Заявление приоритета
В настоящей патентной заявке заявляется приоритет по заявке США с серийным номером 61/670,379, поданной 11 июля 2012 года и по заявке США с серийным номером 61/746,666, поданной 28 декабря 2012 года, каждая из которых включена посредством ссылки во всей их полноте.
Область техники
В настоящем документе описаны соединения, способы получения таких соединений, фармацевтические композиции и способы применения таких соединений и композиций для ингибирования активности тирозинкиназ.
Уровень техники
Рецептор фактора роста фибробластов 4 (FGFR-4) представляет собой белок, который у людей кодируется геном FGFR-4. Этот белок является членом семейства рецепторов фактора роста фибробластов, в котором в ходе эволюции аминокислотная последовательность была высококонсервативной между его членами. Члены семейства FGFR 1-4 отличаются друг от друга по аффиннос/ти их лигандов и распределению в тканях. Иллюстративный белок полной длины состоит из внеклеточной области, состоящей из трех иммуноглобулин-подобных доменов, одного гидрофобного трансмембранного сегмента и домена цитоплазматической тирозинкиназы. Внеклеточная часть белка взаимодействует с факторами роста фибробласта, запуская каскад нисходящих сигналов, что в конечном итоге влияет на митогенез и дифференцировку. Геномная организация гена FGFR-4 охватывает 18 экзонов. Хотя наблюдали альтернативный сплайсинг, нет данных о том, что С-концевая половина домена IgIII этого белка варьируется между тремя альтернативными формами, как отмечено для FGFR 1-3.
Эктопическую минерализацию, которая характеризуется ненадлежащим отложением кальция-фосфора в мягкой ткани, наблюдали у крыс, обработанных ингибитором FGFR-1 (Brown, АР et al. (2005), Toxicol. Pathol., с. 449-455). Это позволяет предположить, что селективное ингибирование FGFR-4 без ингибирования других изоформ FGFR, включая FGFR-1, может быть желательным для предотвращения некоторых видов токсичности. FGFR-4 предпочтительно связывается с фактором роста фибробластов 19 (FGF19) и недавно был сопоставлен с развитием некоторых сарком, рака почечных клеток, рака молочной железы и рака печени.
Краткое описание изобретения
В настоящем документе описаны ингибиторы FGFR-4. В настоящем документе дополнительно описаны фармацевтические композиции, которые содержат ингибитор FGFR-4.
В одном аспекте настоящего изобретения описано соединение Формулы 1 или его фармацевтически приемлемая соль:
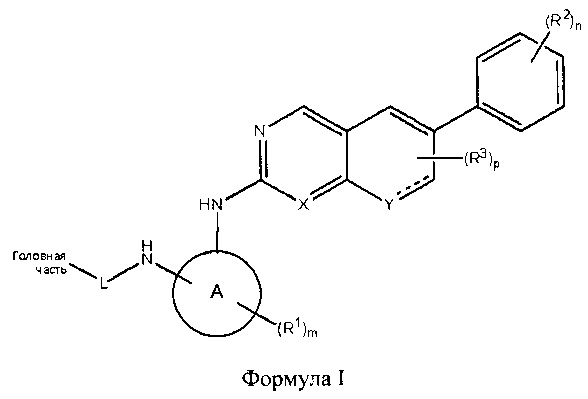
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; кольцо А представляет собой 3-8-членную арильную, гетероарильную, гетероциклическую или алициклическую группу; X представляет собой СН или N; Y представляет собой СН или N-R4, где R4 представляет собой Н или C1-6 алкил; L представляет собой -[C(R5)(R6)]q-, где каждый из R5 и R6 независимо представляет собой Н или C1-6 алкил; и q равен 0-4; каждый из R1-R3 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, оксо, амино, амидо, алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный C1-6 гетероциклил; m равен 0-3; n равен 0-4; и р равен 0-2. В некоторых вариантах реализации кольцо А представляет собой фенил, например, 1,2-дизамещенный фенил; R2 представляет собой галоген или метокси; n равен 2 или 4; X представляет собой N; R1 представляет собой метил; и/или m равен 1.
В другом аспекте настоящего изобретения описано соединение Формулы II или его фармацевтически приемлемая соль:
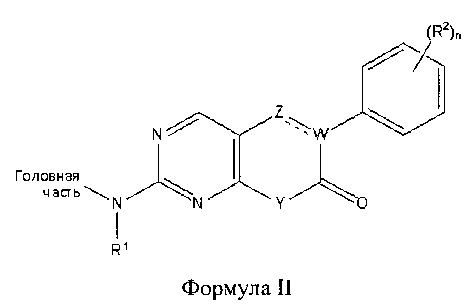
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; W представляет собой С или N; Z представляет собой СН или N; Y представляет собой СН или N-R4, где R4 представляет собой Н или C1-6 алкил; R1 представляет собой Н или C1-6 алкил; каждый из R2 и R3 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, амино, необязательно замещенную алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный C1-6 гетероциклил; n равен 0-4; и р равен 0-2. В некоторых вариантах реализации R2 представляет собой галоген или метокси; n равен 2 или 4; Y представляет собой N-R4, где R4 представляет собой метил; и/или R1 представляет собой метил.
В другом аспекте настоящего изобретения описано соединение Формулы III или его фармацевтически приемлемая соль:
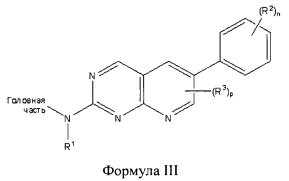
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; R1 представляет собой Н или необязательно замещенный C1-6 алкил, включая диалкиламиноалкил, каждый из R2 и R3 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, амино, амидо, необязательно замещенную алкилмочевину, необязательно замещенный С1-6 алкил, необязательно замещенный C1-6 гетероциклил; n равен 0-4; и р равен 0-2. В некоторых вариантах реализации R2 представляет собой галоген или метокси; n равен 2 или 4. В некоторых вариантах реализации R1 представляет собой метил; в других вариантах реализации R1 представляет собой диэтиламинобутил.
В другом аспекте настоящего изобретения описано соединение Формулы IV или его фармацевтически приемлемая соль:
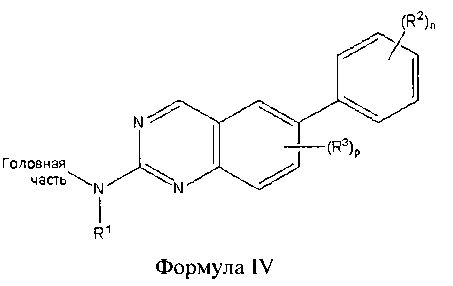
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; R1 представляет собой Н или необязательно замещенный С1-6 алкил; каждый из R2 и R3 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, амино, амидо, необязательно замещенную алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный C1-6 гетероциклил; n равен 0-4; и р равен 0-2. В некоторых вариантах реализации R2 представляет собой галоген или метокси; n равен 2 или 4; и/или R1 представляет собой метил.
В другом аспекте настоящего изобретения описано соединение Формулы V или его фармацевтически приемлемая соль:
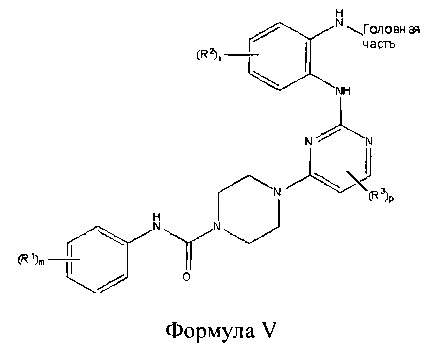
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; каждый из R1-R3 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, амино, амидо, необязательно замещенную алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный C1-6 гетероциклил, необязательно замещенный C1-6 гетероциклиламидо; m равен 0-3; n равен 0-4; и р равен 0-2.
В другом аспекте настоящего изобретения описано соединение Формулы VI или его фармацевтически приемлемая соль:
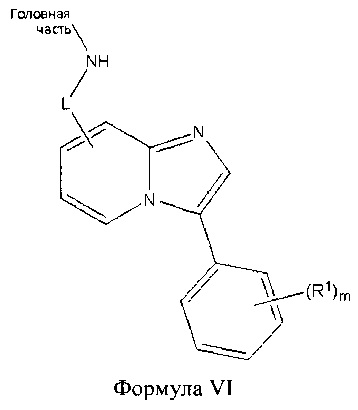
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; L представляет собой арил, гетероарил или -[C(R5)(R6)]q-, где каждый из R5 и R6 независимо представляет собой Н или C1-6 алкил; и q равен 0-4; каждый из R1 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, оксо, амино, амидо, необязательно замещенную алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный гетероциклил; и m равен 0-3. В некоторых вариантах реализации L представляет собой алкилен; в других вариантах реализации L представляет собой фенил. В некоторых вариантах реализации R1 представляет собой трифторэтилмочевину.
В другом аспекте настоящего изобретения описано соединение Формулы VII или его фармацевтически приемлемая соль:
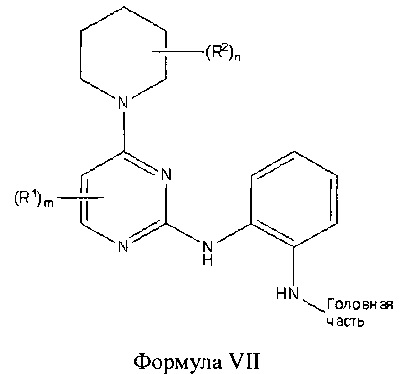
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; каждый из R1 и R2 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, оксо, амино, амидо, необязательно замещенный алкилсульфонамидо, необязательно замещенную алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный С1-6 гетероциклил; m равен 0-3; и n равен 0-4.
В другом аспекте настоящего изобретения описано соединение Формулы VIII или его фармацевтически приемлемая соль:
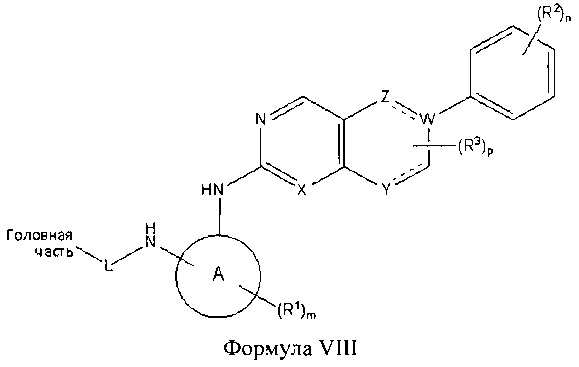
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; кольцо А представляет собой 3-8-членную арильную, гетероарильную, гетероциклическую или алициклическую группу; W представляет собой С или N, каждый из X и Z независимо представляет собой СН или N; Y представляет собой СН или N-R4, где R4 представляет собой Н или C1-6 алкил; L представляет собой -[C(R5)(R6)]q-, где каждый из R5 и R6 независимо представляет собой Н или С1-6 алкил; и q равен 0-4; каждый из R1-R3 независимо представляет собой галоген, циано, необязательно замещенный C1-6 алкокси, гидрокси, оксо, амино, амидо, алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный С1-6 гетероциклил; m равен 0-3; n равен 0-4; и р равен 0-2. В некоторых вариантах реализации кольцо А представляет собой фенил; R2 представляет собой галоген или метокси; n равен 2 или 4; X представляет собой N; R1 представляет собой метил; и/или m равен 1.
В других аспектах соединение представляет собой соединение Формулы IX или его фармацевтически приемлемую соль:
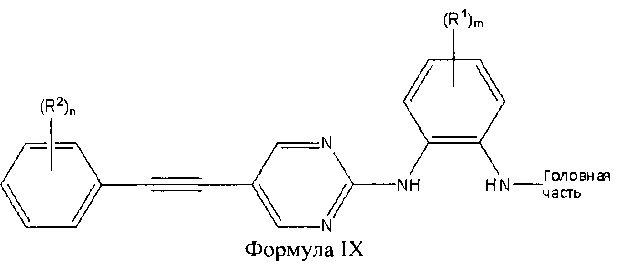
где Головная часть представляет собой фрагмент, способный образовывать ковалентную связь с нуклеофилом; каждый из R1 и R2 независимо представляет собой галоген, циано, необязательно замещенный С1-6 алкокси, гидрокси, оксо, амино, амидо, необязательно замещенную алкилмочевину, необязательно замещенный C1-6 алкил, необязательно замещенный гетероциклил; m равен 0-3; и n равен 0-4.
В других аспектах настоящего изобретения описано соединение Формулы X или его фармацевтически приемлемая соль:
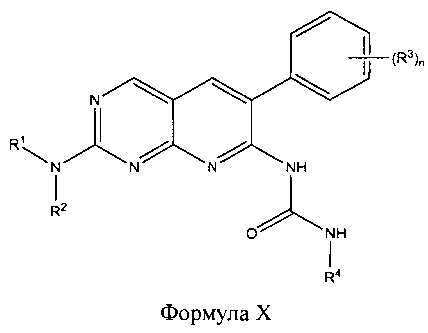
где R1 представляет собой головной фрагмент; R2 представляет собой С1-6 алкил, который необязательно замещен галогеном, амино, гидрокси или циано; каждый R3 независимо представляет собой галоген, амино, циано, С1-6 алкил или С1-6 алкокси, и n равен 2-5; и R4 представляет собой необязательно замещенный C1-6 алкил.
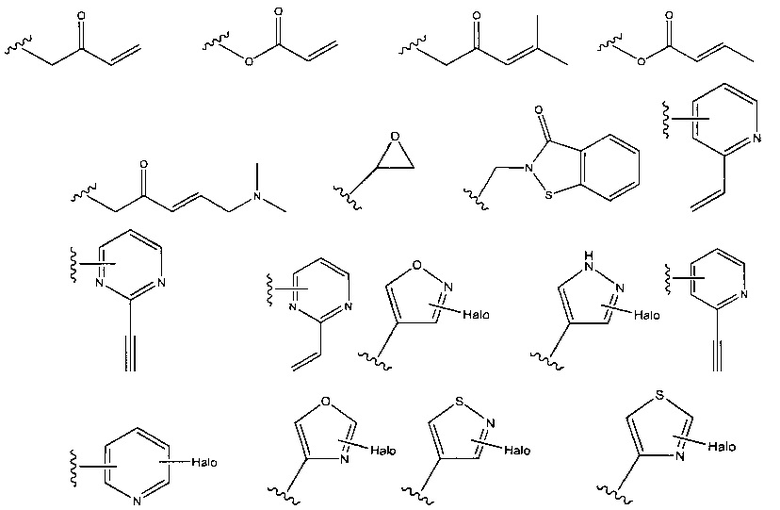
В соединениях, описанных в настоящем документе, головная часть представляет собой фрагмент, который химически активен с нуклеофилом, например, способен образовывать ковалентную связь с нуклеофилом. Примеры головных частей включают, без ограничения, алкилгалогениды, алкилсульфонаты, гетероарилгалогениды, эпоксиды, галоацетамиды, малеимиды, сульфонатные сложные эфиры, альфа-бета ненасыщенные кетоны, альфа-бета ненасыщенные сложные эфиры, винилсульфоны, пропаргиламиды, акриламиды. В некоторых из этих случаев, например, в акриламиде и пропаргиламиде, N в головной части находится рядом с N в формулах, представленных выше. Структуры иллюстративных головных частей изображены ниже:


где X представляет собой уходящую группу, такую как галоген или активированный гидроксильный фрагмент (например, трифлат); и каждый из Ra, Rb и Rc независимо представляет собой Н, замещенный или незамещенный С1-4алкил, замещенный или незамещенный С1-4 циклоалкил или циано.
В формулах, показанных выше, головные части обычно присоединены к атому N ингибитора. В других вариантах реализации головная часть альтернативно может быть присоединена к атому, отличному от N. Примеры иллюстративных головных частей включают, без ограничения,
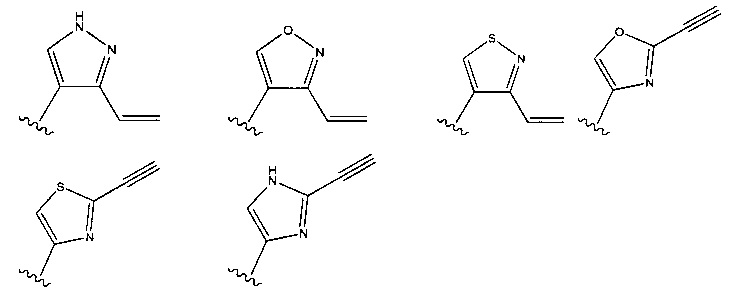
Другие примеры головных частей представлены, например, в WO 2010/028236 и WO 2011/034907.
В некоторых вариантах реализации ингибиторы FGFR-4 по настоящему изобретению ингибируют активность FGFR-4 более эффективно, чем они ингибируют активность FGFR-1. Например, ингибиторы FGFR-4 по настоящему изобретению могут ингибировать активность FGFR-4 по меньшей мере в 10 раз, по меньшей мере в 50 раз, по меньшей мере в 100 раз, по меньшей мере в 200 раз или по меньшей мере в 500 раз более эффективно, чем они ингибируют активность FGFR-1.
В одном аспекте селективность измеряют сравнением ингибирования FGFR-1 и FGFR-4, обусловленного соединением по настоящему изобретению, в анализе одного типа. В одном варианте реализации анализы, используемые для измерения ингибирования FGFR-1 и FGFR-4, представляют собой любые из описанных в настоящем документе анализов. Как правило, ингибирование выражают как IC50 (концентрация ингибитора, при которой подавляется 50% активности фермента) и, следовательно, кратность селективности измеряют по уравнению: (IC50 FGFR-1)/ (IC50 FGFR-4). Такие же измерения и расчеты могут быть использованы также для измерения селективности по сравнению с FGFR-2 и FGFR-3.
Для определения относительного ингибирования FGFR-1 и FGFR-4 под действием соединений по настоящему изобретению могут быть использованы другие анализы активности FGFR, настолько, насколько в таких анализах, по мнению специалистов в данной области техники, используются такие же параметры измерения активности FGFR.
В другом аспекте настоящего изобретения описана фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и соединение, описанное в настоящем документе.
В другом аспекте настоящего изобретения описан ковалентный ингибитор FGFR-4. В некоторых вариантах реализации ковалентный ингибитор FGFR-4, при измерении в биохимическом анализе, ингибирует активность FGFR-4 более эффективно, чем он ингибирует активность FGFR-1. Ингибитор также может содержать головную часть.
В другом аспекте настоящего изобретения описано соединение, которое, при измерении в биохимическом анализе, ингибирует активность FGFR-4 более эффективно, чем оно ингибирует активность FGFR-1, при этом указанное соединение имеет молекулярную массу менее 1500 дальтон. Например, указанное соединение может ингибировать активность FGFR-4 по меньшей мере в 10, 50, 100, 200 или 500 раз более эффективно, при измерении в биохимическом анализе, чем оно ингибирует активность FGFR-1. В некоторых случаях это соединение может образовывать ковалентную связь с FGFR-4, например, с Cys 522 в FGFR-4.
В другом аспекте настоящего изобретения описан ингибированный белок FGFR-4, имеющий ковалентную связь с цистеиновым остатком FGFR-4. Эта ковалентная связь может быть образована между частью фрагмента головной части в ингибиторе и частью цистеинового остатка в FGFR-4, например, цистеинового остатка 552 в белке. Головная часть может представлять собой  .
.
В другом аспекте настоящего изобретения описан способ лечения состояния, опосредованного FGFR-4, состояния, характеризующегося сверхэкспрессией FGFR-4, состояния, характеризующегося амплификацией FGFR4, состояния, опосредованного FGF19, состояния, характеризующегося амплифицированным FGF-19, или состояния, характеризующегося сверхэкспрессией FGF19, любой из этих способов включает введение субъекту терапевтически эффективного количества соединения, описанного в настоящем документе.
В другом аспекте настоящего изобретения описан способ лечения любого из следующих состояний посредством введения субъекту терапевтически эффективного количества соединения, описанного в настоящем документе: гепатоцеллюлярной карциномы, рака молочной железы, рака яичников, рака легких, рака печени, саркомы или гиперлипидемии.
Настоящее изобретение включает все возможные комбинации вариантов реализации, описанных выше.
Краткое описание чертежей
Фиг. 1 представляет собой спектр, демонстрирующий массы для белка FGFR4 без связанного ингибитора и со связанным ингибитором.
Фиг. 2 представляет собой спектр, демонстрирующий массы для белка FGFR4 без связанных ингибиторов и со связанными ингибиторами.
Фиг. 3 представляет собой график, демонстрирующий активность каспазы для Соединения 25.
Фиг. 4 представляет собой схему кристаллической структуры Соединения 52, связанного с белком FGFR4.
Фиг. 5 представляет собой схему кристаллической структуры Соединения 25, связанного с белком FGFR4.
Фиг. 6 представляет собой линейный график, изображающий противоопухолевое действие Соединения 25.
Фиг. 7 представляет собой столбчатую диаграмму, изображающую массы опухоли «голых» мышей с Нер3В.
Фиг. 8 представляет собой линейный график, изображающий изменение массы тела (%) «голых» мышей с Нер3В.
Подробное описание
Известны ингибиторы пан-FGFR, такие как BGJ398 и AZD4547.
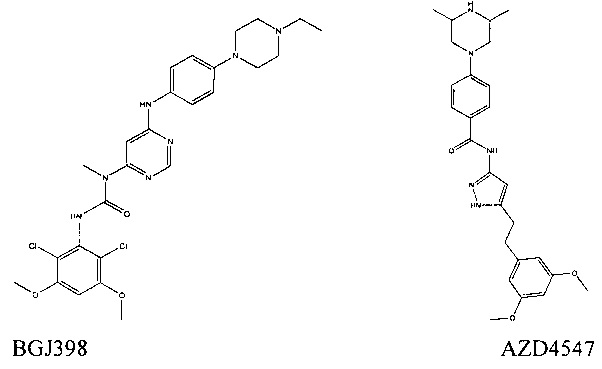
Не было описано, что эти соединения (т.е. ингибиторы пан-FGFR) являются более эффективными против FGFR4, чем против других изоформ FGFR, т.е., FGFR1, FGFR2 и FGFR3. В действительности, AZD4547 менее эффективен против FGFR4, чем против других трех изоформ.
В отличие от BGJ398 и AZD4547, соединения, описанные ниже, могут образовывать ковалентную связь с белком FGFR4; например, эти соединения могут образовывать ковалентную связь с цистеиновым остатком FGFR4, например, цистеином у 552 остатка. FGFR 1-3 не содержат указанного цистеина. Следовательно, способность образовывать ковалентную связь между соединением и FGFR4 представляет собой важный фактор для селективности соединений, описанных в настоящем документе, в отношении FGFR4.
Подробности построения и расположения компонентов, представленных далее в подробном описании или изображенных на чертежах, не являются ограничивающими. В явной форме включены другие варианты реализации и различные пути для практического осуществления изобретения. Также фразеология и терминология, используемая в настоящем документе, предназначены для целей описания, и их не следует толковать как ограничение. Использование терминов «включая», «включает», «включают», «содержащий» или «имеющий», «включающий» и их вариантов охватывает объекты, перечисленные после них, и их эквиваленты, а также дополнительные объекты.
Определения
«Алифатическая группа», при использовании в настоящем документе, относится к прямой, разветвленной или циклической углеводородной группе и включает насыщенные и ненасыщенные группы, такие как алкильная группа, алкенильная группа и алкинильная группа.
«Алкенил», при использовании в настоящем документе, относится к алифатической группе, содержащей по меньшей мере одну двойную связь.
«Алкоксил» или «алкокси», при использовании в настоящем документе, относится к алкильной группе, имеющей кислородный радикал, присоединенный к ней. Иллюстративные алкоксильные группы включают метокси, этокси, пропилокси, трет-бутокси и т.п.
«Алкил», при использовании в настоящем документе, относится к радикалу насыщенных алифатических групп, включая прямые алкильные группы, разветвленные алкильные группы, циклоалкильные (алициклические) группы, алкил-замещенные циклоалкильные группы и циклоалкил-замещенные алкильные группы. «Алкилен» относится к двойному радикалу, который представляет собой алифатическую группу, замещенную с двух концов. В некоторых вариантах реализации прямой или разветвленный алкил имеет 30 или менее атомов углерода в скелете (например, С1-С30 для прямых цепей, С3-С30 для разветвленных цепей), а в других вариантах реализации имеет 20 или менее, или 10 или менее. Точно так же, некоторые циклоалкилы могут иметь 3-10 атомов углерода в их кольцевой структуре, и в некоторых вариантах реализации могут иметь 5, 6 или 7 атомов углерода в кольцевой структуре. Термин «алкенил», используемый в настоящем документе, относится к алифатической группе, содержащей по меньшей мере одну двойную связь; термин «алкинил», используемый в настоящем документе, относится к алифатической группе, содержащей по меньшей мере одну тройную связь.
«Алкилтио», при использовании в настоящем документе, относится к углеводородной группе, имеющей серный радикал, присоединенный к ней. В некоторых вариантах реализации «алкилтио» фрагмент представлен одним из -S-алкила, -S-алкенила или -S-алкинила. Иллюстративные алкилтио-группы включают метилтио, этилтио и т.п.
«Амидо», при использовании в настоящем документе, относится к -С(=O)-N(R1)(R2) или -N(R1)-C(=O)-R2, где каждый из R1 и R2 представляет собой Н или алкил.
«Амино», при использовании в настоящем документе, относится к -NH2, -NH(алкил) или -N(алкил)(алкил).
«Амплифицированный», при использовании в настоящем документе, означает, что в раковых клетках выработаны дополнительные копии гена или хромосомного сегмента, которые могут сообщать преимущество роста или выживания.
«Аралкил», при использовании в настоящем документе, относится к алкильной группе, замещенной арильной группой (например, ароматической или гетероароматической группой).
«Арил», при использовании в настоящем документе, относится к 5-, 6- и 7-членным однокольцевым ароматическим группам, которые могут содержать от нуля до четырех гетероатомов, например, фенил, пирролил, фуранил, тиофенил, имидазолил, оксазолил, тиазолил, триазолил, пиразолил, пиридинил, пиразинил, пиридазинил и пиримидинил и т.п. Арильные группы, содержащие гетероатомы в кольцевой структуре, также могут быть упомянуты как «арильные гетероциклы» или «гетероароматические». Ароматическое кольцо может быть замещено в одном или нескольких кольцевых положениях такими заместителями, как описаны выше, например, галоген, азид, алкил, аралкил, алкенил, алкинил, циклоалкил, полициклил, гидроксил, алкокси, амино, нитро, сульфгидрил, имино, амидо, фосфат, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, сульфонамидо, кетон, альдегид, сложный эфир, гетероциклил, ароматические или гетероароматические фрагменты, -CF3, -CN или т.п. Термин «арил» включает также полициклические кольцевые системы, имеющие два или более циклических колец, в которых два или более атомов углерода являются общими для двух соседних колец (такие кольца являются «конденсированными кольцами»), при этом по меньшей мере одно из указанных колец является ароматическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы и/или гетероциклилы. Каждое кольцо может содержать, например, 5-7 членов.
Термин «карбоцикл» или «циклоалкил», используемый в настоящем документе, относится к ароматическому или неароматическому кольцу, в котором каждый атом в кольце представляет собой атом углерода.
«Ковалентный ингибитор», при использовании в настоящем документе, означает ингибитор, который может образовывать ковалентную связь с белком.
«Энантиомерный избыток» или «% энантиомерный избыток» композиции может быть рассчитан по представленному ниже уравнению. В примере, представленном ниже, композиция содержит 90% одного энантиомера, например, S-энантиомера, и 10% другого энантиомера, т.е. R-энантиомера.
э.и.=(90-10)/100=80%.
Следовательно, в отношении композиции, содержащей 90% одного энантиомера и 10% другого энантиомера, говорят, что она имеет энантиомерный избыток 80%. Некоторые из композиций, описанных в настоящем документе, содержат энантиомерный избыток по меньшей мере 50%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 99% Соединения 1 (S-энантиомера). Другими словами, композиции содержат энантиомерный избыток S-энантиомера, по сравнению с R-энантиомером.
«FGFR-4» или «белок FGFR-4» относится к любой форме белка FGFR-4, включая дикий тип и все вариантные формы (включая, без ограничения, мутантные формы и сплайс-варианты). Белок FGFR-4 представляет собой продукт гена FGFR-4, а белок FGFR-4, следовательно, включает любой белок, кодируемый любой формой гена FGFR-4, включая все аберрации, например, точечные мутации, вставки-делеции, транслокационные гибридизации и фокальные амплификации.
«Гетероарилалкил» относится к алкильной группе, замещенной гетероарильной группой.
«Гетероциклил» или «гетероциклическая группа» относится к кольцевой структуре, такой как 3-7-членная кольцевая структура, кольцо (-а) которой содержит один или более гетероатомов. Гетероциклы также могут представлять собой полициклы, в которых каждая группа имеет, например, 3-7 кольцевых членов. Термин «гетероциклил» или «гетероциклическая группа» включает «гетероарильные» и «насыщенные или частично насыщенные гетероциклильные» структуры. «Гетероарил» относится к ароматической 5-8-членной моноциклической, 8-12-членной бициклической или 11-14-членной трициклической кольцевой системе, имеющей один или более гетероатомов, выбранных из О, N или S. Любой кольцевой атом может быть замещенным (например, одним или более заместителями). Термин «насыщенный или частично насыщенный гетероциклил» относится к неароматической циклической структуре, которая содержит по меньшей мере один гетероатом. Гетероциклильные группы включают, например, тиофенил, тиантренил, фуранил, пиранил, изобензофуранил, хроменил, ксантенил, феноксантин, пирролил, имидазолил, пиразолил, изотиазолил, изоксазолил, пиридинил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, индолил, индазолил, пуринил, хинолизинил, изохинолинил, хинолинил, фталазинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, птеридинил, карбазолил, карболин, фенантридин, акридин, пиримидин, фенантролин, феназин, фенарсазин, фенотиазин, фуразан, феноксазин, пирролидин, оксолан, тиолан, оксазол, пиперидин, пиперазин, морфолин, лактоны, лактамы, такие как азетидиноны и пирролидиноны, сультамы, сультоны и т.п. Гетероциклическое кольцо может быть замещено в одном или нескольких положениях такими заместителями, как описаны выше, например, галоген, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, амино, нитро, сульфгидрил, имино, амидо, фосфат, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, кетон, альдегид, простой эфир, гетероциклил, ароматический или гетероароматический фрагмент, -CF3, -CN или т.п.
«Гетероциклилалкил» относится к алкильной группе, замещенной гетероциклической группой.
«Ингибитор» относится к соединению, которое ингибирует фермент, так что можно наблюдать снижение активности фермента, например, в биохимическом анализе. В некоторых вариантах реализации ингибитор имеет IC50 менее, чем около 1 мкМ, менее, чем около 500 нМ, менее, чем около 250 нМ, менее, чем около 100 нМ, менее, чем около 50 нМ или менее, чем около 10 нМ. Ингибитор FGFR-4 относится к соединению, которое ингибирует FGFR-4.
«Свехрэкспрессированный», при использовании в настоящем документе, означает генный продукт в образце, которого значительно больше, чем наблюдается в группе контрольных образцов (например, нормальной ткани).
«Селективное» относится к соединению, которое ингибирует активность белка-мишени, например, FGFR-4, более эффективно, чем оно ингибирует активность других белков. В этом случае все изоформы FGFR-1, FGFR-2, FGFR-3 и FGFR-4 считаются разными белками. В некоторых вариантах реализации соединение может ингибировать активность белка-мишени, например, FGFR-4, по меньшей мере в 1,5, по меньшей мере в 2, по меньшей мере в 5, по меньшей мере в 10, по меньшей мере в 20, по меньшей мере в 30, по меньшей мере в 40, по меньшей мере в 50, по меньшей мере в 60, по меньшей мере в 70, по меньшей мере в 80, по меньшей мере в 90, по меньшей мере в 100, по меньшей мере в 200, по меньшей мере в 500 или по меньшей мере в 1000 или более раз более эффективно, чем оно ингибирует активность нецелевого белка.
«Замещенный» относится к фрагментам, имеющим заместители, заменяющие водород у одного или более атомов углерода скелета. Следует понимать, что «замещение» или «замещенный» включает подразумеваемое условие, что такое замещение соответствует допустимой валентности замещенного атома и заместителя, и что такое замещение приводит к образованию устойчивого соединения, например, которое самопроизвольно не подвергается преобразованию, такому как перегруппировка, циклизация, элиминирование и т.д. Используемый в настоящем документе термин «замещенный» включает все допустимые заместители органических соединений. В широком аспекте, допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Допустимые заместители могут представлять собой один или несколько одинаковых или разных заместителей для соответствующих органических соединений. Для целей настоящего изобретения, гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные в настоящем документе, которые заполняют валентности указанных гетероатомов. Заместители могут включать любые заместители, описанные в настоящем документе, например, галоген, гидроксил, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как сложный тиоэфир, тиоацетат или тиоформиат), алкоксил, фосфорил, фосфат, фосфонат, фосфинат, амино, амидо, амидин, имин, циано, нитро, азидо, сульгидрил, алкилтио, сульфат, сульфонат, сульфамоил, сульфонамидо, сульфонил, гетероциклический, аралкильный или ароматический, или гетероароматический фрагмент. Специалистам в данной области техники понятно, что фрагменты, замещенные в углеводородной цепи, могут быть сами замещены, если это уместно. Например, заместители замещенного алкила могут включать замещенные и незамещенные формы амино, азидо, имино, амидо, фосфорила (включая фосфонат и фосфинат), сульфонила (включая сульфат, сульфонамидо, сульфамоил и сульфонат) и силильные группы, а также простые эфиры, алкилтио, карбонилы (включая кетоны, альдегиды, карбоксилаты и сложные эфиры), -CF3, -CN и т.п. Иллюстративные замещенные алкилы описаны ниже. Циклоалкилы могут быть дополнительно замещены алкилами, алкенилами, алкокси, алкилтио, аминоалкилами, карбонил-замещенными алкилами, -CF3, -CN и т.п. Аналогичные замещения могут быть сделаны для алкенильных и алкинильньгх групп с образованием, например, аминоалкенилов, аминоалкинилов, амидоалкенилов, амидоалкинилов, иминоалкенилов, иминоалкинилов, тиоалкенилов, тиоалкинилов, карбонил-замещенньгх алкенилов или алкинилов.
При использовании в настоящем документе, определение каждого выражения, например, алкила, m, n и т.д., если он встречается в какой-либо структуре более одного раза, следует считать не зависящим от его определения в ином месте той же структуры.
«Головной фрагмент» или «головная часть» относится к фрагменту ингибитора, который участвует, обратимо или необратимо, в реакции донора, например, белка, с субстратом. Головные части могут, например, образовывать ковалентные связи с белком или могут создавать устойчивые переходные состояния, или могут быть обратимыми или необратимыми алкилирующими агентами. Например, головной фрагмент может представлять собой функциональную группу ингибитора, которая участвует в реакции образования связи, при этом новая ковалентная связь образуется между частью головной части и донором, например, аминокислотным остатком белка. В различных вариантах реализации головная часть представляет собой электрофил, а «донор» представляет собой нуклеофил, такой как боковая цепь цистеинового остатка. Примеры подходящих головных частей включают, без ограничения, группы, изображенные ниже:
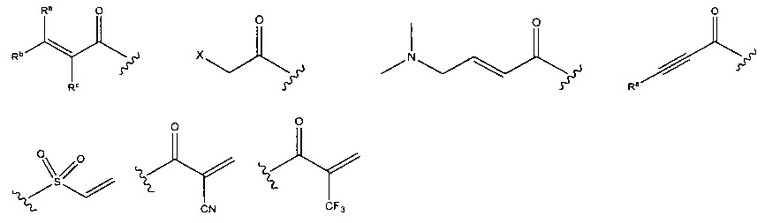
где X представляет собой уходящую группу, такую как галоген, или активированный гидроксильный фрагмент (например, трифлат); и каждый из Ra, Rb и Rc независимо представляет собой Н, замещенный или незамещенный С1-4 алкил, замещенный или незамещенный С1-4 циклоалкил или циано.
Соединения, описанные в настоящем документе, могут содержать неестественные пропорции атомных изотопов у одного или более атомов, которые составляют такие соединения. Например, соединения могут быть меченными радиоактивными изотопами, такими как, например, тритий (3Н) или углерод -14 (14С). Все изотопные варианты соединений, описанных в настоящем документе, радиоактивные или нет, входят в рамки настоящего изобретения. Например, дейтерированыне соединения или соединения, содержащие 13С, считаются входящими в рамки настоящего изобретения.
Некоторые соединения могут существовать в различных таутомерных формах, и все возможные таутомерные формы всех соединений, описанных в настоящем документе, считаются входящими в рамки настоящего изобретения.
Если не указано иное, то структуры, изображенные в настоящем документе, включают также все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационыне)) формы указанной структуры; например, R и S конфигурации для каждого асимметричного центра, Z и Е изомеры двойной связи и Z и Е конформационные изомеры. Следовательно, в рамки настоящего изобретения входят как одиночные стереохимические изомеры, так и энантиомерные, диастереомерные и геометрические (или конформационные) смеси представленных соединений. Если не указано иное, все таутомерные формы соединений настоящего изобретения входят в рамки настоящего изобретения.
Соединения, описанные в настоящем документе, могут быть пригодны в виде свободного основания или в виде соли. Иллюстративные соли включают гидробромидные, гидрохлоридные, сульфатные, бисульфатные, фосфатные, нитратные, ацетатные, валератные, олеатные, сукцинатные, тартратные, нафтилатные, мезилатные, глюкогептонатные, лактобионатные и лаурилсульфонатные соли и т.п. (см., например, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66: 1-19).
Некоторые соединения, описанные в настоящем документе, могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. Как правило, сольватированные формы эквивалентны несольватированным формам и входят в рамки настоящего изобретения. Некоторые соединения, описанные в настоящем документе, могут существовать в многочисленных кристаллических или аморфных формах. Как правило, все физические формы эквивалентны по их применению, предусмотренному настоящим изобретением, и считаются входящими в рамки настоящего изобретения.
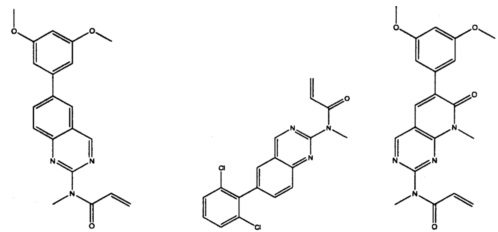
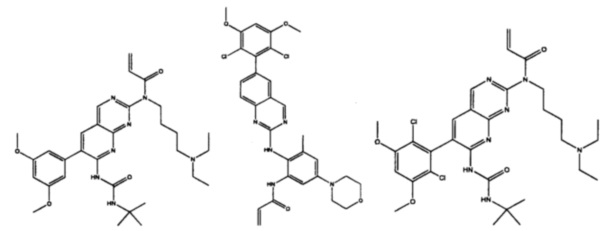
Иллюстративные соединения включают следующие:
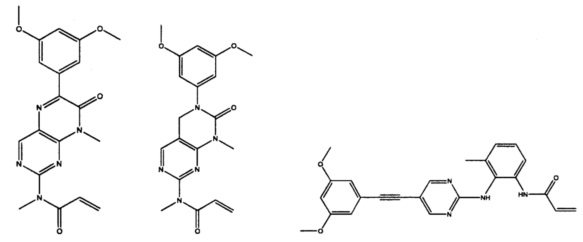
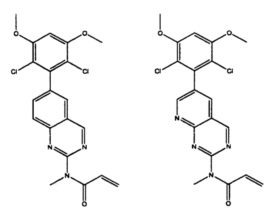
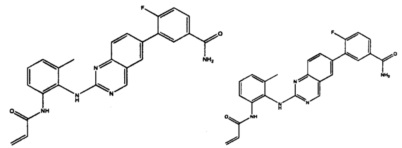
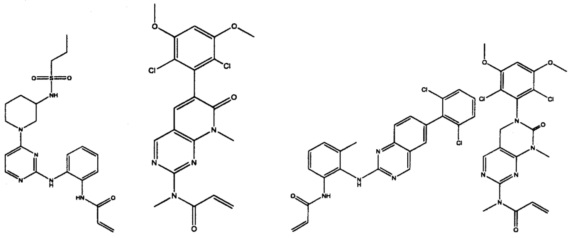
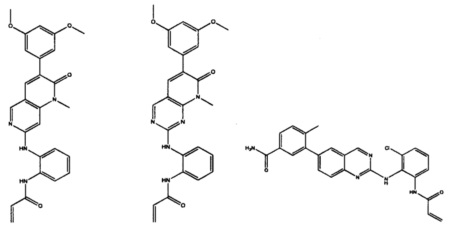
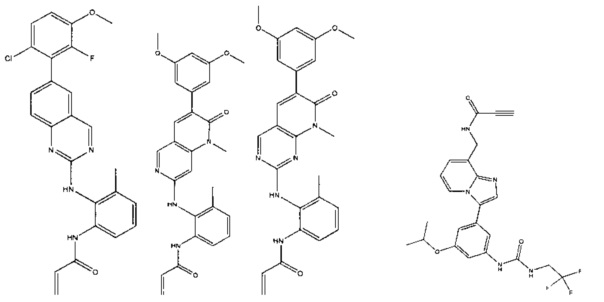
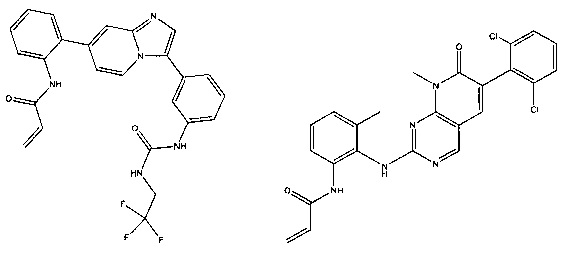
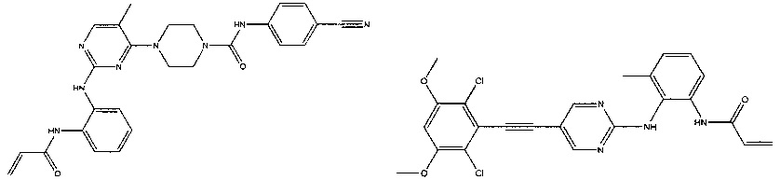
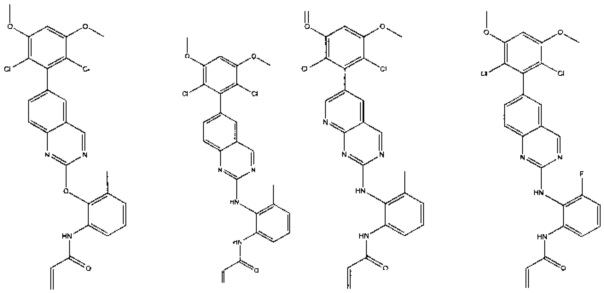
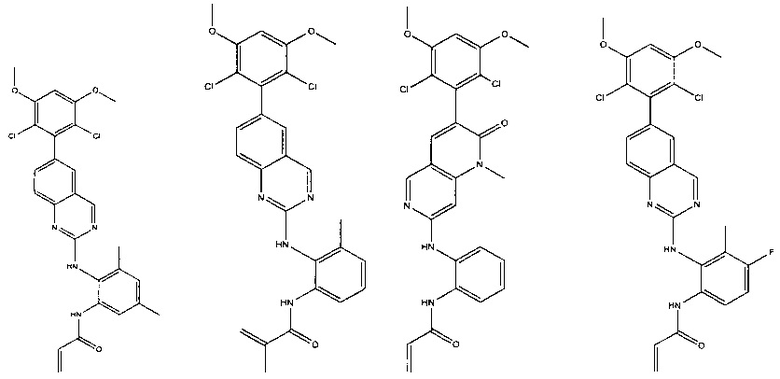
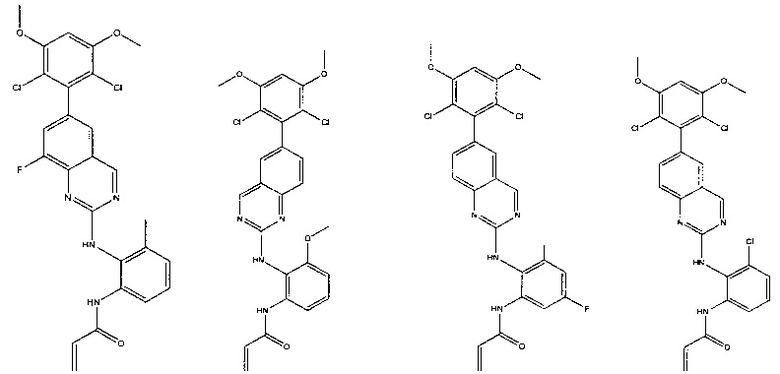
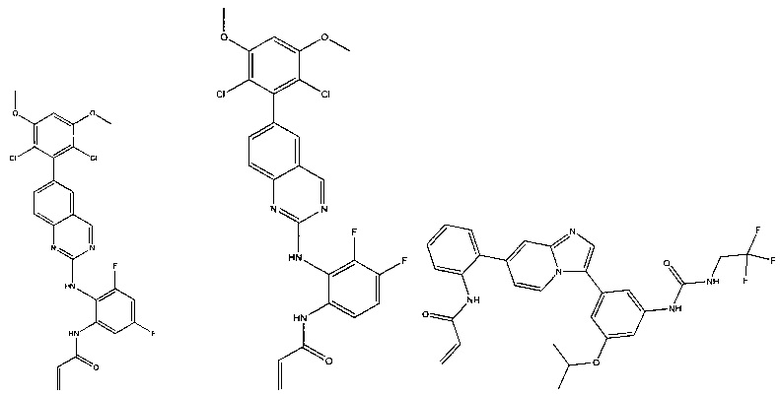
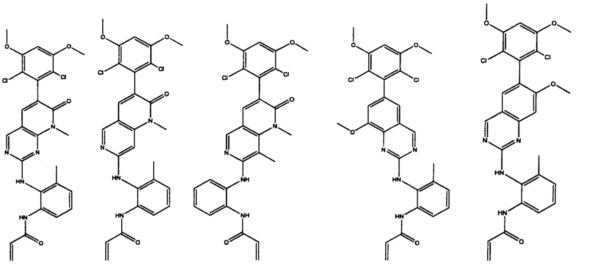
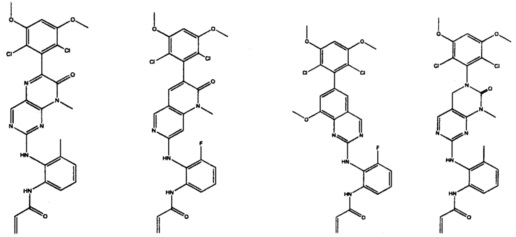
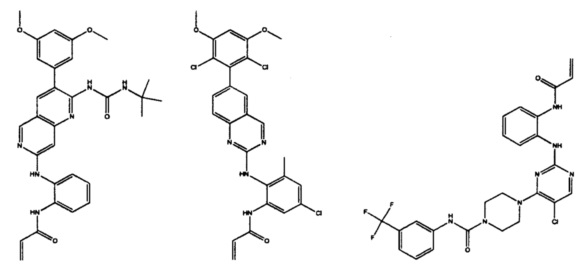
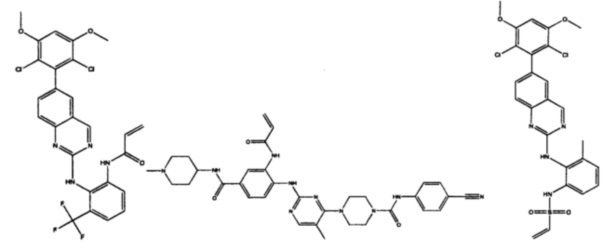
Фармацевтические композиции
Несмотря на возможность отдельного введения соединения, описанного в настоящем документе, указанное соединение предпочтительно вводят в виде фармацевтической композиции, в которой это соединение смешано с одним или более фармацевтически приемлемыми вспомогательными веществами или носителями.
Соединения, описанные в настоящем документе, могут быть составлены в композицию для введения любым удобным способом для применения в медицине или ветеринарии. В некоторых вариантах реализации соединение, входящее в состав фармацевтического препарата, может быть активно само по себе или может представлять собой пролекарство, например, способное преобразовываться в активное соединение в физиологических условиях. В некоторых вариантах реализации соединения, представленные в настоящем документе, включают их гидраты.
Выражение «фармацевтически приемлемый» используется в настоящем документе для обозначения тех соединений, материалов, композиций и/или лекарственных форм, которые, по результатам тщательной медицинской оценки, считаются подходящими для применения в контакте с тканями человека и животных без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерно с целесообразным соотношением пользы/риска.
Примеры фармацевтически приемлемых солей соединений, описанных в настоящем документе, включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих солей кислот включают ацетат, адипинат, бензоат, бензолсульфонат, бутират, цитрат, диглюконат, додецилсульфат, формиат, фумарат, гликолят, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, памоат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тозилат и ундеканоат. Соли, полученные из соответствующих оснований, включают соли щелочных металлов (например, натрия), щелочноземельных металлов (например, магния), аммония и N-(алкил)4+-соли. В настоящем изобретении предусмотрена также кватернизация любых основных азотсодержащих групп соединений, описанных в настоящем документе. При помощи такой кватернизации могут быть получены водо- или маслорастворимые или диспергируемые продукты.
Примеры фармацевтически приемлемых носителей включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) вспомогательные вещества, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатно-солевые буферные растворы; (21) циклодекстрины, такие как Captisol®; направляющие лиганды, присоединенные к наночастицам, такие как Accurins™; и (22) другие нетоксичные совместимые вещества, такие как полимерные композиции, используемые в фармацевтических составах.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п.; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) металл-хелатирующие агенты, такие как лимонная кислота,
этилендиаминтетрауксусная кислота (EDTA), сорбит, винная кислота, фосфорная кислота и т.п. Твердые лекарственные формы (например, капсулы, таблетки, пилюли, драже, порошки, гранулы и т.п.) могут содержать один или более фармацевтически приемлемых носителей, таких как цитрат натрия или фосфат дикальция и/или любые из следующих: (1) наполнители и сухие разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующие вещества, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или гуммиарабик; (3) смачивающие вещества, такие как глицерин; (4) агенты для улучшения распадаемости таблеток, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) агенты для замедления растворения, такие как парафин; (6) ускорители абсорбции, такие как четвертичные аммониевые соединения; (7) увлажняющие агенты, такие как, например, цетиловый спирт и моностеарат глицерина; (8) абсорбенты, такие как каолин и бентонитовая глина; (9) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, натрия лаурилсульфат и их смеси; и (10) красящие агенты. Жидкие лекарственные формы могут включать фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активного ингредиента, жидкие лекарственные формы могут содержать инертные разбавители, которые обычно используют в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, масло проростков, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита, а также их смеси.
Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и эфиры сорбита, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также их смеси.
Мази, пасты, кремы и гели могут содержать, помимо активного соединения, вспомогательные вещества, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевую кислоту, тальк и оксид цинка, или их смеси.
Порошки и спреи могут содержать, помимо активного соединения, вспомогательные вещества, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и полиамидный порошок или смеси этих веществ. Спреи могут дополнительно содержать обычные газы-вытеснители, такие как хлорфтор-углеводороды и летучие незамещенные углеводороды, такие как бутан и пропан.
Препараты могут быть для удобства представлены в единичной дозированной форме и могут быть получены любыми способами, общеизвестными в области фармацевтики. Количество активного ингредиента, которое может быть смешано с материалом носителя для получения единичной дозированной формы, варьируется в зависимости от реципиента, подлежащего лечению, конкретного способа введения. Количество активного ингредиента, которое может быть смешано с материалом носителя для получения единичной дозированной формы, обычно представляет собой такое количество соединения, которое обеспечивает терапевтический эффект.
Лекарственные формы для местного или трансдермального введения соединения по настоящему изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и средства для ингаляции. Активное соединение может быть смешано в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферными агентами или газами-вытеснителями, которые могут быть необходимы.
Если соединения, описанные в настоящем документе, вводят в виде фармацевтических препаратов людям и животным, то они могут быть введены per se или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (более предпочтительно от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Препараты могут быть введены местно, перорально, трансдермально, ректально, вагинально, парентерально, интраназально, интрапульмонально, интраокулярно, внутривенно, внутримышечно, внутриартериально, интратекально, интракапсулярно, интрадермально, внутрибрюшинно, подкожно, субкутикулярно или ингаляцией.
Показания
FGFR-4 регулирует пролиферацию, выживание и альфа-фетопротеиновую секрецию при прогрессировании гепатоцеллюлярной карциномы (НСС); следовательно, ингибиторы FGFR-4 представляют собой перспективные потенциальные терапевтические агенты для этой нерешенной медицинской проблемы (Но et al., Journal of Hepatology, 2009, 50: 118-27). НСС ежегодно поражает более 550000 людей в мире и имеет один из худших 1-летних показателей выживания среди всех типов рака.
Дополнительное свидетельство взаимосвязи между FGFR-4 и НСС демонстрируется участием FGF19, члена семейства факторов роста фибробластов (FGF), которое состоит из гормонов, регулирующих глюкозный, липидный и энергетический гомеостаз. Повышенную пролиферацию гепатоцитов и образование опухоли в печени наблюдали у FGF19 трансгенных мышей. FGF19 активирует FGFR-4, его преобладающий рецептор в печени, и предполагается, что активация FGFR-4 представляет собой механизм, посредством которого FGF19 может увеличивать пролиферацию гепатоцитов и вызывать образование гепатоцеллюлярной карциномы (Wu et al., J Biol Chem (2010) 285(8): 5165-5170). Другими учеными FGF19 также был идентифицирован как ведущий ген в НСС (Sawey et al., Cancer Cell (2011) 19: 347-358). Поэтому предполагается, что соединения, описанные в настоящем документе, которые представляют собой эффективные и селективные ингибиторы FGFR-4, также могут быть использованы для лечения НСС и других раковых заболеваний печени.
В онкогеномном скрининге определили мутацию Y367C, активирующую рецептор 4 фактора роста фибробластов (FGFR-4) в человеческой клеточной линии рака молочной железы MDA-MB-453. Было показано, что такая мутация вызывает конститутивное фосфорилирование, что приводит к активации митоген-активируемого протеинкиназного каскада. Соответственно, было предположено, что FGFR-4 может быть двигателем роста опухоли при раке молочной железы (Roidl et al., Oncogene (2010) 29(10): 1543-1552). Поэтому предполагается, что соединения, описанные в настоящем документе, которые представляют собой эффективные и селективные ингибиторы FGFR-4, могут быть использованы для лечения рака молочной железы, модулируемого FGFR-4.
Молекулярные изменения (например, транслокации) в генах, находящихся перед FGFR-4, могут приводить к активации/сверхэкспрессии FGFR-4. Например, транслокация/слияние генов PAX3-FKHR может приводить к сверхэкспрессии FGFR-4. Сверхэкспрессию FGFR-4 по этому механизму связывают с рабдомиосаркомой (RMS) (Cao et al., Cancer Res (2010) 70(16): 6497-6508). Мутации в самом FGFR-4 (например, мутации в домене киназы) могут приводить к сверхактивации белка; этот механизм связывают с подгруппой RMS (Taylor et al., J Clin Invest (2009) 119: 3395-3407). Поэтому предполагается, что соединения, описанные в настоящем документе, которые представляют собой эффективные и селективные ингибиторы FGFR-4, могут быть использованы для лечения RMS, модулируемых FGFR-4, и других сарком.
С изменениями в генах, расположенных перед FGFR-4, или с мутациями в самом FGFR-4 связывают и другие заболевания. Например, мутации в домене киназы FGFR-4 приводят к сверхактивации, которую связывают с аденокарциномой легких (Ding et al., Nature (2008) 455(7216): 1069-1075). Амплификацию FGFR-4 связывают с такими состояниями как почечно-клеточная карцинома (предварительные данные TCGA). Кроме того, сайленсинг FGFR4 и ингибирование связывания лиганда-рецептора существенно снижает рост опухоли яичников, что позволяет предположить, что ингибиторы FGFR4 могут быть пригодны при лечении рака яичников (Zaid et al., Clin. Cancer Res. (2013) 809).
Патогенное повышение уровней желчной кислоты связывают с изменениями концентраций FGF19 (Vergnes et al., Cell Metabolism (2013) 17, 916-28). Поэтому снижение концентрации FGF 19 может иметь преимущество для ускорения синтеза желчной кислоты и, следовательно, при лечении гиперлипидемии.
Уровни доз
Фактические уровни доз активных ингредиентов в фармацевтических композициях по настоящему изобретению могут варьироваться так, чтобы получать такое количество активного ингредиента, которое эффективно для достижения требуемого терапевтического ответа для конкретного пациента, композиции и способа введения, без токсичности для пациента.
Выбранный уровень доз зависит от различных факторов, включая активность конкретного используемого соединения, описанного в настоящем документе, или его сложного эфира, соли или амида, способа введения, времени введения, скорости экскреции конкретного используемого соединения, продолжительности лечения, других лекарств, соединений и/или материалов, используемых в комбинации с конкретным используемым соединением, возраст, пол, вес, состояние, общее состояние здоровья и медицинский анамнез пациента, подлежащего лечению, и подобных факторов, общеизвестных в области медицины.
Врач или ветеринар, имеющий стандартные навыки в данной области техники, может легко определить и предписать эффективное количество необходимой фармацевтической композиции. Например, врач или ветеринар может начать дозы соединений по настоящему изобретению, используемых в фармацевтической композиции, с уровней ниже, чем необходимы для достижения требуемого терапевтического эффекта, с постепенным повышением дозы до достижения требуемого эффекта.
Как правило, подходящая суточная доза соединения по настоящему изобретению представляет собой количество соединения, которое представляет собой самую низкую дозу, эффективную для получения терапевтического эффекта. Такая эффективная доза обычно зависит от факторов, описанных выше. Как правило, дозы соединений по настоящему изобретению для пациента варьируются от около 0,0001 до около 100 мг на килограмм массы тела в сутки. Например, доза может составлять от 0,1 до 10 г в сутки; от 0,5 до 5 г в сутки; или 1-2 г в сутки. При необходимости эффективная суточная доза активного соединения может быть введена в виде одной, двух, трех, четырех, пяти, шести или более субдоз, введенных по отдельности с подходящими интервалами в течение суток, необязательно в единичных дозированных формах.
Комплексная и таргетная терапия
Введение ингибиторов FGFR-4, описанных в настоящем документе, может быть комбинировано с другими способами лечения рака. Например, эти ингибиторы могут быть введены в комбинации с хирургическими средствами лечения, облучением или другими терапевтическими агентами, такими как антитела, другие селективные ингибиторы киназы или химиотерапевтические средства. Указанные ингибиторы также могут быть введены в комбинации с РНКи терапией или антисмысловой терапией. Ингибиторы FGFR-4, описанные в настоящем документе, могут быть комбинированы с одним, двумя или большим количеством других терапевтических агентов. В примерах, представленных ниже, следует понимать, что «второй терапевтический агент» включает также более одного терапевтического агента, отличного от ингибитора FGFR-4. Ингибитор FGFR-4, описанный в настоящем документе, может быть введен с одним, двумя или большим количеством других терапевтических агентов.
Ингибиторы FGFR-4, описанные в настоящем документе, и второй терапевтический агент не обязательно должны быть введены в виде одной фармацевтической композиции, и из-за различных физических и химических характеристик могут быть введены разными способами. Например, ингибитор FGFR-4 может быть введен перорально, тогда как второй терапевтический агент вводят внутривенно. Определение способа введения и целесообразности введения, если это возможно, в составе одной фармацевтической композиции является общеизвестным для квалифицированных клиницистов. Первоначальное введение может быть выполнено в соответствии с общепринятыми протоколами, известными в данной области техники, а затем, на основании наблюдаемых результатов, дозы, способы введения и время введения могут быть изменены квалифицированным клиницистом.
Ингибитор FGFR-4 и второй терапевтический агент могут быть введены параллельно (например, одновременно, по существу одновременно или в рамках одного протокола лечения) или последовательно (т.е. один после другого, с необязательным временным интервалами между ними), в зависимости от природы пролиферативного заболевания, состояния пациента и фактического выбора второго терапевтического агента для введения.
Кроме того, ингибиторы FGFR-4, описанные в настоящем документе, могут быть введены как часть конъюгата антитела-лекарства, где ингибитор FGFR-4 представляет собой «полезную» часть конъюгата.
Аналитические приборы и способы характеристики соединений:
ЖХМС: Если не указано иное, все данные жидкостной хроматомасс-спектрометрии (ЖХМС) (образцы анализировали на чистоту и идентичность) получали на ЖХ системе Agilent модели 1260 LC, используя масс-спектрометр Agilent модели 6120 с применением электроспрей ионизации под атмосферным давлением (ЭС-IАД), оснащенный обращенно-фазовой колонкой Agilent Poroshel 120 (ЕС-С18, размер частиц 2,7 мкм, размеры 3,0×50 мм) при 22,4 градусах Цельсия. Подвижная фаза состояла из смеси растворителей 0,1% муравьиной кислоты в воде и 0,1%) муравьиной кислоты в ацетонитриле. Использовали постоянный градиент подвижной фазы от 95% водного в 5% органического до 5% водного в 95%) органического (растворителя) за 4 минуты. Скорость потока была постоянной 1 мл/мин.
Протонный ЯМР: Если не указано иное, все спектры 1Н ЯМР были получены при помощи ЯМР прибора Varian 400 МГц Unity Inova 400 МГц ЯМР (время сбора = 3,5 секунды с задержкой в 1 секунду; от 16 до 64 сканов). При наличии описания, все протоны записаны в растворителе ДМСО-d6 в миллионных долях (м.д.) относительно остаточного ДМСО (2,50 м.д.). Препаративные приборы для очистки соединений: Силикагелевую хроматографию выполняли либо на установке Teledyne Isco CombiFlash® Rf, либо на установке Biotage® Isolera Four.
Препаративная ЖХМС: Препаративную ВЭЖХ выполняли на препаративной системе Shimadzu Discovery VP®, оснащенной 5 мкм обращенно-фазовой колонкой Luna 5u С 18(2) 100А, упакованной AXIA, размером 250×21,2 мм при 22,4 градусах Цельсия. Подвижная фаза состояла из растворителей 0,1% муравьиной кислоты в воде и 0,1% муравьиной кислоты в ацетонитриле. Использовали постоянный градиент подвижной фазы от 95% водного в 5% органического до 5% водного в 95% органического (растворителя) за 25 минут. Скорость потока была постоянной при 20 мл/мин. Реакции, выполненные в микроволновом реакторе, проводили в микроволновом устройстве Biotage Initiator.
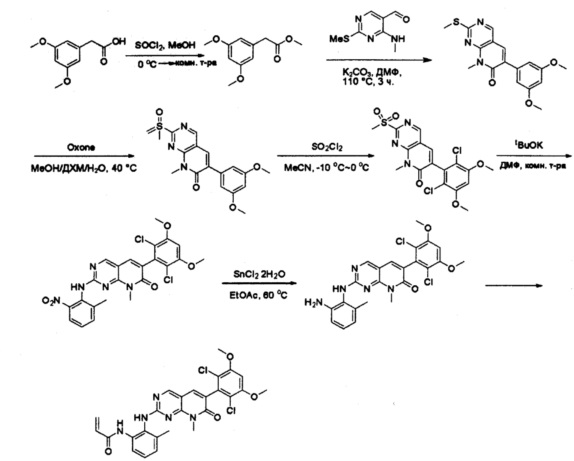
Пример 1: Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)-3-метилфенил)акриламида СОЕДИНЕНИЯ

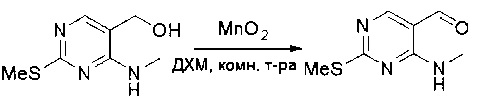
Стадия 1: Синтез этил 4-(метиламино)-2-(метилтио)пиримидин-5-карбоксилата
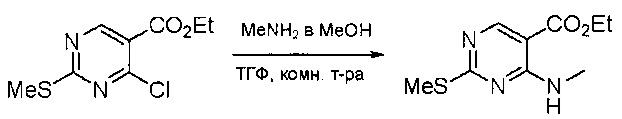
Смесь этил 4-хлор-2-(метилтио)пиримидин-5-карбоксилата (5,0 г, 21,5 ммоль) и 29% метиламина (5,75 г, 53,72 ммоль, раствор в метаноле (МеОН)) в тетрагидрофуране (ТГФ) (100 мл) перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь концентрировали, затем добавляли бикарбонат натрия (NaHCO3) (водный, 20 мл), а полученный раствор экстрагировали этилацетатом (EtOAc) (3×50 мл). Объединенные органические слои промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали с получением этил 4-(метиламино)-2-(метилтио)пиримидин-5-карбоксилата (4,68 г, 96%) в виде желтоватого твердого вещества. МС (ЭС+) C9H13N3O2S, искомое значение: 227, найдено: 228 [М+Н]+. Стадия 2: Синтез (4-(метиламино)-2-(метилтио)пиримидин-5-ил)метанола
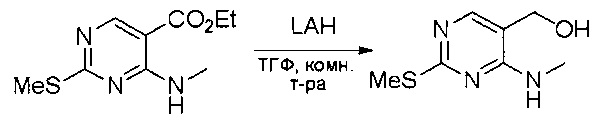
К суспензии алюмогидрида лития (LiAlH4) (1,140 г, 30 ммоль) в ТГФ (100 мл) добавляли этил 4-(метиламино)-2-(метилтио)пиримидин-5-карбоксилат (4,536 г, 20 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. Раствор осторожно гасили при помощи Н2О (2 мл), гидроксида натрия (NaOH) (водный, 15%, 2 мл) и дополнительного количества воды Н2О (7 мл), а затем перемешивали в течение 1 часа. Смесь экстригировали EtOAc (2×100 мл), а объединенные органические слои промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия и концентрировали с получением (4-(метиламино)-2-(метилтио)пиримидин-5-ил)метанола (3,2 г, 85%) в виде желтоватого твердого вещества. МС (ЭС+) C7H11N3OS, искомое значение: 185, найдено: 186 [М+Н]+.
Стадия 3: Синтез 4-(метиламино)-2-(метилтио)пиримидин-5-карбальдегида
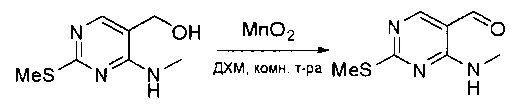
Суспензию (4-(метиламино)-2-(метилтио)пиримидин-5-ил)метанола (3,1 г, 16,73 ммоль) и оксида марганца (7,27 г, 83,67 ммоль) в ДХМ (40 мл) перемешивали при комнатной температуре в течение 12 часов. Полученный осадок фильтровали, а фильтрат концентрировали с получением 4-(метиламино)-2-(метилтио)пиримидин-5-карбальдегида (2,8 г, 91%) в виде желтоватого твердого вещества. МС (ЭС+) C7H9N3OS, искомое значение: 183, найдено: 184 [М+Н]+.
Стадия 4: Синтез метил 2-(3,5-диметоксифенил)ацетата
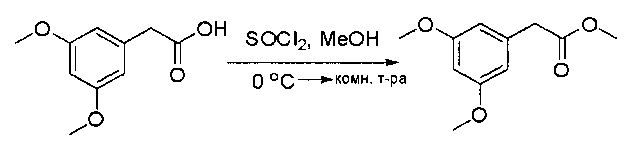
К раствору 2-(3,5-диметоксифенил)уксусной кислоты (5) (600 мг, 3,06 ммоль) в МеОН (30 мл) по каплям добавляли тионилхлорид (3 мл) при 0°С и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакцию контролировали при помощи жидкостной хроматомасс-спектрометрии (ЖХМС). Смесь разбавляли насыщенным раствором бикарбоната натрия (водный, 20 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали с получением метил 2-(3,5-диметоксифенил)ацетата (неочищенный, 700 мг) в виде желтого маслянистого вещества. МС (ЭС+) С11Н14О4, искомое значение: 210, найдено: 211 [М+Н]+.
Стадия 5: Синтез 6-(3,5-диметоксифенил)-8-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8Н)-она
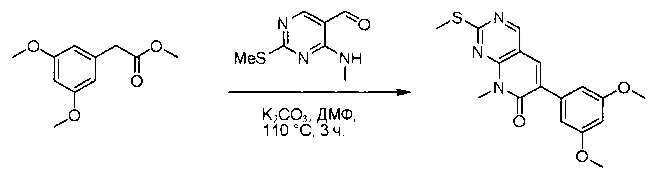
Раствор 2-(3,5-диметоксифенил)ацетата (6) (440 мг, 2,40 ммоль), 4-амино-2-(метилтио)пиримидин-5-карбальдегида (4) (605 мг, 2,88 ммоль) и карбоната калия (662 мг, 4,8 ммоль) в ДМФ (30 мл) перемешивали при 110°С в течение 3 часов. Реакцию контролировали по ЖХМС. Реакционную смесь разбавляли Н2О (30 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (силикагель, петролейный эфир/EtOAc = 2:1) с получением 6-(3,5-диметоксифенил)-8-метил-2-(метилтио)пиридо[2,3-е]пиримидин-7(8Н)-она (7)(683 мг, 83%) в виде белого твердого вещества. МС (ЭС+) C17H17N3O5S, искомое значение: 343, найдено: 344 [М+Н]+.
Стадия 6: Синтез 6-(3,5-диметоксифенил)-8-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8Н)-она
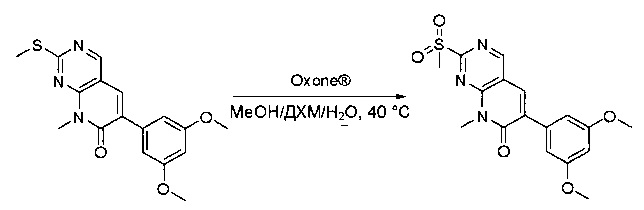
К раствору 6-(3,5-диметоксифенил)-8-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8Н)-она (1,05 г, 3,1 ммоль) в метаноле/дихлорметане (МеОН/ДХМ) (20 мл/20 мл) добавляли раствор Oxone® (пероксимоносульфат калия) (11,3 г, 18,4 ммоль) в Н2О (20 мл) при комнатной температуре и перемешивали реакционную смесь при 40°С в течение 18 часов. Реакцию контролировали по ЖХМС. Реакционную смесь разбавляли Н2О/ДХМ (150 мл/100 мл), а водную фазу экстрагировали ДХМ (100 мл). Объединенные органические слои промывали водой (200 мл) и насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт перекристаллизовали из EtOAc с получением 6-(3,5-диметоксифенил)-8-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8Н)-она (8) (910 мг, выход 78%) в виде желтого твердого вещества. МС (ЭС+) С17H17N3O5S, искомое значение: 375, найдено: 376 [М+Н]+.
Стадия 7: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8Н)-она
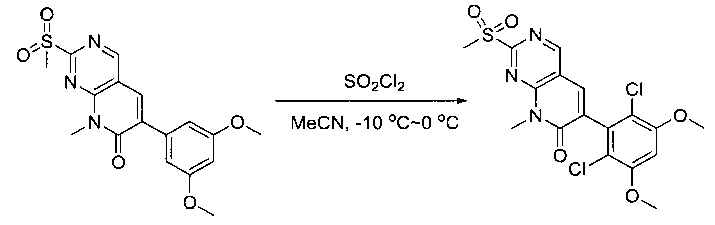
К раствору 6-(3,5-диметоксифенил)-8-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8Н)-она (8) (938 мг, 2,5 ммоль) в ацетонитриле (50 мл) медленно добавляли раствор сульфурилхлорида (1,34 г, 10,0 ммоль) в ацетонитриле (25 мл) на протяжении 0,5 часа при температуре в диапазоне от -10°С до 0°С. Реакцию контролировали тонкослойной хроматографией (ТСХ). Реакционную смесь гасили добавлением Н2О (10 мл). Полученный реакционный раствор концентрировали при пониженном давлении, а остаток перекристаллизовали из смеси EtOAc/петролейного эфира = 1:2 с получением 6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8Н)-она (9) (760 мг, выход 69%) в виде желтого твердого вещества. МС (ЭС+) C17H15Cl2N3O5S, искомое значение: 443, 445, найдено: 444, 446 [М+Н]+.
Стадия 8: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-2-(2-метил-6-нитрофениламино)пиридо[2,3-d]пиримидин-7(8Н)-она
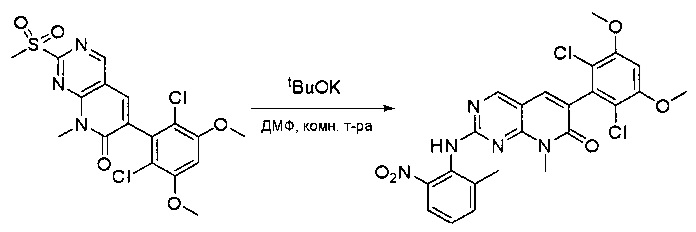
К смеси 6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8Н)-она (9) (1,0 г, 2,26 ммоль) и 2-метил-6-нитробензоламина (684 мг, 4,5 ммоль) в ДМФ (20 мл) добавляли трет-бутоксид калия (756 мг, 6,75 ммоль) при ~10°С и перемешивали реакционную смесь при комнатной температуре в течение 5 минут.Реакционную смесь разбавляли EtOAc (150 мл), а органическую фазу отделяли, промывали водой (2×150 мл), а затем насыщенным солевым раствором (150 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток перекристаллизовали с EtOAc с получением 2-(2-амино-6-метилфениламино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилпиридо[2,3-d]пиримидин-7(8Н)-она (10) (810 мг, выход 70%) в виде желтого твердого вещества. МС (ЭС+) C23H19Cl2N5O5, искомое значение: 515, 517, найдено: 516, 518 [М+Н]+.
Стадия 9: Синтез 2-(2-амино-6-метилфениламино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилпиридо[2,3-d]пиримидин-7(8Н)-она
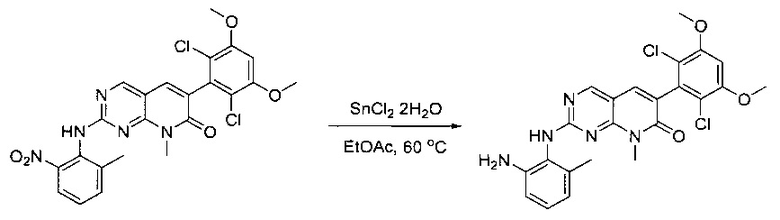
Смесь 2-(2-нитро-6-метилфениламино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилпиридо[2,3-d]пиримидин-7(8Н)-она (10) (810 мг, 1,57 ммоль) и гидрата хлорида олова (II) (1,77 г, 7,86 ммоль) в EtOAc (50 мл) перемешивали при 60°С в течение 2 часов. Реакцию контролировали по ЖХМС. Реакционную смесь подщелочили насыщенным водным раствором бикарбоната натрия до рН=8~9, разбавляли H2O (100 мл), а затем экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (150 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток перекристаллизовали из дихлорметана/этилацетата/петролейного эфира (ДХМ /EtOAc/РЕ)=1/1/2 с получением 2-(2-амино-6-метилфениламино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилпиридо[2,3-d]пиримидин-7(8Н)-она (11) (640 мг, выход 83%) в виде серого твердого вещества. МС (ЭС+) C23H21Cl2N5O3, искомое значение: 485, 487, найдено: 486, 488 [М+Н]+; 1Н-ЯМР (500 МГц, CDCl3) δ м.д. 8,54 (с, 1Н), 7,45 (с, 1Н), 7,08 (t,J=7,5 Гц, 1H), 6,71 (дд, J=3,5, 7,5 Гц, 2Н), 6,65 (ш с, 1H), 6,62 (с, 1Н), 3,94 (с, 6Н), 3,88 (ш с, 2Н), 3,62 (ш с, 3Н), 2,24 (с, 3Н).
Стадия 10: Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)-3-метилфенил)акриламида СОЕДИНЕНИЯ
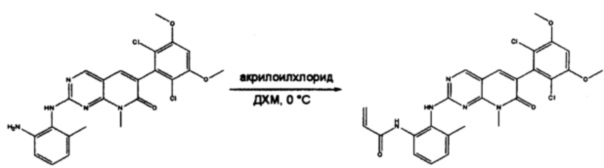
2-(2-Амино-6-метилфениламино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилпиридо[2,3-d]пиримидин-7(8Н)-он (11) растворяли в ДХМ (2 мл) и охлаждали до 0°С, затем добавляли акрилоилхлорид (0,010 мл, 0,13 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Смесь загружали непосредственно на силикагель и очищали флэш-хроматографией с использованием градиента 0-100% EtOAc/гексаны с получением продукта, N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)-3-метилфенил)акриламида (Соединение Е). Продукт получили в виде грязновато-белого твердого вещества (10 мг; выход 19%). МС (ЭС+) C26H23Cl2N5O4, 540 [М+Н]+.
Пример 2: Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-3-метоксифенил)акриламида СОЕДИНЕНИЯ 30
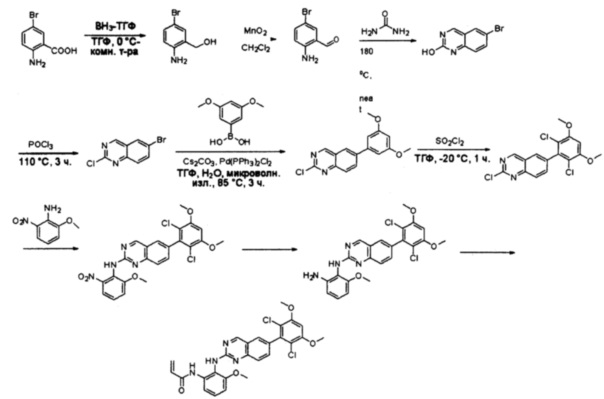
Стадия 1: Синтез (2-амино-5-бромфенил)метанола
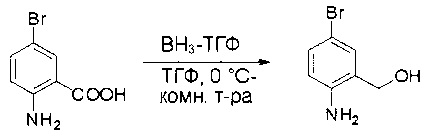
К раствору 2-амино-5-бромбензойной кислоты (10,0 г, 46,3 ммоль) в ТГФ (150 мл) добавляли ВН3-ТГФ (1 М, 231 мл) при комнатной температуре и перемешивали реакционную смесь в течение ночи. Аликвоту реакционной смеси анализировали при помощи ЖХМС, результаты которой показали, что реакция завершена. Реакционную смесь гасили водой (150 мл) и экстрагировали EtOAc (3×500 мл). Органические слои отделяли, объединяли, промывали водой (200 мл) и насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (10 г, неочищенное), которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C7H8BrNO, искомое значение: 201, найдено: 202, 204 [М+Н]+.
Стадия 2: Синтез 2-амино-5-бромбензальдегида
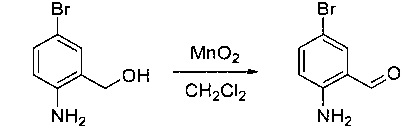
Смесь (2-амино-5-бромфенил)метанола (10 г, 49,5 ммоль) и MnO2 (25,8 г, 296,6 ммоль) в CH2Cl2 (400 мл) перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Твердое вещество фильтровали, а фильтрат концентрировали с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (8 г, 81%), которое использовали напрямую на следующей стадии без дополнительной очистки. МС (ЭС+) C7H6BrNO, искомое значение: 199, найдено: 200, 20 [М+Н]+.
Стадия 3: Синтез 6-бромхиназолин-2-ола
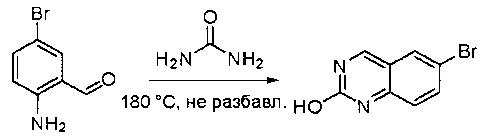
Смесь 2-амино-5-бромбензальдегида (29) (6 г, 30,0 ммоль) и мочевины (30) (27 г, 450,0 ммоль) нагревали до 180°С и перемешивали в течение 5 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, а полученный осадок промывали Н2О (3×500 мл) и три раза совместно упаривали с толуолом для полного удаления захваченной влаги. 6-Бромхиназолин-2-ол (31) (6 г, 89%) получили в виде желтого твердого вещества. МС (ЭС+) C8H5BrN2O, искомое значение: 224, найдено: 225, 227 [М+Н]+.
Стадия 4: Синтез 6-бром-2-хлорхиназолина
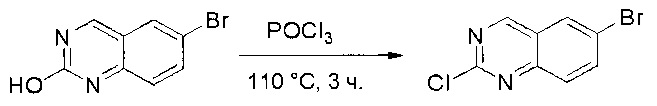
Раствор 6-бромхиназолин-2-ола (31) (6,0 г, 26,7 ммоль) в РОСl3 (80 мл) кипятили с обратным холодильником при 110°С в течение 5 часов. Аликвоту реакционной смеси анализировали по ЖХМС, результаты которой показали, что реакция завершена. Большую часть POCl3 удаляли при пониженном давлении, а остаток по каплям добавляли к ледяной воде (500 мл). Полученный осадок собрали фильтрацией в виде желтого твердого вещества (3,5 г, 54%). МС (ЭС+) C8H4BrClN2, искомое значение: 242, найдено: 243, 245 [М+Н]+.
Стадия 5: Синтез 2-хлор-6-(3,5-диметоксифенил)хиназолина
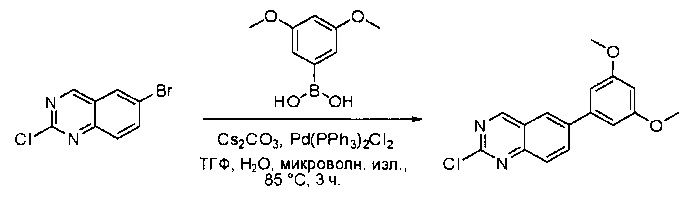
Смесь 6-бром-2-хлорхиназолина (32) (5,0 г, 20,5 ммоль), 3,5-диметоксифенилбороновой кислоты (33) (3,7 г, 20,5 ммоль), Cs2CO3 (20,0 г, 61,5 ммоль) и Pd(PPh3)2Cl2 (1,4 г, 2,1 ммоль) в ТГФ (50 мл), диоксане (50 мл) и воде (10 мл) дегазировали при помощи N2 три раза и перемешивали при 80°С в течение 3 часов. Аликвоту реакционной смеси анализировали при помощи ТСХ и ЖХМС, результаты которых показали, что реакция завершена. Смесь охлаждали до комнатной температуры и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали силикагелевой хроматографией (петролейный эфир/EtOAc = 8:1) с получением 2-хлор-6-(3,5-диметоксифенил)хиназолина (34) в виде светло-желтого твердого вещества (2,4 г, 38%). МС (ЭС+) C16H13ClN2O2, искомое значение: 300, найдено: 301, 303 [М+Н]+.
Стадия 6: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина
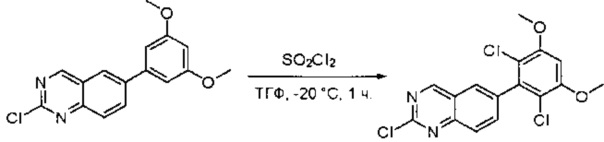
К раствору 2-хлор-6-(3,5-диметоксифенил)хиназолина (34) (2,7 г, 8,9 ммоль) в сухом ТГФ (80 мл) по каплям добавляли SO2Cl2 (3,0 г, 22,3 ммоль) при -20°С и перемешивали реакционную смесь в течение еще одного часа. Аликвоту реакционной смеси анализировали ТСХ и ЖХМС, которые показали, что реакция завершена. Реакционную смесь гасили водой (1 мл), а растворители удаляли под пониженынм давлением. Осадок промывали CH3CN и высушивали с получением 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина (35) (2,6 г, 79%) в виде белого твердого вещества. МС (ЭС+) C16H11C13N2O2, искомое значение: 368, найдено: 369, 371 [М+Н]+; 1Н-ЯМР (500 МГц, ДМСО) δ м.д. 9,67 (с, 1Н), 8,168 (д, J=1,5 Гц, 1Н), 8,10 (д, J=8,5 Гц, 1Н), 7,56 (дд, J=2,0, 8,5 Гц, 1Н), 7,07 (с, 1H), 4,00 (с, 6Н).
Стадия 7: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метокси-6-нитрофенил)хиназолин-2-амина
2-Хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (35) (100 мг, 0,27 ммоль), 2-метокси-6-нитроанилин (36) (57 мг, 0,40 ммоль), Cs2CO3(176 мг, 0,54 ммоль), Pd2(dba)3 (25 мг, 0,027 ммоль) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (Xphos) (26 мг, 0,054 ммоль) растворяли в ДМФ (3 мл) в пробирке для микроволновки и продували N2 в течение 5 минут. Пробирку закрывали и нагревали до 115°С в микроволновом реакторе в течение 30 минут. После охлаждения до комнатной температуры реакционную смесь разбавляли ДХМ и промывали насыщенным солевым раствором три раза. Органическую смесь высушивали над сульфатом натрия и напрямую загружали на силикагель, и очищали, используя градиент 0-100% EtOAc в гексанах. 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метокси-6-нитрофенил)хиназолин-2-амин (37) выделяли в виде желтого твердого вещества (100 мг, выход 73%). МС (ЭС+) C23H18O2N4O5, 501 [М+Н]+.
Стадия 8: Синтез N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-6-метоксибензол-1,2-диамина

6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метокси-6-нитрофенил)хиназолин-2-амин (38) (100 мг, 0,14 ммоль) растворяли в метаноле (10 мл), добавляли 10% Pd/C (15 мг). Смесь перемешивали под H2 из баллона в течение 4 часов. Реакционную смесь фильтровали через целит и удаляли растворитель с получением N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-6-метоксибензол-1,2-диамин (38) с количественным выходом. Соединение 38 использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C23H20Cl2N4O3, 471 [М+Н]+
Стадия 9: Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-3-метоксифенил)акриламида
N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-6-метоксибензол-1,2-диамин (38) (96 мг, 0,20 ммоль) растворяли в ДХМ (2 мл) и охлаждали до 0°С, затем добавляли акрилоилхлорид (0,018 мл, 0,24 ммоль) и перемешивали при 0°С в течение 2 часов. Смесь загружали непосредственно на силикагель и очищали флэш-хроматографией, используя градиент 0-100%) EtOAc в гексанах. N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-3-метоксифенил)акриламид (39) выделяли в виде грязновато-белого твердого вещества (30 мг, выход 28%). МС (ЭС+) C26H22Cl2N4O4, 525 [М+Н]+.
Пример 3: Синтез СОЕДИНЕНИЯ 25
Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)хиназолин-2-амина
2-Хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (35) (5 г, 13,5 ммоль), 2-метил-6-нитроанилин (3,09 г, 20,3 ммоль), Cs2CO3(13,2 г, 40,6 ммоль), Pd2(dba)3(1,24 г, 1,35 ммоль) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (Xphos) (1,29 г, 2,71 ммоль) растворяли в DMA (100 мл) и продували N2 в течение 5 минут. Реакционную смесь нагревали до 110°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли ДХМ (500 мл) и промывали 10% раствором HCl три раза (3×300 мл) и три раза - насыщенным солевым раствором. Органическую смесь высушивали над сульфатом натрия и загружали непосредственно на силикагель, и очищали, используя градиент 0-100% EtOAc в гексанах. 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)хиназолин-2-амин выделяли в виде желтого твердого вещества (5,5 г, выход 81%). МС (ЭС+) C23H18Cl2N4O4, 485 [М+Н]+.
Синтез N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-6-метилбензол-1,2-диамина
6-(2,6-Дихлор-3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)хиназолин-2-амин (5,5 г, 11,33 ммоль) растворяли в метаноле (200 мл) и этилацетате (100 мл), добавляли 10% Pd/C (650 мг). Смесь перемешивали под Н2 из баллона в течение ночи. Реакционную смесь фильтровали через целит и удаляли растворитель с получением N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-6-метилбензол-1,2-диамина с количественным выходом. Его использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C23H20Cl2N4O2, 455 [М+Н]+
Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-3-метилфенил)акриламида
N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-6-метилбензол-1,2-диамин (5,16 г, 11,33 ммоль) растворяли в ДХМ (100 мл) и охлаждали до 0°С, затем добавляли DIEA (1,781 мл, 10,20 ммоль) и акрилоилхлорид (1,013 мл, 12,47 ммоль) и перемешивали при 0°С в течение 2 часов. Смесь загружали непосредственно на силикагель и очищали флэш-хроматографией, используя градиент 0-100%) EtOAc в гексанах. N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-3-метилфенил)акриламид выделяли в виде грязновато-белого твердого вещества (3,5 г, выход 61%). МС (ЭС+) C26H22Cl2N4O3, 509 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,53 (с, 1Н), 9,23 (с, 1Н), 8,68 (с, 1Н), 7,82-7,65 (м,2Н), 7,51 (с, 2Н), 7,21 (м, 1H), 7,12 (д, J=6,8 Гц, 1Н), 7,01 (с, 1Н), 6,49 (дд, J=17,0, 10,2 Гц, 1Н), 6,28-6,15 (м, 1H), 5,68 (дд, J=10,2, 2,0 Гц, 1Н), 3,97 (с, 6Н), 2,19 (с, 3Н).
Пример 4: Синтез СОЕДИНЕНИЯ 26 И СОЕДИНЕНИЯ 10

Синтез 6-бромпиридо[2,3-d]пиримидин-2-амина
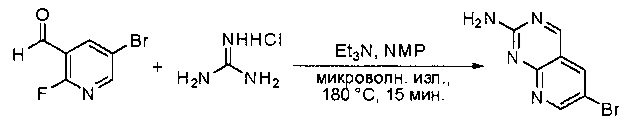
5-бром-2-фторникотинальдегид (3,0 г, 14,78 ммоль), гидрохлорид гуанидина (1,69 г, 17,74 ммоль) и триэтиламин (4,48 г, 44,35 ммоль) растворяли в 1-метил-2-пирролидиноне (15 мл) и перемешивали реакционную смесь при 180°С в течение 15 минут под микроволновым излучением. Смесь охлаждали до комнатной температуры, гасили водой (200 мл) и экстрагировали этилацетатом (2×300 мл). Органические слои объединяли, промывали водой (3×50 мл) и насыщенным солевым раствором (3×50 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали силикагелевой колоночной хроматографией (этилацетат:петролейный эфир = 3:1) с получением 6-бромпиридо[2,3-d]пиримидин-2-амина (2,0 г, 60%) в виде желтого твердого вещества. МС (ЭС+) C7H5BrN4, искомое значение: 224, 226, найдено: 225, 227 [М+Н]+.
Синтез 6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-амина
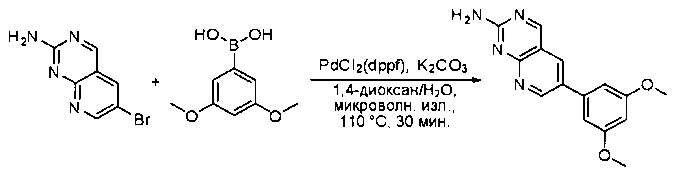
Смесь 6-бромпиридо[2,3-d]пиримидин-2-амина (1,0 г, 4,46 ммоль), 3,5-диметоксифенилбороновой кислоты (1,2 г, 6,70 ммоль), PdCl2(dppf) (364 мг, 0,446 ммоль) и карбоната калия (1,8 г, 13,39 ммоль) в 1,4-диоксане/воде (4 мл/1 мл) дегазировали азотом в течение 5 минут и перемешивали при 110°С в течение 30 минут под микроволновым излучением. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением неочищенного продукта, который очищали силикагелевой колоночной хроматографией (этилацетат:петролейный эфир = 4:1) с получением 6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-амина в виде желтого твердого вещества (400 мг, 31%). МС (ЭС+) C15H14N4O2, искомое значение: 282, найдено: 283 [М+Н]+.
Синтез 6-(3,5-диметоксифенил)-К-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина
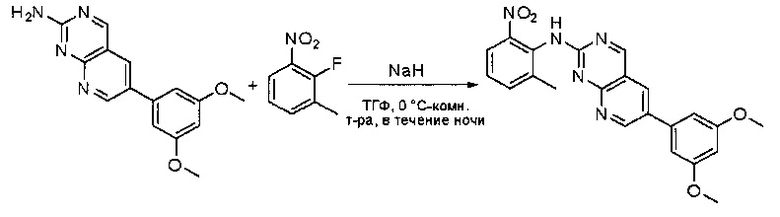
К раствору 6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-амина (400 мг, 1,42 ммоль) в ТГФ (20 мл) при 0°С добавляли гидрид натрия (102 мг, 4,25 ммоль). Раствор перемешивали в течение 20 минут, затем добавляли 2-фтор-1-метил-3-нитробензол (440 мг, 2,84 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, гасили водой (20 мл) и экстрагировали этилацетатом (3×30 мл). Органические слои объединяли, промывали насыщенным солевым раствором (50 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали силикагелевой колоночной хроматографией (этилацетат:петролейный эфир = 4:1) с получением 6-(3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина (310 мг, 51%) в виде коричневого твердого вещества. МС (ЭС+) C22H19N5O4, искомое значение: 417, найдено: 418 [М+Н]+.
Синтез N1-(6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)-6-метилбензол-l,2-диамина
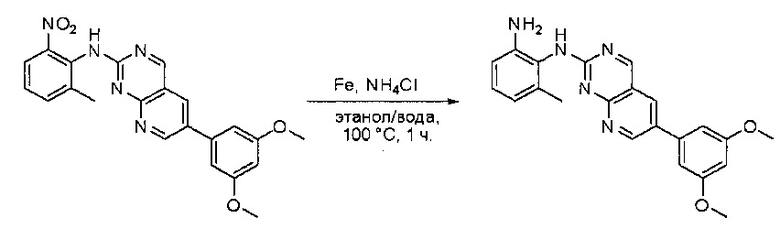
К раствору 6-(3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина (100 мг, 0,24 ммоль) в этаноле (5 мл) и воде (5 мл) добавляли порошок железа (110 мг, 1,92 ммоль) и хлорид аммония (100 мг, 1,920 ммоль). Смесь перемешивали при 100°С в течение 1 часа, охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток очищали препаративной ВЭЖХ с получением N1-(6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)-6-метилбензол-1,2-диамина (29,5 мг, 32%) в виде желтого твердого вещества. МС (ЭС+) C22H21N5O2, искомое значение: 387, найдено: 388 [М+Н]+; 1Н-ЯМР (500 МГц, ДМСО-d6) δ м.д. 9,30, 9,21 (ш, ш, 2Н), 8,95 (с, 1Н), 8,60 (д, 1H, J=3,0 Гц), 6,96-6,92 (м, 3Н), 6,63 (д, 1Н, J=5,5 Гц), 6,55 (т, 1Н, J=2,0 Гц), 6,50-6,48 (м, 1Н), 4,79 (с, 2Н), 3,84 (с, 6Н), 2,08 (с, 3Н).
Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина
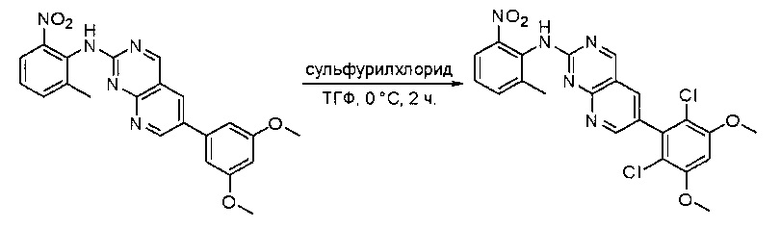
К перемешанному раствору 6-(3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина (100 мг, 0,24 ммоль) в ТГФ (10 мл) при 0°С по каплям добавляли раствор сульфурилхлорида (0,06 мл, 0,72 ммоль) в ТГФ (2 мл). После перемешивания при 0°С в течение 2 часов, реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (3×20 мл). Органические слои объединяли, промывали насыщенным солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали силикагелевой колоночной хроматографией (этилацетат:петролейный эфир = 3:1) с получением 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина (110 мг, 95%) в виде желтого твердого вещества. МС (ЭС+) C22H17Cl2N5O4, искомое значение: 485, 487 найдено: 486, 488 [М+Н]+.
Синтез N1-(6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)-6-метилбензол-1,2-диамина
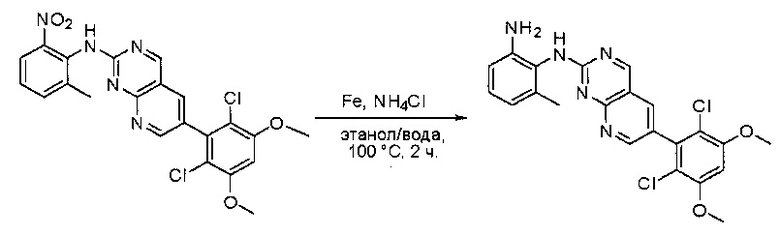
К раствору 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-метил-6-нитрофенил)пиридо[2,3-d]пиримидин-2-амина (80 мг, 0,168 ммоль) в этаноле (4 мл) и воде (4 мл) добавляли порошок железа (75 мг, 1,344 ммоль) и хлорид аммония (74 мг, 1,344 ммоль). Смесь перемешивали при 100°С в течение 2 часов, охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток очищали силикагелевой колоночной хроматографией (этилацетат:петролейный эфир = 4:1) с получением N1-(6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)-6-метилбензол-1,2-диамина (40 мг, 53%) в виде желтого твердого вещества. МС (ЭС+) C22H19Cl2N5O2, искомое значение: 455, 457, найдено: 456, 458 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,33 (ш. с, 1Н), 9,01 (с, 1Н), 9,65 (ш. с, 1H), 8,23 (с, 1Н), 7,05 (с, 1Н), 6,93 (ш. с, 1Н), 6,64-6,63 (м, 1Н), 6,50-6,49 (м, 1Н), 4,80 (с, 2Н), 3,99 (с, 6Н), 2,09 (с, 3Н).
Синтез Н-(2-((6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)амино)-3-метил фенил)акриламида
Н-(2-((6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-50% EtOAc/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C25H23N5O3, искомое значение: 441, найдено: 442.
Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)амино)-3-метилфенил)акриламида
N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-10% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C25H21O2N5O3, искомое значение: 510, найдено: 511 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,53 (с, 1Н), 9,35 (с, 1Н), 9,06 (с, 1Н), 8,70 (с, 1Н), 8,27 (д, J=2,6 Гц, 1Н), 7,78 (с, 1Н), 7,23 (д, J=7,9 Гц, 1Н), 7,15 (с, 1Н), 7,06 (с, 1Н), 6,52 (дд, J=17,0, 10,1 Гц, 1Н), 6,22 (дд, J=17,0, 2,0 Гц, 1Н), 5,69 (д, J=10,6 Гц, 1Н), 3,98 (с, 6Н), 2,20 (с, 3Н).
Пример 4: Синтез СОЕДИНЕНИЯ 45
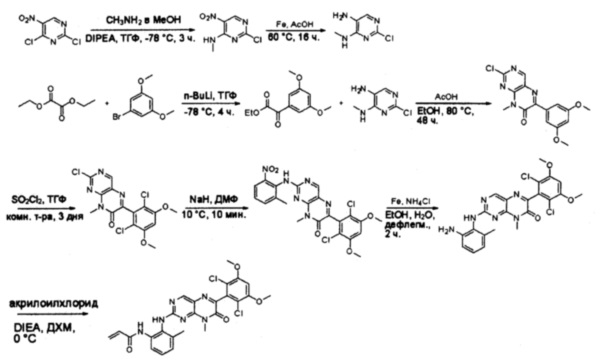
Синтез 2-хлор-N-метил-5-нитропиримидин-4-амина
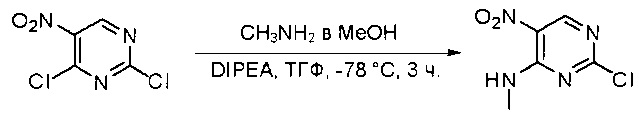
К раствору 2,4-дихлор-5-нитропиримидина (5 г, 26 ммоль) в ТГФ (50 мл) добавляли диизопропилэтиламин (3,36 г, 26 ммоль) при -78°С, затем по каплям добавляли метиламин (13 мл, 2 моль/л в метаноле, 26 ммоль). После добавления смесь нагрели до комнатной температуры и перемешивали в течение 3 часов. Затем реакционную смесь разбавляли этилацетатом и промывали насыщенным солевым раствором (50 мл * 3). Органический слой высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (4,4 г, 100%) в виде желтого твердого вещества. МС (ЭС+) C5H5ClN4O2, искомое значение: 188, 190, найдено: 189, 191 [М+Н]+. Синтез 2-хлор-N4-метилпиримидин-4,5-диамина
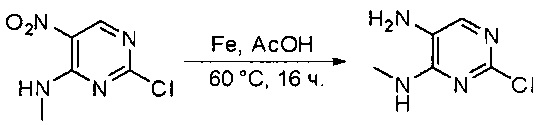
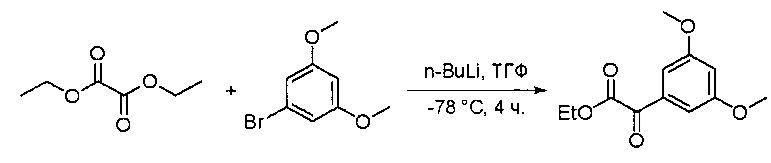
К перемешанному раствору 2-хлор-N-метил-5-нитропиримидин-4-амина (1,9 г, 10 ммоль) в уксусной кислоте (30 мл) добавляли порошок железа (30 мл) (4 г, 71 ммоль) и нагревали смесь в виде суспензии до 60°С в течение 16 часов. Растворитель удаляли при пониженном давлении, а остаток разбавляли насыщенным солевым раствором и этилацетатом. Твердое вещество фильтровали, а фильтрат экстрагировали этилацетатом (50 мл * 12). Органические слои отделяли, объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (1,1 г, 69%). МС (ЭС+) C5H7ClN4, искомое значение: 159, 161, найдено: 160, 162 [М+Н]+. Синтез этил 2-(3,5-диметоксифенил)-2-оксоацетата
К раствору 1-бром-3,5-диметоксибензола (2,17 г, 10 ммоль) в ТГФ (15 мл) по каплям добавляли н-бутиллитий (8 мл, 2,5 моль/л в гексане) при -78°С. После перемешивания в течение 50 минут при -78°С, добавляли раствор диэтилоксалата (4 г, 27 ммоль) в ТГФ (10 мл). Смесь перемешивали при -78°С еще 4 часа, затем гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (50 мл * 3). Органические слои объединяли, промывали насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (1,7 г, 71%). МС (ЭС+) С12Н14О5, искомое значение: 238, найдено: 239 [М+Н]+. Синтез 2-хлор-6-(3,5-диметоксифенил)-8-метилптеридин-7(8Н)-она
Смесь этил 2-(3,5-диметоксифенил)-2-оксоацетата (1 г, 4,2 ммоль) и 2-хлор-N4-метилпиримидин-4,5-диамина (600 мг, 3,8 ммоль) в этаноле (100 мл) и уксусной кислоте (2,5 мл) перемешивали при 80°С в течение 48 часов и охлаждали до комнатной температуры (5°С). Смесь разбавляли дихлорметаном и промывали насыщенным солевым раствором. Органический слой напрямую концентрировали и очищали хроматографией на силикагеле с получением указанного в заголовке соединения (700 мг, 50%). МС (ЭС+) C15H13ClN4O3, искомое значение: 332, 334, найдено: 333, 335 [М+Н]+. Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилптеридин-7(8Н)-она
К раствору 2-хлор-6-(3,5-диметоксифенил)-8-метилптеридин-7(8Н)-она (300 мг, 0,9 ммоль) в ТГФ (5 мл) по каплям добавляли сульфурилхлорид (300 мг) и перемешивали смесь при комнатной температуре в течение 4 часов. Добавляли дополнительное количество сульфурилхлорида (300 мг) и перемешивали при комнатной температуре в течение 3 дней. Реакцию гасили 5 каплями воды, а затем перемешивали в течение 5 минут. Осадок собрали фильтрацией и высушивали с получением указанного в заголовке соединения (240 мг, 67%) в виде желтого твердого вещества. МС (ЭС+) C15H11Cl3N4O3, искомое значение: 400, 402, найдено: 400, 403 [М+Н]+.
Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-2-(2-метил-6-нитрофениламино)птеридин-7(8Н)-она
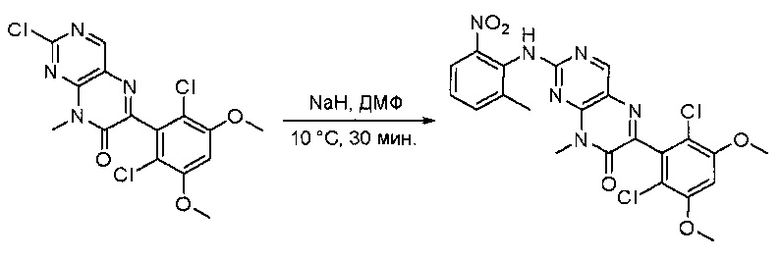
К раствору 2-метил-6-нитробензоламина (100 мг, 1 ммоль) в N,N-диметилформамиде (5 мл) добавляли гидрид натрия (53 мг, 1,3 ммоль) и перемешивали смесь при комнатной температуре (10°С) в течение 10 минут, затем добавляли 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-8-метилптеридин-7(8Н)-он (322 мг, 1 ммоль). Смесь перемешивали при комнатной температуре (10°С) еще 30 минут, а затем гасили водой. Осадок собрали фильтрацией, промывали холодной водой и высушивали с получением указанного в заголовке соединения (180 мг, 75%) в виде желтого порошка. МС (ЭС+) C22H18Cl2N6O5, искомое значение: 516, 518, найдено: 517, 519 [М+Н]+.
Синтез 2-(2-амино-6-метилфениламино)-6-(3,5-диметоксифенил)-8-метилптеридин-7(8Н)-она
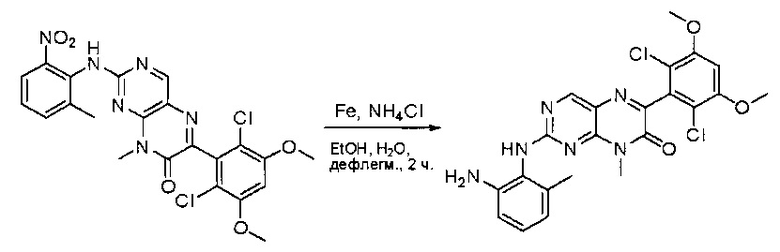
К раствору 6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-2-(2-метил-6-нитрофениламино)птеридин-7(8Н)-она (200 мг, 0,38 ммоль) в этаноле (50 мл) и воде (2 мл) добавляли порошок железа (210 мг, 3,8 ммоль) и хлорид аммония (450 мг, 8 ммоль). Смесь кипятили с обратным холодильником в течение 2 часов. Растворители упаривали, а остаток разбавляли насыщенным солевым раствором и дихлорметаном. Твердое вещество фильтровали, а фильтрат экстрагировали дихлорметаном (50 мл * 6). Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (70 мг, 38%). МС (ЭС+) C22H20Cl2N6O3, искомое значение: 486, 488, найдено: 487, 489 [М+Н]+. 1Н-ЯМР (500 МГц, CDCl3) δ м.д. 8,83 (с, 1Н), 7,09 (т, 1H, J=8,0 Гц), 6,74-6,71 (м, 2Н), 6,65 (с, 1Н), 3,94 (с, 6Н), 3,85 (ш. с., 2Н), 3,63-3,59 (ш, 3Н), 2,25 (с, 3Н).
Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-7,8-дигидроптеридин-2-ил)амино)-3-метилфенил)акриламида
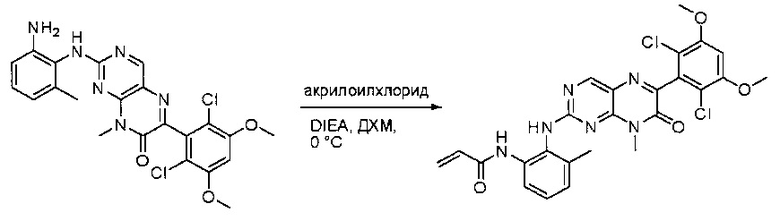
N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-7,8-дигидроптеридин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-10% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C25H22Cl2N6O4, искомое значение: 540, найдено: 541 [М+Н]+.
Пример 5: Синтез СОЕДИНЕНИЯ 39
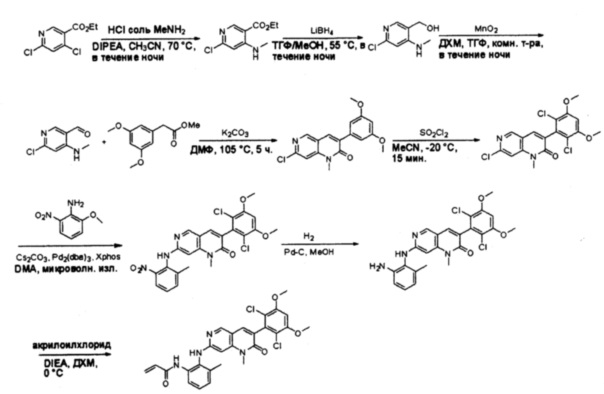
Синтез этил 6-хлор-4-(метиламино)никотината
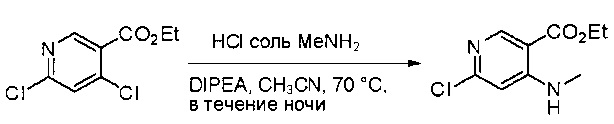
К раствору этил 4,6-дихлорникотината (5,0 г, 22,7 ммоль) в ацетонитриле (50 мл) добавляли гидрохлоридную соль метиламина (1,84 г, 27,2 ммоль) и диизопропилэтиламии (14,6 г, 113,6 ммоль), и нагревали реакционную смесь при 70°С в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, гасили водой (50 мл) и экстрагировали этилацетатом (3×100 мл). Органические слои отделяли, объединяли, промывали водой (50 мл) и насыщенным солевым раствором (100 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (4,7 г, неочищенное), которое использовали непосредственно на следующей стадии без дополнительной очистки. МС (ЭС+) C9H11ClN2O2, искомое значение: 214, 216, найдено: 215, 217 [М+Н]+.
Синтез (6-хлор-4-(метиламино)пиридин-3-ил)метанола
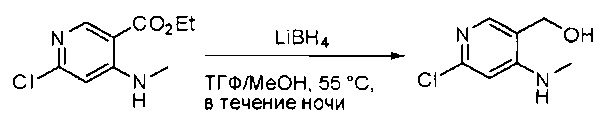
К раствору этил 6-хлор-4-(метиламино)никотината (4,7 г, 21,9 ммоль) в ТГФ (30 мл) и метаноле (30 мл) добавляли боргидрид лития (2,4 г, 109,8 ммоль) и нагревали реакционную смесь при 55°С в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, гасили водой (1 мл) и фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения (4,2 г, неочищенное) в виде белого твердого вещества, которое использовали напрямую на следующей стадии без дополнительной очистки. МС (ЭС+) C7H9ClN2O, искомое значение: 172, 174, найдено: 173, 175 [М+Н]+. Синтез 6-хлор-4-(метиламино)никотинальдегида
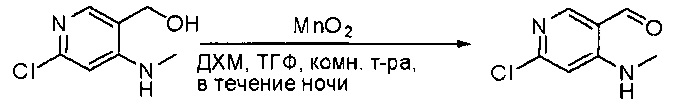
Смесь (6-хлор-4-(метиламино)пиридин-3-ил)метанола (4,2 г, 24,7 ммоль) и оксида марганца (IV) (активный, 25,8 г, 296,6 ммоль) в дихлорметане (50 мл) и ТГФ (50 мл) перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Твердое вещество фильтровали, а фильтрат концентрировали с получением указанного в заголовке соединения (3,7 г, неочищенное) в виде светло-желтого твердого вещества, которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C7H7ClN2O, искомое значение: 170, 172, найдено: 171, 173 [М+Н]+. Синтез 7-хлор-3-(3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-она
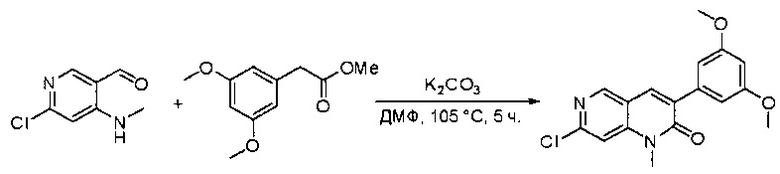
Смесь 6-хлор-4-(метиламино)никотинальдегида (3,7 г, 21,7 ммоль), метил 2-(3,5-диметоксифенил)ацетата (4,5 г, 21,7 ммоль) и карбоната калия (9,0 г, 65,1 ммоль) в N,N-диметилформамиде (30 мл) нагревали при 105°С в течение 5 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, гасили водой (200 мл) и фильтровали. Осадок на фильтре промывали петролейным эфиром (50 мл) и этилацетатом (50 мл) с получением указанного в заголовке соединения (5,8 г, 77%) в виде желтого твердого вещества. МС (ЭС+) C18H19ClN2O3, искомое значение: 346, 348, найдено: 347, 349 [М+Н]+.
Синтез 7-хлор-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-она
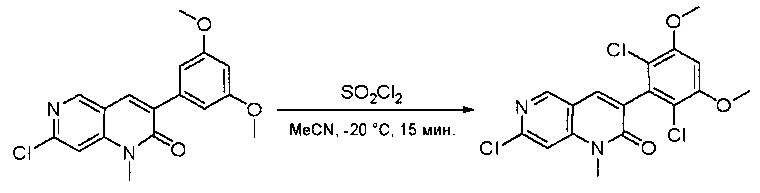
К раствору 7-хлор-3-(3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-она (5,6 г, 16,9 ммоль) в ацетонитриле (30 мл) по каплям добавляли сульфурилхлорид (3,36 мл, 42,2 ммоль) при -20°С и перемешивали смесь еще 15 минут. ЖХМС показала, что реакция завершена. Реакцию гасили водой (1 мл), а растворители удаляли при пониженном давлении. Осадок промывали ацетонитрилом и высушивали с получением указанного в заголовке соединения (5,01 г, 75%) в виде белого твердого вещества. МС (ЭС+) C17H13Cl3N2O3, искомое значение: 399, 401, найдено: 400, 402 [М+Н]+; 1Н-ЯМР (500 МГц, ДМСО-d6) δ м.д. 8,82 (с, 1Н), 8,01 (с, 1H), 7,71 (с, 1Н), 7,04 (с, 1Н), 3,98 (с, 6Н), 3,66 (с, 3Н).
Синтез 3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-7-((2-метил-6-нитрофенил)амино)-1,6-нафтиридин-2(1Н)-она
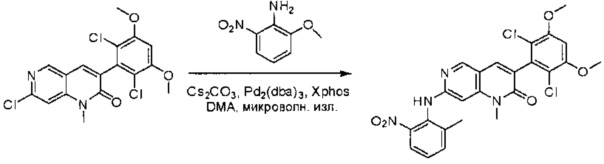
3-(2,6-Дихлор-3,5-диметоксифенил)-1-метил-7-((2-метил-6-нитрофенил)амино)-1,6-нафтиридин-2(1Н)-она получили по такому же способу, как СОЕДИНЕНИЕ 30.
Синтез 7-((2-амино-6-метилфенил)амино)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-она
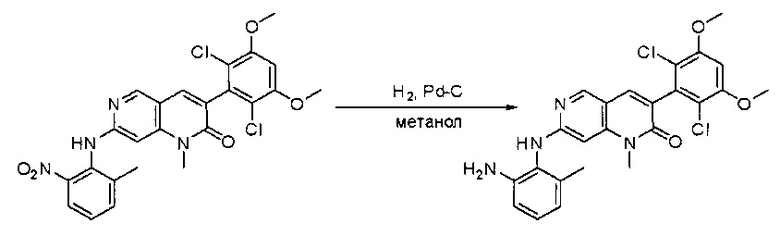
7-((2-Амино-6-метил фенил)амино)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-он получили по такому же способу, как СОЕДИНЕНИЕ 30.
Синтез 7-((2-амино-6-метилфенил)амино)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-она

7-((2-Амино-6-метилфенил)амино)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-1,6-нафтиридин-2(1Н)-он получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-100% EtOAc/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C27H24Cl2N4O4, искомое значение: 538, найдено: 539 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,47 (с, 1Н), 8,43 (д, J=10,0 Гц, 2Н), 7,70 (д, J=12,6 Гц, 2Н), 7,22 (т, J=7,8 Гц, 1Н), 7,14 (д, J=7,6 Гц, 1Н), 6,97 (с, 1Н), 6,46 (дд, J=17,0, 10,2 Гц, 1Н), 6,18 (дд, J=17,0, 2,1 Гц, 1Н), 6,09 (с, 1Н), 5,65 (дд, J=10,2, 2,1 Гц, 1Н), 3,95 (с, 6Н), 3,39 (с, 3Н), 2,20 (с, 3Н).
Пример 6: Синтез СОЕДИНЕНИЯ 48
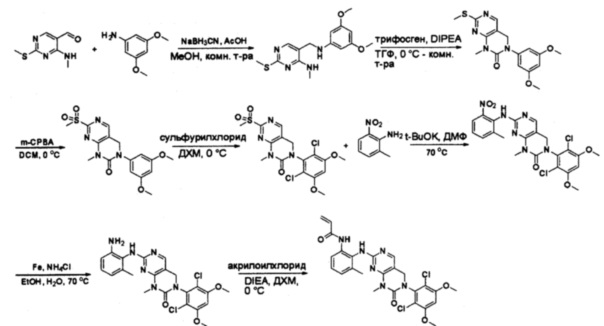
Синтез 5-((3,5-диметоксифениламино)метил)-N-метил-2-(метилтио)пиримидин-4-амина
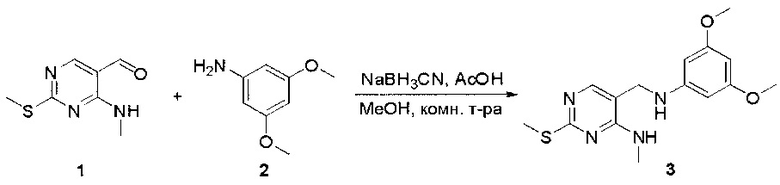
Смесь 4-(метиламино)-2-(метилтио)пиримидин-5-карбальдегида (1,0 г, 5,46 ммоль) и 3,5-диметоксианилина (840 мг, 5,46 ммоль) в метаноле (60 мл) перемешивали при комнатной температуре в течение 3 часов, затем добавляли цианоборгидрид натрия (520 мг, 8,20 ммоль) и 1 мл уксусной кислоты. Затем реакционную смесь перемешивали при комнатной температуре еще 4 часа. ЖХМС показала, что реакция завершена. Реакцию гасили при помощи 30 мл 1 н. HCl, затем перемешивали в течение 0,5 часа и экстрагировали этилцетатом (3×50 мл). Органические слои отделяли, объединяли, промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (неочищенное, 1,2 г, 69%) в виде белого твердого вещества, которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C15H20N4O2S, искомое значение: 320, найдено: 321 [М+Н]+. Синтез 3-(3,5-диметоксифенил)-1-метил-7-(метилтио)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она
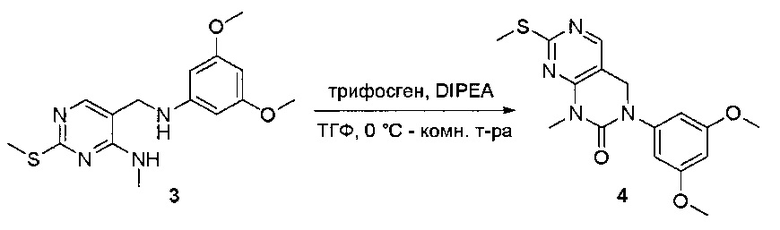
К смеси 5-((3,5-диметоксифениламино)метил)-N-метил-2-(метилтио)пиримидин-4-амина (1,1 г, 3,43 ммоль) и N-этил-N-изопропилпропан-2-амина (1,33 г, 10,30 ммоль) в 10 мл ТГФ добавляли раствор трифосгена (357 мг, 1,20 ммоль) в 5 мл ТГФ при 0°С и перемешивали в течение 1 часа. Затем реакционную смесь нагрели до комнатной температуры и перемешивали еще 5 часов. ЖХМС показала, что реакция завершена. Реакционную смесь гасили водой и экстрагировали этилацетатом (3×15 мл). Органические слои отделяли, объединяли, промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (неочищенное, 1,1 г, 92%) в виде белого твердого вещества, которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C16H18N4O3S, искомое значение: 346, найдено: 347 [М+Н]+.
Синтез 3-(3,5-диметоксифенил)-1-метил-7-(метилсульфонил)-3,4-дигидропиримидо [4,5-d]пиримидин-2(1Н)-она
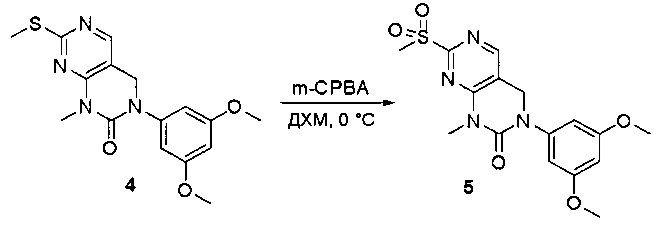
К раствору 3-(3,5-диметоксифенил)-1-метил-7-(метилтио)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она (1,0 г, 2,89 ммоль) в 20 мл дихлорметана добавляли 3-хлорпероксибензойную кислоту (1,50 г, 8,66 ммоль) при 0°С и перемешивали раствор в течение 0,5 часа при 0°С. Смесь нагрели до комнатной температуры и перемешивали в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь разбавляли 30 мл дихлорметана, промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (800 мг, 73%) в виде желтого твердого вещества, которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C16H18N4O5S, искомое значение: 378, найдено: 379 [М+Н]+.
Синтез 3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-7-(метилсульфонил)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она
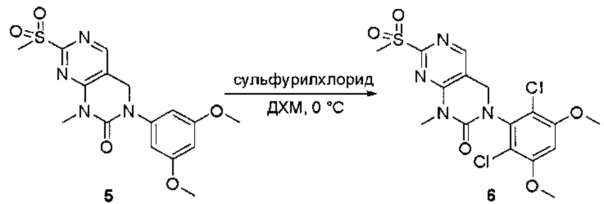
К раствору 3-(3,5-диметоксифенил)-1-метил-7-(метилсульфонил)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она (400 мг, 1,06 ммоль) в 15 мл дихлорметана добавляли сульфурилхлорид (285 мг, 2,12 ммоль) при 0°С, а затем перемешивали при 0°С в течение 3 часов. ЖХМС показала, что реакция завершена. Реакционную смесь разбавляли 20 мл дихлорметана, промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (450 мг, 96%) в виде желтого твердого вещества, которое использовали напрямую на следующей стадии без дополнительной очистки. МС (ЭС+) C16H16Cl2N4O5S, искомое значение: 446, 448, найдено: 447, 449 [М+Н]+. Синтез 3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-7-(2-метил-6-нитрофениламино)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она
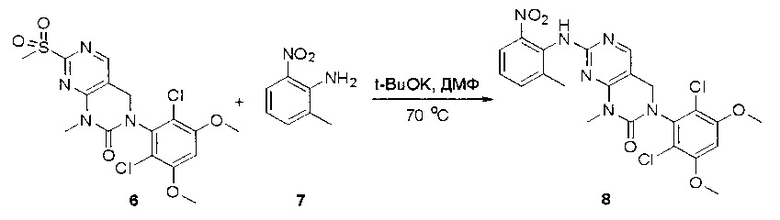
К смеси 3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-7-(метилсульфонил)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она (450 мг, 1,01 ммоль) и 2-метил-6-нитроанилина (230 мг, 1,51 ммоль) в 5 мл N,N-диметилформамида добавляли трет-бутанолат натрия (339 мг, 3,02 ммоль) при комнатной температуре и перемешивали в течение 0,5 часа. ЖХМС показала, что реакция завершена. Смесь гасили 80 мл воды, осадок собрали фильтрацией и высушивали с получением указанного в заголовке соединения (290 мг, 56%) в виде желтого твердого вещества, которое использовали напрямую на следующей стадии без дополнительной очистки. МС (ЭС+) C22H20Cl2N6O5, искомое значение: 518, 520, найдено: 519, 521 [М+Н]+.
Синтез (7-(2-амино-6-метилфениламино)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она
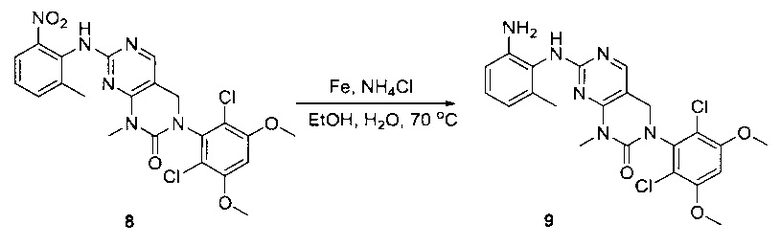
Смесь 3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-7-(2-метил-6-нитрофениламино)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1H)-она (290 мг, 0,56 ммоль) в этаноле (10 мл) и воде (2 мл) перемешивали при 70°С в течение 20 минут, затем добавляли порошок железа (320 мг, 5,60 ммоль) и хлорид аммония (250 мг, 2,79 ммоль). Реакционную смесь перемешивали при 70°С еще 6 часов. ЖХМС показала, что реакция завершена. Твердое вещество фильтровали, а фильтрат концентрировали. Остаток растворяли в этилацетате (30 мл), промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали препаративной ВЖЖХ с получением указанного в заголовке соединения (27 мг, 10%) в виде белого твердого вещества. МС (ЭС+) C22H22Cl2N6O3, искомое значение: 488, 490, найдено: 489, 491 [М+Н]+. 1Н-ЯМР (500 МГц, CDCl2) δ м.д. 7,89 (с, 1Н), 7,04 (т, 1Н, J=8,0 Гц), 6,69 (д, 2Н, J=7,5 Гц), 6,60 (с, 1Н), 4,53 (с, 2Н), 3,94 (с, 6Н), 3,34 (с, 3Н), 2,24 (с, 3Н).
Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-3-метил фенил)акриламида
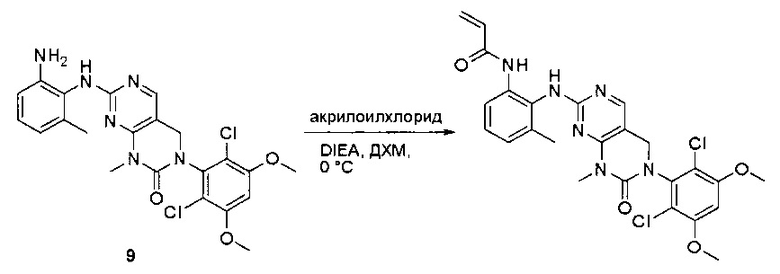
N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-метил-7-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-10% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C25H24Cl2N6O4, искомое значение: 542, найдено: 543 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,48 (с, 1Н), 8,35 (с, 1Н), 7,99 (с, 1H), 7,66 (с, 1H), 7,16 (т, J=7,8 Гц, 1Н), 7,10-7,06 (м, 1Н), 6,99 (с, 1Н), 6,53 (дд, J=17,0, 10,2 Гц, 1Н), 6,22 (дд, J=16,9, 2,1 Гц, 1Н), 5,71 (дд, J=10,2, 2,0 Гц, 1H), 4,48 (с, 2Н), 3,96 (с, 6Н), 3,44 (с, 3Н), 2,17 (с, 3Н).
Пример 7: Синтез СОЕДИНЕНИЯ 24 и СОЕДИНЕНИЯ 6
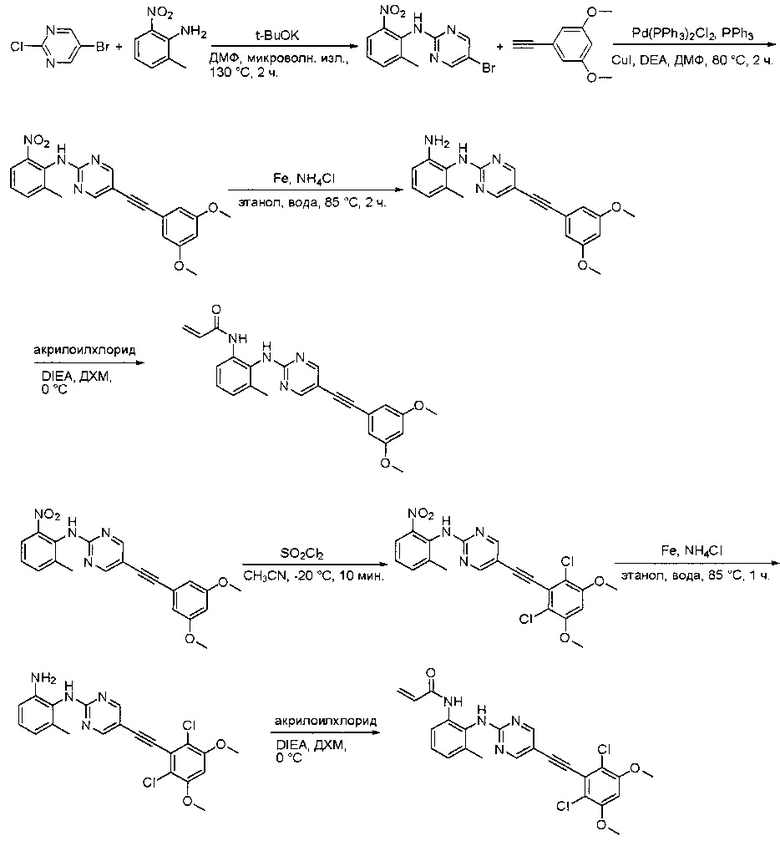
Синтез 5-бром-N-(2-метил-6-нитрофенил)пиримидин-2-амина
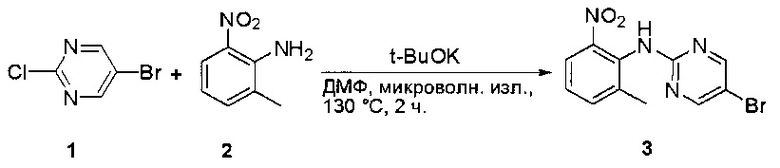
К раствору 5-бром-2-хлорпиримидина (1,5 г, 7,89 ммоль) и 2-метил-6-нитроанилина (800 мг, 5,26 ммоль) в N,N-диметилформамиде (10 мл) в закрытой пробирке добавляли трет-бутоксид калия (1,76 г, 15,78 ммоль) и нагревали смесь под микроволновым излучением при 130°С в течение 2 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, гасили водой (20 мл) и экстрагировали этилацетатом (3×100 мл). Органические слои отделяли, объединяли, промывали водой (50 мл) и насыщенным солевым раствором (100 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали силикагелевой хроматографией (петролейный эфир: этилацетат = 10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества (500 мг, 31%). МС (ЭС+) C11H9BrN4O2, искомое значение 309, 311, найдено: 310, 312 [М+Н]+.
Синтез 5-((3,5-диметоксифенил)этинил)-N-(2-метил-6-нитрофенил)пиримидин-2-амина
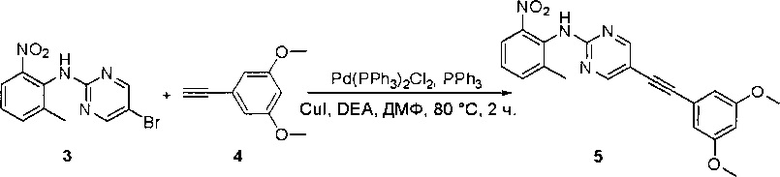
Смесь 5-бром-N-(2-метил-6-нитрофенил)пиримидин-2-амина (573 мг, 3,0 ммоль), 1-этинил-3,5-диметоксибензола (483 мг, 3,0 ммоль), трифенилфосфина (157 мг, 0,60 ммоль), хлорида бис(трифенилфосфин)палладия (II) (210 мг, 0,30 ммоль), йодида меди (I) (57 мг, 0,30 ммоль) и диэтиламина (1,50 мл, 15,0 ммоль) в N,N-диметилформамиде (10 мл) три раза дегазировали азотом, а затем перемешивали при 80°С в течение 2 часов. ЖХМС показала, что реакция завершена. Смесь охлаждали до комнатной температуры, гасили водой (20 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические слои отделяли, промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали силикагелевой хроматографией (петролейный эфир: этилацетат = 4:1) с получением указанного в заголовке соединения в виде желтого твердого вещества (460 мг, 39%). МС (ЭС+) C21H18N4O4, искомое значение: 390, найдено: 391 [М+Н]+.
Синтез N1-(5-((3,5-диметоксифенил)этинил)пиримидин-2-ил)-6-метилбензол-1,2-диамина
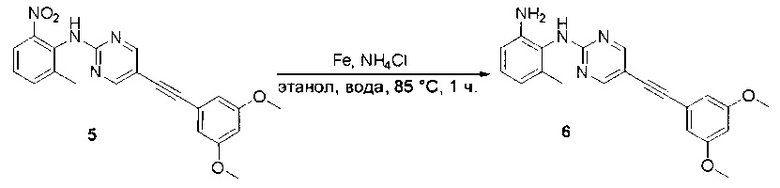
Смесь 5-((3,5-диметоксифенил)этинил)-N-(2-метил-6-нитрофенил)пиримидин-2-амина (150 мг, 0,38 ммоль), железа (171 мг, 3,04 ммоль) и хлорида аммония (246 мг, 4,56 ммоль) в этаноле (20 мл) и воде (2 мл) перемешивали при 85°С в течение 1 часа. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, а твердое вещество фильтровали. Фильтрат концентрировали, а остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (55 мг, 44%). МС (ЭС+) C21H20N4O2, искомое значение: 360, найдено: 361 [М+Н]+. 1Н-ЯМР (500 МГц, ДМСО-d6) д м.д. 8,76 (с, 1Н), 8,50-8,46 (ш, 2Н), 6,88 (т, 1Н, J=7,0 Гц), 6,66 (с, 2Н), 6,57 (д, 1Н, J=7,5 Гц), 6,54 (с, 1Н), 6,44 (д, 1Н, J-6,5 Гц), 4,74 (с, 2Н), 3,76 (с, 6Н), 2,01 (с, 3Н).
Синтез N-(2-((5-((3,5-диметоксифенил)этинил)пиримидин-2-ил)амино)-3-метил фенил)акриламида
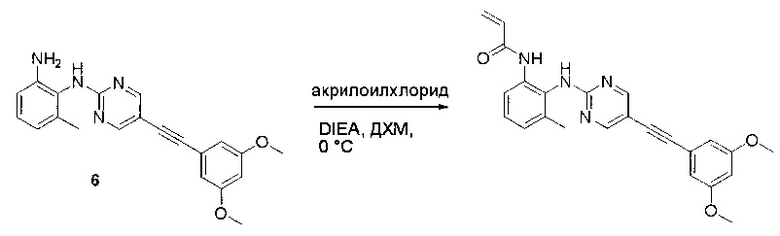
N-(5-((3,5-диметоксифенил)этинил)пиримидин-2-ил)-6-метилбензол-1,2-диамин получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-100% EtOAc в гексанах, с получением указанного в заголовке соединения. МС (ЭС+) C24H22N4O3, искомое значение: 414, найдено: 415 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,60-9,38 (м, 1H), 8,79 (с, 1H), 8,51 (с, 2Н), 7,69 (д, J=8,1 Гц, 1Н), 7,19 (т, J=7,8 Гц, 1Н), 7,15-7,06 (м, 1Н), 6,67 (д, J=2,3 Гц, 2Н), 6,60-6,45 (м, 2Н), 6,22 (дд, J=17,0, 2,1 Гц, 1Н), 5,71 (дд, J=10,2, 2,1 Гц, 1Н), 3,76 (с, 6Н), 2,12 (с, 3Н).
Синтез 5-((2,6-дихлор-3,5-диметоксифенил)этинил)-N-(2-метил-6-нитрофенил)пиримидин-2-амина
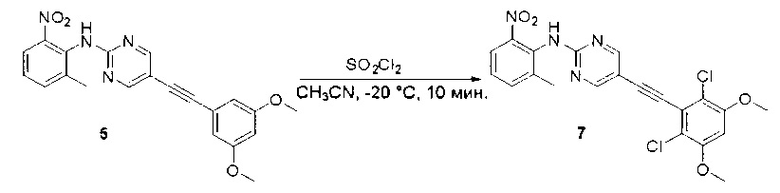
К раствору 5-((3,5-диметоксифенил)этинил)-N-(2-метил-6-нитрофенил)пиримидин-2-амина (50 мг, 0,13 ммоль) в ацетонитриле (5 мл) по каплям добавляли сульфурилхлорид (44 мг, 0,33 ммоль) при -20°С и перемешивали смесь еще 10 минут. ЖХМС показала, что реакция завершена, и реакцию гасили водой (0,5 мл). Растворители упаривали, а остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения в виде желтого твердого вещества (30 мг, 50%). МС (ЭС+) C21H16Cl2N4O4, искомое значение: 459, 461, найдено: 460, 462 [М+Н]+.
Синтез N1-(5-((2,6-дихлор-3,5-диметоксифенил)этинил)пиримидин-2-ил)-6-метилбензол-1,2-диамина
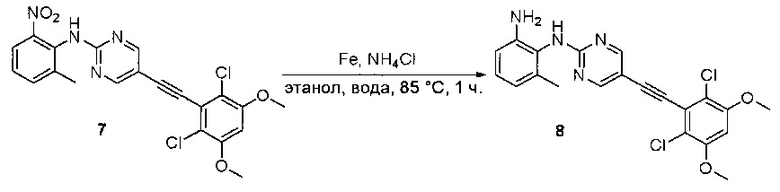
Смесь 5-((2,6-дихлор-3,5-диметоксифенил)этинил)-N-(2-метил-6-нитрофенил)пиримидин-2-амина (150 мг, 0,33 ммоль), железа (147 мг, 2,64 ммоль) и хлорида аммония (214 мг, 3,96 ммоль) в этаноле (20 мл) и воде (2 мл) перемешивали при 85°С в течение 1 часа. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и фильтровали твердое вещество. Фильтрат концентрировали, а остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (58 мг, 35%). МС (ЭС+) C21H18Cl2N4O2, искомое значение: 429, 431, найдено: 430, 432 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ м.д. 8,90 (с, 1Н), 8,55-8,44 (ш, 2Н), 6,97 (с, 1H), 6,89-6,86 (м, 1Н), 6,57 (д, 1H, J=7,6 Гц), 6,44 (д, 1H, J=7,6 Гц), 4,75 (с, 2Н),3,94 (с, 6Н), 2,01 (с, 3Н).
Синтез N-(2-((5-((2,6-дихлор-3,5-диметоксифенил)этинил)пиримидин-2-ил)амино)-3-метилфенил)акриламида
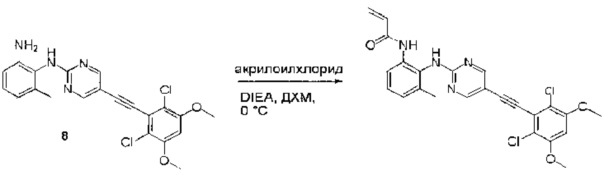
N-(2-((5-((2,6-дихлор-3,5-диметоксифенил)этинил)пиримидин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-100%) EtOAc в гексанах, с получением указанного в заголовке соединения. МС (ЭС+) C24H20Cl2N4O3, искомое значение: 482, найдено: 483 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,47 (с, 1Н), 8,93 (с, 1Н), 8,54 (с, 2Н), 7,71 (д, J=8,1 Гц, 1Н), 7,19 (т, J=7,8 Гц, 1Н), 7,09 (д, J=7,4 Гц, 1Н), 6,98 (с, 1H), 6,53 (дд, J=17,0, 10,2 Гц, 1H), 6,22 (дд, J=17,0, 2,1 Гц, 1Н), 5,70 (дд, J=10,2, 2,1 Гц, 1Н), 3,94 (с, 6Н), 2,13 (с, 3Н).
Пример 8: Синтез СОЕДИНЕНИЯ 40
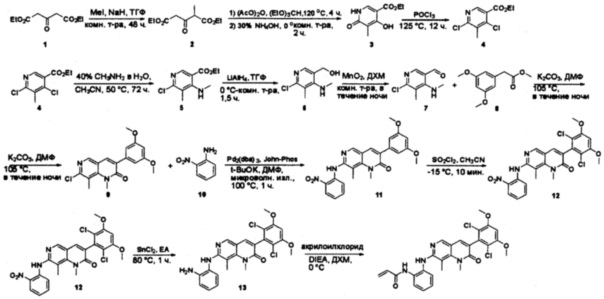
Синтез диэтил 2-метил-3-оксопентандиоата
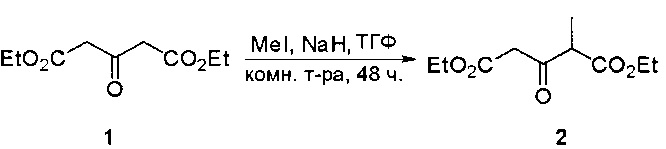
К раствору диэтил 3-оксопентандиоата (23,2 г, 114,8 ммоль) в тетрагидрофуране (100 мл) добавляли гидрид натрия (60%, 4,8 г, 120,5 ммоль) при 0°С и перемешивали реакционную смесь при комнатной температуре в течение 20 минут, затем добавляли йодметан (7,15 мл, 114,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов, гасили водой (500 мл) и экстрагировали этилацетатом (500 мл × 3). Органические слои отделяли, объединяли, промывали водой (200 мл) и насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали силикагелевой колоночной хроматографией (петролейный эфир: этилацетат = 20:1) с получением указанного в заголовке соединения в виде бесцветного маслянистого вещества (9 г, 36%). МС (ЭС+) С10Н16О5, искомое значение: 216, найдено: 217 [М+Н]+. Синтез этил 4-гидрокси-5-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
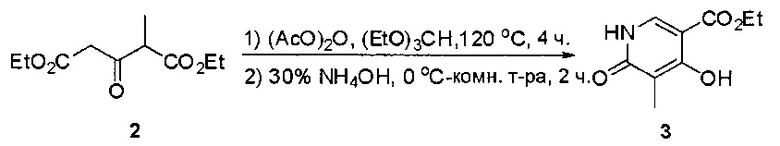
К раствору диэтил 2-метил-3-оксопентандиоата (10 г, 46,25 ммоль) в 1,1'-триоксидандиилдипропан-1-оне (400 мл) добавляли триэтоксиметан (38 мл, 231,25 ммоль) и нагревали смесь при 120°С в течение 4 часов, затем добавляли 30% аммиак (600 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре еще 2 часа. ЖХМС показала, что реакция завершена. Осадок собрали фильтрацией и растворяли в дихлорметане (400 мл). Твердое вещество фильтровали, а фильтрат концентрировали с получением указанного в заголовке соединения (5,5 г, неочищенное) в виде желтого твердого вещества. МС (ЭС+) C9H11NO4, искомое значение: 197, найдено: 198 [М+Н]+. Синтез этил 4,6-дихлор-5-метилникотината
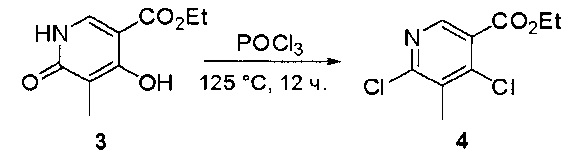
Раствор этил 4-гидрокси-5-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (5,0 г, 21,4 ммоль) в фосфорилтрихлориде (100 мл) кипятили с обратным холодильником при 125°С в течение 12 часов. ЖХМС показала, что реакция завершена. Большую часть фосфорилтрихлорида упаривали, а остаток по каплям добавляли к ледяной воде (100 мл). Полученную смесь нейтрализовали водным раствором карбоната натрия (50 мл) и экстрагировали этилацетатом (200 мл). Органический слой отделяли, объединяли, промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке (петролейный эфир: этилацетат = 15:1) с получением указанного в заголовке соединения (1,6 г, 32%) в виде желтого маслянистого вещества. МС (ЭС+) C9H9Cl2NO2, искомое значение: 232, 234, найдено: 233, 235 [М+Н]+.
Синтез этил 6-хлор-5-метил-4-(метиламино)никотината

К раствору этил 4,6-дихлор-5-метилникотината (2,6 г, 11,1 ммоль) в ацетонитриле (60 мл) по каплям добавляли 40% метиламин в воде (689 мг, 22,2 ммоль, 60 мл) и перемешивали смесь при 50°С в течение 72 часов. ЖХМС показала, что реакция завершена. Реакционную смесь концентрировали и экстрагировали этилацетатом (100 мл). Органический слой отделяли, объединяли, промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке (петролейный эфир: этилацетат = 2:1) с получением указанного в заголовке соединения (2,05 г, 81%) в виде бесцветного маслянистого вещества. МС (ЭС+) C10H13ClN2O2, искомое значение: 228, 230, найдено: 229, 231 [М+Н]+
Синтез (6-хлор-5-метил-4-(метиламино)пиридин-3-ил)метанола
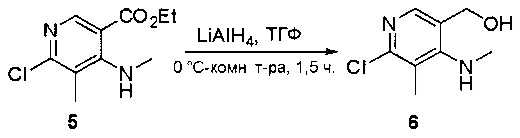
К раствору этил 6-хлор-5-метил-4-(метиламино)никотината (2,0 г, 8,8 ммоль) в тетрагидрофуране (60 мл) добавляли алюмогидрид лития при 0°С и перемешивали смесь при комнатной температуре в течение 1,5 часов. ЖХМС показала, что реакция завершена. Реакцию гасили декагидратом сульфата натрия (1,5 г) и фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения (1,4 г, неочищенное) в виде белого твердого вещества. МС (ЭС+) C8H11ClN2O, искомое значение: 186, 188, найдено: 187, 189 [М+Н]+.
Синтез 6-хлор-5-метил-4-(метиламино)никотинальдегида
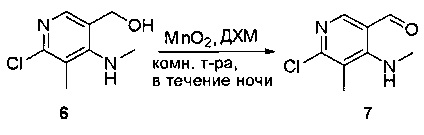
Смесь (6-хлор-5-метил-4-(метиламино)пиридин-3-ил)метанола (1,4 г, 8,0 ммоль) и оксида марганца (2,8 г, 32 ммоль) в дихлорметане (100 мл) перемешивали при комнатной температуре в течение 4 часов. ЖХМС показала, что реакция завершена. Твердое вещество фильтровали, а фильтрат концентрировали с получением указанного в заголовке соедиения (1,2 г, неочищенное) в виде желтого маслянистого вещества. МС (ЭС+) C8H9ClN2O, искомое значение: 184, 186, найдено: 185, 187 [М+Н]+.
Синтез 7-хлор-3-(3,5-диметоксифенил)-1,8-диметил-1,6-нафтиридин-2(1Н)-она
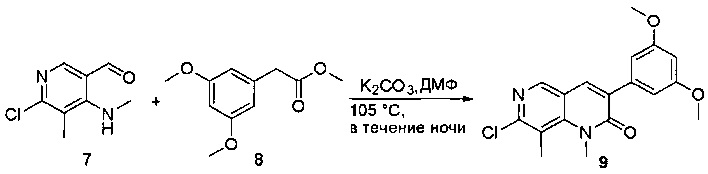
Смесь 6-хлор-5-метил-4-(метиламино)никотинальдегида (3,11 г, 16,8 ммоль), метил 2-(3,5-диметоксифенил)ацетата (4,25 г, 20,2 ммоль) и карбоната калия (2,8 г, 20,3 ммоль) в N,N-диметилформамиде (100 мл) перемешивали при 105°С в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и гасили водой. Осадок фильтровали и высушивали с получением указанного в заголовке соединения (5,5 г, неочищенное) в виде желтого твердого вещества. МС (ЭС+) C18H17ClN2O3, искомое значение: 344, 346, найдено: 345, 347 [М+Н]+. Синтез 3-(3,5-диметоксифенил)-1,8-диметил-7-(2-нитрофениламино)-1,6-нафтиридин-2(1Н)-она
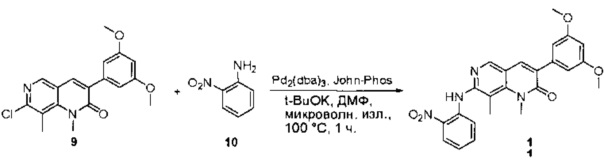
Смесь 7-хлор-3-(3,5-диметоксифенил)-1,8-диметил-1,6-нафтиридин-2(1H)-она (800 мг, 2,32 ммоль), 2-нитробензоламина (320 мг, 2,32 ммоль), Pd2(dba)3 (100 мг), John-Phos(100 мг) и трет-бутанолата калия (480 мг, 4,64 ммоль) в N,N-диметилформамиде (10 мл) нагревали в закрытой пробирке при 100°С под микроволновым излучением в течение 1 часа. ЖХМС показала, что реакция завершена. Смесь концентрировали и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (150 мг, 15%) в виде коричневого твердого вещества. МС (ЭС+) C24H22N4O5, искомое значение: 446, найдено: 447 [М+Н]+.
Синтез 3-(2,6-дихлор-3,5-диметоксифенил)-1,8-диметил-7-(2-нитрофениламино)-1,6-нафтиридин-2(1Н)-она
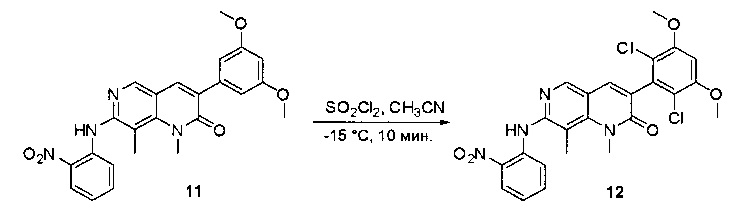
К раствору 3-(3,5-диметоксифенил)-1,8-диметил-7-(2-нитрофениламино)-1,6-нафтиридин-2(1Н)-она (120 мг, 0,27 ммоль) в ацетонитриле (120 мл) добавляли сульфурилхлорид (185 мг, 1,35 ммоль) при -15°С и перемешивали смесь при -15°С в течение 10 минут. ЖХМС показала, что реакция завершена. Реакционную смесь гасили водой (1 мл) и концентрировали. Осадок собрали фильтрацией, промывали ацетоном/петролейным эфиром (1: 5) и высушивали с получением указанного в заголовке соединения (100 мг, 72%) в виде белого твердого вещества. МС (ЭС+) C24H20C12N4O5, искомое значение: 514, 516, найдено: 515, 517 [М+Н]+.
Синтез 7-(2-аминофениламино)-3-(2,6-дихлор-3,5-диметоксифенил)-1,8-диметил-1,6-нафтиридин-2(1Н)-она
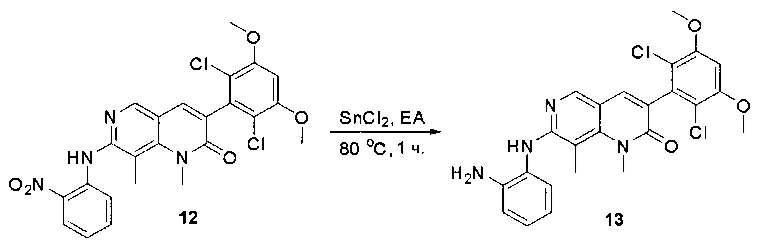
К раствору 3-(2,6-дихлор-3,5-диметоксифенил)-1,8-диметил-7-(2-нитрофениламино)-1,6-нафтиридин-2(1Н)-она (100 мг, 0,2 ммоль) в этилацетате (20 мл) добавляли хлорид олова (II) (150 мг, 0,8 ммоль) и перемешивали смесь при 80°С в течение 1 часа. ЖХМС показала, что реакция завершена. Твердое вещество фильтровали, а фильтрат концентрировали. Остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (38,6 мг, 41%) в виде желтого твердого вещества. МС (ЭС+) C24H22Cl2N4O3, искомое значение: 484, 486, найдено: 485, 487 [М+Н]+; 1Н-ЯМР (500 МГц, ДМСО-d6) δ м.д. 8,24 (с, 1Н), 7,76 (с, 1Н), 7,67 (с, 1Н), 7,03 (д, 1Н, J=7,5 Гц), 6,97 (с, 1Н), 6,92-6,89 (м, 1Н), 6,75-6,73 (м, 1Н), 6,57-6,54 (м, 1H), 4,77 (с, 2Н), 3,95 (с, 6Н), 3,66 (с, 3Н), 2,43 (с, 3Н).
Синтез N-(2-((3-(2,6-дихлор-3,5-диметоксифенил)-1,8-диметил-2-оксо-1,2-дигидро-1,6-нафтиридин-7-ил)амино)фенил)акриламида
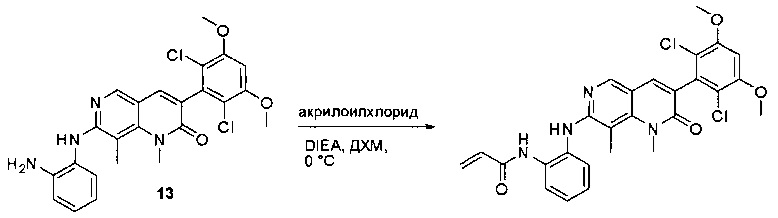
N-(2-((3-(2,6-дихлор-3,5-диметоксифенил)-1,8-диметил-2-оксо-1,2-дигидро-1,6-нафтиридин-7-ил)амино)фенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали препаративной тонкослойной хроматографией, используя градиент 0-5% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C27H24Cl2N4O4, искомое значение: 538, найдено: 539 [М+Н]+.
Пример 9: Синтез СОЕДИНЕНИЯ 42
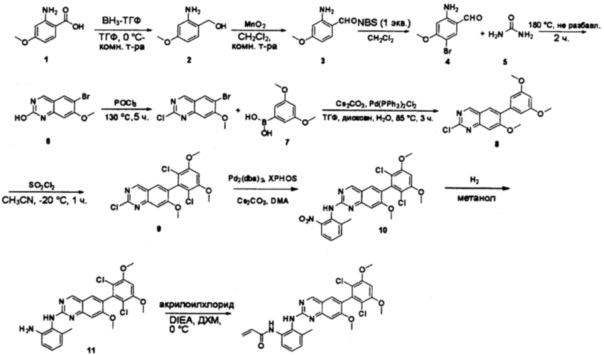
Синтез (2-амино-4-метоксифенил)метанола

К раствору 2-амино-4-метоксибензойной кислоты (15,0 г, 89,8 ммоль) в ТГФ (300 мл) добавляли гидрид бора в ТГФ (450 мл, 450 ммоль) при 0°С и перемешивали реакционную смесь при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь гасили водой (150 мл) и экстрагировали этилацетатом (500 мл ×3). Органические слои отделяли, объединяли, промывали водой (200 мл) и насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения. МС (ЭС+) C8H11NO2, искомое значение: 153, найдено: 154 [М+Н]+.
Синтез 2-амино-4-метоксибензальдегида
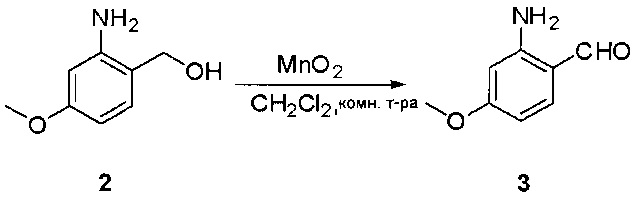
Смесь (2-амино-4-метоксифенил)метанола (20 г, 131,0 ммоль) и оксида марганца (68 г, 786,0 ммоль) в дихлорметане (300 мл) перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Твердое вещество фильтровали, а фильтрат концентрировали. Остаток очищали силикагелевой хроматографией (петролейный эфир: этилацетат = 6:1) с получением указанного в заголовке соединения (7 г, 35%) в виде желтого твердого вещества. МС (ЭС+) C8H9NO2, искомое значение: 151, найдено: 152 [М+Н]+.
Синтез 2-амино-5-бром-4-метоксибензальдегида
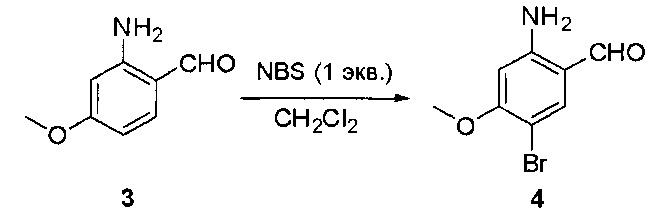
К перемешанному раствору 2-амино-4-метоксибензальдегида (6 г, 39,7 ммоль) в дихлорметане (100 мл) добавляли N-бромсукцинимид (7 г, 39,7 ммоль). Реакцию контролировали с помощью ЖХМС до полного расходования исходного материала. Реакционную смесь разбавляли дихлорметаном и водой. Отделенный органический слой высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (5 г, 56%) в виде желтого твердого вещества. МС (ЭС+) C8H8BrNO2, искомое значение: 229, 231, найдено: 230, 232 [М+Н]+.
Синтез 6-бром-7-метоксихиназолин-2-ола
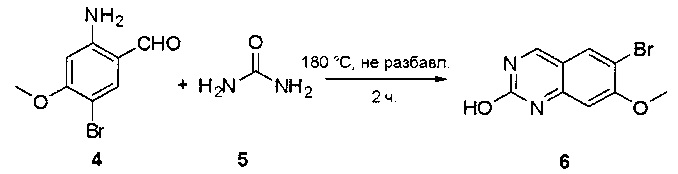
Смесь 2-амино-5-бром-4-метоксибензальдегида (3 г, 13,1 ммоль) и мочевины (12 г, 196,5 ммоль) перемешивали при 180°С в течение 2 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и промывали водой (3×100 мл). Осадок собрали и высушивали с получением указанного в заголовке соединения (3 г, неочищенное) в виде желтого твердого вещества. МС (ЭС+) C8H7BrN2O2, искомое значение: 254, 256, найдено: 255, 257 [М+Н]+.
Синтез 6-бром-2-хлор-7-метоксихиназолина
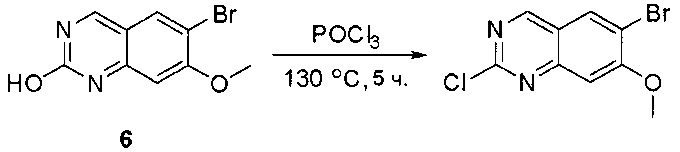
Раствор 6-бром-7-метоксихиназолин-2-ола (3,0 г, 11,8 ммоль) в фосфорилтрихлориде (30 мл) кипятили с обратным холодильником при 130°С в течение 5 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и упаривали большую часть фосфорилтрихлорида. Остаток по каплям добавляли к ледяной воде (100 мл), а полученный осадок собрали фильтрацией с получением указанного в заголовке соединения в виде желтого твердого вещества (2,4 г, 75%). МС (ЭС+) C9H6BrClN2O, искомое значение: 272, 274, найдено: 273, 275 [М+Н]+.
Синтез 2-хлор-6-(3,5-диметоксифенил)-7-метоксихиназолина
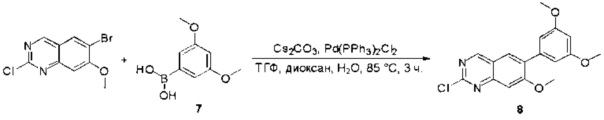
Смесь 6-бром-2-хлор-7-метоксихиназолина (2,4 г, 8,82 ммоль), 3,5-диметоксифенилбороновой кислоты (1,6 г, 8,82 ммоль), карбоната церия (8,6 г, 26,46 ммоль) и Pd(PPh3)2Cl2 (1,4 г, 2,1 ммоль) в ТГФ (10 мл), диоксане (10 мл) и воде (2 мл) три раза дегазировали азотом и перемешивали при 85°С в течение 3 часов. ЖХМС показала, что реакция завершена. Смесь охлаждали до комнатной температуры и экстрагировали дихлорметаном (3×50 мл). Органические слои отделяли, объединяли, промывали водой и насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали силикагелевой хроматографией (петролейный эфир: этилацетат = 1:4) с получением указанного в заголовке соединения (1,1 г, 38%) в виде белого твердого вещества. МС (ЭС+) C17H15ClN2O3, искомое значение: 330, 332, найдено: 331, 333 [М+Н]+.
Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолина
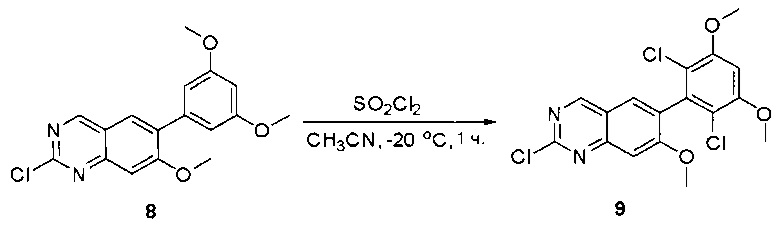
К раствору 2-хлор-6-(3,5-диметоксифенил)-7-метоксихиназолина (200 мг, 0,61 ммоль) в ацетонитриле (5 мл) добавляли сульфурилхлорид (205 мг, 1,52 ммоль) и перемешивали смесь при -20°С в течение 1 часа. Реакцию гасили водой (1 мл) и концентрировали при пониженном давлении. Осадок промывали ацетонитрилом и высушивали с получением указанного в заголовке соединения в виде белого твердого вещества (120 мг, 50%). МС (ЭС+) C17H13Cl3N2O3, искомое значение: 398, найдено: 399, 401 [М+Н]+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,43 (с, 1Н), 8,02 (с, 1Н), 7,55 (с, 1Н), 7,03 (с, 1Н), 3,98 (с, 6Н), 3,93 (с, 3Н).
Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-7-метокси-N-(2-метил-6-нитрофенил)хиназолин-2-амина
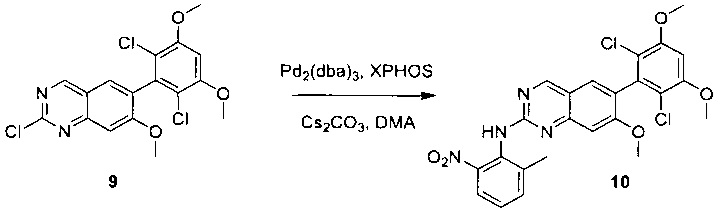
6-(2,6-дихлор-3,5-диметоксифенил)-7-метокси-N-(2-метил-6-нитрофенил)хиназолин-2-амин получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-100% EtOAc в гексанах, с получением указанного в заголовке соединения. МС (ЭС+) C24H20Cl2N4O5, искомое значение: 514, найдено: 515 [М+Н]+.
Синтез N1-(6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолин-2-ил)-6-метилбензол-1,2-диамина
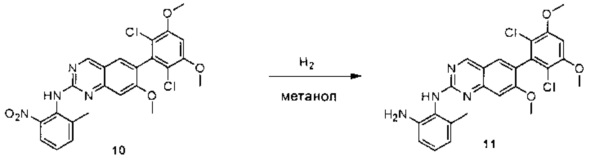
N1-(6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолин-2-ил)-6-метилбензол-1,2-диамин получили по такому же способу, как СОЕДИНЕНИЕ 30. Реакционную смесь фильтровали через целит с получением неочищенного продукта. МС (ЭС+) C24H22Cl2N4O3, искомое значение: 484, найдено: 485 [М+Н]+.
Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолин-2-ил)амино)-3-метилфенил)акриламида
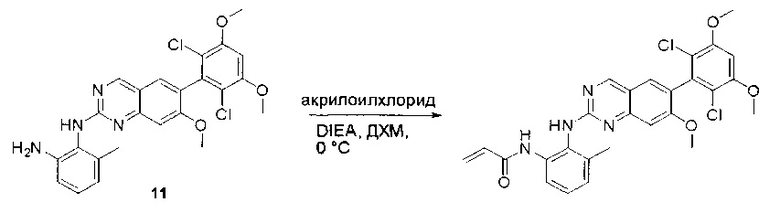
N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-10% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C27H24Cl2N4O4, искомое значение: 538, найдено: 539 [М+Н]+.
Пример 10: Синтез СОЕДИНЕНИЯ 34
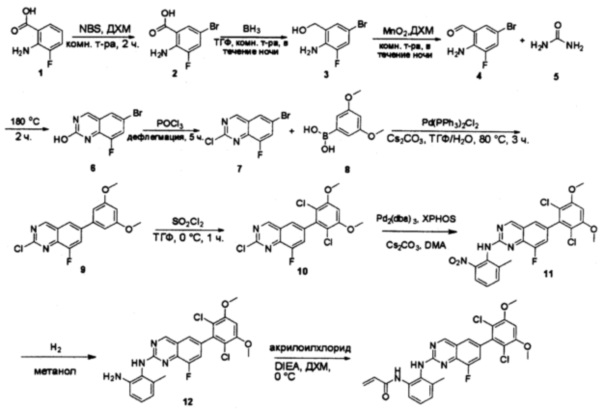
Синтез 2-амино-5-бром-3-фторбензойной кислоты
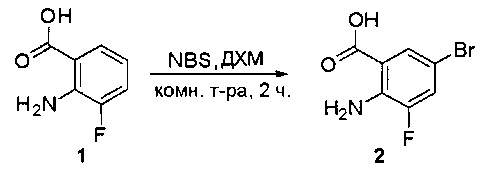
К раствору 2-амино-3-фторбензойной кислоты (10,85 г, 70 ммоль) в дихлорметане (175 мл) добавляли N-бромсукцинимид (12,46 г, 70 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. ЖХМС показала, что реакция завершена. Осадок фильтровали и промывали дихлорметаном (100 мл * 3) с получением указанного в заголовке соединения (12,7 г, 78%) в виде серого твердого вещества, которое использовали напрямую на следующей стадии без дополнительной очистки. МС (ЭС+) C7H5BrFNO2, искомое значение: 233, 235, найдено: 232, 234 [М-Н]-.
Синтез (2-амино-5-бром-3-фторфенил)метанола
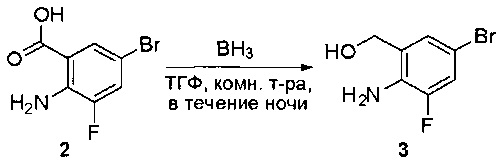
К раствору 2-амино-5-бром-3-фторбензойной кислоты (14,5 г, 62,2 ммоль) в ТГФ (150 мл) при 0°С добавляли гидрид бора в ТГФ (1 М, 310 мл) и перемешивали реакционную смесь при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Реакцию гасили метанолом (150 мл), концентрировали в вакууме, разбавляли водным раствором бикарбоната натрия (400 мл) и экстрагировали этилацетатом (200 мл * 3). Органические слои отделяли, объединяли, промывали водой (200 мл) и насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (13,0 г, неочищенное), которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C7H7BrFNO, искомое значение: 219, 221, найдено: 220, 222 [М+Н]+.
Синтез 2-амино-5-бром-3-фторбензальдегида
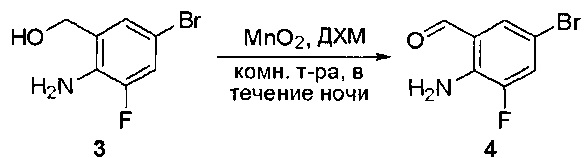
Смесь (2-амино-5-бром-3-фторфенил)метанола (13 г, 59,4 ммоль) и оксида марганца (31 г, 356,4 ммоль) в дихлорметане (400 мл) перемешивали при комнатной температуре в течение ночи. ТСХ показала, что исходный материал полностью израсходован. Твердое вещество фильтровали, а фильтрат концентрировали с получением указанного в заголовке соединения (11 г, 85%) в виде светло-желтого твердого вещества, которое напрямую использовали на следующей стадии без дополнительной очистки.
Синтез 6-бром-8-фторхиназолин-2-ола
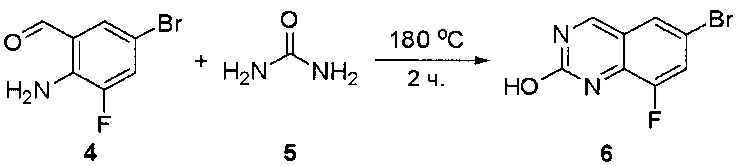
Перемешанную смесь 2-амино-5-бром-3-фторбензальдегида (2,17 г, 10 ммоль) и мочевины (9 г, 150 ммоль) нагревали при 180°С в течение 2 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, а полученный осадок фильтровали и промывали водой (500 мл * 3). Захваченную влагу полностью удаляли трехкратным совместным выпариванием с толуолом. Указанное в заголовке соединение (2 г, 83%) получили в виде желтого твердого вещества. МС (ЭС+) C8H4BrFN2O, искомое значение: 242, 244, найдено: 243, 245 [М+Н]+.
Синтез 6-бром-2-хлорхиназолина
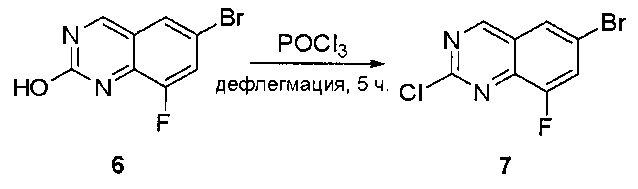
Раствор 6-бромхиназолин-2-ола (9,72 г, 40 ммоль) в оксихлориде фосфора (100 мл) кипятили с обратным холодильником в течение 5 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и удаляли большую часть оксихлорида фосфора при пониженном давлении. Остаток по каплям добавляли к ледяной воде (500 мл), а полученный осадок собрали фильтрацией с получением указанного в заголовке соединения (9 г, 87%) в виде желтого твердого вещества. МС (ЭС+) C8H3BrClFN2, искомое значение: 260, 262, найдено: 261, 263 [М+Н]+.
Синтез 2-хлор-6-(3,5-диметоксифенил)-8-фторхиназолина
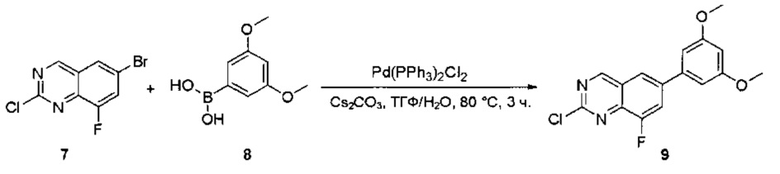
Смесь 6-бром-2-хлор-8-фторхиназолина (4,0 г, 15,4 ммоль), 3,5-диметоксифенилбороновой кислоты (4,47 г, 16,9 ммоль), карбоната цезия (10,0 г, 30,8 ммоль) и Pd(PPh3)2Cl2 (236 мг, 0,77 ммоль) в ТГФ (200 мл) и воде (10 мл) три раза дегазировали азотом и перемешивали при 80°С в течение 3 часов. ТСХ и ЖХМС показали, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и напрямую концентрировали. Остаток очищали силикагелевой хроматографией (петролейный эфир: дихлорметан = от 2:1 до 1:1) с получением указанного в заголовке соединения (2,5 г, 51%) в виде желтого твердого вещества. МС (ЭС+) C16H12ClFN2O2, искомое значение: 318/320, найдено: 319/321 [М+Н]+.
Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолина
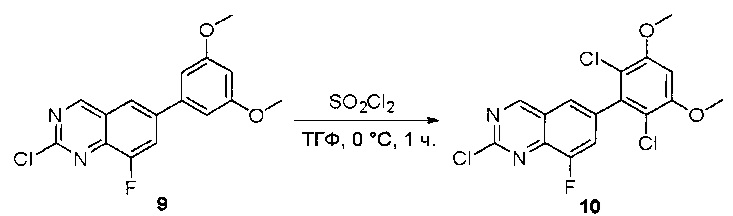
К раствору 2-хлор-6-(3,5-диметоксифенил)-8-фторхиназолина (1,5 г, 4,7 ммоль) в сухом ТГФ (40 мл) по каплям добавляли сульфурилхлорид (1,59 г, 1,75 ммоль) при 0°С и перемешивали смесь в течение 1 часа. ТСХ и ЖХМС показали, что реакция завершена. Реакцию гасили водой (1 мл) и удаляли растворители при пониженном давлении. Остаток промывали ацетонитрилом и высушивали с получением указанного в заголовке соединения (700 мг, 38%) в виде белого твердого вещества. МС (ЭС+) C16H10Cl3FN2O2, искомое значение: 386, 388, найдено: 387, 389 [М+Н]+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,74 (д, 1Н J=1,0 Гц), 8,03-7,99 (м, 2Н), 7,08 (с, 1Н), 4,00 (с, 6Н).
Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-8-фтор-N-(2-метил-6-нитрофенил)хиназолин-2-амина
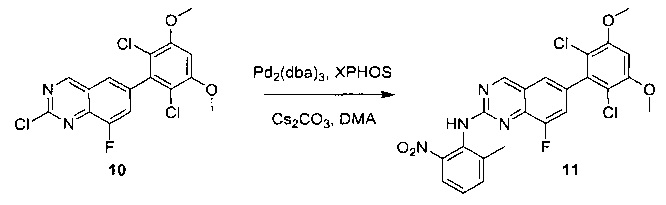
6-(2,6-Дихлор-3,5-диметоксифенил)-8-фтор-N-(2-метил-6-нитрофенил)хиназолин-2-амин получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-100% EtOAc в гексанах, с получением указанного в заголовке соединения. МС (ЭС+) C23H17Cl2FN4O4, искомое значение: 502, найдено: 503 [М+Н]+.
Синтез N1-(6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолин-2-ил)-6-метилбензол-1,2-диамина
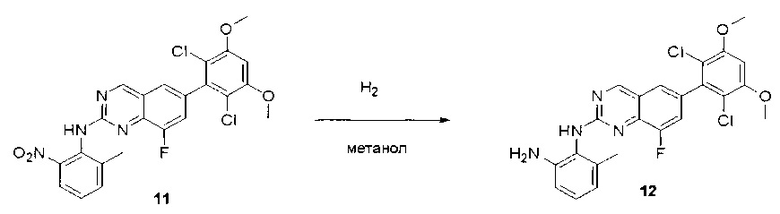
N1-(6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолин-2-ил)-6-метилбензол-1,2-диамин получили по такому же способу, как СОЕДИНЕНИЕ 30. Реакционную смесь фильтровали через целит с получением неочищенного продукта. МС (ЭС+) C23H19Cl2FN4O2, искомое значение: 472, найдено: 473 [М+Н]+.
Синтез N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолин-2-ил)амино)-3-метилфенил)акриламида

N-(2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолин-2-ил)амино)-3-метилфенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали флэш-хроматографией, используя градиент 0-10% МеОН в ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C26H21Cl2FN4O3, искомое значение: 526, найдено: 527 [М+Н]+. 1Н-ЯМР (400 МГц, ДМСО d6) δ 9,53 (д, J=27,9 Гц, 1Н), 9,28 (с, 1Н), 8,96 (с, 1Н), 7,75 (д, J=29,9 Гц, 1Н), 7,59 (д, J=1,7 Гц, 1Н), 7,49 (д, J=10,8 Гц, 1Н), 7,02 (с, 1Н),6,50 (с, 1Н), 6,21 (дд, J=16,9, 2,1 Гц, 1H), 5,75 (с, 1Н),5,68 (дд, J=10,2, 2,0 Гц, 1Н), 3,98 (д, J=4,6 Гц, 6Н), 2,19 (с, 3Н).
Пример 10: Синтез СОЕДИНЕНИЯ 50
Синтез трет-бутил 4-(2,5-дихлорпиримидин-4-ил)пиперазин-1-карбоксилата
К раствору 2,4,5-трихлорпиримидина (0,475 г, 2,6 ммоль) в сухом ДМФ (8,5 мл) добавляли трет-бутил пиперазин-1-карбоксилат (0,51 г, 2,7 ммоль), затем DIEA (0,51 мл, 3,1 ммоль) при 0°С и перемешивали смесь в течение 1 часа. ЖХМС показала, что реакция завершена. Реакционную смесь разбавляли водой (100 мл) и фильтровали белое твердое вещество. Остаток промывали водой и высушивали с получением указанного в заголовке соединения (445 мг, 51%) в виде белого твердого вещества. МС (ЭС+) C13H18Cl2N4O2, искомое значение: 332, найдено: 333 [М+Н]+
Синтез трет-бутил (2-((5-хлор-4-(пиперазин-1-ил)пиримидин-2-ил)амино)фенил)карбамата
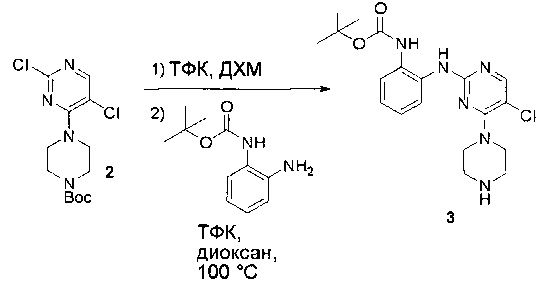
К раствору трет-бутил 4-(2,5-дихлорпиримидин-4-ил)пиперазин-1-карбоксилата (0,1 г, 0,3 ммоль) в ДХМ (1,0 мл) добавляли ТФК (1,0 мл) и перемешивали смесь в течение 1 часа. Аликвоту реакционной смеси анализировали по ЖХМС, которая показала, что реакция завершена. Растворители удаляли, а остаток высушивали под высоким вакуумом. Неочищенный продукт использовали на следующей стадии без дополнительной очистки. К раствору 2,5-дихлор-4-(пиперазин-1-ил)пиримидина (0,3 ммоль) в диоксане (4,0 мл) добавляли ТФК (0,060 мл, 0,75 ммоль) и трет-бутил (2-аминофенил)карбамат (0,094 г, 0,45 ммоль) и перемешивали смесь при 100°С в течение 24 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли EtOAc и промывали насыщенным водным раствором бикарбоната натрия. Органическую смесь высушивали над сульфатом натрия и загружали на силикагель, и очищали, используя градиент 0-10% МеОН в ДХМ, содержащий 10% NH4OH, с получением указанного в заголовке соединения (28 мг, 23%) в виде белого твердого вещества. МС (ЭС+) C19H25ClN6O2, искомое значение: 404, найдено: 405 [М+Н]+
Синтез трет-бутил (2-((5-хлор-4-(4-((3-(трифторметил)фенил)карбамоил)пиперазин-1-ил)пиримидин-2-ил)амино)фенил)карбамата

К раствору трет-бутил (2-((5-хлор-4-(пиперазин-1-ил)пиримидин-2-ил)амино)фенил)карбамата (28 мг, 0,068 ммоль) в ДХМ (0,7 мл) добавляли 1-изоцианато-3-(трифторметил)бензол (0,011 мл, 0,082 ммоль) и триэтиламин (0,015 мл, 0,1 ммоль) и перемешивали смесь при 23°С в течение 16 часов. Неочищенную реакционную смесь загружали на силикагель и очищали, используя градиент 0-50% EtOAc в гексанах, с получением указанного в заголовке соединения (25 мг, 62%). МС (ЭС+) C27H29ClF3N7O3, искомое значение: 591, найдено: 592 [М+Н]+
Синтез 4-(2-((2-акриламидофенил)амино)-5-хлорпиримидин-4-ил)-N-(3-(трифторметил)фенил)пиперазин-1-карбоксамида
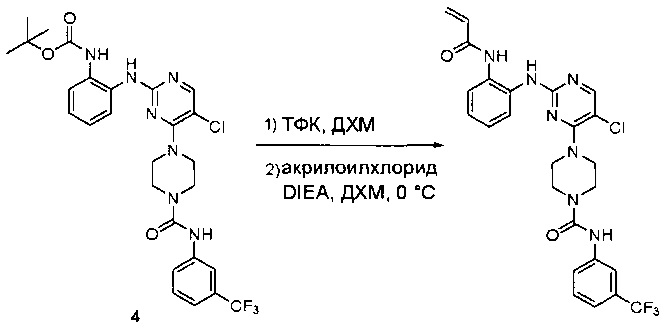
К раствору трет-бутил (2-((5-хлор-4-(4-((3-(трифторметил)фенил)карбамоил)пиперазин-1-ил)пиримидин-2-ил)амино)фенил)карбамата (0,025 г, 0,043 ммоль) в ДХМ (1,0 мл) добавляли ТФК (1,0 мл) и перемешивали смесь в течение 1 часа. Аликвоту реакционной смеси анализировали по ЖХМС, которая показала, что реакция завершена. Растворители удаляли, а остаток высушивали под высоким вакуумом. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
К раствору 4-(2-((2-аминофенил)амино)-5-хлорпиримидин-4-ил)-N-(3-(трифторметил)фенил)пиперазин-1-карбоксамида (0,043 ммоль) в ДХМ (0,5 мл) добавляли акрилоилхлорид (0,004 мл, 0,052 ммоль) и DIEA (0,018 мл, 0,11 ммоль) и перемешивали смесь при 0°С в течение 1 часа. Неочищенную реакционную смесь загружали на силикагель и очищали, используя градиент 0-7% МеОН в ДХМ, с получением указанного в заголовке соединения (10 мг, 43%). МС (ЭС+) C25H23ClF3N7O2, искомое значение: 545, найдено: 546 [М+Н]+
Пример 11: Синтез СОЕДИНЕНИЯ 54
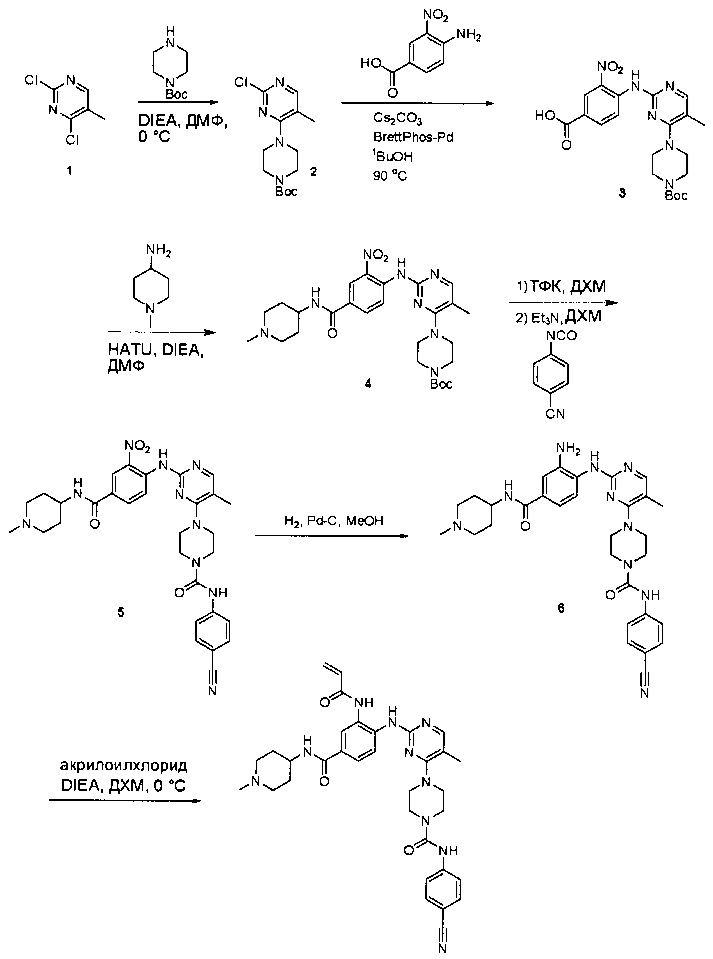
Синтез трет-бутил 4-(2-хлор-5-метилпиримидин-4-ил)пиперазин-1-карбоксилата

К раствору 2,4-дихлор-5-метилпиримидина (0,75 г, 4,6 ммоль) в сухом ДМФ (15,5 мл) добавляли трет-бутил пиперазин-1-карбоксилат (0,9 г, 4,85 ммоль), затем DIEA (0,91 мл, 5,5 ммоль) при 0°С и перемешивали смесь при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь разбавляли водой (120 мл) и фильтровали твердое вещество. Остаток промывали водой и высушивали с получением указанного в заголовке соединения (1,386 г, 96%) в виде белого твердого вещества. МС (ЭС+) C14H21ClN4O2, искомое значение: 312, найдено: 313 [М+Н]+
Синтез 4-((4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-5-метилпиримидин-2-ил)амино)-3-нитробензойной кислоты
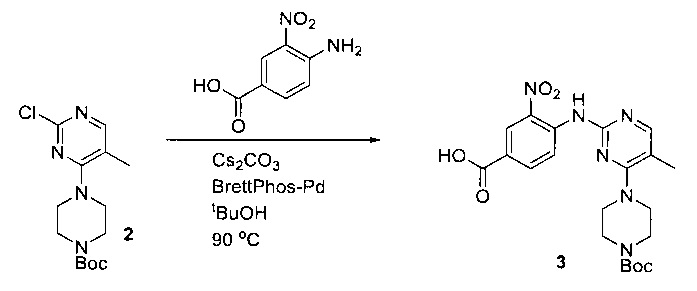
Смесь трет-бутил 4-(2-хлор-5-метилпиримидин-4-ил)пиперазин-1-карбоксилата (0,15 г, 0,48 ммоль), 4-амино-3-нитробензойной кислоты (97 мг, 0,53 ммоль), смеси BrettPhos-Pd (20 мг, 0,015 ммоль) и карбоната цезия (470 мг, 1,44 ммоль) в tBuOH (2,4 мл) нагревали в закрытой пробирке при 100°С в течение ночи. Смесь разбавляли EtOAc, фильтровали через слой целита, загружали на силикагель и очищали, используя градиент 0-10% МеОН в ДХМ, с получением указанного в заголовке соединения (75 мг, 34%). МС (ЭС+) C21H26N6O6, искомое значение: 458, найдено: 459 [М+Н]+ Синтез трет-бутил 4-(5-метил-2-((4-((1-метилпиперидин-4-ил)карбамоил)-2-нитрофенил)амино)пиримидин-4-ил)пиперазин-1-карбоксилата
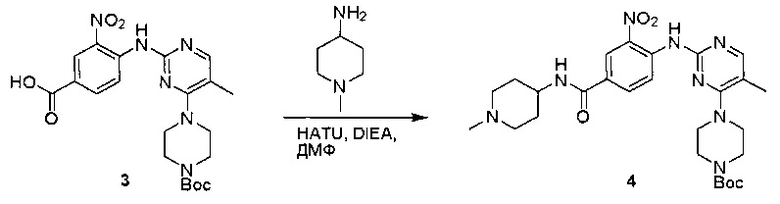
Смесь 4-((4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-5-метилпиримидин-2-ил)амино)-3-нитробензойной кислоты (0,075 г, 0,164 ммоль), 1-метилпиперидин-4-амина (37 мг, 0,33 ммоль), HATU (140 мг, 0,37 ммоль) и DIEA (0,1 мл, 0,6 ммоль) в ДМФ (3,0 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли EtOAc, промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором. Неочищенную смесь загружали на силикагель и очищали, используя градиент 0-10% МеОН в ДХМ, содержащий 10% NH4OH, с получением указанного в заголовке соединения (73 мг, 80%). МС (ЭС+) C27H38N8O5, искомое значение: 554, найдено: 555 [М+Н]+
Синтез N-(4-цианофенил)-4-(5-метил-2-((4-((1-метилпиперидин-4-ил)карбамоил)-2-нитрофенил)амино)пиримидин-4-ил)пиперазин-1-карбоксамида
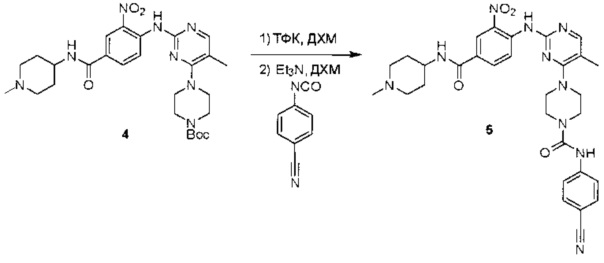
К раствору трет-бутил 4-(5-метил-2-((4-((1-метилпиперидин-4-ил)карбамоил)-2-нитрофенил)амино)пиримидин-4-ил)пиперазин-1-карбоксилата (0,073 г, 0,13 ммоль) в ДХМ (1,0 мл) добавляли ТФК (1,0 мл) и перемешивали смесь в течение 1 часа. Аликвоту реакционной смеси анализировали по ЖХМС, которая показала, что реакция завершена. Растворители удаляли, а остаток высушивали под высоким вакуумом. Неочищенный продукт использовали на следующей стадии без дополнительной очистки. К раствору 4-((5-метил-4-(пиперазин-1-ил)пиримидин-2-ил)амино)-N-(1-метилпиперидин-4-ил)-3-нитробензамида (0,073 ммоль) в ДХМ (1,5 мл) добавляли 4-изоцианатобензонитрил (23 мг, 0,16 ммоль) и триэтиламин (0,055 мл, 0,39 ммоль) и перемешивали смесь при 23°С в течение 16 часов. Неочищенную реакционную смесь фильтровали и промывали минимальным объемом ДХМ, а затем гексанами с получением указанного в заголовке соединения (97 мг, 100%). МС (ЭС+) C30H34N10O4, искомое значение: 598, найдено: 599 [М+Н]+
Синтез 4-(2-((2-амино-4-((1-метилпиперидин-4-ил)карбамоил)фенил)амино)-5-метилпиримидин-4-ил)-N-(4-цианофенил)пиперазин-1-карбоксамида
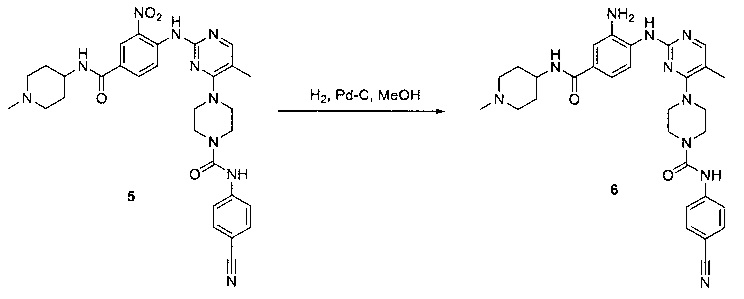
4-(2-((2-Амино-4-((1-метилпиперидин-4-ил)карбамоил)фенил)амино)-5-метилпиримидин-4-ил)-N-(4-цианофенил)пиперазин-1-карбоксамид получили по такому же способу, как СОЕДИНЕНИЕ 30. Реакционную смесь фильтровали через целит с получением неочищенного продукта. МС (ЭС+) C30H36N10O2, искомое значение: 568, найдено: 569 [М+Н]+.
Синтез 4-(2-((2-акриламидо-4-((1-метилпиперидин-4-ил)карбамоил)фенил)амино)-5-метилпиримидин-4-ил)-N-(4-цианофенил)пиперазин-1-карбоксамида
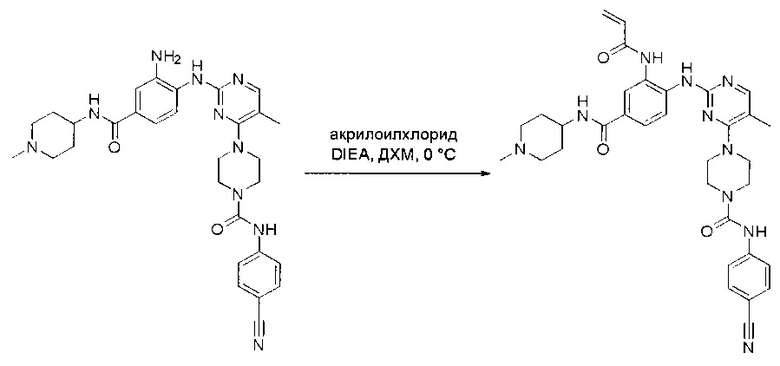
4-(2-((2-Акриламидо-4-((1-метилпиперидин-4-ил)карбамоил)фенил)амино)-5-метилпиримидин-4-ил)-N-(4-цианофенил)пиперазин-1-карбоксамид получили по такому же способу, как СОЕДИНЕНИЕ 30. Реакционную смесь очищали препаративной тонкослойной хроматографией с получением указанного в заголовке продукта. МС (ЭС+) C33H38N10O3, искомое значение: 622, найдено: 623 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,98 (с, 1Н), 9,08 (с, 1H), 8,30 (с, 1Н), 8,21-8,07 (м, 3Н), 7,93 (д, J=10,7 Гц, 2Н), 7,67 (м, 4Н), 6,50 (дд, J=16,9, 10,2 Гц, 1Н), 6,33-6,25 (м, 1Н), 5,83-5,76 (м, 1Н), 3,78 (м, 2Н), 3,59 (м, 4Н), 3,43 (м, 4Н), 2,92 (д, J=11,4 Гц, 2Н), 2,30 (с, 3Н), 2,23 (с, 2Н), 2,14 (с, 3Н), 1,79 (м, 2Н), 1,69- 1,54 (м, 2Н).
Пример 12: Синтез СОЕДИНЕНИЯ 20
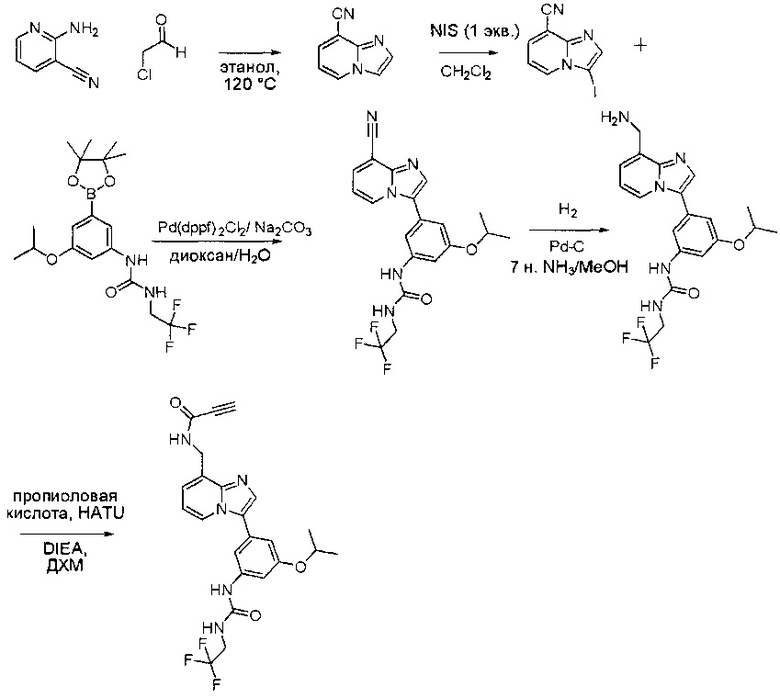
Синтез имидазо[1,2-а]пиридин-8-карбонитрила

К раствору 2-аминоникотинонитрила (1,0 г, 8,39 ммоль) в ЕtOН (10 мл) в 20 мл закрытой пробирке добавляли 2-хлорацетальдегид (1,611 мл, 9,23 ммоль), затем пробирку закрывали и нагревали до 120°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и гасили 2 н. раствором Na2CO3, удаляли EtOH в вакууме и экстрагировали при помощи ДХМ ×3. Объединяли органические слои и промывали их водой, затем насыщенным солевым раствором ×2. Высушивали над сульфатом натрия и удаляли растворитель с получением указанного в заголовке соединения в виде желто-коричневого твердого вещества (1,2 г, 8,38 ммоль, выход 100%), проверили по МС (ЭС+) C8H5N3, искомое значение: 143 найдено: 144 [М+Н]+
Синтез 3-йодимидазо[1,2-а]пиридин-8-карбонитрила
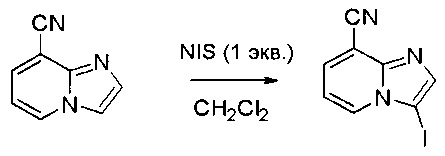
К перемешанному раствору имидазо[1,2-а]пиридин-8-карбонитрила (1,2 г, 8,38 ммоль) в дихлорметане (10 мл) добавляли N-йодсукцинимид (1,89 г, 8,38 ммоль). Контролировали по ЖХМС до полного расходования исходного материала. Реакционную смесь разбавляли дихлорметаном и водой. Отделенный органический слой высушивали сульфатом натрия, фильтровали и концентрировали с получением 3-йодимидазо[1,2-а]пиридин-8-карбонитрила (1,8 г, 6,69 ммоль, выход 80%) в виде коричневого твердого вещества. МС (ЭС+) C8H8IN3, искомое значение: 269, найдено: 270 [М+Н]+.
Синтез 1-(3-(8-цианоимидазо[1,2-а]пиридин-3-ил)-5-изопропоксифенил)-3-(2,2,2-трифторэтил)мочевины
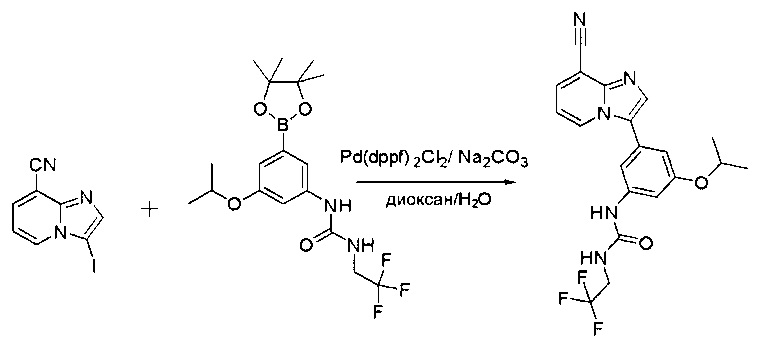
К смеси 3-йодимидазо[1,2-а]пиридин-8-карбонитрила (100 мг, 373 мкмоль), 1-(3-изопропокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-3-(2,2,2-трифторэтил)мочевины (90 мг, 224 мкмоль), аддукта PdCl2(dppf)-CH2Cl2 (30,5 мг, 37,3 мкмоль) в диоксане (3 мл) добавляли 2 М раствор Na2CO3 (0,559 мл, 1119 мкмоль). Пробирку дегазировали в течение 5 минут, затем закрывали и нагревали до 110°С в течение 30 минут в микроволновом реакторе. После охлаждения до комнатной температуры реакционную смесь разделили между EtOAc и насыщенным солевым раствором, отделяли и промывали органический слой насыщенным солевым раствором ×2. Объединенные органические слои напрямую высушивали на силикагеле и очищали флэш-хроматографией (0-100% гексаны/EtOAc; колонка 12 г). Выделяли указанное в заголовке соединение (30 мг, 71,9 мкмоль, выход 32,1%) в виде коричневого твердого вещества. МС (ЭС+) C20H18F3N5O2, искомое значение: 417, найдено: 418 [М+Н]+. Синтез 1-(3-(8-(аминометил)имидазо[1,2-а]пиридин-3-ил)-5-изопропоксифенил)-3-(2,2,2-трифторэтил)мочевины
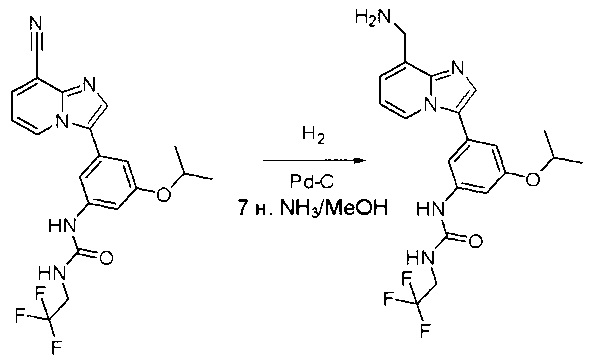
1-(3-(8-Цианоимидазо[1,2-а]пиридин-3-ил)-5-изопропоксифенил)-3-(2,2,2-трифторэтил)мочевину (0,030 г, 0,072 ммоль) растворяли в 7 н. растворе аммиака в метаноле (20 мл, 140 ммоль) и добавляли Pd-C (10 мг, 0,094 ммоль). Реакционную смесь перемешивали под Н2 из баллона в течение 1 часа. Затем смесь фильтровали через целит и удаляли растворитель. Остаток высушивали под высоким вакуумом в течение ночи с получением указанного в заголовке соединения в виде желтого твердого вещества (0,026 г, 0,062 ммоль, выход 86%). МС (ЭС+) C20H22F3N5O2, искомое значение: 421, найдено: 422 [М+Н]+.
Синтез N-((3-(3-изопропокси-5-(3-(2,2,2-трифторэтил)уреидо)фенил)имидазо[1,2-а]пиридин-8-ил)метил)пропиоламида
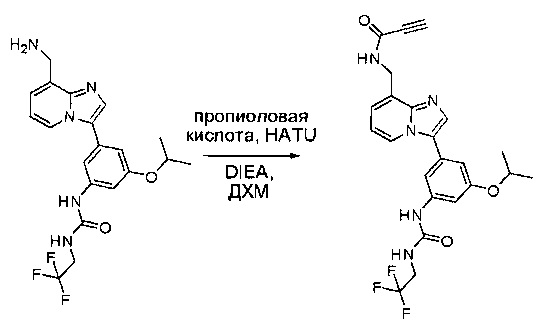
К раствору 1-(3-(8-(аминометил)имидазо[1,2-а]пиридин-3-ил)-5-изопропоксифенил)-3-(2,2,2-трифторэтил)мочевины (26 мг, 0,062 ммоль) в ДХМ (3 мл) добавляли DIEA (0,075 мл, 0,432 ммоль) и HATU (35,2 мг, 0,093 ммоль), а в конце пропиоловую кислоту (4,95 мкл, 0,080 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Реакционную смесь загружали непосредственно на силикагелевую колонку и очищали флэш-хроматографией (0-10% CH2Cl2/МеОН) с получением указанного в заголовке соединения (19 мг, 0,040 ммоль, выход 65,0%) в виде грязновато-белого твердого вещества. МС (ЭС+) C23H22F3N5O3, искомое значение: 473, найдено: 474 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,34 (с, 1Н), 8,92 (с, 1Н), 8,47 (д, J=6,8 Гц, 1Н), 7,74 (с, 1Н), 7,17 (д, J=1,9 Гц, 2Н), 7,10 (с, 1Н), 6,98 (с, 1Н), 6,82 (с, 1Н), 6,74 (с, 1H), 4,69-4,58 (м, 2Н), 3,93 (дд, J=9,7, 6,4 Гц, 2Н), 2,72-2,64 (м, 1Н), 1,30-1,19 (м, 6Н).
Пример 13: Синтез СОЕДИНЕНИЯ 21
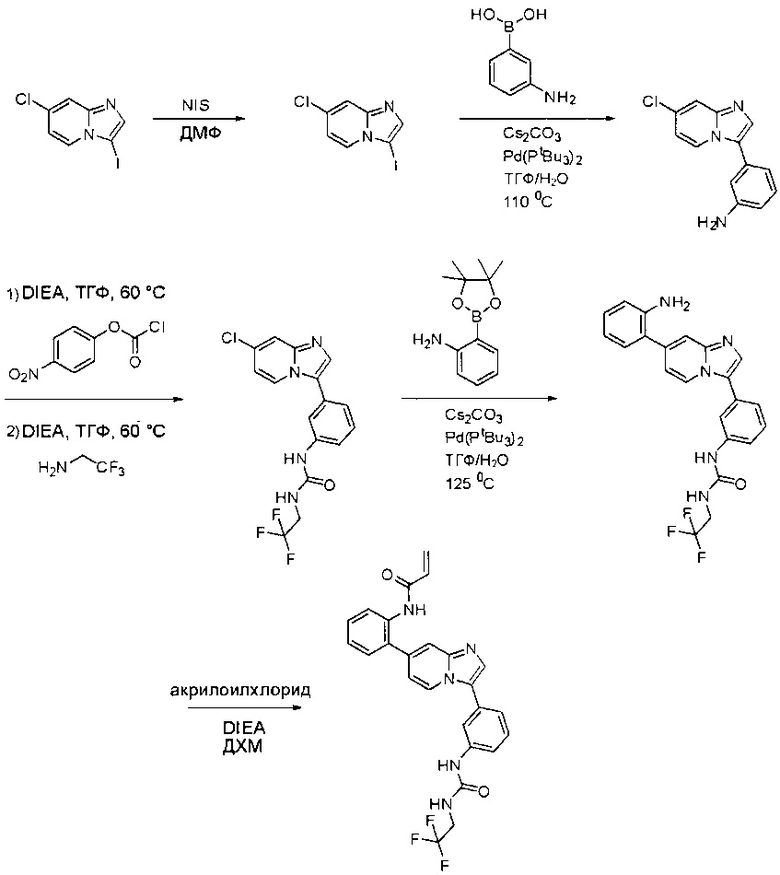
Синтез 7-хлор-3-йодимидазо[1,2-а]пиридина
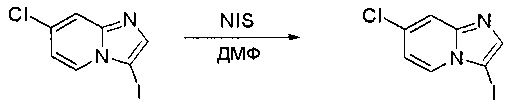
7-Хлор-3-йодимидазо[1,2-а]пиридин получили по способу, описанному в WO 2008078091. МС (ЭС+) C7H4ClIN2, искомое значение: 278, найдено: 279 [М+Н]+.
Синтез 3-(7-хлоримидазо[1,2-а]пиридин-3-ил)анилина
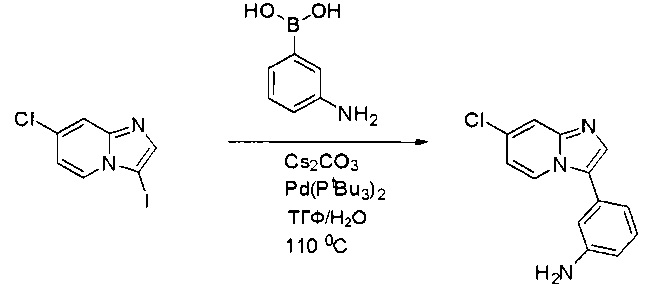
3-(7-Хлоримидазо[1,2-а]пиридин-3-ил)анилин получили по способу, описанному в WO 2008078091. МС (ЭС+) C13H10ClN3, искомое значение: 243, найдено: 244 [М+Н]+. Синтез 1-(3-(7-хлоримидазо[1,2-а]пиридин-3-ил)фенил)-3-(2,2,2-трифторэтил)мочевины
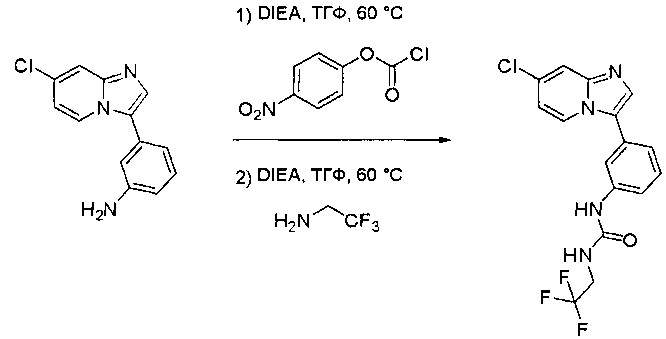
К раствору 3-(7-хлоримидазо[1,2-а]пиридин-3-ил)анилина (0,15 ммоль) в ТГФ (1,5 мл) добавляли 4-нитрофенилкарбонохлоридат (30 мг, 0,15 ммоль) и DIEA (0,036 мл, 0,225 ммоль). Смесь нагревали при 60°С в течение 6 часов. К неочищенному карбамату добавляли DIEA (0,036 мл, 0,225 ммоль) и 2,2,2-трифторэтан-1-амин (0,014 мл, 0,18 ммоль) и нагревали раствор при 60°С в течение ночи. Реакционную смесь разбавляли EtOAc и водой. Отделенный органический слой высушивали сульфатом натрия, фильтровали и концентрировали. Неочищенную смесь очищали флэш-хроматографией (0-6% МеОН/ДХМ) с получением указанного в заголовке соединения (38 мг, выход 69%). МС (ЭС+) C16H12ClF3N4O, искомое значение: 368, найдено: 369 [М+Н]+.
Синтез 1-(3-(7-(2-аминофенил)имидазо[1,2-а]пиридин-3-ил)фенил)-3-(2,2,2-трифторэтил)мочевины
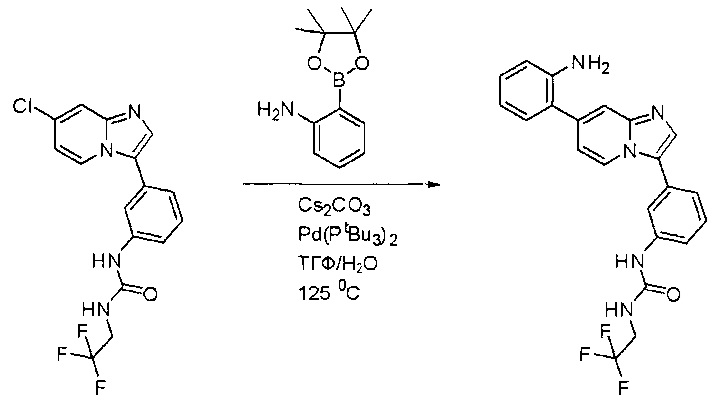
К смеси 1-(3-(7-хлоримидазо[1,2-а]пиридин-3-ил)фенил)-3-(2,2,2-трифторэтил)мочевины (20 мг, 0,052 ммоль), 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (15 мг, 0,066 ммоль) и карбоната цезия (51 мг, 0,156 ммоль) в смеси ТГФ/Н2О (2/1, 0,75 мл) добавляли Pd(PtBu3)2 (3 мг, 0,005 ммоль). Пробирку дегазировали в течение 5 минут, затем закрывали и нагревали до 125°С в течение 20 минут в микроволновом реакторе. После охлаждения до комнатной температуры реакционную смесь фильтровали через слой целита и очищали флэш-хроматографией (градиент 0-10% МеОН/ДХМ, содержащий 10% NH4OH) с получением указанного в заголовке соединения (20 мг, выход 90%). МС (ЭС+) C22H18F3N5O, искомое значение: 425, найдено: 426 [М+Н]+.
Синтез N-(2-(3-(3-(3-(2,2,2-трифторэтил)уреидо)фенил)имидазо[1,2-а]пиридин-7-ил)фенил)акриламида

N-(2-(3-(3-(3-(2,2,2-трифторэтил)уреидо)фенил)имидазо[1,2-а]пиридин-7-ил)фенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали препаративной тонкослойной хроматографией, используя градиент 0-10% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C25H20F3N5O искомое значение: 479, найдено: 480 [М+Н]+.
Пример 14: Синтез СОЕДИНЕНИЯ 38
Синтез N-(2-(3-(3-изопропокси-5-(3-(2,2,2-трифторэтил)уреидо)фенил)имидазо[1,2-а]пиридин-7-ил)фенил)акриламида
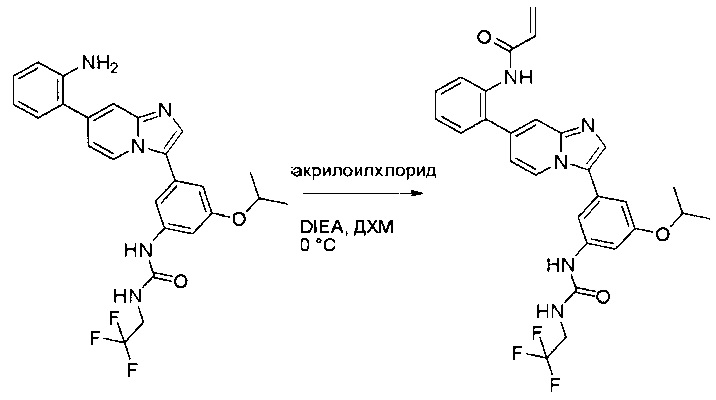
N-(2-(3-(3-изопропокси-5-(3-(2,2,2-трифторэтил)уреидо)фенил)имидазо[l,2-a]пиридин-7-ил)фенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали при помощи ВЭЖХ, используя градиент 5-70% ацетонитрила в воде+0,1% муравьиной кислоты, с получением указанного в заголовке соединения в виде формиатной соли. МС (ЭС+) C28H26F3N5O3, искомое значение: 537, найдено: 538 [М+Н]+.
Пример 15: Синтез СОЕДИНЕНИЯ 11
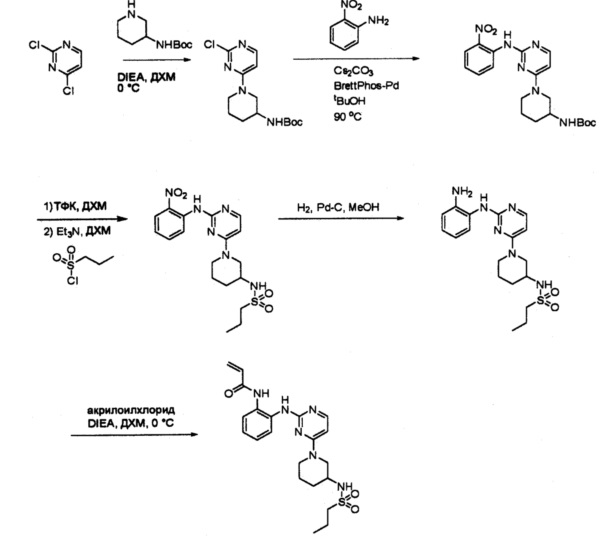
Синтез трет-бутил (1-(2-хлорпиримидин-4-ил)пиперидин-3-ил)карбамата
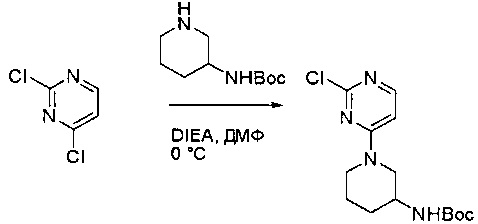
Трет-бутил (1-(2-хлорпиримидин-4-ил)пиперидин-3-ил)карбамат получили по такому же способу, как СОЕДИНЕНИЕ 54, используя 2,4-дихлорпиримидин и трет-бутил пиперидин-3-илкарбамат. МС (ЭС+) C14H21ClN4O2, искомое значение: 312, найдено: 313 [М+Н]+
Синтез трет-бутил (1-(2-((2-нитрофенил)амино)пиримидин-4-ил)пиперидин-3-ил)карбамата
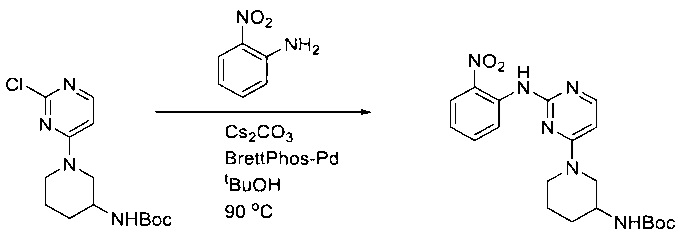
Трет-бутил (1-(2-((2-нитрофенил)амино)пиримидин-4-ил)пиперидин-3-ил)карбамат получили по такому же способу, как СОЕДИНЕНИЕ 54, используя 2-нитроанилин. МС (ЭС+) C20H26N6O4, искомое значение: 414, найдено: 415 [М+Н]+
Синтез N-(1-(2-((2-нитрофенил)амино)пиримидин-4-ил)пиперидин-3-ил)пропан-1-сульфонамида
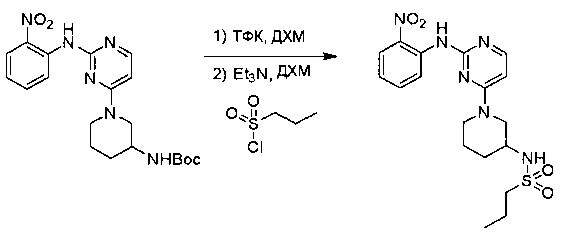
К раствору трет-бутил (I -(2-((2-нитрофенил)амино)пиримидин-4-ил)пиперидин-3-ил)карбамата (0,14 г, 0,34 ммоль) в ДХМ (2,0 мл) добавляли ТФК (1,0 мл) и перемешивали смесь в течение 1 часа. Аликвоту реакционной смеси анализировали по ЖХМС, которая показала, что реакция завершена. Растворители удаляли, а остаток высушивали под высоким вакуумом. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
К раствору 4-(3-аминопиперидин-1-ил)-N-(2-нитрофенил)пиримидин-2-амина (0,34 ммоль) в ДХМ (3,5 мл) при 0°С добавляли пропан-1-сульфонилхлорид (0,045 мл, 0,4 ммоль) и триэтиламин (0,12 мл, 0,85 ммоль) и нагревали смесь до комнатной температуры в течение ночи. Неочищенную реакционную смесь концентрировали и очищали флэш-хроматографией (0-7,5% МеОН/ДХМ) с получением указанного в заголовке соединения (36 мг, выход 24%). МС (ЭС+) C18H24N6O4S, искомое значение: 420, найдено: 421 [М+Н]+. Синтез N-(1-(2-((2-аминофенил)амино)пиримидин-4-ил)пиперидин-3-ил)пропан-1-сульфонамида
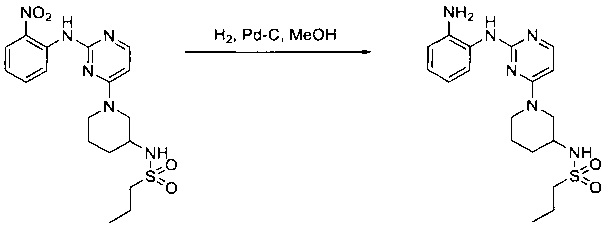
N-(1-(2-((2-аминофенил)амино)пиримидин-4-ил)пиперидин-3-ил)пропан-1-сульфонамид получили по такому же способу, как СОЕДИНЕНИЕ 30. Реакционную смесь фильтровали через целит с получением неочищенного продукта. МС (ЭС+) C18H26N6O2S, искомое значение: 390, найдено: 391 [М+Н]+.
Синтез N-(2-((4-(3-(пропилсульфонамидо)пиперидин-1-ил)пиримидин-2-ил)амино)фенил)акриламида
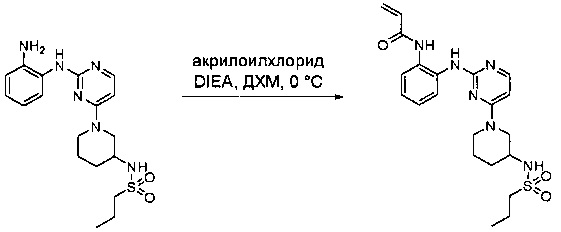
N-(2-((4-(3-(пропилсульфонамидо)пиперидин-1-ил)пиримидин-2-ил)амино)фенил)акриламид получили по такому же способу, как СОЕДИНЕНИЕ 30. Продукт очищали препаративной тонкослойной хроматографией, используя градиент 0-6% МеОН/ДХМ, с получением указанного в заголовке соединения. МС (ЭС+) C21H28N6O3S, искомое значение: 444, найдено: 445 [М+Н]+.
Пример 16: Синтез СОЕДИНЕНИЯ 52
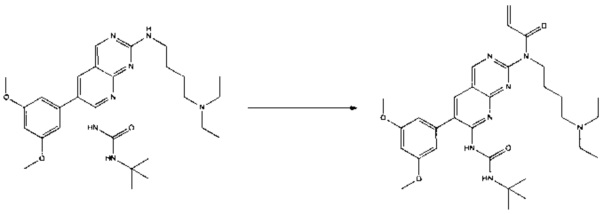
Исходный материал, 1-(трет-бутил)-3-(2-((4-(диэтиламино)бутил)амино)-6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-7-ил)мочевина (PD 173074), может быть приобретена в продаже, например, у компании SelleckChem.com. В высушенном сосуде акрилоилхлорид (2 экв.) и диизопропилэтиламин (4,3 экв.) добавляли к раствору 1-(трет-бутил)-3-(2-((4-(диэтиламино)бутил)амино)-6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-7-ил)мочевины (1 экв.) в безводном дихлорметане при 0°С. После перемешивания при комнатной температуре в течение 2 часов, реакционную смесь концентрировали, разбавляли ДМСО и очищали обращенно-фазовой ВЭЖХ (5-95% вода/ацетонитрил). После концентрирования фракций, продукт, N-(7-(3-(трет-бутил)уреидо)-6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)-N-(4-(диэтиламино)бутил)акриламид получили в виде бледно-желтого пенистого вещества. ЖХМС (М+1)=578,2.
Пример 17: Синтез СОЕДИНЕНИЯ 55
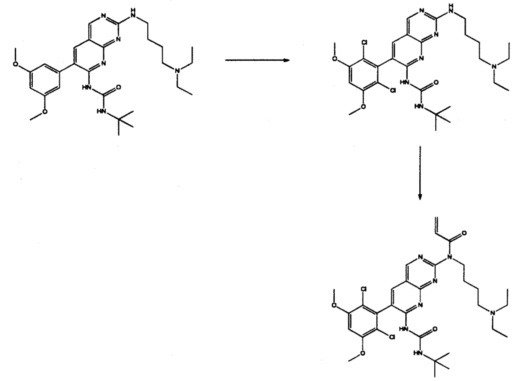
В высушенном сосуде сульфурилхлорид (2 экв.) добавляли к раствору 1-(трет-бутил)-3-(2-((4-(диэтиламино)бутил)амино)-6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидин-7-ил)мочевины (1 экв.) в безводном ацетонитриле при 0°С. После перемешивания в течение 2 часов, реакционную смесь разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Неочищенный продукт, 1-(трет-бутил)-3-(6-(2,6-дихлор-3,5-диметоксифенил)-2-((4-(диэтиламино)бутил)амино)пиридо[2,3-d]пиримидин-7-ил)мочевину, использовали на следующей стадии без дополнительной очистки.
В высушенном сосуде акрилоилхлорид (2 экв.) и диизопропилэтиламин (4,3 экв.) добавляли к раствору продукта, полученного выше (1 экв.) в безводном дихлорметане при 0°С. После перемешивания при комнатной температуре в течение 2 часов, реакционную смесь концентрировали, разбавляли ДМСО и очищали обращенно-фазовой ВЭЖХ (5-95% вода/ацетонитрил). После высушивания под высоким вакуумом, получили продукт, N-(7-(3-(трет-бутил)уреидо)-6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидин-2-ил)- N-(4-(диэтиламино)бутил)акриламид, в виде желтого пенистого вещества. ЖХМС (М+1)=646,3.
Такие же способы, как описаны выше, могут быть использованы для получения других соединений, описанных в настоящем документе.
Данные 1Н ЯМР и ЖХМС для Соединений 1-55 представлены ниже.
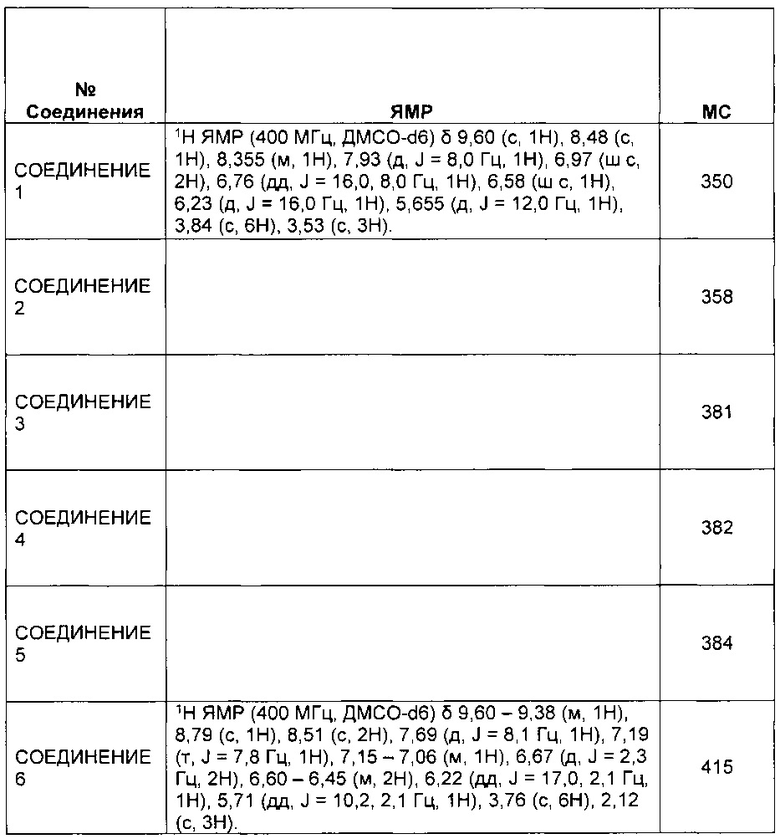
Селективность соединений
Оценка селективности представляет собой объективную меру, которая дает возможность количественного сравнения соединений и подробную дифференциацию, и анализ способов взаимодействия. Один из критериев селективности рассчитывают с использованием процента от контрольных значений в анализах панели киназ. Оценки из первичных проверок (выполненных при одной концентрации) записывают как процент от контроля с ДМСО (РОС) и рассчитывают следующим образом:

где отрицательный контроль представляет собой растворитель, такой как ДМСО (контроль 100%), а положительный контроль представляет собой известное контрольное соединение, которое связывается с высокой аффинностью (контроль 0%).
Оценка селективности (S) каждого проверенного соединения может быть рассчитана делением количества киназ с РОС меньше выбранного значения, например, 10, 20 или 35, при проверке при определенной концентрации, например, 1 мкМ, 3 мкМ, 5 мкМ или 10 мкМ, на общее количество отдельных проверенных киназ (за исключением мутантных вариантов). Например, оценка селективности (S) может быть рассчитана делением количества киназ со значением РОС менее 10, при проверке при 3 мкМ, на общее количество отдельных испытанных киназ (за исключением мутантных вариантов); такую оценку записывают в виде [S(10) при 3 мкМ]. Определили селективность соединений: СОЕДИНЕНИЯ 9; СОЕДИНЕНИЯ 9; СОЕДИНЕНИЯ 11; СОЕДИНЕНИЯ 15; СОЕДИНЕНИЯ 16; СОЕДИНЕНИЯ 20; СОЕДИНЕНИЯ 21; СОЕДИНЕНИЯ 23; СОЕДИНЕНИЯ 24; СОЕДИНЕНИЯ 25; СОЕДИНЕНИЯ 26; СОЕДИНЕНИЯ 27; СОЕДИНЕНИЯ 30; СОЕДИНЕНИЯ 32; СОЕДИНЕНИЯ 35; СОЕДИНЕНИЯ 60; СОЕДИНЕНИЯ 38; СОЕДИНЕНИЯ 39; СОЕДИНЕНИЯ 41; СОЕДИНЕНИЯ 45; СОЕДИНЕНИЯ 48; СОЕДИНЕНИЯ 50; СОЕДИНЕНИЯ 52; СОЕДИНЕНИЯ 54; СОЕДИНЕНИЯ 55; все они имели оценки селективности [S(10) при 3 мкМ], равные 0,030 или менее.
СОЕДИНЕНИЕ 9; СОЕДИНЕНИЕ 11; СОЕДИНЕНИЕ 15; СОЕДИНЕНИЕ 16; СОЕДИНЕНИЕ 20; СОЕДИНЕНИЕ 21; СОЕДИНЕНИЕ 23; СОЕДИНЕНИЕ 24; СОЕДИНЕНИЕ 25; СОЕДИНЕНИЕ 26; СОЕДИНЕНИЕ 32; СОЕДИНЕНИЕ 35; СОЕДИНЕНИЕ 60; СОЕДИНЕНИЕ 38; СОЕДИНЕНИЕ 39; СОЕДИНЕНИЕ 45; СОЕДИНЕНИЕ 48; СОЕДИНЕНИЕ 50; СОЕДИНЕНИЕ 52 имели оценки селективности [S(10) при 3 мкМ], равные 0,010 или менее.
Оценка биохимической активности
Для оценки активности химических соединений против релевантных рассматриваемых киназ использовали технологическую платформу изменения электрофоретической подвижности. Пептид субстрата с флуоресцентной меткой инкубировали в присутствии дозированных концентраций соединений, определенной концентрации киназы и АТФ, чтобы фосфорилировать соответствующую часть пептида. В конце реакции смесь фосфорилированного (продукта) и не фосфорилированного (субстрата) пептидов пропускали через микрожидкостную систему прибора Caliper LabChip® EZ Reader II, при приложенной разности потенциалов. Наличие фосфатной группы на пептиде продукта обеспечивает разность массы и заряда между пептидом продукта и пептидом субстрата, что приводит к разделению пулов субстрата и продукта в образце. При прохождении пулов через светодиодные элементы, расположенные в приборе, эти пулы обнаруживают и определяют по отдельным пикам. Следовательно, соотношение между этими пиками отражает активность химического вещества при определенной концентрации, в определенной лунке, в определенных условиях.
Анализ FGFR-1 дикого типа при Km: В каждой лунке 384-луночного планшета инкубировали 0,1 нг/мкл FGFR-1 дикого типа (Carna Biosciences, Inc.) в общем объеме 12,5 мкл буфера (100 мМ HEPES рН 7,5, 0,015% Brij 35, 10 мМ MgCl2, 1 мМ DTT) с 1 мМ CSKtide (5-FAM- KKKKEEIYFFFG-NH2) и 400 мкМ АТФ при 25°С в течение 90 минут в присутствии или в отсутствие дозированных серий концентраций соединения (конечная концентрация в ДМСО 1%). Реакцию останавливали добавлением 70 мкл стоп-буфера (100 мМ HEPES рН 7,5, 0,015% Brij 35, 35 мМ ЭДТК и 0,2% покрывающего реагента 3 (Caliper Lifesciences)). Затем планшет считывали на приборе Caliper EZ Reader 2 (настройки протокола: -1,9 фунтов/дюйм2, повышающее напряжение -700, понижающее напряжение -3000, время выдержки после образца 35 секунд).
Анализ FGFR-4 дикого типа при Km: В каждой лунке 384-луночного планшета инкубировали 0,5 нг/мкл FGFR-4 дикого типа (Carna Biosciences, Inc.) в общем объеме 12,5 мкл буфера (100 мМ HEPES рН 7,5, 0,015% Brij 35, 10 мМ MgCl2, 1 мМ DTT) с 1 мкМ CSKtide (5-FAM- KKKKEEIYFFFG-NH2) и 400 мкМ АТФ при 25°С в течение 90 минут в присутствии или в отсутствие дозированных серий концентраций соединения (конечная концентрация в ДМСО 1%). Реакцию останавливали добавлением 70 мкл стоп-буфера (100 мМ HEPES рН 7,5, 0,015% Brij 35, 35 мМ ЭДТК и 0,2% покрывающего реагента 3 (Caliper Lifesciences)). Затем планшет считывали на приборе Caliper LabChip® EZ Reader II (настройки протокола: -1,9 фунтов/дюйм2, повышающее напряжение -700, понижающее напряжение -3000, время выдержки после образца 35 секунд).
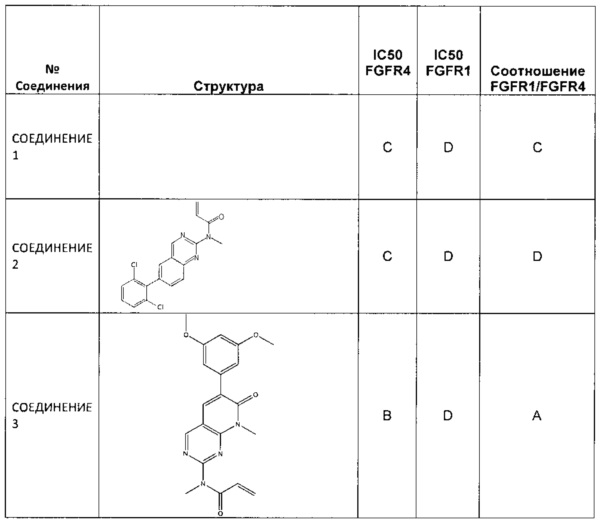
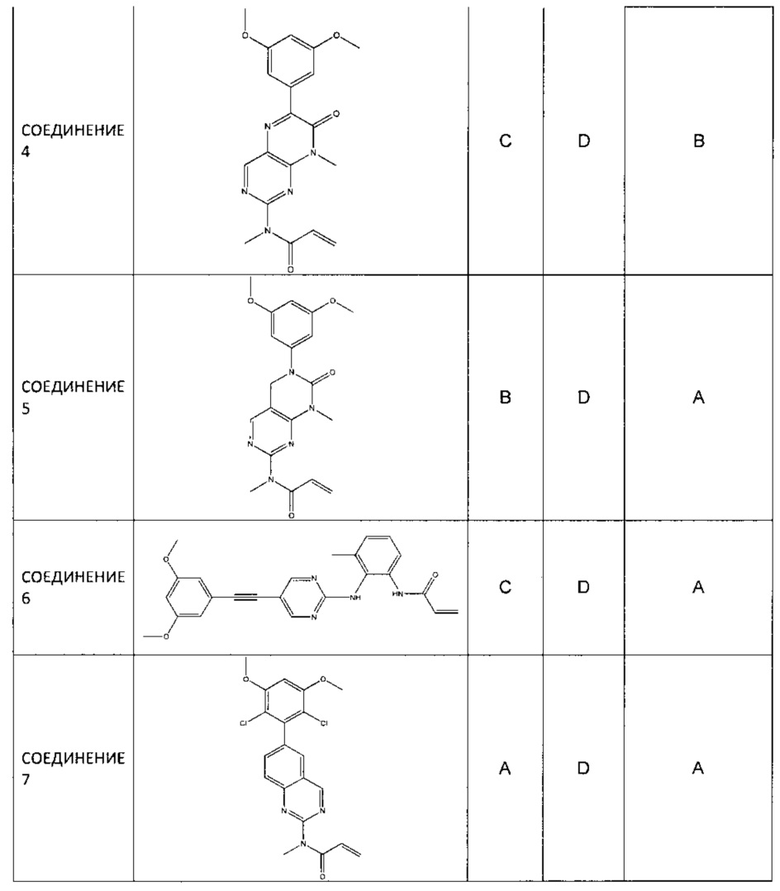
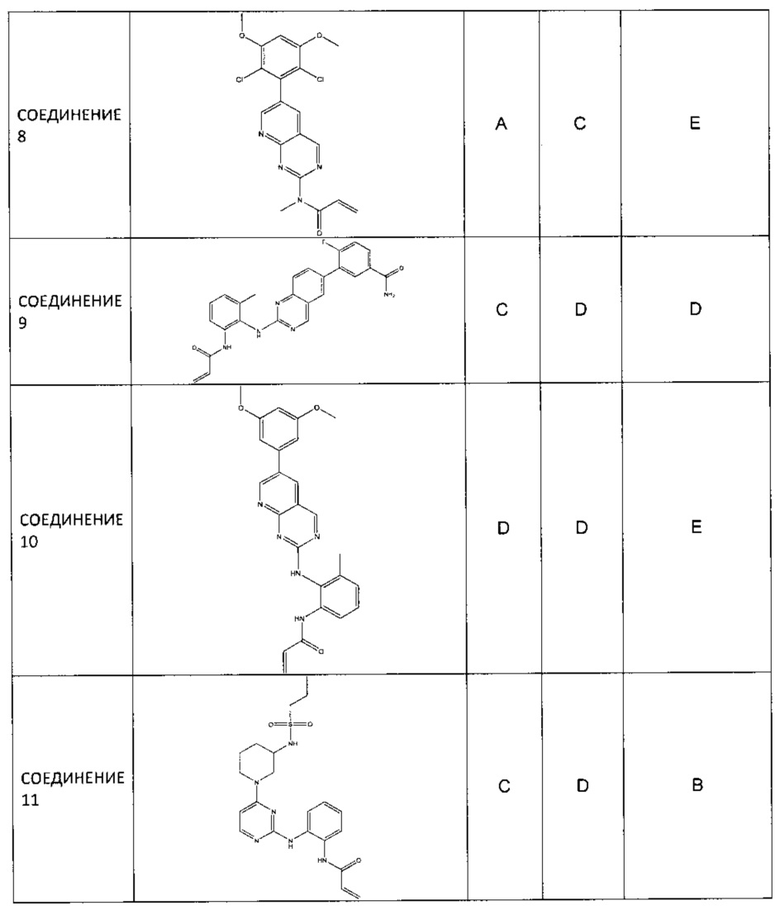
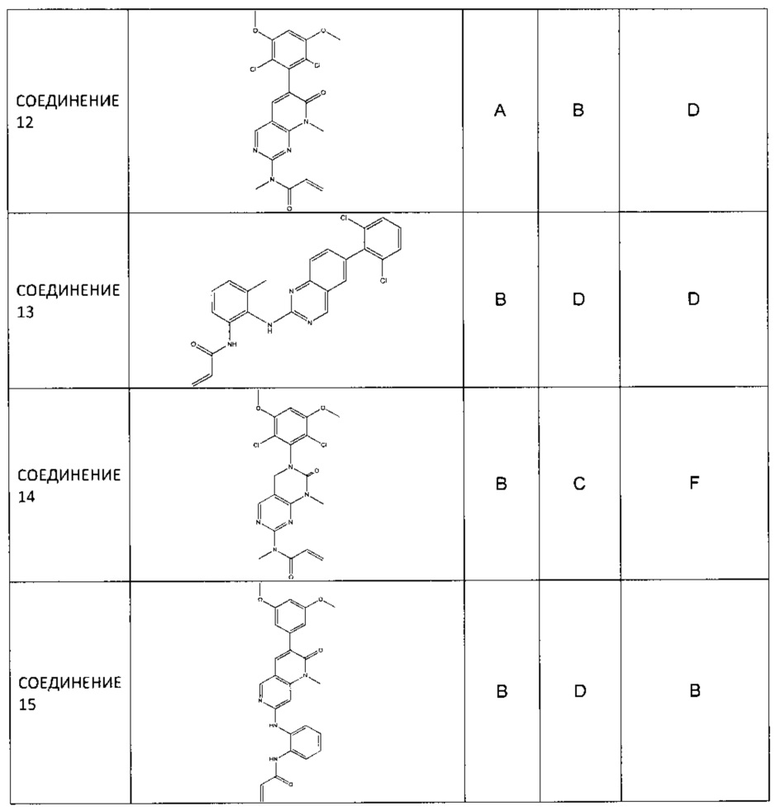
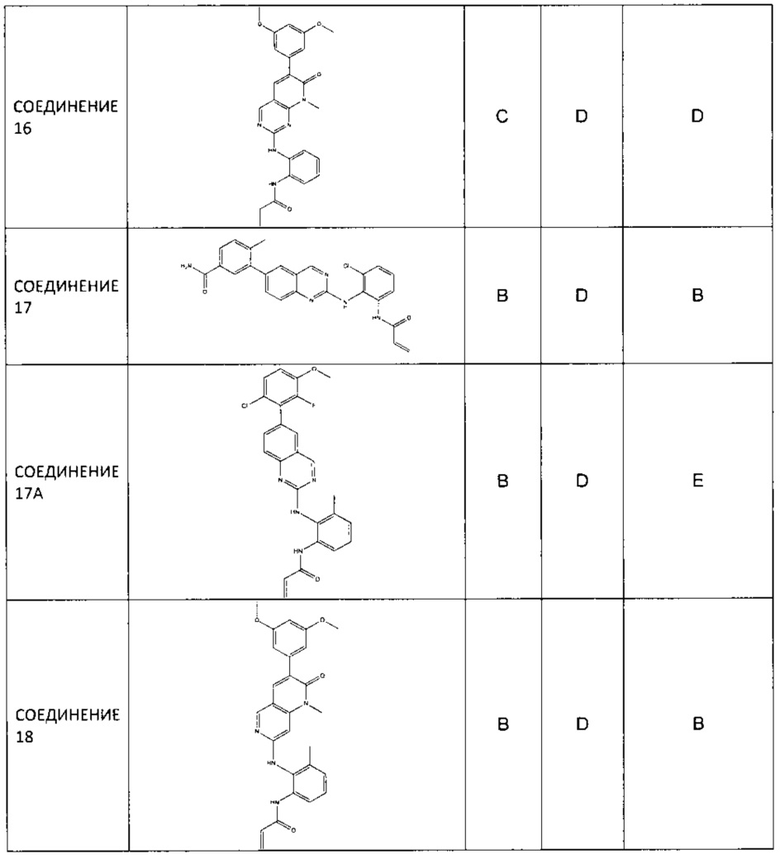
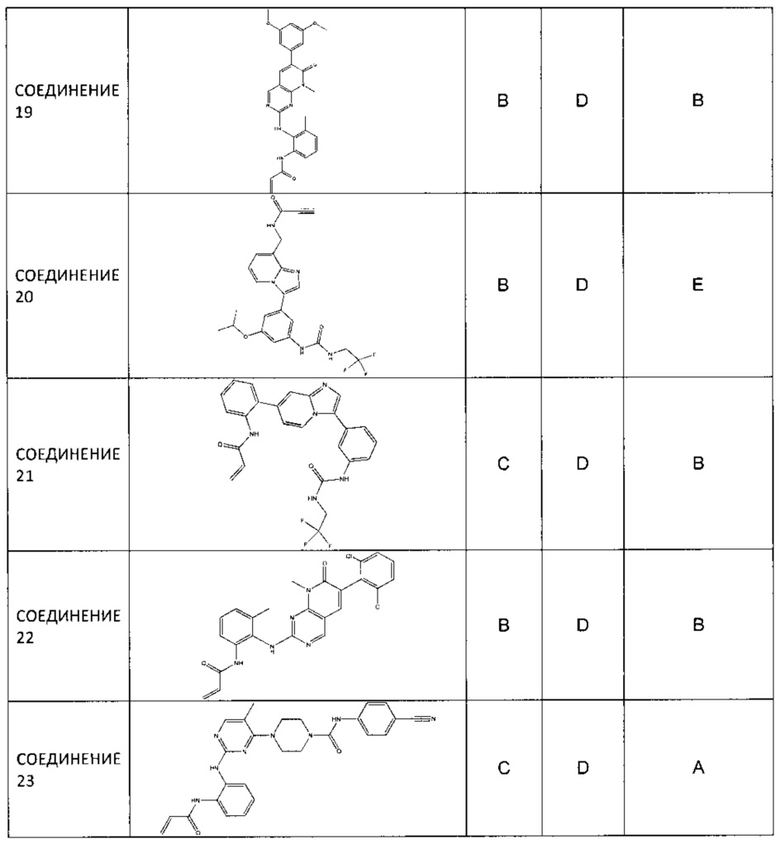
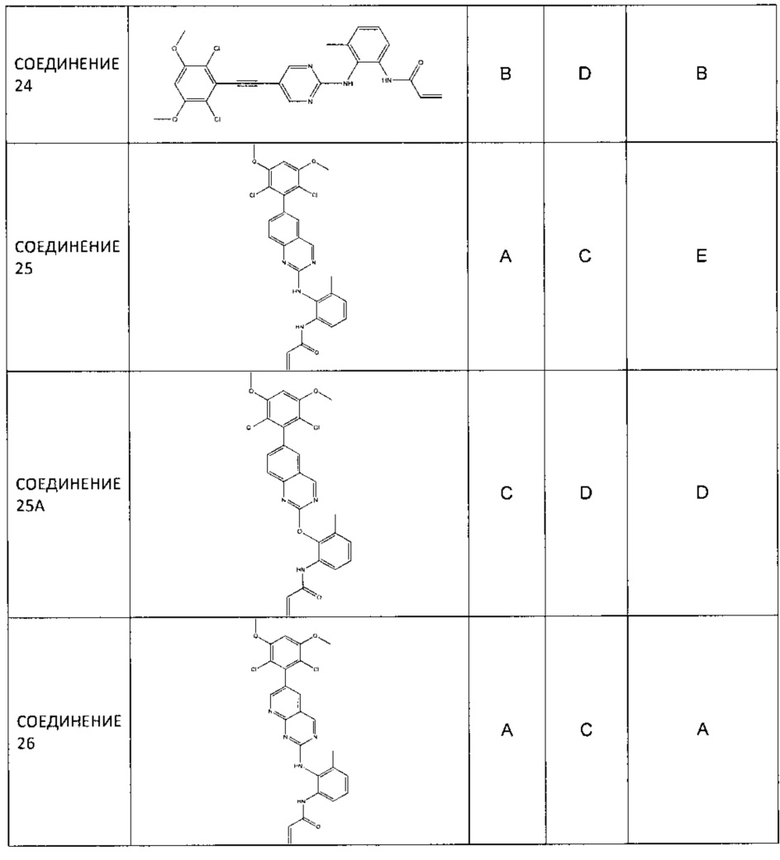
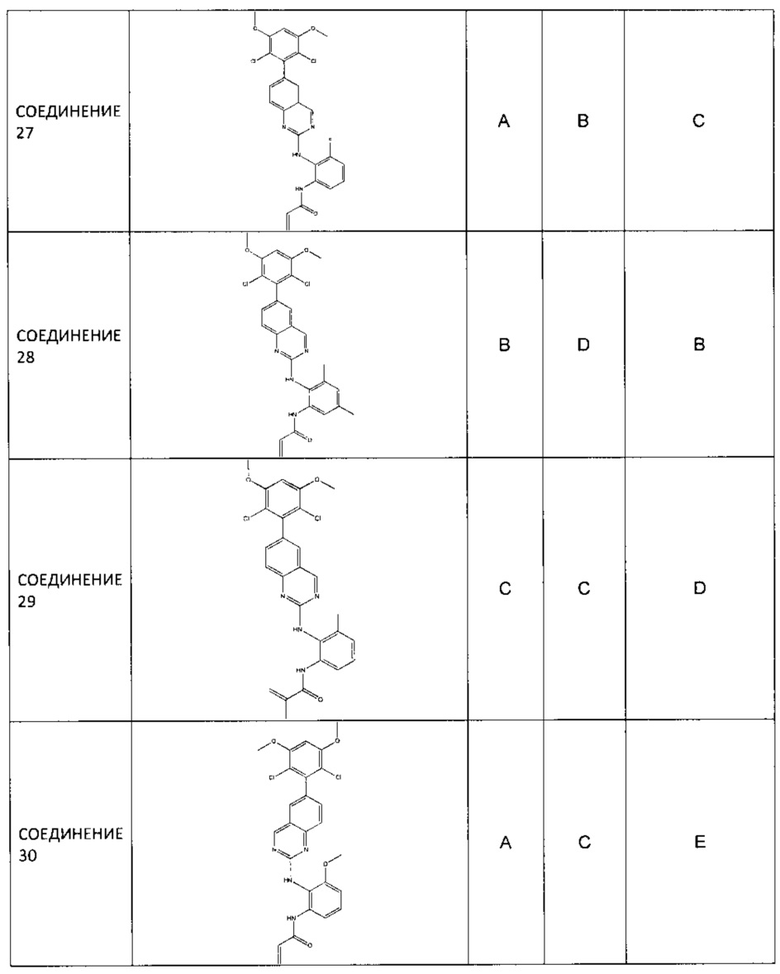


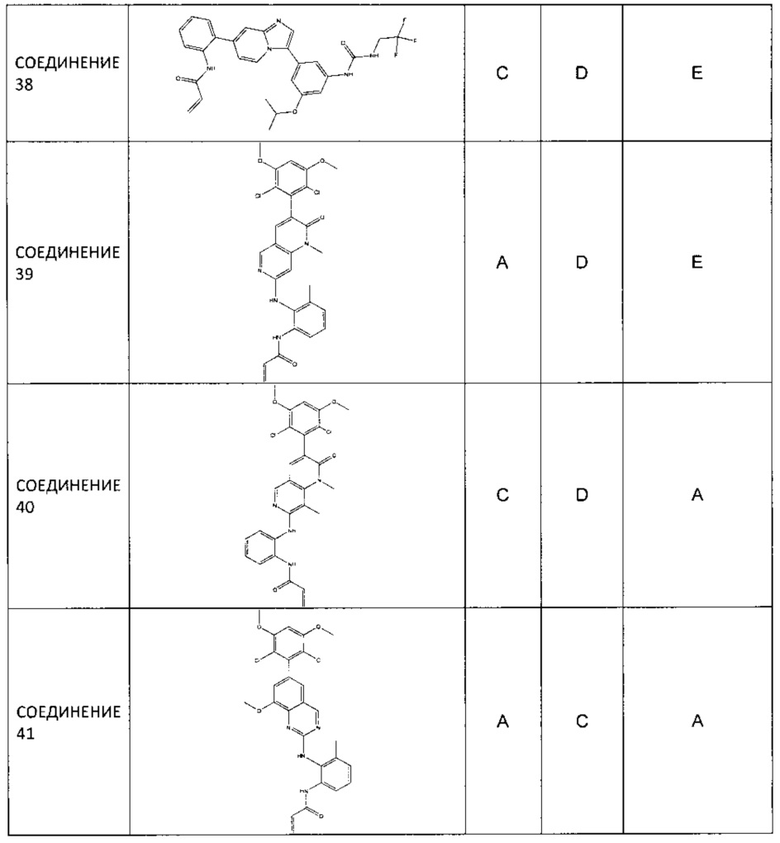
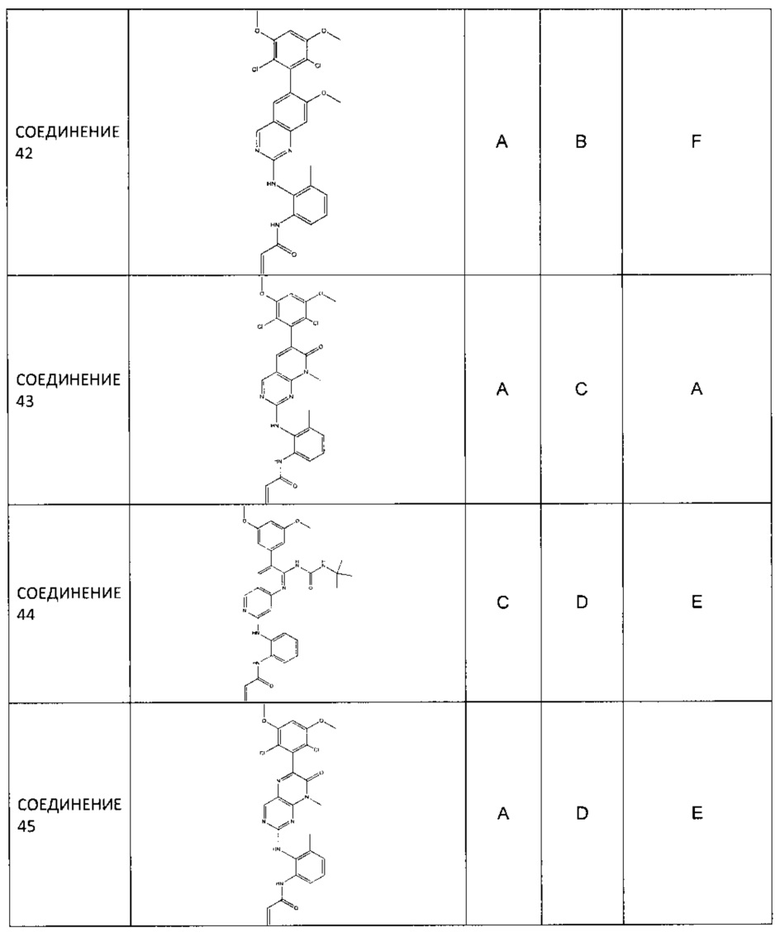
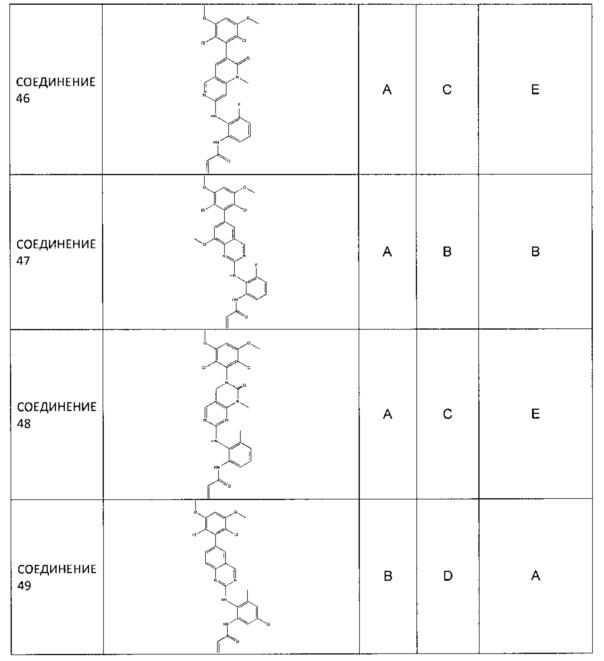

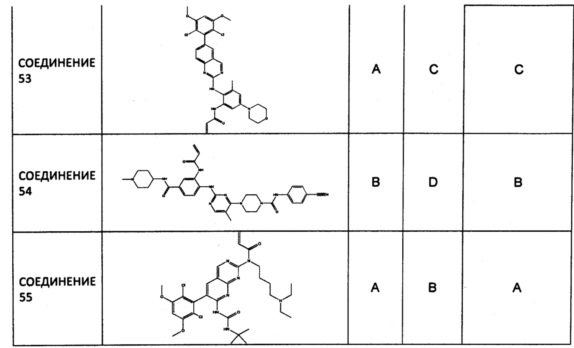
В представленной выше таблице, для FGFR1 и FGFR4: «А» означает, что IC50 меньше 10 нМ; «В» означает, что IC50 больше или равна 10 и меньше 100 нМ; «С» означает, что IC50 больше или равна 100 и меньше 1000 нМ; «D» означает, что IC50 больше, чем 1000 нМ.
Для соотношения: «F» означает, что соотношение [IC50 для FGFR1]/[IC50 для FGFR4] меньше 10; «Е» означает, что это соотношение ≥10 и <50; «D» означает, что это соотношение ≥50 и <100; «С» означает, что это соотношение ≥100 и <200; «В» означает, что это соотношение >200 и <500; «А» означает, что это соотношение >500. Чем выше указанное соотношение, тем более селективно соединение для FGFR4, по сравнению с FGFR1.
Клеточная эффективность
Эффект дозы в клетках MDA-MB-453, которые несут мутации активации FGFR4, 15 измеряли следующим образом. Вкратце, клетки MDA-MB-453 высеивали при концентрации 2,5×106 клеток на 6 лунок и истощали в течение ночи. Соединения добавляли в различных концентрациях (3000, 1000, 300, 100 и 30 нМ) на 1 час. Образцы собирали и лизировали для иммуноблот-анализа. Измеряли фосфорилирование Erk, а среднее для трех повторов значение pErk наносили на график с трехпараметрическим построением кривой зависимости эффекта (ингибирования) от дозы с помощью программы Prism GraphPad, которую использовали для определения значений IC50. Данные представлены ниже в таблице.
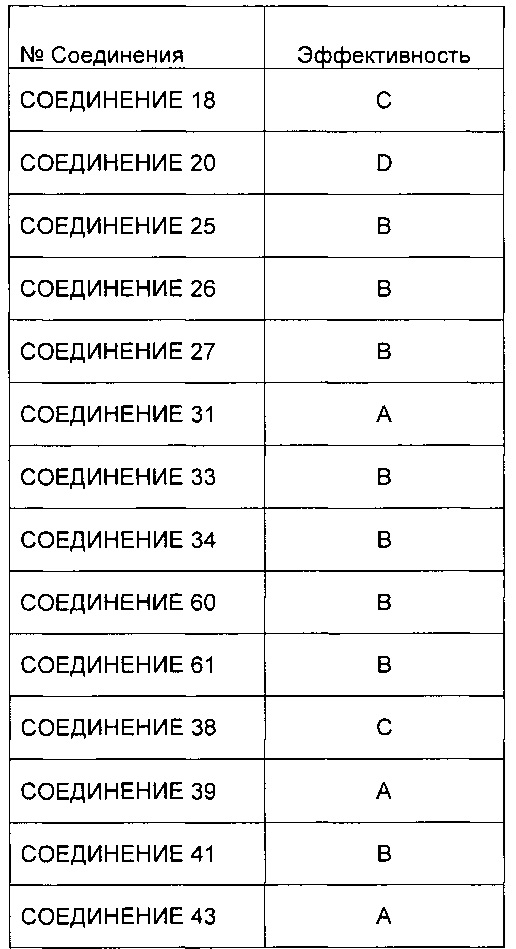
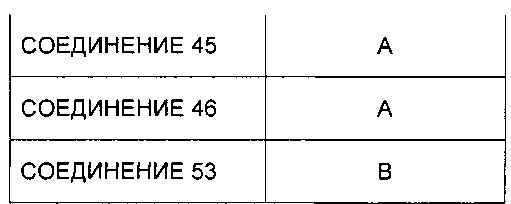
В этой таблице «А» означает IC50<1 нМ; «В» означает IC50≥1 и <10 нМ; «С» означает IC50≥10 и <100 нМ; «D» означает IC50≥100 нМ.
Эти данные указывают, что ингибирование FGFR-4 указанными соединениями приводит к блокированию нисходящей передачи онкогенных сигналов.
Инициация апоптоза при помощи ингибитора FGFR4
Клетки Нер3В высеивали в количестве 20 тыс.на лунку в 96-луночных белых планшетах в 200 мкл DMEM/5% FBS на ночь. На следующий день добавляли соединение с конечной концентрацией в ДМСО 0,1% и инкубировали в течение 6 часов. Активность каспазы измеряли по инструкциям производителя (анализ Caspase-Glo3/7 (Promega)). Вкратце, 100 мкл реагента Caspase-Glo3/7 добавляли в каждую лунку и инкубировали в течение 1 часа в темноте. Люминесценцию измеряли при помощи EnVision. Среднюю для 2 повторов активность каспазы наносили на график с трехпараметрическим построением кривой зависимости ответа (ингибирования) от дозы при помощи программы Prism GraphPad, которую использовали для определения значений IC50. Как показано на Фиг. 3, в клетках Нер3В обработка СОЕДИНЕНИЕМ 25 в течение 6 часов привела к эффективной инициации апоптоза. BGJ398, ингибитор пан-FGFR, также приводит к инициации апоптоза, хотя и при более высокой концентрации.
Ковалентность
Данные о том, что СОЕДИНЕНИЕ 52 ковалентно связывается с FGFR-4, представлены масс-спектрометрическими данными, изображенными на Фиг. 1. В 60 мкл буфера 300 мкМ Соединения 1 инкубировали с 50 мкг (75 мкМ) рекомбинантного FGFR-4 дикого типа с GST-меткой (Carna Biosciences) в течение 3 часов при комнатной температуре, а затем при 4°С в течение 13 часов. Затем комплекс белка-ингибитора обессоливали при помощи колонок для удаления детергентов Pierce (Thermo Pierce). Не модифицированный белок и комплекс белка-ингибитора анализировали электроспрей масс-спектрометрией для определения их соответствующих молекулярных масс. На Фиг. 1а показана масса не модифицированного белка. Как показано, основной релевантный пик имеет массу 65468,371 дальтон. На Фиг. 1b показана масса комплекса белка-ингибитора. Как показано, основной релевантный пик имеет массу 66043,5123 дальтон. Разность между этими массами составляет 575,1252, что находится в пределах инструментальной погрешности массы Соединения 1, составляющей 577,34 дальтон.
Массы комплексов белка-ингибитора, FGFR-4 и СОЕДИНЕНИЯ 11, СОЕДИНЕНИЯ 20 и СОЕДИНЕНИЯ 54, представлены на Фиг. 2. CR9 представляет собой пик для белка FGFR4. Как показывает пик CR3, указанный комплекс демонстрирует сдвиг +441 дальтон, тогда как молярная масса соединения (СОЕДИНЕНИЯ 11) составляла 444,6 (в пределах инструментальной погрешности). В другом примере указанный комплекс продемонстрировал сдвиг +470 дальтон (пик CR2), при этом молярная масса соединения (СОЕДИНЕНИЯ 20) составляла 473,4. В другом примере комплекс продемонстрировал сдвиг +631 дальтон (пик CR1), когда молярная масса соединения (СОЕДИНЕНИЯ 54) составляла 622,7.
Это демонстрирует, что соединения в широком диапазоне структур способны образовывать ковалентные комплексы с FGFR4.
Связывание с Cvs552
Кристаллическая структура СОЕДИНЕНИЯ 52, связанного с FGFR-4, представлена на Фиг. 4. Как показано, СОЕДИНЕНИЕ 52 связывается с цистеином у 552 остатка FGFR-4.
Кристаллическая структура СОЕДИНЕНИЯ 25, связанного с FGFR-4, представлена на Фиг. 5. Как показано, СОЕДИНЕНИЕ 25 также связывается с цистеином у 552 остатка FGFR-4.
Данные об эффективности in vivo
Исследовали влияние СОЕДИНЕНИЯ 25, BGJ398 (ингибитор пан-FGFR) и сорафениба на подавление роста опухоли в модели подкожного ксенотрансплантата клеток рака печени Нер3В с различными дозами.
Использовали шесть самок «голых» мышей (Mus Musculus) возрастом от 6 до 8 недель. Выращивание и инокуляция культуры опухолевых клеток: клетки Нер3В выращивали в среде ЕМЕМ (Invitrogen, США) с добавлением 10% FBS (Gibco, Австралия). Клетки собрали при 90% слиянии и жизнеспособности не менее 90%. В начале исследования мышам подкожно (s.c.) имплантировали 200 мкл 10×106 клеток Нер3В в 50% матригеля в правый бок.
Группирование животных и схема дозирования: через десять дней после имплантации клеток, когда опухоли достигли среднего объема 199 мм3, выбрали 45 мышей по объему опухоли и случайным образом разделили на 5 экспериментальных групп (n=9). День рандомизации обозначили как D0 и с этого дня начали лечение.
Измерение объема опухоли и массы тела: размер опухоли измеряли два раза в неделю в двух направлениях при помощи штангенциркуля, а объем выражали в мм3 по формуле: V=0,5 а×b2, где а и b представляют собой длинный и короткий диаметры опухоли, соответственно. Массу тела измеряли по меньшей мере дважды в неделю.
Окончание части in vivo: в каждой экспериментальной группе у 3 мышей собрали кровь, опухоли и печени через 4, 12 и 24 часа после последней дозы. Левую долю печени собрали для фармакодинамических (ФД) испытаний, остальную часть печени хранили в формалине для гистологии. Небольшие опухоли были приоритетными для использования в фармакокинетических испытаниях. Все остальные опухоли фиксировали для первоначального выполнения гистологического анализа, а затем выполнили быструю заморозку для ФД исследования.
Объемы опухолей для «голых» мышей с Нер3В: Фиг. 5 представляет собой линейный график, изображающий подавление роста в группах, обработанных СОЕДИНЕНИЕМ 25 (100 мг/кг РО BID (перорально, дважды в день)), СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID), BGJ398 (20 мг/кг РО QD (перорально, один раз в день)) и сорафенибом (30 мг/кг РО QD), по сравнению с ксенотрансплантированными опухолями Нер3В у «голых» мышей. Статистически значимое снижение объемов опухоли наблюдали в экспериментальных группах с СОЕДИНЕНИЕМ 25 (100 мг/кг РО BID), СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID) и сорафенибом (30 мг/кг РО QD), по сравнению с группой носителя, все испытания выполнены начиная с 4 дня после первого введения соединений и до конца (19 дней). Однако во время всего исследования не наблюдали существенного различия объема опухоли между группами с BGJ398 (20 мг/кг РО QD) и с носителем. Повышение дозы СОЕДИНЕНИЯ 25 с 100 мг/кг до 300 мг/кг увеличило эффективность подавления опухоли. Опухоли в обеих группах, обработанных СОЕДИНЕНИЕМ 25 (100 мг/кг РО BID) и СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID), регрессировали, а опухоли в группе, обработанной СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID) почти исчезли. В этом исследовании группы, обработанные СОЕДИНЕНИЕМ 25 (100 мг/кг РО BID) и СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID), показали превосходство при подавлении роста опухоли.
Изменение массы тела (%) «голых» мышей с Нер3В: Фиг. 7 представляет собой линейный график, изображающий изменение массы тела (%) во время всего экспериментального периода. Все мыши, за исключением мышей в группе, обработанной СОЕДИНЕНИЕМ 25, продемонстрировали значительное снижение массы тела. Масса тела мышей в группе носителя снизилась приблизительно на 10% на 10 день после введения опухоли. Этот результат показывает, что СОЕДИНЕНИЕ 25 хорошо переносится в рассматриваемых дозах и при указанной схеме введения у «голых» мышей, а также что СОЕДИНЕНИЕ 25 может облегчать снижение массы тела за счет подавления роста опухоли.
Мыши, обработанные СОЕДИНЕНИЕМ 25 (100 мг/кг РО BID), СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID) и сорафенибом (30 мг/кг РО QD), в течение всего исследования демонстрировали существенное снижение объема опухоли, по сравнению с группой носителя. Увеличение дозы СОЕДИНЕНИЯ 25 с 100 мг/кг до 300 мг/кг увеличило эффективность подавления опухоли. Опухоли у мышей в обеих группах, обработанных СОЕДИНЕНИЕМ 25 (100 мг/кг РО BID) и СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID), регрессировали, а опухоли в группе, обработанной СОЕДИНЕНИЕМ 25 (300 мг/кг РО BID) почти исчезли. Все мыши, за исключением мышей в группе, обработанной СОЕДИНЕНИЕМ 25, потеряли значительную часть массы тела. Масса тела мышей в группе с носителем снизилась приблизительно на 10% на 10 день после введения опухоли. Эти результаты показывают, что СОЕДИНЕНИЕ 25 хорошо переносится в рассматриваемых дозах и при указанной схеме введения у «голых» мышей, а также что СОЕДИНЕНИЕ 25 может облегчать снижение массы тела за счет подавления роста опухоли.
Включение посредством ссылки
Все публикации и патенты, упомянутые в настоящем документе, включены в настоящий документ посредством ссылки в полном объеме, как если бы каждая отдельная публикация или патент был специально и отдельно указан как включенный посредством ссылки.
Эквиваленты
Специалисты в данной области техники при помощи не более чем стандартных экспериментов могут определить или выявить многочисленные эквиваленты конкретных вариантов реализации настоящего изобретения, описанных в настоящем документе. Такие эквиваленты считаются входящими в рамки следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2014 |
|
RU2704112C2 |
| ИНГИБИТОРЫ FGFR4 | 2014 |
|
RU2715708C2 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2732127C1 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
| КОМПОЗИЦИИ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С KIT | 2014 |
|
RU2706235C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |

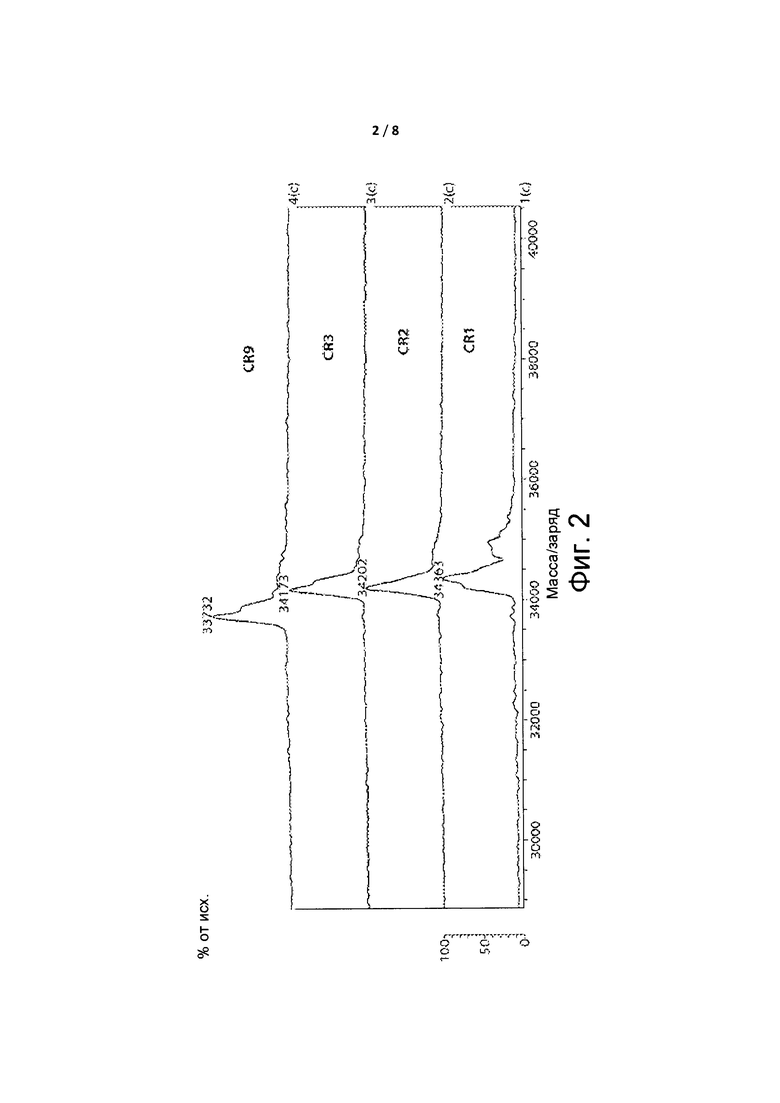
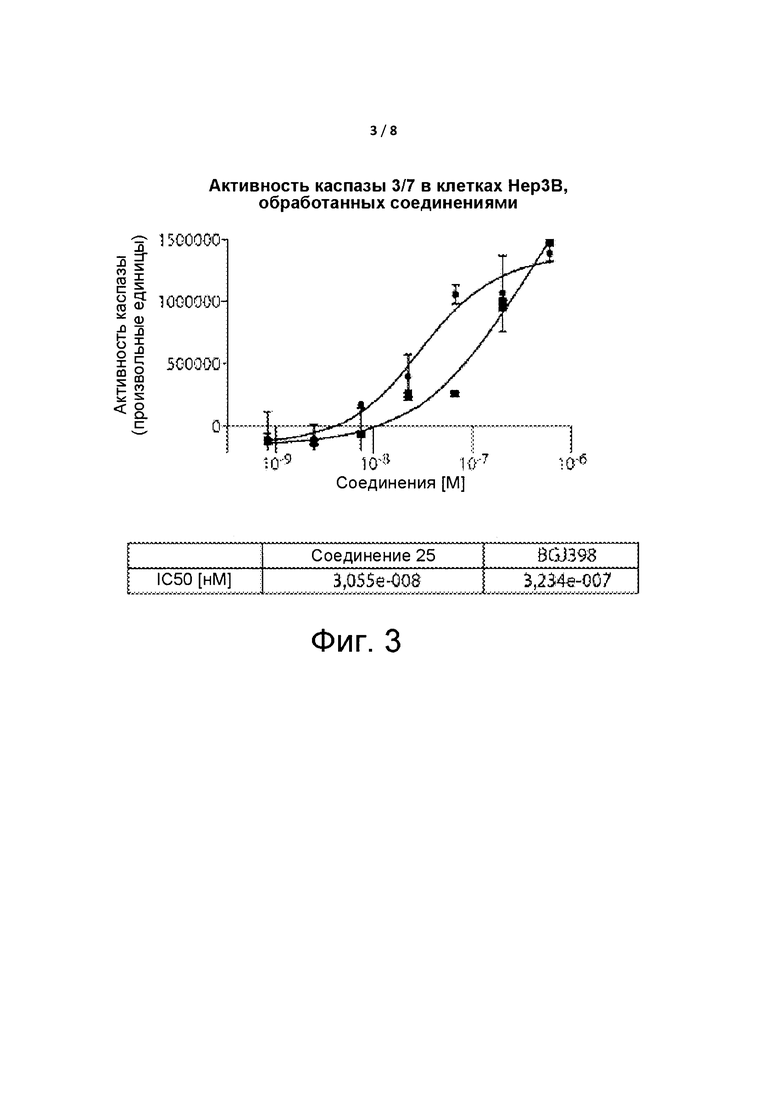

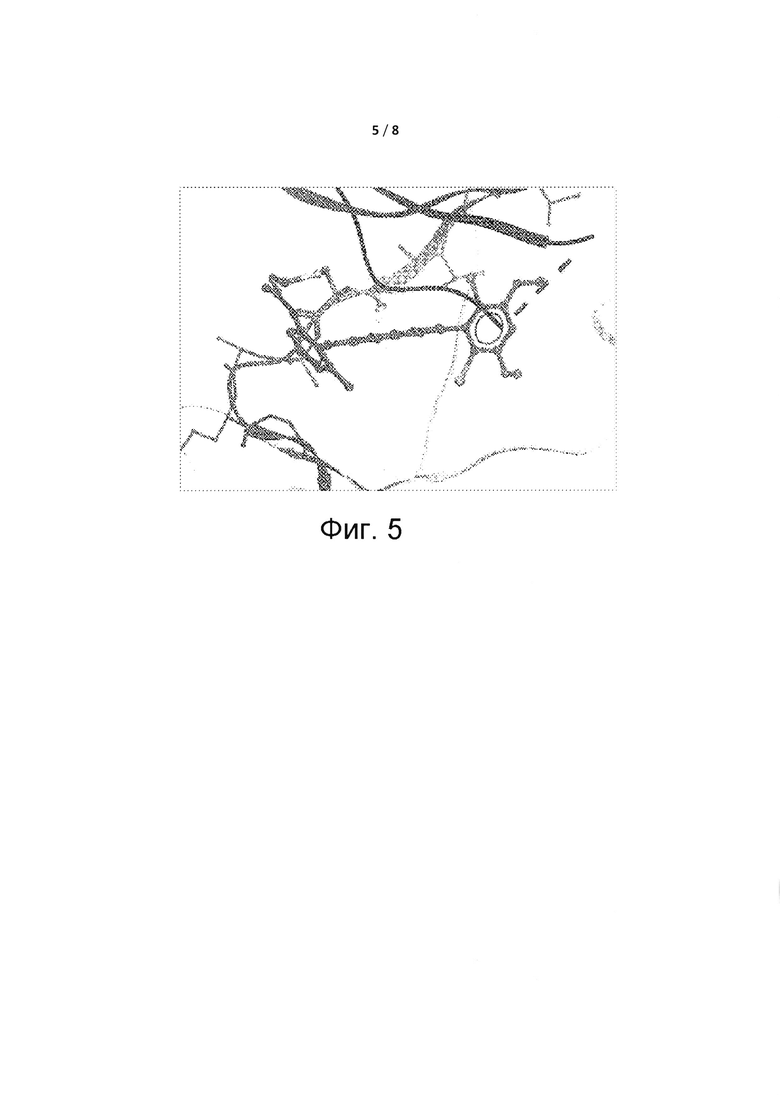
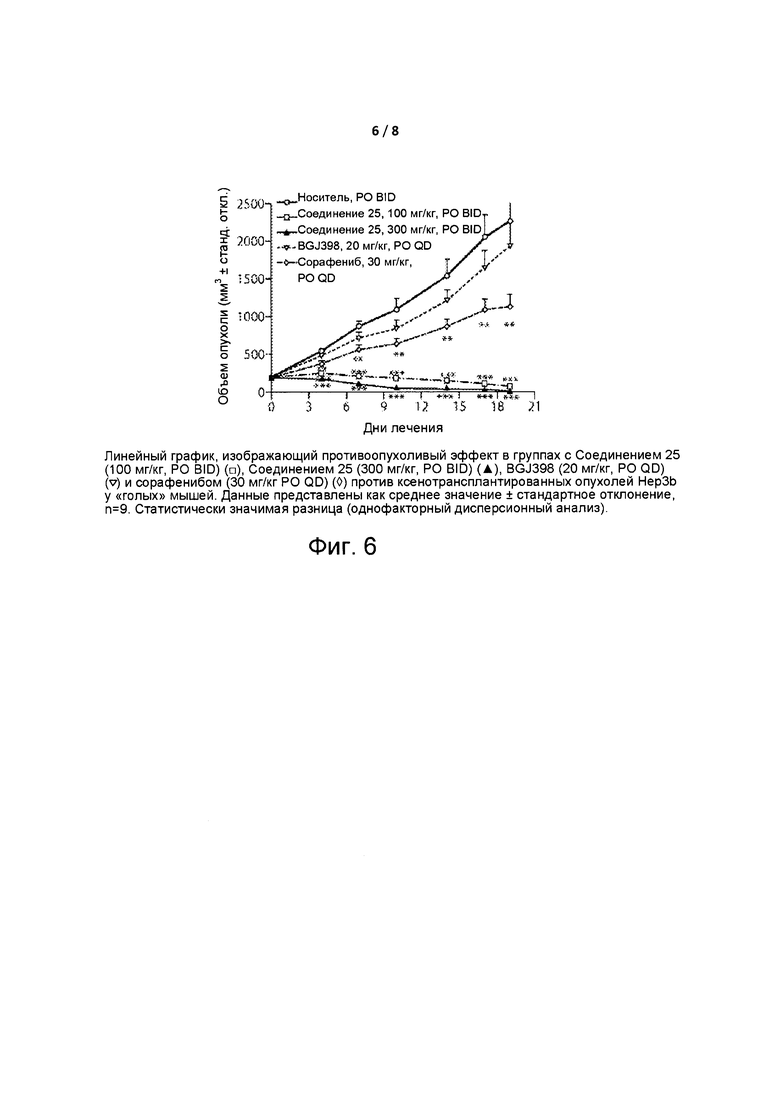
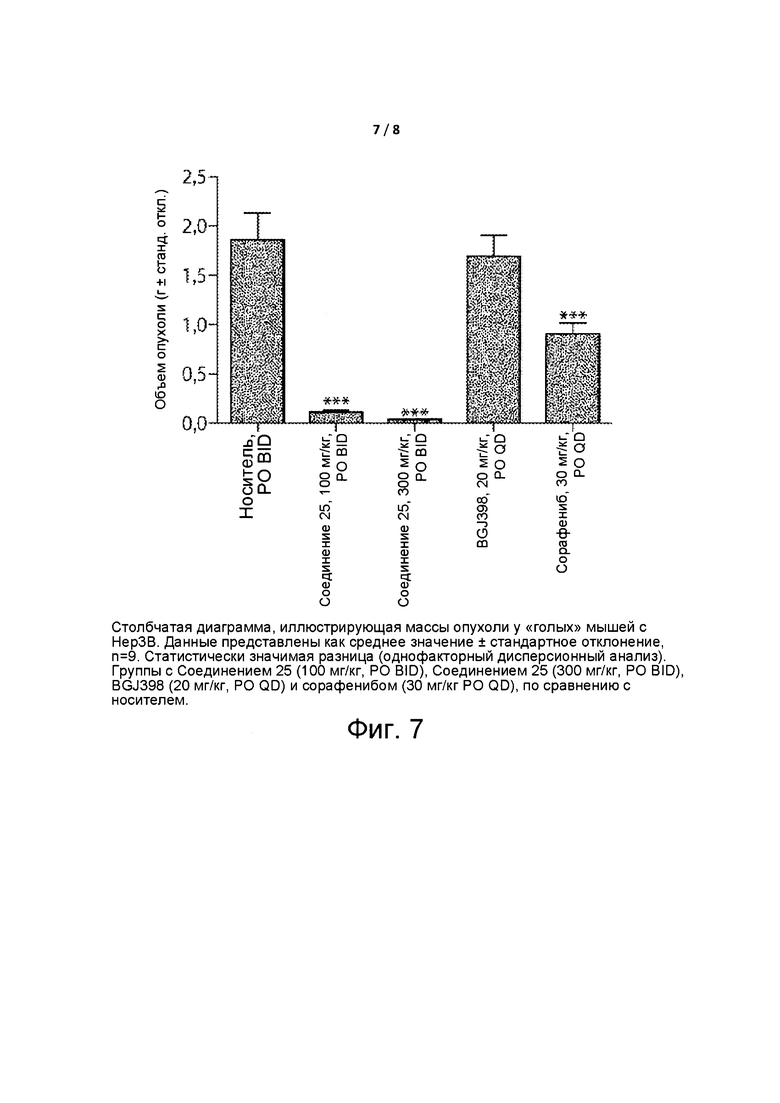
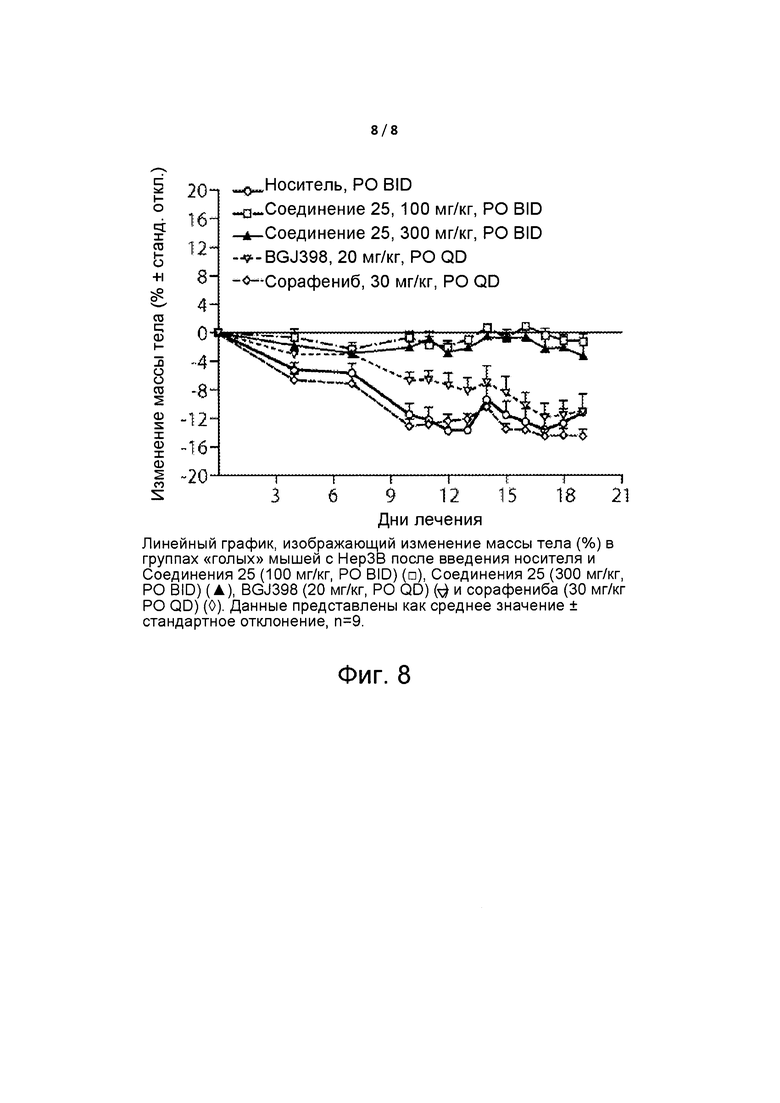
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы I, где кольцо A представляет собой фенил; X представляет собой CH или N; Y представляет собой CH, N или N-R4, где R4 представляет собой метил; L представляет собой связь; каждый из R1-R3 независимо представляет собой галоген, метил, метокси, оксо, CF3 или незамещенный морфолин; m равен 0-2; n равен 4; p равен 0-2; и головная часть представляет собой 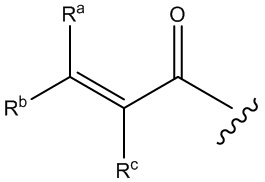 , где каждый из Ra, Rb и Rc независимо представляет собой H или метил. Также изобретение относится к конкретным соединениям, фармацевтической композиции на основе соединения формулы I или конкретного соединения, их применению и способу лечения указанных заболеваний. Технический результат: получены новые гетероциклические соединения, обладающие ингибирующей активностью в отношении рецептора фактора роста фибробластов 4 (FGFR-4). 11 н. и 9 з.п. ф-лы, 9 ил., 17 пр.
, где каждый из Ra, Rb и Rc независимо представляет собой H или метил. Также изобретение относится к конкретным соединениям, фармацевтической композиции на основе соединения формулы I или конкретного соединения, их применению и способу лечения указанных заболеваний. Технический результат: получены новые гетероциклические соединения, обладающие ингибирующей активностью в отношении рецептора фактора роста фибробластов 4 (FGFR-4). 11 н. и 9 з.п. ф-лы, 9 ил., 17 пр.

1. Соединение Формулы I:
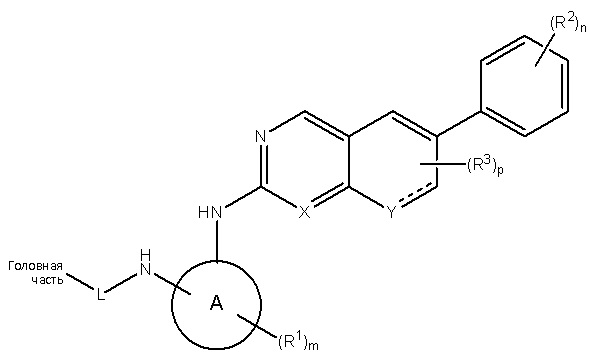
Формула I
где
кольцо A представляет собой фенил;
X представляет собой CH или N;
Y представляет собой CH, N или N-R4, где R4 представляет собой метил;
L представляет собой связь;
каждый из R1-R3 независимо представляет собой галоген, метил, метокси, оксо, CF3 или незамещенный морфолин;
m равен 0-2;
n равен 4;
p равен 0-2; и
головная часть представляет собой
где
каждый из Ra, Rb и Rc независимо представляет собой H или метил.
2. Соединение по п. 1, отличающееся тем, что X представляет собой N, и Y представляет собой CH.
3. Соединение по п. 1, отличающееся тем, что два R2 представляют собой хлор, и два R2 представляют собой метокси.
4. Соединение по п. 1, отличающееся тем, что каждый R2 независимо представляет собой галоген или метокси.
5. Соединение по п. 1, отличающееся тем, что R1 представляет собой метил.
6. Соединение по п. 1, отличающееся тем, что головная часть представляет собой  .
.
7. Соединение, выбранное из группы:
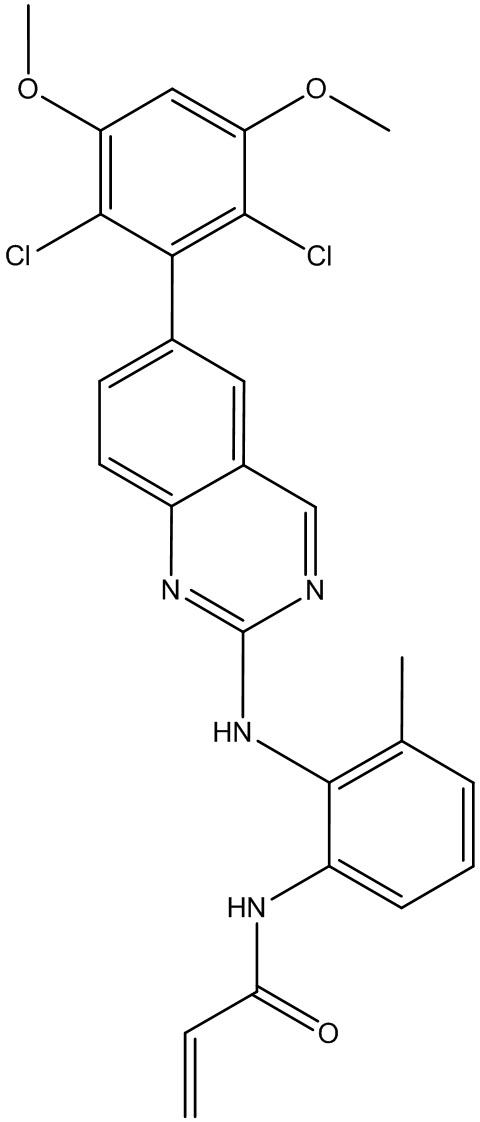
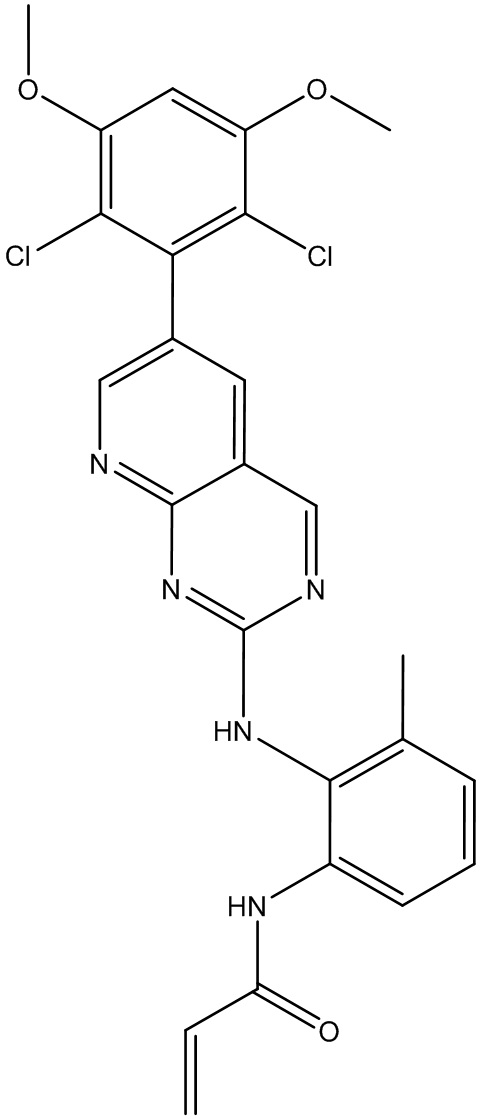
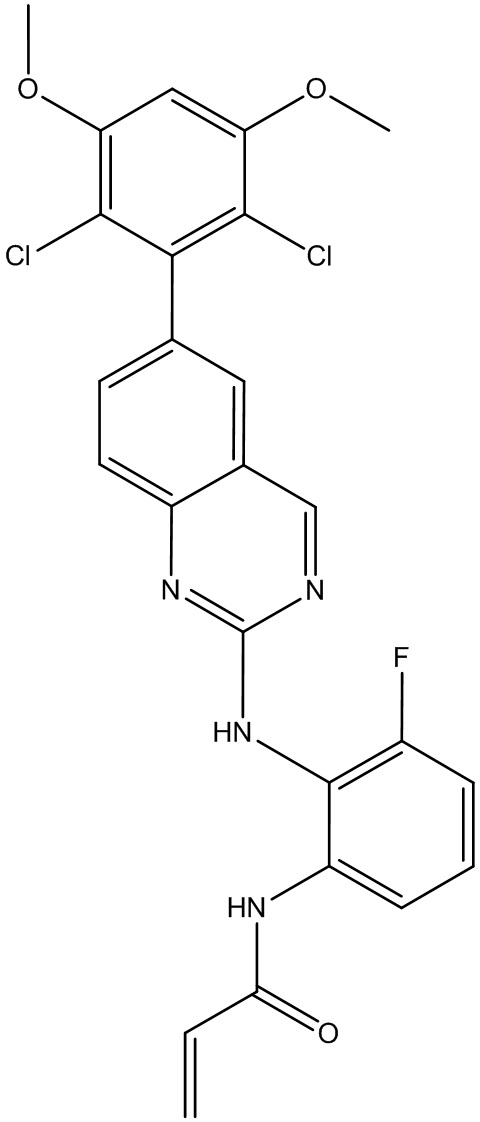
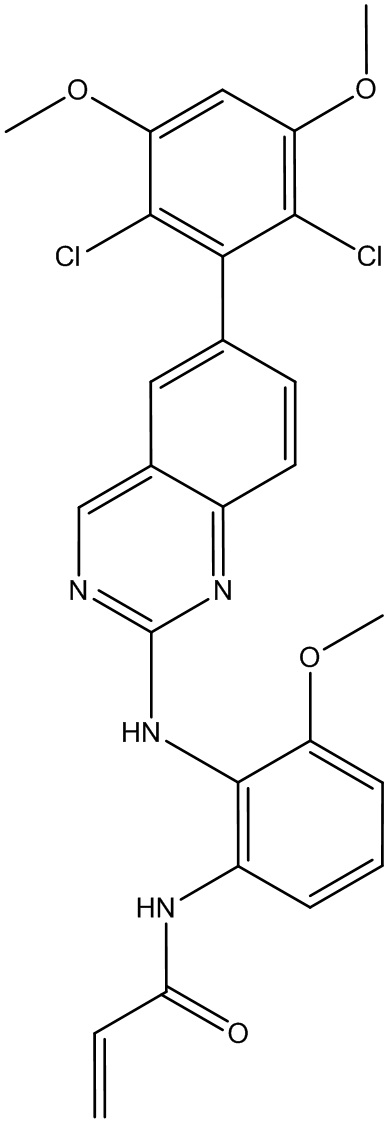
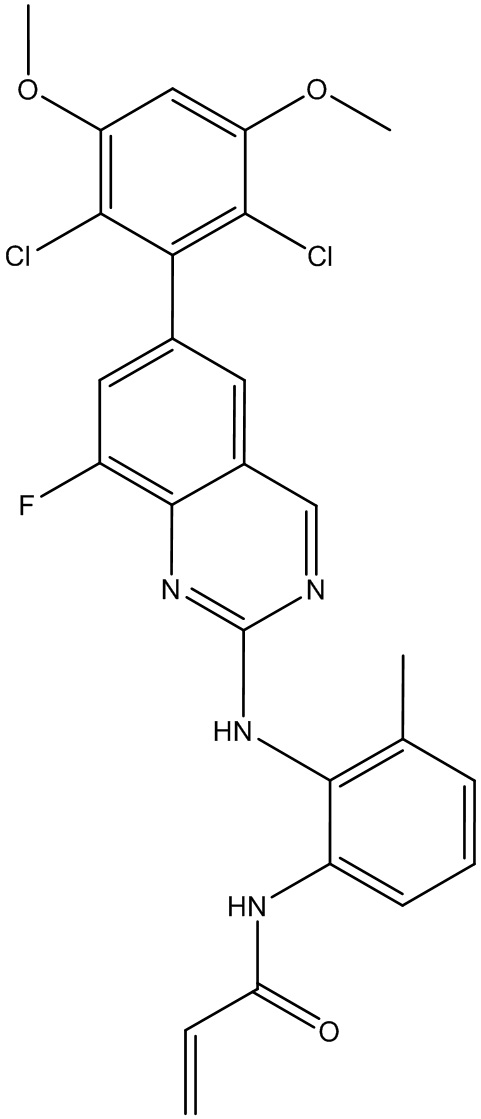
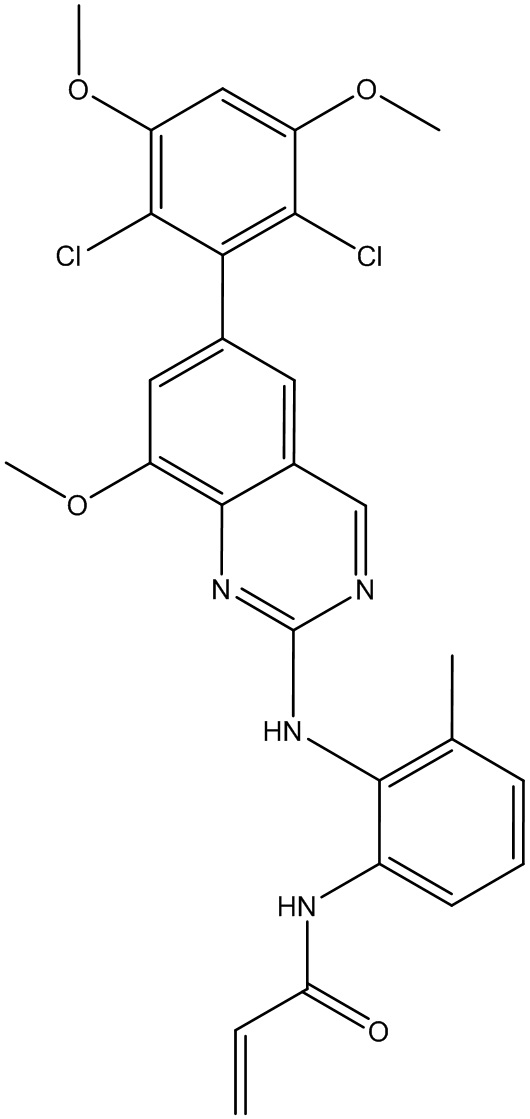

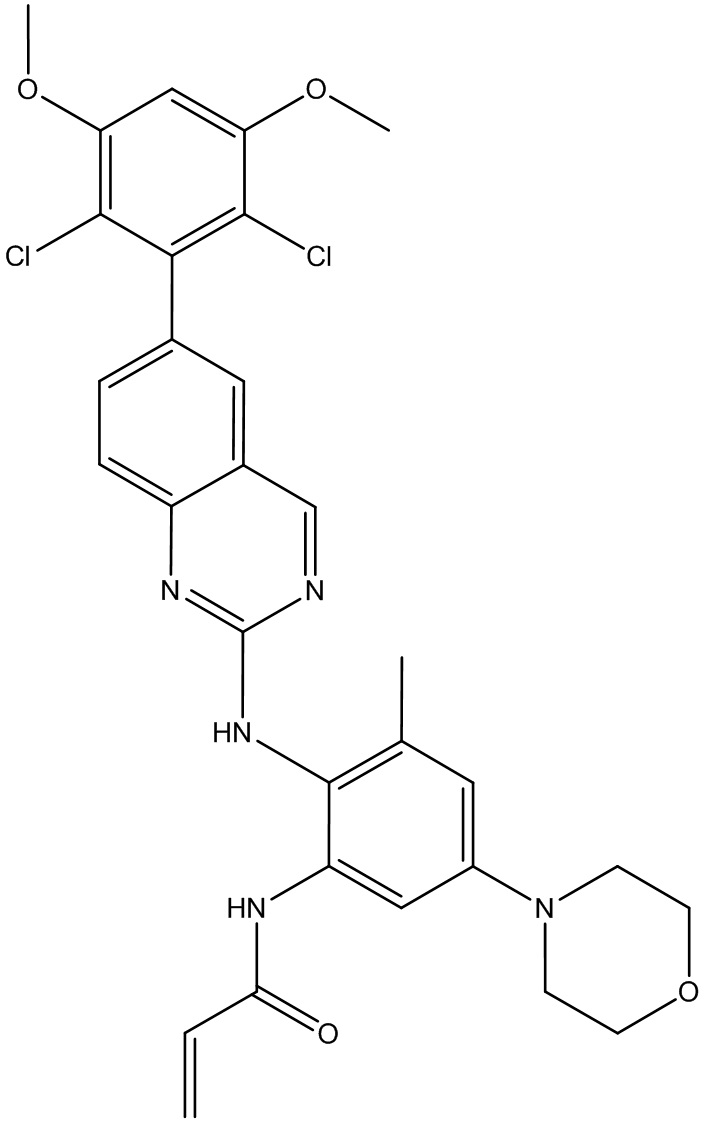
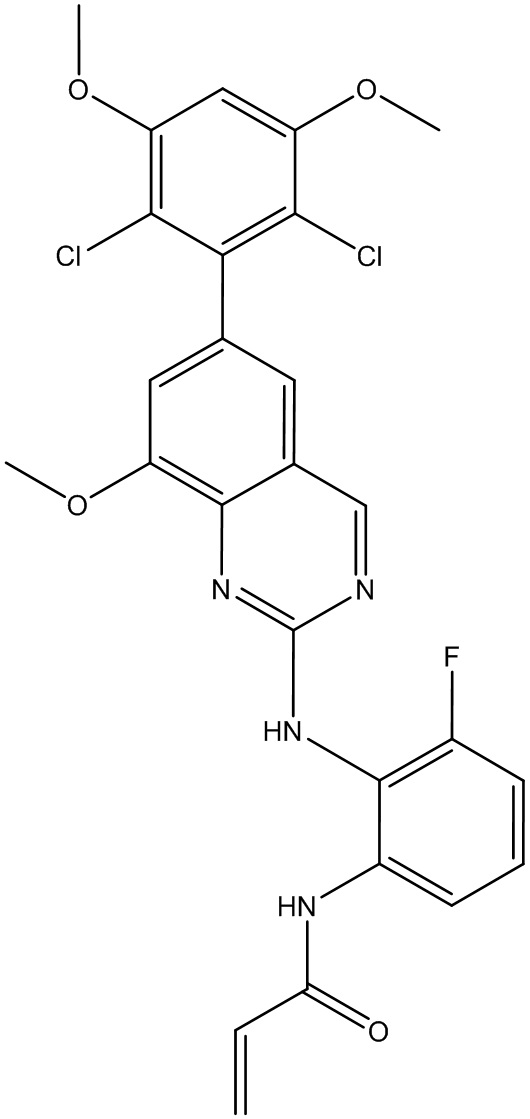
 и
и  .
.
8. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении рецептора фактора роста фибробластов 4 (FGFR-4), содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп. 1-7.
9. Способ лечения состояния, опосредованного FGFR-4, где способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7.
10. Способ лечения состояния, характеризующегося сверхэкспрессией FGFR-4, где способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7.
11. Способ лечения гепатоцеллюлярной карциномы, где способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7.
12. Способ лечения рака молочной железы, рака яичников, рака легких, саркомы, рака печени или гиперлипидемии, где способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7.
13. Соединение по любому одному из пп.1-7 для применения в способе лечения состояния, опосредованного FGFR-4.
14. Соединение по любому одному из пп.1-7 для применения в способе лечения состояния, характеризующегося сверхэкспрессией FGFR-4.
15. Соединение по любому одному из пп.1-7 для применения в способе лечения гепатоцеллюлярной карциномы.
16. Соединение по любому одному из пп.1-7 для применения в способе лечения рака молочной железы, рака яичников, рака легких, саркомы, рака печени или гиперлипидемии.
17. Применение соединения по любому из пп. 1-7 для производства лекарственного средства для лечения состояния, опосредованного FGFR-4.
18. Применение соединения по любому из пп. 1-7 для производства лекарственного средства для лечения состояния, характеризующегося сверхэкспрессией FGFR-4.
19. Применение соединения по любому из пп. 1-7 для производства лекарственного средства для лечения состояния, представляющего собой гепатоцеллюлярную карциному.
20. Применение соединения по любому из пп. 1-14 для производства лекарственного средства для лечения рака молочной железы, рака яичников, рака легких, саркомы, рака печени или гиперлипидемии.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Zhou W | |||
| и др.: "A structure-guided approach to creating covalent FGFR inhibitors", CHEMISTRY & BIOLOGY, 17(3), стр.285-295, 2010 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| ПРОИЗВОДНЫЕ 6-АРИЛПИРИДО[2,3-D]-ПИРИМИДИНЫ И -НАФТИРИДИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ, ВЫЗЫВАЕМОЙ ПРОТЕИНОВОЙ ТИРОЗИНКИНАЗОЙ, И СПОСОБ ИНГИБИРОВАНИЯ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ | 1995 |
|
RU2191188C2 |
| EA 200900819 A1, 26.02.2010. | |||

