Перекрестная ссылка на родственную заявку
Для данной заявки испрашивается приоритет, в соответствии с 35 U.S.C. §119(e), по предварительной заявке США № 61/040911, поданной 31 марта 2008 г., полное содержание которой включено в данный документ путем ссылки.
Государственная поддержка
Объект настоящего изобретения был произведен при поддержке Национальных институтов здравоохранения GM058448. Правительство США обладает определенными правами в отношении настоящего изобретения.
Область техники, к которой относится изобретение
Настоящее изобретение относится к аналогам этомидата, которые обладают улучшенными фармакокинетическими и фармакодинамическими свойствами, и к их применению в качестве анестезирующих средств.
Уровень техники
Каждый год только в США вводится около 30 миллионов общих анестетиков. При концентрациях, необходимых для обеспечения анестезии, все общие анестетики дают потенциальные значительные и, иногда, смертельные побочные эффекты. Особенно важным является подавление сердечно-сосудистой и дыхательной функций, что может угрожать жизни, в частности, пожилым, больным и травмированным пациентам. Такие губительные побочные эффекты вызываются почти всеми общими анестетиками, и это объясняет, почему в целом анестетики обладают наименьшими терапевтическими индексами (LD50/ED50) по сравнению с любым классом терапевтических лекарственных средств. Следовательно, очень большое значение имеет разработка более безопасных анестезирующих средств с меньшими побочными эффектами.
Этомидат (этил 3-(1-фенил)имидазол-4-карбоксилат) является быстро действующим седативным и снотворным внутривенным средством на основе имидазола, который может быть использован для индицирования и поддержания общей анестезии или седации при сохраненном сознании. Он существует в виде двух энантиомеров, однако (R)-энантиомер в 10 раз более активен в качестве анестетика чем (S)-энантиомер. Именно (R)-энантиомер применяется клинически (см. ниже Структуру 1). (R)-энантиомер индуцирует потерю выпрямительного рефлекса у головастиков (Husain, S.S., и др., J Med Chem, 46:1257-1265 (2003)) и потерю восприимчивости у людей (Arden, J.R., и др., Anesthesiology, 65:19-27 (1986) при водной свободной концентрации ~2 мкМ.
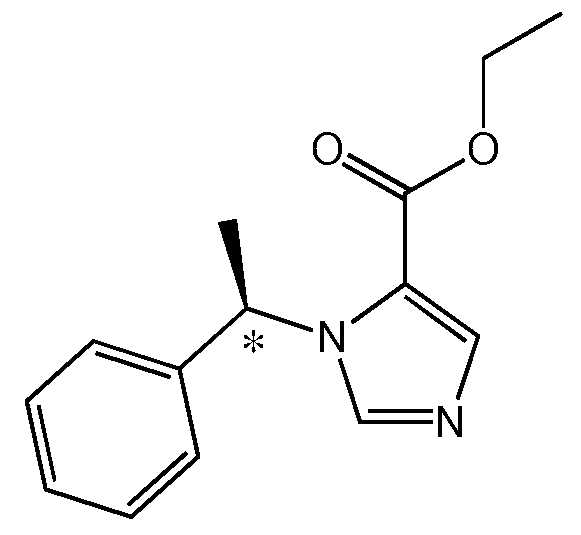
Структура 1 - (R)-этомидат (этил 3-(1-фенил)имидазол-4-карбоксилат)
На молекулярном уровне существуют убедительные доказательства, что этомидат индуцирует анестезирующий эффект за счет усиления функции ГАМКА рецепторов, содержащих β2 или β3 субъединицы. Этомидат усиливает ГАМК рецептор-опосредованные токи, индуцированные низкими концентрациями агониста, но минимально усиливает токи, индуцированные высокой концентрацией агониста. Это сдвигает кривую зависимости ответа от концентрации агониста влево (снижает значение ЕС50 агониста). Этомидат также напрямую активирует ГАМКА рецепторы в отсутствии агониста.
По сравнению с другими общими анестетиками, этомидат имеет необычно высокий терапевтический индекс; терапевтический индекс (R)-этомидата для животных составляет 26,4 по сравнению с 4,6 и 3,1 для тиопенталя и пропофола, соответственно (Janssen, P. A., Arzneimittelforschung, 21:1234- 1243 (1971), Glen, J.B., Br J Anaesth, 52:731-742 (1980), и Zhou, Anesth Analg, 102:129- 134 (2006)). Относительно большая широта терапевтического диапазона, обеспечиваемая этомидатом, по-видимому, отражает его меньшее влияние на сердечно-сосудистую и дыхательную функции. Гемодинамическая стабильность, обеспечиваемая этомидатом, по крайней мере, частично связана с отсутствием у него депрессантного эффекта в отношении симпатических нервов и рефлексов в сфере вегетативной нервной системы (Ebert, TJ., et al., Anesthesiology 76:725-733 (1992)). С другой стороны, пропофол и тиопенталь снижают симпатическую активность, притупляют рефлексы в сфере вегетативной нервной системы и напрямую ухудшают сократимость миокарда (Mazerolles, M., Fundam Clin Pharmacol, 10:298-303 (1996)). Данные действия приводят к подавлению сердечно-сосудистой и дыхательной функций даже у здоровых пациентов. Из-за меньшего влияния (R)-этомидата на сердечно-сосудистую и дыхательную функции, он стал предпочтительным анестезирующим средством для анестезиологов, врачей-реаниматологов и врачей неотложной помощи для введения больным, пожилым или травмированным пациентам. Однако оптимум в отношении данного препарата является сдержанным, и его применение ограничено из-за сильнодействующего и продолжительного ингибирования синтеза адренокортикостероидов.
Ингибирование синтеза стероидов является потенциально смертельным побочным эффектом продолжительного применения (R)-этомидата, особенно для тех пациентов, которые, в другом случае, могли бы получить большую пользу от его благоприятных свойств, касающихся сердечно-сосудистой и дыхательной функций: тяжелобольных. Такое ингибирование крайне сильно и возникает при значительно меньших концентрациях (R)-этомидата, которые используются для обеспечения седативного эффекта или анестезии. При уровнях дозы, необходимых для обеспечения общей анестезии, (R)-этомидат вызывает недостаточность функции коры надпочечников, которая может сохраняться в течение 4 дней после прекращения продолжительного вливания (Wagner, R.L., и White, P.F., Anesthesiology, 61:647-651 (1984)), приводя к значительному повышению уровня смертности среди больных пациентов, находящихся в критическом состоянии (Watt, L, и Ledingham, I.M., Anaesthesia, 39:973-981 (1984) и Ledingham, LM., и Watt, L, Lancet, 1:1270 (1983)). По-видимому, смертность может быть снижена опытным путем за счет введения экзогенных стероидов; однако, данный подход не является оптимальным, так как дозировка, время и продолжительность терапии стероидами для каждого взятого пациента будут спекулятивными. Более того, само по себе введение экзогенных стероидов может вызвать значительные осложнения, включая нарушения гомеостаза глюкозы и заживления ран, подавление иммунитета и задержку жидкости. Из-за глубокого влияния (R)-этомидата на функцию коры надпочечников в его упаковку был добавлен специальный вкладыш, предупреждающий об опасности его длительного вливания, и использование (R)-этомидата для продолжительного обезболивания или анестезии было прекращено.
Клиническая значимость подавления функции коры надпочечников после однократного внутривенного болюсного введения является неоднозначной. См. Morris, C, и McAllister, C, Anaesthesia, 60:737-740 (2005); Jackson, W.L., Jr., Chest, 127:1031-1038 (2005); Murray, H., и Marik, P.E., Chest, 127:707-709 (2005); Zed, P.J., и др., Chem., 8:347-350 (2006); и Bloomfield, R., и Noble, D.W., Crit Care, 10:161 (2006). Исторически считается, что подавление функции надпочечников после однократного болюсного введения является коротким (<8 часов) и клинически не существенно. Однако данное предположение главным образом основано на небольшом объеме исследований пациентов, подвергающихся плановым операциям, которые не являлись тяжелобольными. См. Wagner, R.L., и White, P.F., Anesthesiology, 61:647-651 (1984); Wagner, R.L. и др., N Engl J Med, 310:1415-1421 (1984); Fragen, R.J. и др., Anesthesiology 61:652-656 (1984); и Duthie, D.J. и др., Br J Anaesth, 57:156-159 (1985). Ряд недавних исследований и сообщений о тяжелобольных пациентах показывает, что даже после однократного болюсного введения дозы (R)-этомидата подавление надпочечников может продолжаться в течение 24 часов или дольше, и некоторые считают, что это увеличивает риск смерти, особенно при возникновении сепсиса. См. Absalom A. и др., Anaesthesia, 54:861-867 (1999); Malerba, G. и др., Intensive Care Med, 31:388-392 (2005); den Brinker, M. и др., Intensive Care Med (2007); den Brinker, M. и др.,/Clin Endocrinol Metab, 90:5110-5117 (2005); Lundy, J.B. и др.,/Intensive Care Med, 22:111-117 (2007); Lipiner-Friedman, D. и др., Crit Care Med, 35:1012-1018 (2007); Vinclair, M. и др., Intensive Care Med., (2007); и Cotton, B.A. и др., Arch Surg, 143:62-67 (2008).
(R)-этомидат ингибирует синтез адренокортикостероидов главным образом за счет ингибирования 11β-гидроксилазы, важного фермента синтетического пути, приводящего к получению кортизола, кортикостерона и альдостерона (см. de Jong, F.H. и др.,/Clin Endocrinol Metab, 59:1143-1147 (1984)). Сообщалось, что полумаксимальная ингибирующая концентрация (R)-этомидата (IC50) составляет 0,5-30 нМ (см. Lamberts, S.W. и др., J Pharmacol Exp Ther, 240:259-264 (1987)), при этом диапазон концентраций на порядки ниже, чем его анестезирующая концентрация. Если рассматривать крайне высокую ингибирующую активность (R)-этомидата по отношению к 11β-гидроксилазе наряду с его длительным (несколько часов) периодом полувыведения (см. Van Hamme, MJ. и др., Anesthesiology, 49:274-277 (1978)), то мы предположили, что логическое объяснение выходит за рамки объяснения длительным временем подавления коры надпочечников после введения (R)-этомидата: для того чтобы после введения анестезирующей дозы концентрация (R)-этомидата в сыворотке снизилась за счет метаболизма до достаточного уровня, при котором активность 11β-гидроксилазы больше не ингибируется, должно пройти много периодов полувыведения. Это ведет к прогнозированию того, что период подавления коры надпочечников может быть снижен за счет разработки аналогов (R)-этомидата, которые быстро подвергаются метаболизму. Также можно спрогнозировать, что такие быстро метаболизирующиеся аналоги могут иметь чрезвычайно короткие периоды анестезирующего действия. Это является другим чрезвычайно желаемым свойством анестетика, так как это позволяет более точно корректировать глубину наркоза во время операции и быстро выходить из анестезии в конце операции.
Существует большая потребность в более безопасных общих анестетиках, в частности, для применения тяжелобольными. (R)-этомидат обладает многими свойствами, которые делают его идеальным общим анестезирующим средством, однако его способность подавлять синтез адренокортикостероидов существенно ограничивает его клиническую применимость и безопасность.
Как отмечалось выше, в области техники существует необходимость в разработке аналогов (R)-этомидата, которые сохраняют его полезные свойства (например, быстрое начало действия, небольшой эффект на кровяное давление, высокий терапевтический индекс), но не вызывают потенциально опасное ингибирование функции коры надпочечников. Такие аналоги позволят более безопасно проводить анестезию тяжелобольным пациентам. Настоящее изобретение отвечает этому требованию.
Сущность изобретения
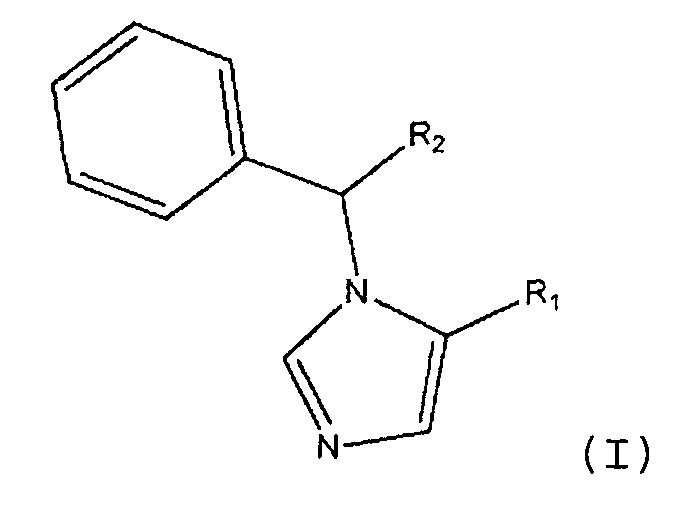
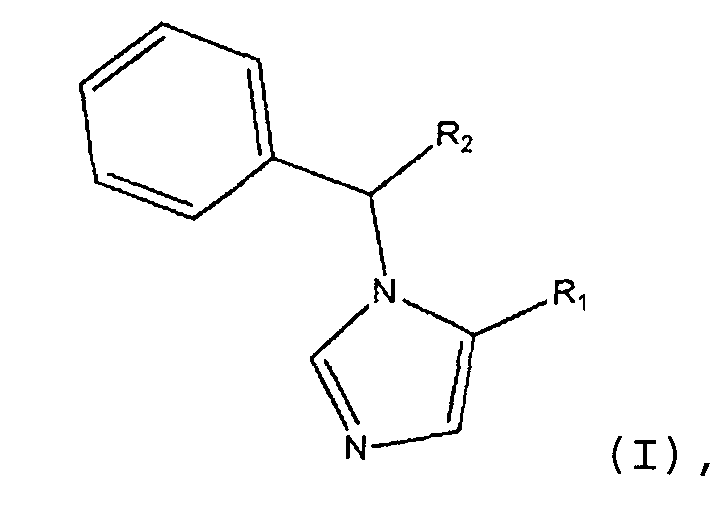
Изобретение направлено на соединения формулы (I):
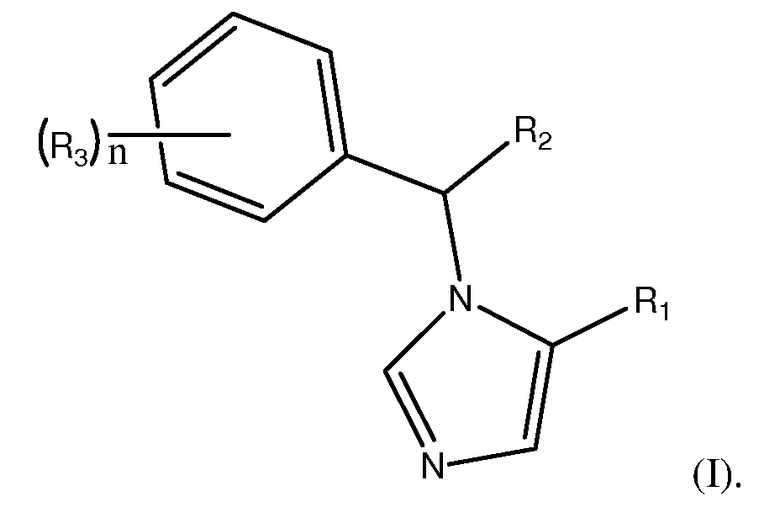
Соединения формулы (I) обладают улучшенными фармакокинетическими и фармакодинамическими свойствами по сравнению с (R)-этомидатом, что предусматривает эквивалентные или улучшенные анестезирующие свойства наряду со снижением нежелательных побочных эффектов. Соединения формулы (I) являются аналогами этомидата, сохраняя полезные анестезирующие свойства (R)-этомидата, но не вызывают клинически значимого ингибирования функции коры надпочечников.
В формуле (I) R1 является L1C(O)OT или L1C(O)OL2C(O)OT. R2 является замещенным или незамещенным C1-C10алкилом, C2-C10алкенилом или C2-C10алкинилом, или R1. Каждый R3 независимо является галогеном или R2. n является целым числом от 0 до 5. L1 и L2 каждый независимо являются связью или замещенным или незамещенным C1-C10алкиленом, C2-C10алкениленом или C2-C10алкиниленом. T является H, замещенным или незамещенным C1-C10алкилом, C2-C10алкенилом или C2-C10алкинилом, нитрофенолом или циклопропилом. Соединения формулы (I) включают их фармацевтически приемлемые соли, смеси стереоизомеров и энантиомеры. Соединения формулы (I) являются объектом настоящего изобретения при условии, что если R1 является L1C(O)OT, R2 является CH3, R3 является фтором, n равно 1 и T является CH2CH3, L1 не является связью.
Другой аспект изобретения направлен на фармацевтическую анестезирующую композицию, содержащую эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель.
Еще один аспект изобретения направлен на способ обеспечения анестезии у млекопитающего, включающий введение млекопитающему эффективного анестезирующего соединения формулы (I) или фармацевтической композиции.
Другой аспект изобретения заключается в применении соединений формулы (I), главным образом, как описано здесь, для приготовления лекарственной формы, или при производстве лекарственной формы для обеспечения анестезии у нуждающегося в ней субъекта.
Краткое описание чертежей
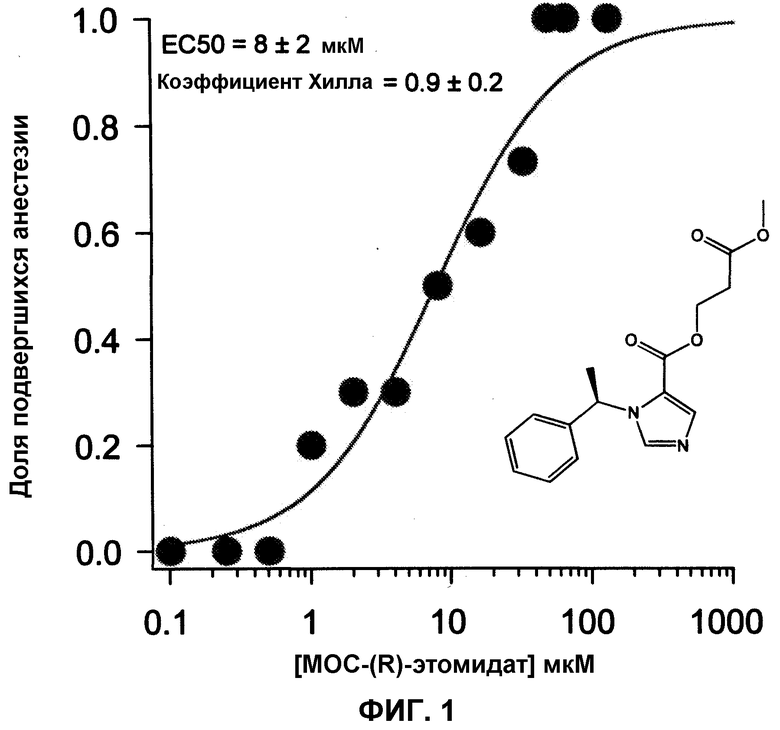
Фиг.1 является графиком, показывающим зависимость ответа анестезии от концентрации MOC-(R)-этомидата (измерение потери выпрямительного рефлекса; LORR) у головастиков. Для построения данной кривой концентрация-ответ всего было использовано 100 головастиков. Анестезирующая ЕС50 составила 8±2 мкМ. Данные результаты показывают что, MOC-(R)-этомидат является сильнодействующим общим анестетиком. Для сравнения, ЕС50 (R)-этомидата составляет 2 мкМ (см. Husain SS и др. J Med Chem (2003).
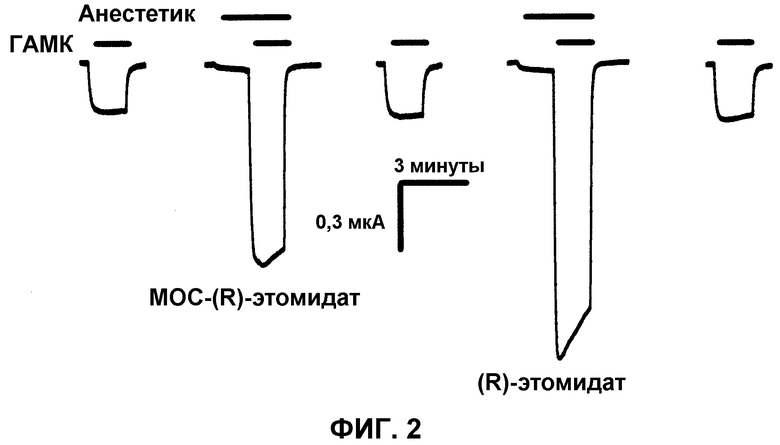
На фиг.2 приведены электрофизиологические кривые, показывающие усиление токов, опосредованных α1β2γ2L ГАМКA рецепторами, экспрессированными в ооцитах Xenopus, за счет MOC-(R)-этомидата и (R)-этомидата при их относительных анестезирующих концентрациях. Первая, третья и пятая кривые являются контролем. Вторая и четвертая кривые показывают аналогичные эффекты усиления 8 мкМ MOC-(R)-этомидата и 2 мкМ (R)-этомидата, соответственно. Данные результаты показывают, что аналогично (R)-этомидату, MOC-(R)-этомидат усиливает субмаксимальные ГАМКA рецепторные токи, индуцированные ГАМК.
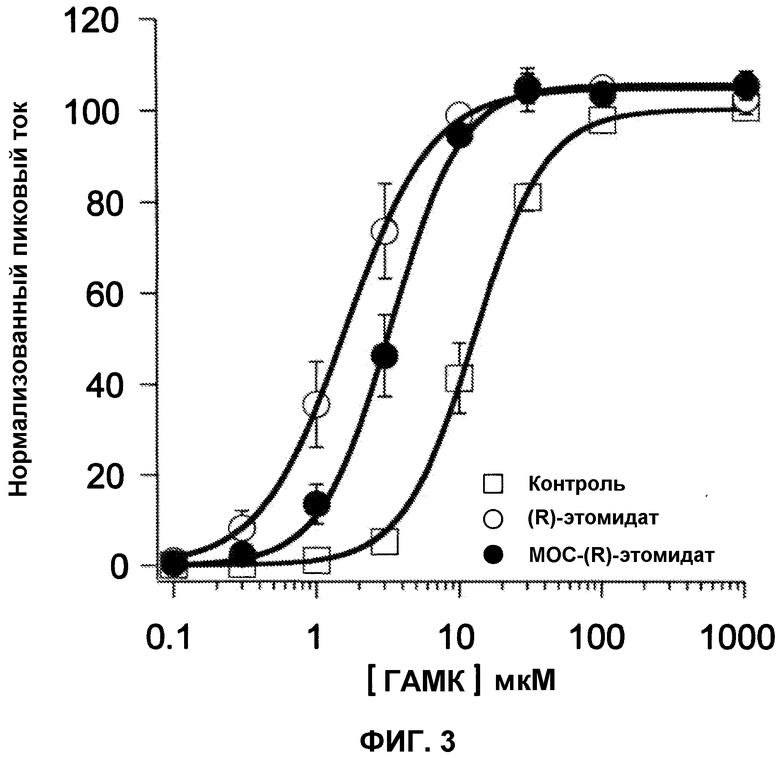
На фиг.3 приведен график зависимости ответа от концентрации ГАМК в отсутствии или в присутствии либо 8 мкМ MOC-(R)-этомидат, либо 2 мкМ (R)-этомидата. Данные результаты показывают, что, как и (R)-этомидат, MOC-(R)-этомидат сдвигает кривую для ГАМКA рецепторов, показывающую зависимость ответа от концентрации ГАМК, влево. Каждая точка кривой отображает значение измерений трех различных ооцитов. Показатель ошибок отражает стандартное отклонение.
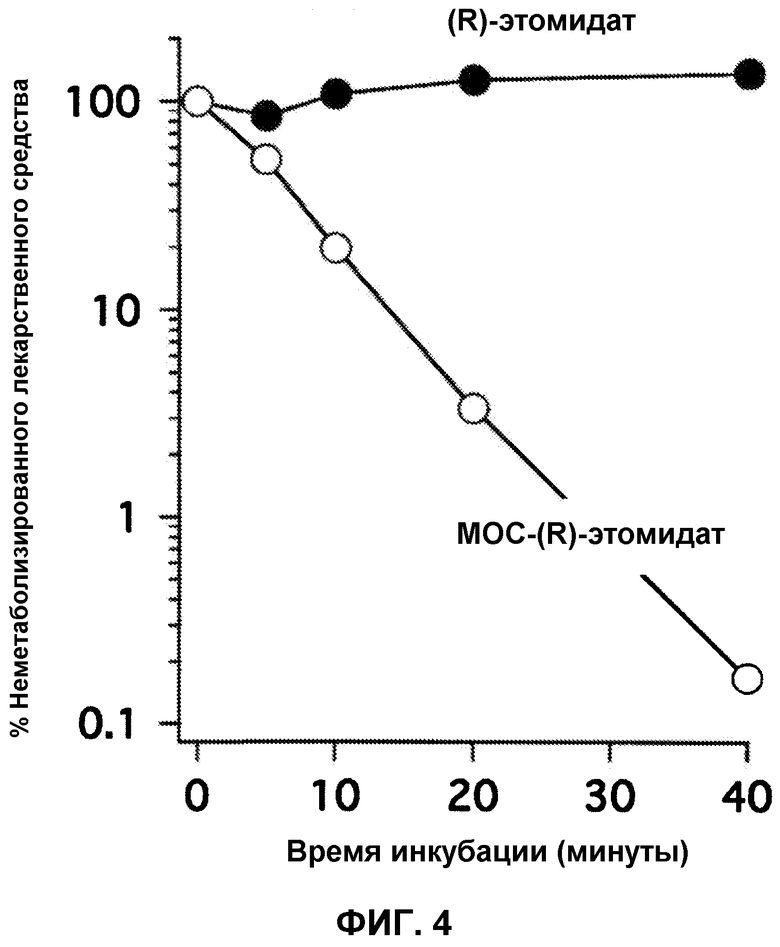
На фиг.4 приведено процентное содержание неметаболизированного (R)-этомидата или неметаболизированного MOC-(R)-этомидата, как функция времени инкубации, при инкубации с фракцией печени человека S9 (+НАДФН) при 37°С. За 40 минут метаболизму подвергалось более 99% MOC-(R)-этомидата, тогда как в данном временном интервале (R)-этомидат не подвергался заметному метаболизму. Данные результаты показывают, что MOC-(R)-этомидат метаболизируется ферментами печени, по крайней мере, в 100 раз быстрее, чем (R)-этомидат.
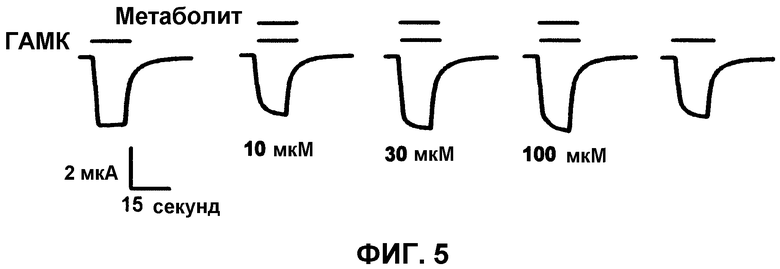
На фиг.5 приведены электрофизиологические кривые, показывающие отсутствие усиления токов, опосредованных α1β2γ2L ГАМКA рецепторами, экспрессированными в ооцитах Xenopus, под действием карбоновой кислоты, являющейся метаболитом MOC-(R)-этомидата. Первая и последняя кривые являются контролем (т.е. без метаболита). Вторая, третья и четвертая кривые показывают отсутствие эффекта 10, 30 и 100 мкМ метаболита.
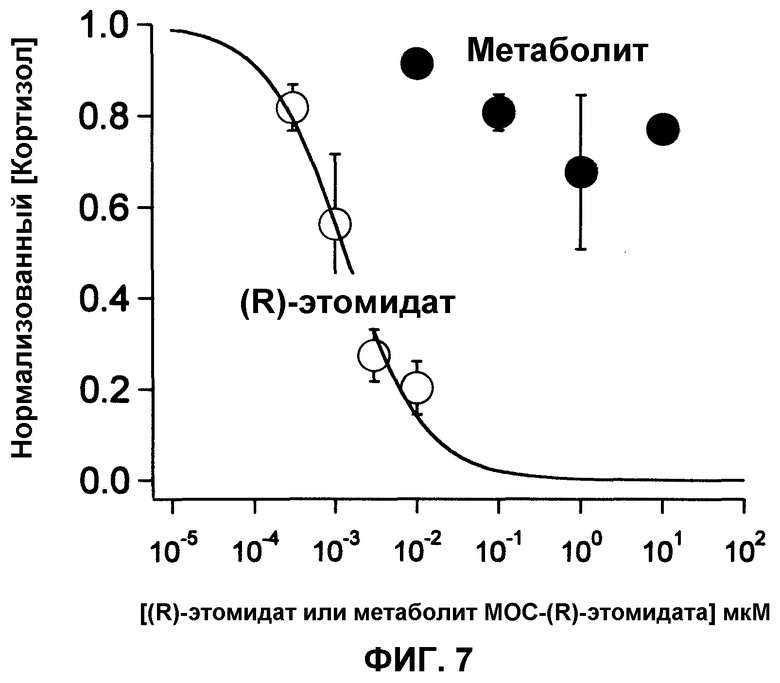
Фиг.6 является графиком, показывающим, что (R)-этомидат ингибирует синтез кортизола адренокортикальных клеток H295R даже при наномолярных концентрациях (IC50=1,3±0,22 нМ), тогда как метаболит MOC-(R)-этомидата обладает относительно малой ингибирующей активностью даже при микромолярных концентрациях. Каждая точка отражает среднюю концентрацию кортизола в 3 лунках. Показатели ошибок являются стандартными отклонениями.
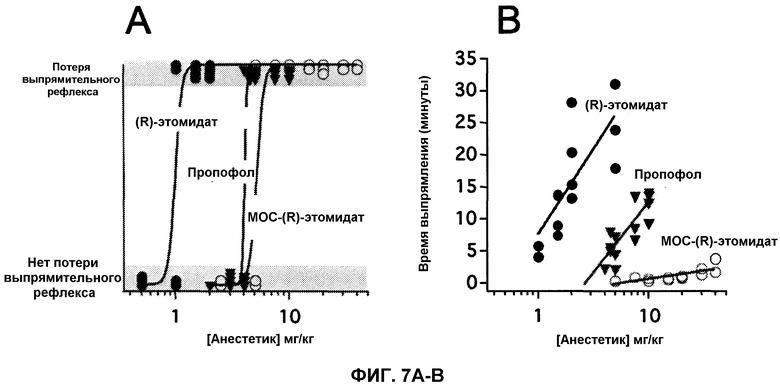
На фиг.7А приведены зависимости ответа от дозы по анестезии (измерено как для LORR) у крыс для пропофола, (R)-этомидата и MOC-(R)-этомидата. На фиг.7В показано, что продолжительность анестезии увеличивается практически линейно с ростом логарифма анестезирующей дозы, и что эта продолжительность значительно короче для MOC-(R)-этомидата по сравнению как с (R)-этомидатом, так и пропофолом.
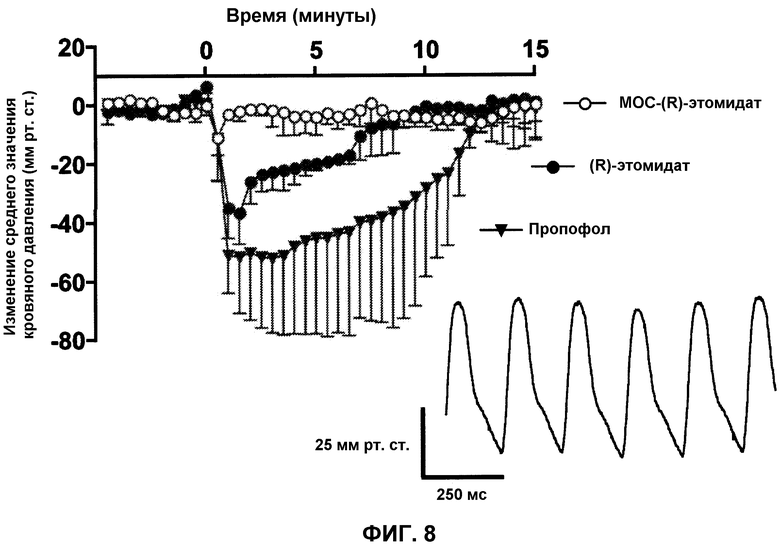
На фиг.8 приведена временная диаграмма значения кровяного давления у крыс после введения эквивалентных анестезирующих доз пропофола, (R)-этомидата и MOC-(R)-этомидата, и показано, что MOC-(R)-этомидат значительно меньше понижает кровяное давление по сравнению с пропофолом или (R)-этомидатом. Каждая точка показывает значение в течение 30 секундного периода. Показатели ошибок являются стандартными отклонениями. На вставке приведена типичная кривая артериального кровяного давления перед введением анестетика.
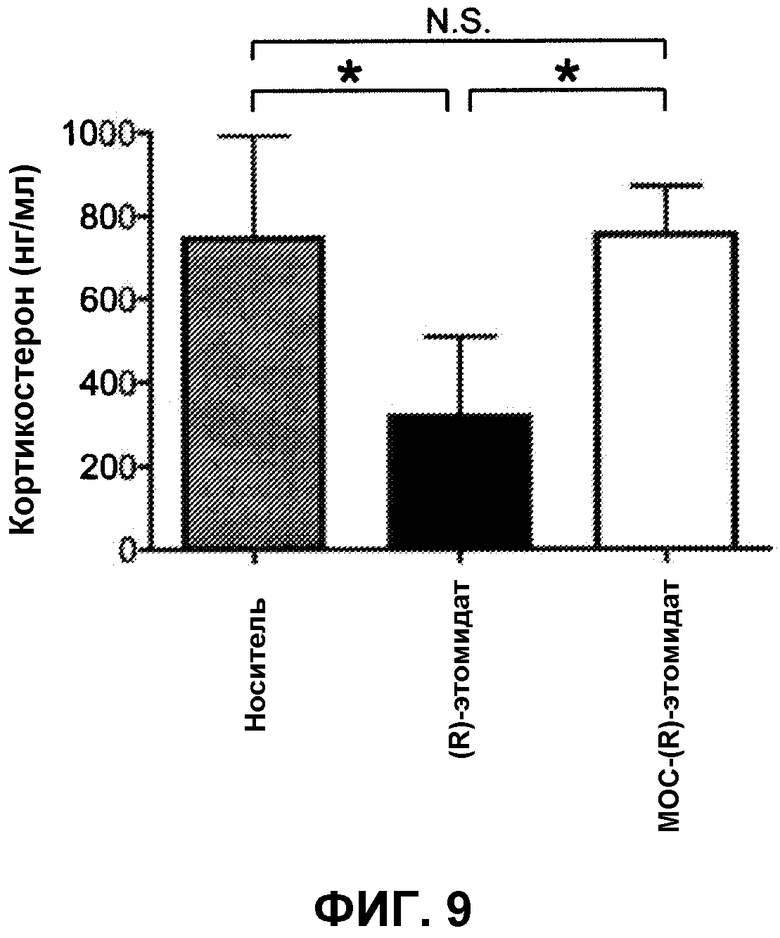
На фиг.9 показано, что концентрация кортикостерона в плазме (адренокортикального стероида) не изменяется в сравнении с контролем (пропиленгликолевый носитель) в течение 30 минут после введения MOC-(R)-этомидата, тогда как после введения эквивалентной анестезирующей дозы (R)-этомидата она значительно снижалась. Образование у крыс кортикостерона стимулировалось с использованием АСТН1-24 спустя 15 минут после введения анестетика или носителя, и затем через 15 минут измерялась концентрация кортикостерона в плазме. Показатели ошибок являются стандартными отклонениями.
Подробное описание изобретения
Настоящее изобретение относится к более безопасным аналогам (R)-этомидата, которые сохраняют его полезные характеристики (например, сильные анестезирующие свойства, быстрое начало действия, небольшой эффект на кровяное давление), но чье влияние на синтез адренокортикальных стероидов и/или длительность анестезирующего действия значительно снижена. Некоторые варианты осуществления включают аналоги этомидата (как R-, так и S-изомера), которые так быстро метаболизируются до менее активного метаболита (например, метаболита, который незначительно ингибирует 11β-гидроксилазу, усиливает функцию ГАМКA рецепторов и/или обеспечивает анестезию), что подавление функции коры надпочечников и/или анестезирующее действие прекращается сразу после прекращения введения анестетика.
Соединения настоящего изобретения могут быть представлены как аналоги этоимдата (как R-, так и S-изомера), дополненные одним или несколькими дополнительными лабильными метаболизирующимися сложноэфирными остатками, непосредственно присоединенными к различным положениям ядра молекулы или через различные линкерные группы (например, -CH2CH2-). На конце сложноэфирного остатка может находиться «концевая группа» (например, -СН3). Далее раскрыты различные варианты осуществления настоящего изобретения.
Изобретение направлено на соединения формулы (I):
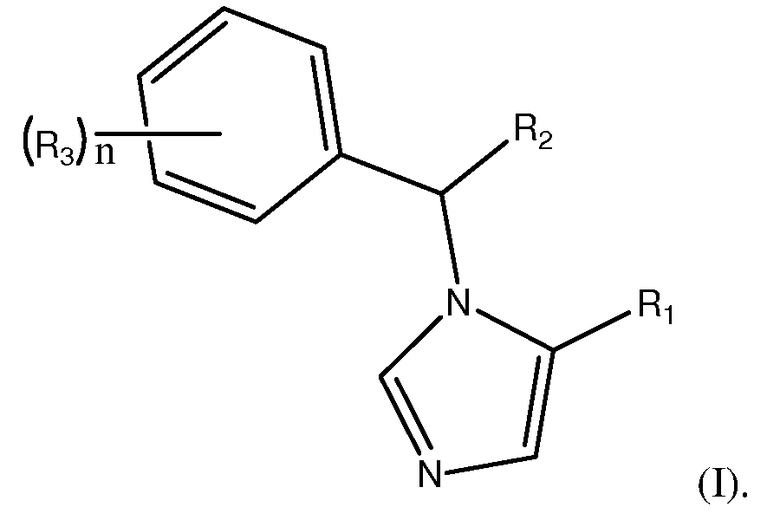
R1 является L1C(O)OT или L1C(O)OL2C(O)OT. В предпочтительном варианте осуществления, R1 является L1C(O)OL2C(O)OT.
R2 является замещенным или незамещенным C1-C10алкилом, C2-C10алкенилом, или C2-C10алкинилом, или R1. Предпочтительно, R2 является алкилом, таким как CH3 или сложным эфиром R1, таким как CH2CH2C(O)OCH3. В наиболее предпочтительном варианте осуществления, R2 является CH3.
Каждый R3 независимо является галогеном или R2. Предпочтительные галогены включают фтор и хлор. Переменная n является целым числом от 0 до 5. В предпочтительном варианте осуществления, n находится в диапазоне 0-3 и наиболее предпочтительно равно 0.
Линкеры L1 и L2 каждый независимо являются связью, замещенной или незамещенной C1-C10алкиленильной, C2-C10алкениленильной или C2-C10алкиниленильной группой. Главная цепь алкилена может содержать один или несколько гетероатомов, таких как O, N или S. Предпочтительно, каждый L1 и L2 независимо является связью или линейной C1-C4алкиленильной группой. Наиболее предпочтительно, L1 является связью или CH2CH2, и L2 является CH2CH2, CH2(CH2)4CH2 или CH2CH2O(CH2)3. В наиболее предпочтительном варианте осуществления, L2 является CH2CH2.
Концевая группа T может быть H, замещенным или незамещенным C1-C10алкилом, C2-C10алкенилом или C2-C10алкинилом. Главная цепь алкила может содержать один или несколько гетероатомов, таких как O, N или S. Концевая группа также может быть циклопропилом, нитрофенолом или любой другой электроноакцепторной группой. Предпочтительно, T является C1-C4алкильной группой. Наиболее предпочтительно, T является CH3, CH2CH3, CH2CH2CH2CH3 или CH2CH2OCH3. В наиболее предпочтительном варианте осуществления, T является CH3. В другом наиболее предпочтительном варианте осуществления, T является нитрофенолом.
Соединение формулы (I) включает их фармацевтически приемлемые соли, смеси стереоизомеров и энантиомеры. Соединения изобретения также включают физиологически приемлемые соли соединений формулы (I). Предпочтительные физиологически приемлемые соли являются солями, полученными добавлением кислоты, известными специалисту в данной области техники. Традиционные физиологически приемлемые соли, полученные добавлением кислоты, включают, но не ограничиваются ими, гидрохлориды, оксалаты и тартраты.
В предпочтительном варианте соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, n равно 0, L1 является связью, L2 является CH2CH2 и T является CH3.
В другом варианте соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, n равно 0, L1 является связью, L2 является CH2(CH2)4CH2 и T является CH2CH2CH2CH3.
В еще одном варианте соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, n равно 0, L1 является связью, L2 является CH2CH2O(CH2)3 и T является CH2CH2OCH3.
В некоторых вариантах соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, каждый R3 независимо является галогеном, n равно 1-5, L1 является связью, L2 является CH2CH2 и T является CH3.
В других вариантах соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, каждый R3 является фтором, n равно 1-5, L1 является связью, L2 является CH2CH2 и T является CH3.
В еще других вариантах соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, каждый R3 является фтором, L1 является связью, L2 является CH2CH2 и T является CH3.
В других вариантах соединения, R1 является L1C(O)OT, R2 является CH3, по крайней мере, один R3 является CH2CH2C(O)OCH3, L1 является связью и T является CH2CH3.
В следующих вариантах соединения, R1 является L1C(O)OL2C(O)OT, R2 является CH3, по крайней мере, один R3 является CH2CH2C(O)OCH3, L1 является связью, L2 является CH2CH2 и T является CH3.
В предпочтительном варианте соединения, R1 является L1C(O)OT, R2 является CH2CH2C(O)OCH3, n равно 0, L1 является связью и T является CH2CH3.
В другом предпочтительном варианте соединения, R1 является L1C(O)OT, R2 является CH3, n равно 0, L1 является CH2CH2 и T является CH2CH3.
Мостиковый атом углерода, соединяющий 6-членное кольцо и 5-членное кольцо, является хиральным центром. Следовательно, соединение может быть в форме чистого энантиомера. В предпочтительном варианте, энантиомер является R энантиомером.
Предпочтительно, соединения формулы (I) обладают такой же стереохимией, как и (R)-этомидат. R2, R3, L1, L2 и T могут быть разветвленными углеводородными цепями, однако так, чтобы стерические затруднения или сопряжение не препятствовали достижению требуемой активности.
В некоторых вариантах осуществления, соединение включает две или более сложноэфирные группы. Подходящие группы, содержащие сложный эфир (например, линкер - сложный эфир - концевая группа или сложный эфир - концевая группа), могут быть введены по мостиковому углероду, или по различным положениям фенильного кольца, или ядра молекулы.
Предпочтительными являются быстро метаболизирующиеся аналоги этомидата, содержащие в структуре (R)-этомидата новые сложноэфирные остатки, которые стерически не затруднены, и/или чья электронная система изолирована от π-электронной системы имидазольного и фенильного кольца. Считается, что такие сложноэфирные остатки, аналогичные тем, которые используются в других лекарствах с ультракоротким временем действия, таких как ремифентанил и эсмолол, высокочувствительны к гидролизу за счет эстераз. См. патент США № 3354173; патент США № 5466700; патент США № 5019583; и патентная заявка США № US 2003/0055023.
Заместители R2, T, L1 и L2 каждый могут быть независимо замещены одним или несколькими электроноакцепторными группами. В некоторых вариантах осуществления, электроноакцепторные группы являются галогеном, нитрофенолом или циклопропилом. Также могут быть использованы другие электроноакцепторные группы, такие как гидроксильные группы, аминогруппы, нитрогруппы, нитрильные группы, сульфонатные группы, карбоксилатные группы, галогеновые группы, меркаптогруппы и ненасыщенные алкильные группы. Наличие электроноакцепторных групп служит для увеличения частичного положительного заряда на карбонильном атоме сложного эфира, что увеличивает активность по отношению к нуклеофильной атаке эстеразами, и также увеличивает скорость гидролиза эстеразами.
Другой аспект изобретения направлен на фармацевтическую композицию, содержащую соединение формулы (I) и фармацевтически приемлемый носитель.
Еще один аспект изобретения направлен на способ обеспечения анестезии у млекопитающего, включающий введение млекопитающему фармацевтической композиции, в значительной степени аналогичной описанной выше.
В некоторых вариантах осуществления, способ включает введение эффективной дозы соединения. Эффективная доза составляет от 0,01 до 100 мг/кг соединения.
В предпочтительном варианте осуществления, способ включает введение инъекции разовой эффективной дозы соединения, после чего может следовать или может не следовать непрерывная инфузия соединения.
В некоторых вариантах осуществления, способ включает введение непрерывной инфузией эффективной дозы соединения формулы (I).
В некоторых вариантах осуществления, способ также включает введение млекопитающему эффективного количества терапевтического агента, выбранного из другого седативного снотворного средства, аналгезирующего средства и миорелаксанта. Неограничивающие примеры седативных снотворных средств включают бензодиазепины, барбитураты, кетамин, пропофол, изофлуран и десфлуран. Неограничивающие примеры аналгезирующих средств включают нестероидные противовоспалительные препараты (NSAID), парацетомол/ацетаминофен, ингибиторы СОХ-2 и опиаты. Неограничивающие примеры миорелаксантов включают рапакуроний, мивакуроний, сукцинилхолин, векуроний и цисатракурий.
Соединения изобретения показали анестезирующую активность и улучшенную активность ГАМКА рецепторов. Концентрации, исследованные в анализах in vitro, находились в диапазоне от 4,32×10-5 до 3,39×10-8 г/мл и от 0,01 до 0,02 г/кг для анализов in vivo. Соединения изобретения проявили сильные анестезирующий и ГАМКА рецепторный эффекты in vitro и in vivo. Данные результаты показывают, что соединения изобретения являются высокоактивными агентами с сильнодействующими in vitro и in vivo эффектами. Важно отметить, что соединения обладают сниженной ингибирующей активностью в отношении синтеза адренокортикостероидов in vitro и in vivo и/или короткой продолжительностью анестезирующего действия.
Описанные выше соединения могут быть введены как сами по себе, так и в виде смесей с другим соединением, или в комбинации с приемлемыми фармацевтическими носителями. Таким образом, изобретение также относится к фармацевтическим композициям, которые содержат эффективное количество, по крайней мере, одного соединения изобретения с фармацевтически или физиологически приемлемым носителем или без него. При необходимости соединение может быть введено в виде физиологически приемлемой соли, например в виде соли, полученной добавлением кислоты.
Изобретение также включает способ лечения животных или человека. Данный способ включает введение животному или человеку эффективного количества, по крайней мере, одного из соединений изобретения, или его физиологически приемлемой соли, с фармацевтически приемлемым носителем или без него. Предпочтительным является внутривенное введение. См. патент США № 4289783, полное содержание которого включено в виде ссылки.
Изобретением является седативное снотворное средство, которое быстро метаболизируется и может быть использовано для достижения и/или подержания анестезии, седативного эффекта или, иначе, понижения чувствительности центральной нервной системы. По сравнению с альтернативными агентами оно обладает одним или несколькими следующими полезными свойствами: более высокая активность, меньшая продолжительность терапевтического действия, меньшая продолжительность действия побочных эффектов, пониженное адренокортикальное подавление, более высокий терапевтический индекс, меньшая токсичность, пониженное подавление сердечно-сосудистой системы и более легкий подбор требуемого эффекта. Изобретение может быть введено путем однократного внутривенного болюсного введения или непрерывной внутривенной капельной инфузии. Другие способы доставки могут включать пероральный, ректальный, трансмукозальный, подкожный или ингаляцию.
Соединения изобретения могут быть получены с использованием способов, описанных в патенте США № 3354173, полное содержание которого включено в виде ссылки. Могут быть применены подходящие модификации исходных соединений с использованием хорошо известных способов в данной области техники. Также соединения изобретения могут быть получены согласно общей синтетической процедуре, которая может быть описана следующим образом. Вначале гидролизуется сложноэфирная связь этомидата или аналога этомидата, давая имидазол-5-карбоновую кислоту. Далее, карбоновая кислота конденсируется с подходящей группой, содержащей сложный эфир (например, линкер - сложный эфир - концевая группа). Конденсация может быть осуществлена с использованием карбодимиидного метода или с использованием других способов, известных в данной области техники. Предпочтительно начинать синтез исходя из (R)-этомидата или его аналога с фиксированной стереохимией.
Приведенные ниже примеры показывают общую синтетическую процедуру, а также конкретное получение соединений настоящего изобретения. Следующие примеры показывают получение соединений настоящего изобретения. Примеры являются пояснительными и не предназначены для того, чтобы каким-либо образом ограничить изобретение, описанное в формуле изобретения
ПРИМЕРЫ
Пример 1
Синтез (R)-1-(1-фенилэтил)-1Н-имидазол-5-карбоновой кислоты (1)
Раствор R-этил-1-(1-фенилэтил)-1Н-имидазол-5-карбоксилата. HCl (R-этомидат.HCl), (281 мг, 1 ммоль) в метаноле (5 мл) и 10% водного NaOH (1,7 мл) кипятили с обратным холодильником в течение 30 минут. После охлаждения раствор нейтрализовали 12,1М HCl (0,351 мл). Смесь сушили с использованием роторного испарителя, осадок суспендировали в системе метанол-дихлорметан 1:4 v/v, и хлорид натрия отфильтровывали. 1-(1-фенилэтил)-1Н-имидазол-5-карбоновую кислоту 1 получали с использованием хроматографии на колонке с силикагелем, уравновешенной системой метанол-дихлорметан 1:4 v/v. 1H ЯМР спектр: (CD3OD) δ 9,30(д, 1H, имидазол CH), 8,23 (д, 1H, имидазол CH), 7,37 (м, 5H, фенил), 6,64 (кв, 1H, метин), 1,97 (д, 3H, метил). См. Схему 1.
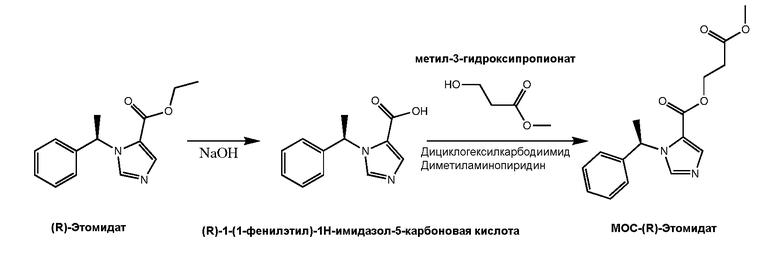
Схема 1
Пример 2
Синтез Метил-3-гидроксипропионата (2)
Соединение получали, как описано у Bartlett и Rylander (см. Bartlett, P.D.И Rylander, P.N., J. Amer. Chem. Soc, 73: 4273-4274 (1951), чье полное содержание включали в виде ссылки). β-Пропионлактон (4,36 г, 60,5 ммоль) по каплям добавляли к перемешиваемому раствору метоксида натрия (121 мг, 2,24 ммоль) в безводном метаноле (15 мл) при - 78°С. Смесь нейтрализовали добавлением эквивалентного количества HCl (2,24 мл 1М HCl). Смесь фильтровали, упаривали на роторном испарителе для удаления метанола, и маслянистый остаток перегоняли при пониженном давлении, получая метил-3-гидроксипропионат 2 (2,7 г, 43%). 1H ЯМР спектр: (CDCl3) δ 3,88 (т, 2H, метилен), 3,73 (с, 3H, метил), 2,59 (д, 2H, метилен).
Пример 3
Синтез (R)-3-Метокси-3-оксипропил-1-(1-фенилэтил)-1Н-имидазол-5-карбоксилата (МОС-(R)-Этомидат, 3)
К смеси (R)-1-(1-фенилэтил)-1Н-имидазол-5-карбоновой кислоты 1 (1 ммоль) и метил-3-гидроксипропионата (115 мг, 1,1 ммоль) в безводном дихлорметане (3,5 мл) добавляли дициклогексилкарбодиимид (139 мг, 1,1 ммоль) и п-диметиламинопиридин (134 мг, 1,1 ммоль). Раствор перемешивали при комнатной температуре в течение 48 часов. Осадок отфильтровывали, и прозрачный раствор наносили на колонку с силикагелем, уравновешенную дихлорметаном. Элюирование 10% эфиром в дихрометане давал продукт, который далее очищали с использованием препаративной тонкослойной хроматографии в системе гексан-этилацетат 1:1 v/v на 1 мм толщины силикагельной пластины. Маслянистый продукт обрабатывали HCl в безводном этилацетате, получая белый кристаллический 3-метокси-3-оксипропил-1-(1-фенилэтил)-1Н-имидазол-5-карбоксилат. HCl (МОС-(R)-этомидат. Гидрохлорид) (200 мг, 59 %). 1H ЯМР спектр: (CDCl3) δ 8,92 (д, 1H, имидазол CH), 7,76 (д, 1H, имидазол CH), 7,36 (м, 5H, фенил), 6,49 (кв, 1H, метин), 4,60 (м, 2Н, метилен), 3,73 (с, 3Н, метил), 2,76 (т, 2Н, метилен) 2,01 (д, 3H, метил).
Пример 4
МОС-(R)-этомидат является сильнодействующим общим анестетиком для головастиков
Для исследования анестезирующей активности использовали тест на потерю выпрямительного рефлекса у головастиков. Группы 5 головастиков Xenopus laevis, находящиеся на ранней стадии зачатка конечности, помещали в 100 мл насыщенного кислородом водного буферного раствора 2,5 мМ буфера Трис HCl (рН=7) и содержащего концентрацию МОС-(R)-этомидата в диапазоне 0,1-128 мкМ. Для структуры МОС-(R)-этомидата см. Схему 1.
Головастиков вручную переворачивали через каждые 5 минут пипеткой, обработанной на пламени. Подразумевается, что головастики находились под действием анестезии, если они не смогли выпрямиться в течение 5 секунд. При всех концентрациях, данная потеря ответа выпрямительного рефлекса стабилизировалась в течение 30 минут после воздействия МОС-(R)-этомидатом. Никакие данные по токсичности не наблюдались; все подвергшиеся анестезии головастики восстанавливали свои выпрямительные рефлексы при возвращении в свежую воду, насыщенную кислородом.
На фиг.1 показана кривая зависимости ответа от концентрации для анестезии.
Доля головастиков, находящаяся под действием анестезии, в каждой группе увеличивалась с ростом концентрации MOC-(R)-этомидата, и при наибольших концентрациях MOC-(R)-этомидата (48-128 мкМ) все головастики находились под действием анестезии. Исходя из этих данных, анестезирующая ЕС50 MOC-(R)-этомидата (т.е. концентрация, при которой 50% головастиков находились под анестезией) была определена как 8±2 мкМ.
Пример 5
МОС-(R)-Этомидат значительно усиливает функцию ГАМК А рецептора
МОС-(R)-Этомидат разработан для индуцирования анестезии за счет молекулярного механизма, аналогичного (R)-этомидату: за счет усиления функции ГАМКА рецептора. ГАМКА рецепторы человека, состоящие из α1β2γ2L субъединиц, экспрессировали в Xenopus laevis ооцитах и использовали для сравнения эффектов МОС-(R)-этомидата и (R)-этомидата на ГАМКА рецептор-опосредованные токи с использованием двухмикроэлектродного метода фиксации напряжения. Данная комбинация субъединиц была выбрана из-за того, что она образует наиболее распространенный подтип ГАМКА рецептора в мозге, и известно, что она является чувствительной к этомидату.
В каждом ооците определяли концентрацию ГАМК, которая индуцирует ответ на ток, чья максимальная амплитуда составляет 5-10% от той, которая индуцируется за счет 1 мМ ГАМК (ГАМК концентрация насыщения рецептора). Данная субмаксимальная концентрация называется ЕС5-10 концентрацией ГАМК. Для оценки и сравнения эффектов МОС-(R)-этомидата и (R)-этомидата на ГАМК-эргические токи измеряли «контрольный» ток, индуцированный ЕС5-10 ГАМК в чистом виде. После 5 минутного периода восстановления измеряли «тестовое» пиковое значение тока, воздействуя на ооциты анестезирующим средством в течение 90 секунд и затем как анестезирующим средством, так и ЕС5-10 ГАМК в течение 90 секунд. После следующего 5 минутного периода восстановления повторяли контрольный эксперимент для оценки обратимости. На фиг.2 приведен типичный контроль и кривые анализов, полученные в отсутствии и в присутствии анестезирующего средства, соответственно для одинаковых ооцитов. Было обнаружено, что при анестезирующей ЕС50 (т.е. 8 мкМ) МОС-(R)-этомидат усиливает амплитуды токов, вызванные ГАМК, на 450±130% (n=6 ооцитов). Данные результаты аналогичны усилению, индуцированному (R)-этомидатом (660±240%) при его анестезирующей ЕС50 (т.е. 2 мкМ) для такого же набора ооцитов. Прямая активация также наблюдалась для малых токов, вызванных как МОС-(R)-этомидатом, так и (R)-этомидатом, даже до применения ГАМК.
Далее была исследована способность МОС-(R)-этомидата и (R)-этомидата сдвигать кривую зависимости ответа от концентрации ГАМК влево (см. фиг.3). В данных экспериментах пиковый ток ответа, полученный при каждой концентрации ГАМК, был нормализован к максимальному ответу, вызванному 1 мМ ГАМК. При их анестезирующих ЕС50 концентрациях, МОС-(R)-этомидат и (R)-этомидат усиливали токи, вызванные низкими концентрациями ГАМК, но обладают относительно малым эффектом на токи, вызванные высокими концентрациями ГАМК. Это сдвигало кривые зависимости ответа от концентрации ГАМК влево, снижая ГАМК ЕС50 (т.е. концентрацию ГАМК, которая вызывала 50% максимального ответа) от 12,7±0,4 мкМ в отсутствии анестетика до 3,3±0,1 мкМ в присутствии МОС-(R)-этомидата и 1,6±0,1 в присутствии (R)-этомидата, соответственно. Коэффициент Хилла находился в диапазоне 1,5-1,8.
Пример 6
In vitro метаболизм МОС-(R)-этомидата в >100 быстрее, чем (R)-этомидата
Скорость метаболизма in vitro (в объединенной фракции печени человека S9) МОС-(R)-этомидата сравнивали со скоростью метаболизма (R)-этомидата. Фракция печени человека S9 была выбрана из-за того, что она богата большим разнообразием ферментов, метаболизирущих лекарства (включая эстеразы), и она традиционно используется при анализах стабильности лекарств по отношению к метаболизму. Так как вероятно, что печень будет являться органом, ответственным за метаболизм МОС-(R)-этомидата in vivo, то она также представляет соответствующий источник ферментов для исследований метаболизма in vitro.
10 мкМ МОС-(R)-этомидата или (R)-этомидата инкубировали при 37°С с 0,3 мг/мл объединенной фракцией печени человека S9, содержащей 1 мМ НАДФН. В различные временные точки (0, 5, 10, 20 и 40 минут) отбирали 100 мкл аликвоты реакционной смеси, и их метаболизм останавливали добавлением 200 мкл ацетонитрила. Аликвоту центрифугировали и концентрацию (неметаболизированного) анестезирующего средства в надосадочной жидкости измеряли количественно с использованием ВЭЖХ с масс-спектрометрическим детектированием.
Фиг.4 является полулогарифмическим графиком зависимости процента неметаболизированного оставшегося анестетика как функция времени инкубации во фракции печени человека S9. Даже спустя 40 минут детектировался неметаболизированный (R)-этомидат, показывая, что его метаболический период полураспада намного больше 40 минут. Совершенно иначе, МОС-(R)-этомидат быстро подвергался метаболизму во фракции печени человека S9. Концентрация МОС-(R)-этомидата снижалась как процесс первого порядка, достигая <1% исходной концентрации (т.е. <0,1 мкМ) за 40 минут. Метаболический период полураспада МОС-(R)-этомидата был рассчитан как 4,2 минуты. В данных исследованиях в качестве внешнего стандарта был использован буспирон для подтверждения метаболической активности фракции печени. Его метаболический период полураспада составлял 15,4 минуты.
Структуру метаболита, образующегося спустя 40 минут инкубации в объединенной фракцией печени человека S9 (+ никатинамидадениндинуклеотидфосфат), анализировали с использованием высокоэффективной жидкостной хроматографии/тандемной масс-спектрометрии. Ионная хроматограмма зарегистрировала наличие лишь одного метаболита. Он имел молекулярный вес 288, что соответствует карбоновой кислоте, образующейся при гидролизе удаленного сложноэфирного остатка МОС-(R)-этомидата. Основываясь на данных результатах, мы заключили, что быстрый метаболизм МОС-(R)-этомидата происходил исключительно за счет предложенного пути, показанного на Схеме 2, в котором удаленный сложноэфирный остаток МОС-(R)-этомидата гидролизуется, давая соответствующий аналог карбоновую кислоту наряду с метанолом в виде уходящей группы.
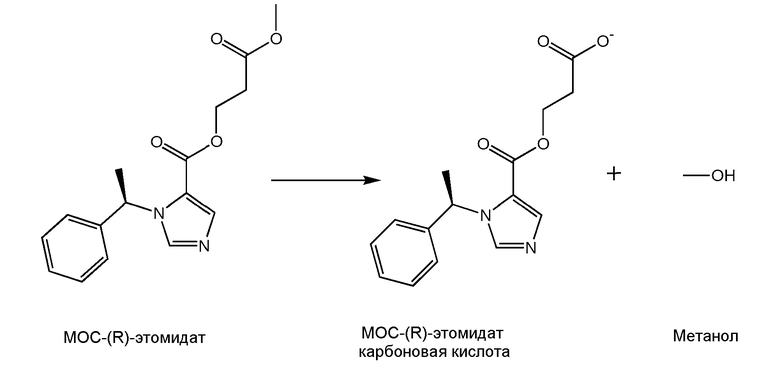
Схема 2
Пример 7
Метаболит МОС-(R)-этомидата обладает малым или не обладает анестезирующим действием
Метаболит МОС-(R)-этомидата (т.е. МОС-(R)-этомидат карбоновая кислота) получали за счет гидролиза МОС-(R)-этомидата в фосфатном буферном растворе, содержащем ~ 1 единицу/мл эстеразы печени свиньи. В ходе гидролиза рН поддерживали при 8,4 за счет добавления NaOH. Затем продукт реакции очищали на ТСХ пластине. ЯМР спектроскопия подтверждает >99% МОС-(R)-этомидата подвергался гидролизу до ожидаемого метаболита карбоновой кислоты.
Метаболит исследовали на анестезирующую активность с использованием теста на потерю выпрямительного рефлекса у головастиков. В данном тесте 5 головастиков помещали в 20 мл стаканы, содержащие метаболит при концентрации 1000 мкМ. Даже спустя 60 минут никакой потери выпрямительного рефлекса у головастиков не наблюдалось, показывая, что метаболит не обладает значительной анестезирующей активностью.
Пример 8
Метаболит МОС-(R)-этомидата обладает малым или не обладает эффектом на функцию ГМКА А рецептора
Усиление активности ГАМКА рецептора метаболитом МОС-(R)-этомидата было оценено с использованием двухмикроэлектродного метода фиксации напряжения. На фиг.5 показано, что даже при концентрациях до 100 мкМ, метаболит МОС-(R)-этомидата не обладает значительным влиянием на ГАМКА рецептор-опосредованные токи.
Пример 9
Метаболит МОС-(R)-этомидата обладает малым или не обладает эффектом на синтез стероидов in vitro
Способность ингибирования синтеза стероидов in vitro метаболитом МОС-(R)-этомидата была оценена с использованием адренокортикальной линии клеток человека H295R (NCI - H295R; ATCC #CRL-2128). Клетки H295R экспрессировали большинство ключевых ферментов, необходимых для биосинтеза стероидных гормонов, включая все ферменты, требуемые для биосинтеза кортизола (например, 11β-гидроксилаза). При стимулировании форсколином данные клетки продуцируют кортизол и секретируют его в среду, где он может быть легко измерен. Ингибирование 11β-гидроксилазы блокирует синтез кортизола, снижая концентрацию кортизола в анализируемой среде.
Клетки H295R выращивали почти до конфлюэнтности в среде для выращивания (DMEM/F12, дополненной 1% ITS, содержащей инсулин, трансферрин, селен и линолевую кислоту, 2,5% NuSerum и Pen/Strep). Среду для выращивания помещали в анализируемую среду, которая обеспечивает синтез кортизола (DMEM/F12, дополненную 1% ITS и 20 мкМ форсколина) наряду либо с (R)-этомидатом, или МОС-(R)-этомидатом, или их метаболитами (или без ничего для контроля). После 48 часов синтеза кортизола, вызванного форсколином, 1,2 мл анализируемой среды отбирали, центрифугировали (для удаления клеток и дебриса) и концентрацию кортизола в надосадочной жидкости измеряли с помощью иммуноферментного анализа.
На фиг.6 приведено сравнение ингибирующего действия (R)-этомидата и метаболита МОС-(R)-этомидата на синтез кортизола клетками H295R. Концентрация (R)-этомидата, необходимая для снижения концентрации кортизола в анализируемой среде на 50% (т.е. IC50), составила 1,3±0,2 нМ, тогда как данная концентрация для метаболита МОС-(R)-этомидата была, по крайней мере, в 1000 раз больше, так как даже при концентрации 1 мкМ не наблюдалось снижение концентрации кортизола в анализируемой среде на 50%. Данные результаты показывали, что метаболит МОС-(R)-этомидата не обладает ингибирующим действием на синтеза кортизола клетками H295R.
Пример 10
МОС-(R)-этомидат является сильнодействующим общим анестетиком с ультракоротким временем действия на крысах
В течение короткого времени крыс удерживали в акриловой камере диаметром 3 дюйма и длиной 9 дюймов с выходным отверстием в концевой части. Необходимую дозу анестетика вводили через катетер в боковой хвостовой вене с последующим промыванием примерно 1 мл изотонического раствора. Сразу же после инъекции крыс удаляли из ограничивающего устройства и переворачивали навзничь. Считается, что у крыс происходит потеря выпрямительного рефлекса, если им не удается перевернуться (на все четыре лапы) в течение 5 секунд после ведения лекарства. Для измерения продолжительности потери выпрямительного рефлекса, что определяется как время от введения лекарства до того момента, когда животное самостоятельно переворачивается, использовался секундомер. ED50 потери выпрямительного рефлекса при болюсном введении анестетика определялся исходя из анестезирующего дозозависимого эффекта потери выпрямительного рефлекса.
На фиг.7 приведены зависимости ответа от концентрации пропофола, этомидата и МОС-(R)-этомидата для потери выпрямительного рефлекса у крыс. Количество крыс, у которых наблюдалась потеря выпрямительного рефлекса, увеличивалось с ростом дозы анестетика. При более высоких уровнях дозы, все крысы находились под анестезией, и явной анестезирующей токсичности не наблюдалось. Из этих данных следует, что значения ED50 для потери выпрямительного рефлекса, наблюдаемого после болюсного введения этомидата, пропофола и МОС-(R)-этомидата, были определены как 1,00±0,03 мг/кг (n=18), 4,1±0,3 мг/кг (n=20) и 5,2±1 мг/кг (n=20), соответственно. При уровнях дозы, достаточных для индуцирования потери выпрямительного рефлекса у крыс, все три анестетика индуцировали потерю выпрямительного рефлекса в течение нескольких секунд после внутривенного болюсного введения. Длительность потери выпрямительного рефлекса (измеряется как время, необходимое крысам для того, чтобы придти в сознание и перевернуться на все четыре лапы) увеличивалась практически линейно от логарифма дозы анестетика (фиг.7В); однако, тангенс угла наклона данной зависимости, который зависит от периода полувыведения в мозге, для МОС-(R)-этомидата был на порядок меньше (2,8±0,4), чем для этомидата (27±7) или пропофола (22±4). Тангенсы угла наклона для этомидата и пропофола незначительно отличались друг от друга. Из полученных данных очевидно, что при эквивалентных анестезирующих дозах, продолжительность потери выпрямительного рефлекса в ~10 раз короче для МОС-(R)-этомидата, чем для пропофола или (R)-этомидата.
Пример 13
МОС-(R)-этомидат обладает лучшей гемодинамической стабильностью по сравнению с пропофолом и (R)-этомидатом
Зачастую этомидат предпочитают другим агентам для обеспечения анестезии у тяжелобольных пациентов, так как он лучше сохраняет гемодинамическую стабильность. Для определения будет ли МОС-(R)-этомидат аналогично сохранять гемодинамическую стабильность, мы измеряли и сравнивали действие пропофола, этомидата и МОС-(R)-этомидата и носителя (35% v/v пропиленгликоля в воде) на частоту сердечных сокращений и кровяное давление у крыс. Для сравнения данных лекарственных средств при эквивалентных анестезирующих уровнях дозы, каждое средство вводили внутривенно при их двойной концентрации ED50 потери выпрямительного рефлекса (т.е. 2 мг/кг этомидата, 10 мг/кг МОС-(R)-этомидата и 8 мг/кг пропофола). Объем введенного пропиленгликоля был одинаковым для групп носителя, этомидата и МОС-(R)-этомидата. После адаптации животных данные регистрировались в течение 5 минут до (базовая линия) и в течение 15 минут после введения лекарства/носителя (фиг.8). Крысы в каждой группе имели аналогичные средние значения частоты сердечных сокращений и кровяного давления базовой линии в течение первых 5 минут (391±49 ударов в минуту (BPM), 118±9 мм рт.ст.). Носитель не вызывал значительных изменений среднего значения кровяного давления по сравнению с базовой линией (5±11 мм рт.ст., n=3, при 90 секунд); для упрощения на фиг.9 данные не приведены. Однако МОС-(R)-этомидат, этомидат и пропофол (n=3 животного для каждого), каждый вызывал значительное снижение среднего значения кровяного давления по сравнению с базовой линией и к друг другу в данном ряду как по максимальной величине (-11±15 мм рт.ст., -36±11 мм рт.ст. и -51±19 мм рт.ст.), так и по длительности значимого эффекта (30 секунд, 6,5 минут и 7 минут, соответственно). Вскоре после инъекции, для всех групп, носитель (36±14 BPM), МОС-(R)-этомидат (24±33 BPM), этомидат (49±67 BPM) и пропофол (64±56 BPM) давали малое, неустойчивое и изменчивое увеличение частоты сердечных сокращений.
Пример 14
В отличие от (R)-этомидата, МОС-(R)-этомидат не подавляет функцию коры надпочечников в течение 30 минут после приема.
В ранее опубликованных сообщениях были адаптированы и оптимизированы способы изучения функции надпочечников у крыс. Сразу же после взвешивания и установки катетера для внутривенного введения каждой крысе вводили дексаметазон (0,2 мг/кг внутривенно; American Regent, Shirley, NY) для ингибирования высвобождения эндогенного адренокортикотропного гормона (АКТГ) с целью подавления образования кортикостерона для базовой линии и для ингибирования изменяющейся стрессовой реакции на удержание и манипуляцию. Катетер хвостовой вены, используемый как для введения лекарственного средства, так и для заборов крови, каждый раз после использования заполняли гепарином, используя 10 ед/мл гепарина, для поддержания его в открытом состоянии; для минимизации гепаринизации крысы и образца перед введением лекарственного средства и забором крови раствор гепарина для заполнения удаляли, впитывая тампоном. Все заборы крови по объему составляли примерно 0,3 мл. После введения всех лекарственных средств проводили промывку 1 мл изотонического раствора для обеспечения полной доставки лекарственного средства.
Спустя два часа после обработки дексаметазоном, кровь забирали (для регистрации базовой концентрации кортикостерона в сыворотке), и вводили вторую дозу дексаметазона либо вместе с внутривенным введением анестетика, либо носителя (35% пропиленгликоль v/v в воде) для контроля. Для стимулирования образования кортикостерона спустя пятнадцать минут внутривенно давали АСТН1-24 (25 мкг/кг; Sigma-Aldrich Chemical Co, St. Louis, MO). Спустя пятнадцать минут после введения АСТН1-24 (т.е. спустя 30 минут после введения анестезирующего средства или носителя) забирали второй образец крови для измерения концентрации кортикостерона в сыворотке, стимулированного АСТН1-24. АСТН1-24 растворяли 1 мг/мл в дезоксигенированной воде в качестве исходного раствора, отбирали аликвоты и замораживали (-20°С); свежую аликвоту размораживали непосредственно перед каждым применением. Крысы во всех трех группах (носитель, этомидат и МОС-(R)-этомидат) получали одинаковый объем пропиленгликоля.
Перед центрифугированием при 3500 g в течение 5 минут образцам крови позволяли свернуться при комнатной температуре (от 10 до 60 минут). Перед вторым центрифугированием при 3500 g в течение 5 минут сыворотку тщательно отделяли от любого полученного поверхностного фибринового сгустка, используя прозрачный микродозатор. После второго центрифугирования полученный слой сыворотки соломенного цвета, не содержащий сгустков, переносили в новую виалу для конечного центрифугирования при высоких скоростях (16000 g, в течение 5 минут) для осаждения каких-либо частиц или красных кровяных клеток. Сыворотку переносили в чистую виалу и быстро замораживали (-20°С), оставляя до измерения кортикостерона на 1-2 дня. После оттаивания и инактивации при нагревании (65°С в течение 20 минут) глобулинов, связывающих кортикостерон, измеряли базовую линию сыворотки и концентрацию кортикостерона, стимулированного АСТН1-24 с использованием иммуноферментного анализа (ELISA) (Diagnostic Systems Laboratories, Webster, TX) и 96-луночного планшет-ридера (Molecular Devices, Sunnyvale, CA).
Инъекция АСТН1-24 стимулировала образование адренокортикостероидов, так как у всех крыс наблюдались большие концентрации кортикостерона в сыворотке спустя пятнадцать минут после введения АСТН1-24. Однако на фиг.9 показано, что крысы, получавшие (R)-этомидат за 15 минут перед стимулированием АСТН1-24, имели значительно меньшие концентрации кортикостерона в сыворотке, чем те, которые получали или носитель, или эквивалентную анестезирующую дозу МОС-(R)-этомидата. С другой стороны, крысы, которые получали МОС-(R)-этомидат, имели концентрации кортикостерона в сыворотке, которые не отличались от концентраций у крыс, которые получали только носитель.
Пример 15
Заключение по МОС-(R)-этомидату
МОС-(R)-этомидат является хорошо переносимым аналогом (R)-этомидата, который сохраняет важные благоприятные фармакологические свойства (R)-этомидата, включая быстрое начало действия, высокую эффективность анестезирующего действия и гемодинамическую стабильность. Как и (R)-этомидат, он эффективно усиливает функцию ГАМКА рецепторов, что является предполагаемым механизмом индуцирования анестезии. Однако в противоположность (R)-этомидату, МОС-(R)-этомидат чрезвычайно быстро метаболизируется, обладает ультракоротким временем действия и не вызывает продолжительное адренокортикальное подавление после внутривенного болюсного введения.
МОС-(R)-этомидат является «мягким аналогом» (R)-этомидата. Мягкий аналог является производным исходного соединения, который был специально разработан для того, что бы он подвергался быстрому и предсказуемому метаболизму после оказания своих терапевтических воздействий. Традиционно используемые мягкие аналоги включают опиат ремифентанил и β-блокатор эсмолол. Оба этих соединения содержат лабильные сложноэфирные остатки, быстро гидролизующиеся до карбоновых кислот за счет эстераз, которые находятся в различных органах и/или в крови. Период полувыведения этих двух лекарственных средств у человека на 1-2 порядка короче, чем для их неэтерифицированных аналогов фенантила и пропранолола. (R)-этомидат также содержит сложноэфирный остаток, который гидролизуется эстеразами печени до карбоновой кислоты, однако он является слабым субстратом для данных эстераз, что отражает его период полувыведения, составляющий несколько часов. По сравнению со структурами ремифентанила и эсмолола, структура (R)-этомидата предполагает две причины низкой скорости гидролиза сложного эфира (R)-этомидата. Во-первых, сложноэфирный остаток (R)-этомидата присоединен непосредственно к его имидазольному кольцу, тогда как лабильные сложноэфирные остатки в ремифентаниле и эсмололе присоединены к циклическим структурам через спейсер, состоящий из двух СН2 групп. Данный спейсер может иметь решающее значение, так как он снижает стерическое препятствие, давая эстеразам свободный доступ к карбонильной группе. В подтверждение этого предположения, при уменьшении длины спейсера эсмолола снижается скорость гидролиза его сложного эфира. Во-вторых, электроны карбонильной группы (R)-этомидата вносят вклад в π-электронную систему, которая распространяется на имидазольное кольцо. Это снижает частичный положительный заряд на карбонильном атоме углерода, что делает его более слабым субстратом для нуклеофильной атаки эстеразами. Основываясь на данных причинах, мы разработали стратегию добавления нового сложноэфирного остатка к (R)-этомидату, который является и стерически незатрудненным, и изолированным от π-электронных систем имидазольного кольца, для получения аналога (R)-этомидата, который будет быстро подвергаться метаболизму. Мы ожидали, что данный сложноэфирный остаток, аналогично остаткам ремифентанила и эсмолола, будет быстро гидролизоваться эстеразами, находящимися в различных тканях и/или в крови. Это было подтверждено нашими исследованиями метаболизма МОС-(R)-этомидата in vitro, которые показали, что данный остаток быстро метаболизируется до карбоновой кислоты в объединенной фракции печени человека S9, используя традиционный анализ биотрансформации in vitro.
Наши исследования показали, что МОС-(R)-этомидат является общим анестетиком в двух видах. Он обладает анестезирующим действием, которое составляет 1/4-1/5 часть от силы действия (R)-этомидата, и также индуцирует анестезию через аналогичный рецепторный механизм (т.е. усиление функции ГАМКА рецепторов). Наши исследования на крысах показали, что МОС-(R)-этомидат является анестетиком с ультракоротким временем действия даже при многократных ED50 потери выпрямительного рефлекса. Считается, что восстановление сознания после анестезии от внутривенного болюсного введения пропофола и (R)-этомидата отражает перераспределение лекарственного средства от мозга к другим тканям, а не происходит за счет метаболизма. Следовательно, аналогичный тангенс угла наклона в зависимости между длительностью потери выпрямительного рефлекса и логарифма анестезирующей дозы (фиг.7В) означает, что пропофол и (R)-этомидат перераспределяются из мозга с близкими скоростями. Намного более быстрое восстановление сознания после анестезии и слабый наклон данной зависимости для МОС-(R)-этомидата означает, что для прекращения анестезирующего действия МОС-(R)-этомидата значительный вклад вносит сверхбыстрый метаболизм.
МОС-(R)-этомидат соответственно индуцирует короткое (30 секунд) снижение кровяного давления, позволяя предположить, что гемодинамические эффекты МОС-(R)-этомидата также прекращаются за счет метаболизма. Кроме этого мы обнаружили, что максимальная величина данного снижения значительно меньше после введения МОС-(R)-этомидата, чем после введения эквивалентных анестезирующих доз (R)-этомидата и пропопофола.
Наряду с другими гидрофобными соединениями, содержащими имидазол, (R)-этомидат подавляет образование адренокортикостероидов. Основной механизм, лежащий в основе данного подавления, состоит в ингибировании 11β-гидроксилазы, важного фермента биосинтетического пути, приводящего к адренокортикальному синтезу кортизола, кортикостерона и альдостерона. Была выдвинута гипотеза, что (R)-этомидат ингибирует 11β-гидроксилазу за счет конкурентного связывания со стероидным предшественником в возможном гидрофобном каталитическом сайте фермента. Так как МОС-(R)-этомидат был разработан для того, чтобы он быстро подвергался метаболизму эстеразами, давая сильнополярную карбоновую кислоту, мы ожидали, что МОС-(R)-этомидат не будет индуцировать после введения продолжительное адренокортикальное подавление. Данное предположение было подтверждено, в течение тридцати минут после введения МОС-(R)-этомидат не дает снижения концентрации кортикостерона в сыворотке, стимулированного АСТН1-24, тогда как эквивалентная анестезирующая доза (R)-этомидата значительно ее снижает. Наши результаты также подразумевают, что после введения однократной внутривенной дозы какой-либо эффект быстро образующегося метаболита(ов) МОС-(R)-этомидата на синтез кортикостерона является пренебрежимо малым.
Несмотря на то что здесь подробно представлены и описаны варианты осуществления, специалисту в данной области техники будет понятно, что могут быть сделаны различные модификации, добавления, замещения и т.п. без отклонения от сущности изобретения, и, следовательно, считается, что они находятся в рамках изобретения, как определено в прилагаемой формуле изобретения, приведенной далее.
Специалист в данной области техники также легко оценит, что настоящее изобретение хорошо приспособлено для выполнения целей и получения конечных целей и указанных преимуществ, а также того, что из этого следует. Описанные здесь молекулярные комплексы и способы, процедуры, обработка, молекулы, конкретные соединения представляют предпочтительные варианты осуществления, являются иллюстративными и не предназначены для ограничений объема изобретения. Изменения в нем и другие применения, которые будут сделаны специалистом в данной области техники, охватываемые сущностью изобретения, ограничены формулой изобретения.
Специалисту в данной области техники также будет очевидно, что изменение замен и модификаций описанного здесь изобретения может быть сделано без отклонения от объема и сущности изобретения.
Все указанные в описании патенты и публикации указывают на уровни специалистов области техники, к которой относится изобретение. Все патенты и публикации включены здесь в виде ссылки в той же мере, как если бы для каждой индивидуальной публикации было специально и индивидуально указано, что она включена в виде ссылки.
Наглядно описанное здесь изобретение надлежащим образом может быть применено на практике в отсутствии какого-либо элемента или элементов, ограничения или ограничений, которые здесь специально не описаны. Таким образом, здесь в каждом случае любой из терминов «включающий», «главным образом состоящий из» и «состоящий из» может быть заменен любым из двух других терминов. Использованные термины и выражения применяются в качестве терминов описания, а не ограничения, и не направлены на то, чтобы при использовании таких терминов и выражений исключались любые эквиваленты показанных и описанных признаков или их частей, но следует понимать, что возможны различные модификации в рамках заявленной формулы изобретения. Таким образом, следует понимать, что хотя настоящее изобретение было конкретным образом описано с помощью предпочтительных вариантов осуществления и возможных признаков, модификации и изменения описанных здесь идей могут быть пересмотрены специалистом в данной области техники, и такие модификации и изменения рассматриваются как подпадающие под объем настоящего изобретения, как определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ ЭТОМИДАТА, КОТОРЫЕ НЕ ИНГИБИРУЮТ СИНТЕЗ АДРЕНОКОРТИКОСТЕРОИДОВ | 2010 |
|
RU2559888C2 |
| СЛОЖНОЭФИРНОЕ ХИРАЛЬНОЕ СОЕДИНЕНИЕ (N-ЗАМЕЩЕННЫЙ ИМИДАЗОЛ)-КАРБОНОВОЙ КИСЛОТЫ, СОДЕРЖАЩЕЕ ПРОСТУЮ ЭФИРНУЮ БОКОВУЮ ЦЕПЬ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2014 |
|
RU2659784C2 |
| АНЕСТЕЗИРУЮЩИЙ СОСТАВ | 2011 |
|
RU2574022C2 |
| СПОСОБ СОЧЕТАННОЙ АНЕСТЕЗИИ ПРИ ЭНДОСКОПИЧЕСКОЙ РИНОСИНУСОХИРУРГИИ | 2021 |
|
RU2779576C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2470907C2 |
| СРЕДСТВО ДЛЯ НАРКОЗА НА ОСНОВЕ 3-АМИНОМЕТИЛЕНИНДОЛИНОНА-2 | 2006 |
|
RU2326113C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2470908C2 |
| МИКРОКОЛЛОИДНЫЙ РАСТВОР ПРОПОФОЛА ДЛЯ АНЕСТЕЗИИ | 2013 |
|
RU2535001C1 |
| Наносуспензия на основе альгинат-хитозанового полиэлектролитного комплекса с гидрохлоридом артикаина для местной анестезии | 2023 |
|
RU2806363C1 |
| ЗАМЕЩЕННЫЕ ЭФИРЫ ФЕНИЛУКСУСНОЙ КИСЛОТЫ В КАЧЕСТВЕ КОРОТКОДЕЙСТВУЮЩИХ СЕДАТИВНЫХ СНОТВОРНЫХ АГЕНТОВ ДЛЯ КРАТКОВРЕМЕННОЙ АНЕСТЕЗИИ И СОЗДАНИЯ СЕДАТИВНОГО ЭФФЕКТА, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2315037C2 |
Изобретение относится к области органической химии, а именно к производным бензимидазола общей формулы (I) и к их фармацевтически приемлемым солям, смесям стереоизомеров и энантиомерам, где R1 является L1C(O)OL2C(O)OT; R2 является незамещенным C1-С10алкилом; L1 является связью; L2 является незамещенным С2-С10алкиленом; Т является C1-С10алкилом. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I) и способу обеспечения анестезии, основанному на использовании соединения формулы (I). Технический результат: получены новые производные имидазола, полезные в качестве анестезирующего агента. 4 н. и 11 з.п. ф-лы, 9 ил., 15 пр.
1. Соединение формулы (I)
где
R1 является L1C(O)OL2C(O)OT;
R2 является незамещенным C1-С10алкилом;
L1 является связью;
L2 является незамещенным С2-С10алкиленом;
Т является C1-С10алкилом; и
его фармацевтически приемлемые соли, смеси стереоизомеров и энантиомеры.
2. Соединение по п.1, где указанное соединение присутствует в виде чистого энантиомера.
3. Соединение по п.2, где указанный энантиомер является R энантиомером.
4. Соединение по п.1, где соединение содержит две сложноэфирных группы.
5. Соединение по п.1, где R2 является СН3, L1 является связью, L2 является СН2СН2 и Т является СН3.
6. Соединение по п.1, где R2 является СН3, L1 является связью, L2 является СН2(СН2)4СН2 и Т является СН2СН2СН2СН3.
7. Соединение по п.1, где R2 является СН3 и L1 является связью.
8. Фармацевтическая композиция для анестезии, содержащая фармацевтически эффективное количество соединения формулы (I)
где
R1 является L1C(O)OL2C(O)OT;
R2 является метилом;
L1 является связью;
L2 является незамещенным С2-С10алкиленом;
Т является C1-С10алкилом; и
фармацевтически приемлемый носитель.
9. Способ обеспечения анестезии у млекопитающего, включающий введение указанному млекопитающему соединения формулы (I) по п.1.
10. Способ обеспечения анестезии у млекопитающего, включающий введение указанному млекопитающему фармацевтической композиции по п.8.
11. Способ по п.9, где композиция содержит соединение формулы (I), где R2 является СН3, L1 является связью, L2 является СН2СН3 и Т является СН3.
12. Способ по п.9, где на стадии введения вводят от 0,01 мг/кг до 100 мг/кг соединения формулы (I).
13. Способ по п.9, где указанное введение включает: инъекцию однократной эффективной дозы соединения формулы (I).
14. Способ по п.9, где указанное введение включает: непрерывную инфузию эффективной дозы соединения формулы (I).
15. Соединение по любому из пп.1-7 для применения для обеспечения анестезии у млекопитающего.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

