Область техники
Изобретение относится к химии органических соединений, фармакологии и медицине и касается новых химических соединений, ингибирующих серин/треониновые киназы, которые, в частности, могут использоваться для профилактики и лечения туберкулеза и онкологических заболеваний.
Уровень техники
Серин-треониновые (серин/треониновые) протеинкиназы (СТПК) играют важную роль в передаче сигналов как в эукариотических, так и в прокариотических клетках, участвуют в регуляции клеточной пролиферации и программируемой гибели клеток (апоптозе), дифференциации клеток и эмбриональном развитии. Из более чем 500 известных протеинкиназ человека, по крайней мере, 125 представляют собой серин/треониновые киназы. Нарушение функционирования киназ ассоциировано с развитием многих заболеваний человека, таких, как диабет, шизофрения, онкологические заболевания, сердечно-сосудистые расстройства, а также с патологическими состояниями, например канцерогенезом и нарушением иммунитета. В последние десятилетия интенсивное развитие получил биомишень-направленный поиск модуляторов (ингибиторов) протеинкиназ как потенциальных лекарственных препаратов нового поколения. Ингибиторы серин/треониновых киназ могут иметь широкое терапевтическое применение: от онкологии до иммунных заболеваний (Tian Q and Wang J (2002) Role of serine/threonine protein phosphatase in Alzheimer's disease. Neurosignals 11:262-269; Fiedler В and Wollert KC (2005) Targeting calcineurin and associated pathways in cardiac hypertrophy and failure. Expert Opin Ther Targets 9:963-973; Parameswara VK, Sule AJ, and Esser V (2005) Have we overlooked the importance of serine/threonine protein phosphatases in pancreatic beta-cells? Role played by protein phosphatase 2A in insulin secretion. JOP 6:303-315).
Циклин-зависимые киназы (CDK) представляют собой семейство серин/треониновых киназ, играющих важнейшую роль в контроле клеточного цикла и пролиферации. Нарушение функционирования этих киназ часто наблюдается при онкологических заболеваниях, вирусных инфекциях, нейродегенеративных заболеваниях и ишемии (J. Cicenas, K. Kalyan, A. Sorokinas, A. Jatulyte, D. Valiunas, A. Kaupinis and M. Valius Highlights of the Latest Advances in Research on CDK Inhibitors // Cancers 2014, 6, 2224-2242; doi:10.3390/cancers6042224). CDK9 фосфорилирует С-терминальный домен РНК полимеразы II и является ключевым фактором элонгации для транскрипции ДНК. Существуют прямые доказательства того, что CDK9 киназа необходима для выживания раковых клеток (Walsby Е, Pratt G, Shao Н, Abbas AY, Fischer РМ, Bradshaw TD, Brennan P, Fegan C, Wang S, Pepper C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. // Oncotarget. 2014 Jan 30; 5(2):375-85). В проведенных клинических исследованиях была показана противораковая активность некоторых неселективных ингибиторов киназ CDK семейства, например, флавопиридола и росковитина, и было обнаружено, что такая активность связана именно с ингибированием CDK9 (V. Kry tof, S. Baumli and  . Perspective of Cyclin-dependent kinase 9 (CDK9) as a Drug Target. // Current Pharmaceutical Design, 2012, 18, 2883-2890). Таким образом, CDK9 является важной мишенью в поиске противораковых препаратов нового поколения. В настоящее время ведутся исследования ингибиторов CDK9: AZD5438, Dinaciclib (SCH-727965), SNS-02, флавопиридол (алвоцидиб), FIT-039, CDKI-73, LY2857785, АТ7519, P276-00m ZK 304709.
. Perspective of Cyclin-dependent kinase 9 (CDK9) as a Drug Target. // Current Pharmaceutical Design, 2012, 18, 2883-2890). Таким образом, CDK9 является важной мишенью в поиске противораковых препаратов нового поколения. В настоящее время ведутся исследования ингибиторов CDK9: AZD5438, Dinaciclib (SCH-727965), SNS-02, флавопиридол (алвоцидиб), FIT-039, CDKI-73, LY2857785, АТ7519, P276-00m ZK 304709.
Серин/треониновые протеинкиназы эукариотического типа обнаружены также у бактерий, включая патогенные для человека. Установлено, что СТПК участвуют в формировании вирулентности, бактериальных биопленок, поддержании толерантности, персистировании патогенных микроорганизмов. Показано ключевое значение СТПК в формировании вирулентности Streptococcus pneumonia, Mycobacterium tuberculosis, Staphylococcus aureus, Pseudomonas aeruginosa и ряда других патогенных бактерий. Установлено их участие в модуляции устойчивости к антибиотикам у М. Tuberculosis. Ингибиторы микобактериальных киназ PknB, PknA, PknG, PknF, отвечающих за рост микобактериальных клеток и их выживание в макрофагах человека (Елизаров С.М., Алексеева М.Г., Новиков Ф.Н., Чилов Г.Г., Маслов Д.А., Штиль А.А., Даниленко В.Н. Идентификация сайтов фосфорилирования аминогликозидфосфотрансферазы VIII // Биохимия, 2012, том 77, №11, с. 1504-1512), могут быть использованы для лечения бактериальных инфекций, в том числе туберкулеза (Sophie Magnet, Ruben С. Hartkoorn а, Rita  et al. Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis 90 (2010) 354e360).
et al. Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis 90 (2010) 354e360).
Все большее значение приобретает проблема лечения туберкулеза, характеризующегося множественной лекарственной устойчивостью. Около 10-15% всех новых случаев туберкулеза в России представляют собой случаи заболевания туберкулезом именно с множественной или даже расширенной лекарственной устойчивостью.
Положение обостряется в связи с тем, что появление и распространение новых лекарственных препаратов через некоторое время регулярно сопровождается последовательным возникновением устойчивых к ним форм микобактерий; новая устойчивость часто "наслаивается" на предыдущую, в результате приобретая множественный характер. Достаточно часто осуществляется заражение de novo штаммами, которые уже приобрели множественную лекарственную устойчивость (MDR - multiple drug resistance). По последним данным ВОЗ, частота выявления MDR-изолятов у всех больных с первичным обращением колебалась от 0 до 22%. В России число случаев заражения MDR-штаммами туберкулеза составляет до 40%. Кроме того, около 6-7 лет назад появились сообщения о случаях туберкулеза, вызванных штаммами с так называемой широкой лекарственной устойчивостью (XDR, extensive drug resistance). XDR-штаммы устойчивы не менее чем к 4-5 препаратам (обычно к 7-8 препаратам); но описаны изоляты, устойчивые даже к 9 препаратам. Из-за трудностей терапии, особенностей эпидемиологии и высокой смертности туберкулез, вызванный XDR-штаммами, выделяют в особую нозологическую единицу (с марта 2005 г). Доля выявления изолятов XDR от общего количества больных с выделением MDR-штаммов составляла от 4 до 30%, с большим разбросом по разным странам. В России доля XDR-туберкулеза составляет до 13% от числа выявленных случаев заболевания, вызванных MDR-штаммами. При систематическом лечении при этой форме туберкулеза выздоравливает до 55-65% больных. В целом, случаи туберкулеза, вызванного XDR-штаммами, требуют длительной госпитализации, долгого лечения и систематической бактериологической диагностики в течение всех курсов терапии. Большие надежды при этом возлагаются на лекарства нового поколения. В связи с этим высокую степень актуальности приобретают поиски новых противотуберкулезных лекарств, подавляющих лекарственную устойчивость либо ингибирующих ранее не задействованные пути метаболизма микобактерий.
Поэтому киназы PknB, PknA, PknG, PknF микобактерий туберкулеза представляются перспективными мишенями для разработки лекарств нового поколения для лечения туберкулеза, способных преодолеть лекарственную устойчивость, а их модулирование (ингибирование) - крайне перспективной стратегией борьбы.
Раскрытие изобретения
Задачей данного изобретения является разработка новых эффективных ингибиторов серин/треониновых протеинкиназ, в частности, CDK9, PknB или PknA, киназы, и являющихся перспективными для применения в терапии различных заболеваний, связанных с аберрантной активностью серин/треониновых киназ, в том числе онкологических заболеваний, туберкулеза, включая резистентные формы туберкулеза.
Техническим результатом данного изобретения является разработка и получение новых ингибиторов серин/треониновых протеинкиназ, в частности, CDK9, PknB или PknA киназы, обладающих повышенной эффективностью в ингибировании этих киназ. Данные соединения являются перспективными для применения в терапии различных заболеваний, связанных с аберрантной активностью серин/треониновых киназ, в том числе онкологических заболеваний, туберкулеза, а также резистентных форм туберкулеза.
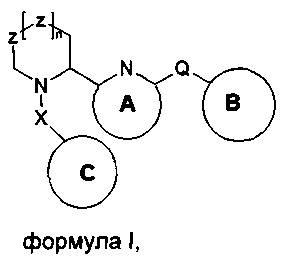
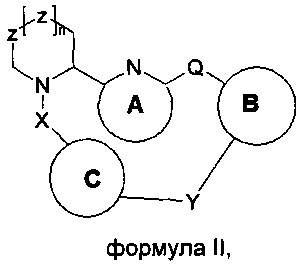
Указанный технический результат достигается путем получения соединений общей формулы (I) или (II):
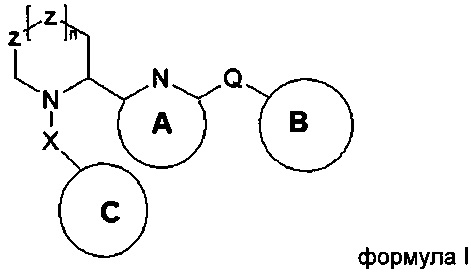 ,
,
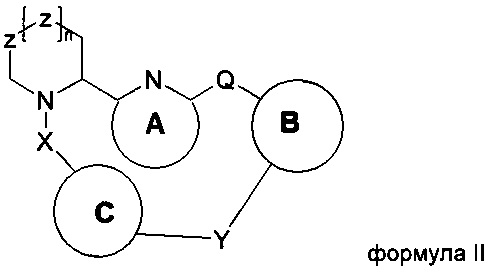
или их таутомеров, стереоизомеров, энантиомеров или фармацевтически приемлемых солей, сольватов или гидратов, где:
n=0-2;
А выбирается независимо и представляет собой 5-7-членный ароматический гетероцикл, содержащий 1-2 атома N и 0-1 атом S; А содержит 0-2 заместителя R;
R выбирается независимо и представляет собой метил или этил;
В выбирается независимо и представляет собой фенил, или 5-6-членный гетероарильный цикл, содержащий 0-2 атома N и 0-1 атом S, или 5-6-членный циклоалкил, содержащий 0-2 атома N и 0-1 атом S; В содержит 0-3 заместителя R1;
С выбирается независимо и представляет собой фенил, -NH2, -NH-C1-3-алкил, -NH(С1-3-алкил)С1-3-алкил, 5-6-членный гетероарильный цикл, содержащий 0-2 атома N и 0-1 атом S, или 5-6-членный циклоалкил, содержащий 0-2 атома N и 0-1 атом S; С содержит 0-3 заместителя R1;
R1 выбирается независимо и представляет собой -C1-6-алкил, галоген, фенил, -С5-7-гетероарил, содержащий 1-2 атома N и 0-1 атом S, -СООН, -CONH2, -NH2 или -NHR2;
R2 выбирается независимо и представляет собой -С1-6-алкил, -C(O)-C1-8-алкил;
линкер X выбирается независимо и представляет собой -СН2-, -С(=O)-СН2- или -СН2-O- группу;
линкер Q выбирается независимо и представляет собой -NH- или -NH-C(O)- группу;
линкер Y выбирается независимо и представляет собой -O-(СН2)m- или -C(O)-NH-(CH2)m-, где m=1-3, образуя 16-20-членную макроциклическую систему;
Z выбирается независимо и представляет собой -СН2- группу или атом кислорода.
Отдельный интерес представляют собой соединения по изобретению, в которых:
n=0-2;
А выбирается независимо и представляет собой тиазол или пиримидин, А содержит 0-2 заместителя R;
R выбирается независимо и представляет собой метил или этил;
В и С выбираются независимо и представляют собой фенил, тиазол, пиридин, пиримидин или пиразин, опционально замещенные 0-3 заместителями R1;
R1 выбирается независимо и представляет собой -С1-6-алкил, галоген, фенил, -С5-7-гетероарил, содержащий 1-2 атома N и 0-1 атом S, -COOH, -CONH2, -NH2 или -NHR2;
R2 выбирается независимо и представляет собой -С1-3-алкил или -С(O)-С1-3-алкил;
линкер X выбирается независимо и представляет собой -СН2-, -С(=O)-СН2- или -СН2-O- группу;
линкер Q выбирается независимо и представляет собой -NH- или -NH-C(O)- группу;
линкер Y выбирается независимо и представляет собой -O-(СН2)- группу или отсутствует;
Z выбирается независимо и представляет собой -СН2- группу или атом кислорода.
Отдельный подкласс соединений, представляющих интерес, включает соединения по изобретению, выбранных из группы:
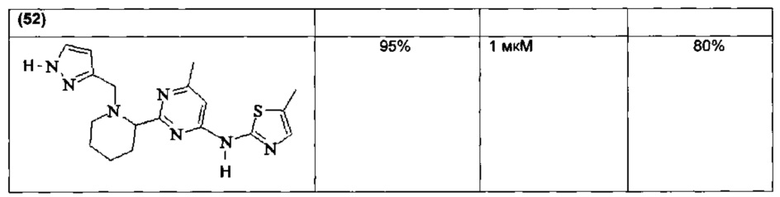
2-(2-(4-Метил-6-(5-метилтиазол-2-иламино)пиримидин-2-ил)пиперидин-1-ил)-1-(пиперидин-1-ил)этанон;
1-(2-(4-Метил-6-(5-метилтиазол2-иламино)пиримидин-2-ил)пиперидин-1-ил)-2-(метиламино)этанон;
N-(2-(1-((5-Хлорпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин;
N-(2-(1-((5-Фторпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин;
{2-[1-(3-Метил-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{6-Метил-2-[1-(2Н-пиразол-3-илметил)-пиперидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{6-Метил-2-[1-(2-метил-тиазол-4-илметил)-пирролидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{2-[1-(5-Фтор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-тиазол-2-ил-амин;
{2-[1-(5-Хлор-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-пиридин-2-ил-амин;
{2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
[6-Метил-2-(1-тиазол-2-илметил-пиперидин-2-ил)-пиримидин-4-ил]-тиазол-2-ил-амин;
{2-[4-(5-Фтор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{2-[4-(5-Хлор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-2Н-пиразол-3-ил)-амин;
(5-Метил-пиридин-2-ил)-[4-(1-тиазол-4-ил-метил-пирролидин-2-ил)-тиазол-2-ил]-амин.
Данное изобретение также относится к применению соединений, являющихся предметом изобретения, в качестве ингибиторов серин/треониновых киназ, в частности, PnkA, PnkB, CDK9, ERK7, LIMK1 или S6k-beta киназы.
Данное изобретение также относится к применению соединений, являющихся предметом изобретения, для получения фармацевтической композиции для лечения и/или предотвращения заболевания, связанного с аберрантной активностью серин/треониновой киназы. В некоторых частных вариантах воплощения изобретения серин/треониновая киназа представляет собой PnkA, PnkB, CDK9, ERK7, LIMK1 или S6k-beta киназу. Заболевание, связанное с аберрантной активностью серин/треониновых киназ, в частности, представляет собой онкологическое заболевание или туберкулез. В некоторых частных вариантах воплощения изобретения, туберкулез имеет резистентную форму.
Кроме того, изобретение предусматривает фармацевтические композиции для лечения и/или предотвращения заболевания, связанного с аберрантной активностью серин/треониновой киназы, содержащих эффективное количество, по меньшей мере, одного соединения по изобретению и, по меньшей мере, один фармацевтически приемлемый носитель, растворитель и/или наполнитель.
Кроме того, изобретение предусматривает фармацевтические композиции, которые характеризуются тем, что обладают бактерицидным действием, а также фармацевтические композиции, которые обладают бактериостатическим действием.
Изобретение также включает получение соединений общих формул (I) и (II).
Подробное описание изобретения
Определения (термины)
Следующие определения применяются в данном документе, если иное не указано явно. Кроме того, если не указано иное, все вхождения функциональных групп выбираются независимо, два вхождения могут быть как одинаковыми, так и разными.
Термин «алкил» сам по себе или как часть другого заместителя, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, имеющие указанное число атомов углерода (то есть, С1-6 подразумевает от одного до шести атомов углерода). Примеры алкилов включают метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил, но не ограничиваются ими.
Термин «гетероцикл» («гетероциклил», «гетероциклический») означает циклическую систему, имеющую от пяти до семи, как правило, от пяти до шести кольцевых атомов углерода, в состав которой наряду с углеродом входят и атомы других элементов. Такими гетероатомами согласно изобретению могут быть N, О или S, которые выбираются независимо. Термин «гетероцикл» относится к насыщенным, частично ненасыщенным или ароматическим циклам (гетероарилам). Степень насыщенности гетероцикла указывается отдельно. В некоторых случаях гетероцикл, предпочтительно, представляет собой 5-7-членное неароматическое кольцо, необязательно содержащее 1-2 двойные связи и, возможно, содержащее 1-2 гетероатома, независимо выбранные из S и N.
Термин «арил», если иное не оговорено, обозначает ароматическую углеводородную группу, которая может представлять собой одну циклическую структуру. Термин «гетероарил» относится к арильным группам, которые содержат от одного до трех гетероатомов, независимо выбранных из N или S. В некоторых случаях гетероарил, предпочтительно, представляет собой 5-6-членное моноциклическое ароматическое кольцо, необязательно содержащее от 1 до 3-х гетероатомов, независимо выбранных из N или S. В некоторых других случаях гетероарил, предпочтительно, представляет собой 5-6-членное гетероарильное кольцо, содержащее 1-2 гетероатома, независимо выбранные из S или N. Неограничивающими примерами арилов и гетероарилов по изобретению являются фенил, пиридинил, пиримидинил, пиразинил, тиазолил и другие.
Термин «галоген» сам по себе или в части другого термина относится к атому фтора, хлора, брома или йода.
Термин «амино» обозначает группу -NH2.
Вышеуказанные группы, согласно изобретению, могут также содержать один или несколько заместителей. Наглядные, но не ограничивающие примеры заместителей представляют собой галоген, алкил, алкилкарбонил. Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40°С в отсутствии химически активных условий в течение, по крайней мере, одной недели.
Некоторые соединения данного изобретения могут существовать в таутомерных формах, и это изобретение включает в себя все такие таутомерные формы таких соединений, если не указано иное.
Если не указано иначе, приведенные в материалах заявки структуры соединений также подразумевают и все стереоизомеры, то есть R- и S-изомеры для каждого ассиметричного центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси настоящих соединений, также являются предметом данного изобретения. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%; более предпочтительно, >95% мольной чистоты), так же как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например путем получения диастереоизомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Кроме того, другой метод разделения включает синтез ковалентных диастереомерных молекул путем реакции соединений изобретения с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Полученные диастереомеры можно разделить обычными способами, например, хроматографией, дистилляцией, кристаллизаций или сублимацией, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания, эфира или соли.
Термин «сольват» относится к ассоциации или комплексу из одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин.
Термин «гидрат» относится к комплексу, где молекулами растворителя является вода.
Соединения настоящего изобретения могут существовать в свободной форме или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, валериат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкил сульфонаты и арил сульфонаты.
Термин «пациент» охватывает все виды млекопитающих, предпочтительно человека.
Термины «лечение», «терапия» охватывают лечение патологических состояний у млекопитающих, предпочтительно у человека, и включают: а) блокирование (приостановку) течения заболевания, б) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания.
Термин «профилактика», «предотвращение», «превентивная терапия» охватывает устранение факторов риска, а также профилактическое лечение субклинических стадий заболевания у млекопитающих, предпочтительно у человека, направленное на уменьшение вероятности возникновения клинических стадий заболевания. Пациенты для профилактической терапии отбираются на основе факторов, которые, на основании известных данных, влекут увеличение риска возникновения клинических стадий заболевания по сравнению с общим населением. К профилактической терапии относится а) первичная профилактика и б) вторичная профилактика. Первичная профилактика определяется как профилактическое лечение у пациентов, клиническая стадия заболевания у которых еще не наступила. Вторичная профилактика - это предотвращение повторного наступления того же или близкого клинического состояния заболевания.
Термин «уменьшение риска» охватывает терапию, которая снижает частоту возникновения клинической стадии заболевания. Примерами уменьшения риска заболевания является первичная и вторичная профилактика заболевания.
Способ терапевтического применения соединений
Предмет данного изобретения также относится к способу лечения и/или предотвращения заболеваний, связанных с аберрантной активностью, по меньшей мере, одной серин/треониновой киназы, включающему введение пациенту соединения по изобретению. В частности, к способу лечения и/или предотвращения заболеваний, связанных с аберрантной активностью PknB, PknA, PknG, PknF и/или CDK9 киназы.
В предпочтительных случаях, предмет данного изобретения относится к способу лечения и/или предотвращения туберкулеза, в том числе резистентных форм туберкулеза; в других вариантах предмет данного изобретения относится к способу лечения и/или предотвращения онкологических заболеваний (злокачественных новообразований), например, таких как немелкоклеточный рак легкого, колоректальный рак, тройной негативный рак молочной железы, остеосаркому, рак поджелудочной железы, гепатоцеллюлярную карциному, аденокарциному поджелудочной железы, множественную миелому, ангиоиммунобластную Т-клеточную лимфому, рак яичника, рак простаты, меланому, но не ограничиваясь ими.
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения общей формулы (I) и/или общей формулы (II). Под терапевтически эффективным количеством подразумевается количество одного или нескольких описанных соединений, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение (профилактику). Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Вещество, или фармацевтическая композиция, содержащая вещество, может быть введена в организм пациента в любом количестве и любым путем введения, эффективным для лечения или профилактики заболевания.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, парентерально, местно и т.п.
Эффективная системная дозировка соединения, вводимая разово или в виде нескольких отдельных доз, как правило, лежит в диапазоне от 0,01 до 500 мг/кг, предпочтительно от 0,1 до 125 мг/кг, еще более предпочтительно, от 1 до 100 мг/кг. Обычно соединение вводится пациенту, нуждающемуся в таком лечении, в дневной дозировке ориентировочно от 50 до 2000 мг на пациента. Введение может осуществляться как разово, так и несколько раз в день, неделю (или любой другой временной интервал), или время от времени. Кроме того, соединение может вводиться в организм пациента ежедневно в течение определенного периода дней (например, 2-10 дней), а затем следует период без приема вещества (например, 1-30 дней).
В том случае, когда соединение данного изобретения используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов.
Фармацевтические композиции
Изобретение также относится с фармацевтическим композициям, которые содержат, по меньшей мере, одно из описанных здесь соединений (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, таких, которые могут быть введены в организм пациента совместно с соединением, составляющем суть данного изобретения, и которые не разрушают фармакологической активности этого соединения, и являются нетоксичным при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахариды, а также их производные; желатин; тальк; эксципиенты, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе, например, для введения в организм орально, местно, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, подкожно, внутримышечно, а также инфузионным способом, в рекомендованных дозировках.
Лекарственная форма данного изобретения может содержать соединение описанной здесь формулы или его фармацевтически приемлемую соль, и дополнительный препарат, например, выбранный из числа следующих: антидепрессант, противоопухолевый препарат, противовирусный препарат, противовоспалительный препарат, и любой фармацевтически приемлемый носитель, адъювант или растворитель. Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом или микрокапсуляционные методы, методами приготовления наноформ препарата, и прочие примеры, известные в фармацевтике.
При получении композиции, например, в форме таблетки активное начало смешивают с одним или несколькими фармацевтическими эксципиентами, такими как желатин, крахмал, лактоза, стеарат магния, тальк, кремнезем, аравийская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки можно покрыть сахарозой, целлюлозным производным или другими веществами, подходящими для нанесения оболочки. Таблетки могут быть получены различными способами, такими как непосредственное сжатие, сухое или влажное гранулирование или горячее сплавление в горячем состоянии.
Фармацевтическую композицию в форме желатиновой капсулы можно получить, смешивая активное начало с растворителем и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным путем используются водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат фармакологически совместимые агенты, например пропиленгликоль или бутиленгликоль.
Для примера, стандартная лекарственная форма, содержащая соединение по изобретению в форме таблетки может содержать следующие ингредиенты:
Обзор методов получения соединений изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Следует понимать, что эти и все приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Получение соединений по изобретению
Некоторые соединения по изобретению могут быть получены согласно общим схемам синтеза 1 и 2 (схема 1 - синтез тиазол-содержащих соединений, схема 2 - синтез пиримидин-содержащих соединений):
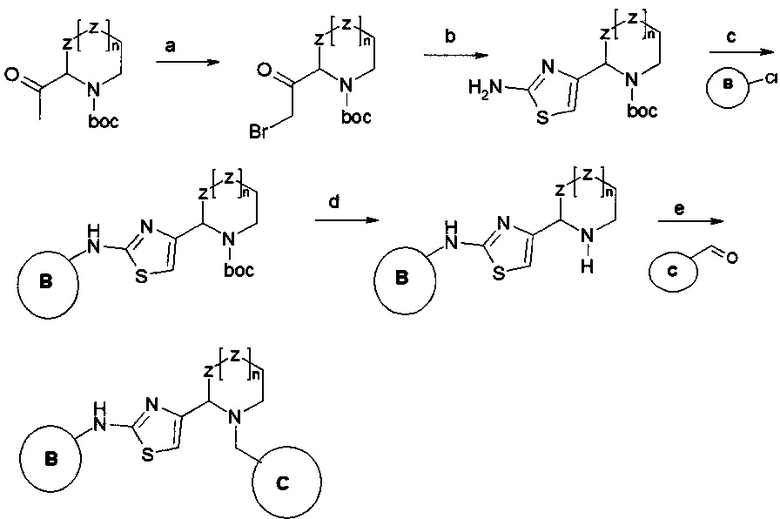
Схема 1. Условия синтеза: а) бис(триметилсилил)амид лития, Me3SiCl, затем Br2; b) тиомочевина; c) 9,9-диметил-4,5-бис(дифенилфосфино)ксантен, трис(дибензилиденоацетон)дипалладий (0)-хлороформ, tBuONa, микроволновой реактор; d) HCl, этанол; e) NaHB(OAc)3, CH2Cl2.
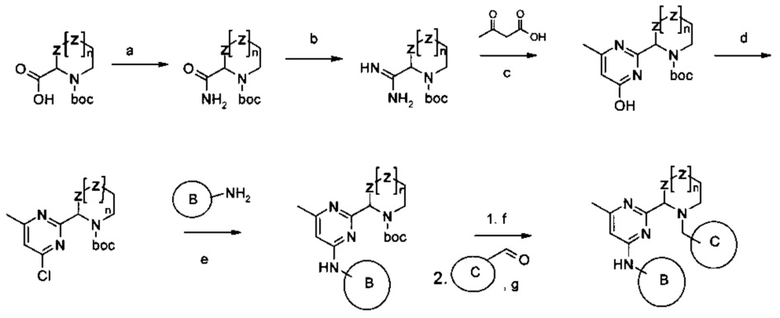
Схема 2. Условия синтеза: a) CDI (карбондиимидазол), 60°С, (NH4)2CO3, Et3N в ацетонитриле, затем K2CO3; b) Et3O+BF4-, CH2Cl2, затем 10% NH3 в МеОН; c. t-BuONa, EtOH; d) POCl3, диметиланилин; e) 9,9-диметил-4,5-бис(дифенилфосфино)ксантен, трис(дибензилиденоацетон)дипалладий (0)-хлороформ, Na2CO3, толуол - вода, микроволновой реактор; f) 16% HCl в диоксане, 5 ч, комн. темп.; g) NaHB(OAc)3, CH2Cl2.
Ниже приводятся примеры синтеза некоторых соединений по изобретению.
Пример 1. 2-(2-(4-Метил-6-(5-метилтиазол-2-иламино)пиримидин-2-ил)пиперидин-1-ил)-1-(пиперидин-1-ил)этанон (10)
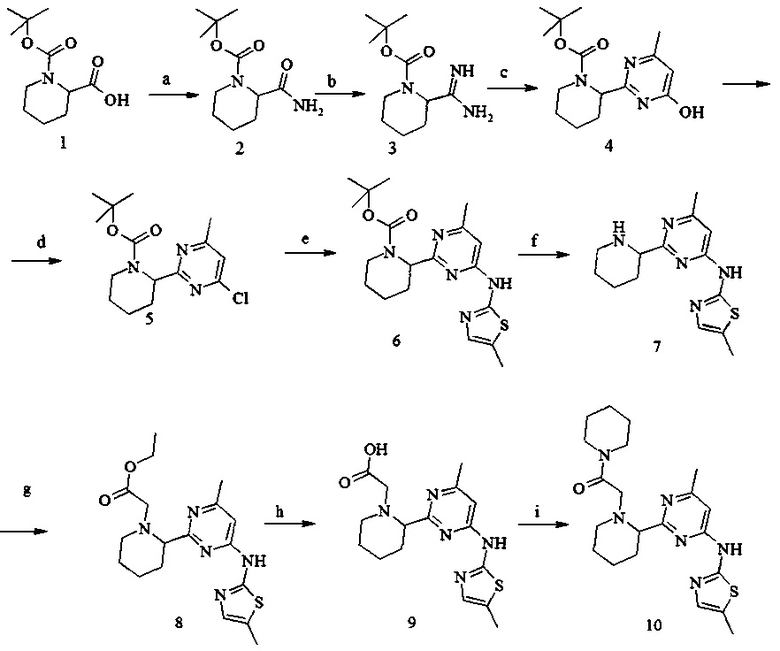
Условия синтеза: a) (NH4)2CO3, TBTU, Et3N, CH3CN, при комнатной температуре; b) Et3OBF4/DCM, NH3/MeOH, при комнатной температуре; c) СН3СОСН2СООС2Н5, NaOtBu, EtOH, кипячение; d) POCl3, Me2N-Ph, толуол, кипячение; e) 5-метил-2-аминотиазол, Pd2dba3, xantphos, Na2CO3, толуол-H2O, микроволновой реактор, 140°С; f) 16% HCl/диоксан, комн. темп.; g) этиловый эфир бромуксусной кислоты, NaHCO3, ДМФА; h) NaOH, МеОН-H2O, комн. темп; i) пиперидин, TBTU, Et3N, CH3CN, при комнатной температуре.
2-Карбамоил-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (2)
К раствору Вос-пипеколин-2-карбоновой кислоты (1) (23.0 г, 100 ммоль) в 150 мл безводного ацетонитрила добавили TBTU (38.5 г, 120 ммоль), триэтиламин (21 мл, 150 ммоль) и карбонат аммония (19.2 г, 200 ммоль). Реакционную массу перемешивали при комнатной температуре 24 ч. По окончании реакции смесь выливали в насыщенный раствор карбоната калия (поташ) (250 мл), экстрагировали дихлорметаном (3×250 мл). Объединенные органические экстракты промывали водой (2×150 мл), сушили над сульфатом натрия и растворитель отгоняли в вакууме. Продукт очищали с помощью флэш-хроматографии на силикагеле, используя гексан-этилацетат. Выход соединения (2) 17.6 г (77%).
2-Карбамимидоил-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (3)
К раствору амида (2) (17.6 г, 77 ммоль) в 200 мл безводного дихлорметана добавляли при перемешивании триэтилоксоний тетрафторборат (14.6 г, 77 ммоль). Реакционную массу перемешивали в течение 3 ч (контроль по ТСХ, этилацетат - дихлорметан, 1:2). Растворитель отгоняли в вакууме при температуре не выше 40°С, маслообразный остаток обрабатывали 10% аммиаком в метаноле (150 мл) в течение 24 час при комнатной температуре. Метанол отгоняли в вакууме при температуре не выше 40°С, остаток растирали с диэтиловым эфиром, эфир декантировали, остаток сушили при комнатной температуре. Выход соединения (3) 19.2 г (79%), амидин (3) использовали на следующей стадии без дополнительной очистки.
2-(4-Гидрокси-6-метил-пиримидин-2-ил)-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (4)
Трет-бутилат натрия (5.9 г, 61 ммоль) растворяли в 150 мл абсолютного этанола, затем добавляли к раствору 19.2 г (60.8 ммоль) амидина (3). После перемешивания в течение 10 мин добавляли этиловый эфир ацетоуксусной кислоты (11.7 г, 90 ммоль). Полученную смесь кипятили в течение 10 час, затем охлаждали до комнатной температуры, выливали в насыщенный раствор хлорида аммония (300 мл), экстрагировали дихлорметаном (2×200 мл), объединенные экстракты сушили над сульфатом натрия, растворитель отгоняли в вакууме. Продукт выделяли на колонке с силикагелем, элюируя смесью этилацетат - дихлорметан, 1:1. Выход соединения (4) 11.4 г (64%).
2-(4-Хлоро-6-метил-пиримидин-2-ил)-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (5)
Интермедиат (4) (10 г, 34.1 ммоль) растворяли в безводном толуоле (200 мл), добавляли N,N-диметиланилин (37 г, 306 ммоль), затем прикапывали хлорокись фосфора (15.6 г, 102 ммоль). Раствор кипятили в течение 3 ч, охлаждали до комнатной температуры и оставляли на ночь. Реакционную массу выливали в холодную воду (300 мл), органический слой отделяли, промывали 1N HCl (2×70 мл), затем водой (1×70 мл), сушили над сульфатом натрия и упаривали. Продукт выделяли на колонке с силикагелем, элюируя смесью этилацетат - гексан, 1:3. Выход соединения (5) 5.7 г (54%).
2-[4-Метил-6-(5-метил-тиазол-2-иламино)-пиримидин-2-ил]-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (6)
Смесь хлорида (5) (2 г, 6.4 ммоль), 5-метил-тиазол-2-иламин (0.68 г, 6 ммоль), 9,9-диметил-4,5-бис(дифенилфосфино)ксантена (174 мг, 0.30 ммоль, 5% моль), комплексного катализатора трис(дибензилиденоацетон)дипалладий (0)-хлороформ (150 мг, 0.15 ммоль, 2.5% моль), карбоната натрия (0.95 г, 9 ммоль) в смеси толуол - вода (10:1, 15 мл) перемешивали в атмосфере аргона в микроволновой печи при 140°С в течение 2 час. После охлаждения реакционной смеси до комнатной температуры добавляли воду (30 мл), экстрагировали этилацетатом (2×30 мл). Объединенные органические экстракты сушили над сульфатом натрия, растворитель отгоняли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент этилацетат - гексан, 1:2). Выход 1.87 г (80%).
(6-Метил-2-пиперидин-2-ил-пиримидин-4-ил)-(5-метил-тиазол-2-ил)-амин (7)
Растворяли Вос-производное (6) (1.87 г, 4.8 ммоль) в 15 мл этанола, добавляли 5 мл 16% HCl в диоксане и перемешивали в течение 4 ч при комнатной температуре. Затем растворитель отгоняли в вакууме, остаток подщелачивали 10% раствором гидроксида натрия до pH 9, экстрагировали дихлорметаном (3×100 мл), сушили Na2SO4, упаривали. Выход соединения (7) количественный.
{2-[4-Метил-6-(5-метил-тиазол-2-иламино)-пиримидин-2-ил]-пиперидин-1-ил}-уксусной кислоты этиловый эфир (8)
К раствору амина (7) (0.87 г, 3 ммоль) в 25 мл безводного ДМФА добавляли NaHCO3 (1 г, 12 ммоль) и этиловый эфир бромуксусной кислоты (0.345 мл, 3.1 ммоль). Реакционную массу перемешивали при 50°С в течение 8 ч. По окончании реакции смесь разбавляли водой (100 мл), экстрагировали этилацетатом (2×100 мл), объединенный органический экстракт промывали водой (2×50 мл), сушили над сульфатом натрия и упаривали. Очищали с помощью флэш-хроматографии на силикагеле, элюируя смесью этилацетат - гексан 1:1. Выход соединения (8) 1.03 г (90%).
{2-[4-Метил-6-(5-метил-тиазол-2-иламино)-пиримидин-2-ил]-пиперидин-1-ил}-уксусная кислота (9)
Соединение (8) (1 г, 2.7 ммоль) растворяли в 30 мл метанола, добавляли раствор NaOH (0.48 г, 12 ммоль) в 10 мл воды и перемешивали в течение 12 ч при комнатной температуре. По окончании реакции смесь нейтрализовали конц. HCl и упаривали досуха. К остатку добавляли 100 мл смеси дихлорметан - метанол (1:1), выпавший NaCl отфильтровывали, остаток упаривали досуха. Выход соединения (9) 0.92 г (98%).
2-(2-(4-Метил-6-(5-метилтиазол-2-иламино)пиримидин-2-ил)пиперидин-1-ил)-1-(пиперидин-1-ил)этанон (10)
К раствору кислоты (9) (173 мг, 0.5 ммоль) в 10 мл сухого ацетонитрила добавляли TBTU (192 мг, 0.6 ммоль), триэтиламин (0.14 мл, 1 ммоль) и пиперидин (51 мг, 0.6 ммоль). Перемешивали при комнатной температуре в течение 16 ч, затем выливали в насыщенный раствор карбоната калия (поташ) (20 мл), экстрагировали дихлорметаном (3×25 мл), сушили над сульфатом натрия и упаривали. Продукт выделяли на колонке с силикагелем, элюируя этилацетатом. Выход соединения (10) 124 мг (60%).
APCI-MS (m/z): 415.23 ([М+Н]+, 100%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.26-1.65 (м, 12Н), 2.26 (м, 6Н), 2.85 (м, 5Н), 3.32 (м, 2Н), 4.21 (м, 1Н), 4.60 (м, 1Н), 6.74 (м, 1Н), 7.11 (с, 1Н), 11.24 (уш.с., 1Н).
Пример 2. 1-(2-(4-Метил-6-(5-метилтиазол2-иламино)пиримидин-2-ил)пиперидин-1-ил)-2-(метиламино)этанон (12)
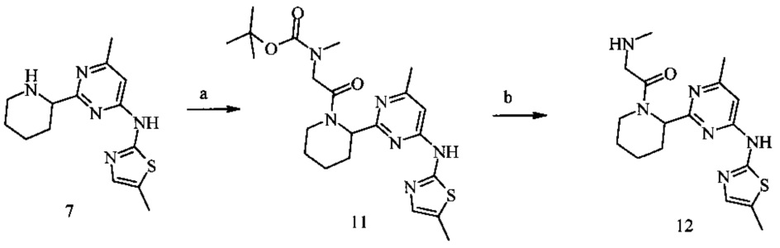
Условия синтеза: a) (трет-бутилоксикарбонил-метиламино)уксусная кислота, TBTU, Et3N, CH3CN, при комнатной температуре; b) 16% HCl/диоксан, при комнатной температуре.
Метил-(2-{2-[4-метил-6-(5-метил-тиазол-2-иламино)-пиримидин-2-ил]-пиперидин-1-ил}-2-оксо-этил)-карбаминовой кислоты трет-бутиловый эфир (11)
К раствору амина (7) (145 мг, 0.5 ммоль) в 10 мл сухого ацетонитрила добавляли TBTU (192 мг, 0.6 ммоль), триэтиламин (0.14 мл, 1 ммоль) и N-Boc-N-метил глицин (113 мг, 0.6 ммоль). Перемешивали при комнатной температуре в течение 16 ч, затем выливали в насыщенный раствор карбоната калия (поташа) (20 мл), экстрагировали дихлорметаном (3×25 мл), сушили над сульфатом натрия и упаривали. Продукт выделяли на колонке с силикагелем, элюируя этилацетатом. Выход соединения (11) 150 мг (65%).
1-(2-(4-Метил-6-(5-метилтиазол2-иламино)пиримидин-2-ил)пиперидин-1-ил)-2-(метиламино)этанон (12)
Растворяли Вос-производное (11) (150 мг, 0.325 ммоль) в 5 мл этанола, добавляли 2 мл 16% HCl в диоксане и перемешивали в течение 4 ч при комнатной температуре. Затем растворитель отгоняли в вакууме досуха, остаток растирали с диэтиловым эфиром, отфильтровывали и сушили. Выход соединения (12) количественный.
APCI-MS (m/z): 361.40 ([М+Н]+, 100%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.21-1.35 (м, 2Н), 1.63 (м, 2Н), 1.77-1.91 (м, 1Н), 2.31 (с, 3Н), 2.35 (д, 3Н), 2.38 (с, 3Н), 2.55 (м, 2Н), 2.67-2.83 (м, 2Н), 4.20-4.42 (м, 3Н), 6.81 (м, 1Н), 7.15 (м, 1Н), 8.76-9.0 (м, 2Н).
Пример 3. N-(2-(1-((5-Хлорпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин (13)
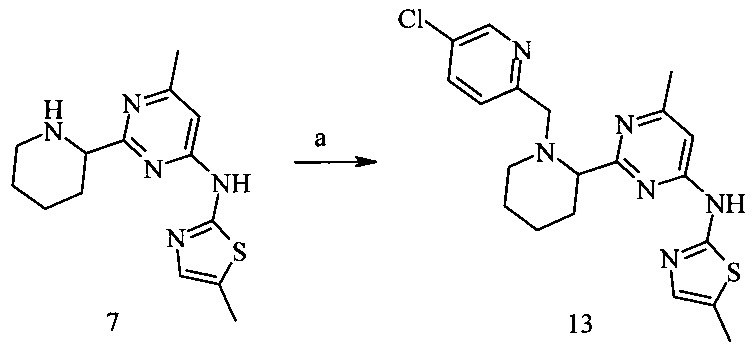
Условия синтеза: а) 5-хлорпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
N-(2-(1-((5-Хлорпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин (13)
К раствору амина (7) (145 мг, 0.5 ммоль) в 10 мл сухого дихлорметана добавляли 5-хлор-пиридин-2-карбальдегид (64 мг, 0.6 ммоль) и триацетоксиборгидрид натрия (127 мг, 0.6 ммоль). Перемешивали при комнатной температуре в течение 16 ч, затем выливали в насыщенный раствор карбоната калия (поташ) (20 мл), экстрагировали дихлорметаном (3×25 мл), сушили над сульфатом натрия и упаривали. Продукт выделяли на колонке с силикагелем, элюируя этилацетатом. Выход соединения (13) 106 мг (56%).
APCI-MS (m/z): 415.19 ([М+Н]+, 100%), 417.14([М+Н]+, 42%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.33-1.42 (м, 1Н), 1.58 (м, 2Н), 1.76 (м, 2Н), 1.91-2.05 (м, 1Н), 2.16-2.22 (м, 1Н), 2.30 (с, 3Н), 2.36 (с, 3Н), 2.92 (м, 1Н), 3.30 (д, 1Н), 3.47 (м, 1Н), 3.63 (д, 1Н), 6.63 (с, 1Н), 7.20 (с, 1Н), 7.60 (м 1Н), 7.73 (м, 1Н), 11.26 (уш.с, 1Н).
Пример 4. N-(2-(1-((5-Фторпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин (14)
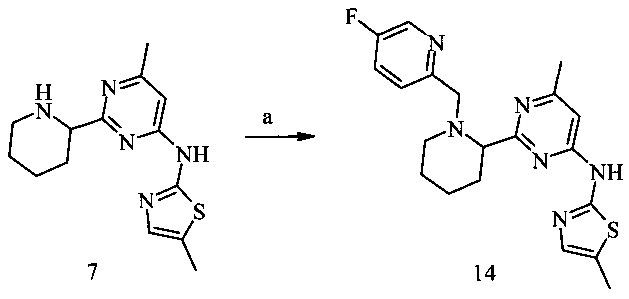
Условия синтеза: а) 5-фторпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
N-(2-(1-((5-Фторпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин (14)
Получали аналогично соединению (13) из амина (7) (145 мг, 0.5 ммоль) и 5-фтор-пиридин-2-карбальдегида. Выход соединения (14) 122 мг (61%)
APCI-MS (m/z): 399.20 ([М+Н]+, 100%).
1Н ЯМР δН (400 МГц, D6-ДМСО): 1.35-1.41 (м, 1Н), 1.59 (м, 2Н), 1.76 (м, 2Н), 1.94-2.11 (м, 1Н), 2.17-2.21 (м, 1Н), 2.31 (с, 3Н), 2.36 (с, 3Н), 2.91 (м, 1Н), 3.28 (д, 1Н), 3.46 (м, 1Н), 3.63 (д, 1Н), 6.61 (с, 1Н), 7.15 (с, 1Н), 7.51-7.64 (м, 2Н), 8.29 (м, 1Н), 11.24 (уш.с, 1Н).
Пример 5. {2-[1-(3-Метил-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (15)
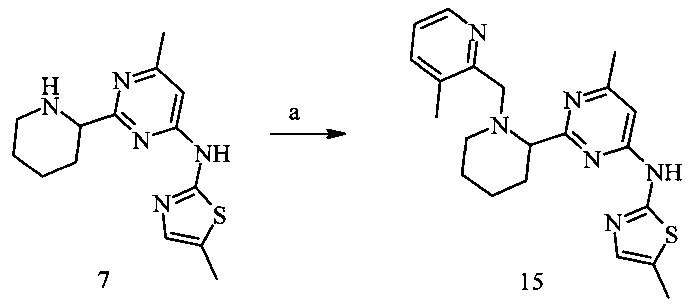
Условия синтеза: а) 3-метилпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
{2-[1-(3-Метил-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (15)
Получали аналогично соединению (13) из амина (7) (145 мг, 0.5 ммоль) и 3-метил-пиридин-2-карбальдегида. Выход соединения (15) 124 мг (63%)
APCI-MS (m/z): 395.20 ([М+Н]+, 100%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.32-1.55 (м, 3Н), 1.72 (м, 2Н), 2.08 (м, 2Н), 2.16 (с, 3Н), 2.33 (с, 3Н), 2.48 (с, 3Н), 2.80 (м, 1Н), 3.19 (д, 1Н), 3.37 (м, 1Н), 3.67 (д, 1Н), 6.71 (с, 1Н).
Пример 6. {6-Метил-2-[1-{2Н-пиразол-3-илметил)-пиперидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)- (16)
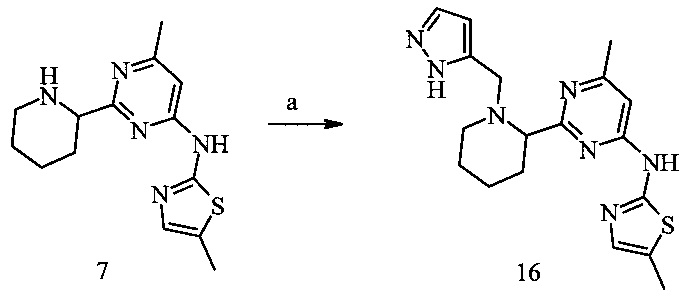
Условия синтеза: а) 2Н-Пиразол-3-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
{6-Метил-2-[1-(2Н-пиразол-3-илметил)-пиперидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (16)
Получали аналогично соединению (13) из амина (7) (145 мг, 0.5 ммоль) и 2Н-пиразол-3-карбальдегида. Выход соединения (16) 96 мг (52%).
APCI-MS (m/z): 370.10 ([М+Н]+, 100%).
Пример 7. {6-Метил-2-[1-(2-метил-тиазол-4-илметил)-пирролидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (24)
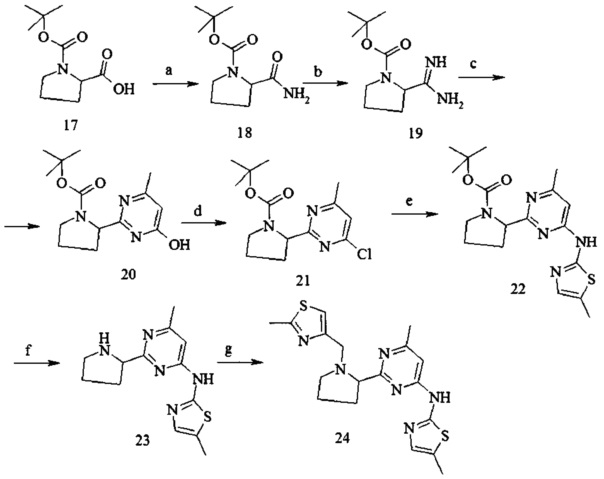
Условия синтеза: a) (NH4)2CO3, TBTU, Et3N, CH3CN, при комнатной температуре; b) Et3OBF4/дихлорметан, NH3/MeOH, при комнатной температуре; c) CH3COCH2COOC2H5, NaOtBu, EtOH, кипячение; d) POCl3, Me2N-Ph, толуол, кипячение; e) 5-метил-2-аминотиазол, Pd2dba3, xantphos, Na2CO3, толуол-H2O, микроволновой реактор, 140°С; f)16% HCl/диоксан, при комнатной температуре; g) 2-метилтиазол-4-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
2-Карбамоил-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (18)
Получали аналогично соединению (2) из Вос-пролина (17) (20 г, 93 ммоль). Выход соединения (18) 12.3 г (62%).
2-Карбамимидоил-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (19)
Получали аналогично соединению (3) из амида (18) (12.3 г, 57,4 ммоль). Выход соединения (19) 16.3 г (95%), амидин (19) использовали на следующей стадии без дополнительной очистки.
2-(4-Гидрокси-6-метил-пиримидин-2-ил)-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (20)
Получали аналогично соединению (4) из 16.3 г (54.5 ммоль) амидина (19). Выход соединения (20) 8.7 г (57%).
2-(4-Хлоро-6-метил-пиримидин-2-ил)-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (21)
Получали аналогично соединению (5) из интермедиата (20) (8.7 г, 31 ммоль). Выход соединения (21) 6.2 г (67%).
2-[4-Метил-6-(5-метил-тиазол-2-иламино)-пиримидин-2-ил]-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (22)
Получали аналогично соединению (6) из хлорида (21) (2 г, 6.7 ммоль) и 5-метил-2-амино-тиазола (0.68 г, 6 ммоль). Выход соединения (22) 1.9 г (85%).
(6-Метил-2-пирролидин-2-ил-пиримидин-4-ил)-(5-метил-тиазол-2-ил)-амин (23)
Получали аналогично соединению (7) из Вос-производного (22) (1.9 г, 5 ммоль). Выход соединения (23) количественный.
{6-Метил-2-[1-(2-метил-тиазол-4-илметил)-пирролидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (24)
Получали аналогично соединению (13) из амина (23) (137 мг, 0.5 ммоль) и 2-метил-тиазол-4-карбальдегида. Выход соединения (24) 117 мг (61%).
APCI-MS (m/z): 387.44 ([М+Н]+, 100%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.76-1.83 (м, 1Н), 1.92-2.10 (м, 1Н), 2.06-2.24 (м, 3Н), 2.31 (с, 3Н), 2.32 (с, 3Н), 2.54 (с, 3Н), 3.11-3.17 (м, 1Н), 3.62 (д, 1Н), 3.75 (т, 1Н), 3.88 (д, 1Н), 6.68 (с, 1Н), 7.06 (м, 2Н), 11.23 (уш.с., 1Н).
Пример 8. {2-[1-(5-Фтор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-тиазол-2-ил-амин (27)
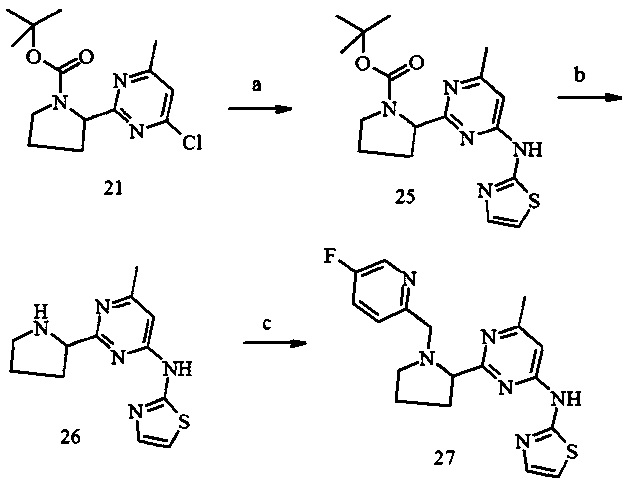
Условия синтеза: a) 2-аминотиазол, Pd2dba3, xantphos, Na2CO3, толуол-H2O, микроволновой реактор, 140°С; b) 16% HCl/диоксан, при комнатной температуре; c) 5-фторпиридин-2-карбальдегид, NaBH(ОАс)3, дихлорметан, при комнатной температуре.
2-[4-Метил-6-(тиазол-2-иламино)-пиримидин-2-ил]-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (25)
Получали аналогично соединению (6) из хлорида (21) (2 г, 6.7 ммоль) и 2-амино-тиазола (0.60 г, 6 ммоль). Выход соединения (25) 1.62 г (75%).
(6-Метил-2-пирролидин-2-ил-пиримидин-4-ил)-тиазол-2-ил-амин (26)
Получали аналогично соединению (7) из Вос-производного (25) (1.62 г, 4.5 ммоль). Выход соединения (26) количественный.
{2-[1-(5-Фтор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-тиазол-2-ил-амин (27)
Получали аналогично соединению (13) из амина (26) (130 мг, 0.5 ммоль) и 5-фтор-пиридин-2-карбальдегида. Выход соединения (27) 120 мг (65%).
APCI-MS (m/z): 371.41 ([М+Н]+, 100%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.78-1.86 (м, 1Н), 1.96-2.04 (м, 1Н), 2.12-2.28 (м, 3Н), 2.30 (с, 3Н), 3.08 (м, 1Н), 3.63 (м, 1Н), 3.77 (м, 1Н), 3.94 (м, 1Н), 6.69 (с, 1Н), 7.12 (с, 1Н), 7.38 (м, 2Н), 7.49 (м, 1Н), 8.32 (м, 1Н), 11.36 (уш.с., 1Н).
Пример 9. {2-[1-(5-Хлор-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-пиридин-2-ил-амин (30)
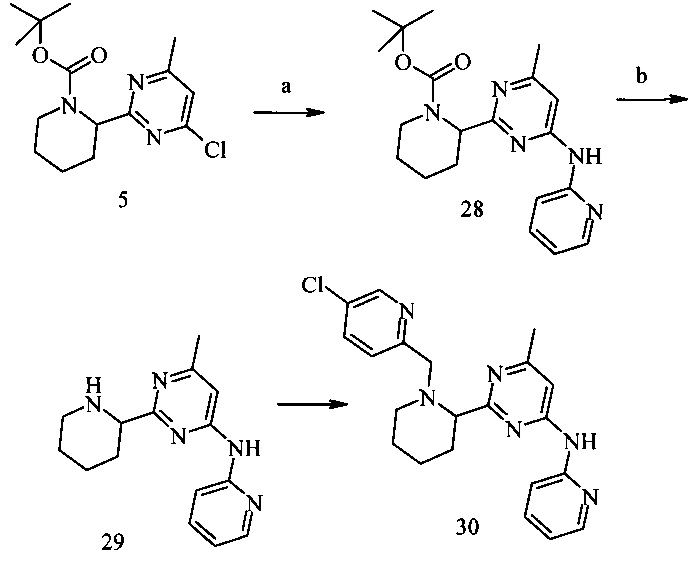
Условия синтеза: a) 2-аминопиридин, Pd2dba3, xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен, Na2CO3, толуол-H2O, микроволновой реактор, 140°С; b) 16% HCl/диоксан, при комнатной температуре; c) 5-хлорпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
2-[4-Метил-6-(пиридин-2-иламино)-пиримидин-2-ил]-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (28)
Получали аналогично соединению (6) из хлорида (5) (2 г, 6.4 ммоль) и 2-аминопиридина (0.564 г, 6 ммоль). Выход соединения (28) 1.21 г (55%).
(6-Метил-2-пиперидин-2-ил-пиримидин-4-ил)-пиридин-2-ил-амин (29)
Получали аналогично соединению (7) из Вос-производного (28) (1.21 г, 3.3 ммоль) с количественным выходом.
{2-[1-(5-Хлор-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-пиридин-2-ил-амин (30)
Получали аналогично соединению (13) из амина (29) (134 мг, 0.5 ммоль) и 5-хлор-пиридин-2-карбальдегида. Выход соединения (30) 126 мг (64%).
APCI-MS (m/z): 395.19 ([M+H]+, 100%), 397.17 ([М+Н]+, 30%).
1Н ЯМР δН (400 МГц, D6-ДМСО): 1.32-1.39 (м, 1Н), 1.51-1.59 (м, 2Н), 1.72-1.87 (м, 3Н), 2.16 (м, 1Н), 2.31 (с, 3Н), 2.89 (м, 1Н), 3.27 (м, 1Н), 3.43 (м, 1Н), 3.63 (м, 1Н), 6.96 (м, 1Н), 7.41 (с, 1Н), 7.59 (м, 1Н), 7.68-7.78 (м, 3Н), 8.25 (м, 1Н), 8.37 (м, 1Н), 9.91 (уш.с., 1Н).
Пример 10. {2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (31)
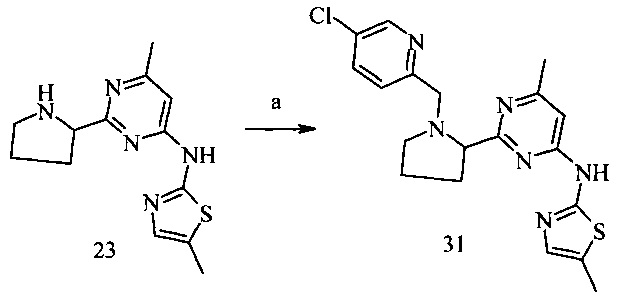
Условия синтеза: a) 5-хлорпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
{2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (31)
Получали аналогично соединению (13) из амина (23) (137 мг, 0.5 ммоль) и 5-хлор-пиридин-2-карбальдегида. Выход соединения (31) 136 мг (68%).
APCI-MS (m/z): 401.13 ([М+Н]+, 100%), 402.86 ([М+Н]+, 42%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.78-1.87 (м, 1Н), 1.95-2.03 (м, 1Н), 2.11-2.24 (м, 3Н), 2.28 (с, 3Н), 2.31 (с, 3Н), 3.09 (м, 1Н), 3.63 (м 1Н), 3.73 (м, 1Н), 3.96 (м, 1Н), 6.67 (с, 1Н), 7.05 (с, 1Н), 7.35 (м, 1Н), 7.70 (м, 1Н), 8.36 (м, 1Н), 11.17 (уш.с., 1Н).
Пример 11. [6-Метил-2-(1-тиазол-2-илметил-пиперидин-2-ил)-пиримидин-4-ил]-тиазол-2-ил-амин (34)
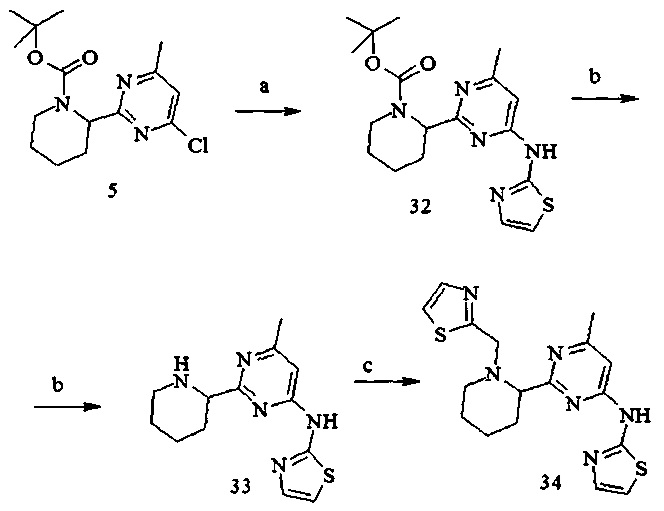
Условия синтеза: a) 2-аминотиазол, Pd2dba3, xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен), Na2CO3, толуол-H2O, микроволновой реактор, 140°С; b) 16% HCl/диоксан, при комнатной температуре; c) тиазол-2-карбальдегид, NaBH(ОАс)3, дихлорметан, при комнатной температуре.
2-[4-Метил-6-(тиазол-2-иламино)-пиримидин-2-ил]-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (32)
Получали аналогично соединению (6) из хлорида (5) (2 г, 6.4 ммоль) и 2-аминотиазола (0.60 г, 6 ммоль). Выход соединения (32) 1.35 г (60%).
(6-Метил-2-пиперидин-2-ил-пиримидин-4-ил)-тиазол-2-ил-амин (33)
Получали аналогично соединению (7) из Вос-производного (32) (1.35 г, 3.6 ммоль) с количественным выходом соединения (33).
[6-Метил-2-(1-тиазол-2-илметил-пиперидин-2-ил)-пиримидин-4-ил]-тиазол-2-ил-амин (34)
Получали аналогично соединению (13) из амина (33) (137 мг, 0.5 ммоль) и тиазол-2-карбальдегида. Выход соединения (34) 115 мг (62%).
APCI-MS (m/z): 373.09 ([М+Н]+, 100%).
1Н ЯМР δH(400 МГц, D6-ДМСО): 1.37-1.43 (м, 1Н), 1.61 (м, 1Н), 1.68-1.83 (м, 2Н), 2.03-2.10 (м, 1Н), 2.29 (м, 1Н), 2.32 (с, 3Н), 3.05 (м, 1Н), 3.60 (м, 1Н), 3.64 (м, 1Н), 3.82 (м, 1Н), 6.72 (с, 1Н), 7.14 (м, 1Н), 7.40 (м, 1Н), 7.49 (м, 1Н), 7.57 (м, 1Н), 11.40 (с, 1Н).
Пример 12. {2-[4-(5-Фтор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (42)
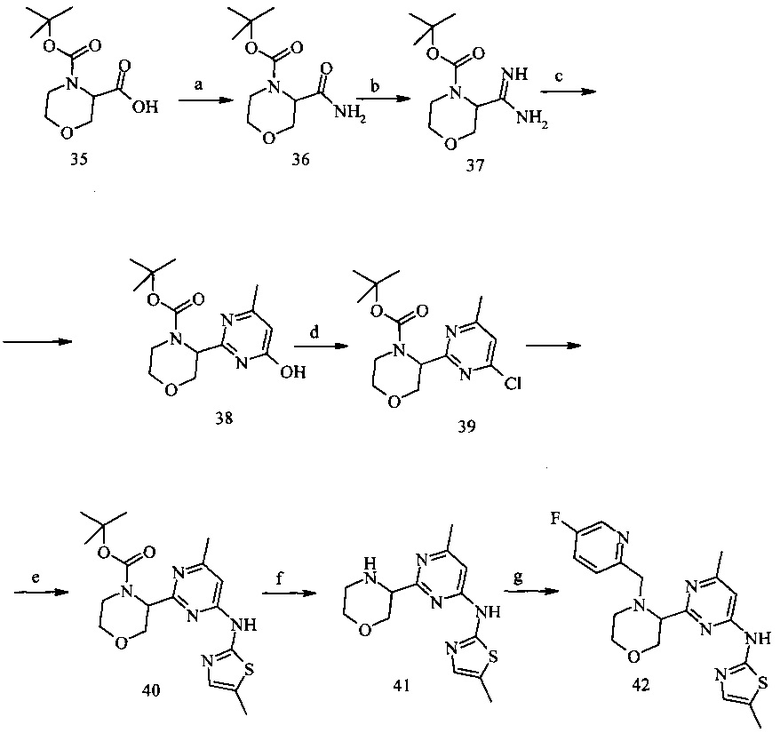
Условия синтеза: a) (NH4)2CO3, TBTU, Et3N, CH3CN, при комнатной температуре; b) Et3OBF4/дихлорметан, NH3/МеОН, при комнатной температуре; c) CH3COCH2COOC2H5, NaOtBu, EtOH, кипячение; d) POCl3, Me2N-Ph, толуол, кипячение; e) 5-метил-2-аминотиазол, Pd2dba3, xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен), Na2CO3, толуол-H2O, микроволновой реактор, 140°С; f) 16% HCl/диоксан, при комнатной температуре; g) 5-фторпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
3-Карбамоил-морфолин-4-карбоновой кислоты трет-бутиловый эфир (36)
Получали аналогично соединению (2) из кислоты (35) (23.1 г, 100 ммоль). Выход соединения (36) 17.25 г (75%).
3-Карбамимидоил-морфолин-4-карбоновой кислоты трет-бутиловый эфир (37)
Получали аналогично соединению (3) из амида (36) (17.25 г, 75 ммоль). Выход соединения (37) 16.38 г (95%).
3-(4-Гидрокси-6-метил-пиримидин-2-ил)-морфолин-4-карбоновой кислоты трет-бутиловый эфир (38)
Получали аналогично соединению (4) из амидина (37) (16.38 г, 71.25 ммоль). Выход соединения (38) 11.35 г (54%).
3-(4-Хлор-6-метил-пиримидин-2-ил)-морфолин-4-карбоновой кислоты трет-бутиловый эфир (39)
Получали аналогично соединению (5) из интермедиата (38) (11.35 г, 38.48 ммоль). Выход соединения (39) 7.0 г (58%).
3-[4-Метил-6-(5-метил-тиазол-2-иламино)-пиримидин-2-ил]-морфолин-4-карбоновой кислоты трет-бутиловый эфир (40)
Получали аналогично соединению (6) из хлорида (39) (2 г, 6.4 ммоль) и 5-метил-тиазол-2-иламин (0.68 г, 6 ммоль). Выход соединения (40) 1.62 г (69%).
(6-Метил-2-морфолин-3-ил-пиримидин-4-ил)-(5-метил-тиазол-2-ил)-амин (41)
Получали аналогично соединению (7) из Вос-производного (40) (1.62 г, 4.1 ммоль) с количественным выходом соединения (41).
{2-[4-(5-Фтор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (42)
Получали аналогично соединению (13) из амина (41) (145 мг, 0.5 ммоль) и 5-фтор-пиридин-2-карбальдегида. Выход соединения (42) 140 мг (70%).
APCI-MS (m/z): 401.15 ([М+Н]+, 100%).
1Н ЯМР δH (400 МГц, D6-ДМСО): 2.32 (с, 3Н), 2.36 (с, 3Н), 2.41 (м 1Н), 2.83 (м, 1Н), 3.41 (м, 1Н), 3.59-3.66 (м, 2Н), 3.75-3.81 (м, 2Н), 3.84 (м, 2Н), 6.69 (с, 1Н), 7.06 (м, 1Н), 7.54-7.62 (м, 2Н), 8.33 (м, 1Н), 11.25 (с, 1Н).
Пример 13. {2-[4-(5-Хлор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (43)
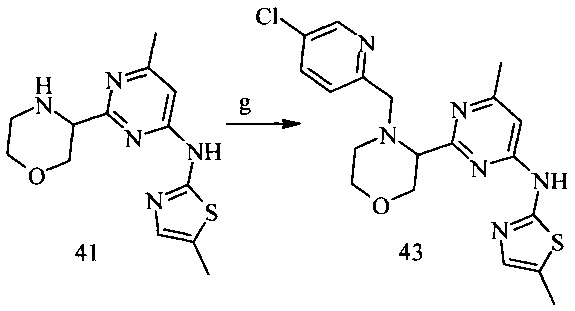
Условия синтеза: a) 5-хлорпиридин-2-карбальдегид, NaBH(ОАс)3, дихлорметан, при комнатной температуре.
{2-[4-(5-Хлор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин (43)
Получали аналогично соединению (13) из амина (41) (145 мг, 0.5 ммоль) и 5-хлор-пиридин-2-карбальдегида. Выход соединения (43) 129 мг (62%).
APCI-MS (m/z): 417.12 ([М+Н]+, 100%), 419.10 ([М+Н]+, 32%),
1Н ЯМР δН (400 МГц, D6-ДМСО): 2.31 (с, 3Н), 2.36 (с, 3Н), 2.42 (м, 1Н), 2.84 (м, 1Н), 3.42 (м, 1Н), 3.59-3.67 (м, 2Н), 3.74-3.81 (м, 2Н), 3.84 (м, 2Н), 6.68 (с, 1Н), 7.10 (м, 1Н), 7.55 (м, 1Н), 7.75 (м, 1Н), 8.38 (м, 1Н), 11.25 (уш.с., 1Н).
Пример 14. {2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-2Н-пиразол-3-ил)-амин (46)
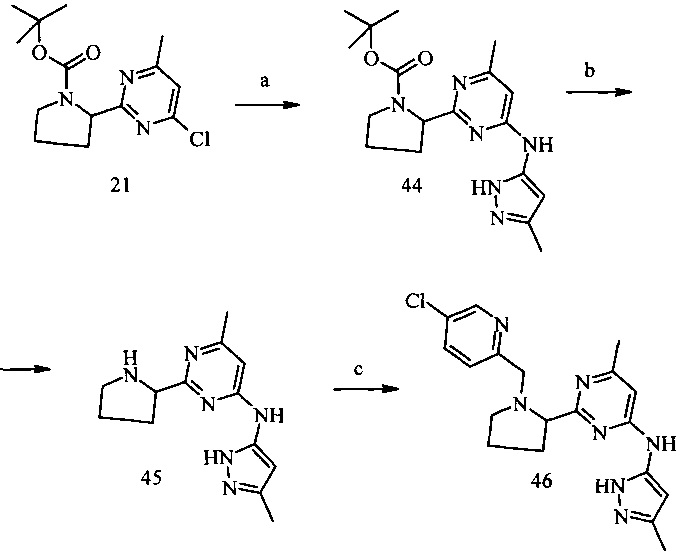
Условия синтеза: a) 5-метил-2Н-пиразол-3-иламин, Pd2dba3, xantphos, Na2CO3, толуол-H2O, микроволновой реактор, 140°С; b) 16% HCl/диоксан, при комнатной температуре; c) 5-хлорпиридин-2-карбальдегид, NaBH(OAc)3, дихлорметан, при комнатной температуре.
2-[4-Метил-6-(5-метил-2Н-пиразол-3-иламино)-пиримидин-2-ил]-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (44)
Получали аналогично соединению (6) из хлорида (21) (2 г, 6.7 ммоль) и 5-метил-2Н-пиразол-3-иламина (0.58 г, 6 ммоль). Выход соединения (44) 0.71 г (33%).
(5-Метил-2Н-пиразол-3-ил)-(6-метил-2-пирролидин-2-ил-пиримидин-4-ил)-амин (45)
Получали аналогично соединению (7) из Вос-производного (44) (0.71 г, 1.98 моль) с количественным выходом соединения (45).
{2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-2Н-пиразол-3-ил)-амин (46)
Получали аналогично соединению (13) из амина (45) (129 мг, 0.5 ммоль) и 5-хлор-пиридин-2-карбальдегида. Выход соединения (46) 98 мг (51%).
APCI-MS (m/z): 384.12 ([М+Н]+, 100%), 386.12 ([М+Н]+, 33%),
1Н ЯМР δH (400 МГц, D6-ДМСО): 1.76-1.81 (м, 1Н), 1.85-1.94 (м, 1Н), 2.04-2.10 (м, 2Н), 2.16 (с, 3Н), 2.22 (с, 3Н), 2.43 (м, 1Н), 3.03 (м, 1Н), 3.49-3.66 (м, 3Н), 3.93 (м, 1Н), 6.02 (уш.с., 1Н), 6.77 (уш.с., 1Н), 7.41 (м, 1Н), 7.73 (м, 1Н), 8.41 (м, 1Н), 9.35 (уш.с., 1Н), 11.80 (уш.с., 1Н).
Пример 15. (5-Метил-пиридин-2-ил)-[4-(1-тиазол-4-ил-метил-пирролидин-2-ил)-тиазол-2-ил]-амин (52).
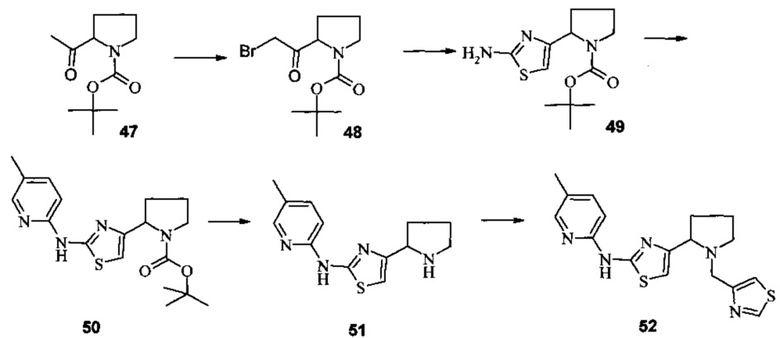
2-(2-Бромо-ацетил)-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (48)
К охлажденному до -78°С раствору N-Boc-2-ацетил-пирролидина (47) (18.7 г, 87.8 ммоль) в сухом ТГФ (150 мл) прикапывают 1М раствор бис(триметилсилил)амида лития в ТГФ (93 мл, 93.0 ммоль) в течение 20 мин. Раствор перемешивают при этой температуре 1 ч, добавляют триметилхлорсилан (12.1 мл, 95.5 ммоль) и перемешивают при 0°С 30 мин. Затем раствор снова охлаждают до -78°С и добавляют бром (4.50 мл, 87.8 ммоль). Реакционную массу перемешивают 30 мин, дают нагреться до комнатной температуры и выливают в 10% раствор Na2S2O3 (100 мл). Суспензию экстрагируют этилацетатом (2×300 мл). Объединенные органические экстракты промывают насыщенным раствором NH4Cl (100 мл), сушат Na2SO4 и концентрируют в вакууме. Продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан/этилацетат, 7:1) Выход соединения (48) 17.2 g (67%).
2-(2-Амино-тиазол-4-ил)-пирролидин-1 -карбоновой кислоты трет-бутиловый эфир (49).
Суспензию соединения (48) (8.1 г, 27.8 ммоль) и тиомочевины (2.11 г, 27.8 ммоль) в этаноле (60 мл) кипятят 1 ч. Растворитель упаривают, остаток суспендируют в 2М NaOH (100 мл) и экстрагируют дихлорметаном (2×200 мл). Объединенные органические экстракты промывают насыщенным раствором NaCl (50 мл), сушат MgSO4 и концентрируют в вакууме. Продукт очищают с помощью колоночной хроматографии на силикагеле (элюент этилацетат). Выход соединения (49) 4.58 г (61%). APCI-MS (m/z): 270.3 ([М+Н]+, 100%), 214.1 ([M-tBu+H]+, 35%).
2-[2-(5-Метил-пиридин-2-ил-амино)-тиазол-4-ил]-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (50).
Смесь соединения (49) (1.62 г, 6.0 ммоль), 2-бром-5-метилпиридина (1.1 г, 6.4 ммоль), 9,9-диметил-4,5-бис(дифенилфосфино)ксантена (174 мг, 0.30 ммоль), комплексного катализатора трис(дибензилиденоацетон)дипалладий (0)-хлороформ (150 мг, 0.15 ммоль), трет-бутилата натрия (0.86 г, 9.0 ммоль) в толуоле (15 мл) перемешивали в атмосфере аргона в микроволновой печи при 130°С в течение 2 час. После охлаждения реакционной смеси до комнатной температуры добавляли воду (30 мл), экстрагировали этилацетатом (2×30 мл). Объединенные органические экстракты сушили над сульфатом натрия, растворитель отгоняли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент этилацетат-гексан, 1:1). Выход соединения (50) 1.34 г (62%). APCI-MS (m/z): 361.4 ([М+Н]+, 100%).
(5-Метил-пиридин-2-ил)-(4-пирролидин-2-ил-тиазол-2-ил)-амин дигидрохлорид (51)
Соединение (50) (1.08 г, 3.0 ммоль) растворяли в 15 мл этанола, добавляли 3 мл HCl и перемешивали при комнатной температуре 4 ч. Растворители упаривали досуха, остаток растирали с этилацетатом. Выход дигидрохлорида (51) 850 мг (85%).
APCI-MS (m/z): 261.1 ([М+Н]+, 100%).
(5-Метил-пиридин-2-ил)-[4-(1-тиазол-4-ил-метил-пирролидин-2-ил)-тиазол-2-ил]-амин (52).
К суспензии соединения (51) (94 мг, 0.28 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (116 мкл, 0.83 ммоль) и тиазол-4-карбальдегид (40 мг, 0.42 ммоль), перемешивали 10-15 мин и добавляли триацетоксиборогидрид натрия (88 мг, 0.42 ммоль). Реакционную массу перемешивали при комнатной температуре 12 ч, выливали в насыщенный раствор карбоната калия (поташ) (30 мл) и экстрагировали дихлорметаном (30 мл). Органический слой отделяли, сушили над сульфатом натрия, упаривали и очищали с помощью колоночной хроматографии на силикагеле (элюент - этилацетат). Выход соединения (52) 77 мг (77%).
APCI-MS (m/z): 358.2 ([М+Н]+, 100%).
Пример 16. Макроциклическое соединение (59)
Синтез интермедиата 4-пирролидин-2-ил-тиазол-2-иламина (53)
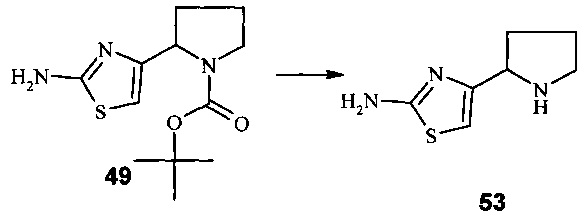
Интермедиат (49) (2.70 г, 10.04 ммоль) растворяли в 30 мл этанола, добавляли 20 мл 16%-HCl в диоксане и перемешивали при комнатной температуре 5 ч. Растворители упаривали, остаток нейтрализовали насыщенным водным раствором карбоната калия (поташ) и экстрагировали несколько раз дихлорметаном. Органические фазы объединяли, сушили над карбонатом калия и упаривали. Выход соединения (53) 1.49 г (88%).
APCI-MS (m/z): 170.2 ([М+Н]+, 100%).
Синтез макроциклического соединения (59)
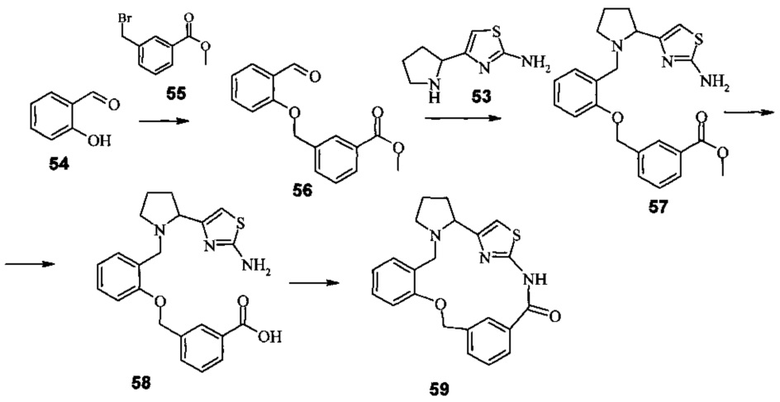
3-(2-Формил-феноксиметил)-бензойной кислоты метиловый эфир (56)
Смесь 2-гидроксибензальдегида (54) (0.73 г, 6 ммоль), бензилбромида (55) (1.37 г, 6 ммоль) и карбоната калия (1 г, 7,2 ммоль) в безводном ацетонитриле (100 мл) кипятили в течение 5 ч (контроль по ТСХ, гексан/этилацетат 9:1). По окончании реакции смесь выливали в воду (150 мл), экстрагировали дихлорметаном (2×100 мл), сушили над сульфатом натрия и упаривали. Выход соединения (56) 1.62 г (100%).
3-{2-[2-(2-Аминотиазол-4-ил)-пирролидин-1-метил]-феноксиметил}- бензойной кислоты метиловый эфир (57)
К раствору амина (53) (169 мг, 1 ммоль) и альдегида (56)(270 мг, 1 ммоль) в дихлорметане (50 мл) добавили NaBH(OAc)3 (254 мг, 1.2 ммоль) и перемешивали при комнатной температуре в течение 8 ч. По окончании реакции смесь выливали в насыщенный раствор карбоната калия (50 мл), экстрагировали дихлорметаном (2×30 мл), сушили над сульфатом натрия и упаривали. Выделяли с помощью колоночной хроматографии на силикагеле (элюент этилацетат). Выход соединения (57)180 мг (42%).
3-{2-[2-(2-Аминотиазол-4-ил)-пирролидин-1-метил]-феноксиметил}-бензойная кислота (58)
Эфир (57) (180 мг, 0.42 ммоль) растворяли в метаноле (10 мл) и добавляли раствор NaOH (84 мг, 2.1 ммоль). Перемешивали при комнатной температуре 6 ч (ТСХ контроль гексан/этилацетат 9:1). Смесь нейтрализовали HCl (конц.) до pH 6 и упаривали досуха. Использовали полученную смесь на следующей стадии без очистки. Выход соединения (58) количественный.
Синтез макроциклического соединения 59
К раствору аминокислоты (58) (174 мг, 0,42 ммоль) в безводном тетрагидрофуране (100 мл) добавляли триэтиламин (0.168 мл, 1.2 ммоль) и TBTU (N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)уроний тетрафторборат) (160 мг, 0.5 ммоль) и перемешивали при комнатной температуре 12 ч. По окончании реакции смесь выливали в насыщенный раствор карбоната калия (150 мл), экстрагировали дихлорметаном (2×100 мл), сушили над сульфатом натрия и упаривали. Выделяли с помощью колоночной хроматографии на силикагеле (элюент этилацетат). Выход соединения (59) 21 мг (12%).
APCI-MS (m/z): 392.4 ([М+Н]+, 100%).
Биологическая активность полученных соединений. Исследование ингибирования серин/треониновых киназ
Для соединений, являющихся предметом данного изобретения, была изучена способность ингибировать киназы, представляющие интерес для терапии различных заболеваний, в частности, онкологических заболеваний, туберкулеза, в том числе его резистентных форм. Список киназ, изучению ингибирования которых, в том числе в соответствии с описанными ниже методиками, уделялось наибольшее внимание, включал, (но принципиально не ограничен) киназами CDK9, ERK7, LIMK1, S6k-beta, PknA, PknB.
Соединения, описанные в данном изобретении, имеют наномолярные или микромолярные значения IC50 в отношении ингибирования различных киназ.
Определение ингибирующей способности соединений по изобретению по отношению к CDK9 серин/треониновой киназе
Исследование выполнялось в компании ProQuinase
Методика
Для измерения киназной активности протеинкиназы CDK9/CycT1 использовался радиометрический метод (33PanQinase® Activity Assay), который проводили в 96-луночных платах объемом 50 мкл. Реакционная смесь состояла из следующих компонентов: 70 мМ HEPES-NaOH pH 7.5, 3 мМ MgCl2, 3 мМ MnCl2, 3 мкМ Na-ортованадат, 1.2 мМ DTT, 50 мкг/мл PEG20000, АТФ 1 мкМ [γ-33Р]-АТФ (около 4×1005 импульств/мин), протеинкиназа 1,9 нМ и субстрат 1 мкг/50 мл, в которую добавляли 5 мкл исследуемого соединения в 10% ДМСО.
Реакционную смесь инкубировали при 30°С в течение 60 минут. Реакцию остнавливали добавлением 50 мкл 2% (по объему) H3PO4, платы отсасывали и промывали два раза 200 мкл 0.9% (вес/объем) NaCl. Содержание 33Pi определяли с помощью сцинтилляционного счетчика для микроплат (Микробета, Валлак).
Для каждой лунки платы вычисляли остаточную активность, для каждой концентрации вычисляли остаточную активность и значение IC50 с использованием программы Quattro Workflow V3.1.0 (www.quattro-research.com). Результаты исследований представлены в таблице 1.
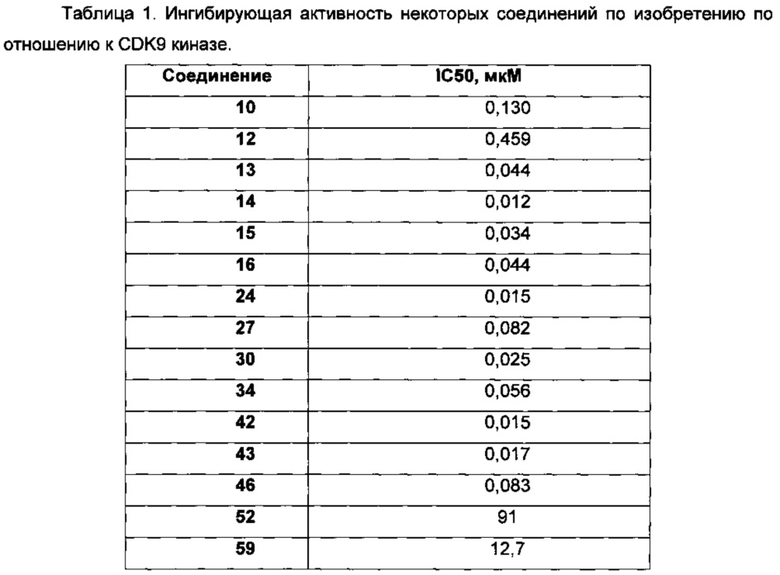
Кроме того, была обнаружена высокая эффективность соединений по изобретению в отношении киназ ERK7, LIMK, S6k-beta, к примеру, соединение 31 по изобретению характеризуется значениями IC50 в отношении этих киназ равными соответственно 0.26, 1, 0.42 мкМ.
Определение ингибирующей способности соединений по изобретению по отношению к микобактериальной PknB серин/треониновой киназе.
Исследование выполнялось в компании DiscoveRx.
Методика
Исследование проводили на высокотехнологичной системе для киназного скрининга соединений KINOMEscan™, принадлежащей компании DiscoveRx.
KINOMEscan™ основана на определении способности соединения конкурентно связываться с ДНК-меченой киназой в присутствии иммобилизованного лиганда. Способность конкурировать с иммобилизованным лигандом количественно измеряется при помощи ПЦР ДНК метки.
Е. coli выращивали до лог-фазы и инфицировали Т7 фагом, инкубировали при встряхивании при 32°С до лизиса. Лизаты центрифугировали и отфильтровывали. Оставшиеся киназы продуцировали в клетки HEK-293 и метили ДНК для количественного ПЦР обнаружения. Магнитные шарики, покрытые стрептавидином, обрабатывали биотинилированным низкомолекулярным лигандом в течение 30 минут при комнатной температуре для получения иммобилизованного лиганда. Полученную смолу блокировали избытком биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% бычьего сывороточного альбумина, 0.05% Твин 20, 1 мМ DTT) для удаления несвязанного лиганда и уменьшения неспецифического связывания. Все реакции проводили в 96-луночных полистирольных планшетах с финальным объемом 0,135 мл. Планшеты, содержащие иммобилизованный лиганд, киназу и исследуемое соединение в блокирующем буфере, инкубировали при встряхивании при комнатной температуре 1 час, смолу промывали промывочным буфером (1×PBS, 0.05% Твин 20), ресуспендировали в новом буфере (1×PBS, 0.05% Твин 20, 0.5 мкМ небиотинилированного лиганда) и инкубировали при комнатной температуре при встряхивании в течение 30 минут. Концентрацию киназы в элюате измеряли методом ПЦР.
Константу связывания Kd определяли с использованием наивысшей концентрации исследуемых соединений 30000 нМ. Константу связывания Kd вычисляли с использованием стандартной кривой доза - ответ по уравнению Хилла:
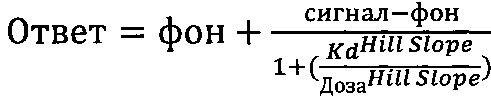 ,
,
где Hill Slope принимали за -1.
Проведенные исследования показали, что соединения по изобретению обладают высокой активностью в отношению киназы PknB. Так, в частности, соединение 52 по изобретению характеризуется Kd равной 29 мкМ.
Изучение противотуберкулезной активности соединений по изобретению.
Соединения по изобретению исследовали на способность подавлять рост вирулентного штамма Mycobacterium Tuberculosis (H37Rv) в жидкой среде в рамках программы Open Innovation компании Eli Lilly. Методика исследования подробно описана в статье A Dual Read-Out Assay to Evaluate the Potency of Compounds Active against Mycobacterium tuberculosis; Ollinger J, Bailey MA, Moraski GC, Casey A, Florio S, et al. (2013) PLoS ONE 8(4): e60531. doi: 10.1371/journal.pone.0060531.
В первичном исследовании соединения испытывали на способность предотвращать рост генетически модифицированного репортерного штамма Mycobacterium Tuberculosis в концентрации 20 мкМ.
Во вторичном исследовании определялась минимальная концентрация ингибирования (MIC), требуемая для полного ингибирования пролиферации Mycobacterium Tuberculosis в жидкой среде. Одновременно соединения исследовали на ингибирование пролиферации клеток HeLa с использованием CellTiter-Glo® Luminescent Cell Viability Assay.
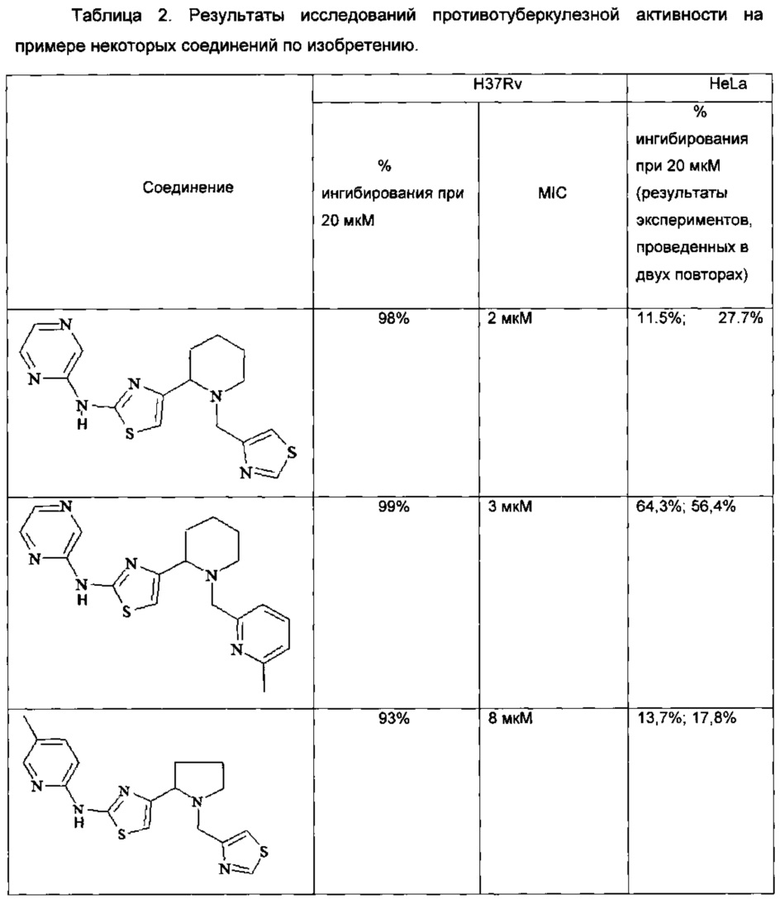
Как показывают данные, представленные в таблице 2, соединений по изобретению эффективно предотвращать рост генетически модифицированного репортерного штамма Mycobacterium Tuberculosis, а также эффективно ингибируют пролиферацию клеток HeLa.
Результаты проведенных исследований свидетельствуют о высокой (ингибирующая концентрация имеет значения в наномолярном диапазоне) активности соединений по изобретению как ингибиторов CDK9 киназы и возможности их применения в качестве эффективных противоопухолевых препаратов.
Кроме того, для соединений по изобретению было также обнаружено, что они эффективно ингибируют микобактериальную киназу PknB (в микромолярной концентрации), Учитывая сложность мишени и малое количество известных ингибиторов этой киназы, соединения по изобретению являются перспективными для создания противотуберкулезных препаратов нового типа.
Таким образом, в результате проведенных исследований было неожиданно обнаружено, что соединения по изобретению общей формулы (I) и общей формулы (II) являются эффективными ингибиторами серин/треониновых киназ и являются перспективными для применения в терапии различных заболеваний, ассоциированных с активностью серин/треониновых киназ, в том числе онкологических заболеваний и туберкулеза.
Несмотря на то что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2422449C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2024 |
|
RU2838180C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ Haspin киназы | 2012 |
|
RU2548363C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| АМИНОМЕТИЛХИНОЛОНЫ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2629111C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| НОВЫЙ ИНГИБИТОР ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ CDK9 | 2018 |
|
RU2738654C1 |
| ИНГИБИТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2468027C2 |
| СЕРУСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ НА ИХ ОСНОВЕ | 2000 |
|
RU2244708C2 |
Изобретение относится к соединениям общей формулы (I) или (II):
n=0-2;
A выбирается независимо и представляет собой 5-7-членный ароматический гетероцикл, содержащий 1-2 атома N и 0-1 атом S; А содержит 0-2 заместителя R; R выбирается независимо и представляет собой метил или этил; B выбирается независимо и представляет собой фенил, 5-6-членный гетероарильный цикл, содержащий 0-2 атома N и 0-1 атом S, или 5-6-членный циклоалкил, содержащий 0-2 атома N и 0-1 атом S; B содержит 0-3 заместителя R1; C выбирается независимо и представляет собой фенил, -NH2, -NH-C1-3-алкил, -NH(С1-3-алкил)С1-3-алкил, 5-6-членный гетероарильный цикл, содержащий 0-2 атома N и 0-1 атом S, или 5-6-членный циклоалкил, содержащий 0-2 атома N и 0-1 атом S; C содержит 0-3 заместителя R1; R1 выбирается независимо и представляет собой -C1-6-алкил, галоген, фенил, -C5-7-гетероарил, содержащий 1-2 атома N и 0-1 атом S, -COOH, -CONH2, -NH2 или -NHR2; R2 выбирается независимо и представляет собой -C1-6-алкил, -C(O)-C1-8-алкил; линкер X выбирается независимо и представляет собой -CH2-, -С(=O)-СН2- или -CH2-O-группу; линкер Q выбирается независимо и представляет собой -NH- или -NH-C(O)-группу; линкер Y выбирается независимо и представляет собой -O-(СН2)m или -С(O)-NH-(СН2)m, где m=1-3; Z выбирается независимо и представляет собой -СН2- группу или атом кислорода. Соединения являются перспективными для применения в терапии заболеваний, ассоциированных с активностью серин/треониновых киназ, в частности, PnkA, PnkB или CDK9 киназы. 3 н. и 10 з.п. ф-лы, 2 табл., 16 пр.
1. Соединение общей формулы I или общей формулы II:
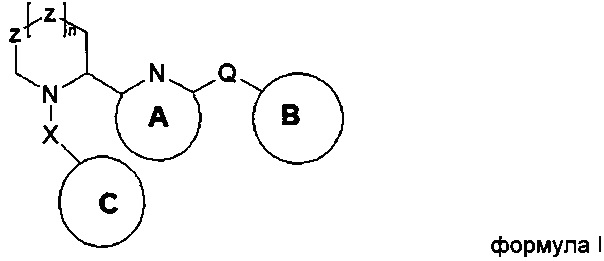 ,
,
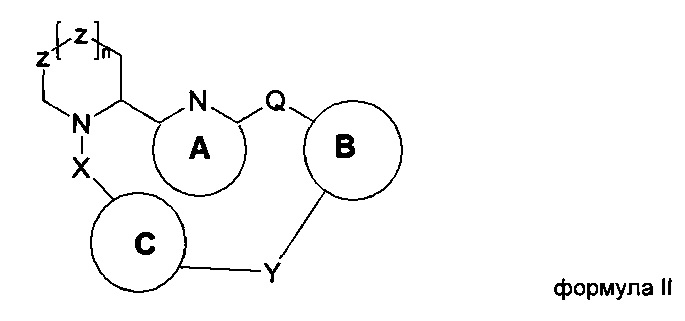
или его таутомер, стереоизомер, энантиомер или фармацевтически приемлемая соль, сольват или гидрат, где:
n=0-2;
A выбирается независимо и представляет собой 5-7-членный ароматический гетероцикл, содержащий 1-2 атома N и 0-1 атом S; А содержит 0-2 заместителя R;
R выбирается независимо и представляет собой метил или этил;
B выбирается независимо и представляет собой фенил, 5-6-членный гетероарильный цикл, содержащий 0-2 атома N и 0-1 атом S, или 5-6-членный циклоалкил, содержащий 0-2 атома N и 0-1 атом S; B содержит 0-3 заместителя R1;
C выбирается независимо и представляет собой фенил, -NH2, -NH-C1-3-алкил, -NH(С1-3-алкил)С1-3-алкил, 5-6-членный гетероарильный цикл, содержащий 0-2 атома N и 0-1 атом S, или 5-6-членный циклоалкил, содержащий 0-2 атома N и 0-1 атом S; C содержит 0-3 заместителя R1;
R1 выбирается независимо и представляет собой -C1-6-алкил, галоген, фенил, -C5-7-гетероарил, содержащий 1-2 атома N и 0-1 атом S, -COOH, -CONH2, -NH2 или -NHR2;
R2 выбирается независимо и представляет собой -C1-6-алкил, -C(O)-C1-8-алкил;
линкер X выбирается независимо и представляет собой -CH2-, -С(=O)-СН2- или -CH2-O-группу;
линкер Q выбирается независимо и представляет собой -NH- или -NH-C(O)-группу;
линкер Y выбирается независимо и представляет собой -O-(СН2)m или -С(O)-NH-(СН2)m, где m=1-3;
Z выбирается независимо и представляет собой -СН2- группу или атом кислорода.
2. Соединение по п. 1, в котором:
n=0-2;
A выбирается независимо и представляет собой тиазол или пиримидин, А содержит 0-2 заместителя R;
R выбирается независимо и представляет собой метил или этил;
B и C выбираются независимо и представляют собой фенил, тиазол, пиридин, пиримидин или пиразин, опционально замещенные 0-3 заместителями R1;
R1 выбирается независимо и представляет собой -C1-6-алкил, галоген, фенил, -C5-7-гетероарил, содержащий 1-2 атома N, 0-1 атом S, -COOH, -CONH2, NH2 или NHR2;
R2 выбирается независимо и представляет собой -C1-3-алкил, -C(O)-C1-3-алкил;
линкер X выбирается независимо и представляет собой -CH2-, -С(=O)-CH2- или -CH2-O-группу;
линкер Q выбирается независимо и представляет собой -NH- или -NH-C(O)-группу;
линкер Y представляет собой -O-(CH2)- группу или отсутствует;
Z выбирается независимо и представляет собой -CH2- группу или атом кислорода.
3. Соединение по п. 1, выбранное из группы:
2-(2-(4-Метил-6-(5-метилтиазол-2-иламино)пиримидин-2-ил)пиперидин-1-ил)-1-(пиперидин-1-ил)этанон;
1-(2-(4-Метил-6-(5-метилтиазол2-иламино)пиримидин-2-ил)пиперидин-1-ил)-2-(метиламино)этанон;
N-(2-(1-((5-Хлорпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин;
N-(2-(1-((5-Фторпиридин-2-ил)метил)пиперидин-2-ил)-6-метилпиримидин-4-ил)-5-метилтиазол-2-амин;
{2-[1-(3-Метил-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{6-Метил-2-[1-(2Н-пиразол-3-илметил)-пиперидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{6-Метил-2-[1-(2-метил-тиазол-4-илметил)-пирролидин-2-ил]-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{2-[1-(5-Фтор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-тиазол-2-ил-амин;
{2-[1-(5-Хлор-пиридин-2-илметил)-пиперидин-2-ил]-6-метил-пиримидин-4-ил}-пиридин-2-ил-амин;
{2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
[6-Метил-2-(1-тиазол-2-илметил-пиперидин-2-ил)-пиримидин-4-ил]-тиазол-2-ил-амин;
{2-[4-(5-Фтор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{2-[4-(5-Хлор-пиридин-2-илметил)-морфолин-3-ил]-6-метил-пиримидин-4-ил}-(5-метил-тиазол-2-ил)-амин;
{2-[1-(5-Хлор-пиридин-2-илметил)-пирролидин-2-ил]-6-метил-пиримидин-4-ил}-(5-метил-2Н-пиразол-3-ил)-амин;
(5-Метил-пиридин-2-ил)-[4-(1-тиазол-4-ил-метил-пирролидин-2-ил)-тиазол-2-ил]-амин.
4. Применение соединения по любому из пп. 1-3 для получения фармацевтической композиции для лечения и/или предотвращения заболевания, связанного с аберрантной активностью серин/треониновой киназы.
5. Применение по п. 4, в котором серин/треониновая киназа представляет собой PnkA, PnkB, CDK9, ERK7, LIMK1 или S6k-beta киназу.
6. Применение по п. 4, в котором заболевание представляет собой туберкулез или онкологическое заболевание.
7. Применение по п. 6, характеризующееся тем, что туберкулез имеет резистентную форму.
8. Фармацевтическая композиция для лечения и/или предотвращения заболевания, связанного с аберрантной активностью серин/треониновой киназы, содержащая эффективное количество, по меньшей мере, одного соединения по любому из пп. 1-3 и, по меньшей мере, один фармацевтически приемлемый носитель, растворитель и/или наполнитель.
9. Фармацевтическая композиция по п. 8, характеризующаяся тем, что серин/треониновая киназа представляет собой PnkA, PnkB, CDK9, ERK7, LIMK1 или S6k-beta киназу.
10. Фармацевтическая композиция по п. 8, характеризующаяся тем, что заболевание представляет собой туберкулез или онкологическое заболевание.
11. Фармацевтическая композиция по п. 10, характеризующаяся тем, что туберкулез имеет резистентную форму.
12. Фармацевтическая композиция по п. 8, характеризующаяся тем, что обладает бактерицидным действием.
13. Фармацевтическая композиция по п. 8, характеризующаяся тем, что обладает бактериостатическим действием.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 2007139732 A1, 06.12 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

