Область техники, к которой относится изобретение
Настоящее изобретение относится к способу, позволяющему очищать уран из концентрата природного урана.
Способ находит применение в области очистки концентратов природного урана, которые производят на урановых рудниках, а содержащийся в них уран после извлечения предназначен, в частности, для превращения или в металлический уран, или в соединения урана, такие как, например, гексафторид урана (или UF6), тетрафторид урана (или UF4), диоксид урана (или UO2) или триуран-октоксид (U3O8), представляющие собой промежуточные продукты в производстве ядерного топлива.
Уровень техники
На предприятиях по очистке природных урановых концентратов с целью доведения урана, содержащегося в этих концентратах, до так называемой «ядерной» чистоты, применяют способ, который после растворения указанных концентратов в азотной кислоте для получения водного раствора неочищенного нитрата уранила включает:
- экстракцию этого нитрата органической фазой, содержащей экстрагирующее средство, имеющее высокое сродство к этому нитрату, в органическом растворителе, и затем
- промывание органической фазы, полученной после завершения экстракции, водной фазой для удаления из органической фазы нежелательных химических соединений, которые могут быть экстрагированы вместе с нитратом уранила, и
- извлечение нитрата уранила из промытой таким образом органической фазы для того, чтобы перевести нитрат уранила в водную фазу.
Эти три операции проводят в аппаратах для жидкость-жидкостной экстракции, например, в колоннах с перемешиванием.
Применяемое экстрагирующее средство представляет собой три-н-бутил-фосфат (или ТВР), который также применяют в качестве экстрагирующего средства при переработке облученного ядерного топлива и который был выбран для всех очистительных установок как наиболее подходящий в отношении ограничений, налагаемых этим способом по селективности, растворимости в воде, химической стабильности, плотности, коррозионному разрушению, токсичности и безопасности.
Тем не менее, этот способ очистки имеет несколько недостатков, которые связаны с применением экстрагирующего средства.
Первый из этих недостатков заключается в том, что в аппарате, в котором проводят операцию экстракции, происходит накопление тория (примеси, присутствующей в урановых рудах), которое связано с циклом экстракция/извлечение этого элемента в этом аппарате. Действительно, поскольку сам торий также можно экстрагировать с помощью ТВР, он экстрагируется в той области аппарата, где органическая фаза имеет низкую концентрацию нитрата уранила, и затем удаляется из этой фазы в той области аппарата, где органическая фаза насыщена нитратом уранила.
Это накопление тория приводит к возникновению пика мощности дозы в результате излучения, генерируемого метастабильным протактинием 234 (234mРа) после распада тория 234, присутствующего в растворе неочищенного нитрата уранила, который представляет собой прямой продукт урана 238.
Известно, что для того чтобы избежать появления такого пика мощности дозы, к раствору неочищенного нитрата уранила можно добавить средства, способные в ходе процедуры экстракции образовывать комплексы с торием, которые могут представлять собой фториды или фосфаты. Однако присутствие таких комплексообразующих агентов в получаемых водных эффлюентах налагает дополнительные ограничения при применении способа и этих эффлюентов, вызванные такими факторами, как: загрязнение регенерированной азотной кислоты и дистиллятов, коррозия используемого оборудования, образование осадка и т.п.
Другой недостаток заключается в том, что операцию извлечения относительно трудно выполнять из-за экстракции урана ТВР, которой нельзя пренебречь даже при низкой кислотности. Следовательно, для количественного извлечение нитрата уранила необходимо нагревать водную фазу, которую применяют для выполнения этой операции (обычно дистиллированную воду), по меньшей мере до 50°С и использовать значительные скорости потоков жидкостей.
В дополнение к тому, что использование водной фазы, имеющей температуру равную по меньшей мере 50°С, приводит к снижению безопасности установки из-за риска возгорания органической фазы, использование значительных потоков жидкостей приводит к существенному разбавлению нитрата уранила. Так, если концентрация нитрата уранила исходно была равна величине порядка 400 г/л в неочищенном растворе нитрата уранила, то в водной фазе, вытекающей после операции извлечения, она в лучшем случае составляет 130 г/л. Насыщенная ураном органическая фаза содержит примерно 150 г/л урана на выходе со стадии экстракции. Для того чтобы избежать потерь урана в этой органической фазе, максимальное отношение О/А (органической к водной) скорости потока при извлечении должно быть меньше 1 (порядка 0,8) даже при 60°С.
Если в процессе извлечения концентрирование оказалось недостаточным, то затем, как следствие, перед последующей обработкой путем денитрирования необходимо подвергнуть эту водную фазу операции концентрирования, которая требует большого расхода энергии.
Наконец, из-за растворимости ТВР в кислой водной фазе, которой нельзя пренебречь, может возникнуть необходимость в промывании водных эффлюентов, полученных при экстракции, органическим растворителем для извлечения ТВР, присутствующего в этих эффлюентах, чтобы таким образом избежать перерасхода экстрагирующего средства.
Следовательно, учитывая вышеизложенное, авторы изобретения поставили перед собой цель создать способ, который, позволяя проводить высокоэффективную очистку урана, содержащегося в природном урановом концентрате, в то же время был бы лишен вышеупомянутых недостатков.
Раскрытие изобретения
Эту цель и другие дополнительные цели достигают с помощью настоящего изобретения, в котором раскрыт способ, позволяющий очищать уран из природного уранового концентрата, содержащего по меньшей мере одну из следующих примесей: торий, молибден, цирконий, железо, кальций и ванадий, включающий:
a) экстракцию урана, присутствующего в виде нитрата уранила в водной фазе А1, полученной в результате растворения указанного природного уранового концентрата в азотной кислоте, путем приведения этой водной фазы в контакт с органической фазой, несмешиваемой с водой, которая содержит экстрагирующее средство в органическом растворителе, и последующего разделения указанных водной и органической фаз;
b) промывание органической фазы, полученной после завершения стадии а), путем приведения этой органической фазы в контакт с водной фазой А2 и последующего разделения указанных органической и водной фаз; и
c) извлечение нитрата уранила из органической фазы, полученной после завершения стадии b), выполняемое путем циркуляции этой органической фазы в аппарате в виде противотока по отношению к водной фазе A3, и разделения указанных органической и водной фаз;
и характеризующийся тем, что, с одной стороны, экстрагирующее средство, содержащееся в органической фазе, представляет собой N,N-диалкиламид, и, с другой стороны, отношение между скоростями потоков, при которых органическая фаза, полученная в конце стадии Ь), и водная фаза A3 циркулируют в аппарате, где протекает стадия с), больше 1, так что извлечение нитрата уранила сопровождается концентрированием этого нитрата.
Таким образом, в способе по изобретению повторяют три основные операции (экстракция, промывание, извлечение) известного из уровня техники способа извлечения, но применяют N,N-диалкиламид в качестве экстрагирующего средства, вместо ТВР.
Следует отметить, что применение органической фазы, содержащей N,N-диалкиламид, для экстракции урана из водной фазы, в которой он был обнаружен, не представляет собой новое применение per se. Так, например, такое применение было предложено для экстракции урана и/или плутония, присутствующих в водных растворах, полученных в результате растворения облученного ядерного топлива (Французская заявка на патент, опубликованная под номером 2 642 562), а также для экстракции урана, присутствующего в водных растворах с концентрированным торием, поскольку его получают облучением этого тория (Французская заявка на патент, опубликованная под номером 2 642 561).
С другой стороны, новым является применение органической фазы, содержащей N,N-диалкиламид для очистки урана из уранового концентрата, и является неожиданным, что это применение одновременно дает возможность для:
- того, чтобы избежать накопления тория тогда, когда операцию экстракции осуществляют с использованием содержащей ТВР органической фазы, что приводит к снижению необходимости использования комплексообразующих веществ со всеми вытекающими преимуществами, в особенности с точки зрения упрощения способа применения и обработки полученных водных эффлюентов;
- осуществления операции извлечения при температурах ниже тех, которые применяют в известном из уровня техники способе очистки из, даже если такое извлечение, тем не менее, может выполняться при температурах от 50 до 60°С, и применения более низкой скорости потока водной фазы, по сравнению со скоростями, которые требуются в случае, когда органическая фаза содержит ТВР, что, как следствие, облегчает последующую операцию концентрирования также со всеми вытекающими преимуществами, в особенности, с точки зрения экономии энергии;
- ограничения потерь экстрагирующего средства в водной фазе из-за низкой растворимости N,N-диалкиламидов в водной фазе, и облегчения переработки экстрагированных рафинатов для повторного использования азотной кислоты в способе.
Что касается N,N-диалкиламидов, можно в особенности применять те, которые соответствуют формуле (I), приведенной ниже:
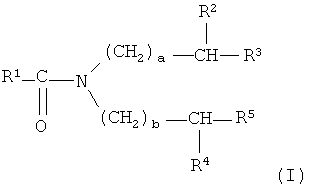
в которой:
- R1 представляет собой алкильную группу, разветвленную в альфа или бета положении от карбонильной группы и включающую от 3 до 12 атомов углерода;
- R2 и R4, которые могут быть одинаковыми или разными, представляют собой линейную или разветвленную алкильную группу, включающую от 2 до 4 атомов углерода;
- R3 и R5, которые могут быть одинаковыми или разными, представляют собой линейную или разветвленную алкильную группу, включающую от 1 до 6 атомов углерода; и
- а и b, которые могут быть одинаковыми или разными, представляют собой целые числа в диапазоне от 1 до 6.
Среди этих N,N-диалкиламидов предпочтительно применять те, в которых оба а и b равны 1 и в которых оба R2 и R4 представляют собой этильные группы, подобные N,N-ди- (2-этилгексил) изобутирамид (или DEHiBA, где R1=-СН(СН3)2 и R3=R5=-(СН2)3СН3), N,N-ди-(2-этилгексил)-2,2-ди-метилпропанамид (или DEHDMPA, где R1=-С(СН3)3 и R3=R5=-(СН2)3СН3) или дополнительно N,N-ди-(2-этилгексил)-3,3-диметилбутанамид (или DEHDMBA, где R1=-СН2С(СН3)3 и R3=R5=-(СН2)3СН3), где DEHiBA наиболее предпочтителен.
Что касается органического растворителя, то предпочтителен изопарафин или смесь изопарафинов, углеродная цепь которых включает от 9 до 13 атомов углерода, подобных тем, которые поставляет на рынок компания «TOTAL» под коммерческим наименованием Isane IP 185.
Однако также можно применять другие алифатические органические растворители, такие как керосин или линейный или разветвленный додекан, такой как n-додекан и гидрогенизированный тетрапропилен (или ТРН).
Во всех случаях, концентрация N,N-диалкиламида в органическом растворителе предпочтительно равна от 1 до 2 моль/л.
Водная фаза А1 предпочтительно содержит от 0,5 до 4 моль/л азотной кислоты.
Водная фаза А2 предпочтительно представляет собой воду, преимущественно дистиллированную воду, или водный раствор, содержащий от 0,01 до 1,5 моль/л азотной кислоты, или еще дополнительно часть водной фазы, полученной после завершения стадии с).
Что касается водной фазы A3, то она может быть или водой, преимущественно дистиллированной водой, или водным раствором азотной кислоты низкой концентрации, т.е. предпочтительно содержащей не более 0,01 моль/л азотной кислоты.
В соответствии с изобретением эта водной фаза A3 может быть применена при комнатной температуре, т.е. при температуре, обычно варьирующейся от 20 до 25°С, но она также может быть нагрета до температуры, равной от 50 до 60°С, как в известном из уровня техники способе извлечения.
Как отмечалось ранее на стадии с), отношение между скоростями потока, при которых органическая фаза, полученная в конце стадии Ь), и водная фаза A3 циркулируют в аппарате, где протекает стадия с), больше 1 и, предпочтительно, равно или больше чем 1,5.
Способ по изобретению может быть применен в аппаратах всех типов, традиционно применяемых в области жидкость-жидкостных экстракций, таких как батареи смесителей-декантаторов, пульсационные колонны или колонны с перемешиванием, отжимные центрифуги и т.п.
В дополнение к упомянутым выше преимуществам, способ изобретения дополнительно имеет другие преимущества, такие как тот факт, что, с одной стороны, продукты деградации N,N-диалкиламидов гораздо менее вредны, чем продукты деградации ТВР, и, в особенности, ди-n-бутил-фосфат, который образует прочные комплексы с определенными катионами металлов, и, с другой стороны, N,N-диалкиламиды могут быть полностью сожжены, поскольку они состоят только из атомов углерода, кислорода, азота и водорода, чего нет в случае ТВР.
Другие признаки и преимущества изобретения станут понятны из последующего дополнительного описания, которое относится к экспериментальным тестам и с помощью которых способ изобретения может быть подтвержден.
Очевидно, что это дополнительное описание дано только в качестве иллюстрации цели изобретения и, ни каким образом, не должно быть интерпретировано как ограничение этой цели.
Краткое описание чертежей
Фиг.1 схематически иллюстрирует первое испытание способа по изобретению в батареях смесителей-декантаторов.
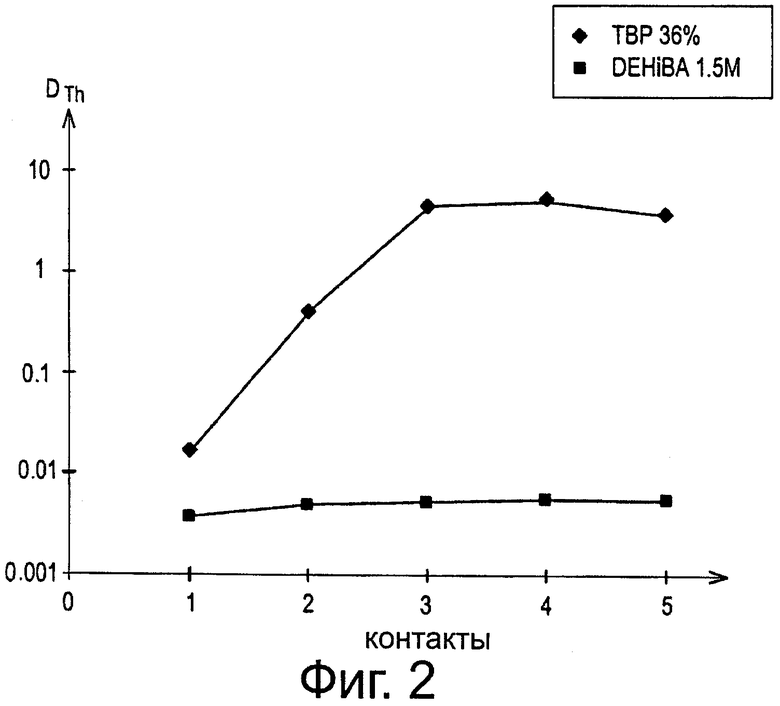
Фиг.2 показывает величины коэффициента распределения тория 234, обозначенного как DTh, наблюдаемые в ходе испытания в трубах, заключающегося в извлечении урана из водного раствора, содержащего 2 моль/л азотной кислоты, с помощью пяти органических фаз, содержащих или DEHiBA, или ТВР в Isane IP 185.
Фиг.3 схематически иллюстрирует другое испытание применения способа изобретения в батареях смесителей-декантаторов.
Фиг.4 показывает профили концентраций урана в водной и органической фазах, полученных экспериментально и по расчетам на разных стадиях в смесителях-декантаторах, примененных в испытании, проиллюстрированном на фиг.3.
Осуществление изобретения Пример 1
Прежде всего, следует рассмотреть фиг.1, которая схематически иллюстрирует первое применение способа по изобретению в батареях смесителей-декантаторов. Это испытание проводят в установке 10, включающей:
- первую батарею, обозначенную как 11, состоящую из 16 смесителей-декантаторов, предназначенных для экстракции нитрата уранила из водной фазы А1;
- вторую батарею, обозначенную как 12, состоящую из 8 смесителей-декантаторов, предназначенных для промывания органической фазы, полученной после завершения экстракции; и
- третью батарею, обозначенную как 13, состоящую из 16 смесителей-декантаторов, предназначенных для извлечения нитрата уранила из органической фазы, полученной после завершения промывания в этой фазе.
Были применены:
- в качестве органической фазы: фаза, содержащая 1 моль/л DEHiBA в ТРН и циркулирующая со скоростью потока, равной 100 мл/час, в трех батареях смесителей-декантаторов;
- в качестве водной фазы А1: раствор 4 моль/л азотной кислоты и 120 г/л урана (в форме нитрата уранила), циркулирующий со скоростью потока 45 мл/час в батарее 11;
- в качестве водной фазы А2: раствор азотной кислоты в концентрации 0,5 моль/л, циркулирующий со скоростью потока 10 мл/час в батарее 12, который добавляют с той же скоростью потока к водной фазе А1 в батарее 11; и
- в качестве водной фазы A3: раствор азотной кислоты в концентрации 0,01 моль/л, циркулирующий со скоростью потока, равной 31,5 мл/час, в батарее 13.
Отношения О/А (органическая по отношению к водной) скоростей потоков в батареях 11,12 и 13 равны 1,8,10 и 3, соответственно.
Все фазы, включая водную фазу A3, использовали при 25°С.
Через 700 часов работы установки тест показал, что в водной фазе, выходящей из батареи 13, возможно извлечь более 99,9% нитрата уранила, исходно присутствовавшего в водной фазе А1, в концентрации, значительно превышающей концентрацию, в которой он находился в водной фазе А1, т.е. 160 г/л по сравнению с 120 г/л, т.е. коэффициент концентрации равен 1,3.
Органическая фаза, выходящая из батареи 12, содержит примерно 54 г/л урана, который количественно извлекают в батарее 13, используя отношение О/А скоростей потоков, равное 3, для получения раствора с 160 г/л урана.
Низкая экстрагируемость урана с DEHiBA при низкой концентрации азотной кислоты, таким образом, дает возможность осуществлять извлечение «с концентрированием» нитрата уранила при температуре 25°С, тогда как этого достичь невозможно, если органическая фаза содержит ТВР, и это даже если для этого извлечения применяют нагревание водной фазы.
Пример 2
Растворимость DEHiBA в водной фазе была оценена путем проведения испытания в смесителях-декантаторах, аналогичного тому, что было описано выше в примере 1, и путем измерения общего количества органического углерода (или ТОС), присутствующего в выходящей из батареи 11 водной фазе (называемой дальше в этом документе «очищенный продукт после экстракции»), с одной стороны, и в водной фазе, выходящей из батареи 13 и содержащей извлеченный нитрат уранила (называемой в этом документе «выработка U»), с другой стороны.
Общее количество органического углерода измеряют с помощью термического ТОС-метра в водных фазах, полученных после декантации, т.е. не подвергая эти фазы какому-либо центрифугированию.
Приведенная ниже таблица 1 показывает обе испытанные водные фазы, их кислотность, полученную величину ТОС и преобразование этой величины в эквивалент DEHiBA, это преобразование, выполняют исходя из предположения, что весь органический углерод присутствует в потоках водных фазах из этого экстрагирующего средства.
Эта таблица показывает, что примерно от 40 до 60 мг/л DEHiBA может быть растворено в водной фазе в условиях испытания.
Эти величины растворимости больше, чем растворимость DEHiBA в водной фазе, согласно результатам работы Al-Jallo et al. (J. Chem. Eng. Data 1984, 29, 479-481) и работы Gasparini и Grossi (Separation Science and Technology 1980, 15(4), 825-844), но они включают растворимость самого экстрагирующего средства, растворимость органического растворителя (ТРН), а также явление вынесения органической фазы в водные растворы на выходе батареи смесителей-декантаторов, которым нельзя пренебречь.
Эти величины растворимости, тем не менее, остаются меньшими, по сравнению с присущей ТВР растворимостью в тех же условиях, которая равна от 200 до 300 мг/л, и подтверждают тот факт, что потери экстрагирующего средства в водной фазе значительно меньше в случае, когда органическая фаза содержит DEHiBA.
Пример 3
Селективность DEHiBA для урана по отношению к примесям, которые в основном присутствуют в природных концентратах урана или которые приводят к издержкам на последующих стадиях переработки и обогащения урана, была также оценена путем проведения двух серий испытаний.
Первые серии испытаний состояли из определения коэффициентов распределения тория (IV), молибдена (VI), циркония (IV), железа (III), кальция (II) и ванадия (V) в конце единичного контакта в трубах между фазой растворителя, содержащей 1,5 моль/л DEHiBA в Isane IP 185, и растворами азотной кислоты, содержащими эти катионы, как в присутствии 30 г/л урана, так и без него, и имеющие различную кислотность (от 0,5 до 4 М).
Для каждого испытания водную и органическую фазы вводят в контакт, объем к объему, и оставляют перемешиваться в течение 1-го часа при постоянной температуре 25°С.Затем, после декантации и разделения этих фаз в водной фазе и в органической фазе определяют концентрацию катионов с помощью атомно-эмиссионной спектрометрии (или ICP-AES).
Этот первый набор испытаний показал, что с ураном или без него в исследуемом диапазоне концентраций азотной кислоты коэффициенты распределения молибдена, циркония, железа, кальция и ванадий были меньше 10-3 и привели к факторам разделения U/примеси, равным больше 10000, т.е. в значительной степени достаточны для достижения технических требований в соответствии со стандартом ASTM С 788, с точки зрения извлечения нитрата уранила.
Коэффициент распределения тория (IV) также был очень мал (менее 5·10-3) для концентрации азотной кислоты 4 моль/л, и это, независимо от концентрации урана в водной фазе, подтверждает высокую селективность DEHiBA для урана по отношению к торию (IV).
Вторая серия испытаний состояла из истощения водного раствора, содержащего 360 г/л урана и 2 моль/л азотной кислоты, путем введения этого водного раствора в трубах в контакт, со следующими одна за другой пятью органическими фазами, содержащими или 1,5 моль/л DEHiBA, или 36% (объем/объем) ТВР в Isane IP 185, и следующего за каждым контактом определения коэффициента распределения тория 234, образующегося из-за распада урана 238.
Для первых двух контактов водную и органическую фазу применяли в количестве одного объема водной фазы на два объема органической фазы, тогда как для последних трех контактов водную и органическую фазу применяли в объемном соотношении 1 к 1. Во всех случаях водную и органическую фазу оставляли для перемешивания в течение 10 минут при постоянной температуре 25°С.
После декантации и разделения водной и органической фаз определяли активность тория 234 в каждой из этих фаз с помощью γ-спектрометрии.
Результаты этой второй серии испытаний проиллюстрированы на фиг.2, которая показывает величины коэффициента распределения тория 234, обозначенного как DTh, полученные после завершения каждого контакта для обоих типов органических фаз.
Эта фигура показывает, что в случае, если органические фазы содержат ТРВ, DTh, становится больше 1 после 3-его контакта, т.е. после истощения урана водной фазы, тогда как, в случае, когда органические фазы содержат DEHiBA, DTh поддерживается на уровне менее 10" даже после истощения урана в фазе.
Таким образом, накопление тория, которое происходит в аппарате экстракции в известном из уровня техники способе извлечения и имитация которого стала возможной благодаря описанным выше испытаниям, не должно происходить в способе по изобретению.
Пример 4
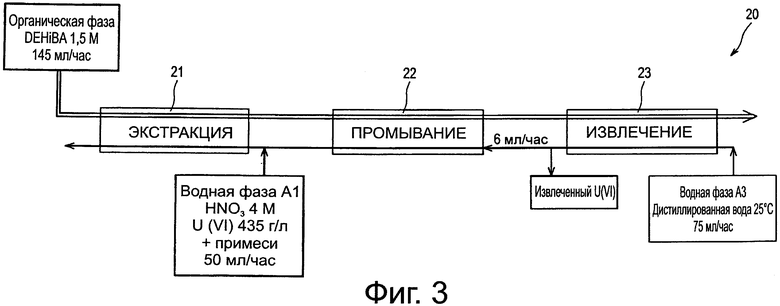
Теперь обратимся к фиг.3, которая схематически иллюстрирует другое испытание применения способа по изобретению в батареях смесителей-декантаторов. Это испытание проводят в установке 20, включающей:
- первую батарею, обозначенную как 21, состоящую из 8 смесителей-декантаторов, предназначенных для экстракции нитрата уранила из водной фазы А1;
- вторую батарею, обозначенную как 22, состоящую из 8 смесителей декантаторов, предназначенных для промывания органической фазы, полученной после завершения экстракции; и
- третью батарею, обозначенную как 23, нагретую до 50°С и состоящую из 8 смесителей-декантаторов, предназначенных для извлечения нитрата уранила из органической фазы, полученной после завершения промывания этой фазы.
Использовали:
- в качестве органической фазы: фазу, содержащую 1,5 моль/л DEHiBA в ТРН и циркулирующую со скоростью потока 145 мл/ч в трех батареях смесителей-декантаторов;
- в качестве водной фазы А1: раствор, содержащий 1,5 моль/л азотной кислоты, 435 г/л урана (в виде нитрата уранила), примерно 5400 кБк/л (т.е. 6 нг/л) тория 234, появляющегося в результате распада урана 238, а также характерные примеси (736 мг/л железа, 359 мг/л молибдена, 258 мг/л циркония, 34 мг/л ванадия и 106 мг/л кальция), и циркулирующий со скоростью потока 50 мл/ч в батарее 21; и
- в качестве водной фазы А2: часть раствора, выходящего из батареи 23, циркулирующую со скоростью потока 6 мл/ч в батарее 22 и добавляемую с той же скоростью потока к водной фазе А1 в батарее 21;
- в качестве водной фазы A3: раствор нагретой дистиллированной воды, циркулирующий со скоростью потока 75 мл/ч в батарее 23.
Отношения О/А (органическая к водной) скоростей потоков в батареях 21, 22 и 23 были, таким образом, равны 2, 6, 24 и 1,9, соответственно.
Через 24 часа работы установки тест показал, что в водной фазе, выходящей из батареи 23, можно извлечь более чем 99% нитрата уранила, исходно присутствовавшего в водной фазе А1, в концентрации, равной 281,5 г/л, т.е. в значительно большей, чем концентрация извлеченного урана в известном из уровня техники способе извлечения (130 г/л). По оценкам потери урана в водной фазе, выходящей из батареи 21, равны 0,2 мг/л.
Торий 234, отслеживаемый методом γ-спектрометрии, снова был количественно обнаружен в водной фазе, выходящей из батареи 21 (т.е. в конце теста извлекли 100% исходного тория), и не накопился в этой батарее, и это без применения специфического комплексообразующего вещества, что, следовательно, подтверждает селективность DEHiBA для урана по сравнению с торием.
Органическая фаза, выходящая из батареи 22, содержит примерно 156 г/л урана, который количественно извлекают в батарее 23, применяя отношение скоростей потоков О/А 1,9 для получения раствора с 281,5 г/л урана.
Кроме того, испытание показало, что нитрат уранила был в значительной степени очищен от основных примесей, присутствующих в рудных концентратах. Результаты показали присутствие в извлеченном растворе нитрата уранила следов железа, молибдена, циркония и кальция в количестве менее 1 мг/л, а присутствие ванадия обнаружить вовсе не удалось. Присутствие тория, концентрация которого в извлеченном нитрате уранила согласно измерениям была равна 8-10-14 г/л, вероятно связано с регенерацией тория в образце урана в результате распада этого урана, что подтверждают очень низкие коэффициенты распределения тория, полученные с органической фазой, содержащей DEHiBA.
Эти величины приведены в нижеприведенной таблице 2 в сравнении с техническими требованиями стандарта ASTM С 788.
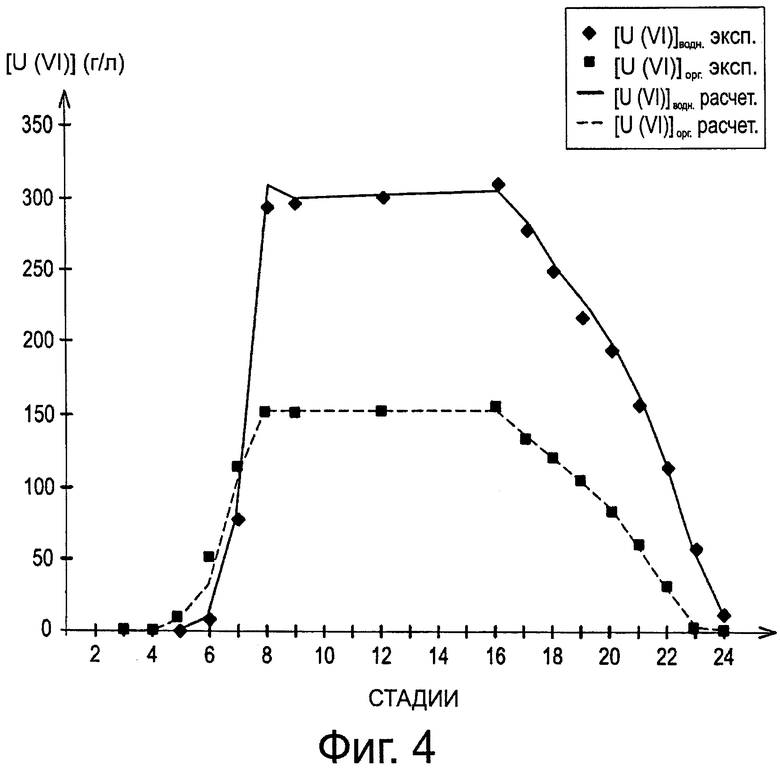
Кроме того, на фиг.4 приведены выраженные в г/л профили концентраций урана в водной и органической фазе, полученные экспериментально и расчетным путем, на разных стадиях в смесителях-декантаторах.
На этой фигуре [U(VI)]ВОДН эксп. соответствует экспериментальным концентрациям урана в водной фазе; [U(VI)]opг. эксп. соответствует экспериментальным концентрациям урана в органической фазе; [U(VI)]ВОДН расчет соответствует рассчитанным концентрациям урана в водной фазе, тогда как [U(VI)]opг. расчет соответствует рассчитанным концентрациям урана в органической фазе.
Хорошее соответствие между экспериментальными и рассчитанными величинами подтверждает обоснованность модели экстракции урана с помощью DEHiBA, разработанной с целью моделирования способа изобретения.
Следовательно, применение DEHiBA позволяет извлекать весь уран, присутствующий в растворе, с концентрацией в два раза более высокой по сравнению с той, которую в настоящее время получают при промышленном производстве с ТВР, а также в достаточном объеме очищать его от основных примесных катионов, присутствующих в рудных концентратах и представляющих собой помеху при последующих операциях обогащения, и в то же время не допускать накопления тория на стадии экстракции.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА, ВКЛЮЧАЮЩИЙ СТАДИЮ ОЧИСТКИ УРАНА (VI) ОТ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО АКТИНИДА (IV) ПУТЕМ ПОЛУЧЕНИЯ КОМПЛЕКСА ДАННОГО АКТИНИДА (IV) | 2014 |
|
RU2663882C1 |
| НЕСИММЕТРИЧНЫЕ N,N-ДИАЛКИЛАМИДЫ, В ЧАСТНОСТИ ИСПОЛЬЗУЕМЫЕ ДЛЯ ОТДЕЛЕНИЯ УРАНА(VI) ОТ ПЛУТОНИЯ(IV), ИХ СИНТЕЗ И ПРИМЕНЕНИЕ | 2018 |
|
RU2762634C2 |
| СПОСОБ ОБРАБОТКИ ВОДНОГО АЗОТНОКИСЛОГО РАСТВОРА, ПОЛУЧЕННОГО ПРИ РАСТВОРЕНИИ ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА, ВЫПОЛНЯЕМЫЙ В ОДНОМ ЦИКЛЕ И НЕ ТРЕБУЮЩИЙ КАКОЙ-ЛИБО ОПЕРАЦИИ, ВКЛЮЧАЮЩЕЙ ВОССТАНОВИТЕЛЬНУЮ РЕЭКСТРАКЦИЮ ПЛУТОНИЯ | 2016 |
|
RU2706954C2 |
| СПОСОБ ПЕРЕРАБОТКИ УРАНОВЫХ РУД | 2010 |
|
RU2434961C1 |
| СПОСОБ ОТДЕЛЕНИЯ ХИМИЧЕСКОГО ЭЛЕМЕНТА ОТ УРАНА ( VI ) И СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА | 2007 |
|
RU2429041C2 |
| СПОСОБ ПЕРЕРАБОТКИ КРЕМНИЙСОДЕРЖАЩИХ ОТХОДОВ УРАНОВОГО ПРОИЗВОДСТВА | 2014 |
|
RU2576819C1 |
| НОВЫЕ АСИММЕТРИЧНЫЕ N,N-ДИАЛКИЛАМИДЫ, ИХ СИНТЕЗ И ПРИМЕНЕНИЕ | 2016 |
|
RU2702739C2 |
| СПОСОБ ЭКСТРАКЦИОННОГО АФФИНАЖА УРАНА | 2005 |
|
RU2295168C1 |
| УЛУЧШЕННЫЙ СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА | 2010 |
|
RU2537952C2 |
| СПОСОБ ЭКСТРАКЦИОННОГО АФФИНАЖА УРАНА | 2013 |
|
RU2554830C2 |
Изобретение относится к способу, с помощью которого можно очищать уран из природного уранового концентрата. Этот способ включает экстракцию урана, присутствующего в виде нитрата уранила в водной фазе А1, полученной в результате растворения природного уранового концентрата в азотной кислоте, с помощью органической фазы, которая содержит экстрагирующее средство в органическом растворителе. Затем ведут промывание органической фазы, полученной после завершения стадии а), водной фазой А2 и извлечение нитрата уранила из органической фазы, полученной в конце стадии b), путем циркуляции этой органической фазы в аппарате в виде противотока водной фазе A3. При этом экстрагирующее средство представляет собой N,N-диалкиламид, а отношение между скоростями потоков, с которыми полученная в конце стадии b) органическая фаза и водная фаза A3 циркулируют в аппарате, где протекает стадия с), больше 1. Техническим результатом является повышение эффективности очистки урана за счет исключения потерь урана в органической фазе. 14 з.п. ф-лы, 2 табл., 4 пр.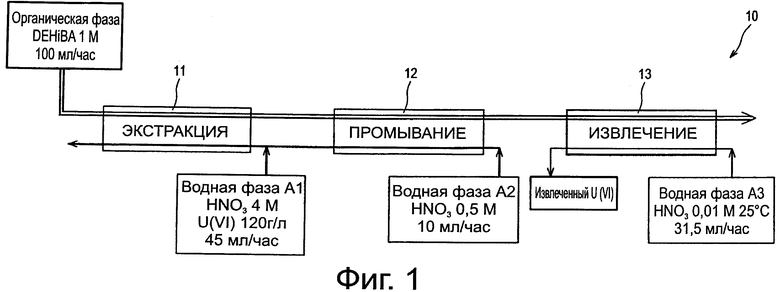
1. Способ очистки урана из природного уранового концентрата, содержащего по меньшей мере одну примесь, выбранную из тория, молибдена, циркония, железа, кальция и ванадия, включающий следующие стадии, на которых:
a) экстрагируют уран, присутствующий в виде нитрата уранила, из водной фазы А1, полученной в результате растворения природного уранового концентрата в азотной кислоте, причем экстракция включает приведение водной фазы А1 в контакт с несмешиваемой с водой органической фазой, которая содержит экстрагирующее средство в органическом растворителе, и последующее разделение указанных водной и органической фаз;
b) промывают органическую фазу, полученную после завершения стадии а), причем промывание включает приведение полученной после завершения стадии а) органической фазы в контакт с водной фазой А2 и последующее разделение указанных водной и органической фаз; и
c) извлекают нитрат уранила из полученной в конце стадии b) органической фазы, причем извлечение включает циркуляцию полученной после завершения стадии b) органической фазы в аппарате в противотоке с водной фазой A3 и последующее разделение указанных органической и водной фаз;
в котором экстрагирующее средство, содержащееся в органической фазе, представляет собой N,N-диалкиламид и отношение между скоростями потока, с которыми полученная в конце стадии b) органическая фаза и водная фаза A3 циркулируют в аппарате стадии с), больше 1.
2. Способ по п.1, в котором N,N-диалкиламид выбирают из N,N-диалкиламидов, соответствующих приведенной ниже формуле (I)
в которой:
- R1 представляет собой алкильную группу, которая разветвлена в альфа или бета положении от карбонильной группы и которая включает от 3 до 12 атомов углерода;
- R2 и R4, которые могут быть одинаковыми или разными, представляют собой линейную или разветвленную алкильную группу, включающую от 2 до 4 атомов углерода;
- R3 и R5, которые могут быть одинаковыми или разными, представляют собой линейную или разветвленную алкильную группу, включающую от 1 до 6 атомов углерода; и
- а и b, которые могут быть одинаковыми или разными, представляют собой целые числа в диапазоне от 1 до 6.
3. Способ по п.2, в котором N,N-диалкиламид соответствует формуле (I), в которой оба а и b равны 1 и оба R2 и R4 представляют собой этильные группы.
4. Способ по п.3, в котором N,N-диалкиламид представляет собой N,N-ди-(2-этилгексил)-изобутирамид.
5. Способ по п.1, в котором органический растворитель представляет собой С9-С13-изопарафин или смесь С9-С13-парафинов.
6. Способ по п.1, в котором концентрация N,N-диалкиламидов в органическом растворителе равна от 1 до 2 моль/л.
7. Способ по п.1, в котором водная фаза А1 включает от 0,5 до 4 моль/л азотной кислоты.
8. Способ по п.1, в котором водная фаза А2 представляет собой воду.
9. Способ по п.8, в котором водная фаза А2 представляет собой дистиллированную воду.
10. Способ по п.1, в котором водная фаза А2 представляет собой водный раствор, включающий от 0,01 до 1,5 моль/л азотной кислоты.
11. Способ по п.1, в котором водная фаза А2 представляет собой часть водной фазы, полученной после завершения стадии с).
12. Способ по п.1, в котором водная фаза A3 представляет собой воду.
13. Способ по п.12, в котором водная фаза A3 представляет собой дистиллированную воду.
14. Способ по п.1, в котором водная фаза A3 представляет собой водный раствор, включающий самое большее 0,01 моль/л азотной кислоты.
15. Способ по п.1, в котором отношение между скоростями потоков, с которыми полученная в конце стадии b) органическая фаза и водная фаза A3 циркулируют в аппарате стадии с), равно или больше 1,5.
| СПОСОБ СЕЛЕКЦИОННОЙ ОЦЕНКИ ГИДРАТЦЕЛЛЮЛОЗНЫХ ВОЛОКОН КАК ПРЕКУРСОРА ПРИ ПОЛУЧЕНИИ УГЛЕРОДНЫХ ВОЛОКОН | 2016 |
|
RU2642561C1 |
| ГРУЗОВАЯ ТЕЛЕЖКА ПОДВЕСНОГО ТОЛКАЮЩЕГО | 0 |
|
SU381579A1 |
| JP 2005214706 A, 11.08.2005 | |||
| СПОСОБ ПЕРЕРАБОТКИ ХИМИЧЕСКОГО КОНЦЕНТРАТА ПРИРОДНОГО УРАНА | 2003 |
|
RU2315716C2 |
| СПОСОБ ПЕРЕРАБОТКИ КОНЦЕНТРАТОВ ПРИРОДНОГО УРАНА | 2007 |
|
RU2360988C2 |
| US 4832924 А, 23.05.1989 | |||
| Способ изготовления ремешковой застежки | 1983 |
|
SU1118337A1 |

