Область техники, к которой относится изобретение
Настоящее изобретение относится к композициям жидкого топлива, содержащим компонент, произведенный из растворимого в воде оксигенированного углеводорода.
Уровень техники
Разработке новых технологий получения энергии из ресурсов, отличных от ископаемых топлив, было уделено большое внимание. Ресурс, который демонстрирует перспективы в качестве альтернативы ископаемому топливу, представляет собой биомасса. В противоположность ископаемому топливу биомасса также является и возобновляемой.
Один тип биомассы представляет собой биомассу растительного происхождения. Биомасса растительного происхождения представляет собой наиболее изобильный источник углевода в мире благодаря лигноцеллюлозным материалам, образующим оболочки клеток у высших растений. Оболочки клеток растений делятся на две секции - первичные оболочки клеток и вторичные оболочки клеток. Первичная оболочка клетки формирует структуру для растущих клеток и образована из трех основных полисахаридов (целлюлозы, пектина и гемицеллюлозы) и одной группы гликопротеинов. Вторичная оболочка клетки, которая образуется после завершения роста клетки, также содержит полисахариды и упрочняется благодаря полимерному лигнину, ковалентно сшитому с гемицеллюлозой. Гемицеллюлоза и пектин обычно встречаются в изобилии, но целлюлоза представляет собой преобладающий полисахарид и наиболее изобильный источник углеводов.
Большинство транспортных средств, будь то суда, поезда, самолеты или автомобили, требуют использования высокой удельной мощности, генерируемой двигателями внутреннего сгорания и/или реактивными двигателями. Данные двигатели требуют использования полностью сгорающих топлив, которые в общем случае имеют жидкую форму или, в меньшей степени, представляют собой сжатые газы. Жидкие топлива являются более пригодными для транспортирования вследствие их высокой удельной энергоемкости и их способности перекачиваться насосом, что делает обращение с ними более легким.
В настоящее время биомасса представляет собой единственную возобновляемую альтернативу жидким топливам транспортных средств. К сожалению, развитие прогресса в разработке новых технологий получения жидких биотоплив происходило медленно, в особенности для жидких топливных продуктов, которые соответствуют современной инфраструктуре. Несмотря на возможность получения из ресурсов, образуемых биомассой, широкого ассортимента топлив, таких как этанол, метанол, биодизельное топливо, дизельное топливо Фишера-Тропша и керосин и газообразные топлива, такие как водород и метан, данные топлива могут потребовать использования или новых технологий распределения и/или технологий сжигания, соответствующих их характеристикам. Получение данных топлив также имеет тенденцию к дороговизне.
Этанол, например, получают в результате превращения углевода из биомассы в сахар, который после этого превращают в этанол по способу ферментации. Этанол представляет собой наиболее широко использующееся биотопливо на сегодняшний день при современном объеме выработки 4,3 миллиарда галлонов (16,3 миллиарда дм3) в год в расчете на крахмалоносную культуру, такую как кукуруза. Однако, этанолу свойственны очень существенные недостатки в отношении его теплоты сгорания в качестве топлива в сопоставлении с величиной энергии, необходимой для его производства. Этанол, полученный в результате ферментации, содержит большие количества воды, обычно при наличии только приблизительно 5 процентов этанола в расчете на объем водно/спиртового продукта ферментации. Удаление данной воды является высокоэнергозатратным и зачастую требует использования в качестве источника тепла природного газа. Этанол также характеризуется меньшим энергосодержанием в сопоставлении с бензином, что означает необходимость использования большего количества топлива для прохождения идентичного расстояния. Этанол является очень коррозионно-активным по отношению к топливным системам и не может транспортироваться в нефтепроводах. В результате этанол транспортируют между городами в бензовозах, что увеличивает для него общую стоимость и энергозатраты. С учетом полной энергии, затрачиваемой в сельскохозяйственных машинах и оборудовании, при культивации, посадках, в удобрениях, пестицидах, гербицидах, фунгицидах на нефтяной основе, системах орошения, при сборе урожая, транспортировании на перерабатывающие предприятия, ферментации, перегонке, высушивании, транспортировании на топливные базы и к насосам автозаправочных станций, и меньшего энергосодержания этанольного топлива, результирующая добавленная и доставленная потребителям величина энергосодержания очень невелика.
Биодизельное топливо представляет собой еще один потенциальный источник энергии. Биодизельное топливо может быть получено из растительных масел, животных жиров, отработанных растительных масел, масел из микроводорослей или повторно используемых ресторанных жиров, и его получают по способу, в котором масла, произведенные из органики, объединяют со спиртом (этанолом или метанолом) в присутствии катализатора для получения этилового или метилового сложного эфира. Произведенные из биомассы этиловый или метиловый сложные эфиры после этого могут быть перемешаны с обычным дизельным топливом или использованы в качестве беспримесного топлива (100%-ное био дизельное топливо). Биодизельное топливо также является дорогим в производстве и создает различные проблемы при своем использовании и сжигании. Например, во избежание гелеобразования при низких температурах могут потребоваться специальные приемы работы.
Биомасса также может быть газифицирована для получения синтез-газа, образованного, в первую очередь, из водорода и монооксида углерода, также называемого синтетическим газом или биосинтез-газом. Синтез-газ, получаемый на сегодняшний день, используют непосредственно для выработки тепла и электрической энергии, но из синтез-газа может быть произведено несколько типов биотоплив. Из синтез-газа может быть извлечен водород, или первый может быть каталитически превращен в метанол. Газ также может быть пропущен через биологический реактор для получения этанола или при использовании катализатора Фишера-Тропша превращен в жидкий поток, обладающий свойствами, подобными свойствам дизельного топлива, что называют дизельным топливом Фишера-Тропша. Однако, данные способы имеют тенденцию к дороговизне.
Существует потребность в композициях жидкого топлива, которые содержат компонент, который может быть произведен из биомассы, и который способен использоваться в современной инфраструктуре, а именно, в той же самой системе распределения и в тех же самых двигателях без возникновения потребности в специальных модификациях. Также существует и потребность в композициях жидкого топлива, которые содержат компонент, который может быть произведен из биомассы, и которые не зависят от микроорганизмов, ферментов или других дорогостоящих и тонких производственных способов.
Краткое изложение изобретения
Настоящее изобретение предлагает композицию жидкого топлива, содержащую фракцию перегонки компонента, содержащего, по меньшей мере, одно С4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, и полученного по способу, включающему;
подачу воды и растворимого в воде оксигенированного углеводорода, включающего C1+O1+ углеводород, в водной жидкой фазе и/или паровой фазе;
подачу Н2;
проведение каталитической реакции в жидкой и/или паровой фазе между оксигенированным углеводородом и Нз в присутствии катализатора деоксигенирования при температуре деоксигенирования и давлении деоксигенирования для получения оксигената, содержащего в реакционном потоке C1+O1-3 углеводород; и
проведение каталитической реакции в жидкой и/или паровой фазе для оксигената в присутствии катализатора конденсации при температуре конденсации и давлении конденсации для получения C4+ соединения,
где С4+ соединение включает представителя, выбираемого из группы, состоящей из С4+ спирта, С4+ кетона, С4+ алкана, С4+ алкена, C5+ циклоалкана, С5+ циклоалкена, арила, конденсированного арила и их смеси;
где композицию жидкого топлива выбирают из:
композиции бензина, характеризующейся температурой начала кипения в диапазоне от 15°С до 70°С (IP123), температурой конца кипения, равной, самое большее, 230°С, (IP123), значением исследовательского октанового числа ИОЧ в диапазоне от 85 до 110 (ASTM D2699) и значением моторного октанового числа МОЧ в диапазоне от 75 до 100 (ASTMD 2700);
композиции дизельного топлива, характеризующейся температурой начала кипения в диапазоне от 130°С до 230°С (IP123), температурой конца кипения, равной, самое большее, 410°С, (IP123) и цетановым числом в диапазоне от 35 до 120 (ASTM Р613); и
композиции керосина, характеризующейся температурой начала кипения в диапазоне от 80 до 150°С, температурой конца кипения в диапазоне от 200 до 320°С и вязкостью при - 20°С в диапазоне от 0,8 до 10 мм2/сек (ASTM D445).
Настоящее изобретение также предлагает композицию бензина, содержащую компонент, содержащий, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, характеризующийся температурой конца кипения в диапазоне от 150 до 220°С, плотностью при 15°С в диапазоне от 700 до 890 кг/м3, уровнем содержания серы, равным, самое большее, 5 мг/кг, уровнем содержания кислорода, равным, самое большее, 3,5% (масс.), значением ИОЧ в диапазоне от 80 до 110 и значением МОЧ в диапазоне от 70 до 100, где упомянутая композиция бензина характеризуется температурой начала кипения в диапазоне от 15°С до 70°С (IP123), температурой конца кипения, равной, самое большее, 220°С, (IP123), значением ИОЧ в диапазоне от 85 до 110 (ASTM D2699) и значением МОЧ в диапазоне от 75 до 100 (ASTM D2700).
Настоящее изобретение также предлагает композицию дизельного топлива, содержащую компонент, содержащий, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующийся значением Т95 в диапазоне от 220 до 380°С, температурой вспышки в диапазоне от 30 до 70°С, плотностью при 15°С в диапазоне от 700 до 900 кг/м3, уровнем содержания серы, равным, самое большее, 5 мг/кг, уровнем содержания кислорода, равным, самое большее, 10% (масс.), и вязкостью при 40°С в диапазоне от 0,5 до 6 сСт, где упомянутая композиция дизельного топлива характеризуется температурой начала кипения в диапазоне от 130°С до 230°С (IP123), температурой конца кипения, равной, самое большее, 410°С, (IP123) и цетановым числом в диапазоне от 35 до 120 (ASTM D613).
Настоящее изобретение также предлагает композицию керосина, содержащую компонент, содержащий, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующийся температурой начала кипения в диапазоне от 120 до 215°С, температурой конца кипения в диапазоне от 220 до 320°С, плотностью при 15°С в диапазоне от 700 до 890 кг/м3, уровнем содержания серы, равным, самое большее, 0,1% (масс.), уровнем содержания совокупной ароматики, равным, самое большее, 30% (об.), температурой замерзания, равной - 40°С и менее, максимальной высотой некоптящего пламени, равной, по меньшей мере, 18 мм, вязкостью при - 20°С в диапазоне от 1 до 10 сСт и удельным энергосодержанием в диапазоне от 40 до 47 МДж/кг, где упомянутая композиция керосина характеризуется температурой начала кипения в диапазоне от 80 до 150°С, температурой конца кипения в диапазоне от 200 до 320°С и вязкостью при - 20°С в диапазоне от 0,8 до 10 мм2/сек (ASTM D445).
Настоящее изобретение также предлагает способ получения композиции жидкого топлива, соответствующей настоящему изобретению, включающий перемешивание:
(а) фракции перегонки компонента, содержащего, по меньшей мере, одно С4+соединение, произведенное из растворимого в воде оксигенированного углеводорода, полученного по способу, включающему:
подачу воды и растворимого в воде оксигенированного углеводорода, содержащего C1+О1+ углеводород, в водной жидкой фазе и/или паровой фазе;
подачу H2;
проведение каталитической реакции в жидкой и/или паровой фазе между оксигенированным углеводородом и H2 в присутствии катализатора деоксигенирования при температуре деоксигенирования и давлении деоксигенирования для получения оксигената, содержащего в реакционном потоке С1+O1-3 углеводород; и
проведение каталитической реакции в жидкой и/или паровой фазе для оксигената в присутствии катализатора конденсации при температуре конденсации и давлении конденсации для получения С4+соединения,
где С4+соединение включает представителя, выбираемого из группы, состоящей из С4+ спирта, С4+ кетона, С4+ алкана, С4+ алкена, С5+ циклоалкана, С5+ циклоалкена, арила, конденсированного арила и их смеси, и (b) по меньшей мере, одного компонента топлива.
Краткое описание чертежей
Фигура 1 представляет собой блок-схему, иллюстрирующую различные пути получения, связанные с настоящим изобретением.
Фигура 2 иллюстрирует потенциальные химические пути, которые делают возможным превращение углеводов, таких как сахара, в неоксигенированные углеводороды.
Фигура 3 представляет собой иллюстрацию различных путей реакции, включенных в деоксигенирование сорбита для получения оксигенатов и водорода риформинга водной фазы (РВФ).
Фигура 4 представляет собой иллюстрацию термодинамического равновесия по ходу пути реакции для превращения ацетона в 2-метилпентан при 100°С и 400°С.
Фигура 5 представляет собой график, иллюстрирующий константы равновесия, связанные с промежуточными продуктами реакции и совокупным превращением в случае реакции между 2 молями ацетона и 3 молями водорода для получения 1 моля 2-метилпентана и 2 молей воды.
Фигура 6 представляет собой блок-схему, иллюстрирующую реакторную систему, сконфигурированную для обеспечения отправления на рецикл водорода, оксигенатов и оксигенированных углеводородов.
Фигура 7 представляет собой блок-схему, иллюстрирующую реакторную систему, сконфигурированную для обеспечения использования воздуха или масла в качестве элемента регулирования температуры.
Фигура 8 представляет собой блок-схему, иллюстрирующую реакторную систему для настоящего изобретения.
Фигура 9 представляет собой блок-схему, иллюстрирующую реакторную систему, использующую два реактора.
Фигура 10 представляет собой блок-схему, иллюстрирующую реакторную систему, использующую две линии исходного сырья.
Фигура 11 представляет собой иллюстрацию реактора, подходящего для использования при осуществлении настоящего изобретения на практике.
Фигура 12 представляет собой график, иллюстрирующий распределение атомов углерода для монооксигенатов, полученных из глицерина.
Фигура 13 представляет собой график, иллюстрирующий осевой температурный профиль для реактора при его использовании для получения соединений из исходного сырья в виде оксигенированных углеводородов.
Фигура 14 представляет собой график, иллюстрирующий зависимость от времени процентной доли атомов углерода исходного сырья, выходящих в виде оксигенатов после превращения потока оксигенатного исходного сырья в C5+ соединения.
Фигура 15 представляет собой график, иллюстрирующий зависимость от времени процентной доли атомов углерода исходного сырья, выходящих в виде С5+ углеводородов после превращения потока оксигенатного исходного сырья.
Фигура 16 представляет собой график, иллюстрирующий зависимость от времени процентной доли атомов углерода исходного сырья, выходящих в виде С5+ ароматических углеводородов после превращения потока оксигенатного исходного сырья.
Фигура 17 представляет собой график, демонстрирующий совокупную массовую процентную долю парафина и ароматических соединений, произведенных после превращения потока исходного сырья в виде сахарозы и ксилозы.
Фигура 18 представляет собой график, иллюстрирующий теплотворную способность С5+ углеводородов, произведенных после получения бензина из сорбита, в виде процентной доли от теплотворной способности исходного сырья.
Фигура 19 представляет собой график, иллюстрирующий процентную долю атомов углерода, извлеченных в виде ароматических углеводородов после получения бензина из сорбита, продемонстрированную в виде процентной доли от атомов углерода, присутствующих в исходном сырье. Подробное описание изобретения
Композиции жидкого топлива настоящего изобретения содержат компонент, содержащий, по меньшей мере, одно С4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода. Предпочтительно растворимый в воде оксигенированный углеводород производят из биомассы.
Обычно способ получения компонента, содержащего, по меньшей мере, одно С4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, приводит к получению углеводородов, кетонов и спиртов из произведенных из биомассы оксигенированных углеводородов, таких как сахара, сахароспирты, целлюлозные полимеры, лигноцеллюлозы, гемицеллюлозы, сахариды и тому подобное.
Компонент, произведенный из растворимого в воде оксигенированного углеводорода, содержит С4+ алканы, С4+алкены, С5+ циклоалканы, C5+ циклоалкены, арилы, конденсированные арилы, С4+ спирты, С4+ кетоны и их смеси (коллективно называемые в настоящем документе «С4+ соединениями»). С4+ углеводороды обычно содержат от 4 до 30 атомов углерода и могут представлять собой алканы или алкены с разветвленной или прямой цепью или незамещенные, монозамещенные или полизамещенные ароматику (арилы) или циклоалканы. С4+ спирты и С4+ кетоны могут иметь циклическую, разветвленную или прямую цепь и содержать от 4 до 30 атомов углерода.
Легкие фракции, в первую очередь, С4-С9, могут быть отделены для использования в бензине. Средние фракции, такие как C7-C14, могут быть отделены в качестве керосина, например, для использования в реактивном топливе, в то время как тяжелые фракции, то есть, C12-C24, могут быть отделены для использования в дизельном топливе. Наиболее тяжелые фракции могут быть использованы в качестве смазок или подвергнуты крекингу для получения дополнительных фракций бензина и/или дизельного топлива. С4+ соединения, произведенные из растворимых в воде оксигенированных углеводородов, также могут найти себе применение в качестве промышленных химических реагентов, таких как ксилол, вне зависимости от того, будет ли это промежуточный или конечный продукт.
Способ получения компонента, произведенного из растворимого в воде оксигенированного углеводорода
Общий способ получения компонента, произведенного из растворимого в воде оксигенированного углеводорода, проиллюстрирован на фигуре 1. Раствор исходного сырья, содержащий растворимый в воде оксигенированный углеводород, содержащий один или несколько атомов углерода, вводят в реакцию с водородом на катализаторе деоксигенирования для получения оксигенатов, а после этого оксигенаты вводят в реакцию на катализаторе конденсации в условиях по температуре и давлению, эффективных для прохождения реакции конденсации, которая приводит к получению С4+ соединений. Водород может иметь своим происхождением любой источник, но предпочтительно его производят из биомассы «по месту» или параллельно при использовании риформинга в водной фазе. Водород и оксигенированные углеводороды также могут быть дополнены отправляемыми на рецикл водородом и оксигенированными углеводородами, произведенными в способе. Оксигенированный углеводород может представлять собой моносахарид, дисахарид, полисахарид, целлюлозу, гемицеллюлозу, лигнин, сахар, сахароспирт или другие многоатомные спирты или может быть произведен при гидрировании сахара, фурфураля, карбоновой кислоты, кетона или фурана или гидрогенолизе сахара, сахароспирта, полисахарида, моносахарида, дисахарида или многоатомного спирта.
Один уникальный аспект способа получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретении, заключается в том, что C4+ соединения производят из компонентов биомассы при использовании каталитических способов вместо способов с применением микроорганизмов, ферментов, высокотемпературной газификации или переэтерификации. Способ получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретения, также может приводить к получению водорода «по месту», что позволяет избегать расчетов на внешние источники водорода, такие как водород, полученный при паровом риформинге природного газа или электролизе или термолизе воды. Способ получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретении, также приводит и к получению воды, которая может быть отправлена на рецикл и использована в способах, расположенных по ходу технологического потока раньше, или возвращена в окружающую среду. Способ получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретении, также способен приводить и к получению неконденсируемых газообразных топлив в целях получения источника тепла в реакторной системе или для внешних способов.
Углеводы на Земле представляют собой наиболее широко распространенные встречающиеся в природе органические соединения. Углеводы образуются во время фотосинтеза - процесса, по которому энергия от солнца превращается в химическую энергию в результате объединения диоксида углерода с водой с образованием углеводов и кислорода:

Sunlight = Солнечный свет.
Энергия солнечного света благодаря данному процессу запасается в растениях в виде химической энергии в форме углеводов. Углеводы, в особенности при нахождении в форме Сахаров, представляют собой высокореакционно-способные соединения, которые легко окисляются живым веществом, что приводит к получению энергии, диоксида углерода и воды. В материалах растений данные углеводы запасаются в виде или Сахаров, или крахмалов, или полимерной целлюлозы и/или гемицеллюлозы.
Присутствие кислорода в молекулярной структуре углеводов вносит свой вклад в реакционную способность Сахаров в биологических системах. Технология этанольной ферментации использует данную высокореакционно-способную природу, обеспечивая получение этанола при температурах окружающей среды. Технология ферментации, по существу, дефункционализует высокореакционно-способный сахар для получения частично окисленного углеводорода - этанола. Однако этанолу свойственны очень существенные недостатки в отношении его теплоты сгорания, как это подчеркивалось выше.
Фигура 2 демонстрирует потенциальные химические пути, которые позволяют углеводы, такие как сахара, превращать в неоксигенированные углеводороды. Растворимые в воде углеводы, как известно, вступают в реакцию с водородом на катализаторе (катализаторах) с образованием многоатомных спиртов в результате либо гидрирования, либо гидрогенолиза. Исторически водород получали извне, то есть, из природного газа или по другим способам, но в соответствии с настоящим изобретением теперь он может быть получен «по месту» или параллельно благодаря риформингу в водной фазе для многоатомного спирта.
Риформинг в водной фазе (РВФ) для многоатомного спирта протекает через образование альдегида (что продемонстрировано на фигуре 2), где альдегид вступает в реакцию с водой на катализаторе с образованием водорода, диоксида углерода и меньшего многоатомного спирта. Многоатомный спирт может дополнительно вступать в реакцию с водородом на катализаторе в ходе последовательности из реакций деоксигенирования с образованием либо спиртовых, либо кетонных, либо альдегидных веществ, которые могут претерпевать реакции конденсации с образованием содержащих большее количество атомов углерода либо соединений с прямой цепью, либо соединений с разветвленной цепью, либо циклических соединений. Реакции конденсации могут быть либо катализированы кислотой, либо катализированы основанием, либо катализированы как кислотой, так и основанием. Получающиеся в результате соединения могут представлять собой углеводороды или углеводороды, содержащие кислород, кислород которых может быть удален по реакции с водородом на катализаторе. Получающиеся в результате конденсированные продукты включают C4+ спирты, C4+ кетоны, C4+ алканы, С4+ алкены, C5+ циклоалканы, C5+ циклоалкены, арилы, конденсированные арилы и их смеси. Смеси могут быть фракционированы и перемешаны для получения надлежащих смесей молекул, обычно использующихся в бензиновых, керосиновых (например, в качестве реактивного топлива) или дизельных топливах.
Дефункционализация начинается в результате проведения реакции между глюкозой и водородом либо по реакции гидрирования, либо по реакции гидрогенолиза для превращения молекулы циклического сахара в его соответствующий линейный спирт, сорбит или низшие многоатомные спирты, такие как, помимо прочего, глицерин, пропиленгликоль, этиленгликоль, ксилит. Как указывалось выше, водород может поступать из любого источника, но предпочтительно им являются водород, полученный «по месту» в результате риформинга в водной фазе, или избыточный водород, отправленный на рецикл из реакторной системы.
Во время процесса риформинга в водной фазе углевод перед расщеплением связей С-С или С-O сначала подвергается дегидрированию с образованием адсорбированных промежуточных соединений. Последующее расщепление связей С-С приводит к образованию СО и Н2, причем затем СО вступает в реакцию с водой с образованием СО; и Н2 по реакции конверсии водяного газа. Различные способы и методики РВФ описываются в патентах США №№6699457; 6964757 и 6964758; и патентной заявке США №11234727 (у всех из которых авторами являются Cortright et al., а заглавие представляет собой «Low-Temperature Hydrogen Production from Oxygenated Hydrocarbons»); и патенте США №6953873 (у которого авторами являются Cortright et al., а заглавие представляет собой «Low Temperature Hydrocarbon Production from Oxygenated Hydrocarbons»); и публикации WO 2007/075476 A2 (у которой авторами являются Cortright et al., а заглавие представляет собой «Catalyst and Methods for Reforming Oxygenated Compounds»), все из которых посредством ссылки включаются в настоящий документ. Термины «риформинг в водной фазе» и «РВФ» в общем случае должны обозначать риформинг для оксигенированных углеводородов и воды с образованием водорода и диоксида углерода вне зависимости от прохождения реакций в газовой фазе или в конденсированной жидкой фазе. Термин «Н2 РВФ» в общем случае должен обозначать водород, полученный по способу РВФ.
Получающийся в результате оксигенированный углеводород, а именно, сорбит или глицерин, пропиленгликоль, этиленгликоль, ксилит и тому подобное, дополнительно подвергают дефункционализации по реакциям деоксигенирования для получения оксигенатов, таких как спирты, кетоны, альдегиды, фураны, диолы, триолы, гидроксикарбоновые кислоты и карбоновые кислоты и предназначенных для использования в последующих реакциях конденсации. Фигура 3 иллюстрирует различные пути реакций, вовлеченные в деоксигенирование сорбита для получения оксигенатов и водорода РВФ. Как можно себе представить в общем случае без ограничения какой-либо конкретной теорией, реакции деоксигенирования включают комбинацию всевозможных различных путей реакции, в том числе без ограничения: реакций гидродеоксигенирования, последовательных дегидратации-гидрирования, гидрогенолиза, гидрирования и дегидратации, что в результате приводит к удалению кислорода из оксигенированного углеводорода с образованием углеводородной молекулы, описывающейся общей формулой C1+O1-3.
После этого полученные оксигенаты превращают в С4+ соединения в результате конденсации. Как можно себе представить без ограничения какими-либо конкретными теориями, реакции кислотной конденсации в общем случае состоят из последовательности стадий, включающих: (а) дегидратацию оксигенатов с образованием олефинов; (b) олигомеризацию олефинов; (с) реакции крекинга; (d) циклизацию более крупных олефинов с образованием ароматики; (е) изомеризацию парафина; и (f) реакции с переносом атома водорода с образованием парафинов. Как можно себе представить, реакции основной конденсации в общем случае состоят из последовательности стадий, включающей: (1) альдольную конденсацию с образованием β-гидроксикетона или β-гидроксиальдегида; (2) дегидратацию β-гидроксикетона или β-гидроксиальдегида с образованием сопряженного енона; (3) гидрирование сопряженного енона с образованием кетона или альдегида, который может принимать участие в последующих реакциях конденсации или превращения в спирт или углеводород; и (4) гидрирование карбонилов с образованием спиртов, или наоборот. Как можно себе представить, реакции кислотно-основной конденсации в общем случае включают любые из предшествующих стадий кислотных и/или основных реакций.
В определенных вариантах осуществления реакции конденсации протекают при типичных температурах и давлениях конденсации. Однако в различных вариантах осуществления более благоприятным также может быть и проведение реакций конденсации в условиях по температуре и/или давлению, которые являются повышенными в сопоставлении с тем, что имеет место в типичных способах конденсации. В общем случае проведение реакций конденсации при повышенных условиях в результате приводит к получению неблагоприятной термодинамики, что ограничивает степень превращения в продукты конденсации. Как выявило настоящее изобретение, проведение реакции при использовании катализаторов конденсации и при описывающихся ниже температурах и давлениях устраняет данные ограничения и неожиданно промотирует непосредственное превращение продуктов конденсации в углеводороды, кетоны и спирты. Превращение, в свою очередь, приводит к удалению продуктов конденсации из реакционной смеси, что, тем самым, устраняет ограничения по термодинамике в системе, делая возможным прохождение дополнительных реакций конденсации. Повышенные условия по температуре и/или давлению также предотвращают избыточное превращение оксигенатов непосредственно в их соответствующие углеводороды. Способу также свойственно и дополнительное преимущество, заключающееся в обеспечении прохождения реакций конденсации, реакций деоксигенирования и реакций РВФ в одном реакторе и при стационарном равновесии.
Для любой заданной реакции показателем благоприятности прохождения прямой реакции является изменение свободной энергии. Чем более отрицательным будет изменение свободной энергии, тем более благоприятной будет реакции. В результате реакции, связанные с большим отрицательным изменением свободной энергии, в общем случае являются благоприятными и имеют потенциал по демонстрации высоких степеней превращения в продукты реакции. Наоборот, реакции, связанные с положительными изменениями свободной энергии, не являются предпочтительными и по самой своей природе являются ограниченными в том, что касается степени, в которой реагенты превращаются в продукты. В порядке иллюстрации фигура 4 демонстрирует изменения свободной энергии, связанные со стадиями по ходу пути реакции для превращения ацетона и водорода в С6 углеводород (2-метилпентан) и воду при 100°С и 400°С. Известные уровни свободной энергии для стабильных промежуточных соединений, произведенных по ходу данного пути, продемонстрированы сплошной линией. Первая стадия на пути реакции представляет собой альдольную конденсацию двух молекул ацетона с образованием одной молекулы диацетонового спирта. Реакция при меньшей температуре (100°С) характеризуется изменением свободной энергии - 53 кДж/моль и является термодинамически благоприятной, в то время как реакция при большей температуре (400°С) является менее благоприятной вследствие изменения свободной энергии - 10 кДж/моль. Имеется в виду то, что максимальная степень превращения чистого ацетона в диацетоновый спирт для данной стадии уменьшается по мере увеличения температуры (от более, чем 99% от теоретической максимальной степени превращения при 100°С и атмосферном давлении до только лишь 15% при 400°С и атмосферном давлении). В соответствии с этим, ограничение по термодинамическому равновесию формирует абсолютный предел для количества диацетонового спирта, которое может быть получено при заданных условиях и в отсутствие других реакций. Это дополнительно проиллюстрировано на фигуре 5, которая представляет константы равновесия, связанные с промежуточными продуктами реакции и совокупным превращением в случае реакции между 2 молями ацетона и 3 молями водорода для получения 1 моля 2-метилпентана и 2 молей воды. Как можно видеть, константа равновесия для превращения ацетона в диацетоновый спирт при увеличении температуры уменьшается.
Настоящее изобретение обходит данную проблему благодаря непосредственному превращению продукта конденсации в соединение, которое обеспечивает получение более благоприятной реакционной среды. В вышеупомянутом случае в результате удаления диацетонового спирта из реакционной смеси по реакции дегидратации, которая образует мезитилоксид, может быть получено дополнительное количество диацетонового спирта. В частности, комбинация в виде стадии конденсации и дегидратации для получения мезитилоксида и воды из ацетона приводит к получению несколько более благоприятной реакционной среды. Как проиллюстрировано на фигуре 5, степень превращения ацетона в мезитилоксид и воду является несколько более благоприятной при более высоких температурах.
Полное давление реакционной системы также оказывает благоприятное воздействие на максимальную теоретическую степень, в которой реагент может образовывать продукт. Принимая во внимание приведенный выше пример реакции конденсации, степень превращения ацетона в диацетоновый спирт ограничивается величиной 15% при 400°С и атмосферном давлении для исходного сырья в виде чистого ацетона. В результате увеличения давления системы до давления 600 фунт/дюйм2 (изб.) (4140 кПа (изб.)) равновесная степень превращения смещается таким образом, чтобы при той же самой температуре могла бы быть достигнута степень превращения, доходящая вплоть до 76%. В случае реакций, демонстрирующих результирующее уменьшение количества молей продукта в сопоставлении с количеством молей реагента, увеличение давления системы (при выдерживании всех других условий постоянными) будет приводить к увеличению равновесной степени превращения в продукт. Для совокупного превращения кетонов в углеводороды обычно имеет место результирующее уменьшение количества молей продукта в сопоставлении с количеством молей реагента, таким образом, более высокие давления реакции будут приводить к более высоким потенциальным равновесным степеням превращения.
Способ получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретении, добивается баланса с вышеупомянутыми термодинамическими ограничениями в результате проведения операции при использовании катализаторов конденсации и в условиях по температуре и давлению, которые компенсируют любое уменьшение выработки продуктов конденсации увеличением степени превращения в другие продукты для процессов, расположенных по ходу технологического потока дальше. Кинетика совокупной системы также является более благоприятной, так что продукты можно будет получать непрерывно и с более желательной скоростью. В том, что касается увеличения масштаба производства, после запуска реакторные системы могут контролироваться самим способом, и реакции могли бы протекать при стационарном равновесии.
Оксигенаты С4+ соединения производят из оксигенатов. В соответствии с использованием в настоящем документе в отношении способа получения компонента, произведенного из растворимого в воде оксигенированного углеводорода, «оксигенаты» в общем случае обозначают углеводородные соединения, содержащие 1 и более атомов углерода и от 1 до 3 атомов кислорода, (что в настоящем документе обозначают как C1+O1-3 углеводороды), такие как спирты, кетоны, альдегиды, фураны, гидроксикарбоновые кислоты, карбоновые кислоты, диолы и триолы. Предпочтительно оксигенаты содержат от 1 до 6 атомов углерода или от 2 до 6 атомов углерода или от 3 до 6 атомов углерода. Спирты могут включать без ограничения первичные, вторичные, линейные, разветвленные или циклические C1+ спирты, такие как метанол, этанол, н-пропиловый спирт, изопропиловый спирт, бутиловый спирт, изобутиловый спирт, бутанол, пентанол, циклопентанол, гексанол, циклогексанол, 2-метилциклопентанонол, гептанол, октанол, нонанол, деканол, ундеканол, додеканол и их изомеры. Кетоны могут включать без ограничения гидроксикетоны, циклические кетоны, дикетоны, ацетон, пропанон, 2-оксопропаналь, бутанон, бутан-2,3-дион, 3-гидроксибутан-2-он, пентанон, циклопентанон, пентан-2,3-дион, пентан-2,4-дион, гексанон, циклогексанон, 2-метилциклопентанон, гептанон, октанон, нонанон, деканон, ундеканон, додеканон, метилглиоксаль, бутандион, пентандион, дикетогексан и их изомеры. Альдегиды могут включать без ограничения гидроксиальдегиды, ацетальдегид, пропиональдегид, бутиральдегид, пентаналь, гексаналь, гептаналь, октаналь, нональ, деканаль, ундеканаль, додеканаль и их изомеры. Карбоновые кислоты могут включать без ограничения муравьиную кислоту, уксусную кислоту, пропионовую кислоту, бутановую кислоту, пентановую кислоту, гексановую кислоту, гептановую кислоту, их изомеры и производные, в том числе гидроксилированные производные, такие как 2-гидроксибутановая кислота и молочная кислота. Диолы могут включать без ограничения этиленгликоль, пропиленгликоль, 1,3-пропандиол, бутандиол, пентандиол, гександиол, гептандиол, октандиол, нонандиол, декандиол, ундекандиол, додекандиол и их изомеры. Триоды могут включать без ограничения глицерин, 1,1,1-трис(гидроксиметил)этан(триметилолэтан), триметилолпропан, гексантриол и их изомеры. Фураны и фурфурали включают без ограничения фуран, тетрагидрофуран, дигидрофуран, 2-фуранметанол, 2-метилтетрагидрофуран, 2,5-диметилтетрагидрофуран, 2-метилфуран, 2-этилтетрагидрофуран, 2-этил фуран, гидроксилметилфурфураль, 3-гидрокситетрагидрофуран, тетрагидро-3-фуранол, 2,5-диметилфуран, 5-гидроксиметил-2(5Н)-фуранон, дигидро-5-(гидроксиметил)-2(3Н)-фуранон, тетрагидропирослизевую кислоту, дигидро-5-(гидроксиметил)-2(3Н)-фуранон, тетрагидрофурфуриловый спирт, 1-(2-фурил)зтанол, гидроксиметилтетрагидрофурфураль и их изомеры.
Оксигенаты могут иметь своим происхождением любой источник, но предпочтительно их производят из биомассы. В соответствии с использованием в настоящем документе термин «биомасса» относится без ограничения к органическим материалам, производимым растениями, (таким как листья, корни, семена и стебли) и конечным продуктам обмена веществ у микробов и животных. Обычные источники биомассы включают: (1) сельскохозяйственные отходы, такие как стебли кукурузы, солома, кожура семян, остатки сахарного тростника, багасса, ореховая скорлупа и навоз от крупного рогатого скота, домашней птицы и свиней; (2) древесные материалы, такие как древесина или кора, древесные опилки, порубочные остатки пиломатериалов и заводской скрап; (3) муниципальные отходы, такие как бумажная макулатура и дворовый мусор; и (4) растения для энергетического использования, такие как тополя, ивы, просо прутьевидное, люцерна, луговой бородач, кукуруза, соевые бобы и тому подобное. Данный термин также относится к основным структурным элементам вышеупомянутых источников, а именно, помимо прочего, к сахаридам, лигнину, целлюлозным полимерам, гемицеллюлозе и крахмалам.
Оксигенаты из биомассы могут быть получены по любому известному способу. Такие способы включают, помимо прочего, технологии ферментации, использующие ферменты и микроорганизмы, реакции Фишера-Тропша для получения С2-10 альфа-спиртов и технологии пиролиза для получения спиртов из нефти. В одном варианте осуществления оксигенаты получают при использовании технологий каталитического риформинга, таких как технология BioForming™, разработанная компанией Virent Energy Systems, Inc. (Мэдисон, Висконсин). Оксигенированные углеводороды
В одном варианте осуществления оксигенаты производят по способу каталитического риформинга оксигенированных углеводородов. Оксигенированные углеводороды могут быть любым растворимым в воде оксигенированным углеводородом, содержащим один или несколько атомов углерода и, по меньшей мере, один атом кислорода, (что в настоящем документе обозначают как C1+O1+ углеводороды). Предпочтительно оксигенированный углеводород содержит от 2 до 12 атомов углерода (C1-12O1-11 и углеводород), а более предпочтительно от 2 до 6 атомов углерода (C1-6O1-6 углеводород). Оксигенированный углеводород также может характеризоваться количественным соотношением между кислородом и углеродом в диапазоне от 0,5:1 до 1,5:1, включая соотношения 0,75:1,0, 1,0:1,0, 1,25:1,0, 1,5:1,0 и другие промежуточные соотношения. В одном примере оксигенированный углеводород характеризуется количественным соотношением между кислородом и углеродом 1:1. Неограничивающие примеры предпочтительных растворимых в воде оксигенированных углеводородов включают моносахариды, дисахариды, полисахариды, сахар, сахароспирты, альдиты, этандиол, этандион, уксусную кислоту, пропанол, пропандиол, пропионовую кислоту, глицерин, глицеральдегид, дигидроксиацетон, молочную кислоту, пировиноградную кислоту, малоновую кислоту, бутандиолы, бутановую кислоту, альдотетрозы, винную кислоту, альдопентозы, альдогексозы, кетотетрозы, кетопентозы, кетогексозы, альдиты, гемицеллюлозы, целлюлозные производные, лигноцеллюлозные производные, крахмалы, полиолы и тому подобное. Предпочтительно оксигенированный углеводород включает сахар, сахароспирты, сахариды и другие многоатомные спирты. Более предпочтительно оксигенированный углеводород представляет собой сахар, такой как глюкоза, фруктоза, сахароза, мальтоза, лактоза, манноза или ксилоза, или сахароспирт, такой как арабит, эритрит, глицерин, изомальт, лактит, мальтит, маннит, сорбит, ксилит, рибит или гликоль.
Оксигенированные углеводороды также должны обозначать и включать спирты, произведенные в результате гидрирования или гидрогенолиза любых представителей из вышеупомянутых. В определенных вариантах осуществления предпочтительным может оказаться превращение исходного оксигенированного углеводорода в другую форму оксигенированного углеводорода, которая может быть легче превращена в желательные оксигенаты (например, первичные, вторичные, третичные или многоатомные спирты). Например, некоторые сахара могут не превращаться в оксигенаты настолько же эффективно, как и их соответствующие производные сахароспиртов. Поэтому желательным может оказаться превращение исходного материала, такого как сахар, фурфураль, карбоновая кислота, кетон или фуран, в его соответствующее спиртовое производное, например в результате гидрирования, или в молекулы меньших спиртов, например в результате гидрогенолиза.
Что касается гидрирования Сахаров, фурфуралей, карбоновых кислот, кетонов и фуранов для получения их соответствующей спиртовой формы, то известны различные его способы, в том числе те, которые описываются авторами: В.S.Kwak et al. (публикации WO 2006/093364 A1 и WO 2005/021475A1) - включение получения производных Сахаров альдитов из моносахаридов в результате гидрирования над рутениевым катализатором; и Elliot et al. (патенты США №№6253797 и 6570043) - описание использования не содержащего никеля и рения рутениевого катализатора на носителе, более, чем 75%, состоящего из рутилового диоксида титана, для превращения Сахаров в сахароспирты, все из которых посредством ссылки включаются в настоящий документ. Другие подходящие рутениевые катализаторы описываются авторами Arndt et al. в опубликованной патентной заявке США 2006/0009661 (подана 3 декабря 2003 года) и Arena в патентах США №№4380679 (подана 12 апреля 1982 года), 4380680 (подана 21 мая 1982 года), 4503274 (подана 8 августа 1983 года), 4382150 (подана 19 января 1982 года) и 4487980 (подана 29 апреля 1983 года), все из которых посредством ссылки включаются в настоящий документ.Катализатор гидрирования в общем случае включает Cu, Re, Ni, Fe, Со, Ru, Pd, Rh, Pt, Os, Ir и их сплавы или комбинации, либо индивидуально, либо совместно с промоторами, такими как W, Mo, Au, Ag, Cr, Zn, Mn, Sn, В, Р, Bi и их сплавы или комбинации. Катализатор гидрирования также может включать любой один из носителей, дополнительно описывающихся ниже и зависящих от желательной функциональности катализатора. Другие эффективные материалы катализаторов гидрирования включают либо нанесенный на носитель никель, либо рутений, модифицированный рением. В общем случае реакцию гидрирования проводят при температурах гидрирования в диапазоне приблизительно от 80°С до 250°С и давлениях гидрирования в диапазоне приблизительно от 100 фунт/дюйм2 (изб.) (689 кПа (изб.)) до 2000 фунт/дюйм2 (изб.) (13800 кПа (изб.)). Водород, использующийся в реакции, может включать Н2, полученный «по месту», внешний H2, Н2, отправляемый на рецикл, или их комбинацию.
Катализатор гидрирования также может включать нанесенный на носитель катализатор на основе металла из группы VIII и металлический губчатый материал, такой как губчатый никелевый катализатор. Хорошо известный класс материалов, эффективных для различных реакций гидрирования, образуют активированные губчатые никелевые катализаторы (например, никель Ренея). Одним типом губчатого никелевого катализатора является катализатор типа А7063, доступный в компании Activated Metals and Chemicals, Inc., Севиервилл, Теннеси. Катализатором типа А7063 является промотированный молибденом катализатор, обычно содержащий приблизительно 1,5% молибдена и 85% никеля. Использование губчатого никелевого катализатора совместно с исходным сырьем, содержащим ксилозу и декстрозу, описывается авторами М.L.Cunningham et al. в публикации US 6,498,248, поданной 9 сентября 1999 года и посредством ссылки включенной в настоящий документ. Использование катализатора на основе никеля Ренея совместно с гидролизованным кукурузным крахмалом также описывается в публикации US 4,694,113, поданной 4 июня 1986 года и посредством ссылки включенной в настоящий документ.
Получение подходящих катализаторов гидрирования на основе никеля Ренея описывается авторами A. Yoshino et al. в опубликованной патентной заявке США 2004/0143024, поданной 7 ноября 2003 года и посредством ссылки включенной в настоящий документ. Катализатор на основе никеля Ренея может быть получен в результате обработки сплава приблизительно равных массовых количеств никеля и алюминия водным щелочным раствором, например, содержащим приблизительно 25% (масс.) гидроксида натрия. Алюминий под действием водного щелочного раствора селективно растворяется, что оставляет частицы, имеющие губчатую структуру и преимущественно образованные из никеля с неосновным количеством алюминия. В первоначальный сплав также могут быть включены промоторные металлы, такие как молибден или хром, в количестве, таком чтобы в губчатом никелевом катализаторе оставалось бы приблизительно 1-2% (масс.).
В еще одном варианте осуществления катализатор гидрирования получают в результате импрегнирования подходящего материала носителя раствором нитрозилнитрата рутения (III), нитрозилнитрата рутения (III) или хлорида рутения (III) в воде для получения твердого вещества, которое после этого высушивают в течение 13 часов при 120°С во вращающейся шаровой печи (остаточный уровень содержания воды составляет менее, чем 1% (масс.)). После этого твердое вещество восстанавливают при атмосферном давлении в потоке водорода при 300°С (без прокаливания) или 400°С (с прокаливанием) в течение 4 часов во вращающейся шаровой топке. После охлаждения и придания инертности азотом катализатор затем может быть пассивирован в результате перепускания над ним 5% (об.) кислорода в азоте в течение периода времени продолжительностью 120 минут.
В еще одном другом варианте осуществления реакцию гидрирования проводят при использовании катализатора, включающего никель-рениевый катализатор или никелевый катализатор, модифицированный вольфрамом. Одним примером подходящего катализатора гидрирования является композиция никель-рениевого катализатора, нанесенного на углеродный носитель, описывающаяся авторами Werpy et al. в публикации US 7,038,094, поданной 30 сентября 2003 года и посредством ссылки включенной в настоящий документ.
В других вариантах осуществления желательным также может оказаться превращение исходного оксигенированного углеводорода, такого как сахар, сахароспирт или другой многоатомный спирт, в меньшую молекулу, которая легче может быть превращена в желательные оксигенаты, так как в результате гидрогенолиза. Такие меньшие молекулы могут включать первичные, вторичные, третичные или многоатомные спирты, содержащие меньше атомов углерода, чем исходный оксигенированный углеводород. Что касается таких реакций гидрогенолиза, то известны различные их способы, в том числе те, которые описываются: авторами Werpy et al. в патентах США №№6479713 (подан 23 октября 2001 года), 6677385 (подан 6 августа 2002 года), 66841085 (подан 23 октября 2001 года) и 7083094 (подан 30 сентября 2003 года), все из которых посредством ссылки включаются в настоящий документ и описывают гидрогенолиз содержащих 5 и 6 атомов углерода Сахаров и сахароспиртов для получения пропиленгликоля, этиленгликоля и глицерина при использовании ренийсодержащего полиметаллического катализатора. Другие системы включают те, которые описываются автором Arena в патенте США №4401823 (подан 18 мая 1981 года), относящемся к использованию углеродистого пирополимерного катализатора, содержащего переходные металлы (такие как хром, молибден, вольфрам, рений, марганец, медь, кадмий) или металлы из группы VIII (такие как железо, кобальт, никель, платина, палладий, родий, рутений, иридий и осмий), для получения спиртов, кислот, кетонов и простых эфиров из полигидроксилированных соединений, таких как сахара и сахароспирты, и в патенте США №4496780 (подан 22 июня 1983 года), относящемся к использованию системы катализатора, содержащей благородный металл из группы VIII на твердом носителе совместно с оксидом щелочноземельного металла, для получения глицерина, этиленгликоля и 1,2-пропандиола из углеводов, при этом каждый из них посредством ссылки включается в настоящий документ. Еще одна система включает ту, которая описывается авторами Dubeck et al. в патенте США №4476331 (подан 6 сентября 1983 года), относящемся к использованию модифицированного сульфидом рутениевого катализатора для получения этиленгликоля и пропиленгликоля из более крупных многоатомных спиртов, таких как сорбит, и также посредством ссылки включенном в настоящий документ.Другие системы включают те, которые описываются в публикации Saxena et al., «Effect of Catalyst Constituents on (Ni, Mo and Cu)/Kieselguhr-Catalyzed Sucrose Hydrogenolysis», Ind. Eng. Chem. Res. 44, 1466-1473 (2005), описывающей использование Ni, W и Cu на кизельгуровом носителе и посредством ссылки включенной в настоящий документ.
В одном варианте осуществления катализатор гидрогенолиза включает Cr, Mo, W, Re, Mn, Cu, Cd, Fe, Co, Ni, Pt, Pd, Rh, Ru, Ir или Os и их сплавы или комбинации, либо индивидуально, либо совместно с промоторами, такими как Au, Ag, Cr, Zn, Mn, Sn, Bi, В, О и их сплавы или комбинации. Другие эффективные материалы катализаторов гидрогенолиза могут включать вышеупомянутые металлы, объединенные с оксидом щелочноземельного металла или приставшие к каталитически активному носителю, такому как кизельгур, или любому одному из носителей, дополнительно описывающихся ниже.
Технологические условия для проведения реакции гидрогенолиза будут варьироваться в зависимости от типа исходного сырья и желательных продуктов. В общем случае реакцию гидрогенолиза проводят при температурах, равных, по меньшей мере, 110°С или находящихся в диапазоне от 110°С до 300°С или от 170°С до 240°С. Реакция должна быть проведена в основных условиях, предпочтительно при значении рН в диапазоне от приблизительно 8 до приблизительно 13 или при значении рН в диапазоне от приблизительно 10 до приблизительно 12. Реакция также должна быть проведена при давлениях в диапазоне приблизительно от 10 фунт/дюйм2 (изб.) (68,9 кПа (изб.)) до 2400 фунт/дюйм2 (изб.) (16500 кПа (изб.)) или приблизительно от 250 фунт/дюйм2 (изб.) (1720 кПа (изб.)) до 2000 фунт/дюйм2 (изб.) (13800 кПа (изб.)) или приблизительно от 700 фунт/дюйм2 (изб.) (4830 кПа (изб.)) до 1600 фунт/дюйм2 (изб.) (11000 кПа (изб.)).
Водород, использующийся в реакции, может включать Hz, полученный «по месту», внешний H2, Н2, отправляемый на рецикл, или их комбинацию.
Получение оксигенатов
Оксигенаты синтезируют в результате проведения реакции между водным раствором исходного сырья, содержащим воду и растворимые в воде оксигенированные углеводороды, и водородом на каталитическом материале для получения желательных оксигенатов. Предпочтительно водород получают «по месту» при использовании риформинга в водной фазе (H2, полученный «по месту», или H2 РВФ), или он представляет собой комбинацию из Н2 РВФ, внешнего H2 или H2, отправляемого на рецикл, или просто лишь внешний H2 или H2, отправляемый на рецикл. Термин «внешний H2» относится к водороду, который не имеет своим происхождением раствор исходного сырья, но который добавляют в реакторную систему из внешнего источника. Термин «H2, отправляемый на рецикл» относится к неизрасходованному водороду, который своим происхождением имеет раствор исходного сырья, и который собирают, а после этого отправляют обратно на рецикл в реакторную систему для последующего использования. Внешний Н2 и H2, отправляемый на рецикл, также могут быть коллективно или индивидуально названы «дополнительным H2». В общем случае дополнительный H2 может быть добавлен в целях дополнения водорода РВФ или для замещения включения стадии получения водорода РВФ или для увеличения давления реакции в системе или для увеличения молярного соотношения между водородом и углеродом и/или кислородом в целях улучшения выхода продукции в виде определенных типов продуктов реакции, таких как кетоны и спирты.
В способах, использующих H2 РВФ, оксигенаты получают в результате проведения каталитической реакции для части водного раствора исходного сырья, содержащего воду и растворимые в воде оксигенированные углеводороды, в присутствии катализатора РВФ при температуре риформинга и давлении риформинга для получения H2 РВФ и каталитической реакции между H2 РВФ (и H2, отправляемым на рецикл, и/или внешним H2) и частью раствора исходного сырья в присутствии катализатора деоксигенирования при температуре деоксигенирования и давлении деоксигенирования для получения желательных оксигенатов. В системах, использующих в качестве источника водорода H2, отправляемый на рецикл, или внешний H2, оксигенаты просто получают в результате проведения каталитической реакции между H2, отправляемым на рецикл, и/или внешним H2 и раствором исходного сырья в присутствии катализатора деоксигенирования при температурах и давлениях деоксигенирования. В каждом из вышеупомянутых случаев оксигенаты также могут включать осигенаты, отправляемые на рецикл, (С1+O1-3 углеводороды, отправляемые на рецикл). Если только не будет указано другого, то любые обсуждения катализаторов РВФ и катализаторов деоксигенирования будут представлять неограничивающие примеры подходящих каталитических материалов.
Катализатором деоксигенирования предпочтительно является гетерогенный катализатор, включающий один или несколько материалов, способных катализировать прохождение реакции между водородом и оксигенированным углеводородом в целях удаления одного или нескольких атомов кислорода из оксигенированного углеводорода для получения спиртов, кетонов, альдегидов, фуранов, карбоновых кислот, гидроксикарбоновых кислот, диодов и триолов. В общем случае материалы будут приставать к носителю и могут включать без ограничения Cu, Re, Fe, Ru, Ir, Co, Rh, Pt, Pd, Ni, W, Os, Mo, Ag, Au, их сплавы и комбинации. Катализатор деоксигенирования может включать данные элементы индивидуально или в комбинации с одним или несколькими представителями из Mn, Cr, Mo, W, V, Nb, Та, Ti, Zr, Y, La, Sc, Zn, Cd, Ag, Au, Sn, Ge, P, Al, Ga, In, Tl и их комбинаций. В одном варианте осуществления катализатор деоксигенирования включает Pt, Ru, Си, Re, Со, Fe, Ni, W или Mo. В еще одном другом варианте осуществления катализатор деоксигенирования включает Fe или Re и, по меньшей мере, один переходный металл, выбираемый из Ir, Ni, Pd, P, Rh и Ru. В еще одном варианте осуществления катализатор включает Fe, Re и, по меньшей мере, Cu или один переходный металл из группы VIIIB. Носителем может быть любой один из носителей, дополнительно описывающихся ниже, в том числе нитрид, углерод, диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана, оксид ванадия, диоксид церия, оксид цинка, оксид хрома, нитрид бора, гетерополикислоты, кизельгур, гидроксиапатит и их смеси. Катализатор деоксигенирования также может быть атомарно идентичным катализатору РВФ или катализатору конденсации.
Катализатором деоксигенирования также может быть бифункциональный катализатор. Например, кислотные носители (например, носители, характеризующиеся низкими изоэлектрическими точками) способны катализировать прохождение реакций дегидратации оксигенированных соединений с последующим прохождением реакций гидрирования на активных центрах металлических катализаторов в присутствии Н2, что опять-таки приводит к получению атомов углерода, которые не связаны с атомами кислорода. Бифункциональный путь дегидратации/гидрирования расходует H2 и приводит к последующему образованию различных полиолов, диолов, кетонов, альдегидов, спиртов и циклических простых эфиров, таких как фураны и пираны. Примеры катализаторов включают вольфрамированный диоксид циркония, диоксид титана-диоксид циркония, сульфатированный диоксид циркония, кислый оксид алюминия, диоксид кремния-оксид алюминия, цеолиты и гетерополикислотные носители. Гетерополикислоты представляют собой класс твердофазных кислот, примером которых являются такие вещества, как Н3+xPMo12-xVxO40, H4SiW12O40, H6PW12O40 и H6P2W18O62. Гетерополикислоты представляют собой твердофазные кислоты, обладающие хорошо определенной локальной структурой, наиболее частой из которых является структура Кеггина на вольфрамовой основе.
Уровень загрузки первого элемента (то есть, Си, Re, Fe, Ru, Ir, Co, Rh, Pt, Pd, Ni, W, Os, Mo, Ag, Au, их сплавов и комбинаций) находится в диапазоне от 0,25% (масс.) до 25% (масс.) на углероде при включении промежуточных массовых процентных долей с приращениями 0,10°/о и 0,05%, является таким как 1,00%, 1,10%, 1,15%, 2,00%, 2,50%, 5,00%, 10,00%, 12,50%, 15,00% и 20,00%. Предпочтительное количественное атомарное соотношение для второго элемента (то есть, Mn, Cr, Mo, W, V, Nb, Та, Ti, Zr, Y, La, Sc, Zn, Cd, Ag, Au, Sn, Ge, P, Al, Ga, In, Tl и их комбинаций) находится в диапазоне от 0,25 к 1 до 10 к 1, включая любые промежуточные соотношения, является таким как 0,50, 1,00, 2,50, 5,00 и 7,50 к 1. В случае приставания катализатора к носителю комбинация из катализатора и носителя будет включать от 0,25% (масс.) до 10% (масс.) первичного элемента.
При получении оксигенатов оксигенированный углеводород объединяют с водой для получения водного раствора исходного сырья, имеющего концентрацию, эффективную для стимулирования образования желательных продуктов реакции. Соотношение между водой и углеродом в расчете на моли предпочтительно находится в диапазоне от приблизительно 0,5:1 до приблизительно 100: 1, включая соотношения, такие как 1:1,2:1,3:1,4:1,5:1,6:1,7:1,8:1,9:1,10:1,15:1,25:1,50:1,75:1,100:1 и любые промежуточные соотношения. Раствор исходного сырья также может быть охарактеризован как раствор, включающий, по меньшей мере, 1,0 массового процента (% (масс.)) от совокупного раствора в качестве оксигенированного углеводорода. Например, раствор может включать один или несколько оксигенированных углеводородов, при этом совокупная концентрация оксигенированных углеводородов в растворе составляет, по меньшей мере, приблизительно 1%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% (масс.) и более, при включении любых промежуточных процентных долей и в зависимости от использующихся оксигенированных углеводородов. В одном варианте осуществления раствор исходного сырья включает, по меньшей мере, приблизительно 10%, 20%, 30%, 40%, 50% или 60% (масс.) сахара, такого как глюкоза, фруктоза, сахароза или ксилоза, или сахароспирта, такого как сорбит, маннит, глицерин или ксилит. Также включаются и количественные соотношения между водой и углеродом и процентные доли вне указанных выше диапазонов. Предпочтительно балансовый компонент раствора исходного сырья представляет собой воду. В некоторых вариантах осуществления раствор исходного сырья, по существу, состоит из воды, одного или нескольких оксигенированных углеводородов и необязательно одного или нескольких модификаторов исходного сырья, описывающихся в настоящем документе, таких как гидроксиды щелочных металлов или соли щелочных или щелочноземельных металлов или кислоты. Раствор исходного сырья также может включать отправленные на рецикл оксигенированные углеводороды, отправляемые на рецикл из реакторной системы. Раствор исходного сырья также может содержать пренебрежимо малые количества водорода, предпочтительно меньшие, чем приблизительно 1,5 моля водорода на один моль исходного сырья. В предпочтительных вариантах осуществления водород в раствор исходного сырья не добавляют.
Раствор исходного сырья вводят в реакцию с водородом в присутствии катализатора деоксигенирования в условиях по температуре и давлению деоксигенирования и при массовой часовой объемной скорости, эффективных для получения желательных оксигенатов. Конкретные полученные оксигенаты будут зависеть от различных факторов, в том числе от раствора исходного сырья, температуры реакции, давления реакции, концентрации воды, концентрации водорода, реакционной способности катализатора и расхода раствора исходного сырья, поскольку это оказывает воздействие на объемную скорость (массу/объем реагента на единицу катализатора в единицу времени), газовую часовую объемную скорость (ГЧОС) и массовую часовую объемную скорость (МЧОС). Например, увеличение с течением времени расхода и, тем самым, уменьшение воздействия на исходное сырье катализаторов будут уменьшать степень прохождения реакций, которые могут протекать, что, тем самым, приведет к получению повышенного выхода для высших диодов и триолов при уменьшении выходов для кетонов и спиртов.
Температуру и давление деоксигенирования предпочтительно выбирают обеспечивающими выдерживание, по меньшей мере, части исходного сырья в состоянии жидкой фазы на впускном отверстии реактора. Однако, признается то, что условия по температуре и давлению также могут быть выбраны и для более благоприятного получения желательных продуктов в паровой фазе. В общем случае реакция должна быть проведена в технологических условиях, когда термодинамика предложенной реакции является благоприятной. Например, минимальное давление, необходимое для выдерживания части исходного сырья в состоянии жидкой фазы, вероятно, будет варьироваться в зависимости от температуры реакции. По мере увеличения температур для выдерживания исходного сырья в состоянии жидкой фазы в общем случае при желании потребуются более высокие давления. Подходящими условиями эксплуатации также являются и давления, большие, чем то, которое потребуется для выдерживания исходного сырья в состоянии жидкой фазы (то есть, паровой фазы).
В жидкостных реакциях в конденсированной фазе давление в реакторе должно быть достаточным для выдерживания реагентов в состоянии конденсированной жидкой фазы на впускном отверстии реактора. Для жидкофазных реакций температура реакции может находиться в диапазоне от приблизительно 80°С до 300°С, а давление реакции - от приблизительно 72 фунт/дюйм2 (изб.) (496 кПа (изб.)) до 1300 фунт/дюйм2 (изб.) (8960 кПа (изб.)). В одном варианте осуществления температура реакции находится в диапазоне приблизительно от 120°С до 300°С или приблизительно от 200°С до 280°С или приблизительно от 220°С до 260°С, а давление реакции предпочтительно находится в диапазоне приблизительно от 72 до 1200 фунт/дюйм2 (изб.) (от 496 до 8270 кПа (изб.)) или приблизительно от 145 до 1200 фунт/дюйм2 (изб.) (от 1000 до 8270 кПа (изб.)) или приблизительно от 200 до 725 фунт/дюйм2 (изб.) (от 1380 до 5000 кПа (изб.)) или приблизительно от 365 до 700 фунт/дюйм2 (изб.) (от 2520 до 4830 кПа (изб.)) или приблизительно от 600 до 650 фунт/дюйм (изб.) (от 4140 до 4480 кПа (изб.)).
В случае парофазных реакций реакция должна быть проведена при температуре, при которой давление паров оксигенированного углеводорода составляет, по меньшей мере, приблизительно 0,1 атм (а предпочтительно значительно больше), и термодинамика реакции является благоприятной. Данная температура будет варьироваться в зависимости от конкретного использующегося оксигенированного углеводородного соединения, но в общем случае для парофазных реакций находится в диапазоне от приблизительно 100°С до 600°С. Предпочтительно температура реакции находится в диапазоне от приблизительно 120°С до приблизительно 300°С или от приблизительно 200°С до приблизительно 280°С или от приблизительно 220°С до приблизительно 260°С.
В еще одном варианте осуществления температура деоксигенирования находится в диапазоне приблизительно от 100°С до 400°С или приблизительно от 120°С до 300°С или приблизительно от 200°С до 280°С, а давление реакции предпочтительно находится в диапазоне приблизительно от 72 до 1300 фунт/дюйм2 (изб.) (от 496 до 8960 кПа (изб.)) или приблизительно от 72 до 1200 фунт/дюйм (изб.) (от 496 до 8270 кПа (изб.)) или приблизительно от 200 до 725 фунт/дюйм2 (изб.) (от 1380 до 5000 кПа (изб.)) или приблизительно от 365 до 700 фунт/дюйм2 (изб.) (от 2520 до 4830 кПа (изб.)).
Способ с использованием конденсированной жидкой фазы также может быть реализован при использовании модификатора, который увеличивает активность и/или стабильность системы катализатора. Предпочитается, чтобы вода и оксигенированный углеводород вступали бы в реакцию при подходящем значении рН в диапазоне от приблизительно 1,0 до приблизительно 10,0, включая промежуточные значения рН с приращениями 0,1 и 0,05, а более предпочтительно при значении рН в диапазоне от приблизительно 4,0 до приблизительно 10,0. В общем случае модификатор к раствору исходного сырья добавляют в количестве в диапазоне от приблизительно 0,1% до приблизительно 10% (масс.) в сопоставлении с совокупной массой использующейся системы катализатора, хотя в настоящее изобретение включаются и количества за пределами данного диапазона.
В общем случае реакцию необходимо проводить в условиях, при которых время пребывания раствора исходного сырья на катализаторе будет надлежащим для получения желательных продуктов. Например, значение МЧОС для реакции может составлять, по меньшей мере, приблизительно 0,1 грамма оксигенированного углеводорода на один грамм катализатора в час, а более предпочтительно значение МЧОС находится в диапазоне приблизительно от 0,1 до 40,0 г/г-час, включая значение МЧОС, равное приблизительно 0,25, 0,5, 0,75, 1,0, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,1, 3,2, 3,3, 3,4, 3,5, 3,6, 3,7, 3,8, 3,9, 4,0, 4,1, 4,2, 4,3, 4,4, 4,5, 4,6, 4,7, 4,8, 4,9, 5,0, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 20, 25, 30, 35, 40 г/г-час.
Водород, использующийся в реакции деоксигенирования, предпочтительно представляет собой Н2, полученный «по месту», но также может представлять собой и внешний Н2 или Н2, отправляемый на рецикл. При наличии внешнего H2 его количество предпочтительно подают экономно. Наиболее предпочтительно количество внешнего H2 подают в виде количеств, которые обеспечивают наличие менее, чем одного атома водорода, на один атом кислорода во всех оксигенированных углеводородах в потоке исходного сырья перед введением в контакт с катализатором деоксигенирования. Например, молярное соотношение между внешним H2 и совокупными растворимыми в воде оксигенированными углеводородами в растворе исходного сырья предпочтительно выбирают обеспечивающим наличие не более, чем одного атома водорода, на один атом кислорода в оксигенированном углеводороде. Молярное соотношение между оксигенированными углеводородами в исходном сырье и внешним H2, введенным в исходное сырье, также предпочтительно является не большим, чем 1:1, или более предпочтительно доходящим вплоть до 2:1,3:1,5:1,10:1,20:1 и более (включая 4:1,6:1,7:1,8:1,9:1,11:1,12:1,13:1,14:1,15:1,16:1,17:1,18:1 и 19:1). Количество (моли) внешнего H2, введенного в исходное сырье, составляет 0-100%, 0-95%, 0-90%, 0-85%, 0-80%, 0-75%, 0-70%, 0-65%, 0-60%, 0-55%, 0-50%, 0-45%, 0-40%, 0-35%, 0-30%, 0-25%, 0-20%, 0-15%, 0-10%, 0-5%, 0-2% или 0-1% от совокупного количества молей оксигенированного углеводорода (углеводородов) в исходном сырье, включая все промежуточные интервалы. В случае введения раствора исходного сырья или любой его части в реакцию с водородом РВФ и внешним Hz молярное соотношение между водородом РВФ и внешним H2 будет составлять, по меньшей мере, 1:20,1:15,1:10,1:5,1:3,1:2,1:1,2:1,3:1,5:1,10:1,15:1,20:1 и промежуточные соотношения (включая 4:1,6:1,7:1,8:1,9:1,11:1,12:1,13:1,14:1,15:1,16:1,17:1,18;1 и 19:1 и обращенные соотношения). Предпочтительно оксигенированный углеводород вводят в реакцию с H2 в присутствии незначительного эффективного количества внешнего H2.
Количество добавленного внешнего H2 (или дополнительного H2) может быть рассчитано при учете концентрации оксигенированных углеводородов в растворе исходного сырья. Предпочтительно количество добавленного внешнего H2 должно обеспечивать получение молярного соотношения между атомами кислорода в оксигенированных углеводородах и атомами водорода (то есть, 2 атома кислорода на одну молекулу газообразного H2), меньшего или равного 1,0. Например, в случае исходного сырья в виде водного раствора, состоящего из глицерина (3 атома кислорода), количество дополнительного На, добавленного к исходному сырью, предпочтительно будет не большим, чем приблизительно 1,5 моля H2 на один моль глицерина (С3Н8О3), а предпочтительно не большим, чем приблизительно 1,25, 1,0, 0,75, 0,50 или 0,25. В общем случае количество добавленного дополнительного H2 является меньшим, чем количество, соответствующее с кратностью 0,75, а более предпочтительно не большим, чем количество, соответствующее с кратностью 0,67, 0,50, 0,33, 0,30, 0,25, 0,20, 0,15, 0,10, 0,05, 0,01 количеству совокупного H2 (H2 РВФ и внешний H2), которое обеспечивало бы получение количественного атомарного соотношения между атомами кислорода и водорода 1:1.
Количество H2 РВФ в реакторе может быть идентифицировано или обнаружено по любому подходящему способу. Количество H2 РВФ может быть определено исходя из композиции потока продуктов в зависимости от композиции потока исходного сырья, композиции (композиций) катализатора и условий проведения реакции, вне зависимости от фактического механизма реакции, наблюдаемого в потоке исходного сырья. Количество H2 РВФ может быть рассчитано исходя из катализатора, условий проведения реакции (например, расхода, температуры, давления и тому подобного) и содержимого исходного сырья и продуктов реакции. Например, исходное сырье может быть введено в контакт с катализатором РВФ (например, платиной) для получения H2 РВФ «по месту» и первого потока продуктов реакции в отсутствие катализатора деоксигенирования. Исходное сырье также может быть введено в контакт как с катализатором РВФ, так и с катализатором деоксигенирования для получения второго потока продуктов реакции. В результате сравнения композиций первого потока продуктов реакций и второго потока продуктов реакции при сопоставимых условиях проведения реакции можно идентифицировать присутствие H2 РВФ и рассчитать количество полученного H2 РВФ, Например, увеличение количества оксигенированных соединений, характеризующихся повышенными степенями гидрирования, в продукте реакции в сопоставлении с компонентами исходного сырья может указывать на присутствие H2 РВФ.
Получение водорода «по месту»
Одно преимущество способа получения компонента, произведенного из растворимого в воде оксигенированного углеводорода, в настоящем изобретении заключается в том, что он обеспечивает получение и использование H2, полученного «по месту». H2 РВФ получают из исходного сырья в условиях риформинга в водной фазе при использовании катализатора риформинга в водной фазе (катализатора РВФ). Катализатором РВФ предпочтительно является гетерогенный катализатор, способный катализировать прохождение реакции для воды и оксигенированных углеводородов с образованием H2 в описывающихся ниже условиях. В одном варианте осуществления катализатор РВФ включает носитель и, по меньшей мере, один металл из группы VIIIB, Fe, Ru, Os, Ir, Co, Rh, Pt, Pd, Ni, их сплавы и комбинации. Катализатор РВФ также может включать, по меньшей мере, один дополнительный материал из металлов из группы VIIIB, группы VIIB, группы VIB, группы VB, группы IVB, группы ИВ, группы IB, группы IVA или группы VA, таких как Cu, В, Mn, Re, Cr, Mo, Bi, W, V, Nb, Та, Ti, Zr, Y, La, Sc, Zn, Cd, Ag, Au, Sn, Ge, P, Al, Ga, In, Tl, их сплавы и комбинации. Предпочтительный металл из группы VIIB включает Re, Mn или их комбинации. Предпочтительный металл из группы VIB включает Cr, Mo, W или их комбинацию. Предпочтительные металлы из группы VIIIB включают Pt, Rh, Ru, Pd, Ni или их комбинацию. Носители могут включать любой один из описывающихся ниже носителей катализаторов в зависимости от желательной активности системы катализатора.
Катализатор РВФ также может быть атомарно идентичным катализатору деоксигенирования или катализатору конденсации. Например, катализатор РВФ и деоксигенирования может включать Pt, сплавленный или смешанный с Ni, Ru, Cu, Fe, Rh, Re, их сплавами и комбинациями. Катализатор РВФ и катализатор деоксигенирования также могут включать Ru, сплавленный или смешанный с Ge, Bi, В, Ni, Sn, Cu, Fe, Rh, Pt, их сплавами и комбинациями. Катализатор РВФ также может включать Ni, сплавленный или смешанный с Sn, Ge, Bi, В, Cu, Re, Ru, Fe, их сплавами и комбинациями.
Предпочтительный уровень загрузки первичного металла из группы VIIIB находится в диапазоне от 0,25% (масс.) до 25% (масс.) на углероде при включении промежуточных массовых процентных долей с приращениями 0,10% и 0,05%, является таким как 1,00%, 1,10%, 1,15%, 2,00%, 2,50%, 5,00%, 10,00%, 12,50%, 15,00% и 20,00%. Предпочтительное количественное атомарное соотношение для второго материала находится в диапазоне от 0,25 к 1 до 10 к 1, включая промежуточные соотношения, является таким как 0,50, 1,00, 2,50, 5,00 и 7,50 к 1.
Получения одной предпочтительной композиции катализатора дополнительно добиваются в результате добавления оксидов элементов из группы IIIB и соответствующих редкоземельных оксидов. В таком случае предпочтительные компоненты будут представлять собой оксиды либо лантана, либо церия. Предпочтительное количественное атомарное соотношение между соединениями элементов из группы IIIB и первичным металлом из группы VIIIB находится в диапазоне от 0,25 к 1 до 10 к 1, включая промежуточные соотношения, является таким как 0,50, 1,00, 2,50, 5,00 и 7,50 к 1.
Еще одной предпочтительной композицией катализатора является та, которая содержит платину и рений. Предпочтительное количественное атомарное соотношение между Pt и Re находится в диапазоне от 0,25 к 1 до 10 к 1, включая промежуточные соотношения, является таким как 0,50, 1,00, 2,50, 5,00 и 7,00 к 1. Предпочтительный уровень загрузки Pt находится в диапазоне от 0,25% (масс.) до 5,0% (масс.) при включении промежуточных массовых процентных долей с приращениями 0,10% и 0,05%. является таким как 0,35%, 0,45%, 0,75%, 1,10%, 1,15%, 2,00%, 2,50%, 3,0% и 4,0%.
Предпочтительно катализатор РВФ и катализатор деоксигенирования имеют один и тот же атомный состав. Катализаторы также могут иметь различные составы. В таком случае предпочтительное количественное атомарное соотношение между катализатором РВФ и катализатором деоксигенирования находится в диапазоне от 5:1 до 1:5, например, но не ограничиваясь этим, 4,5:1,4,0:1,3,5:1,3,0:1,2,5:1,2,0:1,1,5:1,1:1,1:1,5,1:2,0,1:2,5,1:3,0,1:3,5,1:4,0,1:4,5 и любые промежуточные величины.
Подобно реакциям деоксигенирования условия по температуре и давлению предпочтительно выбирают обеспечивающими выдерживание, по меньшей мере, части исходного сырья в состоянии жидкой фазы на впускном отверстии реактора. Условия по температуре и давлению риформинга также могут быть выбраны обеспечивающими более благоприятное получение желательных продуктов в паровой фазе. В общем случае реакция РВФ должна быть проведена при температуре, при которой термодинамика является благоприятной. Например, минимальное давление, необходимое для выдерживания части исходного сырья в состоянии жидкой фазы, будет варьироваться в зависимости от температуры реакции. По мере увеличения температур для выдерживания исходного сырья в состоянии жидкой фазы в общем случае будут требоваться более высокие давления. Подходящим рабочим давлением также является любое давление, превышающее давление, которое необходимо для выдерживания исходного сырья в состоянии жидкой фазы (то есть, паровой фазы). В случае парофазных реакций реакция должна быть проведена при температуре риформинга, когда давление паров оксигенированного углеводородного соединения составляет, по меньшей мере, приблизительно 0,1 атм (а предпочтительно значительно больше), и термодинамика реакции является благоприятной. Температура будет варьироваться в зависимости от конкретного использующегося оксигенированного углеводородного соединения, но в общем случае находится в диапазоне от приблизительно 100°С до 450°С или от приблизительно 100°С до 300°С, для реакций, протекающих в паровой фазе. В случае жидкофазных реакций температура реакции может находиться в диапазоне от приблизительно 80°С до 400°С, а давление реакции - от приблизительно 72 фунт/дюйм (изб.) (496 кПа (изб.)) до 1300 фунт/дюйм2 (изб.) (8960 кПа (изб.)).
В одном варианте осуществления температура реакции находится в диапазоне приблизительно от 100°С до 400°С или приблизительно от 120°С до 300°С или приблизительно от 200°С до 280°С или приблизительно от 150°С до 270°С. Давление реакции предпочтительно находится в диапазоне приблизительно от 72 до 1300 фунт/дюйм2 (изб.) (от 496 до 8960 кПа (изб.)) или приблизительно от 72 до 1200 фунт/дюйм2 (изб.) (от 496 до 8270 кПа (изб.)) или приблизительно от 145 до 1200 фунт/дюйм2 (изб.) (от 1000 до 8270 кПа (изб.)) или приблизительно от 200 до 725 фунт/дюйм2 (изб.) (от 1380 до 5000 кПа (изб.)) или приблизительно от 365 до 700 фунт/дюйм2 (изб.) (от 2520 до 4830 кПа (изб.)) или приблизительно от 600 до 650 фунт/дюйм2 (изб.) (от 4140 до 4480 кПа (изб.)).
Способ с использованием конденсированной жидкой фазы также может быть реализован при использовании модификатора, который увеличивает активность и/или стабильность системы катализатора РВФ. Предпочитается, чтобы вода и оксигенированный углеводород вступали бы в реакцию при подходящем значении рН в диапазоне от приблизительно 1,0 до 10,0 или при значении рН в диапазоне от приблизительно 4,0 до 10,0, включая промежуточные значения рН с приращениями 0,1 и 0,05. В общем случае модификатор к раствору исходного сырья добавляют в количестве в диапазоне от приблизительно 0,1% до приблизительно 10% (масс.) в сопоставлении с совокупной массой использующейся системы катализатора, хотя в настоящее изобретение включаются и количества за пределами данного диапазона.
Для оптимизации доли водорода в продуктах реакции в раствор исходного сырья также могут быть добавлены и соли щелочных или щелочноземельных металлов. Примеры подходящих растворимых в воде солей включают одного или нескольких представителей, выбираемых из группы, состоящей из гидроксида, соли угольной кислоты, азотной кислоты или хлористо-водородной кислоты щелочного или щелочноземельного металла. Например, добавление (основных) солей щелочных металлов для получения значения рН в диапазоне от приблизительно рН 4,0 до приблизительно рН 10,0 может улучшить селективность реакций риформинга по водороду.
Добавление кислотных соединений также может обеспечить получение в описывающихся ниже реакциях гидрирования повышенной селективности по желательным продуктам реакции. Растворимую в воде соль предпочитается выбирать из группы, состоящей из солей азотной кислоты, фосфорной кислоты, серной кислоты, хлористо-водородной кислоты и их смесей. В случае использования кислотного модификатора предпочитается, чтобы он присутствовал бы в количестве, достаточном для уменьшения значения рН потока водного исходного сырья до значения в диапазоне от приблизительно рН 1,0 до приблизительно рН 4,0. Уменьшение значения рН потока исходного сырья данным образом может увеличить долю оксигенатов в конечных продуктах реакции.
В общем случае реакция должна быть проведена в условиях, в которых время пребывания раствора исходного сырья на катализаторе РВФ будет надлежащим для получения количества водорода РВФ, достаточного для проведения реакции со второй частью раствора исходного сырья на катализаторе деоксигенирования и получения желательных оксигенатов. Например, значение МЧОС для реакции может составлять, по меньшей мере, приблизительно 0,1 грамма оксигенированного углеводорода на один грамм катализатора РВФ, а предпочтительно находится в диапазоне приблизительно от 1,0 до 40,0 грамма оксигенированного углеводорода на один грамм катализатора РВФ, а более предпочтительно приблизительно от 0,5 до 8,0 грамма оксигенированного углеводорода на один грамм катализатора РВФ. В том, что касается увеличения масштаба производства, после запуска реакторная система РВФ должна контролироваться самим способом, так что реакции будут протекать при стационарном равновесии.
Стадия конденсации
После этого полученные оксигенаты превращают в C4+ соединения в результате конденсации. Как можно себе представить без ограничения какими-либо конкретными теориями, реакции кислотной конденсации в общем случае состоят из последовательности стадий, включающих (а) дегидратацию оксигенатов с образованием олефинов; (b) олигомеризацию олефинов; (с) реакции крекинга; (d) циклизацию более крупных олефинов с образованием ароматики; (е) изомеризацию парафина; и (f) реакции с переносом атома водорода с образованием парафинов. Как можно себе представить, реакции основной конденсации в общем случае состоят из последовательности стадий, включающих: (1) альдольную конденсацию с образованием β-гидроксикетона или β-гидроксиальдегида; (2) дегидратацию β-гидроксикетона или β-гидроксиальдегида с образованием сопряженного енона; (3) гидрирование сопряженного енона с образованием кетона или альдегида, который может принимать участие в последующих реакциях конденсации или превращения в спирт или углеводород; и (4) гидрирование карбонилов с образованием спиртов, или наоборот. Как можно себе представить, реакции кислотно-основной конденсации в общем случае включают любые из предшествующих стадий кислотных и/или основных реакций.
Получение С4+ соединений проходит в результате конденсации оксигенатов в присутствии катализатора конденсации. Катализатором конденсации в общем случае будет являться катализатор, способный образовывать более длинноцепные соединения в результате соединения двух кислородсодержащих веществ через новую связь углерод-углерод и превращения получающегося в результате соединения в углеводород, спирт или кетон, такой как кислотный катализатор, основный катализатор или многофункциональный катализатор, содержащий как кислотную, так и основную функциональность. Катализатор конденсации может включать без ограничения карбиды, нитриды, диоксид циркония, оксид алюминия, диоксид кремния, алюмосиликаты, фосфаты, цеолиты, оксиды титана, оксиды цинка, оксиды ванадия, оксиды лантана, оксиды иттрия, оксиды скандия, оксиды магния, оксиды церия, оксиды бария, оксиды кальция, гидроксиды, гетерополикислоты, неорганические кислоты, кислотно-модифицированные смолы, основно-модифицированные смолы и их комбинации. Катализатор конденсации может включать вышеупомянутые соединения индивидуально или в комбинации с модификатором, таким как Се, La, Y, Sc, Р, В, Bi, Li, Na, К, Rb, Cs, Mg, Ca, Sr, Ba и их комбинации. Катализатор конденсации для получения функциональности металла также может включать металл, такой как Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os, их сплавы и комбинации. Катализатор конденсации также может быть атомарно идентичным катализатору РВФ и/или катализатору деоксигенирования.
Катализатор конденсации может быть самонесущим (то есть, катализатор не требует использования другого материала для исполнения функции носителя) или может потребовать наличия отдельного носителя, подходящего для суспендирования катализатора в потоке реагента. Один в особенности выгодный носитель представляет собой диоксид кремния, в особенности диоксид кремния, характеризующийся большой площадью удельной поверхности (большей, чем 100 квадратных метров на один грамм) и получаемый по способу золь-гель-синтеза, способу осаждения или пирогенному способу. В других вариантах осуществления, в частности в случае катализатора конденсации в виде порошка, система катализатора может включать связующее для содействия придания катализатору желательной формы катализатора. Применимые способы получения включают экструдирование, гранулирование, скалывание в масло или другие известные способы. Для получения формованного материала также могут быть примешаны друг с другом и экструдированы оксид цинка, оксид алюминия и пептизатор. После высушивания данный материал прокаливают при температуре, надлежащей для получения каталитически активной фазы, что обычно требует использования температур, превышающих 450°С. Другие носители катализатора могут включать те, которые описываются более подробно ниже.
Кислотные катализаторы
Реакцию кислотной конденсации проводят при использовании кислотных катализаторов. Кислотные катализаторы могут включать без ограничения алюмосиликаты (цеолиты), силикаалюмофосфаты (SAPO), фосфаты алюминия (ALPO), аморфный диоксид кремния-оксид алюминия, диоксид циркония, сульфатированный диоксид циркония, вольфрамированный диоксид циркония, карбид вольфрама, карбид молибдена, диоксид титана, кислый оксид алюминия, фосфатированный оксид алюминия, фосфатированный диоксид кремния, сульфатированные разновидности углерода, фосфатированные разновидности углерода, кислотные смолы, гетерополикислоты, неорганические кислоты и их комбинации. В одном варианте осуществления катализатор также может включать модификатор, такой как Се, Y, Sc, La, Р, В, Bi, Li, Na, К, Rb, Cs, Mg, Ca, Sr, Ba и их комбинации. Катализатор для получения функциональности металла также может быть модифицирован в результате добавления металла, такого как Cu, Ag, Аи, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os, их сплавы и комбинации, и/или сульфидов и оксида Ti, Zr, V, Nb, Та, Mo, Cr, W, Mn, Re, Al, Ga, In, Fe, Co, Ir, Ni, Si, Cu, Zn, Sn, Cd, Р и их комбинаций. В особенности подходящим для использования в качестве промотора для настоящего способа также был признан галлий. Кислотный катализатор может быть гомогенным, самонесущим или приставшим к любому одному из носителей, дополнительно описывающихся ниже, включая носители, содержащие углерод, диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана, оксид ванадия, диоксид церия, нитрид, нитрид бора, гетерополикислоты, их сплавы и смеси.
Ga, In, Zn, Fe, Mo, Ag, Au, Ni, Р, Sc, Y, Та и лантаноиды также могут быть в результате обмена введены в цеолиты для получения цеолитного катализатора, обладающего активностью. Термин «цеолит» в соответствии с использованием в настоящем документе относится не только к микропористому кристаллическому алюмосиликату, но также и к микропористым кристаллическим металлсодержащим алюмосиликатным структурам, таким как галлоалюмосиликаты и галлосиликаты. Функциональность металла может быть образована металлами, такими как Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os, их сплавы и комбинации.
Примеры подходящих цеолитных катализаторов включают ZSM-5, ZSM-11, ZSM-12, ZSM-22, ZSM-23, ZSM-35 и ZSM-48. Цеолит ZSM-5 и обычное его получение описываются в патентах США №№3702886; Re. 29,948 (высококремнеземистый ZSM-5); 4100262 и 4139600, все из которых посредством ссылки включаются в настоящий документ. Цеолит ZSM-11 и обычное его получение описываются в патенте США №3709979, который также посредством ссылки включается в настоящий документ. Цеолит ZSM-12 и обычное его получение описываются в патенте США №3832449, посредством ссылки включенном в настоящий документ. Цеолит ZSM-23 и обычное его получение описываются в патенте США №4076842, посредством ссылки включенном в настоящий документ. Цеолит ZSM-35 и обычное его получение описываются в патенте США №4016245, посредством ссылки включенном в настоящий документ. Еще одно получение ZSM-35 описывается в патенте США №4107195, описание которого посредством ссылки включается в настоящий документ.ZSM-48 и обычное его получение описываются в патенте США №4375573, посредством ссылки включенном в настоящий документ. Другие примеры цеолитных катализаторов описываются в патенте США 5019663 и патенте США 7022888, также посредством ссылки включенных в настоящий документ.
В соответствии с описанием в патенте США 7022888 кислотным катализатором может быть бифункциональный пентасильный цеолитный катализатор, включающий, по меньшей мере, один металлический элемент из группы Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os, их сплавов и комбинаций и модификатор из группы Ga, In, Zn, Fe, Mo, Au, Ag, Y, Sc, Ni, P, Та, лантаноидов и их комбинаций. Цеолит предпочтительно содержит сильные кислотные и дегидрирующие центры и может быть использован совместно с потоками реагентов, содержащими оксигенированный углеводород, при температуре, меньшей, чем 500°С. Бифункциональный пентасильный цеолит может обладать кристаллической структурой типа ZSM-5, ZSM-8 или ZSM-11, состоящей из большого количества 5-членных кислородсодержащих колец, то есть, пентасильных колец. Цеолит, обладающий структурой типа ZSM-5, представляет собой в особенности предпочтительный катализатор. Бифункциональный пентасильный цеолитный катализатор предпочтительно представляет собой цеолиты типа ZSM-5, модифицированные при использовании Ga и/или In, такие как H-ZSM-5, импрегнированный при использовании Ga и/или In, H-ZSM-5, содержащий Ga и/или In, введенные в результате обмена, Н-галлосиликат, обладающий структурой типа ZSM-5, и Н-галлоалюмосиликат, обладающий структурой типа ZSM-5. Бифункциональный пентасильный цеолит типа ZSM-5 может содержать тетраэдрический алюминий и/или галлий, находящиеся в цеолитных каркасе или решетке, и октаэдрический галлий или индий. Октаэдрические центры предпочтительно не присутствуют в цеолитном каркасе, но присутствуют в цеолитных каналах в близком соседстве с цеолитными протонными кислотными центрами, которые приписываются присутствию в цеолите тетраэдрического алюминия и галлия. Тетраэдрический или каркасный Al и/или Ga, как представляется, несет ответственность за кислотную функцию цеолита, а октаэдрический или некаркасный Ga и/или In, как представляется, несет ответственность за дегидрирующую функцию цеолита.
В одном варианте осуществления катализатор конденсации может представлять собой Н-галлоалюмосиликат бифункционального пентасильного цеолита типа ZSM-5, характеризующегося молярным соотношением каркасных (тетраэдрических) Si/Al и Si/Ga, равным приблизительно 10-100 и 15-150, соответственно, и уровнем содержания некаркасного (октаэдрического) Ga, равным приблизительно 0,5-5,0% (масс.). В случае использования данных пентасильных Н-галлоалюмосиликатных цеолитов в качестве катализатора конденсации плотность сильных кислотных центров можно регулировать при использовании молярного соотношения каркасных Al/Si: чем большим будет соотношение Al/Si, тем большей будет плотность сильных кислотных центров. Высокодиспергированное вещество некаркасного оксида галлия может быть получено в результате дегаллиирования цеолита при его предварительной обработке при использовании H2 и водяного пара. Предпочтительным является цеолит, содержащий сильные кислотные центры с высокой плотностью, а также высокодиспергированное вещество некаркасного оксида галлия в тесном соседстве с цеолитным кислотным центром. Катализатор необязательно может содержать любое связующее, такое как материал оксида алюминия, диоксида кремния или глины. Катализатор может быть использован в виде гранул, экструдатов и частиц различных форм и размеров.
Кислотные катализаторы могут включать одну или несколько цеолитных структур, включающих клеткоподобные структуры диоксида кремния-оксида алюминия. Цеолиты представляют собой кристаллические микропористые материалы, обладающие хорошо определенной пористой структурой. Цеолиты содержат активные центры, обычно кислотные центры, которые могут быть получены в цеолитном каркасе. Силу и концентрацию активных центров можно подстраивать к конкретным областям применения. Примеры цеолитов, подходящих для конденсации вторичных спиртов и алканов, могут содержать алюмосиликаты, необязательно модифицированные катионами, такими как в случае Ga, In, Zn, Mo и смесей таких катионов, как это описывается, например, в патенте США №3702886, который посредством ссылки включается в настоящий документ. Как это признано на современном уровне техники, структура конкретных цеолита или цеолитов может быть изменена для получения в смеси продуктов других количеств различных углеводородных веществ. В зависимости от структуры цеолитного катализатора смесь продуктов может содержать различные количества ароматических и циклических углеводородов.
В альтернативном варианте, при осуществлении на практике настоящего изобретения могли бы быть использованы твердые кислотные катализаторы, такие как оксид алюминия, модифицированный фосфатами, хлоридом, диоксид кремния и другие кислотные оксиды. Кроме того, необходимую кислотность могут обеспечить либо сульфатированный диоксид циркония, либо вольфрамированный диоксид циркония. При промотировании конденсации оксигенатов для получения C5+ углеводородов и/или C5+ монооксигенатов подходящими также являются катализаторы на основе Re и Pt/Re. Re является достаточно кислым для промотирования кислотно-катализируемой конденсации. Кислотность также может быть увеличена у активированного угля в результате добавления либо сульфатов, либо фосфатов.
Основные катализаторы
Реакцию основной конденсации проводят при использовании основного катализатора. Основный катализатор включает, по меньшей мере, Li, Na, К, Cs, В, Rb, Mg, Са, Sr, Si, Ba, Al, Zn, Се, La, Y, Sc, Y, Zr, Ti, гидроталькит, алюминат цинка, фосфат, подвергнутый обработке основанием алюмосиликатный цеолит, основную смолу, основный нитрид, их сплавы или комбинации. Основный катализатор также может включать оксид Ti, Zr, V, Nb, Та, Мо, Cr, W, Mn, Re, Al, Ga, In, Co, Ni, Si, Cu, Zn, Sn, Cd, Mg, P, Fe и их комбинаций. В одном варианте осуществления катализатор конденсации дополнительно включает металл, такой как Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Мо, W, Sn, Os, их сплавы и комбинации. Предпочтительные материалы элементов из группы IA включают Li, Na, К, Cs и Rb. Предпочтительные материалы элементов из группы НА включают Mg, Са, Sr и Ba. Предпочтительные материалы элементов из группы ПВ включают Zn и Cd. Предпочтительные материалы элементов из группы IIIB включают Y и La. Основные смолы включают смолы, которые демонстрируют наличие основной функциональности, такие как Amberlyst. Основный катализатор может быть самонесущим или приставшим к любому одному из носителей, дополнительно описывающихся ниже, включая носители, содержащие углерод, диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана, оксид ванадия, диоксид церия, нитрид, нитрид бора, гетерополикислоты, их сплавы и смеси.
Основный катализатор также может включать цеолиты и другие микропористые носители, которые содержат соединения элементов из группы IA, таких как Li, Na, К, Cs и Rb. Предпочтительно материал элемента из группы IA присутствует в количестве, большем, чем то, которое требуется для нейтрализации кислотной природы носителя. Данные материалы могут быть использованы в любой комбинации, а также в комбинации с оксидом алюминия или диоксидом кремния. Функция металла также может быть образована и в результате добавления металлов из группы VIIIB или Си, Ga, In, Zn или Sn.
В одном варианте осуществления катализатор конденсации производят из комбинации MgO и Al2O3 для получения материала гидроталькита. Еще один предпочтительный материал содержит ZnO и Al2O3 в форме алюмоцинковой шпинели. Еще один другой предпочтительный материал представляет собой комбинацию ZnO, Al2O3 и CuO. Каждый из данных материалов также может включать дополнительную функцию металла, образуемую металлом из группы VIIIB, таким как Pd или Pt. В одном варианте осуществления основный катализатор представляет собой оксид металла, содержащий Cu, Ni, Zn, V, Zr или их смеси. В еще одном варианте осуществления основный катализатор представляет собой металл алюмината цинка, включающий Pt, Pd, Cu, Ni или их смеси.
Предпочтительный уровень загрузки первичного металла находится в диапазоне от 0,10% (масс.) до 25% (масс.) при включении промежуточных массовых процентных долей с приращениями 0,10% и 0,05%, является таким как 1,00%, 1,10%, 1,15%, 2,00%, 2,50%, 5,00%, 10,00%, 12,50%, 15,00% и 20,00%. Предпочтительное количественное атомарное соотношение для второго металла в случае наличия такового находится в диапазоне от 0,25 к 1 до 10 к 1, включая промежуточные соотношения, является таким как 0,50, 1,00, 2,50, 5,00 и 7,50 к 1.
Кислотно-основные катализаторы
Реакцию кислотно-основной конденсации проводят при использовании многофункционального катализатора, содержащего как кислотную, так и основную функциональность. Кислотно-основный катализатор может включать гидроталькит, алюминат цинка, фосфат, Li, Na, К, Cs, В, Rb, Mg, Si, Ca, Sr, Ba, Al, Ce, La, Sc, Y, Zr, Ti, Zn, Cr и их комбинации. В дополнительных вариантах осуществления кислотно-основный катализатор также может включать один или несколько оксидов элементов из группы Ti, Zr, V, Nb, Та, Мо, Cr, W, Mn, Re, Al, Ga, In, Fe, Co, Ir, Ni, Si, Cu, Zn, Sn, Cd, P и их комбинаций. Кислотно-основный катализатор также может включать функциональность металла, образуемую при использовании Си, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Мо, W, Sn, Os, их сплавов или комбинаций. В одном варианте осуществления катализатор дополнительно включает Zn, Cd или фосфат. В одном варианте осуществления катализатор конденсации представляет собой оксид металла, содержащий Pd, Pt, Си или Ni, а еще более предпочтительно алюминат или оксид металлического циркония, содержащий Mg и Си, Pt, Pd или Ni. Кислотно-основный катализатор также может включать гидроксиапатит (ГАП) в комбинации с любыми одним или несколькими вышеупомянутыми металлами. Кислотно-основный катализатор может быть самонесущим или приставшим к любому одному из носителей, дополнительно описывающихся ниже, включая носители, содержащие углерод, диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана, оксид ванадия, диоксид церия, нитрид, нитрид бора, гетерополикислоты, их сплавы и смеси.
Катализатор конденсации также может включать цеолиты и другие микропористые носители, которые содержат соединения элементов из группы IA, таких как Li, Na, К, Cs и Rb. Предпочтительно материал элемента из группы IA присутствует в количестве, меньшем, чем то, которое требуется для нейтрализации кислотной природы носителя. Функция металла также может быть образована и в результате добавления металлов из группы VIIIB или Си, Ga, In, Zn или Sn.
В одном варианте осуществления катализатор конденсации производят из комбинации MgO и Al2O3 для получения материала гидроталькита. Еще один предпочтительный материал содержит комбинацию MgO и ZrO2 или комбинацию ZnO и Al2O3. Каждый из данных материалов также может включать дополнительную функцию металла, образуемую медью или металлом из группы VIIIB, таким как Ni, Pd, Pt или комбинации вышеупомянутых компонентов.
В случае включения металла из группы IIB, VIB, VIIB, VIIIB, IIA или IVA уровень загрузки металла будет находиться в диапазоне от 0,10% (масс.) до 10% (масс.) при включении промежуточных массовых процентных долей с приращениями 0,10% и 0,05%, будет являться таким как 1,00%, 1,10%, 1,15%, 2,00%, 2,50%, 5,00% и 7,50% и тому подобное. В случае включения второго металла предпочтительное количественное атомарное соотношение для второго металла будет находиться в диапазоне от 0,25 к 1 до 5 к 1, включая промежуточные соотношения, будет являться таким как 0,50, 1,00, 2,50 и 5,00 к 1.
Реакции конденсации
Конкретные полученные С4+ соединения будут зависеть от различных факторов, в том числе без ограничения от типа оксигенатов в потоке реагентов, температуры конденсации, давления конденсации, реакционной способности катализатора и расхода потока реагентов, поскольку это оказывает воздействие на объемную скорость, значение ГЧОС и значение МЧОС. Предпочтительно поток реагентов вводят в контакт с катализатором конденсации при значении МЧОС, которое является надлежащим для получения желательных углеводородных продуктов. Значение МЧОС предпочтительно составляет, по меньшей мере, приблизительно 0,1 грамма оксигената в потоке реагентов в час, более предпочтительно значение МЧОС находится в диапазоне приблизительно от 0,1 до 40,0 г/г-час, включая значение МЧОС, равное приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 20, 25, 30, 35 г/г-час, и промежуточные приращения.
В общем случае реакция конденсации должна быть проведена при температуре, при которой термодинамика предложенной реакции является благоприятной. В случае жидкостных реакций в конденсированной фазе давление в реакторе должно быть достаточным для выдерживания, по меньшей мере, части реагентов в состоянии конденсированной жидкой фазы на впускном отверстии реактора. В случае парофазных реакций реакция должна быть проведена при температуре, при которой давление паров оксигенатов составляет, по меньшей мере, приблизительно 0,1 атм (а предпочтительно значительно больше), и термодинамика реакции является благоприятной. Температура конденсации будет варьироваться в зависимости от конкретного использующегося оксигената, но в общем случае находится в диапазоне от приблизительно 80°С до 500°С для реакций, проходящих в паровой фазе, а более предпочтительно от приблизительно 125°С до 450°С. В случае жидкофазных реакций температура конденсации может находиться в диапазоне от приблизительно 80°С до 500°С, а давление конденсации - от приблизительно 0 фунт/дюйм2 (изб.) (0 кПа (изб.)) до 1200 фунт/дюйм2 (изб.) (8270 кПа (изб.)). Предпочтительно температура конденсации находится в диапазоне приблизительно от 125°С до 300°С или приблизительно от 125°С до 250°С или приблизительно от 250°С до 425°С. Давление реакции предпочтительно составляет, по меньшей мере, приблизительно 0,1 атм или находится в диапазоне приблизительно от 0 до 1200 фунт/дюйм2 (изб.) (от 0 до 8270 кПа (изб.)) или приблизительно от 0 до 1000 фунт/дюйм2 (изб.) (от 0 до 6890 кПа (изб.)) или приблизительно от 0 до 700 фунт/дюйм2 (изб.) (от 0 до 4830 кПа (изб.)).
Варьирование вышеупомянутых, а также и других факторов в общем случае в результате будет приводить к модифицированию конкретных состава и выходов С4+ соединений. Например, варьирование температуры и/или давления реакторной системы или конкретных рецептур катализатора может привести в результате к получению С4+ спиртов и/или кетонов вместо С4+ углеводородов. С4+ углеводородный продукт также может содержать широкий ассортимент олефинов и алканов различных размеров (обычно разветвленные алканы). В зависимости от использующегося катализатора конденсации углеводородный продукт также может включать ароматические и циклические углеводородные соединения. С4+ углеводородный продукт также может характеризоваться нежелательно высокими уровнями содержания олефинов, что может привести к нагарообразованию или образованию отложений в двигателях внутреннего сгорания, или других нежелательных углеводородных продуктов. В таком случае полученные углеводородные молекулы необязательно могут быть подвергнуты гидрированию для восстановления кетонов в спирты и углеводороды, в то время как спирты и ненасыщенный углеводород могут быть восстановлены в алканы, что, тем самым, приводит к образованию более желательного углеводородного продукта, характеризующегося низкими уровнями содержания олефинов, ароматики или спиртов.
Стадия завершающей обработки в общем случае будет представлять собой реакцию гидрирования, которая удаляет оставшуюся карбонильную группу или гидроксильную группу. В таком случае может быть использован любой один из описывавшихся выше катализаторов гидрирования. Такие катализаторы могут включать любые один или несколько из следующих далее металлов: Cu, Ni, Fe, Co, Ru, Pd, Rh, Pt, Ir, Os, их сплавы или комбинации, индивидуально или совместно с промоторами, такими как Au, Ag, Cr, Zn, Mn, Sn, Cu, Bi и их сплавы, могут быть использованы с различными уровнями загрузки в диапазоне от приблизительно 0,01 до приблизительно 20% (масс.) на описывавшемся выше носителе.
В общем случае стадию завершающей обработки проводят при температурах завершающей обработки в диапазоне приблизительно от 80°С до 250°С и давлениях завершающей обработки в диапазоне приблизительно от 100 фунт/дюйм2 (689 кПа (изб.)) до 2000 фунт/дюйм2 (изб.) (13800 кПа (изб.)). Стадия завершающей обработки может быть проведена в паровой фазе или жидкой фазе и по мере надобности может использовать Н2, полученный «по месту», внешний Н2, H2, отправляемый на рецикл, или их комбинации.
На состав и выходы С4+ соединений, а также активность и стабильность катализатора конденсации воздействие могут оказывать также и другие факторы, такие как концентрация воды или нежелательных оксигенатов. В таком случае способ может включать стадию обезвоживания, которая удаляет часть воды перед конденсацией или сепарационную установку для удаления нежелательных оксигенатов. Например, сепарационная установка, такая как фазовый сепаратор, экстрактор, очиститель или перегонная колонна, может быть установлена перед стадией конденсации для удаления части воды из потока реагентов, содержащего оксигенаты. Сепарационная установка также может быть установлена и для удаления конкретных оксигенатов, что обеспечит получение потока желательных продуктов, содержащего углеводороды, характеризующиеся определенным диапазоном количества атомов углерода, или использование в качестве конечных продуктов или в других системах или способах.
C4+ соединения
Практика способа получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретении, в результате приводит к получению С4+ алканов, C4+ алкенов, C5+ циклоалканов, C5+ циклоалкенов, арилов, конденсированных арилов, С4+ спиртов, C4+ кетонов и их смесей. C4+ алканы и C4+ алкены содержат от 4 до 30 атомов углерода (C4-30 алканы и C4-30 алкены) и могут быть алканами или алкенами с разветвленной или прямой цепью. C4+ алканы и C4+ алкены также могут включать фракции C4-9, C7-14, C12-24 алканов и алкенов, соответственно, при этом С4-9 фракция относится к бензину, C7-14 фракция относится к керосину (например, реактивным топливам), а С12-24 фракция относится к дизельному топливу и другим промышленным областям применения. Примеры различных C4+ алканов и С4+ алкенов включают без ограничения бутан, бутан, пентан, пентен, 2-метилбутан, гексан, гексан, 2-метилпентан, 3-метилпентан, 2,2-диметилбутан, 2,3-диметилбутан, гептан, гептен, октан, октен, 2,2,4-триметилпентан, 2,3-диметилгексан, 2,3,4-триметилпентан, 2,3-диметилпентан, нонан, нонен, декан, децен, ундекан, ундецен, додекан, додецен, тридекан, тридецен, тетрадекан, тетрадецен, пентадекан, пентадецен, гексадекан, гексадекан, гептилдекан, гептилдецен, октилдекан, октилдецен, нонилдекан, нонилдецен, эйкозан, эйкозен, унэйкозан, унэйкозен, доэйкозан, доэйкозен, триэйкозан, триэйкозен, тетраэйкозан, тетраэйкозен и их изомеры.
С5+ циклоалканы и C5+ циклоалкены содержат от 5 до 30 атомов углерода и могут быть незамещенными, монозамещенными или полизамещенными. В случае монозамещенных и полизамещенных соединений замещенная группа может включать разветвленный С3+ алкил, С1+ алкил с прямой цепью, разветвленный С3+ алкилен, С1+ алкилен с прямой цепью, C2+ алкилен с прямой цепью, фенил или их комбинацию. В одном варианте осуществления, по меньшей мере, одна из замещенных групп включает разветвленный С3-12 алкил, C1-12 алкил с прямой цепью, разветвленный С3-12 алкилен, C1-12 алкилен с прямой цепью, C2-12 алкилен с прямой цепью, фенил или их комбинацию. В еще одном другом варианте осуществления, по меньшей мере, одна из замещенных групп включает разветвленный С3-4 алкил, C1-4 алкил с прямой цепью, разветвленный С3-4 алкилен, C1-4 алкилен с прямой цепью, C2-4 алкилен с прямой цепью, фенил или их комбинацию. Примеры желательных C5+ циклоалканов и C5+ циклоалкенов включают без ограничения циклопентан, циклопентен, циклогексан, циклогексен, метилциклопентан, метилциклопентен, этилциклопентан, этилциклопентен, этилциклогексан, этилциклогексен и их изомеры.
В общем случае арилы будут состоять из ароматического углеводорода либо в незамещенной (фенил), либо монозамещенной, либо полизамещенной форме. В случае монозамещенных и полизамещенных соединений замещенная группа может включать разветвленный С3+ алкил, С1+ алкил с прямой цепью, разветвленный С3+ алкилен, C2+ алкилен с прямой цепью, фенил или их комбинацию. В одном варианте осуществления, по меньшей мере, одна из замещенных групп включает разветвленный С3-12 алкил, C1-12 алкил с прямой цепью, разветвленный С3-12 алкилен, С2-12 алкилен с прямой цепью, фенил или их комбинацию. В еще одном другом варианте осуществления, по меньшей мере, одна из замещенных групп включает разветвленный С3-4 алкил, C1-4 алкил с прямой цепью, разветвленный С3-4 алкилен, C2-4 алкилен с прямой цепью, фенил или их комбинацию. Примеры различных арилов включают без ограничения бензол, толуол, ксилол (диметилбензол), этилбензол, пара-ксилол, мета-ксилол, орто-ксилол, С9 ароматику.
Конденсированные арилы в общем случае будут состоять из бициклических и полициклических ароматических углеводородов либо в незамещенной, либо в монозамещенной, либо в полизамещенной форме. В случае монозамещенных и полизамещенных соединений замещенные группы могут включать разветвленный С3+ алкил, C1+ алкил с прямой цепью, разветвленный С3+ алкилен, С2+ алкилен с прямой цепью, фенил или их комбинацию. В еще одном варианте осуществления, по меньшей мере, одна из замещенных групп включает разветвленный С3-4 алкил, C1-4 алкил с прямой цепью, разветвленный С3-4 алкилен, C2-4 алкилен с прямой цепью, фенил или их комбинацию. Примеры различных конденсированных арилов включают без ограничения нафталин, антрацен, тетрагидронафталин и декагидронафталин, индан, инден и их изомеры.
С4+ спирты также могут иметь циклическую, разветвленную или прямую цепь и содержать от 4 до 30 атомов углерода. В общем случае С4+ спирты могут представлять собой соединение, описывающееся формулой R1-OH, где R1 является представителем, выбираемым из группы, состоящей из разветвленного C4+ алкила, C4+ алкила с прямой цепью, разветвленного C4+ алкилена, C4+ алкилена с прямой цепью, замещенного C5+ циклоалкана, незамещенного C5+ циклоалкана, замещенного C5+ циклоалкена, незамещенного C5+ циклоалкена, арила, фенила и их комбинаций. Примеры желательных С4+ спиртов включают без ограничения бутанол, пентанол, гексанол, гептанол, октанол, нонанол, деканол, ундеканол, додеканол, тридеканол, тетрадеканол, пентадеканол, гексадеканол, гептилдеканол, октилдеканол, нонилдеканол, эйкозанол, унэйкозанол, доэйкозанол, триэйкозанол, тетраэйкозанол и их изомеры.
С4+ кетоны также могут иметь циклическую, разветвленную или прямую цепь и содержать от 4 до 30 атомов углерода. В общем случае С4+ кетон может представлять собой соединение, описывающееся формулой
где R3 и R4 независимо являются представителем, выбираемым из группы, состоящей из разветвленного С3+ алкила, C1+ алкила с прямой цепью, разветвленного С3+ алкилена, С2+ алкилена с прямой цепью, замещенного C5+ циклоалкана, незамещенного С5+ циклоалкана, замещенного C5+ циклоалкена, незамещенного C5+ циклоалкена, арила, фенила и их комбинации. Примеры желательных C4+ кетонов включают без ограничения бутанон, пентанон, гексанон, гептанон, октанон, нонанон, деканон, ундеканон, додеканон, тридеканон, тетрадеканон, пентадеканон, гексадеканон, гептилдеканон, октилдеканон, нонилдеканон, эйкозанон, унэйкозанон, доэйкозанон, триэйкозанон, тетраэйкозанон и их изомеры.
Легкие фракции из вышеупомянутых, главным образом С4-С9, могут быть отделены для использования в бензине. Средние фракции, такие как C7-C14, могут быть отделены для использования в керосине (например, реактивном топливе), в то время как тяжелые фракции, то есть, С12-С14, могут быть отделены для использования в дизельном топливе. Наиболее тяжелые фракции могут быть использованы в качестве смазок или подвергнуты крекингу для получения дополнительных фракций бензина и/или дизельного топлива.
Носители катализатора
В различных вышеупомянутых вариантах осуществления системы катализатора включают носитель, подходящий для суспендирования катализатора в растворе исходного сырья. Носитель должен быть тем, который обеспечивает наличие стабильной платформы для выбранного катализатора и условий проведения реакций. Носитель может принимать любую форму, которая является стабильной при выбранных условиях проведения реакции с точки зрения исполнения функции на желательных уровнях и особенно стабильной в водных растворах исходного сырья. Такие носители включают без ограничения углерод, диоксид кремния, диоксид кремния-оксид алюминия, оксид алюминия, диоксид циркония, диоксид титана, диоксид церия, оксид ванадия, нитрид, нитрид бора, гетерополикислоты, гидроксиапатит, оксид цинка, оксид хрома и их смеси. Также могут быть использованы и нанопористые носители, такие как цеолиты, углеродные нанотрубки или углеродный фуллерен.
Один в особенности предпочтительный носитель катализатора представляет собой углерод, в особенности углеродные носители, характеризующиеся относительно большими площадями удельной поверхности (большими, чем 100 квадратных метров на один грамм). Такие разновидности углерода включают активированный уголь (гранулированный, порошкообразный или окомкованный), ткань, войлоки или волокна из активированного угля, углеродные нанотрубки или нанорожки, углеродный фуллерен, углеродные ячеистые структуры, характеризующиеся большой площадью удельной поверхности, углеродные пеноматериалы (сетчатые углеродные пеноматериалы) и углеродные блоки. Углерод может быть получен в результате либо химической, либо паровой активации торфа, древесины, лигнита, угля, скорлупы кокосового ореха, оливковых косточек и углерода на нефтяной основе. Еще один предпочтительный носитель представляет собой гранулированный активированный уголь, полученный из кокосовых орехов. В одном варианте осуществления система катализатора РВФ и деоксигенироваания состоит из Pt на углероде, при этом Pt дополнительно сплавляют или смешивают с Ni, Ru, Cu, Fe, Rh, Re, их сплавами и комбинациями.
Еще один предпочтительный носитель катализатора представляет собой диоксид циркония. Диоксид циркония может быть получен в результате осаждения гидроксида циркония из циркониевых солей, при проведении переработки по золь-гель-способу или по любому другому способу. Диоксид циркония предпочтительно присутствует в кристаллической форме, получаемой в результате прокаливания материала предшественника при температурах, превышающих 400°С, и может включать как тетрагональную, так и моноклинную кристаллические фазы. Для улучшения структурообразующих или каталитических свойств диоксида циркония может быть добавлен модификатор. Такие модификаторы включают без ограничения сульфат, вольфрамат, фосфат, диоксид титана, диоксид кремния и оксиды металлов из группы IIIB, в особенности Се, La или Y. В одном варианте осуществления катализатор РВФ и деоксигенирования состоит из Pt на модифицированном диоксидом кремния диоксиде циркония главным образом в тетрагональной фазе, при этом Pt дополнительно сплавляют или смешивают с Ni, Ru, Cu, Fe, Rh, Re, их сплавами и комбинациями.
Еще один другой предпочтительный носитель катализатора представляет собой диоксид титана. Диоксид титана может быть получен в результате осаждения из титановых солей, при проведении переработки по золь-гель-способу или по любому другому способу. Диоксид титана предпочтительно присутствует в кристаллической форме и может включать как анатазную, так и рутиловую кристаллические фазы. Для улучшения структурообразующих или каталитических свойств диоксида титана может быть добавлен модификатор. Такие модификаторы включают без ограничения сульфат, диоксид кремния и оксиды металлов из группы IIIB, в особенности Се, La, или Y. В одном варианте осуществления система катализатора РВФ и получения оксигената состоит из Ru на диоксиде титана главным образом в рутиловой фазе, при этом Ru дополнительно сплавляют или смешивают с Ge, Bi, В, Ni, Sn, Cu, Fe, Re, Rh, Pt, их сплавами и комбинациями.
Еще один предпочтительный носитель катализатора представляет собой диоксид кремния. Диоксид кремния необязательно может быть объединен с оксидом алюминия для получения материала диоксида кремния-оксида алюминия. В одном варианте осуществления система катализатора РВФ представляет собой Pt на диоксиде кремния-оксиде алюминия или диоксиде кремния, при этом Pt дополнительно сплавляют или смешивают с Ni, Ru, Cu, Fe, Rh, Re, их сплавами и комбинациями. В еще одном варианте осуществления система катализатора РВФ представляет собой Ni на диоксиде кремния-оксиде алюминия или диоксиде кремния, при этом никель дополнительно сплавляют или смешивают с Sn, Ge, Bi, Bu, Cu, Re, Ru, Fe, их сплавами и комбинациями.
Носитель также может быть подвергнут обработке или модифицированию для улучшения его свойств. Например, носитель может быть подвергнут обработке, как в результате модифицирования поверхности, для модифицирования поверхностных фрагментов, таких как водород и гидроксил. Поверхностные группы водорода и гидроксила могут приводить к локальным вариациям значений рН, которые оказывают воздействие на каталитическую эффективность. Носитель также может быть модифицирован, например, в результате обработки его сульфатами, фосфатами, вольфраматами, силанами, лантаноидами, соединениями щелочных металлов или соединениями щелочноземельных металлов. В случае углеродных носителей углерод может быть подвергнут предварительной обработке водяным паром, кислородом (из воздуха), неорганическими кислотами или пероксидом водорода для получения большего количества поверхностных кислородных центров. Предпочтительная предварительная обработка заключалась бы в использовании либо кислорода, либо пероксида водорода. Подвергнутый предварительной обработке углерод также может быть модифицирован в результате добавления оксидов элементов из группы IVB и группы VB. Предпочитается использовать оксиды Ti, V, Zr и их смеси.
Системы катализаторов, либо индивидуально, либо в смеси друг с другом, могут быть получены при использовании обычных способов, известных специалистам в соответствующей области техники. Такие способы включают методики достижения начальной влажности, испарительного импрегнирования, химического осаждения из паровой фазы, протравного грунтования, магнетронного распыления и тому подобное. Способ, выбранный для изготовления катализатора, не является особенно критичным моментом для реализации функции изобретения при том условии, что различные катализаторы будут приводить к получению различных результатов в зависимости от факторов, таких как совокупная площадь удельной поверхности, пористость, и тому подобное.
Дополнительные материалы
Для улучшения прохождения реакции или для стимулирования ее прохождения до получения желательных продуктов реакции к раствору исходного сырья на различных этапах способа могут быть добавлены дополнительные материалы и композиции («добавки»). Добавки могут включать без ограничения кислоты, соли и дополнительные количества водорода или исходного сырья. Для прохождения надлежащих реакций такие добавки могут быть добавлены непосредственно в поток исходного сырья перед его введением в контакт с соответствующим катализатором или в сопряжении с данным введением в контакт или непосредственно в реакционный слой.
В одном варианте осуществления добавка может включать дополнительный раствор исходного сырья для подачи дополнительных оксигенированных углеводородов при получении оксигенатов. Исходное сырье может включать любые один или несколько перечисленных выше оксигенированных углеводородов, в том числе любые один или несколько представителей из сахароспиртов, глюкозы, полиолов, глицерина или сахаридов. Например, дополнительный материал может включать глицерин. В данном варианте осуществления неочищенный глицерин используют для инициирования реакции и получения водорода во избежание загрязнения катализатора деоксигенирования загрязнителями из неочищенного глицерина. После этого для увеличения количества оксигенированных углеводородов, доступных для переработки, к раствору исходного сырья добавляют очищенный глицерин до введения первоначального раствора исходного сырья в контакт с катализатором деоксигенирования или одновременно с данным введением. Предположительно в зависимости от характеристик катализатора РВФ и катализатора деоксигенирования обратное может быть использовано для неочищенного глицерина, исполняющего функцию добавки.
В еще одном варианте осуществления добавка может включать дополнительные оксигенаты для реакции конденсации. Оксигенаты могут включать любые один или несколько перечисленных выше оксигенатов. Например, дополнительный материал может включать пропиловый спирт. В данном варианте осуществления пропиловый спирт может быть получен в параллельной системе из глицеринового исходного сырья, а после этого объединен с оксигенатами, полученными в результате переработки сорбитового исходного сырья, для формирования потока реагентов, наиболее эффективного для получения продукта, содержащего комбинацию С6-12 углеводородов.
В еще одном другом варианте осуществления дополнительный материал может включать отправляемые на рецикл оксигенаты и/или оксигенированные углеводороды, не прореагировавшие полностью во время способа получения. Оксигенаты и оксигенированные углеводороды могут включать любые один или несколько перечисленных выше оксигенатов и оксигенированных углеводородов.
В еще одном дополнительном варианте осуществления дополнительный материал может включать кислоты и соли, добавленные в способ. Добавление кислотных соединений может привести к получению повышенной селективности по желательным оксигенатам и, в конечном счете, по С4+ соединениям. Растворимые в воде кислоты могут включать без ограничения соли азотной кислоты, фосфорной кислоты, серной кислоты, хлористо-водородной кислоты и их смеси. В случае использования необязательного кислотного модификатора предпочтительным будет его присутствие в количестве, достаточном для уменьшения значения рН потока водного исходного сырья до значения в диапазоне от приблизительно рН 1,0 до приблизительно рН 4,0. Уменьшение значения рН потока исходного сырья во время получения оксигенатов данным образом может увеличить долю диолов, полиолов, кетонов или спиртов для последующей конденсации.
Реакторная система
Реакции, описывающиеся в настоящем документе, могут быть проведены в любом реакторе подходящей конструкции, в том числе в проточных реакторах непрерывного действия, периодических, полупериодических или многосистемных реакторах без ограничения в отношении конструкции, размера, геометрии, расходов и тому подобного. Реакторная система также может использовать систему с псевдоожиженным каталитическим слоем, систему с короткоцикловыми эксплуатацией и регенерацией слоя, систему с неподвижным слоем, систему с подвижным слоем или комбинацию вышеупомянутых вариантов. Предпочтительно настоящее изобретение осуществляют на практике с использованием проточной системы непрерывного действия при стационарном равновесии.
В проточной системе непрерывного действия реакторная система включает, по меньшей мере, слой риформинга, адаптированный для приема водного раствора исходного сырья и получения водорода, слой деоксигенирования, адаптированный для получения оксигенатов из водорода и части раствора исходного сырья, и слой конденсации для получения С4+ соединений из оксигенатов. Слой риформинга конфигурируют для введения водного раствора исходного сырья в паровой фазе или жидкой фазе в контакт с катализатором РВФ и получения водорода в потоке реагентов. Слой деоксигенирования конфигурируют для приема потока реагентов при введении в контакт с катализатором деоксигенирования и получения желательных оксигенатов. Слой конденсации конфигурируют для приема потока регентов при введении в контакт с катализатором конденсации и получения желательных С4+ соединений. В случае систем, не включающих стадию получения водорода РВФ, может быть удален слой риформинга. В случае систем, не включающих стадию получения водорода или оксигената, могут быть удалены слои риформинга и деоксигенирования. Поскольку катализатор РВФ, катализатор деоксигенирования и катализатор конденсации также могут быть атомарно идентичными, катализаторы могут существовать в виде одного и того же слоя. В случае систем, включающих стадию гидрирования или гидрогенолиза, перед слоем деоксигенирования и/или риформинга может быть включен дополнительный реакционный слой. В случае систем, включающих стадию завершающей обработки, после слоя конденсации может быть включен дополнительный реакционный слой для реализации способа завершающей обработки.
В системах, производящих как водород, так и оксигенаты, слой конденсации может быть размещен в емкости одного и того же реактора совместно со слоем риформинга или в емкости второго реактора, сообщающейся с емкостью первого реактора, включающей слой риформинга. Слой конденсации может находиться в емкости одного и того же реактора совместно со слоем риформинга или деоксигенирования или в емкости отдельного реактора, сообщающейся с емкостью реактора, включающей слой деоксигенирования. Каждая емкость реактора предпочтительно включает выпускное отверстие, адаптированное для удаления потока продуктов из емкости реактора. В системах, включающих стадию гидрирования или стадию гидрогенолиза, слой реакции гидрирования или гидрогенолиза может находиться в емкости одного и того же реактора совместно со слоем риформинга или деоксигенирования или в емкости отдельного реактора, сообщающейся с емкостью реактора, включающей слой риформинга и/или слой деоксигенирования. В случае систем, включающих стадию завершающей обработки, слой реакции завершающей обработки может находиться в емкости одного и того же реактора совместно со слоем конденсации или в емкости отдельного реактора, сообщающейся с емкостью реактора, включающей слой конденсации.
Реакторная система также может включать дополнительные выпускные отверстия для обеспечения удаления частей потока реагентов с целью дополнительных стимулирования или направления прохождения реакции для получения желательных продуктов реакции и для обеспечения сбора и отправления на рецикл побочных продуктов реакции с целью использования в других частях системы. Реакторная система также может включать дополнительные впускные отверстия для обеспечения введения дополнительных материалов с целью дополнительных стимулирования или направления прохождения реакции для получения желательных продуктов реакции и для обеспечения отправления на рецикл побочных продуктов реакции с целью использования в способе риформинга. Например, система может быть сконструирована таким образом, чтобы на катализаторе РВФ был бы получен избыточный водород, при этом часть избыточного водорода удаляют и повторно вводят в способ по ходу технологического потока дальше для дополнения реакции оксигенатов на катализаторе конденсации или завершающей обработки продукта конденсации с целью получения желательных C4+ соединений. В альтернативном варианте, система может быть сконструирована таким образом, чтобы на катализаторе РВФ был бы получен избыточный водород, при этом часть избыточного водорода удаляют и используют в других способах по ходу технологического потока раньше, таких как способы предварительной обработки исходного сырья и реакции гидрирования или гидрогенолиза.
Реакторная система также может включать элементы, которые обеспечивают разделение потока реагентов на различные компоненты, которые могут найти себе применение в различных реакционных схемах, или простое промотирование прохождения желательных реакций. Например, сепарационная установка, такая как фазовый сепаратор, экстрактор, очиститель или перегонная колонна, может быть установлена перед стадией конденсации для удаления воды из потока реагентов с целью стимулирования прохождения реакции конденсации для создания благоприятных условий для получения углеводородов. Сепарационная установка также может быть установлена и для удаления конкретных оксигенатов и обеспечения получения потока желательных продуктов, содержащего углеводороды, характеризующиеся определенным диапазоном количества атомов углерода, или использования в качестве конечных продуктов или в других системах или способах.
В одном варианте осуществления реакционную систему конфигурируют таким образом, чтобы установить направление течения водного раствора исходного сырья обеспечивающим максимальное взаимодействие с Н2, полученным «по месту». Реактор может быть сконструирован таким образом, чтобы поток реагентов протекал бы горизонтально, вертикально или диагонально по отношению к гравитационной плоскости для доведения до максимума эффективности системы. В системах, в которых поток реагентов протекает вертикально или диагонально по отношению к гравитационной плоскости, поток может протекать либо против направления действия силы тяжести (система с восходящим потоком), либо по направлению действия силы тяжести (система с нисходящим потоком), либо по варианту, скомбинированному из обоих. В одном предпочтительном варианте осуществления емкость реактора РВФ и/или деоксигенирования конструируют в виде системы с восходящим потоком, в то время как емкость реактора конденсации конструируют в виде системы с нисходящим потоком. В данном варианте осуществления раствор исходного сырья сначала вводят в контакт со слоем риформинга, содержащим катализатор РВФ, для образования Н2, полученного «по месту». Вследствие данной конфигурации реактора Н2 РВФ затем в определенных условиях будет способен просачиваться через второй реакционный слой, содержащий катализатор деоксигенирования, со скоростью, большей или равной скорости раствора исходного сырья, для доведения до максимума взаимодействия между раствором исходного сырья и Н2 и катализатором деоксигенирования. Получающийся в результате поток реагентов после этого подают для переработки в реактор конденсации в конфигурации с нисходящим потоком.
В случае нахождения катализатора РВФ и катализатора деоксигенирования в одной камере, катализатор РВФ и катализатор деоксигенирования могут быть размещены в конфигурации с укладкой в стопку для обеспечения введения раствора исходного сырья в контакт сначала с катализатором РВФ, а после этого с катализатором деоксигенирования или серией катализаторов деоксигенирования в зависимости от желательных продуктов реакции. Реакционные слои для катализатора РВФ и катализатора или катализаторов деоксигенирования также могут быть размещены бок о бок друг с другом в зависимости от конкретного использующегося механизма течения. В любом случае раствор исходного сырья может быть введен в реакционную емкость через одно или несколько впускных отверстий, а после этого направлен через катализаторы для переработки. В еще одном варианте осуществления раствор исходного сырья направляют через катализатор РВФ для получения Н2 РВФ, а после этого как H2 РВФ, так и оставшийся раствор исходного сырья направляют через катализатор или катализаторы деоксигенирования для получения желательных оксигенатов. В одной параллельной конфигурации раствор исходного сырья может быть разделен для направления первой части раствора исходного сырья в слой риформинга, где получают H2 РВФ, а второй части - в слой деоксигенирования, где получают желательные оксигенаты при использовании H2 РВФ, полученного «по месту». В альтернативном варианте, реактор может быть сконфигурирован для приспосабливания к использованию двух раздельных растворов исходного сырья, при этом первый раствор исходного сырья направляют в емкость реактора РВФ, а второй раствор исходного сырья направляют в емкость реактора деоксигенирования. В одной последовательной конфигурации реактор может быть сконструирован таким образом, чтобы раствор исходного сырья протекал бы через емкость реактора РВФ в емкость реактора деоксигенирования. В вариантах осуществления, использующих объединенный катализатор РВФ/деоксигенирования, получение H2 РВФ и оксигенатов происходит одновременно. В любых из данных систем вследствие получения H2 РВФ «по месту» при использовании механизма перекачивания создают давление, которое также направляет раствор исходного сырья через камеры реактора.
Фигура 6 представляет собой технологическую схему, иллюстрирующую одну потенциальную реакторную систему, подходящую для использования при осуществлении на практике способа получения компонента, произведенного из растворимого в воде оксигенированного углеводорода в настоящем изобретении. Поток исходного сырья в виде оксигенированных углеводородов 1 (содержащий или не содержащий воду) перемешивают с потоком отправляемой на рецикл воды и отправляемых на рецикл оксигенатов в позиции 2 для получения водного раствора исходного сырья 3. Раствор исходного сырья 3 после этого подвергают гидрированию на стадии предварительной обработки 4 для получения раствора исходного сырья 5, который легче превращается в желательные оксигенаты. Н2 для стадии гидрирования может быть произведен из внешнего источника 22 или представлять собой водород, отправленный на рецикл из системы, как это проиллюстрировано ниже для стадий 13-21. Раствор исходного сырья 5 вводят в реакцию в емкости реактора 8, которая содержит катализатор РВФ и катализатор деоксигенирования, для получения потока продуктов 7, содержащего воду, H2, диоксид углерода, углеводороды и оксигенаты. После этого воду в потоке продуктов 7 удаляют в позиции 8 для получения потока продуктов 10, содержащего оксигенаты, водород, диоксид углерода и углеводороды. Затем воду со стадии обезвоживания 8 отправляют на рецикл в позициях 9 и 15 для перемешивания с потоком оксигенированных углеводородов в позиции 2. После этого поток продуктов 10 перепускают через емкость реактора 11, которая включает катализатор конденсации, для получения потока продуктов 12, содержащего C4+ соединения, воду, H2 и диоксид углерода. Затем поток продуктов 12 перепускают через трехфазный сепаратор 13 для отделения неконденсируемых газов 16 (то есть, водорода, диоксида углерода, метана, этана и пропана) от потока углеводородных продуктов 14, содержащих C4+ соединения и воду 15. Вода 15 из сепаратора может быть либо отправлена на рецикл, либо выведена из системы. Неконденсируемый газовый поток 16 может быть перепущен через сепарационную установку 17 для получения потока очищенного Н2 19 и потока рафината 18, содержащего диоксид углерода, метан, этан, пропан и некоторое количество водорода. После этого очищенный H2 19 может быть либо выведен из системы в позиции 20, либо перепущен через компрессор отправления на рецикл 21 для получения потока водорода, отправляемого на рецикл, 23.
В еще одной предпочтительной реакторной системе, проиллюстрированной на фигуре 7, предлагается первая реакторная система для превращения желательного раствора исходного сырья в C4+ соединения. Раствор исходного сырья хранят в резервуаре 1, а после этого перепускают по линии подачи 2 в загрузочный насос 3. Загрузочный насос 3 увеличивает давление раствора исходного сырья до желательного давления реакции, например, 600 фунт/дюйм2 (изб.) (4140 кПа (изб.)), а после этого выгружает раствор по линии 4 в электрический предварительный нагреватель 5, который нагревает исходное сырье до желательной температуры на впускном отверстии. После этого нагретый раствор 6 перепускают в технологическую часть реактора, имеющего, по существу, конфигурацию «труба в трубе» (труба 7 в трубе 8). В зависимости от давления реактора и температур, при которых функционируют несколько ступеней, поток реагентов, протекающий через трубу реактора 7, в общем случае будет все время выдерживаться, по существу, в состоянии жидкой фазы, но может и испаряться вследствие тепла конденсации из удаленной части 7b, так чтобы основная часть продукта, покидающая край реактора с выпускным отверстием по линии 15, имела бы форму пара.
Ступени и зоны ступеней трубы реактора 7 включают (объединенный) катализатор РВФ/деоксигенирования и катализатор конденсации, каждым из которых заполняют последовательные каталитические слои (то есть, один поверх другого). В данном примере труба реактора 7 содержит катализатор РВФ/деоксигенирования в приближенной части 7а трубы реактора 7 и катализатор конденсации в удаленной части 7b. Система катализатора снизу опирается на имеющие малый размер сферы из нержавеющей стали, размещенные на фритте из нержавеющей стали. Сферы из нержавеющей стали также размещаются поверх слоя катализатора. Для облегчения отделения отработанного катализатора при отправлении на рецикл или регенерацию слои катализатора разделяют при помощи пористого материала, такого как стекловата. Реактор также может быть физически разделен на раздельные трубы с каналами, соединяющими трубы для обеспечения непрерывности течения. Такая компоновка может обеспечить лучшее регулирование теплообмена, делая возможной оптимизацию температуры в соответствии с требованиями для реакций на нескольких ступенях реактора.
Реакция РВФ обычно является эндотермической, в то время как реакция конденсации обычно является высокоэкзотермической. Предпочтительно реакторная система делает возможным использование тепла, генерированного в реакции конденсации, для проведения нагревания в реакциях РВФ и деоксигенирования. Одно преимущество проведения обеих данных реакций совместно друг с другом заключается в непосредственной передаче тепла от экзотермической реакции конденсации к эндотермическим реакциям риформинга/деоксигенирования.
Технологическую трубу 7 предпочтительно изготавливают из теплопроводящего материала, сконфигурированного для передачи тепла от удаленной части 7b к приближенной части 7а. В дополнение к этому, технологическая труба может быть обогрета горячим маслом или горячим воздухом, протекающим через кольцевое пространство между технологической трубой 7 и внешней трубой 8. Горячий воздух может быть получен в результате нагревания воздуха окружающей среды из воздуходувки 10 электрическим нагревателем 12 и отправлен в реактор по линии 13. Также может быть использовано и горячее масло, которое получают при использовании нагревателя и насоса (не показаны) и также отправляют в реактор по линии 13. Конфигурация течения для данной системы является такой, чтобы горячий воздух (или масло) в трубе 8 протекал бы в противотоке по отношению к технологической текучей среде в трубе 7. В соответствии с этим, труба реактора 7 предпочтительно является более теплой снизу, чем сверху.
В альтернативном варианте, технологическая труба 7 может быть разделена на две раздельные трубы или зоны для облегчения оптимизации условий проведения реакции отдельно для реакций РВФ и деоксигенирования и для реакции конденсации. Например, данным образом может быть упрощено отделение отработанного катализатора для регенерации. На двухзональной второй ступени в вертикальном реакторе теплу, генерированному в результате конденсации в нижней зоне, может быть обеспечено перемещение в результате конвекции в верхнюю зону для использования в реакции риформинга. Вторая зона также может быть сконфигурирована с целью получения непрерывного или постадийного градиента для перемешанных катализаторов риформинга и конденсации, при этом большее количество катализатора риформинга находится на верхнем краю, а большее количество катализатора конденсации находится на нижнем краю.
Отходящий поток 15 из трубы реактора 7 включает газообразные продукты (такие как водород, СО и СО2), а также водные и органические жидкие продукты. Отходящий поток охлаждают до температуры окружающей среды при использовании охлаждаемой водой трубы в трубчатом конденсаторе 16. После этого отходящий поток 17 из конденсатора 16 направляют в трехфазный сепаратор для разделения фаз продуктов: неконденсируемый газ 18 (верхняя фаза), органическая жидкая фаза меньшей плотности 19 (средняя фаза) и водная жидкая фаза большей плотности 20 (нижняя фаза). Давление системы поддерживают в результате контролируемого выдерживания течения неконденсируемого газа по линии 21. Уровень жидкости поддерживают в результате контролируемого выдерживания течения компонентов водной фазы по линии 23. После этого органическую жидкую фазу снимают с верха водной фазы по линии 22.
Водную фазу 20 отбирают по линии 23. В случае содержания в водной фазе 20 значительных количеств остаточных оксигенатов (то есть, продуктов неполного риформинга) водная фаза 20 может быть отправлена обратно по линии 23 в источник исходного сырья 6, где ее используют в качестве исходного сырья, направляемого обратно в реактор. Таким образом, извлекают уровень содержания углерода и теплоту сгорания на промежуточных способах.
Средняя фаза 19 содержит C5+ соединения. Обычно данная фаза содержит углеводороды и монооксигенаты в диапазоне главным образом от C4 до С30. Легкие фракции, главным образом С4-С9, могут быть отделены для использования в бензине. Средняя фракция, то есть, C7-C14, может быть отделена для использования в качестве керосина (например, в реактивном топливе). Тяжелые фракции, то есть, C12-C24, могут быть отделены для использования в дизельном топливе. Наиболее тяжелые фракции могут быть использованы в качестве смазок или подвергнуты крекингу для получения дополнительных фракций бензина и/или дизельного топлива.
Паровая фаза 18 содержит водород и другие продукты реакции РВФ, такие как газообразные монооксид углерода, диоксид углерода, метан, этан, пропан, бутан, пентан и/или гексан. Часть данного газа выдувают из системы по линии 22 для предотвращения накопления в системе легких углеводородов и CO2. Газы также могут быть использованы в качестве источника топлива с целью получения тепла для реакторной системы. В том, что касается увеличения масштаба производства, после запуска реакторные системы могут контролироваться самим способом, и реакции протекали бы при стационарном равновесии.
Компонент, содержащий, по меньшей мере, одно C4+ соединение
Компонент, содержащий, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде углеводорода по описывавшемуся выше способу, перед его использованием в композициях жидкого топлива настоящего изобретения предпочтительно разделяют на различные фракции перегонки по любому известному способу. Предпочтительно компонент, содержащий, по меньшей мере, одно C4+ соединение, произведенное по описывавшемуся выше способу, разделяют на более, чем одну фракцию перегонки, где, по меньшей мере, одна из фракций перегонки является легкой, средней или тяжелой фракцией, описывающейся в настоящем документе ниже.
Как описывалось выше, легкие фракции, главным образом C4-C9, могут быть отделены для использования в бензине. Средние фракции, например, С7-С14, могут быть отделены для использования в качестве керосина, например, для использования в реактивном топливе. Тяжелые фракции, например, C12-C24, могут быть отделены для использования в дизельном топливе. Наиболее тяжелые фракции могут быть использованы в качестве смазок или могут быть подвергнуты крекингу для получения дополнительных фракций, предназначенных для использования во фракциях бензина, керосина и/или дизельного топлива.
Поскольку компонент, содержащий, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, обычно производят из биомассы, возраст компонента или его фракций является меньшим, чем 100 лет, предпочтительно меньшим, чем 40 лет, более предпочтительно меньшим, чем 20 лет, согласно вычислению по концентрации углерода-14 в компоненте. Легкие фракции
Легкие фракции компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, предпочтительно обладают одним или несколькими из следующих далее свойств (от LF-i до LF-vi):
(LF-i) температура конца кипения в диапазоне от 150 до 220°С, более предпочтительно в диапазоне от 160 до 210°С;
(LF-ii) плотность при 15°С в диапазоне от 700 до 890 кг/м3, более предпочтительно в диапазоне от 720 до 800 кг/м3;
(LF-iii) уровень содержания серы, равный, самое большее, 5 мг/кг, более предпочтительно, самое большее, 1 мг/кг;
(LF-iv) уровень содержания кислорода, равный, самое большее, 3,5% (масс.), более предпочтительно, самое большее, 3,0% (масс.), обычно, самое большее, 2,7% (масс.);
(LF-v) значение ИОЧ в диапазоне от 80 до 110, более предпочтительно в диапазоне от 90 до 100;
(LF-vi) значение МОЧ в диапазоне от 70 до 100, более предпочтительно в диапазоне от 80 до 90.
Обычно легкие фракции компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, обладают свойствами, которые соответствуют каждому из свойств, подробно описывающихся в приведенных выше позициях от LF-i до LF-vi, более часто каждому из предпочтительных значений для каждого из свойств, подробно описывающихся в приведенных выше позициях от LF-i до LF-vi.
Средние фракции
Средние фракции компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, предпочтительно обладают одним или несколькими из следующих далее свойств (от MF-i до MF-vix):
(MF-i) температура начала кипения в диапазоне от 120 до 215°С, более предпочтительно в диапазоне от 130 до 205°С;
(MF-ii) температура конца кипения в диапазоне от 220 до 320°С, более предпочтительно в диапазоне от 230 до 320°С;
(MF-iii) плотность при 15°С в диапазоне от 700 до 890 кг/м, более предпочтительно в диапазоне от 730 до 840 кг/м3;
(MF-iv) уровень содержания серы, равный, самое большее, 0,1% (масс.), более предпочтительно, самое большее, 0,01% (масс.);
(MF-v) совокупный уровень содержания ароматики, равный, самое большее, 30% (об.), более предпочтительно, самое большее, 25% (об.), еще более предпочтительно, самое большее, 20% (об.), наиболее предпочтительно, самое большее, 15% (об.);
(MF-vi) температура замерзания, равная - 40°С и менее, более предпочтительно, по меньшей мере, - 47°С и менее;
(MF-vii) максимальная высота некоптящего пламени, равная, по меньшей мере, 18 мм, более предпочтительно, по меньшей мере, 19 мм, еще более предпочтительно, по меньшей мере, 25 мм;
(MF-viii) вязкость при - 20°С в диапазоне от 1 до 10 сСт, более предпочтительно в диапазоне от 2 до 8 сСт;
(MF-vix) удельное энергосодержание в диапазоне от 40 до 47 МДж/кг, более предпочтительно в диапазоне от 42 до 46 МДж/кг.
Обычно средние фракции компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, обладают свойствами, которые соответствуют каждому из свойств, подробно описывающихся в приведенных выше позициях от MF-i до MF-vix, более часто каждому из предпочтительных значений для каждого из свойств, подробно описывающихся в приведенных выше позициях от MF-i до MF-vix.
Тяжелые фракции
Тяжелые фракции компонента, содержащего, по меньшей мере, одно C4+ соединение, производимое изх растворимого в воде оксигенированного углеводорода, предпочтительно обладают одним или несколькими из следующих далее свойств (от HF-i до HF-vi):
(HF-i) значение Т95 в диапазоне от 220 до 380°С, более предпочтительно в диапазоне от 260 до 360°С;
(HF-ii) температура вспышки в диапазоне от 30 до 70°С, более предпочтительно в диапазоне от 33 до 60°С;
(HF-iii) плотность при 15°С в диапазоне от 700 до 900 кг/м3, более предпочтительно в диапазоне от 750 до 850 кг/м3;
(HF-iv) уровень содержания серы, равный, самое большее, 5 мг/кг, более предпочтительно, самое большее, 1 мг/кг;
(HF-v) уровень содержания кислорода, равный, самое большее, 10% (масс.), более предпочтительно, самое большее, 8% (масс.);
(HF-vi) вязкость при 40°С в диапазоне от 0,5 до 6 сСт, более предпочтительно в диапазоне от 1 до 5 сСт.
Обычно тяжелые фракции компонента, содержащего, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, обладают свойствами, которые соответствуют каждому из свойств, подробно описывающихся в приведенных выше позициях от HF-i до HP-vi, более часто каждому из предпочтительных значений для каждого из свойств, подробно описывающихся в приведенных выше позициях от HF-i до HF-vi.
Композиция жидкого топлива
Количество компонента, содержащего, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и присутствующего в композиции жидкого топлива настоящего изобретения, составляет, по меньшей мере, 0,1% (об.) в расчете на совокупный объем композиции жидкого топлива. Более предпочтительно количество компонента, произведенного из растворимого в воде оксигенированного углеводорода, и присутствующего в композиции жидкого топлива настоящего изобретения, дополнительно соответствует одному или нескольким перечисленным ниже параметрам от (i) до (хх):
(i) по меньшей мере, 0,5% (об.)
(ii) по меньшей мере, 1% (об.)
(in) по меньшей мере, 1,5% (об.)
(iv) по меньшей мере, 2% (об.)
(V) по меньшей мере, 2,5% (об.)
(vi) по меньшей мере, 3% (об.)
(vii) по меньшей мере, 3,5% (об.)
(viii) по меньшей мере, 4% (об.)
(ix) по меньшей мере, 4,5% (об.)
(X) по меньшей мере, 5% (об.)
(xi) самое большее, 99,5% (об.)
(xii) самое большее, 99% (об.)
(xiii) самое большее, 98% (об.)
(xiv) самое большее, 97% (об.)
(XV) самое большее, 96% (об.)
(xvi) самое большее, 95% (об.)
(xvii) самое большее, 90% (об.)
(xviii) самое большее, 85% (об.)
(xix) самое большее, 80% (об.)
(XX) самое большее, 75% (об.)
Обычно количество компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и присутствующего в композиции жидкого топлива настоящего изобретения, соответствует одному параметру, выбираемому из приведенных выше позиций от (i) до (х), и одному параметру, выбираемому из приведенных выше позиций от (xi) до (хх).
Обычно в случае композиций бензина, соответствующих настоящему изобретению, количество компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и присутствующего в композиции бензина, будет находиться в диапазоне от 0,1 до 60% (об.), от 0,5 до 55% (об.) или от 1 до 50% (об.).
Обычно в случае композиций дизельного топлива, соответствующих настоящему изобретению, количество компонента, содержащего, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и присутствующего в композиции дизельного топлива, будет находиться в диапазоне от 0,1 до 60% (об.), от 0,5 до 55% (об.) или от 1 до 50% (об.).
Обычно в случае композиций керосина, соответствующих настоящему изобретению, количество компонента, содержащего, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и присутствующего в композиции керосина, будет находиться в диапазоне от 0,1 до 90% (об.), от 0,5 до 85% (об.) или от 1 до 80% (об.), так как в диапазоне от 0,1 до 60% (об.), от 0,5 до 55% (об.) или от 1 до 50% (об.).
Композицию жидкого топлива настоящего изобретения обычно выбирают из композиции бензина, керосина или дизельного топлива.
В случае композиции жидкого топлива в виде композиции бензина композиция бензина будет характеризоваться температурой начала кипения в диапазоне от 15°С до 70°С (IP123), температурой конца кипения, равной, самое большее, 230°С, (IP123), значением ИОЧ в диапазоне от 85 до 110 (ASTM D2699) и значением МОЧ в диапазоне от 75 до 100 (ASTM D2700).
В случае композиции жидкого топлива в виде композиции керосина композиция керосина будет характеризоваться температурой начала кипения в диапазоне от 110 до 150°С, температурой конца кипения в диапазоне от 200 до 320°С и вязкостью при - 20°С в диапазоне от 0,8 до 10 мм2/сек (ASTM D445).
В случае композиции жидкого топлива в виде композиции дизельного топлива композиция дизельного топлива будет характеризоваться температурой начала кипения в диапазоне от 130°С до 230°С (IP123), температурой конца кипения, равной, самое большее, 410°С, (IP123) и цетановым числом в диапазоне от 35 до 120 (ASTM D613).
Предпочтительно композиция жидкого топлива настоящего изобретения дополнительно содержит одну или несколько топливных присадок.
Композиции бензина
Композиция бензина, соответствующая настоящему изобретению, обычно содержит смеси углеводородов, кипящих в диапазоне от 15 до 230°С, более часто в диапазоне от 25 до 230°С (EN-ISO 3405). Температура начала кипения композиций бензина, соответствующих настоящему изобретению, находится в диапазоне от 15 до 70°С (IP123), предпочтительно в диапазоне от 20 до 60°С, более предпочтительно в диапазоне от 25 до 50°С.Температура конца кипения композиций бензина, соответствующих настоящему изобретению, составляет, самое большее, 230°С, предпочтительно, самое большее, 220°С, более предпочтительно, самое большее, 210°С. Оптимальные диапазоны и кривые перегонки обычно варьируются в соответствии с климатом и сезоном года.
В дополнение к компоненту, содержащему, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, углеводороды в композиции бензина могут быть произведены по любому способу, известному на современном уровне техники, обычно углеводороды могут быть произведены по любому известному способу из прямогонного бензина, синтетически полученных ароматических углеводородных смесей, углеводородов, подвергнутых термическому или каталитическому крекингу, нефтяных фракций, подвергнутых гидрокрекингу, углеводородов, подвергнутых каталитическому риформингу, или их смесей.
Исследовательское октановое число (ИОЧ) композиций бензина, соответствующих настоящему изобретению, находится в диапазоне от 85 до 110 (ASTM D2699). Предпочтительно значение ИОЧ композиции бензина будет составлять, по меньшей мере, 90, например, находиться в диапазоне от 90 до 110, более предпочтительно составлять, по меньшей мере, 91, например, находиться в диапазоне от 91 до 105, еще более предпочтительно составлять, по меньшей мере, 92, например, находиться в диапазоне от 92 до 103, еще более предпочтительно составлять, по меньшей мере, 93, например, находиться в диапазоне от 93 до 102, а наиболее более предпочтительно составлять, по меньшей мере, 94, например, находиться в диапазоне от 94 до 100.
Моторное октановое число (МОЧ) композиций бензина, соответствующих настоящему изобретению, находится в диапазоне от 75 до 100 (ASTM D2699). Предпочтительно значение МОЧ композиции бензина будет составлять, по меньшей мере, 80, например, находиться в диапазоне от 80 до 100, более предпочтительно составлять, по меньшей мере, 81, например, находиться в диапазоне от 81 до 95, еще более предпочтительно составлять, по меньшей мере, 82, например, находиться в диапазоне от 82 до 93, еще более предпочтительно составлять, по меньшей мере, 83, например, находиться в диапазоне от 83 до 92, а наиболее более предпочтительно составлять, по меньшей мере, 84, например, находиться в диапазоне от 84 до 90.
Обычно композиции бензина содержат смесь компонентов, выбираемых из одной или нескольких следующих далее групп: насыщенные углеводороды, олефиновые углеводороды, ароматические углеводороды и оксигенированные углеводороды. Обычно композиция бензина может содержать смесь насыщенных углеводородов, олефиновых углеводородов, ароматических углеводородов и необязательно оксигенированных углеводородов.
Обычно уровень содержания олефиновых углеводородов в композиции бензина находится в диапазоне от 0 до 40% (об.) в расчете на количество бензина (ASTM D1319); предпочтительно уровень содержания олефиновых углеводородов в композиции бензина находится в диапазоне от 0 до 30% (об.) в расчете на количество композиции бензина, более предпочтительно уровень содержания олефиновых углеводородов в композиции бензина находится в диапазоне от 0 до 20% (об.) в расчете на количество композиции бензина.
Обычно уровень содержания ароматических углеводородов в композиции бензина находится в диапазоне от 0 до 70% (об.) в расчете на количество бензина (ASTM D1319), например, уровень содержания ароматических углеводородов в композиции бензина находится в диапазоне от 10 до 60% (об.) в расчете на количество композиции бензина; предпочтительно уровень содержания ароматических углеводородов в композиции бензина находится в диапазоне от 0 до 50% (об.) в расчете на количество композиции бензина, например, уровень содержания ароматических углеводородов в композиции бензина находится в диапазоне от 10 до 50% (об.) в расчете на количество композиции бензина.
Уровень содержания бензола в композиции бензина составляет, самое большее, 10% (об.), более предпочтительно, самое большее, 5% (об.), в особенности, самое большее, 1% (об.), в расчете на количество композиции бензина.
Композиция бензина предпочтительно характеризуется низким или сверхнизким уровнем содержания серы, например, равным, самое большее, 1000 ч./млн. (масс.) (массовых частей на миллион частей), предпочтительно не большим, чем 500 ч./млн. (масс.), более предпочтительно не большим, чем 100, еще более предпочтительно не большим, чем 50, а наиболее предпочтительно не большим, чем даже 10 ч./млн. (масс.).
Композиция бензина также предпочтительно характеризуется низким совокупным уровнем содержания свинца, таким как, самое большее, 0,005 г/л, наиболее предпочтительно является свободной от свинца - не содержит каких-либо добавленных к ней соединений свинца (то есть, является неосвинцованной).
В случае содержания в композиции бензина оксигенированных углеводородов, по меньшей мере, часть неоксигенированных углеводородов будет замещать оксигенированные углеводороды. Уровень содержания кислорода в бензине может доходить вплоть до 30% (масс.) (EN 1601) в расчете на массу композиции бензина. Например, уровень содержания кислорода в бензине может доходить вплоть до 25% (масс.), предпочтительно вплоть до 10% (масс.). Обычно концентрация оксигенатов будет соответствовать минимальной концентрации, выбираемой из любой одной из 0, 0,2, 0,4, 0,6, 0,8, 1,0 и 1,2% (масс.), и максимальной концентрации, выбираемой из любой одной из 5, 4,5, 4,0, 3,5, 3,0 и 2,7 массового процента.
Примеры оксигенированных углеводородов, отличных от оксигенированных углеводородов, которые могут присутствовать в компоненте, содержащем, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, которые могут быть включены в бензин, включают спирты, простые эфиры, сложные эфиры, кетоны, альдегиды, карбоновые кислоты и их производные и кислородсодержащие гетероциклические соединения. Предпочтительно оксигенированные углеводороды, которые могут быть включены в бензин, выбирают из спиртов (таких как метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол и изобутанол), простых эфиров (предпочтительно простых эфиров, содержащих 5 и более атомов углерода на одну молекулу, например, метил-трет-бутилового эфира) и сложных эфиров (предпочтительно сложных эфиров, содержащих 5 и более атомов углерода на одну молекулу); один в особенности предпочтительный оксигенированный углеводород представляет собой этанол.
В случае присутствия в композиции бензина оксигенированных углеводородов количество оксигенированных углеводородов в композиции бензина может варьироваться в широком диапазоне. Например, в странах, таких как Бразилия и США, в настоящее время коммерчески доступны бензины, содержащие основную долю оксигенированных углеводородов, например, Е85, а также бензины, содержащие неосновную долю оксигенированных углеводородов, например, Е10 и Е5. Поэтому количество оксигенированных углеводородов, присутствующих в композиции бензина, предпочтительно выбирают из одного из следующих далее количеств: вплоть до 85% (об.); вплоть до 65% (об.); вплоть до 30% (об.); вплоть до 20% (об.); вплоть до 15% (об.); и вплоть до 10% (об.), в зависимости от желательной конечной рецептуры бензина. Обычно композиция бензина может содержать, по меньшей мере, 0,5, 1,0 или 2,0% (об.)оксигенированных углеводородов.
Примеры подходящих композиций бензина включают бензины, которые характеризуются уровнем содержания олефиновых углеводородов в диапазоне от 0 до 20% (об.) (ASTM D1319), уровнем содержания кислорода в диапазоне от 0 до 5% (масс.) (EN 1601), уровнем содержания ароматических углеводородов в диапазоне от 0 до 50% (об.) (ASTM D1319) и уровнем содержания бензола, равным, самое большее, 1% (об.).
Хотя это и некритично для настоящего изобретения, но композиции бензина настоящего изобретения обычно могут дополнительно включать одну или несколько топливных присадок. Концентрация и природа топливной присадки (присадок), которая может быть включена в композицию бензина настоящего изобретения, не являются критичным моментом. Неограничивающие примеры подходящих типов топливных присадок, которые могут быть включены в композицию бензина настоящего изобретения, включают антиоксиданты, ингибиторы коррозии, моющие присадки, присадки для предотвращения помутнения топлива, антидетонационные присадки, дезактиваторы металлов, соединения, предохраняющие от усиленного износа клапанного седла, красители, модификаторы трения, жидкие носители, разбавители и маркеры. Примеры подходящих таких добавок в общем случае описываются в патенте США №5855629.
Обычно топливные присадки могут быть перемешаны с одним или несколькими разбавителями или жидкими носителями для получения концентрата присадок, после этого концентрат присадок может быть примешан к композиции бензина настоящего изобретения.
Концентрация (активного вещества) любых присадок, присутствующих в композиции бензина настоящего изобретения, предпочтительно доходит вплоть до 1% (масс.), более предпочтительно находится в диапазоне от 5 до 1000 ч./млн. (масс.), в выгодном варианте в диапазоне от 75 до 300 ч./млн. (масс.), таком как от 95 до 150 ч./млн. (масс.).
В альтернативном варианте, композиция бензина настоящего изобретения может представлять собой авиационный бензин. В случае композиции бензина в виде авиационного бензина в зависимости от марки авиационного бензина моторное октановое число обедненной смеси будет составлять, по меньшей мере, 80 (ASTM D2700), а моторное октановое число обогащенной смеси будет составлять, по меньшей мере, 87 (ASTM D 909), или моторное октановое число обедненной смеси будет составлять, по меньшей мере, 99,5 (ASTM D2700), а сортность бензина будет составлять, по меньшей мере, 130 (ASTM D 909). Кроме того, в случае композиции бензина в виде авиационного бензина упругость паров по Рейду при 37,8°С будет находиться в диапазоне от 38,0 до 49,0 кПа (ASTM D323), температура конца кипения будет составлять, самое большее, 170°С (ASTM D 86), а уровень содержания тетраэтилсвинца будет составлять, самое большее, 0,85 г Pb/л, Композиции керосинового топлива
Композиции керосинового топлива настоящего изобретения находят себе применение в авиационных двигателях, таких как реактивные двигатели или авиационные дизельные двигатели, но также и в любом другом подходящем источнике питания или воспламенения.
В дополнение к компоненту, содержащему, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, композиция керосинового топлива может содержать смесь двух и более различных компонентов топлива и/или включать введенные присадки, как это описывается ниже.
Композиции керосинового топлива обычно будут иметь температуры кипения в диапазоне от 80 до 320°С, предпочтительно в диапазоне от 110 до 320°С, более предпочтительно в диапазоне от 130 до 300°С, в зависимости от марки и использования. Обычно они будут иметь плотность в диапазоне от 775 до 845 кг/м3, предпочтительно от 780 до 830 кг/м3, при 15°С (например, ASTM D4502 или IP 365). Обычно они будут характеризоваться температурой начала кипения в диапазоне от 80 до 150°С, предпочтительно в диапазоне от 110 до 150°С, и температурой конца кипения в диапазоне от 200 до 320°С. Их кинематическая вязкость при - 20°С (ASTM D445) обычно находится в диапазоне от 0,8 до 10 мм2/сек, предпочтительно от 1,2 до 8,0 мм2/сек.
В случае композиции керосинового топлива желательным может оказаться содержание топлива, произведенного по способу Фишера-Тропша, если композиция керосинового топлива, действительно, будет содержать топливо, произведенное по способу Фишера-Тропша, то обычно она будет содержать 5% (об.) и более, предпочтительно 10% (об.) и более или более предпочтительно 25% (об.) и более, топлива, произведенного по способу Фишера-Тропша.
Топливо, произведенное по способу Фишера-Тропша, должно быть подходящим для использования в качестве керосинового топлива. Поэтому его компоненты (или основная их часть, например, 95% (масс.) и более) должны иметь температуры кипения в приведенном выше интервале, то есть от 110 до 320°С, предпочтительно от 130 до 300°С. В подходящем случае оно будет характеризоваться температурой перегонки для 90% (об./об.) (Т90) в диапазоне о 180 до 250°С, предпочтительно от 180 до 230°С.
Термин «произведенный по способу Фишера-Тропша» подразумевает то, что топливо представляет собой продукт синтеза по способу конденсации Фишера-Тропша или является его производным. Реакция Фишера-Тропша превращает монооксид углерода и водород в имеющие более длинную цепь, обычно парафиновые, углеводороды:
n(CO+2H2)=(-CH2-)n+nH2O+тепло,
в присутствии надлежащего катализатора и обычно при повышенных температурах (например, в диапазоне от 125 до 300°С, предпочтительно от 175 до 250°С) и/или давлениях (например, в диапазоне от 500 до 10000 кПа, предпочтительно от 1200 до 5000 кПа). При желании могут быть использованы количественные соотношения водород:монооксид углерода, отличные от 2:1.
Сами монооксид углерода и водород могут быть произведены из органических или неорганических, природных или синтетических источников, обычно либо из природного газа, либо из органически произведенного метана.
Керосиновый продукт может быть получен непосредственно в результате проведения данной реакции или опосредованно, например, в результате фракционирования продукта синтеза Фишера-Тропша или из подвергнутого гидрообработке продукта синтеза Фишера-Тропша. Гидрообработка может включать гидрокрекинг для регулирования интервала кипения (смотрите, например, публикации GB-B-2077289 и ЕР-А-0147873) и/или гидроизомеризацию, которая может улучшить характеристики хладотекучести базового топлива в результате увеличения доли разветвленных парафинов. В публикации ЕР-А-0583836 описывается двухстадийный способ гидрообработки, по которому продукт синтеза Фишера-Тропша сначала подвергают гидроконверсии в условиях, таких чтобы он по существу не подвергался бы каким-либо изомеризации или гидрокрекингу (это приводит к гидрированию олефиновых и кислородсодержащих компонентов), а после этого, по меньшей мере, часть получающегося в результате продукта подвергают гидроконверсии в условиях, таких чтобы гидрокрекинг и изомеризация протекали бы для получения, по существу, парафинового углеводородного топлива. Желательная керосиновая фракция (фракции) после этого может быть выделена, например, в результате перегонки.
Для модифицирования свойств продуктов конденсации Фишера-Тропша могут быть использованы другие обработки после синтеза, такие как полимеризация, алкилирование, перегонка, крекинг-декарбоксилирование, изомеризация и гидрориформинг, как это описывается, например, в публикациях US-A-4125566 и US-A-4478955.
Типичные катализаторы для синтеза парафиновых углеводородов по способу Фишера-Тропша содержат в качестве каталитически активного компонента металл из группы VIII периодической таблицы, в частности, рутений, железо, кобальт или никель. Подходящие такие катализаторы описываются, например, в публикации ЕР-А-0583836 (страницы 3 и 4).
Один пример способа на основе способа Фишера-Тропша представляет собой способ SMDS (синтез среднего дистиллята от компании Shell), описывающийся в публикации «The Shell Middle Distillate Synthesis Process», van der Burgt et al. (paper delivered at the 5th Synfuels Worldwide Symposium, Washington DC, November 1985; смотрите также публикацию от ноября 1989 года с тем же самым названием от компании Shell International Petroleum Company Ltd., London, UK). Данный способ (также иногда называемый технологией «превращения газа в жидкость» или «ПГЖ» от компании Shell™) приводит к получению продуктов среднедистиллятного диапазона в результате превращения синтез-газа, произведенного из природного газа (главным образом метана), в тяжелый длинноцепной углеводородный (парафиновый) воск, который после этого может быть подвергнут гидроконверсии и фракционированию для получения жидких топлив для транспортных средств, таких как композиции керосинового топлива. Одна версия способа SMDS, использующая реактор с неподвижным слоем для стадии каталитического превращения, в настоящее время используется в Бинтулу, Малайзия, и ее продукты перемешивали с газойлями, произведенными из нефти, в коммерчески доступных автомобильных топливах.
Газойли и керосины, полученные по способу SDMS, коммерчески доступны в компании Royal Dutch/Shell Group of Companies.
В подходящем случае в соответствии с настоящим изобретением керосиновое топливо, произведенное по способу Фишера-Тропша, будет состоять из, по меньшей мере, 90% (масс.), предпочтительно, по меньшей мере, 95% (масс.), более предпочтительно, по меньшей мере, 98% (масс.), еще более предпочтительно, по меньшей мере, 99% (масс.), наиболее предпочтительно, по меньшей мере, 99,8% (масс.), парафиновых компонентов, обычно нормальных парафинов и изопарафинов. Массовое соотношение между нормальными парафинами и изопарафинами предпочтительно будет находиться в указанных выше диапазонах. Фактическое значение для данного соотношения будет определяться отчасти способом гидроконверсии, использующимся для получения керосина из продукта синтеза Фишера-Тропша. Также могут присутствовать и некоторые циклические парафины.
Благодаря способу Фишера-Тропша керосин, произведенный по способу Фишера-Тропша, характеризуется, по существу, отсутствием серы и азота или необнаружимыми уровнями их содержания. Соединения, содержащие данные гетероатомы, имеют тенденцию к исполнению функции ядов для катализаторов Фишера-Тропша, и поэтому их удаляют из исходного сырья в виде синтез-газа. Кроме того, обычно реализуемый способ не приводит или практически не приводит к получению ароматических компонентов. Уровень содержания ароматики в керосине Фишера-Тропша согласно определению в соответствии с документом ASTM D4629 обычно будут меньшим, чем 5% (масс.), предпочтительно меньшим, чем 2% (масс.), более предпочтительно меньшим, чем 1% (масс.), а наиболее предпочтительно меньшим, чем 0,2% (масс.).
Керосин, произведенный по способу Фишера-Тропша, который может быть использован в композициях керосинового топлива настоящего изобретения, обычно будет характеризоваться плотностью в диапазоне от 730 до 770 кг/м3 при 15°С; кинематической вязкостью в диапазоне от 1,2 до 6, предпочтительно от 2 до 5, более предпочтительно от 2 до 3,5, мм2/сек при - 20°С; и уровнем содержания серы, равным 20 ч./млн. (масс.) (массовых частей на миллион частей) и менее, предпочтительно 5 ч./млн. (масс.) и менее.
Предпочтительно он представляет собой продукт, полученный в результате проведения реакции конденсации метана по способу Фишера-Тропша при использовании количественного соотношения водород/монооксид углерода, меньшего, чем 2,5, предпочтительно меньшего, чем 1,75, более предпочтительно находящегося в диапазоне от 0,4 до 1,5, а в идеальном случае при использовании кобальтсодержащего катализатора. В подходящем случае он будет получен из подвергнутого гидрокрекинга продукта синтеза Фишера-Тропша (например, как это описывается в публикациях GB-B-2077289 и/или ЕР-А-0147873) или более предпочтительно продукта двухступенчатого способа гидроконверсии, такого как тот, который описывается в публикации ЕР-А-0583836 (смотрите выше). В последнем случае предпочтительные признаки способа гидроконверсии могут быть теми, которые описываются на страницах от 4 до 6 и в примерах публикации ЕР-А-0583836.
Композиция керосинового топлива настоящего изобретения предпочтительно содержит не более, чем 3000 ч./млн. (масс.) серы, более предпочтительно не более, чем 2000 ч./млн. (масс.), или не более, чем 1000 ч./млн. (масс.), или не более, чем 500 ч./млн. (масс.), серы.
В композицию керосинового топлива или ее компоненты можно вводить присадки (композиция, содержащая присадки) или не вводить присадки (композиция, свободная от присадок). В случае введения присадок, например, на нефтеперерабатывающем предприятии или на более поздних этапах распределения топлива, композиция будет содержать неосновные количества одной или нескольких присадок, выбираемых, например, из присадок для снятия статических зарядов (например, STADIS™ 450 (от компании Octel)), антиоксидантов (например, замещенных (третичный бутил)фенолов), присадок, дезактивирующих металлы, (например, N,N'-дисалицилиден-1,2-пропандиамина), противообледенительных присадок к топливу (например, диэтиленгликольмонометилового эфира), присадок в виде ингибитора коррозии/улучшителя смазывающей способности (например, APOLLO™ PRI 19 (от компании Apollo), DCI 4A (от компании Octel), NALCO™ 5403 (от компании Naico)) или присадок, улучшающих термостойкость, (например, АРА 101™ (от компании Shell)), которые одобрены в международных гражданских и/или военных технических требованиях к реактивному топливу.
Если только не будет указано другого, то концентрация (активного вещества) каждого такого дополнительного компонента в композиции керосинового топлива, содержащей введенные присадки, будет находиться на уровнях, требуемых или допустимых в международных технических требованиях к реактивному топливу.
В приведенном выше изложении количества (концентрации, % (об.), ч./млн. (масс.), % (масс.)) компонентов относились к активному веществу, то есть, при исключении летучих растворителей/материалов разбавителей, если только не было предусмотрено другого в соответствующем описании.
Композиция керосинового топлива настоящего изобретения будет в особенности применимой в случае использования или намерения использования композиции керосинового топлива в реактивном двигателе.
Композиции дизельного топлива
Композиция дизельного топлива, соответствующая настоящему изобретению, обычно содержит смеси углеводородов, кипящих в диапазоне от 130 до 410°С, более часто в диапазоне от 150 до 400°С. Температура начала кипения композиций дизельного топлива, соответствующих настоящему изобретению, находится в диапазоне от 130 до 230°С (IP123), предпочтительно в диапазоне от 140 до 220°С, более предпочтительно в диапазоне от 150 до 210°С. Температура конца кипения композиций дизельного топлива, ответствующих настоящему изобретению, составляет, самое большее, 410°С, предпочтительно, самое большее, 405°С, более предпочтительно, самое большее, 400°С.
В дополнение к компоненту, содержащему, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, композиция дизельного топлива может содержать смесь двух и более различных компонентов дизельного топлива, и/или в нее могут быть введены присадки, как это описывается ниже.
Такие композиции дизельного топлива будут содержать одно или несколько базовых топлив, которые обычно могут содержать жидкий углеводородный среднедистиллятный газойль (газойли), например, газойли, произведенные из нефти. Такие топлива обычно будут иметь температуры кипения в пределах описывавшегося выше диапазона, в зависимости от марки и использования. Обычно они будут характеризоваться плотностью в диапазоне от 750 до 1000 кг/м3, предпочтительно от 780 до 860 кг/м3, при 15°С (например, ASTM D4502 или IP 365) и цетановым числом (ASTM D613) в диапазоне от 35 до 120, более предпочтительно от 40 до 85. Обычно они будут характеризоваться температурой начала кипения в описывавшемся выше диапазоне и температурой конца кипения, равной, самое большее, 410°С, предпочтительно, самое большее, 405°С, более предпочтительно, самое большее, 400°С, наиболее предпочтительно находящейся в диапазоне от 290 до 400°С. Их кинематическая вязкость при 40°С (ASTM D445) в подходящем случае может находиться в диапазоне от 1,2 до 4,5 мм2/сек.
Один пример газойля, произведенного из нефти, представляет собой базовое топливо Swedish Class 1, которое будет характеризоваться плотностью в диапазоне от 800 до 820 кг/м3 при 15°С (SS-EN ISO 3675, SS-EN ISO 12185), значением Т95, равным 320°С и менее, (SS-EN ISO 3405) и кинематической вязкостью при 40°С (SS-EN ISO 3104) в диапазоне от 1,4 до 4,0 мм2/сек, согласно определению в соответствии с документом Swedish national specification EC1.
Необязательно также дизельное топливо могут образовывать или в дизельном топливе могут присутствовать топлива на основе неминерального масла, такие как биотоплива (отличные от компонента, содержащего, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода) или топлива, произведенные по способу Фишера-Тропша. Такие топлива Фишера-Тропша могут быть, например, произведены из природного газа, газоконденсатных жидкостей, нефти или горючих сланцев, остатков переработки нефти или горючих сланцев, угля или биомассы.
Количество топлива, произведенного по способу Фишера-Тропша и использующегося в композиции дизельного топлива настоящего изобретения, может находиться в диапазоне от 0% до остальной части композиции дизельного топлива (то есть, части композиции дизельного топлива, которая не является компонентом, содержащим, по меньшей мере, одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода), предпочтительно от 5% до остальной части композиции дизельного топлива, более предпочтительно от 5% до 75% (об.) от композиции дизельного топлива. В случае такой композиции дизельного топлива желательным может оказаться содержание 10% (об.) и более, более предпочтительно 20% (об.) и более, еще более предпочтительно 30% (об.) и более, топлива, произведенного по способу Фишера-Тропша. В случае таких дизельных топлив в особенности предпочтительным является содержание от 30 до 75% (об.), а, в частности 30 или 70% (об.), топлива, произведенного по способу Фишера-Тропша. Балансовый компонент дизельного топлива образован компонентом, содержащим, по меньшей мере, одно С4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и необязательно одним или несколькими другими компонентами дизельного топлива.
Такой компонент топлива, произведенный по способу Фишера-Тропша, представляет собой любую фракцию из диапазона среднедистиллятного топлива, которая может быть выделена из (необязательно подвергнутого гидрокрекингу) продукта синтеза Фишера-Тропша. Типичные фракции будут кипеть в интервале кипения лигроина, керосина или газойля. Предпочтительно используют продукт Фишера-Тропша, кипящий в интервале кипения керосина или газойля, потому что с данными продуктами легче работать, например, в жилых помещениях. Такие продукты в подходящем случае будут содержать в количестве, большем, чем 90% (масс.), фракцию, которая кипит в диапазоне от 160 до 400°С, предпочтительно до приблизительно 370°С. Примеры керосина и газойлей, произведенных по способу Фишера-Тропша, описываются в публикациях ЕР-А-0583836, WO-A-97/14768, WO-A-97/14769, WO-A-00/11116, WO-A-00/11117, WO-A-01/83406, WO-A-01/83648, WO-A-01/83 647, WO-A-01/8 3641, WO-A-00/20535, WO-A-00/20534, EP-A-1101813, US-A-5766274, US-A-5378348, US-A-5888376 и US-A-6204426.
Продукт Фишера-Тропша в подходящем случае будет содержать более, чем 80% (масс.), а в более подходящем случае более, чем 95% (масс.), изопарафинов и нормальных параф инов и менее, чем 1% (масс.) ароматики, при этом балансовый компонент представляет собой нафтеновые соединения. Уровень содержания серы и азота будет очень низким и обычно меньшим, чем пределы обнаружения таких соединений. По данной причине уровень содержания серы в композиции дизельного топлива, содержащей продукт Фишера-Тропша, может быть очень низким.
Композиция дизельного топлива предпочтительно содержит не более, чем 5000 ч./млн. (масс.) серы, более предпочтительно не более, чем 500 ч./млн. (масс.), или не более, чем 350 ч./млн. (масс.), или не более, чем 150 ч./млн, (масс.), или не более чем 100 ч./млн. (масс.), или не более, чем 70 ч./млн. (масс.), или не более, чем 50 ч./млн. (масс.), или не более, чем 30 ч./млн. (масс.), или не более, чем 20 ч./млн. (масс.), или наиболее предпочтительно не более, чем 15 ч./млн. (масс.), серы.
Дизельное топливо обычно также включает одну или несколько топливных присадок.
В само топливо на основе дизельного топлива можно вводить присадки (композиция, содержащая присадки) или не вводить присадки (композиция, свободная от присадок). В случае введения присадок, например, на нефтеперерабатывающем предприятии, оно будет содержать неосновные количества одной или нескольких присадок, выбираемых, например, из присадок для снятия статических зарядов, присадок, снижающих сопротивление течению по трубам, присадок, снижающих гидравлические потери, (например, сополимеров этилен/винилацетат или сополимеров акрилат/малеиновый ангидрид), присадок, повышающих смазывающую способность, антиоксидантов и присадок, предохраняющих от осаждения восков.
Известны и коммерчески доступны присадки к дизельным топливам, включающие моющие присадки. Такие присадки могут быть добавлены к дизельным топливам при уровнях содержания, предполагаемых для уменьшения, устранения или замедления накопления отложений в двигателе.
Примеры моющих присадок, подходящих для использования в присадках для дизельного топлива с целями настоящего изобретения, включают полиолефинзамещенные сукцинимиды или сукцинимиды полиаминов, например, полиизобутиленсукцинимиды или полиизобутиленаминсукцинимиды, алифатические амины, основания Манниха или амины и полиолефин- (например, полиизобутилен-) -малеиновые ангидриды. Сукцинимидные диспергирующие присадки описываются, например, в публикациях GB-А-960493, ЕР-А-0147240, ЕР-А-0482253, ЕР-А-0613938, ЕР-А-0557516 и WO-A-98/42808. В особенности предпочтительными являются полиолефинзамещенные сукцинимиды, такие как полиизобутиленсукцинимиды.
Смесь присадок к дизельному топливу может содержать другие компоненты в дополнение к моющей присадке. Примеры представляют собой улучшители смазывающей способности; присадки для предотвращения помутнения топлива, например, алкоксилированные фенолоформальдегидные полимеры; противопенообразователи (например, полисилоксаны, модифицированные простым полиэфиром); присадки, улучшающие воспламенение, (улучшители цетанового числа) (например, 2-этилгексилнитрат (ЭГН), циклогексилнитрат, ди-трет-бутилпероксид и те соединения, которые описываются в публикации US-A-4208190 во фрагменте от колонки 2, строки 27 до колонки 3, сроки 21); антикоррозионные присадки (например, пропан-1,2-диоловый сложный полуэфир тетрапропенилянтарной кислоты или сложные эфиры, полученные из многоатомного спирта и производного янтарной кислоты, при этом производное янтарной кислоты имеет, по меньшей мере, на одном из своих альфа-атомов углерода незамещенную или замещенную алифатическую углеводородную группу, содержащую от 20 до 500 атомов углерода, например, пентаэритритный сложный диэфир полиизобутилензамещенной янтарной кислоты); ингибиторы коррозии; отдушки; противоизносные присадки; антиоксиданты (например, фенольные производные, такие как 2,6-ди-трет-бутилфенол, или фенилендиамины, такие как N,N'-ди-втор-бутил-п-фенилендиамин); дезактиваторы металлов; улучшители сгорания; присадки, снимающие статическое электричество; улучшители хладотекучести; и присадки, предохраняющие от осаждения восков.
Смесь присадок к дизельному топливу может содержать улучшитель смазывающей способности, в особенности в случае малого уровня содержания серы (например, 500 ч./млн. (масс.) и менее) в композиции дизельного топлива. В композиции дизельного топлива, содержащей введенные присадки, улучшитель смазывающей способности обычно присутствует с концентрацией, меньшей, чем 1000 ч./млн. (масс.), предпочтительно находящейся в диапазоне от 50 до 1000 ч./млн. (масс.), более предпочтительно от 70 до 1000 ч./млн. (масс.). Подходящие коммерчески доступные улучшители смазывающей способности включают присадки на основе сложного эфира и кислоты. Другие улучшители смазывающей способности описываются в патентной литературе, в частности, в связи с их использованием в дизельных топливах, характеризующихся низким уровнем содержания серы, например, в:
- статье Danping Wei and H.A.Spikes, «The Lubricity of Diesel Fuels», Wear, III (1986)217-235;
- публикации WO-A-95/33805 - улучшители хладотекучести для улучшения смазывающей способности малосернистых топлив;
- публикации WO-A-94/17160 - определенные сложные эфиры, полученные из карбоновой кислоты и спирта, где кислота содержит от 2 до 50 атомов углерода, а спирт содержит 1 и более атомов углерода, в частности, глицеринмоноолеинат и диизодециладипинат, в качестве топливных присадок для уменьшения износа в системе впрыска дизельного двигателя;
- публикации US-A-5490864 - определенные дитиофосфорные диэфиродиспирты на основе сложных эфиров в качестве противоизносных присадок, повышающих смазывающую способность, для малосернистых дизельных топлив; и
- публикации WO-A-98/01515 - определенные алкилароматические соединения, имеющие, по меньшей мере, одну карбоксильную группу, присоединенную к их ароматическим ядрам, для придания противоизносного действия, повышающего смазывающую способность, в частности, в малосернистых дизельных топливах.
В случае композиции дизельного топлива предпочтительным также может быть содержание противопенообразователя, более предпочтительно в комбинации с антикоррозионной присадкой и/или ингибитором коррозии и/или присадкой, улучшающей смазывающую способность.
Если только не будет указано другого, то концентрация (активного вещества) каждого такого компонента присадок в композиции дизельного топлива, содержащей введенные присадки, предпочтительно будет доходить вплоть до 10000 ч./млн. (масс.), более предпочтительно находиться в диапазоне от 0,1 до 1000 ч./млн. (масс.), в выгодном случае от 0,1 до 300 ч./млн. (масс.), таком как от 0,1 до 150 ч./млн. (масс.).
Концентрация (активного вещества) любой присадки для предотвращения помутнения топлива в композиции дизельного топлива предпочтительно будет находиться в диапазоне от 0,1 до 20 ч./млн. (масс.), более предпочтительно от 1 до 15 ч./млн. (масс.), еще более предпочтительно от 1 до 10 ч./млн. (масс.), в выгодном случае от 1 до 5 ч./млн. (масс.). Концентрация (активного вещества) любой присутствующей присадки, улучшающей воспламенение, предпочтительно будет составлять 2600 ч./млн. (масс.) и менее, более предпочтительно 2000 ч./млн. (масс.) и менее, в удобном случае находиться в диапазоне от 300 до 1500 ч./млн. (масс.). Концентрация (активного вещества) любой моющей присадки в композиции дизельного топлива предпочтительно будет находиться в диапазоне от 5 до 1500 ч./млн. (масс,), более предпочтительно от 10 до 750 ч./млн. (масс.), наиболее предпочтительно от 20 до 500 ч./млн. (масс.).
В случае композиции дизельного топлива, например, смесь топливных присадок обычно будет содержать моющую присадку, необязательно совместно с другими описывавшимися выше компонентами и разбавителем, совместимым с дизельным топливом, который может представлять собой минеральное масло, растворителем, таким как те, которые продаются компанией Shell companies под торговой маркой «SHELLSOL», полярный растворитель, такой как сложный эфир, а, в частности, спирт, например, гексанол, 2-этилгексанол, деканол, изотридеканол и смеси спиртов, такие как те, которые продаются компанией Shell companies под торговой маркой «LINEVOL», в особенности спирт LINEVOL 79, который представляет собой смесь С7-9 первичных спиртов, или смесь С12-14 спиртов, которая коммерчески доступна.
Совокупный уровень содержания присадок в композиции дизельного топлива в подходящем случае может находиться в диапазоне от 0 до 10000 ч./млн. (масс.), а предпочтительно быть меньшим, чем 5000 ч./млн. (масс.).
В приведенном выше изложении количества (концентрации, % (об.), ч./млн. (масс.), % (масс.)) компонентов относятся к активному веществу, то есть, при исключении летучих растворителей/материалов разбавителей.
Способ получения композиции жидкого топлива
Композицию жидкого топлива настоящего изобретения получают в результате перемешивания:
(a) компонента, производимого из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива.
Под термином «компонент топлива» подразумеваются компонент, использующийся при получении композиции жидкого топлива, или композиция жидкого топлива как таковая, которые не представляют собой компонент, произведенный из растворимого в воде оксигенированного углеводорода. Примеры «компонентов топлива» включают компоненты топлива, которые в настоящее время используют при получении бензина, керосина и/или дизельного топлива, такие как потоки продуктов, произведенных из нефти, потоки продуктов, произведенных по способу Фишера-Тропша, оксигенаты и компоненты биотоплива.
Обычно потоками продуктов, произведенных из нефти, являются потоки продуктов, полученные на нефтеперерабатывающем предприятии и также называемые в настоящем документе потоками нефтеперерабатывающего предприятия. Неограничивающие примеры таких потоков нефтеперерабатывающего предприятия включают:
- «C4s» (низкокипящая фракция, обычно содержащая более, чем 80% (об.) парафиновых и олефиновых С4 соединений).
- Прямогонные легкие фракции (фракции ПЛ) или лигроин (низкокипящая фракция С4-С7 углеводородов (температура конца кипения, меньшая, чем приблизительно 140°С). Данные углеводороды в основном представляют собой парафины и характеризуются низким октановым числом. Поток характеризуется низким уровнем содержания ароматики и определенным уровнем содержания нафтенов).
- Изомерат (низкокипящая фракция (температура конца кипения, меньшая, чем приблизительно 110°С), образованная в результате изомеризации C5 и С6 парафинов во фракции ПЛ/лигроине для получения изомеров, характеризующихся более высоким октановым числом).
- Легкий, неочищенный (НО) или тяжелый платформат (или легкий, неочищенный (НО) или тяжелый реформат) (фракция, образованная в результате каталитического риформинга лигроина, подвергнутого гидрообработке, для получения потока, характеризующегося высоким октановым числом, высоким уровнем содержания ароматики и низким уровнем содержания олефинов).
- Алкилат (в том числе авиационный алкилат) (фракция, образованная в результате алкилирования изобутана C4, и С3 олефинами для получения характеризующихся высоким октановым числом парафинов с разветвленной цепью).
- Легкий или тяжелый бензин каталитического крекинга (ЛБКК или ТБКК) (фракция, образованная в результате обработки более тяжелых потоков в установке каталитического крекинга с псевдоожиженным слоем катализатора для получения более легких углеводородов, в том числе олефинов).
- Прямогонный керосин (фракция, характеризующаяся высоким уровнем содержания парафиновых углеводородов и обычно температурой перегонки в диапазоне от приблизительно 150°С +/-10°С до приблизительно 260°С +/- приблизительно 20°С).
- Обессеренный керосин (прямогонный керосин, который подвергли обработке для уменьшения уровней содержания меркаптана, а также уменьшения уровня содержания совокупной кислоты).
- Керосин, подвергнутый гидрообработке или гидроочистке, (прямогонный керосин, подвергнутый мягкой гидропереработке, который характеризуется пониженными уровнями содержания меркаптана, серы, олефинов, кислоты и металлов).
- Керосин, подвергнутый жесткой гидропереработке, (такой как керосин, подвергнутый глубокому гидрированию, керосин, подвергнутый гидрогенизационной сероочистке, керосин, подвергнутый гидрокрекингу, (прямогонный керосин, который подвергли более жесткой гидропереработке, чем керосин, подвергнутый гидрообработке, что обычно приводит к пониженным уровням содержания серы и азота)).
- Прямогонные дистилляты, кипящие в интервале кипения дизельного топлива.
- Газойль, подвергнутый гидрокрекингу, (АГО).
- Легкие масла из установки для коксования, подвергнутые крекингу и гидрообработке.
- Газойль, подвергнутый каталитическому крекингу, (ВГО).
- Газойли, подвергнутые термическому и паровому крекингу.
Оксигенаты и компоненты биотоплива представляют собой любой такой компонент, который является подходящим для использования в композиции жидкого топлива, такой как те, которые описывались выше в настоящем документе.
Способ получения композиции бензина
Для получения композиции бензина настоящего изобретения описывавшийся выше компонент, производимый из растворимого в воде оксигенированного углеводорода, в частности, легкую фракцию компонента, произведенного из растворимого в воде оксигенированного углеводорода, объединяют, по меньшей мере, с одним компонентом топлива.
Типичные компоненты топлива, которые могут быть перемешаны с компонентом, производимым из растворимого в воде оксигенированного углеводорода, для получения композиции бензина, соответствующей настоящему изобретению, включают:
- Композиции бензина как таковые;
- Потоки нефтеперерабатывающего предприятия, такие как C4s, фракции ПЛ, изомерат, легкий платформат, платформат НО, тяжелый платформат, алкилат, ЛБКК и ТБКК;
- Оксигенаты, такие как спирты и простые эфиры. Предпочтительные оксигенатные компоненты включают метанол, этанол, пропанол, бутанол, метил(третичный бутиловый) эфир (МТБЭ) и этил(третичный бутиловый) эфир (ЭТБЭ); и
- Компоненты биотоплива, такие как те, которые описывались выше в настоящем документе.
Термин «композиция бензина» в случае его использования в отношении к компоненту топлива обозначает композицию, определенную в приведенном выше разделе, посвященном композиции бензина.
Обычно композицию бензина настоящего изобретения получают в результате перемешивания:
(a) легкой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива, выбираемого из потока нефтеперерабатывающего предприятия.
Предпочтительно композицию бензина настоящего изобретения получают в результате перемешивания:
(a) легкой фракции произведенного из растворимого в воде оксигенированного углеводорода, как описано выше, и
(b) по меньшей мере, одного компонента топлива, выбираемого из потоков нефтеперерабатывающего предприятия в виде C4s, фракций ПЛ, изомерата, легкого платформата, платформата НО, тяжелого платформата, алкилата, ЛБКК и ТБКК; и необязательно оксигената, выбираемого из этанола, МТБЭ и ЭТБЭ.
Более предпочтительно композицию бензина настоящего изобретения получают в результате перемешивания:
(a) легкой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива, выбираемого из потоков нефтеперерабатывающего предприятия в виде C4s, фракций ПЛ, тяжелого платформата, алкилата и ЛБКК; и необязательно этанола.
В зависимости от количества легкой фракции компонента, произведенного из растворимого в воде оксигенированного углеводорода, который используют, количество использующихся компонентов топлива будут варьировать для получения композиции бензина, обладающей желательными свойствами.
Например, композиция бензина настоящего изобретения может быть получена в результате перемешивания:
(a) по меньшей мере, 0,1% (об.), в расчете на объем совокупной композиции топлива, легкой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива, выбираемого из потоков нефтеперерабатывающего предприятия, в следующих далее количествах: от 1 до 15% (об.) C4s, от 3 до 25% (об.) фракций ПЛ, от 0 до 50% (об.) тяжелого платформата, от 5 до 20% (об.) алкилата и от 10 до 35% (об.) ЛБКК, в расчете на количество совокупной композиции топлива; и необязательно вплоть до 85% этанола, в расчете на количество совокупной композиции топлива.
При получении композиций бензина, соответствующих настоящему изобретению, желательным может оказаться уменьшение относительного количества любых фракций ПЛ и/или тяжелого платформата в композиции бензина, соответствующей настоящему изобретению, при увеличении количеств легкой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода. Поэтому композиция бензина настоящего изобретения может быть получена в результате замещения, по меньшей мере, части любых фракций ПЛ и/или тяжелого платформата, использующихся при получении бензина, легкой фракцией описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода.
В альтернативном варианте, композицию бензина настоящего изобретения получают в результате перемешивания:
(а) легкой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) композиции бензина.
Если получаемой композицией бензина является авиационный бензин, при получении такого авиационного бензина, соответствующего настоящему изобретению, никакие оксигенаты не используются.
Способ получения композиции керосина
Для получения композиции керосина настоящего изобретения описывавшийся выше компонент, производимый из растворимого в воде оксигенированного углеводорода, в частности, средней фракции компонента, произведенного из растворимого в воде оксигенированного углеводорода, объединяют, по меньшей мере, с одним компонентом топлива.
Типичные компоненты топлива, которые могут быть перемешаны с компонентом, производимым из растворимого в воде оксигенированного углеводорода, для получения композиции керосина, соответствующей настоящему изобретению, включают потоки нефтеперерабатывающего предприятия, такие как прямогонный керосин, обессеренный керосин, керосин, подвергнутый гидрообработке или гидроочистке, и керосин, подвергнутый жесткой гидропереработке.
Обычно композицию керосина настоящего изобретения получают в результате перемешивания:
(a) средней фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива, выбираемого из потоков нефтеперерабатывающего предприятия в виде прямогонного керосина, обессеренного керосина, керосина, подвергнутого гидрообработке или гидроочистке, и керосина, подвергнутого жесткой гидропереработке.
Способ получения композиции дизельного топлива
Для получения композиции дизельного топлива настоящего изобретения описывавшийся выше компонент, производимый из растворимого в воде оксигенированного углеводорода, в частности, тяжелую фракцию компонента, произведенного из растворимого в воде оксигенированного углеводорода, объединяют, по меньшей мере, с одним компонентом топлива.
Типичные компоненты топлива, которые могут быть перемешаны с компонентом, производимым из растворимого в воде оксигенированного углеводорода, для получения композиции дизельного топлива, соответствующей настоящему изобретению, включают:
- Композиции дизельного топлива как таковые;
- Потоки нефтеперерабатывающего предприятия, такие как прямогонные дистилляты, кипящие в интервале кипения дизельного топлива, газойль, подвергнутый гидрокрекингу, (АГО), легкие масла из установки для коксования, подвергнутые крекингу и гидрообработке, газойль, подвергнутый каталитическому крекингу, (ВГО), газойли, подвергнутые термическому и паровому крекингу;
- Компоненты биотоплива, такие как те, которые описывались в настоящем документе выше.
Под термином «композиция дизельного топлива» в случае его использования в отношении компонента топлива подразумевается композиция, определенная в приведенном выше разделе, посвященном композиции дизельного топлива.
Обычно композицию дизельного топлива настоящего изобретения получают в результате перемешивания:
(a) тяжелой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива, выбираемого из потока нефтеперерабатывающего предприятия.
Предпочтительно композицию дизельного топлива настоящего изобретения получают в результате перемешивания:
(a) тяжелой фракции описывавшегося выше компонента, произведенного из растворимого в воде оксигенированного углеводорода, и
(b) по меньшей мере, одного компонента топлива, вабираемого из потоков нефтеперерабатывающего предприятия в виде прямогонных дистиллятов, кипящих в интервале кипения дизельного топлива, газойля, подвергнутого гидрокрекингу, (АГО), легких масел из установки для коксования, подвергнутых крекингу и гидрообработке, газойля, подвергнутого каталитическому крекингу, (ВГО), газойлей, подвергнутых термическому и паровому крекингу; и необязательно компонента биотоплива.
Настоящее изобретение дополнительно предлагает способ эксплуатации двигателя внутреннего сгорания, реактивного двигателя или парового котла, где способ включает введение в камеру сгорания двигателя или парового котла композиции жидкого топлива, соответствующей настоящему изобретению.
Настоящее изобретение будет дополнительно понято после ознакомления со следующими далее примерами.
Примеры
Примеры реакторных систем
Пример 1
Фигура 8 демонстрирует технологическую схему, иллюстрирующую одну реакторную систему, подходящую для использования при осуществлении на практике настоящего изобретения. Резервуар для исходного сырья 1 исполняет функцию резервуара хранения растворов исходного сырья. Раствор исходного сырья доставляют из резервуара для исходного сырья 1 в питающий насос 3 по линии подачи 2, где после этого его перепускают по линии выгрузки 4 в предварительный нагреватель 5. Предварительный нагреватель 5 может представлять собой теплообменник, обогреваемый нагревателем с электросопротивлением, или любой другой теплообменник, известный на современном уровне техники. После этого предварительно нагретое исходное сырье перепускают по линии 6, а в некоторых случаях объединяют с водородом 7 перед введением в реактор 9 по линии 8. Одна иллюстрация потенциального реактора 9 представлена на фигуре 11 и более полно описана в приведенном ниже примере 4.
Температуру стенок реактора 9 выдерживают при использовании нагревателей блоков 10а, 10b, 10с и 10d, в данном случае нагревателей с электросопротивлением. После выхода из реактора 9 продукты реакции поступают в линию выпускного отверстия реактора 11 и охлаждаются до температуры, близкой к температуре окружающей среды, в холодильнике продуктов реактора 12, что в результате приводит к получению потенциально трехфазного потока продуктов. Из холодильников продуктов реактора 12 продукты реакции проходят по линии 13 в клапан регулирования давления 14, который при необходимости используют для контроля давления на выпускном отверстии реактора.
После клапана 14 продукты по линии 15 поступают в фазовый сепаратор 16, где они сегрегируются на три раздельные фазы: (1) неконденсируемые газовые компоненты 17, содержащие преимущественно водород, диоксид углерода, метан, этан и пропан; (2) органическая жидкая фракция 18, содержащая как углеводороды, так и С3-30 спирты, кетоны и карбоновые кислоты; и (3) водный слой 19, содержащий главным образом воду и растворимые в воде оксигенированные соединения, такие как этанол, изопропанол, ацетон, пропанол и уксусная кислота. Неконденсируемая газовая фракция 17 может быть направлена по линии газообразных продуктов 20 в клапан уменьшения давления 21. Давление сепаратора 16 выдерживают при использовании клапана уменьшения давления 21. В альтернативном режиме эксплуатации сепаратор 16 можно выдерживать при давлении, приблизительно том же самом, что и давление на выпускном отверстии реактора, в результате открытия или исключения клапана. 14. В альтернативном режиме эксплуатации после этого давление на выпускном отверстии реактора контролируют благодаря действию клапана уменьшения давления 21. Расход и состав газа измеряют после покидания системы по линии 22.
Органическая жидкая фракция 18 покидает сепаратор по линии 23 перед поступлением в клапан спуска органики 24. Уровень органической фазы в сепараторе контролируют в результате регулирования клапана 24. Расход и состав органической фракции определяют после покидания органической фракцией системы по линии 25. Водная жидкая фракция 19 покидает сепаратор по линии 26 перед поступлением в клапан спуска донного отстоя сепаратора 27. Уровень водной фазы в сепараторе контролируют в результате регулирования клапана 27.
Расход и состав водной фракции могут быть определены после покидания водной фракцией системы по линии 28. В одном альтернативном режиме эксплуатации как органическая жидкая фракция 18, так и водная жидкая фракция 19 покидают систему через клапан спуска донного отстоя 27 сепаратора и по линии 28 перед разделением в декантаторе для измерения составов и расходов индивидуальных фаз.
Во всех случаях альтернативные режимы эксплуатации не оказывают воздействия на исследуемые каталитические способы. Альтернативные режимы эксплуатации могут быть использованы в качестве признанной разумной меры для достижения оптимального контроля способа в зависимости от относительных расходов газовой фазы 17, органической жидкой фазы 18 и водной фазы 19.
Перед инициированием течения исходного сырья в реакторы, если только не будет указано другого, катализаторы восстанавливали в потоке проточного водорода при 400°С вне зависимости от завершения восстановления перед загрузкой катализатора в реакторы.
Пример 2
Фигура 9 демонстрирует технологическую схему, иллюстрирующую еще одну реакторную систему, подходящую для использования при осуществлении на практике настоящего изобретения. Данная конфигурация реакторов включает два раздельных реактора при наличии возможности эксплуатации обоих реакторов с последовательным соединением или эксплуатации только первого реактора. В дополнение к этому, данная конфигурация делает возможными выведение катализатора во втором реакторе из эксплуатации и регенерацию его «по месту». После регенерации второй реактор может быть возвращен в эксплуатацию без оказания воздействия на функционирование первого реактора.
Реактор является подобным реактору из примера 1 при том исключении, что продукты реакции из холодильника продуктов реакции 12 могли бы быть направлены во второй реактор по линии 14 или направлены на обход второго реактора по байпасу в результате перепускания по линии 44. В случае использования второго реактора поток будет проходить от линии 14 к клапану регулирования давления 15. Клапан регулирования давления 15 может быть использован для контроля давления на выпускном отверстии первого реактора. От клапана регулирования давления 15 поток проходит к стопорному клапану впускного отверстия второго реактора 17 и в линию 18. Из линии 18 поток проходит в линию 19 и в предварительный нагреватель второго реактора 20. В проиллюстрированном варианте осуществления предварительный нагреватель 20 представляет собой теплообменник, обогреваемый нагревателем с электросопротивлением.
После этого предварительно нагретое исходное сырье перепускают по линии 19 во второй реактор 22, который более полно описывается в примере 4. Температуру стенки реактора 22 выдерживают при использовании нагревателей блоков 23а, 23b, 23с и 24d - в данном случае нагревателей с электросопротивлением. После покидания реактора продукты реакции поступают в линию выпускного отверстия второго реактора 24, а затем охлаждаются в холодильнике продуктов второго реактора 25. Из холодильников продуктов второго реактора 26 технологический поток может быть направлен по линиям 26 и 27 к стопорному клапану выпускного отверстия второго реактора 28, в линии 29, затем 30, а после этого в сепаратор продуктов 31.
В случае желательности эксплуатации второго реактора клапан 17 и клапан 28 будут открытыми, в то время как байпасный клапан второго реактора 45 будет закрыт для предотвращения обхода потоком второго реактора по байпасу. В случае желательности эксплуатации только лишь первого реактора или в случае регенерации второго реактора клапан 17 и клапан 28 будут закрытыми, в то время как клапан 45 будет открыт.В случае обхода второго реактора по байпасу продукт первого реактора перетекает непосредственно из линии 13 в линию 44, через байпасный клапан 45, в линию 46 и затем в линию 30. В любом случае вне зависимости от того, будут ли второй реактор эксплуатировать или обходить по байпасу, поток будет проходить от линии 30 в сепаратор продуктов.
В фазовом сепараторе 31 продукты реакции разделяют на газообразную фракцию 32, органическую фракцию 33 и водную фракцию 34, как это описывалось выше в примере 1. Газообразную фракцию 32 направляют по линии газообразных продуктов 35 к клапану уменьшения давления 36. Давление сепаратора 31 выдерживают при использовании клапана уменьшения давления 36. В случае эксплуатации второго реактора 22 благодаря действию клапана уменьшения давления 36 будут контролировать давление на выпускном отверстии второго реактора 22. В случае обхода второго реактора 22 по байпасу благодаря действию давления уменьшения давления 36 будут контролировать давление на выпускном отверстии первого реактора 9.
Расход и состав газа измеряют при покидании системы по линии 37. Органическая жидкая фракция 33 покидает сепаратор по линии 38 перед поступлением в клапан спуска органики 39. Уровень органической фазы в сепараторе контролируют в результате регулирования клапана 39. Расход и состав органической фракции определяют после покидания органической фракцией системы по линии 40. Водная жидкая фракция 34 покидает сепаратор по линии 41 перед поступлением в клапан спуска донного отстоя сепаратора 42. Уровень водной фазы в сепараторе контролируют в результате регулирования клапана 42. Расход и состав водной фракции определяют после покидания водной фракцией системы по линии 43. В одном альтернативном режиме эксплуатации как органическая жидкая фракция 33, так и водная жидкая фракция 34 покидают систему через клапан спуска донного отстоя сепаратора 42 и линию 43 перед разделением в декантаторе для измерения составов и расходов индивидуальных фаз. Во всех случаях альтернативные режимы эксплуатации не оказывают воздействия на исследуемые каталитические способы. Альтернативные режимы эксплуатации используются в качестве признанной разумной меры для достижения оптимального контроля способа в зависимости от относительных расходов газовой фазы 35, органической жидкой фазы 33 и водной фазы 34.
Пример 3
Фигура 10 демонстрирует технологическую схему, иллюстрирующую реакторную систему двумя питающими насосами, подходящую для использования при осуществлении на практике настоящего изобретения. Систему с двумя питающими насосами будут использовать, если желательная смесь компонентов исходного сырья не будет существовать в виде одной жидкой фазы. Например, два питающих насоса будут использовать в случае желательного исходного сырья в виде смеси 50% (масс.) 2-пентанола и 50% (масс.) воды- один для доставки 2-пентанола, а другой для доставки воды. Подобная система также может быть использована для перемешивания исходного сырья, произведенного из двух раздельных источников, таких как первичное исходное сырье и оксигенированное углеводородное исходное сырье, произведенное из отходящего потока самой реакторной системы.
Первый резервуар для исходного сырья 1 исполняет функцию резервуара для первого раствора исходного сырья, в то время как второй резервуар для исходного сырья 40 исполняет функцию резервуара для второго раствора исходного сырья. Первое исходное сырье доставляют из первого резервуара для исходного сырья 1 к первому питающему насосу 3 по линии подачи первого исходного сырья 2. Первое исходное сырье после этого перепускают по линии выгрузки первого питающего насоса 4 в линию подачи объединенного исходного сырья 44. Второе исходное сырье доставляют из второго резервуара для исходного сырья 40 во второй питающий насос 42 по линии подачи второго исходного сырья 41. Второе исходное сырье после этого перепускают по линии выгрузки второго питающего насоса 43 в линию подачи объединенного исходного сырья 44. Из линии подачи объединенного исходного сырья 44 объединенное исходное сырье перепускают в предварительный нагреватель 5. Все другие элементы представляют собой те, что были приведены в примере 1 при том исключении, что водная фаза 19 может быть отправлена на рецикл в резервуар для исходного сырья 40 в целях последующей переработки или использования в других способах.
Пример 4
Фигура 11 демонстрирует схематическую иллюстрацию одного типа реактора, который может быть использован в реакторных системах, описывавшихся в примерах 1, 2 и 3. Труба реактора 1 образована из нержавеющей стали 316 при наличии у нее либо внутреннего диаметра 8,5 мм, либо внутреннего диаметра 21,2 мм в зависимости от эксперимента. Предусматривают наличие линии впускного отверстия 2 для обеспечения поступления в реактор исходного сырья или промежуточного продукта, такого как оксигенаты. Предусматривают наличие линии выпускного отверстия 3 для удаления продукта из реактора. Функцию фиксации по месту слоев среды предварительного нагревания и катализатора исполняет фритта впускного отверстия 4, образованная из нержавеющей стали. Среда предварительного нагревания 5, состоящая из гранул из нержавеющей стали, исполняет функцию зоны обеспечения передачи тепла от стенок реактора для того, чтобы исходное сырье при поступлении на катализатор 7 имело бы желательную температуру. Для предотвращения перемешивания материалов между средой предварительного нагревания 5 и катализатором 7 может быть размещена сетка из нержавеющей стали. Катализатор 7 в своей позиции может опираться на вторую фритту из нержавеющей стали 8.
В некоторых случаях для обеспечения измерения температур в катализаторе 7 и зоне предварительного нагревания 5 может быть предусмотрен канал для ввода термопар 9. Контролируемое выдерживание температуры на впускном отверстии реактора осуществляют благодаря использованию внешнего предварительного нагревателя перед введением исходного сырья в реактор через линию 2, и может быть проведено дополнительное регулирование в результате контроля теплопередачи, которая имеет место в среде предварительного нагревания. В некоторых случаях для достижения желательного температурного профиля среда предварительного нагревания не требуется. Контролируемого выдерживания температуры стенки реактора добиваются при использовании внешних нагревателей, находящихся в контакте с внешней стенкой реактора. Для контролируемого выдерживания температуры стенки реактора при желании могут быть использованы независимо контролируемые зоны нагревания.
Пример 5 - Методики анализа
Потоки продуктов из описывающихся ниже примеров анализировали следующим образом. Органическую жидкую фазу собирали и анализировали при использовании газового хроматографа либо с масс-спектрометрическим детектированием, либо с пламенно-ионизационным детектированием. Разделения компонентов добивались при использовании колонки, содержащей связанную 100%-ную диметилполисилоксановую неподвижную фазу. Относительные концентрации индивидуальных компонентов оценивали в результате интегрирования пиков и деления их на сумму площадей пиков для всей хроматограммы. Соединения идентифицировали в результате сопоставления со стандартными временами удерживания и/или сопоставления масс-спектров с составленной базой данных по масс-спектрам. Составы газовой фазы определяли по методу газовой хроматографии при использовании детектора теплопроводности и пламенно-ионизационного или масс-спектрометрического детекторов для других компонентов газовой фазы. Водную фракцию анализировали по методу газовой хроматографии с использованием и без использования дериватизации органических компонентов фракции при помощи пламенно-ионизационного детектора. Выходы продуктов представляют количеством атомов углерода, присутствующих в каждой фракции продукта. Массовую часовую объемную скорость (МЧОС) определяли как массу исходного сырья, введенного в систему, на единицу массы катализатора в час и рассчитывали исходя только лишь из массы оксигенированного углеводородного исходного сырья при исключении воды, присутствующей в исходном сырье.
Получение оксигенатов
Пример 6 - Катализатор гидрирования
Катализатор гидрирования получали в результате добавления водного раствора растворенного нитрозилнитрата рутения к углеродному носителю катализатора (UU Carbon, Calgon, при размерах частиц, ограниченных теми, которые удерживались на сите 120 меш после прохождения через сито 60 меш) для получения целевого уровня загрузки 2,5% рутения. Воду добавляли в количестве, превышающем объем пор, и выпаривали в вакууме вплоть до получения сыпучего катализатора. После этого катализатор высушивали в течение ночи при 100°С в вакуумном сушильном шкафу.
Пример 7 - Катализатор РВФ/деоксигенирования
Объединенный катализатор РВФ и деоксигенирования получали в результате растворения гексахлороплатиновой кислоты и рениевой кислоты в воде, а после этого добавления смеси к носителю катализатора в виде моноклинного диоксида циркония (NorPro Saint-Gobain, Product code SZ31164, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) при использовании методики достижения начальной влажности для получения целевого уровня загрузки пластины 1,8% и уровня загрузки рения 6,3% на катализаторе после последующего разложения металлсодержащих предшественников. Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного воздуха при 400°С.
Пример 8 - Превращение сахарозы в оксигенаты
Системы катализаторов, на которые ссылались в примерах 6 и 7, исследовали на предмет превращения сахарозы в промежуточный продукт, содержащий оксигенаты, при использовании реакторной системы, описывавшейся в примере 1. Исследование проводили при использовании трубчатого реактора из нержавеющей стали с внутренним диаметром 21,2 мм, продемонстрированного в примере 4, и проведении анализа так, как это описывалось в примере 5.
В реактор загружали 31 грамм катализатора гидрирования из примера 6 и 76 граммов катализатора РВФ из примера 7, при этом катализатор гидрирования размещали поверх катализатора РВФ при их разделении сеткой из нержавеющей стали. Перед введением исходного сырья в реактор с исходным сырьем объединяли внешний водород. Нагреватели, внешние по отношению к реактору и продемонстрированные на фигуре 8 в позициях 10а, 10b, 10с, 10d, выдерживали при следующих далее температурах стенки реактора: 10а - 125°С, 10b - 200°С, 10с - 265°С, 10d - 265°С, что в результате приводило к получению температур слоев реактора, равных приблизительно ~110-150°С для гидрирования и 150-265°С для катализатора РВФ/деоксигенирования. Данные диапазоны указывают на приблизительные температуры стенки реактора на впускном отверстии и выпускном отверстии каждого слоя катализатора, соответственно. Результаты эксперимента через 39 часов эксплуатации продемонстрированы в таблице 1. Значение МЧОС рассчитывают исходя из массы катализатора РВФ/деоксигенирования. Совокупные монооксигенаты включают спирты, кетоны, тетрагидрофураны и циклические монооксигенаты. Циклические монооксигенаты включают соединения, у которых кольцо не включает кислород, такие как циклопентанон и циклогексанон. Долю атомов углерода исходного сырья, содержащихся в неизвестных компонентах водной фазы, определяли по разнице между количеством атомов углерода, обусловленным известными измеренными компонентами, и совокупным количеством атомов углерода органики.

Пример 9 - Катализатор РВФ/деоксигенирования
Катализатор получали так, как это описывалось в примере 7 за исключением использования носителя катализатора в виде тетрагонального диоксида циркония (NorPro Saint-Gobain, Product code SZ61152) при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш.
Пример 10-Катализатор РВФ/деоксигенирования
Гексахлороплатиновую кислоту и рениевую кислоту, растворенные в воде, добавляли к носителю катализатора в виде моноклинного диоксида циркония (NorPro Saint-Gobain, Product code SZ31164, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) при использовании методики достижения начальной влажности для получения целевых уровня загрузки платины 1,9% и уровня загрузки рения 1,8% на катализаторе после последующего разложения металлсодержащих предшественников. Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного воздуха при 400°С.
Пример 11 - Катализатор РВФ/деоксигенирования
Катализатор получали так, как это описывалось в примере 7 за исключением использования носителя в виде активированного угля, функционализованного пероксидом водорода. Сначала получали носитель в результате медленного добавления активированного угля (углерод Calgon UU, 60×120 меш) к 30%-ному раствору пероксида водорода, после этого смесь оставляли стоять в течение ночи. Водную фазу декантировали и углерод три раза промывали деионизованной водой, а после этого высушивали в вакууме при 100°С. Затем к носителю добавляли раствор гексахлороплатиновой кислоты и рениевой кислоты в воде при использовании методики достижения начальной влажности для получения целевых уровня загрузки платины 1,8% и уровня загрузки рения 6,3% после последующего разложения металлсодержащих предшественников. Препарат высушивали в течение ночи в вакуумном сушильном шкафу при 100°С.
Пример 12 - Превращение сорбита и глицерина
Системы катализаторов, на которые ссылались в примере 9, примере 10 и примере 11, исследовали на предмет превращения сорбита или глицерина в промежуточный продукт, содержащий оксигенаты, при использовании конфигурации реактора, описывавшейся в примере 1, и проведении анализа так, как это описывалось в примере 5. Исследование проводили при использовании трубчатого реактора из нержавеющей стали с внутренним диаметром 8,5 мм, продемонстрированного в примере 4. Во всех случаях давление реактора выдерживали равным 625 фунт/дюйм2 (изб.) (4310 кПа (изб.)). Температуры на впускном отверстии и выпускном отверстии реактора, продемонстрированные в таблице 2, контролируемо выдерживали при использовании нагревателей, внешних по отношению к реактору и продемонстрированных на фигуре 8 в позициях 10а, 10b, 10с, 10d. Результаты данных экспериментов продемонстрированы в таблице 2.
Таблица 2 демонстрирует воздействие состава катализатора, состава исходного сырья и условий эксплуатации на эксплуатационные характеристики по превращению. Фигура 12 демонстрирует распределение количества атомов углерода для монооксигенатов, полученных в эксперименте D и эксперименте Е. Основное различие между данными двумя экспериментами заключалось в температуре реакции. В случае эксперимента D преобладали монооксигенаты, содержащие три и менее атома углерода, в то время как в случае эксперимента Е значительная доля монооксигенатов содержала четыре и более атомов углерода, что свидетельствовало о прохождении реакций конденсации в той же самой реакционной зоне, что и в случае получения водорода и реакций деоксигенирования. Значение МЧОС получают исходя из массы катализатора РВФ/деоксигенирования. Результирующим полученным водородом является водород, присутствующий на выпускном отверстии в виде Н2, который не включает водород, полученный и израсходованный «по месту». Совокупные монооксигенаты включают спирты, кетоны, тетрагидрофураны и циклические монооксигенаты. Циклические монооксигенаты включают соединения, у которых кольцо не включает кислород, такие как циклопентанон и циклогексанон. Долю атомов углерода исходного сырья, содержащихся в неизвестных компонентах в водной фазе, определяли по разнице между количеством атомов углерода, обусловленным известными измеренными компонентами, и совокупным количеством атомов углерода органики.
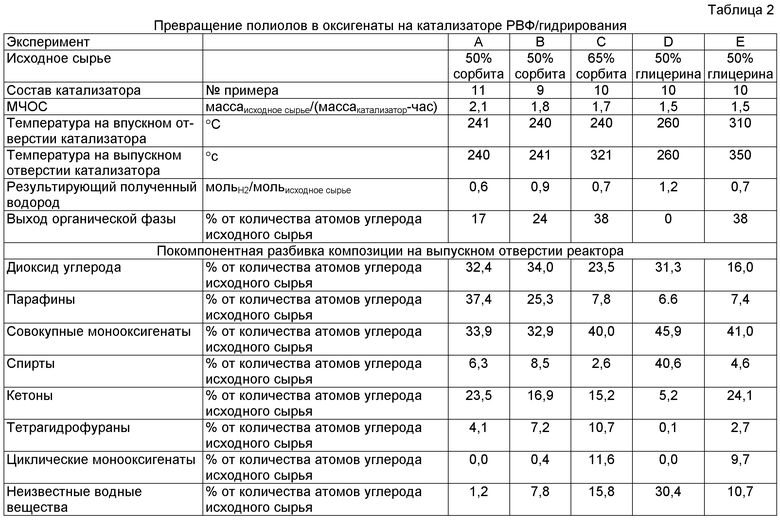
Конденсация оксигенатов при использовании основных катализаторов
Пример 13
Носитель катализатора в виде алюмината цинка получали в результате перемешивания порошкообразного оксида цинка и порошкообразного оксида алюминия (Dispal 18N4-80, Sasol North America, Houston, Texas) для достижения целевого соотношения между 1,0 моля ZnO и 1 молем Al2O3. После этого к оксиду алюминия добавляли разбавленную азотную кислоту при уровне содержания 1% (масс.) HNO3. Консистенцию тестообразной массы смеси регулировали в результате добавления воды для получения удобнообрабатываемой тестообразной массы, которую после этого экструдировали при использовании экструдера лабораторного масштаба. Экструдаты высушивали в течение ночи в вакууме при 100°С, после этого дополнительно высушивали при 200°С в течение одного часа в проточном воздухе, а впоследствии затем прокаливали при 750°С в течение 4 часов в проточном воздухе. Получающийся в результате материал после этого размалывали и просеивали. Извлекали материал, который удерживался на сите 60 меш после прохождения через сито 18 меш.
Пример 14
К прокаленному материалу из примера 13 добавляли гексахлороплатиновую кислоту при использовании методики импрегнирования до достижения начальной влажности для получения целевого уровня загрузки платины 1,0% (масс.). Катализатор высушивали в течение ночи в вакууме при 100°С и прокаливали при 400°С в проточном воздухе.
Пример 15
К прокаленному материалу из примера 13 добавляли нитрат палладия при использовании методики импрегнирования до достижения начальной влажности для получения целевого уровня загрузки палладия 0,5% (масс.). Катализатор высушивали в течение ночи в вакууме при 100°С и прокаливали при 400°С в проточном воздухе.
Пример 16
Катализатор медь-алюминат цинка получали в результате перемешивания оксида цинка, оксида меди (I) и порошкообразного оксида алюминия (Dispal 18N4-80) при целевом соотношении между 0,11 моль CuO и 0,9 моль ZnO с одной стороны и одним молем Al2O3 с другой. После этого к оксиду алюминия добавляли разбавленную азотную кислоту при уровне содержания 1% (масс.) HNO3. Консистенцию тестообразной массы смеси регулировали в результате добавления воды для получения удобнообрабатываемой тестообразной массы, которую после этого экструдировали при использовании экструдера лабораторного масштаба. Экструдаты высушивали в течение ночи в вакууме при 100°С, после этого дополнительно высушивали при 200°С в течение одного часа в проточном воздухе, а впоследствии затем прокаливали при 750°С в течение 4 часов в проточном воздухе. Получающийся в результате материал после этого размалывали и просеивали. Извлекали материал, который удерживался на сите 60 меш после прохождения через сито 18 меш.
Пример 17
Катализатор в виде диоксида кремния-оксида алюминия, модифицированных цезием, получали в результате добавления карбоната цезия, растворенного в воде, к носителю катализатора в виде диоксида кремния-оксида алюминия Siralox (Sasol North America, Houston, Texas). Целевой уровень загрузки цезия составлял 25% (масс.) в расчете на конечную массу катализатора. Данный материал высушивали в течение 24 часов в вакууме при 100°С и прокаливали при 500°С в течение 6 часов в проточном воздухе. После прокаливания добавляли платину при использовании методики импрегнирования до достижения начальной влажности для получения конечного уровня загрузки платины 1% (масс.). После импрегнирования катализатор высушивали, а после этого прокаливали при 500°С в течение 6 часов в проточном воздухе.
Пример 18
Диоксид кремния, модифицированный церием, получали в результате добавления раствора нитрата церия к гелю кремниевой кислоты (Davisil grade 636, WR Grace Company) для достижения конечного уровня загрузки 25% (масс.) CeO2. После этого получающийся в результате материал высушивали при 120°С в течение шести часов и дополнительно прокаливали при 550°С в течение шести часов в проточном воздухе. К прокаленному материалу добавляли нитрат палладия при использовании методики импрегнирования до достижения начальной влажности для получения целевого уровня загрузки палладия 0,5% (масс.). После этого данный материал высушивали при 120°С в течение шести часов и дополнительно прокаливали при 550°С в течение шести часов в проточном воздухе.
Пример 19
Системы катализаторов, на которые ссылались в примерах 14-18, исследовали на предмет конденсации в паровой фазе различных оксигенатов. Исследования проводили при использовании трубчатых реакторов из нержавеющей стали с величинами внутренних диаметров 8,5 мм и 21,2 мм, описывавшихся в примере 4, и в реакторных системах, проиллюстрированных на фигурах 8 и 10. В меньший реактор загружали от 15 до 18 миллилитров катализатора и от 50 до 70 миллилитров катализатора загружали в больший реактор. Во всех случаях катализатор перед использованием восстанавливали при 400°С в проточном водороде.
Органическую жидкую фазу собирали и анализировали так, как это описывалось в примере 5. Таблица 3 демонстрирует выходы и состав органических продуктов в зависимости от условий эксплуатации, состава исходного сырья и добавленного металлсодержащего компонента для катализаторов, описывавшихся в приведенных выше примерах 14-18. Более чем 100%-ные приведенные выходы органической фазы, возникают вследствие экспериментальной погрешности при измерении расходов или состава технологического потока. Неконденсированными компонентами являются те компоненты, которые не требуют образования новых связей углерод-углерод для получения из заданного исходного сырья. Для простоты все соединения, содержащие пять и менее атомов углерода, считаются неконденсированными компонентами. Совокупные продукты конденсации представляют собой те соединения, которые содержат шесть и более атомов углерода, которые требуют образования новых связей углерод-углерод для получения из заданного исходного сырья.
Как демонстрируют эксперименты F и G, на селективность по продукту может оказывать воздействие выбор гидрирующей функции, например, Pt или Pd. Парафины в большей степени образовывались на катализаторе, содержащем 1% платины, в сопоставлении с тем, что имело место для катализатора, содержащего 0,5% палладия. Последний благоприятствовал образованию монооксигенатов, главным образом кетонов. Эксперименты Н и I дополнительно подтверждают данную концепцию. Как демонстрирует эксперимент Н, при использовании в качестве исходного сырья изопропилового спирта с высоким выходом могут быть получены конденсированные монооксигенатные компоненты, составляющие >97% органического продукта и содержащие >90% от совокупного количества атомов углерода на впускном отверстии реактора. В результате увеличения температуры реакции и использования меди для стимулирования прохождения реакций гидрирования селективность может быть смещена к получению значительного выхода олефинов (эксперимент I). Как демонстрируют эксперименты J, К и L, для промотирования конденсации оксигенатов с последующим гидрированием продуктов первоначальной конденсации могут быть использованы несколько других гетерогенных катализаторов. Как демонстрируют эксперименты К и L, по мере уменьшения температуры от 300°С до 250°С скорость конденсации уменьшается, так что в получающейся в результате органической фазе степень превращения в конденсированные продукты понижается от 81% (масс.) до 18% (масс.).
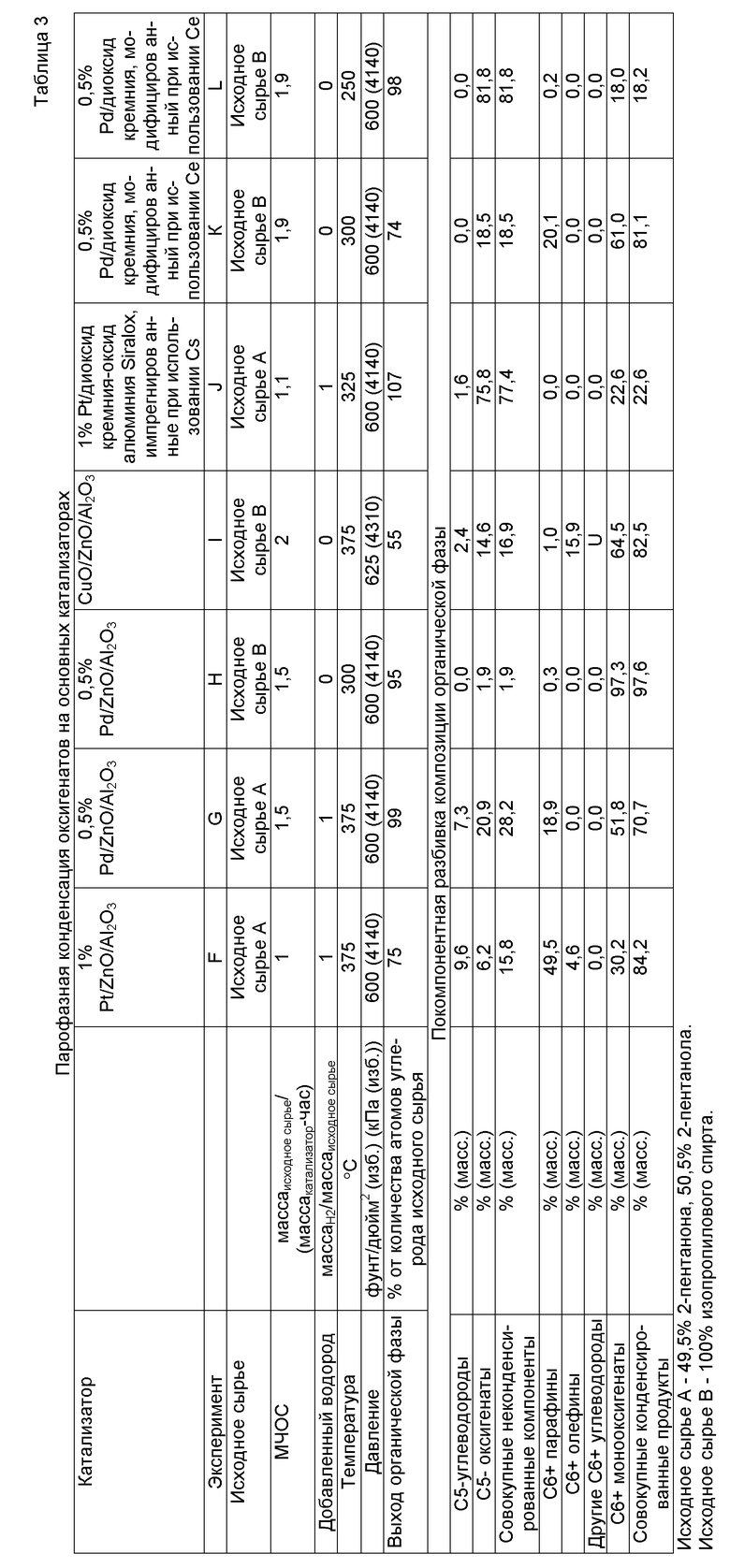
Конденсация оксигенатов при использовании кислотно-основных катализаторов
Пример 20
Гидроталькитовый катализатор получали из коммерчески доступного гидроталькитового носителя (ESM-350, ASM Catalysts, Baton Rouge, LA) в результате размалывания материала и перепускания его через градуированные сита для достижения размеров частиц, больших, чем 60 меш, и меньших, чем 18 меш. После этого материал прокаливали в кварцевом трубчатом реакторе при 450°С в течение 6 часов в проточном азоте.
Пример 21
К гидроталькитовому катализатору из примера 20 добавляли платину при использовании методики импрегнирования до достижения начальной влажности для получения конечного целевого уровня загрузки платины 1% (масс.). Платиносодержащий предшественник представлял собой гексахлороплатиновую кислоту H2PtCl6. Импрегнированный материал высушивали в течение ночи в вакууме при 100°С, а после этого прокаливали при 400°С в течение 2 часов в проточном воздухе.
Пример 22
К гидроталькитовому катализатору из примера 20 добавляли платину и олово при использовании методики импрегнирования до достижения начальной влажности для получения конечных целевых уровней загрузки 1% (масс.) Pt и 0,2% (масс.) Sn. Платиносодержащий предшественник представлял собой гексахлороплатиновую кислоту H2PtCl6, в то время как олово производили из хлорида олова SnCl2·2H2O. Импрегнированный материал высушивали в течение ночи в вакууме при 100°С, а после этого прокаливали при 450°С в течение 8 часов в проточном азоте.
Пример 23
Содержащий 5% оксида магния катализатор, нанесенный на гранулированный диоксид циркония, получали при использовании методики импрегнирования до достижения начальной влажности для получения конечного целевого уровня загрузки 5% (масс.) Mg. Магний добавляли в виде нитрата магния и высушивали в течение ночи в вакууме при 100°С, а после этого прокаливали при 450°С в течение 8 часов в проточном воздухе. К прокаленному материалу добавляли водный раствор нитрата палладия для получения целевого уровня загрузки палладия 0,5% (масс.) при использовании методики импрегнирования до достижения начальной влажности. Катализатор высушивали во второй раз и прокаливали при 400°С в течение шести часов в проточном воздухе.
Пример 24
Носитель катализатора в виде алюмината цинка получали в результате перемешивания порошкообразного оксида цинка и порошкообразного оксида алюминия (Dispal 18N4-80, Sasol North America, Houston, Texas) для достижения целевого соотношения между 0,85 моля ZnO и 1 молем Al2O3. К совокупному твердому веществу добавляли разбавленную азотную кислоту при уровне содержания 1% (масс.) HNO3. Консистенцию тестообразной массы регулировали в результате добавления воды для получения удобнообрабатываемой тестообразной массы, подходящей для экструдирования, и смесь экструдировали при использовании экструдера лабораторного масштаба. Экструдаты высушивали в течение ночи в вакууме при 100°С, а после этого прокаливали при 750°С в течение 8 часов в проточном воздухе. После этого получали материал с размерами 18 на 60 меш. К прокаленному материалу добавляли водный раствор нитрата палладия для получения целевого уровня загрузки палладия 0,5% (масс.) при использовании методики импрегнирования до достижения начальной влажности. После этого данный катализатор высушивали во второй раз и прокаливали при 400°С в течение шести часов в проточном воздухе.
Пример 25
Системы катализаторов, на которые ссылались в примерах 21-24, использовали при проведении реакций парофазной конденсации для различных оксигенатов. Исследования проводили при использовании трубчатых реакторов из нержавеющей стали с величинами внутренних диаметров 8,5 мм и 21,2 мм, описывавшихся в примере 4, и реакторных систем, проиллюстрированных в примерах 1 и 3. В меньший реактор загружали от 15 до 18 миллилитров катализатора и от 50 до 70 миллилитров катализатора загружали в больший реактор. Во всех случаях катализатор перед использованием восстанавливали при 400°С в проточном водороде.
Органическую жидкую фазу собирали и анализировали так, как это описывалось в примере 5. Таблица 4 демонстрирует выходы и состав органических продуктов в зависимости от условий эксплуатации, состава исходного сырья и добавленного металлсодержащего компонента для гидроталькитовых катализаторов, описывавшихся в приведенных выше примерах 21 и 22. Как демонстрируют данные экспериментов, главным образом углеводородный продукт может быть получен из ацетона и изопропилового спирта в отсутствие добавленного металлсодержащего гидрирующего компонента. В эксперименте М продукт органической фазы содержал главным образом девятиуглеродные метилзамещенные циклогексены, отнесенные в таблице 4 к категории других С6+ углеводородов. Добавление к данному катализатору платины (эксперимент N) благоприятствовало образованию конденсированных монооксигенатных продуктов, главным образом кетонов и спиртов, и образованию некоторых парафинов в результате деоксигенирования кетонов и спиртов. Селективность дополнительно смещали в пользу конденсированных монооксигенатов в результате разбавления платины оловом и проведения эксплуатации при более высоком давлении (эксперимент О). Эксперименты Р, Q, R и S иллюстрируют воздействие температуры реакции для конденсации смешанного исходного сырья, содержащего пентанол и пентанон. По мере увеличения температуры реакции от 300°С до 375°С становилось очевидным постепенное изменение состава продуктов при уменьшении селективности по конденсированным монооксигенатам и увеличении селективности по конденсированным парафинам с ростом температуры.
Таблица 5 демонстрирует воздействие компонентов исходного сырья и температуры реакции на выходы и состав органических продуктов для катализаторов из примеров 23 и 24. Эксперименты Т и U сопоставляют конденсацию 2-петанона и 2-метилтетрагидрофурана. В целом конденсация 2-пентанона протекает быстрее, чем конденсация 2-метилтетрагидрофурана. Тем не менее, в данных условиях в продукты конденсации превращалось приблизительно 30% тетрагидрофурана. Эксперименты 10 и 11 демонстрируют воздействие температуры реакции в случае использования исходного сырья в виде чистого изопропилового спирта. При 300°С (эксперимент V) преобладают монооксигенированные продукты конденсации, в то время как при 400°С (эксперимент W) значительная часть продуктов состояла из углеводородов. В сопоставлении с другими экспериментами, перечисленными в таблицах 4 и 5, эксперимент W примечателен тем, что органический продукт характеризовался повышенным уровнем содержания олефинов. Добавление к исходному сырью валериановой кислоты (эксперимент X) подавляло совокупные скорости конденсации и смещало селективность от парафинов к другим углеводородам, главным образом замещенным арильным соединениям.
Более чем 100%-ные приведенные выходы органической фазы, возникают вследствие экспериментальной погрешности при измерении расходов или состава технологического потока. Неконденсированными компонентами являются те компоненты, которые не требуют образования новых связей углерод-углерод для получения из заданного исходного сырья. Для простоты все соединения, содержащие пять и менее атомов углерода, считаются неконденсированными компонентами. Совокупные продукты конденсации представляют собой те соединения, которые содержат шесть и более атомов углерода, которые требуют образования новых связей углерод-углерод для получения из заданного исходного сырья.
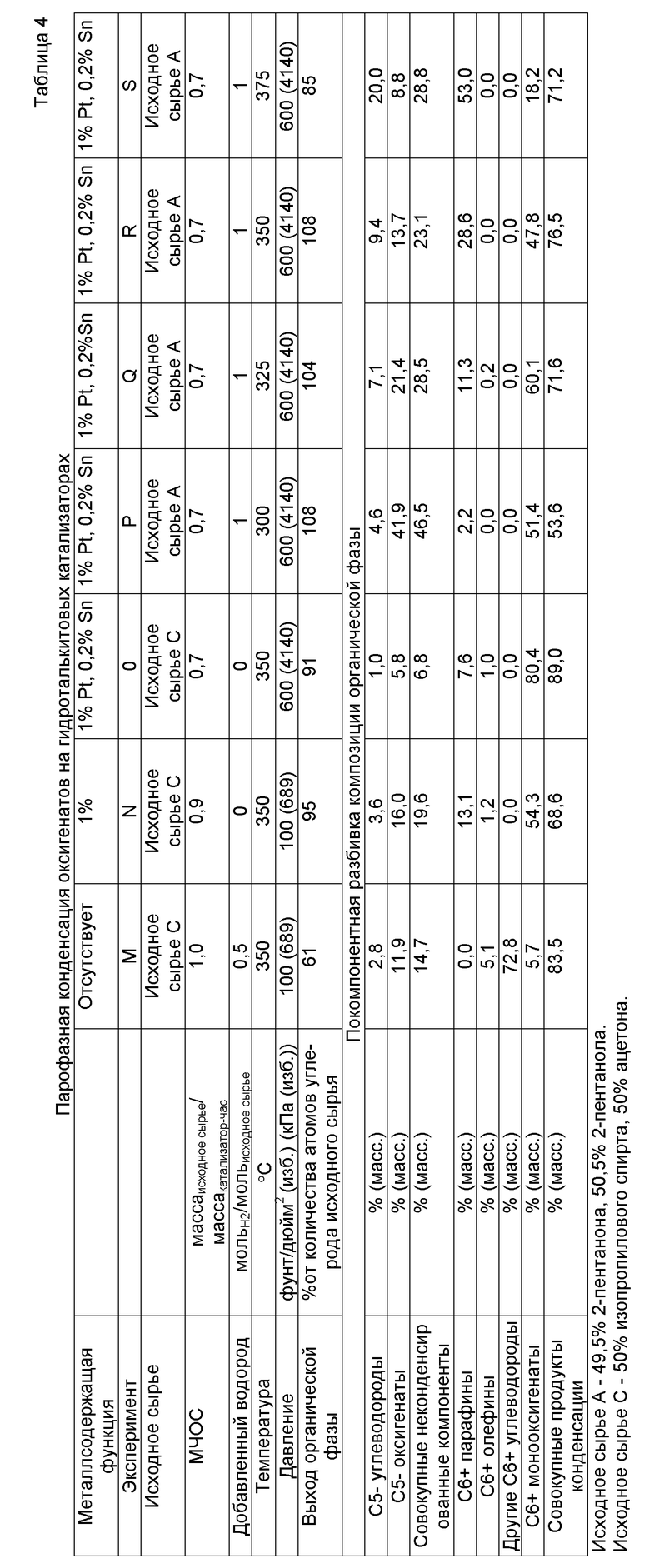
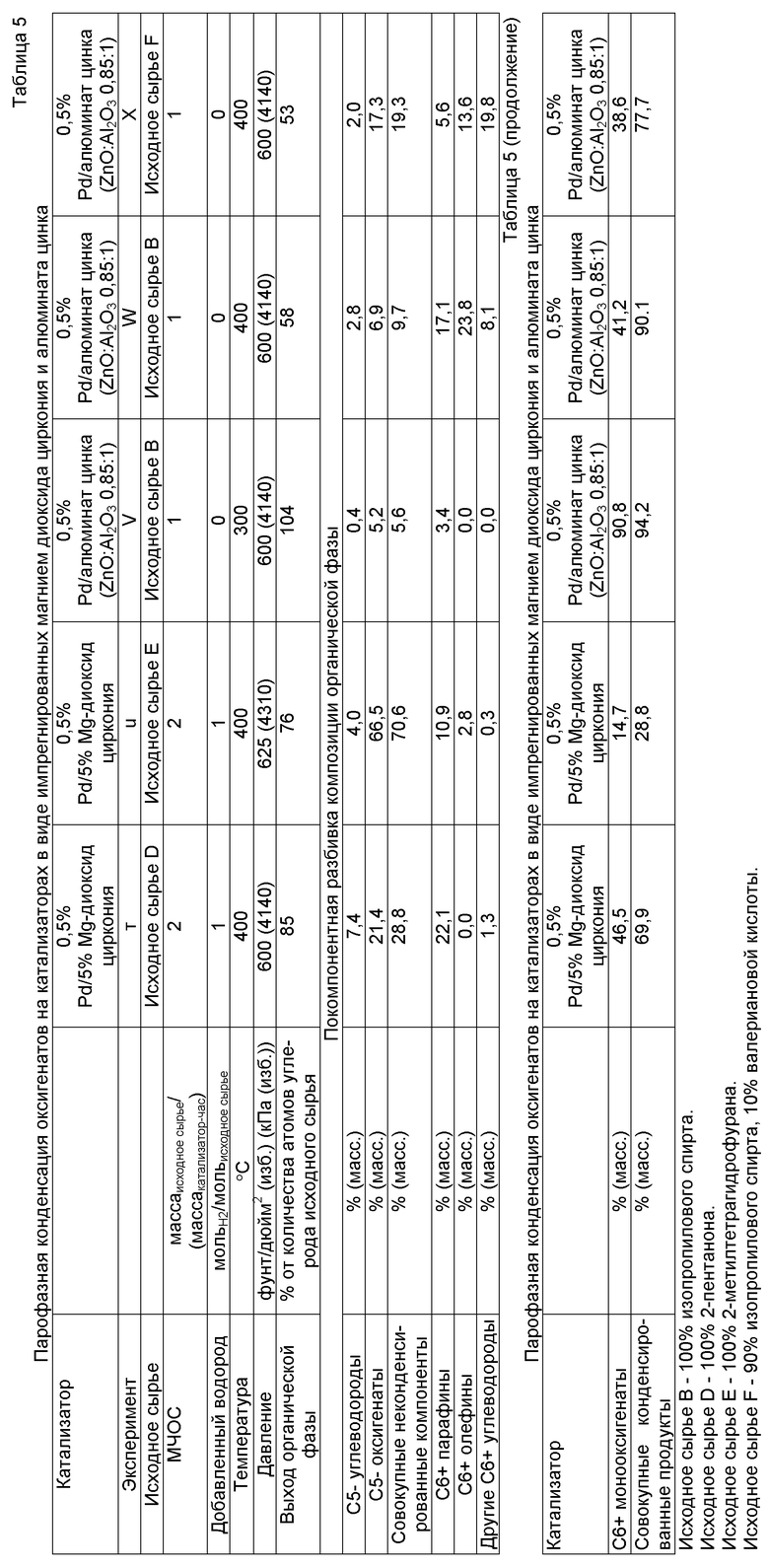
Основная конденсация оксигенатов с последующим деоксигенированием
Пример 26
Носитель катализатора в виде алюмината цинка получали подобно тому, что имеет место в примере 13 за исключением уменьшения количества оксида цинка для получения целевого соотношения между 0,85 моля ZnO и 1 молем Al2O3.
Пример 27
К прокаленному материалу из примера 26 добавляли гексахлороплатиновую кислоту при использовании методики импрегнирования до достижения начальной влажности для получения целевого уровня загрузки платины 1,0% (масс.). Катализатор высушивали в течение ночи в вакууме при 100°С и прокаливали при 400°С в проточном воздухе.
Пример 28
Системы катализаторов, на которые ссылались в примерах 27 и 15, исследовали на предмет парофазной конденсации различных оксигенатов и последующего их превращения в углеводороды. Исследования проводили при использовании трубчатых реакторов из нержавеющей стали с величиной внутреннего диаметра 21,2 мм, описывавшихся в примере 4, и реакторных систем, проиллюстрированных в примерах 2 и 3. В два раздельных реактора загружали приблизительно 100 миллилитров каждого катализатора. Два реактора компоновали таким образом, чтобы отходящий поток из первого реактора проходил бы во второй реактор. Первый реактор содержал катализатор из примера 15, а второй реактор содержал катализатор из примера 27. Катализатор перед использованием восстанавливали при 400°С в проточном водороде. Во всех случаях водород перед введением в реактор объединяли с исходным сырьем.
Продукты разделяли и анализировали так, как это описывалось в примере 5. Таблица 6 демонстрирует выходы и состав органических продуктов в зависимости от условий эксплуатации и состава исходного сырья при получении по последовательным реакциям. Неконденсированными компонентами являются те компоненты, которые не требуют образования новых связей углерод-углерод для получения из заданного исходного сырья. Для простоты все соединения, содержащие пять и менее атомов углерода, считаются неконденсированными компонентами. Совокупные продукты конденсации представляют собой те соединения, которые содержат шесть и более атомов углерода, которые требуют образования новых связей углерод-углерод для получения из заданного исходного сырья.
Как демонстрируют эксперименты АА, ВВ, СС и DD, в последовательных реакциях конденсации и деоксигенирования для получения продукта, содержащего главным образом С6+ алканы, могут быть использованы различные оксигенаты. Продукты характеризовались содержанием большей фракции алканов и низкими уровнями содержания оксигенированных соединений в сопоставлении с результатами, продемонстрированными в таблице 3. Это демонстрирует то, что использование катализаторов, характеризующихся различными функциональностями, (то есть, основный+гидрирующий катализатор в первом реакторе с последующим кислотным+основным+гидрирующим катализатором во втором реакторе) может быть более эффективным для получения углеводородов из оксигенированных соединений в сопоставлении с использованием катализатора, который содержит только основную и гидрирующую функциональность. В эксперименте ЕЕ органический продукт, полученный в экспериментах от АА до DD, отправляли на рецикл через реакционную систему. После данной обработки конечный продукт содержал главным образом алканы при всего лишь следовых количествах кислородсодержащих компонентов. Таким образом полученные углеводороды были бы ценны для использования в качестве жидких топлив, таких как бензин, дизельное топливо и реактивное топливо.
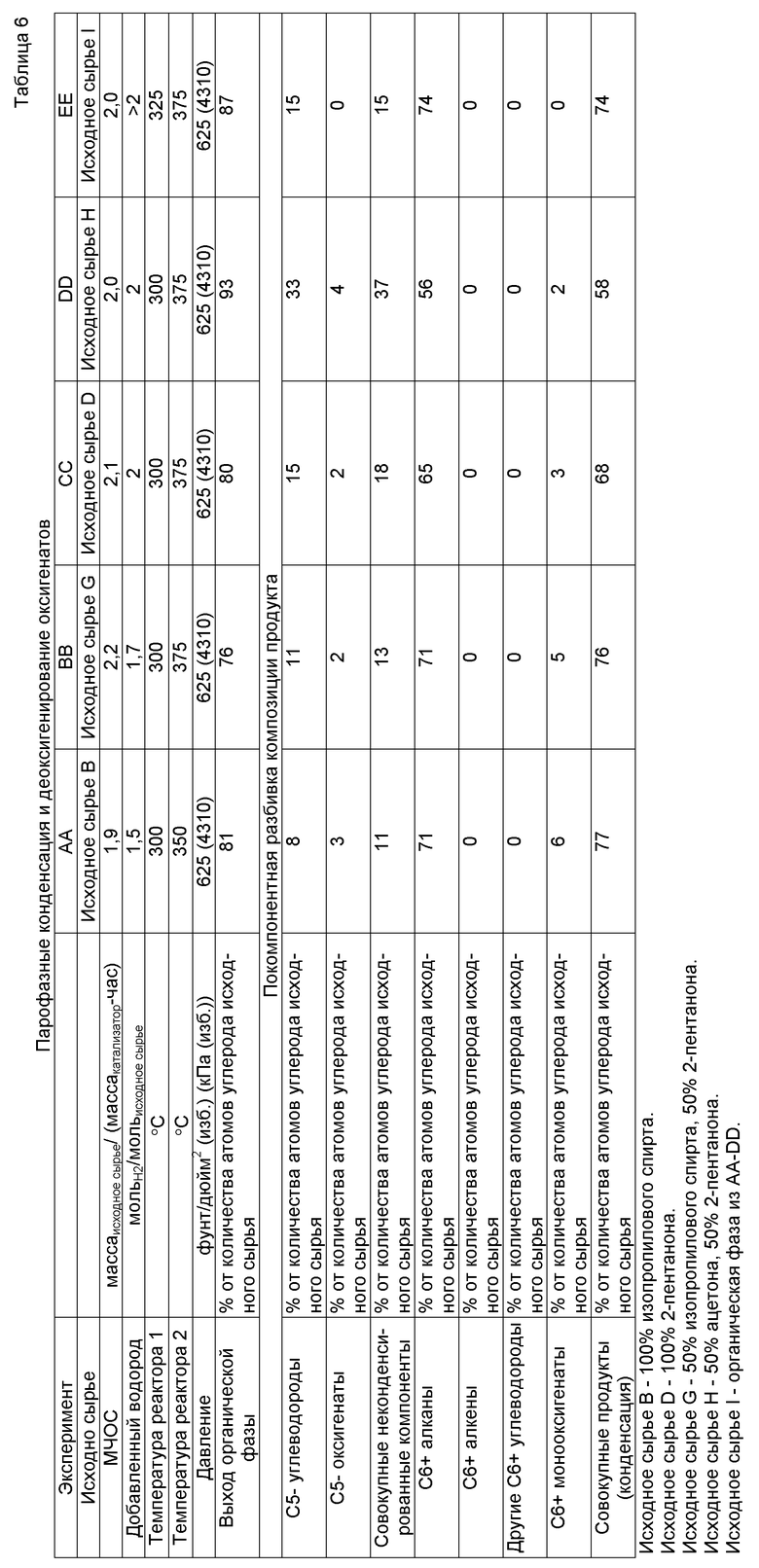
Фракционирование продуктов
Пример 29
Материал из эксперимента ЕЕ из примера 28 собирали и подвергали воздействию стадии перегонки. Перегонку проводили при атмосферном давлении с использованием простого одноступенчатого лабораторного периодического перегонного аппарата. 2,950 литра жидкого продукта добавляли в обогреваемую круглодонную колбу, которая исполняла функцию кипятильника в начале эксперимента. Верхний продукт конденсировали и сегрегировали на раздельные образцы на основании температуры паровой фазы, находящейся в равновесии с кипящей жидкостью, при проведении анализа фракций так, как это описывалось в примере 5. Распределение количеств атомов углерода для фракций продуктов продемонстрировано в таблице 7. Все фракции содержали главным образом алканы.
Фракции, извлеченные при температуре кипения, меньшей, чем 150°С, содержат алканы главным образом в С5-10 диапазоне и были бы подходящими для использования в качестве компонента бензиновой смеси. Материалы из диапазона более высоких температур кипения потенциально могли бы быть подходящими для введения в дистиллятные топлива, керосин и дизельное топливо.
Пример 30
Перегнанный продукт, кипящий в диапазоне от 150°С до 250°С, анализировали на предмет пригодности в качестве реактивного топлива в коммерческой службе по испытаниям (Intertek Testing Services, Illinois) в соответствии с методом испытания ASTM D1655. Образец успешно прошел испытания по всем необходимым техническим условиям за исключением технических условий по температуре вспышки и плотности. Вероятно, технические условия по температуре вспышки могли бы быть удовлетворены благодаря использованию улучшенной перегонки продуктов, в то время как низкая плотность могла бы быть приписана высоким уровням содержания алканов в образце.
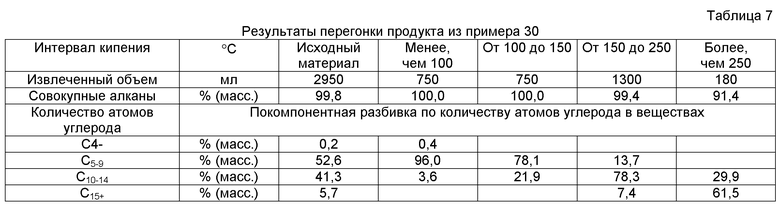
Получение С5+ соединений из глицерина при использовании одной каталитической системы
Пример 31
Систему биметаллического катализатора, содержащую платину и рений (5% (масс.) платины при молярном соотношении Pt:Re 1:2,5) и нанесенную на носитель в виде активированного угля (углерод Calgon UU, 60×120 меш), получали при использовании методик достижения начальной влажности. Активированный уголь медленно добавляли 30%-ному раствору пероксида водорода. После завершения добавления углерода смесь оставляли стоять в течение ночи. Водную фазу декантировали и углерод три раза промывали деионизованной водой, а после этого высушивали в вакууме при 100°С. При перемешивании по каплям на углерод, функционализованный пероксидом водорода, наносили водный раствор, имеющий объем, равный объему достижения начальной влажности для импрегнируемого углерода 10,4 мл и содержащий раствор гексагидрата гексахлороплатината (IV) диводорода (Alfa Aesar, 39,85% Pt) и рениевой кислоты (Alfa Aesar, 76,41% HReO4). Увлажненный углерод высушивали при 100°С в вакууме.
Пример 32
104,4 грамма катализатора на основе Pt/Re, 1:2,5 загружали в трубку реактора длиной 63,5 см, описывавшуюся в примере 4 и примере 1 при том исключении, что температурный профиль контролируемо выдерживали в результате теплообмена с потоком горячего воздуха, создаваемым воздуходувкой и нагревателем так, как это проиллюстрировано на фигуре 7. Перед введением жидкого исходного сырья в слой катализатора катализатор восстанавливали в проточном водороде при 350°С в течение двух часов. 50% (масс.) глицерина (Colgate Palmolive USP Grade), содержащего приблизительно 20 ч./млн. сульфата в водном растворе, сверху вниз подавали по реактору после предварительного нагревания до 182°С при массовой часовой объемной скорости 0,97 грамма глицерина на один грамм катализатора в час. Снизу вверх через кольцевое пространство при 409°С подавали горячий воздух. Осевой температурный профиль в центре слоя катализатора измеряли при использовании выдвижной термопары так, как это продемонстрировано в примере 4 и проиллюстрировано на фигуре 13. Давление сепаратора выдерживали равным 600 фунт/дюйм2 (изб.) (4140 кПа (изб.)). Отходящий поток из реактора охлаждали при использовании конденсатора с водяным охлаждением и разделяли в трехфазном сепараторе. Продукты газовой фазы анализировали при использовании газового хроматографа, который позволял проводить анализ по водороду, диоксиду углерода, метану, этану, пропану, бутану, пентану и гексану. Органическую фазу собирали, взвешивали и отправляли в компанию Southwest Research Institute (San Antonio, Texas) для анализа бензина. Водную фазу собирали и взвешивали, а после этого анализировали при использовании методов как ГХМС, так и ГХ-ПИД. В данной системе имело место полное превращение глицерина. Приведенная ниже таблица 8 демонстрирует выходы водорода, а также выходы соединений углеродсодержащих продуктов.

Получение С5+ соединений из сахароспиртов
Пример 33
Эксперименты проводили при использовании водных растворов оксигенированных углеводородов (например, смесь 50% (масс.) глицерин/вода или смесь 50% (масс.) сорбит/вода), введенных в реакторную систему из примера 1. Исходное сырье дополнительно модифицировали в результате добавления K2SO4 при различных концентрациях (1, 20 или 50 ч./млн.).
Пример 34
В трубу реактора из нержавеющей стали на 8,5 мм, описывавшуюся в примере 4, загружали в совокупности 10,61 грамма катализатора на основе Pt/Re, 1:2,5. Перед введением жидкого исходного сырья в слой катализатора катализатор восстанавливали в проточном водороде при 350°С в течение двух часов. Раствор глицерина с концентрацией 50% (масс.), содержащий приблизительно 1 ч./млн. сульфата в водном растворе, сверху вниз подавали по реактору при значении МЧОС 1,24 грамма глицерина на один грамм катализатора в час. Последующие испытания проводили при добавлении 20 ч./млн. и 50 ч./млн. сульфата в виде K2SO4. Нагреватели блоков контролируемо выдерживали при 260°С, а давление сепаратора выдерживали равным 600 фунт/дюйм2 (изб.) (4140 кПа (изб.)).
Органическую фазу собирали из сепаратора, взвешивали и анализировали по методу ГХ-МС так, как это описывалось в примере 5. Приведенная ниже таблица 9 демонстрирует выходы водорода, а также выходы соединений углеродсодержащих продуктов при различных количествах сульфата, добавленного в систему. В данной системе имело место полное превращение глицерина. Как демонстрирует таблица, жидкая органическая фаза образовывалась при добавлении сульфата в количестве, большем, чем 20 ч./млн.
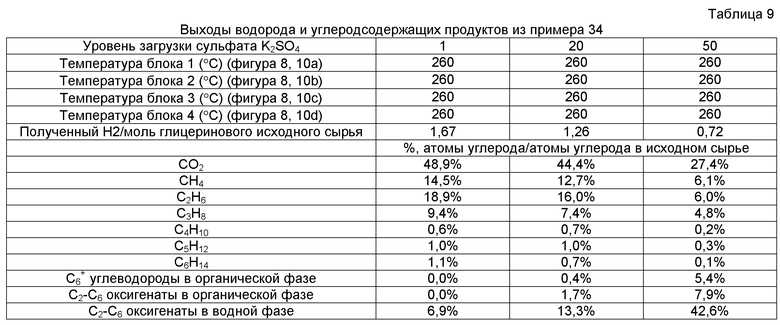
Пример 35
В трубу реактора из нержавеющей стали на 8,5 мм, описывавшуюся в примере 4, и реакторную систему, проиллюстрированную в примере 1, загружали в совокупности 10,61 грамма катализатора на основе Pt/Re, 1:2,5. Перед введением жидкого исходного сырья в слой катализатора катализатор восстанавливали в проточном водороде при 350°С в течение двух часов. Раствор глицерина с концентрацией 50% (масс.), содержащий либо 1 ч./млн., либо 20 ч./млн. сульфата в воде, сверху вниз подавали по реактору при значении МЧОС 1,24 грамма глицерина на один грамм катализатора в час. Нагреватели блоков контролировали таким образом, чтобы первые 10,1 см реактора выдерживались бы при 260°С, вторые 10,1 см реактора выдерживались бы при приблизительно 306°С, последующие 10,1 см реактора выдерживались бы при приблизительно 355°С, а последние 10,1 см реактора выдерживались бы при 400°С. Давление сепаратора выдерживали равным 600 фунт/дюйм2 (изб.) (4140 кПа (изб.)).
Отходящий поток из реактора охлаждали при использовании охлаждаемого водой конденсатора, разделяли в трехфазном сепараторе, а после этого анализировали так, как это описывалось в примере 5. В данной системе имело место полное превращение глицерина. Приведенная ниже таблица 10 демонстрирует выходы водорода, а также выходы соединений углеродсодержащих продуктов.
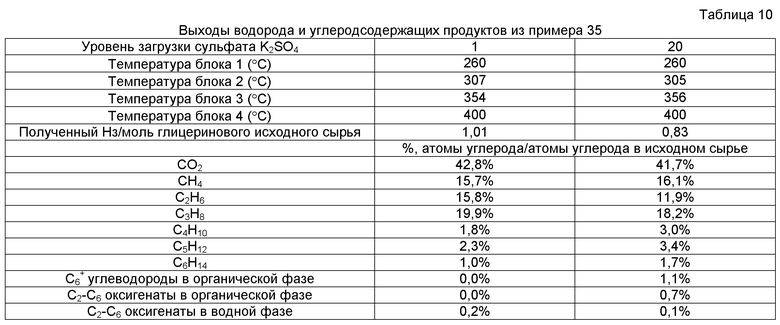
Пример 36
Систему биметаллического катализатора, содержащую платину и рений (5% (масс.) платины при молярном соотношении Pt: Re I: 5) и нанесенную на носитель в виде активированного угля (углерод Calgon UU, 60×120 меш), получали при использовании методики достижения начальной влажности. Активированный уголь медленно добавляли 30%-ному раствору пероксида водорода. После завершения добавления углерода смесь оставляли стоять в течение ночи. Водную фазу декантировали и углерод три раза промывали деионизованной водой, а после этого высушивали в вакууме при 100°С. При перемешивании по каплям на углерод, функционализованный пероксидом водорода, наносили водный раствор, имеющий объем, равный объему достижения начальной влажности для импрегнируемого углерода, и содержащий раствор гексагидрата гексахлороплатината (IV) диводорода (Alfa Aesar, 39,85% Pt) и рениевой кислоты (Alfa Aesar, 76,41% HReO4). Увлажненный углерод после этого высушивали при 100°С в вакууме.
Пример 37
11,97 грамма катализатора на основе Pt/Re, 1:5, описывавшегося в примере 36, загружали в трубку из нержавеющей стали с диаметром 8,5 мм, описывавшуюся в примере 4, и реакторную систему, проиллюстрированную в примере 1. Перед введением жидкого исходного сырья в слой катализатора катализатор восстанавливали в проточном водороде при 350°С в течение двух часов. Раствор сорбита с концентрацией 57,2% (масс.), содержащий 0 ч./млн. сульфата в водном растворе, сверху вниз подавали по реактору при значении МЧОС 1,20 грамма сорбита на один грамм катализатора в час. Нагреватели блоков контролировали таким образом, чтобы первые 10,1 см реактора выдерживались бы при 260°С, вторые 10,1 см реактора выдерживались бы при приблизительно 260°С, последующие 10,1 см реактора выдерживались бы при приблизительно 360°С, а последние 10,1 см реактора выдерживались бы при 410°С. Давление сепаратора выдерживали равным 600 фунт/дюйм2 (изб.) (4140 кПа (изб.)). Отходящий поток из реактора охлаждали при использовании охлаждаемого водой конденсатора и разделяли в трехфазном сепараторе. Фракции продукта анализировали так, как это описывалось в примере 5. В дополнение к этому, органическую фазу собирали, разделяли и взвешивали для отправления образца в компанию Southwest Research Institute (San Antonio, Texas) для анализа бензина. В данной системе имело место полное превращение глицерина. Приведенная ниже таблица 11 демонстрирует выходы водорода, а также выходы соединений углеродсодержащих продуктов.
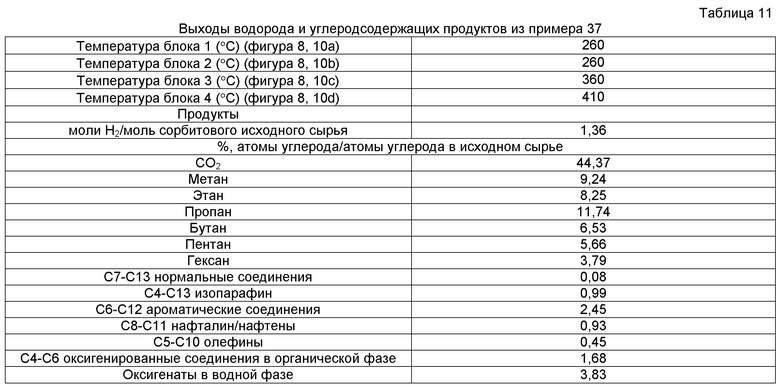
Превращение оксигенатов в С5+ соединения при использовании кислотных соединений
Пример 38
Получали водный 1,0-молярный раствор нитрата лантана и добавляли его к экструдатам Н-морденита (BASF 712A-5-2641-1) для получения целевого уровня загрузки 3% (масс.) La на катализаторе после последующего разложения металлсодержащего предшественника. Раствор La в течение короткого времени перемешивали с катализатором, а после этого добивались пропитывания при 80°С в течение 6 часов. После этого избыточную жидкость удаляли, а катализатор промывали деионизованной водой. Затем катализатор высушивали в вакуумном сушильном шкафу и прокаливали на воздухе при 550°С. После этого катализатор размалывали и просеивали для ограничения размеров частиц теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш.
Пример 39
К экструдатам Н-морденита (BASF 712A-5-2641-1, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) добавляли деионизованную воду вплоть до покрытия носителя избыточной водой. После этого к влажному носителю добавляли водный 0,36-молярный раствор нитрата никеля для получения целевого уровня загрузки 1% (масс.) Ni после разложения металлсодержащего предшественника. Катализатор в течение короткого времени перемешивали и оставляли стоять в течение 48 часов. После этого катализатор высушивали в вакуумном сушильном шкафу и прокаливали на воздухе при 400°С.
Пример 40
Получали водный 1,0-молярный раствор хлорида европия и добавляли его к Н-мордениту (BASF 712A-5-2641-1, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) для получения целевого уровня загрузки 3% (масс.) Eu на катализаторе после последующего разложения металлсодержащих предшественников. Раствор Eu в течение короткого времени перемешивали с катализатором, а затем добивались пропитывания при 80°С в течение 6 часов. После этого избыточную жидкость удаляли, а катализатор промывали деионизованной водой. Затем катализатор высушивали в вакуумном сушильном шкафу и прокаливали на воздухе при 550°С. После этого катализатор размалывали и просеивали для ограничения размеров частиц теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш.
Пример 41
Экструдаты цеолита Н-бета (экструдаты с диаметром 1,6 мм) размалывали и просеивали для ограничения размеров частиц теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш. Добавляли водный раствор нитрата галлия до достижения начальной влажности для получения целевого уровня загрузки 1,2% (масс.) Ga на катализаторе после разложения металлсодержащего предшественника. После этого катализатор высушивали в вакуумном сушильном шкафу и прокаливали на воздухе при 400°С.
Пример 42
Фосфорную кислоту разбавляли деионизованной водой и добавляли до достижения начальной влажности к носителю SiO2/Al2O3 Davicat (Grace-Davis, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) для достижения целевого уровня содержания 5% (масс.) фосфора на катализаторе. После этого катализатор высушивали в течение ночи в вакуумном сушильном шкафу, а затем прокаливали в потоке проточного воздуха при 500°С.
Пример 43
Водный раствор нитрата никеля добавляли к препарату цеолита ZSM-5, связанного оксидом алюминия, (SiO2:Al2O3, 30:1, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) при использовании методики достижения начальной влажности для получения целевого уровня загрузки никеля 1,0% (масс.). Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного воздуха при 400°С.
Пример 44
Водный раствор нитрата галлия добавляли к препарату цеолита ZSM-5, связанного оксидом алюминия, (SiO2:Al2O3, 80:1, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) при использовании методики достижения начальной влажности для получения целевого уровня загрузки галлия 1,2% (масс.). Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного воздуха при 400°С.
Пример 45
Системы катализаторов, полученные при использовании способов из примеров от 38 до 44, исследовали на предмет парофазной конденсации различных оксигенатов при температуре в диапазоне от 325°С до 375°С и полном давлении в диапазоне от 200 фунт/дюйм2 (изб.) (1380 кПа (изб.)) до 625 фунт/дюйм2 (изб.) (4310 кПа (изб.)) и значении МЧОС в диапазоне от 1,9 до 42,8. В данных исследованиях использовали реакторы двух различных размеров; в трубчатый реактор из нержавеющей стали с внутренним диаметром 8,5 мм загружали 15 и 18 миллилитров катализатора или от 50 до 70 миллилитров катализатора загружали в трубчатый реактор из нержавеющей стали на 21,2 мм (пример 4). Технологическая цепочка реакций представляла собой ту, что описывалась в примере 1 или примере 3 в зависимости от исходного сырья, при проведении анализа так, как это описывалось в примере 5.
Условия эксплуатации и результаты данных экспериментов продемонстрированы в таблице 12. Там, где составы исходного сырья дают вкупе менее, чем 100%, балансовый компонент представлял собой воду. Как демонстрируют данные результаты, субстраты, которые могут быть превращены в C5+ углеводороды в широком диапазоне условий, представляют собой широкий ассортимент оксигенатов, в том числе спиртов и кетонов, как 3-углеродных, так и 5-углеродных. При данных превращениях в особенности подходящими для использования являются цеолиты, как это продемонстрировано в экспериментах FF, GG, НН, II, JJ, LL и ММ. Как демонстрируют эксперименты FF, GG, НН, II и JJ, основными продуктами превращения спирта на мордените и бета-цеолитах являются продукты олефиновой конденсации. Катализатор в виде импрегнированных фосфором диоксида кремния-оксида алюминия - эксперимент КК - продемонстрировал подобный профиль селективности по продуктам. В противоположность этому, катализаторы на основе ZSM-5 - эксперименты LL и MM - приводили к получению значительных фракций ароматических и парафиновых компонентов.
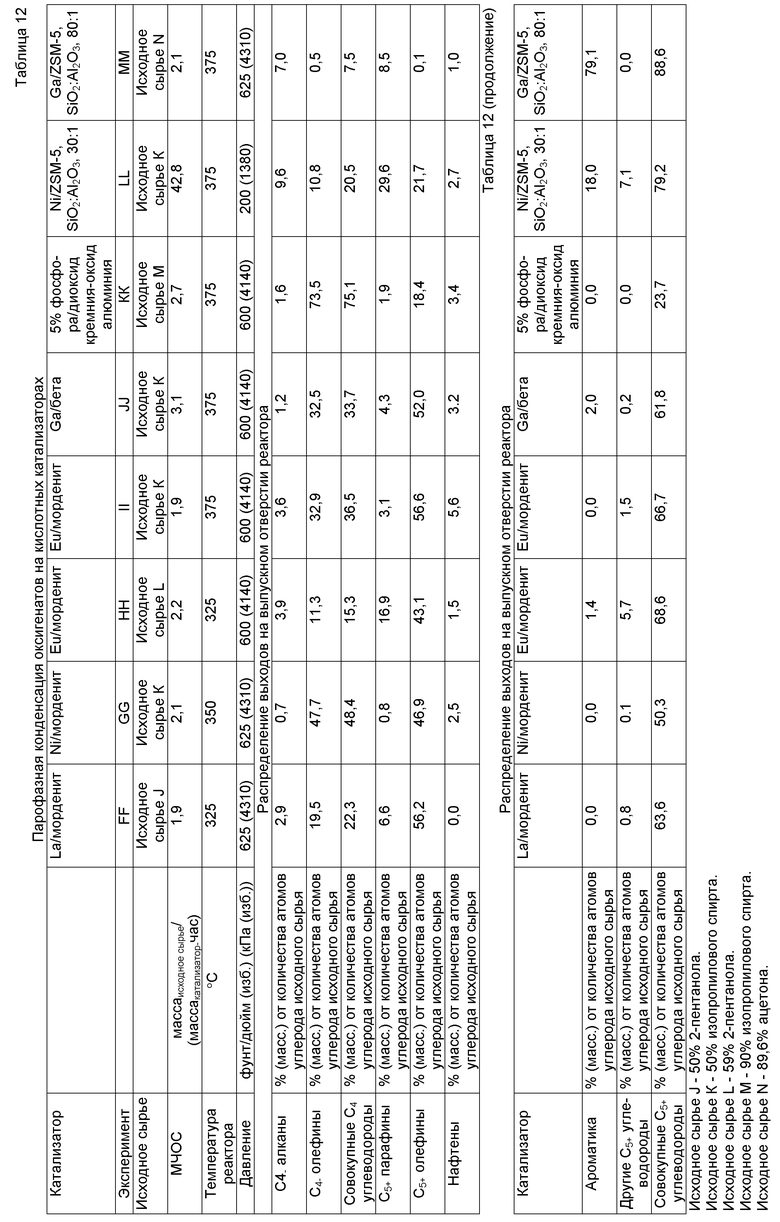
Получение С5+соединений из оксигенированных углеводородов
Пример 46
Следовали методике получения катализатора, идентичной методике из примера 44 при том исключении, что материал ZSM-5, связанный оксидом алюминия, характеризовался количественным соотношением SiO2:Al2O3 30:1.
Пример 47
Катализатор, полученный при использовании способа 46, исследовали на предмет парофазной конденсации смеси оксигенатов при 375°С и 200 фунт/дюйм2 (изб.) (1380 кПа (изб.)). В данном исследовании 11,3 грамма катализатора загружали в трубчатый реактор из нержавеющей стали с внутренним диаметром 8,5 мм, описывавшийся в примере 4. Технологическая цепочка реакций представляла собой ту, что описывалась в примере 3. Смесь оксигенатов включала, в расчете на массу, 25% 2-пентанона, 20% 3-пентанона, 20% 2-пентанола, 10% изопропилового спирта, 10% валериановой кислоты, 5% 2-метилтетрагидрофурана. Данную смесь добавляли при использовании одного насоса в реакторной системе из примера 3, в то время как второй насос добавлял воду таким образом, чтобы совокупное исходное сырье содержало бы 60% (масс.) воды и 40% (масс.) смешанных оксигенатов.
Мониторинг в способе проводили в течение периода времени продолжительностью в 128 часов, при этом образцы периодически удаляли из системы для анализа технологических эксплуатационных характеристик. Каждый анализ проводили так, как это описывалось в примере 5. Фигура 15 демонстрирует зависимость от времени доли атомов углерода исходного сырья, которая покидала реакторную систему в виде C5+ соединений. Фигура 16 демонстрирует зависимость от времени доли атомов углерода исходного сырья, которая покидала реакторную систему в виде ароматического углеводорода. Фигура 14 демонстрирует зависимость от времени доли атомов углерода исходного сырья, которая покидала реакторную систему в виде оксигенатов.
Как демонстрируют фигуры 14, 15 и 16, система катализатора способна функционировать в течение продолжительных периодов времени при использовании смеси оксигенатов, которая включает смесь оксигенатов, включающих спирты, кетоны, кислоту и тетрагидрофуран. С течением времени выработка C5+ соединений остается относительно стабильной, в то время как количество ароматических углеводородов, присутствующих в продукте, уменьшается, а проскок оксигенированных соединений увеличивается (фигура 14). Как можно себе представить, дезактивация катализатора главным образом обусловлена накоплением углеродистых отложений, ограничивающих возможность доступа реагентов к активным центрам.
Пример 48
Водный раствор гексахлороплатиновой кислоты и рениевой кислоты добавляли к углеродному носителю катализатора (OLC-AW, Calgon, при размерах частиц, ограниченных теми, которые удерживались на сите 50 меш после прохождения через сито 120 меш) при использовании методики достижения начальной влажности для получения целевых уровня загрузки платины 1,8% и уровня загрузки рения 6,3% на катализаторе после последующего разложения металлсодержащих предшественников. Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого восстанавливали в потоке проточного водорода при 400°С. После восстановления катализатор хранили в атмосфере азота вплоть до достижения готовности к использованию.
Пример 49
Следовали методике получения катализатора, идентичной методике из примера 44 при том исключении, что материал ZSM-5, связанный оксидом алюминия, характеризовался количественным соотношением SiO2:Al2O3 150:1.
Пример 50
Гексахлороплатиновую кислоту и рениевую кислоту, растворенные в воде, добавляли к носителю катализатора в виде моноклинного диоксида циркония (NorPro Saint Gobain, product code SZ31164, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) при использовании методики достижения начальной влажности для получения целевых уровня загрузки платины 1,8% и уровня загрузки рения 6,3% на катализаторе после последующего разложения металлсодержащих предшественников. Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного воздуха при 400°С.
Пример 51
Следовали той же самой методике, что и использовавшаяся для получения катализатора из примера 50 при том исключении, что целевой уровень загрузки рения составлял 1,8%.
Пример 52
Цеолит ZSM-5, характеризующийся количественным соотношением SiO2:Al2O3 80:1, (Zeolyst International, CBV 8014) перемешивали с порошкообразными ZnO и Al2O3 при молярном соотношении 1:1 таким образом, чтобы объединенные ZnO и Al2O3 (Dispal 18N4-80, Sasol North America, Houston, Texas) составляли бы 30% (масс.) от совокупного количества твердого вещества. К объединенным ZnO и Al2O3 добавляли разбавленную азотную кислоту при уровне содержания 2% (масс.) HNO3. Консистенцию тестообразной массы регулировали в результате добавления воды для получения удобнообрабатываемой тестообразной массы, подходящей для экструдирования, и смесь экструдировали при использовании экструдера лабораторного масштаба. Экструдаты высушивали в течение ночи в вакууме при 100°С, а после этого прокаливали при 600°С в проточном воздухе.
Пример 53
К материалу из примера 52 добавляли водный раствор нитрата галлия при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш, при использовании методики достижения начальной влажности для получения целевого уровня загрузки галлия 1,2% (масс.). Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного водорода при 400°С.
Пример 54
К материалу из примера 52 добавляли водный раствор нитрата никеля при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш, при использовании методики достижения начальной влажности для получения целевого уровня загрузки никеля 1,0% (масс.). Препарат высушивали в течение ночи в вакуумном сушильном шкафу, а после этого прокаливали в потоке проточного водорода при 400°С.
Пример 55
Системы катализаторов, на которые ссылались в примерах 6, 46, 48, 49, 51, 53 и 54, исследовали на предмет превращения глицерина, сорбита, сахарозы и ксилозы в углеводороды при использовании конфигурации реакторов, описывавшейся в примере 2. Исследования проводили при использовании двух трубчатых реакторов из нержавеющей стали с внутренним диаметром 21,2 мм, продемонстрированных в примере 4, и проведении анализа так, как это описывалось в примере 5. Поверх катализатора конденсации, размещенного во втором реакторе, размещали вольфрамированный диоксид циркония (NorPro-Saint Gobain, product code SZ61143, при размерах частиц, ограниченных теми, которые удерживались на сите 60 меш после прохождения через сито 18 меш) для получения зоны испарения отходящего потока первого реактора перед его поступлением на катализатор конденсации.
Таблица 13 демонстрирует результаты данных исследований. В случае эксперимента NN (38% сахарозы+7% ксилозы) с исходным сырьем перед его введением в реактор объединяли поток водорода при целевом расходе, равном 3-кратному значению числа молей сахарозы плюс 1,5-кратное значение числа молей ксилозы. Другие эксперименты проводили без внешней подачи водорода. Нагреватели, внешние по отношению к реактору и продемонстрированные на фигуре 9 в позициях 10а, 10b, 10с, 10d, 23a, 23b, 23с и 23d, использовали для выдерживания температур стенки реактора так, как это указывается в таблице 13. Углеводородные продукты данных исследований, описывающиеся в таблице 13, группировали в C4 - фракцию, которая преимущественно находилась в газовой фазе при температуре и давлении окружающей среды, и C5+ фракцию, которая в общем случае является подходящей для включения в жидкие топлива. Как демонстрируют результаты, широкий ассортимент Сахаров и многоатомных спиртов легко может быть превращен в C5+ углеводороды по способам, описывающимся в настоящем документе. Распределение парафинов и ароматических соединений по данному примеру представлено на фиг.17.
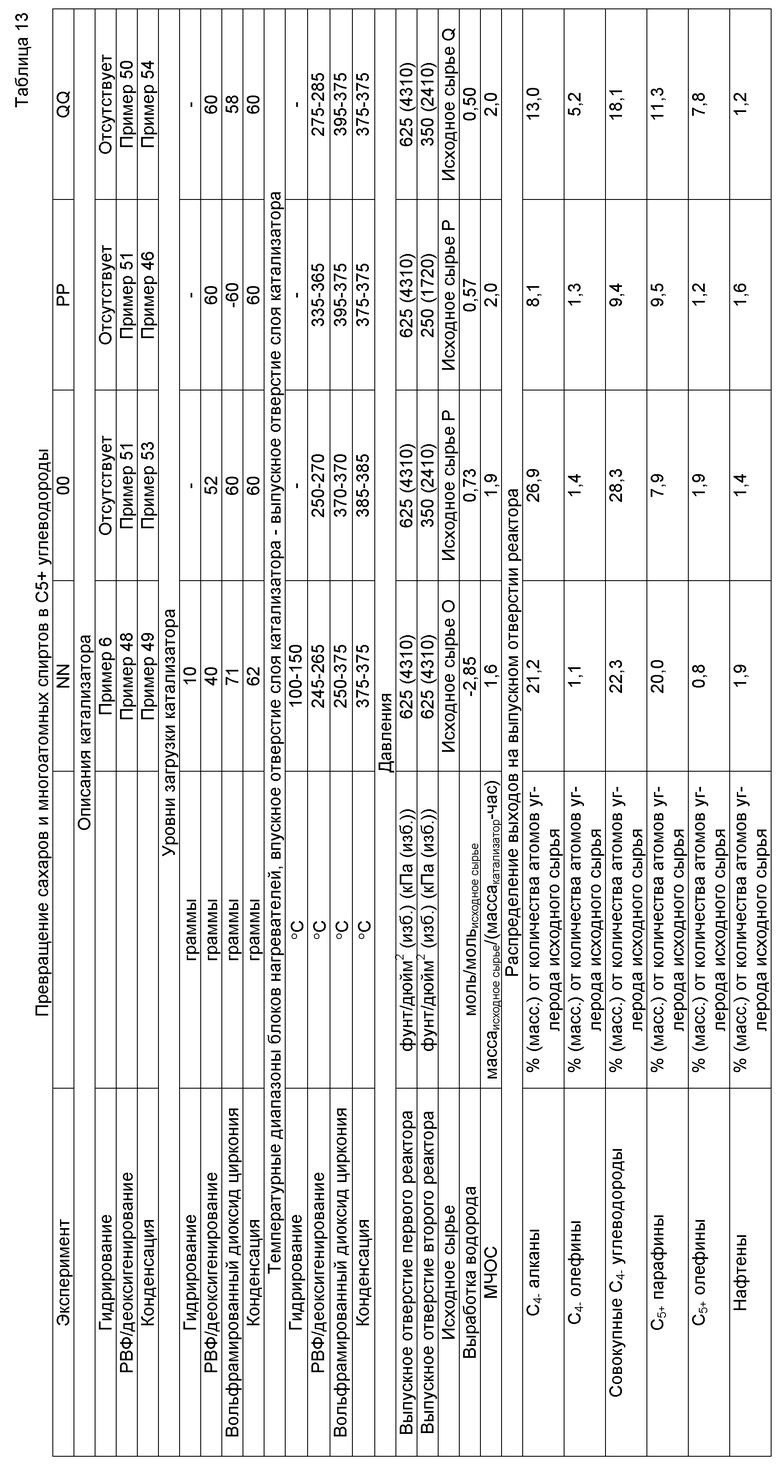

Пример 56
Способ, описывавшийся в примере 55 и в качестве примера проиллюстрированный в эксперименте QQ из таблицы 13, реализовали в течение периода времени продолжительностью, большей, чем 400 часов. По истечении начального периода времени эксплуатации степень превращения в ароматические компоненты и выход углеводородов уменьшались, что продемонстрировано на фигурах 18 и 19 в виде цикла 1. На фигуре 18 продемонстрирована теплотворная способность Cf+углеводородов, присутствующих на выпускном отверстии второго реактора, в виде процентной доли от теплотворной способности исходного сырья. На фигуре 19 продемонстрировано количество атомов углерода, присутствующих в виде ароматических углеводородов на выпускном отверстии второго реактора, в виде процентной доли от количества атомов углерода, присутствующих в исходном сырье. По истечении приблизительно 120 часов в эксплуатации второй реактор обходили по байпасу, в то время как первый реактор продолжал функционировать. После этого проводили окислительную регенерацию катализатора во втором реакторе. Во время регенерации инициировали прохождение потока азота и воздуха таким образом, чтобы целевая концентрация кислорода на впускном отверстии второго реактора составляла бы 1% (моль.). После этого температуры блоков второго реактора увеличивали до 500°С и подачу потока азота и кислорода продолжали вплоть до тех пор, пока диоксид углерода на выпускном отверстии второго реактора больше уже не обнаруживался. Затем концентрацию кислорода увеличивали до целевого уровня 5% (моль.). Подачу данного потока продолжали вплоть до тех пор, пока диоксид углерода на выпускном отверстии второго реактора больше уже не обнаруживался. В данный момент подачу потока кислорода прекращали, в то время как подачу потока азота продолжали. После этого температуры блоков второго реактора уменьшали до 400°С, в то время как композицию газа, протекающего через слой катализатора, изменяли на водород. Затем температуры блоков второго реактора, регулируя, доводили до тех, которые продемонстрированы для эксперимента QQ в таблице 13. После этого второй реактор возвращали обратно в эксплуатацию, ставя себе целью получение условий, продемонстрированных для эксперимента QQ в таблице 13. Затем второй реактор подвергали воздействию нескольких циклов эксплуатации и регенерации, при этом результаты для периода времени в эксплуатации продемонстрированы на фигурах 18 и 19. Как демонстрируют данные результаты, регенерация катализатора конденсации приводила в результате к восстановлению активности, что согласуется с теорией о том, что основной причиной ухудшения эксплуатационных характеристик катализатора с течением времени являлось образование отложений углеродистых материалов. Кроме того, как демонстрируют результаты, катализатор конденсации может быть регенерирован несколько раз без значительной потери эксплуатационных характеристик.
Получение композиций бензина
Пример 57
Композицию бензина (композицию бензина КБ1) получали в результате перемешивания базового бензина с 5% (об.), в расчете на объем конечной композиции бензина, продукта способа, описывавшегося в примере 55 и в качестве примера проиллюстрированного в эксперименте РР из таблицы 13.
Использующийся базовый бензин и полученная композиция бензина подробно описываются в таблице 14.
Пример 58
Композицию бензина (композицию бензина КБ2) получали в результате перемешивания базового бензина с 10% (об.), в расчете на объем конечной композиции бензина, продукта способа, описывавшегося в примере 55 и в качестве примера проиллюстрированного в эксперименте 00 из таблицы 13.
Использующийся базовый бензин и полученная композиция бензина подробно описываются в таблице 14.
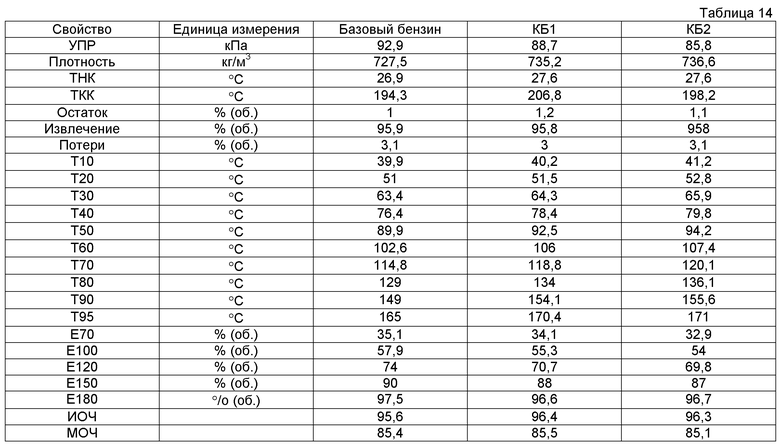
Как можно видеть из информации, представленной в приведенной выше таблице 14, композиции бензина, содержащие продукты, полученные по способу, описывавшемуся в примере 55, демонстрируют характеристики по перегонке, исключительно подобные тому, что имеет место для базового бензина, и в выгодном случае характеризуются большим исследовательским октановым числом (ИОЧ) в сопоставлении с базовым бензином. Как также можно видеть из информации, представленной в приведенной выше таблице 14, композиции бензина, содержащие продукты, полученные по способу, описывавшемуся в примере 55, характеризуются меньшей упругостью паров по Рейду (УПР) в сопоставлении с тем, что имеет место для базового бензина, что в выгодном случае может быть использовано для уменьшения значения УПР бензина в целях удовлетворения конкретных требований к композиции бензина или в альтернативном варианте обеспечения добавления более значительных количеств компонентов бензина, добавление которых к композициям бензина могло бы быть ограниченным вследствие летучести таких компонентов.
Пример 59
При использовании в качестве исходного сырья сорбита и сахарозы получали две различные композиции бензина, а после этого их анализировали на предмет концентрации углерода-14 (14С) в них в сопоставлении с тем, что имеет место для бензина на нефтяной основе.
Первый образец бензина (V-18510) получали из 50%-ного раствора сорбита в воде при использовании реакторной системы, подобной той, что описывалась в примере 55 при том исключении, что система включала второй реактор конденсации в конфигурации опережения/запаздывания. Катализатор РВФ/деоксигенирования получали так, как это описывалось в примере 50, в то время как катализатор конденсации получали так, как это описывалось в примере 43. При различных температурах и в различных условиях провели несколько экспериментов с использованием сорбитового исходного сырья. Реакции РВФ/деоксигенирования проводили в одном реакторе по температурному профилю в диапазоне от низкой до высокой температуры с варьированием температур в диапазоне от 110°С до 130°С на впускном отверстии и в диапазоне от 235°С до 250°С на выпускном отверстии. Реакции конденсации проводили в двухреакторной системе при варьировании температуры в опережающем реакторе в диапазоне от 345°С до 385°С и в запаздывающем реакторе в диапазоне от 260°С до 385°С. Условия по давлению также варьировали в диапазоне от 620 фунт/дюйм2 (изб.) (4270 кПа (изб.)) до 630 фунт/дюйм2 (изб.) (4340 кПа (изб.)) для реакций РВФ/деоксигенирования и в диапазоне от 95 фунт/дюйм2 (изб.) (655 кПа (изб.)) до 105 фунт/дюйм2 (изб.) (724 кПа (изб.)) для реакций конденсации. Значение МЧОС варьировали в диапазоне о 0,9 до 1,1 час-1 для катализатора РВФ/деоксигенирования.
Второй образец бензина (V-18512) получали из совокупного образца бензина, полученного из сахарозы при использовании реакторной системы, проиллюстрированной на фигуре 6. Сахарозу подавали при различных температурах, давлениях и значениях МЧОС на катализатор гидрирования, содержащий 2,5% рутения на углероде UU (Calgon, при размерах частиц, ограниченных теми, которые удерживались на сите 120 меш после прохождения через сито 60 меш), и катализатор РВФ/деоксигенирования и катализатор конденсации, описывавшиеся выше для сорбитового исходного сырья. Условия для реакции гидрирования варьировались в зависимости от температур в диапазоне от 115°С до 140°С и давлений в диапазоне от 620 фунт/дюйм2 (изб.) (4270 кПа (изб.)) до 680 фунт/дюйм2 (изб.) (4690 кПа (изб.)). Условия по значению МЧОС, температуре и давлению для РВФ/деоксигенирования представляли собой то же самое, что и описывавшиеся для сорбита. Реакцию конденсации проводили в одном реакторе конденсации в условиях по значению МЧОС, температуре и давлению, идентичных тем, что и описывавшиеся выше для опережающего реактора.
Потоки продуктов из различных экспериментов с сорбитом объединяли в один образец (V-18510), а после этого подвергали воздействию стадии перегонки так, как это описывалось в примере 29. Потоки продуктов из различных экспериментов с сахарозой также объединяли в один образец (V-18512), а после этого подвергали воздействию перегонки так, как это описывалось в примере 29. Фракции перегонки, имеющие температуру кипения, меньшую, чем 210°С, объединяли для последующего примешивания к конечным композициям топлива. Образец каждой фракции также собирали для проведения испытания на наличие 14С.
Испытание на наличие углерода-14 проводили в компании Beta Analytical Inc (Miami, Florida USA) при использовании документа ASTM-D6866 «Test Methods for Determining the Biobased Content of Natural Range Materials Using Radiocarbon and Isotope Ratio Mass Spectrometry Analysis». В дополнение к испытанию на наличие 14С в образцах V-18510 и V-18152 также проводили и определение уровня содержания материала, полученного из биологического сырья, для двух дополнительных образцов, отобранных на различных автогазозаправочных станциях в Мэдисоне, Висконсин. Первый образец (V-RRGWE) представлял собой обычный неосвинцованный бензин, идентифицированный как содержащий вплоть до 10%, в то время как второй образец (V-SVPNE) представлял собой высокосортный бензин. Результаты исследования приведены в представленной ниже таблице 15.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ ЖИДКОГО ТОПЛИВА И ХИМИЧЕСКИХ РЕАКТИВОВ ИЗ КИСЛОРОДСОДЕРЖАЩИХ УГЛЕВОДОРОДОВ | 2008 |
|
RU2472840C2 |
| Производство химических веществ и топлив из биомассы | 2012 |
|
RU2616620C2 |
| ГИДРОПИРОЛИЗ БИОМАССЫ ДЛЯ ПОЛУЧЕНИЯ ВЫСОКОКАЧЕСТВЕННЫХ ЖИДКИХ ТОПЛИВ | 2010 |
|
RU2539598C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАЗВЕТВЛЕННЫХ УГЛЕВОДОРОДОВ | 2008 |
|
RU2456330C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЖИДКОГО УГЛЕВОДОРОДНОГО ТОПЛИВА ИЗ БИОМАССЫ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ | 2020 |
|
RU2733394C1 |
| КАТАЛИЗАТОР И СПОСОБ РИФОРМИНГА КИСЛОРОДСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2006 |
|
RU2438968C2 |
| СПОСОБЫ И СИСТЕМЫ ДЛЯ ПОЛУЧЕНИЯ МНОГОАТОМНЫХ СПИРТОВ | 2007 |
|
RU2454391C2 |
| ДЕГИДРИРОВАНИЕ АЛКАНОЛОВ ДЛЯ УВЕЛИЧЕНИЯ ВЫХОДА АРОМАТИЧЕСКИХ ВЕЩЕСТВ | 2011 |
|
RU2577855C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ ТРУДНО КОНВЕРТИРУЕМЫХ ОКСИГЕНАТОВ В БЕНЗИН | 2006 |
|
RU2428455C2 |
| СПОСОБ ОЛИГОМЕРИЗАЦИИ БЕНЗИНА БЕЗ ДОПОЛНИТЕЛЬНОГО ОБЛАГОРАЖИВАНИЯ | 2013 |
|
RU2639160C2 |
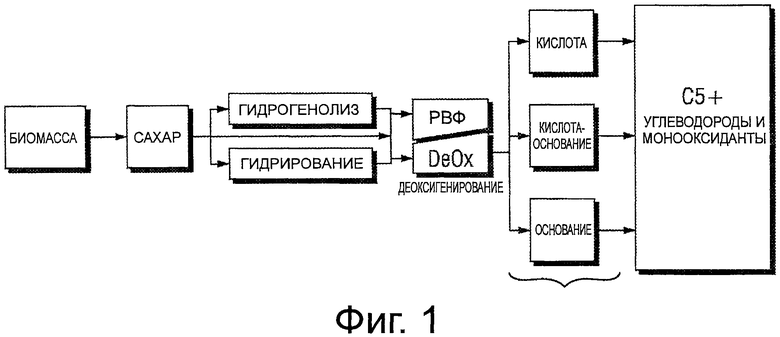
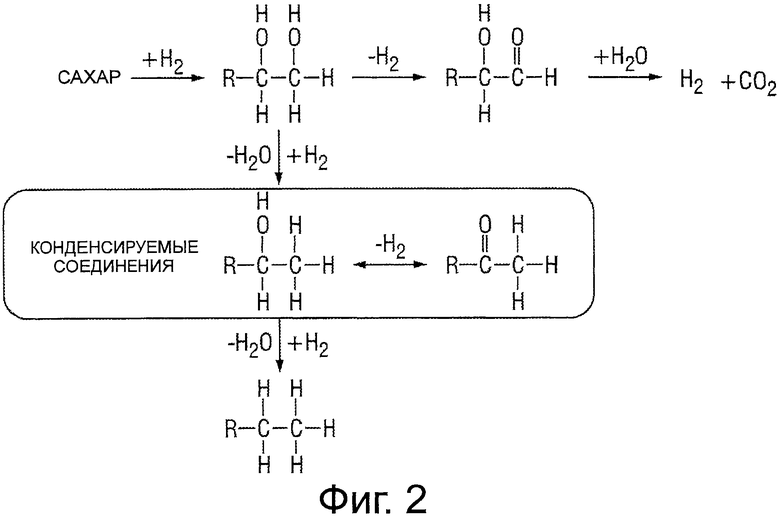

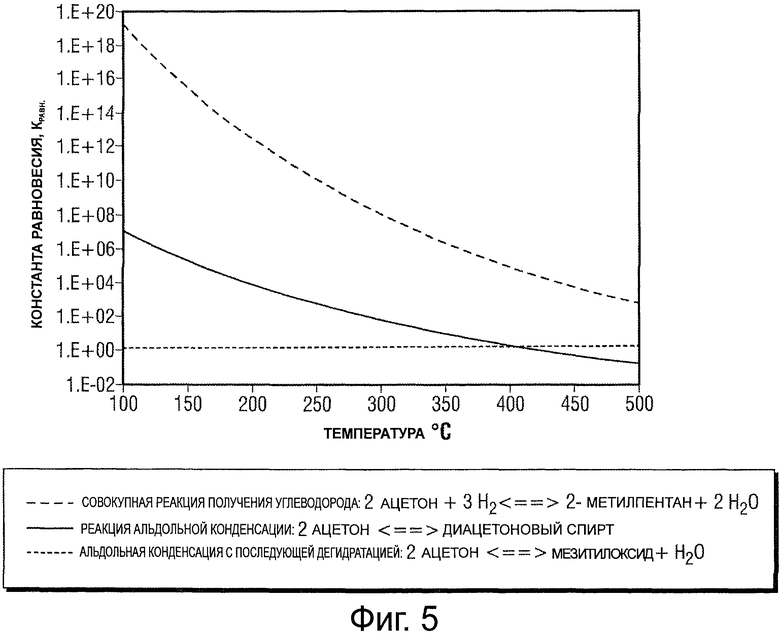

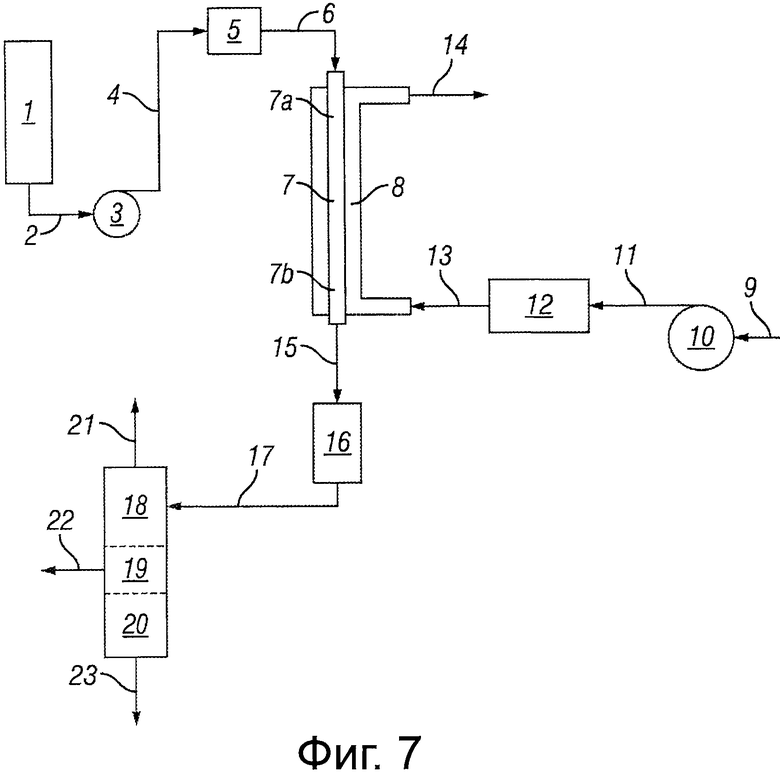
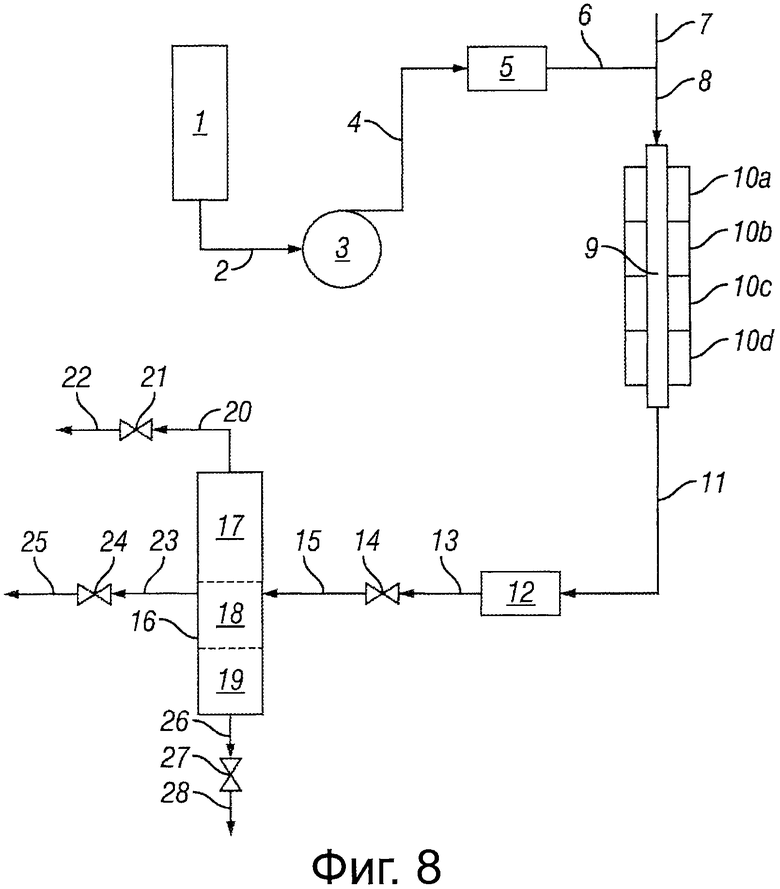
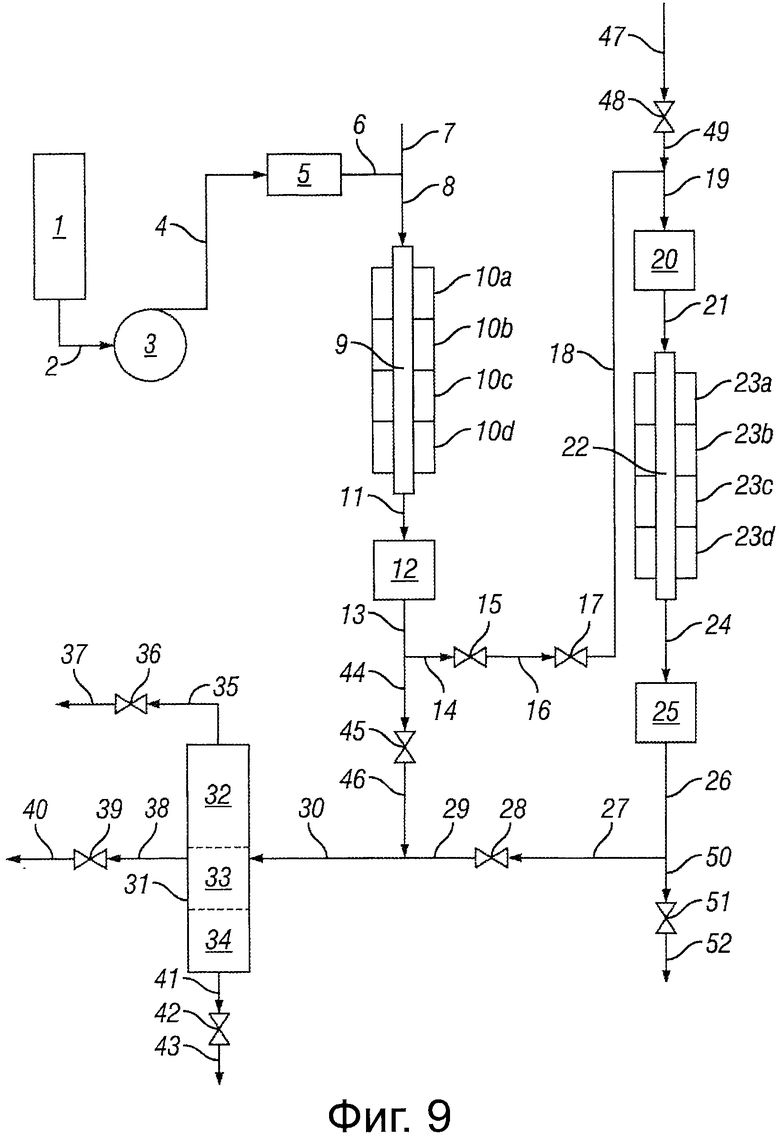
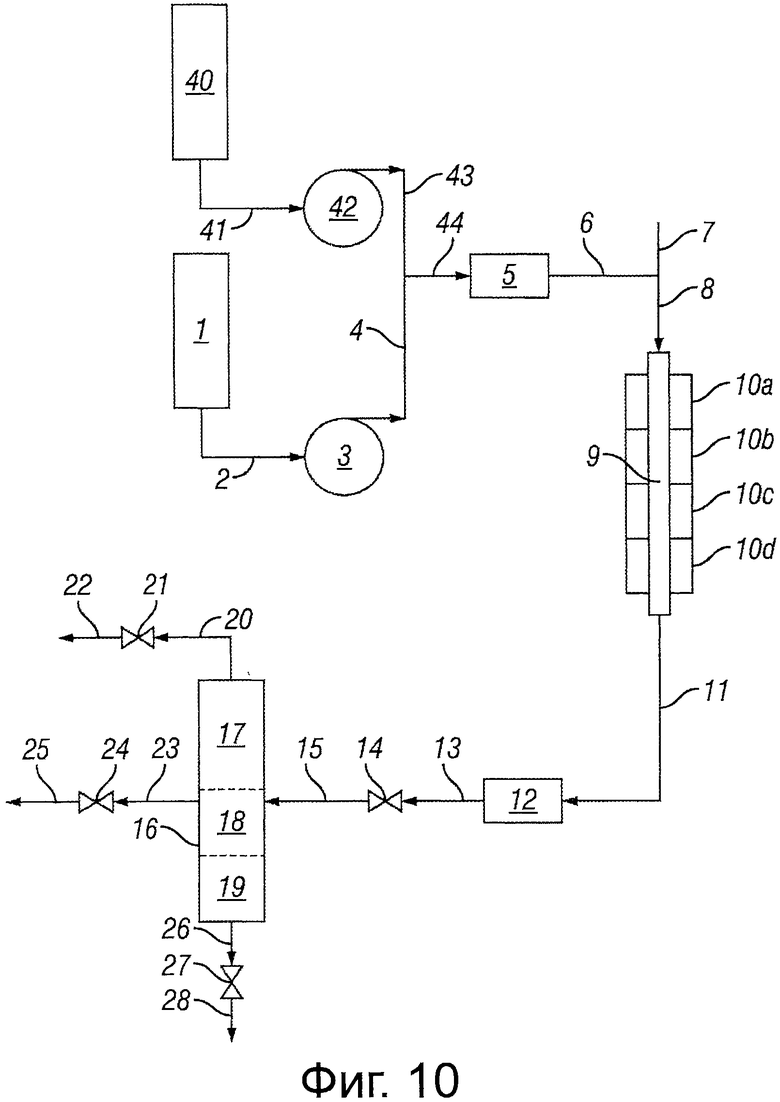
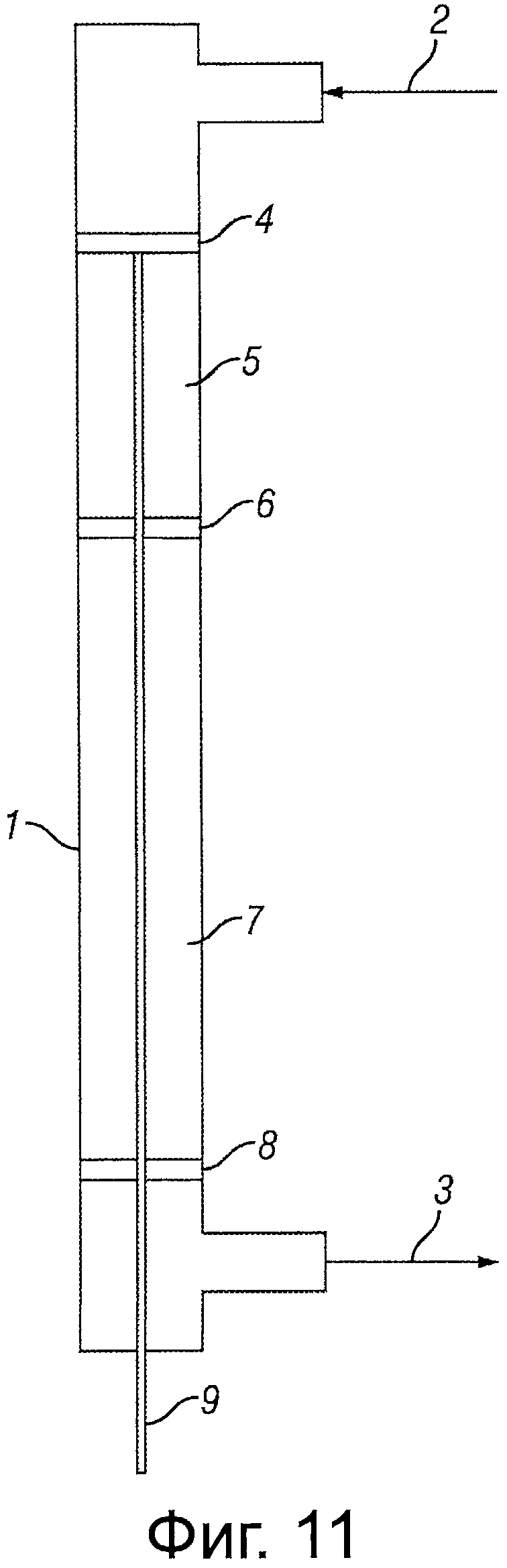
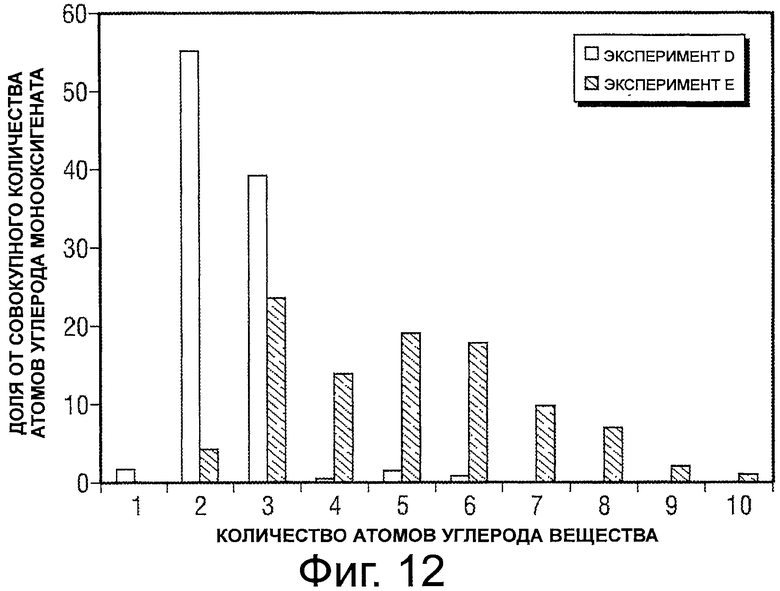

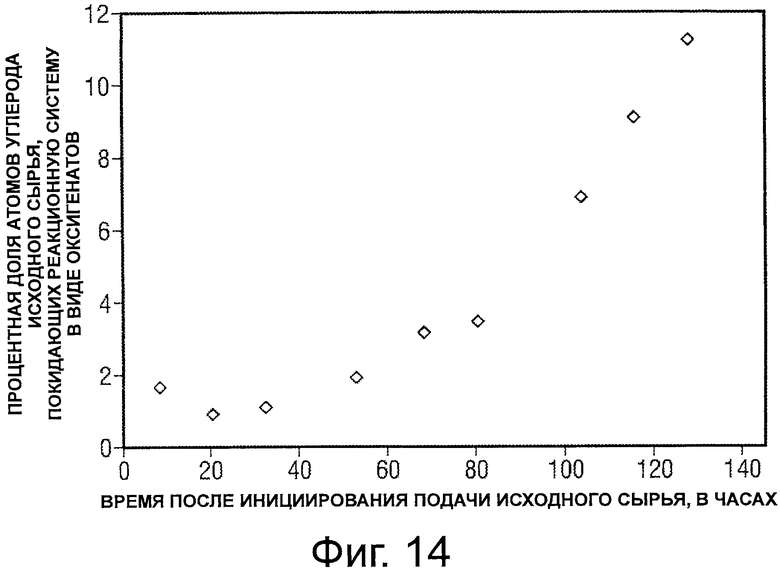
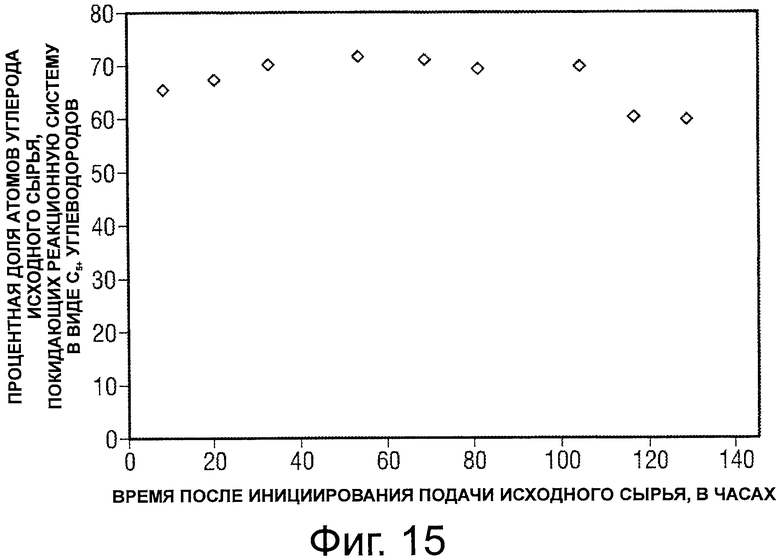
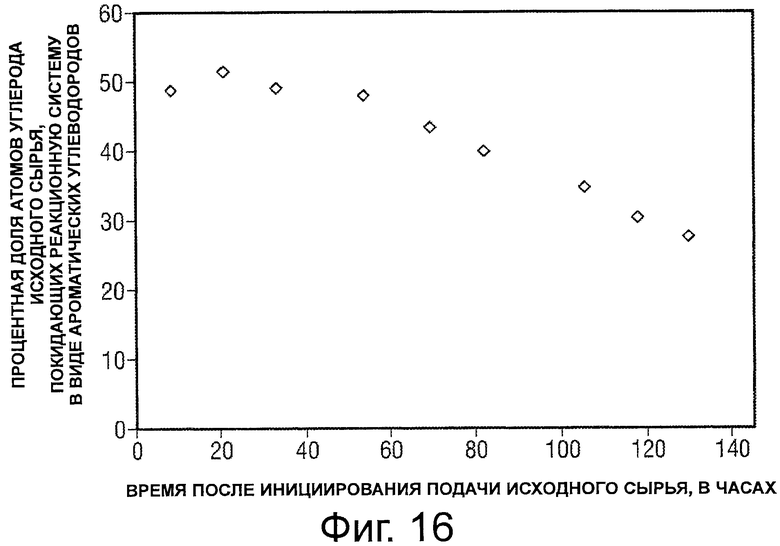
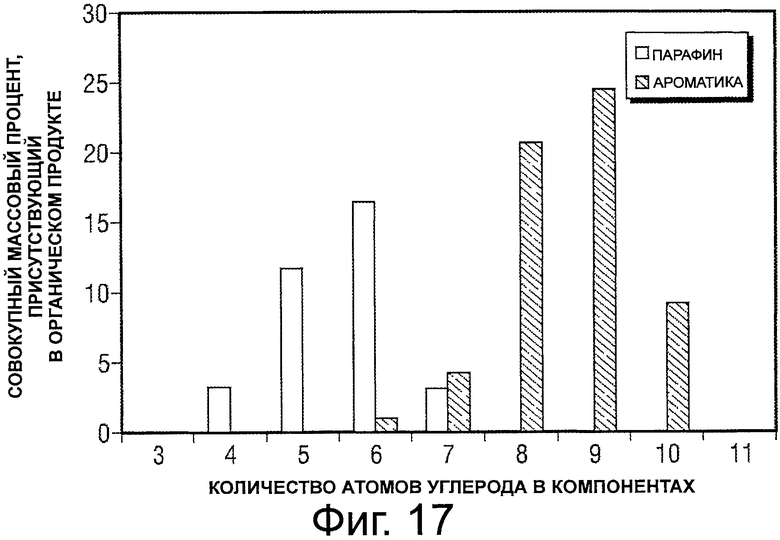
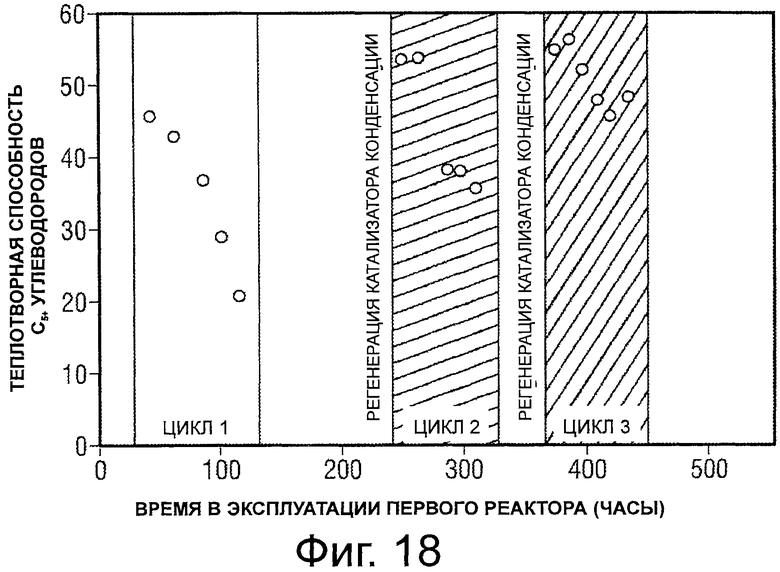

Настоящее изобретение относится к композициям жидкого топлива. Изобретение касается композицию жидкого топлива, содержащей по меньшей мере один компонент топлива и от 0,1%(об.) до 99,5% (об.) фракции перегонки компонента, содержащего, по меньшей мере, одно С4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода. Способ включает подачу воды и растворимого в воде оксигенированного углеводорода, включающего C1+O1+ углеводород, в водной жидкой фазе и/или паровой фазе; подачу Н2; проведение каталитической реакции в жидкой и/или паровой фазе между оксигенированным углеводородом и Н2 в присутствии катализатора деоксигенирования при температуре деоксигенирования и давлении деоксигенирования для получения оксигената, содержащего в реакционном потоке C1+O1-3 углеводород; и проведение каталитической реакции в жидкой и/или паровой фазе для оксигената в присутствии катализатора конденсации при температуре конденсации и давлении конденсации для получения С4+ соединения, где С4+ соединение включает представителя, выбираемого из группы, состоящей из С4+ спирта, С4+ кетона, С4+ алкана, С4+ алкена, C5+ циклоалкана, C5+ циклоалкена, арила, конденсированного арила и их смеси. Изобретение также касается композиции бензина, композиции дизельного топлива, композиции керосина, и способов их получения. Технический результат - улучшенные характеристики композиции топлива, содержащей компонент, произведенный из биомассы. 8 н. и 1 з.п. ф-лы, 19 ил., 14 табл., 59 пр.
1. Композиция жидкого топлива, содержащая по меньшей мере один компонент топлива и от 0,1% (об.) до 99,5% (об.) фракции перегонки компонента, содержащего по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, и полученного по способу, включающему:
подачу воды и растворимого в воде оксигенированного углеводорода, содержащего C1+O1+ углеводород, в водной жидкой фазе и/или паровой фазе;
подачу H2;
проведение каталитической реакции в жидкой и/или паровой фазе между оксигенированным углеводородом и H2 в присутствии катализатора деоксигенирования при температуре деоксигенирования и давлении деоксигенирования для получения оксигената, содержащего в реакционном потоке C1+O1-3углеводород;
проведение каталитической реакции в жидкой и/или паровой фазе для оксигената в присутствии катализатора конденсации при температуре конденсации и давлении конденсации для получения C4+ соединения, и
отделение указанной фракции перегонки;
где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте; и
где C4+ соединение включает представителя, выбираемого из группы, состоящей из C4+спирта, C4+кетона, C4+алкана, C4+алкена, C5+циклоалкана, C5+циклоалкена, арила, конденсированного арила и их смеси;
где композицию жидкого топлива выбирают из:
композиции бензина, характеризующейся температурой начала кипения в диапазоне от 15°C до 70°C (IP123), температурой конца кипения, равной, самое большее, 230°C, (IP123), значением исследовательского октанового числа (ИОЧ) в диапазоне от 85 до 110 (ASTM D2699) и значением моторного октанового числа (МОЧ) в диапазоне от 75 до 100 (ASTM D2700);
композиции дизельного топлива, характеризующейся температурой начала кипения в диапазоне от 130°C до 230°C (IP123), температурой конца кипения, равной, самое большее, 410°C, (IP123) и цетановым числом в диапазоне от 35 до 120 (ASTM D613); и
композиции керосина, характеризующейся температурой начала кипения в диапазоне от 80 до 150°C, температурой конца кипения в диапазоне от 200 до 320°C и вязкостью при - 20°C в диапазоне от 0,8 до 10 мм2/сек (ASTM D445);
где катализатор деоксигенирования содержит элемент, выбранный из группы, состоящей из Cu, Re, Fe, Ru, Ir, Co, Rh, Pt, Pd, Ni, W, Os, Mo, Ag, Au, их сплавов и комбинаций, и носитель, выбранный из группы, состоящей из нитрида, углерода, диоксида кремния, оксида алюминия, диоксида циркония, диоксида титана, оксида ванадия, диоксида церия, оксида цинка, оксида хрома, нитрида бора, гетерополикислот, кизельгура, гидроксиапатита и их смесей;
где катализатор конденсации содержит соединение, выбранное из группы: карбиды, нитриды, диоксид циркония, оксид алюминия, диоксид кремния, алюмосиликаты, фосфаты, цеолиты, оксиды титана, оксиды цинка, оксиды ванадия, оксиды лантана, оксиды иттрия, оксиды скандия, оксиды магния, оксиды церия, оксиды бария, оксиды кальция, гидроксиды, гетерополикислоты, неорганические кислоты, кислотно-модифицированные смолы, основно-модифицированные смолы и их комбинации;
где температуру деоксигенирования и давление деоксигенирования выбирают так, чтобы поддерживать по крайней мере часть оксигенированного углеводорода в состоянии жидкой фазы на впускном отверстии реактора; и
где температуру конденсации и давление конденсации выбирают так, чтобы поддерживать по крайней мере часть оксигената в состоянии жидкой фазы на впускном отверстии реактора.
2. Композиция жидкого топлива по п. 1, где композиция жидкого топлива дополнительно содержит одну или несколько топливных присадок.
3. Композиция бензина, содержащая по меньшей мере один компонент топлива и от 0,1% (об.) до 99,5% (об.) компонента, содержащего по меньшей мере одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующегося температурой конца кипения в диапазоне от 150 до 220°C, плотностью при 15°C в диапазоне от 700 до 890 кг/м3, уровнем содержания серы, равным, самое большее, 5 мг/кг, уровнем содержания кислорода, равным, самое большее, 3,5% (масс.), значением ИОЧ в диапазоне от 80 до 110 и значением МОЧ в диапазоне от 70 до 100, где упомянутая композиция бензина характеризуется температурой начала кипения в диапазоне от 15°C до 70°C (IP123), температурой конца кипения, равной, самое большее, 220°C, (IP123), значением ИОЧ в диапазоне от 85 до 110 (ASTM D2699) и значением МОЧ в диапазоне от 75 до 100 (ASTM D2700); и где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте.
4. Композиция дизельного топлива, содержащая по меньшей мере один компонент топлива и от 0,1% (об.) до 99,5% (об.) компонента, содержащего по меньшей мере одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующегося значением T95 в диапазоне от 220 до 380°C, температурой вспышки в диапазоне от 30 до 70°C, плотностью при 15°C в диапазоне от 700 до 900 кг/м3, уровнем содержания серы, равным, самое большее, 5 мг/кг, уровнем содержания кислорода, равным, самое большее, 10% (масс.), и вязкостью при 40°C в диапазоне от 0,5 до 6 сСт, где упомянутая композиция дизельного топлива характеризуется температурой начала кипения в диапазоне от 130°C до 230°C (IP123), температурой конца кипения, равной, самое большее, 410°C, (IP123) и цетановым числом в диапазоне от 35 до 120 (ASTM D613); и где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте.
5. Композиция керосина, содержащая по меньшей мере один компонент топлива и от 0,1% (об.) до 99,5% (об.) компонента, содержащего по меньшей мере одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующегося температурой начала кипения в диапазоне от 120 до 215°C, температурой конца кипения в диапазоне от 220 до 320°C, плотностью при 15°C в диапазоне от 700 до 890 кг/м3, уровнем содержания серы, равным, самое большее, 0,1% (масс.), уровнем содержания совокупной ароматики, равным, самое большее, 30% (об.), температурой замерзания, равной - 40°C и менее, максимальной высотой некоптящего пламени, равной, по меньшей мере, 18 мм, вязкостью при - 20°C в диапазоне от 1 до 10 сСт и удельным энергосодержанием в диапазоне от 40 до 47 МДж/кг, где упомянутая композиция керосина характеризуется температурой начала кипения в диапазоне от 80 до 150°C, температурой конца кипения в диапазоне от 200 до 320°C и вязкостью при - 20°C в диапазоне от 0,8 до 10 мм2/сек (ASTM D445); и где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте.
6. Способ получения композиции жидкого топлива по п. 1, включающий перемешивание:
(a) от 0,1% (об.) до 99,5% (об.) фракции перегонки компонента, содержащего по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, полученного по способу, включающему:
подачу воды и растворимого в воде оксигенированного углеводорода, содержащего C1+O1+ углеводород, в водной жидкой фазе и/или паровой фазе;
подачу H2;
проведение каталитической реакции в жидкой и/или паровой фазе между оксигенированным углеводородом и H2 в присутствии катализатора деоксигенирования при температуре деоксигенирования и давлении деоксигенирования для получения оксигената, содержащего в реакционном потоке C1+O1-3 углеводород; и
проведение каталитической реакции в жидкой и/или паровой фазе для оксигената в присутствии катализатора конденсации при температуре конденсации и давлении конденсации для получения C4+ соединения,
отделение указанной фракции перегонки;
где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте;
где C4+ соединение включает представителя, выбираемого из группы, состоящей из C4+спирта, C4+кетона, C4+алкана, C4+алкена, C5+циклоалкана, C5+циклоалкена, арила, конденсированного арила и их смеси;
где катализатор деоксигенирования содержит элемент, выбранный из группы, состоящей из Cu, Re, Fe, Ru, Ir, Co, Rh, Pt, Pd, Ni, W, Os, Mo, Ag, Au, их сплавов и комбинаций, и носитель, выбранный из группы, состоящей из нитрида, углерода, диоксида кремния, оксида алюминия, диоксида циркония, диоксида титана, оксида ванадия, диоксида церия, оксида цинка, оксида хрома, нитрида бора, гетерополикислот, кизельгура, гидроксиапатита и их смесей;
где катализатор конденсации содержит соединение, выбранное из группы: карбиды, нитриды, диоксид циркония, оксид алюминия, диоксид кремния, алюмосиликаты, фосфаты, цеолиты, оксиды титана, оксиды цинка, оксиды ванадия, оксиды лантана, оксиды иттрия, оксиды скандия, оксиды магния, оксиды церия, оксиды бария, оксиды кальция, гидроксиды, гетерополикиелоты, неорганические кислоты, кислотно-модифицированные смолы, основно-модифицированные смолы и их комбинации;
где температуру деоксигенирования и давление деоксигенирования выбирают так, чтобы поддерживать по крайней мере часть оксигенированного углеводорода в состоянии жидкой фазы на впускном отверстии реактора; и
где температуру конденсации и давление конденсации выбирают так, чтобы поддерживать по крайней мере часть оксигената в состоянии жидкой фазы на впускном отверстии реактора; и
(b) по меньшей мере, одного компонента топлива.
7. Способ получения композиции бензина по п. 3, включающий перемешивание:
(a) от 0,1% (об.) до 99,5% (об.) компонента, содержащего по меньшей мере одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующегося температурой конца кипения в диапазоне от 150 до 250°C, плотностью при 15°C в диапазоне от 700 до 890 кг/м3, уровнем содержания серы, равным, самое большее, 5 мг/кг, уровнем содержания кислорода, равным, самое большее, 3,5% (масс.), значением ИОЧ в диапазоне от 80 до 110 и значением МОЧ в диапазоне от 70 до 100, где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте; и
(b) по меньшей мере, одного компонента топлива.
8. Способ получения композиции дизельного топлива по п. 4, включающий перемешивание:
(a) от 0,1% (об.) до 99,5% (об.) компонента, содержащего по меньшей мере одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующегося значением T95 в диапазоне от 220 до 380°C, температурой вспышки в диапазоне от 30 до 70°C, плотностью при 15°C в диапазоне от 700 до 900 кг/м3, уровнем содержания серы, равным, самое большее, 5 мг/кг, уровнем содержания кислорода, равным, самое большее, 10% (масс.), и вязкостью при 40°C в диапазоне от 0,5 до 6 сСт, где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте; и
(b) по меньшей мере, одного компонента топлива.
9. Способ получения композиции керосина по п. 5, включающий перемешивание:
(a) от 0,1% (об.) до 99,5% (об.) компонента, содержащего по меньшей мере одно C4+ соединение, производимое из растворимого в воде оксигенированного углеводорода, и характеризующегося температурой начала кипения в диапазоне от 120 до 215°C, температурой конца кипения в диапазоне от 220 до 340°C, плотностью при 15°C в диапазоне от 700 до 890 кг/м3, уровнем содержания серы, равным, самое большее, 0,1% (масс.), уровнем содержания совокупной ароматики, равным, самое большее, 30% (об.), температурой замерзания, равной - 40°C и менее, максимальной высотой некоптящего пламени, равной, по меньшей мере, 18 мм, вязкостью при - 20°C в диапазоне от 1 до 10 сСт и удельным энергосодержанием в диапазоне от 40 до 47 МДж/кг, где компонент, содержащий по меньшей мере одно C4+ соединение, произведенное из растворимого в воде оксигенированного углеводорода, имеет возраст, меньший, чем 100 лет, согласно вычислению по концентрации углерода-14 в компоненте; и
(b) по меньшей мере, одного компонента топлива.
| US 20070142633 A1, 21.06.2007 | |||
| George W | |||
| Huber, Juben N | |||
| Chheda, Cristopher J | |||
| Barret, James A | |||
| Dumesic, Production of liquid alkanes by aqueos-phase processing of biomass-derived carbohydrates, 03.06.2005, Science, American association for the advancement of science , volume 308, issue 5227, 1446-1450 | |||
| W 2007075476 A, 05.07.2007 | |||
| Расцепляющее приспособление для автоматических центральных сцепок, в особенности для железнодорожных повозок | 1927 |
|
SU9812A1 |

