1. Притязание на приоритет
По настоящей заявке испрашивается приоритет предварительной заявки США № 61/675235, озаглавленной "Способ получения 4-амино-5-Фтор-3-галоген-6-(замещенных)пиколинатов", поданной 24 июля 2012. Приведенная выше заявка включена в настоящее описание посредством ссылки во всей своей полноте.
2. Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к способам получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов. Более конкретно, настоящее изобретение относится к способам получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, включающим стадию преобразования хлорпиколиноилхлоридов в фторпиколиноилфториды.
3. Предпосылки создания настоящего изобретения
В патенте США 6297197 B1 описаны, в том числе, определенные 6-(алкокси или арилокси)-4-амино-3-хлор-5-фторпиколинатные соединения и их применение в качестве гербицидов. В патентах США 6784137 B2 и 7314849 B2 описаны, в том числе, определенные 6-(арил)-4-амино-3-хлор-5-фторпиколинатные соединения и их применение в качестве гербицидов. В патенте США 7432227 B2 описаны, в том числе, определенные 6-(алкил)-4-амино-3-хлор-5-фторпиколинатные соединения и их применение в качестве гербицидов. В каждом из этих патентов описано получение 4-амино-3-хлор-5-фторпиколинатных исходных соединений фторированием соответствующих 5-незамещенных пиридинов бис(тетрафторборатом) 1-(хлорметил)-4-фтор-1,4-диазонийбицикло[2,2,2]октана. Преимущественным будет получение 4-амино-5-фтор-3-галоген-6-(замещенные)пиколинатов, без необходимости прямого фторирования положения 5 пиридинового кольца дорогим фторирующим агентом, подобным бис(тетрафторборату) 1-(хлорметил)-4-фтор-1,4-диазонийбицикло[2,2,2]октана.
В заявке США № 13/356691 описаны, в том числе, способы получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, включающие фторирование 5-хлорпиколинатного эфира источником фторидных ионов. В заявке США № 13/356686 описаны, в том числе, способы получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, включающие фторирование 5-хлор-пиколинонитрильного соединения источником фторидных ионов. Из-за относительно слабой электроноакцепторной способности эфирных функциональных групп, высокореакционный источник фторидных ионов, такой как CsF, является предпочтительным для способа фторирования, описанного в заявках США № 13/356691 и 13/356686. При применении менее реакционноспособного источника фторидных ионов, такого как KF, фторирование хлорпиколинатных соединений может приводить к низким-умеренным выходам требуемого продукта из-за неполного фторирования и разрушения исходных соединений и продуктов в форсированных условиях. Будет полезно обеспечить улучшенные и более рентабельные способы получения 4-амино-5-фтор-3-галоген-6-(замещенные)пиколинатов, вне зависимости от дорогих химических реагентов, таких как CsF.
4. Сущность изобретения
Настоящее изобретение относится к способам получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов. Более конкретно, настоящее изобретение относится к способам получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, включающим стадию преобразования хлорпиколиноилхлоридов в фторпиколиноилфториды.
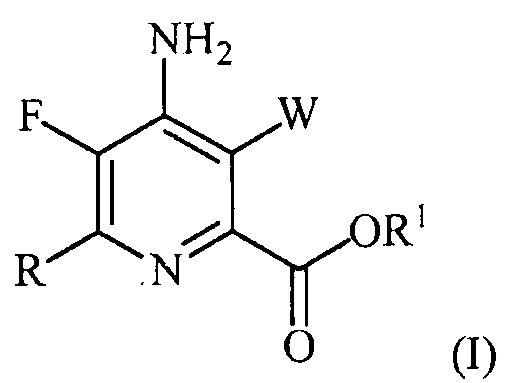
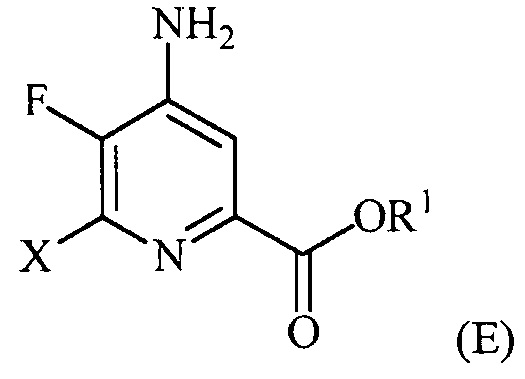
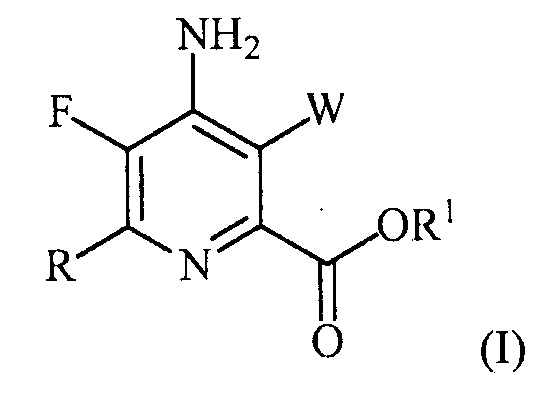
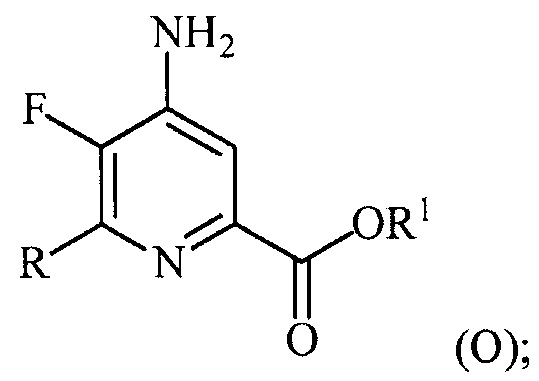
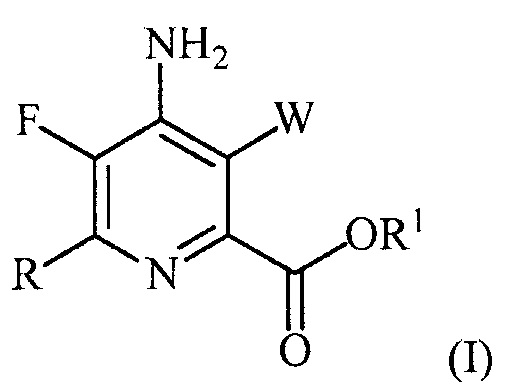
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
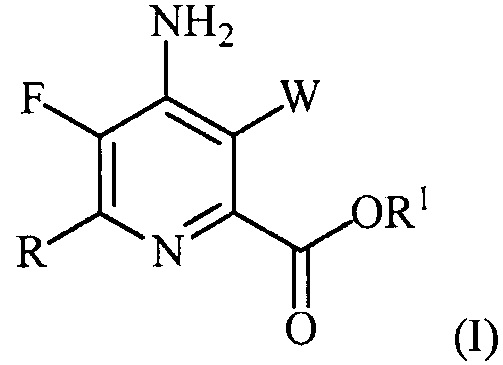
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
введение фтор заместителя в положение 5 пиридиновой структуры в формуле (I) фторированием 5-Cl-пиколиноилхлоридного соединения источником фторидного иона с получением 5-F-пиколиноилфторидного соединения;
введение аминозаместителя в положение 4 пиридиновой структуры в формуле (I) аминированием 4-галогенпиридинового соединения источником аммиака;
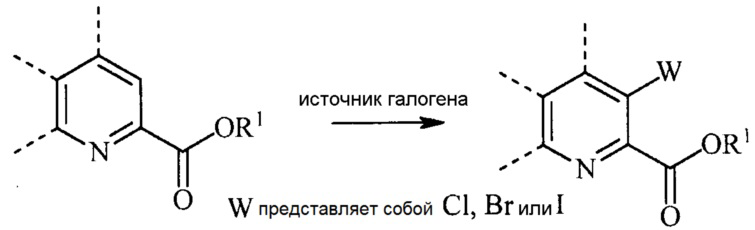
введение W заместителя в положение 3 пиридиновой структуры в формуле (I) галогенированием 3-незамещенного пиридинового соединения источником галогена; и
введение R заместителя в положение 6 пиридиновой структуры в формуле (I) конденсацией 6-галогенпиридинового соединения с R-Met соединением, где R-Met соединение определено в настоящем описании и в другом месте.
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
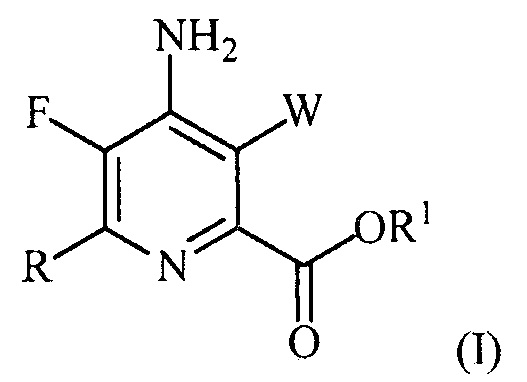
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
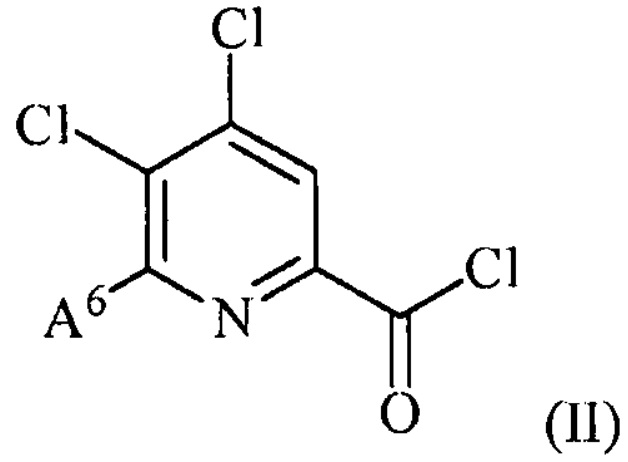
(a) фторирование соединения формулы (II):
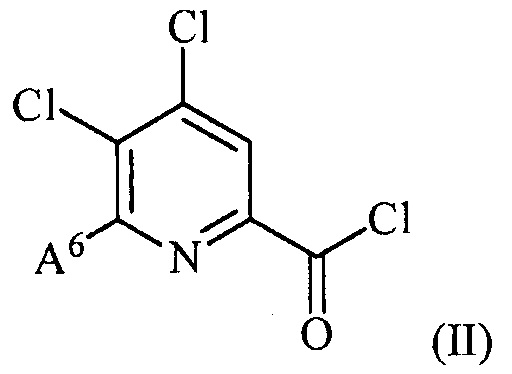 ,
,
где A6 представляет собой галоген или R,
источником фторидного иона с получением соединения формулы (III):
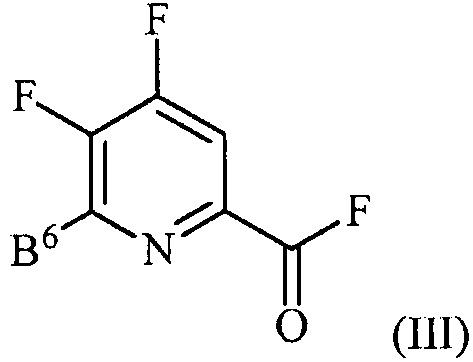 ,
,
где B6 представляет собой F или R;
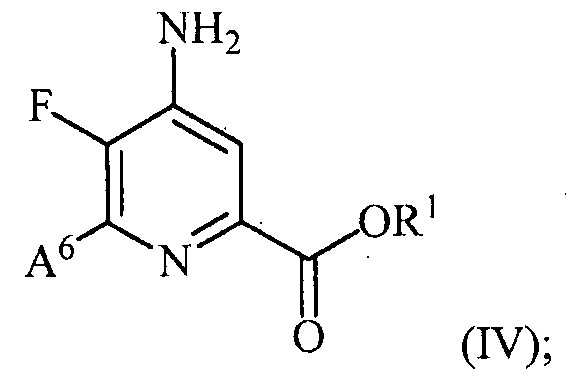
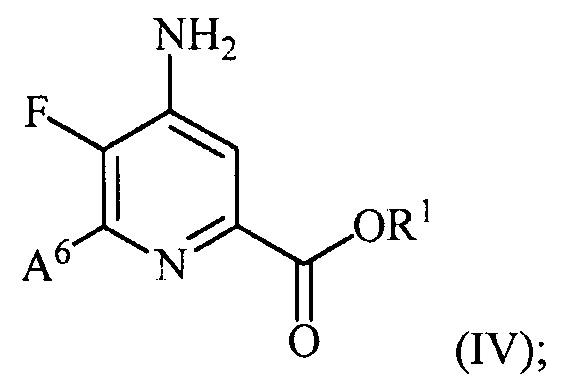
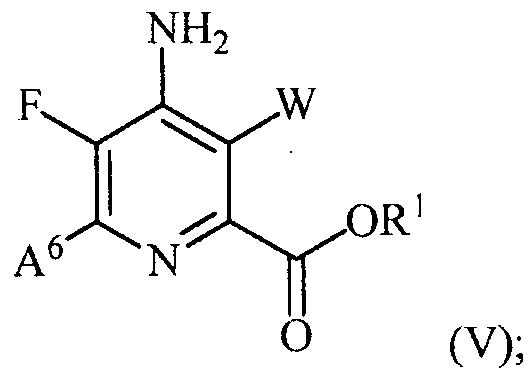
и преобразование соединения формулы (III) в соединение формулы (IV):
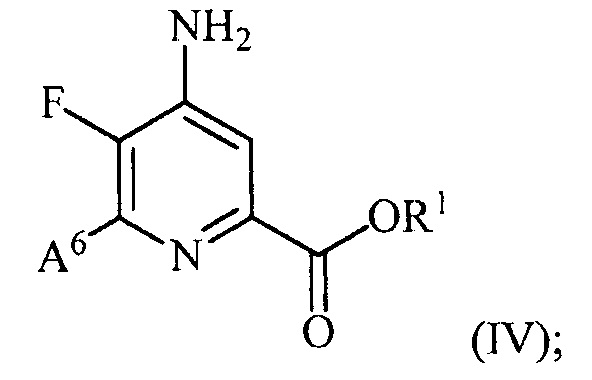
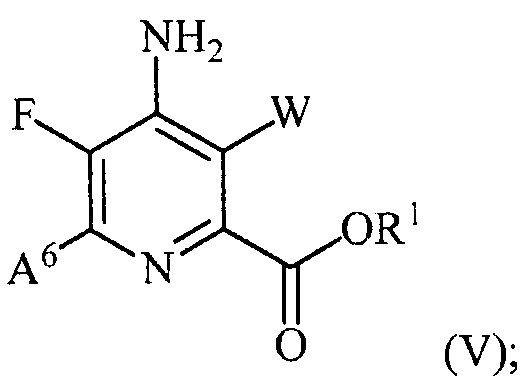
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
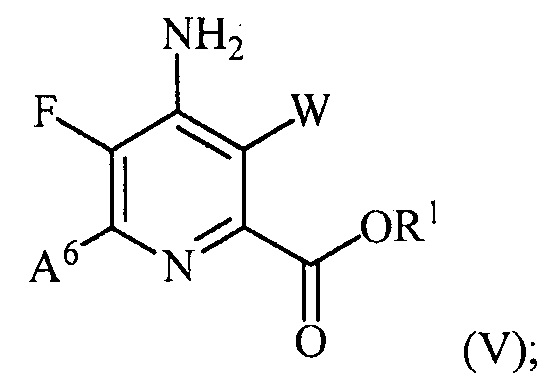 и
и
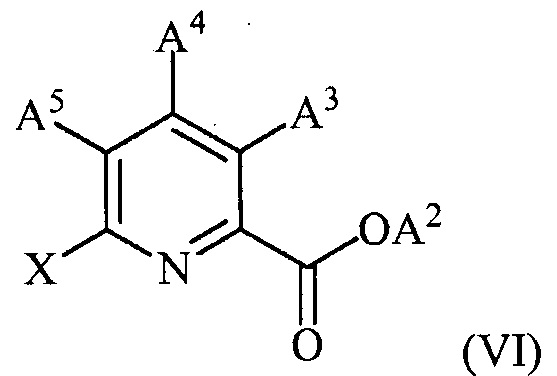
(c) конденсацию соединения формулы (VI):
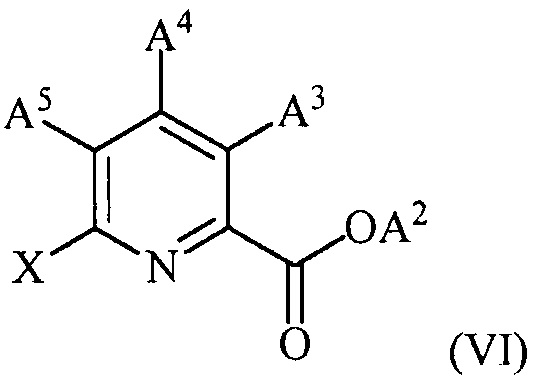 ,
,
где X представляет собой Cl, Br или I;
A2 представляет собой водород или R1;
A3 представляет собой водород или W;
A4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
A5 представляет собой F или Cl;
с соединением формулы (VII):
R-Met (VII),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
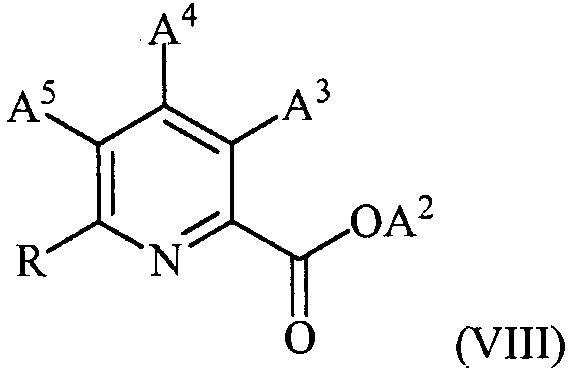 ,
,
где преобразование (c) можно осуществлять перед, между или после преобразований (a) и (b).
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
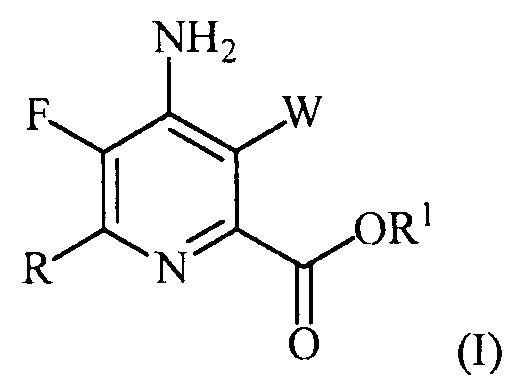
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
введение фтор заместителя в положение 5 пиридиновой структуры в формуле (I) фторированием 5-Cl-пиколиноилхлоридного соединения источником фторидного иона с получением 5-F-пиколиноилфторидного соединения;
введение аминозаместителя в положение 4 пиридиновой структуры в формуле (I) аминированием 4-галогенпиридинового соединения источником аммиака;
введение W заместителя в положение 3 пиридиновой структуры в формуле (I) галогенированием 3-незамещенного пиридинового соединения источником галогена; и
введение R заместителя в положение 6 пиридиновой структуры в формуле (I) конденсацией 6-галогенпиридинового соединения с R-Met соединением, где R-Met соединение определено в настоящем описании и в другом месте.
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
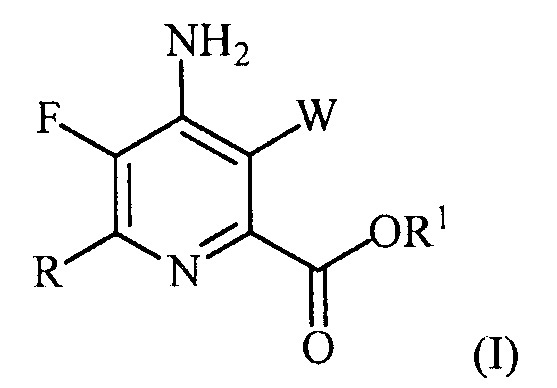
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
(a) фторирование соединения формулы (II):
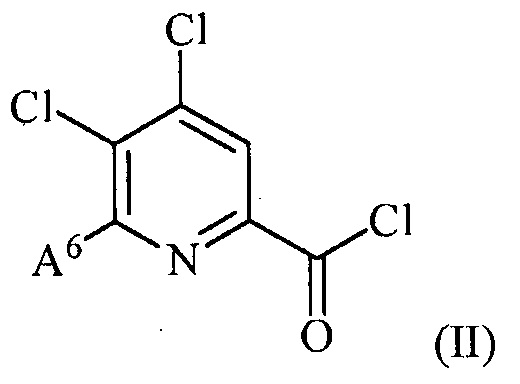 ,
,
где A6 представляет собой галоген или R;
источником фторидного иона с получением соединения формулы (III):
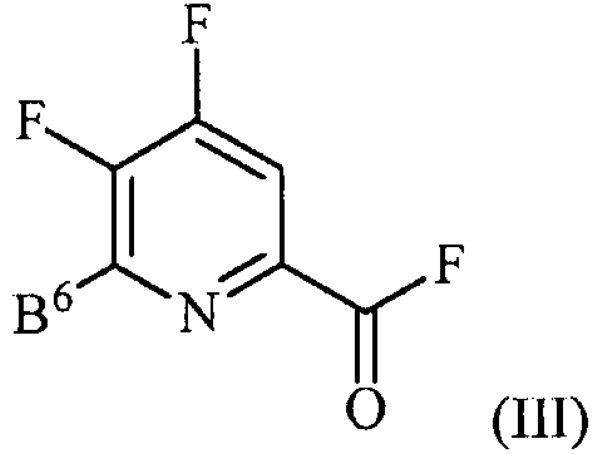 ,
,
где B6 представляет собой F или R;
и преобразование соединения формулы (III) в соединение формулы (IV):
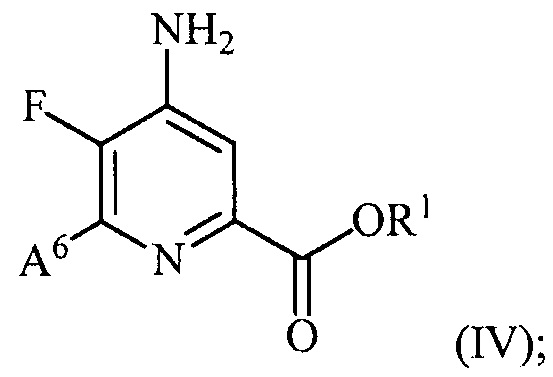
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
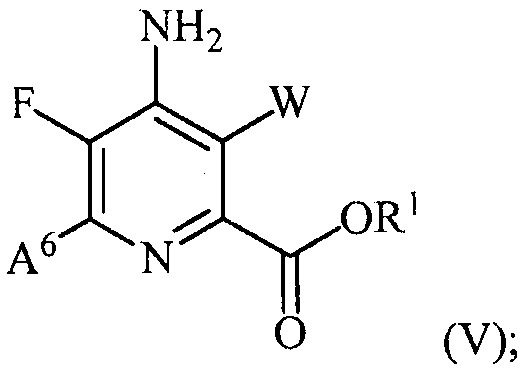
(c) конденсацию соединения формулы (VI):
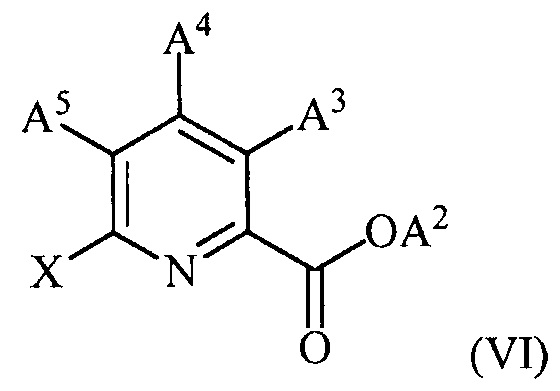 ,
,
где X представляет собой Cl, Br или I;
A2 представляет собой водород или R1;
A3 представляет собой водород или W;
A4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
A5 представляет собой F или Cl;
с соединением формулы (VII):
R-Met (VII),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
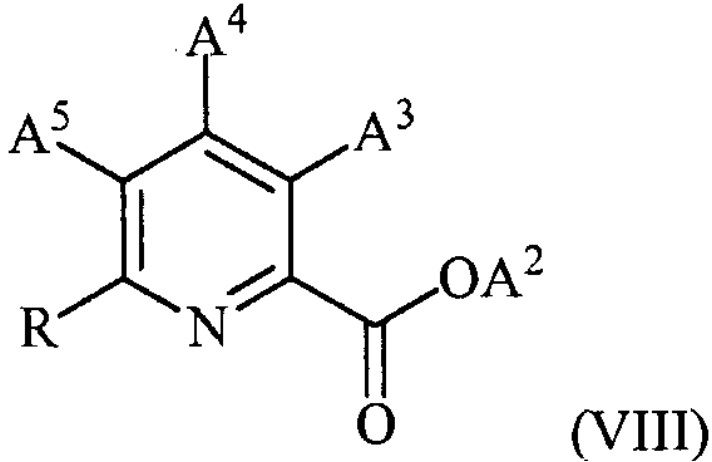 ,
,
где преобразование (c) можно осуществлять перед, между или после преобразований (a) и (b).
5. Подробное описание
5.1 Определения
Как использовано в настоящем описании и если не указано иное, термин "способ(бы)", представленный(ые) в настоящем описании, относится к способам, описанным в настоящем описании, которые являются пригодными для получения соединения, представленного в настоящем описании. Модификации способов, описанных в настоящем описании (например, исходные соединения, реагенты, защитные группы, растворители, температуры, продолжительность реакций, очистка) могут также быть включены в настоящее изобретение.
Как использовано в настоящем описании и если не указано иное, термин "добавление", "взаимодействие", "обработка" или подобные означает контакт одного взаимодействующего вещества, реагента, растворителя, катализатора, реакционноспособной группы или подобного с другим взаимодействующим веществом, реагентом, растворителем, катализатором, реакционноспособной группой или подобным. Взаимодействующие вещества, реагенты, растворители, катализаторы, реакционноспособные группы или подобные можно добавлять отдельно, одновременно или отдельно и можно добавлять в любом порядке. Их можно добавлять в присутствии или отсутствии нагревания и можно необязательно добавлять в инертной атмосфере. "Взаимодействие" может относиться к in situ образованию или внутримолекулярной реакции, когда реакционноспособные группы находятся в одной молекуле.
Как использовано в настоящем описании и если не указано иное, реакция, которая является "по существу завершенной" или доведенной "по существу до завершения", означает, что реакция составляет более чем приблизительно 80% выход, в одном из вариантов осуществления более чем приблизительно 90% выход, в другом варианте осуществления более чем приблизительно 95% выход и в другом варианте осуществления более чем приблизительно 97% выход требуемого продукта.
Как использовано в настоящем описании и если не указано иное, термин "соль" включает, но не ограничиваясь ими, соли кислых или основных групп, которые могут присутствовать в соединениях, описанных в настоящем описании. Соединения, которые являются основными по свойствам, способны образовывать большой набор солей с различными неорганическими и органическими кислотами. Кислоты, которые можно использовать для получения солей таких основных соединений, представляют собой кислоты, которые образуют соли, содержащие анионы, включая, но не ограничиваясь ими, ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, эдетат кальция, камзилат, карбонат, хлорид, бромид, йодид, цитрат, дигидрохлорид, эдетат, эдизилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидроксинафтоат, изотионат, лактат, лактобионат, малат, малеат, манделат, мезилат, метилсульфат, мускат, напсилат, нитрат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сукцинат, сульфат, таннат, тартрат, теоклат, триэтиодид и памоат. Соединения, которые содержат аминогруппу, могут также образовывать соли с различными аминокислотами, в добавление к кислотам, приведенным выше. Соединения, которые являются кислыми по свойствам, способны образовывать основные соли с различными катионами. Неограничивающие примеры таких солей включают соли щелочных или щелочноземельных металлов и в некоторых вариантах осуществления соли кальция, магния, натрия, лития, цинка, калия и железа. Соединения, которые являются кислыми по свойствам, способны образовывать основные соли с соединениями, которые содержат аминогруппу.
Как использовано в настоящем описании и если не указано иное, термин "гидрат" означает соединение или его соль, которое дополнительно содержит стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными силами.
Как использовано в настоящем описании и если не указано иное, термин "сольват" означает сольват, образованный в результате ассоциации одной или более молекул растворителя с соединением. Термин "сольват" включает гидраты (например, моногидрат, дигидрат, тригидрат, тетрагидрат и подобные).
Как использовано в настоящем описании и если не указано иное, термин "полиморф" означает твердые кристаллические формы соединения или его комплекса. Различные полиморфы одного соединения могут обладать различными физическими, химическими и/или спектроскопическими свойствами.
Термины "алкил", "алкенил" и "алкинил", а также производные термины, такие как "алкокси", "ацил", "алкилтио" и "алкилсульфонил", как использовано в настоящем описании, включают в свой объем группы с нормальной, разветвленной цепью или циклические группы. Если специально не указано иное, каждая может быть незамещенной или замещенной одним или более заместителями, выбранными, но не ограничиваясь ими, из галогена, гидрокси, алкокси, алкилтио, C1-C6ацила, формила, циано, арилокси или арила, при условии, что заместители являются стерически совместимыми и они удовлетворяют правилам химического связывания и энергии напряжения. Предполагается, что термины "алкенил" и "алкинил" включают одну или более ненасыщенных связей.
Термин "арилалкил", как использовано в настоящем описании, относится к замещенной фенилом алкильной группе, содержащей в сумме 7-11 атомов углерода, такой как бензил (-CH2C6H5), 2-метилнафтил (-CH2C10H7) и 1- или 2-фенетил (-CH2CH2C6H5 или -CH(CH3)C6H5). Фенильная группа может быть сама по себе незамещенной или замещенной одним или более заместителями, независимо выбранными из галогена, нитро, циано, C1-C6алкила, C1-C6алкокси, галогенированного C1-C6алкила, галогенированного C1-C6алкокси, C1-C6алкилтио, C(O)OC1-C6алкила, или где два соседних заместителя взяты вместе в виде -O(CH2)nO-, где n=1 или 2, при условии, что заместители являются стерически совместимыми и они удовлетворяют правилам химического связывания и энергии напряжения.
Если специально не указано иное, термин "галоген", а также производные термины, такие как "гало", относится к фтору, хлору, брому и йоду.
Как использовано в настоящем описании и если не указано иное, термин "амино" или "аминогруппа" означает одновалентную группу формулы -NH2, -NH(алкил), -NH(арил), -N(алкил)2, -N(арил)2 или -N(алкил)(арил).
Аминозащитные группы, такие как амидные группы (например, -C(=O)Raa), включают, но не ограничиваясь ими, формамид,
ацетамид, хлорацетамид, трихлорацетамид, трифторацетамид,
фенилацетамид, 3-фенилпропанамид, пиколинамид,
3-пиридилкарбоксамид, N-бензоилфенилаланильное производное,
бензамид, п-фенилбензамид, o-нитрофенилацетамид,
o-нитрофеноксиацетамид, ацетоацетамид,
(N-дитиобензилоксикарбониламино)ацетамид,
3-(п-гидроксифенил)пропанамид, 3-(o-нитрофенил)пропанамид,
2-метил-2-(o-нитрофенокси)пропанамид,
2-метил-2-(o-фенилазофенокси)пропанамид, 4-хлорбутанамид,
3-метил-3-нитробутанамид, o-нитроциннамид,
N-ацетилметиониновое производное, o-нитробензамид и
o-(бензоилоксиметил)бензамид.
Аминозащитные группы, такие как карбаматные группы (например, -C(=O)ORaa), включают, но не ограничиваясь ими,
метилкарбамат, этилкарбамат, 9-флуоренилметилкарбамат (Fmoc),
9-(2-сульфо)флуоренилметилкарбамат,
9-(2,7-дибром)флуороенилметилкарбамат,
2,7-ди-трет-бутил-[9-(10,10-диоксо-10,10,10,10-тетрагидротиоксантил)]метилкарбамат (DBD-Tmoc),
4-метоксифенацилкарбамат (Phenoc),
2,2,2-трихлорэтилкарбамат (Troc),
2-триметилсилилэтилкарбамат (Teoc), 2-фенилэтилкарбамат (hZ),
1-(1-адамантил)-1-метилэтилкарбамат (Adpoc),
1,1-диметил-2-галогенэтилкарбамат,
1,1-диметил-2,2-дибромэтилкарбамат (DB-t-BOC),
1,1-диметил-2,2,2-трихлорэтилкарбамат (TCBOC),
1-метил-1-(4-бифенилил)этилкарбамат (Bpoc),
1-(3,5-ди-трет-бутилфенил)-1-метилэтилкарбамат (t-Bumeoc),
2-(2'- и 4'-пиридил)этилкарбамат (Pyoc),
2-(N,N-дициклогексилкарбоксамидо)этилкарбамат,
трет-бутилкарбамат (BOC), 1-адамантилкарбамат (Adoc),
винилкарбамат (Voc), аллилкарбамат (Alloc),
1-изопропилаллилкарбамат (Ipaoc), циннамилкарбамат (Coc),
4-нитроциннамилкарбамат (Noc), 8-хинолилкарбамат,
N-гидроксипиперидинилкарбамат, алкилдитиокарбамат,
бензилкарбамат (Cbz), п-метоксибензилкарбамат (Moz),
п-нитробензилкарбамат, п-бромбензилкарбамат,
п-хлорбензилкарбамат, 2,4-дихлорбензилкарбамат,
4-метилсульфинилбензилкарбамат (Msz), 9-антрилметилкарбамат,
дифенилметилкарбамат, 2-метилтиоэтилкарбамат,
2-метилсульфонилэтилкарбамат, 2-(п-толуолсульфонил)этилкарбамат,
[2-(1,3-дитианил)]метилкарбамат (Dmoc),
4-метилтиофенилкарбамат (Mtpc),
2,4-диметилтиофенилкарбамат (Bmpc),
2-фосфониоэтилкарбамат (Peoc),
2-трифенилфосфониоизопропилкарбамат (Ppoc),
1,1-диметил-2-цианоэтилкарбамат,
м-хлор-п-ацилоксибензилкарбамат,
п-(дигидроксиборил)бензилкарбамат,
5-бензизоксазолилметилкарбамат,
2-(трифторметил)-6-хромонилметилкарбамат (Tcroc),
м-нитрофенилкарбамат, 3,5-диметоксибензилкарбамат,
o-нитробензилкарбамат, 3,4-диметокси-6-нитробензилкарбамат,
фенил(o-нитрофенил)метилкарбамат, трет-амилкарбамат,
S-бензилтиокарбамат, п-цианобензилкарбамат, циклобутилкарбамат,
циклогексилкарбамат, циклопентилкарбамат,
циклопропилметилкарбамат, п-децилоксибензилкарбамат,
2,2-диметоксикарбонилвинилкарбамат,
o-(N,N-диметилкарбоксамидо)бензилкарбамат,
1,1-диметил-3-(N,N-диметилкарбоксамидо)пропилкарбамат,
1,1-диметилпропинилкарбамат, ди(2-пиридил)метилкарбамат,
2-фуранилметилкарбамат, 2-йодэтилкарбамат, изоборнилкарбамат,
изобутилкарбамат, изоникотинилкарбамат,
п-(п-метоксифенилазо)бензилкарбамат, 1-метилциклобутилкарбамат,
1-метилциклогексилкарбамат, 1-метил-1-циклопропилметилкарбамат,
1-метил-1-(3,5-диметоксифенил)этилкарбамат,
1-метил-1-(п-фенилазофенил)этилкарбамат,
1-метил-1-фенилэтилкарбамат, 1-метил-1-(4-пиридил)этилкарбамат,
фенилкарбамат, п-(фенилазо)бензилкарбамат,
2,4,6-три-трет-бутилфенилкарбамат,
4-(триметиламмоний)бензилкарбамат и
2,4,6-триметилбензилкарбамат.
Аминозащитные группы, такие как сульфамидные группы (например, -S(=O)2Raa), включают, но не ограничиваясь ими, п-толуолсульфамид (Ts), бензолсульфамид, 2,3,6-триметил-4-метоксибензолсульфамид (Mtr), 2,4,6-триметоксибензолсульфамид (Mtb), 2,6-диметил-4-метоксибензолсульфамид (Pme), 2,3,5,6-тетраметил-4-метоксибензолсульфамид (Mte), 4-метоксибензолсульфамид (Mbs), 2,4,6-триметилбензолсульфамид (Mts), 2,6-диметокси-4-метилбензолсульфамид (iMds), 2,2,5,7,8-пентаметилхроман-6-сульфамид (Pmc), метансульфамид (Ms), β-триметилсилилэтансульфамид (SES), 9-антраценсульфамид, 4-(4',8'-диметоксинафтилметил)бензолсульфамид (DNMBS), бензилсульфамид, трифторметилсульфамид и фенацилсульфамид.
Другие аминозащитные группы включают, но не ограничиваясь ими, фенотиазинил-(10)-карбонильное производное, N'-п-толуолсульфониламинокарбонильное производное, N'-фениламинотиокарбонильное производное, N-бензоилфенилаланильное производное, производное N-ацетилметионина, 4,5-дифенил-3-оксазолин-2-он, N-фталимид, N-дитиасукцинимид (Dts), N-2,3-дифенилмалеимид, N-2,5-диметилпиррол, аддукт N-1,1,4,4-тетраметилдисилилазациклопентана (STABASE), 5-замещенный 1,3-диметил-1,3,5-триазациклогексан-2-он, 5-замещенный 1,3-дибензил-1,3,5-триазациклогексан-2-он, 1-замещенный 3,5-динитро-4-пиридон, N-метиламин, N-аллиламин, N-[2-(триметилсилил)этокси]метиламин (SEM), N-3-ацетоксипропиламин, N-(1-изопропил-4-нитро-2-оксо-3-пирролин-3-ил)амин, четвертичные аммониевые соли, N-бензиламин, N-ди(4-метоксифенил)метиламин, N-5-дибензосубериламин, N-трифенилметиламин (Tr), N-[(4-метоксифенил)дифенилметил]амин (MMTr), N-9-фенилфлуорениламин (PhF), N-2,7-дихлор-9-флуоренилметиленамин, N-ферроценилметиламино (Fcm), N-оксид N-2-пиколиламина, N-1,1-диметилтиометиленамин, N-бензилиденамин, N-п-метоксибензилиденамин, N-дифенилметиленамин, N-[(2-пиридил)мезитил]метиленамин, N-(N',N'-диметиламинометилен)амин, Ν,Ν'-изопропилидендиамин, N-п-нитробензилиденамин, N-салицилиденамин, N-5-хлорсалицилиденамин, N-(5-хлор-2-гидроксифенил)фенилметиленамин, N-циклогексилиденамин, N-(5,5-диметил-3-оксо-1-циклогексенил)амин, производное N-борана, производное N-дифенилбориновой кислоты, N-[фенил(пентакарбонилхром- или вольфрам)карбонил]амин, N-медный хелат, N-цинковый хелат, N-нитроамин, N-нитрозоамин, N-оксидамин, дифенилфосфинамид (Dpp), диметилтиофосфинамид (Mpt), дифенилтиофосфинамид (Ppt), диалкилфосфорамидаты, дибензилфосфорамидат, дифенилфосфорамидат, бензолсульфенамид, o-нитробензолсульфенамид (Nps), 2,4-динитробензолсульфенамид, пентахлорбензолсульфенамид, 2-нитро-4-метоксибензолсульфенамид, трифенилметилсульфенамид и 3-нитропиридинсульфенамид (Npys).
Как использовано в настоящем описании и если не указано иное, термин "замещенный" или "замещение", при применении для описания химической структуры или группы, относится к производному данной структуры или группы, где один или более его атомов водорода заменены заместителем, таким как, но не ограничиваясь ими: алкил, алкенил, алкинил и циклоалкил; алкоксиалкил; ароил; дейтерий, галоген; галогеналкил (например, трифторметил); гетероциклоалкил; галогеналкокси (например, трифторметокси); гидрокси; алкокси; циклоалкилокси; гетероциклоокси; оксо; алканоил; арил; гетероарил (например, индолил, имидазолил, фурил, тиенил, тиазолил, пирролидил, пиридил и пиримидил); арилалкил; алкиларил; гетероарил; гетероарилалкил; алкилгетероарил; гетероцикло; гетероциклоалкилалкил; арилокси, алканоилокси; амино; алкиламино; ариламино; арилалкиламино; циклоалкиламино; гетероциклоамино; моно- и дизамещенный амино; алканоиламино; ароиламино; аралканоиламино; аминоалкил; карбамил (например, CONH2); замещенный карбамил (например, CONH-алкил, CONH-арил, CONH-арилалкил или примеры, в которых присутствуют два заместителя при азоте); карбонил; алкоксикарбонил; карбокси; циано; сложный эфир; эфир; гуанидино; нитро; сульфонил; алкилсульфонил; арилсульфонил; арилалкилсульфонил; сульфонамидо (например, SO2NH2); замещенный сульфонамидо; тиол; алкилтио; арилтио; арилалкилтио; циклоалкилтио; гетероциклотио; алкилтионо; арилтионо и арилалкилтионо. В некоторых вариантах осуществления заместитель сам может быть замещен одной или более химическими группами, такими как, но не ограничиваясь ими, группы, описанные в настоящем описании.
Как использовано в настоящем описании и если не указано иное, термин "приблизительно" применяют для описания указанных величин, которые являются приблизительными. Например, термин "приблизительно", при применении в сочетании с температурами реакций, означает, что изменения температур в пределах 30%, 25%, 20%, 15%, 10% или 5% охвачены указанной температурой. Аналогично, термин "приблизительно", при его применении в связи с продолжительностью реакции, означает, что изменения продолжительности реакции в пределах 30%, 25%, 20%, 15%, 10% или 5% охвачены указанной продолжительностью реакции.
Настоящее описание будет понятно более полно со ссылкой на следующее подробное описание и иллюстративные примеры, которые предлагаются в качестве примеров неограничивающих вариантов осуществления.
5.2 Способы
Настоящее изобретение относится к способам получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов. Более конкретно, настоящее изобретение относится к способам получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, включающим стадию преобразования хлорпиколиноилхлоридов в фторпиколиноилфториды.
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
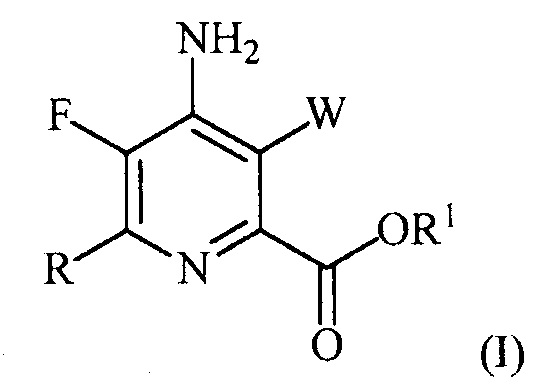
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
введение фтор заместителя в положение 5 пиридиновой структуры в формуле (I) фторированием 5-Cl-пиколиноилхлоридного соединения источником фторидного иона с получением 5-F-пиколиноилфторидного соединения;
введение аминозаместителя в положение 4 пиридиновой структуры в формуле (I) аминированием 4-галогенпиридинового соединения источником аммиака;
введение W заместителя в положение 3 пиридиновой структуры в формуле (I) галогенированием 3-незамещенного пиридинового соединения источником галогена; и
введение R заместителя в положение 6 пиридиновой структуры в формуле (I) конденсацией 6-галогенпиридинового соединения с R-Met соединением, где R-Met соединение определено в настоящем описании и в другом месте.
В одном из вариантов осуществления R представляет собой фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси. В одном из вариантов осуществления R представляет собой 4-замещенный фенил, 2,4-дизамещенный фенил, 2,3,4-тризамещенный фенил, 2,4,5-тризамещенный фенил и 2,3,4,6-тетразамещенный фенил, где каждый заместитель независимо выбран из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси.
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
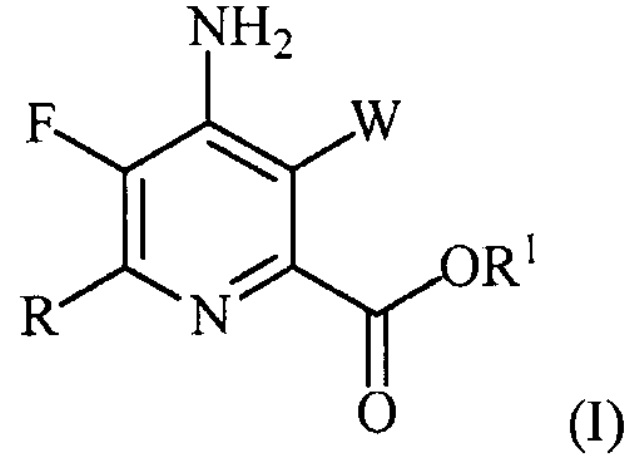
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
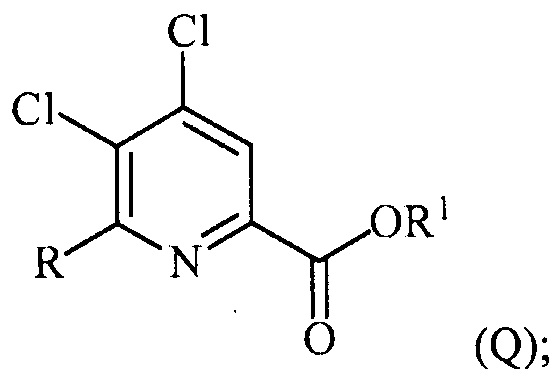
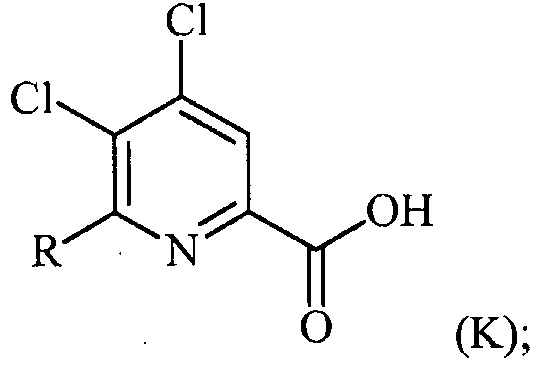
(a) фторирование соединения формулы (II):
 ,
,
где A представляет собой галоген или R;
источником фторидного иона с получением соединения формулы (III):
,
где B6 представляет собой F или R;
и преобразование соединения формулы (III) в соединение формулы (IV):
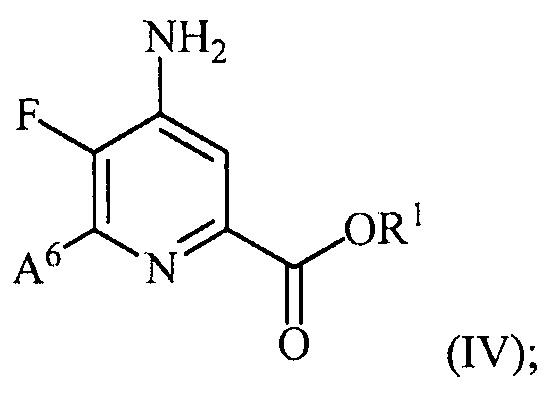
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
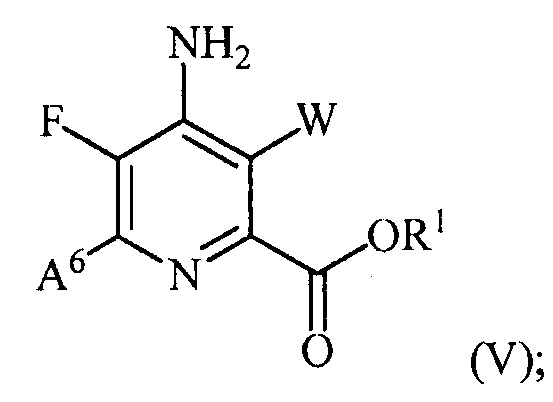 и
и
(c) конденсацию соединение формулы (VI)
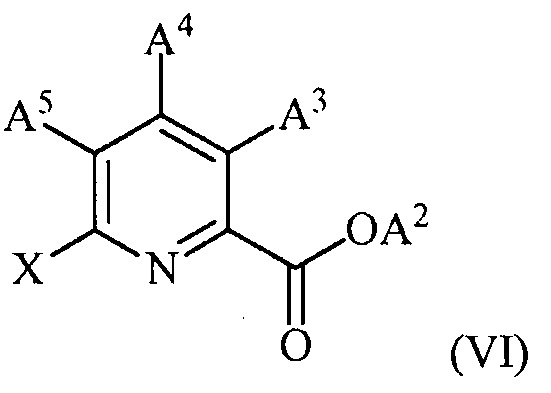 ,
,
где X представляет собой Cl, Br или I;
A2 представляет собой водород или R1;
A3 представляет собой водород или W;
A4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
A5 представляет собой F или Cl;
с соединением формулы (VII):
R-Met (VII),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
,
где преобразование (c) можно осуществлять перед, между или после преобразований (a) и (b).
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
введение фтор заместителя в положение 5 пиридиновой структуры в формуле (I) фторированием 5-Cl-пиколиноилхлоридного соединения источником фторидного иона с получением 5-F-пиколиноилфторидного соединения;
введение аминозаместителя в положение 4 пиридиновой структуры в формуле (I) аминированием 4-галогенпиридинового соединения источником аммиака;
введение W заместителя в положение 3 пиридиновой структуры в формуле (I) галогенированием 3-незамещенного пиридинового соединения источником галогена; и
введение R заместителя в положение 6 пиридиновой структуры в формуле (I) конденсацией 6-галогенпиридинового соединения с R-Met соединением, где R-Met соединение определено в настоящем описании и в другом месте.
В одном из вариантов осуществления R представляет собой фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси. В одном из вариантов осуществления R представляет собой 4-замещенный фенил, 2,4-дизамещенный фенил, 2,3,4-тризамещенный фенил, 2,4,5-тризамещенный фенил и 2,3,4,6-тетразамещенный фенил, где каждый заместитель независимо выбран из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси.
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
(a) фторирование соединения формулы (II):
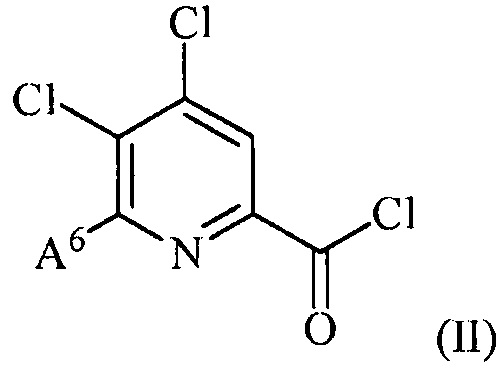 ,
,
где A6 представляет собой галоген или R;
источником фторидного иона с получением соединения формулы (III):
,
где B6 представляет собой F или R;
и преобразование соединения формулы (III) в соединение формулы (IV):
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
 и
и
(c) конденсацию соединения формулы (VI):
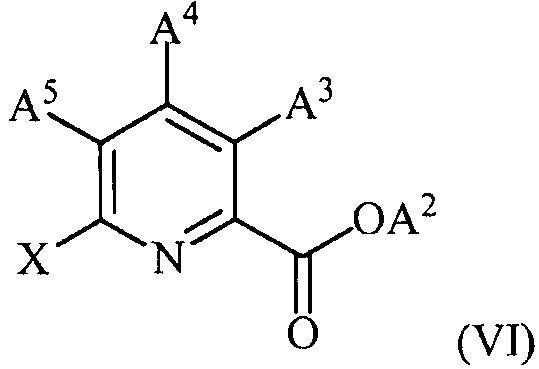 ,
,
где X представляет собой Cl, Br или I;
A2 представляет собой водород или R1;
A3 представляет собой водород или W;
A4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
A5 представляет собой F или Cl;
с соединением формулы (VII):
R-Met (VII),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
,
где преобразование (c) можно осуществлять перед, между или после преобразований (a) и (b).
Порядок, в котором проводят преобразование (c), можно корректировать исходя из условий реакций, известных в данной области техники. Например, в одном из вариантов осуществления преобразование (c) можно осуществлять перед преобразованием (a) и преобразованием (b). В другом варианте осуществления преобразование (c) можно осуществлять между преобразованием (a) и преобразованием (b). В еще другом варианте осуществления стадию (c) можно осуществлять после преобразования (a) и преобразования (b).
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
(a) фторирование соединения формулы (II):
 ,
,
где A6 представляет собой галоген или R;
источником фторидного иона с получением соединения формулы (III):
,
где B6 представляет собой F или R;
и преобразование соединения формулы (III) в соединение формулы (IV):
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
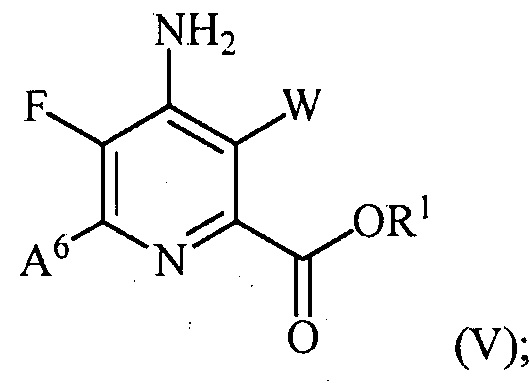 и
и
(c) конденсацию соединения формулы (VI):
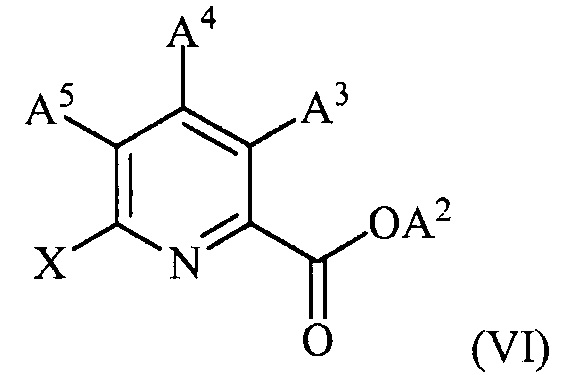 ,
,
где X представляет собой Cl, Br или I;
A2 представляет собой водород или R1;
A3 представляет собой водород или W;
A4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
A5 представляет собой F или Cl;
с соединением формулы (VII):
R-Met (VII),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
,
где преобразования осуществляют в следующем порядке:
(i) преобразование (a) осуществляют перед преобразованием (b), которое осуществляют перед преобразованием (c);
(ii) преобразование (a) осуществляют перед преобразованием (c), которое осуществляют перед преобразованием (b); или
(iii) преобразование (c) осуществляют перед преобразованием (a), которое осуществляют перед преобразованием (b).
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
(a) фторирование соединения формулы (II):
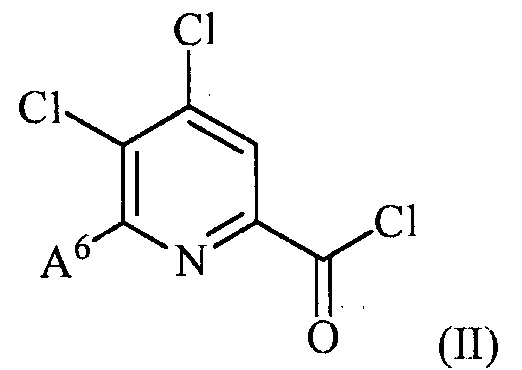 ,
,
где A6 представляет собой галоген или R;
источником фторидного иона с получением соединения формулы (III):
,
где B6 представляет собой F или R;
и преобразование соединения формулы (III) в соединение формулы (IV):
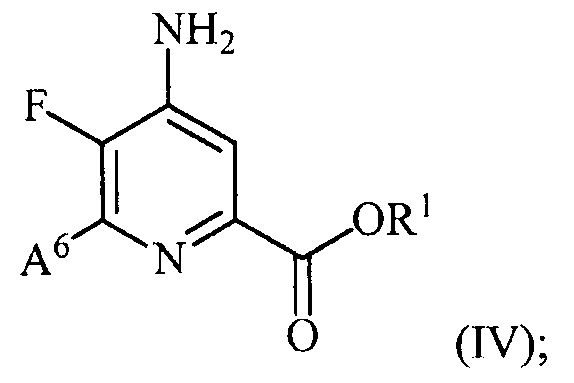
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
 и
и
(c) конденсацию соединения формулы (VI):
 ,
,
где X представляет собой Cl, Br или I;
A2 представляет собой водород или R1;
A3 представляет собой водород или W;
A4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
A5 представляет собой F или Cl;
с соединением формулы (VII):
R-Met (VII),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
,
где преобразования осуществляют в следующем порядке:
(i) преобразование (a) осуществляют перед преобразованием (b), которое осуществляют перед преобразованием (c);
(ii) преобразование (a) осуществляют перед преобразованием (c), которое осуществляют перед преобразованием (b); или
(iii) преобразование (c) осуществляют перед преобразованием (a), которое осуществляют перед преобразованием (b).
Способы, представленные в настоящем описании, могут дополнительно включать дополнительные стадии, включающие манипулирование с функциональными группами в пиридиновой структуре формул, представленных в настоящем описании, (например, этерификацию и/или гидролиз карбоксильной группы в положении 2 пиридиновой структуры, защиту и/или деблокирование аминогруппы в положении 4 пиридиновой структуры; и галогеновый обмен в положении 6 пиридиновой структуры). Такие манипуляции с функциональными группами можно осуществлять в любых подходящих условиях, известных в данной области техники. См., например, заявки США № 13/356691 и 13/356686, содержание которых включено в настоящее изобретение посредством ссылки.
В одном из вариантов осуществления фторирования соединения формулы (II) для получения соединения формулы (III), A6 представляет собой галоген и B6 представляет собой F. В одном из вариантов осуществления фторирования соединения формулы (II) для получения соединения формулы (III), A6 представляет собой Cl и B6 представляет собой F. В другом варианте осуществления фторирования соединения формулы (II) для получения соединения формулы (III), оба A6 и B6 представляют собой R.
Преобразование соединения формулы (III) в соединение формулы (IV) может включать несколько стадий, такие как, но не ограничиваясь ими, этерификация в положении 2 пиридиновой структуры, аминирование в положении 4 пиридиновой структуры и замена фтор заместителя в положении 6 пиридиновой структуры йод, бром или хлор заместителем. Порядок, в котором проводят указанные стадии, не является критическим. Указанные стадии можно осуществлять в условиях, известных в данной области техники, которые являются пригодными для данного преобразования.
В одном варианте осуществления пример преобразования соединения формулы (III) в соединение формулы (IV) показан на схеме 1.
Схема 1
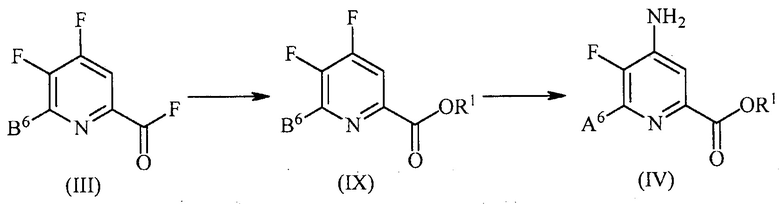
В одном из вариантов осуществления преобразование соединения формулы (III) в соединение формулы (IV) включает:
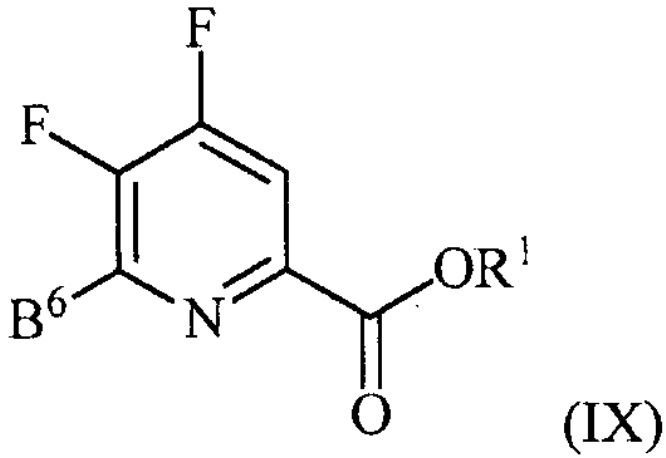
контакт соединения формулы (III) со спиртом R1OH с получением соединения формулы (IX):
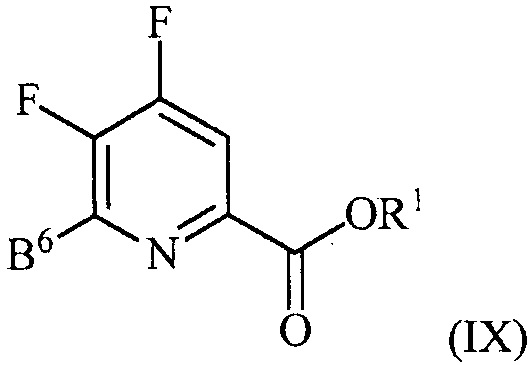 , и
, и
аминирование соединения формулы (IX) источником аммиака с получением соединения формулы (IV).
В одном из вариантов осуществления пример преобразования соединения формулы (III) в соединение формулы (IV) показан на схеме 2.
Схема 2
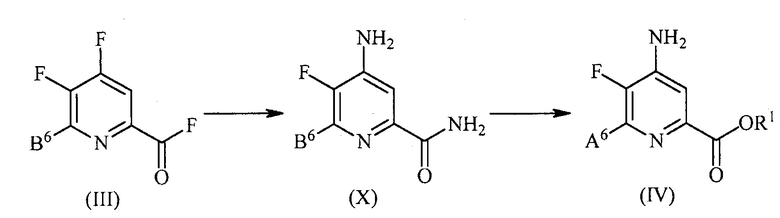
В одном из вариантов осуществления преобразование соединения формулы (III) в соединение формулы (IV) включает:
контакт соединения формулы (III) с источником аммиака с получением соединения формулы (X):
 , и
, и
контакт соединения формулы (X) с кислотой HX и спиртом R1OH с получением соединения формулы (IV), где X представляет собой I, Br или Cl.
В одном из вариантов осуществления галогенирования соединения формулы (IV) для получения соединения формулы (V), A6 представляет собой галоген. В одном из вариантов осуществления галогенирования соединения формулы (IV) для получения соединения формулы (V), A6 представляет собой Cl, Br или I. В другом варианте осуществления галогенирования соединения формулы (IV) для получения соединения формулы (V), A6 представляет собой R.
В одном варианте осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A2 представляет собой водород. В другом варианте осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A2 представляет собой R1.
В одном из вариантов осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A4 представляет собой Cl. В другом варианте осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A4 представляет собой F. В еще другом варианте осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A4 представляет собой NH2. В еще другом варианте осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A4 представляет собой NHCOCH3. В еще другом варианте осуществления конденсации соединения формулы (VI) с R-Met для получения соединения формулы (VIII), A4 представляет собой NHCOPh, NHCOCH2Ph, NHCOOMe, NHCOOEt, NHCOOCMe3, NHCOOPh или NHCOOCH2Ph.
В некоторых вариантах осуществления способы, представленные в настоящем описании, могут дополнительно включать защиту и деблокирование аминозаместителя в положении 4 пиридиновой структуры. В некоторых вариантах осуществления способы, представленные в настоящем описании, могут дополнительно включать защиту NH2 заместителя в положении 4 пиридиновой структуры перед преобразованием (c); и дополнительно включать стадию деблокирования. Подходящие аминозащитные группы хорошо известны специалисту в данной области техники. Выбор и применение защитных групп и условий реакции для введения и удаления защитных групп описаны в T. W. Green, Protective Groups in Organic Synthesis (Third Ed., Wiley, New York, 1999), которая включена в настоящее изобретение посредством ссылки во всей своей полноте.
В одном варианте осуществления способы, представленные в настоящем описании, дополнительно включают защиту NH2 заместителя в положении 4 пиридиновой структуры в формулах, представленных в настоящем описании, в виде NHCOCH3. В одном варианте осуществления указанная защита включает контакт 4-аминопиридинового соединения, представленного в настоящем описании, с Ac2O.
В одном варианте осуществления пример преобразования соединения формулы (VI) в соединение формулы (VIII) показан на схеме 3.
Схема 3
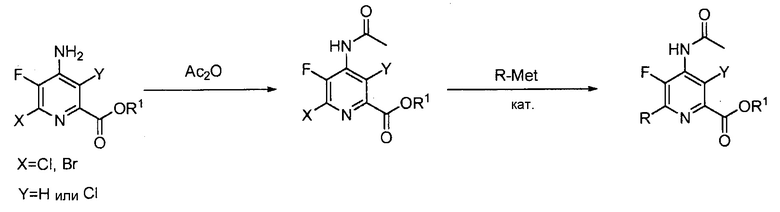
В одном варианте осуществления защита аминозаместителя в положении 4 пиридиновой структуры обеспечивает меньшую концентрацию катализатора в реакциях конденсации, представленных в настоящем описании, между соединением формулы (VI) и R-Met.
В одном варианте осуществления настоящее изобретение относится к способу, как показано на схеме 4.
Схема 4
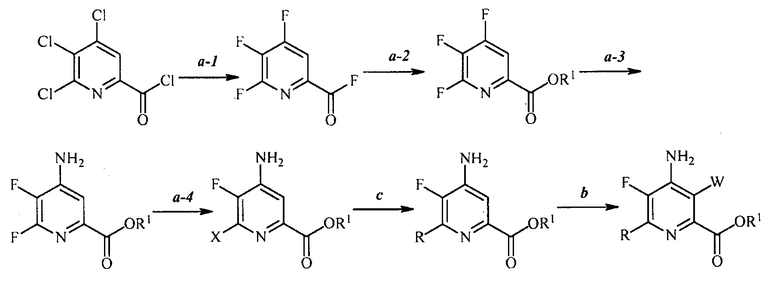
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
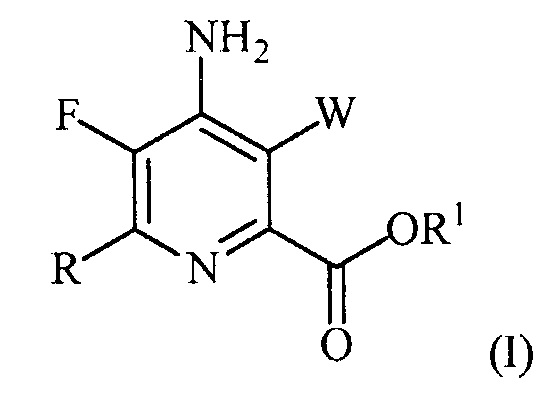
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (a), которое включает:
(a-1) фторирование соединения формулы (A):
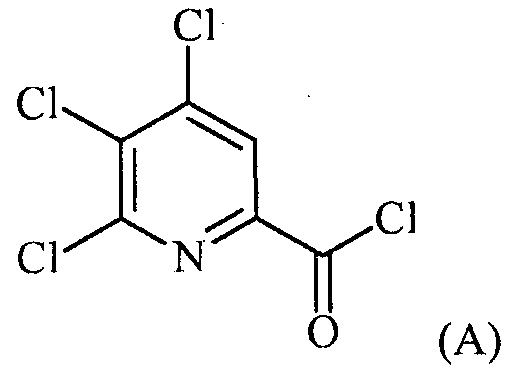
источником фторидного иона с получением соединения формулы (B):
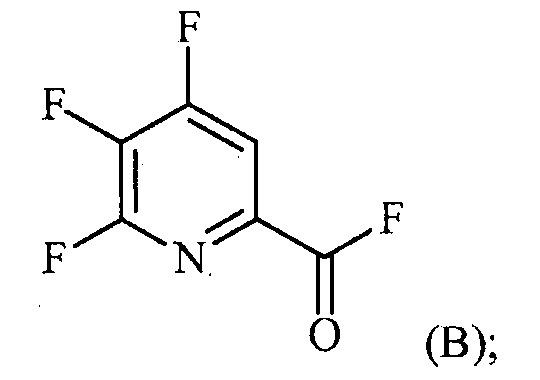
(a-2) контакт соединения формулы (B) со спиртом R1OH с получением соединения формулы (C):
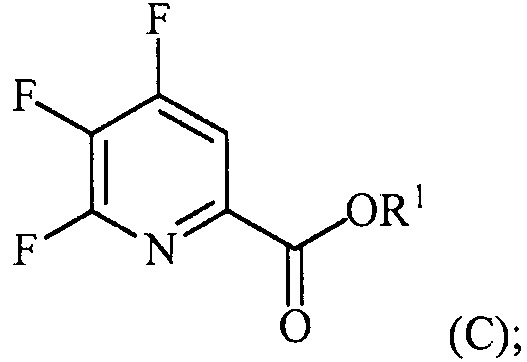
(a-3) аминирование соединения формулы (C) источником аммиака с получением соединения формулы (D):
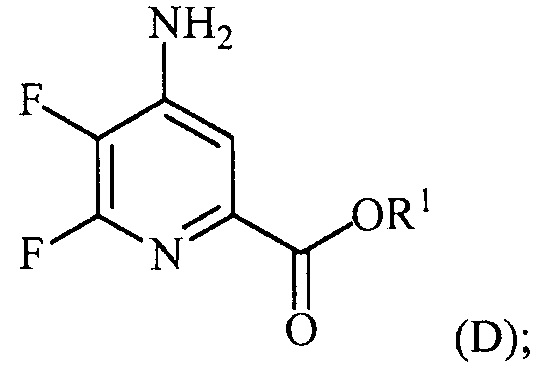 и
и
(a-4) контакт соединения формулы (D) с йодидом, бромидом или хлоридом в условиях, пригодных для галогенового обмена, с получением соединения формулы (E):
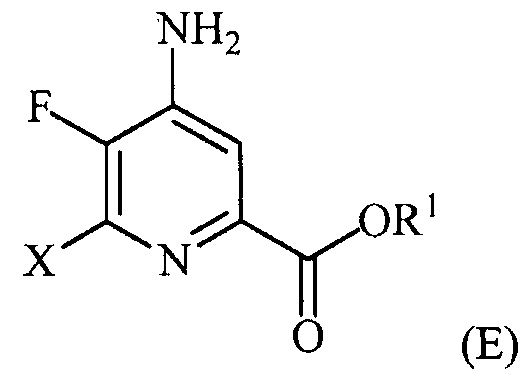 ,
,
где X представляет собой Cl, Br или I;
преобразование (c), которое включает:
конденсацию соединения формулы (E) с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (G):
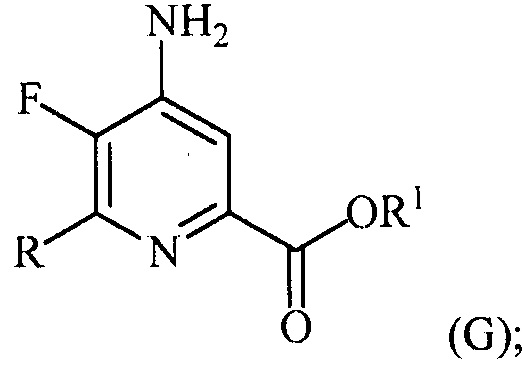 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (G) источником галогена с получением соединения формулы (I).
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (a), которое включает:
(a-1) фторирование соединения формулы (A):
источником фторидного иона с получением соединения формулы (B):
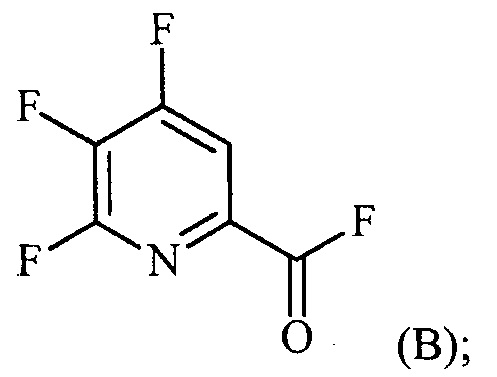
(a-2) контакт соединения формулы (B) со спиртом R1OH с получением соединения формулы (C):
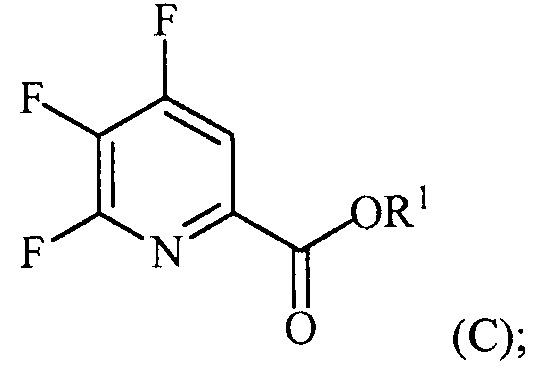
(a-3) аминирование соединения формулы (C) источником аммиака с получением соединения формулы (D):
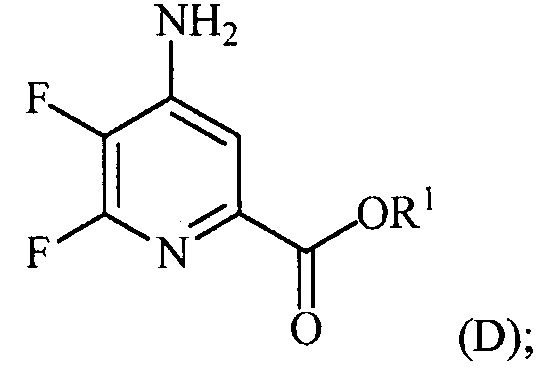 и
и
(a-4) контакт соединения формулы (D) с йодидом, бромидом или хлоридом в условиях, подходящих для галогенового обмена, с получением соединения формулы (E):
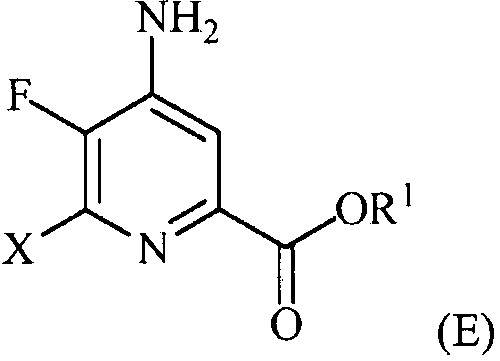 ,
,
где X представляет собой Cl, Br или I;
преобразование (c), которое включает:
конденсацию соединения формулы (E) с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (G):
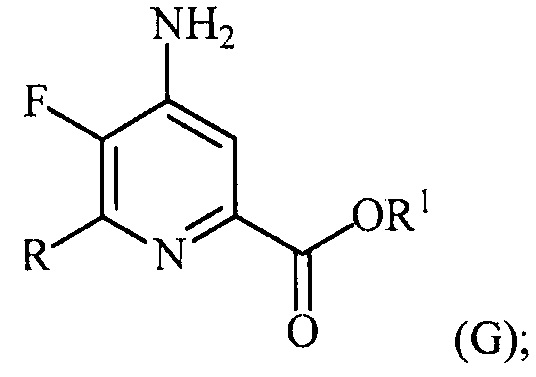 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (G) источником галогена с получением соединения формулы (I).
В одном варианте осуществления настоящее изобретение относится к способу, как показано на схеме 5.
Схема 5
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (a), которое включает:
(a-1) фторирование соединения формулы (A):
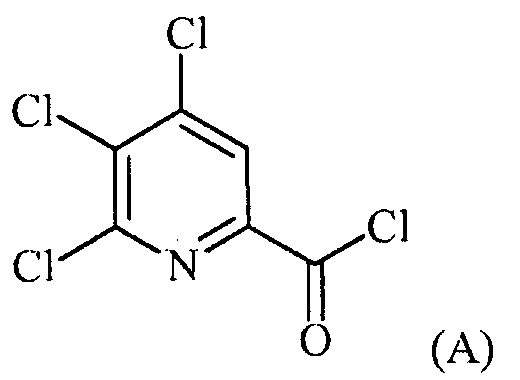
источником фторидного иона с получением соединения формулы (B):
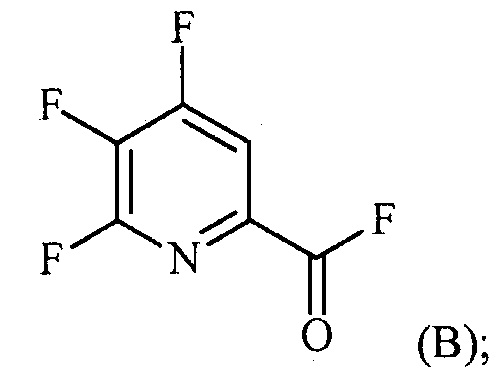
(a-2) контакт соединения формулы (B) со спиртом R1OH с получением соединения формулы (C):
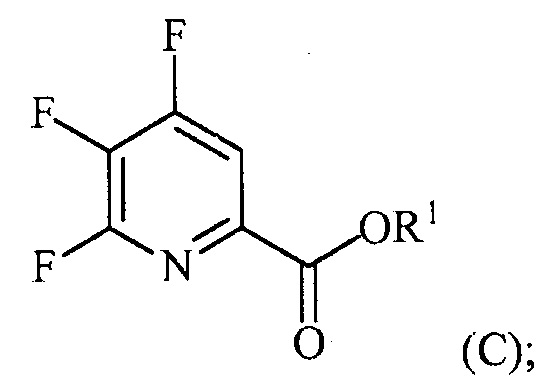
(a-3) аминирование соединения формулы (C) источником аммиака с получением соединения формулы (D):
 и
и
(a-4) контакт соединения формулы (D) с источником йодида, бромида или хлорида в условиях, подходящих для галогенового обмена, с получением соединения формулы (E):
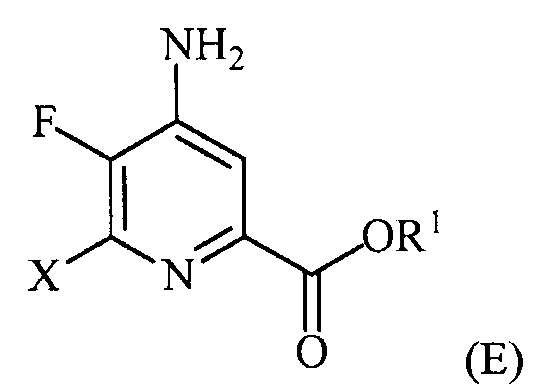 ,
,
где X представляет собой Cl, Br или I;
преобразование (b), которое включает:
галогенирование соединения формулы (E) источником галогена с получением соединения формулы (H):
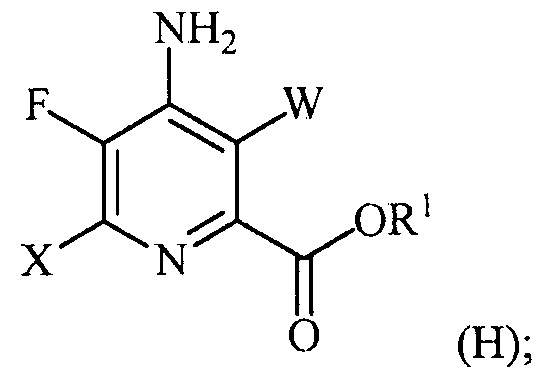 и
и
преобразование (c), которое включает:
конденсацию соединения формулы (H) с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (I).
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
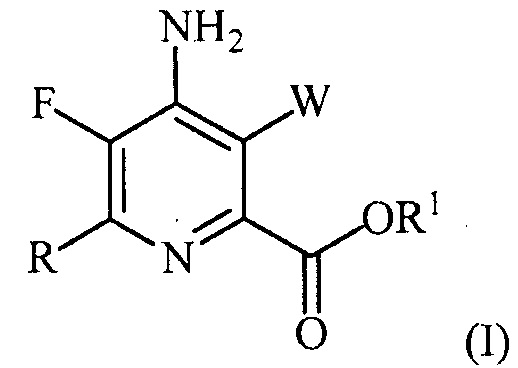
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (a), которое включает:
(a-1) фторирование соединения формулы (A):
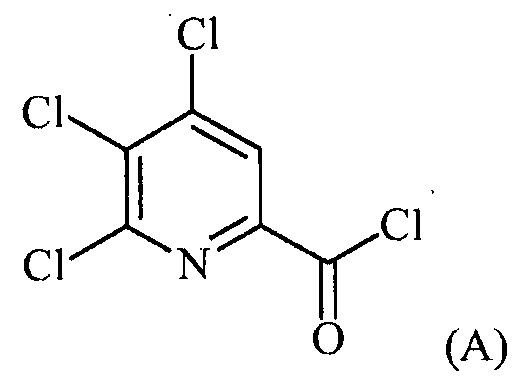
источником фторидного иона с получением соединения формулы (B):
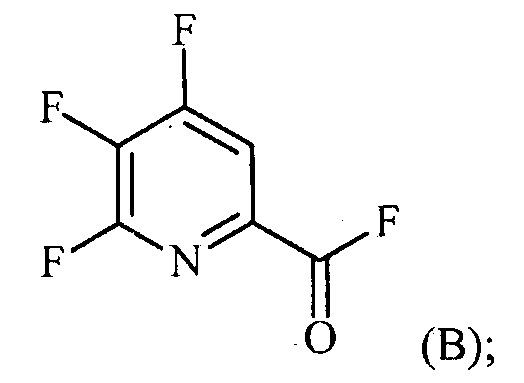
(a-2) контакт соединения формулы (B) со спиртом R1OH с получением соединения формулы (C):
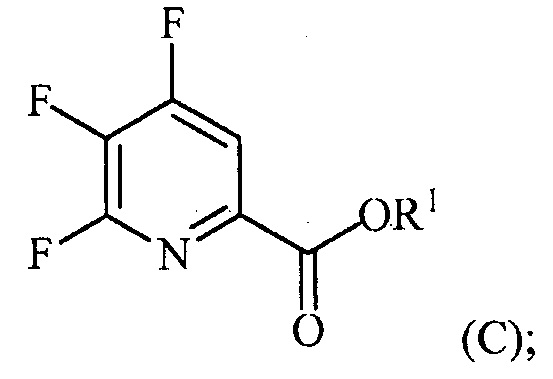
(a-3) аминирование соединения формулы (C) источником аммиака с получением соединения формулы (D):
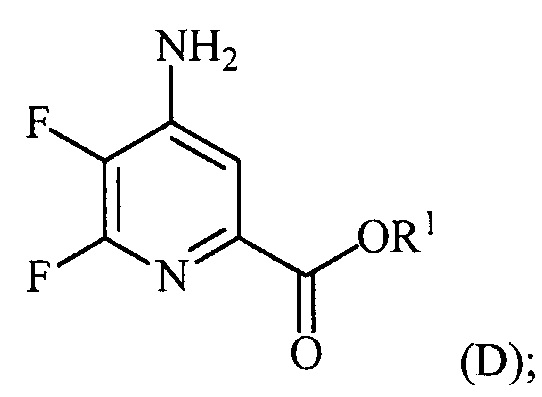 и
и
(a-4) контакт соединения формулы (D) с источником йодида, бромида или хлорида в условиях, подходящих для галогенового обмена, с получением соединения формулы (E):
 ,
,
где X представляет собой Cl, Br или I;
преобразование (b), которое включает:
галогенирование соединения формулы (E) источником галогена с получением соединения формулы (H):
 и
и
преобразование (c), которое включает:
конденсацию соединения формулы (H) с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (I).
В одном из вариантов осуществления способы, представленные в настоящем описании, показаны на схеме 6.
Схема 6
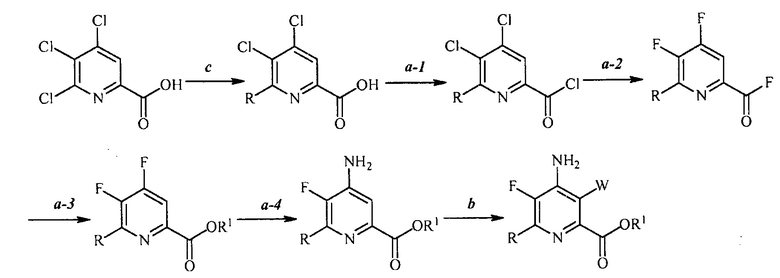
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
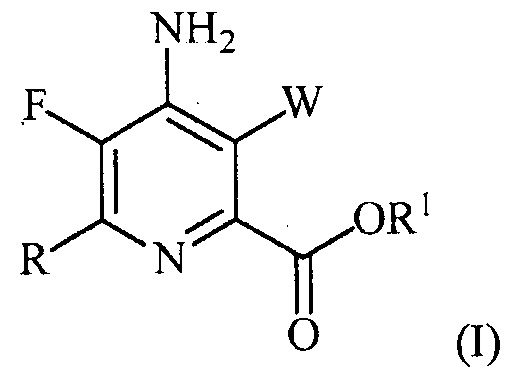
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (c), которое включает:
конденсацию соединения формулы (J):
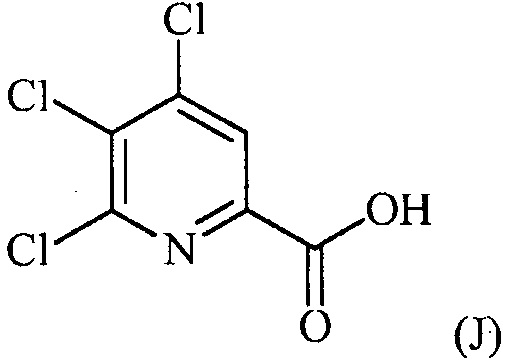
с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (K):
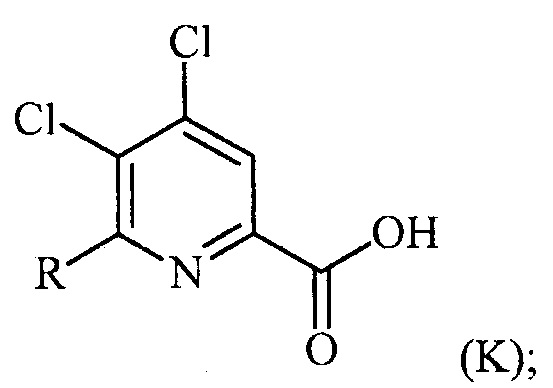
преобразование (a), которое включает:
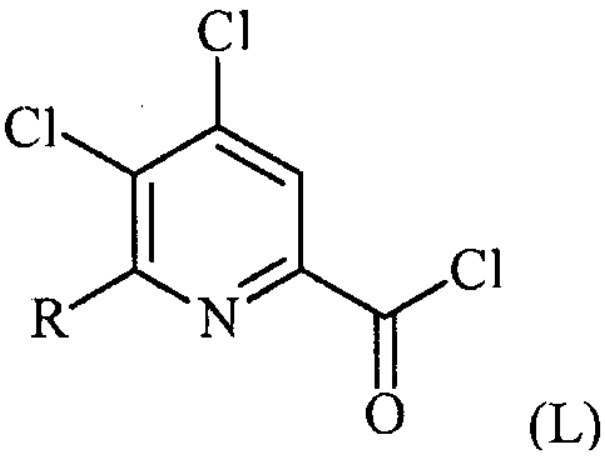
(a-1) преобразование соединения формулы (K) в соединение формулы (L):
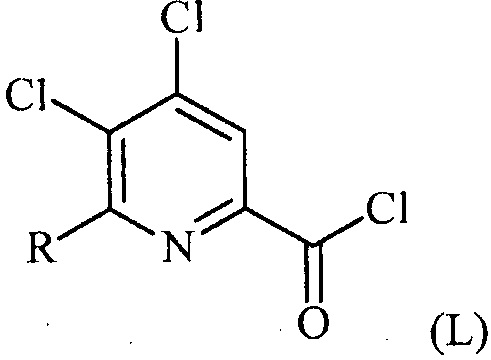
в условиях, подходящих для образования хлорангидрида кислоты;
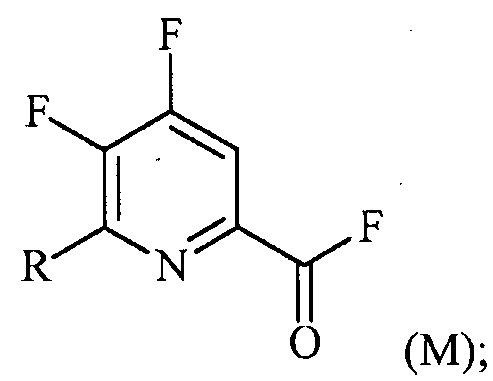
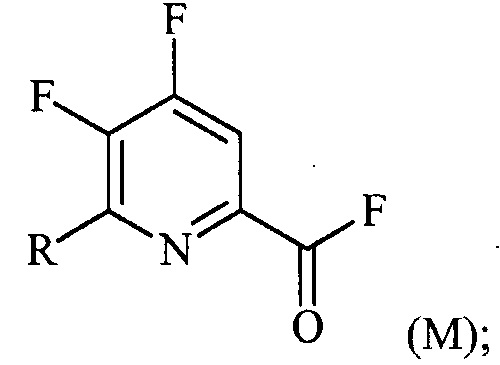
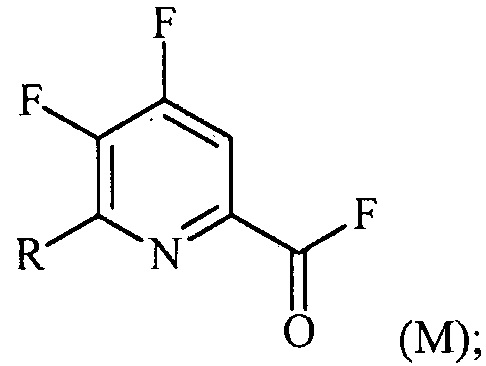
(a-2) фторирование соединения формулы (L) источником фторидного иона с получением соединения формулы (M):
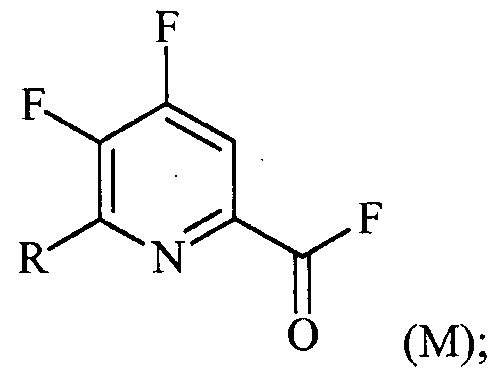
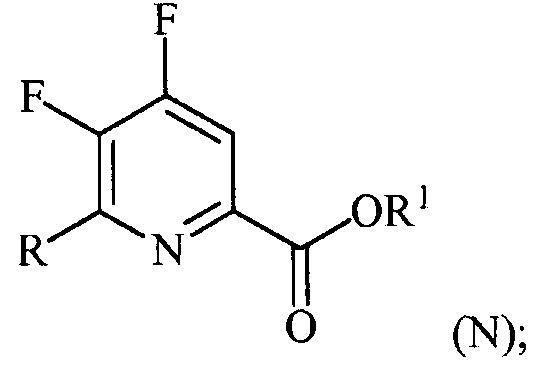
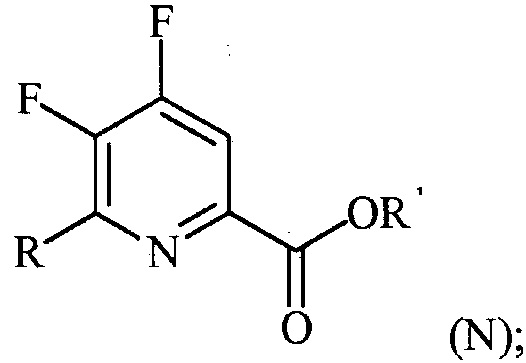
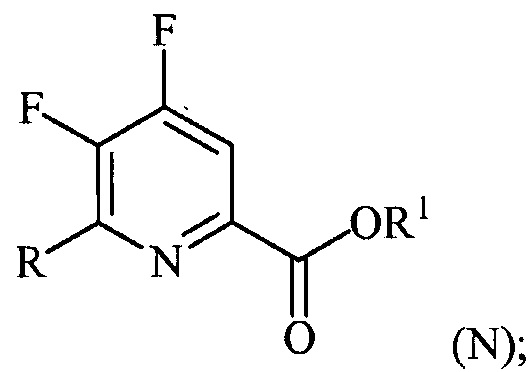
(a-3) контакт соединения формулы (M) со спиртом R1OH с получением соединения формулы (N):
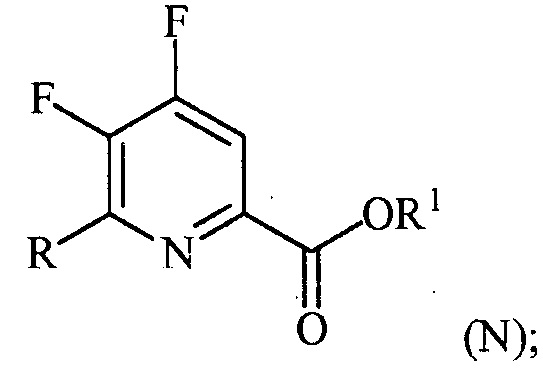 и
и
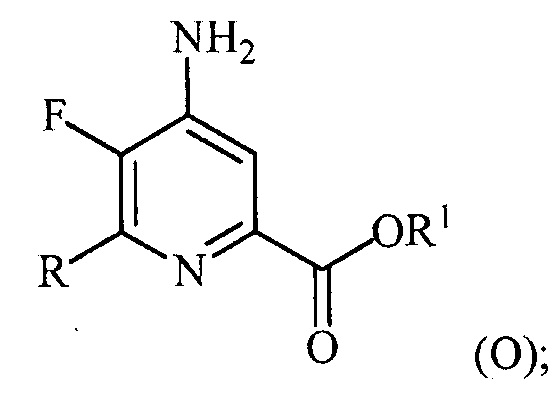
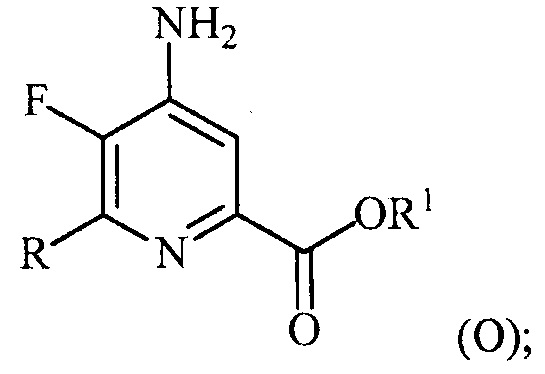
(a-4) аминирование соединения формулы (N) источником аммиака с получением соединения формулы (O):
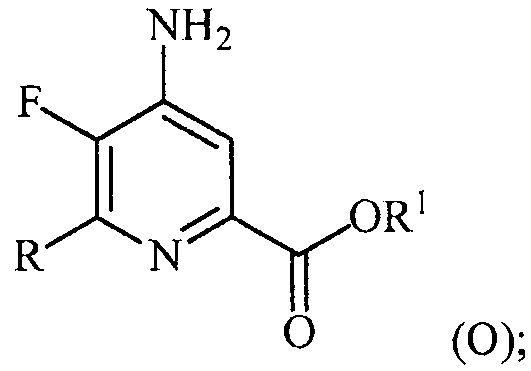 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (O) источником галогена с получением соединения формулы (I).
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
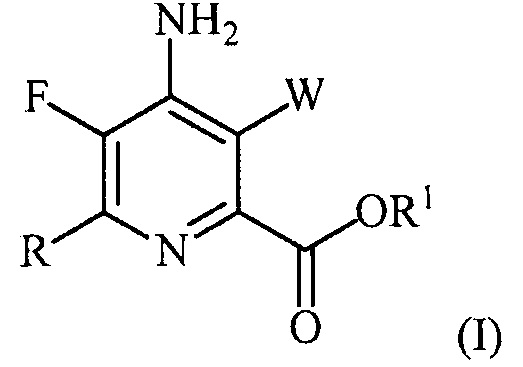
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (c), которое включает:
конденсацию соединение формулы (J):
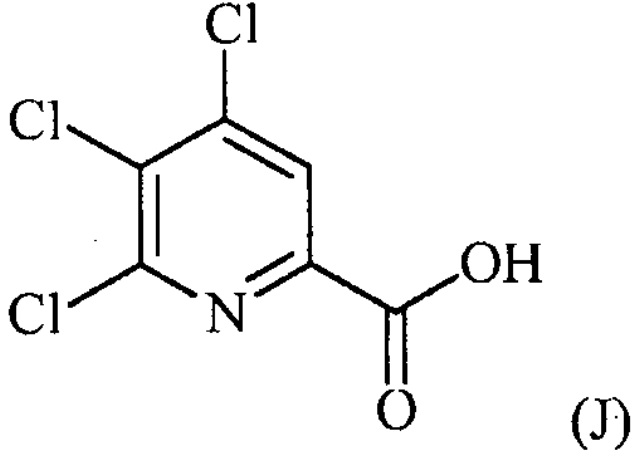
с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь, или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (K):
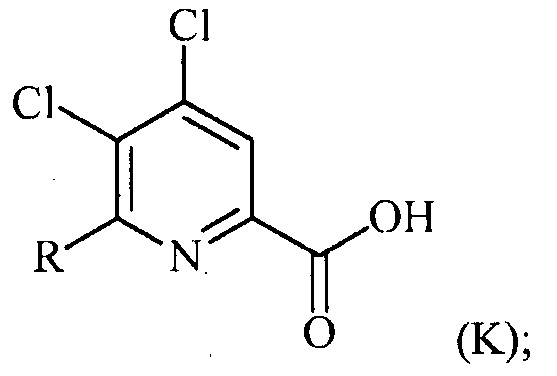
преобразование (a), которое включает:
(a-1) преобразование соединения формулы (K) в соединение формулы (L):
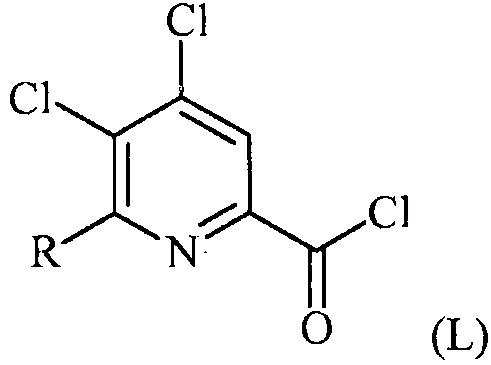
в условиях, подходящих для образования хлорангидрида кислоты;
(a-2) фторирование соединения формулы (L) источником фторидного иона с получением соединения формулы (M):
(a-3) контакт соединения формулы (M) со спиртом R1OH с получением соединения формулы (N):
 и
и
(a-4) аминирование соединения формулы (N) источником аммиака с получением соединения формулы (O):
 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (O) источником галогена с получением соединения формулы (I).
В одном варианте осуществления, способы, представленные в настоящем описании, показаны на схеме 7.
Схема 7
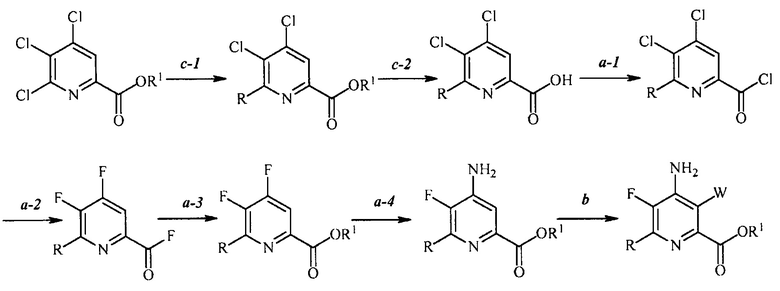
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 1-4 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (c), которое включает:
(c-1) конденсацию соединение формулы (P):
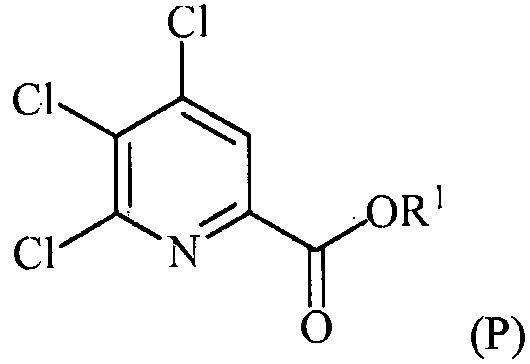
с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (Q):
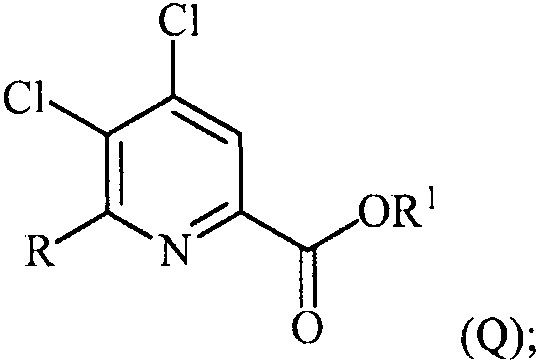 и
и
(c-2) гидролиз соединения формулы (Q) до соединения формулы (K):
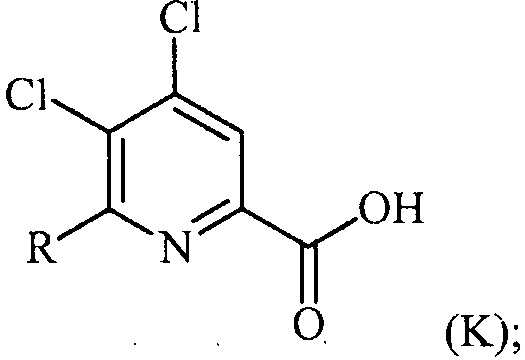
преобразование (a), которое включает:
(a-1) преобразование соединения формулы (K) в соединение формулы (L):
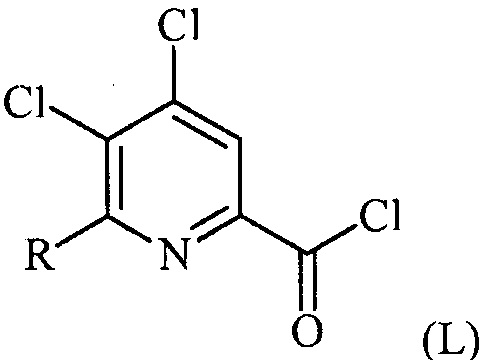
в условиях, подходящих для образования хлорангидрида кислоты;
(a-2) фторирование соединения формулы (L) источником фторидного иона с получением соединения формулы (M):
(a-3) контакт соединения формулы (M) со спиртом R1OH с получением соединения формулы (N):
 и
и
(a-4) аминирование соединения формулы (N) источником аммиака с получением соединения формулы (O):
 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (O) источником галогена с получением соединения формулы (I).
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, сольвата, включая гидрат, изотополога или полиморфа,
где W представляет собой Cl, Br или I;
R представляет собой C1-C4алкил, циклопропил, C2-C4алкенил или фенил, замещенный 0-5 заместителями, независимо выбранными из галогена, C1-C4алкила, C1-C4галогеналкила, C1-C4алкокси или C1-C4галогеналкокси; и
R1 представляет собой C1-C12алкил или незамещенный или замещенный C7-C11арилалкил;
включающему:
преобразование (c), которое включает:
(c-1) конденсацию соединение формулы (P):

с соединением формулы (F):
R-Met (F),
где Met представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (Q):
 и
и
(c-2) гидролиз соединения формулы (Q) до соединения формулы (K):
преобразование (a), которое включает:
(a-1) преобразование соединения формулы (K) в соединение формулы (L):
в условиях, подходящих для образования хлорангидрида кислоты;
(a-2) фторирование соединения формулы (L) источником фторидного иона с получением соединения формулы (M):
(a-3) контакт соединения формулы (M) со спиртом R1OH с получением соединения формулы (N):
 и
и
(a-4) аминирование соединения формулы (N) источником аммиака с получением соединения формулы (O):
 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (O) источником галогена с получением соединения формулы (I).
Хлорпиколиноилхлоридные соединения (например, соединение формулы (II)) представляют собой известные соединения, и/или их можно получить из известных хлорпиколинатов, применяя стандартные способы, известные в данной области техники. См., например, патент США 6784137 B2. Две иллюстративные схемы получения 6-арилпиколиноилхлорида показаны ниже:
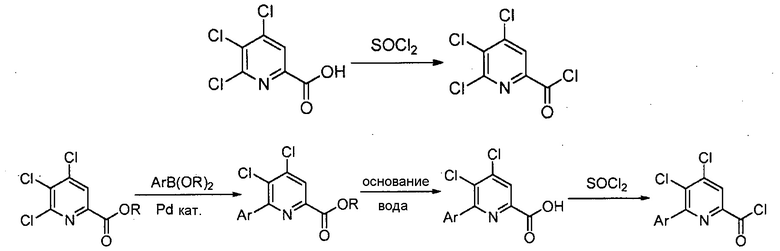
В реакции обмена фтора (например, преобразование соединения формулы (II) в соединение формулы (III)), фторированный пиколинат получают взаимодействием соответствующего хлорированного пиколината, по меньшей мере, с одним эквивалентом источника фторидного иона для каждого хлор заместителя, который будут обменивать.
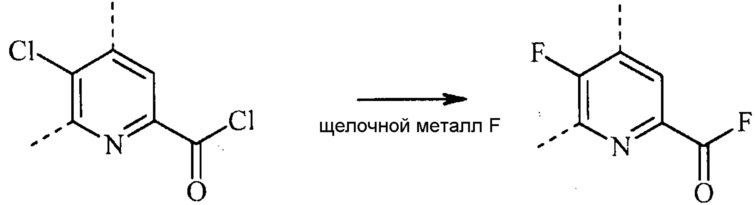
Стандартные источники фторидных ионов включают фториды щелочных металлов ("M-F"), которые включают фторид натрия (NaF), фторид калия (KF) и фторид цезия (CsF). Можно также использовать фторидные соли, такие как фторид тетрабутиламмония (н-Bu4NF).
В некоторых вариантах осуществления, реакции фторного обмена осуществляют в растворителе или реакционной среде, такой как ацетонитрил, сульфолан, алкилнитрилы, полиэфиры или алкилсульфоны, включая их смеси. В определенных вариантах осуществления используемый растворитель представляет собой алкилнитрил или алкилсульфон. В определенных вариантах осуществления используемый растворитель представляет собой ацетонитрил или сульфолан.
Можно также использовать катализаторы, такие как краун-эфиры или агенты фазового переноса, о которых известно, что они увеличивают скорость фторидного обмена. В некоторых вариантах осуществления, катализатор представляет собой краун-эфир, галогенид фосфония, полиэфир, фосфазениевую соль или галогенид тетра-замещенного аммония. В определенных вариантах осуществления катализатор представляет собой краун-эфир, например, 18-краун-6.
Температура, при которой осуществляют реакцию фторного обмена, не является критической, но обычно составляет приблизительно от 50°C до приблизительно 200°C и в некоторых вариантах осуществления приблизительно от 80°C до приблизительно 140°C. В зависимости от используемого растворителя в конкретной реакции, оптимальная температура будет изменяться. Как правило, чем ниже температура, тем медленнее протекает реакция. Реакции настоящего изобретения обычно осуществляют при энергичном перемешивании, достаточном для поддержания по существу диспергированной смеси реагентов.
При проведении реакции обмена фтора, ни скорость, ни порядок добавления реагентов обычно не являются критическими. Обычно растворитель и фторид щелочного металла и необязательно катализатор смешивают перед добавлением пиколиноилхлорида к реакционной смеси. Стандартная реакция обычно требует приблизительно от 2 до приблизительно 100 часов, и ее обычно проводят при атмосферном давлении. В некоторых вариантах осуществления реакцию проводят при давлении вплоть до и включая 500 фунт/кв. дюйм.
Точное количество реагентов не является критическим. В одном из вариантов осуществления используют количество фторида щелочного металла, которое будет соответствовать, по меньшей мере, приблизительно эквимолярному количеству атомов фтора исходя из количества атомов хлора, которые будут обменивать, в исходном соединении, т.е., по меньшей мере, эквимолярное количество фторида щелочного металла.
При аминировании 4-фторпиколинат способен взаимодействовать с аммиаком, для замены атома фтора аминогруппой.
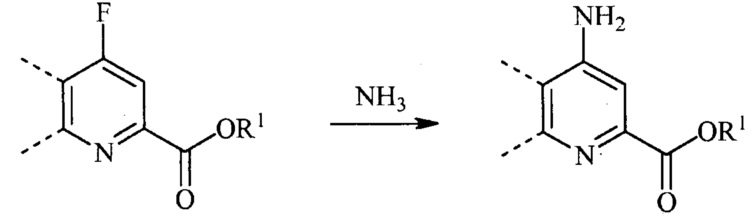
Тогда как требуется только стехиометрическое количество аммиака, часто удобно использовать большой избыток аммиака. Реакцию осуществляют в инертном растворителе, в некоторых вариантах осуществления в полярном апротонном растворителе или реакционной среде, такой как ДМСО, NMP, ДМФА, HMPA или сульфолан. Альтернативно можно использовать водный гидроксид аммония с или без использования органического растворителя. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 0°C до приблизительно 45°C и в некоторых вариантах осуществления, приблизительно от 10°C до приблизительно 30°C.
При проведении реакции аминирования 4-фторпиколинат растворяют в растворителе и добавляют к реакционной смеси аммиак при охлаждении. Избыток газообразного аммиака обычно барботируют через реакционную смесь. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов, и ее обычно проводят при атмосферном давлении. Реакцию можно проводить под давлением аммиака вплоть до 30 фунт/кв. дюйм. Реакцию можно также проводить, используя водный аммиак.
Аминосодержащие продукты или промежуточные соединения, полученные любым из таких способов, можно выделить общепринятыми способами, такими как упаривание или экстракция, и их можно очистить стандартными способами, такими как перекристаллизация или хроматография. Очистка аминосодержащих продуктов или промежуточных соединений можно также осуществлять протонированием кислотой с получением соли, которую выделяют с высокой чистотой кристаллизацией, осаждением или экстракцией. Можно использовать ряд кислот, таких как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, уксусная кислота или серная кислота. В одном из вариантов осуществления кислота представляет собой безводную хлористоводородную кислоту. Затем очищенную соль нейтрализуют основанием с получением нейтрального аминосодержащего продукта или промежуточного соединения. Можно использовать неорганические основания, такие как гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия или бикарбонат натрия. В одном из вариантов осуществления основание представляет собой органическое основание, такое как триэтиламин. Очистку аминосодержащего продукта или промежуточного соединения можно осуществлять данным способом непосредственно после стадии аминирования или после проведения последующих стадий, например галогенирования, конденсации.
В реакции обмена галогена (йода, брома или хлора), 6-йодированный, 6-бромированный или 6-хлорированный пиколинат получают взаимодействием соответствующего 6-фторированного пиколината, по меньшей мере, с одним эквивалентом йодида, бромида или хлорида.
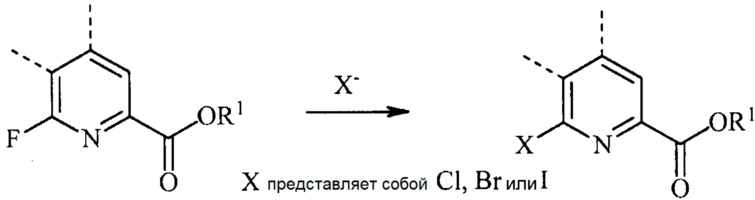
Обычно реакцию галогенового обмена осуществляют в присутствии большого избытка безводного йодоводорода (HI), бромоводорода (HBr) или хлороводорода (HCl). Реакцию обычно осуществляют в отсутствие воды, доводя до минимума образование побочных продуктов. Обмен галогенов обычно требует приблизительно от 5 до приблизительно 50 эквивалентов HI, HBr или HCl, в некоторых вариантах осуществления 10-20 эквивалентов. Реакцию осуществляют в инертном растворителе, в некоторых вариантах осуществления в полярном растворителе, таком как диоксан или уксусная кислота. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 75°C до приблизительно 150°C и в некоторых вариантах осуществления приблизительно от 100°C до приблизительно 125°C. Реакцию обычно осуществляют в герметичном реакторе под давлением, в котором можно держать газообразный HI, HBr или HCl. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов.
В реакции галогенирования атом хлора, брома или йода вводят в положение 3 пиколината взаимодействием 3-незамещенного пиколината с источником галогена в инертном растворителе.
Когда атом галогена в положении 3 представляет собой Cl, источником хлора может быть сам хлор (Cl2) или реагенты, такие как сульфурилхлорид, N-хлорсукцинимид или 1,3-дихлор-5,5-диметилгидантоин. При использовании хлора или сульфурилхлорида, применяют большой избыток хлорирующего агента. При использовании газообразного хлора, реакцию проводят в инертном растворителе, в некоторых вариантах осуществления в растворителе, таком как дихлорметан, дихлорметан-вода или уксусная кислота. Когда используют сульфурилхлорид, реакцию можно проводить в инертном растворителе, таком как дихлорметан или чистый сульфурилхлорид. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 0°C до приблизительно 45°C и в некоторых вариантах осуществления приблизительно от 10°C до приблизительно 30°C. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов. Реакцию хлорирования обычно проводят при атмосферном давлении.
Когда используемым хлорирующим агентом является N-хлорсукцинимид или 1,3-дихлор-5,5-диметилгидантоин, реакцию осуществляют, используя стехиометрическое количество хлорирующего агента. Что касается хлорирования, при использовании 1,3-дихлор-5,5-диметилгидантоина в качестве хлорирующего агента обнаружено, что взаимодействуют оба хлора в гидантоине. Реакцию проводят в инертном полярном растворителе, таком как ДМФА или ацетонитрил. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 20°C до приблизительно 85°C и в некоторых вариантах осуществления приблизительно от 50°C до приблизительно 80°C. При использовании ацетонитрила в качестве растворителя, удобно проводить реакцию при температуре кипячения с обратным холодильником. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов. Реакцию хлорирования обычно проводят при атмосферном давлении.
Когда атом галогена в положении 3 представляет собой Br, источником брома может быть сам бром (Br2) или реагенты, такие как сульфурилбромид, N-бромсукцинимид или 1,3-дибром-5,5-диметилгидантоин. При использовании Br2 в качестве бромирующего агента, можно использовать большой избыток, и реакцию проводят в инертном растворителе, в некоторых вариантах осуществления в растворителе, таком как дихлорметан, дихлорметан-вода или уксусная кислота. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 0°C до приблизительно 45°C и в некоторых вариантах осуществления приблизительно от 10°C до приблизительно 30°C. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов. Реакцию бромирования обычно проводят при атмосферном давлении.
Когда используемый бромирующий агент представляет собой N-бромсукцинимид или 1,3-дибром-5,5-диметилгидантоин, реакцию осуществляют, используя стехиометрическое количество бромирующего агента. Реакцию проводят в инертном полярном растворителе, таком как ДМФА или ацетонитрил. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 20°C до приблизительно 85°C и в некоторых вариантах осуществления приблизительно от 50°C до приблизительно 80°C. Когда ацетонитрил используют в качестве растворителя, удобно проводить реакцию при температуре кипячения с обратным холодильником. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов. Реакцию бромирования обычно проводят при атмосферном давлении.
Когда атом галогена в положении 3 представляет собой I, источником йода может быть сам йод (I2) или реагенты, такие как монохлорид йода или N-йодсукцинимид. Перйодную кислоту можно использовать в сочетании с I2. Когда I2 используют в качестве йодирующего агента, можно использовать большой избыток I2, и реакцию проводят в инертном растворителе, в некоторых вариантах осуществления в растворителе, таком как дихлорметан, дихлорметан-вода, метиловый спирт или уксусная кислота. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 0°C до приблизительно 45°C и в некоторых вариантах осуществления приблизительно от 10°C до приблизительно 30°C. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 5 часов. Реакцию йодирования обычно проводят при атмосферном давлении.
В реакции конденсации 6-йод, бром или хлорпиколинат взаимодействует с арильным, алкильным или алкенильным металлосодержащим соединением, где металл представляет собой Zn-галогенид, Zn-R, три(C1-C4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, C1-C4алкил или взятые вместе образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла.

"Катализатор", как используют в реакции конденсации, представляет собой катализатор, содержащий переходный металл, в частности палладиевый катализатор, такой как диацетат палладия или бис(трифенилфосфин)палладий(II)дихлорид, или никелевый катализатор, такой как ацетилацетонат никеля (II) или бис(трифенилфосфин)никель(II)дихлорид. Кроме того, катализаторы можно получить in situ из солей металлов и лигандов, таких как ацетат палладия и трифенилфосфин или хлорид никеля (II) и трифенилфосфин. Данные in situ катализаторы можно получить предварительным взаимодействием соли металла и лиганда с последующим добавлением к реакционной смеси или отдельным добавлением соли металла и лиганда непосредственно в реакционную смесь.
Обычно реакции конденсации осуществляют в отсутствие кислорода, используя инертный газ, такой как азот или аргон. Способы, применяемые для удаления кислорода из смесей реакции конденсации, такие как барботирование инертного газа, хорошо известны специалисту в данной области техники. Примеры таких способов описаны в The Manipulation of Air-Sensitive Compounds, 2ое издание; Shriver, D. F., Drezdzon, M. A., Eds.; Wiley-Interscience, 1986. Используют субстехиометрическое количество катализатора, обычно приблизительно от 0,0001 эквивалентов до 0,1 эквивалентов. Дополнительное количество лиганда можно необязательно добавлять для увеличения стабильности и активности катализатора. Кроме того, добавки, такие как Na2CO3, K2CO3, KF, CsF, K2HPO4, K3PO4 и NaF, обычно добавляют в реакцию конденсации. Реакция конденсации обычно требует приблизительно от 1 до приблизительно 5 эквивалентов этих добавок, в некоторых вариантах осуществления от 1 до 2 эквивалентов. Воду можно необязательно добавлять к реакции конденсации для увеличения растворимости данных добавок. Реакция конденсации обычно требует от 1 до приблизительно 3 эквивалентов арильного, алкильного или алкенильного металлосодержащего соединения, в некоторых вариантах осуществления 1-1,5 эквивалента. Реакцию осуществляют в инертном растворителе, таком как толуол, тетрагидрофуран (ТГФ), диоксан или ацетонитрил. Температура, при которой проводят реакцию, не является критической, но обычно составляет приблизительно от 25°C до приблизительно 150°C и в некоторых вариантах осуществления приблизительно от 50°C до приблизительно 125°C. Стандартная реакция обычно требует приблизительно от 0,5 до приблизительно 24 часов. Конкретный порядок добавления реагентов обычно не требуется. Часто операционно проще объединять все реагенты, за исключением катализатора, затем удалять кислород из реакционного раствора. После удаления кислорода, можно добавлять катализатор для инициирования реакции конденсации.
Когда Met часть арильного, алкильного или алкенильного металлосодержащего соединения представляет собой Zn-галогенид, Zn-R или медь, может быть необходима защита реакционноспособных функциональных групп. Например, если присутствует аминозаместитель (-NHR или -NH2), может потребоваться защита данных реакционноспособных групп. Ряд групп известен в данной области техники для защиты аминогрупп при реакции с органометаллическими реагентами. Примеры таких защитных групп описаны в Protective Groups in Organic Synthesis, 3ое издание; Greene, T. W.; Wuts, P. G. M., Eds.; Wiley-Interscience, 1999. Выбор того, какой металл будет использоваться в R-Met, зависит от ряда факторов, таких как стоимость, стабильность, реакционная способность и необходимость защиты реакционноспособных функциональных групп.
Продукты, полученные любым из указанных способов, можно выделить общепринятыми способами, такими как упаривание или экстракция, и их можно очистить стандартными способами, такими как перекристаллизация или хроматография.
Все из комбинаций приведенных выше вариантов осуществления включены в настоящее изобретение.
6. Примеры
Как использовано в настоящем описании, символы и условные обозначения, использованные в данных способах, схемах и примерах, несмотря на то, определено специально конкретное сокращение, соответствуют символам и условным обозначениям, применяемым в современной научной литературе, например, the Journal of the American Chemical Society или the Journal of Biological Chemistry. Конкретно, но без ограничения, следующие сокращения можно применять в примерах и во всем описании: г (граммы); мг (миллиграммы); мл (миллилитры); мкл (микролитры); M (молярный); мМ (миллимолярный); мкМ (микромолярный); экв. (эквивалент); ммоль (миллимоли); Гц (герцы); МГц (мегагерцы); час или часы (час или часы); мин (минуты) и МС (масс-спектрометрия).
Для всех приведенных ниже примеров, если не указано иное, можно применять стандартные способы обработки и очистки, известные специалистам в данной области техники. Если не указано иное, все температуры выражены в °C (градусы Цельсия). Все реакции проводят при комнатной температуре, если не указано иное. Предполагается, что способы получения, проиллюстрированные в настоящем описании, поясняют применяемую химию посредством конкретных примеров и не указывают на объем настоящего изобретения.
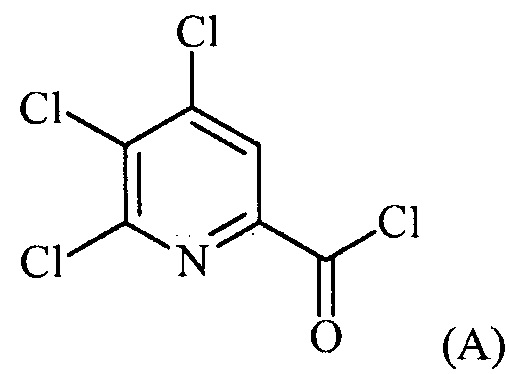
Пример 1. 4,5,6-трихлорпиколиноилхлорид
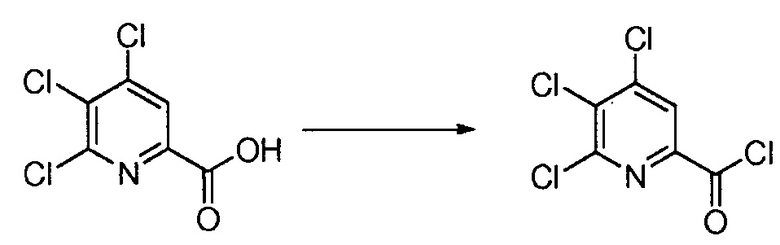
500 мл трехгорлую круглодонную колбу с термокарманом снабжали холодильником, который соединен с ловушкой, затем поглотителем газа с 10% NaOH, якорем магнитной мешалки, двумя пробками и термометром. В сосуд добавляли 4,5,6-трихлорпиколиновую кислоту (86 г, содержащими 8,6 г воды, 77,4 г активного соединения, 0,32 моль), толуол (160 мл), тионилхлорид (85 мл, 1,12 моль) и ДМФА (0,3 мл). Суспензию нагревали до 70-80°C, выдерживали при данной температуре в течение 7 часов, затем охлаждали до комнатной температуры и перемешивали в течение ночи. Приблизительно 0,2 мл бледно-желтого раствора помещали в пробирку и концентрировали до твердого вещества в токе N2. Твердое вещество обрабатывали смесью триэтиламин/метанол (0,3 мл/2 мл) и затем нагревали термофеном в течение приблизительно 1 минуты. ВЭЖХ анализ показал, что осталось менее чем 1% карбоновой кислоты по сравнению с метилэфиным производным. Раствор концентрировали на роторном испарителе с получением бледно-желтого твердого вещества. Твердое вещество сушили (40°C/20 мм рт.ст.) в течение приблизительно 12 часа с получением указанного в заголовке соединения (92 г), Тпл 68-70°C;
1H ЯМР (CDCl3, 400 MГц) δ 8,12 (с), твердое вещество содержало приблизительно 4% масс. толуола, как определено интегрированием протонных сигналов; 13С ЯМР (CDCl3, 101 MГц) δ 167,3, 151,0, 145,82, 145,8, 135,7, 125,5.
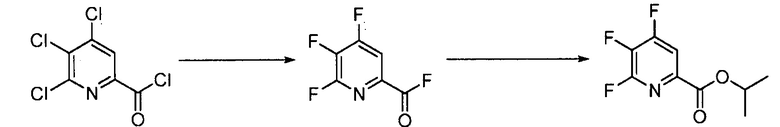
Пример 2. 4,5,6-трифторпиколиноилфторид

1-литровую трехгорлую круглодонную колбу продували N2 и соединяли с холодильником/N2 барботером, механической мешалкой и пробкой. В колбу добавляли CsF (172 г, 1,13 моль), безводный ацетонитрил (400 мл), 18-краун-6 (6,0 г, 0,023 моль) и 4,5,6-трихлорпиколиноилхлорид (55 г, 0,23 моль). Смесь кипятили с обратным холодильником и выдерживали в течение 20 часов. Суспензию охлаждали до комнатной температуры и соли фильтровали в атмосфере N2 под давлением. Солевой осадок на фильтре промывали сухим ацетонитрилом (100 мл) с получением янтарной жидкости (372 г). Трехгорлую, продутую N2, 250 мл круглодонную колбу с термокарманом снабжали двумя пробками, якорем магнитной мешалки и дистилляционной колонкой Vigruex с вакуумной рубашкой (15 см×1 см) с коллектором фракций, соединенным с барботером N2. В колбу добавляли 140 г ацетонитрильного раствора, полученного выше. Дистилляционную колбу нагревали до 82-85°C, при этом собирая прозрачный бесцветный дистиллят (ацетонитрил) с верха колонны при 80-83°C. Когда температура колбы для перегонки начинала возрастать и температура головки начинала снижаться, перегонку прекращали и охлаждали до комнатной температуры в атмосфере N2. Остаток в колбе для перегонки переносили в продутую N2 двугорлую 25 мл круглодонную колбу. Колбу снабжали термометром, якорем магнитной мешалки и той же системой для перегонки, описанной выше. Систему для перегонки можно подключать к вакууму или N2. Получали вакуум (приблизительно 70 мм рт.ст.) и затем начинали нагревание дистилляционной колбы. Продукт собирали в виде прозрачной бесцветной жидкости (6,7 г, Ткип 55-60°C и 55-60 мм рт.ст.). Анализ площади под кривой в процентах ГХ показал, что материал имел 99,1% чистоту:
1H ЯМР (CDCl3, 400 MГц) δ 8,08 (ддд, J=8,4, 4,4, 0,4 Гц); МС (ГХ, 70 эВ электронный удар) 179 (M+, 100%), 160 (8%), 151 (100%), 132 (80%), 82 (63%); 13С ЯМР (101 MГц, CDCl3) δ 157,71 (дт, J=269,0, 6,5 Гц), 152,96 (дд, J=246,1, 13,4 Гц), 152,49 (д, J=348,6 Гц), 138,69 (ддд, J=275,3, 30,2, 12,9 Гц), 135,44 (дддд, J=74,6, 15,1, 7,8 Гц), 117,00 (дт, J=18,2, 4,2 Гц).
В другом варианте осуществления после фильтрования и промывки солевого осадка на фильтре, получали 366 г янтарного раствора. Анализ площади под кривой в процентах ГХ показал, что смесь представляла собой 86,4% 4,5,6-трифторпиколиноилфторида и 13,6% 18-C-6. Способ анализа ГХ с применением внутреннего стандарта разрабатывали, используя диметилфталат в качестве внутреннего стандарта и материал, полученный выше, в качестве чистого компонента. ГХ анализ янтарного раствора показал, что он представляет собой 9,8% масс. продукта, что соответствует выходу 89%.
Пример 3. Изопропил 4,5,6-трифторпиколинат (путем с CsF)
В 250 мл 3-горлую колбу, снабженную механической мешалкой и холодильником, добавляли 4,5,6-трихлорпиколиноилхлорид (23,3 г, 95 ммоль), CsF (72,2 г, 475 ммоль), 18-краун-6 (2,5 г, 9,5 ммоль) и безводный ацетонитрил (150 мл). Колбу заполняли азотом и смесь кипятили с обратным холодильником при энергичном перемешивании в течение 22 часов. Отбирали образец и анализировали ГХ. Результаты показали, что реакция не завершилась, следовательно, добавляли дополнительное количество CsF (14,43 г, 95 ммоль) и смесь кипятили с обратным холодильником в течение дополнительных 24 часов, после которых считали, что реакция завершилась. Затем 6,28 г (104,5 ммоль) безводного 2-пропанола и 9,61 г (95 ммоль) безводного триэтиламина по каплям добавляли в колбу при 6°C. Смесь перемешивали в течение 5-6 часов при комнатной температуре. Реакцию останавливали, когда ГХ показала отсутствие в смеси исходного 4,5,6-трифторпиколиноилфторида. Соли удаляли фильтрованием и промывали некоторым количеством ацетонитрила. После удаления ацетонитрила на роторном испарителе, влажную пасту продукта растворяли в этиловом эфире. Смесь промывали водой и сушили над MgSO4. Большую часть этилового эфира удаляли на роторном испарителе. Концентрированную неочищенную смесь продукта в этиловом эфире фильтровали через слой силикагеля и элюировали некоторым количеством этилового эфира. После удаления растворителя на роторном испарителе получали 17,52 г (84% выход, 94% ВЭЖХ чистота) изопропил-4,5,6-трифторпиколината.
1H ЯМР (CDCl3, 300 MГц) δ 7,90 (ддд, 1H, J=9 Гц, 5 Гц, 1 Гц), 5,30 (м, 1H, J=6 Гц), 1,41 (д, 6H, J=6 Гц); 13C ЯМР (CDCl3, 75 MГц, 1H декуплед) δ 161,2 (д, J=4 Гц), 157,2 (ддд, J=265 Гц, 9 Гц, 6 Гц), 152,1 (ддд, J=241 Гц, 12 Гц, 5 Гц), 140,9 (м), 136,8 (ддд, J=269 Гц, 30 Гц, 13 Гц), 113,6 (м), 70,4 (с), 21,3 (с).
Пример 4. Изопропил 4,5,6-трифторпиколинат (способ с KF)
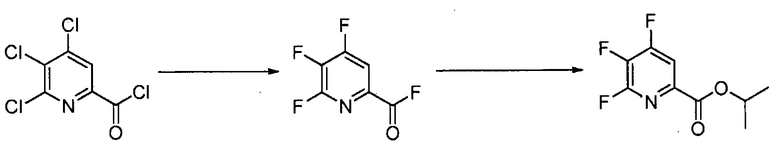
Реактор Парра (100 мл, конструкция Hastelloy C) очищали, сушили и проверяли на герметичность в атмосфере азота. В данный сосуд добавляли 3,673 г (15 ммоль) 4,5,6-трихлорпиколиноилхлорида, 7,844 г (135 ммоль) KF, 0,396 г (1,5 ммоль) 18-краун-6 и 40 мл безводного ацетонитрила. Всю систему продували азотом. Реакционную смесь перемешивали при 135°C в течение 22 часов и затем охлаждали ниже 45°C. Систему медленно вентилировали. Отбирали образец и анализировали его ГХ, ГХ-МС и ЯМР. ГХ показала, что реакция завершилась.
EIMS m/z=179 (M+, 96%), 160 (11%), 151 (100%), 132 (79%), 82 (73%); 19F ЯМР (376 MГц, CD3CN) δ 15,68 (т, J=3,4 Гц), -82,55 (т, J=23,3 Гц), (-119,60)-(-119,82) (м), -154,95 (дд, J=24,3, 17,3 Гц).
Безводный 2-пропанол (1,517 г, 25 ммоль) и Et3N (1,518, 15 ммоль) медленно добавляли к полученной выше реакционной смеси в реакторе Парра при 5-10°C. Смесь перемешивали при комнатной температуре в течение ночи и затем удаляли из реактора. Соли удаляли фильтрованием и промывали некоторым количеством ацетонитрила. Выход в реакторе составлял 77% (ГХ), как определено, используя очищенный изопропил 4,5,6-трифторпиколинат (чистота=97 ЖХ % пощади и 98 ГХ% площади) в качестве стандарта и дипропилфталат в качестве эталона. EIMS m/z=178 (M+, 40%), 160 (100%), 132 (69%), 82 (22%), 43 (35%).
Пример 5. Изопропил 4-амино-5,6-дифторпиколинат
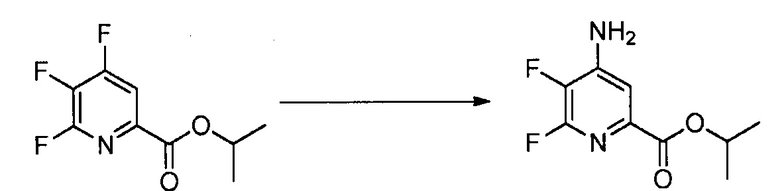
Изопропил 4,5,6-трифторпиколинат (17,52 г, 80 ммоль) растворяли в NMP (175 г) в 500 мл колбе, снабженной магнитным якорем. Смесь продували азотом, затем в колбу по каплям добавляли гидроксид аммония (10,70 г, 176 ммоль, 28% в воде) при комнатной температуре и реакционную смесь перемешивали в течение ночи. К реакционной смеси добавляли воду (190 мл), полученную в результате суспензию охлаждали на бане со льдом и перемешивали в течение 5 часов. Твердое вещество собирали фильтрованием и осадок на фильтре промывали водой. Сушка в вакууме давала изопропил 4-амино-5,6-дифторпиколинат в виде не совсем белого твердого вещества (11,7 г, 68% выход, 96% ВЭЖХ чистота): Тпл 167-168°C;
1H ЯМР (ТГФ-d8, 300 MГц) δ 7,25 (д, 1H, 6 Гц), 6,07 (с, 2H), 5,01 (м, 1H, 6 Гц), 1,20 (д, 6H, J=6,3 Гц); 13C ЯМР (ТГФ-d8, 75 MГц, 1H декуплед) 162,9 (с), 151,5 (дд, J=228 Гц, 12 Гц), 146,1 (дд, J=9 Гц, 6 Гц), 140,4 (дд, J=17 Гц, 5 Гц), 134,5 (дд, 7=251 Гц, 32 Гц), 111,9 (м), 68,4 (с), 21,0 (с).
Пример 6. Изопропил 4-амино-6-хлор-5-фторпиколинат

Реактор Парра (100 мл, конструкция Hastelloy C) очищали, сушили и проверяли на герметичность в атмосфере азота. В сосуд загружали 1,081 г (5 ммоль) изопропил 4-амино-5,6-дифторпиколината и 50 мл HCl (1М в уксусной кислоте) (50 ммоль). Всю систему продували азотом. Реакционную смесь перемешивали при 110°C в течение 3 часов и затем давали охладиться ниже 50°C. Систему медленно вентилировали и продували азотом. Линию продувки погружали в раствор Na2CO3/вода. Реакционную смесь концентрировали почти досуха в токе азота в течение ночи. Отбирали образец и подвергали ЖХ-МС анализу. Результаты показали, что было 4 основных компонента в данной реакционной смеси: изопропил 4-амино-6-хлор-5-фторпиколинат (46% площади), изопропил 4-ацетамидо-6-хлор-5-фторпиколинат (33% площади), 4-ацетамидо-6-хлор-5-фторпиколиновая кислота (10% площади) и 4-амино-6-хлор-5-фторпиколиновая кислота (7% площади).
Затем в колбу добавляли 50 мл безводного 2-пропанола вместе с 6 каплями концентрированной H2SO4. Смесь перемешивали при 100°C в течение 22 часов. После охлаждения до комнатной температуры, смесь удаляли из реактора. Избыток 2-пропанола удаляли на роторном испарителе. Твердую пасту растворяли в этилацетате. Этилацетатную смесь сначала обрабатывали раствором Na2CO3/вода, затем промывали водой и сушили над MgSO4. Растворитель удаляли на роторном испарителе с получением неочищенного продукта (1,00 г, ЖХ чистота=90%). После перекристаллизации из 2-пропанола получали белый твердый продукт (0,7 г, ЖХ чистота=98%, выход=60%).
1H ЯМР (400 MГц, ТГФ-d8) δ 7,44 (д, J=6,1 Гц, 1Н), 6,16 (с, 2H), 5,17 (гепт., J=6,3 Гц, 1Н), 1,35 (д, J=6,3 Гц, 6H); 13C ЯМР (101 MГц, ТГФ-d8) δ 162,97 (с), 145,46 (с), 144,47 (д, J=11,8 Гц), 143,64 (д, J=5,2 Гц), 142,93 (с), 137,06 (д, J=16,4 Гц), 112,57 (д, J=4,0 Гц), 68,51 (с), 21,02 (с); 19F ЯМР (376 MГц, ТГФ-d8) δ -143,12 (с).
Пример 7. Метил 4-амино-6-бром-5-фторпиколинат
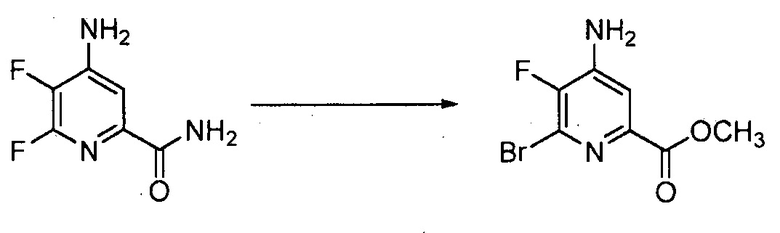
Реактор Парра (100 мл, конструкция Hastelloy C) очищали, сушили и проверяли на герметичность в атмосфере азота. 4-амино-5,6-дифторпиколинамид (5,196 г, 30 ммоль) и HBr (54 мл, 300 ммоль, 33% масс. в уксусной кислоте) загружали в реактор Парра. Всю систему продували азотом. Реакционную смесь перемешивали при 115°C в течение 3 часов и затем охлаждали ниже 45°C. Систему медленно вентилировали и продували азотом. Линию продувки погружали в раствор Na2CO3/вода. Реакционную смесь упаривали досуха в токе азота в течение ночи. Затем добавляли в колбу 55 мл безводного метанола вместе с 5 каплями концентрированной H2SO4. Смесь перемешивали при 95°C в течение 6 часов. После охлаждения до комнатной температуры выпадало твердое вещество, и его отфильтровывали. Неочищенный продукт (3,26 г) (ЖХ чистота=93%) получали в виде первой порции продукта. Фильтрат концентрировали на роторном испарителе, удаляя большую часть растворителя. Затем влажную твердую пасту экстрагировали этилацетатом. Этилацетатную смесь промывали водой и сушили над MgSO4. Растворитель удаляли на роторном испарителе. Дополнительный неочищенный продукт (2,56 г) (ЖХ чистота=81%) получали в виде второй порции. Обе порции неочищенного продукта объединяли и перекристаллизовывали из метанола с получением 4,3 г (57% выход, ЖХ чистота=97%) продукта. Метанольный маточный раствор помещали в холодильник на выходные. Получали дополнительные 0,14 г (96% ЖХ чистота) продукта: Тпл 194-196°C.
1H ЯМР (ТГФ-d8, 300 MГц) δ 7,29 (д, 1H, J=6 Гц), 6,06 (с, 2H), 3,71 (с, 3H); 13C ЯМР (ТГФ-d8, 75 MГц, 1H декуплед) δ 164,0 (с), 147,2 (с), 144,0 (м), 127,9 (д, J=20 Гц), 112,8 (д, J=5 Гц), 51,5 (с); EIMS m/z=250, 248 (M-, 1Br, 9%), 220 (14%), 218 (15%), 192 (95%), 191 (31%), 190 (100%), 189 (27%), 164 (10%), 162 (10%), 137 (7%), 111 (12%), 110 (19%), 109 (10%), 84 (9%), 83 (14%), 82 (10%).
Пример 8. Метил 4-(ацетиламино)-3,6-дихлор-5-фторпиколинат
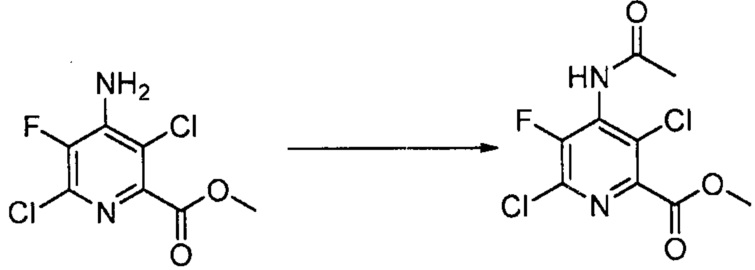
В 50 мл трехгорлую круглодонную колбу загружали метил 4-амино-3,6-дихлор-5-фторпиколинат (12,1 ммоль), толуол (50 мл) и CHCl3 (15 мл). К полученной суспензии по каплям добавляли предварительно смешанный раствор серной кислоты (0,05 ммоль) и уксусного ангидрида (24,2 ммоль) в толуоле (5 мл). Затем реакционную смесь нагревали в атмосфере N2 до 55-60°C и реакцию мониторили ТСХ. Когда считали, что реакция завершена, колбонагревалетель удаляли и реакционный раствор охлаждали до температуры окружающей среды. Затем смесь по каплям добавляли к смеси насыщенный водный NaHCO3/толуол/CHCl3 (180 мл, 3/10/5), перемешивали в течение 20 минут и слои разделяли, используя делительную воронку. К органическому слою добавляли силикагель (14,8 г) и растворитель удаляли, используя роторный испаритель. Затем неочищенный продукт на силикагеле очищали, используя систему очистки Combi-Flash на силикагеле и этилацетат и гексан в качестве элюентов. Чистоту продукта анализировали ВЭЖХ: Тпл=183-186°C;
1Н ЯМР (400 MГц, ДМСО-d6) δ 10,5 (с, 1H, NH), 3,93 (с, 3H), 2,17(с, 3H); 13C ЯМР (100,6 MГц, ДМСО-d6) δ 167,9, 162,8, 151,7, 149,0, 142,8 (д, JF-C=24 Гц), 136,2 (д, JF-C=80 Гц), 134,7 (д, JF-C=56 Гц), 126,6, 53:2, 22,5; 19F ЯМР (376,5 MГц, ДМСО-d6) δ -114,5.
Пример 9. Метил 4-(ацетиламино)-6-бром-3-хлор-5-фторпиколинат
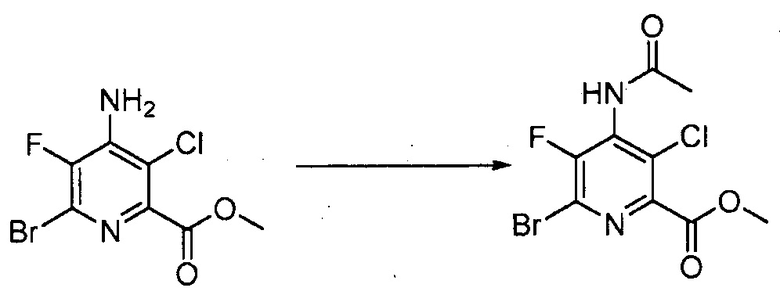
Метил 4-(ацетиламино)-6-бром-3-хлор-5-фторпиколинат получали аналогично метил 4-(ацетиламино)-3,6-дихлор-5-фторпиколинату в примере 8, за исключением того, что метил 4-амино-6-бром-3-хлор-5-фторпиколинат использовали вместо метил 4-амино-3,6-дихлор-5-фторпиколината: Тпл=190-192°C;
1H ЯМР (400 MГц, ДМСО-d6) δ 10,5 (с, 1H), 3,93 (с, 3H), 2,16 (с, 3H); 13C ЯМР (100,6 MГц, ДМСО-d6) δ 168,0, 162,8, 153,1, 150,5, 143,7 (д, JF-C=24 Гц), 134,1 (д, JF-C=56 Гц), 127,3 (д, JF-C=96 Гц), 53,2, 22,5; 19F ЯМР (376,5 MГц, ДМСО-d6) δ -108,3; МС Вычислено для С9Н7ВrСl2N2О3: 323,93, найдено: 289, 226.
Пример 10. Изопропил 4-(амино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат

В 50 мл трехгорлую круглодонную колбу загружали дигидрат фторида калия (12,0 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (5,40 ммоль), изопропил 4-амино-6-бром-5-фторпиколинат (4,0 ммоль), MeCN (15 мл) и H2O (5 мл). Полученную в результате смесь продували N2 в течение 15 минут, затем добавляли Pd(PPh3)2Cl2 (0,006-0,08 ммоль). Полученную в результате суспензию продували в течение 20 минут, затем нагревали при 68-70°C. После перемешивания в течение 1 часа, отбирали аликвоту (1-2 мкл) и разбавляли MeCN (2 мл). Аликвоту анализировали ВЭЖХ, контролируя потребление исходного 4-(ацетиламидо)-5-фторпиколинатного эфира. После того как считали, что реакция завершилась, колбонагревалетель удаляли и смесь охлаждали до температуры окружающей среды и разбавляли смесью MeCN/толуол/H2O (75 мл, 3/1/1); в случае необходимости, дополнительный объем смеси растворителей добавляли для растворения твердого вещества. Затем слои разделяли, используя делительную воронку. Затем анализ продукта осуществляли, используя внутренний стандарт (гексанофенон): Тпл=177-179°C;
1H ЯМР (400 MГц, ДМСО-d6) δ 7,52 (д, JF-H=6,8 Гц, 1H), 7,47 (дд, J=8,4, 1,6 Гц, 1H), 7,29 (дд, J=8,4, 7,2 Гц, 1H), 6,70 (с, 2H), 5,12 (п, J=6,4 Гц, 1H), 3,93 (с, 3H), 1,30 (д, J=6,4 Гц, 6H); 13C ЯМР (100,6 MГц, ДМСО-d6) δ 163,6, 154,4, 151,2, 148,0, 145,5, 144,0 (дд, JF-C=38,3, 4,2 Гц), 143,7 (д, JF-C=2,9 Гц), 139,0 (д, JF-C=13,6 Гц), 128,2 (д, JF-C=3,1Гц), 126,0 (д, JF-C=3,4 Гц), 125,4 (д, JF-C=3,5 Гц), 123,9 (дд, JF-C=14,2, 3,5 Гц), 112,5 (д, JF-C=5,1 Гц), 68,5, 61,5 (д, JF-C=4,0 Гц), 21,6; 18F{1H} ЯМР (376,5 MГц, ДМСО-d6) δ -129,4 (д, JF-F=25,6 Гц), -142,8 (д, JF-F=25,6 Гц); МС Вычислено для С16Н15СlF2N2О3: 356,1, найдено: 356 (M+), 270.
Пример 11. Метил 4-(амино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
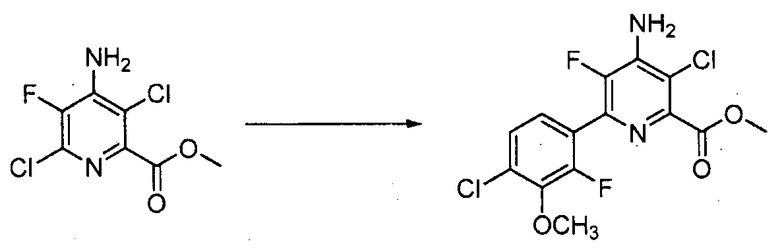
Метил 4-(амино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат получали аналогично изопропил 4-(амино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинату в примере 10, за исключением того, что метил 4-амино-3,6-дихлор-5-фторпиколинат использовали вместо изопропил 4-амино-6-бром-5-фторпиколината: Тпл=169-171°C;
1H ЯМР (400 MГц, ДМСО-d6) δ 7,46 (дд, J=8,8, 1,6 Гц, 1H), 7,29 (дд, 8,8, 7,2 Гц, 1H), 7,10 (с, 2H), 3,93 (с, 3H), 3,87 (с, 3H); 13C ЯМР (100,6 MГц, ДМСО-d6) δ 164,8, 154,3, 151,8, 146,2, 144,5, 143,9-143,7 (м), 141,6 (д, JF-С=14,0 Гц), 136,3 (д, JF-С=13,4 Гц), 128,5 (д, JF-С=2,6 Гц), 125,5 (д, JF-С=3,6 Гц), 122,9 (д, JF-С=13,7, 4,0 Гц), 61,6 (д, JF-С=4,2 Гц), 52,7; 19F{1H} ЯМР (376,5 MГц, ДМСО-d6) δ -129,2 (д, JF-С=27,5 Гц), -137,7 (д, JF-С=27,1 Гц); ИК: 3482, 3381, 2950, 1741, 1612, 1466, 1423, 1366, 1228, 1202, 1045, 970, 905, 815; МС Вычислено для С14Н11Сl2F2N2О3: 362,0. Найдено: 362 (M+), 304.
Пример 12. Метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
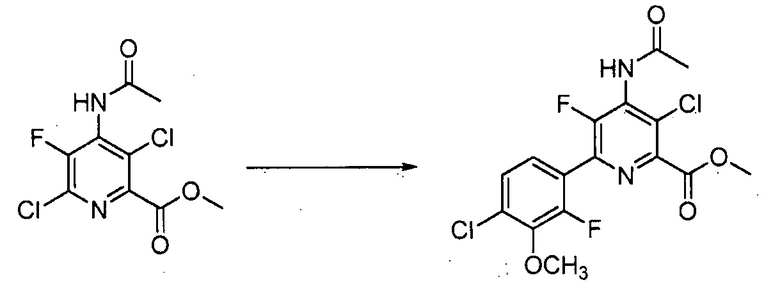
Метил 4-(ацетиламино)-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат получали аналогично изопропил 4-(амино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинату в примере 10, за исключением того, что метил 4-ацетамидо-3,6-дихлор-5-фторпиколинат использовали вместо изопропил 4-амино-6-бром-5-фторпиколината: Тпл=175-177°C;
1H ЯМР (400 MГц, ДМСО-d6) δ 7,52 (дд, J=8,4, 1,6 Гц, 1H), 7,38 (дд, J=8,4, 7 Гц, 1H), 3,95 (д, J=0,8 Гц, 3H), 3,94 (с, 3H), 2,17 (с, 3H); 13С ЯМР (100,6 MГц, ДМСО-d6) δ 168,0, 163,9, 154,4, 153,7, 151,9, 151,0, 144,9 (д, JF-C=4,4 Гц), 143,9 (д, JF-C=13,3 Гц), 139,5 (д, JF-C=16,7 Гц), 133,7 (д, JF-C=14,4 Гц), 129,3 (д, JF-C=3,2 Гц), 126,7, 125,9 (дд, JF-C=14,6, 3,6 Гц), 121,9 (д, JF-C=13,2, 4,3 Гц), 61,7 (д, JF-C=4,4 Гц), 53,1, 22,5; 19F{1H} ЯМР (376,5 MГц, ДМСО-d6) δ -119,0 (д, JF-F=28,6 Гц), -129,0 (д, JF-F=28,6 Гц); ИК: 3182, 3008, 2952, 1736, 1674, 1512, 1464, 1422, 1401, 1372, 1263, 1238, 1213, 1173, 1049, 1026, 977, 904, 687; МС Вычислено для C16H12Cl2F2N2O4: 404,0. Найдено: 404 (М+), 369.
Пример 13. Метил 4-(ацетиламино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
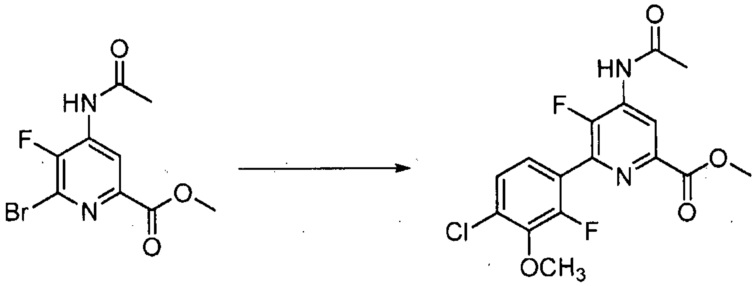
Метил 4-(ацетиламино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат получали аналогично изопропил 4-(амино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинату в примере 10, за исключением того, что метил 4-ацетамидо-6-бром-5-фторпиколинат использовали вместо изопропил 4-амино-6-бром-5-фторпиколината: Тпл=189-191°C;
1H ЯМР (400 MГц, ДМСО-d6) δ 10,5 (с, 1H), 9,04 (д, JF-H=5,6 Гц, 1H), 7,53 (дд, J=8,4, 1,6 Гц, 1H), 7,37 (дд, J=8,4, 7,0 Гц, 1H), 3,95 (с, 3H), 3,89 (с, 3H), 2,22 (с, 3H); 13С ЯМР (100,6 MГц, ДМСО-d6) δ 170,3, 164,1, 154,4, 151,9, 149,4, 146,7, 144,1-143,8 (м), (д, JF-C=15,3 Гц), 135,1 (д, JF-C 10,8 Гц), 128,9 (д, JF-C=3,4 Гц), 125,9 (дд, JF-C=35,0, 3,5 Гц), 122,9 (дд, JF-C=13,7, 3,5 Гц), 61,6, 52,6, 24,1; 19F{1H} ЯМР (376,5 MГц, ДМСО-d6) δ -129,5 (д, JF-F=27,5 Гц), -142,8 (д, JF-F =27,5 Гц); ИК: 3341, 2972, 2944, 1733, 1705, 1599, 1507, 1439, 1416, 1406, 1371, 1283, 1166, 1039, 974, 926, 890, 823, 678; МС Вычислено для C16H13ClF2N2O4: 370,1. Найдено: 370 (M+), 312.
Пример 14. 4,5-дихлор-6-(4-хлор-2-фтор-3-метоксифенил)пиколиновая кислота
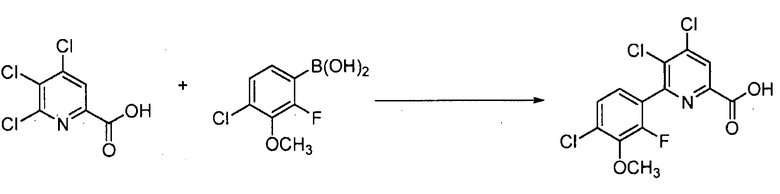
В 50 мл трехгорлую колбу, снабженную магнитным якорем и холодильником, помещали 4,5,6-трихлорпиколиновую кислоту (0,503 г, 90% чистота, 2 ммоль), триэтиламин (0,607, 6 ммоль), ацетонитрил (5 г) и воду (5 г). Смесь превращалась в гомогенный раствор, который продували азотом в течение 15 минут. Добавляли бис(трифенилфосфин)палладий(II)дихлорид (0,042 г, 0,06 ммоль) и смесь нагревали до 50°C. В одногорлую колбу помещали (4-хлор-2-фтор-3-метоксифенил)бороновую кислоту (1,951 г, 22% масс. в смеси метилизобутилкетон/диметоксиэтан, 2,1 ммоль) и ацетонитрил (2,5 г). Раствор продували азотом в течение 15 минут и затем медленно добавляли к раствору трихлорпиколиновой кислоты шприцевым насосом при 50°C. Суммарное время добавления составляло приблизительно часа. Реакционную смесь перемешивали при 50°C в течение следующих часов. Отбирали образец и анализировали его ВЭЖХ. Результаты ЖХ показал, что реакция завершилась. Реакционную смесь охлаждали до комнатной температуры. Для подкисления реакционной смеси, хлористоводородную кислоту (6 г, 1н. HCl в воде) по каплям добавляли в колбу. Полученную в результате суспензию перемешивали в течение ночи. Смесь фильтровали и получали светло-коричневое твердое вещество. Полученное неочищенное вещество перемешивали в ацетонитриле при комнатной температуре в течение 2 часов. После фильтрования и сушки в вакууме, получали белый продукт (0,44 г, 92% ЖХ чистота, 58% выход).
1H ЯМР (400 MГц, ТГФ-d8) δ 8,35 (с, 1Н), 7,40 (дд, J=8,5, 1,7 Гц, 1Н), 7,23 (дд, J=8,5, 6,9 Гц, 1Н), 4,00 (д, J=1,3 Гц, 3H); 13С ЯМР (101 MГц, ТГФ-d8) δ 163,62 (с), 154,81 (с), 153,03 (с), 152,31 (с), 147,36 (с), 144,64 (д, J=13,4 Гц), 143,70 (с), 133,21 (с), 129,55 (д, J=3,5 Гц), 126,34 (д, J=14,4 Гц), 125,71 (с), 125,41-125,1 (м), 60,91 (д, J=4,8 Гц); 13F ЯМР (376 MГц, ТГФ-d8) δ -128,96. ESI/ЖХ/МС/PI (M+H)+=349,9559 (100%), 351,9531 (99,66%), 353,95 (33,59%).
Пример 15. 6-(4-хлор-2-фтор-3-метоксифенил)-4,5-дихлор-2-пиридинкарбонилхлорид
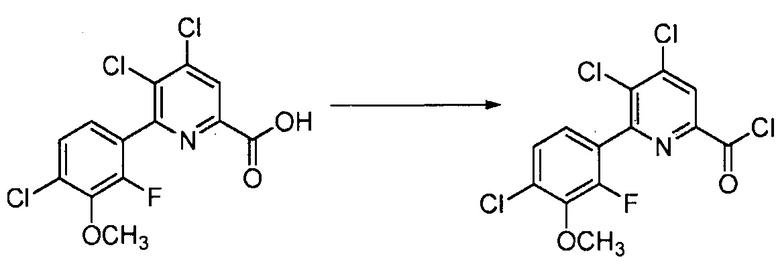
Смесь 33,5 г (~95 ммоль) 6-(4-хлор-2-фтор-3-метоксифенил)-4,5-дихлор-2-пиридинкарбоновой кислоты, 10,2 мл (140 ммоль) тионилхлорида, 0,1 мл N,N-диметилформамида (ДМФА) и 200 мл толуола нагревали при 75°C в течение 5 часов. Протекание реакции мониторили преобразованием хлорангидрида кислоты в ее метиловый эфир (одну каплю реакционной смеси добавляли к 5 каплям 10% масс. раствора метанола, содержащего 4-(диметиламино)пиридин, быстро нагревали до кипения с обратным холодильником, разбавляли ацетонитрилом и вводили в хроматограф). ЖХ анализ показал 8% площади оставшейся карбоновой кислоты и 3% площади неопределенного близко идущего продукта. Добавляли дополнительные 5 мл тионилхлорида и 0,1 мл ДМФА и нагревание продолжали в течение дополнительных 2 часов. ЖХ анализ показал продукт, метиловый эфир карбоновой кислоты, идущий только с оставшимся неопределенным полярным пиком. После перемешивания при комнатной температуре в течение ночи, реакционную смесь фильтровали, удаляя небольшое количество нерастворимого вещества. Фильтрат концентрировали в вакууме, дважды добавляли толуол и концентрировали в вакууме, упаривая остаточный тионилхлорид. Белое твердое вещество (38,6 г) сушили в вакуумном сушильном шкафу при 40°C с получением 33,3 г указанного в заголовке соединения (Тпл 134-136°C). ЖХ анализ с внутренним стандартом (преобразование в его метиловый эфир, как описано выше) показал 98,1% масс.
EIMS m/e (относительная интенсивность) 369 (4Cl, 80), 332 (3Cl, 38), 304 (3Cl, 82), 269 (2Cl, 100), 254 (2Cl, 30), 226 (2Cl, 73), 191 (30), 156 (46); 1H ЯМР (400 MГц, CDCl3) δ 8,23 (с, 1H), 7,32 (дд, J=8, 2 Гц, 1H), 7,15 (дд, J=8, 7 Гц, 1H), 4,02 (дд, J=1 Гц, 3H); 19F ЯМР (376 MГц, 1H декуплед, CDCl3) δ -126,83.
Пример 16. Метил 6-(4-хлор-2-фтор-3-метоксифенил)-4,5-дифтор-2-пиридинкарбоксилат
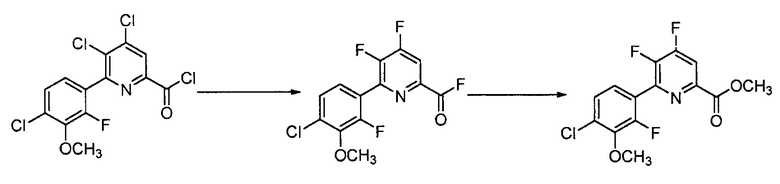
Смесь 1,74 г (30 ммоль, 6 экв.) KF (сушили при 115°C в токе N2 в течение ночи), 1,85 г (5 ммоль) 6-(4-хлор-2-фтор-3-метоксифенил)-4,5-дихлор-2-пиридинкарбонилхлорида и 10 мл сульфолана (сушили 4Å над молекулярными ситами) нагревали при 130°C в течение 10 часов и затем при комнатной температуре в течение ночи. Анализ ЖХ площади показал, что реакция не завершилась (63% продукта, 15% монофторные промежуточные соединения). Смесь нагревали при 130°C в течение следующих 7 часов, после чего анализ ЖХ площади показал 74% продукта и 4% монофторных промежуточных соединений. Данные для промежуточного фторида кислоты:
EIMS m/e (относительная интенсивность) 319 (1Cl, 100), 304 (1Cl, 20), 276 (1Cl, 56), 241 (11), 228 (8), 213 (12), 194 (18), 174 (5). 19F ЯМР (376 MГц, сульфолан) δ 17,84 (с), (-123,40)-(-123,54) (м), -129,29 (дд, J=29, 7 Гц), -137,70 (ддд, J=29, 21, 5 Гц), 19F ЯМР (376 MГц, 1H декуплед, сульфолан) δ 17,84 (ушир.с), (-123,38)-(-123,58) (м), -129,29 (д, J=29 Гц), -137,70 (дд, J=29,21 Гц).
После охлаждения до 50°C, добавляли 0,24 мл (6 ммоль) MeOH и смесь перемешивали при комнатной температуре в течение ночи. К янтарной смеси по каплям добавляли 10 мл H2О в течение 20 минут. Первоначально образовывались вязкие твердые вещества, которые, в конце концов, исчезали, оставляя густую коричневато-серую смесь. После перемешивания при комнатной температуре в течение 15 минут, смесь фильтровали, промывали 4 мл 1:1 сульфолан/H2O и 2× 4 мл H2O с получением 5,44 г коричневого твердого вещества. Твердое вещество сушили на воздухе с получением 1,54 г коричневого порошка. Анализ ЖХ с внутренним стандартом показал чистоту 78,4% масс. для выхода 73,0%.
Материал из эксперимента по фторированию выше (1,8 г, 67% под кривой ЖХ) нагревали и растворяли в 15 мл толуола. Полученный раствор подвергали флэш-хроматографии на силикагеле (500 г, 70-230 меш), элюируя толуолом. После пропускания через колонку 10 л толуола, наблюдали продукт и собирали с последующими 2 л элюента. Толуольные фракции, содержащие продукт, концентрировали в вакууме с получением 647 мг белого твердого вещества, чистота 94% площади ЖХ анализом. Полученное твердое вещество растворяли в 3 мл ацетонитрила, охлаждали в холодильнике, фильтровали и промывали 0,5 мл холодного ацетонитрила с получением 529 мг белого твердого вещества после сушки в шкафу в течение ночи, Тпл 134-134°C, чистота 97% площади ЖХ анализом.
EIMS m/e (относительная интенсивность) 331 (1Cl, 50), 273 (1Cl, 100), 238 (46), 237 (28), 222 (14), 194 (48); 1Н ЯМР (400 MГц, ДМСО) δ 8,5 (дд, J=9,6 Гц, 1H), 7,35-7,27 (м, 2H), 4,01 (с, 3H), 4,00 (д, J=1 Гц, 3H); 19F ЯМР (376 MГц, 1H декуплед, CDCl3) δ -123,64 (д, J=20 Гц), -128,51 (д, J=31 Гц), -139,59 (дд, J=31, 20 Гц); 19F ЯМР (376 MГц, CDCl3) δ -123,64 (дд, J=19, 9 Гц), -128,51 (дд, J=31, 6 Гц), -139,59 (ддд, J=31,19, 6 Гц).
Пример 17. Изопропил 4,5-дихлор-6-фенилпиколинат
В 125 мл трехгорлую круглодонную колбу загружали дигидрат фторида калия (4,52 г, 38,0 ммоль), фенилбороновую кислоту (4,88 г, 40 ммоль), изопропил 4,5,6-трихлорпиколинатный эфир (4,28 г, 16,0 ммоль), MeCN (60 мл) и H2O (20 мл). Полученную в результате суспензию продували N2 в течение 15 минут, затем добавляли бис(трифенилфосфин)палладий(II)хлорид (0,45 г, 0,64 ммоль). Затем полученную в результате желтую суспензию барботировали в течение 15 минут, затем нагревали при 65-68°C. После 1 часа перемешивания отбирали аликвоту (1-2 мкл) и разбавляли MeCN (2 мл). Аликвоту анализировали ВЭЖХ, контролируя потребление исходного соединения, изопропил 4,5,6-трихлорпиколинатного эфира. Через 3 часа считали, что реакция завершилась. Колбонагревалетель удаляли, смесь охлаждали до температуры окружающей среды и разбавляли MeCN/EtOAc/H2O (150 мл, 2/2/1). Затем слои разделяли, используя делительную воронку, и к органическому слою добавляли силикагель (22 г). Растворитель удаляли в вакууме и твердое вещество очищали CombiFlash, используя колонку 220 г. Концентрирование аликвот давало белое твердое вещество массой 4,07 г (82%).
Тпл=94-96°C; 1H ЯМР (400 MГц, CDCl3) δ 8,12 (с, 1H), 7,74-7,71 (м, 2H), 7,49-7,46 (м, 3H), 5,31 (г, J=6,4 Гц, 1H), 1,41 (д, J=6,4 Гц, 6H); 13C ЯМР (100,6 MГц, CDCl3) δ 163,1, 158,6, 146,5, 144,3, 137,5, 132,1, 129,6, 129,4, 128,0, 125,0, 70,2, 21,8; МС Вычислено для C15H13Cl2N2O2: 309,03. Найдено: 309 (M+), 223, 188, 152, 125.
Пример 18. Изопропил 4,5-дихлор-6-(4-метоксифенил)пиколинат
В 125 мл трехгорлую круглодонную колбу загружали дигидрат фторида калия (5,65 г, 60,0 ммоль), 4-метоксифенилбороновый эфир (3,42 г, 22,5 ммоль), изопропил 4,5,6-трихлорпиколинатный эфир (4,00 г, 15,0 ммоль), MeCN (72 мл) и H2O (24 мл). Полученную в результате суспензию продували N2 в течение 15 минут, затем добавляли бис(трифенилфосфин)палладий(II)хлорид (0,42 г, 0,60 ммоль). Затем полученную в результате желтую суспензию барботировали в течение 15 минут, затем нагревали при 60-62°C. После 1 часа перемешивания отбирали аликвоту (1-2 мкл) и разбавляли MeCN (2 мл). Аликвоту анализировали ВЭЖХ, контролируя потребление исходного соединения, изопропил 4,5,6-трихлорпиколинатного эфира. После 3 часов считали, что реакция завершилась. Колбонагревалетель удаляли, смесь охлаждали до температуры окружающей среды и разбавляли MeCN/PhMe/H2O (100 мл, 4/3/3). Слои разделяли, используя делительную воронку, и к органическому слою добавляли силикагель (22 г). Растворитель удаляли в вакууме и твердое вещество очищали CombiFlash с получением белого твердого вещества массой 2,90 г (57%). Тпл=113-116°C;
1H ЯМР (400 MГц, CDCl3) δ 8,07 (с, 1H), 7,74 (дт, J=9,2, 2,8 Гц, 2H), 6,99 (дт, J=8,8, 2,8 Гц, 2H), 5,30 (г, J=6,0 Гц, 1H), 3,87 (с, 3H), 1,41 (д, J=6,0 Гц, 6H); 13C ЯМР (100,6 MГц, CDCl3) δ 163,2, 160,6, 158,1, 146,4, 144,2, 131,7, 131,2, 129,9, 124,4, 113,4, 70,1, 55,3, 21,8; МС Вычислено для C16H15Cl2NO3: 339,04. Найдено: 339 (M+), 253, 218, 203, 182.
Пример 19. Изопропил 4,5-дихлор-6-(4-хлорфенил)пиколинат
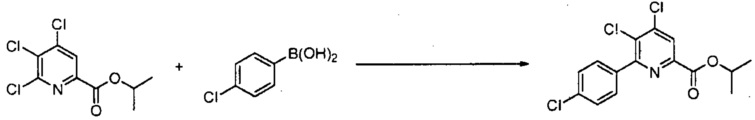
В 125 мл трехгорлую круглодонную колбу загружали дигидрат фторида калия (4,52 г, 38,0 ммоль), 4-хлорфенилбороновый эфир (5,00 г, 32,0 ммоль), изопропил 4,5,6-трихлорпиколинатный эфир (4,28 г, 16,0 ммоль), MeCN (70 мл) и H2O (23 мл). Полученную в результате суспензию продували N2 в течение 15 минут, затем добавляли бис(трифенилфосфин)палладий(II)хлорид (0,45 г, 0,64 ммоль). Затем полученную в результате желтую суспензию барботировали в течение 15 минут, затем нагревали при 65-68°C. После 1 часа перемешивания отбирали аликвоту (1-2 мкл) и разбавляли MeCN (2 мл). Аликвоту анализировали ВЭЖХ, контролируя потребление исходного соединения, изопропил 4,5,6-трихлорпиколинатного эфира. Через 3 часа считали, что реакция завершилась. Колбонагревалетель удаляли, смесь охлаждали до температуры окружающей среды и разбавляли MeCN/PhMe/H2O (80 мл, 2/3/2). Затем слои разделяли, используя делительную воронку, и к органическому слою добавляли силикагель (22,5 г). Растворитель удаляли в вакууме и твердое вещество очищали CombiFlash с получением после концентрирования растворителя белого твердого вещества массой 3,44 г (62%). Тпл=133-135°C;
1H ЯМР (400 MГц, CDCl3) δ 8,13 (с, 1H), 7,69 (дт, J=8,8, 2,0 Гц, 2H), 7,29 (дд, J=8,4, 2,0 Гц, 2H), 5,31 (г, J=6,0 Гц, 1H), 1,41 (д, J=6,0 Гц, 6H); 13C ЯМР (100,6 MГц, CDCl3) δ 162,9, 157,4, 146,6, 144,5, 135,8, 135,7, 132,0, 131,0, 128,3, 125,2, 70,3, 21,8; МС Вычислено для C15H12Cl3NO2: 342,99. Найдено; 343 (M+), 257, 222, 186, 151.
Пример 20. 4,5-дихлор-6-(4-метоксифенил)пиколиновая кислота
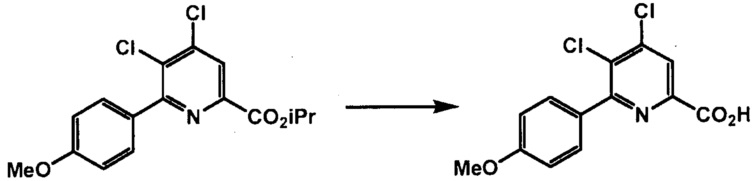
К смеси изопропил 4,5-дихлор-6-(4-метоксифенил)пиколината (5,25 г, 15,4 ммоль) в тетрагидрофуране (40 мл) и воде (10 мл) добавляли гидроксид калия (1,26 г, 22,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После 1 часа перемешивания из смеси выпадал осадок. Добавляли к реакционной смеси HCl (водн.) (2н., 25 мл), и образовывалась прозрачная двухфазная смесь. Смесь добавляли к воде (75 мл) в делительной воронке и экстрагировали EtOAc (2×75 мл). Объединенные органические слои промывали водой (25 мл) и насыщенным NaCl (50 мл) и затем концентрировали при пониженном давлении с получением 4,57 г (99% выход) 4,5-дихлор-6-(4-метоксифенил)пиколиновой кислоты в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,23 (с, 1Н), 7,72-7,64 (м, 2H), 7,07-6,99 (м, 2H), 3,89 (с, 3H); 13C ЯМР (101 MГц, CDCl3) δ 162,78, 161,05, 157,26, 146,30, 143,76, 133,54, 130,98, 128,72, 123,45, 113,77, 55,48; Тпл=164-181°C.
Пример 21. 4,5-дихлор-6-(4-хлорфенил)пиколиновая кислота
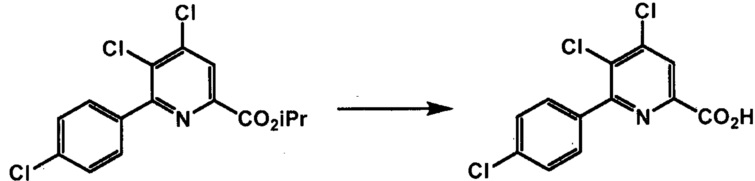
В 125 мл 3-горлую круглодонную колбу, снабженную холодильником, впускным отверстием для азота, верхнеприводной мешалкой, термометром и колбонагревалетелем, загружали изопропил 4,5-дихлор-6-(4-хлорфенил)пиколинат (7,6 г, 22,1 ммоль) и изопропиловый спирт (70 мл). Реакционную смесь нагревали до 40°C и добавляли гидроксид калия (85%, 5,1 г, 77,4 ммоль) и воду (5 мл). Из смеси выпадал осадок, и ее становилось трудно перемешивать. Смесь разбавляли водой (250 мл), растворяя большую часть твердого вещества, и перемешивали при комнатной температуре. По каплям добавляли к реакционной смеси концентрированную HCl (12н., 5,6 мл) с получением pH~2, и из смеси выпадал осадок. Твердое вещество выделяли вакуумным фильтрованием и промывали водой (2×100 мл), затем сушили с получением 7,3 г (108%) выход по массе) 4,5-дихлор-6-(4-хлорфенил)пиколиновой кислоты в виде белого твердого вещества.
1H ЯМР (400 MГц, ТГФ-d8/D2O) δ 8,19 (д, J=11,2 Гц, 1H), 7,84-7,73 (м, 2Н), 7,50 (дд, J=10,3, 3,5 Гц, 2Н); 13С ЯМР (101 MГц, ТГФ-d8/D2O) δ 167,70, 156,03, 152,40, 143,60, 136,49, 134,76, 131,22, 129,24, 128,04, 124,71; Тпл=229°C.
Пример 22. 4,5-дихлор-6-(фенил)пиколиновая кислота
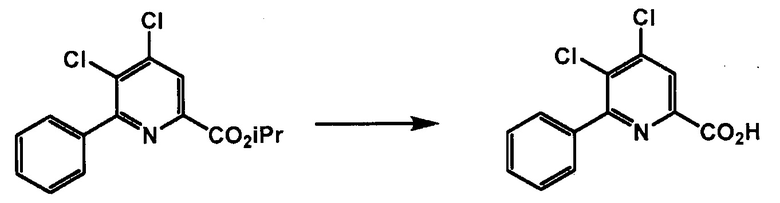
В 125 мл 3-горлую круглодонную колбу, снабженную холодильником, впускным отверстием для азота, верхнеприводной мешалкой, термометром и колбонагревалетелем, загружали изопропил 4,5-дихлор-6-фенилпиколинат (7,0 г, 22,5 ммоль) и изопропиловый спирт (65 мл). Реакционную смесь нагревали до 40°C и добавляли гидроксид калия (85%, 5,1 г, 77,4 ммоль) и воду (5 мл). Из смеси выпадал твердый осадок, и ее становилось трудно перемешивать. Смесь разбавляли водой (250 мл), растворяя большую часть твердого вещества, и перемешивали при комнатной температуре. По каплям добавляли к реакционной смеси концентрированную серную кислоту (5 мл) с получением pH~2, и из смеси выпадал осадок. Твердое вещество выделяли вакуумным фильтрованием и промывали водой (2×100 мл), затем сушили с получением 5,8 г (96% выход) 4,5-дихлор-6-фенилпиколиновой кислоты в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,28 (с, 1Н), 7,74-7,60 (м, 2H), 7,59-7,45 (м, 3H), 5,98 (ушир.с, 1Н); 13C ЯМР (101 MГц, CDCl3) δ 162,97, 157,76, 146,26, 144,00, 136,51, 133,84, 130,02; Тпл=159-160°C.
Пример 23. 4,5-дихлор-6-(4-хлорфенил)пиколиноилхлорид
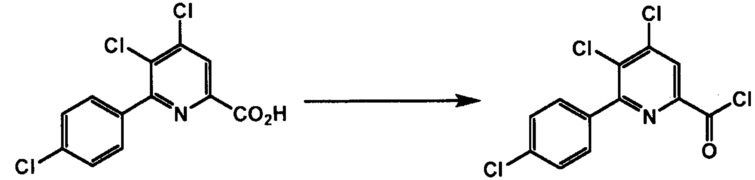
К смеси 4,5-дихлор-6-(4-хлорфенил)пиколиновой кислоты (3,00 г, 9,9 ммоль) в толуоле (25 мл) добавляли тионилхлорид (1,08 мл, 14,9 ммоль) и диметилформамид (0,04 мл, 0,5 ммоль). Реакционную смесь нагревали при 80°C в течение 2,5 часов. ВЭЖХ анализ аликвоты реакционной смеси, обработанной метанолом и диметиламинопиридином, показал, что осталось исходное соединение. Реакционную смесь охлаждали до комнатной температуры и добавляли дополнительные количества тионилхлорида (0,5 мл, 6,9 ммоль) и диметилформамида (0,04 мл, 0,5 ммоль). Реакционную смесь нагревали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением белого твердого вещества. Добавляли толуол (40 мл), растворяя твердое вещество, и концентрировали при пониженном давлении, затем данный способ повторяли второй раз. 4,5-Дихлор-6-(4-хлорфенил)пиколиноилхлорид выделяли в виде белого твердого вещества (3,05 г, 96% выход).
1H ЯМР (400 MГц, CDCl3) δ 8,15 (с, 1Н), 7,79-7,72 (м, 2H), 7,53-7,46 (м, 2H); 13C ЯМР (101 MГц, CDCl3) δ 168,66, 157,63, 146,48, 145,44, 136,35, 135,10, 134,28, 131,04, 128,63, 124,90. МС Вычислено для C12H5Cl4NO; 320,91. Найдено: 257, 222, 207, 186, 151.
Пример 24. 4,5-дихлор-6-(фенил)пиколиноилхлорид
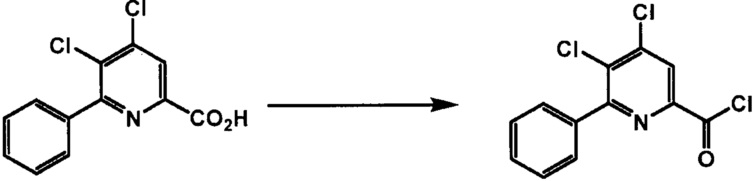
К смеси 4,5-дихлор-6-фенилпиколиновой кислоты (3,00 г, 1 1,2 ммоль) в толуоле (40 мл) добавляли тионилхлорид (1,22 мл, 16,8 ммоль) и диметилформамид (0,04 мл, 0,6 ммоль). Реакционную смесь нагревали при 80°C в течение 3 часов. ВЭЖХ анализ аликвоты реакционной смеси, обработанной метанолом и диметиламинопиридином, показал неполное преобразование исходного соединения. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением белого твердого вещества. Добавляли толуол (40 мл), растворяя твердое вещество, и концентрировали при пониженном давлении, затем данный способ повторяли второй раз. 4,5-Дихлор-6-фенилпиколиноилхлорид выделяли в виде белого твердого вещества (2,84 г, 89% выход).
1H ЯМР (400 MГц, CDCl3) δ 8,14 (с, 1Н), 7,83-7,75 (м, 2H), 7,55-7,47 (м, 3H); 13C ЯМР (101 MГц, CDCl3) δ 168,80, 158,88, 146,42, 145,21, 136,79, 134,40, 129,98, 129,61, 128,31, 124,74; MS: Вычислено для C12H6Cl3NO: 284,95. Найдено: 285 (M+), 250, 222, 187, 152; Тпл=106-111°C
Пример 25. 4,5-дихлор-6-(4-метоксифенил)пиколиноилхлорид
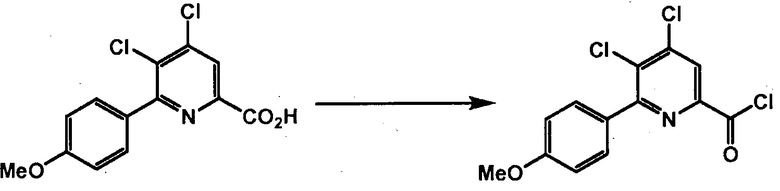
К смеси 4,5-дихлор-6-(4-метоксифенил)пиколиновой кислоты (4,50 г, 15,1 ммоль) в толуоле (40 мл) добавляли тионилхлорид (1,65 мл, 22,6 ммоль) и диметилформамид (0,06 мл, 0,8 ммоль). Реакционную смесь нагревали при 80°C в течение 12 часов. ВЭЖХ анализ аликвоты реакционной смеси, обработанной метанолом и диметиламинопиридином, показал неполное преобразование исходного соединения. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением желтого твердого вещества. Добавляли толуол (40 мл), растворяя твердое вещество, и концентрировали при пониженном давлении, затем данный способ повторяли второй раз. 4,5-Дихлор-6-(4-метоксифенил)пиколиноилхлорид выделяли в виде желтого твердого вещества (4,64 г, 97% выход).
1H ЯМР (400 MГц, CDCl3) δ 8,09 (с, 1Н), 7,85-7,77 (м, 2H), 7,06-6,98 (м, 2H); 13C ЯМР (101 MГц, CDCl3) δ 168,91, 161,06, 158,35, 146,26, 145,13, 133,92, 131,35, 129,16, 124,13, 113,70; МС Вычислено для С13H8Cl3NO2: 314,96. Найдено: 253, 218.
Пример 26. Изопропил 4,5-дифтор-6-(4-хлорфенил)пиколинат
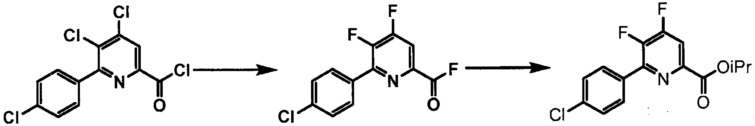
К раствору 4,5-дихлор-6-(4-хлорфенил)пиколиноилхлорида (2,0 г, 6,23 ммоль) в сульфолане (40 мл, сушили над 4Å молекулярными ситами, 100 ч./млн. H2O) добавляли фторид калия (2,2 г, 37,4 ммоль). Реакционную смесь нагревали при 130°C в течение 24 часов. Реакционную смесь анализировали ГХ-МС и 19F ЯМР. Данные для 6-(4-хлорфенил)-4,5-дифторпиколиноилфторида, ГХ-МС: m/z=271, 223; 19F ЯМР (376 МГц, толуол-d8) δ 17,05 (с), -123,81 (д, J=19,1 Гц), -140,17 (д, J=19,1 Гц). Реакционную смесь охлаждали до комнатной температуры и добавляли триэтиламин (1,1 мл, 7,8 ммоль) и изопропанол (0,7 мл, 9,4 ммоль). После перемешивания в течение 1,5 часов, реакционную смесь разбавляли водой (100 мл) и переносили в делительную воронку. Реакционную смесь экстрагировали метил-трет-бутиловым эфиром (MTBE, 2×50 мл). Объединенные органические экстракты промывали водой (3×50 мл) и насыщенным NaCl (50 мл) и концентрировали при пониженном давлении с получением коричневого масла. Неочищенный продукт в виде масла очищали флэш-хроматографией на силикагеле (градиент гексан/этилацетат, 100% гексан→20% гексан/этилацетат) с получением 0,93 г (48% выход) изопропил 6-(4-хлорфенил)-4,5-дифторпиколината в виде белого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,04-7,98 (м, 2H), 7,90 (дд, J=9,4, 5,3 Гц, 1Н), 7,51-7,45 (м, 2H), 5,31 (гепт. 6,3 Гц, 1Н), 1,43 (д, J=6,3 Гц, 6H); 13C ЯМР (101 MГц, CDCl3) δ 162,72 (д, J=3,5 Гц), 158,12 (д, J=12,6 Гц), 155,49 (д, J=12,4 Гц), 149,41 (д, J=11,0 Гц), 147,16 (дд, J=7,9, 1,0 Гц), 146,73 (д, J=10,9 Гц), 136,51 (д, J=0,9 Гц), 130,34 (д, J=6,6 Гц), 128,93 (с), 113,80 (д, J=16,1 Гц), 70,25 (с), 21,85 (с); 19F ЯМР (376 MГц, CDCl3) δ -124,73 (дд, J=17,7, 9,5 Гц), -144,38 (дд, J=17,7, 5,4 Гц); МС Вычислено для C16H15F2NO3: 307,10. Найдено: 307 (М+), 221, 206; Тпл 73-74°C.
Пример 27. Изопропил 4,5-дифтор-6-(фенил)пиколинат
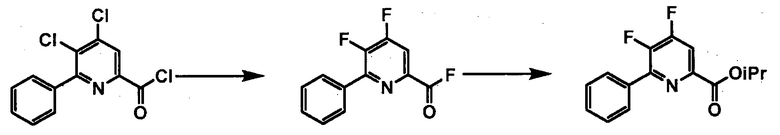
К раствору 4,5-дихлор-6-фенилпиколиноилхлорида (1,76 г, 6,14 ммоль) в сульфолане (40 мл, сушили над 4Å молекулярными ситами, ~100 ч./млн. H2O) добавляли фторид калия (2,14 г, 36,9 ммоль). Реакционную смесь нагревали при 130°C в течение 24 часов. Реакционную смесь анализировали ГХ-МС и 19F ЯМР. Данные для 4,5-дифтор-6-фенилпиколиноилфторида, ГХ-МС: m/z=237, 189; 19F ЯМР (376 МГц, толуол-d8) δ 17,03 (с), -124,14 (д, J=19,1 Гц), -140,76 (д, J=19,1 Гц). Реакционную смесь охлаждали до комнатной температуры и добавляли триэтиламин (1,1 мл, 7,7 ммоль) и изопропанол (0,7 мл, 9,2 ммоль). После перемешивания в течение 1,5 часов, реакционную смесь разбавляли водой (100 мл) и переносили в делительную воронку. Реакционную смесь экстрагировали метил-трет-бутиловым эфиром (MTBE, 2×50 мл). Объединенные органические экстракты промывали водой (3×50 мл) и насыщенным NaCl (50 мл) и концентрировали при пониженном давлении с получением коричневого масла. Неочищенный продукт в виде масла очищали флэш-хроматографией на силикагеле (градиент гексан/этилацетат, 100% гексан→20% гексан/этилацетат) с получением 1,2 г (70% выход) изопропил 4,5-дифтор-6-фенилпиколината в виде желтого масла.
1H ЯМР (400 MГц, CDCl3) δ 8,07-7,99 (м, 2H), 7,89 (дд, 9,4, Гц, 1Н), 7,56-7,42 (м, 3H), 5,31 (гепт. J=6,3 Гц, 1Н), 1,43 (д, J=6,3 Гц, 6H); 13С ЯМР (101 MГц, CDCl3) δ 162,89 (д, J=3,4 Гц), 156,74 (дд, J=264,2, 12,5 Гц), 148,07 (дд, J=268,9, 10,8 Гц), 146,99 (дд, J=309,2, 10,8 Гц), 145,45 (с), 134,12-133,60 (м), 130,20 (с), (д, J=5,9 Гц), 128,64 (с), 113,56 (д, J=16,0 Гц), 70,14 (с), 21,86 (с); 19F ЯМР (376 MГц, CDCl3) δ -125,22 (дд, J=17,7, 9,5 Гц), -144,74 (дд, J=17,7, 5,4 Гц); МС Вычислено для C15H13F2NO2: 277,09, найдено: 277 (M+), 218, 191.
Пример 28. Изопропил 4,5-дифтор-6-(4-метоксифенил)пиколинат
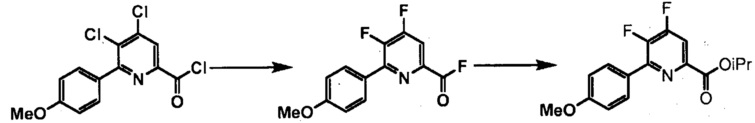
К раствору 4,5-дихлор-6-(4-метоксифенил)пиколиноилхлорида (2,5 г, 7,9 ммоль) в сульфолане (40 мл, сушили над 4Å молекулярными ситами, 100 ч./млн. воды) добавляли фторид калия (2,75 г, 47,4 ммоль). Реакционную смесь нагревали при 150°C в течение 24 часов. Добавляли дополнительное количество фторида калия (1,4 г, 24 ммоль) и реакционную смесь нагревали при 150°C в течение дополнительных 24 часов. Реакционную смесь анализировали ГХ-МС и 19F ЯМР. Данные для 4,5-дифтор-6-(4-метоксифенил)пиколиноилфторида, ГХ-МС: m/z=267, 224, 176; 19F ЯМР (376 МГц, толуол) δ 16,94 (с), -124,65 (д, J=19,1 Гц), -141,23 (д, J=19,1 Гц). Реакционную смесь охлаждали до комнатной температуры и добавляли триэтиламин (1,4 мл, 9,9 ммоль) и изопропанол (0,9 мл, 11,9 ммоль). После перемешивания в течение 1,5 часов, реакционную смесь разбавляли водой (125 мл) и переносили в делительную воронку. Реакционную смесь экстрагировали метил-трет-бутиловым эфиром (MTBE, 2×75 мл). Объединенные органические экстракты промывали водой (3×75 мл) и насыщенным NaCl (75 мл) и концентрировали при пониженном давлении с получением коричневого масла. Неочищенный продукт в виде масла очищали флэш-хроматографией на силикагеле (градиент гексан/этилацетат, 100% гексан 20% гексан/этилацетат) с получением 0,60 г (25% выход) изопропил 4,5-дифтор-6-(4-метоксифенил)пиколината в виде бледно-желтого твердого вещества.
1H ЯМР (400 MГц, CDCl3) δ 8,08-8,01 (м, 2H), 7,82 (дд, J=9,5, 5,2 Гц, 1Н), 7,04-6,97 (м, 2H), 5,30 (гепт. J=6,3 Гц, 1Н), 3,86 (с, 3H), 1,42 (д, J=6,3 Гц, 6H); 13C ЯМР (101 MГц, CDCl3) δ 162,93 (с), 161,22 (с), 156,68 (дд, J=263,5, 12,7 Гц), 147,70 (дд, J=267,9, 10,9 Гц), 146,61 (дд, J=286,4, 10,5 Гц), 145,18 (с), 130,53 (д, J=6,6 Гц), 126,43, 114,02 (с), 112,77 (д, J=16,1 Гц), 69,99 (с), 55,32 (с), 21,82 (с); 19F ЯМР (376 MГц, CDCl3) δ -125,81 (д, J=17,7 Гц), -145,30 (д, J=19,1 Гц); МС Вычислено для C16H15F2NO3: 307,10. Найдено: 307 (M+), 221,206.
Пример 29. Изопропил 4-амино-3-хлор-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат
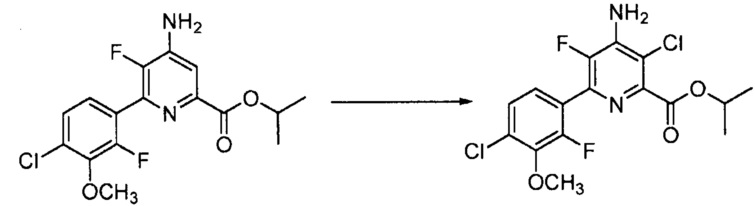
В 50 мл трехгорлую круглодонную колбу загружали изопропил 4-(амино)-6-(4-хлор-2-фтор-3-метоксифенил)-5-фторпиколинат (450 мг, 1,26 ммоль), ацетонитрил (25 мл) и 1,3-дихлор-5,5-диметилгидантоин (137 мг, 0,69 ммоль). Полученную в результате смесь продували N2 в течение 5 минут и затем нагревали при 68-70°C. После 2 часов перемешивания, отбирали аликвоту (1-2 мкл) и разбавляли MeCN (2 мл). Аликвоту анализировали ВЭЖХ, контролируя потребление исходного соединения, пиридилового эфира. После того как считали, что реакция завершилась, колбонагревалетель удаляли, и смесь охлаждали до температуры окружающей среды и разбавляли EtOAc (50 мл) и NaHSO3 (20 мг) в H2O (20 мл). Затем слои разделяли, используя делительную воронку. К органическому слою добавляли силикагель (10,1 г) и растворитель удаляли, используя роторный испаритель. Затем неочищенный продукт на силикагеле очищали, используя Combi-Flash систему градиентной очистки на силикагеле EtOAc/Hex (5/100-70/30). После концентрирования получали желтое твердое вещество массой 466 мг (95%), Тпл=113-115°C;
1H ЯМР (400 MГц, ДМСО-d6) δ 7,47 (дд, 8,8, 1,6 Гц, 1H), 7,29 (дд, J=8,4, 6,8 Гц, 1H), 7,07 (с, 2H), 5,17 (гепт., 7=6,0 Гц, 1H), 3,93 (с, 3H), 1,32 (д, J=6,0 Гц, 6H); 13С ЯМР (100,6 MГц, ДМСО-d6) δ 164,1, 154,3, 151,8 (д, JF-C=3,7 Гц), 146,1, 145,5 (д, JF-C=4,3 Гц), 143,8(д, JF-C=13,7 Гц), 143,5, 141,4 (д, JF-C=14,2 Гц), 136,3 (д, JF-C=13,4 Гц), 128,5 (д, JF-C=3,1 Гц), 125,8 (д, JF-C=2,6 Гц), 125,5 (д, JF-C=3,4 Гц), 122,9 (дд, JF-C=13,9, 3,8 Гц), 69,6, 61,6, 21,4; 19F{1H} ЯМР (376,5 MГц, ДМСО-d6) δ -129,1 (д, JF-F=27,9 Гц), -138,2 (д, JF-F=27,9 Гц).
Предполагается, что варианты осуществления, описанные выше, являются просто иллюстрирующими настоящее изобретение, и специалистам в данной области техники будет очевидно или они смогут установить, применяя не более чем стандартные эксперименты, многочисленные эквиваленты конкретных соединений, материалов и способов. Считают, что все данные эквиваленты включены в объем настоящего изобретения и прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2542985C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545074C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-3-ХЛОР-5-ФТОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545021C1 |
| ФТОРПИКОЛИНОИЛФТОРИДЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2627659C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-3-ХЛОР-5-ФТОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2539578C1 |
| СОЕДИНЕННЫЕ МОСТИКОВОЙ СВЯЗЬЮ N-ЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДО-ИНГИБИТОРЫ ГАММА-СЕКРЕТАЗЫ | 2006 |
|
RU2422443C2 |
| 6-АМИНО-2-ЗАМЕЩЕННЫЕ-5- ВИНИЛСИЛИЛПИРИМИДИН-4-КАРБОНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ И 4-АМИНО-6-ЗАМЕЩЕННЫЕ-3-ВИНИЛСИЛИЛПИРИДИН-ПИКОЛИНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ КАК ГЕРБИЦИДЫ | 2012 |
|
RU2556000C2 |
| СЛОЖНЫЕ АРИЛАЛКИЛОВЫЕ ЭФИРЫ 4-АМИНО-6-(ЗАМЕЩЕННЫЙ ФЕНИЛ)ПИКОЛИНАТОВ И 6-АМИНО-2-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-4-ПИРИМИДИНКАРБОКСИЛАТОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2012 |
|
RU2566760C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРИДИНА ИЛИ 2-АМИНОПИРИМИДИНА В КАЧЕСТВЕ ЦИКЛИНЗАВИСИМЫХ ИНГИБИТОРОВ КИНАЗЫ | 2019 |
|
RU2790006C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРИДИНА ИЛИ 2-АМИНОПИРИМИДИНА В КАЧЕСТВЕ ЦИКЛИНЗАВИСИМЫХ ИНГИБИТОРОВ КИНАЗЫ | 2019 |
|
RU2762557C1 |
Изобретение относится к способу получения соединения формулы (I):
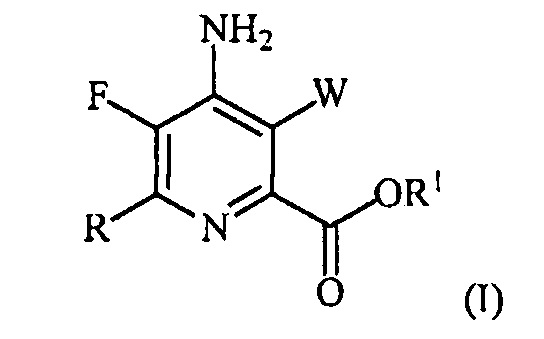
или его соли, где W представляет собой Cl, Br или I; R представляет собой фенил, замещенный 0-5 заместителями, независимо выбранными из галогена и С1-С4алкокси;и R1 представляет собой С1-С12алкил или незамещенный или замещенный С7-С11арилалкил; включающему:
(а) фторирование соединения формулы (II):
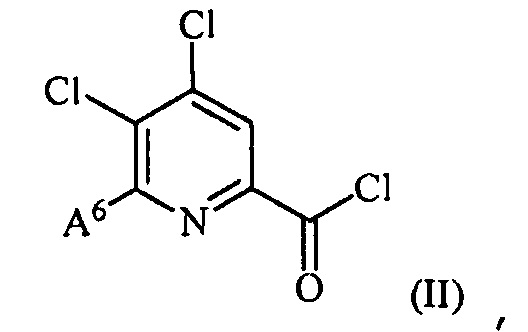
где А6 представляет собой галоген или R;источником фторидного иона с получением соединения формулы (III):
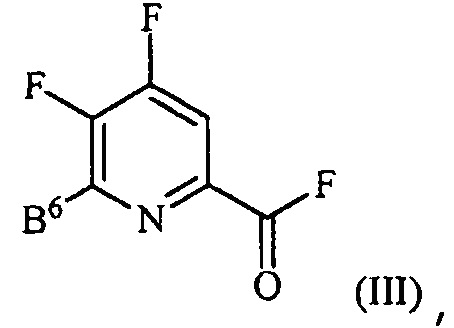
где В6 представляет собой F или R; и преобразование соединения формулы (III) в соединение формулы (IV):
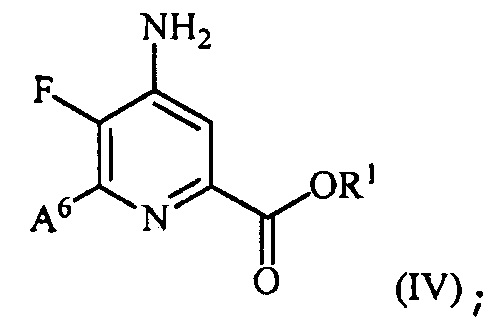
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
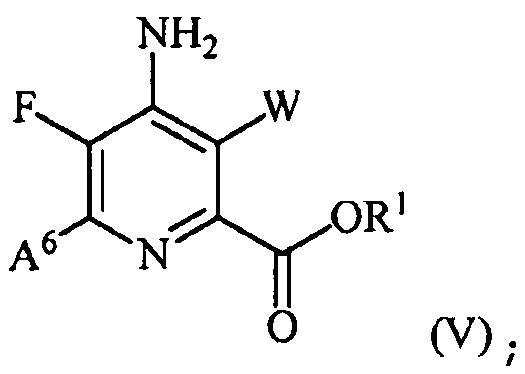 и
и
(с) конденсацию соединения формулы (VI):
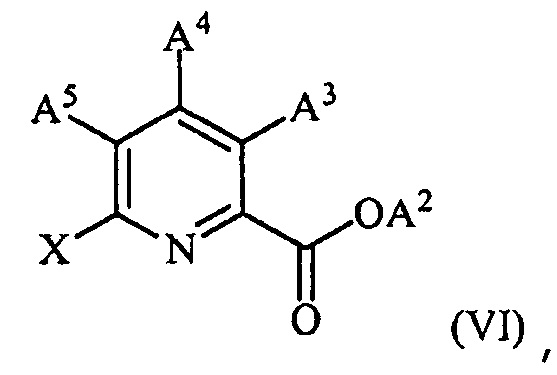
где X представляет собой Cl, Br или I; А2 представляет собой водород или R1; А3 представляет собой водород или W; А4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу; А5 представляет собой F или Cl; с соединением формулы (VII):
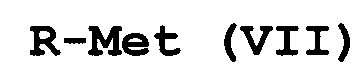 ,
,
где Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь или В(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, С1-С4алкил или взятые вместе образуют этиленовую или пропиленовую группу; в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
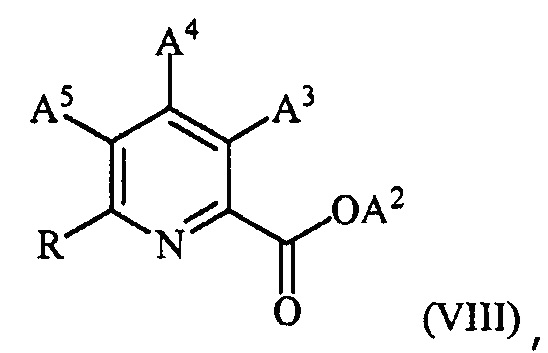
где преобразование (с) можно осуществлять перед, между или после преобразований (а) и (b). 16 з.п. ф-лы, 29 пр.
1. Способ получения соединения формулы (I):
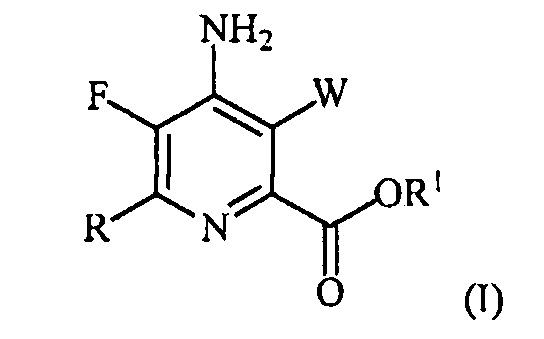
или его соли, где
W представляет собой Cl, Br или I;
R представляет собой фенил, замещенный 0-5 заместителями, независимо выбранными из галогена и С1-С4алкокси; и
R1 представляет собой С1-С12алкил или незамещенный или замещенный С7-С11арилалкил;
включающий:
(а) фторирование соединения формулы (II):
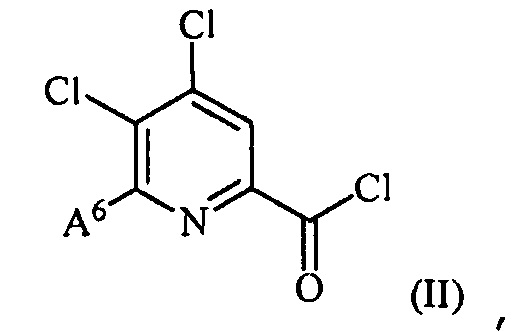
где А6 представляет собой галоген или R;
источником фторидного иона с получением соединения формулы (III):
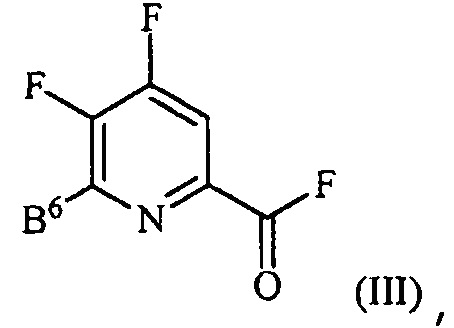
где В6 представляет собой F или R;
и преобразование соединения формулы (III) в соединение формулы (IV):
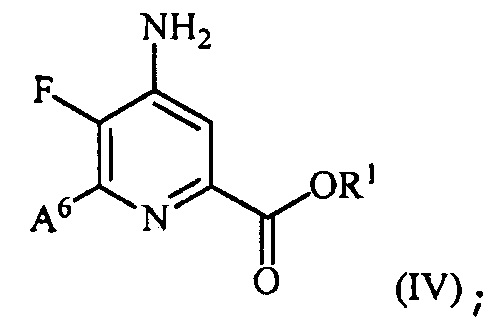
(b) галогенирование соединения формулы (IV) источником галогена с получением соединения формулы (V):
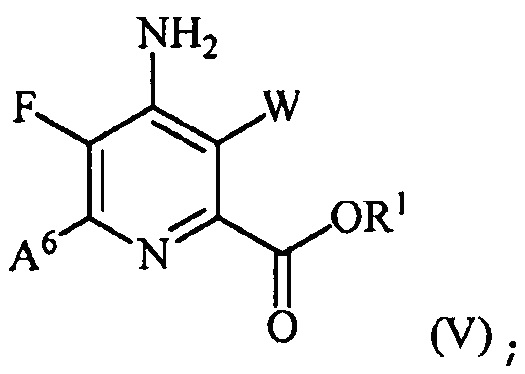 и
и
(с) конденсацию соединения формулы (VI):
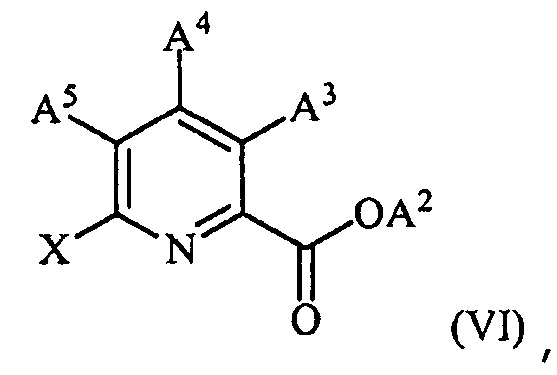
где X представляет собой Cl, Br или I;
А2 представляет собой водород или R1;
А3 представляет собой водород или W;
А4 представляет собой Cl, F, NH2, NHCOCH3 или защищенную аминогруппу;
А5 представляет собой F или Cl;
с соединением формулы (VII):
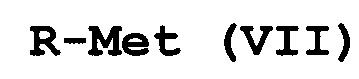
где Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь или В(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, С1-С4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (VIII):
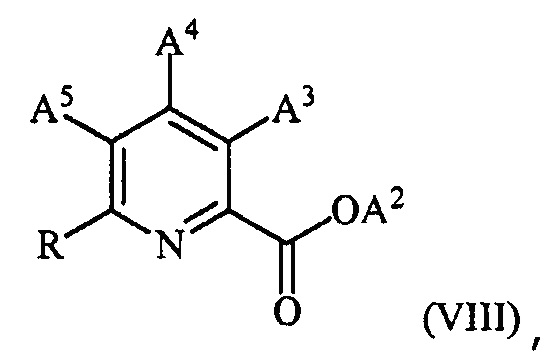
где преобразование (с) можно осуществлять перед, между или после преобразований (а) и (b).
2. Способ по п. 1, где преобразование соединения формулы (III) в соединение формулы (IV) включает:
контакт соединения формулы (III) со спиртом R1OH с получением соединения формулы (IX):
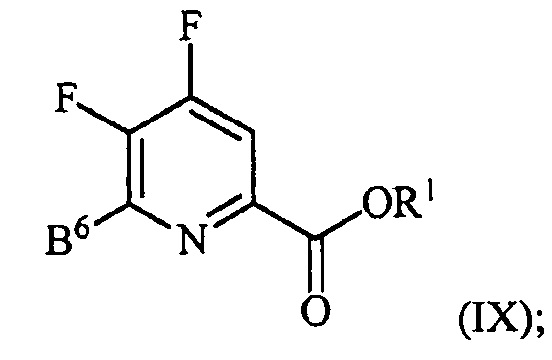 и
и
аминирование соединения формулы (IX) источником аммиака с получением соединения формулы (IV).
3. Способ по п. 1, где преобразование соединения формулы (III) в соединение формулы (IV) включает:
контакт соединения формулы (III) с источником аммиака с получением соединения формулы (X):
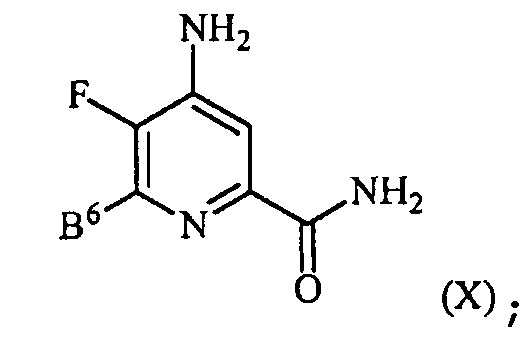 и
и
контакт соединения формулы (X) с кислотой НХ и спиртом R1OH с получением соединения формулы (IV), где X представляет собой I, Br или Cl.
4. Способ по п. 1, включающий:
преобразование (a), которое включает:
(а-1) фторирование соединения формулы (А):
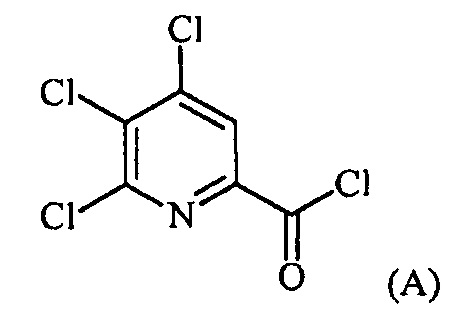
источником фторидного иона с получением соединения формулы (В):
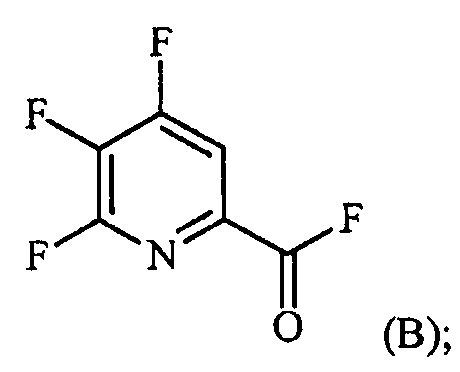
(а-2) контакт соединения формулы (В) со спиртом R1OH с получением соединения формулы (С):
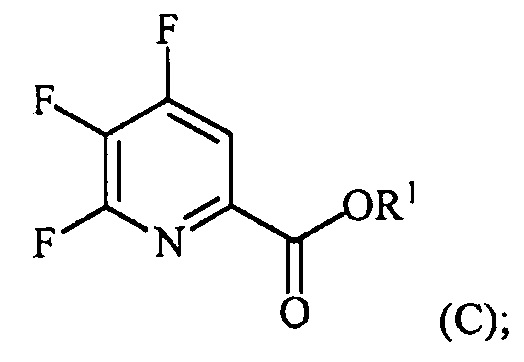
(а-3) аминирование соединения формулы (С) источником аммиака с получением соединения формулы (D):
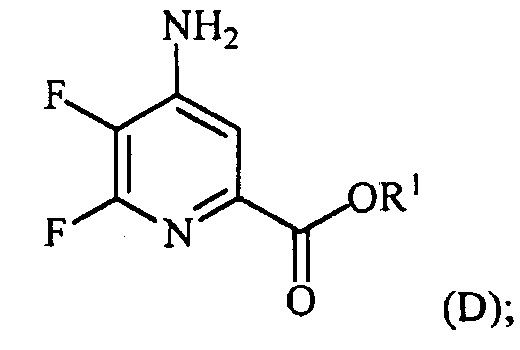 и
и
(а-4) контакт соединения формулы (D) с источником йодида, бромида или хлорида в условиях, подходящих для галогенового обмена, с получением соединения формулы (Е):
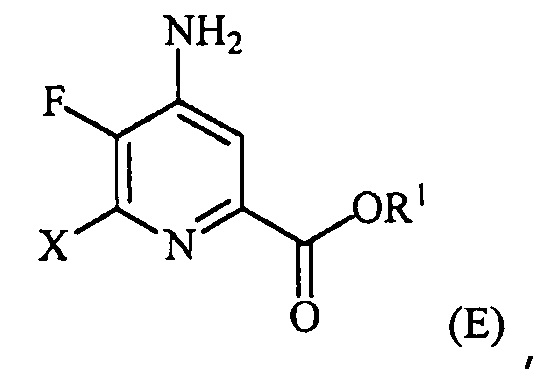
где X представляет собой Cl, Br или I;
преобразование (с), которое включает:
конденсацию соединения формулы (Е) с соединением формулы (F):
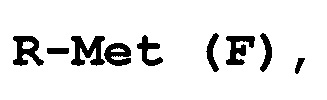
где Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь или В(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, С1-С4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (G):
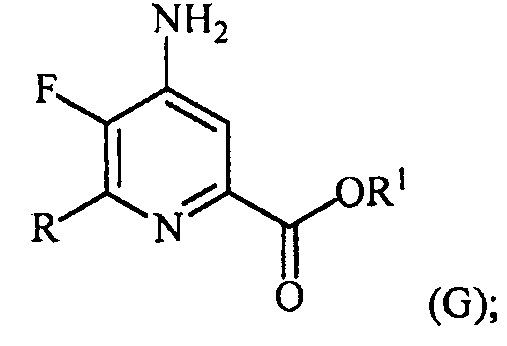 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (G) источником галогена с получением соединения формулы (I).
5. Способ по п. 1, включающий:
преобразование (а), которое включает:
(а-1) фторирование соединения формулы (А):
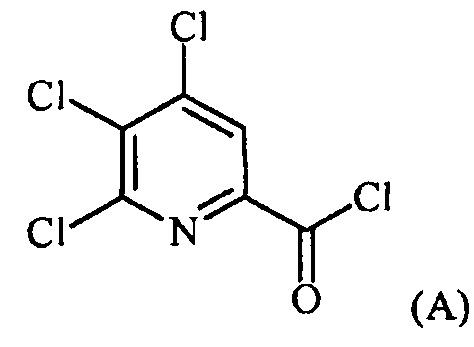
источником фторидного иона с получением соединения формулы (В):
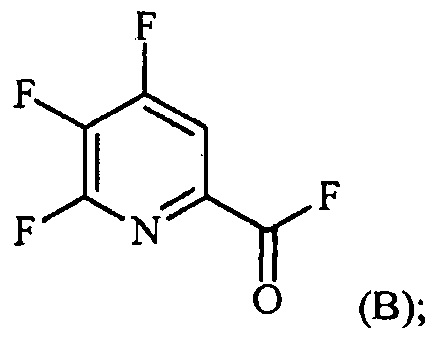
(а-2) контакт соединения формулы (В) со спиртом R1OH с получением соединения формулы (С):
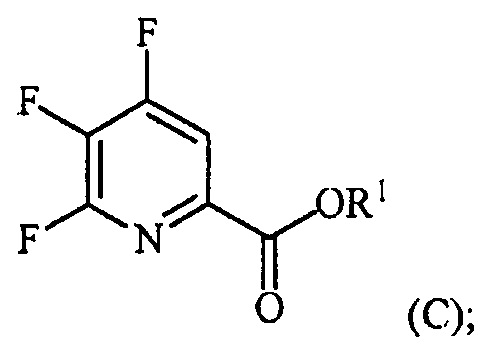
(а-3) аминирование соединения формулы (С) источником аммиака с получением соединения формулы (D):
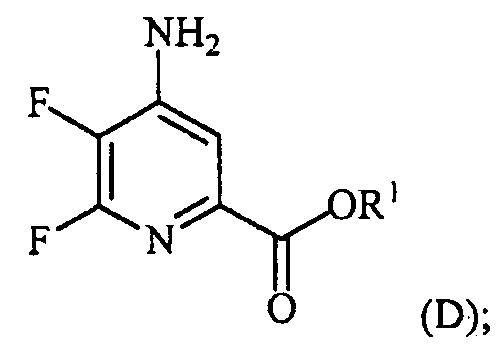 и
и
(а-4) контакт соединения формулы (D) с йодидом, бромидом или хлоридом в условиях, подходящих для галогенового обмена, с получением соединения формулы (Е):
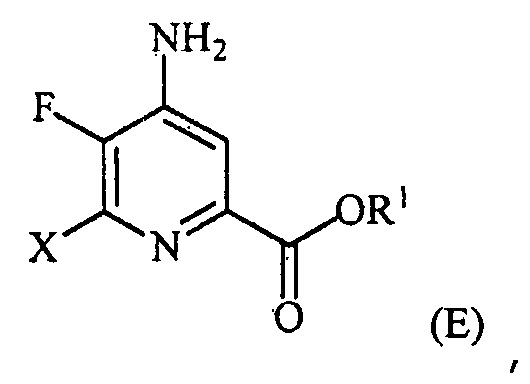
где X представляет собой Cl, Br или I;
преобразование (b), которое включает:
галогенирование соединения формулы (Е) источником галогена с получением соединения формулы (Н):
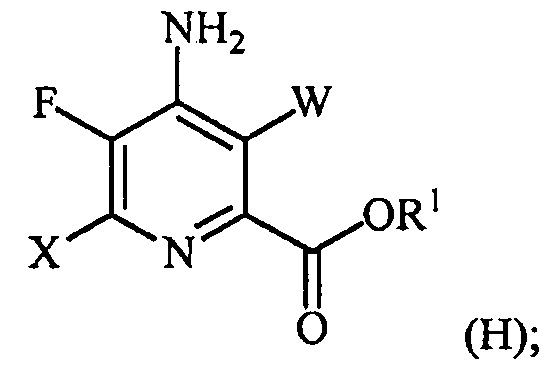 и
и
преобразование (с), которое включает:
конденсацию соединения формулы (Н) с соединением формулы (F):

где Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, С1-С4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (I).
6. Способ по п. 1, включающий:
преобразование (с), которое включает:
конденсацию соединения формулы (J):
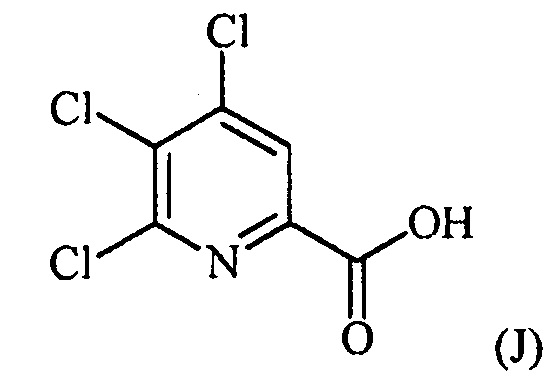
с соединением формулы (F):

где Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, С1-С4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (K):
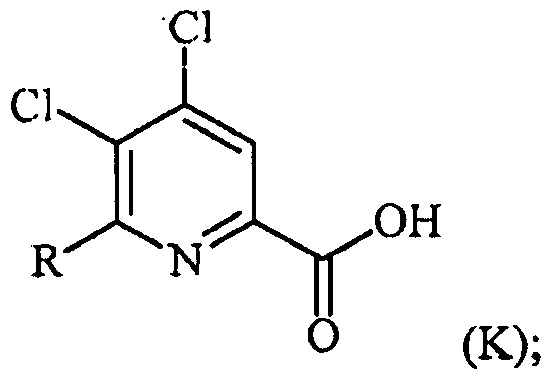
преобразование (а), которое включает:
(а-1) преобразование соединения формулы (K) в соединение формулы (L):
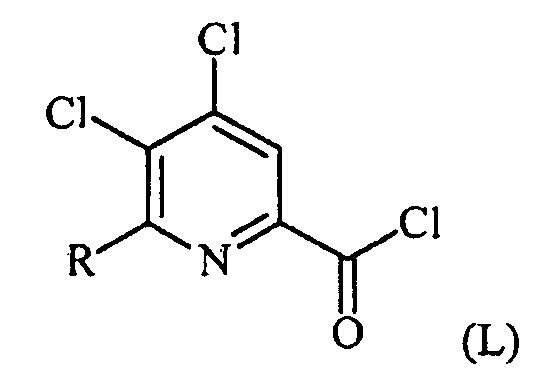
в условиях, подходящих для образования хлорангидрида кислоты;
(а-2) фторирование соединения формулы (L) источником фторидного иона с получением соединения формулы (М):
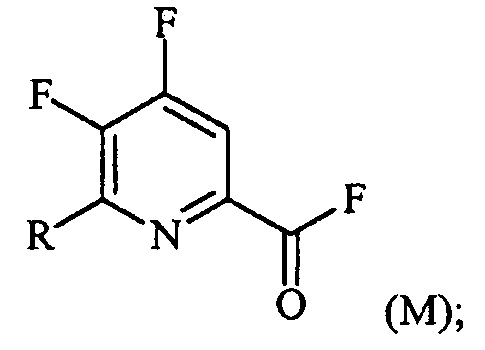
(а-3) контакт соединения формулы (М) со спиртом R1OH с получением соединения формулы (N):
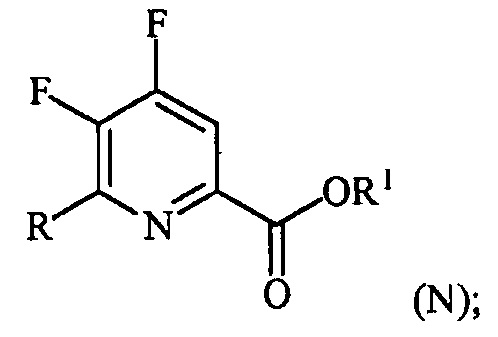 и
и
(а-4) аминирование соединения формулы (N) источником аммиака с получением соединения формулы (О):
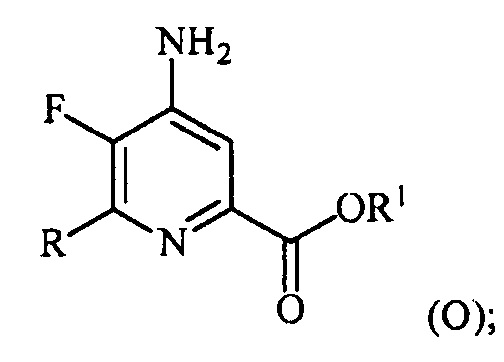 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (О) источником галогена с получением соединения формулы (I).
7. Способ по п. 1, включающий:
преобразование (с), которое включает:
(с-1) конденсацию соединения формулы (Р):
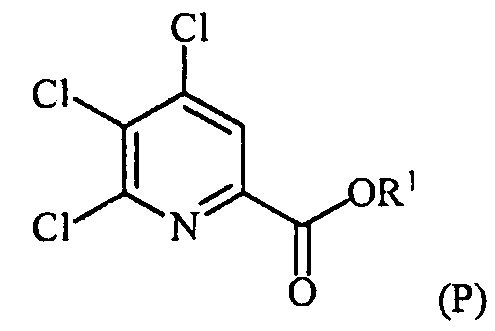
с соединением формулы (F):

где Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь или B(OR2)(OR3), где каждый R2 и R3 независимо представляет собой водород, С1-С4алкил или взятые вместе образуют этиленовую или пропиленовую группу;
в присутствии катализатора на основе переходного металла с получением соединения формулы (Q):
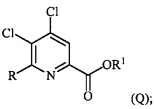 и
и
(с-2) гидролиз соединения формулы (Q) до соединения формулы (K):
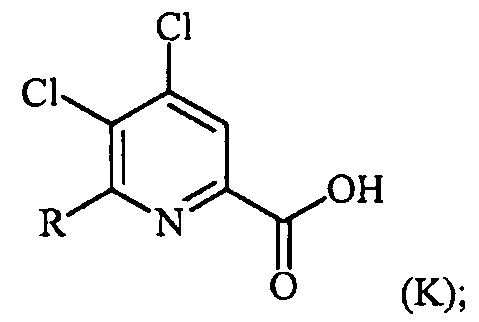
преобразование (а), которое включает:
(а-1) преобразование соединения формулы (K) в соединение формулы (L):
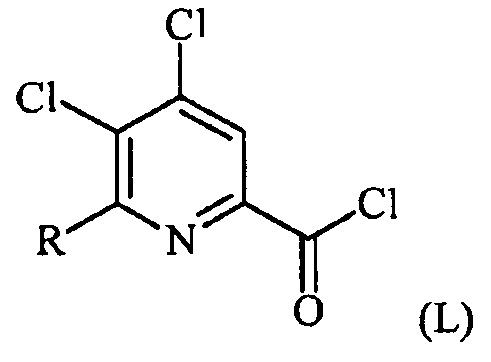
в условиях, подходящих для образования хлорангидрида кислоты;
(а-2) фторирование соединения формулы (L) источником фторидного иона с получением соединения формулы (М):
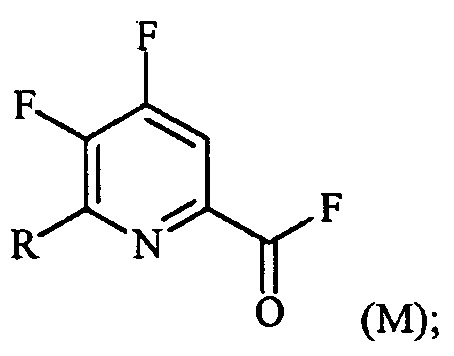
(а-3) контакт соединения формулы (М) с соединением R1OH с получением соединения формулы (N):
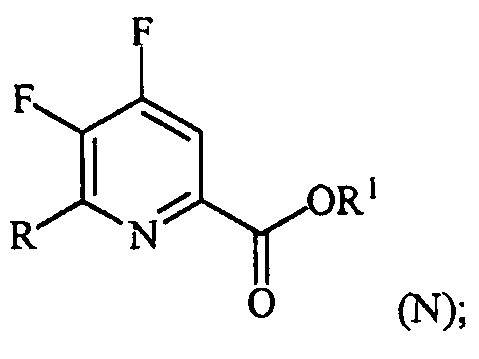 и
и
(а-4) аминирование соединения формулы (N) источником аммиака с получением соединения формулы (О):
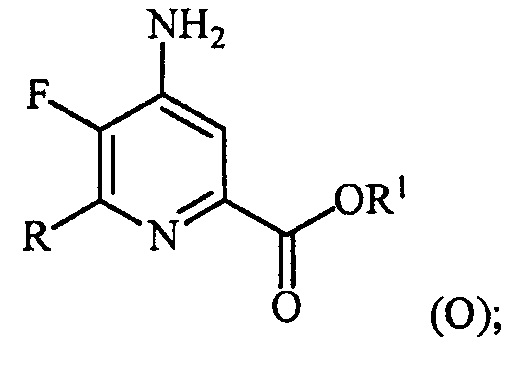 и
и
преобразование (b), которое включает:
галогенирование соединения формулы (О) источником галогена с получением соединения формулы (I).
8. Способ по п. 1, дополнительно включающий защиту NH2 заместителя в положении 4 пиридиновой структуры перед преобразованием (с); и дополнительно включающий стадию удаления защитных групп.
9. Способ по п. 1, где аминосодержащее соединение формул (I), (IV), (V), (VI) или (VIII) очищают: а) протонированием кислотой с получением соли, b) выделением соли с высокой чистотой перекристаллизацией, осаждением или экстракцией, с) нейтрализацией очищенной соли основанием с получением очищенного нейтрального аминосодержащего продукта или промежуточного соединения.
10. Способ по п. 1, где источник фторидного иона представляет собой фторид металла.
11. Способ по п. 10, где фторид металла выбран из фторида натрия, фторида калия и фторида цезия.
12. Способ по п. 11, где фторид металла представляет собой фторид калия.
13. Способ по п. 1, где стадию фторирования в преобразовании (а) осуществляют в присутствии катализатора, выбранного из краун-эфира, галогенида фосфония, полиэфира, фросфазениевой соли и галогенида тетразамещенного аммония.
14. Способ по п. 13, где катализатор представляет собой краун-эфир.
15. Способ по п. 14, где краун-эфир представляет собой 18-краун-6.
16. Способ по п. 1, где стадию фторирования в преобразовании (а) осуществляют в присутствии растворителя, выбранного из алкилнитрила или алкилсульфона.
17. Способ по п. 16, где растворитель представляет собой ацетонитрил или сульфолан.
| US 4847442 A, 11.07.1989 | |||
| Jørn B | |||
| Christensen, et al | |||
| "TFFH as an Excellent Reagent for Acylation of Alcohols, Thiols and Dithiocarbamates" | |||
| SYNTHESIS, vol.15, 2004, p.2485-2492, DOI: 10.1055/s-2004-831250 | |||
| 6-АЛКИЛ ИЛИ АЛКЕНИЛ-4-АМИНОПИКОЛИНАТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2004 |
|
RU2332404C2 |

