Изобретение относится к способу получения 3-пиридилметил аммоний бромидов и дальнейшего превращения этих соединений в гербицидные 5-замещенные-2-(2-имидазолин-2-ил)никотиновые кислоты, такие как имазамокс.
Производные 2-(2-имидазолин-2-ил) никотиновых кислот, такие как имазамокс (2-[(Р8)-4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил]-5-метоксиметилникотиновая кислота),

являются пригодными селективными гербицидами, которые действуют в качестве ALS-ингибиторов и могут использоваться для обработки перед- и после всходов.
Из литературы известны различные процессы для синтеза этих соединений, см., например, EP-A 0322616, EP-A 0747360, EP-A 0933362 или Q. Bi и др. Modern Agrochemicals 6(2) (2007) 10-14.
Несмотря на то что в промышленном масштабе можно осуществить синтез с помощью этих методов, они все еще нуждаются в улучшении, в особенности с учетом экономических и экологических аспектов, таких как улучшение суммарного выхода или избегание применения определенных растворителей или реагентов.
В EP-A 0548532 описано получение 5,6-дизамещенных-3-пиридилметил аммоний галогенидных соединений путем галогенирования соответствующих 5,6-дизамещенных-3-метил-пиридинов и последующей реакции с триалкиламином или циклическим ненасыщенным или насыщенным амином.
Галогенирующие агенты, предложенные в EP-A 0548532, включают N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантоин, бром, хлор, трет-бутилгипохлорит, сульфурил хлорид, сульфурил бромид, N-хлорсукцинимид и другие; однако все примеры осуществляют либо с N-бромсукцинимидом, либо с 1,3-дибром-5,5-диметилгидантоином.
Одной задачей изобретения является обеспечение улучшенного способа галогенирования 5,6-дизамещенных-3-метил-пиридинов. Дальнейшей задачей изобретения является обеспечение улучшенного способа получения 5,6-дизамещенных-3-пиридилметил аммоний бромидов (I) и дальнейшего превращения этих соединений в гербицидные 2-(2-имидазолин-2-ил)никотиновые кислоты или их производные.
Было обнаружено, что бромирование 5,6-дизамещенных-3-метилпиридинов и дальнейшая реакция с амином могут быть существенно улучшены путем применения специфических растворителей и брома в качестве бромирующего средства в двухфазной системе с водой.
В DE-A 3330604 описан процесс получения бромметилтиофеновых эфиров карбоновых кислот путем бромирования метилового соединения с бромом в условиях облучения в двухфазной системе, содержащей воду и фторхлоруглеводород. Тем не менее в этой ссылке не описаны специфические исходные продукты, растворители и условия реакций согласно изобретению.
Таким образом, в одном аспекте изобретения обеспечивается способ получения 5,6-дизамещенных-3-пиридилметил аммоний бромидов (I),

где
Q представляет собой третичный алифатический или циклический, насыщенный, частично ненасыщенный или ароматический амин;
Z представляет собой водород или галоген;
Z1 представляет собой водород, галоген, циано или нитро;
Y и Y1 каждый независимо представляют собой OR1, NR1R2, или где вместе YY1, представляют собой -O-, -S- или NR3-;
R1 и R2 каждый независимо представляют собой водород,
C1-C4 алкил, алкил, необязательно замещенный C1-C4 алкокси или фенилом, необязательно замещенным одной-тремя C1-C4 алкильными группами, C1-C4 алкокси группами или атомами галогена, или
фенил, необязательно замещенный одной-тремя C1-C4 алкильными группами, C1-C4 алкокси группами или атомами галогена;
R3 представляет собой водород или C1-C4 алкил;
который включает стадии:
(i) взаимодействие соединения формулы (II),
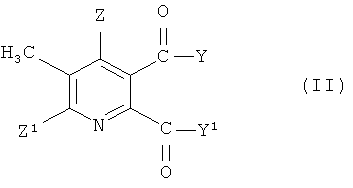
где символы имеют значения, указанные для формулы (I), с бромом в присутствии радикального инициатора в смеси растворителей, содержащей водную фазу и органическую фазу, где органическая фаза содержит растворитель, выбранный из 1,2-дихлорэтана, хлорбензола, 1,2-дихлорбензола, 1,3-дихлорбензола, 1,4-дихлорбензола и тетрахлорметана, и где значение рН водной фазы составляет от 3 до <8, получая 3-бромметил-5,6-дизамещенное пиридиновое соединение (III),

где Y, Y1, Z и Z1 имеют значения, указанные для формулы (I), и
(ii) взаимодействие бром соединения формулы (III) с третичным амином Q в растворителе при температуре в интервале от приблизительно 0°C до 100°C.
В дальнейшем аспекте изобретения обеспечивается способ получения 5,6-дизамещенного-3-метоксиметилпиридина формулы (IV),
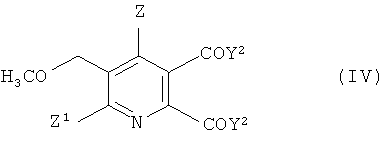
Z1 и Z2 имеют значения, указанные в формуле (I), и
Y2 представляет собой OCH3 или ОМ, и
M представляет собой щелочной металл, щелочноземельный металл
или H, предпочтительно щелочной металл или щелочноземельный металл,
который включает стадии:
(i)/(ii) получение 5,6-дизамещенного-3-пиридилметил аммоний бромида формулы (I), как описано выше, и
(iii) взаимодействие соединения формулы (I) в метаноле, толуоле или смесях метанол/толуол с основанием, выбранным из MOCH3 и МОН (если растворителем является метанол), где M имеет значение, указанное в формуле (IV), и необязательно подкисление полученного продукта (M=Н).
В дальнейшем аспекте изобретения обеспечивается способ получения гербицидного имидазолинонового соединения формулы (V),
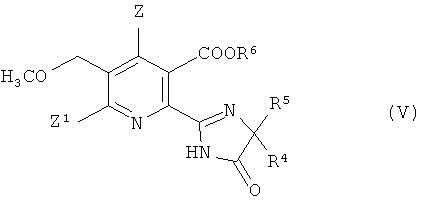
где
Z, Z1 имеют значения, указанные в формуле (I);
R4 представляет собой C1-C4 алкил;
R5 представляет собой C1-C4 алкил, C3-C6 циклоалкил, или R4 и R5, где вместе с атомом, к которому они присоединены, представляют собой C3-C6 циклоалкильную группу, необязательно замещенную метилом, и
R6 представляет собой водород; группу формулы -N=С(низший алкил)2;
C1-C12 алкил, необязательно замещенный одной из следующих групп: C1-С3 алкокси, галоген, гидроксил, C3-C6 циклоалкил, бензилокси, фурил, фенил, галофенил, низший алкилфенил, низший алкоксифенил, нитрофенил, карбоксил, низший алкоксикарбонил, циано или три низший алкиламмоний; С3-C12 алкенил, необязательно замещенный одной из следующих групп: C1-С3 алкокси, фенил, галоген или низший алкоксикарбонил, или двумя C1-С3 алкокси группами, или двумя галогеновыми группами; С3-C6 циклоалкил, необязательно замещенный одной или двумя C1-C3 алкильными группами; или
катион предпочтительно выбирают из группы, включающей щелочные металлы, щелочноземельные металлы, марганец, медь, железо, цинк, кобальт, свинец, серебро, никель, аммоний и органический аммоний;
который включает стадии:
(i)/(ii)/(iii) получение соединения формулы (IV), как описано выше,
(iv) превращение соединения формулы (IV) в гербицидное соединение формулы (V).
Способ в соответствии с изобретением обеспечивает более высокие выходы, более высокую производительность (более высокую объемную производительность, более низкие фиксированные расходы), более низкую стоимость сырьевых материалов (бром дешевле органических бромирующих агентов, таких как DBDMH) и улучшенную селективность для соединений формулы (I). Органическая фаза не содержит органических побочных продуктов из бромирующего агента (например, диметилгидантоина из DBDMH), которые должны быть отделены, например, путем дополнительной щелочной промывки. В частности, более высокая плотность растворителя 1,2-дихлорэтана предоставляет возможность выделения соединений формулы (I) в более высоком концентрированном водном растворе, который не содержит нерастворимых примесей.
Предпочтительными являются соединения формулы (I), в которых символы имеют следующие значения:
Z предпочтительно представляет собой водород или галоген;
Z1 предпочтительно представляет собой водород, галоген, циано или нитро;
Y и Y1 каждый независимо предпочтительно представляет собой OR1 или, где вместе YY1, предпочтительно представляет собой O;
R1 предпочтительно представляет собой независимо водород или C1-С4 алкил;
R3 предпочтительно представляет собой водород или C1-С4 алкил;
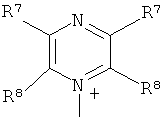
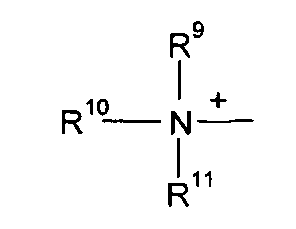
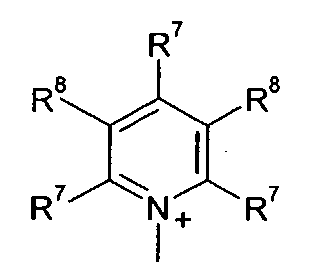
Q+ предпочтительно представляет собой
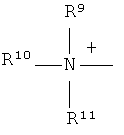 ;
; 
 ;
;  ;
; 
и
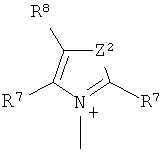
Z2 представляет собой О, S или NR12;
R12 представляет собой C1-C4 алкил;
R7 и R8 каждый независимо представляют собой водород, галоген, C1-C4 алкил или C1-C4 алкокси, или, где вместе, R7 и R8 образуют 5-ти или 6-ти членное насыщенное или ненасыщенное кольцо, необязательно прерванное О, S, или NR12 и необязательно замещенное одним-тремя атомами галогена, C1-C4 алкильными группами или C1-C4 алкокси группами, и
R9, R10 и R11 каждый независимо представляют собой C1-C4 алкил, или R9 и R10, где вместе, образуют 5-ти или 6-ти членное кольцо, в котором R9R10, представлен структурой:
-(CH2)n-, необязательно прерванное O, S или NR9, где n представляет собой целое число 3, 4 или 5, при условии, что R11 представляет собой C1-С4 алкил.
Предпочтительно, все символы в формуле (I) имеют предпочтительные значения.
Более предпочтительными являются соединения формулы (I), в которых символы имеют следующие значения:
Z и Z1 более предпочтительно представляют собой водород;
Y и Y1 более предпочтительно представляют собой OR1;
R1 более предпочтительно представляет собой C1-C4 алкил;
R3 более предпочтительно представляет собой водород или C1-C4 алкил;
Q+ более предпочтительно представляет собой ⊕NR9R10R11 или пиридиний;
R9, R10 и R11 более предпочтительно представляют собой каждый независимо C1-C4 алкил, или R9 и R10, где вместе, образуют 5-ти или 6-ти членное кольцо, в котором R9R10, представлен структурой: -(CH2)n-, необязательно прерванное О, S или NR12, где n представляет собой целое число или 3, 4 или 5, при условии, что R11 представляет собой C1-C4 алкил; более предпочтительно R9, R10 и R11 представляют собой C1-C4 алкил.
Более предпочтительными являются соединения формулы (I), в которых все символы имеют более предпочтительные значения.
Особенно предпочтительными являются формулы (I), в которых символы имеют следующие значения:
Z и Z1 особенно предпочтительно представляют собой водород;
Y и Y1 особенно предпочтительно представляют собой OR1;
R1 особенно предпочтительно представляет собой СН3;
Q особенно предпочтительно представляет собой ⊕NR9R10R11 или пиридиний;
R9, R10, R11 особенно предпочтительно представляют собой метил.
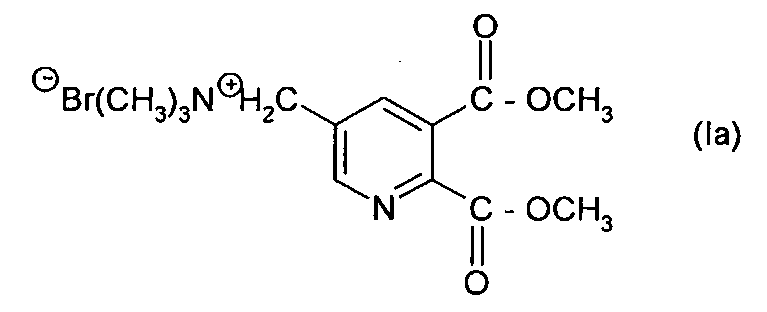
Таким образом, особенно предпочтительное соединение формулы (I) представляет собой соединение формулы (Ia):

Соответствующее пиридиниевое соединение (Ib) также является особенно предпочтительным.
Предпочтительные, более предпочтительные и особенно предпочтительные соединения формулы (II) представляют собой соединения, которые приводят к соответствующим соединениям формулы (I).
Соединения формулы (II) и их получение известно, например, из EP-A 0933362.
Молярное отношение пиридинового соединения (II) к брому обычно находится в интервале 1:0,5-1,2, предпочтительно 1:0,6-1,0, более предпочтительно 1:0,7-0,95.
Также представляется возможным работать с половиной эквивалентов брома и получать бром в реакционной смеси из HBr с окислителем, таким как H2O2.
Подходящими генераторами свободных радикалов для инициации реакции являются такие вещества, которые разлагаются при выбранной температуре реакции. Примерами предпочтительных инициаторов являются генераторы свободных радикалов, такие как азосоединения и пероксиды. Тем не менее также представляется возможным использовать окислительно-восстановительные системы, в особенности те, которые основываются на гидропероксидах, таких как гидропероксид кумена.
Радикальные инициаторы, подходящие для применения в способе согласно изобретению, включают 2,2'-азобисизобутиронитрил, 2,2'-азобис(2-метилбутаннитрил), 2,2'-азобис(2,4-диметил-пентаннитрил), 1,1'-азобис(циклогексанкарбонитрил), органические и неорганические пероксиды, такие как дилауроил пероксид водорода, пероксид, трет-бутилперокси-пивалат, бензоил пероксид и другие, где предпочтительными являются 2,2'-азобисизобутиронитрил, 2,2'-азобис(2-метилбутаннитрил) и дилауроил пероксид и где особенно предпочтительными являются 2,2'-азобисизобутиронитрил и 2,2'-азобис(2-метилбутаннитрил).
Молярное отношение инициатора к брому предпочтительно представляет собой в интервале 0,04-0,15:1, более предпочтительно 0,06-0,10:1.
Органический растворитель выбирают из группы, включающей 1,2-дихлорэтан, хлорбензол 1,2-дихлорбензол, 1,3-дихлорбензол, 1,4-дихлорбензол и тетрахлорметан, предпочтительно 1,2-дихлорэтан и хлорбензол. Особенно предпочтительным является 1,2-дихлорэтан. Также можно применять смеси, в частности дихлорбензолов.
Количество органического растворителя может изменяться в широких пределах. Предпочтительно применяют от 900 г до 2000 г, более предпочтительно от 1000 г до 1300 г, органического растворителя на моль соединения (II).
Реакционная смесь содержит органическую фазу и водную фазу. Количество водной фазы может изменяться в широких пределах. Предпочтительно применяют от 140 г до 500 г, более предпочтительно от 140 г до 300 г, в особенности от 150 г до 200 г воды на моль соединения формулы (II).
При осуществлении реакции значение рН водной фазы поддерживают в интервале от 3 до <8, предпочтительно от 3 до 7, более предпочтительно от 4 до 7. Контроль за значением pH можно осуществлять путем добавления подходящего основания, предпочтительно неорганического основания, такого как гидроксид щелочного металла, например NaOH, или щелочноземельный металл. Предпочтительным основанием является водный NaOH, особенно в разведенной форме (например, содержащий 5-20 мас.% NaOH).
Для осуществления желательного контроля значения рН, основание можно добавлять непрерывно при осуществлении реакции, или значение рН проверяют постоянно и основание добавляют с помощью присоединенного автоматизированного дозированного устройства.
В одном предпочтительном варианте осуществления стадию (i) реакции осуществляют путем растворения соединения (II) в органической фазе и добавления воды с образованием водной фазы.
Инициатор добавляют в виде чистого соединения или в раствор, при комнатной температуре (обычно при температуре в интервале от 22 до 25°C) или при температуре реакции после нагревания. В зависимости от температуры разложения инициатора часть или даже все количество инициатора следует добавлять перед началом дозирования брома. Количество инициатора, которое необходимо добавлять при добавлении брома, также зависит от температуры разложения. Минимальная концентрация свободных радикалов всегда должна быть доступной при реакции бромирования.
Для 2,2'-азобис(2-метилбутаннитрила) добавляют раствор с инициатором в органическом растворителе. Медленное добавление брома, а также основания, контролирование значения рН можно начинать в то же время или несколько позже. Предпочтительным является начало дозирования брома/основания для обеспечения достаточного количества свободных радикалов в смеси на начале реакции бромирования. После завершения реакции смесь охлаждают и фазы разделяют.
Реакцию обычно осуществляют при температуре от приблизительно 50°C до приблизительно 120°C, предпочтительно от приблизительно 60°C до приблизительно 90°C.
Реакцию можно осуществлять при атмосферном давлении или при повышенном давлении вплоть до 6 бар. Предпочтительным является атмосферное давление.
Время реакции (для стадии (i)) зависит от параметров реакции, но в целом находится в диапазоне от 1 часа до 24 часов.
Для улучшения общего выхода и увеличения селективности реакции, то есть для уменьшения образования нежелательных дибромных и трибромных побочных продуктов, предпочтительным является осуществление реакции только вплоть до превращения 5-60% (исходя из количества соединения (II)), предпочтительно 30-55%. В одном предпочтительном варианте осуществления реакцию осуществляют вплоть до превращения до приблизительно 50% (исходя из соединения (II)). Степень превращения можно проверять с помощью стандартных методов, известных специалисту в данной области техники, например, путем ВЭЖХ анализа.
При достижении желательной степени превращения реакцию останавливают и фазы разделяют.
Органическую фазу, содержащую продукт со стадии (i), соединение (III), непрореагировавшее исходное вещество (I) и дибромные и трибромные побочные продукты можно экстрагировать водой для удаления нерастворимых в воде примесей, таких как кислоты и бромид. Продукт (III) можно выделять с помощью известных процедур, тем не менее предпочтительным является использование органической фазы без дальнейшей обработки для реакции с третичным амино Q (стадия (ii)).
Также представляется возможным экстрагировать водную фазу с органическим растворителем и объединять органические фазы для повышения выхода соединения (III).
На стадии (ii) реакции соединение (III) подвергают реакции с третичным амином Q, получая аммонийное соединение (I).
Предпочтительные третичные амины Q следуют из предпочтительных значений Q в формуле (I), то есть пиридин и третичные алкил амины NR9R10R11,
где
R9, R10 и R11 каждый независимо представляют собой C1-C4 алкил, или R9 и R10, где вместе, образуют 5-ти или 6-ти членное кольцо, в котором R9R10 представлен структурой:
-(CH2)n-, необязательно прерванное О, S или NR12, где n представляет собой целое число 3, 4, или 5, при условии, что R11 представляет собой C1-C4 алкил, и
R12 представляет собой C1-C4 алкил.
Особенно предпочтительными являются триметиламин (NMe3), и пиридин.
Как правило, используют избыток третичного амина. Обычно применяют от 1,05 до 2, предпочтительно от 1,05 до 1,5 эквивалентов третичного амина на эквивалент соединения (II).
Как правило, третичный амин или пиридин, необязательно растворенный в растворителе, медленно добавляют к раствору соединения (II), при этом образуется и осаждается соль (I). В случае предпочтительного амина NMe3, который является газообразным при комнатной температуре, предпочтительным является робота в закрытом сосуде и загрузка газообразного амина или превращенного в жидкость амина под давлением в раствор соединения (III).
Стадию (ii) предпочтительно осуществляют при температуре от приблизительно 0°C до 70°C, более предпочтительно от 5°C до 70°C, особенно предпочтительно от 5°C до 55°C. Реакцию можно осуществлять при атмосферном давлении или при повышенном давлении. В предпочтительном варианте осуществления реакцию осуществляют в закрытом сосуде под давлением растворителя и/или амина, повышающемся при температуре реакции.
Обработку реакционной смеси и выделение аммонийного соединения (I) можно осуществлять с помощью общепринятых методов, например соединение (I) можно отфильтровывать.
В предпочтительном варианте осуществления воду добавляют к реакционной смеси для растворения продукта, соединения (I), и водную фазу и органическую фазу разделяют. Водную фазу можно дополнительно экстрагировать с помощью органического растворителя для повышения чистоты продукта (I) и для повышения выхода восстановленного исходного вещества (II) в органической фазе. Количество воды должно быть достаточным для образования водной фазы и предпочтительно выбирают для образования 20 -45% по весу раствора соединения (I) в водной фазе.
Аммонийное соединение (I) может быть выделено из водной фазы с помощью известных методов. В предпочтительном варианте осуществления соединение (I) не выделяют, и водную фазу, полученную со стадии (ii), используют в последующих реакциях без дополнительной обработки. В дальнейшем предпочтительном варианте осуществления водную фазу смешивают с растворителем, который образует азеотроп с водой, например толуолом, и воду удаляют путем азеотропной перегонки. Полученную суспензию соединения (I) можно использовать для дальнейших реакций.
В дальнейшем предпочтительном варианте осуществления изобретения после отделения соединения (I) органическую фазу со стадии (ii), содержащую вплоть до 80% исходного вещества (II) (исходя из первоначального количества, используемого на стадии (i)), восстанавливают и повторно используют в реакционном процессе на стадии (i). Предпочтительно добавляют дополнительное количество исходного вещества (II) для компенсации количества, превращенного на предыдущей стадии (i). Таким образом, в принципе, органическую фазу со стадии (i) можно многократно повторно использовать, однако вследствие накопления побочных продуктов, главным образом ди- и трибромированного продукта соединения (II), вплоть до 20, предпочтительно вплоть до 10 циклов обычно практически осуществимо.
В предпочтительном варианте осуществления циклического реакционного процесса не добавляют дополнительного количества исходного вещества (II) на последнем цикле для улучшения общего выхода и скорости превращения.
В дальнейшем варианте осуществления циклического реакционного процесса определенное количество органической фазы, предпочтительно приблизительно от 5 до 20% по весу, удаляют для уменьшения или подавления накопления побочных продуктов в органической фазе. В этом варианте осуществления изобретения практически не существует ограничения количества циклов, в которых можно использовать органическую фазу. Соединения формулы (I) являются ценными промежуточными продуктами в органическом синтезе. Они чрезвычайно пригодны для превращения в метоксиметильные соединения (IV) и, кроме того, в гербицидные имидазолиноновые соединения (V).
В одном аспекте изобретения обеспечивается способ получения соединений формулы (IV), который включает стадии:
(i)/(ii) получение 5,6-дизамещенного-3-пиридилметил аммоний бромида формулы (I), как описано выше, и
(iii) взаимодействие соединения формулы (I) в метаноле, толуоле или смеси метанол/толуол с основанием, выбранным из МОСН3 и МОН (если растворитель содержит метанол), где М имеет значение, указанное в формуле (IV).
В одном предпочтительном варианте осуществления, где Y2 представляет собой OM, стадию (iii) осуществляют, как описано в EP-A 0747360, то есть путем реакции соответствующего соединения формулы (I) в метаноле с основанием.
Основания, которые пригодны в этом варианте осуществления изобретения, представляют собой гидриды, гидроксиды, карбонаты или C1-C4 алкоголяты щелочных металлов или щелочноземельных металлов, предпочтительно гидроксид или алкоголят натрия или калия. Подходящими щелочными металлами являются натрий или калий. Подходящими щелочноземельными металлами являются кальций, магний и другие. Предпочтительными являются щелочные металлы, такие как натрий или калий.
Подходящими температурами реакции являются приблизительно от 120° до 180°C, предпочтительно приблизительно от 120° до 150°C. Давлениями реакции должны быть такие давления, которые обычно сопровождают нагревание растворителя в замкнутой реакционной системе до температурного диапазона выше его точки кипения.
Соединения формулы (IV), где Y2 представляет собой OH, могут быть получены путем подкисления соответствующих дикарбоксилатов.
В дальнейшем предпочтительном варианте осуществления, где Y2 представляет собой OCH3 или ОН, стадию (iii) осуществляют, как описано в EP-A 0548532, то есть путем взаимодействия соединения (I) с МОСH3, где M представляет собой щелочной металл, такой как Na или K, в присутствии органического растворителя предпочтительно при температуре в интервале от 0°C до 110°C с образованием первой смеси, дальнейшей реакции указанной первой смеси с по меньшей мере 2,0 молярными эквивалентами водного основания предпочтительно при температуре в интервале от приблизительно 20°C до 120°C с образованием второй смеси и доведения pH указанной второй смеси до значения ниже 2,5 с помощью кислоты для образования кислотных соединений формулы (IV).
Водные основания, которые являются подходящими для применения в этом варианте осуществления изобретения, включают водный раствор гидроксида натрия, водный раствор гидроксида калия и другие. Кислоты, которые могут использоваться в способе согласно изобретению, включают минеральные кислоты, такие как серная кислота, соляная кислота и другие.
Органические растворители, которые можно использовать в способе согласно изобретению, включают ацетонитрил, тетрагидрофуран, ароматические углеводороды, метанол и другие. Предпочтительным инертным органическим растворителем является метанол.
В другом предпочтительном варианте осуществления, где Y2 представляет собой JCH3, стадию (iii) осуществляют, как описано в EP-A 0548532, и соединение формулы (I) подвергают реакции с МОСH3, где M представляет собой Na или K, в инертном растворителе, предпочтительно метаноле.
Дальнейшее превращение соединения (IV) в гербицидные имидазолиноны (V) можно осуществлять с помощью методов, известных в данной области техники.
Методы, которые можно использовать для создания имидазолиноновых гербицидов, иллюстрируются в книге "The Imidazolinone Herbicides", под редакцией D.LShaner и S.L.O'Connor, опубликованной в 1991 г. в CRC Press, Boca Raton, Florida с особой ссылкой на раздел 2, озаглавленный "Synthesis of The Imidazolinone Herbicides", страницы 8-14 и ссылки, которые цитируются в этом источнике. Следующие ссылки на патентную литературу также иллюстрируют методы, которые можно использовать для превращения пиридиновых дикислот, сложных эфиров и солей в имидазолиноновые конечные продукты:
US патенты №№5371229; 5250694; 5276157; 5110930; 5122608; 5206368; 4925944; 4921961; 4959476;5103009;4816588; 4757146; 4798619; 4766218; 5001254; 5021078; 4723011; 4709036; 4658030; 4608079; 4719303; 4562257; 4518780; 4474962; 4623726; 4750978; 4638068; 4439607; 4459408; 4459409; 4460776; 4125727 и 4758667, и EP-A-0041623.
В одном варианте осуществления превращение соединения (IV) в гербицидный имидазолинон (V) осуществляют аналогично методу, описанному в EP-A 0233150 или B.I.Quang и др., Modem Agrochemicals 6 (2007), с.14.
В этом варианте осуществления гербицидное имидазолиноновое соединение (V) получают путем
(i)/(ii)/(iii) получения соединения (IV), как описано выше, и
(iv) взаимодействия соединения (IV) в присутствии сильного основания с 2-аминоалкан карбоксамидом формулы (VI),
 ,
,
где R4 и R5 имеют значения, как указано в формуле (V),
и доведения значения рН для получения соединения формулы (V).
Реакцию осуществляют в инертном растворителе, например ароматических углеводородах и галогенированных углеводородах, таких как толуол, спирты, такие как метанол или трет-бутанол. Предпочтительными являются несмешивающиеся с водой растворители. Предпочтительными сильными основаниями являются алкоголяты щелочных металлов и гидроксиды щелочных металлов, такие как NaOCH3 или KO-трет-C4Р9. Реакцию осуществляют в интервале от комнатной температуры (обычно 22°C) до температуры флегмы реакционной смеси, предпочтительно от 50 до 90°C.
В дальнейших вариантах осуществления изобретения превращение соединения (IV) в гербицидный имидазолинон (V) осуществляют аналогично методам, описанным в EP-A 0041623, US 4518780 или EP-A 0144595.
В соответствии с этим вариантом осуществления соединение (IV) сначала превращают в соответствующий ангидрид с помощью известных методов, таких как реакция с уксусным ангидридом.
В одном варианте осуществления соединение (V) получают путем
(i)/(ii)/(iii) получения соединения (IV), как описано выше;
(iv-1) превращения соединения (IV) в ангидрид (VII),

(iv-2) взаимодействия ангидрида (VII) с 2-аминоалкан карбоксамидом формулы (VI),
,
получая амид (VIII),

(iv-3) конденсации амида (VIII), получая гербицидный имидазолинон (V).
Стадии (iv-2) и (iv-3) можно осуществлять в виде реакции в одном сосуде.
В одном варианте осуществления стадию (iv-2) осуществляют аналогично процедуре, описанной в примере 10 в EP-A 0322616. Соединение (IV), замещенный 2-аминоалкан карбоксамид (VI) и третичный амин, предпочтительно триэтиламин, подвергают реакции в полярном апротонном растворителе, таком как ацетонитрил, получая аммонийную соль (VIII) (R6=HNR3), которую можно подкислить с получением кислоты (VIII) (R6=H).
Альтернативные процедуры описаны в US 4518780 и EP-A 0144595. В последнем документе описано добавление азотистого основания, выбранного из пиридина, пиколинов, хинолина и лутидина, для улучшения региоселективности реакции, то есть для увеличения количества продукта 2-присоединения.
В одном варианте осуществления стадии (iv-3) амидо соединение (VIII), предпочтительно в форме аммонийной соли (R6 представляет собой HNR3), подвергают реакции с метилатом щелочного металла, предпочтительно NaOCH3 в метаноле аналогично примеру 11 в EP 0322616. Полученную суспензию поддерживают при флегме до полного превращения. После охлаждения смеси подкисляют, получая соединение (III) либо в виде аммонийной соли (подкисление до рН приблизительно 4), либо свободной кислоты (подкисление до рН≤2).
В дальнейшем предпочтительном варианте осуществления реакционную смесь со стадии (iv-2) подвергают реакции с метанолом (как правило, от 2 до 100 эквивалентов исходя из (VIII)) в присутствии водного основания (как правило, от 3 до 100 эквивалентов исходя из (VIII)), основание предпочтительно выбирают из МОН и МОСH3, где M представляет собой щелочной металл, предпочтительно Na или K, в особенности Na.
Реакцию осуществляют при температуре в интервале от 20 до 120°C, предпочтительно от 40 до 90°C. Реакцию можно осуществлять при атмосферном давлении или при повышенном давлении, предпочтительно давлении, которое образуется при желательной температуре реакции. Время реакции обычно составляет от 1 до 8 часов, предпочтительно от 1 до 5 часов.
Выделение продукта (V) можно осуществить с помощью стандартных методов. В предпочтительном варианте осуществления добавляют воду, и органические растворители отгоняют. Остаток можно ресуспендировать в воде и подкислять, при этом осаждается соединение (V). После фильтрации неочищенный продукт можно дополнительно очищать, например, путем перемешивания с водой или перекристаллизации.
В дальнейшем варианте осуществления соединение (V) получают путем (i)/(ii)/(iii) получения соединения (IV), как описано выше;
(iv-1) превращения соединения (IV) в ангидрид (VII),
(iv-2) взаимодействия ангидрида (VIII) с аминокарбонитрилом (IX),

где R4 и R5 имеют значения, как указано в формуле (V), получая амидонитрильное соединение (X),
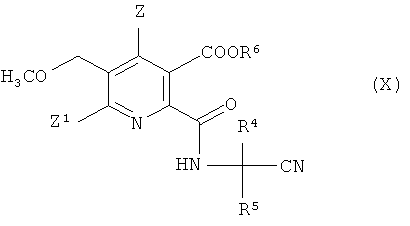
где символы имеют значения, как указано в формуле (V), и R6 предпочтительно представляет собой H,
(iv-3) гидролиза нитрильной группы в соединении (X), получая амид (VIII),
где символы имеют такие же значения, как указано в формуле (V), и R6 предпочтительно представляет собой H,
и
(iv-4) конденсации амида (VIII), получая гербицидный имидазолинон (V).
Получение ангидрида можно осуществлять, как описано выше.
Аминонитрилы (IX), которые используются на стадии (iv-2), являются коммерчески доступными или могут быть получены с помощью методов, известных в данной области техники. Как правило, используют от 0,8 до 1,2 эквивалентов аминонитрила (IX) на эквивалент соединения (IV), предпочтительно от 0,95 до 1,1.
Реакцию осуществляют в растворителе, который предпочтительно выбирают из ароматических углеводородов, предпочтительно толуола, мезитиленов, хлорированных ароматических углеводородов, таких как хлорбензол, дихлорбензол, хлорированных углеводородов, таких как 1,2-дихлорэтан, дихлорметан, уксусной кислоты, и их смесей.
Если уксусную кислоту не используют в качестве основного растворителя, то благоприятным является добавление от 0,5 до 4 эквивалентов, предпочтительно от 1 до 3 эквивалентов (исходя из соединения (I)). Также благоприятными являются вспомогательные вещества, которые улучшают селективность реакции раскрытия цикла (2 относительно 3 положения), которые перечислены в EP-A 0144555, и включают пиридин, 4-пиколин, 2-пиколин и хинолин.
Реакцию обычно осуществляют при температуре в интервале от приблизительно 40 до приблизительно 120°C, предпочтительно от приблизительно 60 до приблизительно 100°C. Время реакции обычно составляет от приблизительно 1 до приблизительно 3 часов.
В предпочтительном варианте осуществления соединение (IV) растворяют в растворителе, и доводят до температуры реакции, и постепенно добавляют аминонитрил (IX). После завершения реакции и охлаждения нитрильное соединение (X) может быть выделено с помощью стандартных методов.
Тем не менее в предпочтительном варианте осуществления соединение (X) не выделяют, а реакционную смесь непосредственно используют на последующей стадии гидролиза нитрила (стадия iv-3).
В типичной процедуре добавляют небольшой избыток (например, от 1,1 до 1,5 эквивалентов исходя из (IX)) сильной минеральной кислоты, предпочтительно серной кислоты (предпочтительно при концентрации от 30 до 98%) и воды (например, от 2 до 10 эквивалентов) при температуре, которая обычно находится в интервале приблизительно от 30°C до 120°C, предпочтительно до 50°C до 90°C. После этого смесь перемешивают до завершения превращения. Время реакции обычно составляет от 1 до 8 часов, предпочтительно от 1 до 5 часов.
Обработку и выделение амида (VIII) можно осуществлять с помощью стандартных методов, таких как в виде водного раствора (например, в виде его аммонийной соли). В предпочтительном варианте осуществления реакционную смесь используют непосредственно на последующей стадии конденсации (iv-4).
В альтернативном варианте осуществления гидролиз нитрильной группы осуществляют путем реакции с водным NaOH/H2O2, как описано, например, в EP-A 0144595 и US 4518780.
Конденсацию амидо соединения (X) до гербицидного имидазолинона можно осуществлять, как описано выше.
Все вышеописанные процессы являются особенно предпочтительными для получения соединения формулы (V), где Z и Z1 представляют собой H, R6 представляет собой H, R4 представляет собой CH3 и R5 представляет собой СН(СН3)2, то есть имазамокса.
Изобретение иллюстрируется с помощью последующих примеров, которые никоим образом его не ограничивают.
Примеры
Примеры 1
Синтез [(5,6-дикарбокси-3-пиридил)метил] триметиламмоний бромида, простой диметиловый эфир (Ia)
a) Синтез диметил 5-(бромметил)-2,3-пиридиндикарбоксилата (IIIa) (50% превращение)
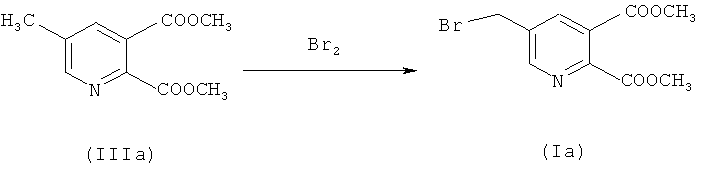
218,4 г (1,0 моль) соединения (IIа) растворяли в 1139,0 г 1,2-дихлорэтана - (EDС), загружали 160,0 г воды и нагревали до 72°C (приблизительно 1-2°C ниже флегмы). 14,4 г (0,075 моль) 2,2'-азобис(2-метилбутиронитрила) (Vazo 67) в 160,0 г EDC добавляли в течение 2 часов при 72°C. Через 30 минут 143,8 г (0,9 моль) добавляли бром в течение 2 часов при контроле значения рН (рН 5-7) путем дозирования приблизительно 375,0 г водного NaOH (15%). Смесь перемешивали в течение 1 часа до завершения реакции (ВЭЖХ анализ). После охлаждения до 40°C фазы разделяли.
b) Синтез соединения (Ia)
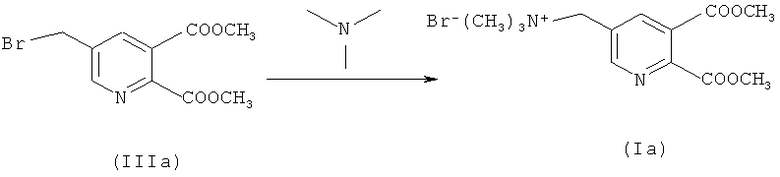
288,1 г (1,0 моль) соединения (IIIа) в смеси с ди- и трибромированными побочными продуктами растворяли в 3359,0 г EDC (органическая фаза со стадии а, включая непрореагировавшее соединение (IIa)) и загружали интенсивно бромированные побочные продукты). Смесь нагревали до 30°C и сосуд откачивали до 200 мбар. 70,9 г (1,2 моль) триметиламина (ТМА) добавляли к газовой фазе в течение 2 часов при 40°C (закрытая система). Смесь перемешивали дополнительно в течение 1 часа (ВЭЖХ проверка превращения: соединение (IIIa) в растворе <0,1%).
Избыток ТМА отгоняли совместно с EDC (масса: 40% EDC массы переносили на стадию 2 (1344 г) при 50-55°C (370-250 мбар). Значение рН дистиллята составляло <9. 630,0 г воды распыляли на стенку таким образом, чтобы твердое вещество растворялось, и смесь переносили в следующий сосуд. После этого смесь перемешивали 0,25 часа и нижнюю органическую фазу отделяли при 40°C. Добавляли 320,4 г EDC. Смесь перемешивали и нижнюю органическую фазу отделяли при 40°C. Обратную экстракцию повторяли (40°C) с 320,4 г EDC. Две органические фазы обратной экстракции объединяли с первой органической фазой и повторно использовали на последующей партии бромирования (после "добавления 50% свежего" соединения "(IIа) для дальнейшего цикла");
Стадии (a) и (b) повторяли шесть раз. В последнем цикле не добавляли соединения (IIIa) на стадии (a) и 0,8 моль ТМА добавляли на стадии (b).
Общая степень превращения соединения (IIа) составила 96,6%. Выход соединения (Ia) (за 7 циклов) составил 77,4% с чистотой >95% (как определено с помощью ВЭЖХ).
Пример 2
Синтез [(5,6-дикарбокси-3-пиридил)метил] триметиламмоний бромида, простой диметиловый эфир (Ia)
Синтез осуществляли, как описано в примере 1, за исключением того, что на стадии (а) все количество 13,65 г (0,071 моль) 2,2'-азобис(2-метилбутиронитрила) (Vazo 67) в 160,0 г EDC добавляли при 72°C и через 5 минут добавляли 135,8 г (0,85 моль) брома в течение 2 часов под контролем рН (рН 5) путем дозирования приблизительно 375,0 г водного NaOH (15%).
Общая степень превращения соединения (IIа) составила 96,6%. Выход соединения (Ia) (за 7 циклов) составил 78,2% с чистотой>95% (как определено с помощью ВЭЖХ).
Пример 3
Синтез [(5,6-дикарбокси-3-пиридил)метил] пиридиний бромида, простой диметиловый эфир
Стадию (а) осуществляли в соответствии с процедурой из примера 1.
Синтез соединения (Ib)

82,4 г (0,286 моль) соединения (IIIa) в смеси с ди- и трибромированными побочными продуктами в EDC (1183,8 г органическая фаза со стадии (а), включая непрореагировавшее соединение (IIа) и интенсивно бромированные побочные продукты) загружали. Смесь нагревали до 40°C. По каплям добавляли 27,88 г (0,292 моль) пиридина в течение 60 минут при 40°C. Смесь перемешивали при 55°C дополнительно в течение одного часа.
Избыток пиридина отгоняли совместно с EDC при 55-60°C в вакууме (~90 торр) до тех пор, пока общий вес смеси не составил 382,7 г. Добавляли 134,8 г воды. Смесь перемешивали в течение 15 минут и затем позволяли осадиться и разделиться в течение 15 минут. Фазы разделяли и анализировали.
Выход соединения (Ib) для одного превращения составил 73,4% (исходя из молей превращенного монобром соединения (IIIa)).
Пример 4
Получение диметил 5-(метоксиметил)-2,3-пиридиндикарбоксилата (в соответствии с примером 7 из EP-A 0548532)
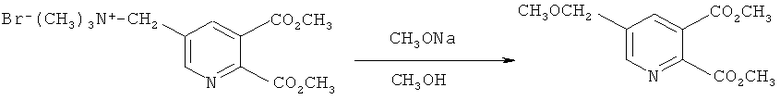
Смесь 25% метилата натрия в метаноле (320,0 г, 1,5 моль) и [(5,6-дикарбокси-3-пиридил)-метил]триметиламмоний бромида, сложный диметиловый эфир (160,0 г, 0,5 моль) в метаноле (630 мл) нагревали в колбе с обратным холодильником в течение 6 часов в атмосфере азота. Реакционную смесь охлаждали до 5°C и добавляли уксусную кислоту (90 г) и воду (200 мл). Метанол удаляли в вакууме, добавляли воду и смесь экстрагировали метиленхлоридом.
Пример 5
Получение 5-(метоксиметил)-2,3-пиридиндикарбоновой кислоты (в соответствии с примером 8 из EP-A 0548532)

Смесь диметил 5-(метоксиметил)-2,3-пиридиндикарбоксилата (60,0 г, 0,25 моль) и 50% раствора гидроксида натрия (50,0 г, 0,63 моль) в воде нагревали при 90-110°C в течение 2 часов в атмосфере азота, в то время как отгоняли метанол и воду. Реакционную смесь охлаждали до 10°C, обрабатывали серной кислотой для доведения pH до 2,0 и фильтровали, получая твердое вещество. Твердое вещество промывали водой и высушивали в вакууме, получая указанный в заглавии продукт в виде белого твердого вещества (44,3 г, tпл=161-162°C).
Пример 6
Получение 5-(метоксиметил)-2,3-пиридиндикарбоновой кислоты (в соответствии с примером 3 из EP-A 0548532)

Смесь 25% метилата натрия в метаноле (270 г, 1,25 моль) и [(5,6-дикарбокси-3-пиридил)-метил]триметиламмоний бромида, сложный диметиловый эфир (Ia) (347 г, 1,00 моль) в метаноле (650 мл) нагревали в колбе с обратным холодильником в течение 1 часа в атмосфере азота. Добавляли воду (1 l) и гидроксид натрия (80,0 г, 2,0 моль) и реакционную смесь дистиллировали до тех пор, пока сосуд не составлял 100-105°C. Реакционную смесь охлаждали до комнатной температуры, обрабатывали серной кислотой для доведения рН до значения от 1,5 до 2 и фильтровали, получая твердое вещество. Твердое вещество промывали водой и высушивали в вакуумном сушильном шкафу, получая указанный в заглавии продукт в виде белого твердого вещества (tпл=161-162°C), который характеризовался чистотой более 99% согласно ВЖЭХ анализу.
Пример 7
Получение динатрий 5-(метоксиметил)пиридин-2,3-дикарбоксилата из динатрий [5,6-(дикарбоксилат-3-пиридил)метил]триметиламмоний бромида (в соответствии с примером 3 из EDP-A 0747360)

Смесь динатрий [(5,6-дикарбоксилат-3-пиридил)метил]триметиламмоний бромида (5,0 г, 13,8 ммоль) и 25 мас.% раствора метилата натрия в метаноле (4,46 г, 20,7 ммоль NaOCH3) в 75 г метанола нагревали при 120°C в течение 21 часа в реакторе под давлением. Реакцию охлаждали до комнатной температуры, обрабатывали водой и концентрировали до конечного веса 55,03 г. Образец 5,0 г анализировали путем ЖХ анализа (30% CH3CN, 0,77 М Н3РO4). Оставшийся реакционный раствор упаривали насухо, получая твердый остаток, который идентифицировали путем ЯМР анализа.
Изобретение относится к способу получения 5,6-дизамещенных-3-пиридилметил аммоний бромидов (I),
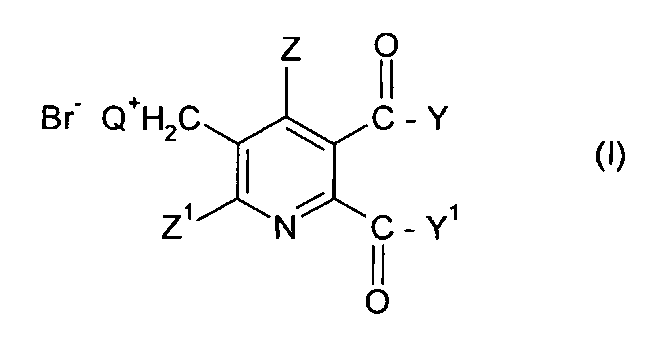
где Q представляет собой третичный алифатический или циклический, насыщенный, частично ненасыщенный или ароматический амин; Z представляет собой водород или галоген; Z1 представляет собой водород, галоген, циано или нитро; Y и Y1 каждый независимо представляют собой OR1, NR1R2, или где вместе YY1, представляют собой -О-, -S- или NR3-; R1 и R2 каждый независимо представляют собой водород, С1-С4 алкил, необязательно замещенный С1-С4 алкокси или фенилом, необязательно замещенным одной-тремя С1-С4 алкильными группами, С1-С4 алкокси группами или атомами галогена, или фенил, необязательно замещенный одной-тремя С1-С4 алкильными группами, С1-С4 алкокси группами или атомами галогена; R3 представляет собой водород или С1-С4 алкил, который включает стадии (i) взаимодействия соединения формулы (II),
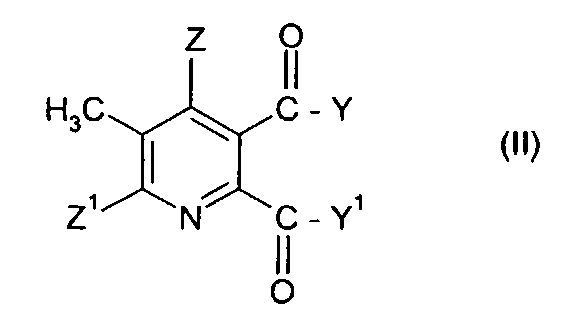
где символы имеют значения, указанные для формулы (I), с бромом в присутствии радикального инициатора в смеси растворителей, содержащей водную фазу и органическую фазу, где органическая фаза содержит растворитель, выбранный из 1,2-дихлорэтана, хлорбензола, 1,2-дихлорбензола, 1,3-дихлорбензола, 1,4-дихлорбензола и тетрахлорметана, и где значение pH водной фазы составляет от 3 до <8, получая 3-бромметил-5,6-дизамещенное пиридиновое соединение (III),

где Y, Y1, Z и Z1 имеют значения, указанные для формулы (I), и (ii) взаимодействия бром соединения формулы (III) с третичным аминовым основанием Q в растворителе при температуре в интервале от 0°C до 100°C. 4 н. и 10 з.п. ф-лы, 7 пр.
1. Способ получения 5,6-дизамещенных-3-пиридилметил аммоний бромидов (I),
где
Q представляет собой третичный алифатический или циклический,
насыщенный, частично ненасыщенный или ароматический амин;
Z представляет собой водород или галоген;
Z1 представляет собой водород, галоген, циано или нитро;
Y и Y1 каждый независимо представляют собой OR1, NR1R2, или где вместе YY1 представляют собой -О-, -S- или NR3-;
R1 и R2 каждый независимо представляют собой водород,
С1-С4 алкил, необязательно замещенный С1-С4 алкокси или фенилом, необязательно замещенным одной-тремя С1-С4 алкильными группами, С1-С4 алкокси группами или атомами галогена, или фенил, необязательно замещенный одной-тремя С1-С4 алкильными группами, С1-С4 алкокси группами или атомами галогена;
R3 представляет собой водород или С1-С4 алкил;
который включает стадии:
(i) взаимодействие соединения формулы (II),
где символы имеют значения, указанные для формулы (I), с бромом в присутствии радикального инициатора в смеси растворителей, содержащей водную фазу и органическую фазу, где органическая фаза содержит растворитель, выбранный из 1,2-дихлорэтана, хлорбензола, 1,2-дихлорбензола, 1,3-дихлорбензола, 1,4-дихлорбензола и тетрахлорметана, и где значение pH водной фазы составляет от 3 до <8, получая 3-бромметил-5,6-дизамещенное пиридиновое соединение (III),
где Y, Y1, Z и Z1 имеют значения, указанные для формулы (I), и
(ii) взаимодействие бром соединения формулы (III) с третичным аминовым основанием Q в растворителе при температуре в интервале от 0°C до 100°C.
2 Способ, как заявлено в п. 1, где органический растворитель на стадии (i) представляет собой 1,2-дихлорэтан.
3. Способ, как заявлено в п. 1 или 2, где Q+ в формуле (I) представляет собой
 ;
; 
и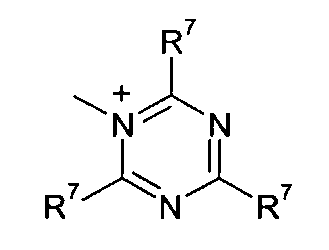 ;
;  ;
; 
 ;
;
Z2 представляет собой О, S или NR12;
R12 представляет собой С1-С4 алкил; и
R7 и R8 каждый независимо представляют собой водород, галоген, С1-С4 алкил или С1-С4 алкокси, или R7 и R8, где вместе, образуют 5- или 6-членное насыщенное или ненасыщенное кольцо, необязательно прерванное О, S, или NR12 и необязательно замещенное одним-тремя атомами галогена, С1-С4 алкильными группами или С1-С4 алкокси группами, и где
R9, R10 и R11 каждый независимо представляют собой С1-С4 алкил, или R9 и R10, где вместе, образуют 5-ти или 6-ти членное кольцо, в котором R9R10, представлен структурой: -(CH2)n-, необязательно прерванное О, S или NR9, где n представляет собой целое число 3, 4 или 5, при условии, что R11 представляет собой С1-С4 алкил.
4. Способ, как заявлено в п. 1 или 2, где соединение формулы (I) представляет собой соединение формулы (Ia):
5. Способ, как заявлено в п. 1 или 2, где реакцию на стадии (i) осуществляют вплоть до превращения соединения (II) до 20-60%.
6. Способ, как заявлено в п. 1 или 2, где органическую фазу на стадии (ii) используют повторно на стадии (i) с необязательным добавлением соединения (II).
7. Способ, как заявлено в п. 1 или 2, где значение pH водной фазы на стадии (i) составляет от 3 до 7.
8. Способ, как заявлено в п. 1 или 2, где инициатор на стадии (i) выбирают из азобисизобутиронитрила, 2,2'-азобис(2-метилбутаннитрила), 2,2'-азобис(2,4-диметил-пентаннитрила и 1,1'-азобис(циклогексанкарбонитрила).
9. Способ, как заявлено в п. 1 или 2, где молярное отношение пиридинового соединения (II) к брому на стадии (i) составляет 1:0,5-1,2.
10. Способ, как заявлено в п. 1 или 2, где стадию (i) осуществляют при температуре от 50°C до 120°C.
11. Способ, как заявлено в п. 1 или 2, где третичный амин Q на стадии (ii) представляет собой триметиламин.
12. Способ получения 5,6-дизамещенного-3-метоксиметилпиридина формулы (IV), который включает стадии:
(i) и (ii) получения соединения формулы (I), способом в соответствии с п. 1,
(iii) взаимодействие соединения формулы (I) в метаноле, толуоле или смеси метанол/толуол с основанием, выбранным из МОСН3 и МОН (если растворитель содержит метанол), где М представляет собой щелочной металл или щелочноземельный металл, с образованием 5,6-дизамещенного-3-метоксиметилпиридина формулы (IV),
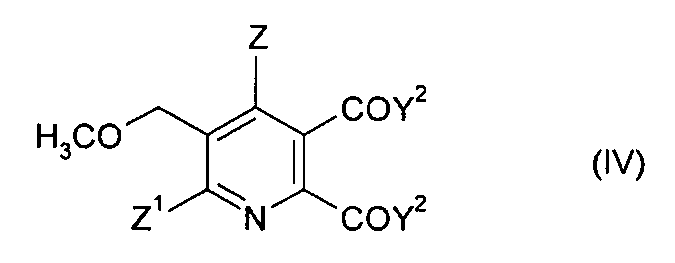
где
Z представляет собой водород или галоген;
Z1 представляет собой водород, галоген, циано или нитро;
Y2 представляет собой ОСН3 или ОМ.
13. Способ получения гербицидного имидазолинона формулы (V) дальнейшими стадиями:
(i), (ii) и (iii) получения соединения формулы (IV), способом в соответствии с п. 12,
(iv) превращение соединения формулы (IV) в гербицидное имидазолиноновое соединение формулы (V) путем
(iv-1) необязательного приготовления ангидрида (VII) соединения (IV);
(iv-2) взаимодействия соединения (IV) или его ангидрида (VII) в присутствии основания с 2-аминоалкан карбоксамидом формулы (VI),
H2N-CR4R5-CONH2 (VI)
где R4 и R5 имеют значения, как указано в формуле (V),
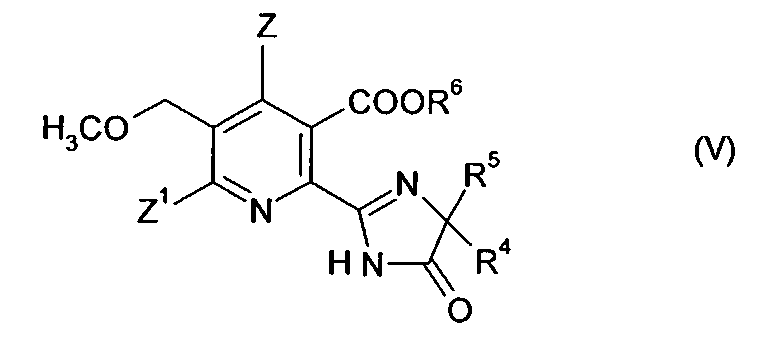
где
Z представляет собой водород или галоген;
Z1 представляет собой водород, галоген, циано или нитро;
R4 представляет собой С1-С4 алкил;
R5 представляет собой С1-С4 алкил, С3-С6 циклоалкил, или R4 и R5;
где вместе с атомом, к которому они присоединены, представляют собой С3-С6 циклоалкильную группу, необязательно замещенную метилом, и
R6 представляет собой водород; группу формулы -N=C (низший алкил)2;
С1-С12 алкил, необязательно замещенный одной из следующих групп: С1-С3 алкокси, галоген, гидроксил, С3-С6 циклоалкил, бензилокси, фурил, фенил, галофенил, низший алкилфенил, низший алкоксифенил, нитрофенил, карбоксил, низший алкоксикарбонил, циано или три низший алкиламмоний; С3-С12 алкенил, необязательно замещенный одной из следующих групп: С1-С3 алкокси, фенил, галоген, или низший алкоксикарбонил, или двумя С1-С3 алкокси группами или двумя галогеновыми группами; С3-С6 циклоалкил, необязательно замещенный одной или двумя С1-С3 алкильными группами; или катион.
14. Способ получения гербицидного имидазолинона формулы (V) дальнейшими стадиями:
(i), (ii) и (iii) получения соединения формулы (IV) способом в соответствии с п. 12,
(iv) превращение соединения формулы (IV) в гербицидное имидазолиноновое соединение формулы (V) путем
(iv-1) превращения соединения (IV) в ангидрид (VII);
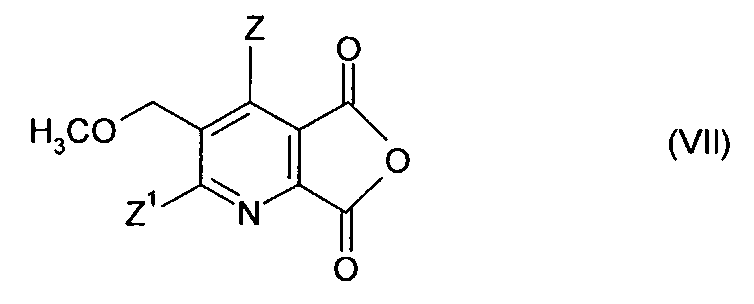
(iv-2) взаимодействия ангидрида (VII) с аминокарбонитрилом (IX),
H2N-CR4R5-CN (IX)
где R4 и R5 имеют значения, как указано в формуле (V), с получением амидонитрильного соединения (X),
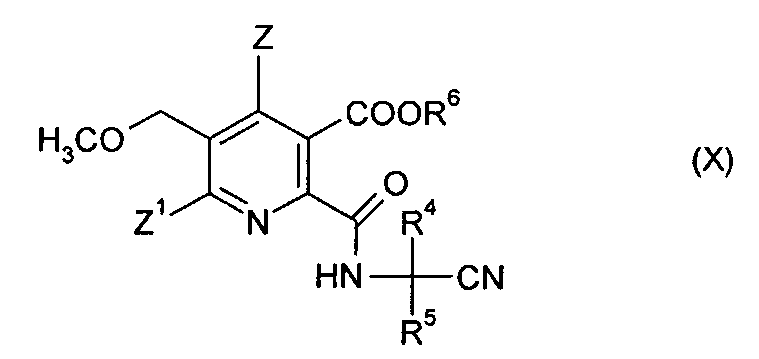
где символы имеют значения, как указано в формуле (V) в п. 13, и R6 представляет собой Н;
(iv-3) гидролиза нитрильной группы в соединении (X) с получением амида (VIII),
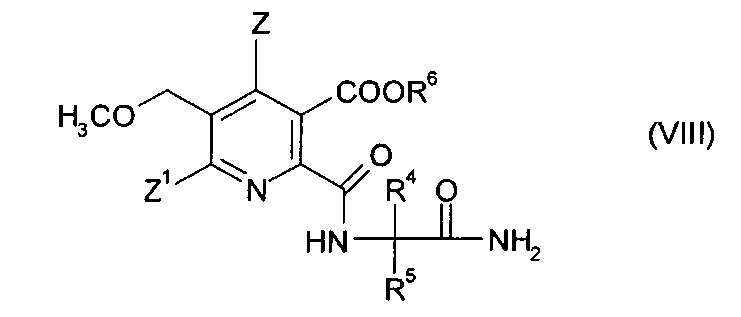
где символы имеют значения, как указано в формуле (V) в п. 13, и R6 представляет собой Н,
и
(iv-4) конденсации амида (VIII) с получением гербицидного имидазолинона (V).

