Область техники, к которой относится изобретение
Изобретение относится к новым асимметричным N,N-диалкиламидам и к способу их синтеза.
Оно также относится к применению этих N,N-диалкиламидов в качестве экстрагентов для экстракции урана и/или плутония из водного раствора кислоты, и в частности, из водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте.
Оно также относится к применению этих N,N-диалкиламидов в качестве экстрагентов для полного или частичного выделения урана и плутония из водного раствора кислоты, и в частности, из водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте.
Оно также относится к способу обработки водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте, позволяющему экстрагировать, разделять и очищать от загрязнителей уран и плутоний, содержащиеся в этом растворе, в одном цикле без необходимости какой-либо операции восстановления плутония, и где один из этих N,N-диалкиламидов или их смесь применяют в качестве экстрагента.
Изобретение может применяться в переработке отработавшего ядерного топлива, включающего уран (в особенности оксиды урана - UOX), или уран и плутоний (в особенности смешанные оксиды урана и плутония - МОХ).
Предшествующий уровень техники
Пурекс-процесс (PUREX), применяющийся на всех заводах по переработке отработавшего ядерного топлива во всем мире (La Hague во Франции, Rokkasho в Японии, Sellafield в Соединенном Королевстве, и т.д.) использует три-н-бутил фосфат (или ТБФ) в качестве экстрагента для извлечения урана и плутония путем жидкостно-жидкостной экстракции из водных растворов, полученных при растворении этих видов топлива в азотной кислоте.
В этом способе ТБФ применяют в 30 об.% растворе в органическом разбавителе (гидрированном тетрапропилене (или ТПГ) или н-додекане). Этот органический раствор обычно называют «растворителем» в области, к которой относится изобретение.
Извлечение урана и плутония в PUREX процессе проводят в несколько циклов:
- первый цикл очистки урана и плутония (называемый «1CUPu», предназначенный для очистки урана и плутония от америция, кюрия и продуктов деления, с разделением урана и плутония на два водных потока в данном первом цикле посредством восстановительной реэкстракции плутония;
- второй цикл очистки урана (называемый «2CU»), предназначенный для полной очистки от загрязнителей урана для достижения требований, изложенных в стандартах ASTM (Американского общества по испытанию материалов) для конечного продукта урана; и
- второй, и на некоторых заводах третий цикл очистки плутония (соответственно, называемый «2CPu» или «3CPu»), предназначенный для полной очистки от загрязнителей плутония для достижения требований, изложенных в стандартах ASTM для конечного продукта урана для его концентрирования перед преобразованием в оксид.
Уровни эффективности PUREX процесса являются удовлетворительными, и отзывы об опыте, приобретенном с момента запуска установок, использующих этот способ, являются положительными.
Однако применение ТБФ имеет пределы, ограничивающие возможность достижения с этим экстрагентом целей простоты, компактности и повышенной безопасности, установленных для будущих заводов по переработке отработавшего ядерного топлива, в частности, направленных на разделение урана и плутония на два водных потока без использования восстанавливающих агентов.
Эти пределы являются следующими:
- коэффициенты очистки урана и плутония от некоторых продуктов деления (технеция и рутения) и трансурановых элементов (Np) являются недостаточными в конце первого цикла очистки, следовательно, невозможно достичь с ТБФ схемы, приводящей к конечным продуктам, удовлетворяющим вышеупомянутым требованиям, в одном цикле;
- разделение урана и плутония на два водных потока требует восстановления плутония (IV) в плутоний (III) (поскольку с ТБФ коэффициент разделения урана (VI) и плутония (IV) является недостаточным, независимо от кислотности водного раствора, используемого для достижения такого разделения), и в результате требует использования высоких количеств восстанавливающих и анти-азотнокислых агентов, которые генерируют нестабильные и активные формы при деградации, и по этой причине ограничивают применение с точки зрения безопасности;
- продукты деградации ТБФ оказывают влияние на уровни производительности процесса; в частности, ди-н-бутил-фосфат (или ДБФ) приводит к образованию комплексов металлов, некоторые из которых являются нерастворимыми и могут вызывать удерживание плутония в растворителе; следовательно, необходимо проводить операцию, известную как «барьер Pu», которая осуществляется после восстановительной реэкстракции плутония, и которая предназначена для завершения этой реэкстракции;
- риск образования 3-й фазы, индуцированной присутствием плутония, ограничивает осуществление схемы концентрирования плутония (риск критичности) или схемы, обеспечивающей переработку отработавшего ядерного топлива с высоким содержанием плутония, такого как МОХ топливо, полученное из легководных реакторов или реакторов на быстрых нейтронах;
- реэкстракция урана из растворителя, в который он был предварительно экстрагирован, является неполной, если её проводят при комнатной температуре, следовательно, необходимо выполнить эту реэкстракцию при температуре 50°С (что соответствует максимальной температуре, допускаемой точкой воспламенения растворителя); однако, даже при этой температуре реэкстракцию урана проводят при разбавлении (отношение органического/водного потоков (О/В) составляет меньше 1);
- растворимость ТБФ, которая является довольно большой в водной фазе (до 300 мг/л, в зависимости от кислотности водной фазы), требует промывания органическим разбавителем водной фазы из-за разницы циклов экстракции для восстановления ТБФ, солюбилизированного в этих водных фазах; и
- сжигание отработанного ТБФ и его продуктов деградации приводит к образованию вторичных отходов, включающих твердые фосфатные остатки.
Таким образом, в перспективе будущих заводов по переработке отработавшего ядерного топлива, которые являются более простыми и более компактными, чем современные заводы, и кроме того, обладают повышенной безопасностью, авторы изобретения разработали способ, который при выполнении также PUREX процесса для извлечения и очистки от загрязнителей урана и плутония, содержащихся в водных азотнокислых растворах, полученных при растворении отработавшего ядерного топлива, позволяет преодолеть все ограничения, связанные с применением ТБФ в качестве экстрагента, и который, в частности, включает только один производственный цикл и не содержит какой-либо операции для восстановительной реэкстракции плутония.
Таким образом, авторы настоящего изобретения вначале сосредоточились на поиске экстрагентов, обладающих необходимыми свойствами, позволяющими разработать такой способ.
N,N-диалкиламиды представляют собой семейство экстрагентов, которые в основном изучались в качестве альтернативы ТБФ для переработки отработавшего ядерного топлива, в частности, потому что они, как правило, обладают хорошей аффинностью к урану и плутонию в условиях высокой кислотности, являются менее растворимыми, чем ТБФ, в водной фазе, полностью сжигаются («принцип CHON»), и имеют продукты деградации, которые создают меньше проблем, чем у ТБФ.
Имеется два типа N,N-диалкиламидов:
- так называемые «симметричные» N,N-диалкиламиды, в которых две алкильных группы, которые несет атом азота, являются идентичными; и
- так называемые «асимметричные» N,N-диалкиламиды, в которых две алкильных группы, которые несет атом азота, являются разными.
Сначала были исследованы симметричные N,N-диалкиламиды. Например, три французских патентных заявки (FR-A-2591213, FR-A-2642561 и FR-A-2642562, далее обозначенные как [1], [2] и [3]), относящиеся к применению симметричных N,N-диалкиламидов в качестве экстрагентов для переработки отработавшего ядерного топлива, были поданы в 1980-х; и две из них, а именно ссылки [1] и [3], предусматривают возможность разделения урана и плутония с применением этих N,N-диалкиламидов без выполнения восстановительной реэкстракции плутония.
Некоторые из симметричных N,N-диалкиламидов, предложенных в ссылках [1] и [3], обеспечивают эффективную совместную экстракцию урана (VI) и плутония (IV) из растворов с высокой кислотностью, с последующим их разделением в условиях низкой кислотности без необходимости восстановления плутония.
Однако доказано, что эти N,N-диалкиламиды обеспечивают меньшую экстракцию плутония из водной фазы с высокой кислотностью, чем ТБФ. В результате для достижения количественной экстракции плутония необходимо увеличить число стадий экстракции, по сравнению с числом, необходимым для ТБФ, что противоречит намеченной цели компактности.
Асимметричные N,N-диалкиламиды позднее стали предметом определенного числа исследований, среди которых можно упомянуть исследования, проведенные в Центре атомных исследований имени Хоми Баба в Бомбее (см., например, публикации Ruikar et al., Journal of Radioanalytical and Nuclear Chemistry 1993, 176(2), 103-111, и Prabhu et al., Radiochimica Acta 1993, 60, 109-114, далее упоминаемые как [4] и[5]), и исследования, проведенные группой под руководством Guo-Xin Sun в Цзинаньском университете (см., например, публикации Cui et al., Radiochimica Acta 2005, 93, 287-290, и Sun et al., Journal of Radioanalytical and Nuclear Chemistry 2005, 264(3), 711-713, далее упоминаемые как [6] и [7]).
Однако, помимо того факта, что результаты этих исследований являются фрагментарными и отчасти противоречивыми, ни в одном из них не предполагается возможность отделения урана от плутония без восстановления плутония.
Раскрытие сущности изобретения
Таким образом, изобретение, во-первых, предлагает новые N,N-диалкиламиды, которые являются асимметричными и соответствуют следующей формуле (I):
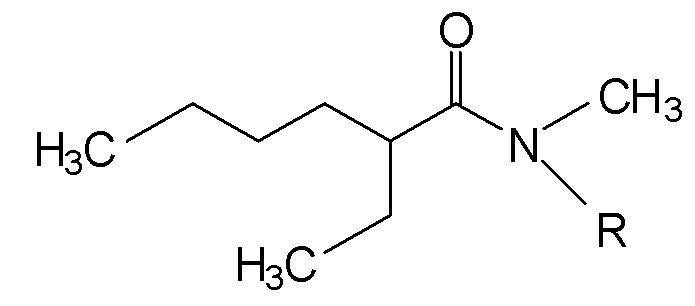 (I),
(I),
где R является линейной или разветвленной алкильной группой, содержащей от 8 до 15 атомов углерода.
В вышеприведенном тексте и далее, выражения «от … до …», «в диапазоне от … до …» и «между … и …» являются эквивалентными и предназначены для того, чтобы показать, что указанные пределы включены.
Таким образом:
- «линейная алкильная группа, содержащая от 8 до 15 атомов углерода», означает любую алкильную группу, выбранную из н-октильной, н-нонильной, н-децильной, н-ундецильной, н-додецильной, н-тридецильной, н-тетрадецильной и н-пентадецильной групп, в то время как
- «разветвленная алкильная группа, содержащая от 8 до 15 атомов углерода», означает любую алкильную группы, содержащую 8, 9, 10, 11, 12, 13, 14 или 15 атомов углерода, и имеющую одну или несколько одинаковых или разных ветвей, таких как н-гептильная, н-октильная, н-нонильная, н-децильная, н-ундецильная, н-додецильная, н-тридецильная или н-тетрадецильная группы, замещенные метильной группой (например, 2- или 4-метилгептильная группа, 2- или 4-метилоктильная группа, 2- или 4-метилоктильная группа, и т.д.); н-гексильная, н-гептильная, н-октильная, н-нонильная, н-децильная, н-ундецильная, н-додецильная или н-тридецильная группа, замещенная этильной группой (например, 2- или 4-этилгексильная группа, 2- или 4-этилоктильная группа, 2- или 4-этилдецильная группа, и т.д.); н-октильная, н-нонильная, н-децильная, н-ундецильная или н-додецильная группа, замещенная н-пропильной или изопропильной группой; н-нонильная, н-децильная или н-ундецильная группа, замещенная н-бутильной, изобутильной, сек-бутильной или трет-бутильной группой; н-гексильная, н-гептильная, н-октильная, н-нонильная, н-децильная, н-ундецильная, н-додецильная или н-тридецильная группа, замещенная двумя метильными группами; н-гексильная, н-гептильная, н-октильная, н-нонильная, н-децильная, н-ундецильная или н-додецильная группа, замещенная метильной группой и этильной группой (например, 3-этил-4-метилгексильная группа, 3-метил-4-этилгексильная группа, 3-этил-4-метилоктильная группа, 3-метил-4-этилоктильная группа); и т.д.
Кроме того, термины «водный раствор» и «водная фаза» являются эквивалентными и применяются взаимозаменяемо, и подобным образом, термины «органический раствор» и «органическая фаза» являются эквивалентными и применяются взаимозаменяемо.
В соответствии с изобретением, линейная или разветвленная алкильная группа, представленная R в вышеприведенной формуле (I), предпочтительно не содержит больше 12 атомов углерода по соображениям вязкости (вязкость N,N-диалкиламидов эффективно повышается с увеличением числа атомов углерода, представленных R).
Более предпочтительно, эта группа выбрана из н-октильной, н-децильной, н-додецильной, 2-этилгексильной и 2-этилоктильной групп, при этом особо предпочтительной является н-октильная группа.
Вышеуказанные N,N-диалкиламиды предпочтительно получают путем реакции галоида следующей формулы (II):
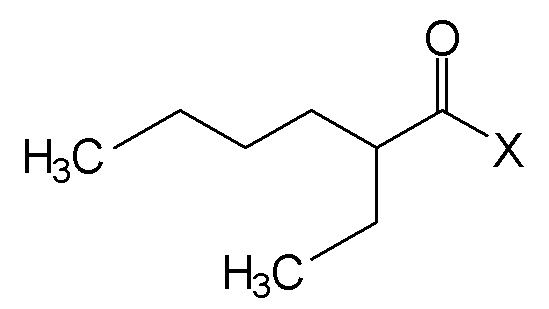 (II),
(II),
где Х является атомом галогена, и предпочтительно, атомом хлора, с амином формулы HN(CH3)R, где R является линейной или разветвленной алкильной группой, содержащей от 8 до 15 атомов углерода, в присутствии основания.
Таким образом, другой задачей изобретения является способ синтеза N,N-диалкиламидов, включающий эту реакцию.
Такую реакцию можно проводить либо в водном растворе, и в этом случае основанием является, например, натрия гидроксид или калия гидроксид, или в органическом растворе, таком как дихлорметан или диэтиловый эфир, и в этом случае основанием является, например, триэтиламин или диизопропилэтиламин.
Доказано, что N,N-диалкиламиды, определенные выше, способны к очень эффективной экстракции урана (VI) и плутония (IV) из водного раствора кислоты, такого как водный раствор азотной кислоты.
Таким образом, другой задачей изобретения является применение N,N-диалкиламида или смеси N,N-диалкиламидов, как определено выше, для экстракции урана (VI) и/или плутония (IV) из водного раствора кислоты.
В соответствии с изобретением, уран и/или плутоний предпочтительно экстрагируют из водного раствора кислоты путем экстракции жидкости жидкостью, т.е. путем контактирования этого водного раствора с органическим раствором, содержащим N,N-диалкиламид или смесь N,N-диалкиламидов в органическом разбавителе, с последующим разделением водного и органического растворов.
В этом случае органический раствор предпочтительно содержит от 1 моль/л до 2 моль/л, и более предпочтительно от 1,3 до 1,5 моль/л N,N-диалкиламида или смеси N,N-диалкиламидов.
Водный раствор кислоты предпочтительно является водным раствором, полученным при растворении отработавшего ядерного топлива в азотной кислоте, т.е. водным раствором, как правило, включающим от 3 моль/мл до 6 моль/мл азотной кислоты.
Доказано, что в дополнение к способности к количественной экстракции урана (VI) и плутония (IV) из водного раствора кислоты, вышеуказанные N,N-диалкиламиды обеспечивают дополнительное разделение урана и плутония, экстрагированных таким способом, без восстановления плутония, где это разделение может быть:
- либо полным разделением урана и плутония, т.е. где получают два водных раствора, где один из них содержит плутоний без урана, а другой содержит уран без плутония;
- либо частичным разделением урана и плутония, т.е. где получают два водных раствора, где один из них содержит смесь плутония и урана, а другой содержит уран без плутония.
Таким образом, другой задачей изобретения является применение N,N-диалкиламида или смеси N,N-диалкиламидов, как определено выше, для полного или частичного разделения урана (VI) и плутония (IV) из водного раствора кислоты, где применение включает:
(а) совместную экстракцию урана и плутония из водного раствора, включающую по меньшей мере одно контактирование водного раствора с органическим раствором, включающим N,N-диалкиламид или смесь N,N-диалкиламидов в качестве экстрагента в растворе в органическом разбавителе, с последующим разделением водного и органического растворов;
(b) реэкстракцию плутония в степени окисления +IV из органического раствора, полученного на этапе (а), включающую по меньшей мере одно контактирование органического раствора с водным раствором, содержащим от 0,1 моль/л до 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов; и
(с) экстракцию всей или части урановой фракции, содержащейся в водном растворе, полученном на этапе (b), где экстракция включает по меньшей мере одно контактирование водного раствора с органическим раствором, идентичным органическому раствору, использованному на этапе (а), с последующим разделением водного и органического растворов;
где получают водный раствор, содержащий либо плутоний без урана, либо смесь плутония и урана, и органический раствор, содержащий уран без плутония.
Органический раствор, используемый на этапе (а), и следовательно, используемый на этапе (с), предпочтительно содержит от 1 моль/л до 2 моль/л, и предпочтительно от 1,3 моль/л до 1,5 моль/л N,N-диалкиламида или смеси N,N-диалкиламидов.
Что касается водного раствора кислоты, из которого осуществляют совместную экстракцию урана и плутония, он предпочтительно является водным раствором, полученным при растворении отработавшего ядерного топлива в азотной кислоте, т.е. водным раствором, как правило, содержащим от 3 моль/л до 6 моль/л азотной кислоты.
Уран, содержащийся в органическом растворе, полученном с этапа (с), может быть реэкстрагирован из этой фазы путем контактирования органического раствора с водным раствором, содержащим не более 0,05 моль/л азотной кислоты, с последующим разделением органического и водного растворов.
В дополнение к проявлению вышеупомянутых свойств, было доказано, что вышеуказанные N,N-диалкиламиды обеспечивают экстракцию урана (VI) и плутония (IV) из водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте, с очень высокими коэффициентами разделения в отношении основных продуктов деления, содержащихся в этом растворе.
Что касается этой совокупности свойств, эти N,N-диалкиламиды обеспечивают разработку способа переработки водного азотнокислого раствора, полученного при растворении отработавшего ядерного топлива, который при такой же хорошей эффективности, как у PUREX процесса, с точки зрения извлечения и очистки от загрязнителей урана и плутония, содержащихся в этом растворе, не включает какой-либо операции восстановительной реэкстракции плутония, и включает только один производственный цикл (цикл обработки).
Таким образом, дополнительной задачей изобретения является способ из одного цикла для обработки водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте, где водный раствор содержит уран, плутоний, америций, кюрий и продукты деления, включая технеций, где этот цикл включает:
(а) совместную экстракцию урана и плутония из водного раствора, включающую по меньшей мере одно контактирование в экстракторе водного раствора с органическим раствором, включающим N,N-диалкиламид или смесь N,N-диалкиламидов, как определено выше, в качестве экстрагента в растворе в органическом разбавителе, с последующим разделением водного и органического растворов;
(b) очистку органического раствора, полученного на этапе (а), от америция, кюрия и продуктов деления, включающую по меньшей мере одно контактирование в экстракторе указанного органического раствора с водным раствором, включающим от 0,5 моль/л до 6 моль/л азотной кислоты, с последующим разделением органического и водного растворов;
(с) разделение урана и плутония, содержащихся в органическом растворе, полученном на этапе (b), на водный раствор и органический раствор, где водный раствор содержит либо плутоний без урана, либо смесь плутония и урана, а органический раствор содержит уран без плутония, где это разделение включает:
(с1) реэкстракцию плутония в степени окисления +IV и фракции урана из органического раствора, полученного на этапе (b), включающую по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, содержащим от 0,1 моль/л до 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов;
(с2) экстракцию всей или части урановой фракции, содержащейся в водном растворе, полученном на этапе (с1), при этом экстракция включает по меньшей мере одно контактирование в экстракторе водного раствора с органическим раствором, идентичным органическому раствору, используемому на этапе (а), с последующим разделением водного и органического растворов;
(d) очистку органического раствора, полученного на этапе (с1), от технеция, включающую:
(d1) реэкстракцию технеция в степени окисления +IV из органического раствора, полученного на этапе (с1), включающую по меньшей мере одно контактирование в экстракторе этого органического раствора с водным раствором, содержащим от 0,1 моль/л до 3 моль/л азотной кислоты и по меньшей мере один восстанавливающий агент, способный восстанавливать технеций из степени окисления +VII до степени окисления +IV, с последующим разделением органического и водного растворов;
(d2) экстракцию урановой фракции, содержащейся в водном растворе, полученном на этапе (d1), включающую по меньшей мере одно контактирование в экстракторе водного раствора с органическим раствором, идентичным органическому раствору, используемому на этапе (а), с последующим разделением водного и органического растворов;
(е) реэкстракцию урана из органического раствора, полученного на этапе (d1), включающую по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, содержащим не более 0,05 моль/л азотной кислоты, с последующим разделением органического и водного растворов; и
(f) регенерацию органической фазы, полученной на этапе (е);
где получают первый и второй водные растворы, очищенные от америция, кюрия и продуктов деления, включая технеций, где первый водный раствор включает либо плутоний без урана, либо смесь плутония и урана, а второй водный раствор включает уран без плутония.
В соответствии с изобретением, органический раствор, используемый на этапе (а), и следовательно, используемый на этапах (c2) и (d2), поскольку органические растворы, используемые на этапах (a), (c2) и (d2), имеют один и тот же состав, предпочтительно содержит от 1 моль/л до 2 моль/л, и более предпочтительно от 1,3 моль/л до 1,5 моль/л N,N-диалкиламида или смеси N,N-диалкиламидов.
Как указывалось ранее, водный раствор, используемый на этапе (b), может включать от 0,5 моль/л до 6 моль/л азотной кислоты.
Однако предпочтительно, чтобы этот водный раствор содержал от 4 моль/л до 6 моль/л азотной кислоты для облегчения реэкстракции рутения и технеция из органического раствора, полученного на этапе (а). В этом случае этап (b) предпочтительно также включает нейтрализацию (уменьшение кислотности) органического раствора, включающую по меньшей мере одно контактирование органического раствора с водным раствором, содержащим от 0,1 моль/л до 1 моль/л, и предпочтительно 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов.
В соответствии с изобретением, контактирование органического и водного растворов в экстракторе, в котором осуществляют этап (с1), включает циркуляцию этих растворов при отношении потоков О/В, предпочтительно составляющем больше 1, предпочтительно 3 или больше, и более предпочтительно 5 или больше, для достижения концентрирующей реэкстракции плутония, т.е. реэкстракции плутония, приводящей к водному раствору, в котором концентрация плутония превышает концентрацию этого элемента в органическом растворе, из которого его реэкстрагируют.
Восстанавливающий агент (агенты) в водном растворе, используемом на этапе (d1), предпочтительно выбирают из нитрата урана (также называемого «U (IV)», гидразиния нитрата (также называемого «гидразина нитрат»), гидроксиламмония нитрата (также называемого «гидроксиламина нитрат»), ацетальдоксима и их смесей, таких как смесь урана нитрата и гидразиния нитрата, смесь урана нитрата и гидроксиламмония нитрата, или смесь урана нитрата и ацетальдоксима, предпочтительно смесь урана нитрата и гидразиния нитрата, или смесь урана нитрата и гидроксиламмония нитрата, которые предпочтительно применяются в концентрации в диапазоне от 0,1 моль/л до 0,3 моль/л, и обычно 0,2 моль/л.
Кроме того, этап (d1), который можно проводить при комнатной температуре, предпочтительно проводят, однако, при температуре в диапазоне от 30 до 40°С, и предпочтительно при 32°С для стимуляции кинетики реэкстракции технеция, при этом способствуя ограничению феномена повторного окисления данного элемента в водной фазе. Таким образом, экстрактор, в котором осуществляют этап (d1), предпочтительно нагревают до температуры от 30°С до 40°С.
В соответствии с изобретением, предпочтительно этап (d2) дополнительно включает подкисление водного раствора, полученного на этапе (d1), где это подкисление включает добавление азотной кислоты в экстрактор, в котором проводят этап (d2), для доведения концентрации азотной кислоты до значения по меньшей мере 2,5 моль/л.
Этап (е) можно проводить при комнатной температуре. Однако его предпочтительно проводить при температуре в диапазоне от 40°С до 50°С, также для стимуляции реэкстракции урана. Таким образом, экстрактор, в котором осуществляют этап (е), предпочтительно нагревают до температуры от 40°С до 50°С.
Независимо от температуры, при которой проводят этап (е), контактирование органического и водного растворов в экстракторе, в котором осуществляют этап (е), включает циркуляцию этих растворов с отношением потоков О/В больше 1, так чтобы достичь концентрирующей реэкстракции урана, т.е. реэкстракции урана, приводящей к водному раствору, в котором концентрация урана превышает концентрацию этого элемента в органическом растворе, из которого его реэкстрагируют.
Как указано ранее, способ из настоящего изобретения дополнительно включает этап (f) для регенерации органического раствора, полученного на этапе (е), где эта регенерация предпочтительно включает по меньшей мере одно промывание органического раствора основным водным раствором, с последующим по меньшей мере одним промыванием органического раствора водным раствором азотной кислоты.
Способ из настоящего изобретения, в дополнение к тому, что уже упомянуто, имеет следующие преимущества:
- реэкстракцию урана проще проводить, чем с помощью PUREX процесса, поскольку её можно выполнять как при комнатной температуре, так и при нагревании, с применением отношения потоков О/В больше 1, таким образом, обеспечивая концентрирующую реэкстракцию урана, которая невозможна для PUREX процесса;
- благодаря тому, что не проводится какой-либо реакции восстановления плутония, и таким образом, устраняется какой-либо риск повторного окисления плутония, реэкстракцию плутония также легче осуществлять, чем при PUREX процессе, и можно проводить с большим концентрированием, чем для последнего способа; важность этих преимуществ еще более велика, так как в будущем заводам по переработке отработавшего ядерного топлива придется перерабатывать топливо с более высоким содержанием плутония (такое, как МОХ топливо из легководных реакторов или реакторов на быстрых нейтронах), чем топливо, перерабатываемое в настоящее время;
- продукты деградации (посредством гидролиза и радиолиза) N,N-диалкиламидов являются менее проблематичными, чем продукты деградации ТБФ, поскольку они являются водорастворимыми и не образуют комплексов, которые могут удерживать плутоний;
- N,N-диалкиламиды, как правило, имеют растворимость в водной фазе в 100-200 раз ниже растворимости ТБФ, и таким образом, можно предусмотреть отмену или по меньшей мере снижение числа промываний в органическом разбавителе водных растворов, полученных в результате способа по настоящему изобретения, по сравнению числом, представленным в PUREX процессе;
- поскольку N,N-диалкиламиды и продукты их деградации включают только атомы углерода, водорода, кислорода и азота, они полностью поддаются сжиганию, и таким образом, не производят обременительных вторичных отходов, в отличие от ТБФ и его продуктов деградации.
Другие характеристики и преимущества изобретения станут понятными из следующего дополнительного описания, со ссылкой на прилагаемые фигуры.
Однако это дополнительное описание приведено только для иллюстрации предмета обсуждения изобретения, и ни при каких обстоятельствах не должно рассматриваться как ограничивающее этот предмет обсуждения.
Краткое описание фигур
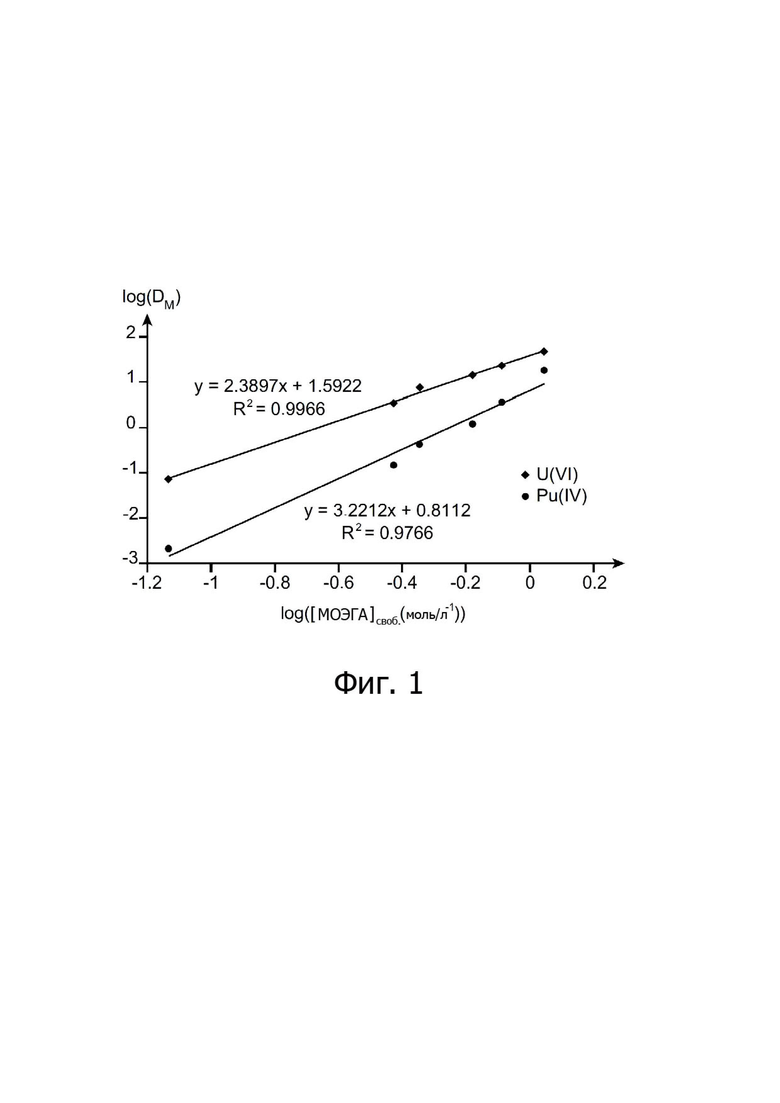
Фигура 1 иллюстрирует двумя прямыми линиями изменение логарифма коэффициентов распределения, обозначенных DM, урана и плутония, полученных в тестах по экстракции, проведенных с N,N-диалкиламидом для настоящего изобретения, в зависимости от логарифма свободной концентрации (в моль/л) этого свободного N,N-диалкиламида в органической фазе, используемой в этих тестах по экстракции.
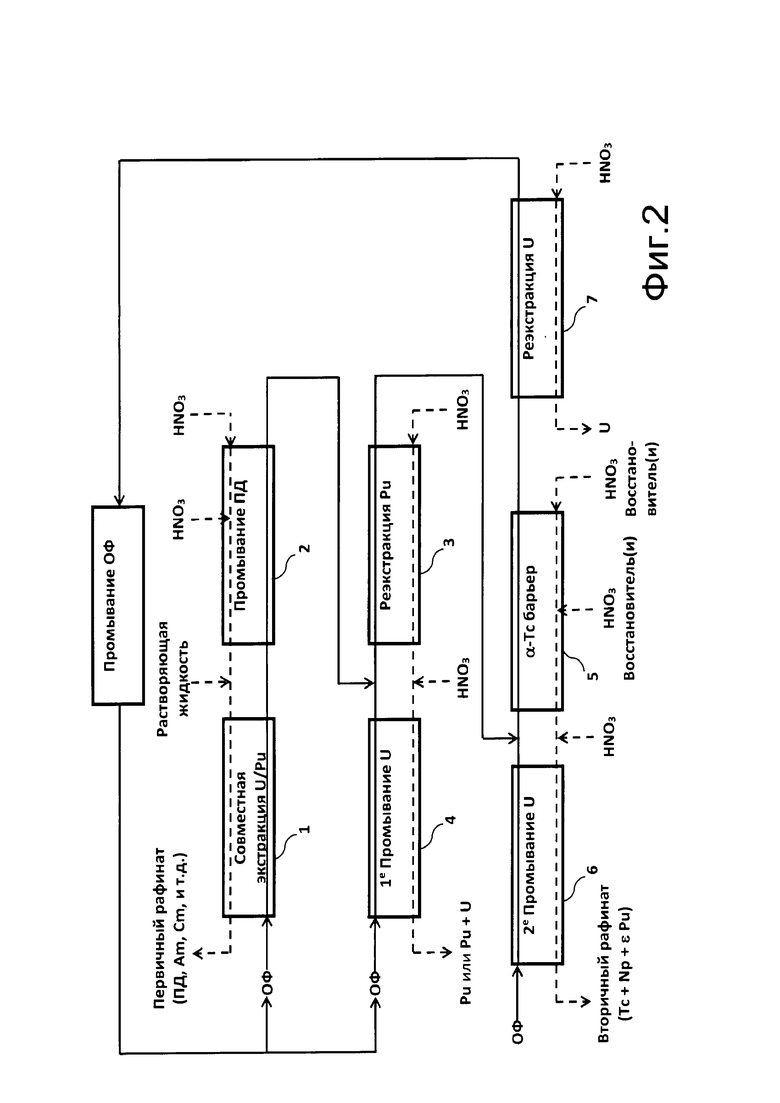
Фигура 2 демонстрирует поточную диаграмму способа из настоящего изобретения для обработки водного азотнокислого раствора, полученного при растворении отработавшего ядерного топлива; на этой фигуре прямоугольники 1-7 представляют многоступенчатые экстракторы, такие как те, которые обычно применяются для переработки отработавшего ядерного топлива (смесители-отстойники, пульсационные колонны или отжимные центрифуги); органические фазы условно обозначены сплошными линиями, а водные фазы - пунктирными линиями.
Подробное описание частных вариантов осуществления
I - Синтез N,N-диалкиламидов из настоящего изобретения
Как упоминалось выше, N,N-диалкиламиды из настоящего изобретения можно получить с помощью следующей схемы реакции А:
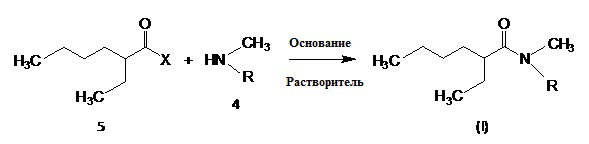
где X = атом галогена, а R = линейная или разветвленная C8 - C15 алкильная группа.
Когда амин, обозначенный как 4 в вышеуказанной схеме, не является коммерчески доступным, его можно получить по следующей схеме реакции В:
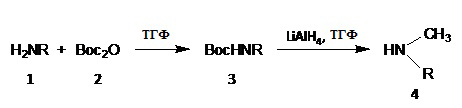
где R = линейная или разветвленная C8 - C15 алкильная группа.
I.1. Синтез N-метил-N-октил-2-этилгексанамида или МОЭГА.
МОЭГА, который соответствует вышеуказанной формуле (I), где R является н-октильной группой, синтезируют из 2-этилгексаноил хлорида и N-метил-N-октиламина, в присутствии натрия гидроксида в воде (схема реакции А).
С этой целью натрия гидроксид (30% NaOH - 112 г - 0,839 моль - 1,19 экв.), воду (100 г) и N-метил-N-октиламин (100 г - 0,698 моль - 1 экв.) помещают в полностью оснащенный 500 мл реактор. В системе устанавливают перемешивание и температуру 4°C. Загружают 2-этилгексаноил хлорид (136,5 г - 0,839 моль - 1,19 экв.) при температуре загрузки от 14°C до 17°C (продолжительность загрузки: 90 минут). Протекание реакции контролируют, и показывают присутствие 0,6% остаточного амина. Получают 90% МОЭГА и 8,8% неизвестной примеси. Среду нагревают до 50°С в течение 30 минут для потребления остаточного амина. Затем среду охлаждают до 20°С и отделяют декантацией. Органическую фазу дважды промывают 100 мл воды до получения 208 г продукта реакции.
Затем получают МОЭГА с чистотой 98,3% (при измерении посредством газовой хроматографии в сочетании с детектором пламенной ионизации ГХ-ДПИ) после двух дистилляций под давлением (2 и 8 мбар, соответственно).
13C ЯМР (100 МГц, CDCl3, 25°C) δ (ч./млн.): 3,37 (t, J= 7,0, 2H, 2HαA); 3,28 (t, J= 7,0, 2H, 2HαB); 3,00 (s, 3H, CH3A); 2,92 (s, 3H, CH3B); 2,61 - 2,42 (m, 2H, H2A и H2B); 1,90 - 1,77 (m, 2H, CH2A); 1,71 - 1,35 (m, 10H, 2CH2A и 3CH2B); 1,34 - 1,11 (m, 28H, 7CH2A и 7CH2B); 0,94 -0,77 (m, 18H, 3CH3A и CH3B)
1H ЯМР (400 МГц, CDCl3, 25°C) δ (ч./млн.): 176,2; 176,0 (COA и COB); 50,1; 48,1 (CαA и CαB); 43,2; 42,9 (C2A и C2B); 35,6; 33,8 (CH3A и CH3B); 32,8; 32,7; 31,9; 31,9; 30,1; 30,0; 29,5; 29,4; 29,4; 29,3; 29,2; 27,5; 27,5; 27,0; 26,9; 26,2; 23,1; 23,0; 22,8 (20CH2); 14,2; 14,2; 14,1; 14,1 (2CH3A и 2CH3B); 12,3; 12,2 (CH3A и CH3B).
МС (ионизация электронным ударом), m/z (I%): 269 (5%) [M]+, 240 (22%) [M -C2H5]+, 226 (22%) [M -C3H7]+, 212 (40%) [M -C4H9]+, 198 (25%) [M -C5H11]+, 170 (100%) [M -C7H15]+, 142 (5%) [C9H20N]+, 127 (5%) [C8H15O]+
МСВР (ионизация электронным ударом): m/z при расчете по [MH]+ (C17H35NO) 269,2714; установлено 269,2672.
I.2. Синтез N-децил-N-метил-2-этилгексанамида или МДЭГА.
МДЭГА, который соответствует вышеуказанной формуле (I), где R является н-децильной группой, синтезируют в соответствии со следующей схемой реакции А из 2-этилгексаноил хлорида и N-децил-N-метиламина, в присутствии триэтиламина (Et3N) в безводном дихлорметане (ДХМ) (схема реакции А).
С этой целью дихлорметан (100 мл), Et3N (21,2 г - 0,21 моль - 1,48 экв.) и N-децил-N-метиламин (24 г - 0,14 моль - 1 экв.) помещают в полностью оснащенный 500 мл реактор. В системе устанавливают перемешивание и охлаждение до 0°C. Добавляют 2-этилгексаноил хлорид (25 г - 0,15 моль - 1,1 экв.) при температуре загрузки от 5°C до 16°C (продолжительность загрузки: 45 минут). При перемешивании температура загрузки постепенно повышается до комнатной температуры. Спустя 90 минут контролируют прогресс реакции, и показывают отсутствие любого остаточного амина, но остается 5% 2-этилгексаноил хлорида. Получают 88,9% МДЭГА и 4,6% неизвестной примеси. Затем среду последовательно промывают дважды 100 мл 10% раствора натрия гидроксида, затем дважды 100 мл 1 н раствора соляной кислоты и 100 мл 5% раствора натрия карбоната. Органическую фазу концентрируют при сниженном давлении до получения 43,6 г масла. Это масло содержит 89% МДЭГА и 9,46% неизвестной примеси.
Затем получают МДЭГА с чистотой 99,4% (при измерении посредством газовой хроматографии в сочетании с детектором пламенной ионизации ГХ-ДПИ) после двух дистилляций под давлением (1,5 мбар).
МС (ионизация электронным ударом), m/z (I%): 297 (3%) [M]+, 268 (13%) [M -C2H5]+, 254 (15%) [M -C3H7]+, 240(12%) [M -C4H9]+, 226 (12%) [M -C5H11]+, 198 (100%) [M -C7H15]+
МСВР (ионизация электронным ударом): m/z при расчете [M]+ (C19H39NO) 297,3026; установлено 297,3000.
I.3. Синтез N-додецил-N-метил-2-этилгексанамида или МДдЭГА
МДдЭГА, который соответствует вышеуказанной формуле (I), где R является н-додецильной группой, синтезируют из 2-этилгексаноил хлорида и N-додецил-N-метиламина, в присутствии Et3N в безводном ДХМ (схема реакции А).
С этой целью ДХМ (150 мл), Et3N (22,7 г - 0,223 моль - 1,49 экв.) и N-додецил-N-метиламин (30 г - 0,15 моль - 1 экв.) помещают в полностью оснащенный 500 мл реактор. В системе устанавливают перемешивание и охлаждение до примерно 0°C. Затем добавляют 2-этилгексаноил хлорид (26 г - 0,16 моль - 1,106 экв.) при температуре загрузки от 0°C до 2°C (продолжительность загрузки: 40 минут). При перемешивании температура загрузки постепенно повышается до комнатной температуры. Спустя 4 часа контролируют прогресс реакции, и показывают отсутствие какого-либо исходного амина. Отмечается образование 97% МДдЭГА. Затем среду последовательно промывают дважды 100 мл 10% раствора натрия гидроксида, затем один раз 100 мл 5% раствора натрия карбоната. Затем концентрируют органическую фазу при сниженном давлении до получения 55,5 г масла.
Затем получают МДдЭГА с чистотой 99,5% (при измерении посредством газовой хроматографии в сочетании с детектором пламенной ионизации ГХ-ДПИ) после одной дистилляции при сниженном давлении (0,7 мбар).
МС (ионизация электронным ударом), m/z (I%): 325 (3%) [M]+, 296 (11%) [M -C2H5]+, 282 (12%) [M -C3H7]+, 268 (15%) [M -C4H9]+, 254 (10%) [M -C5H11]+, 226 (100%) [M -C7H15]+
МСВР (ионизация электронным ударом): m/z при расчете по [M]+ (C21H43NO) 325,3339; установлено 325,3325.
I.4. Синтез N-2-этилгексил-N-метил-2-этилгексанамида или М(2-ЭГ)ЭГА.
М(2-ЭГ)ЭГА, который соответствует вышеуказанной формуле (I), где R является 2-этилгексильной группой, синтезируют из 2-этилгексаноил хлорида и N-метил-2-этилгексанамина, в присутствии Et3N в безводном ДХМ (схема реакции А), где N-метил-2-этилгексанамин предварительно синтезируют из 2-этилгексиламина (схема реакции В).
* Синтез N-метил-2-этилгексанамина
К раствору 2-этилгексамина (15,0 мл - 90,5 ммоль - 1 экв.) в безводном тетрагидрофуране (ТГФ) (70 мл) добавляют по каплям при 0°C, с применением воронки, раствор ди-трет-бутил дикарбоната (Boc2O- 23,7 г- 108,0 ммоль- 1,2 экв.) в безводном ТГФ (30 мл). Смесь оставляют при перемешивании на 20 часов при комнатной температуре, а затем концентрируют под контролируемым вакуумом (0,150 мбар/20°C). Полученное неочищенное масло очищают посредством хроматографии на силикагеле (элюция: 10% ДХМ), и после контролируемого концентрирования под вакуумом (0,035 мбар/40°C) получают соединение 3 из схемы реакции В, где R является 2-этилгексильной группой (21 г), в форме бесцветного масла.
К раствору соединения 3 (5,0 г - 21,8 ммоль - 1 экв.) в безводном ТГФ (100 мл), охлажденном до 0°C, добавляют по каплям в течение 10 минут раствор алюминия гидрида и лития (LiAlH4) в концентрации 2,4 моль/л в ТГФ (13,6 мл - 32,7 ммоль - 1,5 экв.). Смесь оставляют для возврата к комнатной температуре, а затем нагревают до 50°C в течение 16 часов. После возврата к комнатной температуре реакционную смесь осторожно гидролизуют постепенным добавлением по каплям этилацетата (1 мл), воды (1,2 мл), 12 н натрия гидроксида (2,5 мл), а затем воды (2,4 мл). Спустя 20 минут при интенсивном перемешивании смесь фильтруют на воронке Бюхнера. Фильтрат концентрируют под контролируемым вакуумом (0,035 мбар, 15°C) до получения N-метил-2-этилгексанамина (3,0 г) в форме бесцветного масла. Это масло применяют на следующем этапе без дополнительной очистки.
* Синтез М(2-ЭГ)ЭГА
К раствору N-метил-2-этилгексанамина (3,0 г -20,9 ммоль - 1 экв.) в безводном ДХМ (40 мл), охлажденном до 0°C, добавляют по каплям Et3N (4,4 мл; 31,4 ммоль; 1,5 экв.), затем 2-этилгексаноил хлорид (3,6 мл - 20,9 ммоль - 1 экв.). Смесь оставляют при перемешивании на 20 часов при комнатной температуре, после чего добавляют воду (40 мл). Фазы отделяют декантацией, и водную фазу экстрагируют ДХМ (2 х 40 мл). Органические фазы объединяют, сушат над натрия сульфатом, фильтруют и концентрируют при сниженном давлении. Полученное неочищенное масло очищают посредством хроматографии на силикагеле (элюция: 100% ДХМ) до получения 98.5% чистой фракции (ВЭЖХ при 210 нм) М(2-ЭГ)ЭГА (2,6 г) в форме бледно-желтого масла. С извлеченными неочищенными фракциями выход от синтеза из 2-этилгексанамина составляет 71%.
13C ЯМР, DEPT (CDCl3) δ (ч./млн.): 176,6 (C=O), 53,8 (CH2), 51,5 (CH2), 43,1 (CH3), 42,7 (CH3), 38,9 (CH), 37,3 (CH), 36,1 (CH), 34,6 (CH), 32,5 (CH2), 30,5(CH2), 29,9(CH2), 28,7 (CH2), 25,9(CH2), 23,6(CH2), 23,1(CH2), 22,9(CH2), 14,0(CH3), 12,1(CH3), 10,8(CH3), 10,5(CH3).
1H ЯМР (CDCl3) δ (ч./млн.): 3,16 (d, 2H, 1CH2, ротамер 1), 3,30 (d, 2H, 1CH2, ротамер 2), 2,90 (s, 3H, 1CH3, ротамер 1), 2,98 (s, 3H, 1CH3, ротамер 2), 2,54 (m, 1H, 1CH), 1,61 (m, 3H, 1CH2 + 1CH), 1,42 (m, 2H,CH2), 1,25 (m, 12H, 6CH2), 0,84 (m, 12H, 4CH3).
MS (ESI+), m/z: 270,3 [MH]+, 292,3 [MNa]+
I.5. Синтез N-2-этилоктил-N-метил-2-этилгексанамида или М(2-ЭО)ЭГА
М(2-ЭО)ЭГА, который соответствует вышеуказанной формуле (I), где R является 2-этилоктильной группой, синтезируют из 2-этилгексаноил хлорида и N-метил-N-2-этилоктанамина в присутствии Et3N в безводном ДХМ (схема реакции А), где N-метил-N-2-этилоктанамин предварительно синтезируют из 2-этилоктиламина (схема реакции В), который получают путем связывания 1-бромгексана и бутиронитрила, с последующим восстановлением полученного 2-этил-октаннитрила.
* Синтез N-метил-N-2-этилоктанамина
К раствору диизопропиламина (11,3 мл - 80,0 ммоль - 1 экв.) в безводном ТГФ (42 мл), охлажденном до -78°C, добавляют н-бутиллитий (н-BuLi - 2,5 M в гексане; 32,0 мл - 80,0 ммоль - 1 экв.). Перемешивание осуществляют в течение 10 минут при -78°C, после чего добавляют бутиронитрил (7,0 мл - 80,0 ммоль - 1 экв.) по каплям. Перемешивание осуществляют в течение 10 минут при -78°C, после чего добавляют 1-бромгексан (11,3 мл - 80,0 ммоль - 1 экв.) по каплям. Затем смесь оставляют при перемешивании на 20 часов с постепенным возвращением к комнатной температуре. Добавляют насыщенный раствор аммония хлорида (40 мл), а затем диэтиловый эфир (Et2O - 50 мл). Фазы отделяют декантацией, и водную фазу экстрагируют Et2O (2 x 50 мл). Объединенные органические фазы сушат над натрия сульфатом, фильтруют и выпаривают под контролируемым давлением (15°C/0,050 мбар). Неочищенное масло получают посредством хроматографии на силикагеле (элюция: циклогексан/ДХМ) до получения 2-этилоктан-нитрила (60 масс.% раствора 24 г с ДХМ/циклогексаном, т.е. при подсчете 9,8 г) в форме бледно-желтого раствора.
К раствору 2-этилоктаннитрила (7,18 г - 45,6 ммоль - 1экв.) в безводном ТГФ (100 мл), охлажденному до 0°C, добавляют по каплям в течение 10 минут раствор LiAlH4 (2,4 M в ТГФ; 38,1 мл - 91,3 ммоль - 2 экв.). Смесь возвращают к комнатной температуре, затем нагревают до 50°C в течение 16 часов. После возвращения к 0°C реакционную смесь осторожно гидролизуют путем последовательного добавления по каплям этилацетата (3 мл), воды (3,3 мл), 12 н натрия гидроксида (7 мл), затем воды (6,7 мл). Спустя 20 минут при интенсивном перемешивании смесь фильтруют на воронке Бюхнера. Фильтрат концентрируют при контролируемом вакууме (0,150 мбар, 35°С) до получения 2-этилоктиламина (7,0 г в 55 масс.% растворе в ТГФ) в форме бесцветного масла. Это масло применяют на следующем этапе без дополнительной очистки.
Синтез N-метил-N-2-этилоктанамина из 2-этилоктиламина, полученного таким способом, выполняют с помощью протокола операции, подобного тому, который ранее описан для синтеза N-метил-N-этилгексанамина.
* Синтез М(2-ЭО)ЭГА
К раствору N-метил-N-2-этилоктанамина (5,6 г- 33,0 ммоль- 1 экв.) в безводном ДХМ (60 мл), охлажденному до 0°C, добавляют по каплям Et3N (6,8 мл- 49,5 ммоль- 1,5 экв.), а затем 2-этилгексаноил хлорид (5,6 мл - 33,0 ммоль - 1 экв.). Смесь оставляют при перемешивании на 20 часов при комнатной температуре, после чего добавляют воду (60 мл). Фазы отделяют декантацией, и водную фазу экстрагируют с ДХМ (2 х 60 мл). Объединенные органические фазы сушат над натрия сульфатом, фильтруют и концентрируют при сниженном давлении. Полученное неочищенное масло очищают хроматографией на силикагеле (элюция: 100% ДХМ) до получения 98,3% чистой фракции (ВЭЖХ при 210 нм) М(2-ЭО)ЭГА (2,6 г) в форме бледно-желтого масла. С извлеченными неочищенными фракциями выход с этапа составляет 64%.
13C ЯМР, DEPT (CDCl3) δ (ч./млн.): 176,2 (C=O), 53,8 (CH2), 51,5 (CH2), 43,2 (CH3), 42,7 (CH3), 39,0 (CH), 37,3 (CH), 36,1 (CH), 34,7 (CH), 32,5 (CH2), 31,8 (CH2), 30,8 (CH2), 29,8 (CH2), 29,7 (CH2), 26,5 (CH2), 26,0 (CH2), 23,7 (CH2), 22,9 (CH2), 22,6 (CH2), 14,0(CH3), 12,1(CH3), 10,8(CH3), 10,5(CH3)
1H ЯМР (CDCl3) δ(ч./млн.): 3,34 (m, 2H, 1CH2, ротамер 1), 3,22 (m, 2H, 1CH2, ротамер 2), 3,03 (s, 3H, 1CH3, ротамер 1), 2,95 (s, 3H, 1CH3, ротамер 2), 2,59 (m, 1H, 1CH), 1,65 (m, 3H, 1CH2 + 1CH), 1,49 (m, 2H, 1CH2), 1,29 (m, 16H, 8CH2), 0,90 (m, 12H, 4CH3)
МС (ионизация электронным ударом +), m/z: 298,4 [MH]+, 320,4 [MNa]+
II - Экстракционные свойства N,N-диалкиламидов из настоящего изобретения
II.1 - Получение коэффициентов распределения урана и плутония и коэффициентов разделения FSU/Pu, из синтетического водного раствора урана и плутония для МОЭГА, МДЭГА и МДдЭГА
Во-первых, проводили тесты экстракции с применением:
- органических фаз: растворов, содержащих 1,4 моль/л МОЭГА, МДЭГА или МДдЭГА в ТПГ; и
- водных фаз: аликвот водных растворов, содержащих 90 г/л урана (VI), примерно 70 мг/л плутония (IV) и 4,15 моль/л HNO3.
Тесты реэкстракции затем проводили с использованием:
- органических фаз: органических фаз, полученных после вышеописанных тестов экстракции; и
- водных фаз: аликвот водных растворов, содержащих 0,1 моль/л HNO3.
Каждый из этих тестов проводили, помещая в контакт органическую фазу с аликвотой водного раствора в пробирке при перемешивании в течение 15 минут при 25°C. Использовали объемное отношение О/В 1 для тестов экстракции, и 1 для тестов реэкстракции. Эти фазы отделяли друг от друга после центрифугирования.
Концентрации урана и плутония измеряли в разделенных органических и водных фазах посредством рентгеновской флуоресценции для урана и α-спектрометрии для плутония.
В Таблице 1 внизу для каждого тестируемого N,N-диалкиламида приведены концентрации урана, обозначенные как [U]орг., такие, как получены в органических фазах после тестов экстракции; коэффициенты распределения урана, обозначенные как DU, и плутония, обозначенные как DPu, такие, как получено после тестов экстракции и реэкстракции; концентрации азотной кислоты, обозначенные как [HNO3]вод., как получено в водных фазах после тестов экстракции и реэкстракции; и коэффициенты разделения U/Pu, обозначенные как FSU/Pu, как получено после тестов реэкстракции.
В этой таблице также приведены результаты экспериментов, полученных в тех же рабочих условиях, но с применением растворов, содержащих N,N-диалкиламиды из предшествующего уровня техники в качестве органических фаз, а именно:
- раствора, содержащего 0,9 моль/л N,N-ди(2-этилгексил)-изобутанамида (или ДЭГиБА) и 0,5 моль/л N,N-ди(2-этилгексил)-н-бутанамида (или ДЭГБА) в ТПГ; эти два N,N-диалкиламида представлены в ссылке [3] под наименованиями DOiBA и DOBA; и
- раствора, содержащего 1,4 моль/л N,N-ди(2-этилгексил)-3,3-диметилбутанамида (или ДЭГДМБА) в ТПГ, где этот N,N-диалкиламид представлен в ссылке [1] под наименованием DOTA.
Таблица 1
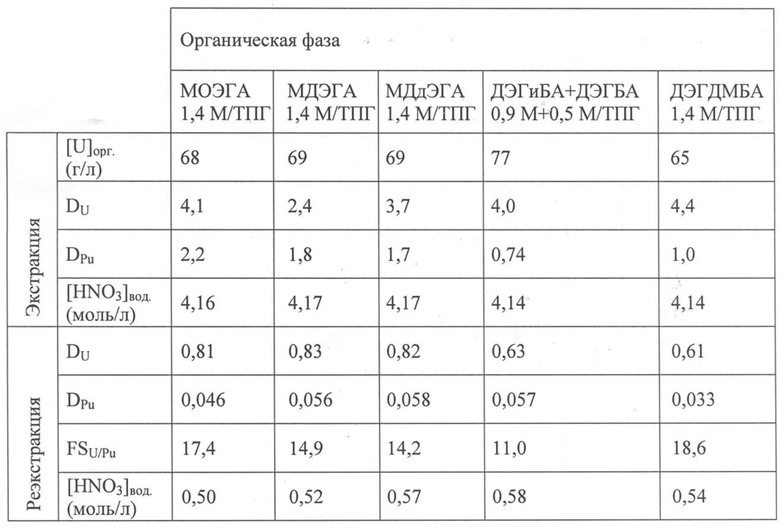
В этой таблице показано, что при сильной кислотности N,N-диалкиламиды из настоящего изобретения экстрагируют уран (VI) (DU(VI) ≥ 2,4) так же, как N,N-диалкиламиды из предшествующего уровня техники, но они гораздо сильнее экстрагируют плутоний (IV) (DPu(IV) ≥ 1,7), чем последние.
Таблица также показывает, что плутоний (IV) можно затем легко реэкстрагировать из органической фазы с применением водного раствора азотной кислоты с низкой кислотностью ([HNO3] =0,5 M), в то время как уран преимущественно удерживается в этой органической фазе (FSU/Pu > 14).
II-2. Исследование стехиометрии комплексов, образованных МОЭГА с ураном и плутонием.
Тесты экстракции проводили с использованием:
- органических фаз: растворов, содержащих, соответственно, 0,1 моль/л, 0,5 моль/л, 0,75 моль/л, 1,0 моль/л, 1,25 моль/л, 1,5 моль/л и 2 моль/л МОЭГА в ТПГ; и
- водных фаз: аликвот водного раствора, содержащего 2 г/л урана (VI), 1 моль/л HNO3 и 2 моль/л LiNO3, и аликвот водного раствора, содержащего 1,7x10-4 моль/л плутония (IV), 1 моль/л HNO3 и 2 моль/л LiNO3.
Для этих тестов каждую органическую фазу помещали в контакт с аликвотой водного раствора в пробирке при перемешивании в течение 15 минут при 25°C, с объемным отношением О/В 1. Эти фазы затем отделяли друг от друга после центрифугирования.
Концентрации урана в водных фазах измеряли посредством атомно-эмиссионной спектроскопии с индуктивно-связанной плазмой (или ICP-AES), а концентрации урана в органических фазах определяли путем реэкстракции этих элементов в воде и измерения их концентрации путем ICP-AES в водных фазах, полученных при этой реэкстракции. Концентрации плутония измеряли в водных и органических фазах посредством α-спектрометрии.
Результаты иллюстрированы на Фигуре 1, где в форме двух прямых линий показано изменение логарифма коэффициентов распределения, обозначенных DM, для урана и плутония, в зависимости от логарифма свободной концентрации (в моль/л) МОЭГА в органической фазе (общая концентрация МОЭГА скорректирована на фракцию азотной кислоты, экстрагированной в органической фазе).
Эта Фигура показывает, что угловой коэффициент прямой линии, соответствующей экстракции урана (VI), близок к 2, что подтверждает образование комплекса UO2(NO3)2(МОЭГА)2, соответствующего комплексам, обычно наблюдаемым с N,N-диалкиламидами.
С другой стороны, в соответствии с этими результатами, комплекс, образованный МОЭГА с плутонием (IV), как кажется, включает три молекулы МОЭГА на один катион Pu4+, таким образом, обеспечивая Pu:МОЭГА стехиометрию 1:3 [Pu(NO3)4(МОЭГА)3], уже наблюдаемую с другими асимметричными N,N-диалкиламидами (ссылка [5]). Таким образом, экстракционное равновесие плутония (IV) посредством МОЭГА может быть описано следующим образом:
Pu4+ + 4NO3- + 3МОЭГА ↔ Pu(NO3)4МОЭГА3
II.3. Получение коэффициентов распределения урана, плутония и продуктов деления в водном растворе, полученном при растворении гранул ядерного топлива в HNO3, для МОЭГА.
Тесты экстракции проводили с использованием:
- органической фазы: раствора, содержащего 1,4 моль/л МОЭГА в ТПГ; и
- водной фазы: водного раствора, предварительно полученного путем растворения гранул, полученных из различных видов облученного топлива UOX-BWR типа (ядерного реактора на кипящей воде) и UOX-PRW типа (водо-водяного энергетического реактора) в 5 М азотной кислоте.
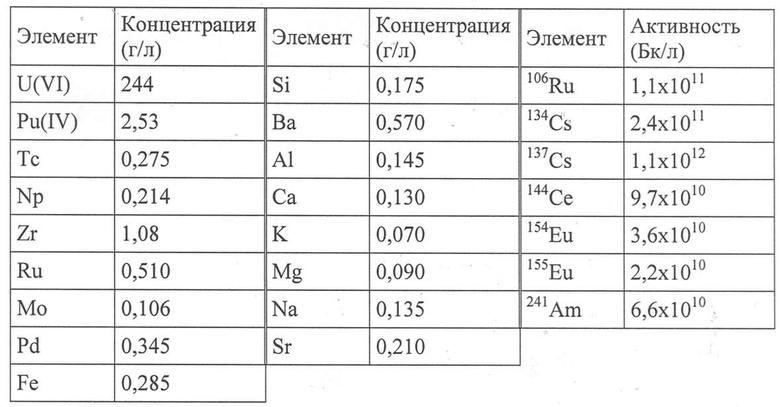
Этот водный раствор содержит 4,3 моль/л HNO3, и его элементарный состав приведен в Таблице 2 ниже.
Таблица 2
Органическую фазу, предварительно уравновешенную 6 моль/л HNO3, помещали в контакте с водной фазой в пробирке при перемешивании в течение 15 минут при 25°C, с объемным отношением О/В 2,5.
Затем эти фазы отделяли друг от друга после центрифугирования.
Концентрации урана и плутония и активности β-γ изотопов измеряли в каждой из органических и водных фаз, разделенных таким способом, посредством рентгеновской флуоресценции для урана и плутония, и γ-спектрометрии для β-γ изотопов.
Концентрации Tc, Np, Zr, Mo и Fe можно было выявить только в водной фазе посредством ICP-AES, а концентрации этих элементов в органической фазе были установлены по разнице между исходными концентрациями указанных элементов в водной фазе и концентрациями, измеренными при равновесии после экстракции.
Результаты, полученные в контексте кислотности водной фазы, обозначенной как [H+]вод., для концентраций урана и плутония в водной и органических фазах, соответственно обозначенных как [U]вод., [U]орг., [Pu]вод. и [Pu]орг., и коэффициенты распределения, обозначенные D, приведены в Таблице 3 ниже.
В этой Таблице также приведены результаты экспериментов, полученные при тех же самых рабочих условиях, но с применением в качестве органической фазы раствора, содержащего 30 об.% ТБФ и ТПГ.
Таблица 3
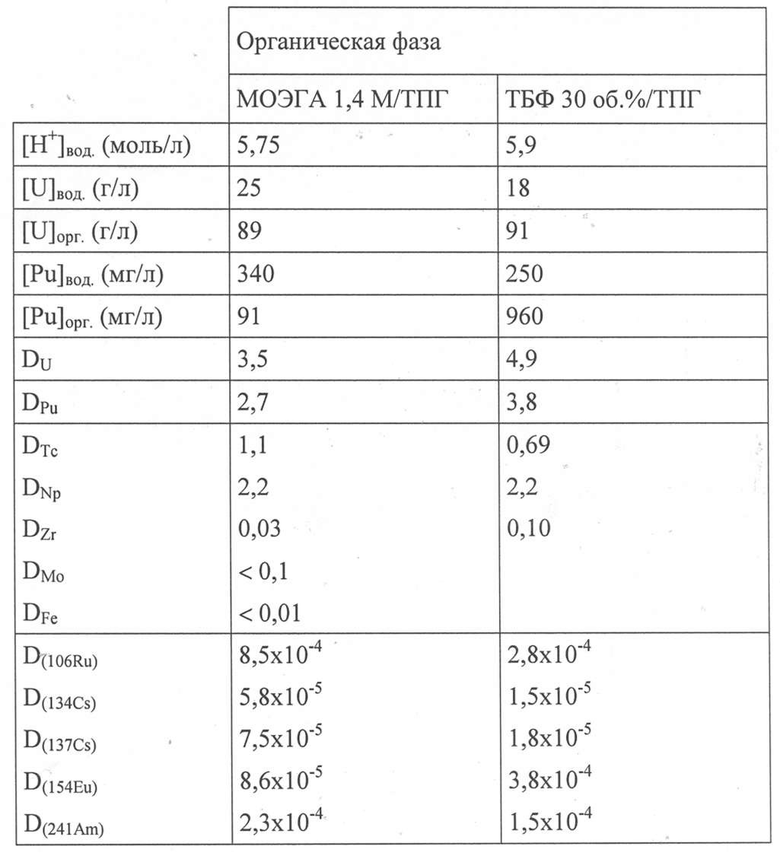
В этой Таблице показано, что применение МОЭГА в качестве экстрагента приводит к высоким коэффициентам распределения (>>1) для урана (VI) и плутония (IV) при кислотности 5,75 моль/л HNO3, несмотря на сильное насыщение ураном органической фазы (89 г урана на литр).
Было также показано, что МОЭГА в качестве экстрагента также приводит к высоким коэффициентам разделения FSU/PF и FSPu/PF, в частности, по отношению к рутению-106, поскольку они всегда выше 3000. Коэффициенты разделения FSU/Am и FSPu/Am также являются очень высокими.
Эти результаты, которые очень близки к тем, которые наблюдались в идентичных условиях, но с применением ТБФ в качестве экстрагента, подтверждают, что N,N-диалкиламиды из настоящего изобретения обеспечивают количественную и избирательную экстракцию урана и плутония по отношению к америцию, кюрию и основным продуктам деления, потенциально содержащимся в водном растворе, полученном при растворении отработавшего ядерного топлива в азотной кислоте, при этом впоследствии позволяя разделять уран и плутоний на два водных потока, где первый из них содержит уран без плутония, а второй содержит плутоний с ураном или без урана, без необходимости восстановления плутония, в отличие от применения ТБФ.
II.4 - Получение коэффициентов распределения урана и плутония и коэффициентов разделения FSU/Pu из синтетического водного раствора урана и плутония для М(2-ЭГ)ЭГА и М(2-ЭО)ЭГА.
Экстракционные тесты проводили с использованием:
- органических фаз: растворов, содержащих 0,5 моль/л М(2-ЭГ)ЭГА или М(2-ЭО)ЭГА в ТПГ; и
- водных фаз: аликвот водных растворов урана (VI) (≈ 11,5 г/л) с примесью плутония (IV) (≈ 0,4 МБк/мл) и включающих либо 4 моль/л HNO3, либо 0,5 моль/л HNO3 (для имитации водной фазы с низкой кислотностью, которую обычно применяют для реэкстракции плутония на этапе разделения U/Pu на два водных потока).
Каждый из этих тестов проводили, помещая в контакт органическую фазу в пробирке при перемешивании с аликвотой водного раствора в течение 15 минут при 25°С. Использовали объемное отношение О/В 1. Эти фазы затем отделяли друг от друга после центрифугирования.
Концентрацию урана и активность плутония (239+240Pu) измеряли в органической и водной фазах, разделенных таким способом, с применением ICP-AES и α-спектрометрии, соответственно.
В Таблице 4 ниже для каждого тестируемого N,N-диалкиламида приведены коэффициенты распределения урана, обозначенные как DU, и плутония, обозначенные как DPu, полученные таким способом, и коэффициенты разделения U/Pu, обозначенные как FSU/Pu, полученные с кислотностью 0,5 моль HNO3/л.
В этой Таблице также приведены результаты экспериментов, полученных в тех же самых рабочих условиях, но с применением в качестве органических фаз аликвот раствора, содержащего 0,5 моль/л МОЭГА в ТПГ.
Таблица 4
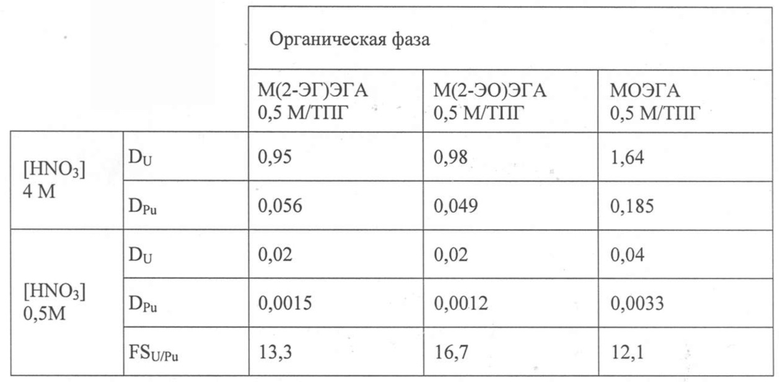
Эта Таблица показывает, что присутствие разветвления в алкильной группе, представленной R в вышеуказанной формуле (I), дает соединения, имеющие отличную избирательность U(VI)/Pu(IV) при низкой кислотности (FSU/Pu > 13), обеспечивая избирательную реэкстракцию плутония из слабокислой водной фазы, такой, как обычно применяется на этапе разделения U/Pu, без необходимости восстановления плутония.
Коэффициенты распределения DU и DPu слегка ниже тех, которые получают с МОЭГА, в особенности из-за стерического ограничения, вызванного разветвлением. Эти коэффициенты распределения, однако, можно сильно повысить путем увеличения содержания экстрагента. С учетом образования комплекса Pu(NO3)4L3, как показано в вышеприведенном Примере II.2 с МОЭГА, повышение содержания М(2-ЭГ)ЭГА от 0,5 моль/л до 1,5 моль/л позволяет повысить коэффициент распределения плутония в 27 раз, т.е. от 0,056 до 1,5, с 4 моль/л HNO3, до значения, достаточного в процессе для совместной экстракции плутония при сильной кислотности.
III - Поточная диаграмма способа из настоящего изобретения для обработки водного раствора азотной кислоты, полученного при растворении отработавшего ядерного топлива.
Ссылка делается на Фигуру 2, где изображена поточная диаграмма способа из настоящего изобретения для переработки водного азотнокислого раствора, полученного при растворении отработавшего ядерного топлива.
Как показано на этой Фигуре 2, способ включает 8 этапов.
Первый из этих этапов, обозначенный как «совместная экстракция U/Pu» на Фигуре 2, предназначен для достижения объединенной экстракции урана и плутония, где первый из них находится в степени окисления +VI, а второй в степени окисления +IV, из водного азотнокислого раствора, полученного при растворении отработавшего ядерного топлива.
Такой раствор, как правило, включает от 3 до 6 моль/л HNO3, уран, плутоний, минорные актиноиды (америций, кюрий и нептуний), продукты деления (La, Ce, Pr, Nd, Sm, Eu, Gd, Mo, Zr, Ru, Tc, Rh, Pd, Y, Cs, Ba, и т.д.) и несколько продуктов коррозии, таких как железо.
«Этап совместной экстракции U/Pu» проводят путем циркуляции растворяющей жидкости в экстракторе 1, в противоточном потоке к органической фазе (обозначенной как «ОФ» на Фигуре 2), включающей от 1 моль/л до 2 моль/л, и предпочтительно от 1,3 моль/л до 1,5 моль/л N,N-диалкиламида из настоящего изобретения, или смеси N,N-диалкиламидов из настоящего изобретения, в растворе в органическом разбавителе.
Этот органический разбавитель является алифатическим, линейным или разветвленным углеводородом, таким как н-додекан, ТПГ; изопарафинный разбавитель, выпускаемый TOTAL под торговой маркой Isane IP185T; предпочтение отдается ТПГ.
Второй этап способа, обозначенный как «промывание ПД» на Фигуре 2, предназначен для извлечения из органической фазы, полученной от «совместной экстракции U/Pu», фракции продуктов деления, которые были экстрагированы из растворяющей жидкости совместно с ураном и плутонием.
Для этой цели этап «промывания ПД» включает одну или несколько операций промывания органической фазы, полученной при «совместной экстракции U/Pu», где каждую операцию промывания выполняют путем циркуляции этой органической фазы в экстракторе 2, в противоточном потоке к водному азотнокислому раствору, имеющему концентрацию, по возможности, в диапазоне от 0,5 моль/л до 6 моль/л HNO3, но предпочтительно от 4 моль/л до 6 моль/л HNO3, и более предпочтительно от 4 до 5 моль HNO3, для облегчения реэкстракции рутения и технеция.
Если этап «промывания ПД» проводят с одним или несколькими водными растворами с сильной кислотностью, т.е. как правило, 3 моль/л HNO3 или больше, то этот этап дополнительно включает уменьшение кислотности органической фазы, которую проводят путем циркуляции этой органической фазы в противоточном потоке к слабокислому водному азотнокислому раствору, т.е. включающему от 0,1 до 1 моль/л HNO3, например, водному раствору, включающему 0,5 моль/л HNO3, для предотвращения отведения слишком большого количества кислоты к экстрактору, предназначенному для третьего этапа, обозначенного как «реэкстракция Pu» на Фигуре 2, что может нарушать производительность этого третьего этапа.
Этап «реэкстракции Pu», который представляет первый этап разделения U/Pu, предназначен для извлечения плутония в степени окисления +IV, и таким образом, без восстановления этого плутония, из органической фазы, полученной при «промывании ПД».
Этот этап проводят путем циркуляции этой органической фазы в экстракторе 3 в противоточном потоке к водному раствору, содержащему от 0,1 моль/л до 0,5 моль/л HNO3, и предпочтительно с применением отношения потоков О/В больше 1, предпочтительно 3 или больше, и более предпочтительно 5 или больше, так чтобы плутоний (IV) реэкстрагировался концентрирующим образом.
Реэкстракция плутония (IV), выполняемая на этапе «реэкстракции Pu», сопровождается реэкстракцией фракции урана (VI), который также содержится в органической фазе, полученной при «промывании ПД».
Четвертый этап способа, обозначенный как «1-е промывание U» на Фигуре 2, и представляющий второй этап разделения U/Pu, таким образом, предназначен для экстракции из водной фазы, полученной при «реэкстракции Pu»:
- либо всего урана, содержащегося в водной фазе, если необходимо, чтобы разделение U/Pu приводило к водному раствору, содержащему плутоний без урана, и к органическому раствору, содержащему уран без плутония;
- либо количества урана, которое после «1-го промывания U» позволяет получать водный раствор, содержащий уран и плутоний в предварительно выбранном отношении, если необходимо, чтобы разделение U/Pu приводило к водному раствору, содержащему смесь плутония и урана в этом отношении, и к органическому раствору, содержащему уран без плутония.
В обоих случаях «1-е промывание U» проводят путем циркуляции водной фазы, полученной при «реэкстракции Pu» в экстракторе 4, в противоточном потоке к органической фазе, имеющей идентичный состав с органической фазой, используемой при «совместной экстракции U/Pu». Количество экстрагируемого урана вначале подводят путем действия на отношение потоков О/В, а затем на кислотность водной фазы; экстракция урана увеличивается при повышении отношения потока органической фазы/водной фазы и усилении кислотности водной фазы. Таким образом, добавление HNO3 большей или меньшей концентрации к водной фазе, циркулирующей в экстракторе 4, можно проводить в зависимости от кислотности, которую необходимо обеспечить в этой водной фазе.
Пятый этап, обозначенный как «α-барьер Тс» на Фигуре 2, предназначен для извлечения из органической фазы, полученной при «реэкстракции Pu», фракции технеция, который был экстрагирован при «совместной экстракции U/Pu», но не был реэкстрагирован при «промывании ПД», с целью очистки этой органической фазы от технеция.
Это также позволяет проводить реэкстракцию из органической фазы, полученной при «реэкстракции Pu», фракции нептуния, которая была экстрагирована при «совместной экстракции U/Pu», и которая следует за технецием до «α-Тс барьера», а также следов плутония, которые все еще может содержать эта органическая фаза.
Этот этап проводят путем циркуляции органической фазы, полученной при «реэкстракции Pu» в экстракторе 5, в противоточном потоке к водному азотнокислому раствору с низкой кислотностью, т.е. содержащему от 0,1 моль/л до 3 моль/л HNO3, и предпочтительно 1 моль/л HNO3, и содержащему один или несколько восстанавливающих агентов, обеспечивающих восстановление технеция, который присутствует в органической фазе в степени окисления +VII, до технеция (IV), не экстрагируемого N,N-диалкиламидами; нептуния (VI) до нептуния (IV) или нептуния (V), не экстрагируемого N,N-диалкиламидами при низкой кислотности; и плутония (IV) до плутония (III), который меньше экстрагируется N,N-диалкиламидами при низкой кислотности, чем плутоний (IV), без восстановления урана (VI).
В качестве восстанавливающих агентов можно применять урана нитрат (или U(IV)), гидразиния нитрат (или HN), гидроксиламмония нитрат (или HAN), ацетальдоксим, или их смесь, такую как смесь U(IV)/HN, U(IV)/HAN или U(IV)/ацетальдоксима, предпочтительной является смесь U(IV)/HN или U(VI)/HAN. Можно добавлять глюконовую кислоту к водному раствору для уменьшения явления повторного окисления технеция в водной фазе, и таким образом, ограничения потребления восстанавливающего агента (агентов).
Этот этап можно проводить при комнатной температуре (т.е. 20-25°С), но предпочтительно его проводят при температуре в диапазоне от 30°С до 40°С, и более предпочтительно при 32°С для стимуляции кинетики реэкстракции технеция при ограничении явления повторного окисления технеция в водной фазе, и таким образом, для ограничения риска обратной экстракции реэкстрагированного технеция в органическую фазу.
Шестой этап, обозначенный как «2-е промывание U» на Фигуре 2, предназначен для экстракции из водной фазы, полученной от «α-Тс барьера», урана, который был реэкстрагирован вместе с технецием на предыдущем этапе, так чтобы избежать получения на этапе «α-Тс барьера» слишком большой потери урана в водной фазе.
Его проводят путем циркуляции водной фазы, полученной от «α-Тс барьера» в экстракторе 6, в противоточном потоке к органической фазе, имеющей идентичный состав с органическими фазами, используемыми для «совместной экстракции U/Pu» и «1-го промывания U», после подкисления этой водной фазы путем добавления концентрированной азотной кислоты, например, 10 М, для того чтобы стимулировать экстракцию урана.
Седьмой этап, обозначенный как «реэкстракция U» на Фигуре 2, предназначен для извлечения урана (VI) из органической фазы, полученной на этапе «α-Тс барьера».
Его проводят путем циркуляции органической фазы, полученной от «α-Тс барьера» в экстракторе 7, в противоточном потоке к водному азотнокислому раствору низкой кислотности, т.е. содержащему не более 0,05 моль/л HNO3, например, водному раствору, содержащему 0,01 моль/л HNO3. Этот этап можно проводить при комнатной температуре (т.е. 20-25°С), но предпочтительно его проводят при нагревании (т.е., как правило, при температуре 40-50°С) с применением отношения потоков О/В больше 1, так чтобы уран (VI) реэкстрагировался с концентрированием.
После этих 7 этапов мы получаем:
- два рафината, соответствующих водным фазам, соответственно выходящим из экстракторов 1 и 6, где первый включает продукты деления и америций и кюрий (Первичный рафинат» на Фигуре 2), а второй включает технеций, нептуний, и возможно, следы плутония («Вторичный рафинат» на Фигуре 2);
- водную фазу, выходящую из экстрактора 4, включающую либо очищенный от загрязнителей (дезактивированный) плутоний, либо смесь очищенного от загрязнителей (дезактивированного) плутония и очищенного от загрязнителей (дезактивированного) урана, называемую «поток Pu» или «поток Pu+U», соответственно;
- водную фазу, выходящую из экстрактора 7, включающую очищенный от загрязнителей (дезактивированный уран), называемую «поток U»; и
- органическую фазу, выходящую из экстрактора 7, которая больше не содержит какого-либо плутония или урана, но может содержать определенный ряд примесей и продуктов распада (образованных при гидролизе и радиолизе) экстрагента, которые могут накапливаться в предыдущих этапах.
Таким образом, восьмой этап, обозначенный как «промывание ОФ» на Фигуре 2, предназначен для регенерации этой органической фазы путем её обработки посредством одного или нескольких промываний основным водным раствором, например, первого промывания водным раствором 0,3 моль/л натрия карбоната, с последующим вторым промыванием водным раствором 0,1 моль/л натрия гидроксида, затем с одним или несколькими промываниями водным раствором азотной кислоты, обеспечивающим повторное подкисление органической фазы, например, водным раствором, содержащим 2 моль/л HNO3, где каждое промывание выполняют путем циркуляции указанной органической фазы в экстракторе в противоточном потоке к водному промывающему раствору.
Как можно видеть на Фигуре 2, органическая фаза, регенерированная таким способом, может быть возвращена обратно к экстракторам 1 и 4 для повторного ввода в цикл переработки.
Цитированные ссылки
[1] FR-A-2 591 213
[2] FR-A-2 642 561
[3] FR-A-2 642 562
[4] Ruikar et al., Journal of Radioanalytical and Nuclear Chemistry 1993, 176(2), 103-111
[5] Prabhu et al., Radiochimica Acta 1993, 60, 109-114
[6] Cui et al., Radiochimica Acta 2005, 93, 287-290
[7] Sun et al., Journal of Radioanalytical and Nuclear Chemistry 2005, 264(3), 711-713.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОБРАБОТКИ ВОДНОГО АЗОТНОКИСЛОГО РАСТВОРА, ПОЛУЧЕННОГО ПРИ РАСТВОРЕНИИ ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА, ВЫПОЛНЯЕМЫЙ В ОДНОМ ЦИКЛЕ И НЕ ТРЕБУЮЩИЙ КАКОЙ-ЛИБО ОПЕРАЦИИ, ВКЛЮЧАЮЩЕЙ ВОССТАНОВИТЕЛЬНУЮ РЕЭКСТРАКЦИЮ ПЛУТОНИЯ | 2016 |
|
RU2706954C2 |
| НЕСИММЕТРИЧНЫЕ N,N-ДИАЛКИЛАМИДЫ, В ЧАСТНОСТИ ИСПОЛЬЗУЕМЫЕ ДЛЯ ОТДЕЛЕНИЯ УРАНА(VI) ОТ ПЛУТОНИЯ(IV), ИХ СИНТЕЗ И ПРИМЕНЕНИЕ | 2018 |
|
RU2762634C2 |
| ПРИМЕНЕНИЕ АЛЬДОКСИМОВ, СОДЕРЖАЩИХ ПО МЕНЬШЕЙ МЕРЕ ПЯТЬ АТОМОВ УГЛЕРОДА, В КАЧЕСТВЕ АГЕНТОВ ПРОТИВОАЗОТИСТОГО ДЕЙСТВИЯ В ОПЕРАЦИЯХ ВОССТАНОВИТЕЛЬНОЙ РЕЭКСТРАКЦИИ ПЛУТОНИЯ | 2016 |
|
RU2718437C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА, НЕ ТРЕБУЮЩИЙ ВОССТАНОВИТЕЛЬНОЙ РЕЭКСТРАКЦИИ ПЛУТОНИЯ | 2011 |
|
RU2558332C9 |
| КАРБАМИДЫ ДЛЯ РАЗДЕЛЕНИЯ УРАНА (VI) И ПЛУТОНИЯ (IV) БЕЗ ВОССТАНОВЛЕНИЯ ПЛУТОНИЯ (IV) | 2018 |
|
RU2782220C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА (ОЯТ) АЭС | 1997 |
|
RU2132578C1 |
| СПОСОБ РЕГЕНЕРАЦИИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА И ПОЛУЧЕНИЯ СМЕШАННОГО УРАН-ПЛУТОНИЕВОГО ОКСИДА | 2007 |
|
RU2431896C2 |
| СПОСОБ ВЫВЕДЕНИЯ НЕПТУНИЯ ПРИ ФРАКЦИОНИРОВАНИИ ДОЛГОЖИВУЩИХ РАДИОНУКЛИДОВ | 2010 |
|
RU2454740C1 |
| ПРИМЕНЕНИЕ ГИДРОКСИИМИНОАЛКАНОВЫХ КИСЛОТ В КАЧЕСТВЕ АГЕНТОВ ПРОТИВОАЗОТИСТОГО ДЕЙСТВИЯ ПРИ ОПЕРАЦИЯХ ВОССТАНОВИТЕЛЬНОЙ РЕЭКСТРАКЦИИ ПЛУТОНИЯ | 2016 |
|
RU2713495C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОВМЕСТНОГО РАСТВОРА U И Pu | 2014 |
|
RU2561065C1 |
Изобретение относится к новым асимметричным N,N-диалкиламидам формулы (I):
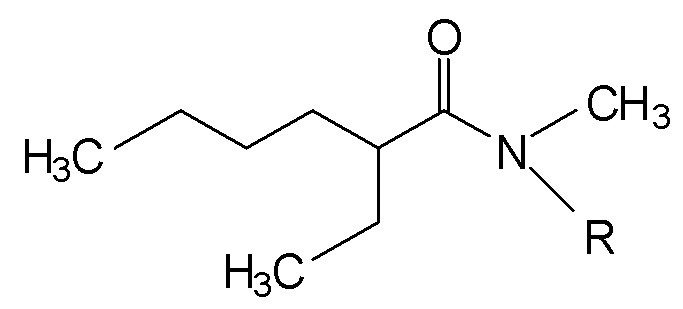 (I),
(I),
где R является линейной или разветвленной алкильной группой, имеющей от 8 до 15 атомов углерода. Изобретение также относится к способу синтеза N,N-диалкиламидов формулы (I), к их применению для экстракции урана и/или плутония из водного раствора кислоты, к их применению для полного или частичного отделения урана от плутония из водного раствора кислоты, и к способу из одного цикла для обработки водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте. Изобретение позволяет экстрагировать, разделять и очищать от загрязнителей уран и плутоний, содержащиеся в растворе, в одном цикле без необходимости какой-либо операции восстановления плутония. 5 н. и 18 з.п. ф-лы, 2 ил., 4 табл.
1. N,N-диалкиламид формулы (I), приведенной ниже:
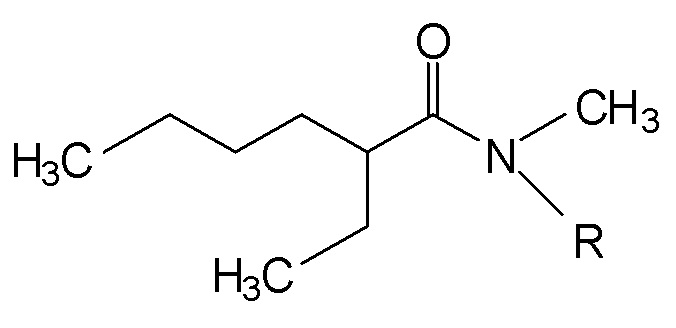 (I),
(I),
где R является линейной или разветвленной алкильной группой, имеющей от 8 до 15 атомов углерода.
2. N,N-диалкиламид по п.1, где алкильная группа содержит не более 12 атомов углерода.
3. N,N-диалкиламид по п.2, где алкильная группа является н-октильной, н-децильной, н-додецильной, 2-этилгексильной или 2-этилоктильной группой.
4. Способ синтеза N,N-диалкиламида по любому из пп.1-3, включающий реакцию галоида формулы (II), приведенной ниже:
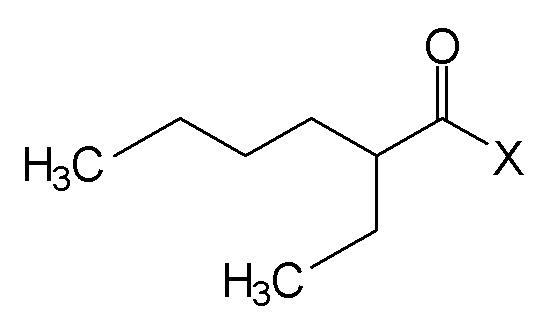 (II),
(II),
где Х является атомом галогена, с амином формулы HN(CH3)R, где R является линейной или разветвленной алкильной группой, имеющей от 8 до 15 атомов углерода.
5. Применение N,N-диалкиламида или смеси N,N-диалкиламидов по любому из пп.1-3 для экстракции урана (VI) и/или плутония (IV) из водного раствора кислоты.
6. Применение по п.5, включающее контактирование водного раствора кислоты с органическим раствором, включающим N,N-диалкиламид или смесь N,N-диалкиламидов в органическом разбавителе, с последующим разделением водного и органического растворов.
7. Применение по п.6, где органический раствор содержит от 1 моль/л до 2 моль/л N,N-диалкиламида или смеси N,N-диалкиламидов.
8. Применение по п.5, где водный раствор представляет собой водный раствор, полученный при растворении отработавшего ядерного топлива в азотной кислоте.
9. Применение N,N-диалкиламида или смеси N,N-диалкиламидов по любому из пп.1-3 для полного или частичного отделения урана (VI) от плутония (IV) из водного раствора кислоты, включающее следующие этапы:
(а) совместную экстракцию урана и плутония из водного раствора, включающую по меньшей мере одно контактирование указанного водного раствора с органическим раствором, включающим N,N-диалкиламид или смесь N,N-диалкиламидов в качестве экстрагента в растворе в органическом разбавителе, с последующим разделением водного и органического растворов;
(b) реэкстракцию плутония в степени окисления +IV и фракции урана из органического раствора, полученного на этапе (а), где реэкстракция включает по меньшей мере одно контактирование органического раствора с водным раствором, содержащим от 0,1 моль/л до 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов; и
(с) экстракцию всей или части урановой фракции, содержащейся в водном растворе, полученном на этапе (b), где экстракция включает по меньшей мере одно контактирование водного раствора с органическим раствором, идентичным органическому раствору этапа (а), с последующим разделением водного и органического растворов;
где получают водный раствор, содержащий либо плутоний без урана, либо смесь плутония и урана, и органический раствор, содержащий уран без плутония.
10. Применение по п.9, где органический раствор с этапа (а) включает от 1 моль/л до 2 моль/л N,N-диалкиламида или смеси N,N-диалкиламидов.
11. Применение по п.10, где водный раствор кислоты является водным раствором, полученным при растворении отработавшего ядерного топлива в азотной кислоте.
12. Способ из одного цикла для обработки водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте, где водный раствор содержит уран, плутоний, америций, кюрий и продукты деления, включая технеций; где цикл включает следующие этапы:
(а) совместную экстракцию урана и плутония из водного раствора, включающую по меньшей мере одно контактирование в экстракторе водного раствора с органическим раствором, включающим N,N-диалкиламид или смесь N,N-диалкиламидов по любому из пп.1-3 в качестве экстрагента в растворе в органическом разбавителе, с последующим разделением водного и органического растворов;
(b) очистку органического раствора, полученного на этапе (а), от америция, кюрия и продуктов деления, включающую по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, включающим от 0,5 моль/л до 6 моль/л азотной кислоты, с последующим разделением органического и водного растворов;
(с) разделение урана и плутония, содержащихся в органическом растворе, полученном на этапе (b), на водный раствор, содержащий либо плутоний без урана, либо смесь плутония и урана, и органический раствор, содержащий уран без плутония, где это разделение включает:
(с1) реэкстракцию плутония в степени окисления +IV и фракции урана из органического раствора, полученного на этапе (b), где реэкстракция включает по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, содержащим от 0,1 моль/л до 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов;
(с2) экстракцию всей или части урановой фракции, содержащейся в водном растворе, полученном с (с1), где экстракция включает по меньшей мере одно контактирование в экстракторе водного раствора с органическим раствором, идентичным органическому раствору, используемому на этапе (а), с последующим разделением водного и органического растворов;
(d) очистку органического раствора, полученного с (с1), от технеция, включающую:
(d1) реэкстракцию технеция в степени окисления +IV из органического раствора, полученного с (с1), где реэкстракция включает по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, содержащим от 0,1 моль/л до 3 моль/л азотной кислоты и по меньшей мере один восстанавливающий агент, способный восстанавливать технеций из степени окисления +VII до степени окисления +IV, с последующим разделением органической и водной фаз;
(d2) экстракцию урановой фракции, содержащейся в водном растворе, полученном с (d1), где экстракция включает по меньшей мере одно контактирование в экстракторе водного раствора с органическим раствором, идентичным органическому раствору этапа (а), с последующим разделением водного и органического растворов;
(е) реэкстракцию урана из органического раствора, полученного с (d1), где реэкстракция включает по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, содержащим не более 0,05 моль/л азотной кислоты, с последующим разделением органического и водного растворов; и
(f) регенерацию органической фазы, полученной на этапе (е);
в результате чего получают первый и второй водные растворы, очищенные от америция, кюрия и продуктов деления, включая технеций, где первый водный раствор включает либо плутоний без урана, либо смесь плутония и урана, а второй водный раствор включает уран без плутония.
13. Способ по п.12, где органический раствор на этапе (а) содержит от 1 моль/л до 2 моль/л N,N-диалкиламида или смеси N,N-диалкиламидов.
14. Способ по п.12, где водный раствор с этапа (b) содержит от 4 моль/л до 6 моль/л азотной кислоты.
15. Способ по п.14, где этап (b) дополнительно включает уменьшение кислотности органического раствора, включающее по меньшей мере одно контактирование органического раствора с водным раствором, содержащим от 0,1 моль/л до 1 моль/л азотной кислоты, с последующим разделением органического и водного растворов.
16. Способ по п.12, где контактирование органического и водного растворов в экстракторе с (c1) включает циркуляцию органического и водного растворов при отношении потока органического раствора к потоку водного раствора больше 1.
17. Способ по п.12, где восстанавливающий агент является урана нитратом, гидразиния нитратом, гидроксиламмония нитратом, ацетальдоксимом или их смесью.
18. Способ по п.12, где экстрактор на этапе (d1) нагревают до температуры от 30°C до 40°C.
19. Способ по п.12, где этап (d2) включает подкисление водного раствора, полученного с (d1), для доведения концентрации азотной кислоты в водном растворе до значения по меньшей мере 2,5 моль/л, где подкисление включает добавление азотной кислоты в экстрактор с (d2).
20. Способ по п.12, где экстрактор на этапе (е) нагревают до температуры от 40°C до 50°C.
21. Способ по любому из пп.11-19, где контактирование органического и водного растворов в экстракторе на этапе (е) включает циркуляцию органического и водного растворов при отношении потока органического раствора к потоку водного раствора больше 1.
22. Способ по п.12, где регенерация органического раствора, полученного на этапе (е), включает по меньшей мере одно промывание органического раствора основным раствором, с последующим по меньшей мере одним промыванием органического раствора водным раствором азотной кислоты.
23. Способ по п.12, где органический раствор, полученный на этапе (f), разделяют на первую и вторую фракцию, где первая фракция образует органический раствор с этапа (а), а вторая фракция образует органический раствор с этапа (c2).
| RUIKAR P.B | |||
| et al, Extraction of uranium, plutonium and some fission product with γ-irradiated unsymetrical and branched chain diallylamides introduction, J | |||
| RADIOANAL | |||
| NUCL | |||
| CHEM., LETTERS, 1993, v | |||
| Приспособление для удаления таянием снега с железнодорожных путей | 1920 |
|
SU176A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Клапанный регулятор для паровозов | 1919 |
|
SU103A1 |
| PRABHU B.R | |||
| et al, Extraction of Uranium(VI) and Plutonium(IV) with Unsymmetrical Monoamides, Radiochimica Acta, 1993, v | |||
| Способ получения молочной кислоты | 1922 |
|
SU60A1 |
| Шкив для канатной передачи | 1920 |
|
SU109A1 |
| SUN G | |||
| et al, Extraction of U(VI) with unsymmetrical N-methyl-N-octyl alkylamides in toluene, Journal of Radioanalytical and Nuclear Chemistry, 2005, v | |||
| Железнодорожный снегоочиститель | 1920 |
|
SU264A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ШТАНГЕН-ЦИРКУЛЬ С ВЫДВИЖНОЮ НОЖКОЮ | 1922 |
|
SU711A1 |
| УСТРОЙСТВО ДЛЯ ИСПЫТАНИЙ МОЩНОГО ЧАСТОТНО-УПРАВЛЯЕМОГО ГРЕБНОГО ЭЛЕКТРОПРИВОДА СИСТЕМЫ ЭЛЕКТРОДВИЖЕНИЯ В УСЛОВИЯХ СТЕНДА | 2014 |
|
RU2591213C1 |
| СПОСОБ СЕЛЕКЦИОННОЙ ОЦЕНКИ ГИДРАТЦЕЛЛЮЛОЗНЫХ ВОЛОКОН КАК ПРЕКУРСОРА ПРИ ПОЛУЧЕНИИ УГЛЕРОДНЫХ ВОЛОКОН | 2016 |
|
RU2642561C1 |
| СПОСОБ СЕЛЕКЦИОННОЙ ОЦЕНКИ ГИДРАТЦЕЛЛЮЛОЗНЫХ ВОЛОКОН КАК ПРЕКУРСОРА ПРИ ПОЛУЧЕНИИ УГЛЕРОДНЫХ ВОЛОКОН | 2016 |
|
RU2642561C1 |
| УСОВЕРШЕНСТВОВАНИЕ СПОСОБА PUREX И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2400841C2 |

