Изобретение относится к области противоопухолевой биомолекулярной фармакологии, в частности к векторному полипептиду, его противоопухолевому конъюгату и фармацевтической композиции на основе этого конъюгата.
Низкая избирательность действия, а также первичная и приобретенная в процессе лечения резистентность опухолевых клеток к химиопрепаратам являются одними из главных причин, существенно ограничивающих эффективность противоопухолевой химиотерапии. Повышение селективности химиопрепаратов может быть достигнуто посредством их направленного транспорта к клеткам-мишеням с помощью различных белковых векторов. При этом химиопрепараты попадают внутрь клетки в результате рецептор-опосредованного эндоцитоза конъюгата, представляющего собой ковалентно связанные векторный белок и химиопрепарат. Избирательность действия конъюгатов достигается либо за счет наличия на поверхности опухолевых клеток специфических рецепторов, «узнаваемых» векторным белком или антителом, либо за счет значительно более высокого уровня рецепторов векторного белка на поверхности опухолевых клеток по сравнению с нормальными. В ряде работ опубликованы данные об успешном применении цитотоксических конъюгатов, созданных на основе цитотоксических антибиотиков и векторных молекул, осуществляющих направленную доставку антибиотика в опухолевые клетки или эндотелиальные клетки сосудов опухоли. В качестве векторов активно используют онкофетальные белки, трансферрин, моноклональные антитела к специфическим опухолевым антигенам, гормоноподобные пептиды, а также циклические пептиды, содержащие RGD-последовательность, избирательно связывающиеся с αvβ3-интегринами эндотелия опухоли [1-4].
В последние годы усилия ряда научных коллективов направлены на разработку препаратов, мишенью которых является рецептор эпидермального фактора роста (РЭФР, ErbB1). РЭФР играет важную роль не только в регуляции пролиферативных процессов нормальных эпителиальных клеток, но и в индукции многих опухолевых заболеваний (рак молочной железы, желудка, предстательной железы и др.) [5, 6]. Одним из путей ухода опухолевых клеток из-под рост-регулирующего контроля организма может служить гиперпродукция этими или окружающими стромальными клетками эпидермального фактора роста (ЭФР), что при наличии на поверхности опухолевых клеток РЭФР обеспечивает возможность осуществления ауто- или паракринной регуляции клеточной пролиферации.
В связи с этим вмешательство в процессы восприятия и рецептор-опосредованной передачи рост-стимулирующего сигнала может оказаться перспективным подходом к терапии широкого спектра опухолей.
Существует несколько возможных вариантов воздействия на РЭФР и/или регулируемые им клеточные процессы при разработке новых подходов к противоопухолевой терапии, среди которых одно из первых мест занимает применение цитотоксических препаратов, направленных к РЭФР, таких как гибридные белки, содержащие комбинации векторов-лигандов РЭФР и бактериальных токсинов, а также химические конъюгаты на основе факторов роста и цитотоксических агентов [7]. В данном случае система лиганд-рецептор служит направленным переносчиком цитотоксического агента в клетку. Подобные препараты показывают высокую эффективность в предварительных экспериментах [6]. Мишенью для воздействия подобных препаратов является довольно широкий спектр опухолей, характеризующихся высокой экспрессией соответствующих рецепторов. Так, наиболее часто гиперэкспрессия РЭФР обнаруживается при раке легкого (89%) и пищевода (63%), а также раке яичников, тела матки, молочной железы, саркоме костей и др.
Трансформирующий фактор роста α (ТФРα) - белок семейства лигандов РЭФР - был впервые очищен из культуральной среды человеческой меланомы. ТФРα обнаруживается в организме в виде нескольких растворимых и трансмембранных форм. Основная секретируемая форма представлена небольшим термостабильным белком, состоящим из 50 аминокислот, с молекулярной массой 6 кДа. Зрелый ТФРα характеризуется 40%-ной структурной гомологией с ЭФР и представляет собой кислый негликозилированный белок, стабилизированный тремя дисульфидными связями.
В настоящее время считается, что ТФРα является эмбриональным эквивалентом ЭФР, а во взрослом организме представляет собой неопластическую форму эпидермального фактора роста. Главной мишенью действия основной зрелой формы ТФРα является рецептор ЭФР, активация которого вызывает митогенный эффект. ТФРα является сильным стимулятором пролиферации, играющим важную роль в физиологической регуляции нормального роста и дифференцировки клеток как в процессе эмбрионального развития, так и во взрослом организме. ТФРα играет также важную роль в злокачественной трансформации клеток различных типов. Коэкспрессия ТФРα и рецептора ЭФР в клетках карцином (легких, желудка, простаты и др.) является важным условием обеспечения паракринной или аутокринной стимуляции опухолевого роста.
Как и ЭФР, ТФРα является ангиогенным фактором, стимулирующим развитие капиллярной сети, однако ангиогенное действие ТФРα значительно более выражено [8]. Помимо непосредственного действия на эндотелиальные клетки через рецептор ЭФР оба фактора роста обладают способностью к стимуляции экспрессии ангиогенных факторов неэндотелиальными клетками, такими как фибробласты. Поскольку ангиогенез является не только ключевым физиологическим процессом, но и процессом, вовлеченным в прогрессию злокачественных новообразований и другие патологии, роль ТФРα в этих процессах представляется чрезвычайно важной.
Известно использование для химиотерапии рака смесей (композиций) противоопухолевых цитотоксических препаратов различных классов как с целым ТФРα, так и его пептидными фрагментами различной длины [пат. США №4863902]. Усиление противоопухолевого действия таких препаратов объясняется более высокой чувствительностью опухолевых клеток к действию цитотоксического агента на фоне стимуляции клеточной пролиферации, опосредованной фактором роста или его активными фрагментами. Однако нековалентная иммобилизация противоопухолевого агента с векторными пептидами не гарантирует его целенаправленную доставку к опухолевым клеткам-мишеням.
Существует также ряд патентов, посвященных различным конъюгатам на основе ТФРα с различными противоопухолевыми агентами и способам их получения [пат. США №5622929, 1997 г.; №5606017, 1997 г.]. Однако в данных изобретениях не рассматривается использование селективных пептидных фрагментов ТФРα в качестве векторной составляющей направленного препарата.
Предметом изобретения патента США №5969099, 1999 г. являются два коротких пептидных фрагмента, состоящих из 12 и 13 аминокислот, соответствующие последовательности рецептор-связывающего фрагмента молекулы зрелого ТФРα и проявляющие биологическую активность, выражающуюся в стимуляции клеточной пролиферации. Однако в каждом пептиде присутствуют по два остатка цистеина, вследствие чего оба они имеют замкнутую структуру, и возможность химического присоединения к ним молекулы цитотоксического агента с целью получения направленного противоопухолевого препарата весьма ограничена.
Известен также патент РФ №2236251, сент., 2004 г., в котором ТФРα использован в полном объеме как цитокиновый компонент.
Близким аналогом заявленного изобретения является полипептид - аналог фрагмента эпидермального фактора роста (ЭФР), способный связываться с рецептором ЭФР и вызывать пролиферацию клеток, конъюгат его с доксорубицином и фармкомпозиция на его основе, отличающиеся от предыдущих изменением аминокислотной последовательности векторной части препарата с целью увеличения способности его связывания с рецептором на опухолевых клетках (пат. РФ №2196604, 2003 г.).
Технической задачей настоящего изобретения является расширение арсенала препаратов на основе векторных полипептидов, предназначенных для направления непосредственно в опухолевые клетки и способных химически связывать и доставлять в опухоли широкий спектр цитотоксических средств с избирательным противоопухолевым действием.
Технический результат достигается за счет создания векторного декапептида, являющегося аналогом фрагмента трансформирующего фактора роста α (ТФРα) с 22 по 31 аминокислоту, способного эффективно связываться с рецепторами опухолевых клеток, выполнять функцию вектора для селективной доставки цитотоксических агентов в опухолевые клетки. Декапептид имеет следующую структуру:

В отличие от нативного фрагмента человеческого ТФРα, в патентуемом декапептиде 8-й аминокислотный остаток Lys заменен на Ser. Такая замена (Lys на Ser) позволяет предотвратить нежелательное конъюгирование по ε-аминогруппам лизина в активном центре рецептор-связывающего фрагмента, что в свою очередь позволяет избежать ингибирования связывания декапептида с рецептором опухолевой клетки.
Другим объектом изобретения является конъюгат декапептида с цитотоксическим агентом, обладающий избирательным действием в отношении злокачественных опухолей и способный значительно снижать резистентность раковых клеток к противоопухолевым препаратам, в котором обе части связаны кислотолабильной химической связью, в которой задействованы NH2-группы цитотоксического компонента и первого аминокислотного остатка (Arg) векторного декапептида. В качестве сшивающего агента по аминогруппам полипептида и цитотоксического агента может быть использован глутаровый альдегид, а также другие бифункциональные реагенты.
В качестве цитотоксического агента могут быть использованы препараты, обладающие противоопухолевой активностью и несущие свободные аминогруппы, например дауномицин, карминомицин, мелфалан, метотрексат, цитарабин, доксорубицин, а также цитотоксические препараты белковой природы, такие как токсины (дифтерийный или рицин) и др.
Предметом настоящего изобретения является также фармацевтическая композиция, обладающая противоопухолевым действием, на основе конъюгата декапептида с цитотоксическим агентом в эффективном количестве (0.05-0.1%) и приемлемых носителей для внутривенного введения.
При этом носителем может быть физиологический раствор на воде для инъекций, фосфатно-солевой раствор на воде для инъекций, водные растворы обезболивающих препаратов.
Композиция также может содержать дополнительно противомикробные, противовирусные и противопаразитарные средства, а также другие вспомогательные вещества и препараты, применяемые для лечения онкологических больных, в количестве не более 50% (вес.) от общего веса композиции.
Ниже приведены примеры, иллюстрирующие предлагаемое изобретение и доказывающие возможность использования полипептида, его конъюгатов с цитотоксическими агентами и композиций на их основе по указанному назначению.
Пример 1. Получение декапептида
Пептид был получен методом твердофазной пептидной химии с использованием Fmoc стратегии синтеза. В качестве полимерного носителя была выбрана смола Wang-resin. На каждом шаге пептидной конденсации карбоксильную группу Fmoc-аминокислот активировали превращением в гидроксибензотриазоловый эфир с помощью HoBt и N.N-диизопропилкарбодиимида. Fmoc-аминокислоты, HoBt и N.N-диизопропилкарбодиимид вводили в реакцию в 10-кратных молярных избытках по отношению к аминогруппам полимера. За ходом реакции следили по изменению окраски бромфенолового синего, который добавляли в реактор в виде раствора в диметилсульфоксиде (ДМСО) в соотношении 1/1000 (мол.) к количеству аминогрупп. Реакция длилась 1,5-2 час. После завершения конденсации полученную смолу промывали 4 раза диметилформамидом (ДМФ) и обрабатывали 50%-ным раствором пиперидина в ДМФ. Отщепление полипептида от полимерного носителя проводили смесью трифторуксусной кислоты 95%; воды 2.5%; 1,2-этандитиола - 2.5%. После того, как смола была отфильтрована, пептид высаживали из раствора диэтиловым эфиром. Осадок отделяли центрифугированием, высушивали в вакууме и анализировали методом высокоэффективной жидкостной хроматографии (ВЭЖХ) на колонке Phenomenex Luna C18 250×3. Градиентное элюирование полипептида с колонки проводили в системе растворителей: ацетонитрил/0.1% трифторуксусная кислота/вода. Полупрепаративная ОФ ВЭЖХ проводилась в тех же условиях на колонке Nucleosil С18 250×8. Чистота полипептида после хроматографического разделения составила >96%.
Полипептид был охарактеризован методом масс-спектрометрии (молекулярный ион - 1265). Структура полипептида приведена в табл.1.

Пример 2. Биологическая активность декапептида
Определение пролиферативной активности пептида проводили на клетках фибробластов мыши линии NIH 3T3. Интенсивность биосинтеза ДНК определяли по включению 3Н-тимидина в кислоторастворимую фракцию клеток.
Клетки линии фибробластов мыши линии NIH 3T3 культивировали в пластиковых флаконах (Costar) в среде RPMI (Sigma), содержащей 10% эмбриональной бычьей сыворотки (Sigma), 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина, в увлажненной атмосфере 5% CO2 при 37°С. Пролиферацию синхронизированных фибробластов линии NIH 3T3 индуцировали, добавляя к суспензии клеток полипептид или ЭФР в различных концентрациях (0.1-1000 нг/мл). Фибробласты культивировали в 96-луночных планшетах в 200 мкл среды по 200-800 тысяч/лунку в полной среде, 3Н-тимидин (40 мКи/моль) вносили за 2 ч до окончания инкубации. Радиоактивность клеток измеряли на жидкостном сцинтилляционном счетчике (Rackbeta, LKB) с использованием сцинтилляционной жидкости ЖС-8 на основе толуола. Интенсивность биосинтеза ДНК выражали в виде индекса стимуляции (отношение стимулированных клеток к контрольным) по формуле I=N/No×100%, где I - индекс стимуляции, N - число стимулированных ЭФР клеток, No - число контрольных клеток.
На фиг.1 представлены сравнительные данные о влиянии предлагаемого декапептида и нативного человеческого ЭФР на пролиферацию клеток фибробластов мыши линии NIH 3Т3. Уровень стимуляции пролиферации фибробластов декапептидом сравним с действием ЭФР. Полученный результат свидетельствует о выраженной биологической активности и высокой способности к регуляции процесса пролиферации исследуемого пептида.
Пример 3. Получение конъюгата декапептида с карминомицином (КМ)
Полипептид растворяли в 5 мл 0.05 М фосфатного буфера (рН 7) (концентрация 1 мг/мл) и добавляли карминомицин, предварительно растворенный в 5 мл дистиллированной воды (рН 5.5) (концентрация 1.2 мг/мл). Синтез конъюгата осуществляли по схеме 1. К реакционной смеси при перемешивании добавляли по каплям глутаровый альдегид (ГА) в 0.05 М фосфатном буфере (рН 7.2) (концентрация ГА 0.5 мг/мл). Объем добавленного раствора ГА составил 3 мл. После добавления ГА смесь инкубировали в течение 1 часа при 50°С. Синтезированный конъюгат отделяли от исходных веществ на колонке PD-10 Sephadex G-25 (Pharmacia, Sweden). Конъюгат анализировали методом обращеннофазовой ВЭЖХ (ОФ ВЭЖХ) на колонке Nucleosil С18 250×4.6. Градиентное элюирование конъюгата с колонки проводили в системе растворителей: ацетонитрил/0.1% трифторуксусная кислота/вода.
Концентрацию полипептида в конъюгате определяли спектрофотометрически (длина волны λ=280 нм). Содержание КМ в конъюгате определяли спектрофотометрически при длине волны λ=495 нм. Молярное соотношение полипептид:КМ в конъюгате составляет 1:1.
Схема 1
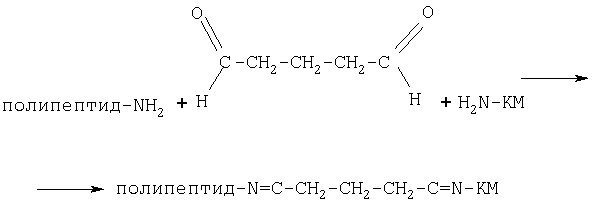
Пример 4. Эффективность конъюгата декапептида с карминомицином в отношении раковых клеток различных линий, оцениваемая по выживаемости
Действие конъюгата было исследовано на следующих линиях клеток: карцинома молочной железы человека линии (MCF-7Wt) и резистентная линия карциномы молочной железы человека (MCF-7AdrR), карцинома яичника человека линии (SKOV3) и резистентная линия карциномы яичника человека (SKVLB); мышиная меланома линии В16.
Протокол испытаний был общим для всех проведенных испытаний. Клетки культивировали в пластиковых флаконах ("Costar") в среде RPMI ("Sigma"), содержащей 10% эмбриональной бычьей сыворотки ("Sigma"), 100 ед/мл пенициллина и 100 мкг/мл стрептомицина, в увлажненной атмосфере 5% CO2 при 37°С. Клетки рассевали в 96-луночные планшеты (Costar) по 10 тысяч клеток/лунку, добавляя исследуемые препараты в различных концентрациях, и инкубировали 72 ч. За 2-4 ч до окончания инкубации в каждую лунку добавляли по 50 мкл раствора (1 мг/мл) МТТ (3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия бромид, Sigma) в среде для культивирования клеток. После развития окраски среду удаляли, растворяли кристаллы формазана в 150 мкл диметилсульфоксида и спектрофотометрически измеряли интенсивность окраски по поглощению при 540 нм. Выживаемость клеток, подвергшихся воздействию карминомицина и конъюгата, оценивали в процентах, принимая за 100% выживаемость контрольной культуры.
Данные о выживаемости клеток после их инкубации с конъюгатом декапептид-КМ и свободным карминомицином приведены в табл.2. Из представленных данных следует, что предлагаемый конъюгат пептида с карминомицином оказывает значительно более высокое токсическое действие в отношении опухолевых клеток человека линий MCF-7Wt и SKOV3, чувствительных к карминомицину, а также в отношении клеток мышиной меланомы В16, чем свободный антибиотик. Конъюгат пептид-КМ также проявлял высокую цитотоксическую активность в отношении клеток линий MCF-7AdrR и SKVLB, обладающих резистентностью к антрациклиновым антибиотикам. Кроме того, новый конъюгат демонстрирует высокую цитотоксическую активность в отношении пролиферирующих эндотелиальных клеток (HUVEC), моделирующих эндотелий опухолевых сосудов, и проявляет значительно более низкую цитотоксичность в отношении покоящихся клеток той же линии, характерных для нормальных сосудов организма.
Пример 5. Получение фармкомпозиции на основе конъюгата
Фармацевтические композиции для инъекций получают растворением конъюгата в физрастворе или фосфатно-солевом растворе, приготовленном на воде для инъекций. рН растворов около 7,4. Концентрация конъюгата в растворе составляет 0,05-0.1%. При необходимости в раствор добавляют другие компоненты.
Пример 6. Противоопухолевая активность композиции конъюгата декапептид-карминомицин с физиологическим раствором (по примеру 5) на модели перевиваемых опухолей мышей
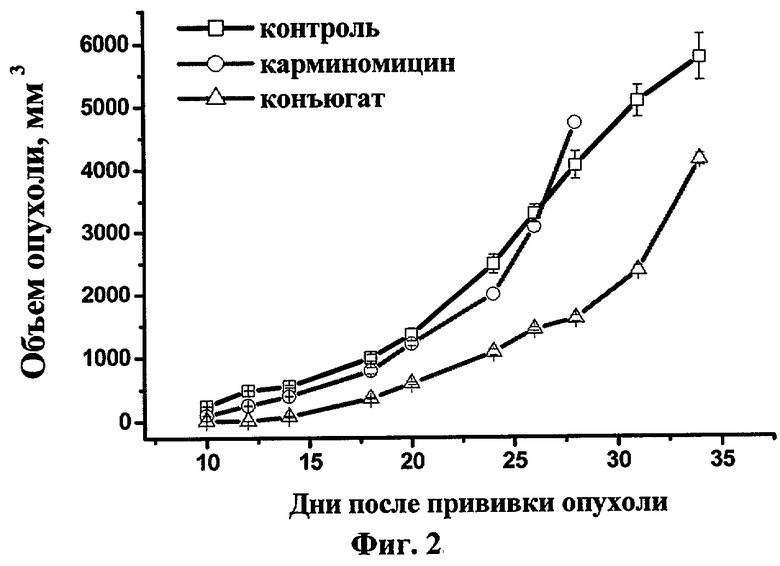
Для сравнительной оценки терапевтической активности нового конъюгата в отношении солидных опухолей, полученных подкожным введением мышам опухолевых клеток линии В 16, использовали внутривенный способ введения композиций, описанных в примере 5. Полученные результаты представлены на фиг.2. Препараты вводили в дозах 0,5 мг/кг по КМ по схеме: один раз в 4 дня, всего три инъекции, начиная с 3-го дня после прививки опухоли.
Из фиг.2 следует, что новый конъюгат, в отличие от свободного антибиотика, значительно замедлял время появления опухолей и скорость их роста. Торможение роста опухолей составило 51%.
Таким образом разработана новая система препаратов на основе векторного декапептида, способного присоединять химическим путем цитотоксические агенты, а полученный конъюгат положен в основу новых фармацевтических противоопухолевых композиций.
ЛИТЕРАТУРА
1. Munns J. et al., Br. J. Urology 82: 284-289, 1998.
2. Schally A.V. et al., Eur J Endocrinology 141: 1-14, 1999.
3. Агар W. et al., Science 279: 377-380, 1998.
4. Severin S.E. et al., Tumor Targeting 2: 299-306, 1996.
5. Hunter T. et al., Biochem. Soc. Trans. 12: 757-759, 1984.
6. Modjtahedi H. and Dean Ch., Intern. J. Oncol. 4: 277-296, 1995.
7. TokunadaA. et al., Cancer 65: 1418-1425, 1995.
8. Schreiber A.B. et al., Science 232: 1250-1253, 1986.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИПЕПТИД, ЯВЛЯЮЩИЙСЯ АНАЛОГОМ РЕЦЕПТОРСВЯЗЫВАЮЩЕГО ФРАГМЕНТА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА С 21-Й ПО 31-Ю АМИНОКИСЛОТУ, ЕГО КОНЪЮГАТ С ДОКСОРУБИЦИНОМ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2001 |
|
RU2196604C1 |
| ПРОТИВООПУХОЛЕВЫЙ ПЕПТИДНЫЙ ПРЕПАРАТ НА ОСНОВЕ ФРАГМЕНТА АЛЬФА-ФЕТОПРОТЕИНА, ЕГО КОНЪЮГАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ГОРМОНЗАВИСИМЫХ ОПУХОЛЕЙ | 2005 |
|
RU2285537C1 |
| КОНЪЮГАТ, ОБЛАДАЮЩИЙ ИЗБИРАТЕЛЬНЫМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К РАКОВЫМ ОПУХОЛЯМ | 2005 |
|
RU2303997C2 |
| БЕЛКОВОПЕПТИДНЫЙ ПРОТИВООПУХОЛЕВЫЙ КОМПОЗИТ, КЛЕТОЧНЫЙ ПРЕПАРАТ, АКТИВИРОВАННЫЙ ЭТИМ КОМПОЗИТОМ, И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ | 2004 |
|
RU2283129C1 |
| ПЕПТИД, ЯВЛЯЮЩИЙСЯ АНАЛОГОМ ФРАГМЕНТА АЛЬФА-ФЕТОПРОТЕИНА, КОНЪЮГАТ ПЕПТИДА С ДОКСОРУБИЦИНОМ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2317102C1 |
| КОНЪЮГАТ ПРОТИВООПУХОЛЕВЫХ ПРЕПАРАТОВ С РЕКОМБИНАНТНЫМ АЛЬФА-ФЕТОПРОТЕИНОМ И ЕГО ФУНКЦИОНАЛЬНЫМИ ФРАГМЕНТАМИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2630974C1 |
| ПРОТИВООПУХОЛЕВЫЙ ЛИПОСОМНЫЙ ПРЕПАРАТ НАПРАВЛЕННОГО ДЕЙСТВИЯ | 2005 |
|
RU2292898C1 |
| ПРОТИВООПУХОЛЕВЫЙ ПРЕПАРАТ | 2011 |
|
RU2451509C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЪЮГАТА ФРАГМЕНТА АЛЬФА-ФЕТОПРОТЕИНА ЧЕЛОВЕКА С ЦИПРОФЛОКСАЦИНОМ | 2010 |
|
RU2431639C1 |
| СЛИТЫЙ БЕЛОК, ИМЕЮЩИЙ АКТИВНОСТЬ ИНГИБИТОРА АНГИОГЕНЕЗА | 2008 |
|
RU2372354C1 |
Изобретение относится к области биомолекулярной фармакологии. Сущность изобретения составляет декапептид, являющийся аналогом рецепторсвязывающего фрагмента ТФРα с 22 по 31 аминокислоту, в котором 8-й аминокистотный остаток Lys заменен на остаток Ser, аналог способен связываться с широким спектром цитотоксических агентов и работать в качестве вектора для направленной доставки противоопухолевых агентов в опухолевые клетки. Изобретение содержит также конъюгат декапептида с цитотоксическим агентом, обладающий избирательным действием в отношении опухолей и способный снижать резистентность опухолевых клеток к цитотоксическим препаратам, в котором конъюгированные части соединены неустойчивой к кислотному гидролизу химической связью. Другим объектом изобретения является фармацевтическая композиция, которая содержит эффективное количество конъюгата и носитель, пригодный для внутривенного введения. Технический результат - высокая селективность противоопухолевого действия. 3 н. и 3 з.п. ф-лы, 2 табл., 2 ил.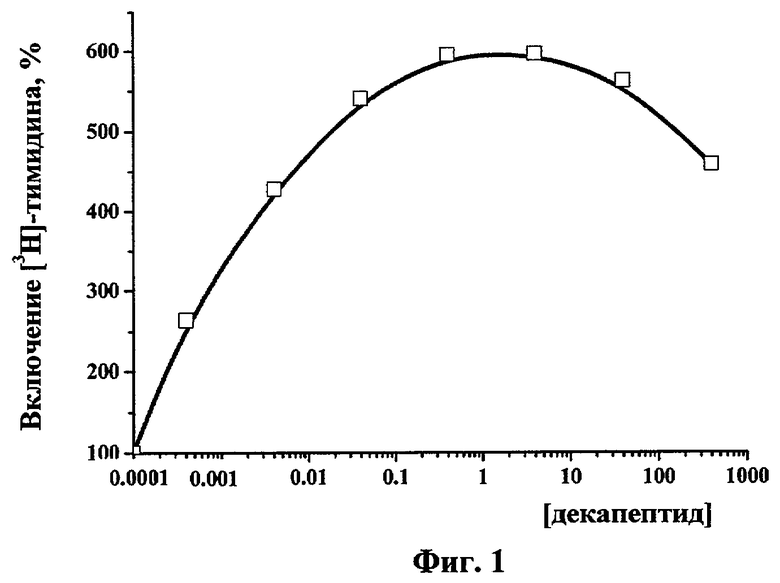
| ПОЛИПЕПТИД, ЯВЛЯЮЩИЙСЯ АНАЛОГОМ РЕЦЕПТОРСВЯЗЫВАЮЩЕГО ФРАГМЕНТА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА С 21-Й ПО 31-Ю АМИНОКИСЛОТУ, ЕГО КОНЪЮГАТ С ДОКСОРУБИЦИНОМ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2001 |
|
RU2196604C1 |
| СПОСОБЫ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА, БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ ТИРОЗИНКИНАЗУ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА АКТИВНОСТЬЮ, И КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ КОНТРАЦЕПТИВНЫМ ДЕЙСТВИЕМ | 1995 |
|
RU2174980C2 |
| WO 9220642, 26.11.1992. | |||

