Настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов. Более конкретно, настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, в которых заместитель 5-фтор вводится путем замены галогена в начале схемы способа.
Патент США 6297197 B1 описывают inter alia конкретные соединения 6-(алкокси или арилокси)-4-амино-3-хлор-5-фторпиколинатов и их применение в качестве гербицидов. Патенты США 6784137 B2 и 7314849 B2 описывают inter alia конкретные соединения 6-(арил)-4-амино-3-хлор-5-фторпиколинатов и их применение в качестве гербицидов. Патент США 7432227 B2 описывает inter alia конкретные соединения 6-(алкил)-4-амино-3-хлор-5-фторпиколинатов и их применение в качестве гербицидов. Каждый из этих патентов описывает получение исходных веществ для 4-амино-3-хлор-5-фторпиколината путем фторирования соответствующих 5-незамещенных пиридинов 1-(хлорметил)-4-фтор-1,4-диазонийбицикло[2.2.2]октан бис(тетрафторборатом). Было бы выгодно получать 4-амино-5-фтор-3-галоген-6-(замещенные)пиколинаты, без необходимости прямого фторирования положения 5 пиридинового кольца дорогим агентом фторирования, таким как 1-(хлорметил)-4-фтор-1,4-диазонийбицикло[2.2.2]октан бис(тетрафторборатом).
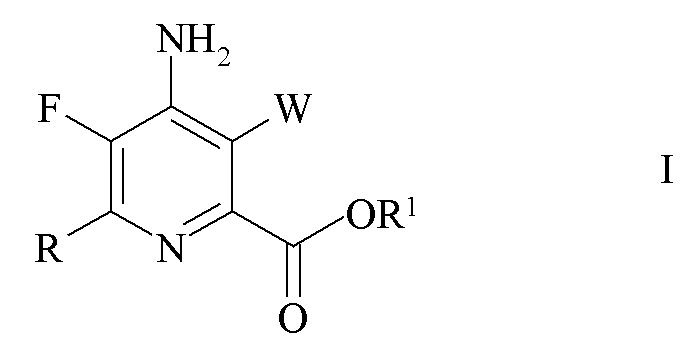
Настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов из 4,5,6-трихлорпиколинатов. Более конкретно, настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
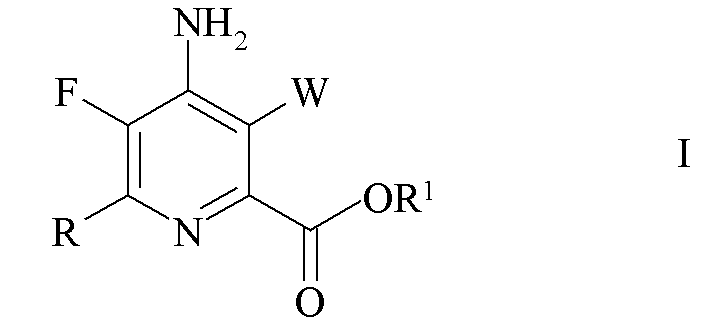
где
W представляет собой Cl, Br или I;
R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и
R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
который включает следующие стадии:
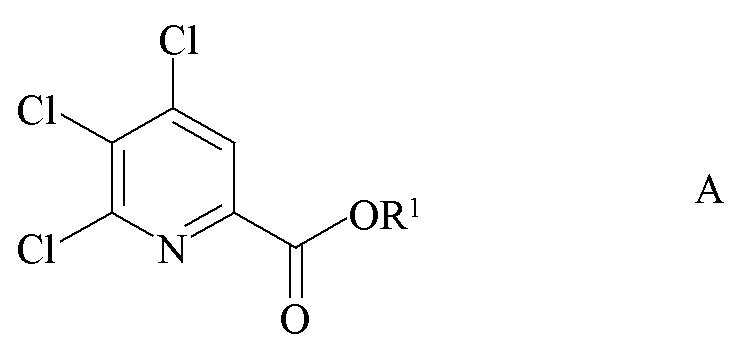
a) фторирование 4,5,6-трихлорпиколината формулы A
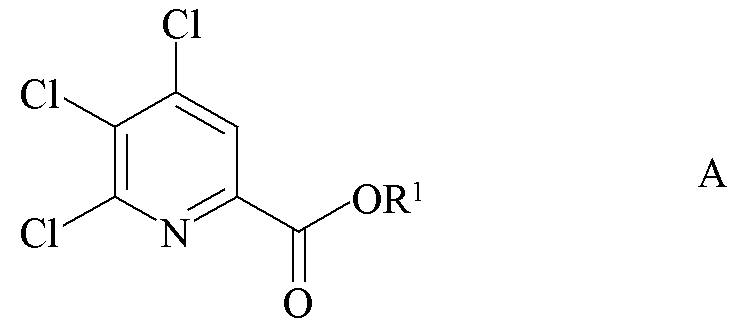
где R1 является таким, как определено ранее;
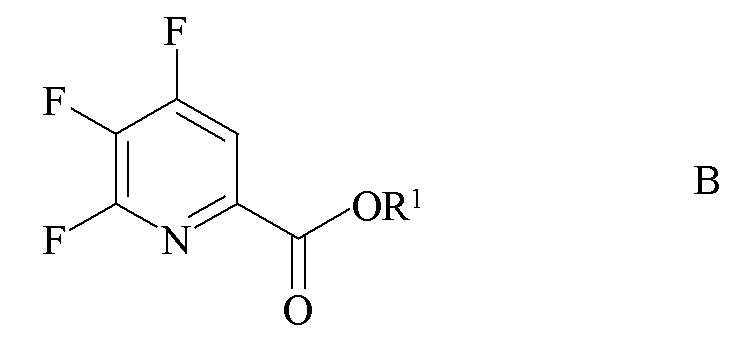
источником фторид-иона с получением 4,5,6-трифторпиколината формулы B
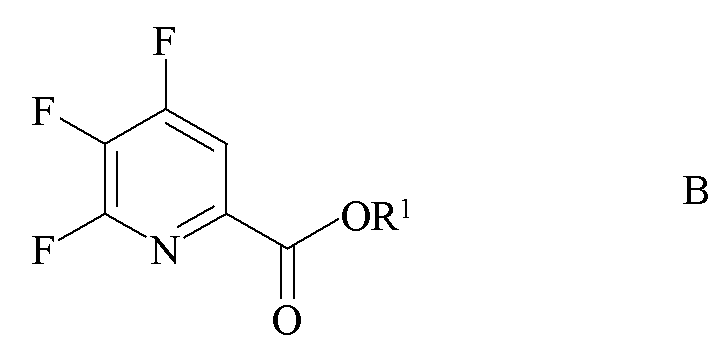
где R1 является таким, как определено ранее;
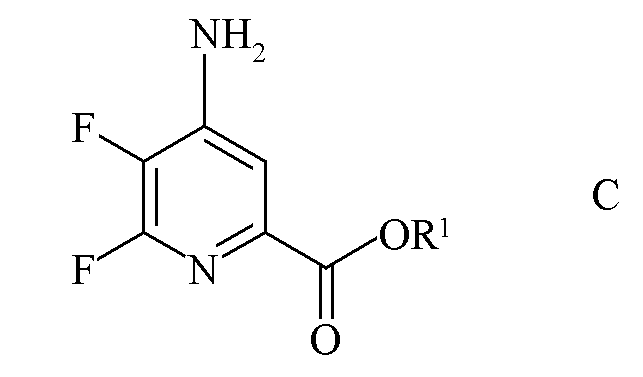
b) аминирование 4,5,6-трифторпиколината формулы B аммиаком с получением 4-амино-5,6-дифторпиколината формулы C
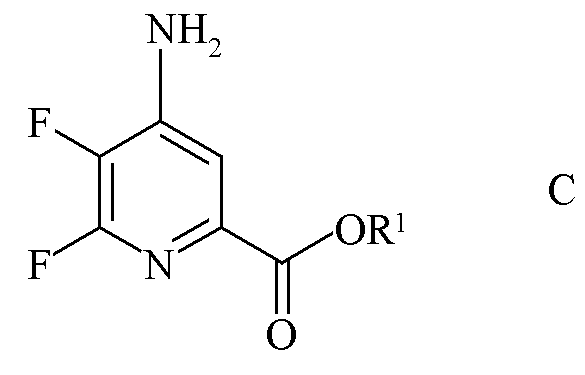
где R1 является таким, как определено ранее;
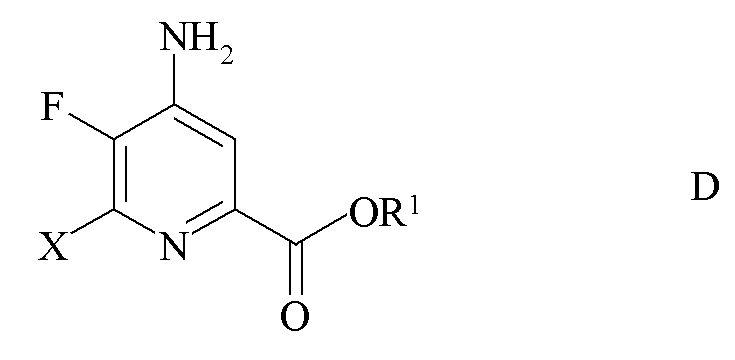
c) замена фтор-заместителя в положении 6 4-амино-5,6-дифторпиколината формулы C на йод, бром или хлор-заместитель путем обработки источником ионов йода, брома или хлора с получением 4-амино-5-фтор-6-галогенпиколината формулы D
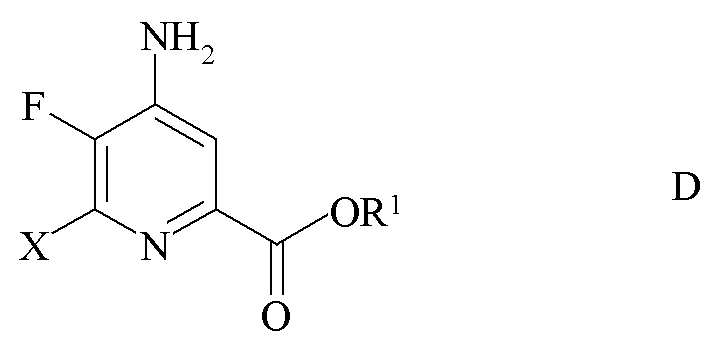
где X представляет собой Cl, Br или I; и
R1 является таким, как определено ранее;
d) галогенирование 4-амино-5-фтор-6-галогенпиколината формулы D источником галогена с получением 4-амино-3,6-дигалоген-5-фторпиколината формулы E
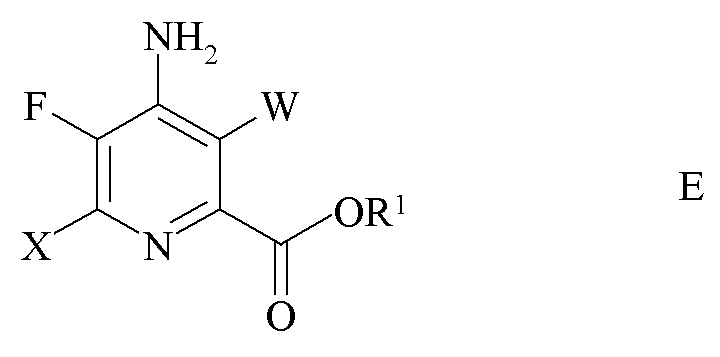
где W и X независимо представляют собой Cl, Br или I и
R1 является таким, как определено ранее; и
e) связывание 4-амино-3,6-дигалоген-5-фторпиколината формулы E с арильным, алкильным или алкенильным металлическим соединением формулы F

где R является таким, как определено ранее, и Met представляет собой Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо представляют собой водород, C1-C4 алкил или, взятые вместе, образуют этиленовую или пропиленовую группу в присутствии катализатора на основе переходного металла с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
Стадии a)-e) могут быть выполнены в перечисленном порядке, как изображено на схеме I.
Схема I
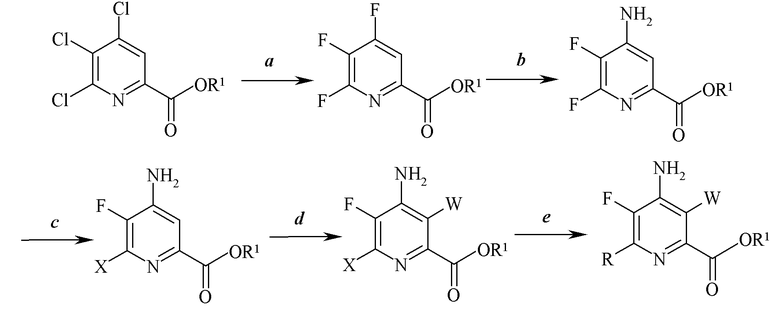
Альтернативно, порядок, в котором выполняют стадии, может быть перегруппирован так, как проиллюстрировано, например, на схемах II и III.
Схема II
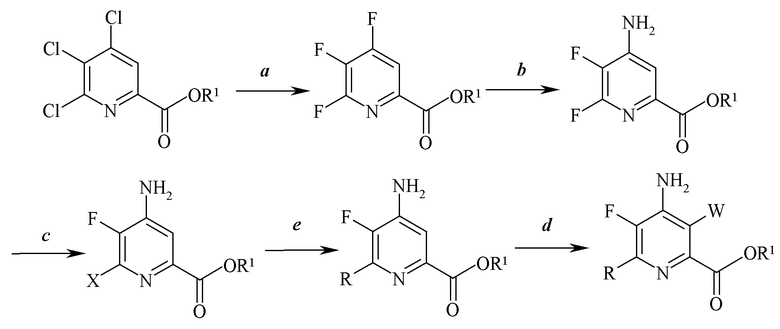
В соответствии со схемой II настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
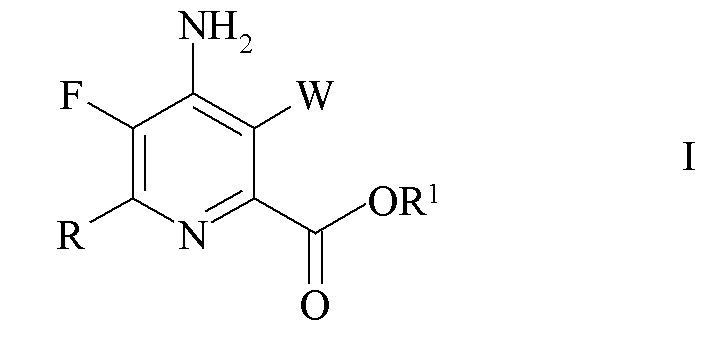
где
W представляет собой Cl, Br или I;
R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и
R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
который включает следующие стадии:
a) фторирование 4,5,6-трихлорпиколината формулы A

где R1 является таким, как определено ранее;
источником фторид-иона с получением 4,5,6-трифторпиколината формулы B
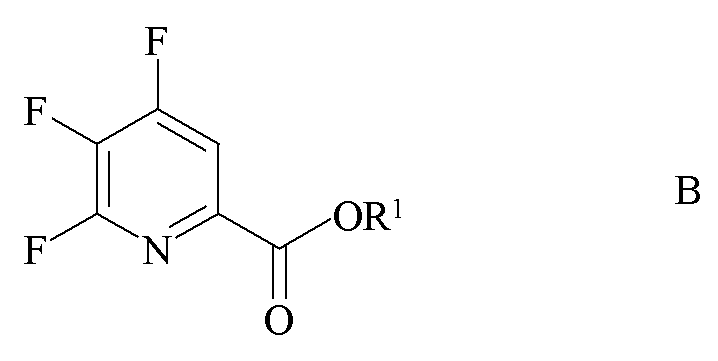
где R1 является таким, как определено ранее;
b) аминирование 4,5,6-трифторпиколината формулы B аммиаком с получением 4-амино-5,6-дифторпиколината формулы C
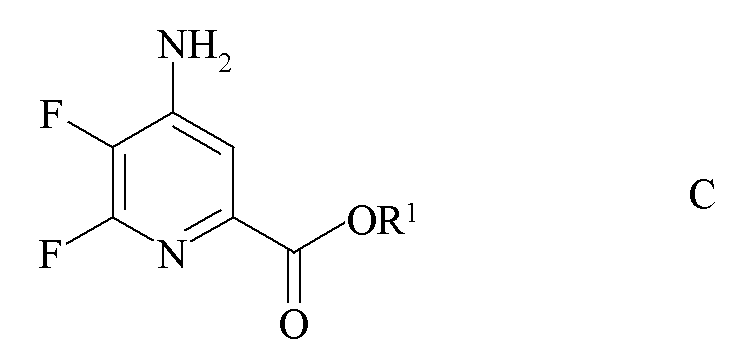
где R1 является таким, как определено ранее;
c) замена фтор-заместителя в положении 6 4-амино-5,6-дифторпиколината формулы C на йод, бром или хлор-заместитель путем обработки источником ионов йода, брома или хлора с получением 4-амино-5-фтор-6-галогенпиколината формулы D
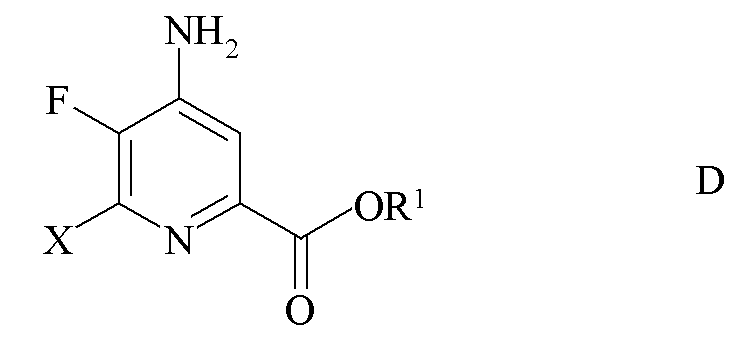
где X представляет собой Cl, Br или I и
R1 является таким, как определено ранее;
d) связывание 4-амино-5-фтор-6-галогенпиколината формулы D с арильным, алкильным или алкенильным металлическим соединением формулы F

где R является таким, как определено ранее, и Met представляет собой Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, C1-C4 алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу в присутствии катализатора на основе переходного металла с получением 4-амино-5-фтор-6-(замещенного)пиколината формулы G.
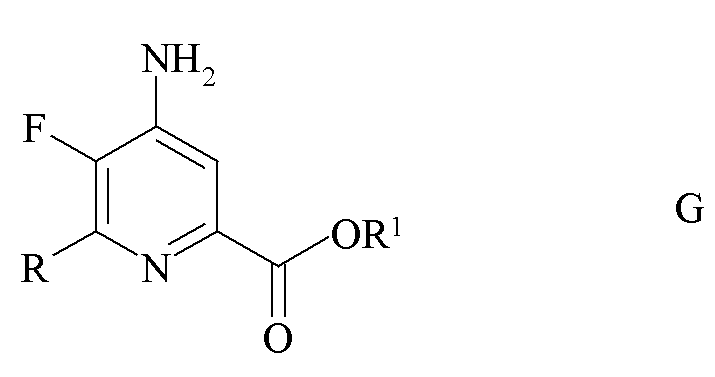
где R и R1 являются такими, как определено ранее; и
e) галогенирование 4-амино-5-фтор-6-(замещенного)пиколината формулы G источником галогена с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
Схема III
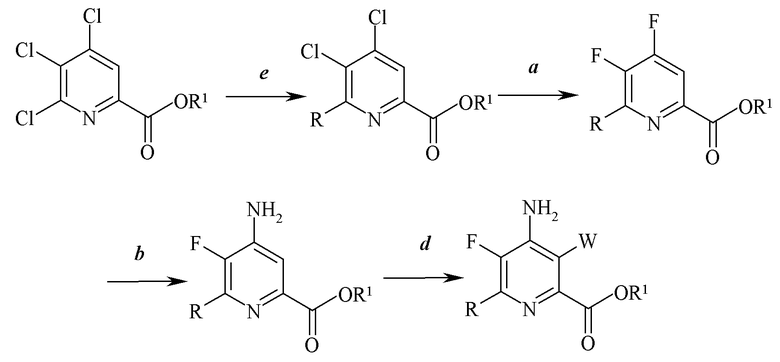
На схеме III стадия c) замены йода, брома или хлора не является необходимой. Таким образом, настоящее изобретение также относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
где
W представляет собой Cl, Br или I;
R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и
R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
который включает следующие стадии:
a) связывание 4,5,6-трихлорпиколината формулы A
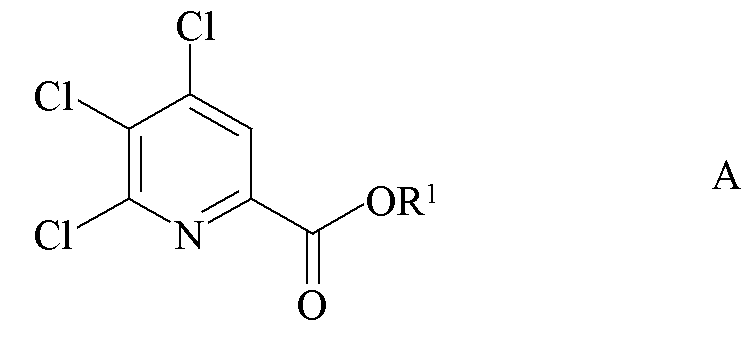
где R1 является таким, как определено ранее;
с арильным, алкильным или алкенильным металлическим соединением формулы F

где R является таким, как определено ранее, и Met представляет собой Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, C1-C4 алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу в присутствии катализатора на основе переходного металла с получением 4,5-дихлор-6-(замещенного)пиколината формулы H
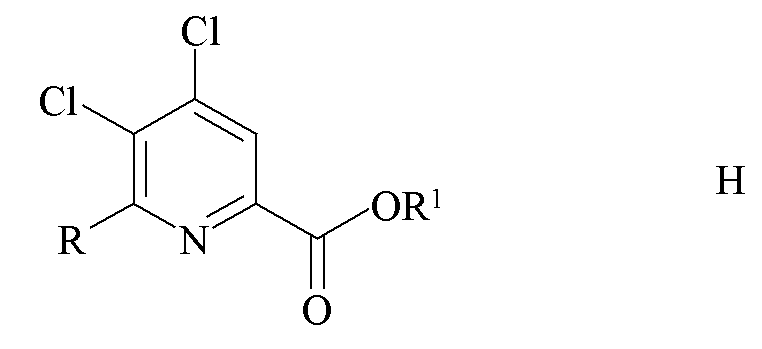
где R и R1 являются такими, как определено ранее;
b) фторирование 4,5-дихлор-6-(замещенного)пиколината формулы H источником фторид-ионов с получением 4,5-дифтор-6-(замещенного)пиколината формулы J

где R1 является таким, как определено ранее;
c) аминирование 4,5-дифтор-6-(замещенного)пиколината формулы J аммиаком с получением 4-амино-5-фтор-6-(замещенного)пиколината формулы K
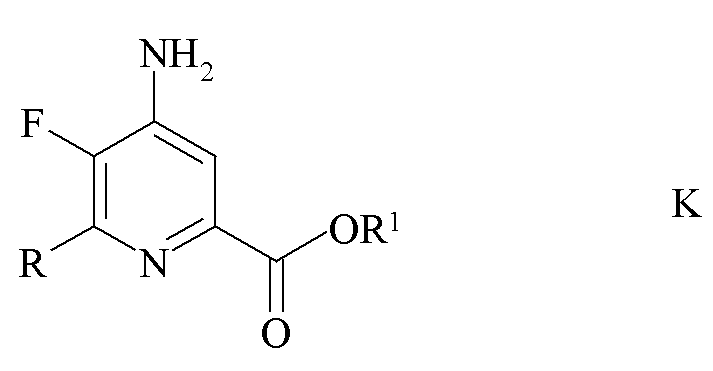
где R и R1 являются такими, как определено ранее; и
d) галогенирование 4-амино-5-фтор-6-(замещенного)пиколината формулы K источником галогена с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
На любой стадии на схемах I-III заместитель сложного эфира R1 может необязательно быть заменен на другой заместитель R1. Эти сложные эфиры, включая незамещенные или замещенные сложные C7-C11 арилалкиловые эфиры, могут быть получены этерификацией без катализатора или посредством реакций переэтерификации с применением способов, известных в области техники.
Другим аспектом настоящего изобретения являются новые промежуточные продукты, получаемые в настоящем способе, то есть соединения, выбранные из группы, состоящей из:
a)
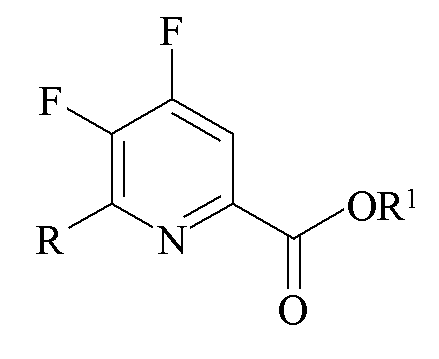
где R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
b)
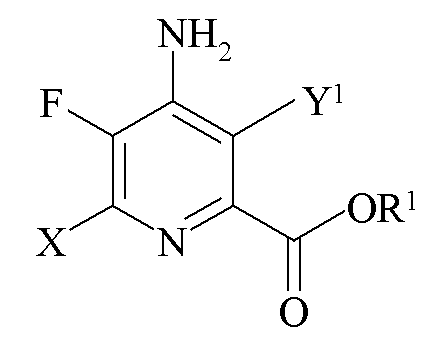
где X представляет собой I, Br, Cl или F, Y1 представляет собой H, Cl, Br или I, при условии, что, когда X представляет собой Cl, Y1 представляет собой H, Br или I, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
c)
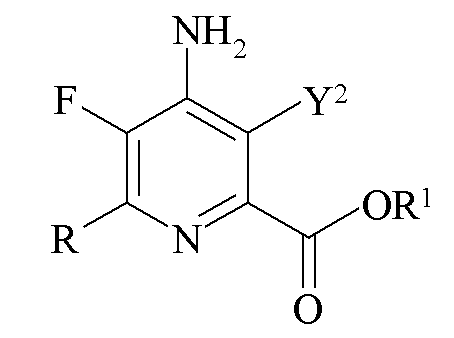
где Y2 представляет собой H, Br или I и R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил; и
d)
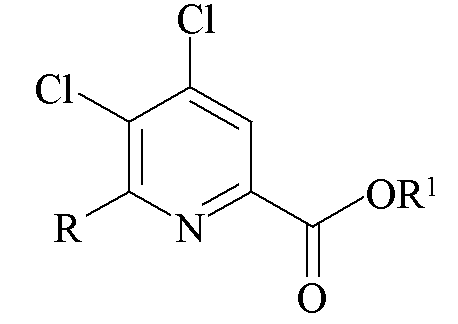
где R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил.
Термины «алкил», «алкенил» и «алкинил», а также производные термины, такие как «алкокси», «ацил», «алкилтио» и «алкилсульфонил», как используется в настоящем описании, охватывают фрагменты с неразветвленной, разветвленной цепью и циклические фрагменты. Если конкретно не указано иначе, каждый может быть незамещенным или замещенным одним или более заместителями, выбранными из, но не ограничиваясь ими, галогена, гидрокси, алкокси, алкилтио, C1-C6 ацила, формила, циано, арилокси или арила, при условии, что заместители являются стерически совместимыми и соблюдаются правила образования химической связи и энергии деформации. Термины «алкенил» и «алкинил» предназначены для обозначения включения одной или более ненасыщенных связей.
Термин «арилалкил», как используется в настоящем описании, относится к фенилзамещенной алкильной группе, содержащей в общей сложности 7-11 углеродных атомов, такой как бензил (-CH2C6H5), 2-метилнафтил(-CH2C10H7) и 1- или 2-фенэтил (-CH2CH2C6H5 или -CH(CH3)C6H5). Фенильная группа может быть сама по себе незамещенной или замещенной одним или более заместителями, независимо выбранными из галогена, нитро, циано, C1-C6 алкила, C1-C6 алкокси, галогенированного C1-C6 алкила, галогенированной C1-C6 алкокси, C1-C6 алкилтио, C(O)OC1-С6алкил, или где два смежных заместителя, взятые вместе, представляют собой O(CH2)nO-, где n=1 или 2, при условии, что заместители являются стерически совместимы и соблюдаются условия образования химической связи и энергии деформации.
Если конкретно не указано иначе, термин «галоген», а также производные термины, такие как «гало», относятся к фтору, хлору, брому и йоду.
6-Фенильные группы, замещенные 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, могут иметь любую ориентацию, но предпочтительны изомеры 4-замещенный фенил, 2,4-двузамещенный фенил, 2,3,4-трехзамещенный фенил, 2,4,5-трехзамещенный фенил и 2,3,4,6-четырехзамещенный фенил.
4-Амино-5-фтор-3-галоген-6-(замещенные)пиколинаты получают из 4,5,6-трихлорпиколинатов серией стадий, включающих замену фтора, аминирование, замену галогена, галогенирование и связывание при помощи переходного металла. Индивидуальные стадии могут быть выполнены в различных последовательностях.
Исходные вещества для 4,5,6-трихлорпиколината являются известными соединениями; см., например, пример 3 в патенте США 6784137 B2. Высшие сложные эфиры, включая незамещенные или замещенные сложные C7-C11 арилалкиловые эфиры, могут быть получены реакциями прямой этерификацией или переэтерификации с применением способов, известных в области техники.
В реакции замены фтора фторированный пиколинат получают взаимодействием соответствующего хлорированного пиколината с по меньшей мере одним эквивалентом источника фторид-ионов для замены каждого заменяемого хлора-заместителя в кольце.
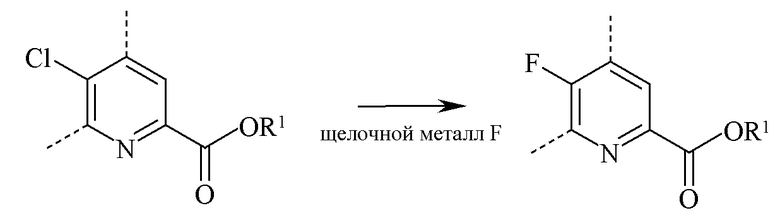
Типичные источники фторид-ионов представляют собой фториды щелочных металлов, включающие фторид натрия (NaF), фторид калия (KF) и фторид цезия (CsF), где предпочтительными являются KF и CsF. Также могут быть применены фторидные соли, такие как фторид тетрабутиламмония (н-Bu4NF). Предпочтительно реакцию проводят в полярном апротонном растворителе или такой реакционной среде, как диметилсульфоксид (ДМСО), N-метилпирролидинон (ΝΜΡ), NN-диметилформамид (ДМФ), гексаметилфосфорамид (HMPA) или сульфолан. Также могут быть применены добавки, такие как краун-эфиры или агенты фазового переноса, которые, как известно, увеличивают скорость обмена фторидом. Температура, при которой проводят реакцию, не является важной, но обычно составляет от 70°C до 180°C и предпочтительно от 80°C до 120°C. Оптимальная температура будет меняться в зависимости от того, какой применяют растворитель в конкретной реакции. В общем, чем ниже температура, тем медленнее будет протекать реакция. Реакция по настоящему изобретению, как правило, проводится при энергичном перемешивании, достаточном для поддержания по существу однородной дисперсной смеси реагентов.
При проведении реакции фторирования ни скорость, ни порядок добавления реагентов не являются важными. Обычно растворитель и фторид щелочного металла смешивают до того, как в реакционную смесь добавляют хлорированный пиколинат. Типовая реакция, как правило, занимает от 2 до 100 часов и обычно проводится при атмосферном давлении.
В то время как точные количества реагентов не важны, предпочтительно применение такого количества фторида щелочного металла, которое обеспечит по меньшей мере эквимолярное количество атомов фтора по отношению к количеству замещаемых атомов хлора в исходном веществе, то есть по меньшей мере эквимолярного количества фторида щелочного металла. После завершения реакции требуемый продукт регенерируют путем применения стандартных способов разделения и очистки.
При аминировании 4-фторпиколинату позволяют реагировать с аммиаком для замещения атома фтора аминогруппой:
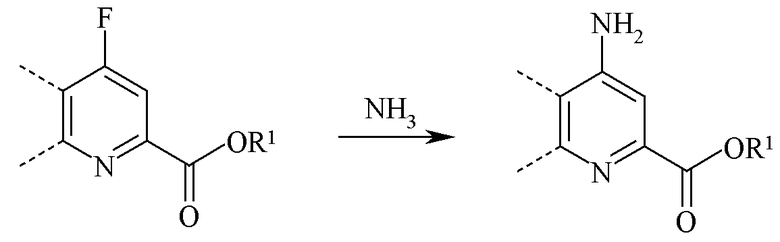
В то время как необходимо только стехиометрическое количество аммиака, часто удобно применять большой избыток аммиака. Реакцию проводят в инертном растворителе, предпочтительно полярном апротонном растворителе или в реакционной среде, такой как ДМСО, NMP, ДМФ, HMPA или сульфолан. Альтернативно, может быть применен водный гидроксид аммония с использованием или без использования органического растворителя. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 0°C до 45°C и предпочтительно от 10°C до 30°C.
При проведении реакции аминирования 4-фторпиколинат растворяют в растворителе, и в реакционную смесь добавляют аммиак с охлаждением. Избыток газа аммиака, как правило, барботируют через реакционную смесь. Типичная реакция, как правило, занимает от 0,5 до 5 часов и обычно проводится при атмосферном давлении.
Аминсодержащие продукты или промежуточные продукты, получаемые любым из этих способов, могут быть регенерированы обычными способами, такими как выпаривание или экстракция, и могут быть очищены стандартными способами, такими как перекристаллизация или хроматография. На очистку аминсодержащих продуктов или промежуточных продуктов может также влиять протонирование кислотой с образованием соли, которую выделяют с высокой чистотой путем кристаллизации, осаждения или экстракции. Может быть применено множество кислот, таких как соляная кислота, бромистоводородная кислота, азотная кислоты, уксусная кислота или серная кислота. Безводная соляная кислота является предпочтительной кислотой. Очищенную соль затем нейтрализуют основанием с образованием нейтрального аминсодержащего продукта или промежуточного продукта. Могут быть применены неорганические основания, такие как гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия или гидрокарбонат натрия. Предпочтительны органические основания, такие как триэтиламин. Очистка аминсодержащего продукта или промежуточного продукта может быть выполнена таким образом незамедлительно после стадии аминирования или после проведения последующих реакций, например, галогенирования, связывания.
В реакции обмена галогеном (йодом, бромом или хлором), 6-йодированный, 6-бромированный или 6-хлорированный пиколинат получают взаимодействием соответствующего 6-фторированного пиколината с по меньшей мере одним эквивалентом иона йодида, бромида или хлорида.
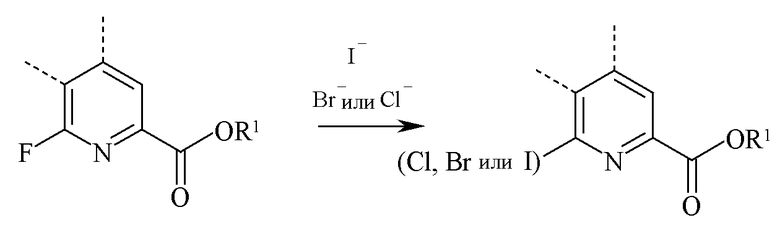
Как правило, реакцию обмена галогеном проводят в присутствии большого избытка безводного йодоводорода (HI), бромводорода (HBr) или хлороводорода (HCl). Реакцию, как правило, проводят в отсутствие воды для снижения образования побочных продуктов. Для обмена галогеном, как правило, требуется от 5 до 50 эквивалентов HI, HBr или HCl, предпочтительно от 10 до 20 эквивалентов. Реакция проводят в инертном растворителе, предпочтительно полярном растворителе, таком как диоксан или уксусная кислота. Температура, при которой проводят реакции, не является важной, но, как правило, составляет от 75°C до 150°C и предпочтительно от 100°C до 125°C. Реакцию, как правило, проводят в герметично закрытом реакторе под давлением, в котором могут удерживаться газы HI, HBr или HCl. Типичная реакция занимает, как правило, от 0,5 до 5 часов.
В реакции галогенирования атом хлора, брома или йода вводят в положение 3 молекулы пиколината путем реакции 3-незамещенного пиколината источником галогена в инертном растворителе:
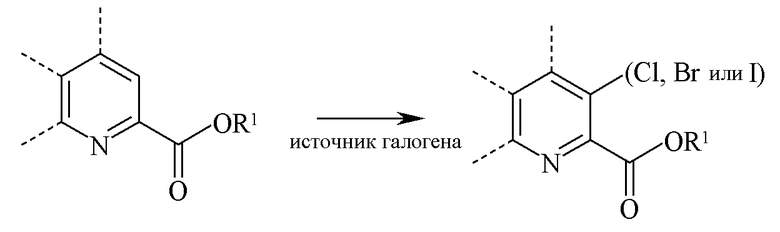
Когда атомом галогена в положении 3 является Cl, источником хлора может быть собственно хлор (Cl2) или реагенты, такие как сульфурилхлорид, N-хлорсукцинимид или 1,3-дихлор-5,5-диметилгидантоин. При использовании хлора или сульфурилхлорида применяют большой избыток хлорирующего агента. При применении хлоргаза реакцию проводят в инертном растворителе, предпочтительно таком растворителе, как дихлорметан, дихлорметан-вода или уксусная кислота. При использовании сульфурилхлорида реакция можно проводить в инертном растворителе, таком как дихлорметан или беспримесный сульфурилхлорид. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 0°C до 45°C и предпочтительно от 10°C до 30°C. Типичная реакция, как правило, занимает от 0,5 до 5 часов. Реакцию хлорирования, как правило, проводят при атмосферном давлении.
Когда применяемый хлорирующий агент представляет собой N-хлорсукцинимид или 1,3-дихлор-5,5-диметилгидантоин, реакцию проводят с применением стехиометрического количества хлорирующего реагента. Обнаружено, что в реакциях хлорирования с применением 1,3-дихлор-5,5-диметилгидантоина в качестве хлорирующего агента реагируют оба хлора в гидантоине. Реакцию проводят в инертном полярном растворителе, таком как ДМФ или ацетонитрил. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 20°C до 85°C и предпочтительно от 50°C до 80°C. Когда ацетонитрил применяют в качестве растворителя, удобно проводить реакцию при температуре кипения с обратным холодильником. Типичная реакция, как правило, занимает от 0,5 до 5 часов. Реакцию хлорирования, как правило, проводят при атмосферном давлении.
Когда атом галогена в положении 3 является бромом, источником брома может быть собственно бром (Br2) или реагенты, такие как сульфурилбромид, N-бромсукцинимид или 1,3-дибром-5,5-диметилгидантоин. Когда Br2 применяют в качестве агента бромирования, может быть использован большой избыток, и реакцию проводят в инертном растворителе, предпочтительно таком растворителе, как дихлорметан, дихлорметан-вода или уксусная кислота. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 0°C до 45°C и предпочтительно от 10°C до 30°C. Типичная реакция, как правило, занимает от 0,5 до 5 часов. Реакцию бромирования обычно проводят при атмосферном давлении.
Когда применяемый агент бромирования представляет собой N-бромсукцинимид или 1,3-дибром-5,5-диметилгидантоин, реакцию проводят с применением стехиометрического количества реагента бромирования. Реакцию проводят в инертном полярном растворителе, таком как ДМФ или ацетонитрил. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 20°C до 85°C и предпочтительно от 50°C до 80°C. Когда ацетонитрил применяют в качестве растворителя, удобно проводить реакцию при температуре флегмы. Типичная реакция, как правило, занимает от 0,5 до 5 часов. Реакцию бромирования, как правило, проводят при атмосферном давлении.
Когда атом галогена в положении 3 представляет собой I, источником йода может быть собственно йод (I2) или реагенты, такие как монохлорид йода или N-йодсукцинимид. Йодная кислота может быть использована вместе с I2. Когда I2 используют в качестве йодирующего реагента, может быть использован большой избыток I2, и реакцию проводят в инертном растворителе, предпочтительно, таком растворителе, как дихлорметан, дихлорметан-вода, метиловый спирт или уксусная кислота. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 0°C до 45°C и предпочтительно от 10°C до 30°C. Типичная реакция, как правило, занимает от 0,5 до 5 часов. Реакцию йодирования, как правило, проводят при атмосферном давлении.
В реакции связывания 6-йод, бром или хлорпиколинат реагирует с арильным, алкильным или алкенильным металлическим соединением, где металлом является Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, C1-C4 алкил или, взятые вместе, образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла:
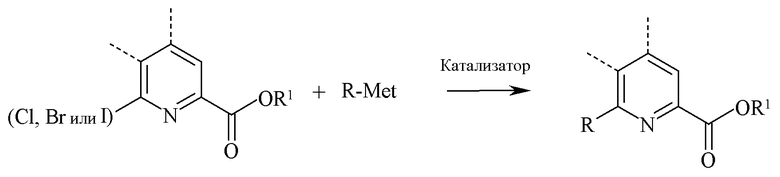
«Катализатор» представляет собой катализатор на основе переходного металла, в частности, палладиевый катализатор, такой как диацетат палладия или дихлоридбис(трифенилфосфин)палладия(II) или никелевый катализатор, такой как никель(II), ацетилацетонат или дихлорид бис(трифенилфосфин)никеля(II). Кроме того, катализаторы могут быть получены in situ из солей металлов и лигандов, таких как ацетат палладия и трифенилфосфин или хлорид никеля(II) и трифенилфосфин. Эти in situ катализаторы могут быть получены предварительным взаимодействием соли металла и лиганда с последующим добавлением к реакционной смеси, или отдельным добавлением соли металла и лиганда непосредственно в реакционную смесь.
Как правило, реакции связывания проводят в отсутствие кислорода с применением инертного газа, такого как азот или аргон. Способы, применяемые для удаления кислорода из реакционных смесей, где происходит связывание, такие как барботирование инертным газом, известны специалистам в области техники. Примеры таких способов описаны в The Manipulation of Air-Sensitive Compounds, 2-е издание; Редакторы Shriver, D. F., Drezdzon, М. A.; Wiley-Interscience, 1986. Применяют субстехиометрические количества катализатора, как правило, от 0,0001 эквивалента до 0,1 эквивалента. Дополнительные количества лиганда могут быть необязательно добавлены для увеличения устойчивости катализатора и его действия. Дополнительно, добавки, такие как Na2CO3, K2CO3, KF, CsF и NaF, как правило, добавляют к реакции связывания. Реакция связывания, как правило, требует от 1 до 5 эквивалентов такой добавки, предпочтительно от 1 до 2 эквивалентов. Вода может быть необязательно добавлена к реакции связывания для увеличения растворимости этих добавок. Реакция связывания, как правило, требует от 1 до 3 эквивалентов арильного, алкильного или алкенильного металлического соединения, предпочтительно от 1 до 1,5 эквивалентов. Реакцию проводят в инертном растворителе, таком как толуол, тетрагидрофуран (ТГФ), диоксан или ацетонитрил. Температура, при которой проводят реакцию, не является важной, но, как правило, составляет от 25°C до 150°C и предпочтительно от 50°C до 125°C. Типичная реакция, как правило, занимает от 0,5 до 24 часов. Никакого специфического порядка добавления реагентов, как правило, не требуется. На практике часто проще объединять все реагенты кроме катализатора и затем удалять кислород из реакционной смеси, в которой будет протекать реакция связывания. После удаления кислорода может быть добавлен катализатор для инициирования реакции связывания.
Когда Met часть арильного, алкильного или алкенильного металлического соединения представляет собой Zn-галогенид, Zn-R или медь, может быть необходимой защита реакционноспособных функциональных групп. Например, если присутствует амино-заместитель (-NHR или -NH2), может быть необходимой защита этих реакционноспособных групп. В области техники известно множество групп для защиты аминогрупп от реакции с металлорганическими реагентами. Примеры таких блокирующих групп описаны в Protective Groups in Organic Synthesis, 3-е издание; Редакторы Greene, T. W.; Wuts, P. G. M.; Wiley-Interscience, 1999. Выбором применяемого металла в R-Met зависит от многих факторов, таких как стоимость, устойчивость, реакционная способность и необходимость в защите реакционноспособных функциональных групп.
Продукты, полученные любым из этих способов, могут быть регенерированы обычными способами, такими как выпаривание или экстракция, и могут быть очищены стандартными способами, такими как перекристаллизация или хроматография.
Следующие примеры представлены для иллюстрации настоящего изобретения.
Примеры
Получение исходного вещества
Пример A. Пропан-2-ил 4,5,6-трихлорпиколинат
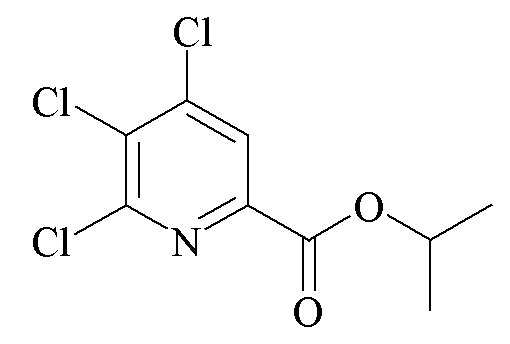
Метил 4,5,6-трихлорпиколинат (14,19 граммов (г), 59,0 миллимоль (ммоль)) суспендировали в 2-пропаноле (150 миллилитров (мл)) в круглодонной колбе объемом 250 мл, оборудованной ловушкой Дина-Старка и обратным холодильником. Добавляли серную кислоту (98% H2SO4; 8,07 г, 82 ммоль) и реакционную смесь нагревали с обратным холодильником. Через 20 часов (ч) нагревания с обратным холодильником, большая часть 2-пропанола (100 мл) отгонялась в первую фракцию дистиллята. Содержимое резервуара отвердевало при охлаждении до комнатной температуры. Полученное твердое вещество перемешивали с этилацетатом (EtOAc; 500 мл) и насыщенным (насыщ.) водным (водн.) раствором гидрокарбоната натрия (NaHCO3; 500 мл). Органический слой отделяли, промывали солевым раствором и затем фильтровали через целит. Органический экстракт концентрировали до 150 мл на роторном испарителе. Добавляли гексан (100 мл) и раствор оставляли при -20°C в течение ночи. Кристаллы собирали, промывали гексаном и высушивали на воздухе (7,58 г, температура плавления 10,6-105,7°C). Второй продукт получали путем концентрации фильтрата с получением в общей сложности 10,36 г (65%) 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (с, 1H, пиридин H), 5,16 (септет, J=6,3 Гц, 1H, CHMe2), 1,34 (д, J=6,3 Гц, 6H, CHMe2); 13C{1H} ЯМР (101 МГц, CDCl3) δ 161,9 (CO2R), 150,6, 145,9, 145,0, 133,1, 125,4 (C3), 70,7 (CHMe2), 21,7 (Me). Аналитически рассчитано для C9H8Cl3NO2: C, 40,26; H, 3,00; N, 5,22. Обнаружено: C, 40,25; H, 3,02; N, 5,22.
Пример B. Бензил 4,5,6-трихлорпиколинат
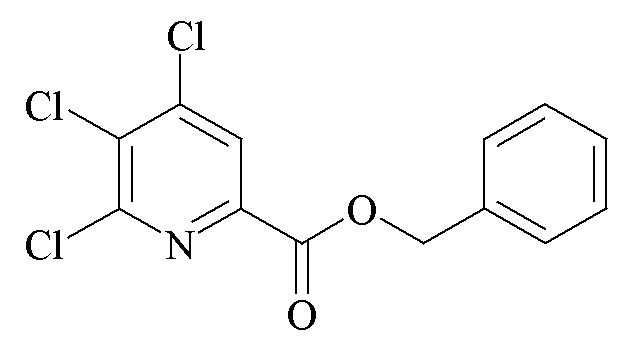
Смесь метил 4,5,6-трихлорпиколината (25 г, 0,10 моль (моль)) и бензилового спирта (100 г, 0,2 моль) в трехгорлой круглодонной колбе объемом 250 мл нагревали в атмосфере азота до 100°C. Добавляли изопропилат титана (0,6 г, 0,02 моль). Через 4 ч при 100°C почти бесцветный раствор охлаждали и переносили в одногорловую круглодонную колбу объемом 250 мл. Избыток бензилового спирта удаляли под вакуумом с получением почти белого твердого вещества (31 г, 94%): температура плавления 125-126,5°C; 1H ЯМР (400 МГц, CDCl3) δ 8,08 (с, 1H, пиридин H), 7,42 (м, 2H, фенил), 7,31 (м, 3H, фенил), 5,40 (с, 2H, CH2Ph); 13C{1H} ЯМР (101 МГц, CDCl3) δ 162,0 (CO2R), 150,4, 145,0, 144,9, 134,7, 133,1, 128,3 (фенил CH), 125,4 (пиридин CH), 67,88 (CH2Ph).
Пример C. Бензил 4,5,6-трихлорпиколинат
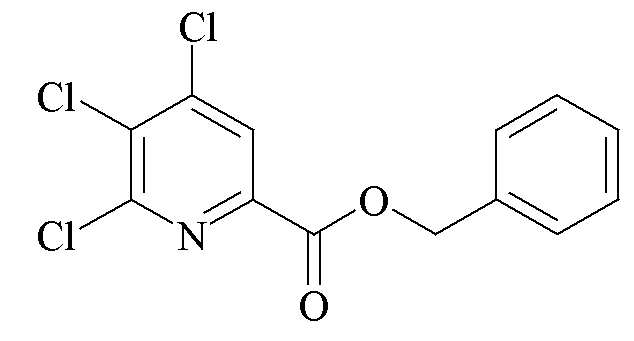
Круглодонная колба на 22 л была оснащена термопарой, механической мешалкой и ловушкой Дина-Старка, связанной с азотным барботером. Сосуд продували азотом и затем добавляли 4,5,6-трихлорпиколинат (2547 г, 10,07 моль), п-толуолсульфоната пиридиния (PPTS; 130 г, 0,52 моль), бензиловый спирт (2249 г, 20,8 моль) и ксилолы (10278 г). Начинали перемешивание и резервуар нагревали до 140-145°C. Азеотропную смесь ксилолы/вода собирали в ловушке Дина-Старка в течение 5 ч. Общее количество дистиллята составляло 4750 г (415 г составляла вода). После того как вода прекращала переходить в первую фракцию дистиллята, из реактора отбирали образец и анализировали высокоэффективной жидкостной хроматографией (ВЭЖХ), чтобы гарантировать остаток исходной карбоновой кислоты менее 1,5% площади хроматограммы. Реакционную смесь оставляли охлаждаться до комнатной температуры и перемешивали в течение ночи. Ксилолы (4000 г) удаляли перегонкой под вакуумом. Раствор охлаждали до 85-100°C и затем вакуумно переносили в сосуд с рубашкой для кристаллизации объемом 30 л, оснащенный механической мешалкой и термопарой. Вакуум наполняли азотом и азотный барботер помещали на сосуд для кристаллизации. В течение 15 минут (мин) к раствору кслилола добавляли изопропиловый спирт (IPA; 6200 г). Полученной взвеси позволяли медленно охладиться до комнатной температуры и затем охлаждали дополнительно до 5°C. Твердое вещество собирали путем фильтрации и фильтровальный осадок промывали холодным (5-10°C) IPA (3731 г). Твердое вещество высушивали на воздухе до постоянной массы с получением белых кристаллов (2765 г, газовая хроматография (ГХ) внутренний стандарт чистоты 96,5%, 84,3%).
Обмен фтором
Пример 1a. Пропан-2-ил 4,5,6-трифторпиколинат
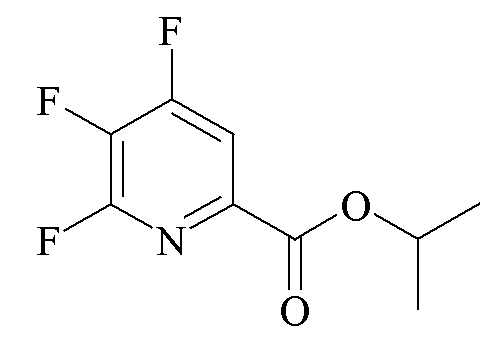
Трехгорлая колба объемом 250 мл была оборудована механической мешалкой, ловушкой Дина-Старка со входом для азота и термопарой. Колбу продували азотом и добавляли CsF (23,38 г, 154 ммоль). Добавляли безводный ДМСО (124 мл) и атмосферу над суспензией разрежали при помощи вакуумного насоса/заполняли (5×) азотом. Суспензию нагревали при 80°C в течение 30 минут. ДМСО (20 мл) отгоняли под вакуумом при 75°C для удаления любой остаточной воды. Добавляли пропан-2-ил 4,5,6-трихлорпиколинат (13,45 г, 50,1 ммоль) на фоне продувки азотом. Атмосферу над реакционной смесью разрежали при помощи вакуумного насоса/заполняли (3×) азотом и нагревали при 100°C в течение 1 ч при энергичном перемешивании.
Вторая трехгорлая колба объемом 250 мл была оборудована механической мешалкой, ловушкой Дина-Старка со входом для азота и термопарой. Колбу продували азотом и добавляли CsF (24,41 г, 0,160 ммоль). Добавляли безводный ДМСО (30 мл) и атмосферу над суспензией разрежали при помощи вакуумного насоса/заполняли (5×) азотом. Суспензию нагревали при 80°C в течение 30 минут. ДМСО (22 мл) отгоняли под вакуумом при 75°C для удаления остаточной воды. Охлажденную реакционную смесь из первой колбы фильтровали через канюлю во вторую колбу в атмосфере азота. Атмосферу над реакционной смесью разрежали при помощи вакуумного насоса/заполняли азотом (5×) и затем нагревали при 100°C в течение 1 ч и затем в течение еще 90 минут при 110°C. ГХ аликвоту образца анализировали при помощи ГХ, анализ показал присутствие 96% пропан-2-ил 4,5,6-трифторпиколината с примесью только 1,4% пропан-2-ил 5-хлор-4,6-дифторпиколината. Раствор неочищенного продукта применяли непосредственно на стадии аминирования без дополнительной очистки. Альтернативно, продукт может быть выделен путем обработки водой, экстракцией при помощи EtOAc и высушиванием с получением светлого желто-коричневого масла: 1H ЯМР (400 МГц, CDCl3) δ 7,94 (дд, JF-H=4,5, 8,7 Гц, 1H, H3), 5,30 (септет, JH-H=6,3 Гц, 1H, CHMe2), 1,44 (д, JΗ-Η=6,3 Гц, 6H, CHMe2); 13C {1H} ЯМР (101 МГц, CDCl3) δ 161,2 (с, CO2iPr), 157,3 (ддд, JF-C=266, 8, 6 Гц, C4/C6), 152,2 (ддд, JF-C=241, 12, 5 Гц, C4/C6), 141,1 (дт, JF-C=14, 7 Гц, C2), 137,0 (ддд, JF-C=270, 31, 13 Гц, C5), 113,8 (дд, JF-C=17, 4 Гц, C3), 70,4 (с, CHMe2), 21,33 (с, ME); 19F ЯМР (376 МГц, CDCl3) δ -74,29 (дд, JF-F=24, 22 Гц, F6), -112,67 (ддд, JF-F=22, 19, JF-H=8,3 Гц, F4), -151,58 (ддд, JF-F=24, 19, JF-H=4,7 Гц, F5).
Пример 1b. Пропан-2-ил 4,5-дифтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
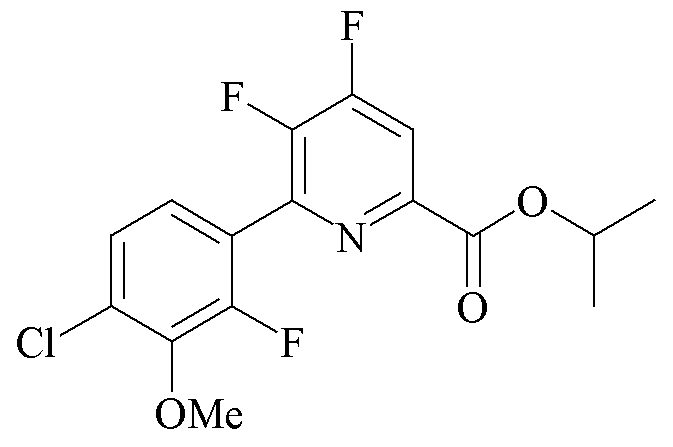
Трехгорлая колба объемом 250 мл была оборудована дефлегматором, входом для азота, механической мешалкой и термопарой. Колбу заполняли CsF (14,2 г, 93,0 ммоль), добавляли безводный ДМСО (80 мл) и атмосферу над суспензией разрежали при помощи вакуумного насоса/заполняли (5×) азотом. Суспензию нагревали при 80°C в течение 30 минут. ДМСО (20 мл) отгоняли под вакуумом для удаления любой остаточной воды. Добавляли твердый пропан-2-ил 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (10,44 г, 26,6 ммоль), и атмосферу над раствором разрежали при помощи вакуумного насоса/заполняли (5×) азотом. Реакционную смесь нагревали до 105°C в атмосфере азота. Через 4 ч при 105°C ГХ анализ аликвоты образца показал отношение 91:6 дифторзамещенных к монофторзамещенным продуктам. Реакционную смесь оставляли охлаждаться до комнатной температуры.
Вторая трехгорлая колба объемом 250 мл была оборудована механической мешалкой, дефлегматором с входом для азота и термопарой. Колбу продували азотом и добавляли CsF (7,5 г, 49,4 ммоль). Добавляли безводный ДМСО (20 мл) и атмосферу над суспензией разрежали при помощи вакуумного насоса/заполняли (5×) азотом. Суспензию нагревали при 80°C в течение 30 минут. ДМСО (15 мл) отгоняли под вакуумом для удаления остаточной воды. Охлажденную реакционную смесь из первой колбы фильтровали через канюлю во вторую колбу в атмосфере азота. Атмосферу над реакционной смесью разрежали при помощи вакуумного насоса/заполняли азотом (5×) и затем нагревали при 100°C в течение 2 ч. ГХ анализ аликвоты образца показал отношение 93:2 желаемого продукта к монофторозамещенному промежуточному продукту. Реакционную смесь выливали в воду со льдом (550 г) и экстрагировали при помощи EtOAc (3×200 мл). Объединенные органические экстракты промывали водой (5×100 мл) и солевым раствором, высушивали над сульфатом магния (MgSO4) и концентрировали при пониженном давлении с получением коричневого масла (8,57 г), которое кристаллизовалось при отстаивании. Твердое вещество очищали хроматографией на силикагеле (330 г колонка с силикагелем; градиент EtOAc-гексан 0-50%) с получением белого твердого вещества (4,98 г, 52%): температура плавления 98,4-100,0°C; 1H ЯМР (400 МГц, ацетон-d6) δ 8,16 (дд, JF-H=10,0, 5,6 Гц, 1H, пиридин H), 7,43 (м, 2H, фенил), 5,24 (гептет, JH-H=6,3 Гц, 1H, CHMe2), 4,01 (д, JF-H=1,1 Гц, 3H, OMe), 1,37 (д, JH-H=6,3 Гц, 6H, CHMe2); 13С{1H} ЯМР (101 МГц, ацетон-d6) δ 163,1 (CO2R), 157,1 (дд, JF-C=264, 12 Гц, C4/C5), 154,8 (д, JF-C=254 Гц, C2' фенил), 148,6 (дд, JF-C=267, 11 Гц, C4/C5), 147,4 (т, JF-C=6 Гц), 145,5 (д, JF-C=13 Гц), 144,6 (д, JF-C=13 Гц), 131,0, 126,8, 126,6 (д, JF-C=3,7 Гц), 123,2, 115,8 (д, JF-C=16 Гц), 70,6 (CHMe2), 62,1 (д, JF-C=4 Гц, OMe), 21,9 (CHMe2); 19F ЯМР (376 МГц, CDCl3) δ -124,82 (дд, JF-F=21 Гц, JF-H=9,9 Гц, F4), -129,45 (дд, JF-F=27,8 Гц, JF-H=6,9 Гц, фенил F), -141,81 (м, F5). Аналитически рассчитано для C16H13ClF3NO3: C, 53,42; H, 3,64; N, 3,89. Обнаружено: C, 53,77; H, 3,70; N, 3,95. ГХ анализ аликвоты образца показал, что продукт был на 95,5% чистым с примесью 1,7% монофторозамещенного продукта.
Пример 1c. Бензил 4,5-дифтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
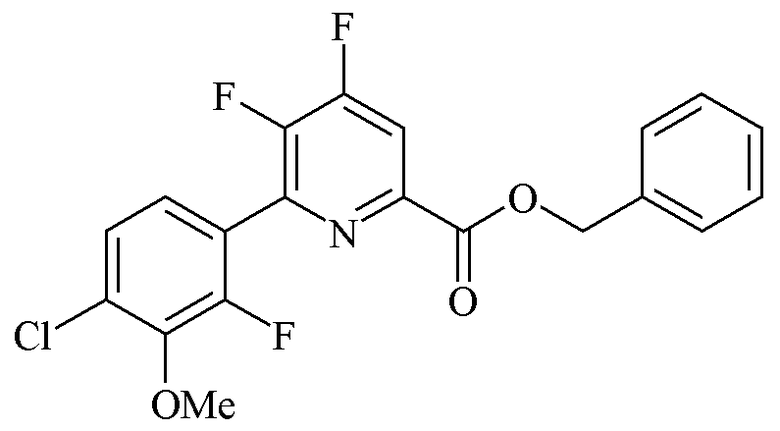
Трехгорлая колба объемом 250 мл была оборудована дефлегматором, входом для азота, механической мешалкой и термопарой. Колбу заполняли CsF (21,07 г, 139,0 ммоль). Добавляли безводный ДМСО (100 мл) и атмосферу над суспензией разрежали при помощи вакуумного насоса/заполняли (5×) азотом. Суспензию нагревали при 80°C в течение 30 минут. ДМСО (30 мл) отгоняли под вакуумом для удаления любой остаточной воды. Добавляли твердый бензил 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (15,34 г, 34,8 ммоль), и атмосферу над раствором разрежали при помощи вакуумного насоса/заполняли азотом (5×). Реакционную смесь нагревали до 105°C в атмосфере азота. Через 6 ч при 105°C ГХ анализ аликвоты образца показал отсутствие пика монофторозамещенного промежуточного продукта на хроматограмме. Реакционную смесь оставляли охлаждаться до комнатной температуры. Реакционную смесь выливали в воду со льдом (400 г) и экстрагировали при помощи EtOAc (3×200 мл). Объединенные органические экстракты промывали насыщ. раствором NaHCO3, водой (5×100 мл) и солевым раствором. Экстракты высушивали (MgSO4) и концентрировали при пониженном давлении с получением желто-коричневого твердого вещества (12,97 г). Твердое вещество очищали при помощи флэш-хроматографии (330 г колонка с силикагелем; EtOAc-градиент 0-20%) с получением белого твердого вещества (9,95 г; 70%): температура плавления 114-116°C; 1H ЯМР (400 МГц, CDCl3) δ 8,01 (дд, JF-H=9,4, 5,5 Гц, 1H, пиридин H), 7,53-7,20 (м, 7H, фенил), 5,44 (с, 2H, CH2Ph), 3,99 (д, JF-H=1,2 Гц, 3H, OMe); 13C ЯМР (101 МГц, CDCl3) δ 162,8 (д, JF-C=3 Гц, CO2Bn, 156,2 (дд, JF-C=267, 12 Гц), 153,9 (д, JF-C=255 Гц), 148,0 (дд, JF-C=269, 11 Гц), 145,4 (т, JF-C=7 Гц), 144,7 (д, JF-C=13 Гц), 144,6 (дд, JF-C=13, 2 Гц), 135,2 (c), 130,6 (д, JF-C=3 Гц), 125,6 (д, JF-C=4 Гц), 125,4 (д, JF-C=2 Гц), 122,0 (д, JF-C=14 Гц), 115,0 (д, JF-C=16 Гц), 67,9 (с, CH2Ph), 61,6 (д, JF-C=5 Гц, OMe); 19F{1H} ЯМР (376 МГц, CDCl3) δ -123,90 (д, JF-F=19,7 Гц, F4), -128,37 (д, JF-F=33,5 Гц, F2'), -139,64 (дд, JF-F=33,5, 19,7 Гц, F5). Аналитически рассчитано для C20H13ClF3NO3: C, 58,91; H, 3,21; N, 3,43. Обнаружено: C, 59,03; H, 3,20; N, 3,39.
Пример 1d. Бензил 4,5-дифтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
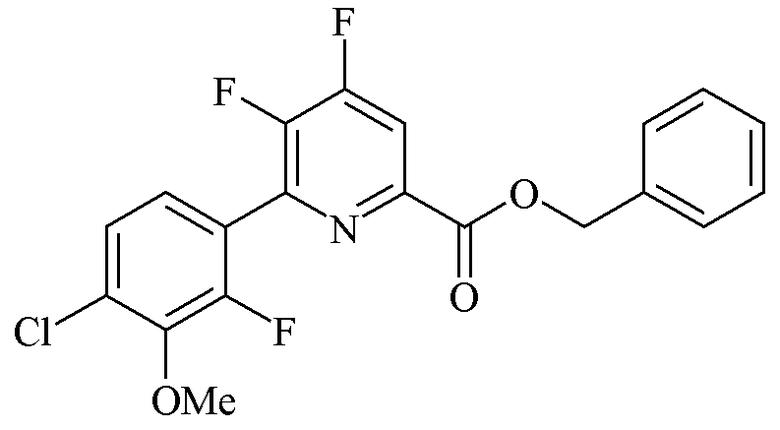
Прямостенный реактор объемом 22 л с рубашкой был оснащен верхней мешалкой, конденсатором, входом для азота и выходом и закрывающимся пробкой отверстием для загрузки твердых веществ. Реактор продували азотом в течение 2 дней. Отверстие для загрузки открывали, в реактор быстро загружали бензил 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (2032 г, 4,12 моль, чистота 89,3%). CsF (2500 г, 16,46 моль) быстро выливали в реактор. Затем реактор загружали безводным (<100 ppm воды) ДМСО (8869 г). Смесь нагревали при 110°C в течение 2 ч. Смесь охлаждали до 35°C и затем фильтровали. Фильтрованные соли промывали ДМСО (2×1108 г). Объединенный фильтрат охлаждали до 15-20°C и добавляли воду (3023 г) с перемешиванием в течение 1 ч. Смесь охлаждали до 10-12°C и затем фильтровали. Собранный твердый осадок промывали ДМСО/вода 3:1 (1814 г) и затем водой (2000 г). Полученное твердое желто-коричневое вещество высушивали с получением соединения, указанного в заголовке (1626 г, чистота ВЭЖХ 85,7 масс.% (внутренний стандарт гексанофенон), 83%).
Аминирование
Пример 2a. Пропан-2-ил 4-амино-5,6-трифторпиколинат
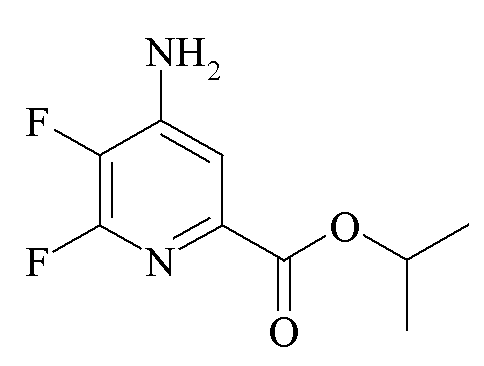
Реакционную смесь из примера 1a фильтровали для удаления солей Cs и соли промывали при помощи ДМСО (50 мл). Промывной раствор ДМСО добавляли к раствору ДМСО (150 мл), который насыщали аммиаком в течение 15 минут. Колбу выдерживали в холодной ванне, в которой поддерживали температуру около 16°C. Аммиак барботировали через реакционную смесь в течение 30 минут, в это время образовывался белый осадок. Через 90 минут ГХ анализ аликвоты образца показал единственный главный пик для 4-аминозамещенного продукта. Реакционную смесь гасили добавлением насыщ. водн. раствора (100 мл) хлорида аммония (NH4C1), затем водой (400 мл). Водный раствор экстрагировали простым эфиром (Et2O 3×150 мл) и затем EtOAc (3×150 мл). Объединенные органические экстракты промывали водой (5×150 мл) и затем солевым раствором. Экстракты высушивали (MgSO4) и выпаривали до получения желто-коричневого твердого вещества, который промывали смесью простой эфир-гексан 1:1 с получением светлого желто-коричневого порошка (5,57 г, в целом 51,4%): температура плавления 168-170°C; 1H ЯМР (400 МГц, CDCl3) δ 7,42 (д, JF-H=5,5 Гц, 1H, пиридин H), 5,22 (септет, J=6,2 Гц, 1H, CHMe2), 4,75 (с, 2Η, NH2), 1,35 (д, J=6,2 Гц, 6H, CHMe2); 13C{1H} ЯМР (101 МГц, ДМСО-d6) δ 162,8 (CO2R), 151,2 (дд, JF-C=228, 12 Гц, C6), 146,5 (дд, JF-C=9, 6 Гц, C2/C4), 139,3 (дд, JF-C=16, 5 Гц, C2/C4), 133,8 (дд, JF-C=252, 31 Гц, C5), 112,3 (C3), 68,8 (CHMe2), 21,5 (Me); 19F ЯМР (376 МГц, ДМСО-d6) δ -91,9 (д, JF-F=26,6 Гц, F6), -163,9 (дд, JF-F=26,6, JH-F=5,6 Гц, F5). Аналитически рассчитано для C9H10F2N2O2: C, 50,00; H, 4,66; N, 12,96. Обнаружено: C, 49,96; H, 4,65; N, 12,91.
Пример 2b. Пропан-2-ил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
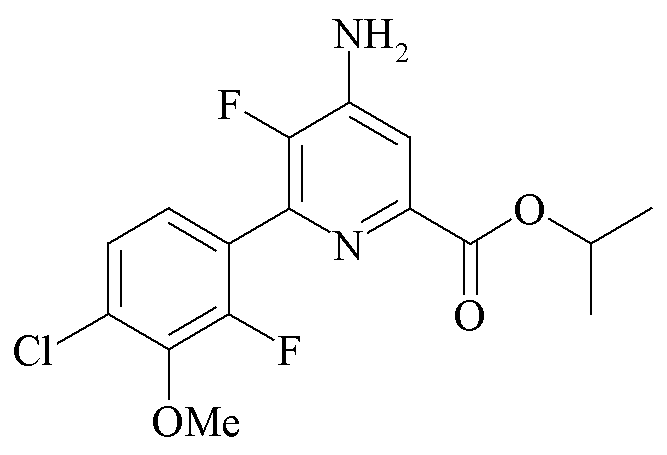
Пропан-2-ил 4,5-дифтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (4,89 г, 13,9 ммоль) растворяли в ДМСО (100 мл). Аммиак барботировали через раствор в течение в общей сложности 100 минут в течение 48 ч. Реакционную смесь выливали в воду со льдом (500 мл). Продукт экстрагировали в EtOAc (3×250 мл). Объединенные органические экстракты промывали водой (5×100 мл) и затем солевым раствором, высушивали (MgSO4) и концентрировали при пониженном давлении с получением белого твердого вещества (4,36 г, 88%): температура плавления 180,2-181,9°C; 1H ЯМР (400 МГц, CDCl3) δ 7,54 (д, JF-H=6,5 Гц, 1H, пиридин H), 7,27 (м, 2H, фенил), 5,27 (гептет, JH-H=6,3 Гц, 1H, CHMe2), 4,69 (с, 2Η, NH2), 3,96 (д, JF-H=0,9 Гц, 3H, OMe), 1,38 (д, JH-H=6,3 Гц, 6H, CHMe2); 13C{1H}ЯМР (101 МГц, ДМСО-d6) δ 163,7 (CO2R), 153,2 (д, JF-C=252 Гц), 146,8 (д, JF-C=254 Гц), 144,2 (д, JF-C=4 Гц), 143,9, 143,7, 139,0 (д, JF-C=14 Гц), 128,2 (д, JF-C=3 Гц), 126,0 (д, J=3 Гц), 125,4 (д, JF-C=3 Гц), 123,9 (дд, JF-C=14, 3 Гц), 112,5 (д, JF-C=5 Гц), 68,5 (CHMe2), 61,5 (д, JF-C=4 Гц, OMe), 21,56 (CHMe2); 19F ЯМР (376 МГц, CDCl3) δ -128,43 (дд, JF-F=32,0, JF-H=6,6 Гц), -142,27 (дд, дд, JF-F=32,0, JF-H=6,3 Гц). Аналитически рассчитано для C16H15ClF2N2O3: C, 53,87; H, 4,24; N, 7,85. Обнаружено: C, 53,65; H, 4,28; N, 7,75.
Пример 2c. Бензил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
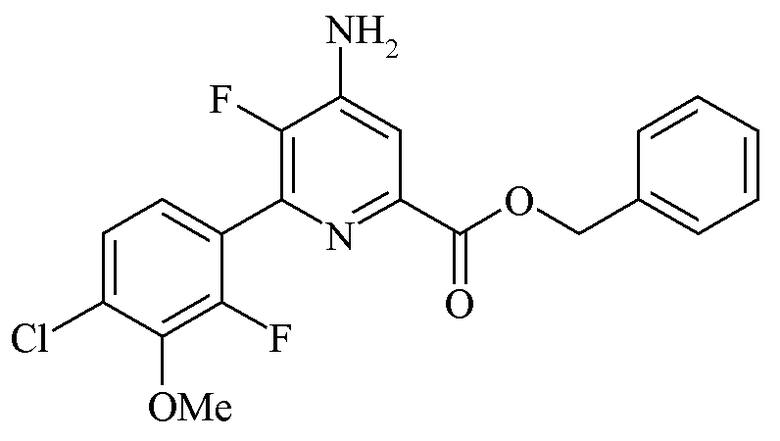
Бензил 4,5-дифтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (4,99 г, 12,2 ммоль) суспендировали в ДМСО (100 мл). Аммиак барботировали через раствор в течение 30 минут. После перемешивания в течение ночи реакционную смесь выливали в воду со льдом (500 мл). Продукт экстрагировали в EtOAc (3×150 мл). Объединенные органические экстракты промывали водой (5×100 мл) и солевым раствором, высушивали (MgSO4) и концентрировали при пониженном давлении с получением белого твердого вещества (4,99 г, 101%); 1H ЯМР (400 МГц, CDCl3) δ 7,52 (д, JF-H=6,5 Гц, 1H, пиридин H3), 7,45-7,38 (м, 2H), 7,37-7,17 (м, 5H), 5,38 (с, 2H, CH2Ph), 4,67 (ушир.с, 2Η, NH2), 3,94 (д, JF-H=1,1 Гц, 3H, OMe); 13C{1H} ЯМР (101 МГц, CDCl3) δ 164,4 (CO2R), 153,9 (д, JF-C=254 Гц), 147,6 (д, JF-C=256 Гц), 144,4 (д, JF-C=14 Гц), 144,0 (д, JF-C=5 Гц), 142,2 (д, JF-C=12 Гц), 140,4 (д, JF-C=15 Гц), 135,6 (c), 129,5 (д, JF-c=3 Гц), 128,5 (CH), 128,3 (CH), 128,3 (CH), 125,6 (д, JF-C=3 Гц, CH), 125,2 (д, JF-C=4 Гц, CH), 123,3 (дд, JF-C=14, 4 Гц), 113,1 (д, JF-C=4 Гц, C3), 67,3 (с, CH2Ph), 61,5 (д, JF-C=4 Гц, OMe); 19F{1H} ЯМР (376 МГц, CDCl3) δ -128,54 (дд, J=30,7, 5,2 Гц, F2'), -141,84 (дд, J=30,8, 6,5 Гц, F5), HRMS-ESI (m/z): [М]+ рассчитано для C20H15ClF2N2O3, 404,0739; обнаружено 404,0757.
Пример 2d. Бензил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
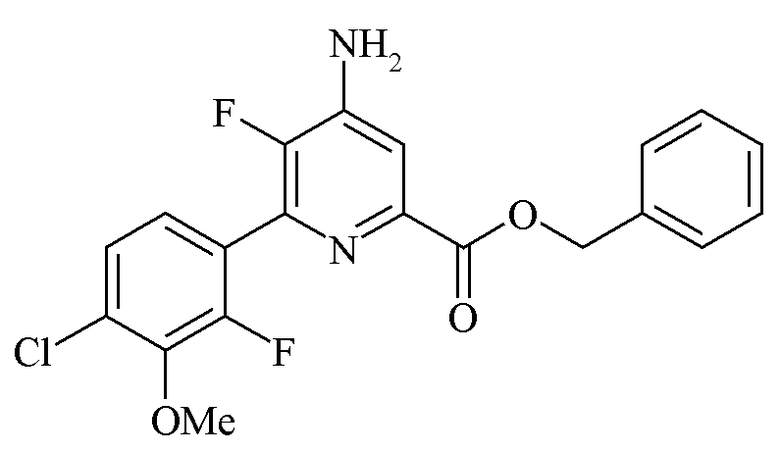
Раствор бензил 4,5-дифтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколината (65,0 г, 0,16 моль, чистота 87%) в ДМСО (400 мл) получали в четырехгорлой колбе объемом 1 л, оборудованной механической мешалкой, термометром, входом для аммиака и газоотводом. Аммиачный газ (8,1 г, 0,48 моль, 3 экв.) добавляли путем барботирования через тефлоновую трубку под поверхностью раствора ДМСО в течение 20 минут. Во время введения аммиака цвет реакционной смеси изменялся на розовый/светло-красный, и внутренняя температура повысилась до 30°C. Через 5 ч перемешивания добавляли дополнительный аммиачный газ (6,1 г, 0,36 моль, 2,25 экв.) в течение 20 минут. После перемешивания еще в течение 2,5 ч анализ ВЭЖХ показал полный расход исходного вещества. Азот барботировали в реакционную смесь и реакцию оставляли с перемешиванием на ночь. Реакционную смесь фильтровали с удалением солей, образованных во время реакции, и твердые частицы промывали ДМСО (50 мл). К ДМСО раствор добавляли воду (225 мл) по каплям в течение 1 ч. Полученный осадок фильтровали и затем промывали смесью ДМСО/вода (2:1, 2×40 мл), затем водой (2×50 мл). Твердое вещество высушивали с получением соединения, указанного в заголовке (55,35 г, 87%, чистота ВЭЖХ 87% (гексанофенон внутренний стандарт)).
Обмен галогеном
Пример 3a. Пропан-2-ил 4-амино-6-хлор-5-фторпиколинат
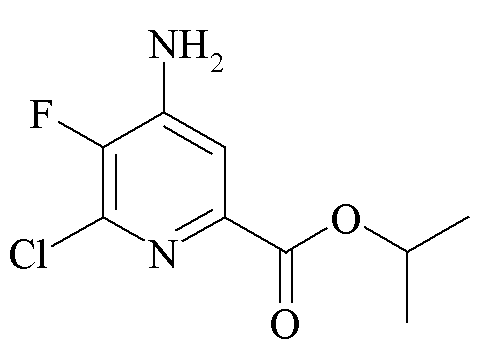
Пропан-2-ил 4-амино-5,6-дифторпиколинат (4,25 г, 19,7 ммоль) растворяли в соляной кислоте (HCl, 4 М в диоксане; 65 мл) с перемешиванием в реакторе Parr из сплава «Хастеллой» объемом 100 мл. Реактор нагревали до 100°C в течение 2 ч. При отстаивании при комнатной температуре в течение ночи образовывалось желтое кристаллическое твердое вещество. Это твердое вещество было нерастворимо в EtOAc, но при этом растворялось при встряхивании с насыщ. водн. раствором NaHCO3 (500 мл) и EtOAc (300 мл). Водный слой экстрагировали в EtOAc (2×250 мл). Объединенные органические экстракты промывали водой (5×50 мл) и затем солевым раствором. Экстракты высушивали (MgSO4) и концентрировали под вакуумом с получением не совсем белого твердого вещества. Неочищенный продукт очищали колоночной хроматографией (120 г колонка с силикагелем; градиент гексан-EtOAc 0-100%) с получением белого твердого вещества (2,11 г, 46%): температура плавления 190,7-192,4°C; 1H ЯМР (400 МГц, ДМСО-d6) δ 7,543 (д, JF-H=5,7 Гц, 1H), 6,91 (ушир.с, 2H, NH2), 5,09 (септет, J=6 Гц, 1H, CHMe2), 1,29 (д, J=6 Гц, 6H, CHMe2); 13C{1H}ЯМР (101 МГц, ДМСО-d6) δ 162,8 (CO2R), 144,8 (д, JF-C=12 Гц, C2/C4), 143,4 (д, JF-C=254 Гц, C5), 142,7 (д, JF-C=4,8 Гц, C2/C4), 136,5 (д, JF-C=17 Гц, C6), 112,8 (д, JF-C=5 Гц, C3), 68,9 (CHMe2), 21,6 (Me); 19F ЯМР (376 МГц, ДМСО-d6) δ -141,0 (д, JF-H=6 Гц). Аналитически рассчитано для C9H10ClFN2O2: C, 46,47; H, 4,33; N, 12,04. Обнаружено: C, 46,50; H, 4,33; N, 11,96.
Галогенирование
Пример 4a. Пропан-2-ил 4-амино-3,6-дихлор-5-фторпиколинат
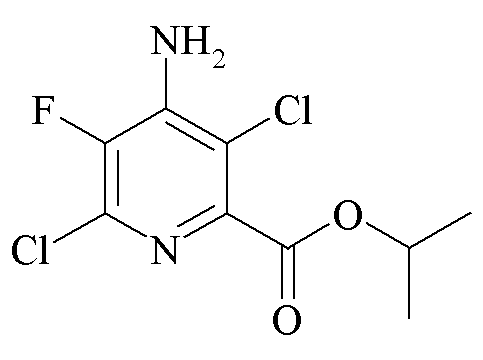
Пропан-2-ил 4-амино-6-хлор-5-фторпиколинат (1,191 г, 5,12 ммоль) практически полностью растворяли в CH2Cl2 (40 мл). Добавляли воду (40 мл). Хлор барботировали через раствор в течение 5 минут. Через 30 минут аликвоту реакционной смеси анализировали при помощи ГХ, которая показывала желаемый продукт и только 1,7% исходного вещества. Водный слой отделяли и экстрагировали в CH2Cl2 (50 мл). Объединенные органические экстракты промывали насыщ. водн. раствором NaHCO3 и затем солевым раствором. Экстракты высушивали (MgSO4) и концентрировали при пониженном давлении с получением оранжевого масла. При помощи флэш-хроматографии (120 г колонка с силикагелем; градиент EtOAc-гексан 0-50%) выделяли ярко-желтое кристаллическое тверое вещество (394 мг, 28%): 1H ЯМР (400 МГц, CDCl3) δ 5,29 (септет, J=6,3 Гц, 1H, CHMe2), 5,19 (ушир.с, 2Η, NH2), 1,40 (д, J=6,3 Гц, 6H, CHMe2); 13C{1H}ЯМР (101 МГц, CDCl3) δ 163,2 (CO2R), 143,3 (д, JF-C=5 Гц, C2), 142,8 (д, JF-C=270 Гц, C5), 141,0 (д, JF-C=26 Гц, C4), 135,3 (д, JF-C=17 Гц, C6), 114,9 (с, C3), 70,6 (CHMe2), 21,6 (с, ME); 19F ЯМР (376 МГц, CDCl3) δ -136,5.
Пример 4b. Пропан-2-ил 4-амино-3,6-дихлор-5-фторпиколинат
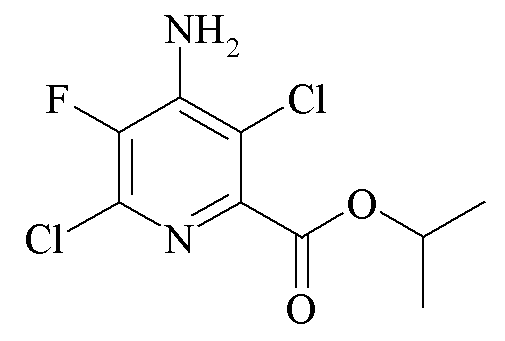
Пропан-2-ил 4-амино-6-хлор-5-фторпиколинат (634 миллиграмма (мг), 2,73 ммоль) суспендировали в ацетонитриле (11 мл). 1,3-Дихлор-5,5-диметилгидантоин (303 мг, 1,54 ммоль) добавляли в виде твердого вещества и реакционную смесь перемешивали при нагревании с обратным холодильником в течение 2,5 ч. Дополнительный 1,3-дихлор-5,5-диметилгидантоин (50 мг, 0,25 ммоль) и реакционную смесь перемешивали при нагревании с обратным холодильником в течение дополнительного часа. Добавляли воду (20 мл). Затем ацетонитрил удаляли путем испарения на роторном испарителе с получением маслянистого, желтого твердого вещества, которое экстрагировали в EtOAc (2×20 мл). Объединенные органические экстракты промывали 10% раствором бисульфита натрия (NaHSO3), насыщ. водн. раствором NaHCO3 и солевым раствором, высушивали (MgSO4) и концентрировали при пониженном давлении с получением бледно-оранжевого твердого вещества (671 мг, 92%): 1H ЯМР (400 МГц, CDCl3) δ 5,29 (септет, J=6,3 Гц, 1H, CHMe2), 5,19 (ушир.с, 2Η, NH2), 1,40 (д, J=6,3 Гц, 6H, CHMe2); 19F ЯМР (376 МГц, CDCl3) δ -136,5.
Пример 4c. Метил 4-амино-3-бром-6-хлор-5-фторпиколинат
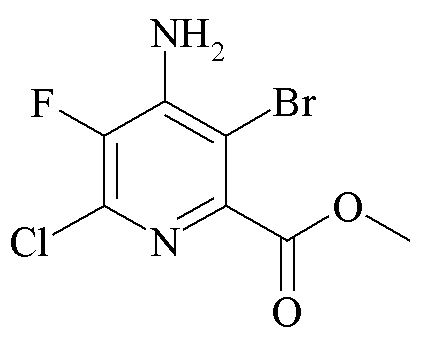
Метил 4-амино-6-хлор-5-фторпиколинат (1,0 г, 4,9 ммоль) объединяли с 1,3-дибром-5,5-диметилгидантоином (1,7 г, 5,9 ммоль) в 1,2-дихлорэтане (15 мл) и нагревали с обратным холодильником (83°C) в течение 4 ч. Охлажденную смесь перемешивали с 10% раствором NaHSO3 и EtOAc (30 мл). Органическую фазу отделяли, промывали водой (2×20 мл), солевым раствором (10 мл), высушивали (Na2SO4) и концентрировали. Остаток очищали хроматографией на силикагеле (EtOAc-гексан градиент 5-50%) с получением оранжевого твердого вещества (840 мг, 61%): температура плавления 138-139°C; EIMS m/z 282, 284; 1H ЯМР (400 МГц, CDCl3) δ 5,09 (с, 2H, NH2), 3,97 (с, 3H, Me); 19F ЯМР (376 МГц, CDCl3) δ -135,55 (с).
Пример 4d. Метил 4-амино-6-хлор-5-фтор-3-йодопиколинат
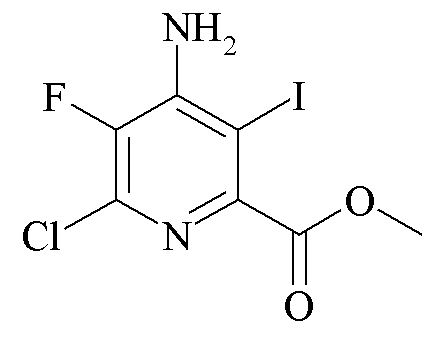
Метил 4-амино-6-хлор-5-фторпиколинат (2,2 г, 10,8 ммоль) растворяли в метиловом спирте (CH3OH; 20 мл). Раствор обработали йодной кислотой (880 мг, 3,9 ммоль) и йодом (2,2 г, 8,6 ммоль) и затем нагревали с обратным холодильником в течение 20 ч. Смесь охлаждали и летучие компоненты удаляли под вакуумом. Остаток растворяли в EtOAc (50 мл) и затем перемешивали с 10% раствором NaHSO3 (20 мл) в течение 10 минут. Органическую фазу отделяли и промывали солевым раствором (10 мл), высушивали (Na2SO4) и выпаривали. Остаток очищали хроматографией на силикагеле (градиент EtOAc-гексан 5-50%) с получением соединения, указанного в заголовке в виде светло-оранжевого твердого вещества (2,5 г, 70%): температура плавления 149-151°C; ESIMS m/z 330 ([М]+); 1H ЯМР (400 МГц, CDCl3) δ 5,17 (с, 2H, NH2), 3,97 (с, 3H, OMe); 19F ЯМР (376 МГц, CDCl3) δ -135,79 (с).
Пример 4e. Пропан-2-ил 4-амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
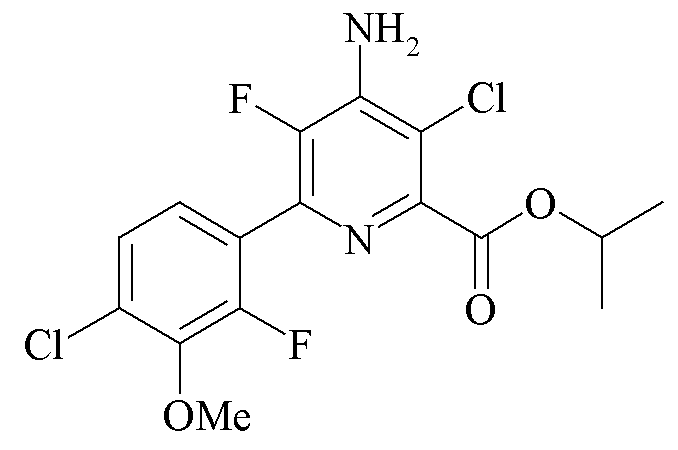
Пропан-2-ил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (3,065 г, 8,59 ммоль) растворяли в сульфурилхлориде (150 мл). Раствор перемешивали в атмосфере азота при комнатной температуре в течение 8 ч. В это время образовывался белый осадок. Добавляли гексан (100 мл) и смесь оставляли на ночь при температуре -20°C. Продукт фильтровали, промывали гексаном и затем суспендировали в EtOAc (100 мл). Органическую суспензию нейтрализовали насыщ. водн. раствором NaHCO3, в результате чего все твердые частицы растворялись. Органический слой отделяли и промывали 10% водн. раствором NaHSO3 и солевым раствором, высушивали (MgSO4) и концентрировали при пониженном давлении с получением белого твердого вещества (1,962 г, 58%): температура плавления 109-111°C; 1H ЯМР (400 МГц, CDCl3) δ 7,25 (м, 2H), 5,32 (септет, J=6,3 Гц, 1H, CHMe2), 5,07 (ушир.с, 2Η), 3,97 (д, JF-H=1,0 Гц, 3H, OMe), 1,40 (д, J=6,3 Гц, 6H, CHMe2), 19F ЯМР (376 МГц, CDCl3) δ -128,16 (дд, JF-F=33,3 Гц, JF-H=2,5 Гц, фенил F), -138,35 (д, JF-F=33,4 Гц, пиридин F5).
Пример 4f. Бензил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
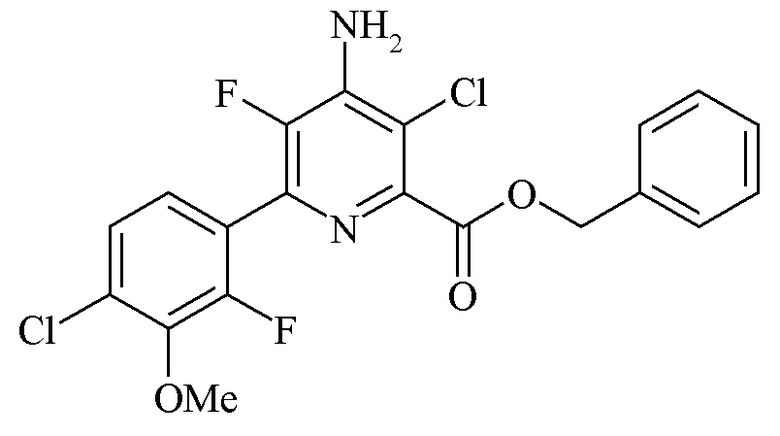
Бензил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат (2,07 г, 5,12 ммоль) суспендировали в ацетонитриле (20 мл) в сцинтилляционном флаконе. Добавляли 1,3-дихлор-5,5-диметилгидантоин (554 мг, 2,181 ммоль) в виде твердого вещества и реакционную смесь перемешивали при нагревании с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры добавляли воду (40 мл) для осаждения продукта. Твердое вещество собирали на воронке Бюхнера и промывали водой. В результате сушки под вакуумом при 55°C получали белое твердое вещество (2,187 г, 97%): 1H ЯМР (400 МГц, CDCl3) δ 7,50-7,41 (м, 2H, ароматического ряда), 7,41-7,20 (м, 5H, ароматического ряда), 5,42 (с, 2H, CH2Ph), 4,92 (ушир.с, 2Η, NH2), 3,97 (д, J=1,2 Гц, 3H, OMe); 19F {1H} ЯМР (CDCl3) δ -128,19 (д, J=33,9 Гц, F2'), -137,79 (д, J=33,8 Гц, F5).
Пример 4g. Бензил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
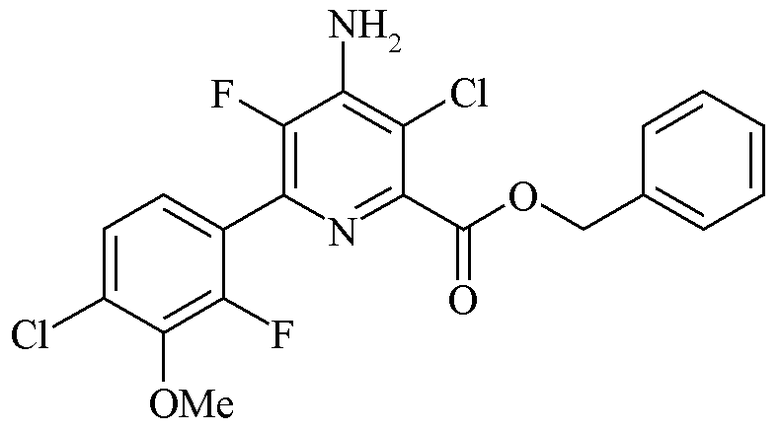
Раствор бензил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината (53,0 г, 0,131 моль, чистота 95%) в ацетонитриле (450 мл) получали в трехгорлой круглодонной колбе объемом 1 л, оборудованной механической мешалкой и термометром. Добавляли 1,3-дихлор-5,5-диметилгидантоин (14,2 г, 0,072 моль, 0,55 экв.). Реакционную смесь нагревали при 80°C в течение 3 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем по каплям добавляли к разбавленному раствору бисульфита натрия (NaHSO3) (990 мл, 7,5 г NaHSO3) в течение 1 ч. Полученный осадок отделяли путем фильтрации, промывали смесью ацетонитрил-вода (1:1 об./об., 2×50 мл) и затем водой (2×50 мл). Твердое вещество высушивали с получением бледно-желтого порошка (53,44 г, 94%, чистота ВЭЖХ 96,1% (октанофенон внутренний стандарт)).
Пример 4h. Пропан-2-ил 4-амино-3-йодо-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
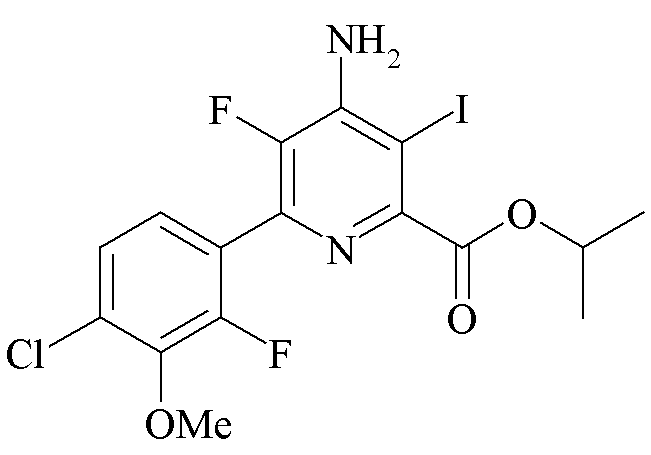
Пропан-2-ил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (600 мг, 1,682 ммоль) растворяли в уксусной кислоте (5,6 мл). Добавляли ацетат натрия (1,5 г, 18,50 ммоль), затем монохлорид йода (2,2 г, 13,45 ммоль). Во время добавления наблюдали экзотермический эффект приблизительно в 10°C. Реакционную смесь нагревали при 80°C в течение 20 ч. Затем реакционную смесь разбавляли водой и экстрагировали при помощи EtOAc. Органический слой промывали водой, насыщ. водн. раствором NaHCO3, высушивали (MgSO4) и затем концентрировали до сухого состояния. Остаток помещали на колонку из силикагеля (80 г) и затем элюировали (градиент ацетон-гексаны 0-70%) с получением оранжевого твердого вещества (343 мг, 42%): температура плавления 134-135°C; 1H ЯМР (400 МГц, ацетон-d6) δ 7,41 (дд, J=8,5, 1,6 Гц, 1H), 7,33 (дд, J=8,5, 6,8 Гц, 1H), 6,29 (с, 2H), 5,29-5,14 (септет, 1H), 3,98 (д, J=1,1 Гц, 3H), 1,37 (д, J=6,3 Гц, 6H); EIMS m/z 396.
Пример 4i. Пропан-2-ил 4-амино-3-бром-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
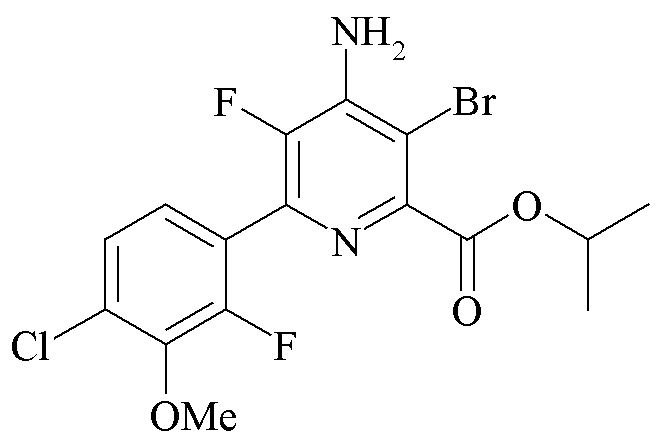
Пропан-2-ил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (500 мг, 1,402 ммоль) растворяли в дихлорметане (3,2 мл). Добавляли N-бромсукцинимид (299 мг, 1,682 ммоль) и раствор перемешивали при температуре окружающей среды в течение 20 ч. Затем реакционную смесь концентрировали до сухого состояния. Остаток очищали хроматографией (колонка силикагеля 40 г; градиент EtOAc-гексаны 0-70%) с получением желто-коричневого твердого вещества (504 мг, 83%): 1H ЯМР (400 МГц, ДМСО-d6) δ 7,47 (дд, J=8,5, 1,6 Гц, 1H), 7,29 (дд, J=8,5, 7,1 Гц, 1H), 7,01 (с, 2H), 5,22-5,10 (м, 1H), 3,93 (д, J=0,9 Гц, 3H), 1,32 (д, J=6,3 Гц, 6H); EIMS m/z 350.
Связывание
Пример 5a. Пропан-2-ил 4-амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
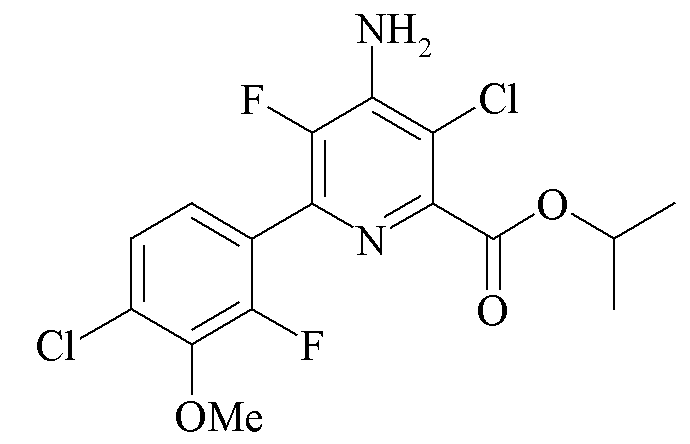
В колбу Шленка объемом 50 мл помещали пропан-2-ил 4-амино-3,6-дихлор-5-фторпиколинат (2,162 г, 8,09 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (2,775 г, 11,35 ммоль) и CsF (2,601 г, 17,12 ммоль). Добавляли ацетонитрил (20 мл) и воду (7 мл). Атмосферу над раствором разрежали при помощи вакуумного насоса/заполняли азотом (5×). Добавляли твердый дихлорид бис(трифенилфосфин)палладия (II) (Pd(PPh3)2Cl2; 281 мг, 4,9% моль). Атмосферу над раствором разрежали при помощи вакуумного насоса/заполняли азотом (5×) и затем нагревали при 70°C в атмосфере азота в течение 3 ч. Через 3 ч реакционную смесь оставляли охлаждаться до комнатной температуры. Водный слой отделяли. К органическому слою добавляли воду (20 мл). Полученный темно-коричневый осадок фильтровали и промывали водой. Твердое вещество растворяли в EtOAc (60 мл) и фильтровали для удаления небольшого количества черного твердого вещества. Раствор EtOAc обрабатывали активированным углем (175 мг) и фильтровали с получением раствора винного цвета. В результате выпаривания при пониженном давлении получали темно-красное твердое вещество. В результате очистки (120 г колонка с силикагелем; градиент EtOAc-гексан 0-50%) получали белое твердое вещество (2,59 г, 82%): температура плавления 110,6-112,1°C; 1H ЯМР (400 МГц, CDCl3) δ 7,25 (м, 2H), 5,32 (септет, J=6,3 Гц, 1H, CHMe2), 5,07 (ушир.с, 2Η), 3,97 (д, JF-H=1,0 Гц, 3H, OMe), 1,40 (д, J=6,3 Гц, 6H, CHMe2); 13C{1H}ЯМР (101 МГц, CDCl3) δ 164,2 (CO2R), 153,8 (д, JF-C=254 Гц, C5/C2'), 145,5 (д, JF-C=258 Гц, C5/C2'), 145,0 (д, JF-C=5 Гц), 144,4 (д, JF-C=14 Гц), 140,0 (д, JF-C=13 Гц), 137,5 (д, JF-C=14 Гц), 129,7 (д, JF-C=3 Гц), 125,4 (д, JF-C=2 Гц, C5'/C6'), 125,2 (д, JF-C=3 Гц, C5'C6'), 122,7 (дд, JF-C=14, 4 Гц, Cl'), 114,6 (C3), 70,2 (CHMe2), 61,5 (д, JF-С=4 Гц, OMe), 21,6 (CHMe2); 19F ЯМР (376 МГц, CDCl3) δ -128,16 (дд, JF-F=33,3 Гц, JF-H=2,5 Гц, фенил F), -138,35 (д, JF-F=33,4 Гц, пиридин F5). Аналитически рассчитано для C16H14Cl2F2N2O3: C, 49,12; H, 3,61; N, 7,16. Обнаружено: C, 49,30; H, 3,69; N, 7,08. Чистота продукт, как обнаружено при помощи ВЭЖХ, составила 97,5%.
Пример 5b. Пропан-2-ил 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
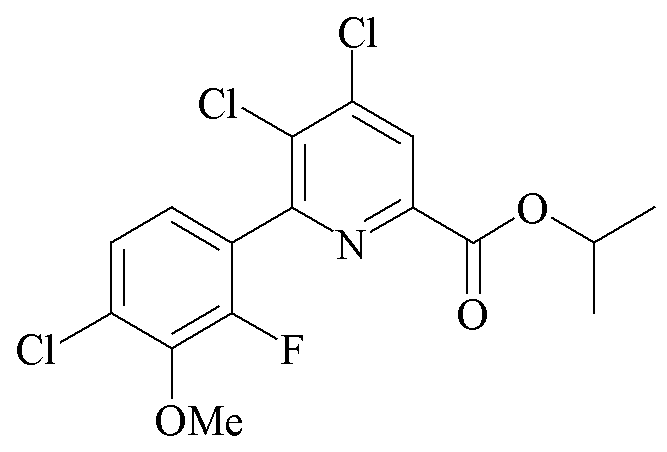
В колбу Шленка объемом 100 мл помещали пропан-2-ил 4,5,6-трихлорпиколинат (10,46 г, 39,0 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (13,27 г, 54,3 ммоль) и CsF (11,76 г, 77,0 ммоль). Добавляли ацетонитрил (75 мл) и воду (25 мл). Атмосферу над реакционной смесью разрежали при помощи вакуумного насоса/заполняли N2 (5×). Добавляли твердый Pd(PPh3)2Cl2 (1,331 г, 1,896 ммоль). Атмосферу над раствором разрежали при помощи вакуумного насоса/заполняли (5×) N2 и затем перемешивали с обратным холодильником в течение 2 ч. При охлаждении до комнатной температуры выпадал белый твердый осадок. Твердое вещество фильтровали, промывали водой и сушили на воздухе (10,56 г, 69%): температура плавления 123,8-127,7°C; 1H ЯМР (400 МГц, CDCl3) δ 8,20 (с, 1H, пиридин), 7,28 (дд, JH-H=8,5 Гц, JF-H=1,6 Гц, 1H), 7,13 (дд, JH-H=8,5 Гц, JF-H=6,8 Гц, 1H), 5,32 (септет, J=6,3 Гц, 1H, CHMe2), 3,99 (д, J=1,2 Гц, 3H, OMe), 1,41 (д, J=6,3 Гц, 6H, CHMe2); 13C{1H} ЯМР (101 МГц, CDCl3) δ 162,7 (CO2R), 153,8, 153,6 (д, JF-С=253 Гц, C2'), 146,7, 144,5 (д, JF-С=13 Гц), 144,0, 134,0, 130,0 (д, JF-С=3,4 Гц), 125,9, 125,3 (д, JF-С=3 Гц), 125,1 (д, JF-С=3 Гц), 70,4 (CHMe2), 61,6 (д, JF-C=4 Гц, OMe), 21,7 (CHMe2). Аналитически рассчитано для C16H13Cl3NO3: C, 48,94; H, 3,34; N, 3,57. Обнаружено: C, 48,91; H, 3,50; N, 3,51.
Пример 5c. Метил 4,5-дихлор-6-этилпиколинат
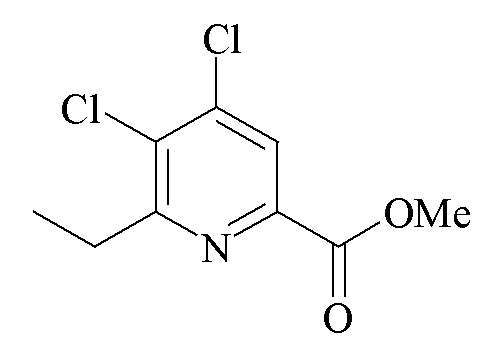
Трехгорлую колбу объемом 100 мл, оборудованную дефлегматором и входом для азота, загружали метил 4,5,6-трихлорпиколинатом (2,40 г, 9,98 ммоль). Добавляли безводный ТГФ (50 мл) с последующим добавлением N,N-диметилэтаноламина (0,20 г). Реакционную смесь барботировали азотом в течение 15 минут. Добавляли твердый Pd(PPh3)2Cl2 (140 мг, 0,2 ммоль). Реакционную смесь перемешивали в атмосфере азота в течение 20 минут. Добавляли диэтилцинк (1 М раствор в гексанах; 10 мл, 10 ммоль) порциями по 2 мл. Когда исходное вещество больше не наблюдали посредством ГХ анализа, реакционную смесь гасили водой и экстрагировали в EtOAc. Объединенные органические экстракты промывали солевым раствором, высушивали (MgSO4) и концентрировали при пониженном давлении до белого твердого вещества (2,34 г). Анализ ГХ-МС показал, что твердое вещество содержало 11% исходного метил 4,5,6-трихлорпиколината. 1H ЯМР (400 МГц, CDCl3) δ 8,06 (с, 1H, пиридин H), 4,01 (с, 3H, CO2Me), 3,10 (кв, J=8 Гц, 2H, CH2), 1,33 (т, J=8 Гц, 3H, CH2CH3); 13C{1H} ЯМР (101 МГц, CDCl3) δ 164,4, 162,8, 145,4, 143,3, 132,9, 124,5 (C3), 53,1 (CO2Me), 30,0 (CH2), 12,3 (CH2CH3).
Пример 5d. Бензил 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
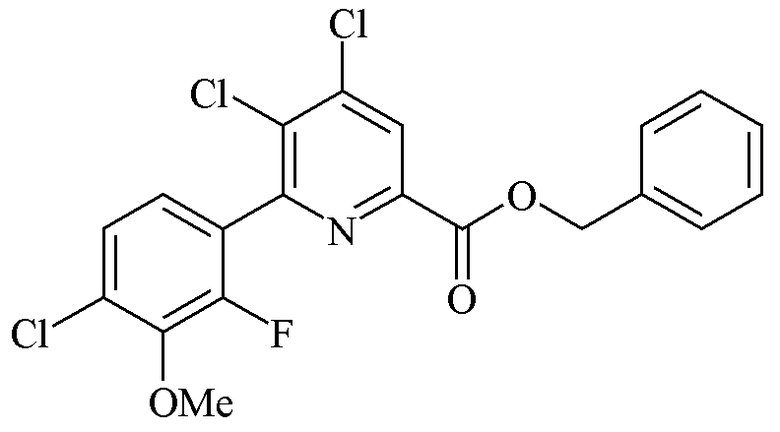
Трехгорлую колбу объемом 250 мл, оборудованную дефлегматором и входом для азота, загружали бензил 4,5,6-трихлорпиколинатом (17,77 г, 56,10 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинаном (19,20 г, 79,0 ммоль) и CsF (17,04 г, 112,0 ммоль). Добавляли ацетонитрил (100 мл) и воду (30 мл). Атмосферу над реакционной смесью разрежали при помощи вакуумного насоса/заполняли азотом (5×). Добавляли твердый Pd(PPh3)2Cl2 (1,724 г, 2,456 ммоль). Атмосферу над раствором разрежали при помощи вакуумного насоса/заполняли азотом (5×) и затем перемешивали с обратным холодильником в течение 90 минут. При охлаждении до комнатной температуры выпадал белый твердый осадок. Твердое вещество фильтровали, промывали водой и сушили на воздухе (18,66 г, 75%): 1H ЯМР (400 МГц, CDCl3) δ 8,23 (с, 1H, пиридин H), 7,52-7,32 (м, 5H, фенил), 7,27 (дд, JH-H=8,4 Гц, JF-H=1,7 Гц, 1H, ароматического ряда), 7,10 (дд, JH-H=8,4 Гц, JF-H=6,8 Гц, 1H, ароматического ряда), 5,44 (с, 2H, CH2Ph), 3,98 (д, JF-H=1-3 Гц, 3H, OMe); 13C{1H} ЯМР (101 МГц, CDCl3) δ 163,0, 153,7, 153,5 (д, JF-C=253 Гц, C2'), 146,0, 144,5 (д, JF-C=13 Гц), 144,1, 135,0, 134,2, 129,9 (д, JF-C=3 Гц), 128,5, 126,1, 125,8 (д, JF-C=14 Гц), 125,3 (д, JF-C=3 Гц), 124,9 (д, JF-C=2 Гц), 67,9 (CH2), 61,5 (д, JF-C=4 Гц, OMe). Аналитически рассчитано для C20H13Cl3FNO3: C, 54,51; H, 2,97; N, 3,18. Обнаружено: C, 54,60; H, 3,08; N, 3,16.
Пример 5e. Бензил 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
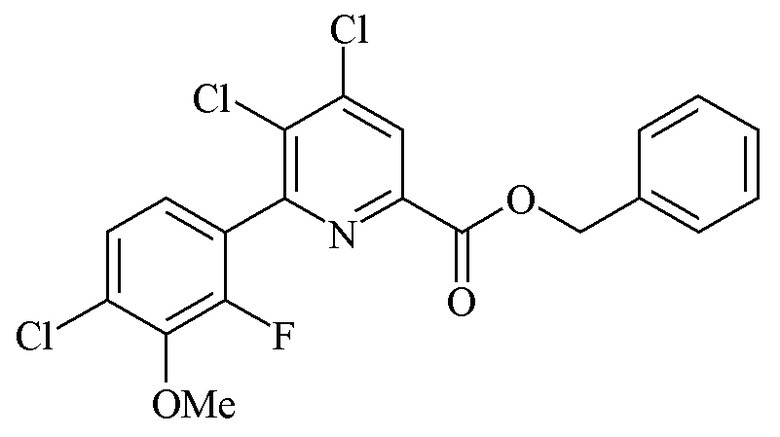
В прямостенный реактор объемом 22 л, оборудованный механической мешалкой, заливали водопроводную воду (3403 г). Затем добавляли фосфат дикалия (K2HPO4; 2596 г, 14,9 моль) и смесь перемешивали вплоть до растворения всех твердых частиц с одновременным барботированием потоком азота. После растворения всех твердых частиц в реактор загружали ацетонитрил (8173 г). В отдельный прямостенный реактор с рубашкой объемом 30 л, оборудованный нижним выпуском и механической верхней мешалкой, загружали 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (1724 г, 7,01 моль) и бензил 4,5,6-трихлорпиколинат (1630 г, 4,97 моль). Атмосферу в реакторе разрежали при помощи вакуумного насоса и наполняли азотом (3×). Затем в реактор объемом 30 л переносили смесь ацетонитрил/H2O, содержащую K2HPO4, и линии промывали ацетонитрилом (1434 г). Взвесь барботировали азотом в течение 30 минут и затем добавляли трифенилфосфин (114,8 г, 0,44 моль). Затем взвесь барботировали азотом в течение 15 минут с последующим добавлением хлорида бис(бензонитрил)палладия (II) (83,8 г, 0,22 моль). Ярко-желтую взвесь барботировали азотом в течение 15 минут и затем смесь нагревали до 74-75°C. После перемешивания при 74°C в течение 3,3 ч реакцию считали завершенной по результатам анализа ВЭЖХ. На данной стадии температуру охлаждения в реакторе устанавливали на 5°C и сразу же заливали в реактор холодную воду (4448 г, приблизительно 3°C). Полученный осадок фильтровали с получением фильтровального осадка бело-кремового цвета. Фильтровальный осадок промывали холодной смесью ацетонитрил/H2O (3345 г, 1,4:1, 8-10°C) с получением не совсем белого влажного фильтровального осадка. Фильтровальный осадок высушивали в потоке азота до постоянной массы 2044 г. Анализ ВЭЖХ с спользованием внутреннего стандарта (тетрафенилэтилена) показал, что продукт имел чистому 90,0% и содержал 1840 г (84,0%) продукта.
Очистка солей аммония
Пример 6a. Пропан-2-ил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
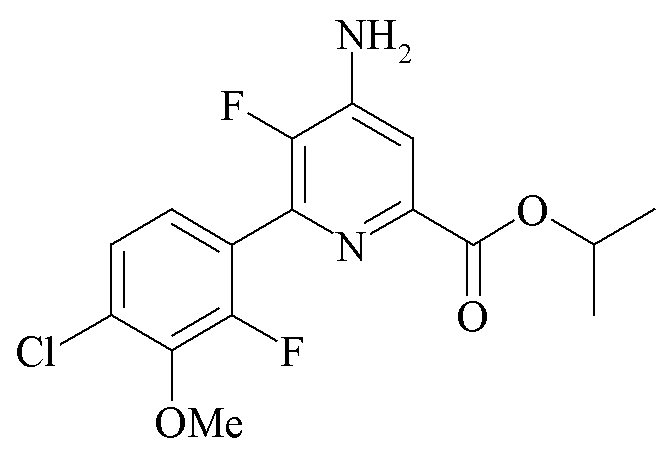
Пропан-2-ил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (1,50 г, чистота по площади хроматограммы ЖХ 71%) добавляли к тетрагидрофурану (ТГФ; 10 мл) и нагревали до 40°C с получением прозрачного желтого раствора. Раствор оставляли охлаждаться до комнатной температуры и добавляли HCl (4 M в диоксане; 1,3 мл, 5,2 ммоль). После добавления HCl твердые частицы осаждались из раствора, и реакционную смесь охлаждали до 0°C. Твердые частицы отделяли путем вакуумной фильтрации и промывали холодным ТГФ (5 мл). Соленый влажный фильтровальный осадок добавляли к ТГФ (10 мл) и воде (5 мл). К смеси добавляли триэтиламин (Et3N; 0,8 мл, 5,7 ммоль), и реакционная смесь образовывала прозрачный двухфазный раствор. Реакционную смесь переносили в делительную воронку и отделяли органический слой. К органическому слою добавляли гексаны (20 мл), и твердые частицы выпадали в осадок из раствора. Реакционную смесь охлаждали до 0°C и оставляли перемешиваться в течение 30 минут. Твердые частицы выделяли путем вакуумной фильтрации, промывали гексанами (10 мл) и высушивали в вакуумном сушильном шкафу при 40°C с получением пропан-2-ил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината в виде белого твердого вещества (0,90 г, чистота по площади хроматограммы ЖХ 89%): температура плавления 174-176°C; 1H ЯМР (400 МГц, CDCl3) δ 7,54 (д, J=6,4 Гц, 1H), 7,34-7,22 (м, 2H), 5,27 (септет, J=6,3 Гц, 1H), 4,56 (ушир.с, 2H), 3,97 (д, J=1,0 Гц, 3H), 1,39 (д, J=6,3 Гц, 6H); 13C ЯМР (101 МГц, CDCl3) δ 164,03, 154,07 (д, J=253,7 Гц), 147,70 (д, J=256,1 Гц), 144,92 (д, J=5,0 Гц), 144,48 (д, J=13,9 Гц), 142,01 (д, J=12,2 Гц), 140,58 (д, J=14,4 Гц), 129,59 (д, J=3,4 Гц), 125,85 (д, J=3,7 Гц), 125,29 (д, J=3,8 Гц), 123,57 (дд, J=14,1, 3,7 Гц), 112,85 (д, J=3,7 Гц), 69,58 (с), 61,63 (д, J=4,5 Гц), 21,86 (с); 19F ЯМР (376 МГц, CDCl3) δ -128,44 (д, J=32,7 Гц), -142,30 (д, J=31,3 Гц).
Пример 6b. Пропан-2-ил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
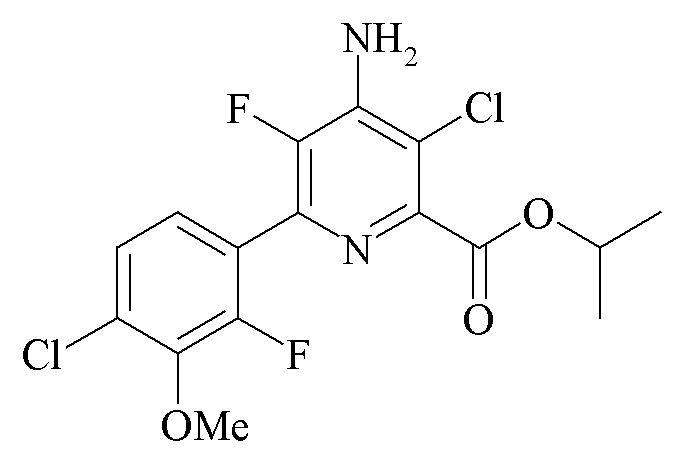
Пропан-2-ил 4-амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (1,75 г, чистота по площади хроматограммы ЖХ 95%) добавляли к дихлорметану (15 мл) и смесь нагревали до 40°C с получением прозрачного желтого раствора. Раствор оставляли охлаждаться до комнатной температуры и добавляли HCl (4 М в диоксане; 1,25 мл, 5 ммоль). После добавления HC1 твердые частицы выпадали в осадок из раствора и реакционную смесь охлаждали до 0°C. Твердое вещество отделяли путем вакуумной фильтрации и промывали холодным дихлорметаном (5 мл). Соленый влажный фильтровальный осадок добавляли к дихлорметану (10 мл) и воде (5 мл). К смеси добавляли Et3N (0,6 мл, 4,3 ммоль), и реакционная смесь образовывала прозрачный двухфазный раствор. Реакционную смесь переносили в делительную воронку и органический слой отделяли. К органическому слою добавляли гексаны (20 мл), и из раствора осаждались твердые частицы. Реакционную смесь охлаждали до 0°C и твердые частицы отделяли путем вакуумной фильтрации и промывали гексанами (10 мл). Твердое вещество высушивали в вакуумном сушильном шкафу при 40°C с получением пропан-2-ил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината в виде белого твердого вещества. (1,02 г, чистота по площади хроматограммы ЖХ 99%): температура плавления 115-117°C; 1H ЯМР (400 МГц, CDCl3) δ 7,31-7,22 (м, 2H), 5,32 (септет, J=6,3 Гц, 1H), 4,93 (ушир.с, 2H), 3,98 (д, J=1,1 Гц, 3H), 1,41 (д, J=6,3 Гц, 6H); 13C ЯМР (101 МГц, CDCl3) δ 164,28, 153,93 (д, J=254,4 Гц), 145,79 (д, J=230,6 Гц), 145,23 (д, J=4,9 Гц), 144,44 (д, J=13,8 Гц), 139,97 (д, J=13,5 Гц), 137,72 (д, J=13,8 Гц), 129,90 (д, J=3,4 Гц), 125,59 (д, J=3,2 Гц), 125,41 (д, J=3,7 Гц), 122,82 (дд, J=14,0, 4,4 Гц), 114,82, 70,36, 61,66 (д, J=4,7 Гц), 21,76; 19F ЯМР (376 МГц, CDCl3) δ -128,15 (д, J=34,1 Гц), 138,44 (д, J=34,1 Гц).
Пример 6c. Метил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
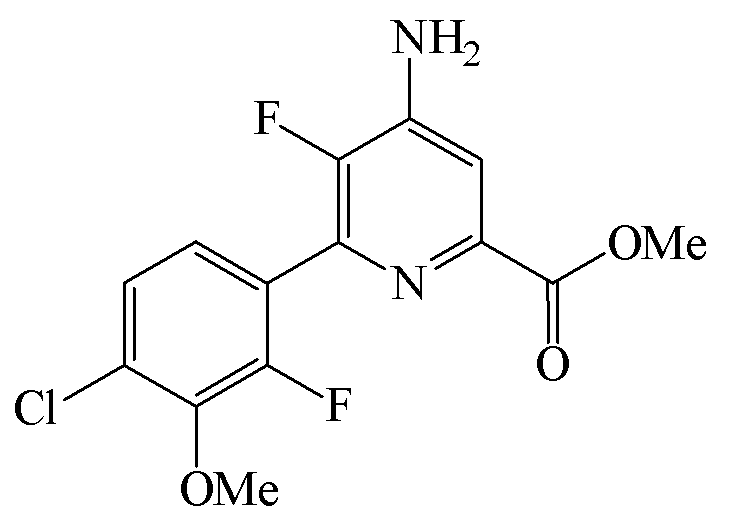
Бензил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (3,00 г, 7,41 ммоль) добавляли к CH3OH (35 мл). В реакционную смесь добавляли метилат натрия (25 масс.% в CH3OH; 2,0 мл, 8,9 ммоль) и оставляли перемешиваться в течение 24 ч. В реакционную смесь добавляли воду (50 мл) и смесь концентрировали при пониженном давлении с удалением большинства CH3OH. Смесь экстрагировали при помощи EtOAc (2×40 мл), объединенные органические слои промывали водой (40 мл) и насыщ. хлористым натрием (40 мл) и концентрировали при пониженном давлении с получением метил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината в виде бледно-желтого твердого вещества (1,86 г; чистота по площади хроматограммы ЖХ 67%).
Метил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат (1,50 г, чистота по площади хроматограммы ЖХ 67%) добавляли к ТГФ (10 мл) и дихлорметану (5 мл) и смесь нагревали до 40°C с получением прозрачного желтого раствора. Раствор оставляли охлаждаться до комнатной температуры и добавляли HCl (4 М в диоксане; 1,4 мл, 5,6 ммоль). После добавления HCl из раствора в осадок выпадали твердые частицы, и реакционную смесь охлаждали до 0°C. Твердое вещество отделяли путем вакуумной фильтрации и промывали холодным ТГФ (5 мл). Соленый влажный фильтровальный осадок добавляли к ТГФ (25 мл) и воде (10 мл). К смеси добавляли Et3N (0,8 мл, 5,7 ммоль), и реакционная смесь образовывала прозрачный двухфазный раствор. Реакционную смесь переносили в делительную воронку и органический слой отделяли и концентрировали до раствора объемом ~10 мл. К органическому слою добавляли гексаны (20 мл), и твердые частицы осаждались из раствора. Реакционную смесь охлаждали до 0°C, и твердые частицы отделяли путем вакуумной фильтрации и промывали гексанами (10 мл). Твердое вещество высушивали в вакуумном сушильном шкафу при 40°C с получением метил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината в виде белого твердого вещества (0,53 г, чистота по площади хроматограммы ЖХ 89%): температура плавления 203-204°C; 1H ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=6,4 Гц, 1H), 7,28-7,25 (м, 2H), 4,58 (ушир.с, 2H), 3,97 (д, J=1,1 Гц, 3H), 3,96 (с, 3H); 13C ЯМР (101 МГц, CDCl3) δ 165,27, 154,05 (д, J=254,1 Гц), 147,83 (д, J=256,3 Гц), 144,56 (д, J=13,7 Гц), 144,31 (д, J=5,1 Гц), 142,16 (д, J=12,4 Гц), 140,63 (д, J=14,5 Гц), 129,69 (д, J=3,5 Гц), 125,63 (д, J=3,1 Гц), 125,41 (д, J=3,7 Гц), 123,43 (дд, J=14,1, 3,6 Гц), 113,05 (д, J=3,7 Гц), 61,64 (д, J=4,5 Гц), 52,98; 19F ЯМР (376 МГц, CDCl3) δ -128,71 (д, J=28,6 Гц), -141,94 (д, J=28,6 Гц).
Пример 6d. Метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
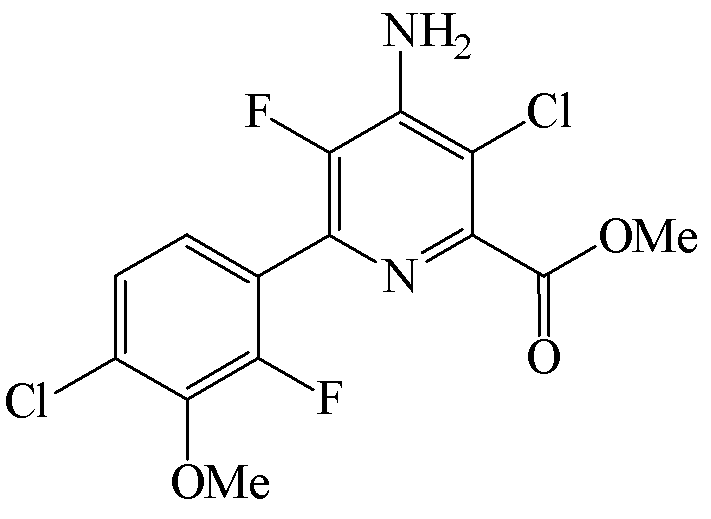
Бензил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат (3,00 г, 6,83 ммоль) добавляли к CH3OH (35 мл). В реакционную смесь добавляли метилат натрия (25 масс.% в CH3OH; 1,9 мл, 8,2 ммоль) и реакционную смесь оставляли перемешиваться в течение 24 ч. В реакционную смесь добавляли воду (50 мл) и смесь концентрировали при пониженном давлении с удалением большей части CH3OH. Смесь экстрагировали при помощи EtOAc (2×40 мл) и объединенные органические слои промывали водой (40 мл) и насыщ. раствором хлорида натрия (40 мл). Концентрирование летучих компонентов при пониженном давлении приводило к получению метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината в виде бледно-желтого твердого вещества (2,04 г, чистота по площади хроматограммы ЖХ 88%).
Метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат (1,25 г, чистота по площади хроматограммы ЖХ 88%) добавляли к дихлорметану (15 мл) и ТГФ (3 мл) и смесь нагревали до 40°C с получением прозрачного желтого раствора. Раствор оставляли охлаждаться до комнатной температуры и добавляли HCl (4 M в диоксане; 0,95 мл, 3,8 ммоль). После добавления HCl твердые частицы осаждались из раствора, и реакционную смесь охлаждали до 0°C. Твердое вещество отделяли путем вакуумной фильтрации и промывали холодным дихлорметаном (5 мл). Соленый влажный фильтровальный осадок добавляли к дихлорметану (20 мл) и воде (10 мл). К смеси добавляли Et3N (0,6 мл, 4,3 ммоль), и реакционная смесь образовывала прозрачный двухфазный раствор. Реакционную смесь переносили в делительную воронку, и органический слой отделяли и концентрировали при пониженном давлении до раствора объемом ~10 мл. К органическому слою добавляли гексаны (20 мл), и твердые частицы осаждались из раствора. Реакционную смесь охлаждали до 0°C и твердые частицы отделяли вакуумной фильтрацией и промывали гексанами (10 мл). Твердое вещество высушивали в вакуумном сушильном шкафу при 40°C с получением метил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината в виде белого твердого вещества (0,51 г, чистота по площади хроматограммы ЖХ 96%): температура плавления 170-171°C; 1H ЯМР (400 МГц, CDCl3) δ 7,30-7,20 (м, 2H), 4,98 (ушир.с, 2H), 3,98 (м, 6H); 13C ЯМР (101 МГц, CDCl3) δ 164,67, 153,95 (д, J=254,7 Гц), 145,93 (д, J=245,8 Гц), 144,57 (с), 143,65 (д, J=4,6 Гц), 140,21 (д, J=13,3 Гц), 137,71 (д, J=13,9 Гц), 130,02 (д, J=3,5 Гц), 125,49 (д, J=7,1 Гц), 125,49, 122,70 (дд, J=14,1, 4,3 Гц), 115,89 (д, J=1,5 Гц), 61,67 (д, J=4,5 Гц), 53,06; 19F ЯМР (376 МГц, CDCl3) δ -128,34 (д, J=31,3 Гц), -137,60 (д, J=32,7 Гц).
Пример 6e. Бензил 4-амино-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
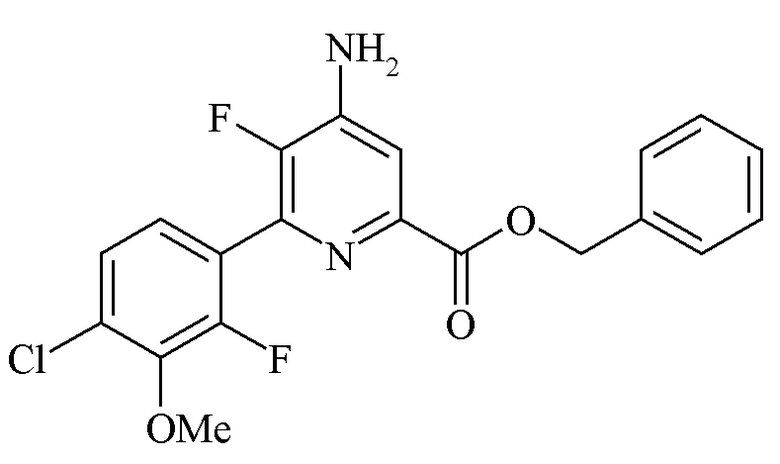
Взвесь бензил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)-2-пиридинкарбоксилата (98 г, 0,209 моль, чистота 86,5%) и ТГФ (300 мл) медленно нагревали до 28°C с получением прозрачного раствора янтарного цвета. Раствору позволяли охладиться до 20°C и в течение минуты посредством шприца добавляли HC1 (4 М в 1,4-диоксане; 55 мл, 0,219 моль, 1,05 экв.). Раствор быстро становился мутным с образованием твердого осадка, и температура смеси достигала 28°C. Смесь охлаждали до менее 10°C. Осадок отфильтровывали и промывали холодным ТГФ (2×40 мл) с получением белого твердого вещества (120,4 г). Это твердое вещество перемешивали с ТГФ (300 мл) и водой (100 мл). К этой взвеси в течение 1 минуты добавляли Et3N (30,5 мл, 0,219 моль) посредством шприца, и твердые частицы растворялись с образованием мутной смеси. Органический слой отделяли. При перемешивании добавляли гексаны (450 мл) и раствор охлаждали до менее 10°C. Полученный осадок отфильтровывали и промывали гексанами (2×40 мл) с получением белого твердого вещества (76,1 г, чистота по ВЭЖХ 95,1 масс.% (гексанофенон внутренний стандарт)). Дополнительные 7,56 г чистого продукта с чистотой 93,9% получали путем концентрации фильтрата. 1H и 19F спектры ЯМР выделенного продукта были идентичны наблюдаемым в примере 2c.
Пример 6f. Бензил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
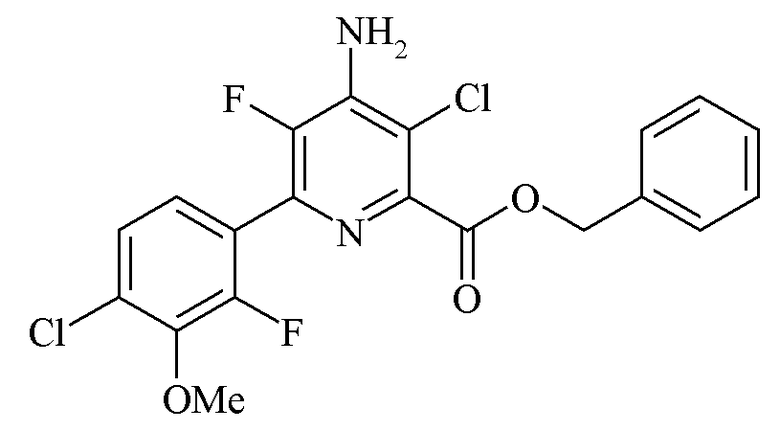
К раствору бензил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколината (5,00 г, чистота по площади хроматограммы ЖХ 87%, по ЖХ анализу 88 масс.%) в дихлорметане (25 мл) добавляли HCl (4 М в 1,4-диоксане; 3,1 мл, 12,4 ммоль) посредством шприца. Раствор становился мутным с образованием твердых частиц через 1 минуту после перемешивания. Смесь охлаждали в ванне со льдом до менее 10°C, фильтровали и промывали холодным дихлорметаном (5 мл) с получением белого твердого вещества. Это твердое вещество суспендировали с дихлорметаном (30 мл) и водой (15 мл) и добавляли Et3N (1,98 мл, 14,2 ммоль). Твердые частицы растворялись с образованием двухфазной смеси. После перемешивания в течение 15 минут смесь переносили в делительную воронку и фазам позволяли разделяться в течение более 15 минут. Органический слой отделяли, добавляли гексаны (60 мл) и смесь охлаждали до менее 10°C. Раствор в течение ночи становился мутным, и из смеси выпадал твердый осадок. В результате вакуумной фильтрации смеси получали белое твердое вещество, высушивали в вакуумном сушильном шкафу при 40°C с получением бензил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фтор-2-пиридинкарбоксилата в виде белого твердого вещества (3,11 г, 70%, чистота по площади хроматограммы ЖХ 97%, чистота по ЖХ анализу 97 вес.%). 1H и 19F спектры ЯМР изолированного продукта были идентичны наблюдаемым в примере 4f.
| название | год | авторы | номер документа |
|---|---|---|---|
| 6-АМИНО-2-ЗАМЕЩЕННЫЕ-5- ВИНИЛСИЛИЛПИРИМИДИН-4-КАРБОНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ И 4-АМИНО-6-ЗАМЕЩЕННЫЕ-3-ВИНИЛСИЛИЛПИРИДИН-ПИКОЛИНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ КАК ГЕРБИЦИДЫ | 2012 |
|
RU2556000C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-3-ХЛОР-5-ФТОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545021C1 |
| СЛОЖНЫЕ АРИЛАЛКИЛОВЫЕ ЭФИРЫ 4-АМИНО-6-(ЗАМЕЩЕННЫЙ ФЕНИЛ)ПИКОЛИНАТОВ И 6-АМИНО-2-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-4-ПИРИМИДИНКАРБОКСИЛАТОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2012 |
|
RU2566760C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-3-ХЛОР-5-ФТОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2539578C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2542985C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 4-АМИНО-3-ГАЛОГЕН-6(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ И 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2658825C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2632203C2 |
| ФТОРПИКОЛИНОИЛФТОРИДЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2627659C2 |
| 4-АМИНО-6-(ГЕТЕРОЦИКЛИЧЕСКИЕ)ПИКОЛИНАТЫ И 6-АМИНО-2-(ГЕТЕРОЦИКЛИЧЕСКИЕ)ПИРИМИДИН-4-КАРБОКСИЛАТЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2014 |
|
RU2672587C2 |
| 4-АМИНО-6-(ГЕТЕРОЦИКЛИЛ)ПИКОЛИНАТЫ И 6-АМИНО-2-(ГЕТЕРОЦИКЛИЛ)ПИРИМИДИН-4-КАРБОКСИЛАТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2014 |
|
RU2672584C2 |
Изобретение относится способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
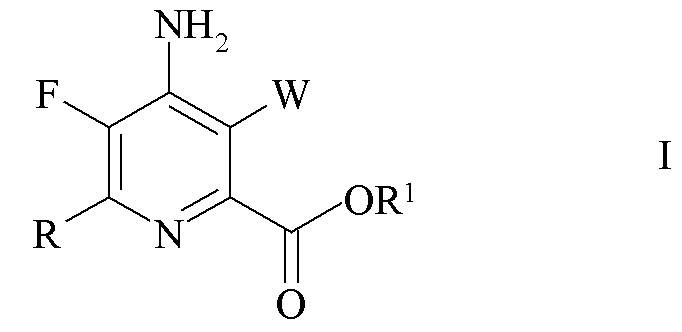
где W представляет собой Cl, Br или I; R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил; который включает следующие стадии: обмен фтором, аминирование, обмен галогеном, галогенирование и связывание при помощи переходного металла. 4 н. и 3 з.п. ф-лы, 6 пр.
1. Способ получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
где W представляет собой Cl, Br или I;
R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и
R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
который включает следующие стадии:
a) фторирование 4,5,6-трихлорпиколината формулы A
где R1 является таким, как определено ранее;
источником фторид-иона с получением 4,5,6-трифторпиколината формулы B
где R1 является таким, как определено ранее;
b) аминирование 4,5,6-трифторпиколината формулы B аммиаком с получением 4-амино-5,6-дифторпиколината формулы C
где R1 является таким, как определено ранее;
c) замена фтор-заместителя в положении 6 4-амино-5,6-дифторпиколината формулы C на йод, бром или хлор-заместитель путем обработки источником ионов йода, брома или хлора с получением 4-амино-5-фтор-6-галогенпиколината формулы D
где X представляет собой Cl, Br или I
и R1 является таким, как определено ранее;
d) галогенирование 4-амино-5-фтор-6-галогенпиколината формулы D источником галогена с получением 4-амино-3,6-дигалоген-5-фторпиколината формулы E
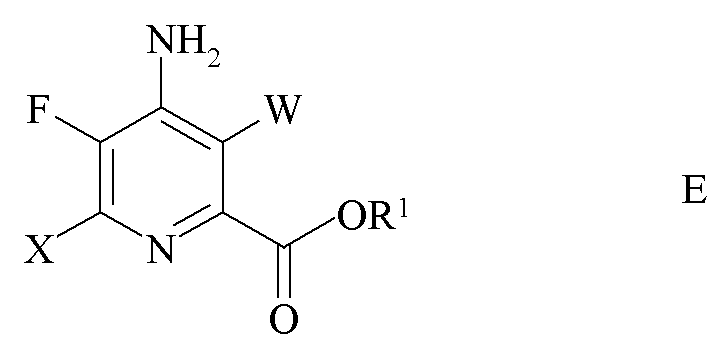
где W и X независимо представляют собой Cl, Br или I и
R1 является таким, как определено ранее; и
e) связывание 4-амино-3,6-дигалоген-5-фторпиколината формулы E с арильным, алкильным или алкенильным металлическим соединением формулы F

где R является таким, как определено ранее, и Met представляет собой Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, C1-C4 алкил или, взятые вместе, образуют этиленовую или пропиленовую группу в присутствии катализатора на основе переходного металла с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
2. Способ по п.1, в котором аминсодержащий продукт или промежуточный продукт очищены путем: a) протонирования кислотой с образованием соли, b) выделения соли с высокой чистотой путем кристаллизации, осаждения или экстракции и c) нейтрализации очищенной соли основанием с образованием очищенного нейтрального аминсодержащего продукта или промежуточного соединения.
3. Способ получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
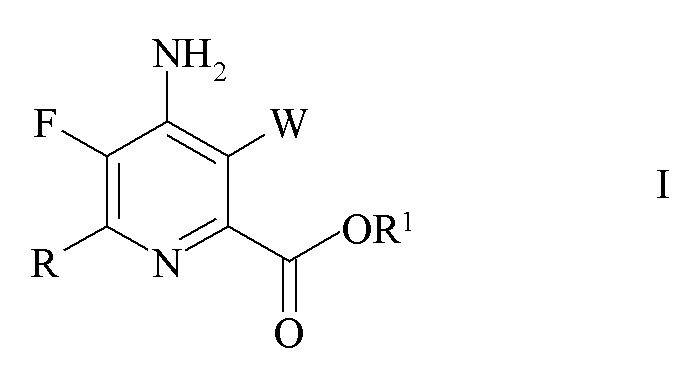
где W представляет собой Cl, Br или I;
R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и
R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
который включает следующие стадии:
a) фторирование 4,5,6-трихлорпиколината формулы A
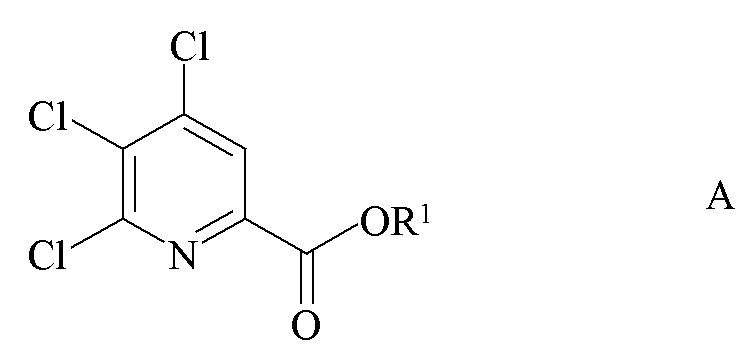
где R1 является таким, как определено ранее;
источником фторид-иона с получением 4,5,6-трифторпиколината формулы B
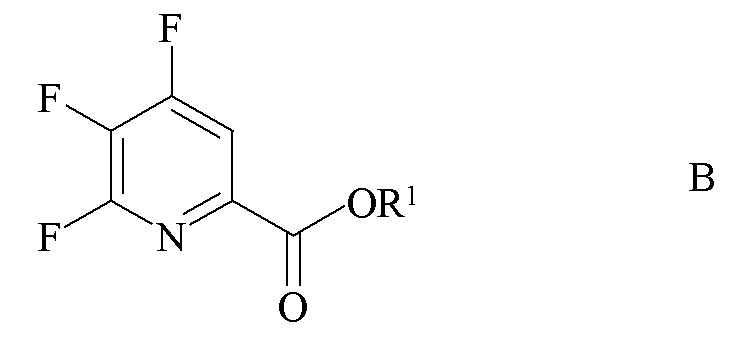
где R1 является таким, как определено ранее;
b) аминирование 4,5,6-трифторпиколината формулы B аммиаком с получением 4-амино-5,6-дифторпиколината формулы C
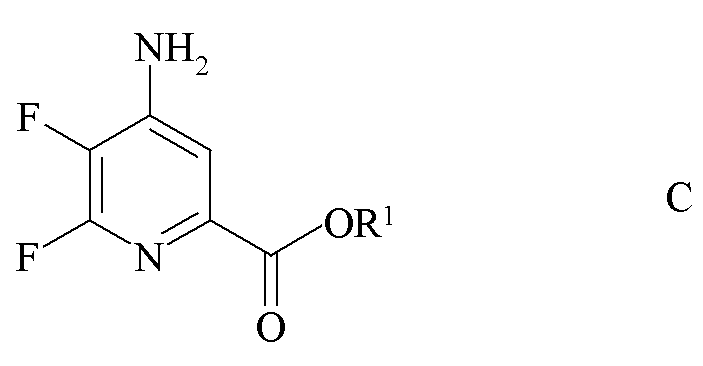
где R1 является таким, как определено ранее;
c) замена фтор-заместителя в положении 6 4-амино-5,6-дифторпиколината формулы C на йод, бром или хлор-заместитель путем обработки источником йода, брома или хлора с получением 4-амино-5-фтор-6-галогенпиколината формулы D
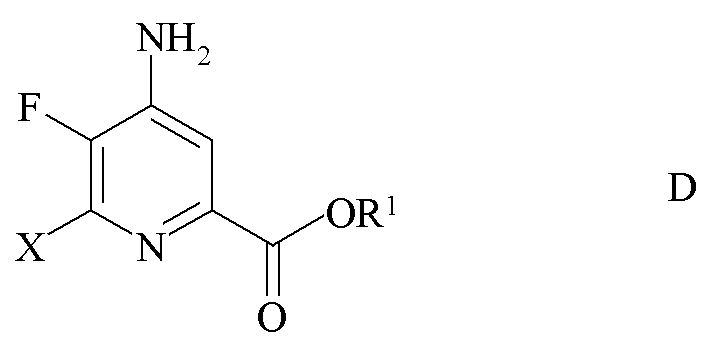
где X представляет собой Cl, Br или I и
R1 является таким, как определено ранее;
d) связывание 4-амино-5-фтор-6-галогенпиколината формулы D с арильным, алкильным или алкенильным металлическим соединением формулы F

где R является таким, как определено ранее, и Met представляет собой Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, C1-C4 алкил или, взятые вместе, образуют этиленовую или пропиленовую группу в присутствии катализатора на основе переходного металла с получением 4-амино-5-фтор-6-(замещенного)пиколината формулы G,
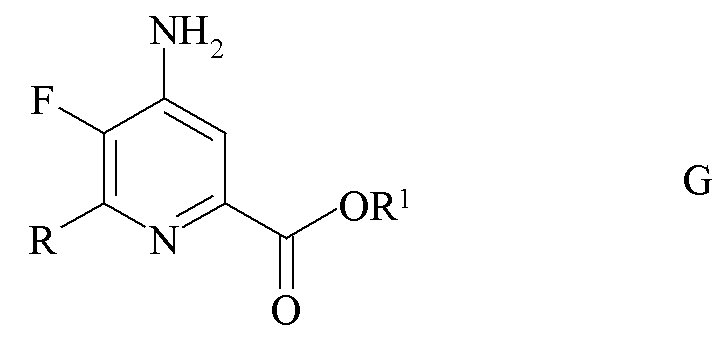
где R, R1 являются такими, как определено ранее; и
e) галогенирование 4-амино-5-фтор-6-(замещенного)пиколината формулы G источником галогена с получением 4-амино-5-фтор-3-гало-6-(замещенного)пиколината формулы I.
4. Способ по п.3, в котором аминсодержащий продукт или промежуточный продукт очищены путем: a) протонирования кислотой с образованием соли, b) выделения соли с высокой чистотой путем кристаллизации, осаждения или экстракции и c) нейтрализации очищенной соли основанием с образованием очищенного нейтрального аминсодержащего продукта или промежуточного соединения.
5. Способ получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
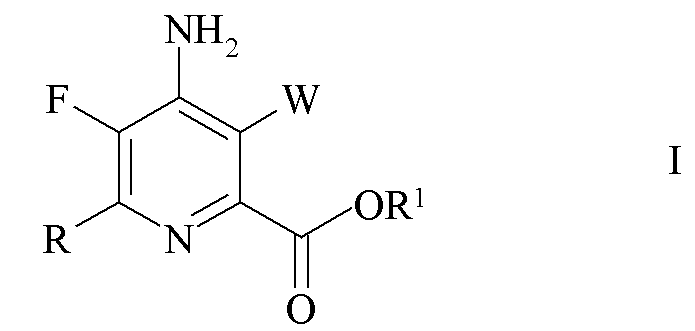
где W представляет собой Cl, Br или I;
R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси; и
R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
который включает следующие стадии:
a) связывание 4,5,6-трихлорпиколината формулы A
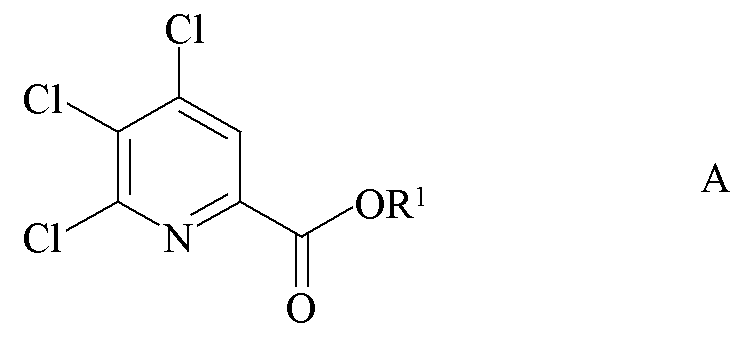
где R1 является таким, как определено ранее;
с арильным, алкильным или алкенильным металлическим соединением формулы F

где R является таким, как определено ранее, и Met представляет собой Zn-галогенид, Zn-R, три-(C1-C4 алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, C1-C4 алкил или, взятые вместе, образуют этиленовую или пропиленовую группу в присутствии катализатора на основе переходного металла с получением 4,5-дихлор-6-(замещенного)пиколината формулы H
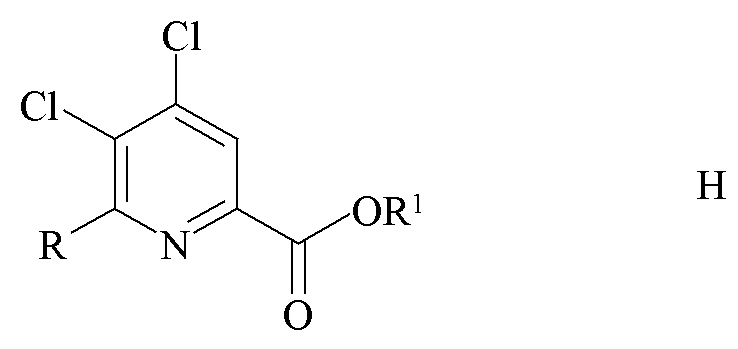
где R и R1 являются такими, как определено ранее;
b) фторирование 4,5-дихлор-6-(замещенного)пиколината формулы H источником фторид-ионов с получением 4,5-дифтор-6-(замещенного)пиколината формулы J
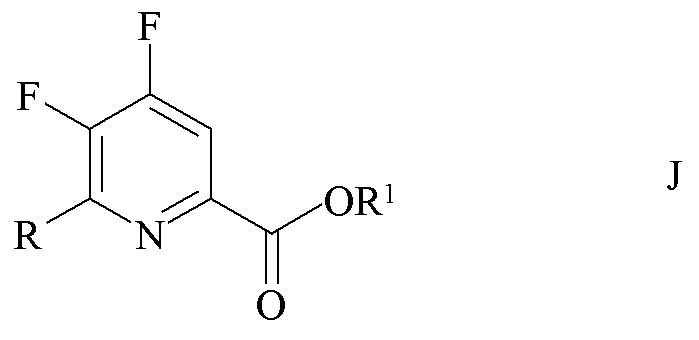
где R1 является таким, как определено ранее;
c) аминирование 4,5-дифтор-6-(замещенного)пиколината формулы J аммиаком с получением 4-амино-5-фтор-6-(замещенного)пиколината формулы K
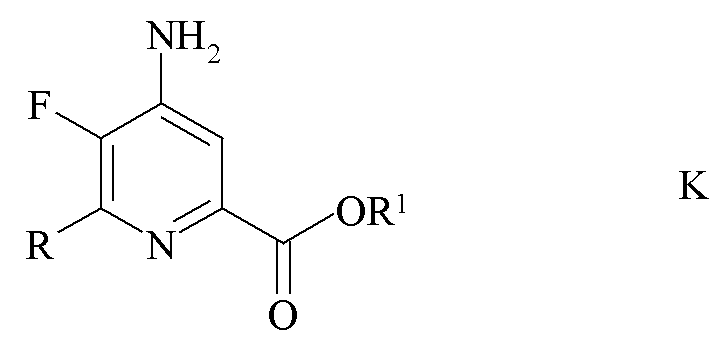
где R и R1 являются такими, как определено ранее; и
d) галогенирование 4-амино-5-фтор-6-(замещенного)пиколината формулы K источником галогена с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
6. Способ по п.5, в котором аминсодержащий продукт или промежуточное соединение очищены путем: a) протонирования кислотой с образованием соли, b) выделения соли с высокой чистотой путем кристаллизации, осаждения или экстракции и c) нейтрализации очищенной соли основанием с образованием очищенного нейтрального аминсодержащего продукта или промежуточного соединения.
7. Соединение, выбранное из группы, состоящей из:
a)
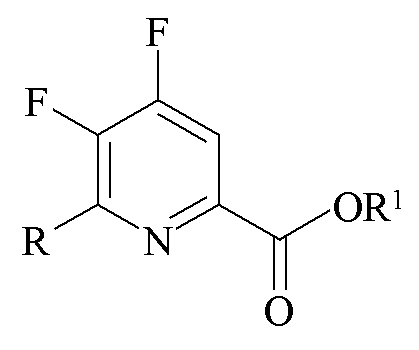
где R представляет собой С1-С4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
b)
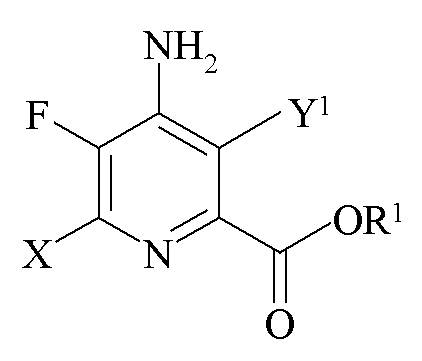
где X представляет собой I, Br, Cl или F, Y1 представляет собой H, Cl, Br или I, при условии, что, когда X представляет собой Cl, Y1 представляет собой H, Br или I, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил;
c)
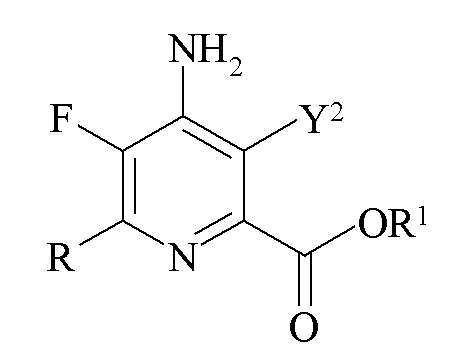
где Y2 представляет собой H, Br или I и R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил; и
d)
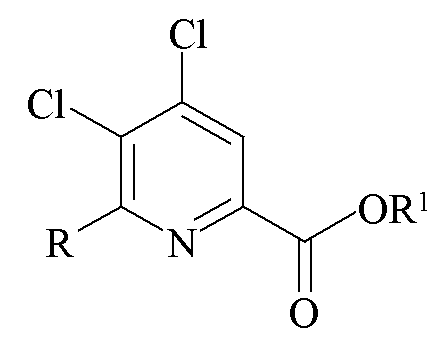
где R представляет собой C1-C4 алкил, циклопропил, C2-C4 алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкокси или C1-C4 галогеналкокси, и R1 представляет собой C1-C12 алкил или незамещенный или замещенный C7-C11 арилалкил.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| 6-АЛКИЛ ИЛИ АЛКЕНИЛ-4-АМИНОПИКОЛИНАТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2004 |
|
RU2332404C2 |
| RU 2002120924, А, 10.01.2004 | |||
| СЕЛЕКТИВНОЕ ЭЛЕКТРОХИМИЧЕСКОЕ ВОССТАНОВЛЕНИЕ ГАЛОГЕНИРОВАННЫХ 4-АМИНОПИКОЛИНОВЫХ КИСЛОТ | 2001 |
|
RU2254401C2 |

