Область изобретения
Настоящее изобретение описывает производные аннелированных бициклических ароматических [6+5] гетероциклов, содержащих хотя бы один атом азота в пятичленном цикле. Эти соединения представляют собой селективные ингибиторы серин-треониновой Haspin протеинкиназы, включающие соединения, способные вызывать блокировку митоза (простого деления) в пролиферирующих (делящихся) клетках, в том числе опухолевых клетках, и могут применяться при лечении заболеваний, включающих рак.
Уровень техники
Протеинкиназы (киназы) представляют собой большой класс клеточных ферментов, выполняющих такие ключевые клеточные функции, как регуляция деления клетки, ее пролиферация, метаболиз и, очевидно, играют ключевую роль во многих болезненных состояниях, таких как заболевания, характеризующиеся неконтролируемыми пролиферацией и дифференциацией клеток. Эти заболевания вовлекают разнообразные типы клеток и включают такие недуги, как рак, атеросклероз и рестеноз.
Препараты, воздействующие на киназы, а особенно препараты на основе низкомолекулярных химических соединений, которые ингибируют активность киназ, представляют собой важные лекарства, которые могут значительно улучшить здоровье, увеличить срок жизни или улучшить качество жизни для людей во всем мире, включая пациентов, страдающих от таких изнурительных заболеваний, как рак. Действительно, препарат «иматиниб» (GLEEVEC/GLIVEC; Novartis) представляет собой вещество, ингибирующее киназы, и применяется для успешного лечения определенных типов рака, включая, в частности, лейкемию (хроническая миелоидная лейкемия, ХМЛ) и некоторые виды рака желудочно-кишечного тракта (стромальные опухоли ЖКТ). Отмечается высокая эффективность этого препарата по сравнению с применяемым ранее стандартным лечением ХМЛ, а также достоверно лучшая его переносимость, что проявляется в менее выраженных побочных эффектах.
В тоже время лекарства, которые прерывают процесс простого деления (митоза) раковой клетки, представляют собой эффективные противораковые препараты. Хорошо известны примеры таких лекарственных препаратов, воздействующих на аппарат веретена деления, например, таксаны винкаалкалоиды, эпотилоны. Несмотря на то, упомянутые выше препараты эффективны для терапии ряда раковых заболеваний, они не лишены ряда серьезных недостатков. В частности, у данного класса препаратов отмечаются серьезные побочные эффекты, такие как нейтропения (снижение в крови количества белых кровяных телец), утомляемость и инфекционные процессы. Они также вызывают чрезмерное раздражение кожи при введении, что может затруднять их применение. Для решения указанной проблемы были предприняты поиски соединений с новым механизмом действия, способных блокировать процесс митоза и запускать процесс апоптоза (программируемой гибели) раковых клеток, но не обладающих токсическим действием на покоящиеся клетки. Относительно недавно были открыты новые классы соединений, действующие на уникальные молекулярные мишени в процессе митоза, в том числе и ряд серин-треониновых протеинкиназ. К протеинкиназам, играющим ключевую роль в процессе митоза относятся, например, семейство киназ Aurora (включает в себя три представителя Aurora A, Aurora В и Aurora С), большое семейство киназ CDK, СНК.1,2 киназы, Nek2 киназа, Pik киназы [Susanne М.A.Lens, Emile E. Voest and Rene H.Medema, Nature Reviews Cancer, volume 10, 2010, p.825], а также Haspin киназа.
Haspin киназа - это серин-треониновая протеинкиназа, которая фосфорилирует остаток треонина Thr-3 на аминосодержащем концевом фрагменте гистона НЗ в процессе митоза [Dai J, Sultan S, Taylor SS, Higgins JMG, Genes Dev 2005; 19:472-488; Dai J, Higgins JMG, Cell Cycle 2005;4:665-668; Dai J, Sullivan BA, Higgins JMG; Dev Cell 2006,11:741-750]. Множество факторов свидетельствует о том, что специфические (селективные) ингибиторы Haspin киназы могут представлять собой интересный и перспективный класс соединений для изучения и лечения онкологических заболеваний. Первый, наиболее важный фактор: Haspin киназа играет важную роль в регуляции митоза (простое клеточное деление). Как показали эксперименты с Haspin RNAi in vitro, блокирование функции Haspin киназы нарушает выравнивание хромосом на ранних стадиях митоза. Это приводит к невозможности нормального завершения митоза, что свидетельствует о том, что ингибиторы Haspin киназы могут быть новыми антимитотическими агентами, способными предотвращать избыточную пролиферацию раковых клеток.
Второе, Haspin киназа экспрессируется только в пролиферирующих, но не в покоящихся клетках [Higgins, Gene 2001; 267:55-69]. Данный факт свидетельствует о том, что селективные ингибиторы Haspin киназы могут выступить в качестве менее токсичной альтернативы традиционным антимитотическим препаратам. И третье, основываясь на уникальной первичной структуре Haspin киназы, можно рассчитывать на то, что создание селективных ингибиторов данной мишени является вполне решаемой задачей [Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW, Higgins JM, Knapp S. Proc Nati Acad Sci USA. (2009) 106(48): 20198-20203].
В последнее время в научной литературе появилось несколько публикаций, сообщающих об идентификации первых ингибиторов Haspin киназы [Debasis Patnaik, J Biomol Screen 2008; 13; 1025; Gregory D.Cuny, Bioorganic & Medicinal Chemistry Letters, 20, 491], а также о методе связывания некоторых малых молекул с активным (АТФ-связывающим) сайтом Haspin киназы на основании данных рентгено структурного анализа. Однако анализ современной научной и патентной литературы показывает, что к настоящему времени нет данных о действительно активных и селективных ингибиторах Haspin киназы.
В соответствии с изложенным выше, несмотря на достигнутый определенный прогресс, продолжается поиск соединений с низким молекулярным весом, являющихся киназными ингибиторами, особенно соединений, ингибирующих Haspin киназу, которые могут применяться в лечении разнообразных заболеваний, включая рак, и другие пролиферативные заболевания или расстройства, включая рестеноз, ангиогенез, диабетическую ретинопатию, псориаз, хирургические спайки, макулярную дистрофию и атеросклероз, или другие заболевания и расстройства, упомянутые выше. Таким образом, существует насущная потребность в создании композиций, фармацевтических и/или медикаментозных, обладающих киназной ингибиторной активностью, в частности, ингибиторной активностью по отношению к Haspin киназе и антипролиферативной активностью по отношению к таким клеткам, как клетки опухоли. Такие композиции, фармацевтические и/или медикаментозные, могут обладать не только указанной активностью, но и вызывать допустимые, приемлемые или ограниченные побочные эффекты по сравнению с другими антипролиферативными веществами. Более того, диапазон опухолей или других заболеваний, восприимчивых к лечению такими соединениями, может быть широким. Активные ингредиенты в таких композициях, лекарствах и медикаментах могут применяться по вышеупомянутым показаниям в качестве монотерапии и/или в комбинированной терапии вместе с другими терапевтическими агентами, с облучением, с оперативными/хирургическими процедурами, с тепловым лечением или с любым другим лечением, известным для лечения упомянутых показаний.
Известно, что некоторые классы бициклических аннелированных ароматических [6+5] гетероциклических соединений, например, определенным образом замещенные имидазопиридины, обладают полезными фармакологическими свойствами, такими как антипролиферативная или противовоспалительная активность. Однако, на данный момент, приведенные в научной и патентной литературе соединения структурно отличаются от соединений данного изобретения.
В статье компании UCB [Bioorganic & Medicinal Chemistry Letters 18, 3291] описываются замещенные имидазо[1,2-а]пиридины, которые, как было показано, ингибируют IRAK 1,4 киназы и подавляют образование известного медиатора воспаления цитокина TNF-α in vitro.
В недавней статье [Yoshiyuki Sato, Bioorganic & Medicinal Chemistry Letters, Volume 19, 4673] был описан новый класс селективных ингибиторов Plk1 киназы на основе замещенных имидазопиридинов, проявляющих противоопухолевую активность in vivo на ксенографтных моделях.
Также необходимо упомянуть соединение SGI-1776, которое является неселективным ингибитором PIM-1,2,3 киназ, разработанное американской компанией SuperGene для лечения гематологических пролиферативных заболеваний (в частности, устойчивой к терапии острой миелоидной лейкемии) и ряда солидных опухолей. Соединение SGI-1776 представляет собой производное имидазо[1,2-b]пиридазина. Следует отметить, что наряду с ингибированием PIM-киназ, SGI-1776 также проявляет ингибирующую активность по отношению к Haspin киназе.
Краткое описание изобретения
Нами открыт класс специфически замещенных аннелированных бициклических [6+5] гетероароматических соединений, которые являются селективными ингибиторами серин-треониновой протеинкиназы Haspin, а также способны блокировать процесс митоза раковых клеток, вызывая тем самым, их гибель. Предложенные соединения дают возможность разрабатывать новые и эффективные методы лечения заболеваний, которые связанны с неконтролируемой пролиферацией.
Соединения, согласно настоящему изобретению, представляют собой определенным образом замещенные аннелированные бициклические [6+5] гетероароматические соединения, как детально описывается ниже, и могут применяться для дальнейших доклинических или клинических исследований и разработок с целью лечения различных заболеваний и расстройств, включая онкологические, пролиферативные, дегенеративные, воспалительные и другие заболевания и расстройства.
В одном аспекте, настоящее изобретение описывает определенным образом замещенные аннелированные бициклические [6+5] гетероароматические соединения, имеющие структуру, которая описывается общей формулой (I), представленной ниже, или ее таутомерной или стереоизомерной формами, которые могут применяться в качестве ингибиторов Haspin киназы и, следовательно, могут применяться для лечения заболеваний и расстройств, упоминаемых в данном документе, включая пролиферативные заболевания и расстройства, такие как рак, воспалительные заболевания или расстройства.
В другом аспекте, настоящее изобретение относится к фармацевтическим композициям, которые включают фармацевтически приемлемые разбавители, наполнители или носители, и описываемые ингибиторы Haspin киназы в количестве, составляющем их терапевтически эффективную дозу, например, дозу, которая, как ожидается, облегчит симптомы заболевания или расстройства, упомянутого в заявке, включая пролиферативные заболевания и расстройства, такие как рак и воспалительные заболевания или расстройства.
В другом аспекте, настоящее изобретение относится к фармацевтической упаковке, которая включает данную фармацевтическую композицию и инструкции, в которых отмечается, что данная фармацевтическая композиция может применяться для лечения пациента, страдающего заболеванием или расстройством, упомянутым в заявке, включая пролиферативные заболевания и расстройства, такие как рак и воспалительные заболевания или расстройства.
С другой стороны, изобретение относится к способам, с помощью которых терапевтически эффективное количество соединения или описываемая в заявке фармацевтическая композиция применяется субъектом или приводится в контакт с субъектом, клеткой, тканью, органом или организмом. Эти способы включают профилактику и/или лечение заболевания или расстройства, упомянутых в заявке, включая пролиферативные заболевания и расстройства, такие как рак и воспалительные заболевания или расстройства, но не ограничиваются ими.
С другой стороны, настоящее изобретение относится к применению соединений настоящего изобретения для приготовления лекарственных средств, применяемых для лечения заболевания или расстройства, упомянутого в заявке, включая пролиферативные заболевания и расстройства, такие как рак.
С другой стороны, изобретение относится к применению соединений настоящего изобретения для приготовления лекарственных средств, применяемых для лечения воспалительного заболевания или расстройства.
С другой стороны, изобретение относится к методам синтеза как конечных соединений по настоящему изобретению, так и интермедиатов для получения упомянутых соединений.
Таким образом, настоящее изобретение предлагает соединения, имеющие структуру, представленную общей формулой (I)
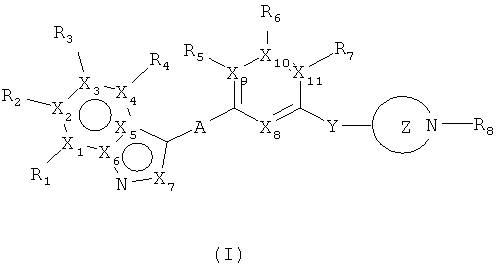
или любые их таутомерные формы, или их фармацевтически приемлемые соли, где
каждый из Х1, Х2, Х3, Х4, Х5, Х6 выбран из N или С;
Х7 выбран из N или СН;
каждый из Х8, Х9, X10 и Х11 независимо выбран из N или СН при условии, что фрагмент может одновременно содержать один или два атома азота;
R1, R2, R3 и R4 выбраны из Н, С1-12алкила, 6-14-членного арила, ОС1-12алкила, CF3, галогена, СООН, СООС1-12алкил, CONH2, CON(С1-12алкил)2, CN, N(C1-12алкил)2, 4-14-членного гетероарила, содержащего, по крайней мере один гетероатом, выбранный из азота и кислорода;
R5, R6, R7 выбраны из С1-12алкила, галогена, CN при условии, что Х9, Х10 или Х11 в этом случае соответственно равен С;
"А" может представлять собой простую связь или мостиковый этиновый фрагмент;
Y может представлять собой простую связь, или независимо выбран из метиленового или этиленового мостиковых фрагментов;
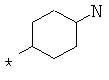
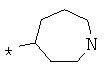
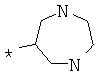
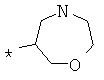
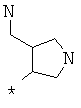
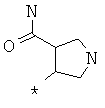
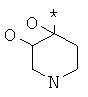
Фрагмент Z независимо выбран из незамещенного или замещенного по атому азота гетероциклоалкила, или является незамещенным или замещенным циклоалкилом при условии, что N (азот) равно С (углерод):
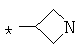

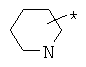
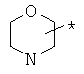
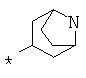
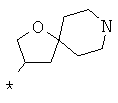

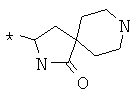

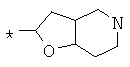
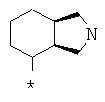
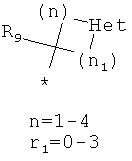
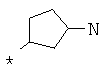
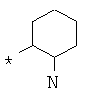
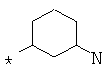
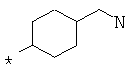
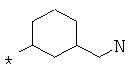

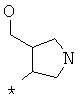


где R9 выбран из ОН, F, ОС1-12алкила, NH2, NH(С1-12алкил), N(С1-12алкил)2, СН2ОН, CON(R15R16), где R15, R16 могут независимо представлять собой Н, C1-12 алкил, C3-12циклоалкил, 6-14-членный арил, 4-14-членный гетероарил, содержащий, по крайней мере один гетероатом, выбранный из азота и кислорода, или
R9 может представлять собой следующие гетероциклические заместители:


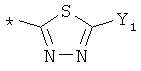

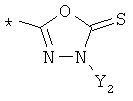
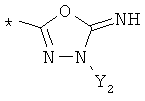
где Y1, Y2=Н или С1-С5алкил.
R8 выбран из Н, С1-12алкила,
циклоалкила и гетероциклоалкила общего строения:

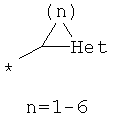 ,
,
С1-12алкилкарбонила, С3-12циклоалкалкарбонила,
гетероциклоалкилкарбонила общего строения:
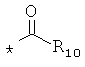
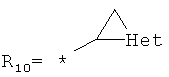

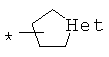

арилкарбонилов общего строения:

гетероарилкарбонилов общего строения:

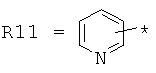
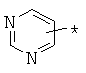
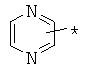
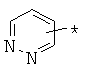
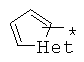

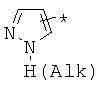
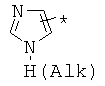
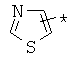
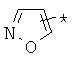
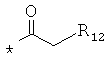
производных алкил-, циклоалкил-, гетероциклоалкил-, арил-, или гетероарилуксусной кислоты общего строения:
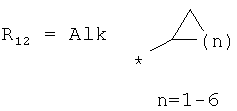
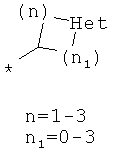

производных метилциклоалкилов, метилгетероциклоалкилов, метиларилов (бензилов), метилгетероарилов общего строения:
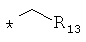
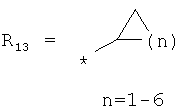
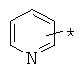

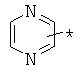

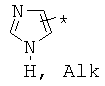 ,
,
производных алкилсульфонилов, арилсульфонилов и диалкиламиносульфонилов общего строения:
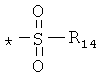
 , ,
, ,
где во всех случаях где Het, Het1 представляют собой гетероатомы и могут быть независимо выбраны из N или О.
Другие аспекты и преимущества изобретения будут очевидны из следующего детального описания и из формулы изобретения.
Подробное описание изобретения
Общие термины
Термин «Н» обозначает водород.
Термин «алкил» или «Alk» относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, содержащие от 1 до 12 атомов углерода. В определенных вариантах осуществления алкильные заместители могут представлять собой низшие алкильные заместители. Термин «низший алкил» относится к алкильным группам, содержащим от 1 до 6 атомов углерода. Примеры низших алкилов включают метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил, но не ограничиваются ими.
При упоминании в заявке термин «циклоалкил» следует понимать как углеводородный цикл, являющийся частью любой стабильной моноциклической или полициклической системы, в которой этот цикл содержит от 3 до 12 атомов углерода, но не содержит гетероатотомов. Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, адамантил, циклооктил, бициклоалкилы, включая бициклооктаны, такие как [2.2.2]бициклооктан, бициклононаны, такие как [4.3.0]бициклононан и бициклодеканы, такие как [4.4.0]бициклодекан (декалин) или спиросоединения, но не ограничиваются ими.
Термин «гетероциклоалкил» обозначает цикл, который является стабильной моноциклической или полициклической системы, или является частью полициклической системы, в которой этот цикл содержит от 3 до 8 атомов, и в которой этот цикл состоит из атомов углерода и, по крайней мере, одного гетероатома, независимо выбранного из группы, состоящей из N, О и S, при этом гетероциклоалкил обозначает систему, где этот цикл полностью насыщен. Для ясности, если заместитель представляет собой полициклическую систему, в которой один цикл содержит, по крайней мере, один гетероатом, как описано в заявке, то такой заместитель будет упоминаться как «гетероциклоалкил». Гетероциклоалкильные группы могут быть связаны с другими группами через циклический атом углерода («гетероциклоалкил, связанный через С-атом»), или через циклический атом азота («гетероциклоалкил, связанный через N-атом»). В определенных вариантах осуществления азот в гетероцикле может быть кватернизован.
Примеры гетероциклоалкилов включают пирролидинил, тетрагидрофуранил, морфолинил, тиоморфолинил, пиперазинил, пиперидинил и декагидрохинолинил (присоединенный через пиперидинильный фрагмент), но не ограничиваются ими. Также охватываются конденсированные циклические системы и спиро-соединения, содержащие, например, любой из вышеназванных гетероциклов, в тех случаях, когда они присоединены через цикл, содержащий гетероатом.
Термин «арил» обозначает цикл или циклическую систему, являющуюся частью любой стабильной моноциклической или полициклической системы, в которой упомянутый цикл или циклическая система содержит от 6 до около 14 атомов углерода, но не содержит ни одного гетероатома, и в которой цикл или циклическая система представляет собой ароматический фрагмент, что определяется правилом «(4n+2)» π-электронов». Для ясности, если заместитель является полициклической системой, в которой в указанную полициклическую систему включается один цикл или циклическая система, представляющая собой ароматический фрагмент, как определено в заявке, то заместитель будет определяться как «арил», если замещение происходит через упомянутый ароматический фрагмент. Такие системы включают фенил и циклические конденсированные системы бензола, например, нафталиновые, антраценовые или фенантреновые циклические системы, или, например, бензольное ядро, конденсированное с одним или более циклоалкильным фрагментом с образованием, например, инданила, флуоренила или тетрагидронафтила, или конденсированное с одним или более гетероциклоалкильным фрагментом, как, например, в индолиниле, однако, при условии, что во всех этих случаях эта конденсиованная система присоединена в качестве заместителя через ароматический фрагмент, но не ограничиваются ими.
При упоминании в заявке термин «гетероарил» обозначает цикл или циклическую систему, являющуюся частью любой стабильной моно- или полициклической системы, где этот цикл или циклическая система содержит от 4 до примерно 14 атомов, и где этот цикл или циклическая система представляет собой ароматический фрагмент, что определяется правилом «(4n+2)» π-электронов», и состоит из атомов углерода и одного или более гетероатомов, которые выбраны из азота или кислорода. Для ясности, если заместитель представляет собой полициклическую систему, в которую включен цикл или циклическая система, представляющая собой ароматический фрагмент, содержащий гетероатом, как определено в заявке, то такой заместитель будет определяться как «гетероарил», если замещение происходит через ароматический фрагмент, содержащий гетероатом. В определенных вариантах осуществления азот в гетероцикле может быть кватернизован или окислен в N-оксид. Примеры гетероарилов включают пирролил, пиразолил, имидазолил, индолил, бензимидазолил, фуранил, бензофуранил, пиридинил, пиримидинил, пиразинил, триазинил, хинолинил, хиназолинил, но не ограничиваются ими.
В термин гетероарил также включаются конденсированные гетероарилы, содержащие, например, вышеупомянутые гетероарилы, конденсированные с циклоалкилами или гетероциклоалкилами (при условии, что во всех случаях конденсированная система присоединяется в качестве заместителя через ароматический фрагмент, содержащий, по крайней мере, один гетероатом).
Фрагмент -С(O)- обозначает карбонильную группу. Примерами карбонилсодержащих фрагментов, не ограничивая объем данного изобретения, являются алкилкарбонилы, циклоалкилкарбонилы, арилкарбонилы и т.д.
Термины «алкилсульфонил» и «арилсульфонил» обозначают соответственно «алкил» или «арил», определение которых приведено выше, присоединенные к соответствующему фрагменту молекулы через сульфонильную группу -SO2. Примерами алкилсульфонилов, не ограничивая объем данного изобретения, являются метилсульфонил, этилсульфонил и т.д. Примерами арилсульфонилов, не ограничивая объем данного изобретения, являются фенилсульфонил, п-метилфенилсульфонил и т.д.
Во всем тексте заявки алкильная, циклоалкильная, гетероциклоалкильная, арильная и гетероарильная группы, а также любые другие подструктуры, содержащие в своем составе, по крайней мере, один атом водорода, могут замещаться одним или более заместителями.
Заместители могут быть независимо выбраны из следующего списка:
- «гетероатомы»: О, N, S, Р, (а также СО, SO2, SO включенные в цикл гетероциклоалкильной структуры),
- «галогены»: F, Cl, Br, I,
- «функциональные группы»: СНО, CN, СООН, NO2, NAlkz, SO2Alk, SO2арил SO2NH2, SO2NHAlk, SO2NAlk2, CONH2, CONHAlk, CONAlk2, P(O)(OAlk)2, PO(OH)2,
- «арил»,
- «алкил»
- «гетероарил»
- «гетероциклоалкил»
Пример: любой атом водорода может быть замещен на гетероциклоалкильный заместитель (N-морфолинил), арил или карбоксильную группу, если химическая структура образованных при таком замещении соединений не противоречит общим принципам теории строения органических соединений
Термин «замещенный» указывает на то, что один или более атомов водорода на атоме или группе, упоминаемой как «замещенный», заменен на любую из перечисленных групп, при условии, что упоминаемый атом обладает нормальной валентностью, или что валентность замещаемого соответствующего атома группы не является избыточной, и что замещение приводит к стабильному соединению.
Термин «стереоизомер» и «таутомер», при упоминании в заявке, включает все возможные стереоизомерные и таутомерные формы соединений настоящего изобретения. Там, где соединения настоящего изобретения содержат один или более хиральный центр, включаются все возможные энантиомерные и диастереомерные формы, если не указано иначе.
Настоящее изобретение подразумевает включенными все изотопы атомов, содержащихся в соединениях изобретения. Изотопы представляют собой атомы, имеющие одинаковый атомный номер, но различные массовые числа. В качестве общего примера и не ограничиваясь этим, изотопы водорода включают тритий и дейтерий.
Изотопы углерода включают 12С, 13С и 14С.
Термин «метаболит», используемый в заявке, относится к любому веществу, образующемуся в результате метаболизма или метаболического процесса in vitro и in vivo. Метаболизм, при упоминании в заявке, относится к различным физическим/химическим/биохимическим/фармакологическим реакциям, участвующим в трансформации молекул или химических соединений, которые происходят в клетке, ткани, системе, теле, животном, индивиде, пациенте или человеке.
Термин «IC50», при упоминании в заявке, относится к концентрациям, при которых измеряемая активность, фенотип или отклик, например, рост или пролиферация клеток, таких как опухолевые клетки, ингибируется на 50%. Значения 1050 могут оцениваться из соответствующих кривых зависимости ответа от дозы, например, на глаз или с использованием подходящих программ для обработки кривых или статистических программ. Более точно значения IC50 могут быть определены с помощью нелинейного регрессионного анализа.
При упоминании в заявке термины «пролиферативное заболевание» или «пролиферативное расстройство» понимают заболевания или расстройства, на которые оказывают влияние процессы клеточного роста, дифференциации или пролиферации.
При упоминании в заявке «онкогенные заболевания и расстройства» понимают заболевания и расстройства, которые характеризуются нарушенной регуляцией клеточного роста, пролиферации, дифференциации, сращения или миграции, которая может приводит к возникновению опухолей или к возникновению склонности к опухоли. При упоминании в заявке «опухоль» понимают доброкачественное или злокачественное разрастание ткани. Примеры заболеваний клеточного роста или пролиферации включают опухоли, рак, аутоиммунные заболевания, вирусные заболевания, грибковые заболевания, нейродегенеративные заболевания и кардиоваскулярные заболевания, но не ограничиваются ими.
При упоминании в заявке терминов «противораковый препарат» или «антипролиферативный препарат» их относят к соединениям с противораковьми или антипролиферативными свойствами, соответственно. Такие соединения включают алтерамин, бусульфан, хлорамбуцил, циклофосфамид, ифосфамид, мехлорэтамин, мелфалан, тиотепа, кладрибин, флуороурацил, флоксуридин, гемцитабин, тиогуанин, пентостатин, метотрексан, 6-меркаптопурин, цитарабин, кармустин, ломустин, стрептозотоцин, карбоплатин, цисплатин, оксалиплатин, пикоплатин, LA-12, ипроплатин, тетраплатин, лобаплатин, JM216, JM335, сатраплатин, флударабин, аминоглутетимид, флутамид, госерелин, леупролид, мегестрола ацетат, ципротерона ацетат, тамоксифен, анастрозол, бикалутамид, дексаметазон, диэтилстибестрол, преднизон, блеомицин, дактиномицин, даунорубицин, доксирубицин, идарубицин, митоксантрон, митомицин-с, пликамицин, паклитаксел, доцетаксел, топотекан, иринотекан, 9-аминокамптотекан, 9-нитрокамптотекан, GS-211, JM 118, этопозид, тенипозид, винбластин, винкристин, винорелбин, прокарбазин, аспарагиназа, пэгаспарагаза, октреотид, эстрамустин и гидроксимочевина, SAHA, эрлотиниб, сунитиниб, лапатиниб, сорафениб, вемурафениб, азацитадин, децитабин, но не ограничиваются ими. Указанные термины также включают высокомолекулярные препараты, такие как антитела, например, 1D09C3 и другие анти-HLA-DR антитела, как описано в патентах WO 01/87337 и WO 01/97338, ритуксан. как описано в патентах США 5,736,137, 5,776,456, 5,843,437, 4D5, Mab225, C225, даклизумаб (Zenapax), Antegren, CDP 870, CMB-401, MDX-33, MDX-220, MDX-477, CEA-CIDE, AHM, витаксин, 3622W94, Therex, 5G1.1, IDEC-131, HU-901, Mylotarg, Zarnyl (SMART M195), MDX-210, Humicade, EymphoCIDE, ABX-EGF, 17-1 А, трастузумаб (Herceptin®, rhuMAb), Epratuzumab, Cetuximab (Erbitux®), Pertuzumab (Omnitarg®, 2C4), R3, CDP860, Bevacizumab (Avastin®), tositumomab (Bexxar®), Ibritumomab tiuxetan (Zevalin®), M195, 1D10, Hu1D10 (Remitogen®, apolizumab), Danton/DN1924, «HD» антитело, такое как HD4 или HD8, САМРАТН-1 и САМРАТН-1Н или другие их варианты, фрагменты, конъюгаты, производные и другие модификации, или другие эквивалентные композиции с улучшенными или оптимизированными свойствами, и белки или пептиды, например, как те, которые описаны в Trends in Biotechnology (2003), 21(12), р.556-562, но не ограничиваются ими.
При упоминании в заявке, термины «воспалительные болезни» или «воспалительные заболевания» включают болезни или заболевания, которые вызваны или сопровождаются воспалительными процессами. Они включают такие болезни или заболевания, как артриты, включая, ревматоидные артриты, спондилоартропатии, подагрические артриты, остерартриры, системную красную волчанку и хронические полиартриты у детей, остеоартриты, подагрические артриты и другие артритные заболевания, но не ограничиваются ими; легочные заболевания и воспаления легких, включая респираторный дистресс-синдром взрослых, легочный саркодиоз, астму, силикоз и хронические воспалительные заболевания легких; вирусные и бактериальные инфекции, включая сепсис, септический шок, грамм-отрицательный сепсис, малярию, менингиты, кахексию, вызванную инфекцией или злокачественным заболеванием, кахексию, вызванную синдромом приобретенного иммунодефицита (СПИД), СПИД, СПИД-ассоциированный комплекс, пневмонию и вирусный герпес; болезни резорбции кости, такие как остеоартроз, эндотоксивный бактериально-токсический шок, синдром токсического шока, реперфузионное повреждение, аутоиммунное заболевание, включающее реакцию «трансплантант против хозяина» и отторжение аллотрансплантанта, кардиоваскулярные заболевания, включая атеросклероз, тромбоз, застойную сердечную недостаточность и сердечное реперфузионное повреждение, почечное реперфузионное повреждение, заболевание печени и нефриты, и миалгии вследствии инфекции; болезнь Альцгеймера, грипп, множественный склероз, рак, диабет, системная красная волчанка (SLE), заболевания, связанные с кожей, такие как псориаз, экзема, чувство жжения, дерматиты, келоидные образования и образование рубцовой ткани; гастроинтестинальные заболевания, такие как воспалительное заболевание кишечника, болезнь Крона, гастриты, синдром раздраженного кишечника и язвенные колиты; глазные болезни, такие как ретиниты, ретинопатии, увеиты, светобоязнь и острые травмы тканей глаза; ангиогенез, включая неоплазию; метастазирование; офтальмологические заболевания, такие как отторжение ткани роговицы, офтальмологическая неоваскуляризация, ретинальная неоваскуляризация, включая неоваскуляризацию после травмы или инфекции, диабетическую ретинопатию, ретролентальную фиброплазию и неоваскулярную глаукому; язвенные заболевания, такие как язва желудка; патологические, но не злокачественные заболевания, такие как гемангиомы, включая инфантильные гемангиомы, ангиофибромы носоглотки и аваскулярный некроз костей; диабетическая нефропатия и кардиомиопатия; и заболевания женской репродуктивной системы, такие как эндометриоз.
При упоминании в заявке термин «фармацевтически приемлемые соли» относится к производным описываемых соединений, в которых исходное соединение модифицировано получением их кислых или основных солей. Примеры фармацевтически приемлемых солей включают соли неорганических или органических кислот по основным остаткам, таким как амины; соли щелочей или органических оснований по кислотным остаткам, таким как карбоновые кислоты и т.п., но не ограничиваются ими. Фармацевтически приемлемые соли включают обычные нетоксические соли или четвертичные аммониевые соли исходного соединения, которые образуются, например, с нетоксичными неорганическими или органическими кислотами. Например, такие традиционные нетоксичные соли включают соли, образованные такими неорганическими кислотами, как соляная, бромоводородная, серная, сульфаминовая, фосфорная, азотная и т.п.; и соли, полученные с такими органическими кислотами, как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изатиновая и т.п.
Фармацевтически приемлемые соли настоящего изобретения могут быть синтезированы из исходных соединений, которые содержат основной или кислотный фрагмент, с помощью традиционных химических методов. Обычно такие соли могут быть приготовлены путем реакции этих соединений в форме свободных кислот или оснований со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе, или в смеси обоих; обычно предпочтительна неводная среда, такая как эфир, этил ацетат, этанол, изопропанол или ацетонитрил. Перечень подходящих солей можно найти в Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p.1445, содержание которого внесено в заявку посредством ссылки.
Любая соль, сохраняющая желательную биологическую активность соединений, содержащихся в заявке, и проявляющая незначительные или вовсе не проявляющая нежелательных токсических эффектов, считается включенной в заявку. Фармацевтически приемлемые соли включают соли, полученные с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями. Фармацевтически не приемлемые соли и основания также находят применение в заявке, например, при синтезе и/или очистке интересующих соединений. Таким образом, все «соли» также охвачены границами настоящего изобретения.
Неограничивающие примеры подходящих солей включают соли, полученные с неорганическими кислотами, такими как, например, соляная кислота, бромоводородная кислота, серная кислота, фосфорная кислота, азотная кислота, угольная кислота; и соли, полученные с органическими кислотами, такими как, например, муравьиная кислота, уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, малоновая кислота, аскорбиновая кислота, лимонная кислота, бензойная кислота, дубильная кислота, альгиновая кислота, полиглутаминовая кислота, толуолсульфоновая кислота, метансульфоновая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота, альфа-кетоглутаровая кислота, 3-глицерофосфорная кислота и полигалактуроновая кислота. Подходящие соли включают соли, полученные со щелочными металлами, такими как литий, калий и натрий, с щелочноземельными металлами, такими как кальций и магний, а также с другими кислотами, хорошо известными специалисту в области фармации. Другие подходящие соли включают соли, полученные с металлическими катионами, такими как цинк, висмут, барий или алюминий, или с катионами, полученными из аминов, такими как аммоний, N,N-дибензилэтилендиамин, D-глюкозамин, тетраэтиламмоний или этилендиамин. Кроме того, подходящие соли включают соли, полученные с комбинацией кислот и оснований, таких как, например, цинк-таннатная соль.
Фраза «фармацевтически приемлемый» применяется в заявке для обозначения тех соединений, материалов, композиций и/или лекарственных форм, которые, в терминах тщательной медицинской оценки, пригодны для применения при контакте с тканями человеческого организма и животного, не вызывая избыточную токсичность, воспаление, аллергические реакции или другие проблемы или сложности, сопоставимые с разумным соотношением пользы и риска.
Термин «пролекарство», упоминаемый в заявке, относится к веществу, которое превращается в фармакологически активное исходное лекарство in vivo, такое, как соединения, описываемые в заявке. Термин «пролекарство» включает любой ковалентно связанный носитель, который высвобождает активное исходное лекарство настоящего изобретения in vivo при введении пролекарства животным. Поскольку известно, что пролекарства улучшают многие желательные качества препаратов (например, растворимость, биодоступность, производство, транспортировку, фармакодинамику и т.д.), соединения настоящего изобретения могут доставляться в форме пролекарства. Пролекарства, например, могут обладать биодоступностью при пероральном применении, даже если исходное лекарство не обладает ею. Таким образом, настоящее изобретение охватывает пролекарства заявленных соединений, способы доставки соединений и композиций, их содержащие. Пролекарства настоящего изобретения получают путем модификации функциональных групп, представленных в соединении, таким образом, чтобы модифицирующие группы отщеплялись либо при обычном обращении, либо in vivo, высвобождая исходное соединение. Пролекарства включают соединения настоящего изобретения, в которых гидрокси, амино или сульфгидрильная группа связана с любой группой, которая, при введении пролекарства настоящего изобретения млекопитающим субъектам, отщепляется, давая свободную гидроксильную, свободную амино или свободную сульфгидрильную группу, соответственно. Примеры пролекарств включают ацетатные, формиатные и бензоатные производные соединений настоящего изобретения по спиртовой и аминной функциональным группам.
Говоря в целом, пролекарства представляют собой производные лекарств как таковых, которые после употребления претерпевают превращение или метаболизм в физиологически активные соединения. Превращение может быть спонтанным, таким как гидролиз в физиологической среде, или катализируемым ферментами. Пролекарства включают соединения, которые могут окисляться, восстанавливаться, аминироваться, деаминироваться, гидроксилироваться, дегидроксилироваться, гидролизоваться, этерифицироваться, алкилироваться, деалкилироваться, фосфорилироваться и/или дефосфорилироваться, образуя активное соединение.
Из многотомной научной литературы, посвященной в целом пролекарствам, даются ссылки на следующие источники: Gang-war и соавт., «Prodrug, molecular structure and percutaneous delivery», Des. Biopharm. Prop. Prodrugs Analogs, [Symp.] Meeting Date 1976, 409-21. (1977); Nathwani и Wood, «Penicillins: a current review of their clinical pharmacology and therapeutic use», Drags 45(6): 866-94 (1993); Sinhababu and Thakker, «Prodrugs of anticancer agents», Adv. Drag Delivery Rev. 19(2): 241-273 (1996); Stella и соавт., «Prodrugs. Do they have advantages in clinical practice?». Drugs 29(5): 455-73 (1985); Tan и соавт. «Development and optimization of anti-HIV nucleoside analogs and prodrugs: A review of their cellular pharmacology, structure-activity relationships and pharmacokinetics», Adv. Drag Delivery Rev. 39(1-3): 117-151 (1999); Design of Prodrugs (Bundgaard H. ed.) 1985 Elsevier Science Publishers B.V. (Biomedical Division), Chapter 1; Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities (Hans Bundgaard); Bundgaard et и соавт. Int. J. of Pharmaceutics 22 (1984) 45-56 (Elsevier); Bundgaard и соавт. Int. J. of Pharmaceutics 29 (1986) 19-28 (Elsevier); Bundgaard и соавт. J. Med. Chem. 32 (1989) 2503-2507 Chem. Abstracts 93, 137935y (Bundgaard и соавт.); Chem. Abstracts 95, 13 8493 f (Bundgaard и соавт.); Chem. Abstracts 95, 138592n (Bundgaard и соавт.); Chem. Abstracts 110, 57664p (Alminger и соавт.); Chem. Abstracts 115, 64029s (Buur и соавт.); Chem. Abstracts 115, 189582y (Hansen и соавт.); Chem. Abstracts 117, 14347q (Bundgaard и соавт.); Chem. Abstracts 117, 55790x (Jensen и соавт.); и Chem. Abstracts 123, 17593b (Thomsen и соавт.).
Термин «применяемый», «применение», «введение» соединения должен пониматься как предоставление любого соединения изобретения индивиду, включая животное, имеющему показания к лечению, путем приведения такого индивида в контакт с, или применение индивидом такого соединения каким-либо другим способом.
Термин «in vitro» относится к биологическому объекту, биологическому процессу или биологической реакции вне организма, в искусственных условиях. Например, рост клеток in vitro должен пониматься как рост клеток в среде вне организма, например, в пробирке, чашке для культивирования или микропланшете.
Термин «терапевтически эффективное количество» обозначает количество соединения, которое будет вызывать тот биологический, физиологический, фармакологический, терапевтический или медицинский отклик в клетке, ткани, системе, организме, животном, индивидууме, пациенте или человеке, который предполагает исследователь, ученый, фармаколог, фармацевт, ветеринар, доктор или другой клиницист, например, ослабление эффектов/симптомов болезни или заболевания, например, рака или опухоли, или остановка или ингибирование роста пролиферирующих клеток, таких как раковые клетки. Терапевтически эффективное количество может быть определено в соответствии со стандартными процедурами, включая те, которые будут описаны ниже в разделе «Дозировки».
Термин «дополнительное лечение», «дополнительное применение» или «дополнительно применяемый» обозначает, что различные лекарственные вещества или соединения могут применяться вместе, поочередно или периодически. Такое дополнительное применение может быть разделенным во времени или пространстве, например, в разное время, в разные дни или путем различных способов или путей применения.
Некоторые соединения настоящего изобретения
В одном варианте осуществления настоящего изобретения соединения изобретения обладают молекулярным весом от 250 до 1000, в частности, от 400 до 800, более конкретно, от 400 до 600 и еще более конкретно, от 280 до 500. В определенных вариантах осуществления соединения изобретения обладают одной или более из следующих характеристик: (i) не более 5 доноров водородной связи, (11) не более 10 акцепторов водородной связи и (111) не более 10 свободно вращаемых связей (исключая связи с концевыми атомами). В определенных вариантах осуществления соединения формулы (I) находятся в соответствии с «правилом пяти» Липински (Lipinski, Adv. Drug Del. Rev. 1997; 23: 3), поскольку имеют молекулярный вес меньше 500, не более 5 доноров водородных связей, не более 10 акцепторов водородных связей и значение cLogP от -2 до 5.
В одном из вариантов осуществления настоящего изобретения соединения изобретения представляют собой ингибиторы активности Haspin киназы. В определенных вариантах осуществления изобретения соединения ингибируют активность Haspin киназы, со значениями IC50 менее 1 µM, эти значения IC50 определяются в соответствии с анализом ингибирования с помощью метода HTRF, описываемыми в примерах ниже. В определенных вариантах осуществления изобретения IC50 против киназы Haspin составляет менее 0.5 µМ, менее 0.2 µM, менее 0.1 µМ или менее 0.01 µM. В определенных вариантах осуществления изобретения соединения ингибируют активность не только Haspin, то и ряда других, не родственных Haspin киназ: cKIT, FLT3, FLT3 ITD (и другие мутантные формы FLT киназы), IRAK-4, Lyn (и других киназ семейства Src киназ), МЕК1.2 киназы, PDGFR-alfa, PDGFR-beta, VEGFR1,2,3, а также LRRK2 киназу и ее мутантную форму LRRK2(G19S20) со значениями IC50 менее 10 µМ.
Соединение настоящего изобретения может существовать в одной или более кристаллических формах, и может существовать в виде твердого безводного вещества или в виде сольватов, включающих определенные количества растворителей, включая гидраты, включающие определенные количества воды.
В другом варианте осуществления соединения изобретения представляют собой спланированные и обдуманные продукты синтетической химической схемы, т.е. получаются в результате определенных и спланированных химических процессов, проводимых в реакционных сосудах, а не путем деградации, метаболизма или ферментирования, или получаются в виде примесей или побочных продуктов при синтезе других соединений. В определенных вариантах осуществления настоящего изобретения соединения очищаются или выделяются, например, до степени чистоты по крайней мере 80%, в частности, по крайней мере 90%, более конкретно, по крайней мере 95%, еще более конкретно, по крайней мере 97%, по крайней мере 98% или даже по крайней мере 99%. Чистота, при упоминании в заявке, может определяться либо как абсолютная, либо как относительная чистота. Абсолютная чистота определяется как количество соединения изобретения, полученного в качестве продукта синтетической химической схемы, либо до, либо после одной или более ступеней очистки. Относительная чистота обозначает количество соединения изобретения по отношению и одной или более примеси, таких как побочные продукты, продукты разложения (например, метаболиты, продукты окисления или гидролиза и т.п.) и/или соединениям, которые разлагаются с образованием соединения изобретения (например, прекурсоры и пролекарства), например, которая может относиться к продукту синтетической химической схемы. Таким образом, абсолютная чистота относится к количеству соединения по отношению ко всем прочим, в то время как относительная чистота в общем не зависит от наличия неродственных соединений, таких как наполнители, стабилизаторы и другие медикаменты для совместного применения. Чистота может оцениваться по весовым, объемным или молярным отношениям соединения по отношению к другим соединениям. Чистота может определяться различными аналитическими методами, включающими элементный анализ, УФ-спектроскопию в видимой области, ВЭЖХ, ГХ-МС, ЯМР, масс-спектрометрию и тонкослойную хроматографию, предпочтительно, ВЭЖХ, ГХ-МС или ЯМР.
Лекарственные формы, дозировки и применения
Настоящее изобретение дополнительно предусматривает фармацевтическую композицию, включающую соединение, описанное выше, или его пролекарство, и фармацевтически приемлемый наполнитель, разбавитель или носитель, включающую терапевтически эффективное количество такого соединения или пролекарства.
Лекарственные формы
Композиции настоящего изобретения могут быть включены в состав лекарственной формы и применяться для лечения индивида, нуждающегося в лечении, любым способом, который обеспечивает контакт активного ингредиента с местом действия вещества, таким как клетка, в организме индивида. Они могут применяться любым традиционным способом, доступным для применения, совместно с фармацевтическими препаратами, либо в качестве индивидуального терапевтически активного ингредиента, либо в комбинации с другими терапевтически активными ингредиентами. Они могут применяться индивидуально, но обычно применяются вместе с фармацевтически приемлемыми разбавителями, наполнителями или носителями, выбранными на основании избранного пути введения и стандартной фармацевтической практики.
Также может применяться фармацевтическая композиция, включающая меньшее, чем терапевтически эффективное, количество соединения, описанного выше, или его пролекарства, например, при применении в комбинации с другой фармацевтической композицией, такой как противораковый агент, так что такая комбинация является терапевтически эффективной, или может применяться для профилактического лечения.
Фармацевтическая композиция для применения в соответствии с настоящим изобретением может включаться в состав лекарственной формы традиционным способом, с применением одного или более фармацевтически приемлемых разбавителей, наполнителей или носителей. Фармацевтические композиции изобретения могут входить в состав лекарственных форм для различных путей введения, включая системное, местное или локальное введение. Общие методы приготовления и лекарственные формы можно найти в Remington's Pharmaceutical Sciences, Meade Publishing Co., Easton, PA. Как в деталях описывается ниже, фармацевтические композиции настоящего изобретения могут входить в состав лекарственных форм для применения в твердом или жидком виде, включая лекарственные формы, адаптированные для: (1) перорального введения, например, в виде микстур (водных или неводных растворов или суспензий), таблеток, капсул, болюсов, порошков, гранул, паст для применения на язык; (2) парентерального введения, например, в виде подкожных, внутримышечных или внутривенных инъекций, как, например, стерильные растворы или суспензии; (3) местного применения, например, в виде крема, мази или спрея для нанесения на кожу; или (4) интравагинально или интраректально, например, в виде суппозиториев, крема или пены. В определенных вариантах осуществления фармацевтические препараты могут быть апирогенными, т.е. не вызывать повышения температуры тела у пациента.
В композициях также могут присутствовать смачивающие вещества, эмульгаторы и лубриканты, такие как натрия лаурилсульфат и магния стеарат, а также красители, высвобождающие вещества, вещества оболочки, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, цистеина гидрохлорид, натрия бисульфат, натрия метабисульфит, натрия сульфит и т.п.; (2) жирорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (Е320, ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) вещества, хелатирующие металлы, такие как лимонная кислота, этилендиаминотетрауксусная кислота (EDTA), сорбитол, винная кислота, фосфорная кислота и т.п.
Лекарственные формы настоящего изобретения включают лекарственные формы, приемлемые для перорального, назального, местного (включая трансбуккальное и сублингвальное), ректального, вагинального и/или парентерального применения. Лекарственные формы могут быть обычным образом представлены в виде дозированных лекарственных форм и могут быть приготовлены любыми способами, хорошо известными в фармации. Количество активного ингредиента, которое может объединяться с материалом носителя с образованием единой лекарственной формы, может варьироваться в зависимости от организма, который предполагается лечить, а также от конкретного способа введения. Количество активного ингредиента, которое может объединяться с материалом носителя с образованием единой лекарственной формы, обычно составляет такое количество ингибитора, которое вызывает терапевтический эффект. Обычно это количество составляет из ста процентов от примерно 1 до примерно 95 процентов активного ингредиента, предпочтительно, от примерно 5 до примерно 70 процентов, наиболее предпочтительно, от примерно 10 процентов до примерно 30 процентов.
Способы получения этих лекарственных форм или композиций включают стадию объединения соединения настоящего изобретения с носителем и, необязательно, одним или более вспомогательным ингредиентом. В общем, лекарственные формы приготовляют равномерным и тщательным смешением соединения настоящего изобретения с жидким наполнителем, или тщательно измельченным твердым наполнителем, или обоими, а затем, при необходимости, оформлением продукта.
Для системного введения предпочтительными являются инъекции, включая внутримышечные, внутривенные, внутрибрюшинные и подкожные (i.m., i.v., i.p.и s.c. соответственно). Фразы «системное введение», «применяемые системно» и «применяемые периферически» при упоминании в заявке означает применение соединения, лекарства или другого материала иным способом, чем прямо в центральную нервную систему, так что оно попадает в кровеносную систему пациента и, таким образом, подвергается метаболизму и подобным процессам, например, подкожное введение.
Фармацевтические композиции изобретения в лекарственной форме для инъекций могут представлять собой жидкие растворы, предпочтительно, в физиологически совместимой буфферной системе, такой как раствор Хэнкса или раствор Рингера. Кроме того, фармацевтические композиции могут быть в твердой лекарственной форме и растворяться или суспендироваться непосредственно перед употреблением. Также включаются лиофилизированные лекарственные формы.
Фармацевтические композиции изобретения могут быть в лекарственной форме, применимой для перорального введения, в виде капсул, саше, пилюль, такблеток, драже (с использованием ароматизированных основ, обычно сахарозы или аравийской или трагакантовой камеди), порошков, гранул, или в виде раствора или суспензии в водной или неводной среде, или в виде эмульсии масло-в-воде или вода-в-масле, или в виде эликсира или сиропа, или в виде пастилок (с использованием инертной основы, такой как желатин и глицерин, или сахароза или камедь) и/или в виде полосканий для рта и т.п., каждая из которых содержит заранее определенное количество соединения настоящего изобретения в качестве активного ингредиента. Соединение настоящего изобретения может также применяться в виде болюсов, электуария или пасты.
В прописи фармацевтических композиций изобретения в твердых лекарственных формах для перорального (р.о.) введения (капсулы, таблетки, пилюли, драже, порошки, гранулы и т.п.), соединение изобретения в качестве активного ингредиента смешивается с одним или более фармацевтически приемлемым наполнителем, таким как цитрат натрия или или дикальцийфосфатом и/или одним из следующих ингредиентов: (1) наполнителями или добавками, такими как крахмалы, лактоза, сахароза, глюкоза, маннитол и/или кремневая кислота; (2) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; (3) увлажнители, такие как глицерин; (4) вещества для улучшения распадаемости таблеток, такие как агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) замедлитель схватывания раствора, такой как парафин; (6) ускоритель всасывания, такой как четвертичные аммониевые соединения; (7) смачивающие вещества, такие как, например, цетиловый спирт или глицеринмоностеарат; (8) абсорбенты, такие как каолин и бентонитовая глина; (9) лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, натрия лаурилсульфат и их смеси; (10) красители. В случае капсул, таблеток и пилюль фармацевтическая композиция также может включать буферные вещества. Твердые композиции сходного типа также могут применяться в мягких и твердых желатиновых капсулах с использованием таких наполнителей, как лактоза или молочный сахар, высокомолекулярные полиэтиленгликоли и т.п.
Желатиновые капсулы содержат соединение настоящего изобретения в качестве активного ингредиента и порошкообразные наполнители, такие как лактоза, крахмал, производные целлюлозы, стеарат магния, стеариновая кислота и т.п. Аналогичные наполнители могут использоваться для изготовления прессованных таблеток. Как таблетки, так и капсулы могут изготавливаться в виде лекарственных форм с замедленным высвобождением для того, чтобы обеспечить непрерывное высвобождение медикамента за протяженный период. Прессованные таблетки могут покрываться сахарной или пленочной оболочкой для маскировки неприятного вкуса и для защиты таблетки от атмосферы, или кишечнорастворимой оболочкой для селективного переваривания в желудочно-кишечном тракте. Твердые композиции такого типа также применяются в качестве наполнителей в мягких и твердых желатиновых капсулах; предпочтительные для этого материалы также включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Предпочтительная лекарственная форма представляет собой раствор или суспензию в масле, например, оливковом масле, Miglyol или Capmul, в мягкой желатиновой капсуле. При необходимости для предотвращения разложения со временем могут добавляться антиоксиданты.
Таблетки могут изготавливаться прессованием или формовкой, необязательно, с одним или более вспомогательным ингредиентом. Прессованная таблетка может изготавливаться с использованием связующего вещества (например, желатина или гидроксипропилметилцеллюлозы), лубриканта, инертного разбавителя, консерванта, разрыхлителя (например, натриевой соли гликолята крахмала или кросс-сшитой карбоксиметилцеллюлозы натрия), поверхностно-активных или диспергирующих веществ. Формованная таблетка может изготавливаться формованием порошкообразного ингибитора, смоченного инертным жидким разбавителем, в подходящем аппарате.
Таблетки и другие твердые лекарственные формы фармацевтических композиций настоящего изобретения, такие как драже, капсулы, пилюли и гранулы, необязательно, могут содержать метку или покрываться оболочкой, такой как кишечнорастворимые или другие оболочки, что хорошо известно в области изготовления лекарственных форм. Они также могут быть в такой лекарственной форме, которая обеспечивает медленное или контролируемое высвобождение содержащегося в ней активного ингредиента, используя, например, гидроксипропилметилцеллюлозу в различных соотношениях, для обеспечения желаемого профиля высвобождения, а также другие полимерные матрицы, липосомы и/или микросферы. Они также могут стерилизоваться, например, путем фильтрования через фильтр, удерживающий бактерии, или путем введения стерилизующего агента в форму стерильной твердой композиции, которая может быть растворена в стерильной воде или какой-то другой стерильной среде для инъекций непосредственно перед применением. Эти композиции также могут необязательно содержать рентгеноконтрастные вещества и могут представлять собой композицию, которая высвобождает активный ингредиент(ы) только, или предпочтительно, в определенных отделах гастро-интестинального тракта, необязательно, замедленным образом. Примеры включенных композиций, которые могут применяться, включают полимерные вещества и смолы.
Жидкие лекарственные формы для перорального применения фармацевтических композиций изобретения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активного ингредиента жидкие лекарственные формы могут содержать инертные разбавители, известные в данной области, такие как, например, воду или другие растворители, солюбилизирующие вещества и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и эфиры жирных кислот сорбитана и их смеси.
Помимо инертных разбавителей фармацевтические композиции для перорального применения могут также включать такие вспомогательные агенты, как смачивающие вещества, эмульгаторы и суспендирующие вещества, подсластители, ароматизаторы, красители, отдушки и консерванты.
Суспензии, кроме фармацевтической композиции настоящего изобретения, могут содержать суспендирующие вещества, такие как, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбитола и сорбитана, микрокристаллическую целлюлозу, алюминия метагидроксид, бентонит, агар-агар и трагакантовую камедь, и их смеси.
Для трансбуккального применения фармацевтические композиции могут принимать традиционную форму таблеток или таблеток для рассасывания.
Для применения с помощью ингаляции, фармацевтические композиции настоящего изобретения могут предусматриваться в форме распыляемого аэрозоля, находящегося в упаковках под давлением, или в форме небулайзера с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением разовая доза может определяться применением подходящего клапана для доставки отмеренного количества вещества. Капсулы и картриджи из, например, желатина для применения в ингаляторе или аппарате для вдувания могут содержать порошок из смеси терапевтических агентов и подходящей порошкообразной основы, такой как лактоза и крахмал.
Фармацевтические композиции могут включаться в лекарственную форму для парентерального введения при помощи инъекций, например, болюсной инъекции или непрерывной инфузии. Лекарственные формы для инъекции могут быть представлены в виде разовой дозы, например, в ампулах или многоразовых контейнерах, содержащей дополнительный консервант. Фармацевтические композиции также могут быть в форме суспензий, растворов или эмульсий в масляной или водной среде и могут содержать дополнительные вещества, такие как суспендирующие вещества, стабилизаторы и/или диспергирующие вещества. Альтернативно, активный ингредиент может быть в форме порошка, который разводится подходящей средой, например, стерильной апирогенной водой, перед употреблением.
Фраза «парентеральное введение» и «применяемый парентерально» при упоминании в заявке означает способ введения, отличный от энтерального и местного применения, обычно путем инъекции, и включает, без ограничений, внутривенные, внутримышечные, внутриартериальные, интратекальные, интракалсулярные, внутриглазничные, внутрисердечные, внутрикожные, внутрибрюшинные, транстрахеальные, подкожные, подкутикульные, внутрисуставные, подкапсулярные, субарахноидальные, интраспинальные и интрастернальные инъекции и инфузии.
Фармацевтические композиции настоящего изобретения, применимые для парентерального способа введения, включают один или более ингибитор Haspin киназы в комбинации с одним или более фармацевтически приемлемым стерильным изотоническим водным или неводным раствором, дисперсией, суспензией или эмульсией, или представляют собой стерильные порошки, которые могут быть восстановлены в стерильные растворы или дисперсии для инъекций непосредственно перед введением, эти композиции могут содержать антиоксиданты, буферные растворы, бактериостаты, растворы, поддерживающие изотоничность состава с кровью пациента, или суспендирующие вещества, или загустители.
Примеры подходящих водных и неводных наполнителей, которые могут применяться в фармацевтических композициях изобретения, включают воду, этанол, полиспирты (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.) и их приемлемые смеси, растительные масла, такие как оливковое масло, и пригодные для инъекций органические эфиры, такие как этилолеат. Необходимая консистенция может поддерживаться, например, с помощью материалов оболочки, таких как лецитин, с помощью поддерживания необходимого размера частиц в случае дисперсии и с помощью поверхностно-активных веществ.
Заявленные фармацевтические композиции также могут содержать вспомогательные вещества, такие как консерванты, смачивающие вещества, эмульгаторы и диспергирующие вещества. Защита от микроорганизмов может достигаться включением различных антибактериальных и антигрибковых веществ, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и т.п. Также может быть желательным включить в фармацевтическую композицию изотонические вещества, такие как сахара, хлорид натрия и т.п. Кроме того, с помощью включения веществ, задерживающих абсорбцию, таких как моностеарат алюминия и/или желатин, может достигаться пролонгированное всасывание инъекционной лекарственной формы.
Кроме уже описанных лекарственных форм, фармацевтические композиции могут быть представлены в лекарственной форме с замедленным всасыванием. Такие лекарственные формы длительного действия могут применяться с помощью имплантации (например, подкожно или внутримышечно) или с помощью внутримышечной инъекции. Так, например, фармацевтические композиции могут входить в состав лекарственной формы вместе с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в подходящем масле) или ион-обменными смолами, или в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
Системное введение также может осуществляться трансмукозальными или трансдермальными методами. Для трансмукозального или трансдермального применения в прописи используются соответствующие подходящие пенетранты для преодоления тканевого барьера. Такие пенетранты известны в фармакологии и включают, например, соли желчных кислот и производные фузидовой кислоты для трансмукозального применения. Трансмукозальное введение может осуществляться путем назального спрея или с использованием суппозиториев. Фармацевтические композиции изобретения для местного применения входят в состав мазей, притираний, гелей или кремов, что в общем известно в области фармацевтики. Растворы для промываний могут применяться местно для обработки ран или мест воспаления для ускорения заживления.
В некоторых случаях для пролонгации терапевтического действия ингибитора при введении с помощью подкожных или внутримышечных инъекций желательно замедлить всасывание ингибитора. Это может быть достигнуто применением жидкой суспензии кристаллического или аморфного материала, обладающего низкой растворимостью в воде. В этом случае скорость всасывания ингибитора зависит от скорости растворения, которая, в свою очередь, может зависеть от размера и формы кисталлов. Как вариант, замедленная абсорбция парентерально вводимого ингибитора может достигаться растворением или суспендированием ингибитора в жировой среде.
Фармацевтические композиции изобретения могут входить в состав лекарственной формы для ректального или вагинального введения в виде суппозиториев, которые могут приготавливаться путем смешивания одного или более соединения изобретения с одним или более подходящим наполнителем или носителем, не вызывающим раздражения, включающем, например, масло какао, полиэтиленгликоль, воск для суппозиториев или салицилат, который остается в твердом состояниии при комнатной температуре, но становится жидким при температуре тела, и, следовательно, будет плавиться в заднем проходе или вагинальной полости и высвобождать активный ингибитор.
Лекарственные формы фармацевтических композиций настоящего изобретения, пригодные для вагинального применения, также включают маточные кольца, тампоны, кремы, гели, пасты, пены или спрей, содержащие наполнители, пригодные для этого.
Лекарственные формы для местного или трансдермального применения соединения настоящего изобретения включают порошки, спрей, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и лекарственные формы для ингаляций. Такое соединение может смешиваться в стерильных условиях с фармацевтически приемлемым наполнителем и любым необходимым консервантом, буфером или пропеллентом.
Мази, пасты, кремы и гели кроме соединения изобретения могут содержать наполнители, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакантовая камедь, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремневая кислота, тальк и оксид цинка, или их смеси.
Порошки и спреи соединения изобретения могут содержать наполнители, такие как лактоза, тальк, кремневая кислота, гидроксид алюминия, силикаты кальция и полиамидный порошок, или их смеси. Спрей может кроме того содержать общеупотребительные пропелленты, такие как хлорфторуглеводороды и летучие незамещенные углеводороды, такие как бутан и пропан.
Трансдермальные пластыри имеют дополнительное преимущество контролируемой доставки в организм соединения настоящего изобретения. Такая лекарственная форма может быть изготовлены путем растворения или диспергирования ингибитора настоящего изобретения в подходящей среде. Для увеличения проницаемости лекарства через кожу также могут применяться усилители абсорбции. Скорость всасывания при этом можно контролировать либо наличием специальной мембраны, контролирующей скорость, либо диспергированием соединения изобретения в полимерной матрице или геле.
Глазные лекарственные формы, глазные мази, порошки, растворы и т.п. также находятся в рамках настоящего изобретения.
Фармацевтические композиции могут, при желании, находиться в упаковке или в дозирующем устройстве, которые могут содержать одну или более разовую лекарственную дозу, содержащую активный ингредиент. Упаковка может, например, включать металлическую или пластиковую фольгу, такую как в блистерной упаковке. К упаковке или дозирующему устройству могут прилагаться инструкции к применению. В других вариантах осуществления упаковка или дозирующее устройство могут дополнительно упаковываться в картонную коробку.
Фармацевтические композиции настоящего изобретения также могут входить в состав лекарственной формы с замедленным и/или пролонгированным высвобождением. Такие лекарственной формы с замедленным и/или пролонгированным высвобождением могут осуществляться с помощью способов и устройств для доставки для замедленного высвобождения, хорошо известных любому специалисту, например, как описано в Патентах США №№: 3,845,770; 3,916,899; 3,536,809; 3,598,123; 4,008,719; 4,710,384 5,674,533; 5,059,595; 5,591,767; 5,120,548; 5,073,543; 5,639,476; 5,354,556; и 5,733,566, содержание которых вводится в заявку посредством ссылки. Фармацевтические композиции настоящего изобретения могут применяться для того, чтобы обеспечить медленное или пролонгированное высвобождение одного или более активного ингредиента, используя, например, гидроксипропилметилцеллюлозу, другие полимерные матрицы, гели, проницаемые мембраны, осмотические системы, многослойные покрытия, микрочастицы, липосомы, микросферы и т.п., или их комбинации в различных соотношениях для обеспечения желаемого профиля высвобождения. Подходящие лекарственные формы для замедленного высвобождения, известные любому специалисту в области фармакологии, включающие те, которые описаны в заявке, могут быть легко применены для использования с фармацевтическими композициями изобретения. Таким образом, разовые лекарственные формы, пригодные для перорального введения, такие как таблетки, капсулы, желатиновые капсулы, таблетки в виде капсул, порошки и т.п., но не только эти, адаптированные для применения с замедленным высвобождением, считаются охваченными настоящим изобретением.
Инъекционные лекарственные формы с замедленным высвобождением получают с помощью образования микроинкапсулированных частиц упомянутого ингибитора в биоразлагаемых полимерах, таких как полиактид-полигликолид. Скорость высвобождения лекарства можно контролировать с помощью соотношения лекарства и полимера и природы конкретного используемого полимера. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъекционные лекарственные формы с замедленным высвобождением получают путем включения лекарства в липосомы или микроэмульсии, совместимые с тканями организма.
При применении соединений настоящего изобретения в качестве лекарственных препаратов индивидам, таким как человек и животные, они могут употребляться сами по себе или в составе фармацевтической композиции, включающей, например, от 0,1 до 99,5% (в определенных вариантах осуществления от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым наполнителем.
Настоящее изобретение предусматривает новые способы лечения пролиферативных, дегенеративных и других заболеваний и расстройств, включая рак, путем применения терапевтически эффективного количества, по крайней мере, одного из соединений, описываемых в заявке, или его пролекарства, таутомера, фармацевтически приемлемой соли, N-оксида или стереоизомерной формы. Настоящее изобретение дополнительно предусматривает способы лечения пролиферативных, дегенеративных и других заболеваний и расстройств, включая рак, путем применения терапевтической комбинации по крайней мере одного из этих соединений и другого противоракового или антипролиферативного препарата.
Соединение может применяться в виде его соли или пролекарства, которое, при употреблении его индивидуумом, способно, прямо или непрямо, высвобождать исходное соединение, такое как соединение, описанное в заявке, или проявлять активность как таковую. Не ограничивающие примеры включают фармацевтически приемлемую соль, альтернативно упоминаемую как «физиологически приемлемая соль». Кроме того, такие модификации соединения могут повлиять на его биологическую активность, в некоторых случаях увеличивая ее по сравнению с исходным соединением. Эту активность можно оценить путем получения соединения в форме соли или пролекарства и определения его активности, используя способы, описанные в заявке, или другие способы, известные специалисту.
Для специалиста очевидно, что при применении пролекарства определенного соединения индивидуумом, таким как животное, употребляющее или принимающее такое пролекарство, он также будет подвергаться воздействию и, следовательно, непрямо употреблять исходное соединение. Такая процедура может подвергнуть клетки, ассоциированные с заболеванием, таким как пролиферативное заболевание или расстройство, включающее рак, воздействию исходного соединения.
Соединения настоящего изобретения могут содержать асимметрически замещенный атом углерода и могут выделяться в виде оптически активных или рацемических форм. Специалистам хорошо известны способы получения оптически активных форм, такие как разделение рацемических форм или синтез из оптически активных исходных реагентов. Изобретение охватывает все хиральные, диастереомерные, рацемические формы, если специально не обозначены определенная стереохимическая или изомерная формы. Все процессы, используемые для получения соединений настоящего изобретения, и интермедиаты для их получения являются частью настоящего изобретения.
Дозировки
Применяемая дозировка, которая представляет собой терапевтически эффективное количество, достаточное или могущее, по мнению таких медицинских специалистов, как врач, медсестра или провизор, привести к ослаблению симптомов, например, онкологического заболевания, очевидно, будет варьироваться в зависимости от таких известных факторов, как фармакодинамические характеристики конкретного активного ингредиента и его способа и пути введения; возраста, пола, состояния здоровья и веса реципиента; природы и тяжести симптомов; вида параллельного лечения, периодичности лечения и желаемого эффекта.
Выбранные соединения также могут применяться для профилактического лечения. Если соединение применяется до клинического проявления нежелательных симптомов (например, болезни или других нежелательных состояний животного), то такое лечение является профилактическим (т.е. оно защищает индивидуума от начала, развития или дальнейшего развития нежелательных симптомов). Заявленные соединения могут также применяться для предотвращений симптомов, болезней или расстройств, таких как рак, или комплекса симптомов, таких как сердечная недостаточность, или других медицинский заболеваний. Такое лечение включает применение соединения с целью уменьшить частоту или отсрочить наступление симптомов медицинского заболевания у индивидуума по сравнению с индивидом, который не получает этого соединения. Таким образом, профилактика рака включает, например, уменьшение числа обнаруживаемых раковых опухолей, образований или злокачественных новообразований в популяции пациентов, получающих профилактическое лечение по отношению к контрольной популяции, не получающей такого лечения, более позднее возникновение обнаруживаемых раковых опухолей, образований или злокачественных новообразований в популяции пациентов, получающих лечение по отношению к контрольной популяции, не получающей лечения, и/или замедление прогрессирования заболевания и/или улучшение качества жизни пациента, например, статистически достоверное или клинически значимое.
Токсичность и терапевтическая эффективность фармацевтических композиций настоящего изобретения может определяться стандартными фармацевтическими методами на культурах клеток или на экспериментальных животных, например, путем определения LD50 (дозы, летальной для 50% популяции) и ED50 (дозы, терапевтически эффективной для 50% популяции). Отношение токсической и терапевтической дозы представляет собой терапевтический индекс и выражается соотношением LD50/ED50.
Терапевтические препараты, демонстрирующие высокие терапевтические индексы, полезны во многих случаях. В определенных обстоятельствах могут применяться также и терапевтические композиции, проявляющие незначительный терапевтический эффект или имеющие токсические эффекты, включая случаи, когда лечение предпринимается для того, чтобы разработать систему доставки к участкам поврежденной ткани для того, чтобы минимизировать потенциальную возможность повреждения незатронутых клеток и, таким образом, уменьшить или локализовать побочные эффекты.
Данные, полученные из экспериментов на клетках тканей и на моделях животных, могут применяться для формулирования дозировки для применения людям. Предпочтительно, дозировка лежит в диапазоне циркулирующих концентраций, которые включают ED50 и не вызывают или вызывают незначительную токсичность. Дозировка
может варьироваться в этом диапазоне в зависимости от применяемой лекарственной формы и применяемых путей введения. Для любых препаратов, применяемых в способе изобретения, терапевтически эффективная доза может первоначально оцениваться исходя из клеточных экспериментов. Дозировка может подбираться в моделях животных до достижения концентрации в циркулирующей плазме, включающей IC50 (т.е. такой концентрации исследуемого терапевтического агента, которая обеспечивает половину от максимального ингибирования симптомов или ингибирования биохимической активности), которая определяется на культурах клеток. Эта информация может применяться для более точного определения дозировки на людях. Уровень в плазме может определяться, например, методом высокоэффективной жидкостной хроматографии.
Следует понимать, что подходящая дозировка терапевтических веществ зависит от ряда факторов, известных специалисту, например, врачам. Дозировка (дозировки) конкретных соединений будет варьироваться, например, в зависимости от индивидуальности, размера и состояния употребляющего их субъекта или обрабатываемого образца, кроме того, от пути введения композиции, и эффекта препарата, который желает получить врач, на терапевтическую мишень или мишени, такие как клетки, нуклеиновые кислоты или полипептиды, являющиеся причиной заболевания или опосредующие заболевание или симптомы.
Примерные дозировки включают миллиграммовые или микрограммовые количества соединений настоящего изобретения на килограмм веса субъекта или образца, например, от примерно 1 микрограмма на килограмм до примерно 500 миллиграмм на килограмм, от примерно 100 микрограмм на килограмм до примерно 50 миллиграмм на килограмм или от примерно 1 миллиграмма на килограмм до примерно 5 миллиграмм на килограмм.
Специалисту будет понятно, что дозировка также может вычисляться на основании поверхности тела. Человек весом 70 кг имеет поверхность тела около 1,8 кв. метров, и дозировки могут выражаться как миллиграммовые или микрограммовые количества соединения на площадь поверхности тела субъекта или образца, например, от примерно 50 микрограмм на кв. метр до примерно 15 грамм на кв. метр, от примерно 5 миллиграмм на кв. метр до примерно 1,5 грамм на кв. метр, или от примерно 50 миллиграмм на кв. метр до примерно 150 миллиграмм на кв. метр.
Применения
Настоящее изобретение дополнительно предусматривает соединения, описываемые выше, для лечения. В других аспектах изобретение предлагает соединения настоящего изобретения для профилактического применения.
В определенных вариантах осуществления указанное лечение или профилактическое применение представляет собой лечение или профилактику пролиферативных заболеваний или расстройств, таких как опухоль или рак. В определенных вариантах осуществления указанное лечение представляет собой лечение рака, который можно лечить путем ингибирования активности протеинкиназы или ее мутантной формы.
Таким образом, настоящее изобретение дополнительно предлагает способ лечения индивидуума, такого как млекопитающего, страдающего болезненным состоянием, выбранным из группы пролиферативных заболеваний и расстройств, или воспалительных заболеваний и расстройств, включающий применение указанным индивидуумом терапевтически эффективного количества соединения, пролекарства или фармацевтической композиции, как описано выше. В определенных вариантах осуществления указанный индивидуум представляет собой человека. В определенных вариантах осуществления указанное пролиферативное заболевание или расстройство представляет собой рак. В определенных вариантах осуществления указанное лечение представляет собой лечение рака, который можно лечить путем ингибирования активности протеинкиназы или ее мутантной формы.
Настоящее изобретение также предлагает способ профилактического лечения индивида, такого как животное, включая млекопитающее, в частности, человека, с целью уменьшения частоты или отсрочки наступления симптомов медицинского заболевания, такого как рак, у субъекта по сравнению с субъектом, не получающим композиции.
В дополнительном аспекте изобретение предлагает способы лечения или профилактики таких индивидов, страдающих от болезни, как млекопитающие, включая домашних млекопитающих, кошек, собак, лошадей, овец, коров, грызунов и человека, включающие стадию воздействия на указанных индивид некоторого количества, включая терапевтически эффективное количество, заявленного соединения. В определенных вариантах осуществления болезнь представляет собой пролиферативное заболевание или расстройство, такое как рак или опухоль. В еще одном варианте осуществления клетки, ассоциированные с указанным пролиферативным заболеванием или расстройствам, включающим клетки раковой опухоли, подвергаются воздействию заявленного соединения. В определенных вариантах осуществления указанное соединение, или его пролекарство, вводится указанному индивиду. В определенных вариантах осуществления указанное лечение представляет собой лечение рака, которое заключается в ингибировании активности протеинкиназы или ее мутантной формы. В определенных вариантах осуществления указанное соединение, или его пролекарство, вводится указанному индивиду.
В дополнительном аспекте изобретение предлагает способ подавления или ингибирования пролиферации или роста клеток, включающий контактирование клетки с соединением изобретения. В одном варианте осуществления клетки выращивают in vitro, в то время как в альтернативном варианте осуществления клетка находится в индивиде. В определенном варианте осуществления клетка представляет собой раковую клетку, например, клетки раковой клеточной линии или клетки, включенные в опухоль, включая раковые клетки опухоли, которую можно лечить путем ингибирования активности протеинкиназы или ее мутанта (мутантной формы).
Еще один аспект изобретения относится к применению описанного выше соединения, или его пролекарства, для изготовления медицинского препарата для лечения или профилактики пролиферативного заболевания или расстройства, включающего рак, включая те виды рака, которые можно лечить путем ингибирования активности протеинкиназы или ее мутанта. Кроме того, изобретение относится к фармацевтической композиции, включающей описанное выше соединение, или его пролекарство, и фармацевтически приемлемый разбавитель, наполнитель или носитель, для лечения или профилактики пролиферативного заболевания или расстройства, включающего рак, включая те виды рака, которые можно лечить путем ингибирования активности протеинкиназы или ее мутанта.
Заявленные соединения могут быть полезны при лечении различных заболеваний и расстройств, включая пролиферативные заболевания и расстройства и воспалительные заболевания и расстройства. Термин «пролиферативные заболевания и расстройства» также признается специалистами и включает заболевания и расстройства, поражающие индивидов, таких как животные, таким образом, который характеризуется нарушенной, или как-то иначе нежелательной, пролиферацией группы клеток индивида. Рак и опухоли представляют собой пролиферативные заболевания или расстройства. Клетки, выделенные или полученные из опухоли, обычно считаются пролиферирующими клетками, типично, гиперпролиферирующими клетками, и, при других обстоятельствах, опухолевая клетка может быть диспластической или пролиферирующей. В определенных вариантах осуществления указанное лечение представляет собой лечение рака, который можно лечить путем ингибирования активности протеинкиназы или ее мутанта.
При чтении описания настоящего изобретения специалисту будет очевидно, что способы, фармацевтические композиции и упакованные фармацевтические препараты, включающие заявленные соединения, могут быть полезны для лечения других пролиферативных заболеваний или расстройств, или для подавления или ингибирования пролиферации клеток, включая опухолевые клетки.
Соединения настоящего изобретения могут применяться в лечении болезненных процессов, которые характеризуются нарушенной пролиферацией клеток, таких как гиперпролиферативные заболевания, включающие рак, доброкачественную гиперплазию простаты, семейный аденоматоз толстой кишки, нейрофиброматоз, псориаз, грибковые инфекции, эндотоксический шок, образование гипертрофического рубца, воспалительные заболевания кишечника, отторжение трансплантанта, пролиферация гладкомышечных клеток сосуда, ассоциированная с атеросклерозом, псориаз, фиброз легких, артриты, гломерунефриты, рестеноз после ангиопластики или сосудистой хирургии и другие послеоперационные стенозы и рестенозы. См., например. Патенты США№№. 6,114,365 и 6,107,305.
Ожидается, что описанные в заявке соединения могут применяться в лечении пролиферативных или гиперпролиферативных заболеваний или расстройств, таких как рак, аутоиммунные заболевания, вирусные заболевания, грибковые заболевания, нейродегенеративные расстройства и кардиоваскулярные заболевания.
В определенных вариантах осуществления опухоль может представлять собой солидную опухоль, которая представляет собой рак тканей организма, отличных от крови, костного мозга или лимфатической системы. В определенных вариантах осуществления опухоли могут представлять собой гематологические опухоли, такие как лейкемия и лимфома. Лейкемия обозначает собирательный термин для злокачественных заболеваний, характеризующихся пролиферацией злокачественно измененных белых кровяных клеток. Заболевания, проистекающие в лимфатической ткани, называются лимфомами.
Солидные опухоли могут быть выбраны из: рака печени, рака желудка, рака толстой кишки, рака груди, рака поджелудочной железы, рака простаты, рака кожи, рака почек, рака костной ткани, рака щитовидной железы, рака кожи, включая эпидермоидный рак, рак пищевода, рак почки, рак мочевого пузыря, рак желчного пузыря, рак шейки матки, рак яичников, рак легких, рак бронхов, мелкоклеточный и немелкоклеточный рак легкого, рак желудка, рак головы и шеи.
Гематологические опухоли могут представлять собой лейкемию, такую как острая миелоидная лейкемия (ОМЛ), острый лимфобластозный лейкоз (ОЛЛ), острый лимфолейкоз, острая лейкемия, острый промиелоцитарный лейкоз, хроническая гранулоцитарная лейкемия (ХГЛ), хроническая лейкемия, хроническая лимфоцитарная лейкемия (ХЛЛ), хроническая миелоидная лейкемия (ХМЛ), хронический миеломоноцитарный лейкоз, обыкновенный острый лимфобластозный лейкоз, эозинофильная лейкемия, эритролейкемия, экстранодальная лимфома, фолликулярная лимфома, волосатоклеточный лейкоз, моноцитарная лейкемия, пролимфоцитарный лейкоз.
Гематологические опухоли могут также представлять собой лимфому, такую как лимфомы В-клеток, лимфома Беркитта, кожная Т-клеточная лимфома, лимфома высокой степени злокачественности, лимфома Ходжкина, неходжкинская лимфома, лимфома низкой степени злокачественности, лимфобластозная лимфома, лимфома из клеток зоны мантии, лимфома краевой зоны лимфоузлов, лимфома лимфоидных тканей слизистых оболочек (MALT), Т-клеточные лимфомы, периферическая Т-клеточная лимфома, множественная миелома, эссенциальный тромбоцитоз, волосатоклеточая лимфома, экстрамедуллярная миелома, гранулоцитарная саркома.
Гематологические опухоли могут представлять собой также опухоли миелоцитарного происхождения, включая острую и хроническую миелоцитарную лейкемию, миелодиспластический синдром и промиелоцитарный лейкоз.
Опухоли могут также быть мезенхимальной природы, такие как фибросаркома и рабдомиосаркома. Более того, опухоли могут представлять собой опухоли в центральной и периферической нервной системе, такие как астроцитома, глиобластома, медулобластома, нейробластома, глиома и шваннома; опухоли могут представлять собой другие опухоли, такие как меланома, семинома, тератокарцинома, остеосаркома, пигментная ксеродерма, каратоакантома, фоликулярный рак щитовидной железы и саркома Калоши.
Опухоли, которые устойчивы или плохо поддаются лечению другими противораковыми или антипролиферативными препаратами, могут также выиграть от лечения способами и фармацевтическими композициями настоящего изобретения.
Описываемые в заявке соединения также могут быть полезны в химиопрофилактике рака. Химиопрофилактика определяется как ингибирование развития инвазивного рака либо путем блокирования начальных мутагенных процессов, либо путем блокирования развития пред-раковых клеток, например, блокированием роста опухоли, которой уже страдает пациент или ингибированием метастаз.
Описываемые в заявке соединения могут также быть полезны в ингибировании опухолевого ангиогенеза и метастаз.
Соединения настоящего изобретения также могут применяться в комбинации (назначаемой одновременно или последовательно) с известным противораковым лечением, таким как лучевая терапия, или с противораковыми, антипролиферативными, цитостатическими или цитотоксическими препаратами. Другие противораковые и антипролиферативные препараты, которые могут применяться в комбинации с соединениями настоящего изобретения, включают препараты, описанные в заявке. В комбинированном лечении соединения настоящего изобретения могут применяться дополнительно с любым другим противораковьм и антипролиферативным препаратом, описанным в заявке.
При применении в составе единой лекарственной формы такая комбинация продуктов включает соединения настоящего изобретения в диапазоне дозировок, описанном в заявке, и другой фармацевтически активный препарат или медикамент в утвержденном для него диапазоне дозировки. Например, было обнаружено, что ингибитор cdc2 оломуцин (olomucine) действует синергетически с известными цитотоксическими препаратами, индуцируя апоптоз (J. Cell Sci., 108, 2897 (1995)). Описываемые в заявке соединения также могут применяться последовательно с известными противораковыми или антипролиферативными препаратами, если их единая лекарственная форма неприемлема. Изобретение не ограничивает последовательность употребления; описанные в заявке соединения могут применяться до или после приема известных противораковых или антипролиферативных препаратов. Например, цитотоксическая активность ингибитора циклин-зависимой киназы флавопиридола (flavopindol) зависит от последовательности его применения с противораковыми препаратами (Cancer Research, 57, 3375 (1997)).
Дополнительные аспекты изобретения
Другой аспект изобретения предусматривает фармацевтическую упаковку, в которой указанная упаковка включает соединение с любой из формул настоящего изобретения. В определенных вариантах осуществления упаковка включает инструкции, которые указывают, что указанная композиция может применяться в лечении индивида, нуждающегося в таком лечении, включая человека. В определенных вариантах осуществления упаковка включает лекарственную форму, содержащую одно или более соединение настоящего изобретения вместе с другим фармацевтическим ингредиентом, таким как противораковое или антипролиферативное вещество. В этом случае соединение (соединения) настоящего изобретения и другой фармацевтический ингредиент могут входить в отдельные лекарственные формы в индивидуальной дозировке.
Другие фармацевтические ингредиенты, которые могут входить в состав лекарственной формы вместе или раздельно с соединениями настоящего изобретения, включают другие противораковые и антипролиферативные препараты, такие как те, которые описаны выше, но не ограничиваются ими. В определенных дополнительных вариантах осуществления фармацевтическая упаковка включает инструкции для лечения пациента, нуждающегося в таком лечении. Еще в одном аспекте изобретение предусматривает фармацевтическую упаковку для лечения индивида, страдающего пролиферативными заболеваниями и расстройствами, такими как опухоль или рак, в которой указанная упаковка включает, по крайней мере, одно соединение настоящего изобретенияю В определенных дополнительных вариантах осуществления фармацевтическая упаковка включает инструкции для лечения заболевания.
При упоминании в заявке термин «фармацевтическая упаковка» или «фармацевтическая фасовка» относится к любой упаковочной системе для хранения и фасовки индивидуальных дозировок медикамента. Предпочтительно, фармацевтическая упаковка содержит количество единиц дозирования, достаточное для периода лечения, или количество, удобное для режима пациента. В определенных вариантах осуществления фармацевтическая упаковка включает одну или более емкость включающую активный ингредиент, например, соединение настоящего изобретения. Такая емкость может представлять собой контейнер, такой как бутылка, сосуд, шприц или капсула, или может представлять собой стандартную лекарственную форму, такую как пилюля. Активный ингредиент может предусматриваться в емкости в своей фармацевтически приемлемой форме или может предусматриваться, например, в виде лиофилизированного порошка. В дополнительных вариантах осуществления, фармацевтическая упаковка может дополнительно включать растворитель для приготовления активного ингредиента для введения. В определенных вариантах осуществления активный ингредиент может содержаться уже в системе доставки, таком как шприц, или подходящее устройство для доставки может быть включено в упаковку. Фармацевтическая упаковка может включать пилюли, жидкости, гели, таблетки, драже или фармацевтические препараты в любой другой подходящей форме. Упаковка может содержать любое число дневных фармацевтических единиц дозировки. Упаковка может иметь любую форму, и стандартные лекарственные формы могут располагаться любым образом, таким как круговая, треугольная, трапециидальная гексагональная или другая конфигурация. Одна или более дозировка или субъединица может выделяться, например, в помощь доктору, фармацевту или пациенту, например, применением окрашивания, метками, отпечатками, рельефом, нанесением линий или композиций. Фармацевтическая упаковка может также включать инструкции для пациента, доктора, фармацевта или любого другого имеющего отношение к лечению лица.
Некоторые варианты осуществления включают применение более одного активного ингредиента, включая соединения, описанные в заявке. Такое применение может осуществляться одновременно или последовательно. Активные ингредиенты могут входить в состав единой лекарственной формы, так что одно введение обеспечивает доставку обоих компонентов. Альтернативно, активные ингредиенты могут входить в разные лекарственные формы. Фармацевтическая упаковка может включать соединение настоящего изобретения и другой фармацевтический ингредиент в единой прописи, т.е. они входят в состав одной лекарственной формы, или соединение настоящего изобретения и другой фармацевтический ингредиент могут входить в состав разных лекарственных форм. Каждая лекарственная форма может включать соединение настоящего изобретения и другой фармацевтический ингредиент в количестве индивидуальных дозировок (в примерно равных или неравных количествах). Применение соединения настоящего изобретения и другого фармацевтического ингредиента создает концентрацию, дающую в комбинации терапевтически эффективное количество.
При упоминании в заявке термин «инструкция» означает этикетку на продукте и/или документы или другую информацию, описывающие релевантные материалы и методологии, относящиеся к комплектации, приготовлению или применению набора или упаковки препарата. Эти материалы могут включать любую комбинацию следующего: справочную информацию, шаги или правила применения, перечень компонентов, подходящие дозировки, предостережения относительно возможных побочных эффектов, инструкция по применению лекарства, справочные материалы или любые другие родственные документы. Инструкции могут быть в печатной форме, такие как этикетка на упаковке или вкладыш в упаковку. Инструкции для упакованного препарата или фармацевтической композиции могут быть вложены в картонный футляр или в финишную упаковку, например, в виде вкладыша в упаковку, а текст вкладыша должен быть одобрен компетентным органом контроля, таким как Управление по контролю за качеством пищевых продуктов и медикаментов (FDA) США. Альтернативно или дополнительно, инструкция может также храниться в электронной форме, например, на читаемом компьютером носителе, таком как устройства компьютерной памяти, централизованная база данных, магнитный носитель, такой как жесткий диск, дискета и магнитная лента; оптический носитель, такой как компакт-диск, CD-ROM и голографические устройства; магнитно-оптические носители, такие как флоптический диск; и аппаратное оборудование, которое специально конфигурируется для хранения и обработки программного кода, такие как интегральные схемы прикладной ориентации (ASICs), программируемое логическое устройство (PLDs) и ROM (постоянное запоминающее устройство) и RAM (оперативное запоминающее устройство) устройства. Инструкции могут включать вэб-адрес соответствующего вэб-сайта в Интернете, с которого можно загрузить более подробные инструкции, или записанную презентацию. Инструкции могут содержать один или несколько документов для будущего обновления.
Таким образом, в одном аспекте изобретение относится к фармацевтической упаковке, включающей фармацевтическую композицию настоящего изобретения и инструкции, которые указывают, что указанная фармацевтическая композиция может применяться для лечения индивида, нуждающегося в нем.
В определенных вариантах осуществления такой фармацевтической упаковки, указанная инструкция указывает, что данная фармацевтическая композиция может применяться для лечения человека.
В определенных вариантах осуществления такой фармацевтической упаковки, указанная инструкция указывает, что данная фармацевтическая композиция может применяться для лечения индивида, страдающего заболеванием или расстройством.
В определенных вариантах осуществления такой фармацевтической упаковки, указанная инструкция указывает, что данная фармацевтическая композиция может применяться для лечения человека, страдающего заболеванием или расстройством.
В другом аспекте изобретение относится к способу лечения заболевания или расстройства у индивидуума, включающему воздействие на клетки, вовлеченные в указанное заболевание или расстройство, соединения настоящего изобретения.
В определенных вариантах осуществления этого способа указанное соединение, или его пролекарство, вводится указанному индивиду.
В определенных вариантах осуществления этого способа указанный индивид представляет собой млекопитающее, выбранное из: домашнего млекопитающего, собаки, кошки, лошади, овцы, коровы, грызунов и человека.
В определенных вариантах осуществления этого способа указанное млекопитающее представляет собой человека.
В другом аспекте изобретение относится к способам ингибирования пролиферации клеток, включающим контактирование клетки с соединением настоящего изобретения.
В другом аспекте настоящее изобретение относится к применению соединения настоящего изобретения для изготовления медикамента для лечения заболевания или расстройства.
В другом аспекте настоящее изобретение относится к применению фармацевтической композиции, включающей соединение настоящего изобретения и фармацевтически приемлемый наполнитель, разбавитель или носитель, для лечения заболевания или расстройства.
В определенных вариантах осуществления такой фармацевтической композиции, фармацевтической упаковки, способа или применения настоящего изобретения заболевание или расстройство представляет собой пролиферативное заболевание или расстройство.
В определенных вариантах осуществление указанное пролиферативное заболевание или расстройство представляет собой рак.
В определенных вариантах осуществление указанное заболевание или расстройство представляет собой воспалительное заболевание или расстройство.
Для получения по данному изобретению соединений общей формулы (1) было разработано несколько синтетических схем.
Синтез необходимых производных имидазоазинов (4), как и их азааналогов осуществляют рядом методов (схема I):
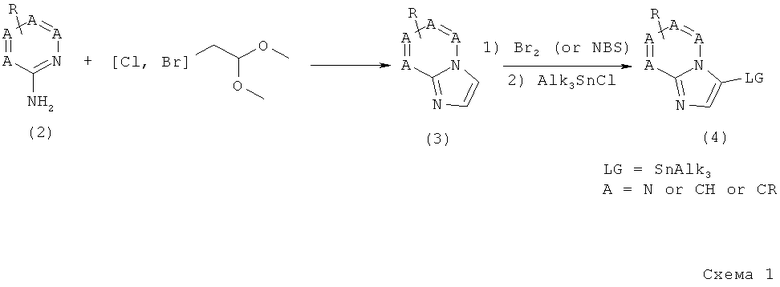
При взаимодействии производных 2-аминоазинов (и ряда других аминоазинов, таких как 2-аминопиримидин, 2-аминопиразин) общей формулы (2) с диалкилацеталями хлоро(бром)уксусного альдегида с хорошим выходом получают производные имидазоазинов (3). Эти соединения пригодны в качестве субстрата для катализируемой палладием реакции кросс-сочетания Хека. Полученные имидазоазины (3) легко бромируются (иодируются) в имидазольное кольцо действием различных галогенирующих реагентов. Синтезированные, таким образом, галогенсодержащие имидазоазины переводятся в триалкилоловопроизводные в результате взаимодействия соответствующих галогенсодержащих имидазоазинов с гексаалкилдиоловом (или триалкиоловомгалогенидом) в присутствии различных катализаторов на основе палладия (как правило (тетракис)трифенилфосфин палладия(0) и др.). В результате получают соединения общей формулы (4), которые являются подходящим субстратом для реакции кросс-сочетания по Стиле.
Синтез моно-галоген производных пиридинов и пиразинов (схема 2):
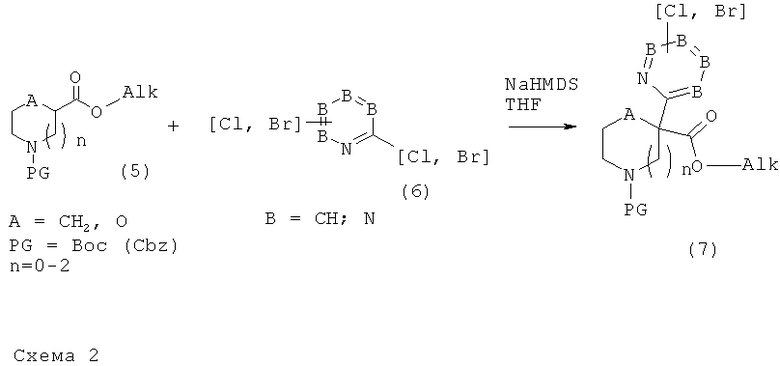
Взаимодействием натриевой соли гексаметилдисилазана (1.1 экв.) с соответствующими эфирами карбоновых кислот (1 экв.) (соединения общей структуры 5), которые содержат в α-положении к карбонильной группе С-Н кислый атом водорода, получают натривую соль, которая реагирует далее с дигалогенпроизводными пиридинов (а также других диналогеназинов, содержащих активированный атом галогена). Реакцию проводят при умеренном охлаждении (-50-0 С°) в сухом тетрагидрофуране в течение 1-4 часов.
Полученный продукт (7) выделяют путем разбавления реакционной массы водой с последующей экстракцией дихлорметаном и концентрированием при пониженном давлении. Далее, сложноэфирную группу омыляют до карбоновой кислоты действием 10% водного раствора гидроксида калия при умеренном нагревании (60-70 С°). Полученную кислоту (9) выделяют, подкисляя реакционную смесь водной хлороводородной кислотой до рН=4 и экстрагируруя дихлорметаном. Органический экстракт кислоты (9) концентрируют при пониженном давлении и используют в дальнейших превращениях без дополнительной очистки. Галогенпроизводные (10) получают декарбоксилированием соответствующих кислот (9) в диметилсульфоксиде при 100 С° или в смеси при 120-130 С° (схема 3):
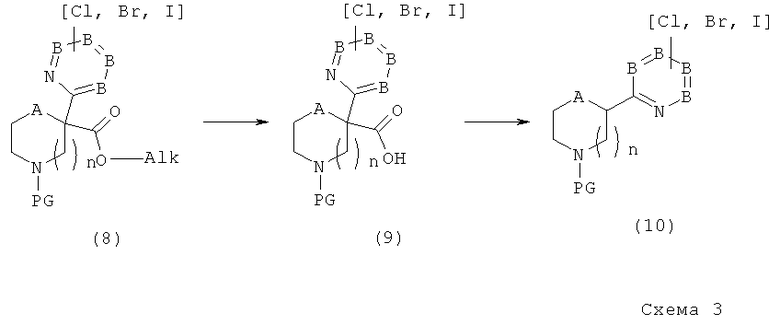
Получение моно-галоген производных пиримидинов проводили согласно схеме 4:

На основе карбоновых кислот общей формулы (9а) синтезируют кетоэфиры (11). Последние получают в сухом ацетонитриле действием метилового эфира малоновой кислоты мононатриевой соли на соответствующий имидазолид (промежуточный продукт образуется при взаимодействии кислоты (9а) и карбонилдиимидазола) в присутствии хлорида магния. Выделенные кетоэфиры (11) легко реагирует с амидинами (гуанидинами) в присутствии метилата натрия в различных растворителях (спирт, диоксан и др.) и образуют производные гидроксипиримидинов (13).
На основе карбоновых кислот (9а) синтезируют амидины (12) действием триэтилоксония тетрафторбората на первичный амид кислоты (9а) в сухом дихлорметане с последующей обработкой образовавшегося имидоэфира насыщенным раствором аммиака в спирте (метанол, этанол) в течение 12-24 часов при комнатной температуре. После удаления растворителя при пониженном давлении из реакционной смеси выделяют соответствующие амидины (12), которые используют в дальнейших превращениях без дополнительной очистки. Выделенные амидины (12) легко реагируют с кетоэфирами (производными ацетоуксусного эфира) в присутствии оснований (карбонаты калия (цезия), фосфаты калия, диазобициклоундецен и др.) в различных растворителях (спирт, диоксан и др.), образуя производные пиримидинов (14).
Производные пиримидинов (13-14) при реакции с оксихлоридом фосфора (V) в присутствии N,N-диметиланилина (4-5 экв.) в бензоле (толуоле) при комнатной температуре с высоким выходом образуют хлорпиримидины (15), которые используют в дальнейших реакциях кросс-сочетания для получения конечных целевых соединений.
Ситез моно-галоген производного пиридинов (пиразинов, пиримидинов), связанных метиленовым (этиленовым) «мостиком»:
Синтез указанных производных осуществляют путем взаимодействия дигалогенпроизводных пиридинов (пиразинов, пиримидинов) и экзоциклических алкенов.
Для синтеза алкенов применяют 2 подхода (Схема 5):
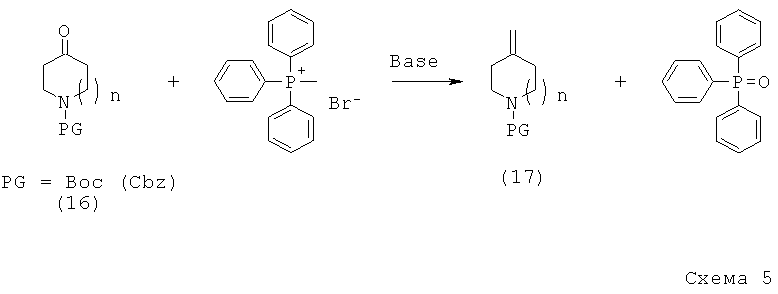
Первый подход основан на использовании реакции Виттига (реакция карбонильных соединений с илидами фосфора).
Указанную реакцию обычно проводят в тетрагидрофуране, диоксане или диметилсульфоксиде. Для этого суспендируют фосфониевую соль в соответствующем растворителе. Далее добавляют основание (как правило - гексаметидисилазид натрия (или лития) или гидрид натрия (1,1-1,2 эквивалента). После выдержки в течение 1-2 часа (генерация фосфониевого илида) прибавляют порциями (или по каплям) раствор соответствующего кетона. Далее реакционную смесь постепенно доводят до комнатной температуры и выдерживают несколько часов (от 3 до 24 часов). Выделение продукта реакции стандартное (обработывают реакционную смесь водой, далее целевой продукт экстрагируют дихлорметаном, экстракт промывают водой, затем насыщенным раствором соли, сушат над сульфатом натрия и концентрируют в вакууме). Вещества выделяют методом хроматографии на силикагеле (Flash-хроматография) с использованием в качестве растворителя смесей гексана и этилацетата. Выходы целевых продуктов находятся в диапазоне - 60-85%.
Второй подход основан на внутримолекулярной реакции «двойного» нуклеофильного присоединения путем циклизации производных 1,2- аминоспиртов (или 1,2-диаминов) с дихлорпропеном. В ходе реакции получают соответствующие семичленные карбогетероциклы (Схема 6).

При взаимодействии этаноламина с третбутилоксикарбонилпирокарбонатом в дихлорметане при комнатной температуре получают N-третбутилоксикарбонильное производное. В случае этилендиамина вначале получают моно-Вос-производное (используют 5-6 кратный избыток этилендиамина - чтобы снизить вероятность получения ди-Вос-производного). Далее, синтезированное моно-Вос-производное переводят в Boc, Cbz-производное этилендиамина по реакции ацилирования с Cbz-Cl.
Полученные таким образом, интермедиаты (18а, b) реагируют с дихлорпропеном с образованием семичленного цикла в присутствии подходящего основании по принципу двойной реакции нуклеофильного замещения. Подобную реакцию проводят либо в безводном диметилформамиде (1,4-диоксане) в присутствии поташа, либо в условиях реакций в двухфазной системе (вода-дихлорметан) в присутствии катализатора межфазного переноса (используется реже).

Полученные алкены (17) и (19a, b) при реакции с 9-BBN (9-борабициклононан, (20) (по принципу реакции гидроборирования) образуют алкил-9-BBN производные, которое используется в последующих превращениях без промежуточного выделения в виде раствора в ТГФ (~0.2-0.4 М раствор). Указанное превращение проводят при комнатной температуре в среде тетрагидрофурана в течение 24 часов. Реакцию можно ускорить (время реакции 3-4 часа) если проводить при более высокой температуре (например, при кипячении в ТГФ).
Наиболее важной стадией синтеза ключевых интермедиатов является реакция кросс-сочетания алкил-9-BBN производных, как показано выше, и ароматических гетероциклических галогенидов (или их синтетических эквивалентов) (соединения (4), схема 1) в присутствии палладиевых (никелевых) катализаторов и оснований (обычно поташ, щелочь и др.) в водно-диоксановой (тетрагидрофурановой, диглим и др.) средах.
В качестве источника палладия обычно применяют:
- ацетат (трифторацетат) палладия;
- комплекс палладия(0) с дибензилиденацетоном;
-комплексы хлорида палладия(II) с различными алкенами (пропен, 1,5-циклооктадиен и др.);
В качестве лигандов обычно используют:
- фосфины (реже фосфиты);
- карбены на основе имидазола (и его восстановленных форм);
- дикарбонильные соединения (например, ацетилацетон);
Иногда применяют методику получения катализаторов in-situ. Так используют предварительно приготовленную форму катализатора (тетракис)трифенилфосфина палладия.
Очистку и последующее выделение продуктов реакции обычно осуществляют методом колоночной хроматографии на силикагеле с использованием типичных органических растворителей (н-гексан, петролейный эфир, этилацетат, дихлорметан и прочее).
Реакции кросс-сочетания полученных ранее галоген-производных пиридинов (пиримидина, пиразина) таких как (10), (15), (21), (22) с производными имидазоазинов и их аналогов (4) приводят к ключевым интермедиатам общей формулы (24) (схема 8):

Указанное превращение осуществляется в условиях реакции Хека (LG = H) либо реакции Стилле (LG = SnAlk3) при катализе комплексными соединениями палладия в присутствии основания или же без такового.
В качестве источника палладия обычно применяют:
- ацетат(трифторацетат)палладия(II);
- комплекс палладия(0) с дибензилиденацетоном;
- комплексы хлорида палладия(II) с различными алкенами (пропен, 1,5-циклооктадиен и др.).
В качестве лигандов, входящих в состав комплексов палладия обычно используют:
- фосфины (реже фосфиты);
- карбены на основе имидазола (и его восстановленных форм);
- дикарбонильные соединения (ацетилацетон например).
Для получения целевых соединений необходимо в полученных ранее производных (24) удалить «защитные» группы (например, Boc, Cbz).
В случае Вос-производных процедура сводится к обработке (24) раствором хлороводорода в безводном 1,4-диоксане или смесью трифторуксусная кислота - дихлорметан. Температура может варьировать от комнатной до 100 С° (в случае 1,4-диоксана). Снятие Cbz-группы обычно осуществляется действием 30% раствора бромоводорода в ледяной уксусной кислоте, либо каталитическим гидрогенолизом (в присутствии металлического палладия на активированном угле). Выходы в таких реакциях, как правило, высокие (более 80%) или почти количественные.
Полученные, таким образом, вторичные (или первичные) амины далее переводят в амиды (сульфамиды), карбаматы, третичные амины, карбаматы и другие производные с использованием хорошо разработанных методов органического синтеза.
Синтез амидов осуществляют путем взаимодействия между аминами и кислотами (или их активированных производных) в присутствии конденсирующего агента (весь спектр агентов известный на сегодняшний день в пептидной химии, например TBTU, CDI) и, как правило, органического основания (например, триэтиламина) в инертном растворителе (инертном в условиях реакции ацилирования, например, ацетонитриле).
Получение третичных аминов проводят реакцией между соответствующими аминами и карбонильными соединениями (альдегидами и кетонами) в присутствии восстановителя (комплексные боро- или металлогидриды, молекулярный водород, органические восстановители).
Реакцию N-арилирование осуществляют путем взаимодействия между аминами и ароматическими (гетероароматическими) галогенидами (вариант реакции Бухвальда-Хартвига) или бороновьми кислотами в присутствии солей меди(II) (вариант реакции Чана).
Очистку и последующее выделение продуктов реакции обычно осуществляют методом колоночной хроматографии на силикагеле с использованием типичных органических растворителей (н-гексан, петролейный эфир, этилацетат, дихлорметан, четыреххлористый углерод, метанол) или же методом препаративной ВЭЖХ на обращено-фазовой колонке с использованием в качестве подвижной фазы смеси воды с метанолом (ацетонитрилом).
В ряде специфических случаев, как будет продемонстрировано в разделе «Примеры», часть соединений была получена с помощью синтетических схем, не попадающих под данное выше формальное описание общей методологии синтеза.
Примеры
Выборка соединений, лежащих в границах настоящего изобретения, представлена в Таблице 1. Соединения Таблицы 1 были синтезированы в соответствии с примерами 1-35 ниже, а ингибирующая активность Haspin киназы этих соединений в биохимических экспериментах представлена в Таблице 5. Профиль селективности избранного соединения на панели серин-треониновых и тирозинкиназ представлен в Таблице 6.
Материалы и методы
Все исходные материалы, реагенты и растворители были химически чистыми и использовались без дополнительной очистки. Растворители для хроматографии имели чистоту «для ВЭЖХ» и применялись без дополнительной очистки. Реакции контролировали при помощи тонкослойной хроматографии (ТСХ) с использованием пластин для тонкослойной хроматографии Merck Silica Gel 60 F-254. Колоночную флэш-хроматографию выполняли на силикагеле Merck silica gel 60 (0.015-0.040 мм).
ЖХ/МС анализ соединений выполняли на Surveyor MSQ (Thermo Fisher Scientific) с использованием химической ионизации при атмосферном давлении.
1. Тип ВЭЖХ колонки: ZORBAX Eclipse XDB-C18 2.1×15 мм, 1.8 мкм; Part No: 921700-902
2. Растворитель: 50% ДМСО, 50% ацетонитрил
3. Скорость потока: 750 мкл/мин; температура колонки 25°С.
4. Подвижная фаза: А=0.1% раствор муравьиной кислоты в воде, В = ацетонитрил
5. Градиент:
6. Обнаружение: диодная матрица (PDA), 200-800 нм; фотодиодный матричный детектор. Обнаружение проводилось на всем диапазоне ультрафиолетовой и видимой области от 200 до 800 нм.
APCI (+ и/или - ионы) - химическая ионизация при атмосферном давлении ELSD (испарительный детектор светорассеяния) (PL-ELS 2100)
7. Общее время пробега: 3 мин
8. Объем инъекции: 2.5 мкл.
Спектры 1Н ЯМР были сняты на «MERCURY plus 400 MHz» спектрометре (Varian). Значения химических сдвигов в м.д. даны относительно тетраметилсилана (ТМС), используя резонанс остатков растворителя в качестве внутреннего стандарта.
Препаративная ВЭЖХ очистка выполнялась на хроматографе Agilent 1100 Series, с использованием колонки Luna С18(2) (Phenomenex, USA), 250×21.2 мм, мкм.
Реакции в микроволновом реакторе СЕМ Discovery Unit проводили при мощности излучения 200 W с непрерывным перемешиванием и контролем температуры.
Методика определения ингибирования киназной активности Haspin киназы
Рекомбинантная человеческая Haspin киназа была получена из коммерческих источников.
Взаимодействие тестируемых соединений с киназой Haspin определяли методом иммуноанализа сэндвич-типа (ELISA - enzyme-linked immunosorbent assay).
Краткое описание эксперимента:
Реакцию на определение киназной активности проводили в полипропиленовых планшетах (Costar, 3363) в реакционном буфере (20 мМ HEPES, рН 7.5, 15 мМ MgCl2, 2 мМ DTT, 0.2 мМ Na3VO4, 0.005% Triton Х-100) в течение 60 минут при 30°С и интенсивном встряхивании на горизонтальном шейкере для планшетов. Конечные концентрации компонентов реакции составляли: 0.05 мкг/мл Haspin kinase, 5 нМ субстрата Histon Н3 (1-21) биотинилированного (Anaspec, 61702), 150 мкМ АТР (Sigma, A6419), исследуемых соединений (от 10 мкМ с полулогарифмическим шагом) и 5% ДМСО.
Ферментативную реакцию останавливали буфером, содержащим 20 мМ HEPES (Sigma, Н4034), рН 7.5, и 150 мМ EDTA (Sigma, E5513).
Для детекции фосфорилированного субстрата реакционную смесь переносили в заранее подготовленные планшеты (Nunc, 468667), покрытые нейтравидином (Pierce, 31000) 1 нг/лунку и обработанные бычьим сывороточным альбумином для блокирования мест неспецифического связывания, и инкубировали в течении часа при комнатной температуре. После трехкратной отмывки планшетов фосфатно-солевым буфером с Tween-20, последовательно проводили инкубацию с anti-phospho-Histon НЗ антителами (Millipore, 04-746) 0.3 нг/мкл, и инкубацию со специфическими антителами, конъюгированными ферментной меткой Anti-rabbit IgG, HRP-lmked Antibody (Cell Signaling, 7074) титр 1/5000. По завершении каждой стадии инкубации (60 минут при комнатной температуре и постоянном перемешивании) платы трижды отмывали от несвязавшихся молекул антител.
Перед измерением оптической плотности проводили остановку цветной реакции с помощью 0.5 М серной кислоты.
Оптическую плотность раствора окрашенного продукта измеряли при λ=450 нм с использованием планшетного спектрофотометра (TECAN Safire).
Значения IC50 ингибирования активности Haspin киназы вычисляли с использованием программы GraphPad Prism 5.0
Для расчета значений IC50 использовались данные не менее чем двух независимых экспериментов, причем стандартное отклонение не превышало 20%.
Определение профиля селективности для избранного соединения
Определение профиля селективности для избранного соединения проводили методом радиолигандного анализа в компании ProQinase (Германия)






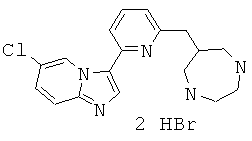
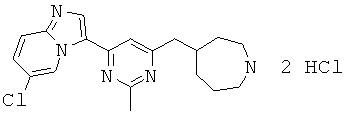
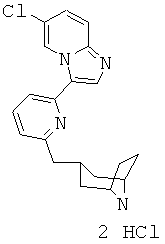
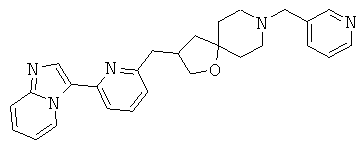
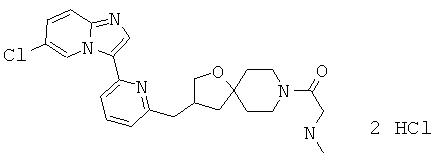
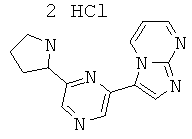
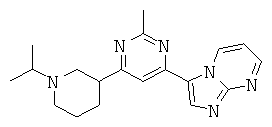
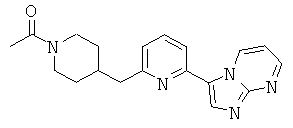

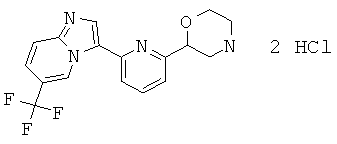
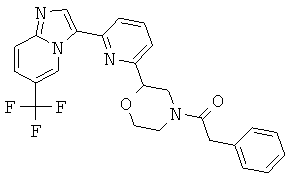
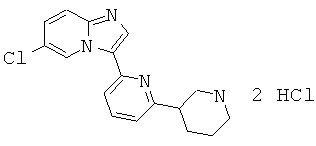

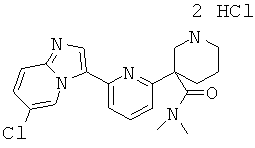

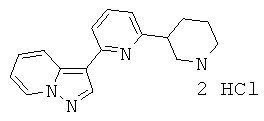
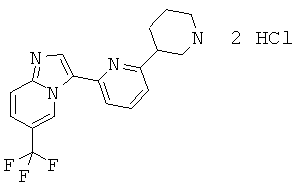
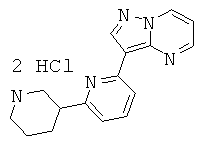
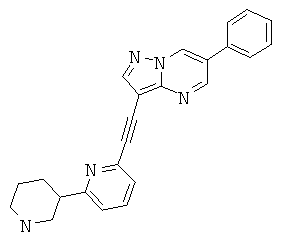


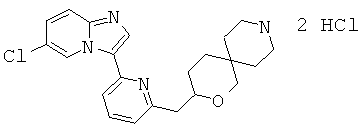
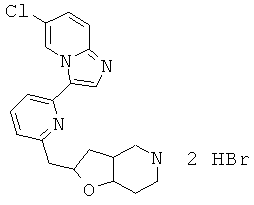
Синтез ключевых интермедиатов
Интермедиаты 4a-k получали с использованием коммерчески доступных дигалоген-азинов (3) и алкенов общей формулы (2) по следующей схеме в три стадии (схема 1):
Схема 1
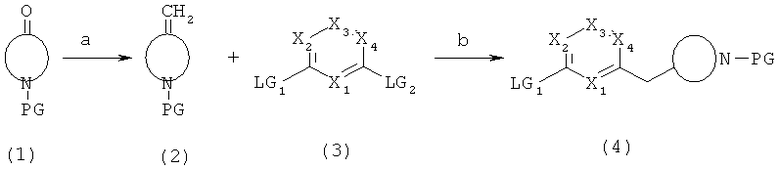
Общая методика синтеза замещенных алкенов (2), стадия (а), схема 1:
Для синтезов алкенов использовали коммерчески доступные карбонильные соединения. Часть карбонильных соединений была синтезирована по описанным в литературе методам.
К суспензии метилтрифенилфосфонийиодида (50 ммоль) в безводном ТГФ (100 мл) постепенно добавили гидрид натрия (75 ммоль) под инертной атмосферой при 5°С. Реакционную смесь перемешивали 1 ч при 10°С. После этого к реакционной смеси при 5°С добавили раствор соответствующего карбонильного соединения (1) (45 ммоль) в безводном ТГФ (20 мл). Полученную реакционную смесь перемешивали при комнатной температуре 12 часов. После окончания реакции реакционную смесь вылили в воду (150 мл) и экстрагировали дихлорметаном (2×70 мл). Органический слой отделяли, сушили над безводным сульфатом натрия и упаривали в вакууме роторного испарителя. Остаток очищали методом колоночной хроматографии на силикагеле. Выходы полученных замещенных алкенов указаны в Таблице 2.

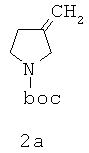
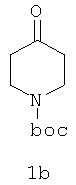
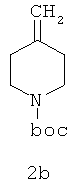

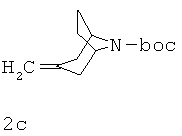
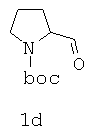

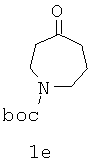

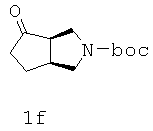
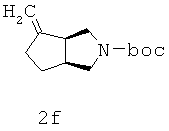



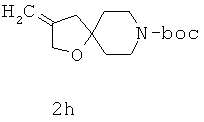

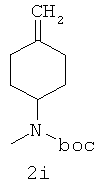
Синтез интермедиатов (4a-k). Стадия (b), схема 1
Алкены 2a-i были синтезированы, как описано выше. Алкены (2j) и (2k) (Таблица 3) были синтезированы по методике, приведенной в патентной литературе (WO 2004/74291 А1, стр.67-68).
К раствору соответствующего алкена (2) (0.1 моль) в 80 мл безводного ТГФ под атмосферой аргона прибавили 9-борабицикло[3.3.1]нонан (0.1 моль, 200 мл, 0.5М раствор в ТГФ). Полученный раствор перемешивали при комнатной температуре 24 ч. После этого добавили соответствующий галогенид (3) (0.12 моль), карбонат калия (110.4 г, 0.8 моль), воду (80 мл) и Pd(PPh3)4 (5.5 г, 0.0067 моль). Полученную реакционную смесь кипятили 3 ч, затем охлаждали до комнатной температуры и выливали в воду со льдом. Реакционную смесь несколько раз экстрагировали этилацетатом. Объединенные органические экстракты высушивали над безводным сульфатом натрия в вакууме. Остаток очищали колоночной хроматографией на силикагеле и получили соответствующий ключевой интермедиат (4). Выходы указаны в Таблице 3.
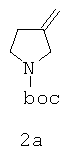
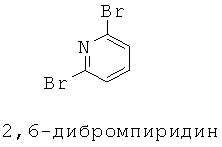
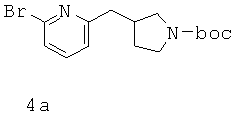
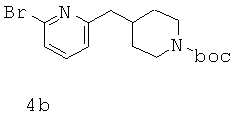
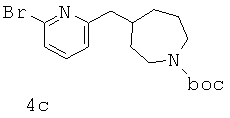

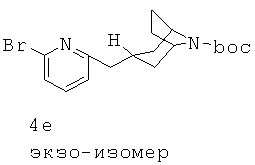
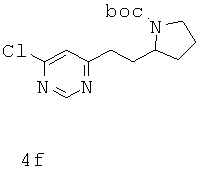
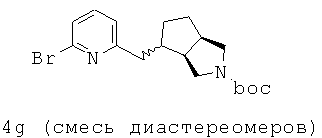



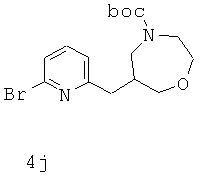

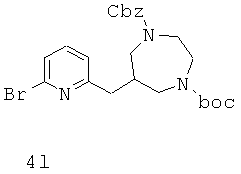
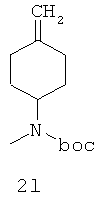
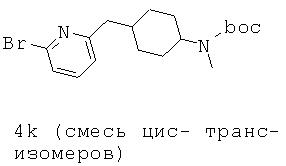
Интермедиаты 8a-d получали с использованием коммерчески доступных метиловых эфиров циклических аминокислот (5) и дигалоген-азинов (3) по Схеме 2 в три стадии:
Схема 2
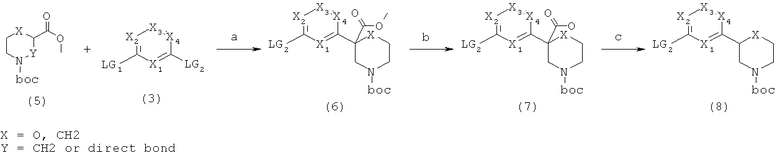
Стадия (а), синтез интермедиатов (6)
К раствору сложного эфира (5) (1.5 моль) в безводном ТГФ (800 мл) по каплям и при интенсивном перемешивании при 0°С добавили раствор натрия бис(триметилсилил)амида (1М в ТГФ, 1950 мл). Смесь перемешивали 1,5 ч при 0°С, а затем по каплям прибавили раствор соответствующего галогенида (3) (1.6 моль) в безводном ТГФ (700 мл). После этого реакционную смесь еще перемешивали 3 ч при 0°С. Затем осторожно добавили раствор аммония хлорида (67 г, 1.1 моль) в воде. Органический слой отделили и концентрировали в вакууме. Продукт (6) использовали в следующей стадии без дополнительной очистки.
Стадия (b), синтез интермедиатов (7)
К раствору сложного эфира (6) в этаноле (300 мл) добавили 10% раствор гидроксида натрия в воде (2000 мл). Смесь кипятили 5 ч (ТСХ контроль). Реакционную смесь охладили, концентрировали в вакууме до половины первоначального объема и экстрагировали МТВЕ (метил-трет-бутиловым эфиром) (2×150 мл). Водный слой подкислили 3М соляной кислотой до рН=4 и экстрагировали дихлорметаном (3×150 мл). Объединенные органические фракции высушивали над безводным сульфатом натрия. Растворитель упаривали в вакууме и получали продукт (7). Кислоты (7) использовали в следующей стадии без дополнительной очистки.
Интермедиаты 8a-d, Стадия с
Раствор кислоты (7) в ДМСО (400 мл) перемешивали при 120°С в течение 5 ч (ТСХ контроль). Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (700 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические экстракты высушивали над сульфатом натрия. Растворитель упаривали в вакууме и получали сырой продукт (8), который очищали колоночной хроматографией на силикагеле. В Таблице 4 приведены выходы интермедиатов 6, 7 и 8
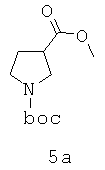

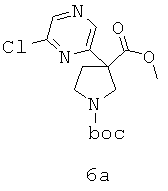
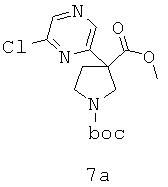

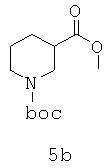
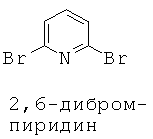

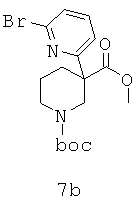
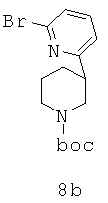
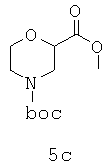
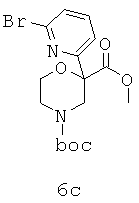

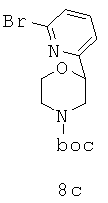
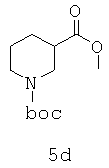
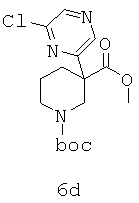
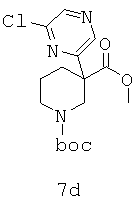
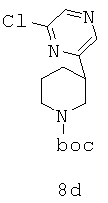
Интермедиат (11) получали по Схеме 3
Схема 3
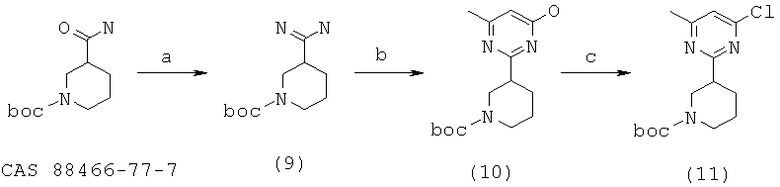
3-Карбамимидоил-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (9)
К раствору трет-бутилового эфира 3-карбамоил-пиперидин-1-карбоновой кислоты (296 г, 1.3 моль) в дихлорметане при постоянном перемешивании добавляли триэтилоксония тетрафторборат (270 г, 1.365 моль); полученную реакционную смесь перемешивали 4 ч при комнатной температуре. После этого реакционную смесь концентрировали при ~45°С.Остаток обработали раствором NH3/CH3OH (рН=10) и оставили на ночь. Реакционную смесь концентрировали в вакууме. Остаток использовали на следующей стадии без дополнительной очистки.
3-(4-Гидрокси-6-метил-пиримидин-2-ил)-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (10)
Соединение (9) (94 г, 0.3 моль) добавили к раствору этилата натрия (0.3 моль) в безводном этаноле (200 мл). Смесь перемешивали при комнатной температуре 10 мин, а затем небольшими порциями добавили метил-3-оксобутират (23.2 г, 0.2 моль). Полученную смесь кипятили 2 ч (ТСХ контроль). Реакционную смесь концентрировали под пониженным давлением, остаток растворяли в воде, подкисляли 1N HCl до рН 5.0 и экстрагировали продукт этилацетатом (2×200 мл). Объединенные экстракты высушивали над сульфатом натрия и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (элюент: н-гексан/этилацетат 1:1) и получили соединение (10) (39 г, 74%).
3-(6-Метил-4-хлор-пиримидин-2-ил)пиперидин-1-карбоновой кислоты трет-бутиловый эфир (11)
Интермедиат (10) (50 г, 0.170 моль) и N,N-диметиланилин (166 г, 1.365 моль) растворяли в безводном толуоле (1000 мл) и по каплям добавляли POCl3 (52 г, 0.34 моль). Реакционную смесь кипятили 3 ч; затем охлаждали до комнатной температуры и оставляли на ночь. После этого смесь выливали в воду, органический слой отделяли и промывали последовательно 1N HCl и водой. Сырой продукт концентрировали в вакууме и очищали флэш-хроматографией на силикагеле (элюент: н-гексан/этилацетат 4:1), получили продукт (11) (34.3 г, 65%).
СИНТЕЗ КОНЕЧНЫХ СОЕДИНЕНИИ
Пример 1
3-(6-(Пиперидин-4-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорид
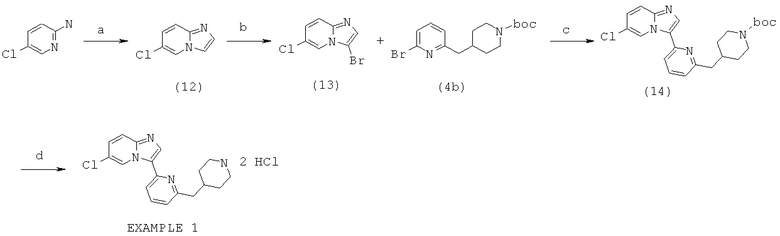
6-хлоримидазо[1,2а]пиридин (12)
Суспензию 5-хлор-2-аминопиридина (0.195 моль), хлорацетальдегида (50% водный раствор) (0.234 моль) и гидрокарбоната натрия (0.195 моль) в 150 мл воды перемешивали 8 часов при комнатной температуре. Затем реакционную смесь подкислили концентрированной хлороводородной кислотой до рН=1, перемешивали реакционную смесь при комнатной температуре в течение часа, а затем подщелочили водным раствором NaOH до рН=9 и экстрагировали дихлорметаном (2 раза по 50 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, затем упарили в вакууме роторного испарителя. Сырой продукт очищали методом флэш-хроматографии на силикагеле. В качестве элюента использовали градиент растворителей н-гексан/этилацетат от 100% н-гексана до 20% этилацетата. Выход: 85%.
3-Бром-6-хлоримидазо [1,2-а] пиридин (13)
К раствору соединения (12) (45 ммоль) в 120 мл безводного дихлорметана при интенсивном перемешивании и комнатной температуре добавили N-бромсукцинимид (50 ммоль) небольшими порциями. Реакционную смесь перемешивали четыре часа при комнатной температуре. Затем упарили на роторном испарителе примерно треть объема растворителя. Выпавший осадок отфильтровали, промыли небольшим количеством метил-трет-бутилового эфира. Объединенные фильтраты промыли водой (дважды по 50 мл), сушили над безводным сульфатом натрия и упарили в вакууме роторного испарителя. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. В качестве элюента использовали градиент растворителей этил н-гексан/этилацетат от 100% н-гекасана до 10% этилацетата. Выход: 70%.
4-[6-((6-хлоримидазо11,2-а]пиридин-3-ил)-пиридин-2ил)метил]-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (14)
К раствору соединения (13) (20 ммоль) в безводном ТГФ (100 мл) в инертной атмосфере при -78°С прибавили через капельную воронку 13 мл (25 ммоль) 2М раствора изопропилмагнийхлорида в ТГФ. Реакционную смесь перемешивали полтора часа при -78°, затем через капельную воронку прибавили раствор три(н-бутил)оловахлорида (25 ммоль) в 30 мл ТГФ. После чего реакционной смеси позволили нагреться до комнатной температуры в течение часа. При интенсивном перемешивании добавили раствор соединения 4b (Таблица 3) (20 ммоль) в 30 мл ТГФ и 1.5 ммоль Pd(PPh3)4. Реакционную смесь кипятили при перемешивании 3 часа. После окончания реакции реакционную смесь охладили до комнатной температуры и вылили в 5% водный раствор фторида калия (500 мл), после чего перемешивали 12 часов, а затем экстрагировали этилацетатом. Экстракт промыли водой, сушили над безводным сульфатом натрия и упарили на роторном испарителе. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. В качестве элюента использовали смесь н-гексан/этилацетат 3:2. Выход: 66%
3-((6-пиперидин-4-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорид
К раствору соединения (14) (1 ммоль) в метаноле (5 мл) добавляли раствор 16% хлороводорода в безводном 1,4-диоксане (1 мл), перемешивали 24 часа при комнатной температуре. Затем растворитель упарили в вакууме. Остаток кристаллизовали из смеси этилацетат/метанол. Выход: 95%.
Масс-спектр (APCI), m/z (Iотн, %): 327.18 [М+Н]+ (100), 329.20 [М+Н]+ (35).
ПРИМЕР 2
3-(6-(Азепан-4-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорид

Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединений (13) и ключевого интермедиата (4 с) (Таблица 3). Выход на стадии с) 51%, на стадии d) 90%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м. д.: 1.32-1.48 (м, 1Н), 1.61-1.97 (м, 5Н), 2.16-2.27 (м, 1H), 2.81-3.27 (м, 6Н), 7.29-7.38 (м, 1Н), 7.82-8.04 (м, 4Н), 8.85 (с, 1Н), 8.98-9.22 (м, 2Н), 10.25 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 341.28 [M+H]+ (100), 343.45 [M+H]+ (35).
ПРИМЕР 3
3-((6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)метил)-1-окса-8-азаспиро[4.5]декана дигидрохлорид

Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединений (13) и ключевого интермедиата (4i) (Таблица 3). Выход на стадии а) 45%, на стадии b) 88%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м. д.: 1.48-1.62 (м, 1Н), 1.65-1.96 (м, 4Н), 1.97-2.09 (м, 1Н), 2.76-3.10 (м, 7Н), 3.50-3.62 (м, 1Н), 3.91-4.05 (м, 1Н), 7.35-7.39 (м, 1Н), 7.90-8.12 (м, 4н0, 8.91-9.25 (м, 3Н), 10.29 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 383.25 [М+Н]+ (100), 385.22 [М+Н]+ (35).
ПРИМЕР 4
3-((6-(6-(Трифторметил)имидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)метил)-1-окса-8-азаспиро[4.5]декана дигидрохлорид
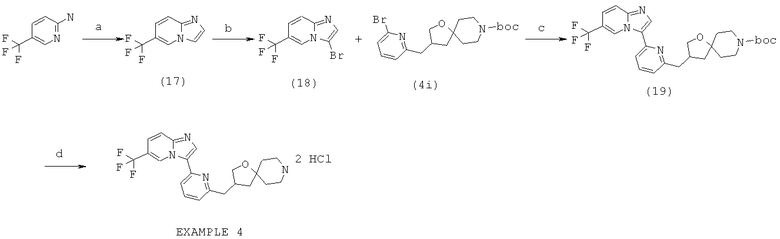
Вышеназванное соединение было получено в четыре стадии способом, аналогичным приведенному ранее в Примере 1, исходя из коммерчески доступного 5-трифторметил-2-аминопиридина. Выход на стадии а) 71%, на стадии b) 88%, на стадии с) 61%, на стадии d) 90%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м. д.: 1.51-1.56 (м, 1Н), 1.71-1.89 (м, 4Н), 1.98-2.03 (м, 1Н), 2.80-2.88 (м, 1Н), 2.95-2.03 (м, 6Н), 3.54 (т, 1Н), 3.94 (т, 1Н), 7.31 (д, 1Н), 7.85-7.96 (м, 3Н), 8.07 (д, 1Н), 8.78 (с, 1Н), 8.93 (уш. с, 2Н), 10.61 (с, 1Н)
Масс-спектр (APCI), m/z (Iотн, %): 417,19 [М+Н]+ (100).
ПРИМЕР 5
3-((6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)метил)-1,4-оксазепана дигидрохлорид
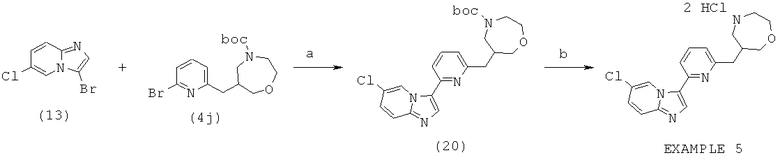
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4j) (Таблица 3). Выход на стадии а) 65%, на стадии b) 98%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м. д.: 2.68-2.71 (м, 1Н), 2.87-2.97(м,2Н), 3.09-3.33 (м, 4Н), 3.51-3.62 (м, 1Н), 3.78-3.92 (м, 3Н), 7.43 (м, 1Н), 7.97-8.10 (м, 2Н), 8.08 (к, 2Н), 8.82-9.09 (м, 3Н), 10.22 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %):343.11 [М+Н]+ (100), 345.14 [М+Н]+ (35).
ПРИМЕР 6
{4-[6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-пиридин-2-илметил]-циклогексил}-метиламина дигидрохлорид
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4k) (Таблица 3). Выход на стадии а) 70%, на стадии b) 78%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.11 (к, 2Н), 1.39 (к, 2Н), 1.48-1.68 (м, 2Н), 1.72-1.93 (м, 3Н), 2.02-2.13 (м, 2Н), 2.72-2.91 (м, 3Н), 3.01-3.12 (м, 1H), 7.34 (д, 1Н), 7.91-7.98 (м, 2Н), 8.01-8.06 (м, 1Н), 8.08-8.12 (м, 1Н), 9.00 (с, 1Н), 9.12-9.20 (м, 2Н), 10.27-10.33 (м, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 355.28 [М+Н]+ (100), 357.25 [M+H]+ (35).
ПРИМЕР 7
3-(6-(((3aS,6aR)-Октагидро-циклопента[с]пиррол-4-ил)метил)пиридин-2-ил)-6-хлоримидазо[1,2-a]пиридина дигидрохлорид (смесь диастереомеров)

Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4g) (Таблица 3). Выход на стадии а) 46%, на стадии b) 97%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.48-1.79 (м, 4Н), 2.66-2,82 (м, 3Н), 2.90-3.08 (м, 3Н), 3.12-3.22 (м, 2Н), 3.32-3.46 (м, 1Н), 7.38-7.41 (м, 1Н), 7.92-8.11 (м, 4Н), 8.98-9.51 (м, 2Н), 10.29 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 353.22 [М+Н]+ (100), 355.23 [М+Н]+ (35).
ПРИМЕР 8
3-(6-(((3aS,7aR)-Октагидро-1H-изоиндол-4-ил)метил)пиридин-2-ил)-6-хлоримидазо[1,2-a]пиридина дигидрохлорид (смесь диастереомеров)
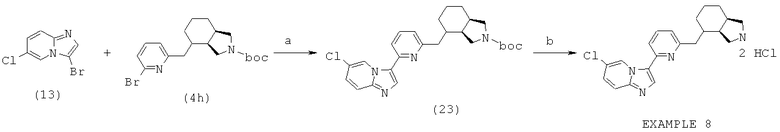
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4h) (Таблица 3). Выход на стадии а) 69%, на стадии b) 93%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.17-1.29 (м, 2Н), 1.42-1.72 (м, 3Н), 1.99-2.43 (м, 3Н), 2.74-2.92 (м, 2Н), 2.94-3.25 (м, 5Н), 7.32-7.51 (м, 1Н), 7.94-8.12 (м, 4H), 8.99 (с, 1H), 9.41-9.78 (м, 2Н), 10.29 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 367.24 [М+H]+ (100), 369.24 [М+Н]+ (35).
ПРИМЕР 9
3-(6-((1,4-Диазепан-6-ил)метил)пиридин-2-ил)-6-хлоримидазо[1,2-a]пиридина дигидробромид
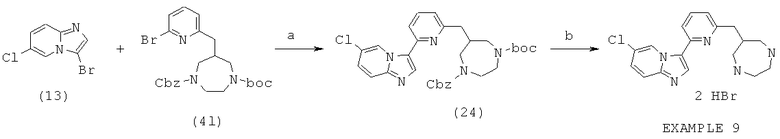
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4l) (Таблица 3). Выход на стадии а) 29%. Стадию b) проводили следующим образом: соединение (24) (1 ммоль) нагревали при 60°С в 30% растворе HBr в уксусной кислоте в течение 30 минут. После этого реакционную смесь упарили в вакууме роторного испарителя. Остаток обработали диэтиловым эфиром, отфильтровали, промыли на фильтре несколько раз эфиром и сушили на воздухе. Выход: 83%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 2.86-3.03 (м, 3Н), 3.18-3.57 (м, 8Н), 7.42-7.51 (м, 1н), 7.92-8.11 (м, 3Н), 8.92 (с, 1н), 9.02-9.30 (м, 3Н), 10.12 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 342.25 [М+Н]+ (100), 344.16 [М+Н]+ (35).
ПРИМЕР 10
3-(6-(Азепан-4-илметил)-2-метилпиримидин-4-ил)-6-хлоримидазо[1,2-a]пиридина дигидрохлорид

Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4d) (Таблица 3). Выход на стадии а) 90%, на стадии b) 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.32-1.43 (м, 1Н), 1.58-1.92 (м, 5Н), 2,22-2,33 (м, 1Н), 2.71-2.86 (м, 5Н), 2.92-3.22 (м, 4Н), 7.81-7.86 (м, 1Н), 7.96-8.01 (м, 1Н), 8.07 (с, 1Н), 9.04 (с, 1Н), 9.11-9.13 (м, 2Н), 10.12 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 356.25 [М+H]+ (100), 358.25 [М+Н]+ (35).
ПРИМЕР 11
3-(6-((1R,5S)-8-Азабицикло[3.2.1]окт-3-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-a]пиридина дигидрохлорид
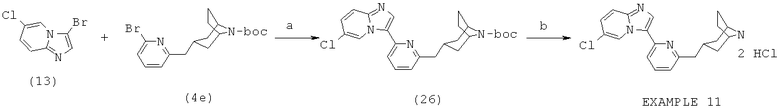
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединения (13) и ключевого интермедиата (4e) (Таблица 3). Выход на стадии а) 79%, на стадии b) 87%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.60-1.78 (м, 3Н), 1.91-2.01 (м, 1Н), 2.21-2.43 (м, 3Н), 2.76-2.84 (м, 1Н), 3.06-3.19 (м, 3Н), 4.85-4.96 (м, 2Н), 7.35-7.45 (м, 1Н), 7.93-8.03 (м, 3Н), 8.07-8.12 (м, 1Н), 8.93-9.11 (м, 2Н), 9.28-9.43 (м, 1Н), 10.29 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 353.23 [М+Н]+ (100), 355.21 [М+Н]+ (35).
ПРИМЕР 12
3-((6-(Имидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)метил)-8-(пиридин-3-илметил)-1-окса-8-азаспиро[4.5]декан
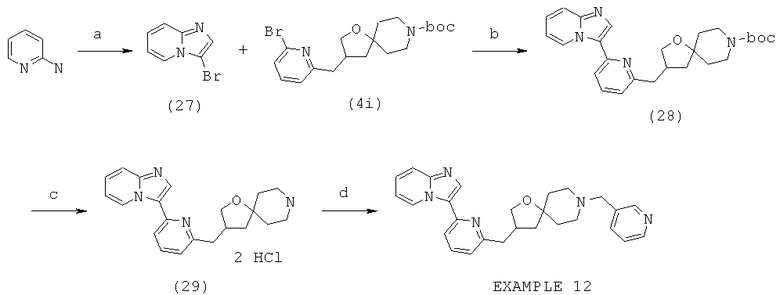
Вышеназванное соединение было получено в четыре стадии способом, аналогичным приведенному ранее в Примере 1, исходя из коммерчески доступного 2-аминопиридина. Выход на стадии а) 90%, на стадии b) 89%, на стадии с) 51%. Стадию d) проводили следующим образом: смесь соединения (29) (0.5 ммоль), 3-пиридинкарбальдегида (0.55 ммоль), триацетоксиборгидрида натрия (1 ммоль) в ацетонитриле (5 мл) перемешивали 24 часа при комнатной температуре. После реакционную смесь разбавили водой, экстрагировали этилацетатом. Экстракт промыли водой, высушили над безводным сульфатом натрия и упарили на роторном испарителе. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. Выход: 79%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.20-1.31 (м, 1Н), 1.32-1.66 (м, 5Н), 1.85-1.98 (м, 1Н), 2.21-2.40 (м, 3Н), 2.72-2.98 (м, 3Н), 3.35-3.52 (м, 3Н), 3.86-3.92 (м, 1Н), 7.07 (т, 1Н), 7.14 (д, 1Н), 7.31 (м, 1Н), 7.38 (т, 1Н), 7.63-7.72 (м, 1Н), 7.74-7.83 (м, 1Н), 8.34 (с, 1Н), 8.40-8.46 (м, 2Н), 9.95 (д, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 440.27 [М+Н]+ (100).
ПРИМЕР 13
2-Метиламино-1-{3-[6-(6-хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-этанона дигидрохлорид
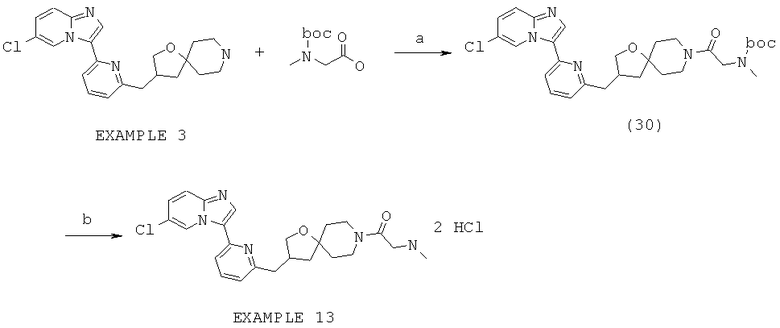
(2-{3-[6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-пирвдин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-2-оксоэтил)-метилкарбаминовой кислоты трет-бутиловый эфир (30)
Смесь 3-((6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-1-окса-8-азаспиро[4.5]декана (ПРИМЕР 3) (0. 5 ммоль), N-Boc-саркозина (0.55 ммоль), TBTU и триэтиламина (2 ммоль) в ацетонитриле (4 мл) перемешивали 24 часа при комнатной температуре. После окончания реакции реакционную смесь вылили в воду и экстрагировали ДХМ. Экстракт промыли водой, высушили над безводным сульфатом натрия и упарили на роторном испарителе. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. Выход: 90%
2-Метиламино-1-{3-[6-(6-хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-этанона дигидрохлорид (ПРИМЕР 13)
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, (стадия d) исходя из соединения (30). Выход: 98%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.51-1.72 (м, 4Н), 1.97-2.05 (м, 1Н), 2.82-2.87 (м, 1Н), 2.97-3.02 (м, 2Н), 3.23-3.37 (м, 3Н), 3.55-3.58 (м, 2Н), 3.73-3.78 (м, 1Н), 3.97-3.98 (м, 3Н), 7.34 (д, 1Н), 7.90-7.96 (м, 3Н), 8.01-8.05 (м, 1Н), 8.9 (м, 3Н), 10.28 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 454.21 [М+Н]+ (100), 456.20 [М+Н]+ (35)
ПРИМЕР 14
3-(6-(Пирролидин-2-ил)пиразин-2-ил)имидазо[1,2-a]пиримидин дигидрохлорид
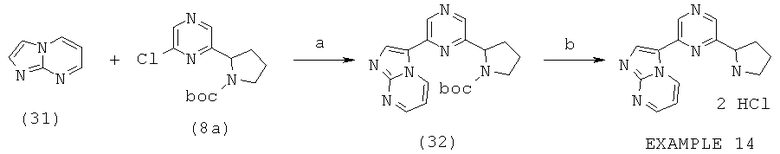
Имидазо[1,2-а]пиримидин (31) был синтезирован в соответствии с литературным методом [Loeber, Stefan; Huebner, Harald; Gmeiner, Peter Bioorganic & Medicinal Chemistry Letters, 1999, vol.9, #1 p.97-102]
2-(6-Имидазо[1,2-а]пиримидин-3-илпиразин-2-ил)-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (32)
Имидазо[1,2-а]пиримидин (1 ммоль), соединение (8a) (Таблица 4) (1 ммоль), карбонат калия (3 ммоль), ацетат палладия(II), (5% мольных), трис-(циклогексил)фосфин (10% мольных) растворили в безводном ДМФА (4 мл). Реакционную смесь продули аргоном и нагревали в микроволновой печи при 150°С в течение 30 минут (мощность излучения 150 Вт). Реакционную смесь охладили, вылили в воду, экстрагировали этилацетатом. Экстракт промыли водой, сушили над безводным сульфатом натрия и упарили на роторном испарителе. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. В качестве элюента использовали смесь хлороформ/метанол 20:1. Выход: 34%
3-(6-(Пирролидин-2-ил)пиразин-2-ил)имидазо[1,2-a]пиримидина дигидрохлорид (ПРИМЕР 14)
К раствору соединения (32) (0.34 ммоль) в метаноле (1 мл) прибавляли 16% HCl в диоксане (1 мл), перемешивали 24 часа при комнатной температуре, затем растворитель удаляли в вакууме. Выход 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 2.07-2.24 (м, 4Н), 2.57-2.59 (м, 1H), 3.41-3.44 (м, 2Н), 4.92 (м, 1Н), 7.38 (с, 1Н), 8.71 (с, 1H), 8.81 (с, 1H), 8.87 (с, 1H), 9.34 (с, 1Н), 9.40-9.52 (м, 1Н), 9.23-9.25 (м, 1Н), 10.66-10.77 (м, 1Н)
Масс-спектр (APCI), m/z (Iотн, %): 267.30 [M+H]+ (100).
ПРИМЕР 15
3-(6-(1-Изопропилпиперидин-3-ил)-2-метилпиримидин-4-ил)имидазо[1,2-a]пиримидин
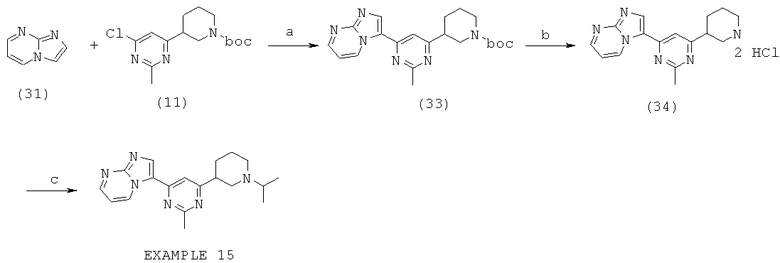
3-(2-Метил-6-пиперидин-3-илпиримидин-4-ил)-имидазо[1,2-а]пиримидина дигидрохлорид (34)
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 14, исходя из соединений (31) (4 ммоль) и (11) Выход: стадия а) 25%, стадия b) 99%
3-(6-(1-Изопропилпиперидин-3-ил)-2-метилпиримидин-4-ил) имидазо[1,2-a]пиримидин (ПРИМЕР 15)
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в ПРИМЕРЕ 13 (стадия d), исходя из соединения (34) (1 ммоль) и ацетона. Выход: 51%
Масс-спектр (APCI), m/z (Iотн, %): 337.40 [М+Н]+ (100).
ПРИМЕР 16
1-(4-((6-(Имидазо[1,2-a]пиримидин-3-ил)пиридин-2-ил)метил)пиперидин-1-ил)этанон
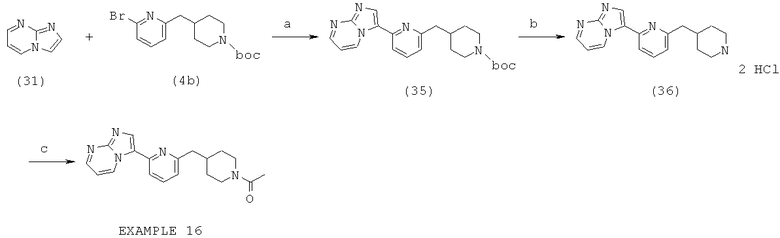
3-(6-Пиперидин-4-илметилпиридин-2-ил)-имидазо[1,2-a]пиримидина дигидрохлорид (36)
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 14, исходя из соединений (31) (2 ммоль) и (4b) (Таблица 3). Выход: стадия а) 49%, стадия b) 99%
1-(4-((6-(Имидазо[1,2-а]пиримидин-3-ил)пиридин-2-ил)метил)пиперидин-1-ил)этанон (ПРИМЕР 16)
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в ПРИМЕРЕ 13 (стадия а), исходя из соединения (36) (1 ммоль) и уксусной кислоты. Выход: 93%
Масс-спектр (APCI), m/z (Iотн, %): 336.40 [М+Н]+ (100).
ПРИМЕР 17
1-(2-(2-(6-(Имидазо[1,2-а]пиримидин-3-ил)пиримидин-4-ил)этил)пирролидин-1-ил)этанон

3-[6-(2-Пирролидин-2-илэтил)-пиримидин-4-ил]-имидазо[1,2-а]пиримидина дигидрохлорид (38)
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенном ранее в ПРИМЕРЕ 14, исходя из соединений (31) (4 ммоль) и (4f) (Таблица 3). Выход: стадия а) 23%, стадия b) 99%, стадия с) 88%
1-(2-(2-(6-(Имидазо[1,2-a]пиримидин-3-ил)пиримидин-4-ил)этил)пирролидин-1-афтанон (ПРИМЕР 17)
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в ПРИМЕРЕ 13 (стадия а), исходя из соединения (36) (1 ммоль) и уксусной кислоты. Выход: 93%
Масс-спектр (APCI), m/z (Iотн, %): 337.39 [М+Н]+ (100).
ПРИМЕР 18
3-(6-Пирролидин-3-илметилпиридин-2-ил)-имидазо[1,2-а]пиримидина дигидрохлорид

Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 14, исходя из соединений (31) (4 ммоль) и (4а) (Таблица 3). Выход: стадия а) 39%, стадия b) 99%
3-(6-((1-(Метилсульфонил)-пирролидин-3-ил)метил)пиридин-2-ил)имидазо[1,2-a]пиримидин (ПРИМЕР 18)
Смесь соединения (40) (0. 5 ммоль), метансульфохлорида (0.52 ммоль), и триэтиламина (2 ммоль) в ацетонитриле (4 мл) перемешивали 24 часа при комнатной температуре. После окончания реакции реакционную смесь вылили в воду и экстрагировали ДХМ. Экстракт промыли водой, высушили над безводным сульфатом натрия и упарили на роторном испарителе. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. Выход: 76%
Масс-спектр (APCI), m/z (Iотн, %): 358.40 [М+Н]+ (100).
ПРИМЕР 19
3-(6-(Пиперидин-3-ил)пиридин-2-ил)-6-хлоримидазо[1,2-a]пиридина дигидрохлорид
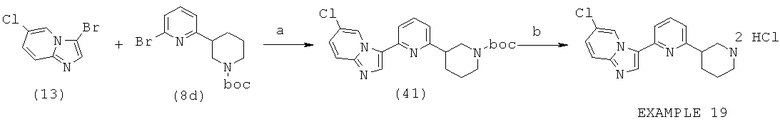
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 2, исходя из соединения (13) (2 ммоль) и (8b) (Таблица 4). Выход: стадия а) 59%, стадия b) 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.72-1.98 (м, 1Н), 2.12-2.30 (м, 1Н), 2.80-2.92 (м, 1Н), 3.09-3.21 (м, 1Н), 3.30-3.43 (м, 2Н), 3.55-3.63 (м, 1Н), 7.35-7.42 (м, 1Н), 7.70-7.81 (м, 1Н), 7.86-8.00 (м, 3Н), 8.71 (с, 1Н), 9.26-9.51 (м, 2Н), 10.09 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %):313.19 [M+Н]+ (100), 315.18 [М+Н]+ (35)
ПРИМЕР 20
3-(6-(Пиперидин-3-ил)пиразин-2-ил)-6-хлоримидазо[1,2-a]пиридина дигидрохлорид
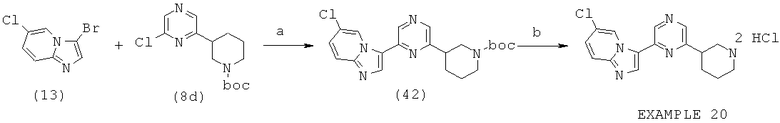
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 2, исходя из соединения (13) (2 ммоль) и (8d) (Таблица 4). Выход: стадия а) 69%, стадия b) 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.71-1.98 (м, 3Н), 2.10-2.18 (м, 1Н), 2.80-2.98 (м, 1Н), 3.12-3.22 (м, 2Н), 3.28-3.40 (м, 2Н), 7.65-7.72 (м, 1Н), 7.88-7.95 (м, 1Н), 8.60 (м, 1Н), 8.75 (м, 1Н), 9.12-9.41 (м, 3Н), 9.72 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 314.20 [М+Н]+ (100), 316.22 [М+Н]+ (35)
ПРИМЕР 21
3-(6-(Пиперидин-3-ил)пиридин-2-ил)-6-(трифторметил)-имидазо[1,2-a]пиридина дигидрохлорид
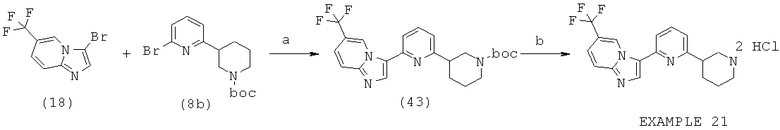
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 2, исходя из соединения (18) (1 ммоль) и (8b) (Таблица 4). Выход: стадия а) 70%, стадия b) 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.75-1.96 (м, 3Н), 2.10-2.21 (м, 1Н), 2.71-2.87 (м, 1Н), 3.03-3.12 (м, 1Н), 3.30-3.50 (м), 7.38-7.42 (м, 1Н), 7.88-8.10 (м, 4Н), 8.75 (с, 1Н), 9.11-9.32 (м, 1Н), 10.52 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %):347.20 [М+Н]+ (100).
ПРИМЕР 22
2-(6-(6-(Трифторметил)имидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)морфолина дигидрохлорид

Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 2, исходя из соединения (18) (4 ммоль) и (8 с) (Таблица 4). Выход: стадия а) 45%, стадия b) 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 3.05-3.28 (м, 2Н), 3.54-3.68 (м, 2Н), 4.01-4.22 (м, 2Н), 5.07 (д, 1Н), 7.52 (д, 1Н), 7.88 (д, 1Н), 8.01-8.15 (м, 3Н), 8.78 (с, 1Н), 9.66 (уш. с, 1Н), 9.82 (уш. с, 1Н), 10.49 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %):349.33 [М+Н]+ (100).
ПРИМЕР 23
1-(2-(6-(6-(Трифторметил)имидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)морфолино)-2-фенил-этанон
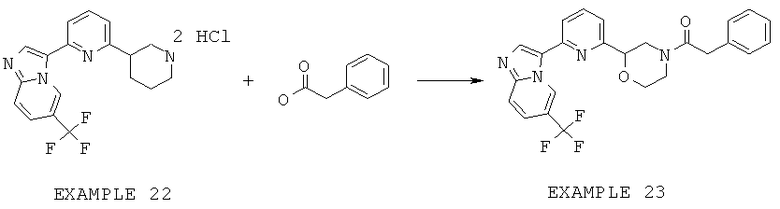
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в ПРИМЕРЕ 13 (стадия а), исходя из соединения (ПРИМЕР 22) (0.5 ммоль) и фенилуксусной кислоты. Выход: 67%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 2.28-3.00 (м, 1Н), 3.52-3.68 (м, 1Н), 3.78 (с, 2Н), 3.95-4.04 (м, 2Н), 4.25-4.41 (м, 1Н), 4.54-4.58 (м, 1Н), 4.61-4.77 (м, 1Н), 7.12-7.45 (м, 6Н), 7.58-7.66 (м, 1Н), 7.89-8.06 (м, 3Н), 8.57 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 467.19 [М+Н]+ (100).
ПРИМЕР 24
3-(6-(Пиперидин-3-ил)пиридин-2-ил)-6-хлоримидазо[1,2-b]пиридазина дигидрохлорид
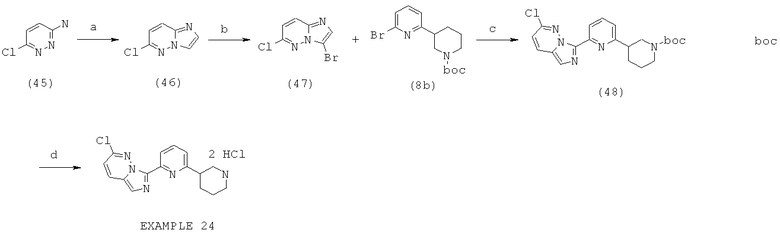
Вышеназванное соединение было получено в четыре стадии способом, аналогичным приведенному ранее в Примере 1, исходя из коммерчески доступного 3-амино-6-хлорпиридазина. Выход на стадии а) 41%, на стадии b) 48%, на стадии с) 29%, на стадии d) 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.72-1.82 (м, 1Н), 1.84-1.89 (м, 2Н), 2.06-2.12 (м, 1Н), 2.94-2.96 (м, 1Н), 3.26-3.34 (м, 3Н), 3.48-3.51 (м, 1Н). 7.40 (д, 1Н), 7.55 (д. 1Н), 7.99 (т, 1Н), 8.29 (д, 1Н), 8.38 (д, 1Н), 8.57 (с, 1Н), 9.30-9.33 (м, 2Н).
Масс-спектр (APCI), m/z (Iотн, %): 314.18 [М+Н]+ (100), 316.16 [M+H]+ (100).
ПРИМЕР 25
6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)-1',4',5',6'-тетрагидро-2'H-[2,3']бипиридинил-3'-карбоновой кислоты дигидрохлорид
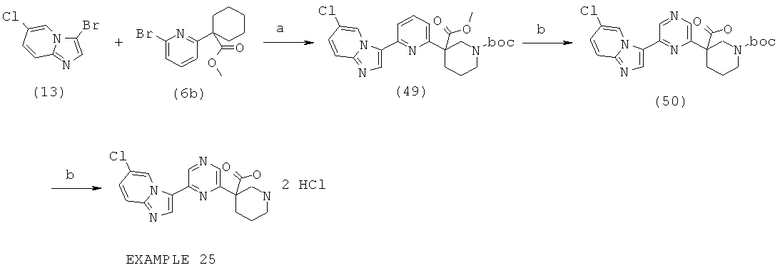
6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-5',6'-дигидро-4'Н-[2,3']бипиридинил-1',3'-дикарбоновой кислоты 1'-трет-бутиловый эфир 3'-метиловый эфир (49)
Вышеназванное соединение было получено в одну стадии способом, аналогичным приведенному ранее в ПРИМЕРЕ 2, исходя из соединения (13) (15 ммоль) и (6b) (Таблица 4). Выход: 65%
6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-5',6'-дигидро-4'Н-[2,3']бипиридинил-1',3'-дикарбоновой кислоты 1'-трет-бутиловый эфир (50)
Соединение (49) (5 ммоль) нагревали при 50°С с гидроксидом калия (10 ммоль) в водно-метанольной смеси (4:1) при перемешивании в течение 8 часов. После окончания реакции половину объема раствора упарили на роторном испарителе. Оставшуюся реакционную смесь профильтровали через стеклянный фильтр и подкислили до рН=4 разбавленной хлороводородной кислотой. Выпавший осадок отфильтровали, промыли на фильтре небольшим количеством ледяной воды и сушили в вакууме в течение 48 часов. Выход: 87%
6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)-1',4',5',6'-тетрагидро-2'H-[2,3']бипиридинил-3'-карбоновой кислоты дигидрохлорид (ПРИМЕР 25)
Вышеназванное соединение было получено в одну стадии способом, аналогичным приведенному ранее в Примере 1, (стадия d). Выход: 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.65-1.79 (м, 1H), 1.89-1.97 (м, 1Н), 2.34 (т, 1H), 2.88-3.011 (м, 1Н), 3.12-3.19 (м, 1Н), 3.35-3.46 (м, 1Н), 3.78-4.12 (м), 7.53-7.56 (м, 1Н), 7.71 (д, 1Н), 7.91 (д, 1Н), 8.03-8.04 (м, 2Н), 8.39-8.52 (м, 1Н), 8.72 (с, 1Н), 9.41-9.52 (м, 1Н), 10.04 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 313.11 [М+H-CO2]+ (100), 315.12 [М+Н-CO2]+ (35).
ПРИМЕР 26
6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-1',4',5',6'-тетрагидро-2'Н-[2,3']бипиридинил-3'-карбоновой кислоты диметиламина дигидрохлорид

6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-3'-диметилкарбамоил-3',4',5',6'-тетрагидро-2'Н-[2,3']бипиридинил-1'-карбоновой кислоты трет-бутиловый эфир (51)
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в ПРИМЕРЕ 13 (стадия а), исходя из соединения (50) (1 ммоль) и диметиламина гидрохлорида (2 ммоль). Выход: 43%
6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-1',4',5',6'-тетрагидро-2'Н-[2,3']бипиридинил-3'-карбоновой кислоты диметиламина дигидрохлорид (ПРИМЕР 26)
Вышеназванное соединение было получено в одну стадии способом, аналогичным приведенному ранее в Примере 1, (стадия d). Выход: 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.57 (к, 1Н), 2.00 (д, 1H), 2.81 (т, 2Н), 2.95 (к, 2Н), 3.22 (д, 1Н), 3.65 (д, 1Н), 7.50 (д, 1Н), 7.76 (д, 1Н), 7.95 (д, 2Н), 8.05-8.11 (м, 2Н), 8.83 (с, 1Н), 9.83-9.85 (м, 1Н), 9.99 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 384.10 [М+Н]+ (100), 386.12 [М+Н]+ (35).
ПРИМЕР 27
3-(6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)пиперидин-3-ил)метанола дигидрохлорид
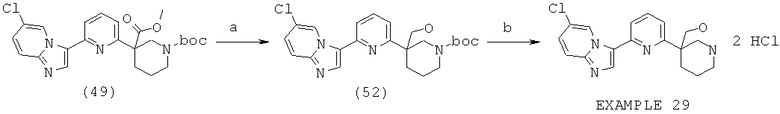
6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)-3'-гидроксиметил-3',4',5',6'-тетрагидро-2'H-[2,3']бипиридинил-1'-карбоновой кислоты трет-бутиловый эфир (52)
К раствору соединения (49) (3 ммоль) в безводном ТГФ (30 мл) при 0°С и интенсивном перемешивании добавили в несколько порций литийалюминийгидрид (3 ммоль). Реакционную смесь перемешивали при 10°С в течение 10 часов. После этого смесь охладили до 0°С и добавили 20% раствор КОН (1 мл). Осторожно декантировали прозрачный раствор с выпавшего плотного осадка и упарили в вакууме роторного испарителя. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. Выход: 66%
3-(6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)пиперидин-3-ил)метанола дигидрохлорид (ПРИМЕР 27)
Вышеназванное соединение было получено в одну стадии способом, аналогичным описанному ранее в Примере 1, (стадия d). Выход: 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.48-1.62 (м, 1Н), 1.80-1.83 (м, 1Н), 1.94-2.03 (м, 1Н), 2.22-2.25 (м, 1Н), 2.98 (уш. с, 2Н), 3.32 (т, 1Н), 3.64-3.71 (м, 2Н), 3.76-3.82 (м, 1Н), 6.78 (уш. с), 7.63 (д, 1Н), 7.92-7.97 (м, 2Н), 8.00-8.06 (м, 2Н), 8.43 (уш. с, 1Н), 8.85 (с, 1Н), 9.49-9.52 (м, 1Н), 10.02 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 343.11 [М+Н]+ (100), 345.10 [М+Н]+ (35).
ПРИМЕР 28
3-(6-(Пиперидин-3-ил)пиридин-2-ил)пиразоло[1,5-a]пиридина дигидрохлорид
6-Этинил-3',4',5',6'-тетрагидро-2'H-[2,3']бипиридинил-1'-карбоновой кислоты трет-бутиловый эфир (53)
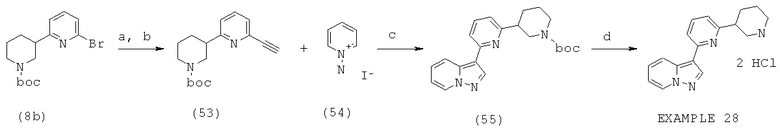
Иодид меди (I) (0.19 г, 1 ммоль), PdCl2(PPh3)2 (0.35 г, 0.5 ммоль), триэтиламин (4 мл) добавили к раствору соединения (8b) (3.5 г, 10 моль) в безводном ТГФ (20 мл) в атмосфере аргона при комнатной температуре и перемешивали в течение 5 минут. Затем к раствору прибавили по каплям триметилсилилацетилен (1.5 г, 15 ммоль). Реакционную массу перемешивали 2 часа при комнатной температуре (контроль за протеканием реакции по ТСХ и LCMS), разбавляли водой (50 мл) и экстрагировали дихлорметаном. Экстракт сушили над безводным сульфатом натрия, пропускали через слой силикагеля и упаривали на роторном испарителе. Полученный коричневый остаток растворили в ТГФ (20 мл), охладили до 5°С и добавили 1 М раствор тетрабутиламмонийфторида в ТГФ (0.5 мл). Реакционную массу перемешивали 1 час при 5°С (контроль по ТСХ), разбавили водой (50 мл) и экстрагировали дихлорметаном. Экстракт сушили над сульфатом натрия, растворитель удаляли в вакууме. Целевое соединение выделяли с помощью колоночной хроматографии на силикагеле. Выход: 59%
6-Пиразоло[1,5-а]пиридин-3-ил-3',4',5',6'-тетрагидро-2'Н-[2,3']бипиридинил-1'-карбоновой кислоты трет-бутиловый эфир (55)
К раствору коммерчески доступного соединения (54) (2 ммоль) в безводном ДМФА (5 мл) прибавили карбонат калия (2.8 ммоль), реакционную массу перемешивали 30 минут при комнатной температуре, при этом раствор изменил окраску с бесцветной на темно-синюю.
Затем прибавили ацетилен (53) (1.5 ммоль) и продолжали перемешивание при 90°С в течение 2 часов (контроль по ТСХ). Реакционную массу разбавили водой (50 мл), экстрагировали дихлорметаном. Экстракт сушили над сульфатом натрия, растворитель удаляли в вакууме. Целевое соединение выделяли с помощью колоночной хроматографии на силикагеле. Выход: 39%
3-(6-(Пиперидин-3-ил)пиридин-2-ил)пиразоло[1,5-a]пиридина дигидрохлорид (ПРИМЕР 28)
К раствору соединения (55) (0.6 ммоль) в метаноле (5 мл) добавляли 16% раствор HCl в 1,4-диоксане (1 мл), перемешивали 3 часа при комнатной температуре, затем растворитель удалили в вакууме. Целевое вещество кристаллизовали из этилацетата. Выход: 97%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.76-1.86 (м, 1Н), 1.88-1.93 (м, 2Н), 2.11-2.14 (м, 1Н), 2.93-3.03 (м, 1Н), 3.19-3.41 (м, 3Н), 3.54-3.61 (м, 1Н), 7.04 (т, 1Н), 7.17 (д, 1Н), 7.45 (т, 1Н), 7.77-7.74 (м, 2Н). 8.52 (д, 1Н), 8.69 (с, 1Н), 8.77 (д, 1Н), 9.37 (уш. с, 2Н).
Масс-спектр (APCI), m/z (Iотн, %): 279.17 [М+Н]+ (100).
ПРИМЕР 29
3-(6-(Пиперидин-3-ил)пиридин-2-ил)-6-(трифторметил)-[1,2,4]триазоло[4,3-a]пиридина дигидрохлорид (5).

1'-трет-Бутилоксикарбонил-6-циано-3',4',5',6'-тетрагидро-2'H-[2,3']-бипиридинил (56)
К раствору соединения (8b) (12 ммоль) в диметилацетамиде (18 мл) прибавили при комнатной температуре цианид цинка(II) (7.5 ммоль), цинковую пыль (1.9 ммоль), Pd2dba3 (0.26 ммоль) и DPPF (0.37 ммоль). Реакционную смесь нагревали при 130-140°С в атмосфере аргона в течение 3 часов, после чего охладили до комнатной температуры, вылили в 100 мл воды и экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия, растворитель удаляли в вакууме. Целевое соединение выделяли с помощью колоночной хроматографии на силикагеле. Выход: 70%
1'-трет-Бутилоксикарбонил-6-формил-3',4',5',6'-тетрагидро-2'Н-[2,3']-бипиридинил (57)
К раствору соединения (56)(8.2 ммоль) в сухом дихлорметане (100 мл) при -78°С в атмосфере аргона через капельную воронку прибавили (14 ммоль) 1М раствора DIBAL-H при температуре не выше -78°С. После окончания прибавления полученную реакционную массу перемешивали при -78°С в течение 3-4 часов. Затем реакционную массу нагрели до -40°С, прибавили к ней 1М раствор соляной кислоты и оставили перемешиваться до достижения комнатной температуры. После этого органический слой отделили, сушили над сульфатом натрия. Растворитель упарили при пониженом давлении. Из полученного остатка целевой продукт выделяют хроматографированием на силикагеле (элюент гексан -этилацетат). Выход: 60%
1'-трет-Бутилоксикарбонил-6-(6-трифторметил-[1,2,4]триазоло[4,3а]пиридин-3-ил)-3',4',5',6'-тетрагидро-2'Н-[2,3']бипиридинил (58)
К раствору соединения (57) (2 ммоль) в ТГФ (5 мл) при комнатной температуре прибавили (5-трифторметил-пиридин-2-ил)-гидразина (2 ммоль). Полученный раствор нагревали при 60-70°С в течение 2 часов. Наличие в реакционной смеси промежуточно образующегося гидразона контролировали с помощью ТСХ. После завершения процесса образования гидразона реакционную смесь охладили на водяной бане до комнатной температуры и прибавили 0.55 г (2.4 ммоль) хлорамина-Т. Полученную суспензию нагревали при 80°С около 1 часа. Затем реакционную массу охладили до комнатной температуры, разбавленным раствором гидроксида калия довели рН до 9-10, экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия, упарили в вакууме роторного испарителя. Целевое соединение выделяли методом флэш-хроматографии на силикагеле (элюент гексан - этилацетат). Выход: 60%
3-(6-(Пиперидин-3-ил)пиридин-2-ил)-6-(трифторметил)-[1,2,4]триазоло[4,3-a]пиридина дигидрохлорид (ПРИМЕР 29).
Вышеназванное соединение было получено в одну стадии способом, аналогичным приведенному ранее в Примере 1, (стадия d). Выход: 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.78-1.89 (м, 1Н), 1.93-1.97 (м, 2Н), 2.17-2.21 (м, 1Н), 2.75-2.87 (м, 1Н), 3.07-3.16 (м, 1Н), 3.34-3.45 (м, 2Н), 7.58 (д, 1Н), 7.76 (д, 1Н), 8.07 (т, 1Н), 8.03 (д, 1Н), 9.42 (уш. с, 2Н), 10.16 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 348.17 [М+Н]+ (100).
ПРИМЕР 30
3-(6-(Пиперидин-3-ил)пиридин-2-ил)пиразоло[1,5-a]пиримидина дигидрохлорид

4-Иод-2H-пиразол-3-иламин (59)
К раствору 3-аминопиразола (0.1 моль) в 100 мл метанола прибавили йод (0.1 моль) и карбонат калия (0.1 моль). Реакционную массу перемешивали при комнатной температуре 3 ч, затем разбавили насыщенным раствором сульфита натрия, экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и упаривали при пониженном давлении. Целевое соединение очищали методом флэш-хроматографии на силикагеле. Выход: 57%
3-Иод-пиразоло[1,5-a]пиримидин (60)
К раствору соединения (59) (10 ммоль) в 20 мл ледяной уксусной кислоты добавили малоновый диальдегид (12 ммоль). Реакционную смесь перемешивали при 70°С в течение 3 ч (контроль по ТСХ), затем охладили и разбавили водой. Образовавшийся осадок отфильтровали, промыли водой. Целевое соединение очищали методом флэш-хроматографии на силикагеле, затем кристаллизовали из смеси дихлорметан/н-гексан (1:10). Выход: 79%
3-(6-(Пиперидин-3-ил)пиридин-2-ил)пиразоло[1,5-a]пиримидина дигидрохлорид (ПРИМЕР 30)
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 1, исходя из соединений (60) и (8b). Выход на стадии с) 31%, на стадии d) 98%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.76-1.83 (м, 3Н), 2.07-2.12 (м, 1Н), 3.07-3.09 (м, 1Н), 3.18-3.24 (м, 1Н), 3.33-3.38 (м, 2Н), 3.42-3.47 (м, 1Н), 7.17-7.23 (м, 2Н), 7.87 (т, 1Н), 8.22 (д, 1Н), 8.77 (д, 1Н), 8.91 (с, 1Н), 9.22 (д, 1Н), 9.28 (уш. с, 1Н), 9.42 (уш. с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 280.15 [М+Н]+ (100).
ПРИМЕР 31
3-(6-(Пиперидин-3-ил)пиридин-2-ил)этинил)-6-фенилпиразоло[1,5-a]пиримидин
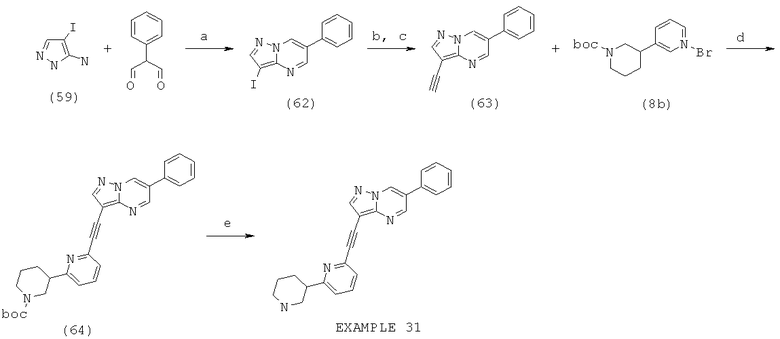
3-Иод-6-фенил-пиразоло[1,5-a]пиримидин (62)
К раствору соединения (59) (10 ммоль) в 20 мл ледяной уксусной кислоты через капельную воронку прибавили 2-фенилмалоновый альдегид (12 ммоль). Смесь перемешивали при 70°С в течение 3 ч (контроль по ТСХ), затем охладили до комнатной температуры и разбавили водой (50 мл). Образовавшийся осадок отфильтровали, промыли водой. Целевое соединение очищали методом флэш-хроматографии на силикагеле (элюент дихлорметан) и кристаллизовали из смеси дихлорметан - н-гексан (1:10). Выход: 69%
6-Фенил-3-этинил-пиразоло[1,5-а]пиримидин (63)
К раствору соединения (62) (4 ммоль) в 10 мл ТГФ в атмосфере аргона прибавили иодид меди(I) (0.4 ммоль), PdCl2(PPh3)2 (0.2 ммоль), триэтиламин (8 ммоль). Образовавшуюся смесь перемешивали 10 минут при комнатной температуре, а затем прибавили триметилсилилацетилен (8 ммоль). Реакционную массу перемешивали 4 часа при 60°С (контроль по ТСХ), затем разбавили водой (50 мл) и экстрагировали дихлорметаном. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Остаток растворили в ТГФ (20 мл), охладили до 5°С и прибавляли 1 М раствор тетрабутиламмонийфторида в ТГФ (1,5 мл). Реакционную массу перемешивали 1 час при +5°С (контроль по ТСХ), разбавили водой и экстрагировали дихлорметаном. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Целевое соединение выделяли с помощью колоночной хроматографии на силикагеле (элюент этилацетат/н-гексан, 1:4). Выход: 61%
6-(6-Фенилпиразоло[1,5-а]пиримидин-3-илэтинил)-3',4',5',6'-тетрагидро-2'Н-[2,3']бипиридинил-1'-карбоновой кислоты трет-бутиловый эфир (64)
Смесь ацетата палладия(II) (0.025 ммоль) и трифенилфосфина (0.05 ммоль) в безводном ТГФ в атмосфере аргона перемешивали в течение 10 минут, затем прибавили соединение (8b) (0.8 ммоль), соединение (63) (0.8 ммоль) и карбонат цезия (1 ммоль). Полученную смесь кипятили в течение 6 ч (контроль по ТСХ), разбавили водой, экстрагировали дихлорметаном. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Целевое соединение выделяли с помощью колоночной хроматографии на силикагеле. Выход: 40%
3-(6-(Пиперидин-3-ил)пиридин-2-ил)этинил)-6-фенилпиразоло [1,5-a]пиримидин (ПРИМЕР 31)
К раствору соединения (64) (0.2 ммоль) в 3 мл 1,4-диоксана добавили 10% водную серную кислоту (4 мл). Перемешивали при комнатной температуре 8 ч (контроль по ТСХ), затем вылили в воду, нейтрализовали карбонатом калия, образовавшийся осадок отфильтровывали, промывали водой на фильтре и сушили на воздухе. Выход: 93%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.29-1.79 (м, 3Н), 1.83-1.98 (м, 1Н), 2.58-2.63 (м, 2Н), 2.71-311 (м, 2Н), 3.97-4.29 (м, 1Н), 7.24 (д, 1Н), 7.36-7.56 (м, 5Н), 7.72 (т, 1Н), 7.85-7.92 (м, 2Н), 8.58 (с, 1Н), 9.07 (с, 1Н), 9.52 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 380.24 [М+Н]+ (100).
ПРИМЕР 32
3-(6-(Пиперидин-3-ил)пиридин-2-ил)-6-хлорпиразоло[1,5-a]пиридина дигидрохлорид
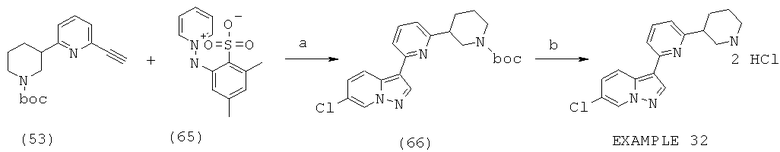
Вышеназванное соединение было получено в две стадии способом, аналогичным приведенному ранее в Примере 28 из соединений (53) и (65). Синтез соединения (65) осуществляли согласно литературному методу [Johnston, Karen A.; Allcock, Robert W.; Jiang, Zhong; Collier, Ian D.; Blakli, Haakon; Rosair, Georgina М.; Bailey, Patrick D.; Morgan, Keith М.; Kohno, Yasushi; Adams, David R. Organic and Biomolecular Chemistry, 2008, vol.6, #1 pp.175-186] Выход: стадия а) 9%, стадия b) 99%
Масс-спектр (APCI), m/z (Iотн, %): 313.15 [M+H]+ (100), 315.13 [М+Н]+ (35).
ПРИМЕР 33
3-((6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)метил)-2-окса-8-азаспиро[4.5]декана дигидрохлорид

4-Аллилпиперидин-1,4-дикарбоновой кислоты 1-трет-бутиловый эфир 4-метиловый эфир (67)
94 К раствору метилового эфира N-Boc-4-пиперединкарбоновой кислоты (0.2 моль) в безводном ТГФ (200 мл) в атмосфере аргона прибавили 2М раствор NaHMDS в ТГФ (0.22 моль) при температуре 0°С. Реакционную смесь перемешивали 1 час при 0°С, затем температуру понизили до -30°С и через капельную воронку при интенсивном перемешивании добавили аллилбромид (0.22 моль). После этого охлаждение реакционной смеси сняли и довели температуру реакционной смеси до комнатной. После окончания реакции реакционную смесь вылили в насыщенный раствор хлорида аммония, экстрагировали дихлорметаном. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Целевое соединение выделяли с помощью флеш-хроматографии на силикагеле. Выход: 91%
4-Аллил-4-гидроксиметилпиперидин-1-карбоновой кислоты трет-бутиловый эфир (68)
К раствору соединения (67) (50 ммоль) в безводном ТГФ (100 мл) при 0°С и интенсивном перемешивании прибавили в несколько приемов литийалюминийгидрид (30 ммоль). Реакционную смесь перемешивали при 10°С в течение 10 часов. После этого смесь охладили до 0°С и прибавили 20% раствор КОН (3 мл). Осторожно декантировали прозрачный раствор с выпавшего плотного осадка и отогнали растворитель в вакууме роторного испарителя. Целевой продукт выделяли методом флэш-хроматографии на силикагеле. Выход: 76%
3-[6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)-пиридин-2-илметил]-2-окса-8-азаспиро[4.5]декан-8-карбоновой кислоты трет-бутиловый эфир (70)
Смесь соединений (67) (3 ммоль), (69) [George M.Buckley и др. Bioorganic & Medicinal Chemistry Letters 18 (2008), pp.3656-3660] (3 ммоль), карбоната цезия (9 ммоль), Pd2dba3 (1% мольный), DPEphos (2% мольный) в безводном ТГФ кипятили в атмосфере аргона 15 часов при интенсивном перемешивании. После окончания реакции реакционную смесь вылили в насыщенный раствор хлорида аммония, экстрагировали дихлорметаном. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Целевое соединение выделяли с помощью флеш-хроматографии на силикагеле. Выход: 21%
3-((6-(6-Хлоримидазо[1,2-a]пиридин-3-ил)пиридин-2-ил)метил)-2-окса-8-азаспиро[4.5]декана дигидрохлорид (ПРИМЕР 33)
Вышеназванное соединение было получено в одну стадии способом, аналогичным приведенному ранее в Примере 1, (стадия d). Выход: 99%
Масс-спектр (APCI), m/z (Iотн, %): 383.22 [М+Н]+ (100), 385.24 [М+Н]+ (35).
ПРИМЕР 34
3-((6-(6-Хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-2-окса-9-азаспиро[5.5]ундекана дигидрохлорид
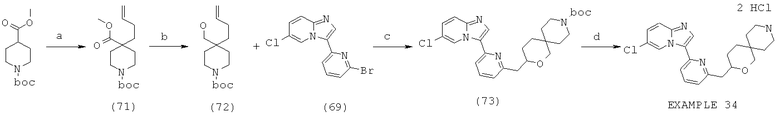
Вышеназванное соединение было получено в четыре стадии способом, аналогичным приведенному ранее в Примере 33, из коммерчески доступных метилового эфира N-Boc-4-пиперединкарбоновой кислоты (0.2 моль) и 4-бром-бут-1-ена. Выход: стадия а) 78%, стадия b) 65%, стадия с) 34%, стадия d) 99%
Масс-спектр (APCI), m/z (Iотн, %): 397.28 [M+H]+ (100), 399.28 [М+Н]+ (35).
ПРИМЕР 35
2-((6-(6-Хлоримидазо[1,2-д]пиридин-3-ил)пиридин-2-ил)метил)октагидрофуро[3,2-c] пиридина дигидробромид
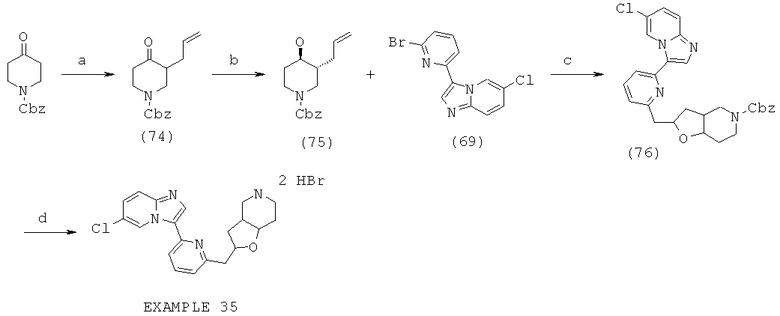
3-Аллил-4-оксопиперидин-1-карбоновой кислоты бензиловый эфир (74)
К раствору N-бензилоксикарбонил-4-пиперидона (100 ммоль) и аллилового спирта (110 ммоль) в ДМСО (100 мл) в атмосфере аргона прибавили димер аллилпалладия (1% мольный процент), BINAP (2% мольных), рацемический пролин (30 ммоль). Реакционную смесь нагревали при 70°С в течение 10 часов (контроль по ТСХ).
После окончания реакции реакционную смесь охладили, вылили в воду, экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Целевое соединение выделяли с помощью флеш-хроматографии на силикагеле. Выход: 69%
транс-3-Аллил-4-гидроксипиперидин-1-карбоновой кислоты бензиловый эфир (75) Соединение (74) (100 ммоль) растворили в безводном ТГФ (100 мл).
Реакционную смесь охладили до 0°С и добавили через капельную воронку при интенсивном перемешивании раствор трет-(втор-бутил(гидридо)бората лития (L-selectride) (150 ммоль). Реакционную смесь перемешивали 12 часов при комнатной температуре.
После завершения реакции к реакционной смеси добавили концентрированный раствор карбоната калия, экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом натрия, пропустили через слой силикагеля, отогнали растворитель при пониженном давлении. Целевое соединение выделяли с помощью флеш-хроматографии на силикагеле. Выход:72%
2-[6-(6-Хлоримидазо[1,2-а]пирвдин-3-ил)-пиридин-2-илметил]-гексагидро-фуро[3,2-с]пиридин-5-карбоновой кислоты бензиловый эфир (76)
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в Примере 33, из соединений (75) (10 ммоль) и (69). Выход: 13%
2-((6-(6-Хлоримидазо[1,2-я]пиридин-3-ил)пиридин-2-ил)метил)октагидрофуро[3,2-с]пиридина дигидробромид (ПРИМЕР 35)
Вышеназванное соединение было получено в одну стадию способом, аналогичным приведенному ранее в Примере 9, из соединения (76) (1 ммоль). Выход: 99%
Спектр ЯМР 1H (400 МГц, D6-ДМСО), δ, м.д.: 1.88-2.00 (м, 4Н), 2.56-2.69 (м, 1Н), 2.79-2.90 (м, 1Н), 3.06-3.24 (м, 4Н), 4,08 (м), 4.66-4.73 (м, 1Н), 7.40 (т, 1Н), 7.90-8.08 (м, 4Н), 8.35-8.43 (м, 1Н), 8.64-8.67 (м, 1Н), 8.94 (с, 1Н), 10.31 (с, 1Н).
Масс-спектр (APCI), m/z (Iотн, %): 369.07 [М+Н]+ (100), 371.06 [М+Н]+ (35).
Специалисту будет понятно, что настоящее изобретение адаптировано для доведения объектов до конца и получения упомянутых и подразумевающихся преимуществ.
Способы и реагенты, описанные в заявке, являются типичными в предпочтительных вариантах осуществления, но взяты в качестве примера и не должны ограничивать возможности изобретения. Их возможные модификации и другие применения известны специалистам. Эти модификации охвачены духом изобретения и включаются в формулу изобретения.
Специалистам известны или могут быть установлены с помощью рутинных экспериментов много эквивалентов конкретных вариантов осуществления описываемого в заявке изобретения. Эти эквиваленты считаются охваченными следующей ниже формулой изобретения. Специалистам также понятно, что все комбинации вариантов осуществления, комбинации аспектов или признаков формулы изобретения, описанной в заявке, лежат в границах изобретения.
Все вышеупомянутые ссылки и публикации включены в заявку посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ N-(1H-ИНДАЗОЛ-4-ИЛ) ИМИДАЗОЛ [1,2-А]ПИРИДИН-3- КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ III ТИПА | 2011 |
|
RU2591195C2 |
| НОВЫЕ ИНГИБИТОРЫ СЕРИН-ТРЕОНИНОВЫХ КИНАЗ, В ТОМ ЧИСЛЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ И ТУБЕРКУЛЕЗА | 2016 |
|
RU2633032C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| ПРОИЗВОДНЫЕ N-(ИМИДАЗОПИРИМИДИН-7-ИЛ)-ГЕТЕРОАРИЛАМИДОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10A | 2011 |
|
RU2562066C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ПИПЕРАЗИНА | 2012 |
|
RU2628616C2 |
| ИНГИБИТОРЫ ТИПА ErbB | 2006 |
|
RU2592703C9 |
| ПРОИЗВОДНЫЕ N4-ФЕНИЛХИНАЗОЛИН-4-АМИНА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ ТИПА ERBB ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2428421C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811403C2 |
| ИМИДАЗОПИРАЗИНЫ | 2012 |
|
RU2600327C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
Изобретение относится к области органической химии, а именно к новым гетероциклическим соединениям общей формулы (I) или к его таутомерной форме, или к его фармацевтически приемлемой соли, где 1-2 из X1, Х2, Х3, Х4 Х5, X6 выбран из N, и другие представляют собой С; X7 выбран из N или СН; каждый из X8, X9, X10 и Х11 независимо выбран из N или СН при условии, что фрагмент может одновременно содержать один или два атома азота; R1, R2, R3 и R4 выбраны из Н, 6-членного арила, CF3, галогена; R5, R6 R7 представляют собой C1-алкил при условии, что Х9, X10 или Х11 в этом случае соответственно равен С; ″А″ может представлять собой простую связь или мостиковый этиновый фрагмент; Y может представлять собой простую связь или независимо выбран из метиленового или этиленового мостиковых фрагментов; фрагмент Z независимо выбран из незамещенного или замещенного по атому азота гетероциклоалкила или является незамещенным или замещенным циклоалкилом при условии, что N (азот) равно С (углерод): 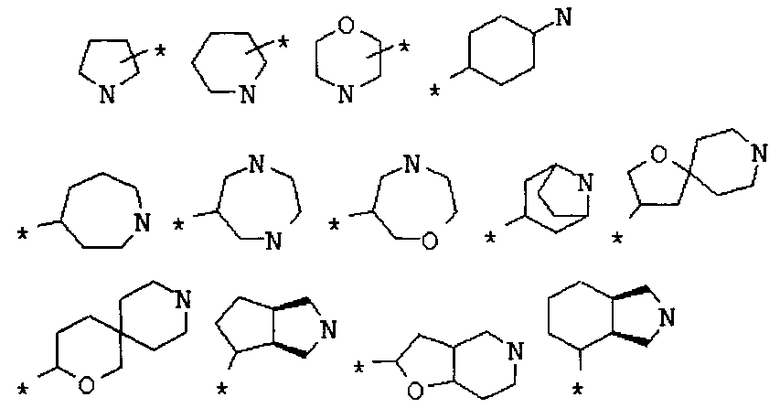 ,
, 
где R9 выбран из СН2ОН, CON(R15 R16), где R15, R16 могут независимо представлять собой Н, C1-алкил, Het представляет собой N, n=1, n1=3; R8 выбран из Н, C1-6-алкила, C1-алкилкарбонила, производных арилуксусной кислоты общего строения:  метилгетероарилов общего строения:
метилгетероарилов общего строения: 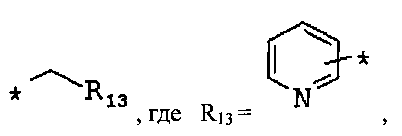
производных алкилсульфонилов общего строения:  где R14=Alk, причем Alk представляет собой С1-алкил, или к 2-метиламино-1-{3-[6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-этанона дигидрохлориду, или к 6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты дигидрохлориду, или к 6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты диметиламина дигидрохлориду. Также изобретение относится к фармацевтической композиции на основе заявленного соединения и к способу ингибирования Haspin киназы. Технический результат: получены новые соединения, обладающие полезными биологическими свойствами. 3 н. и 2 з.п. ф-лы, 6 табл., 35 пр.
где R14=Alk, причем Alk представляет собой С1-алкил, или к 2-метиламино-1-{3-[6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-этанона дигидрохлориду, или к 6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты дигидрохлориду, или к 6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты диметиламина дигидрохлориду. Также изобретение относится к фармацевтической композиции на основе заявленного соединения и к способу ингибирования Haspin киназы. Технический результат: получены новые соединения, обладающие полезными биологическими свойствами. 3 н. и 2 з.п. ф-лы, 6 табл., 35 пр.
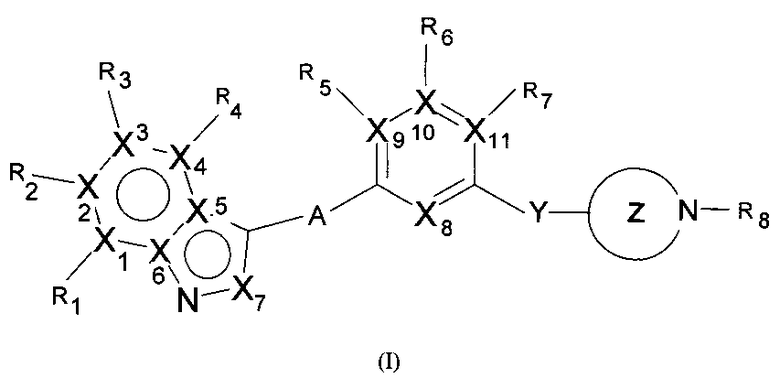
1. Соединение общей формулы (I)
или любая его таутомерная форма, или его фармацевтически приемлемая соль,
где
1-2 из X1, Х2, Х3, Х4 Х5, X6 выбран из N, и другие представляют собой С;
Χ7 выбран из N или СН;
каждый из X8, X9, X10 и Х11 независимо выбран из N или СН при условии, что фрагмент может одновременно содержать один или два атома азота;
R1, R2, R3 и R4 выбраны из Н, 6-членного арила, CF3, галогена;
R5, R6 R7 представляют собой C1-алкил при условии, что Х9, X10 или Х11 в этом случае соответственно равен С;
″А″ может представлять собой простую связь или мостиковый этиновый фрагмент;
Y может представлять собой простую связь или независимо выбран из метиленового или этиленового мостиковых фрагментов;
фрагмент Z независимо выбран из незамещенного или замещенного по атому азота гетероциклоалкила или является незамещенным или замещенным циклоалкилом при условии, что N (азот) равно С (углерод):
где R9 выбран из СН2ОН, CON(R15 R16), где R15, R16 могут независимо представлять собой Н, C1-алкил,
Het представляет собой N,
n=1, n1=3;
R8 выбран из Н, C1-6-алкила, C1-алкилкарбонила,
производных арилуксусной кислоты общего строения:
метилгетероарилов общего строения:
производных алкилсульфонилов общего строения:
где R14=Alk, причем Alk представляет собой С1- алкил,
или соединение 2-метиламино-1-{3-[6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-этанона дигидрохлорид,
или соединение 6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты дигидрохлорид,
или соединение 6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты диметиламина дигидрохлорид,
или любая фармацевтически приемлемая соль указанных соединений.
2. Соединение по п.1, выбранное из:
3-(6-пиперидин-4-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорида,
3-(6-(азепан-4-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорида,
3-((6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-1-окса-8-азаспиро[4.5]декана дигидрохлорида,
3-((6-(6-(трифторметил)имидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-1-окса-8-азаспиро[4.5]декана дигидрохлорида,
3-((6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-1,4-оксазепана дигидрохлорида,
{4-[6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-пиридин-2-илметил]-циклогексил}-метиламина дигидрохлорида,
3-(6-(((3aS,6aR)-октагидро-циклопента[с]пиррол-4-ил)метил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорида (смесь диастереомеров),
3-(6-(((3aS,7aR)-октагидро-1Н-изоиндол-4-ил)метил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорида (смесь диастереомеров),
3-(6-((1,4-диазепан-6-ил)метил)пиридин-2-ил)-6-хлор-имидазо[1,2-а]пиридина дигидробромида,
3-(6-(азепан-4-илметил)-2-метилпиримидин-4-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорида,
3-(6-((1R,5S)-8-азабицикло[3.2.1]окт-3-илметил)пиридин-2-ил)-6-хлоримидазо[1,2-а]пиридина дигидрохлорида,
3-((6-(имидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-8-(пиридин-3-илметил)-1-окса-8-азаспиро[4.5] декана,
2-метиламино-1-{3-[6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-илметил]-1-окса-8-азаспиро[4.5]декан-8-ил}-этанона дигидрохлорида,
3-(6-(пирролидин-2-ил)пиразин-2-ил)имидазо[1,2-а]пиримидина дигидрохлорида,
3-(6-(1-изопропилпиперидин-3-ил)-2-метилпиримидин-4-ил)имидазо[1,2-а]пиримидина,
1-(4-((6-(имидазо[1,2-а]пиримидин-3-ил)пиридин-2-ил)метил)пиперидин-1-ил)этанона,
1-(2-(2-(6-(имидазо[1,2-а]пиримидин-3-ил)пиримидин-4-ил)этил)пирролидин-1-ил)этанона,
3-(6-((1-(метилсульфонил)-пирролидин-3-ил)метил)пиридин-2-ил)имидазо[1,2-а]пиримидина,
3-(6-(пиперидин-3-ил)пиридин-2-ил)-6-хлор-имидазо[1,2-а]пиридина дигидрохлорида,
3-(6-(пиперидин-3-ил)пиразин-2-ил)-6-хлор-имидазо[1,2-а]пиридина дигидрохлорида,
3-(6-(пиперидин-3-ил)пиридин-2-ил)-6-(трифторметил)-имидазо[1,2-а]пиридина дигидрохлорида,
2-(6-(6-(трифторметил)имидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)морфолина дигидрохлорида,
1-(2-(6-(6-(трифторметил)имидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)морфолино)-2-фенил-этанона,
3-(6-(пиперидин-3-ил)пиридин-2-ил)-6-хлоримидазо[1,2-b]пиридазина дигидрохлорида,
6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты дигидрохлорида,
6-(6-хлоримидазо[1,2-а]пиридин-3-ил)-1′,4′,5′,6′-тетрагидро-2′Н-[2,3′]бипиридинил-3′-карбоновой кислоты диметиламина дигидрохлорида,
3-(6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)пиперидин-3-ил)метанола дигидрохлорида,
3-(6-(пиперидин-3-ил)пиридин-2-ил)пиразоло[1,5-а]пиридина дигидрохлорида,
3-(6-(пиперидин-3-ил)пиридин-2-ил)-6-(трифторметил)-[1,2,4]триазоло[4,3-а]пиридина дигидрохлорида,
3-(6-(пиперидин-3-ил)пиридин-2-ил)пиразоло[1,5-а]пиримидина дигидрохлорида,
3-(6-(пиперидин-3-ил)пиридин-2-ил)этинил)-6-фенилпиразоло[1,5-а]пиримидина,
3-(6-(пиперидин-3-ил)пиридин-2-ил)-6-хлорпиразоло[1,5-а]пиридина дигидрохлорида,
3-((6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-2-окса-8-азаспиро[4.5]декана дигидрохлорида,
3-((6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)-2-окса-9-
азаспиро[5.5]ундекана дигидрохлорида,
2-((6-(6-хлоримидазо[1,2-а]пиридин-3-ил)пиридин-2-ил)метил)октагидрофуро[3,2-с]пиридина дигидробромида.
3. Соединение по любому из пп.1 или 2 в качестве лекарственного средства, обладающего способностью ингибировать Haspin киназу.
4. Фармацевтическая композиция, обладающая способностью ингибировать Haspin киназу, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый наполнитель, разбавитель или носитель.
5. Способ ингибирования Haspin киназы, включающий введение терапевтически эффективного количества соединения по п.1.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Ступенчатый шкив для приводных ремней с натяжным роликом | 1928 |
|
SU10261A1 |

