Предпосылки изобретения
По данной заявке испрашивается приоритет согласно предварительной заявке США № 61/106380, поданной 17 октября 2008, и предварительной заявке США № 61/177109, поданной 11 мая, 2009, полное содержание которых включено в настоящее описание посредством ссылки.
1. Область техники, к которой относится изобретение
Изобретение относится к регуляции роста клеток и, более конкретно, к регуляции роста раковых клеток. В частности, было показано, что MUC1-пептиды, полученные на основе последовательности из определенной области цитоплазматического домена белка MUC1, ингибируют олигомеризацию и ядерную транслокацию MUC1, вызывая остановку роста и даже гибель опухолевых клеток, экспрессирующих MUC1.
2. Уровень техники
Муцины представляют собой интенсивно О-гликозилированные белки, которые экспрессируются преимущественно эпителиальными клетками. Секретируемые и мембраносвязанные муцины образуют физический барьер, который защищает апикальные границы эпителиальных клеток от повреждений, вызываемых токсинами, микроорганизмами и другими видами стрессовых воздействий, возникающих на границе с внешней окружающей средой. Трансмембранный муцин 1 (MUC1) также может осуществлять передачу сигнала внутрь клетки через свой цитоплазматический домен. MUC1 не имеет гомологии по последовательности с другими мембраносвязанными муцинами, за исключением домена белка спермы морского ежа/энтерокиназы/агрина (SEA-домена) (Duraisamy et al., 2006). Вследствие наличия этого домена, MUC1 транслируется в виде одиночного полипептида, а затем претерпевает ауторасщепление по SEA-домену (Macao, 2006).
N-концевая субъединица MUC1 (MUC1-N) содержит различное количество тандемных повторов с высоким содержанием остатков серина и треонина, модифицированных О-гликозилированием (Siddiqui, 1988). MUC1-N отвечает за гликокаликс клетки и соединен с ее поверхностью с помощью нековалентного связывания с трансмембранной С-концевой субъединицей MUC1 (MUC1-C) (Merlo, 1989). MUC1-C состоит из 58-аминокислотного внеклеточного домена, 28-аминокислотного трансмембранного домена и 72-аминокислотного цитоплазматического домена, который взаимодействует с различными сигнальными молекулами (Kufe, 2008). Сбрасывание MUC1-N в защитный физический барьер оставляет MUC1-C на клеточной поверхности в качестве потенциального рецептора для передачи внутриклеточных сигналов, которые обуславливают рост и выживание клеток (Ramasamy et al., 2007; Ahmad et al., 2007).
Доступные на сегодняшний момент данные указывают на то, что карциномы человека используют функцию MUC1 для усиления онкогенных свойств. В этом случае при трансформации и потере полярности высокий уровень экспрессии MUC1 наблюдается на всей поверхности клеток эпителия молочных желез и других органов (Kufe, 1984). В другой работе было показано, что сверхэкспрессия MUC1 обуславливает независимый от прикрепления рост и онкогенные свойства (Li et al., 2003a; Raina et al., 2004; Ren et al., 2004; Wei et al., 2005) по меньшей мере частично за счет стабилизации β-катенина (Huang et al., 2005). Более того, в соответствии с функцией, обеспечивающей выживание нормальных эпителиальных клеток, сверхэкспрессия MUC1 придает устойчивость клеткам карциномы к индуцируемому стрессом апоптозу (Ren et al., 2004; Yin and Kufe, 2003; Yin et al., 2004; Yin et al., 2007).
Утрата ограничения на апикальной мембране дает возможность формирования комплексов с рецептором фактора роста эпидермиса (EGFR) и коактивации опосредованных EGFR сигнальных путей (Li et al., 2001; Ramasamy et al., 2007). Сверхэкспрессия MUC1 клетками карциномы также ассоциирована с накоплением MUC1-C в цитозоле и транслокацией этой субъединицы в ядро клетки (Li et al., 2003b; Li et al., 2003c) и митохондрию (Ren et al., 2004; Ren et al., 2006). Важно отметить, что олигомеризация MUC1-C необходима для его переноса в ядро клетки и взаимодействия с различными эффекторами (Leng et al., 2007). Например, цитоплазматический домен MUC1-C (MUC1-CD) функционирует в качестве субстрата для c-Src (Li et al., 2001), c-Abl (Raina et al., 2006), протеин-киназы Cδ (Ren et al., 2002) и гликоген-синтаза-киназы 3β (Li et al., 1998), и напрямую взаимодействует с эффектором Wnt-сигнального пути, β-катенином (Yamamoto et al., 1997; Huang et al., 2005) и опухолевым супрессором, p53 (Wei et al., 2005). Таким образом, хотя олигомеризация, по-видимому, является важной, не существует непосредственных свидетельств о том, что нарушение формирования олигомеров MUC1 имело бы отрицательный эффект на опухолевые клетки, еще меньше известно о способах выполнения этой задачи.
Краткое изложение сущности изобретения
Таким образом, настоящее изобретение относится к способу ингибирования опухолевых клеток, экспрессирующих MUC1 (MUC1-положительных опухолевых клеток), у субъекта, включающему введение указанному субъекту MUC1-пептида длиной по меньшей мере 4 последовательных остатка MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQC, в которой аминоконцевой цистеин из CQC закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1. Пептид может содержать по меньшей мере 5 последовательных остатков MUC1, по меньшей мере 6 последовательных остатков MUC1, по меньшей мере 7 последовательных остатков MUC1, по меньшей мере 8 последовательных остатков MUC1 и, более конкретно, последовательность может включать CQCR (SEQ ID NO:54), CQCRR (SEQ ID NO:50), CQCRRR (SEQ ID NO:51), CQCRRRR (SEQ ID NO:52), CQCRRK (SEQ ID NO:4) или CQCRRKN (SEQ ID NO:53). Пептид может содержать не более 10 последовательных остатков, 11 последовательных остатков, 12 последовательных остатков, 13 последовательных остатков, 14 последовательных остатков, 15 последовательных остатков, 16 последовательных остатков, 17 последовательных остатков, 18 последовательных остатков или 19 последовательных остатков MUC1. Пептид может быть слит с доменом для переноса в клетки, таким как поли-D-R, поли-D-P или поли-D-K. Пептид может содержать все L-аминокислоты, все D-аминокислоты или смесь L- и D-аминокислот.
MUC1-положительной опухолевой клеткой может быть клетка карциномы, клетка лейкемии или клетка миеломы, такая как клетка карциномы простаты или молочной железы. Введение может включать внутривенное, внутриартериальное, внутриопухолевое, подкожное, поверхностное или внутрибрюшинное введение, или местное, зональное, системное или непрерывное введение. Ингибирование может включать остановку роста указанной опухолевой клетки, апоптоз указанной опухолевой клетки и/или некроз опухолевой ткани, включающей указанную опухолевую клетку. Субъектом может быть человек.
Способ может дополнительно включать проведение для указанного субъекта второй противораковой терапии. Второй противораковой терапией может быть хирургическое вмешательство, химиотерапия, радиационная терапия, гормональная терапия, терапия токсинами, иммунотерапия и криотерапия. Вторую противораковую терапию можно осуществлять перед введением указанного пептида, после введения указанного пептида или одновременно с введением указанного пептида. Способ может дополнительно включать стадию оценки экспрессии MUC1 в опухолевой клетке указанного субъекта перед введением указанного пептида, и/или способ может дополнительно включать стадию оценки эффекта указанного пептида на экспрессию MUC1 в опухоли указанного субъекта.
Пептид можно вводить в количестве 0,1-500 мг/кг/день или 10-100 мг/кг/день. Пептид можно вводить ежедневно, например, в течение 7 дней, 2 недель, 3 недель, 4 недель, одного месяца, 6 недель, 8 недель, двух месяцев, 12 недель или 3 месяцев. Пептид можно вводить еженедельно, например, в течение 2 недель, 3 недель, 4 недель, 6 недель, 8 недель, 10 недель или 12 недель.
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей: (а) MUC1-пептид длиной по меньшей мере 4 последовательных остатка MUC1 и не более 20 последовательных остатков MUC1, и содержащий последовательность CQC, в которой аминоконцевой цистеин из CQC закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1; и (b) фармацевтически приемлемый носитель, буфер или разбавитель. Длина пептида может составлять по меньшей мере 5, 6, 7 или 8 последовательных остатков MUC1. Длина пептида может составлять не более 10 последовательных остатков, 11 последовательных остатков, 12 последовательных остатков, 13 последовательных остатков, 14 последовательных остатков, 15 последовательных остатков, 16 последовательных остатков, 17 последовательных остатков, 18 последовательных остатков или 19 последовательных остатков MUC1. Пептид может быть слит с доменом для переноса в клетку, таким как поли-D-R, поли-D-P или поли-D-K, или с доменом для трансдукции клеток, таким как домен для трансдукции клеток белка HIV tat. Длина пептида может составлять по меньшей мере 8 аминокислотных остатков, и по меньшей мере два, не примыкающих друг к другу остатка, образуют мостик через свои боковые цепи. Мостик может включать линкер, химически модифицированные боковые цепи или углеводородную сшивку. Линкеры могут содержать модификации, которые стабилизируют альфа-спиральную структуру указанного пептида. Буфер может содержать β-меркаптоэтанол, глутатион или аскорбиновую кислоту, или другой восстанавливающий агент, который поддерживает пептид в мономерном состоянии.
В еще одном варианте осуществления изобретение относится к способу ингибирования олигомеризации и ядерного транспорта MUC1 в клетке, включающему контактирование клетки, экспрессирующей MUC1, с MUC1-пептидом длиной по меньшей мере 4 последовательных остатка MUC1 и не более 20 последовательных остатков MUC1, и содержащим последовательность CQC, в которой аминоконцевой цистеин из CQC закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1. Пептид может содержать по меньшей мере 5 последовательных остатков MUC1, по меньшей мере 6 последовательных остатков MUC1, по меньшей мере 7 последовательных остатков MUC1, по меньшей мере 8 последовательных остатков MUC1, и, более конкретно, последовательность может содержать CQCR, CQCRR, CQCRRR, CQCRRRR, CQCRRK или CQCRRKN. Пептид может содержать не более 10 последовательных остатков, 11 последовательных остатков, 12 последовательных остатков, 13 последовательных остатков, 14 последовательных остатков, 15 последовательных остатков, 16 последовательных остатков, 17 последовательных остатков, 18 последовательных остатков или 19 последовательных остатков MUC1. Пептид может быть слит с доменом для переноса в клеток, таким как поли-D-R, поли-D-P или поли-D-K. Пептид может содержать все L-аминокислоты, все D-аминокислоты или смесь L- и D-аминокислот.
Клеткой, экспрессирующей MUC1, может быть опухолевая клетка, такая как клетка карциномы, клетка лейкемии или клетка миеломы, такая как клетка карциномы простаты или молочной железы. Опухолевая клетка может находиться в живом субъекте. Субъектом может быть человек.
В еще одном варианте осуществления изобретение относится к пептидомиметику, который имитируют структуру и способность к связыванию MUC1 MUC1-пептида длиной по меньшей мере 4 последовательных остатка MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQC, в которой аминоконцевой цистеин из CQC закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1. Дополнительный вариант осуществления изобретения относится к MUC1-пептиду длиной по меньшей мере от 3 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащему последовательность CQC, причем все аминокислотные остатки указанного пептида являются D-аминокислотами. Пептид может дополнительно содержать последовательность KRRCQC (SEQ ID NO:49).
Раковой клеткой может быть, например, клетка рака молочной железы, рака легких, рака толстой кишки, рака поджелудочной железы, рака почек, рака желудка, рака печени, рака костей, гематологического рака, рака нейрональной ткани, меланомы, рака яичников, рака яичек, рака простаты, рака шейки матки, вагинального рака или рака мочевого пузыря.
Также изобретение охватывает способы уничтожения раковой клетки. Способы могут включать получение субъектом одного или нескольких дополнительных методов лечения перед, после или одновременно с проведением описанных выше способов. Методы лечения могут представлять собой, например, одну или несколько форм ионизирующей радиации и/или один или несколько химиотерапевтических агентов. Одним или несколькими химиотерапевтическими агентами могут быть, например, цисплатин, карбоплатин, прокарбазин, мехлоретамин, циклофосфамид, камптотецин, ифосфамид, мелфалан, хлорамбуцил, бисульфан, нитрозомочевина, дактиномицин, даунорубицин, доксорубицин, блеомицин, пликомицин, митомицин, этопозид, верампил, подофиллотоксин, тамоксифен, таксол, трансплатин, 5-фторурацил, винкристин, винбластин, метотрексат или аналог любого из указанных выше соединений. Также, в качестве комбинированных методов лечения предусмотрены гормональная терапия, иммунотерапия, терапия токсинами, криотерапия и хирургическое вмешательство.
Предусмотрено, что любой способ или композицию, описанную в настоящем описании, можно осуществлять применительно к любому другому способу или композиции, описанной в настоящем описании.
Использование единственного числа в связи с термином «содержащий» («включающий») в формуле изобретения и/или описании изобретения может означать «один», но также соответствует значению «один или несколько», «по меньшей мере один» и «один или более одного». Слово «примерно» означает плюс или минус 5% от указанного числа.
Другие цели, признаки и преимущества настоящего изобретения будут очевидны из приведенного ниже следующего подробного описания. Однако следует понимать, что подробное описание и конкретные примеры, которые указывают на определенные варианты осуществления изобретения, приведены только для иллюстративных целей, поскольку из подробного описания для специалистов в данной области будут очевидны различные изменения и модификации, входящие в сущность и объем изобретения.
Краткое описание фигур
Приведенные ниже фигуры представляют собой часть настоящего описания и включены для дополнительной демонстрации некоторых аспектов настоящего изобретения. Изобретение можно лучше понять с помощью одной или нескольких таких фигур вместе с подробным описанием.
Фиг.1A-D. MUC1/CQC-пептид блокирует олигомеризацию MUC1. (Фиг.1A) Схематично представлена субъединица MUC1-C и показана 72-аминокислотная последовательность MUC1-CD. N-концевой пептид MUC1/CQC (затемненная последовательность) и мутированный N-концевой пептид MUC1/AQA длиной 15 аминокислот были синтезированы с доменом для трансдукции поли-dArg. (Фиг.1B) Пептиды His-MUC1-CD (1,4 мг/мл) иммобилизовали на сенсорном чипе для системы BIAcore. Пептид MUC1/CQC пропускали через чип в концентрации 10 мкМ. Исходные данные по связыванию анализировали с помощью программного обеспечения BIAevaluation software version 3.0 и приводили к модели связывания Лангуира 1:1. (Фиг.1С) Очищенный пептид His-MUC1-CD (1,5 мг/мл) инкубировали с PBS, MUC1/CQC, 200 мкМ, или MUC1/AQA, 200 мкМ, в течение 1 часа при комнатной температуре. Белки разделяли электрофорезом ДСН-полиакриламидном геле в невосстанавливающих условиях и анализировали иммуноблоттингом с антителами к MUC1-C. (Фиг.1D) Клетки 293 транзиентно трансфицировали для экспрессии «пустого» вектора или GFP-MUC1-CD и Flag-MUC1-CD. Через 48 часов после трансфекции клетки обрабатывали MUC1/CQC или MUC1/AQA, 5 мкМ, в течение 3 дней. Затем клетки собирали для иммуноблоттинга с антителами к MUC1-C (левая панель). Также проводили иммуноосаждение общих клеточных лизатов анти-Flag антителами и проводили иммуноблоттинг полученных осадков с указанными антителами (правые панели).
Фиг.2А-С. Пептид MUC1/CQC блокирует ядерную локализацию MUC1-C. (Фиг.2А) Клетки инкубировали с меченным FITC пептидом MUC1/CQC (5 мкМ) в течение указанных периодов времени и затем анализировали с помощью проточной цитометрии. В каждую из панелей включен средний показатель флуоресценции (MFI). (Фиг.2В-С) Клетки ZR-75-1 (фиг.2B) и MCF-7 (фиг.2C) инкубировали в присутствии 5 мкМ пептидов MUC1/CQC или MUC1/AQA в течение 3 дней. Проводили иммуноблоттинг общих клеточных лизатов (ОКЛ) (левые панели) и ядерных лизатов (правые панели) с указанными антителами.
Фиг.3A-D. Пептид MUC1/CQC вызывает торможение в S-фазе и некроз. Клетки ZR-75-1 (фиг.2A-B) и MCF-7 (фиг.2C-D) обрабатывали пептидами MUC1/CQC или MUC1/AQA (5 мкМ) в течение 3 и 4 дней. Клетки фиксировали и анализировали распределение по фазам клеточного цикла с помощью проточной цитометрии (фиг.2А и 2С). Процент клеток в G1-, S- и G2/M-фазах включен в панели. Клетки также окрашивали пропидиййодидом и анализировали с помощью проточной цитометрии на некроз (фиг.2B и 2D). Процент некротических клеток включен в панели.
Фиг.4А-Е. Селективность пептида MUC1/COC в отношении клеток рака молочной железы, экспрессирующих MUC1. (Фиг.4A) Клетки ZR-75-1 стабильно инфицировали «пустым» лентивирусом (вектором) или лентивирусом, экспрессирующим киРНК к MUC1. Иммуноблоттинг лизатов инфицированных клеток проводили с указанными антителами. (Фиг.4В) Клетки ZR-75-1, несущие вектор, оставляли необработанными (ромбы), а клетки ZR-75-1, несущие вектор (квадраты) и клетки ZR-75-1, несущие киРНК к MUC1 (треугольники), обрабатывали пептидом MUC1/CQC в течение указанных периодов времени. Число живых клеток определяли по исключению красителя трипанового синего. (Фиг.4С) Клетки 293 оставляли необработанными (ромбы) и обрабатывали 5 мкМ пептидов MUC1/CQC (квадраты) или MUC1/AQA (треугольники) в течение указанных периодов времени. Число живых клеток определяли по исключению красителя трипанового синего. (Фиг.4D) Клетки MCF-10A оставляли необработанными (левая панель) и обрабатывали 5 мкМ пептидов MUC1/CQC (средняя панель) или MUC1/AQA (правая панель). Через 3 дня анализировали распределение по фазам клеточного цикла. (Фиг.4Е) Клетки MCF-10A оставляли необработанными (ромбы) и обрабатывали 5 мкМ пептидов MUC1/CQC (квадраты) или MUC1/AQA (треугольники) в течение указанных периодов времени. Число живых клеток определяли по исключению красителя трипанового синего.
Фиг.5А-С. Пептид MUC1/CQC блокирует рост ксенографтов опухоли молочной железы, ZR-75-1. (Фиг.5A) 4-6-недельным самкам мышей Balb-c nu/nu имплантировали капсулы с 17-β-эстрадиолом. Через 24 часа в подвздошную область вводили подкожно клетки рака молочной железы, ZR-75-1, заключенные в матригель. Когда объем опухоли достигал ~150 мм3, мышей распределяли соответствующими парами по группам и вводили с помощью внутрибрюшинных инъекций PBS (контроль носителя; закрашенные квадраты), 50 мг/кг пептида MUC1/AQA (контрольный пептид; незакрашенные квадраты) или 10 мг/кг MUC1/CQC-пептида (закрашенные треугольники) ежедневно в течение 21 дня. Другая группа получала 50 мг/кг пептида MUC1/CQC ежедневно в течение 6 дней (незакрашенные треугольники). Мышей взвешивали дважды в неделю, и измерение опухоли проводили каждые 4 дня. (Фиг.5В и 5С). На 24-й день (стрелка) опухоли, собранные у контрольной группы и группы, получавшей 50 мг/кг/день в течение 6 дней, окрашивали H&E (гематоксилином/эозином) (фиг.5B) и антителами к MUC1 (фиг.5C).
Фиг.6А-В. Пролонгированные эффекты MUC1/CQC-пептида на опухоли ZR-75-1. (Фиг.6А) Мышам вводили с помощью инъекций клетки ZR-75-1, как описано в подписях к фиг.5А. Когда объем опухоли достигал ~275 мм3, мышей распределяли соответствующими парами по группам и вводили с помощью внутрибрюшинных инъекций PBS (контроль носителя; сплошные квадраты), 50 мг/кг пептида MUC1/AQA (контрольный пептид; незакрашеные квадраты) или 30 мг/кг пептида MUC1/CQC (закрашенные квадраты) ежедневно в течение 21 дня. Контрольных мышей забивали на 32-й день, когда объем опухоли достигал ~1200 мм3. За получавшими воздействие мышами вели наблюдение до 52-го дня, когда опухоли собирали для окрашивания H&E (Фиг.6B).
Фиг.7. 7-членный пептид MUC1 ингибирует карциному простаты. Клетки DU145 карциномы простаты обрабатывали 5 мкМ короткого CQC-пептида (7-членного) или 5 мкМ длинного CQC-пептида (15-членного) в течение 4 дней. Рост клеток измеряли с помощью МТТ-анализа. Данные представлены как процент ингибирования роста относительно необработанных клеток (контроль).
Фиг.8. Последовательности соединенных углеводородной сшивкой MUC1-CD-пептидов.
Фиг.9А. Эффекты соединенных углеводородной сшивкой MUC1-CD-пептидов на рост клеток немелкоклеточной легочной карциномы H1650. Для оценки чувствительности к ингибированию функции MUC1 клетки немелкоклеточной легочной карциномы, H1650, обрабатывали 1 и 5 мкМ соединенных углеводородной сшивкой MUC1/CQC-пептидов (GO-200-1B) в течение 7 дней. Обработка клеток Н1650 5 мкМ GO-200-1B сопровождалась значительным ингибированием роста и затем снижением числа клеток.
Фиг.9В. Эффект GO-200-2B на пролиферацию клеток. Клеточную линию H-1975 немелкоклеточной легочной карциномы выращивали в среде DMEM с 10% температурно-инактивированной фетальной телячьей сыворотки, 100 ед/мл пенициллина, 100 мкг/мл стрептомицина и 2 ммоль/л L-глутамина. Клетки пересевали через один день после обработки. Клетки обрабатывали 5 мкМ GO-200-2B в течение 3 дней, и жизнеспособность клеток определяли по исключению трипанового синего.
Фиг.10. Эффект различных MUC1-CD-пептидов (содержащих CQC-область) на рост гормонозависимых клеток карциномы молочной железы. Для определения влияния воздействия различных содержащих CQC-область MUC1-CD-пептидов, клетки карциномы молочной железы ZR-75-1 обрабатывали различными пептидами в концентрации 5 мкМ в течение 4 дней и контролировали пролиферацию клеток. Несомненно наблюдалось значительное ингибирование роста клеток по сравнению с необработанными клетками.
Фиг.11. Эффект различных MUC1-CD-пептидов (содержащих CQC-область) на рост клеток немелкоклеточной легочной карциномы. Клетки немелкоклеточной легочной карциномы А549 обрабатывали 5 мкМ GO-203, GO-203-2 или GO-203cyc в течение 7 дней. Число живых клеток определяли на 7-й день по исключению трипанового синего, а процент ингибирования роста вычисляли, сравнивая клеточный рост с необработанными клетками.
Фиг.12. Эффект различных MUC1-CD-пептидов (содержащих CQC-область) на рост клеток немелкоклеточной легочной карциномы Н1975. Клетки немелкоклеточной легочной карциномы Н1975 обрабатывали 5 мкМ различных содержащих CQC-область MUC1-CD-пептидов в течение 6 дней. Число живых клеток определяли на 6-й день по исключению трипанового синего. Результаты демонстрируют, что обработка клеток Н1975 5 мкМ различных пептидов сопровождалась значительным ингибированием роста.
Фиг.13. Эффект различных MUC1-CD-пептидов (содержащих CQC-область) на рост клеток «тройной негативной» карциномы молочной железы. Клетки «тройной негативной» карциномы молочной железы MDA-MB-231 обрабатывали 5 мкМ различных содержащих CQC-область MUC1-CD-пептидов в течение 6 дней. Число живых клеток определяли на 6-й день по исключению трипанового синего. Результаты демонстрируют, что обработка клеток MDA-MB-231 различными пептидами сопровождалась значительным ингибированием роста.
Фиг.14. Эффект более коротких пептидов GO-203 на пролиферацию клеток карциномы молочной железы ZR-75-1. Клетки рака молочной железы человека ZR-75-1 выращивали в среде RPMI1640 с 10% температурно-инактивированной фетальной телячьей сыворотки, 100 ед/мл пенициллина, 100 мкг/мл стрептомицина. Клетки обрабатывали различными пептидами с концентрацией 5 мкМ ежедневно в течение 4 дней, и жизнеспособность клеток определяли по исключению трипанового синего. В отличие от GO-210 обработка клеток карциномы молочной железы ZR-75-1 5 мкМ пептидов GO-203 (SEQ ID NO:53), GO-207 (SEQ ID NO:4), GO-208 (SEQ ID NO:50) и GO-209 (SEQ ID NO:54) ежедневно в течение 4 дней сопровождалось значительным ингибированием роста.
Фиг.15A-D. Эффекты GO-203 в комбинации с различными противораковыми лекарственными средствами. Клетки ZR-75-1 обрабатывали указанными концентрациями цисплатина (фиг.15А), доксорубицина (фиг.15В), rh-TNF-α (фиг.15C) и таксола (фиг.15D) отдельно и в комбинации с GO-203. Для цисплатина, доксорубицина и таксола обработка была последовательной, когда клетки обрабатывали этими агентами в течение 72 часов, с последующим воздействием 5 мкМ GO-203 в течение 72 часов. В случае исследований с rh-TNF на клетки воздействовали различными концентрациями rh-TNF отдельно и в комбинации с 5 мкМ GO-203 в течение 72 часов. Для определения выживаемости клеток использовали MTS-анализ.
Фиг.16. GO-203 обладает аддитивным или синергическим действием относительно противораковых агентов. На графиках показаны комбинационные индексы относительно различных уровней воздействия (фракции пораженных клеток). Уровень воздействия получали, обрабатывая клетки указанными комбинациями противораковых лекарственных средств и GO-203.
Описание иллюстративных вариантов осуществления изобретения
I. Настоящее изобретение
Роль белка MUC1 в раковых заболеваниях интенсивно исследуется авторами изобретения и другими учеными. Как обсуждалось выше, белок MUC1 человека представляет собой гетеродимерный гликопротеин, транслируемый в виде единого полипептида и расщепляемый на N- и С-концевые субъединицы в эндоплазматическом ретикулуме (Ligtenberg et al., 1992; Macao et al., 2006; Levitin et al., 2005). Нарушенная сверхэкспрессия MUC1, обнаруживаемая в большинстве карцином человека (Kufe et al., 1984), обуславливает не зависящий от прикрепления рост и онкогенность (Li et al., 2003a; Huang et al., 2003; Schroeder et al., 2004; Huang et al., 2005). В других исследованиях было показано, что сверхэкспрессия MUC1 придает устойчивость к апоптозу, вызываемому окислительным стрессом и генотоксичными противораковыми агентами (Yin and Kufe, 2003; Ren et al., 2004; Raina et al., 2004; Yin et al., 2004; Raina et al., 2006; Yin et al., 2007).
Семейство связанных и секретируемых муцинов функционирует, обеспечивая защитный барьер поверхности эпителиальных клеток. При повреждении эпителиального слоя, плотные связи между соседними клетками разрушаются, и теряется полярность, поскольку клетки начинают индуцируемую герегулином программу восстановления (Vermeer et al., 2003). MUC1-N сбрасывается с поверхности клеток (Abe and Kufe, 1989), оставляя MUC1-C функционировать в качестве передатчика стрессовых сигналов от окружения в клетку. В этом случае MUC1-C образует комплексы на клеточной поверхности с членами семейства ErbB-рецепторов, и MUC1-C транслоцируется в ядро клетки в ответ на стимуляцию герегулином (Li et al., 2001; Li et al., 2003c). MUC1-C также функционирует, соединяя вместе сигнальные пути ErbB-рецептора и Wnt в результате прямого взаимодействия между цитоплазматическим доменом MUC1 (MUC1-CD) и членами катенинового семейства (Huang et al., 2005; Li et al., 2003c; Yamamoto et al., 1997; Li et al., 1998; Li et al., 2001; Li and Kufe, 2001). В других исследованиях было продемонстрировано, что MUC1-CD фосфорилируется гликоген-синтаза-киназой 3β, c-Src, белковой киназой Cδ и c-Abl (Raina et al., 2006; Li et al., 1998; Li et al., 2001; Ren et al., 2002).
Механизмы, отвечающие за ядерную локализацию MUC1-C, не ясны. Содержащие классический сигнал ядерной локализации (NLS) белки импортируются в ядро клетки, сначала связываясь с импортином α, а затем, в свою очередь, с импортином β (Weis, 2003). Транспортный комплекс импортинов α/β прикрепляется к ядерной поре, связываясь с нуклеопоринами, и переносится через нее с помощью механизма, зависимого от ГТФазы Ran. Классические NLS состоят либо из одного сигнального участка с кластером из 4-5 основных аминокислот или из двух участков с двумя кластерами основных аминокислот, разделенных линкером из 10-12 аминокислот. MUC1-CD содержит RRK-мотив, который не соответствует прототипному NLS с одним сигнальным участком (Hodel et al., 2002). Однако некоторые белки, содержащие неклассические NLS, транспортируются через ядерные поры, напрямую связываясь с импортином β (Kau et al., 2004). Импортин β связывается с несколькими нуклеопоринами (Ryan and Wente, 2000), включая Nup62, который выходит как на цитоплазматическую, так и нуклеоплазматическую стороны комплексов ядерных пор (Percipalle et al., 1997). В других исследованиях было показано, что импорт β-катенина в ядро клетки осуществляется по импортин- и нуклеопорин-независимому механизму (Suh and Gumbiner, 2003).
В 2006 году авторы изобретения опубликовали данные о том, что MUC1 импортируется в ядро клетки с помощью механизма, включающего связывание с Nup62. Авторы также продемонстрировали, что MUC1 образует олигомеры через CQC-мотив в цитоплазматическом домене MUC1, и что олигомеризация MUC1 необходима для ядерного импорта. В этом исследовании авторы продолжили данную работу для дальнейшего понимания роли, которую CQC-мотив играет в образовании олигомеров. Авторы изобретения продемонстрировали, что короткие пептиды, соответствующие этой области, способны нарушать образование олигомеров MUC1, ингибируя его транспорт в ядро опухолевых клеток. Эти пептиды способны ингибировать рост опухолевых клеток, а также вызывать апоптоз в таких клетках и даже некроз в опухолевой ткани. Эти и другие аспекты изобретения подробно описаны ниже.
II. MUC1
A. Структура
MUC1 представляет собой гликопротеин муцинового типа, который экспрессируется на апикальных границах нормальных секреторных эпителиальных клеток (Kufe et al., 1984). MUC1 образует гетеродимер после синтеза в виде единого полипептида и расщепления предшественника на две субъединицы в эндоплазматическом ретикулуме (Ligtenberg et al., 1992). Расщепление может опосредоваться автокаталитическим процессом (Levitan et al., 2005). N-концевая субъединица MUC1 с молекулярной массой, превышающей 250 кДа (MUC1 N-ter, MUC1-N), содержит различное число 20-аминокислотных несовершенных тандемных повторов с высоко консервативными вариациями и модифицирована О-связанными гликанами (Gendler et al., 1988; Siddiqui et al., 1988). MUC1-N прикреплена к клеточной поверхности в результате димеризации с С-концевой субъединицей массой примерно 23 кДа (MUC1 C-ter, MUC1-C), которая содержит 58-аминокислотную внеклеточную область, 28-аминокислотный трансмембранный домен и 72-аминокислотный цитоплазматический домен (CD; SEQ ID NO:1) (Merlo et al., 1989). Последовательность MUC1 человека показана ниже:

Полужирным шрифтом выделена последовательность цитоплазматического домена, и подчеркнутая часть последовательности соответствует пептиду, ингибирующему олигомеризацию (SEQ ID NO:3), описанному в примерах.
При трансформации нормального эпителия в карциному MUC1 аномально экспрессируется в цитозоле и по всей клеточной мембране (Kufe et al., 1984; Perey et al., 1992). Ассоциированный с клеточной мембраной MUC1 направляется в эндосомы путем клатрин-опосредованного эндоцитоза (Kinlough et al., 2004). Кроме того, MUC1-С, но не MUC1-N, транспортируется в ядро клетки (Baldus et al., 2004; Huang et al., 2003; Li et al., 2003a; Li et al., 2003b; Li et al., 2003c; Wei et al., 2005; Wen et al., 2003) и митохондрии (Ren et al., 2004).
В. Функция
MUC1 взаимодействует с членами семейства ErbB-рецепторов (Li et al., 2001b; Li et al., 2003c; Schroeder et al., 2001) и с эффектором Wnt-сигнального пути, β-катенином (Yamamoto et al., 1997). Рецептор фактора роста эпидермиса и c-Src фосфорилируют цитоплазматический домен MUC1 (MUC1-CD) по Y-46 и, тем самым, усиливают связывание MUC1 и β-катенина (Li et al., 2001a; Li et al., 2001b). Связывание MUC1 и β-катенина также регулируется гликоген-синтаза-киназой 3β и белковой киназой Cδ (Li et al., 1998; Ren et al., 2002). MUC1 колокализуется с β-катенином в ядре (Baldus et al., 2004; Li et al., 2003a; Li et al., 2003c; Wen et al., 2003) и совместно с ним активирует транскрипцию генов-мишеней Wnt-сигнального пути (Huang et al., 2003). В других исследованиях было показано, что MUC1 также напрямую связывается с р53 и регулирует транскрипцию генов-мишеней р53 (Wei et al., 2005). Следует отметить, что сверхэкспрессия MUC1 является достаточной для индукции независимого от прикрепления роста и онкогенности (Huang et al., 2003; Li et al., 2003b; Ren et al., 2002; Schroeder et al., 2004).
Большинство митохондриальных белков кодируются ядерной ДНК и импортируются в митохондрии с помощью комплексов-переносчиков во внешней и внутренней митохондриальных мембранах. Некоторые митохондриальные белки содержат N-концевые митохондриальные сигнальные последовательности и взаимодействуют с Tom20 во внешней митохондриальной мембране (Truscott et al., 2003). Другие митохондриальные белки содержат внутренние сигнальные последовательности и взаимодействуют с Tom70-рецептором (Truscott et al., 2003). В недавних исследованиях было показано, что митохондриальные белки без внутренних сигнальных последовательностей доставляются к Tom70 с помощью комплекса, состоящего из HSP70 и HSP90 (Young et al., 2003).
III. MUC1-пептиды
A. Структура
Настоящее изобретение предусматривает разработку, продуцирование и использование различных MUC1-пептидов. Структурные признаки таких пептидов следующие. Во-первых, пептиды имеют не более 20 последовательных аминокислотных остатков из MUC1. Поэтому термин «пептид, имеющий не более 20 последовательных остатков», даже включая термин «содержащий» не может истолковываться, как включающий большее число последовательных аминокислотных остатков MUC1. Во-вторых, пептиды должны содержать CQC-мотив, и могут также включать CQCR-мотив, CQCRR-мотив и CQCRRK-мотив. Поэтому, пептиды будут иметь по меньшей мере эти три последовательных остатка из MUC1-C-домена. В-третьих, пептиды будут иметь по меньшей мере один аминокислотный остаток, присоединенный к NH2-концу первого остатка цистеина в CQC-мотиве, так что первый остаток цистеина будет «закрыт» по меньшей мере одной аминокислотой, присоединенной к нему. Этот остаток может принадлежать MUC1 (например, входить в трансмембранный домен), может представлять собой случайно выбранную аминокислоту (любую из 20 природных аминокислот или их аналогов) или может являться частью другой пептидной последовательности (например, последовательности, используемой для очистки, стабилизирующей последовательности или домена, обеспечивающего доставку в клетку).
Как правило, длина пептида будет составлять 50 аминокислотных остатков или меньше, в свою очередь, содержащих не более 20 последовательных остатков из MUC1. Общая длина может составлять 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 или 50 остатков. Предусмотрены диапазоны размеров пептидов из 4-50 остатков, 5-50 остатков, 6-50 остатков, 7-50 остатков, 7-25 остатков, 4-20 остатков, 5-20 остатков, 6-20 остатков, 7-20 остатков и 7-15 остатков. Число последовательных аминокислотных остатков из MUC1 может составлять 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20. Предусмотрены диапазоны последовательных аминокислотных остатков из 4-20 остатков, 5-20 остатков, 6-20 остатков, 7-20 остатков и 4-15 остатков, 5-15 остатков, 6-15 остатков или 7-15 остатков.
В настоящем изобретении можно использовать L-конфигурацию аминокислот, D-конфигурацию аминокислот или их смесь. Хотя L-аминокислоты представляют подавляющее большинство аминокислот в белках, D-аминокислоты найдены в некоторых белках, продуцируемых редко встречающимися морскими организмами, такими как улитки-конусы. Они также являются распространенными компонентами пептидогликанов клеточной стенки бактерий. D-серин может действовать в качестве нейротрансмиттера в мозге. L и D-обозначения для конфигурации аминокислот относятся не к оптической активности самих аминокислот, а к оптической активности изомера глицеральдегида, из которого эту аминокислоту теоретически можно синтезировать (D-глицеральдегид является правовращающими; L-глицеральдегид является левовращающим).
Одной формой пептида, состоящего только из D-аминокислот, является ретроинвертированный пептид. Ретроинвертированная модификация природных полипептидов включает синтетическую сборку аминокислот со стереохимией α-углерода, противоположной стереохимии соответствующих L-аминокислот, т.е. D-аминокислот в обратном порядке относительно природной пептидной последовательности. Ретроинвертированный аналог, поэтому, будет иметь инвертированные концы и инвертированное (противоположное) направление пептидных связей (NH-CO, а не CO-NH), при этом приблизительно сохраняя топологию боковых цепей, как в природной пептидной последовательности. См. патент США 6261569, включенный в настоящее описание посредством ссылки.
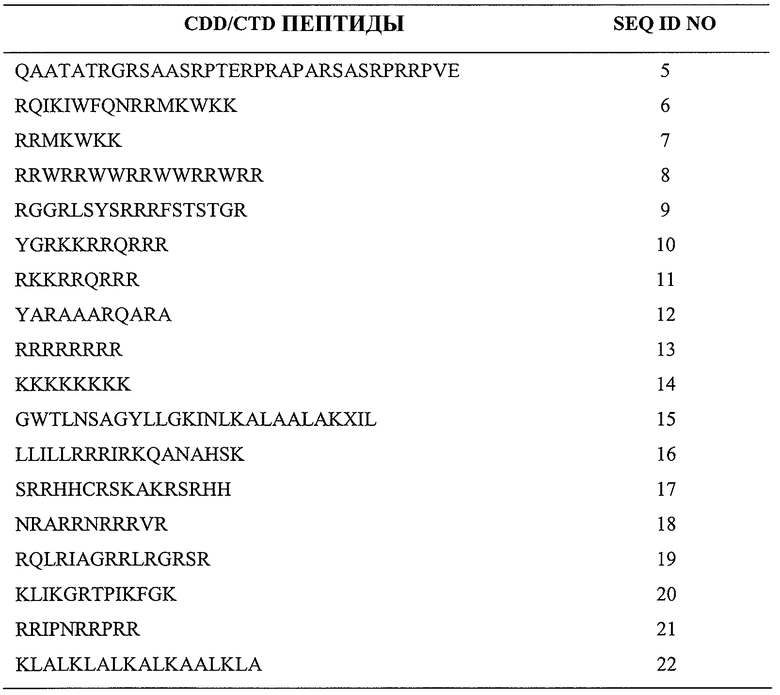
Как указано выше, настоящее изобретение предполагает слияние или конъюгацию домена для доставки в клетку (также называемого вектором для клеточной доставки, или доменом для трансдукции клеток, CDD/CTD). Такие домены хорошо известны в данной области и обычно представляют собой короткие амфипатические или катионные пептиды и пептидные производные, часто содержащие ряд остатков лизина и аргинина (Fischer, 2007). Особый интерес представляют собой последовательности поли-D-Arg и поли-D-Lys (например, правовращающие остатки, восемь аминокислотных остатков в длину), в то время как другие показаны в таблице 1, приведенной ниже.
Таблица 1

Также, как указанно выше, предусмотрены пептиды, модифицированные для использования in vivo путем добавления на амино- и/или карбокси-концы блокирующего агента для увеличения времени жизни пептида in vivo. Это может быть полезным в ситуациях, когда деградация пептидных концов под действием протеаз происходит до введения пептида в клетку. Такие блокирующие агенты могут включать, но без ограничений, дополнительные связанные или несвязанные пептидные последовательности, которые могут быть присоединены к амино- и/или карбокси-концевым остаткам вводимого пептида. Эти агенты могут быть добавлены либо химическим способом в ходе синтеза пептида, либо с помощью технологии рекомбинантных ДНК способами, известными в данной области. Альтернативно, блокирующие агенты, такие как пироглутаминовая кислота или другие молекулы, известные в данной области, могут быть присоединены к амино- и/или карбокси-концевым остаткам.
B. Синтез
Предпочтительно получать пептиды, используя методы твердофазного синтеза (Merrifield, 1963). Специалистам в данной области хорошо известны другие методы пептидного синтеза (Bodanszky et al., 1976; Peptide Synthesis, 1985; Solid Phase Peptide Synthelia, 1984). Соответствующие защитные группы, используемые в таких методах, можно найти в приведенной выше литературе, а также в книге Protective Groups in Organic Chemistry, 1973. Такие способы синтеза включают последовательное добавление одного или нескольких аминокислотных остатков или соответствующим образом защищенных аминокислотных остатков к растущей пептидной цепи. Обычно, либо амино-, либо карбоксильную группу первого аминокислотного остатка защищают соответствующей, селективно удаляемой защитной группой. Другую селективно удаляемую защитную группу используют для аминокислот, содержащих реакционно-способные боковые группы, такие как лизин.
При использовании твердофазного синтеза в качестве примера защищенные аминокислоты или их производные присоединяют к инертной твердой подложке через их незащищенную карбоксильную или аминогруппу. Затем селективно удаляют защитную группу амино или карбоксильной группы, и добавляют следующую аминокислоту в последовательности, имеющую соответственно защищенную комплементарную группу (амино или карбоксильную), которая вступает во взаимодействие с остатком, уже прикрепленным к твердой подложке. Затем удаляют защитную группу амино или карбоксильной группы из вновь добавленного аминокислотного остатка, и затем добавляют следующую аминокислоту (соответствующим образом защищенную), и т.д. После того, как все планируемые аминокислоты были соединены друг с другом в правильном порядке, любые оставшиеся защитные группы концевых и боковых групп (и твердую подложку) удаляют последовательно или одновременно, получая конечный пептид. Предпочтительно, чтобы пептиды по изобретению были свободны от бензилированных или метилбензилированных аминокислот. Такие защитные группы можно использовать в ходе синтеза, но их удаляют до использования пептида. Может быть необходимо проведение дополнительных реакций, описанных в литературе, для образования внутримолекулярных связей, удерживающих конформацию пептида.
Помимо 20 стандартных аминокислот, которые можно использовать, существует широкий ряд «нестандартных» аминокислот. Две из них кодируются генетическим кодом, но в белках встречаются довольно редко. Селеноцистеин включается в некоторые белки по UGA-кодону, который в норме представляет собой стоп-кодон. Пирролизин используется некоторыми метанобразующими археями в ферментах, которые они используют для продукции метана. Он кодируется кодоном UAG. Примеры нестандартных аминокислот, которые не встречаются в белках, включают лантионин, 2-аминоизомасляную кислоту, дегидроаланин и нейротрансмиттер гамма-аминомасляную кислоту. Нестандартные аминокислоты часто встречаются в качестве промежуточных соединений в метаболитических путях для стандартных аминокислот, например, орнитин или цитруллин возникают в цикле мочевины, участка катаболизма аминокислот. Нестандартные аминокислоты обычно образуются в результате модификаций стандартных аминокислот. Например, гомоцистеин образуется в результате транссульфирования или деметилирования метионина через промежуточный метаболит S-аденозилметионин, тогда как гидроксипролин возникает в результате посттрансляционной модификации пролина.
C. Линкеры
Линкеры или агенты для перекрестной сшивки можно использовать для слияния MUC1-пептидов с другими белковыми последовательностями. Бифункциональные реагенты для перекрестной сшивки интенсивно используются для различных целей, включая получение аффинных носителей, модификации и стабилизации различных структур, идентификации сайтов связывания лигандов и рецепторов и структурных исследований. Гомобифункциональные реагенты, которые несут две идентичные функциональные группы, оказались высокоэффективными для образования перекрестных сшивок между одинаковыми и различными макромолекулами или субъединицами макромолекул, и для сшивания полипептидных лигандов с их специфичными сайтами связывания. Гетеробифункциональные реагенты содержат две различные функциональные группы. Используя преимущество разной реакционной способности двух различных функциональных групп, можно контролировать как селективность, так и последовательность перекрестной сшивки. Бифункциональные реагенты для перекрестной сшивки можно разделить по специфичности их функциональных групп, например, амино-, сульфгидрил-, гуанидино-, индол- или карбоксил-специфичные группы. Из них особенно широко используемыми являются реагенты, направленные на свободные аминогруппы, благодаря их коммерческой доступности, простоте синтеза и мягких реакционных условиях, при которых их можно использовать. Большинство гетеробифункциональных реагентов для перекрестной сшивки содержат группу, вступающую во взаимодействие с первичными аминами, и группу, вступающую во взаимодействие с тиолами.
В другом примере гетеробифункциональные реагенты для перекрестной сшивки и способы применения реагентов для перекрестной сшивки описаны в патенте США 5889155, полное содержание которого включено в настоящее описание посредством ссылки. Реагенты для перекрестной сшивки объединяют нуклеофильный гидразидный остаток с электрофильными малеимидным остатком, давая возможность сочетанию в одном примере альдегидов со свободными тиолами. Реагент для перекрестной сшивки может быть модифицирован для перекрестной сшивки различных функциональных групп и, таким образом, является пригодным для перекрестной сшивки полипептидов. В случае, когда конкретный пептид не содержит остаток, подходящий для данного реагента для перекрестной сшивки в своей исходной последовательности, можно использовать консервативные аминокислотные замены в первичной последовательности, введенные генетическими или синтетическими методами.
Другим использованием линкеров в контексте пептидов в качестве терапевтических средств является так называемая технология «пептидов, соединенных углеводородной сшивкой» от компании Aileron Therapeutics. Основополагающим подходом для соединения пептида сшивкой является подход, в котором два ключевых остатка в пептиде модифицируют, присоединяя линкеры через боковые цепи аминокислот. После синтеза линкеры соединяют через катализатор, создавая таким образом мостик, физически удерживающий пептид в его природной α-спиральной форме. В дополнение к поддержке природной структуры, необходимой для взаимодействия с молекулой-мишенью, эта конформация также обеспечивает стабильность в отношении пептидаз, а также в отношении проникновения в клетку. Патенты США 7192713 и 7183059, в которых описана указанная технология, включены в настоящее описание посредством ссылки. См. также статью Schafmeister et al., Journal of the American Chemical Society, 2000, 122(24): p. 5891-5892.
D. Структура, варианты и аналоги
Настоящее изобретение направлено на пептиды, содержащие последовательность CQC. Определив эту ключевую структуру в формировании олигомеров MUC1, авторы изобретения также предусмотрели возможность использования вариантов последовательности CQC. Например, некоторые неприродные аминокислоты, которые удовлетворяют структурным ограничениям последовательности CQC, можно использовать в качестве заместителей без потери и, возможно, с усилением биологической функции. Кроме того, авторы настоящего изобретения также предусмотрели использование структурных аналогов для имитации ключевых участков пептидов или полипептидов настоящего изобретения. Такие соединения, называемые пептидомиметиками, можно использовать аналогично пептидам настоящего изобретения и, следовательно, они также являются их функциональными эквивалентами.
Некоторые миметики, которые имитируют элементы вторичной и третичной структуры белков, описаны у Johnson et al. (1993). Основной аргументацией в пользу применения пептидных миметиков является то, что пептидный каркас белков существует главным образом для поддержания ориентации аминокислотных боковых цепей, способствующей молекулярным взаимодействиям, таким как, например, взаимодействие антитела и/или антигена. Таким образом, пептидные миметики разрабатываются для обеспечения молекулярного взаимодействия, аналогично взаимодействию природных молекул.
Способы создания конкретных структур описаны в данной области. Например, миметики α-спирали раскрыты в патентах США 5446128; 5710245; 5840833 и 5859184. Способы создания конформационного ограниченных β-поворотов и β-выпуклостей описаны, например, в патентах США 5440013; 5618914 и 5670155. Другие типы миметиков поворотов включают реверсивные и γ-повороты. Миметики реверсивных поворотов раскрыты в патентах США 5475085 и 5929237, и миметики γ-поворотов описаны в патентах США 5672681 и 5674976.
Используемый в настоящем описании термин «молекулярное моделирование» означает количественный и/или качественный анализ структуры и функции белок-белкового физического взаимодействия на основе информации о трехмерной структуре и моделях белок-белковых взаимодействий. Этот термин включает обычные числовые модели молекулярной динамики и минимизации энергии, интерактивные компьютерные графические модели, модифицированные модели молекулярной механики, метрическую геометрию и другие ограничивающие модели на основе структуры. Молекулярное моделирование обычно проводят с использованием компьютеров, и его можно дополнительно оптимизировать, используя известные способы. Для разработки таких соединений особенно подходят компьютерные программы, в которых используются данные рентгеноструктурного анализа. Для создания трехмерных моделей можно использовать, например, программы, такие как RasMol. Компьютерные программы, такие как INSIGHT (Accelrys, Burlington, MA), GRASP (Anthony Nicholls, Columbia University), Dock (Molecular Design Institute, University of California at San Francisco) и Auto-Dock (Accelrys), обеспечивают возможность дополнительных манипуляций и способны вводить новые структуры. Способы могут включать дополнительную стадию выведения на внешнее устройство модели 3-мерной структуры соединения. Кроме того, можно провести сравнение 3-мерных данных потенциальных соединений с компьютерной базой данных, например, 3-мерных структур.
Соединения по изобретению также можно разрабатывать интерактивно на основе структурной информации о соединениях, описанных в настоящем описании, используя другие методики разработки/моделирования соединений на основе структуры (см., например, Jackson, 1997; Jones et al., 1996). Потенциальные соединения затем можно тестировать в стандартных методах анализа, известных специалистам в данной области. Примеры методов анализа описаны в настоящем описании.
3-мерную структуру биологических макромолекул (например, белков, нуклеиновых кислот, углеводов и липидов) можно установить, исходя из данных, полученных с помощью ряда методик. Эти методики, которые наиболее часто используются для оценки 3-мерной структуры белков, включают: (а) рентгеноструктурную кристаллографию; (b) спектроскопию ядерного магнитного резонанса (ЯМР); (c) анализ ограничений физических расстояний, образуемых между определенными участками на макромолекуле, например, внутримолекулярных химических перекрестных сшивок между аминокислотными остатками на белке (например, PCT/US00/14667, описание которого полностью включено в настоящее описание посредством ссылки); и (d) методы молекулярного моделирования, основанные на знании первичной структуры целевого белка, например, методы моделирования гомологов, «протягивающие» алгоритмы или ab initio структурное моделирование, с использованием компьютерных программ, таких как MONSSTER (моделирование новых структур на основе вторичных и третичных ограничений) (см., например, международную заявку № PCT/US99/11913, описание которой полностью включено в настоящее описание посредством ссылки). Другие методики молекулярного моделирования также можно использовать согласно настоящему изобретению (например, Cohen et al., 1990; Navia et al., 1992, описание которых полностью включено в настоящее описание посредством ссылки). Все эти способы дают данные, которые поддаются компьютерному анализу. Другие спектроскопические методы, которые также могут подойти для способа настоящего изобретения, но которые не обеспечивают структурной информации об биомолекулах на атомном уровне, включают круговой дихроизм и флуоресценцию и спектроскопию в ультрафиолете/видимом свете. Предпочтительным способом анализа является рентгеноструктурная кристаллография. Описание этой методики и ЯМР-спектроскопии приведены ниже.
Рентгеноструктурная кристаллография
Рентгеноструктурная кристаллография основана на дифракции рентгеновского излучения с длиной волны характеристического спектра электронными облаками, окружающими ядра атомов в кристалле представляющей интерес молекулы или молекулярного комплекса. В методике используют кристаллы очищенных биологических макромолекул или молекулярных комплексов (но они часто включают растворители, кофакторы, субстраты или другие лиганды) для определения с разрешением, близким к атомному, атомов, составляющих конкретную биологическую макромолекулу. Необходимым условием для разрешения 3-мерной структуры с помощью рентгеноструктурной кристаллографии является высоко упорядоченный кристалл, который будет сильно рассеивать рентгеновские лучи. В способе пучок рентгеновских лучей направляют на правильную повторяющуюся решетку из множества идентичных молекул, так что рентгеновские лучи рассеиваются на решетке, давая картину, по которой можно установить структуру индивидуальной молекулы. Высоко упорядоченные кристаллы, например, молекул глобулярных белков, представляют собой крупные, сферические или эллипсоидальные объекты с неоднородной поверхностью. Кристаллы содержат крупные каналы между индивидуальными молекулами. Такие каналы, которые в норме занимают более половины объема кристалла, заполнены неупорядоченными молекулами растворителя, и молекулы белка находятся в контакте друг с другом только в нескольких небольших областях. Это является одной причиной, почему структуры белков в кристаллах обычно идентичны структурам белков в растворе.
Способы получения кристаллов представляющих интерес белков описаны ниже. Формирование кристаллов зависит от ряда различных параметров, включая рН, температуру, концентрацию биологической макромолекулы, природу растворителя и осадителя, а также от присутствия добавленных ионов или лигандов белка. Для подбора всех параметров, которые будут составлять комбинацию, дающую кристалл, подходящий для рентгеноструктурного анализа может быть необходимо проведение множества рутинных экспериментов по кристаллизации. С помощью автоматизированных устройств для кристаллизации можно автоматизировать и ускорить работу по воспроизводимой постановке большого числа экспериментов по кристаллизации (см., например, патент США 5790421, описание которого полностью включено в настоящее описание посредством ссылки).
Кристаллизация полипептидов происходит в растворах, в которых концентрация полипептида превышает его максимальную растворимость (т.е. раствор полипептида является перенасыщенным). Такие растворы можно поддерживать в равновесии, снижая концентрацию полипептида, предпочтительно, в результате осаждения кристаллов полипептида. Часто можно вызывать образование кристаллов полипептидов из перенасыщенных растворов, добавляя агенты, которые меняют заряд поверхности полипептида, или нарушают взаимодействие между полипептидом и окружающей водой, усиливая взаимодействия, приводящие к кристаллизации.
Кристаллизацию обычно проводят при температуре от 4°C до 20°C. Часто используют вещества, известные как «осадители», для снижения растворимости полипептида в концентрированном растворе в результате образования энергетически невыгодного осаждающего обедненного слоя вокруг молекул полипептида (Weber, 1991). Кроме осадителей, в растворы для кристаллизации полипептидов иногда добавляют другие материалы. Они включают буферы для доведения рН раствора и соли для снижения растворимости полипептида. В данной области известны различные осадители, включающие следующие соединения: этанол, 3-этил-2-4-пентандиол и многие полигликоли, такие как полиэтиленгликоль (ПЭГ). Растворы для осаждения могут включать, например, 13-24% ПЭГ 4000, 5-41% сульфата аммония и 1,0-1,5М хлорид натрия, с рН в диапазоне 5,0-7,5. Другие добавки могут включать 0,1М Hepes, 2-4% бутанола, 20-100 мМ ацетата натрия, 50-70 мМ лимонной кислоты, 120-130 мМ фосфата натрия, 1 мМ этилендиаминтетрауксусной кислоты (EDTA) и 1 мМ дитиотреитола (DTT). Эти агенты приготавливают в буферах и добавляют по каплям в различных комбинациях к кристаллизационному буферу. Кристаллизуемые белки могут быть модифицированы, например, фосфорилированием или с использованием имитатора фосфата (например, вольфрамата, какодилата или сульфата).
Обычно используемые способы кристаллизации полипептидов включают следующие методики: в объеме, в висящей капле, с использованием затравки и диализ. В каждом из этих способов, важно сохранение непрерывного процесса кристаллизации после образования зародышей кристаллов путем поддержания перенасыщенного раствора. При кристаллизации в объеме полипептид смешивают с осадителями для достижения перенасыщенного раствора, емкость герметично закрывают и оставляют до возникновения кристаллов. В способе с использованием диализа полипептид оставляют внутри замкнутой диализной мембраны, которую помещают в раствор, содержащий осадитель. Равновесная диффузия через мембрану увеличивает концентрации полипептида и осадителя, тем самым приводя к перенасыщению полипептида.
В предпочтительной методике «висящей капли» (McPherson, 1976) исходную смесь полипептида создают, добавляя осадитель к концентрированному раствору полипептида. Концентрации полипептида и осадителей являются такими, что в этом исходном растворе полипептид не кристаллизуется. Маленькую каплю этой смеси помещают на стеклянную пластинку, которую переворачивают и помещают над резервуаром со вторым раствором. Затем систему герметично закрывают. Обычно, второй раствор содержит более высокую концентрацию осадителя или другого дегидратирующего агента. Разница в концентрациях осадителя служит причиной того, что раствор белка имеет большее давление паров относительно второго раствора. Поскольку система с двумя растворами герметично закрыта, то устанавливается равновесие, и вода из полипептидной смеси переносится во второй раствор. Это равновесие увеличивает концентрацию полипептида и осадителя в растворе полипептида. При достижении критической концентрации полипептида и осадителя может образоваться кристалл полипептида.
В другом способе кристаллизации в концентрированный раствор полипептида вводят затравку. Обычно, приготавливают концентрированный раствор полипептида, и в этот раствор вводят зародыш кристалла полипептида. Если концентрации полипептида и любых осадителей являются правильными, то зародыш кристалла обеспечит затравку, вокруг которой образуется более крупный по размеру кристалл.
Еще одним способом кристаллизации является электрокристаллизация, в которой используются дипольные моменты белковых макромолекул, которые сами по себе укладываются в слой Гельмгольца на поверхности электрода (см., например, патент США 5597457, описание которого полностью включено в настоящее описание посредством ссылки).
Некоторые белки могут не поддаваться кристаллизации. Однако специалисту известны несколько приемов, с помощью которых можно вызвать кристаллизацию. Например, удаление гибких фрагментов полипептида на амино- или карбоксильном конце белка может способствовать получению кристаллов белка. Эти фрагменты можно удалить с помощью методов молекулярной биологии или обработкой белка протеазами, такими как трипсин, химотрипсин или субтилизин.
В дифракционных экспериментах узкий или параллельный пучок рентгеновских лучей получают из источника рентгеновского излучения и направляют на кристалл для получения отраженного пучка. Падающий первичный пучок вызывает повреждение как макромолекулы, так и молекул растворителя. Поэтому, для увеличения времени жизни кристалл охлаждают (например, до температуры в диапазоне от -220°C до -50°C). Для получения всех возможных дифракционных пятен первичный пучок должен попадать на кристалл по разным направлениям, так что в ходе эксперимента кристалл вращают относительно направления пучка. Дифракционные пятна регистрируются на пленке или с помощью электронного детектора. Засвеченную пленку оцифровывают и количественно оценивают в сканирующем устройстве, в то время как электронные детекторы передают сигналы, которые они детектируют, непосредственно на компьютер. Электронные двухкоординатные детекторы значительно снижают время, требуемое для сбора и изменения дифракционных данных. Каждый дифракционный пучок, который детектируется в виде пятна на пленке или детекторной пластинке, имеет три характеристики: амплитуду, которую измеряют по интенсивности пятна; длину волны, которая определяется источником рентгеновского излучения; и фазой, которая теряется в экспериментах рентгеноструктурного анализа. Все эти три характеристики необходимы для всех отражаемых пучков для определения положения атомов, вызывающих отраженные пучки. Один путь определения фаз называется множественным изоморфным замещением (MIR), который требует введения экзогенных рассеивателей рентгеновских лучей (например, тяжелых атомов, таких как атомы металлов) в ячейку кристалла. Более подробное описание MIR приведено в патенте США 6093573 (колонка 15), описание которого полностью включено в настоящее описание посредством ссылки.
Координаты атомов представляют собой декартовы координаты (положения по осям x, y и z), получаемые математической обработкой, включая синтез Фурье, данных, получаемых из картины, возникающей при дифракции монохроматического пучка рентгеновских лучей атомами (центрами рассеяния) представляющих интерес биологических макромолекул в кристаллической форме. Дифракционные данные используют для вычисления карт электронной плотности повторяющихся элементарных единиц кристалла (ячеек). Карты электронной плотности используют для установления положения (координат атомов) индивидуальных атомов в ячейке кристалла. Абсолютные значения координат атомов передают информацию о пространственных взаимодействиях между атомами, поскольку абсолютные значения, приписываемые атомным координатам, можно изменить вращательным и/или поступательным движением по x, y и/или z-осям, совместно или отдельно, одновременно сохраняя такое же относительное пространственное взаимодействие между атомами. Таким образом, можно считать, что биологическая макромолекула (например, белок), чей набор абсолютных значений атомных координат можно с помощью вращательных или поступательных движений подогнать так, чтобы он совпадал с набором ранее определенных значений из анализа другого образца, имеет такие же атомные координаты, как координаты, полученные для другого образца.
Дополнительное подробное описание рентгеноструктурного анализа можно получить из одновременно находящейся на рассмотрении патентной заявки США № 2005/0015232, патента США 6093573 и международных патентных заявок PCT/US99/18441, PCT/US99/11913 и PCT/US00/03745. Полное описание всех указанных патентных документов включено в настоящее описание посредством ссылки.
ЯМР-спектроскопия
В то время как для рентгеноструктурной кристаллографии требуются одиночные кристаллы целевой макромолекулы, ЯМР-исследования проводятся в растворе в условиях, приближенных к физиологическим. Однако получаемые с помощью ЯМР структуры не являются такими подробными, как структуры, полученные на основе кристаллов.
Хотя применение ЯМР-спектроскопии до относительно недавнего времени было ограничено установлением 3-мерной структуры относительно небольших молекул (например, белков, состоящих из 100-150 аминокислотных остатков), последние открытия, включая изотопное мечение представляющей интерес молекулы и спектроскопию с оптимизированной поперечной релаксацией (TROSY), позволили расширить методологию для анализа гораздо более крупных молекул, например, белков с молекулярной массой 110 кДа (Wider, 2000).
В ЯМР используется радиочастотное излучение для определения окружения атомных ядер с ненулевым магнитным моментом в гомогенном магнитом поле, пульсирующем с определенной радиочастотой. Импульсы нарушают ядерную намагниченность этих атомов с ядрами, имеющими отличный от нуля спин. При возврате системы в равновесное состояние детектируется диапазон затухающих сигналов. Фурье-преобразование затухающего сигнала в частотный диапазон дает одномерный ЯМР-спектр. Пики в этом спектре представляют собой химические сдвиги различных активных ядер. Химический сдвиг для атома определяется его локальным электронным окружением. Двухмерные ЯМР-исследования могут предоставить информацию о близости различных атомов в структуре и в трехмерном пространстве. Структуру белков можно определить, проводя ряд двух- (и иногда 3- или 4-)мерных ЯМР-исследований и используя полученную информацию в качестве ограничений в ряде методов моделирования пространственной укладки белка.
Больше информации по ЯМР-спектроскопии, включая подробное описание того, как исходные данные, полученные из ЯМР-эксперимента, можно использовать для определения 3-мерной структуры макромолекул, можно найти в: Protein NMR Spectroscopy, Principles and Practice, (1996); Gronenborn et al. (1990); и Wider (2000), supra., полное описание которых включено в настоящее описание посредством ссылки.
Также интерес представляют соединения пептидомиметиков, которые разработаны на основе аминокислотных последовательностей соединений настоящего изобретения, представляющих собой пептиды. Пептидомиметики являются синтетическими соединениями, имеющими трехмерный конформационный «мотив», который по существу соответствует трехмерной конформации выбранного пептида. Пептидный мотив представляет собой соединение пептидомиметик, которое способно ингибировать олигомеризацию MUC1. Пептидомиметики могут иметь дополнительные характеристики, увеличивающие их пригодность для применения in vivo, такие как лучшее проникновение в клетку и более длительное время полужизни. Пептидомиметики обычно имеют каркас, который частично или полностью не является пептидным, но с боковыми группами, идентичными боковым группам аминокислотных остатков, которые присутствуют в пептиде, на основе которого разработан пептидомиметик. В данной области известны несколько типов химических связей, например, сложноэфирная, тиоэфирная, тиоамидная, ретроамидная, восстановленная карбонильная, диметиленовая и кетометиленовая связи, которые обычно используются для замены пептидных связей при конструировании устойчивых к протеазам пептидомиметиков.
IV. Методы лечения
А. Фармацевтические составы и пути введения
Если предполагается клиническое применение, то будет необходимо получение фармацевтических композиций в форме, подходящей для предполагаемого применения. В общем, этот процесс будет включать приготовление композиций, по существу свободных от пирогенных соединений, а также от других примесей, вредных для человека или животных.
В общем, желательно использовать соответствующие соли и буферы для обеспечения стабильности векторов, используемых для доставки и для их введения в клетки-мишени. Для введения пациенту рекомбинантных клеток также используют буферы. Водные композиции настоящего изобретения содержат эффективное количество вектора относительно клеток, растворенного или диспергированного в фармацевтически приемлемом носителе или водной среде. Такие композиции также называются инокулятом. Фраза «фармацевтически или фармакологически» приемлемый относится к молекулярным соединениям или композициям, которые не дают отрицательных, аллергических или других нежелательных реакций при введении животному или человеку. Используемый в настоящем описании термин «фармацевтически приемлемый носитель» включает все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические агенты и агенты, замедляющие абсорбцию, и т.п. В данной области хорошо известны способы применения таких сред и агентов для фармацевтически активных веществ. За исключением случаев, когда любая обычная среда или агент являются несовместимыми с векторами или клетками согласно настоящему изобретению, предусмотрено их использование в терапевтических композициях. Также в композиции можно включать вспомогательные активные ингредиенты.
Активные композиции настоящего изобретения могут включать классические фармацевтические препараты. Введение композиций настоящего изобретения можно будет проводить любым путем, при условии доступности ткани-мишени при таком введении. Такие пути включают пероральный, назальный, буккальный, ректальный, вагинальный или местный пути ведения. Альтернативно, введение можно осуществлять с помощью ортотопической, внутрикожной, подкожной, внутримышечной, внутрибрюшинной или внутривенной инъекции. Такие композиции обычно вводят в виде фармацевтически приемлемых композиций, описанных выше. Особый интерес представляют прямое введение в опухоль, перфузия опухоли или введение в место или область опухоли, например, в местную сосудистую или лимфатическую систему, или в ложе удаленной опухоли.
Активные соединения можно также вводить парентерально или внутрибрюшинно. Растворы активных соединений в виде свободных оснований или фармакологически приемлемых солей можно приготовить в воде, соответственно смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Также можно приготовить дисперсии в глицерине, жидких полиэтиленгликолях и их смесях, и в маслах. В обычных условиях хранения и использования такие препараты содержат консервант для предупреждения роста микроорганизмов.
Фармацевтические формы, подходящие для инъекций, включают стерильные водные растворы или дисперсии, и стерильные порошки для экстемпорального приготовления стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и достаточно жидкой для легкого введения через шприц. Она должна быть стабильной при изготовлении и хранении, и должна быть защищена от загрязнения микроорганизмами, такими как бактерии и грибки. Носителем может быть растворитель или дисперсионная среда, включающие, например, воду, этанол, многоатомный спирт (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль, и т.п.), их подходящие смеси и растительные масла. Подходящую текучесть можно поддерживать, например, используя покрытие, такое как лецитин, поддерживая требуемый размер частиц в случае дисперсии и используя поверхностно-активные вещества. Действие микроорганизмов можно предупредить с помощью различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и т.п. Во многих случаях предпочтительно включать изотонические агенты, например, сахара или хлорид натрия. Пролонгированную абсорбцию вводимых с помощью инъекции композиций можно обеспечить, используя в композициях агенты, задерживающие абсорбцию, например, моностеарат алюминия и желатин.
Стерильные растворы для инъекций приготавливают, включая активные соединения в требуемом количестве в соответствующий растворитель, в случае необходимости, совместно с рядом других ингредиентов, перечисленных выше, с последующей стерилизацией фильтрованием. Обычно, дисперсии приготавливают, включая различные стерильные активные ингредиенты в стерильный носитель, который содержит базовую дисперсионную среду и необходимые другие ингредиенты из перечисленных выше. В случае стерильных порошков для приготовления стерильных растворов для инъекций предпочтительными способами приготовления являются вакуумная сушка и лиофилизация, дающие порошок действующего ингредиента плюс любой дополнительный желаемый ингредиент, из их раствора, стерилизованного фильтрованием.
Используемый в настоящем описании термин «фармацевтически приемлемый носитель» включает все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические агенты и агенты, задерживающие абсорбцию, и т.п. В данной области хорошо известны способы применения таких сред и агентов для фармацевтически активных веществ. За исключением случаев, когда любые обычные среда или агент являются несовместимыми с активным ингредиентом, предполагается их использование в терапевтических композициях. Также в композиции можно включать вспомогательные активные ингредиенты.
Для перорального введения полипептиды настоящего изобретения можно включить в состав с эксципиентами и использовать в форме ополаскивателей для рта и средств для чистки зубов, не предназначенных для проглатывания. Ополаскиватель для рта можно приготовить, включая активный ингредиент в требуемом количестве в подходящем растворителе, таком как раствор бората натрия (раствор Добелла). Альтернативно, активный ингредиент можно включить в антисептический ополаскиватель, содержащий борат натрия, глицерин и бикарбонат калия. Активный ингредиент также можно диспергировать в средствах для чистки зубов, включая гели, пасты, порошки и суспензии. Активный ингредиент можно добавить в терапевтически эффективном количестве в пасту для чистки зубов, которая может включать воду, связующие вещества, абразивы, отдушки, вспениватели и увлажнители.
Композиции настоящего изобретения можно составить в нейтральной форме или в виде соли. Фармацевтически приемлемые соли включают кислотно-аддитивные соли (образуемые свободными аминогруппами белков), которые образуются с неорганическими кислотами, такими как, например, хлористоводородная или фосфорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, винная, миндальная и т.п. Соли, образуемые со свободными карбоксильными группами, также можно получить с неорганическими основаниями, такими как, например, гидроксиды натрия, калия, аммония, кальция или железа (III), и такими органическими основаниями, как изопропиламин, триметиламин, гистидин, прокаин и т.п.
После составления растворы вводят способом, совместимым с дозированной формой, и в количестве, которое является терапевтически эффективным. Составы легко вводить в ряде дозированных форм, таких как растворы для инъекций, капсулы для высвобождения лекарственных средств и т.п. Для парентерального введения в водном растворе, например, раствор при необходимости должен содержать буфер, и жидкому растворителю сначала необходимо придать изотоничность с помощью достаточного количества соли или глюкозы. Данные конкретные водные растворы особенно подходят для внутривенного, внутримышечного, подкожного и внутрибрюшинного введения. В связи с этим, специалистам в данной области известна стерильная водная среда, которую можно будет использовать для настоящего изобретения. Например, одну дозу можно растворить в 1 мл изотонического раствора NaCl и либо добавить к 1000 мл гиподермоклизисной жидкости, либо ввести с помощью инъекции в предполагаемое место инфузии (см., например, «Remington's Pharmaceutical Sciences», 15-е издание, страницы 1035-1038 и 1570-1580). Некоторые изменения дозировки будут неизбежными в зависимости от состояния субъекта, подвергаемого лечению. Человек, отвечающий за введение препарата, будет в любом случае определять соответствующую дозу для конкретного субъекта. Более того, для введения людям, препараты должны соответствовать стандартам стерильности, пирогенности, общей безопасности и чистоты согласно требованиям стандартов FDA (Управления США по надзору за качеством пищевых продуктов и лекарственных средств) для биологических веществ.
B. Типы раковых заболеваний и субъекты
Раковые клетки, к которым можно применять способы настоящего изобретения, включают в общем любую клетку, которая экспрессирует MUC1 и, более конкретно, сверхэкспрессирует MUC1. Подходящей раковой клеткой может быть клетка рака молочной железы, рака легких, рака толстой кишки, рака поджелудочной железы, рака почек, рака желудка, рака печени, рака кости, гематологического рака (например, лейкемии или лимфомы), рака нервной ткани, меланомы, рака яичников, рака яичек, рака простаты, рака шейки матки, рака влагалища или рака мочевого пузыря. В дополнение, способы настоящего изобретения можно применять у широкого ряда биологических видов, например, у человека, приматов (кроме человека) (например, макак, бабуинов или шимпанзе), лошадей, крупного рогатого скота, свиней, овец, коз, собак, кошек, кроликов, морских свинок, песчанок, хомяков, крыс и мышей.
C. Способы лечения
Пептиды или аналоги, которые ингибируют образование олигомеров MUC1, обычно пригодны в качестве противораковых терапевтических или профилактических средств. Их можно вводить млекопитающим (например, пациентам с раком молочной железы) отдельно или в сочетании с другими лекарственными средствами и/или радиотерапией. Соединения можно также вводить субъектам, которые генетически и/или в результате воздействия окружающей среды (вследствие, например, физиологических факторов и/или факторов окружающей среды) подвержены возникновению ракового заболевания, например, субъектам, которые имеют семейную историю раковых заболеваний (например, рака молочной железы), субъектам с хроническим воспалением или подвергающихся хроническому стрессу, или субъектам, которые подвергались воздействию природных или неприродных канцерогенных условий окружающей среды (например, избыточному воздействию солнечного излучения, промышленных канцерогенов или табачного дыма).
При применении способов настоящего изобретения субъектам с раковыми заболеваниями перед введением соединения необязательно провести тестирование ракового заболевания на экспрессию MUC1 (экспрессию белка MUC1 или мРНК MUC1) способами, известными в данной области. В этом случае субъектов можно классифицировать как имеющих раковое заболевание с экспрессией или сверхэкспрессией MUC1. Такие способы можно провести in vitro на раковых клетках, полученных от субъекта. Альтернативно, можно применять методики визуализации in vivo с использованием, например, радиоактивномеченых антител, специфичных к MUC1. Кроме того, у субъектов с раковыми заболеваниями можно провести тестирование жидкостей организма (например, крови или мочи) на повышение уровня белка MUC1 или фрагментов белка MUC1.
Требуемая дозировка зависит от выбора пути введения; природы состава; природы заболевания пациента; размера, массы, размера обрабатываемой поверхности, возраста и пола; других вводимых лекарственных средств; и решения лечащего врача. Подходящие дозировки находятся в диапазоне от 0,0001 мг/кг до 100 мг/кг. Ожидается значительный разброс необходимых дозировок вследствие разнообразия доступных соединений и различной эффективности разных путей введения. Например, ожидается, что при пероральном введении будут требоваться более высокие дозировки, чем в случае внутривенных инъекций. Как хорошо известно в данной области, изменения уровня дозировок можно подобрать, используя стандартные эмпирические методики для оптимизации. Введение может быть однократным или многократным (например, 2-, 3-, 4-, 5-, 6-, 8-, 10-, 20-, 50-, 100-, 150-кратным или большее число раз). Эффективность доставки пептида может повысить заключение полипептида в подходящий носитель (например, полимерные микрочастицы или имплантируемые устройства), в частности, в случае перорального введения.
V. Комбинированные методы лечения
Опухолевые клетки, устойчивые к действию агентов, повреждающих ДНК, представляют главную проблему в клинической онкологии. Одной целью современных исследований в области раковых заболеваний является поиск путей повышения эффективности химио- и радиотерапии. Одним из путей является объединение таких традиционных методов лечения с генной терапией. В контексте настоящего изобретения предполагается, что терапию MUC1-пептидом можно использовать аналогичным образом совместно с химиотерапевтическим, радиотерапевтическим или иммунотерапевтическим вмешательством.
Для уничтожения клеток, ингибирования клеточного роста, ингибирования метастазирования, ингибирования ангиогенеза или иного обращения или уменьшения злокачественного фенотипа опухолевых клеток, при применении способов и композиций согласно настоящему изобретению, как правило, осуществляется контакт клетки-мишени с MUC1-пептидом и по меньшей мере одним методом лечения. Такие методы лечения будут проводиться с равнозначной эффективностью для уничтожения клетки или ингибирования ее пролиферации. Этот процесс может включать одновременный контакт клетки с агентами/методами лечения. Процесс можно осуществить контактом клетки с одной композицией или фармакологическим составом, содержащим оба агента, или одновременным контактом клетки с двумя различными композициями или составами, причем одна композиция содержит MUC1-пептид, а другая содержит агент.
Альтернативно, воздействие MUC1 может предварять другое воздействие или следовать за ним через интервалы от одной минуты до недель. В вариантах осуществления, в которых клетку по отдельности подвергают другому воздействию и воздействию MUC-1, в общем, следует учитывать, чтобы период времени между каждым воздействием не был слишком большим, так чтобы методы лечения были способны оказывать предпочтительный комбинированный эффект на клетку. В этих случаях предполагается осуществление контакта клетки с двумя типами воздействия в интервале примерно 12-24 часов между ними, в интервале примерно 6-12 часов между ними, или с интервалом только примерно 12 часов. Однако в некоторых ситуациях может быть желательно значительно увеличить временной период лечения; с интервалом в несколько дней (2, 3, 4, 5, 6 или 7) и до нескольких недель (1, 2, 3, 4, 5, 6, 7 или 8) между соответствующими введениями.
Также является возможным, что будет желательно более чем одно введение либо MUC1-пептида или другого метода лечения. Могут быть использованы различные комбинации, в которых MUC1-пептидом является «А», и другим методом лечения является «В», примеры которых приведены ниже:

Другие комбинации также предусмотрены. С другой стороны, для клетку с равнозначной эффективностью для уничтожения клетки.
Агенты или факторы, подходящие для использования в комбинированной терапии, включают любое химическое соединение или способ лечения, которые вызывают повреждение ДНК при воздействии на клетку. Такие агенты и факторы включают радиацию и волны, которые вызывают повреждение ДНК, такие как γ-облучение, рентгеновское облучение, УФ-облучение, микроволны, эмиссия электронов и т.п. Предполагается, что ряд химических соединений, также описываемых как «химиотерапевтические» или «генотоксичные агенты», будут пригодны в комбинированных способах лечения, раскрытых в настоящем описании. При лечении рака согласно изобретению осуществляют контакт опухолевых клеток с агентом в дополнение к экспрессионной конструкции. Это может быть осуществлено облучением локализованного участка опухоли, например рентгеновскими лучами, УФ-светом, γ-лучами или даже микроволнами. Альтернативно, опухолевая клетка может быть подвергнута контакту с агентом путем введения субъекту терапевтически эффективного количества фармацевтической композиции.
В комбинации с пептидами настоящего изобретения предполагается использование различных классов химиотерапевтических агентов. Например, селективные антагонисты эстрогеновых рецепторов («SERM»), такие как тамоксифен, 4-гидрокси-тамоксифен (афимоксфен), фалсодекс, ралоксифен, базедоксифен, кломифен, фемарель, лазоксифен, ормелоксифен и торемифен.
Предполагается, что пригодные химиотерапевтические агенты включают, например, камптотецин, актиномицин-D, митомицин C. Изобретение также охватывает использование комбинации одного или нескольких агентов, повреждающих ДНК, либо радиационных, либо химических соединений, таких как использование рентгеновского облучения с цисплатином или использование цисплатина с этопозидом. Агент можно приготовить и использовать в объединенной терапевтической композиции или наборе путем объединения его с MUC1-пептидом, как описано выше.
Белок теплового шока 90 представляет собой регуляторный белок, обнаруживаемый во многих эукариотических клетках. Было показано, что ингибиторы HSP90 пригодны для лечения рака. Такие ингибиторы включают гелданамицин, 17-(аллиламино)-17-деметоксигелданамицин, PU-H71 и рифабутин.
Также предусмотрены агенты, которые напрямую сшивают ДНК или образуют аддукты с ДНК. Можно использовать агенты, такие как цисплатин, и другие ДНК-алкилирующие агенты. Цисплатин широко используется при лечении рака с эффективными дозами, используемыми в клиническом применении, составляющими 20 мг/м2 в течение 5 дней каждые три недели, всего три курса. Цисплатин не всасывается перорально и поэтому его необходимо вводить с помощью внутривенных, подкожных, внутриопухолевых или внутрибрюшинных инъекций.
Агенты, которые повреждают ДНК, также включают соединения, нарушающие репликацию ДНК, митоз и разделение хромосом. Такие химиотерапевтические соединения включают адриамицин, также известный как доксорубицин, этопозид, верапамил, подофиллотоксин и т.п. Широко используемые в клинике для лечения неоплазм такие соединения вводят болюсными инъекциями внутривенно в дозах, варьирующих в диапазоне от 25-75 мг/м2 с 21-дневными интервалами для доксорубицина, до 35-50 мг/м2 для этопозида внутривенно, или удваивая внутривенную дозу в случае перорального введения. Также предусмотрены ингибиторы образования микротрубочек, такие как таксаны. Эти молекулы представляют собой дитерпены, продуцируемые растениями рода Taxus, и включают паклитаксел и доцетаксел.
Ингибиторы фактора роста эпидермиса, такие как иресса, mTOR, мишень рапамицина у млекопитающих, также известного как FK506-связывающий белок 12-рапамицин-ассоциированный белок 1 (FRAP1), представляет собой сериновую/треониновую протеинкиназу, которая регулирует клеточный рост, клеточную пролиферацию, клеточное движение, клеточное выживание, синтез белка и транскрипцию. Поэтому, рапамицин и его аналоги («рапалоги») предусмотрены для использования в комбинированной противораковой терапии согласно настоящему изобретению.
Другим возможным веществом для комбинированной терапии с заявленными в настоящем описании пептидами является TNF-α (фактор некроза опухолей альфа), цитокин, участвующий в системном воспалении, и член группы цитокинов, которые стимулируют острофазовую реакцию. Первичной ролью TNF является регуляция иммунных клеток. TNF также способен вызывать апоптотическую гибель клеток, вызывать воспаление и ингибировать онкогенез и вирусную репликацию.
Агенты, которые нарушают синтез и точность предшественников и субъединиц нуклеиновых кислот также приводят к повреждению ДНК. Соответственно, был разработан ряд предшественников нуклеиновых кислот. Особенно пригодны агенты, которые прошли интенсивное тестирование и легкодоступны. Соответственно, агенты, такие как 5-фторурацил (5-FU), предпочтительно используются неопластической тканью, что делает этот агент особенно пригодным для проникновения в неопластические клетки. Несмотря на значительную токсичность, 5-FU применяется с широким спектром носителей, включая носители для местного введения, однако обычно используется внутривенное введение с дозировками, варьирующими в диапазоне от 3 до 15 мг/кг/день.
Другие факторы, которые вызывают повреждение ДНК и интенсивно используются, включают общеизвестное гамма-облучение, рентгеновское облучение и/или направленную доставку радиоизотопов в опухолевые клетки. Также предусмотрены другие формы повреждающих ДНК факторов, такие как микроволны и УФ-излучение. Наиболее вероятно, что все эти факторы вызывают разнообразные повреждения ДНК, повреждение предшественников ДНК, нарушение репликации и репарации ДНК, и нарушение сборки и поддержания хромосом. Диапазоны дозировок для рентгеновского облучения варьируют от ежедневных дозировок 50-200 рентген в течение длительных периодов времени (3-4 недель) до разовых доз от 2000 до 6000 рентген. Диапазон дозировок для радиоизотопов варьирует в широких пределах и зависит от времени полужизни изотопа, силы и типа испускаемого радиоактивного излучения и поглощения неопластическими клетками.
Специалист в данной области может обратиться к «Remington's Pharmaceutical Sciences», 15-е издание, глава 33, в частности, страницы 624-652. Обязательно возникнут некоторые изменения дозировки в зависимости от состояния субъекта, подвергаемого лечению. В любом случае, лицо, отвечающее за введение, будет определять соответствующую дозу для конкретного субъекта. Более того, для введения людям, препараты должны соответствовать стандартам стерильности, пирогенности, общей безопасности и чистоты, требуемым стандартами FDA (Управления США по надзору за качеством пищевых продуктов и лекарственных средств) для биологических веществ.
Авторы изобретения предполагают, что локальное или местное введение MUC-1 пептидов пациентам с раком будет представлять собой очень эффективный способ лечения клинически выраженного заболевания. Аналогично, химио- или радиотерапия могут быть направлены на конкретную, пораженную область тела субъекта. Альтернативно, в некоторых обстоятельствах может подходить региональная или системная доставка экспрессионной конструкции и/или агента, например, при интенсивном метастазировании.
В дополнение к комбинации методов лечения с использованием MUC1 с химио- и радиотерапией, также предусмотрена комбинация с иммунотерапией, гормональной терапией, терапией с использованием токсинов и хирургическим вмешательством. В частности, можно использовать целевую терапию, такую как терапия авастином, эрбитуксом, гливеком, герцептином и ритуксаном.
Следует также отметить, что любой из приведенных выше методов лечения может оказаться полезным сам по себе при лечении рака.
VI. Примеры
Следующие примеры включены для демонстрации конкретных вариантов осуществления изобретения. Специалистам в данной области следует принимать во внимание, что методики, описанные в приведенных ниже примерах, представляют собой методики, которые, как было раскрыто авторами изобретения, хорошо работают при применении изобретения на практике, и, поэтому, их можно рассматривать, как практические варианты применения изобретения на практике. Однако специалистам в данной области следует принимать во внимание, что в конкретные варианты осуществления настоящего изобретения можно вносить различные изменения, получая аналогичный результат и не отступая от сущности и объема изобретения.
Пример 1 - Материалы и методы
Культивирование клеток
Клеточные линии рака молочной железы человека, ZR-75-1, ZR-75-1/вектор, ZR-75-1/MUC1siRNA (Ren et al., 2004) выращивали в среде RPMI1640 с 10% инактивированной нагреванием фетальной телячьей сывороткой (HI-FBS), 100 ед/мл пенициллина и 100 мкг/мл стрептомицина (Invitrogen) в инкубаторе с увлажнением в 5% СО2 при 37°С. Клетки рака молочной железы человека MCF-7 и клетки 293 выращивали на модифицированной Дульбекко среде Игла (DMEM) с 10% HI-FBS, антибиотиками и 2 мМ L-глутамина. Клетки эпителия молочной железы человека MCF-10A выращивали в среде для роста эпителиальных клеток молочных желез (MEGM; Lonza). Клетки обрабатывали пептидами MUC1/CQC или MUC1/AQA, синтезированными в MIT Biopolymer Laboratory, Cambridge, MA. Жизнеспособность клеток определяли по исключению трипанового синего.
Иммуноосаждение и иммуноблоттинг
Лизаты цельных клеток и ядер получали, как описано (Leng et al., 2007). Иммуноосаждение растворимых белков проводили с анти-Flag антителами (Sigma, St. Louis, MO). Полученные иммуноосадки и растворимые белки анализировали иммуноблоттингом с анти-His (Cell Signaling Technology, Danvers, MA), анти-GFP (Millipore, Danvers, MA), анти-Flag, анти-MUC1-C (Ab1; NeoMarkers, Fremont, CA), анти-ламин B (EMD, La Jolla, CA) или анти-β-актин (Sigma) антителами. Взаимодействие детектировали с помощью конъюгированных с пероксидазой хрена вторичных антител и хемилюминесцентной реакции.
Трансфекция клеток
Клетки 293 трансфицировали векторами, экспрессирующими GFP, GFP-MUC1-CD или Flag-MUC1-CD в присутствии Lipofectamine, как описано (Leng et al., 2007).
Поглощение пептида клетками
Клетки инкубировали с меченным FITC пептидом MUC1/CQC (MIT Biopolymer Laboratory), промывали холодным PBS, фиксировали в 1%-ом параформальдегиде в PBS и проводили анализ уровня флуоресценции с помощью проточной цитометрии.
Анализ распределения по фазам клеточного цикла, апоптоза и некроза
Клетки собирали, промывали PBS, фиксировали 80%-ым этанолом и инкубировали в PBS, содержащем 40 мкг/мл РНКазы и 40 мкг/мл пропидиййодида в течение 30 мин при 37°С. Анализ клеточного цикла проводили с помощью проточной цитометрии. Содержание ДНК во фракции суб-G1 оценивали, окрашивая фиксированные этанолом и пермеабилизованные цитратным буфером клетки пропидиййодидом и анализируя с помощью проточной цитометрии, как описано (Yin et al., 2007). Для оценки некроза клетки инкубировали с 1 мкг/мл пропидиййодида в PBS в течение 45 мин при комнатной температуре и затем анализировали с помощью проточной цитометрии, как описано (Yin et al., 2007).
Ксенографтная модель опухоли молочной железы человека
4-6 недельным самкам мышей Balb-c nu/nu (Charles River Laboratories, Wilmington, MA) с массой 18-22 граммов имплантировали подкожно капсулы с 17-β-эстрадиолом (0,72 мг; Innovative Research, Sarasota, FL) с помощью троакара. Через 24 часа 1×107 клеток ZR-75-1, заключенных в Matri-Gel (BD Biosciences), вводили подкожно в подвздошную область. Когда опухоли становились детектируемыми ~150 мм3 (группа 1) или 275 мм3 (группа 2) мышей распределяли соответствующими парами по группам «воздействие» и «контроль». Каждая группа содержала 5 мышей, каждая из которых имела метку на ухе и наблюдалась в течение всего исследования. Первую дозу вводили в день распределения по группам (день 1). Забуференный фосфатом солевой раствор (носитель), пептид MUC1/CQC и пептид MUC1/AQA вводили с помощью внутрибрюшинных инъекций ежедневно. Мышей взвешивали дважды в неделю, и измерение опухоли с помощью штангенциркуля проводили каждые 4 дня. Объем опухоли (V) вычисляли, используя формулу V=W2×L/2, где W и L представляют собой меньший и больший диаметры, соответственно. Перед умерщвлением мышей перфузировали солевым раствором и затем в забуференном фосфатом формалине через сердце. Опухоли вырезали, фиксировали иммерсионным способом в течение 4 часов, дегидратировали в серии растворов этанола с возрастающей концентрацией и заключали в парафин. Анализ опухолей проводили с помощью окрашивания H&E (гематоксилином/эозином) и иммунопероксидазного окрашивания с антителами к MUC1, как описано (Kufe, 1984).
Лекарственные средства и цитокины
Цисдиаминхлорплатин (II), доксорубицин (адриамицин), таксол (паклитаксел) были приобретены у компании Sigma (St. Louis, MO). rh-TNF-alpha был приобретен у компании Promega (Madison, WI). Пептид GO-203 был синтезирован Anaspec Inc.
In vitro анализ цитотоксичности и комбинированного действия соединений
Клетки высевали в 96-луночной микротитровальный планшет с плоским дном лунок (Fisher) в количестве 1000 клеток на лунку для 6-дневного эксперимента или 3000 клеток на лунку для 3-дневного эксперимента. Затем клетки культивировали в течение 24 часов. Противораковые лекарственные средства и GO-203 разводили до указанных концентраций и добавляли к клеткам. GO-203 (5 мкмоль/л) добавляли каждые 24 часа в течение 72 часов. Выживаемость клеток определяли, добавляя к клеткам MTS-реагент и измеряя поглощение при 490 нм на микроплашет-ридере.
Анализ данных
Величину IC50 для всех противораковых лекарственных средств определяли нелинейным регрессионным анализом, используя программу Graphpad Prism (GraphPad Software, San Diego CA). Для вычисления комбинационного индекса использовали программу CombiTool (version 2.001, 1MB Jena Biocomputing Group) для диапазона доз в присутствии 5 мкмоль/л GO-203.
Пример 2 - Результаты
Эффекты MUC1/CQC-пептида на образование олигомеров MUC1
Цитоплазматический домен MUC1 (MUC1-CD) содержит CQC-мотив, который необходим для образования его олигомеров и ядерной транслокации (Leng et al., 2007). Для определения возможности ингибирования олигомеризации низкомолекулярными соединениями авторы изобретения синтезировали пептид на основе N-концевой области MUC1-CD, который содержит CQC-мотив (MUC1/CQC-пептид; фиг.1A). При синтезе в пептид был включен трансдуцирующий поли-D-аргининовый домен, способствующий вхождению пептида в клетки (Fischer, 2007) (фиг.1A). В качестве контроля синтезировали аналогичный пептид, в котором CQC-мотив был заменен на AQA (MUC1/AQA-пептид; фиг.1A). Для оценки связывания пептидов с MUC1-CD авторы изобретения иммобилизовали His-меченный MUC1-CD на сенсорном чипе для системы BIAcore. Пептид MUC1/CQC связывался с His-MUC1-CD с константой диссоциации (Kd) 30 нМ (фиг.1B), аналогичной константе диссоциации, полученной для олигомеров MUC1-CD (Leng et al., 2007). В отличие от этого не наблюдалось видимого связывания пептида MUC1/AQA (данные не показаны). Очищенный His-меченный MUC1-CD образовывал олигомеры, детектируемые электрофорезом в полиакриламидном геле (фиг.1С). Инкубация His-MUC1-CD с пептидом MUC1/CQC значительно снижала образование олигомеров и увеличивала содержание мономеров (фиг.1С). Более того, инкубация с пептидом MUC1/AQA давала минимальный эффект или не давала какого-либо эффекта (фиг.1С). Для оценки на олигомеризацию MUC1 in vivo клетки 293 трансфицировали векторами, экспрессирующими GFP-MUC1-CD и Flag-MUC1-CD (фиг.1D, слева). Комплексы GFP-MUC1-CD и Flag-MUC1-CD детектировали методом соосаждения из лизатов клеток, не обработанных пептидом (фиг.1D, справа). Аналогично результатам, полученным in vitro, инкубация трансфицированных клеток 293 с пептидом MUC1/CQC была ассоциирована с разрушением взаимодействия между Flag-MUC1-CD и GFP-MUC1-CD (фиг.1D, справа). Кроме того, пептид MUC1/AQA не имел выраженного эффекта (фиг.1D, справа). Эти результаты указывают на то, что пептид MUC1/CQC связывает MUC1-CD и блокирует образование олигомеров MUC1-CD in vitro и в клетках.
Пептид MUC1/CQC блокирует транслокацию MUC1-C в ядро клетки
Клетки рака молочной железы человека ZR-75-1 и MCF-7 сверхэкспрессируют эндогенный MUC1 и, поэтому, представляют собой потенциальные модели для оценки эффектов пептида MUC1/CQC (Ramasamy et al., 2007). Для оценки поглощения пептида клетки ZR-75-1 инкубировали с 5 мкМ пептида FITC-MUC1/CQC (фиг.2A). Через 2 часа анализ клеток с помощью проточной цитометрии показал значительное увеличение интенсивности флуоресценции со средним значением (MFI) 145 (фиг.2A). Дополнительное повышение MFI детектировалось через 6 и 24 часа (фиг.2A). Олигомеризация MUC1-C необходима для его ядерного импорта (Leng et al., 2007). Обработка клеток ZR-75-1 пептидами MUC1/CQC или MUC1/AQA не давало какого-либо влияния на клеточный уровень MUC1-C (фиг.2B). Однако в соответствии с эффектом в отношении олигомеризации обработка пептидом MUC1/CQC, но не MUС1/AQA, была связана со снижением уровня MUC1-C в ядре (фиг.2B). Аналогичные результаты были получены для клеток MCF-7 со снижением уровня ядерного MUC1-C в ответ на обработку пептидом MUC1/CQC (фиг.2C). Эти данные указывают на то, что пептид MUC1/CQC блокирует олигомеризацию MUC1-C и, таким образом, блокирует перенос MUC1-C в ядро клетки.
Пептид MUC1/CQC блокирует рост и вызывает некроз клеток
Для определения влияния пептида MUC1/CQC на клеточный рост клетки ZR-75-1 обрабатывали 5 мМ MUC1/CQC в течение 72 часов и анализировали распределение по фазам клеточного цикла. Несомненно, наблюдалось выраженное торможение клеточного цикла в S-фазе по сравнению с необработанными клетками или клетками, обработанными пептидом MUC1/AQA (фиг.3A). К 96 часам численность популяции, находящейся в S-фазе, снижалась, возможно, вследствие отсева в результате смерти клеток (фиг.3A). Практически не наблюдалось или наблюдалось очень низкое накопление клеток с содержанием ДНК в суб-G1 фракции, чтобы подтвердить индукцию апоптоза (фиг.3А). Однако обработка клеток ZR-75-1 пептидом MUC1/CQC, а не MUC1/AQA, была ассоциирована с индукцией некроза, который детектировался через 72 часа и был более выражен через 96 часов (фиг.3В). Клетки MCF-7 отвечали аналогичным образом на пептид MUC1/CQC с торможением роста в S-фазе (фиг.3C) и индукцией некроза (фиг.3D). Эти данные указывают на то, что пептид MUC1/CQC ингибирует рост и вызывает некроз клеток рака молочной железы человека.
Специфичность пептида MUC1/CQC к экспрессирующим MUC1 клеткам карциномы
Для определения избирательной активности пептида MUC1/CQC в отношении клеток карциномы молочной железы, которые сверхэкспрессируют эндогенный MUC1, авторы изобретения проводили эксперименты с клетками ZR-75-1, в которых экспрессия MUC1 была стабильно подавлена с помощью коротких интерферирующих РНК к MUC1, MUC1siRNA (фиг.4A). В отличие от торможения роста и смерти контрольных клеток ZR-75-1/вектор, пептид MUC1/CQC оказывал значительно меньший эффект на клетки ZR-75-1/MUC1siRNA (фиг.4B). Кроме того, пептид MUC1/CQC не имел явного воздействия на рост MUC1-отрицательных клеток 293 (фиг.4C). Также проводили исследования на нетрансформированной клеточной линии эпителия молочной железы, MCF-10A (Muthuswamy, 2001; Soule, 1990), в которой экспрессируется MUC1, но на уровне ниже уровня, найденном в клетках ZR-75-1 и MCF-7 (Ahmad et al., 2007). Примечательно, что в отличие от клеток ZR-75-1 и MCF-7 пептид MUC1/CQC не оказывал воздействия на клеточный цикл (фиг.4D) и рост (фиг.4E) клеток MCF-10A. Эти данные указывают на то, что пептид MUC1/CQC обладает избирательной активностью в отношении клеток карциномы молочной железы, которые сверхэкспрессируют эндогенный MUC1.
Пептид MUC1/CQC ингибирует онкогенез in vivo
Для определения связи между введением пептида MUC1/CQC и влиянием на массу тела пяти самкам бестимусных (nu/nu) мышей вводили внутрибрюшинно (IP) однократно каждый день дозу 50 мг/кг. Введение пептида в течение 11 дней не оказывало видимого влияния на массу индивидуальных мышей. Более того, не наблюдалось значительного эффекта на массу тела в течение следующих 28 дней после отмены введения MUC1/CQC (данные не показаны). Для оценки противоопухолевой активности клетки ZR-75-1 (1×107) вводили подкожно в подвздошные области бестимусных мышей. Через 12 дней мышам, имеющим опухоли приблизительно 150 мм3, вводили пептид MUC1/CQC в дозах 10 и 50 мг/кг/день. В качестве контроля мышам вводили только носитель или носитель с пептидом MUC1/AQA. Введение пептида MUC1/CQC в концентрации 10 мг/кг/день в течение 21 дня замедляло рост опухолей по сравнению с результатами, полученными с пептидом MUC1/AQA, вводимом в концентрации 50 мг/кг/день (фиг.5А). Кроме того, введение пептида MUC1/CQC в концентрации 50 мг/кг/день блокировало рост опухолей за первоначальные 7 дней лечения (фиг.5А). После чего воздействие прекращали, и наблюдали повторный рост опухолей у мышей. Несомненно, не наблюдалось детектируемого роста опухолей в течение следующих 17 дней (фиг.5А). Для оценки, в частности, причин активности проводили гистопатологическое исследование опухолей, собранных у контрольных и подвергаемых обработке мышей. Опухоли мышей, обработанных MUC1/CQC (10 и 50 мг/кг), были заметно некротическими по сравнению с опухолями мышей, обработанных носителем или пептидом MUC1/AQA (фиг.5B и данные не показаны). Однако следует отметить, что опухолевые клетки также детектировались вокруг области некроза (фиг.5В). Срезы опухолей также окрашивали антителами к MUC1. Обработка пептидом MUC1/AQA была ассоциирована со значительным снижением экспрессии MUC1 по сравнению с контрольными опухолями и опухолями, обработанными пептидом MUC1/AQA (фиг.5C и данные не показаны).
Также проводили исследования на опухолях большего размера (~275 мм3) (фиг.6A). Введение пептида MUC1/CQC в промежуточной дозе 30 мг/кг/д в течение 21 дня ассоциировалось с торможением роста опухоли (фиг.6А). Более того, не наблюдалось видимого повторного роста в течение следующих 31 дней после воздействия (фиг.6А), что дополнительно указывало на то, что пептид MUC1/CQC эффективен в отношении торможения роста опухолей. Опухоли, собранные на 52-й день, имели обширные некротические участки (фиг.6В) и сниженную экспрессию MUC1. Эти данные указывают на то, что пептид MUC1/CQC снижает экспрессию MUC1 и ассоциирован с индукцией некроза и продолжительным торможением роста опухоли.
MUC1-C-концевые CQC-пептиды, соединенные углеводородными сшивками
Внутриклеточные белок-белковые взаимодействия, которые регулируют множество биологических путей, часто опосредованы α-спиральной структурой белков. Спиральные пептиды могут также нарушать или стабилизировать белок-белковые взаимодействия. Природные спиральные пептиды имеют большое число недостатков в качестве терапевтических агентов вследствие низкой активности, нестабильности и неэффективного проникновения в клетку. В недавних исследованиях было показано, что эти проблемы можно преодолеть с помощью химической модификации α-спиральных пептидов, называемой «соединением с помощью углеводородных сшивок».
Авторы изобретения использовали последовательность эндогенного С-концевого пептида MUC1 (AIVYLIALAVCQCRRKNYG) и создали два α-спиральных пептида, GO-200-1B и GO-200-2B, используя соединение углеводородными сшивками:
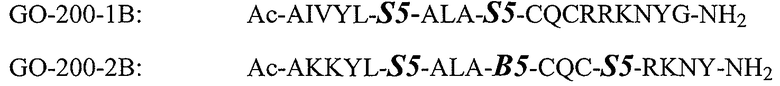
Для определения влияния воздействия GO-200-1B на рост клеток немелкоклеточной карциномы легких, H-1650, клетки обрабатывали 1 и 5 мкМ GO-200-1B в течение 7 дней и контролировали рост клеток. Результаты показали, что обработка клеток 5 мкМ GO-200-1B была ассоциирована со значительным ингибированием роста (фиг.9А). Кроме того, другую клеточную линию немелкоклеточной карциномы легких, H-1975, обрабатывали 5 мкМ GO-200-2B в течение 3 дней и контролировали рост клеток, а также гибель клеток. Результаты демонстрируют, что обработка клеток H-1975 пептидом GO-200-2B в течение 3 дней была ассоциирована с более чем 80%-ым ингибированием пролиферации клеток. Более того, воздействие GO-200-2B также было ассоциировано со значительной индукцией гибели клеток (фиг.9В). Эти данные указывают на то, что пептиды MUC1-C, соединенные углеводородными сшивками, эффективно вызывают торможение роста и гибель MUC1-положительных клеток раковых заболеваний человека.
Аналоги GO-203
В недавних исследованиях авторов изобретения было показано, что С-концевой пептид MUC1 (CQCRRKNYGQLDIFP) ингибирует рост множества карциномных клеточных линий. Авторы также продемонстрировали, что более короткий MUC1-C-концевой пептид, CQCRRKN, также обладает активностью в отношении уничтожения опухолевых клеток. Однако эти MUC1-C-концевые пептиды состоят из L-аминокислот. Важно отметить, что пептиды из L-аминокислот восприимчивы к деградации протеолитическими ферментами, в то время как было показано, что пептиды, содержащие D-аминокислоты, являются более стабильными. Соответственно, авторы изобретения создали полностью правовращающую форму вышеописанного С-концевого пептида MUC1, в которой L-аминокислоты заменены D-аминокислотами (GO-203). Кроме того, для определения минимальной последовательности аминокислотных остатков из С-концевой области MUC1, которая требуется для сохранения цитотоксичной активности, авторы изобретения создали ряд различных вариантов GO-203, описанный на фиг.8.
Набор опухолевых клеточных линий (гормонозависимая карцинома молочных желез ZR-75-1; «тройная негативная» карцинома молочных желез MDA-MB-231; немелкоклеточная карцинома легких А549; немелкоклеточная карцинома легких H-1975) выращивали на среде RPMI-1640 с 10% инактивированной нагреванием фетальной телячьей сывороткой, 100 ед/мл пенициллина, 100 мкг/мл стрептомицина и 2 ммоль/л L-глутамина. Клетки обрабатывали по отдельности различными аналогами GO-203 с концентрацией 5 мкМ (фиг.8) в течение 3-7 дней, и определяли жизнеспособность по исключению трипанового синего. Пролиферацию различных клеточных линий сравнивали с клетками, обработанными только носителем. Результаты демонстрируют, что обработка набора опухолевых клеточных линий 5 мкМ различных аналогов GO-203 ассоциирована со значительным ингибированием роста (фиг.10-14).
Комбинированная терапия
Величины комбинационного индекса (CI), полученные с помощью программы CombiTool, наносили на диаграмму относительно фракции пораженных клеток в результате действия комбинации лекарственных средств. Величины CI были близки к 1 для цисплатина и GO-203, указывая на аддитивное воздействие. Величины CI, полученные для доксорубицина (25, 50, 100, 200 нМ), таксола (25, 50, 100 нМ) и TNF (10,20,40 нг/мл) с GO-203 были меньше 1, указывая на сильный аддитивный эффект или синергизм (фиг.15А-D и 16).
Пример 3 - Обсуждение
Пептид MUC1/CQC блокирует олигомеризацию MUC1
Сверхэкспрессия MUC1 является достаточной для индукции независимого от прикрепления роста и онкогенности (Li et al., 2003a; Huang et al., 2003; Huang et al., 2005). Однако примечательно, что трансформирующая функция MUC1 блокируется мутацией CQC-мотива в цитоплазматическом домене на AQA (Leng et al., 2007). MUC1 образует олигомеры, и CQC-мотив необходим для его олигомеризации (Leng et al., 2007). Более того, образование олигомеров необходимо для транслокации субъединицы MUC1-C в ядро клетки (Leng et al., 2007). Другие функции субъединицы MUC1-C, такие как активация Wnt/β-катенинового и IKKβ->NF-κB сигнальных путей, также зависят от образования олигомеров MUC1-C (неопубликованные данные). На основе этих данных авторы изобретения пришли к выводу, что нарушение олигомеризации MUC1 низкомолекулярным соединением, возможно, будет блокировать трансформирующую функцию MUC1. Для этого они синтезировали пептид на основе последовательности MUC1, которая включает CQC-мотив и поли-Arg-домен для доставки пептида в клетки. Начальные исследования пептида MUC1/CQC-пептида показали, что он ингибирует олигомеризацию MUC1-CD in vitro. Как было показано ранее с помощью BIAcore-анализа, MUC1-CD образует димеры с константой диссоциации (Kd) 33 нМ (Leng et al., 2007). MUC1/CQC-пептид аналогично связывает MUCl-CD с Kd 30 нМ. Кроме того, данные о том, что пептид MUC1/AQA практически не оказывает влияния на олигомеризацию MUC1, поддерживают гипотезу зависимости олигомеризации от CQC-мотива. MUC1/CQC, а не MUC1/AQA, также был эффективен в отношении ингибирования олигомеризации MUC1-CD в клетках. Поэтому, эти данные указывают на то, что пептид MUC1/CQC можно использовать для нарушения олигомеризации MUC1 и, возможно, таким образом, функции MUC1 в клетках карциномы молочной железы человека.
Селективность MUC1/CQC-пептида в отношении сверхэкспрессирующих MUC1 клеток карциномы
Домены для доставки в клетки, такие как поли-D-Arg, и их конъюгаты входят в клетки по меньшей мере частично, за счет эндоцитоза, и затем им необходимо попасть в целевой компартмент клетки (Fischer, 2007). Вхождение MUC1/CQC-пептида в клетки рака молочной железы, ZR-75-1, легко детектировалось, и сохранялось по меньшей мере в течение 24 часов. В значительной степени и в соответствии с зависимостью ядерной транслокации MUC1 от его олигомеризации (Leng et al., 2007), поглощение клетками MUC1/CQC-пептида было ассоциировано со снижением уровня MUC1-C в ядре. Аналогичные результаты были получены на клетках рака молочной железы, MCF-7, указывая на то, что этот ответ на действие пептида MUC1/CQC не был клеточно-специфичным. Кроме того, важно отметить, что воздействие на эти клетки MUC1/CQC, а не MUC1/AQA, было ассоциировано с торможением роста и индукцией некроза. Важно было установить, вызывает ли MUC1/CQC гибель клеток по механизму, зависимому от экспрессии его мишени, или является неспецифичным цитотоксином. Подавление экспрессии MUC1 в клетках ZR-75-1 отменяло цитотоксические эффекты MUC1/CQC. В отличие от этого, воздействие пептида MUC1/CQC на незлокачественные эпителиальные клетки молочной железы, MCF-10A, производило минимальный эффект. Эти данные указывают на то, что чувствительность к MUC1/CQC-пептиду зависит от сверхэкспрессии MUC1 и функции MUC1, ассоциированной со злокачественным фенотипом. Поэтому, MUC1/CQC-пептид, по-видимому, имеет доминантно-негативную активность, избирательную в отношении карциномных клеток, сверхэкспрессирующих MUC1.
Противоопухолевая активность MUC1/CQC-пептида
Домены для доставки в клетки используются для введения в клетки терапевтических соединений (Fischer, 2007). Однако как для любого из этих агентов, важной проблемой является возможность доставки MUC1/CQC-пептида in vivo с эффективным терапевтическим индексом, то есть противоопухолевой активностью и приемлемым профилем токсичности. При изучении этого вопроса авторы изобретения обнаружили, что введение MUC1/CQC-пептида в количестве 10 и 30 мг/кг/день в течение 21 для хорошо переносилось животными без явных острых токсичных эффектов. Авторами также было обнаружено, что воздействие при этих дозах было эффективно для остановки роста опухоли. Эти результаты были противоположными введению пептида MUC1/AQA в количестве 50 мг/кг/день в течение 21 дня, который не обладал противоопухолевой активностью. Помимо этого и достаточно неожиданно, не было получено данных о повторном росте опухоли после введения пептида в дозе 30 мг/кг/день в течение 21 дня. Введение MUC1/CQC-пептида в количестве 50 мг/кг/день в течение 7 дней также продемонстрировало, что рост опухоли прекращался на протяжении длительных периодов времени после воздействия. Эти результаты объясняются, по меньшей мере частично, данными о том, что воздействие MUC1/CQC-пептида ассоциировано с индукцией некроза опухоли.
In vitro активность 7-мерного пептида MUC1/CQC была аналогична активности 15-мерного пептида MUC1/CQC. Исходя из этой информации, можно ожидать, что 7-мерный пептид MUC1/CQC будет таким же активным в качестве противоопухолевого агента на опухолевых моделях in vivo.
Все композиции и/или способы, раскрытые и заявленные в настоящем описании, можно осуществить и выполнить без лишнего экспериментирования, исходя из настоящего описания. Хотя композиции и способы настоящего изобретения были описаны на предпочтительных вариантах осуществления изобретения, для специалистов в данной области будет очевидно, что можно изменять композиции и/или способы и стадии или последовательности стадий способа, описанного в настоящем описании, не отступая от концепции, сущности и объема изобретения. Более конкретно, будет очевидно, что агенты, описанные в настоящем описании, можно заменить некоторыми агентами, аналогичными как химически, так и физиологически, получая одинаковые или аналогичные результаты. Подразумевается, что все такие аналогичные заместители и модификации, очевидные специалистам в данной области, входят в сущность, объем и концепцию изобретения, определенные в приложенной ниже формуле изобретения.
VII. Ссылки
Следующие ссылки, при условии, что в них предоставлены примеры экспериментов или другие подробности, дополняющие описания, приведенные в настоящем описании, в связи с этим включены в настоящее описание посредством ссылки:
Патент США 5440013
Патент США 5446128
Патент США 5475085
Патент США 5597457
Патент США 5618914
Патент США 5670155
Патент США 5672681
Патент США 5674976
Патент США 5710245
Патент США 5790421
Патент США 5840833
Патент США 5859184
Патент США 5889155
Патент США 5929237
Патент США 6093573
Патент США 6261569
Патентная заявка США 2005/0015232
Abe and Kufe, Cancer Res., 49(11):2834-2839, 1989.
Ahmad et al., Nat. Cell Biol., 9:1419-1427, 2007.
Baldus et al., Clin. Cancer Res., 10(8):2790-2796, 2004.
Bodanszky et al., J. Antibiot, 29(5):549-53, 1976.
Cohen et al., J. Med. Chem., 33:883-894, 1990.
Culver et al., Science, 256(5063):1550-1552, 1992.
Duraisamy et al., Gene, 373:28-34, 2006.
Fischer, Med. Res. Rev., 27(6):755-796, 2007.
Gendler et al., J. Biol. Chem., 263:12820-12823, 1988.
Gronenborn et al., Anal Chem., 62(1):2-15, 1990.
Hodel et al., Mol. Cell, 10(2):347-58, 2002.
Huang et al., Cancer Biol. Ther., 2:702-706, 2003.
Huang et al., Cancer Res., 65:10413-10422, 2005.
Jackson, Seminars in Oncology, 24:L164-172, 1997.
Johnson et al., In: Biotechnology And Pharmacy, Pezzuto et al. (Eds.), Chapman and Hall, NY, 1993.
Jones et al., J. Med. Chem., 39:904-917, 1996.
Kau et al., Nat. Rev. Cancer, 4(2): 106-17, 2004.
Kinlough et al., J. Biol. Chem., 279(51):53071-53077, 2004.
Kufe et al., Hybridoma, 3:223-232, 1984.
Kufe, Cancer Biol. Therapy, 7:81-84, 2008.
Leng et al., J. Biol. Chem., 282:19321-19330, 2007.
Levitan et al., J. Biol. Chem., 280:33374-33386, 2005.
Li et al., Cancer Biol. Ther., 2:187-193, 2003b.
Li et al., J. Biol. Chem., 276:35239-35242, 2001.
Li et al., J. Biol. Chem., 276:6061-6064, 2001.
Li et al., Mol. Cancer Res., 1:765-775, 2003c.
Li et al., Mol. Cell Biol., 18:7216-7224, 1998.
Li et al., Oncogene, 22:6107-6110, 2003a.
Ligtenberg et al., J. Biol. Chem., 267, 6171-6177, 1992.
Macao, Nat. Struct. Mol Biol., 13, 71-76, 2006.
McPherson, J. Biol. Chem., 251:6300-6306, 1976.
Merlo et al., Cancer Res., 49, 6966-6971, 1989.
Merrifield, J. Am. Chem. Soc., 85:2149-2154, 1963.
Muthuswamy, Nat. Cell Biol., 3(9):785-92, 2001.
Navia et al., Curr. Opin. Struct. Biol., 2:202-210, 1992.
Заявка PCT/US00/03745
Заявка PCT/US00/14667
Заявка PCT/US99/11913
Заявка PCT/US99/18441
Peptide Synthesis, 1985.
Percipalle et al., J. Mol. Biol., (4):722-32, 1997.
Perey et al., Cancer Res., 52(22):6365-6370, 1992.
Protective Groups in Organic Chemistry, 1973.
Protein NMR Spectroscopy, Principles and Practice, J. Cavanagh et al., Academic Press, San Diego, 1996.
Raina et al., EMBO J., 25:3774-3783, 2006.
Raina et al., J. Biol. Chem., 279:20607-20612, 2004.
Ramasamy et al., Mol. Cell, 27:992-1004, 2007.
Remington's Pharmaceutical Sciences, 15th Ed., 1035-1038 and 1570-1580, 1990.
Remington's Pharmaceutical Sciences, 15th Ed., 3:624-652, 1990.
Ren et al., Cancer Cell, 5:163-175, 2004.
Ren et al., J. Biol. Chem., 277:17616-17622, 2002.
Ren et al., Oncogene, 25:20-31, 2006.
Ryan and Wente, Curr. Opin. Cell Biol., 12(3):361-71, 2000.
Schroeder et al., J. Biol. Chem., 276(16):13057-13064 2001.
Schroeder et al., Oncogene, 23:5739-5747, 2004.
Siddiqui et al., Proc. Natl. Acad. Sci. USA, 85:2320-2323, 1988.
Solid Phase Peptide Synthelia, 1984.
Soule et al., Cancer Res., 50(18):6075-6086, 1990.
Suh and Gumbiner, Exp. Cell Res., 290(2):447-56, 2003.
Truscott et al., J Cell Biol., 163(4):707-713, 2003.
Vermeer et al., Nature, 422(6929):322-6, 2003.
Weber, Advances Protein Chem., 41:1-36, 1991.
Wei et al., Cancer Cell, 7:167-178, 2005.
Weis, Cell, 112(4):441-51, 2003.
Wen et al., J. Biol. Chem., 278:38029-38039, 2003.
Wider, BioTechniques, 29:1278-1294, 2000.
Yamamoto et al., J. Biol. Chem., 272:12492-12494, 1997.
Yin et al., J. Biol. Chem., 278:35458-35464, 2003b.
Yin et al., J. Biol. Chem., 279:45721-45727, 2004.
Yin et al., J. Biol. Chem., 282:257-266, 2007.
Young et al., Cell. 112(l):41-50, 2003.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИММУНОТЕРАПЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ ДРОЖЖЕЙ-MUC1 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2642300C2 |
| ВЕКТОРНЫЙ ПОЛИПЕПТИД - АНАЛОГ ФРАГМЕНТА ТРАНСФОРМИРУЮЩЕГО ФАКТОРА РОСТА АЛЬФА (ТФРАЛЬФА), ЕГО ПРОТИВООПУХОЛЕВЫЙ КОНЪЮГАТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ КОНЪЮГАТА | 2004 |
|
RU2277930C1 |
| КОМПОЗИЦИЯ ДНК ДЛЯ ВЫЗОВА ИММУННОГО ОТВЕТА ПРОТИВ ОПУХОЛЕАССОЦИИРОВАННЫХ МАКРОФАГОВ | 2007 |
|
RU2459631C2 |
| ВАРИАНТЫ ИЛ-21 | 2006 |
|
RU2412199C2 |
| СПЕЦИФИЧЕСКИЕ К ОПУХОЛИ АНТИТЕЛА И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2595403C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕЙ И МЕТАСТАЗОВ С ИСПОЛЬЗОВАНИЕМ КОМБИНАЦИИ АНТИАНГИОГЕННОЙ ТЕРАПИИ И ИММУНОТЕРАПИИ | 2000 |
|
RU2236251C2 |
| НАНОАНТИТЕЛО, СПЕЦИФИЧЕСКИ СВЯЗЫВАЮЩЕЕ БЕЛОК MUC1, СПОСОБ ДЕТЕКЦИИ БЕЛКА MUC1 С ПОМОЩЬЮ НАНОАНТИТЕЛ | 2012 |
|
RU2493165C1 |
| ХИМЕРНЫЕ АНТИГЕННЫЕ РЕЦЕПТОРЫ С MND-ПРОМОТОРОМ | 2015 |
|
RU2708311C2 |
| Полипептиды для лечения онкологических заболеваний | 2017 |
|
RU2728870C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ РАКА С ИСТОЩАЮЩИМИ АРГИНИН И ИММУНООНКОЛОГИЧЕСКИМИ АГЕНТАМИ | 2017 |
|
RU2771313C2 |


















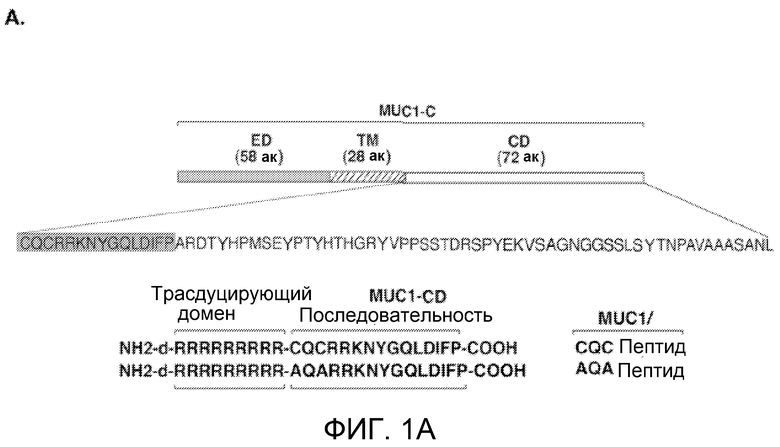
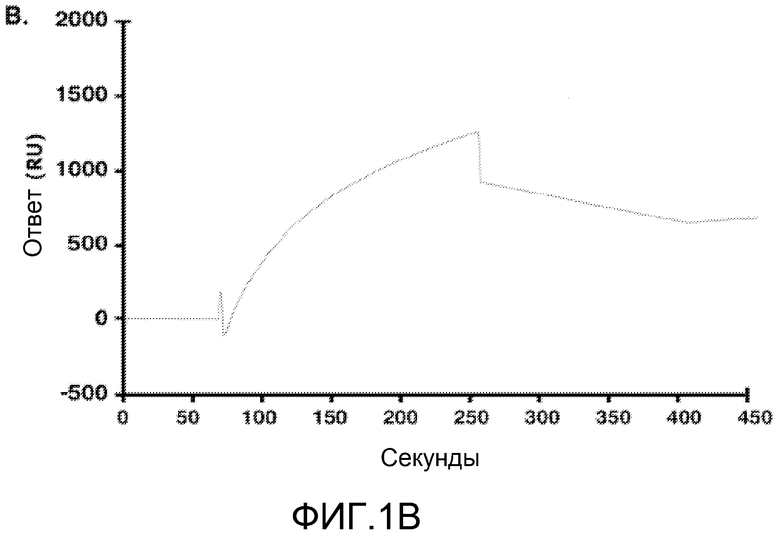
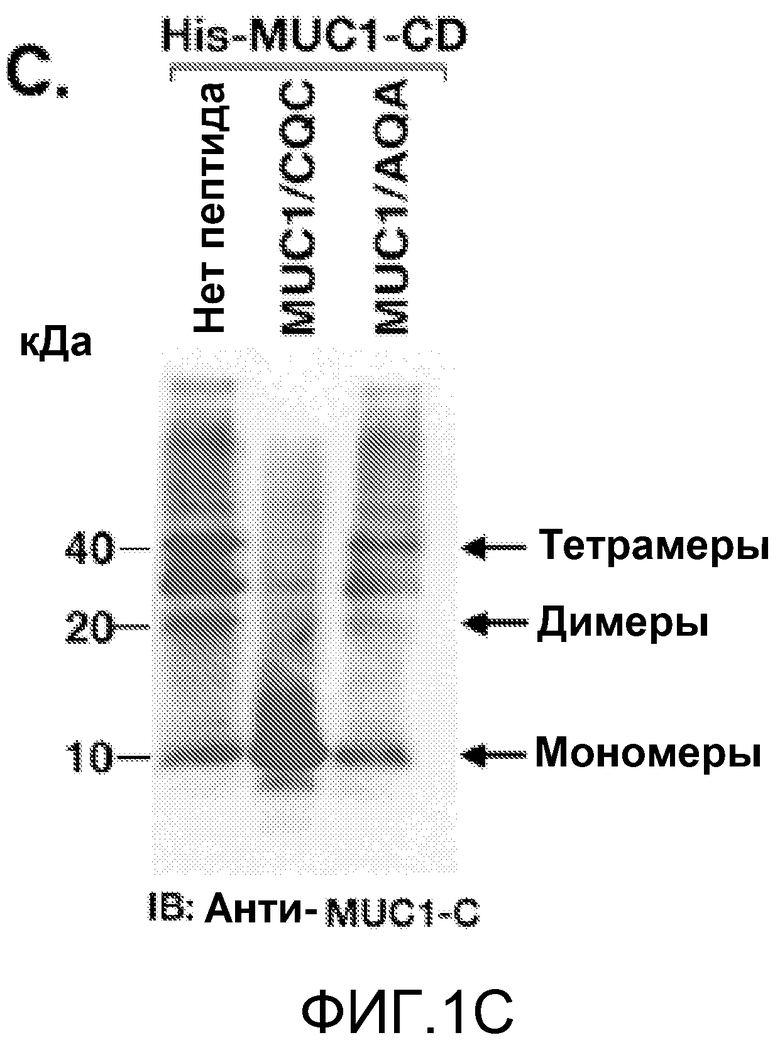
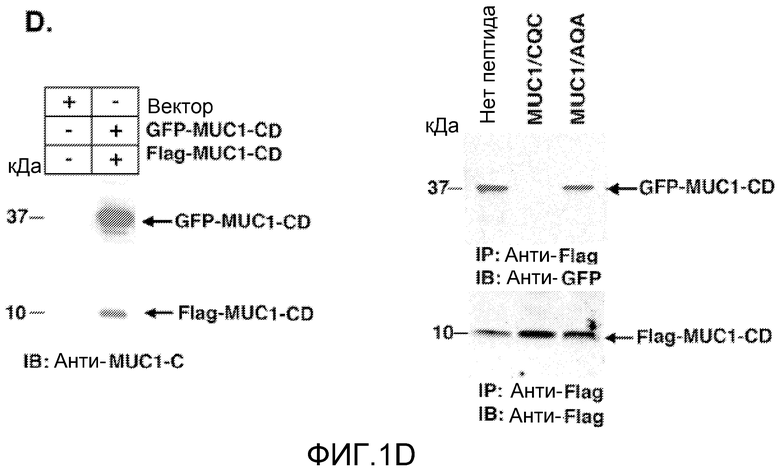
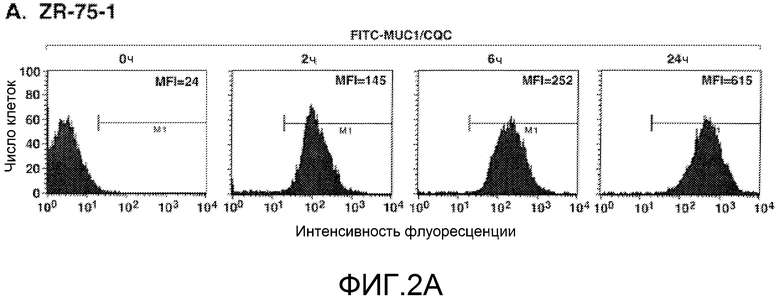
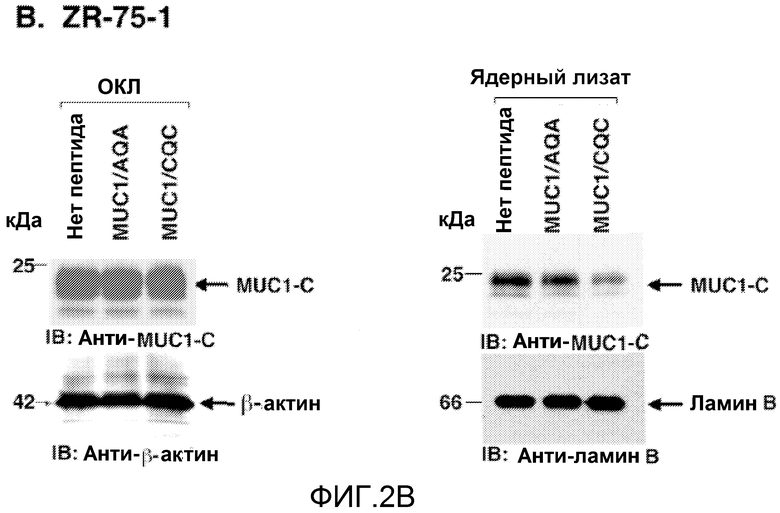
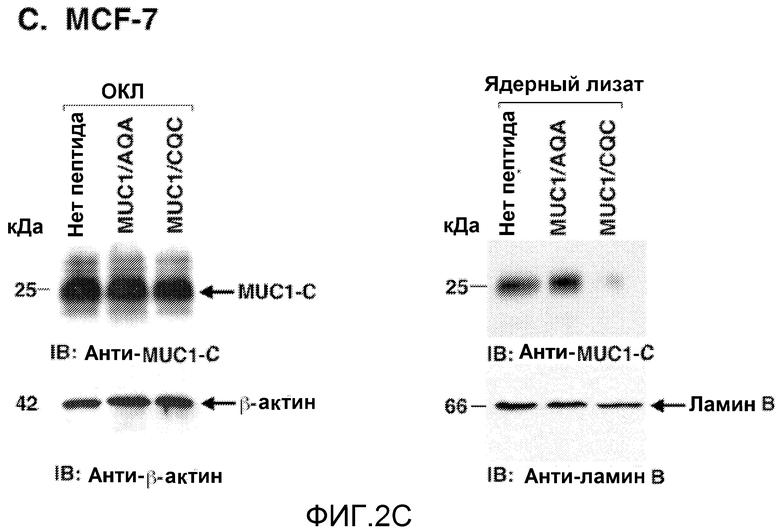
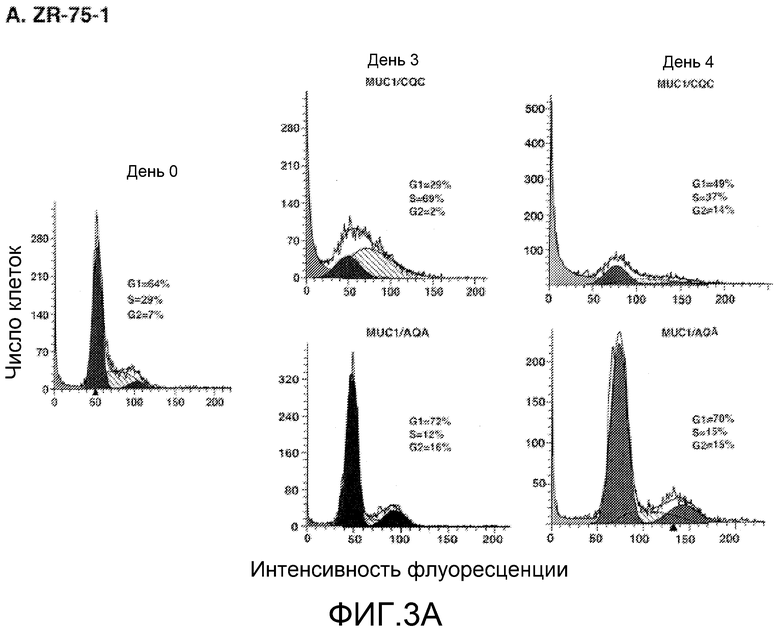
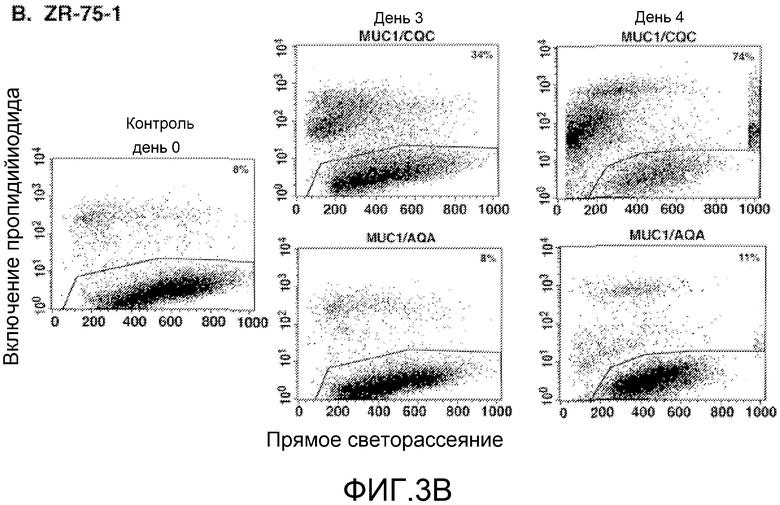
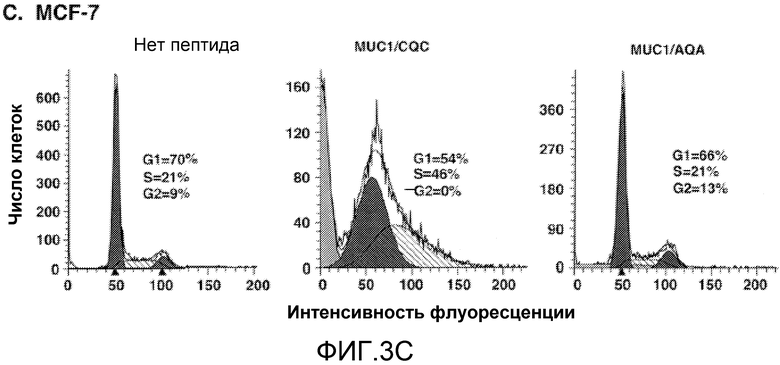
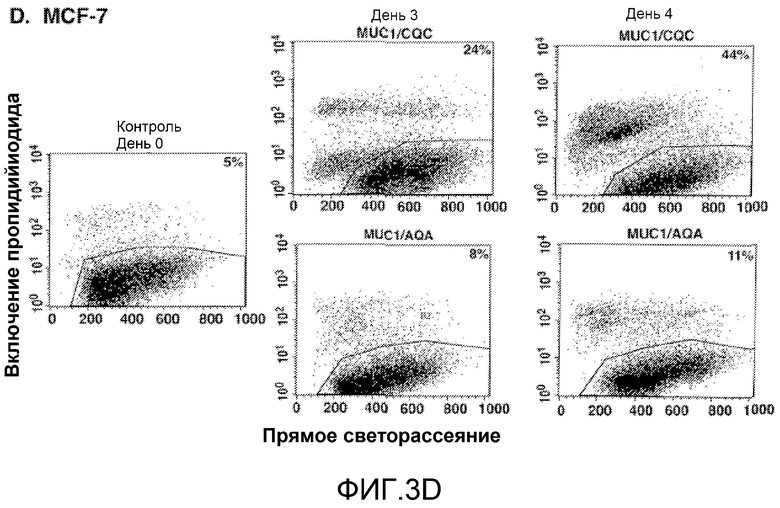
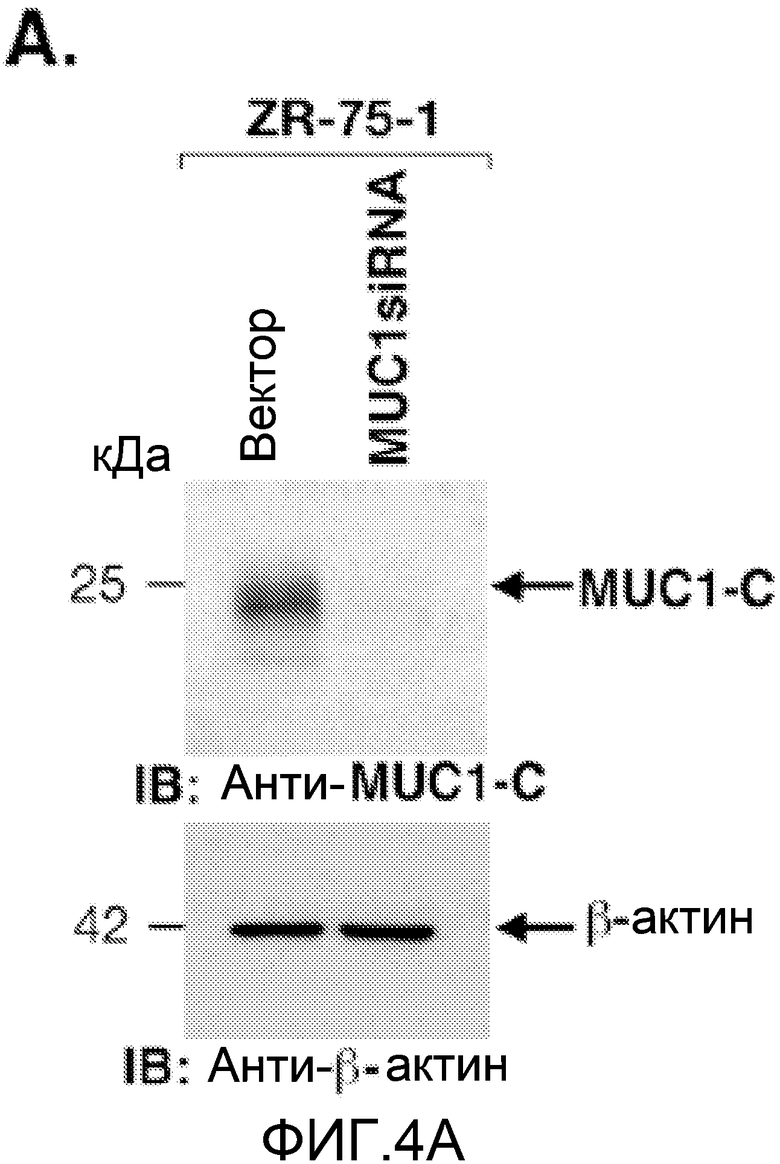
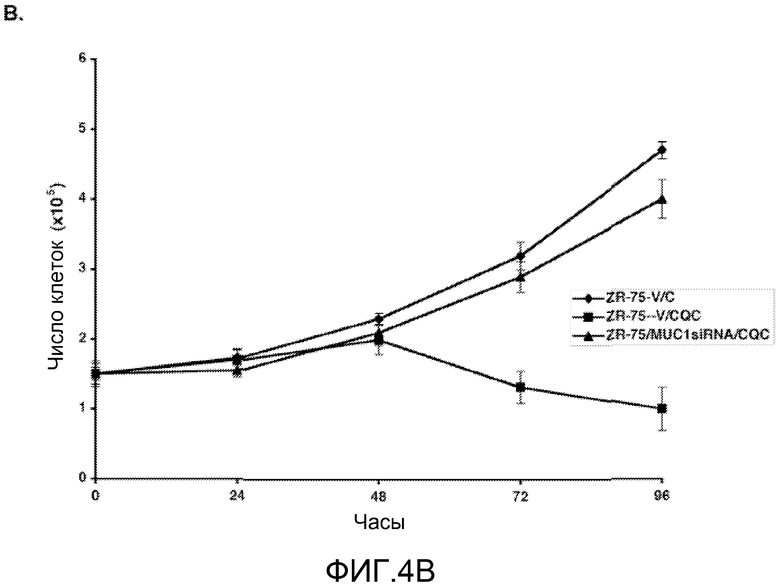
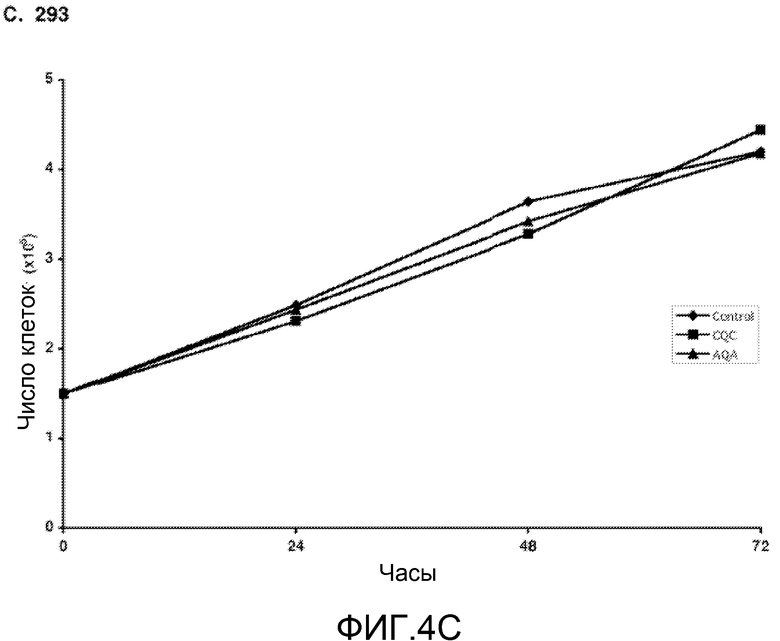
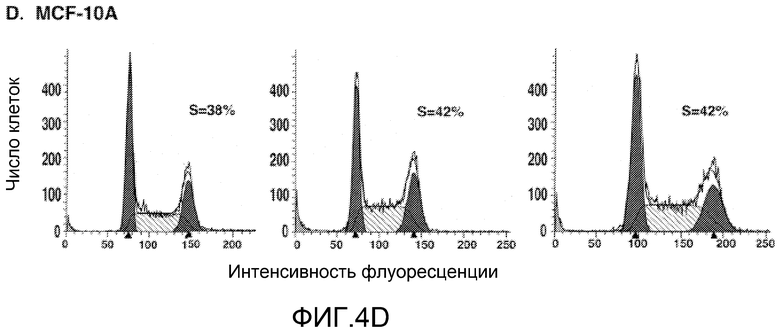
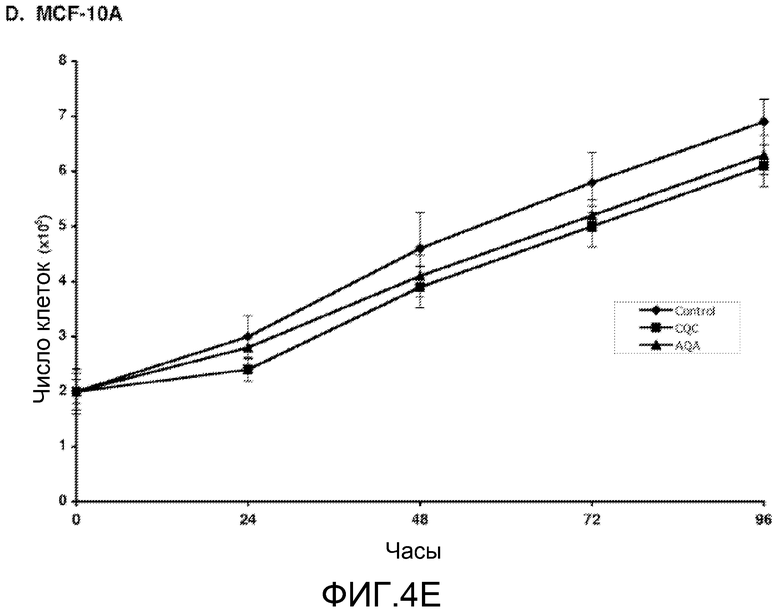
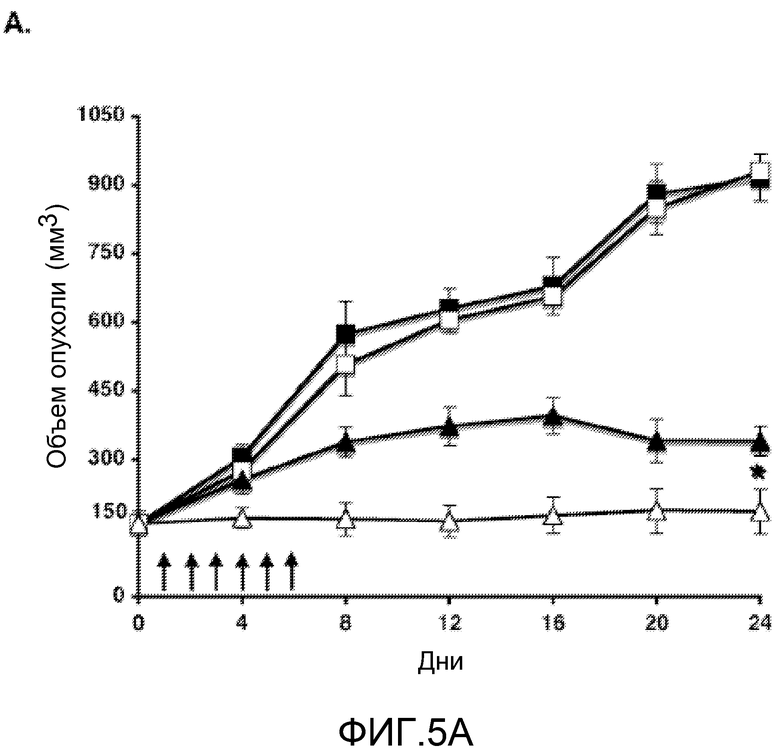
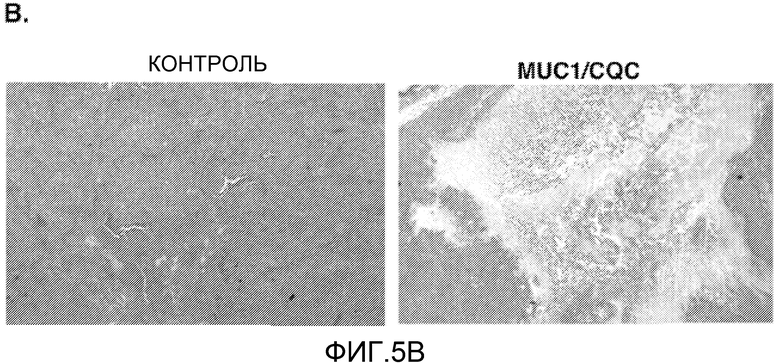
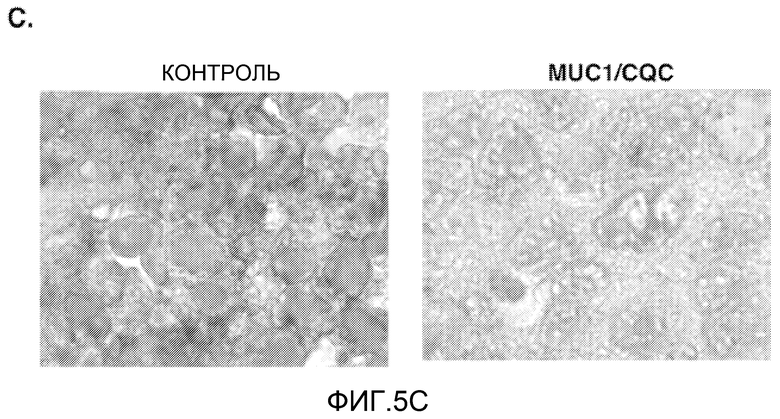
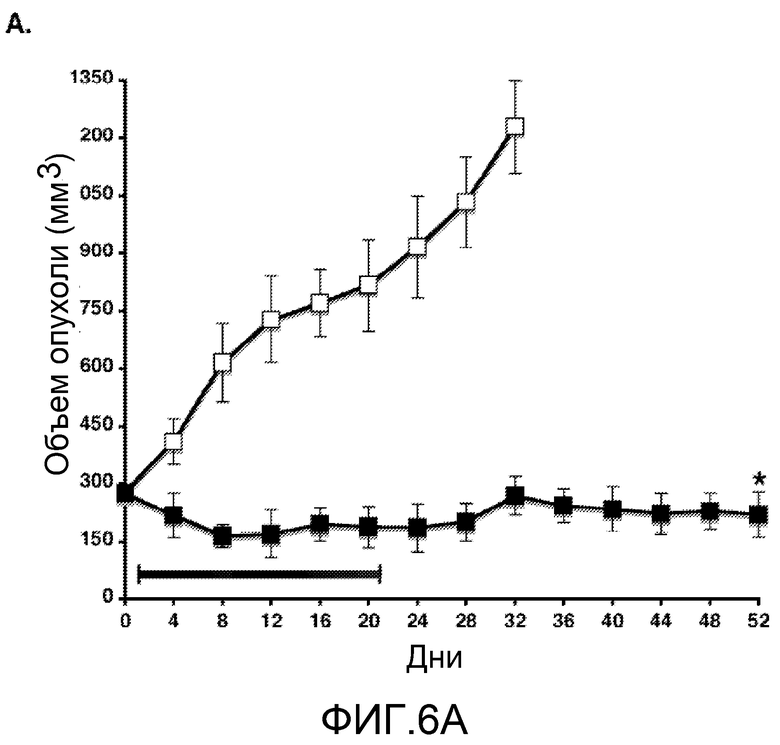
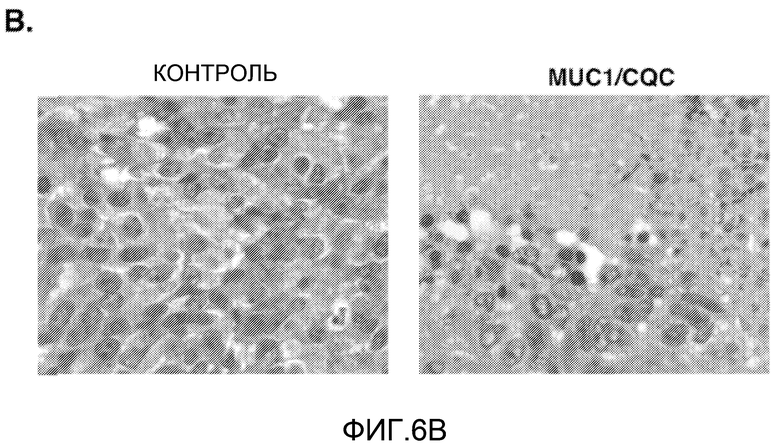
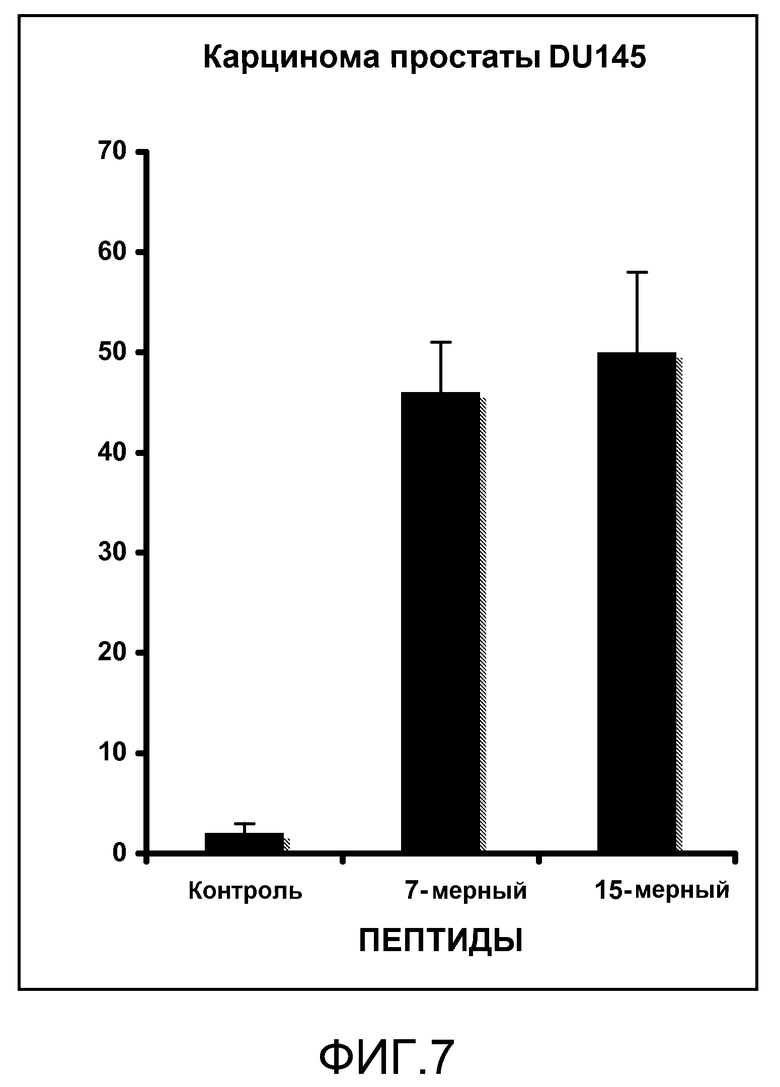

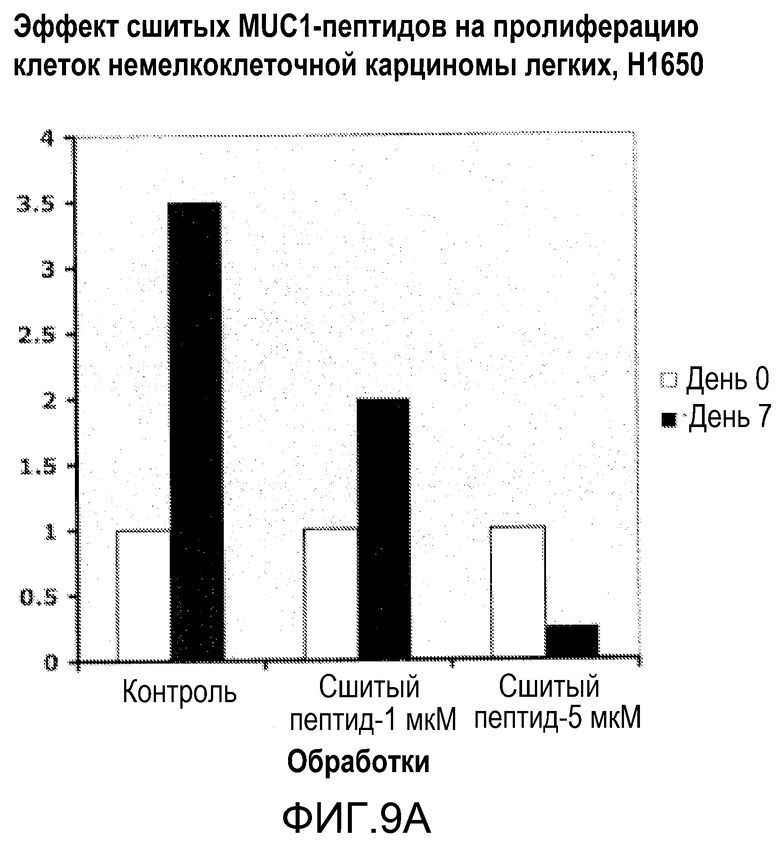
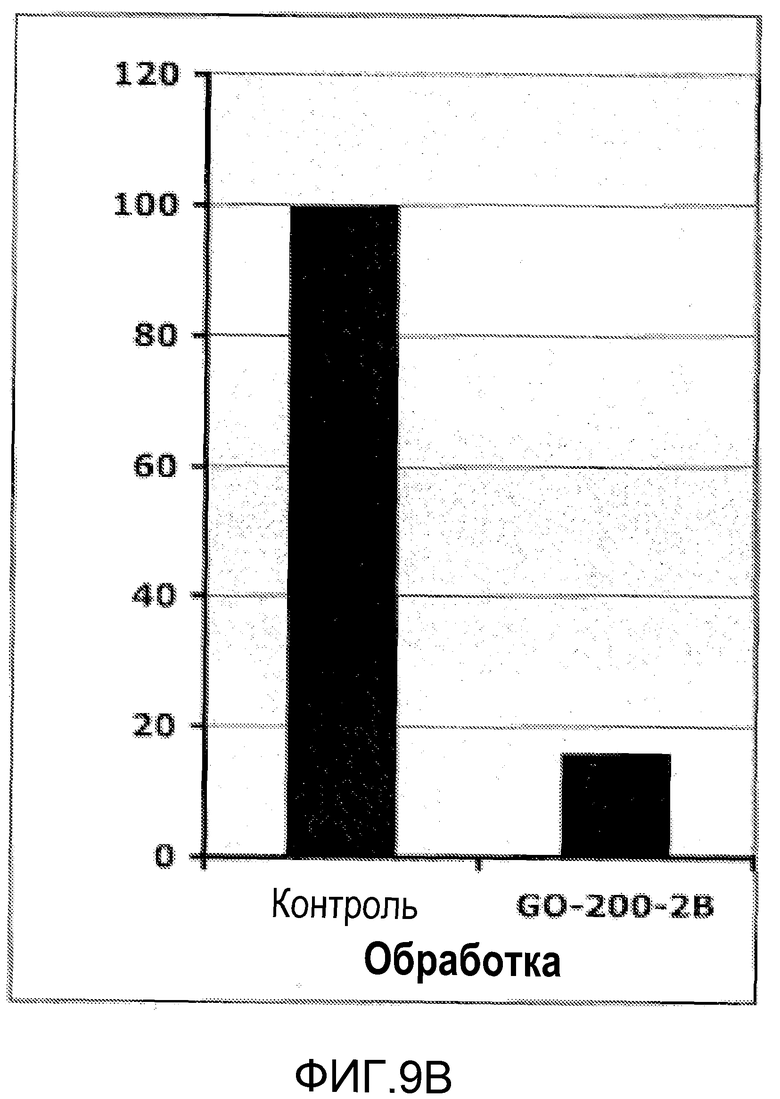
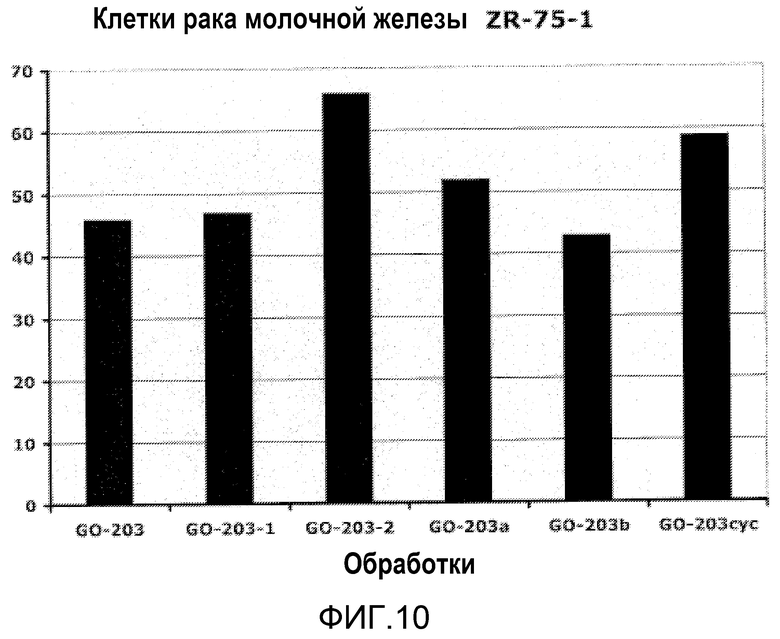
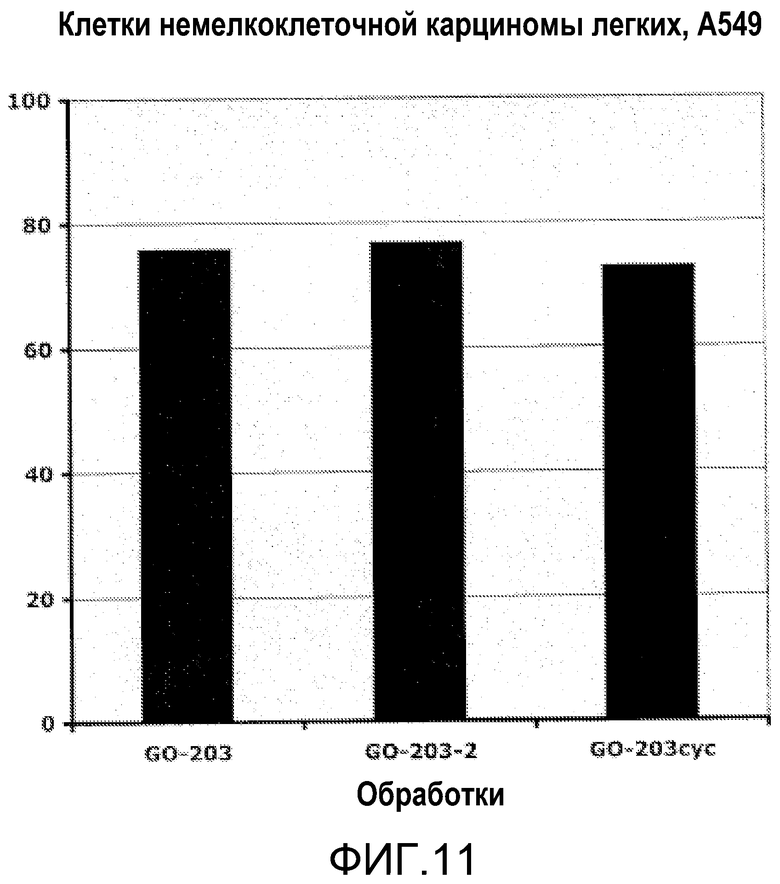
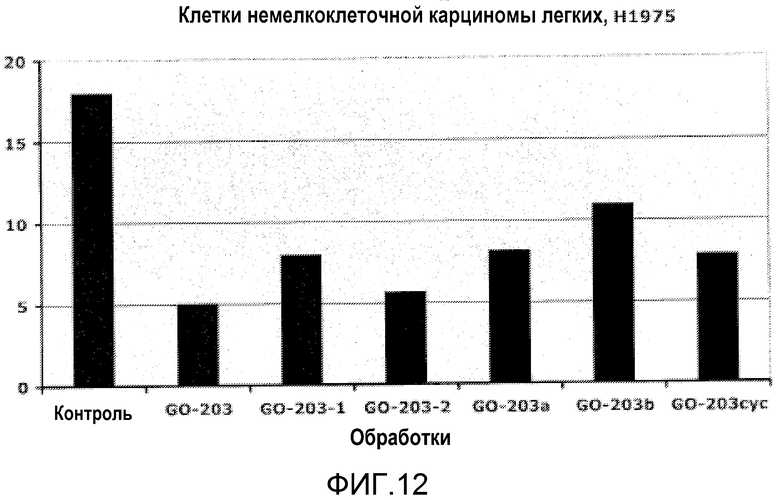
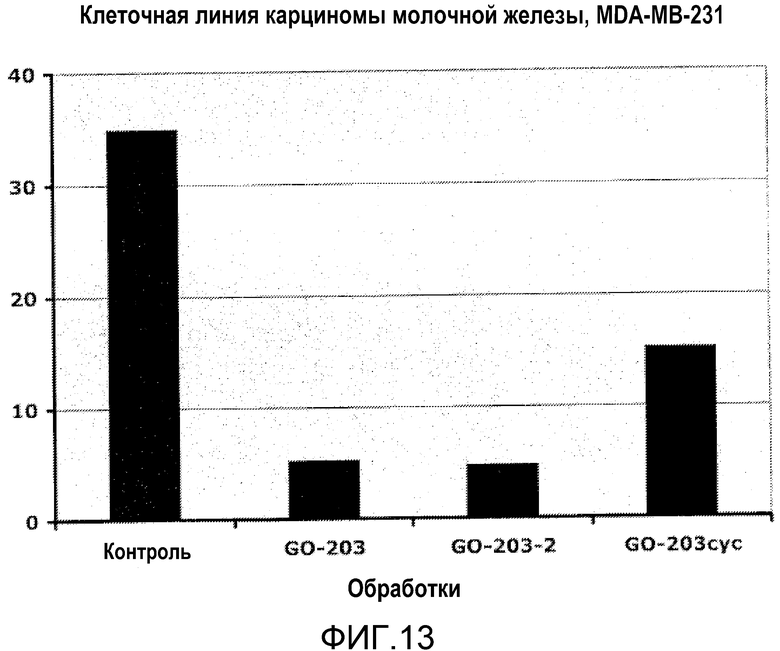
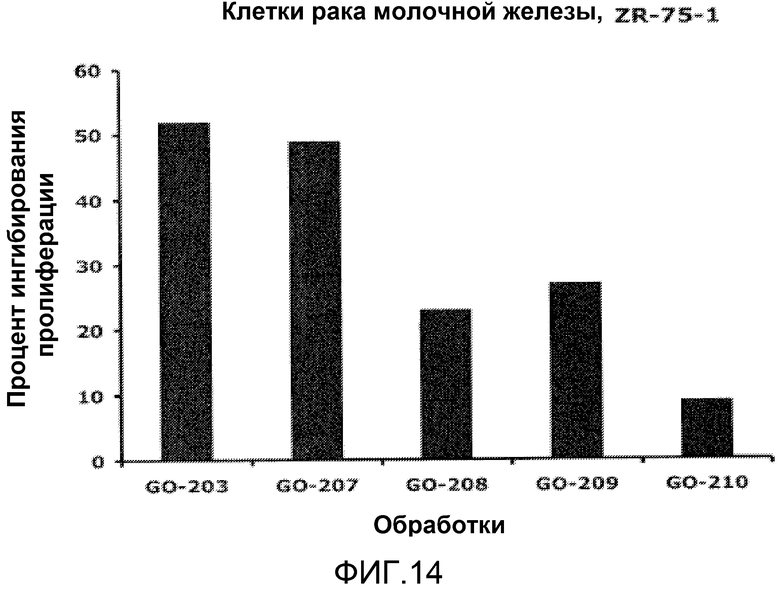
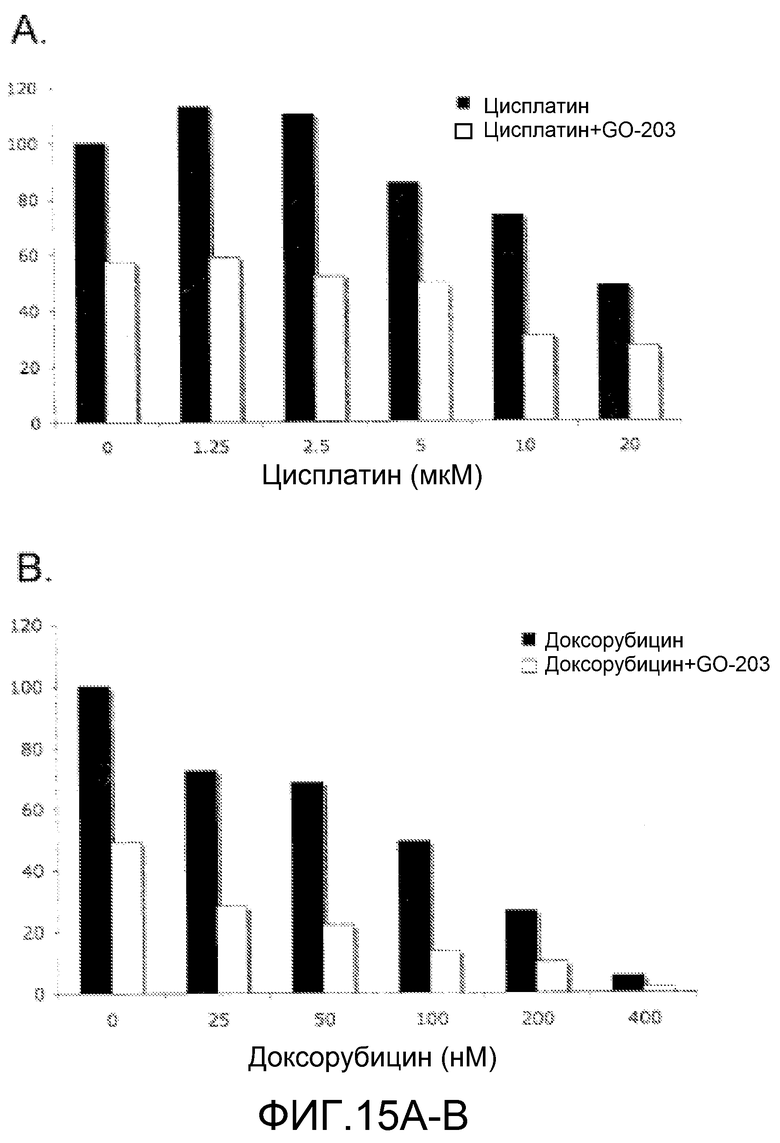
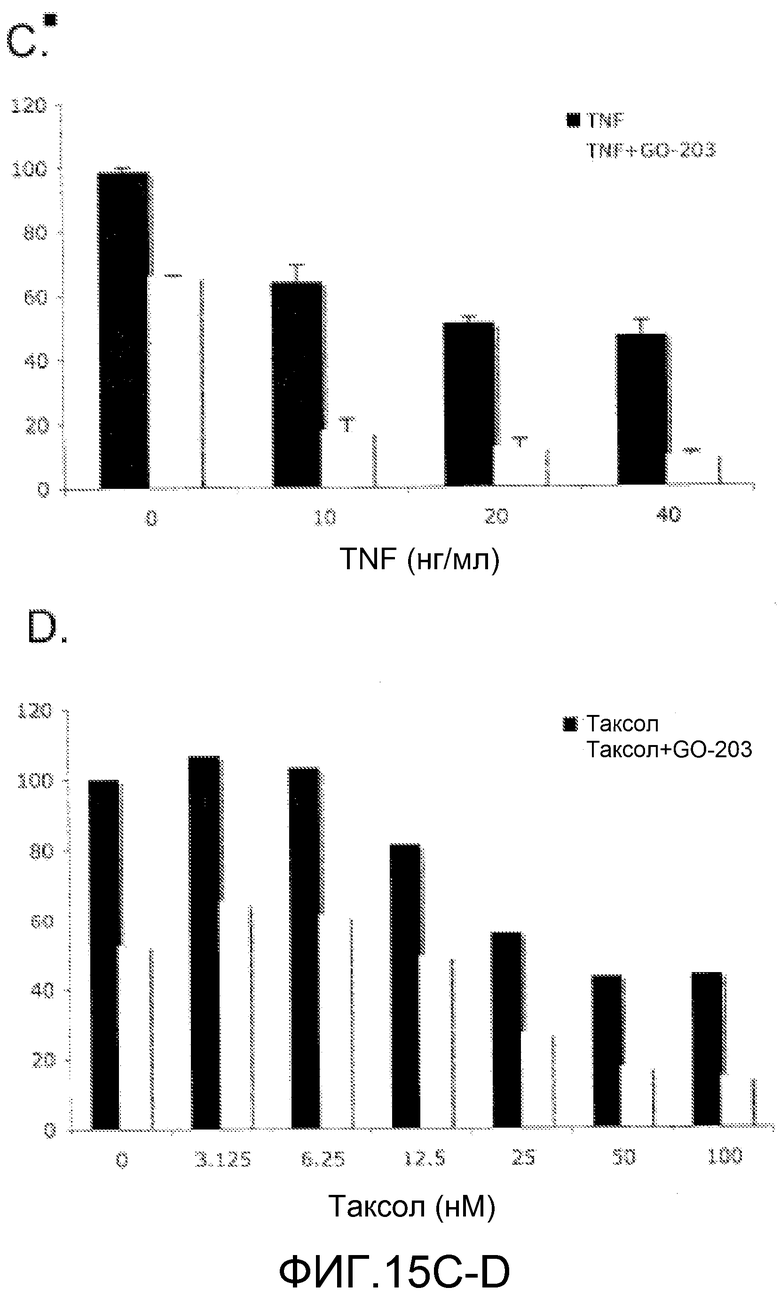
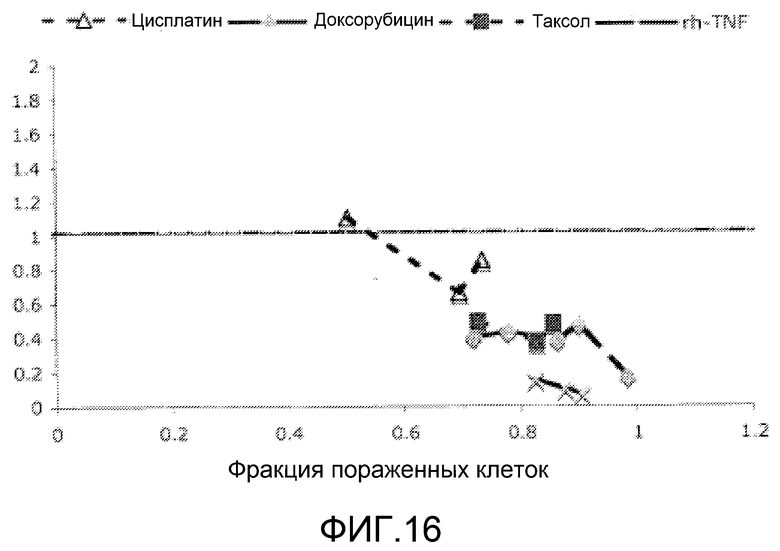
Изобретение относится к области биотехнологии, конкретно к пептидам из цитоплазматического домена MUC1, и может быть использовано в противоопухолевой терапии. Способ ингибирования MUC1-положительной опухолевой клетки у индивидуума включает введение указанному индивидууму MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и, содержащего последовательность CQCRRK, в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1. Альтернативно может быть использован MUC1-пептид длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, который содержит последовательность CQCRRK, причем все аминокислотные остатки указанного пептида являются D-аминокислотами. Изобретение позволяет эффективно ингибировать олигомеризацию MUC1 и индуцировать апоптоз опухолевых клеток и некроз опухолевых тканей in vivo. 11 н. и 69 з.п. ф-лы, 16 ил., 1 табл., 3 пр.
1. Способ ингибирования MUC1-положительной опухолевой клетки у индивидуума, включающий введение указанному индивидууму (а) MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK (SEQ ID NO:4), в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1, или (b) MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK, причем все аминокислотные остатки указанного пептида являются D-аминокислотами.
2. Способ по п.1, где указанный пептид содержит по меньшей мере 7 последовательных остатков MUC1.
3. Способ по п.1, где указанный пептид содержит не более 15 последовательных остатков MUC1.
4. Способ по п.1, где MUC1-положительной опухолевой клеткой является клетка карциномы, клетка лейкемии или клетка миеломы.
5. Способ по п.4, где клеткой карциномы является клетка карциномы простаты или молочной железы.
6. Способ ингибирования MUC1-положительной опухолевой клетки у индивидуума, включающий введение указанному индивидууму (а) MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK (SEQ ID NO:4), в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1, или (b) MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK, причем все аминокислотные остатки указанного пептида являются D-аминокислотами,
где указанный пептид слит с доменом для доставки в клетку.
7. Способ по п.6, где указанным доменом для доставки в клетку является поли-D-R, поли-D-P или поли-D-K.
8. Способ по п.1 или 6, где введение включает внутривенное, внутриартериальное, внутриопухолевое, подкожное, местное или внутрибрюшинное введение.
9. Способ по п.1 или 6, где введение включает местное, зональное, системное или непрерывное введение.
10. Способ по п.1 или 6, где ингибирование включает индукцию торможения роста указанной опухолевой клетки, апоптоз указанной опухолевой клетки и/или некроз опухолевой ткани, содержащей указанную опухолевую клетку.
11. Способ по п.1 или 6, дополнительно включающий проведение для указанного индивидуума второй противораковой терапии.
12. Способ по п.11, где указанной противораковой терапией является хирургическое вмешательство, химиотерапия, радиотерапия, гормональная терапия, терапия токсинами, иммунотерапия и криотерапия.
13. Способ по п.11, где указанную вторую противораковую терапию проводят до введения указанного пептида.
14. Способ по п.11, где указанную противораковую терапию проводят после введения указанного пептида.
15. Способ по п.11, где указанную противораковую терапию проводят одновременно с введением указанного пептида.
16. Способ по п.1 или 6, где указанным индивидуумом является человек.
17. Способ по п.1 или 6, где указанный пептид вводят в количестве 0,1-500 мг/кг/день.
18. Способ по п.1 или 6, где указанный пептид вводят в количестве 10-100 мг/кг/день.
19. Способ по п.1 или 6, где указанный пептид вводят ежедневно.
20. Способ по п.19, где указанный пептид вводят ежедневно в течение 7 дней, 2 недель, 3 недель, 4 недель, одного месяца, 6 недель, 8 недель, двух месяцев, 12 недель или 3 месяцев.
21. Способ по п.1 или 6, где указанный пептид вводят еженедельно.
22. Способ по п.21, где указанный пептид вводят еженедельно в течение 2 недель, 3 недель, 4 недель, 6 недель, 8 недель, 10 недель или 12 недель.
23. Способ по п.1 или 6, где указанный пептид (a) содержит все L-аминокислоты.
24. Способ по п.1 или 6, где указанный пептид (b) содержит все D-аминокислоты.
25. Способ по п.1 или 6, дополнительно включающий стадию оценки экспрессии MUC1 в опухолевой клетке указанного индивидуума до введения указанного пептида.
26. Способ по п.1 или 6, дополнительно включающий стадию оценки эффекта указанного пептида на экспрессию MUC1 в опухолевой клетке указанного индивидуума.
27. Способ по п.1 или 6, где указанный пептид представляет собой пептид GO-201, имеющий аминокислотную последовательность, указанную в SEQ ID NO:3.
28. Фармацевтическая композиция для ингибирования MUC1-положительной опухолевой клетки у индивидуума, содержащая (а) эффективное количество MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK, в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1; и (b) фармацевтически приемлемый носитель, буфер или разбавитель.
29. Композиция по п.28, в которой указанный пептид содержит по меньшей мере 7 последовательных остатков MUC1.
30. Композиция по п.28, в которой указанный пептид содержит не более 15 последовательных остатков MUC1.
31. Фармацевтическая композиция для ингибирования MUC1-положительной опухолевой клетки у индивидуума, содержащая (a) эффективное количество MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK, в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1; и (b) фармацевтически приемлемый носитель, буфер или разбавитель, в которой указанный пептид MUC1 слит с доменом для доставки в клетку или доменом для трансдукции клетки.
32. Композиция по п.31, в которой указанным доменом для доставки в клетку является поли-D-R, поли-D-P или поли-D-K.
33. Композиция по п.28, где указанный пептид представляет собой пептид GO-201, имеющий аминокислотную последовательность, указанную в SEQ ID NO:3.
34. Способ ингибирования олигомеризации и ядерного транспорта MUC1 в раковой клетке, включающий контактирование клетки, экспрессирующей MUC1, с MUC1-пептидом длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащим последовательность CQCRRK, в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1.
35. Способ по п.34, где указанный пептид содержит по меньшей мере 7 последовательных остатков MUC1.
36. Способ по п.34, где указанный пептид содержит не более 15 последовательных остатков MUC1.
37. Способ по п.34, где экспрессирующей MUC1 клеткой является опухолевая клетка.
38. Способ по п.34, где MUC1-положительной опухолевой клеткой является клетка карциномы, клетка лейкемии или клетка миеломы.
39. Способ по п.38, где клеткой карциномы является клетка карциномы простаты или молочной железы.
40. Способ по п.37, где опухолевая клетка находится в живом индивидууме.
41. Способ по п.40, где указанным живым индивидуумом является человек.
42. Способ по п.34, где указанный пептид представляет собой пептид GO-201, имеющий аминокислотную последовательность, указанную в SEQ ID NO:3.
43. MUC1-пептид длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащий последовательность CQCRRK, причем все аминокислотные остатки указанного пептида являются D-аминокислотами, где указанный пептид ингибирует MUC1-положительные опухолевые клетки у индивидуума.
44. Пептид по п. 43, где указанный пептид представляет собой пептид GO-201, имеющий аминокислотную последовательность, указанную в SEQ ID NO:3.
45. Применение MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK (SEQ ID NO:4), в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1, для ингибирования MUC1-положительной опухолевой клетки у индивидуума, путем введения указанному индивидууму указанного пептида.
46. Применение по п.45, где указанный пептид содержит по меньшей мере 7 последовательных остатков MUC1.
47. Применение по п.45, где указанный пептид содержит не более 15 последовательных остатков MUC1.
48. Применение по п.45, где MUC1-положительной опухолевой клеткой является клетка карциномы, клетка лейкемии или клетка миеломы.
49. Применение по п.48, где клеткой карциномы является клетка карциномы простаты или молочной железы.
50. Применение MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK (SEQ ID NO:4), в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1, для ингибирования MUC1-положительной опухолевой клетки у индивидуума, путем введения указанному индивидууму указанного пептида,
где указанный пептид слит с доменом для доставки в клетку.
51. Применение по п.50, где указанным доменом для доставки в клетку является поли-D-R, поли-D-P или поли-D-K.
52. Применение по п.45 или 50, где введение включает внутривенное, внутриартериальное, внутриопухолевое, подкожное, местное или внутрибрюшинное введение.
53. Применение по п.45 или 50, где введение включает местное, зональное, системное или непрерывное введение.
54. Применение по п.45 или 50, где ингибирование включает индукцию торможения роста указанной опухолевой клетки, апоптоз указанной опухолевой клетки и/или некроз опухолевой ткани, содержащей указанную опухолевую клетку.
55. Применение MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK (SEQ ID NO:4), в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1, для ингибирования MUC1-положительной опухолевой клетки у индивидуума, путем введения указанному индивидууму указанного пептида, дополнительно включающее проведение для указанного индивидуума второй противораковой терапии.
56. Применение по п.55, где указанной противораковой терапией является хирургическое вмешательство, химиотерапия, радиотерапия, гормональная терапия, терапия токсинами, иммунотерапия и криотерапия.
57. Применение по п.55, где указанную вторую противораковую терапию проводят до введения указанного пептида.
58. Применение по п.55, где указанную противораковую терапию проводят после введения указанного пептида.
59. Применение по п.55, где указанную противораковую терапию проводят одновременно с введением указанного пептида.
60. Применение по п.45 или 50, где указанным индивидуумом является человек.
61. Применение по п.45 или 50, где указанный пептид вводят в количестве 0,1-500 мг/кг/день.
62. Применение по п.45 или 50, где указанный пептид вводят в количестве 10-100 мг/кг/день.
63. Применение по п.45 или 50, где указанный пептид вводят ежедневно.
64. Применение по п.63, где указанный пептид вводят ежедневно в течение 7 дней, 2 недель, 3 недель, 4 недель, одного месяца, 6 недель, 8 недель, двух месяцев, 12 недель или 3 месяцев.
65. Применение по п.45 или 50, где указанный пептид вводят еженедельно.
66. Применение по п. 65, где указанный пептид вводят еженедельно в течение 2 недель, 3 недель, 4 недель, 6 недель, 8 недель, 10 недель или 12 недель.
67. Применение по п.45 или 50, где указанный пептид содержит все L-аминокислоты.
68. Применение по п.45 или 50, где указанный пептид содержит все D-аминокислоты.
69. Применение по п.45 или 50, где указанный пептид содержит смесь L- и D-аминокислот.
70. Применение MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK (SEQ ID NO:4), в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1, для ингибирования MUC1-положительной опухолевой клетки у индивидуума, путем введения указанному индивидууму указанного пептида,
дополнительно включающий стадию оценки экспрессии MUC1 в опухолевой клетке указанного индивидуума до введения указанного пептида, или стадию оценки эффекта указанного пептида на экспрессию MUC1 в опухолевой клетке указанного индивидуума.
71. Применение по п.45 или 50, где указанный пептид представляет собой пептид GO-201, имеющий аминокислотную последовательность, указанную в SEQ ID NO:3.
72. Применение MUC1-пептида длиной по меньшей мере 6 последовательных остатков MUC1 и не более 20 последовательных остатков MUC1, и содержащего последовательность CQCRRK, в которой аминоконцевой цистеин из CQCRRK закрыт на своем NH2-конце по меньшей мере одним аминокислотным остатком, который не должен соответствовать нативной трансмембранной последовательности MUC-1 для ингибирования олигомеризации и ядерного транспорта MUC1 в раковой клетке, путем контактирования клетки, экспрессирующей MUC1, с указанным пептидом.
73. Применение по п.72, где указанный пептид содержит по меньшей мере 7 последовательных остатков MUC1.
74. Применение по п.72, где указанный пептид содержит не более 15 последовательных остатков MUC1.
75. Применение по п.72, где экспрессирующей MUC1 клеткой является опухолевая клетка.
76. Применение по п.72, где MUC1-положительной опухолевой клеткой является клетка карциномы, клетка лейкемии или клетка миеломы.
77. Применение по п.76, где клеткой карциномы является клетка карциномы простаты или молочной железы.
78. Применение по п.75, где опухолевая клетка находится в живом индивидууме.
79. Применение по п.78, где указанным живым индивидуумом является человек.
80. Применение по п.72, где указанный пептид представляет собой пептид GO-201, имеющий аминокислотную последовательность, указанную в SEQ ID NO:3.

