо (i) в виде жидкости и вновь переведены в пар, либо (ii) в виде пара, засасываемого обратно в нижнюю фракцию дистилляционного аппарата.
Как показано на фиг.2, испарившиеся продукты гидрирования (236еа и 245eb) можно смешать с продуктами рециклизации со стадии дегидрофторирования. При последующем нагревании до температуры реакции их загружают в реактор дегидрофторирования. При желании единственный реактор, представленный на технологической принципиальной схеме, может быть заменен двумя или несколькими реакторами или реакционными зонами, чтобы обеспечить оптимизацию условий для осуществления различных реакций дегидрофторирования.
Как упоминалось выше, форсунки на входе и выходе сырья для реактора(ов) дегидрофторирования могут быть совмещены с продувкой рециклизованного продукта реактора гидрирования перед удалением HF, который образуется в нем и может находиться в виде со-сырья для реактора(в) дегидрофторирования. Удаление HF достигается, как показано на фиг.2, при помощи скруббера, например, промыванием водой. Однако, для удаления HF, могут быть использованы и другие подходящие способы, например, азеотропная перегонка
После удаления HF и сушки (например, при помощи H2SO4 в сушильной башне), неочищенный продукт затем обычно направляют через конденсатор отработанного газа и полость коллектора в перегонный аппарат для дегидрофторирования рециклизованного продукта. Здесь менее летучие непрореагировавшие фторпропаны собираются на дне перегонного аппарата (нижняя фракция) и их возвращают в реактор, а целевой неочищенный фторпропиленовый продукт (содержащий 1225уе и 1234yf) извлекают из перегонного аппарата в виде верхней фракции (дистиллята).
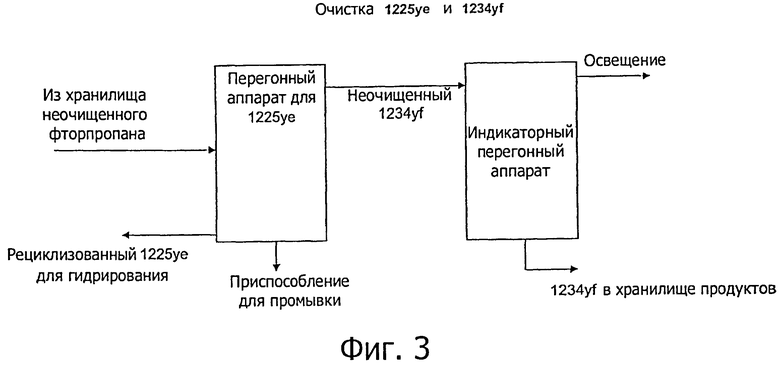
Как показано на фиг.3, неочищенные фторпропилены обычно перекачивают, необязательно через резервуар насоса хранилища фторпропилена, в первую дистилляционную колонну (обозначенную на фиг.3 как 1225уе перегонный аппарат), который удаляет 1225уе как нижнюю фракцию для повторного использования в реакции гидрирования. Верхнюю фракцию (дистиллят), содержащую 1234yf, извлекают из перегонного аппарата и затем могут пропустить через перегонный аппарат для очистки 1234yf, например, удалением любых летучих компонентов.
Настоящее изобретение далее будет дополнительно проиллюстрировано следующими неограничивающими примерами.
Осуществление изобретения
Пример 1. Гидрирование HFP и 2Т-1225уе
В трубчатый реактор, имеющий приблизительно 1,25 см (0,5") в диаметре и 20 см по длине, загружают 10 г влажного 0,5% Pd/C катализатора. Реактор помещают внутри печи, снабженной вентилятором. Во входное и выходное отверстие реактора помещают термопары, находящиеся в тесном контакте с катализатором. Если реактор находится в печи, в него поступает азот, водород и органическое сырье. Потоки этого сырья устанавливаются и контролируются при помощи автоматического регулирующего устройства массового расхода.
Перед использованием катализатор вначале сушат в потоке азота (95 мл/мин) при 110°C. Катализатор считают высохшим, когда обе внутренние термопары показывают приблизительно 110°C. Затем катализатор восстанавливают пропусканием водорода (5 мл/мин) в токе азота и поддерживают температуру при 110°C в течение 2 часов. Затем температуру повышают до 150°C еще в течение 30 минут.
Готовят смесь органического сырья, состоящего из 48,5 моль. % гексафторпропилена (1216) и 51,5 моль. % 2-3,3,3,2,1-пентафторпропилена (Z-1225ye). Затем пропускают смеси этого сырья с водородом и азотом через реактор и приводят в контакт с катализатором. Периодически отбирают образцы газов, выходящих из реактора и анализируют при помощи ГХ и ГХ-МС. Эти приборы калибруют, используя известные стандарты. Результаты ряда экспериментов с различными составами сырья представлены в таблице 1.
* CF3CFHCH3
Пример 2. Дегидрофторирование 236еа
2 г образца аморфного катализатора, содержащего 6 масс.% Zn на хроме загружают в реакционный сосуд Inconel® (15 см х 1,25 мм), помещенный в трубчатую печь. Этот катализатор сушат при 250°C в течение 1 часа, затем предварительно фторируют при соотношении N2."HF, равном 6:1 в течение 1 часа при 250°C перед повышением температуры до 380°C, при которой прекращается поток азотного разбавителя. Приблизительно через 18 часов исходный продукт HF отсоединяют и реактор охлаждают до 220-240°C.
После предварительного фторирования изучают зависимость дегидрофторирования 236еа как функцию температуры и соотношения HF:236. Скорости потока исходного газообразного сырья выбирают так, чтобы время контакта между катализатором и смесью исходного сырья достигало приблизительно 5 секунд. Соотношения HF:236ea рассмотрены в диапазоне 0-10. При каждой температуре системе уравновешивают в течение приблизительно 20 минут прежде, чем начинают отбирать образцы реакторного отработанного газа при каждой температуре для анализа либо ГХ, либо ГХ-МС, как описано в примере 1. Результаты представлены в таблице 2.
Пример 3. Дегидрофторирование 245eb
В реакторную трубку Inconel® (1,25 см (0.5") × 30 см) загружают 6 г 5,2% Zn/хромового катализатора. Этот катализатор перед использованием предварительно обрабатывают следующим образом:
- Сушат при 250°C в течение ночи в токе азота со скоростью 80 мл/мин при 3 бар.
- Нагревают до 300°C и обрабатывают HF при скорости 4 мл/мин и азотом при скорости 80 мл/мин при 3 бар в течение 16 час.
- Уменьшают поток азота до нуля, а поток HF поддерживают постоянным, продолжая нагревание при 300°C еще в течение 4 час.
- Поток HF поддерживают постоянным, а температуру повышают до 380°C при 25°C/час.
- Поток HF и нагревание при 380°C поддерживают постоянным еще в течение 3 часов
В конце предварительного фторирования температуру реактора понижают до 310°C, давление снижают до 5 бар и смесь HF (приблизительно 1-60 мл/мин) и 245eb (приблизительно 30-80 мл/мин) загружают в реактор. Образцы реакторного отработанного газа периодически отбирают для анализа ГХ и ГХ-МС. Эти приборы калибруют, используя известные стандарты. Результаты представлены в нижеприведенной таблице.
(°C)
** CF3CF2CH3
Настоящее изобретение определяется следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,3,3,3-ТЕТРАФТОРПРОПИЛЕНА | 2009 |
|
RU2484079C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,3,3-ТЕТРАФТОРПРОПИЛЕНА | 2009 |
|
RU2535214C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННЫХ ОЛЕФИНОВ (ВАРИАНТЫ) | 2007 |
|
RU2457195C2 |
| СПОСОБ | 2007 |
|
RU2466121C2 |
| СПОСОБЫ КАТАЛИТИЧЕСКОГО ПОЛУЧЕНИЯ ГЕКСАФТОРПРОПЕНОВ | 2007 |
|
RU2451659C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,3,3-ТЕТРАФТОРПРОПЕНА | 2009 |
|
RU2523546C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,3,3-ТЕТРАФТОРПРОПЕНА | 2009 |
|
RU2463285C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ТЕТРАФТОРПРОПЕНА | 2007 |
|
RU2445302C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1,1-ТРИФТОР-2,3-ДИХЛОРПРОПАНА | 2009 |
|
RU2476413C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,3,3-ТЕТРАФТОРПРОПЕНА | 2009 |
|
RU2466122C2 |
Изобретение относится к способу получения 2,3,3,3-тетрафторпропилена (1234yf). Способ включает: (a) взаимодействие 1,1,2,3,3,3-гексафторпропилена (1216) с водородом в присутствии катализатора гидрирования с образованием 1,1,2,3,3,3-гексафторпропана (236еа); (b) дегидрофторирование 236еа с образованием 1,2,3,3,3-пентафторпропилена (1225yе); (c) взаимодействие 1225yе с водородом в присутствии катализатора гидрирования с образованием 1,2,3,3,3-пентафторпропана (245eb); и (d) дегидрофторирование 245eb с образованием 1234yf, где стадии (b) и (d) проводят в присутствии катализатора, содержащего активированный уголь, металл основной группы и/или переходный металл. Использование настоящего способа позволяет избежать использования дорогих реагентов и образования токсичных побочных продуктов. 16 з.п. ф-лы, 3 пр., 4 табл., 3 ил.
1. Способ получения 2,3,3,3-тетрафторпропилена (1234yf), включающий:
(a) взаимодействие 1,1,2,3,3,3-гексафторпропилена (1216) с водородом в присутствии катализатора гидрирования с образованием 1,1,2,3,3,3-гексафторпропана (236еа);
(b) дегидрофторирование 236еа с образованием 1,2,3,3,3-пентафторпропилена (1225yе);
(c) взаимодействие 1225yе с водородом в присутствии катализатора гидрирования с образованием 1,2,3,3,3-пентафторпропана (245eb); и
(d) дегидрофторирование 245eb с образованием 1234yf, где
стадии (b) и (d) проводят в присутствии катализатора, содержащего активированный уголь, металл основной группы и/или переходный металл.
2. Способ по п.1, в котором стадии (а) и (с) проводят в одном и том же реакторе.
3. Способ по п.2, в котором стадии (а) и (с) проводят одновременно.
4. Способ по п.1, в котором стадии (b) и (d) проводят в одном и том же реакторе.
5. Способ по п.4, в котором стадии (b) и (d) проводят одновременно.
6. Способ по п.1, в котором стадии (а) и (с) проводят при температуре в диапазоне приблизительно от -25 приблизительно до 275°C и давлении в диапазоне приблизительно от 0 приблизительно до 40 бар.
7. Способ по п.6, в котором стадии (а) и (с) проводят в паровой фазе при температуре в диапазоне приблизительно от 0 приблизительно до 250°C, предпочтительно в диапазоне приблизительно от 20 приблизительно до 200°C, более предпочтительно в диапазоне приблизительно от 50 приблизительно до 150°C.
8. Способ по п.1, в котором соотношение водород:1216 на стадии (а) и водород: 1225yе на стадии (с) находится в диапазоне приблизительно от 1:1 приблизительно до 40:1.
9. Способ по п.1, в котором катализатор гидрирования на стадии (а) и в котором катализатор гидрирования на стадии (с) содержит нанесенный или ненанесенный переходный металл, выбранный из группы, состоящей из Ni, Pd, Pt, Re, Rh, Ru и их смесей.
10. Способ по п.9, в котором катализатор гидрирования или каждый из катализаторов гидрирования нанесен на алюминий, титан, кремний, цирконий (или фториды вышеописанных элементов), фторид кальция, уголь и/или сульфат бария.
11. Способ по п.9, в котором катализатор гидрирования или каждый из катализаторов гидрирования представляет собой палладий, нанесенный на уголь (Pd/C).
12. Способ по п.1, в котором стадии (b) и (d) проводят при температуре в диапазоне от -70 до 1000°C и под давлением в диапазоне приблизительно от 0 приблизительно до 30 бар.
13. Способ по п.1, в котором стадии (b) и (d) проводят в присутствии катализатора, содержащего переходный металл.
14. Способ по п.13, в котором катализатор, содержащий переходный металл, содержит хром.
15. Способ по п.1, в котором стадии (b) и (d) проводят при температуре в диапазоне приблизительно от 0 приблизительно до 400°C и давлении от 0,01 приблизительно до 25 бар, предпочтительно в диапазоне приблизительно от 200 приблизительно до 360°C и приблизительно от 1 приблизительно до 10 бар.
16. Способ по п.1, в котором стадии (b) и (d) проводят в присутствии фтористого водорода (HF) из отдельного исходного продукта.
17. Способ по п.16, в котором молярное соотношение HF:органические вещества (например, 236еа на стадии (b) и 245eb на стадии (d)) находится в диапазоне приблизительно от 0,01:1 приблизительно до 50:1, предпочтительно в диапазоне приблизительно от 0,5:1 приблизительно до 30:1.
| KNUNYANTS I L; KRASUSKAYA M P; MYSOV E I: “REACTIONS OF FLUORO OLEFINS | |||
| Насос | 1917 |
|
SU13A1 |
| CATALYTIC HYDROGENATION OF PERFLUORO OLEFINS”, BULLETIN OF THE ACADEMY OF SCIENCES OF THE USSR, DIVISION OF CHEMICAL SCIENCES, 01.01.1960, PAGES 1312-1317 | |||
| WO 2007117391 A1, 18.10.2007 | |||
| RU 94046237 A1, 27.10.1996 | |||
| RU 2006107535 A, 27.07.2006 | |||

