Изобретение относится к области техники, касающейся химических способов получения соединений из ряда гербицидных фенилсульфонилмочевин и их промежуточных продуктов.
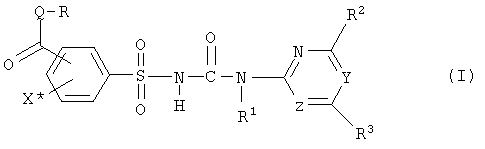
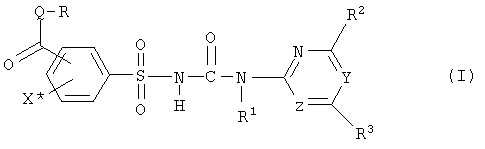
Ряд замещенных фенилсульфонилмочевин был описан в качестве гербицидов и регуляторов роста растений. В отношении групп фенилсульфонилмочевин возникает проблема синтеза соединений с карбоксильной группой или группой производных карбоновых кислот в фенильном кольце. Интерес представляют известные из заявок EP-A-007687 или WO-A-92/13845 соединения формулы (I) и их соли,
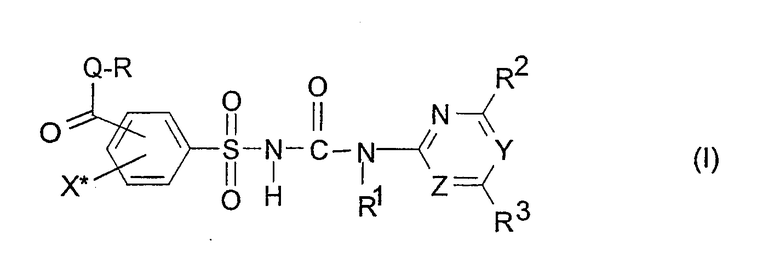
где
Q обозначает кислород или серу,
X* обозначает водород, галоген, циано, нитро,(C1-C3)-алкил или метокси, предпочтительно водород или йод, в особенности йод,
Y, Z независимо друг от друга обозначают CH или N, причем Y и Z не одновременно являются CH,
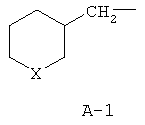
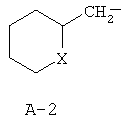
R обозначает водород, (C1-C12)-алкил, (C2-C10)-алкенил, (C2-C10)-алкинил, (C1-C6)-алкил, одно - четырехкратно замещенный остатком из группы: галоген, (C1-C4)-алкокси, (C1-C4)-алкилтио, CN, [(C1-C4)-алкокси]-карбонил и (C2-C6)-алкенил, или (C3-C8)-циклоалкил, незамещенный или замещенный остатком из группы: (C1-C4)-алкил, (C1-C4)-алкокси, (C1-C4)-алкилтио и галоген, (C5-C8)-циклоалкенил, фенил-(C1-C4)-алкил, в фенильном остатке незамещенный или замещенный одним или несколькими остатками из группы: галоген, (C1-C4)-алкил, (C1-C4)-алкокси, (C1-C4)-галогеналкил, (C1-C4)-алкилтио, [(C1-C4)-алкокси]-карбонил, [(C1-C4)-алкил]-карбонилокси, карбамоил, [(C1-C4)-алкил]-карбониламино, [(C1-C4)-алкил]-аминокарбонил, ди-[(C1-C4)-алкил]-аминокарбонил и нитро, или остаток формул A-1 до A-10
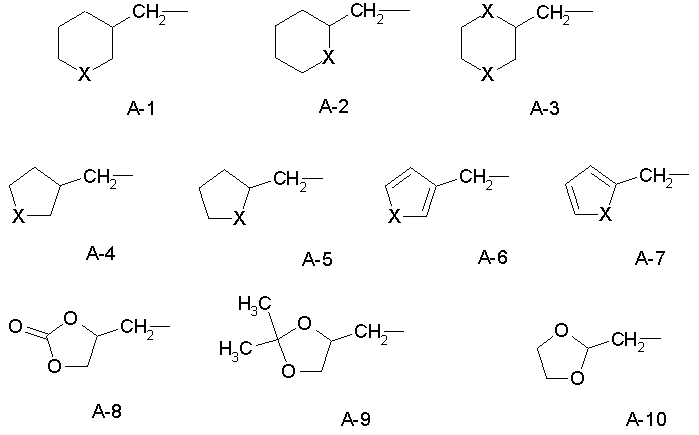
причем в формулах A-1 до A-10
X или оба X независимо друг от друга обозначают O, S, S(O) или SO2,
R1 обозначает водород или (C1-C3)-алкил,
R2 обозначает водород, галоген, (C1-C3)-алкил или (C1-C3)-алкокси, причем каждый из обоих остатков, названных последними, незамещен или одно- или многократно замещен галогеном или (C1-C3)-алкокси,
R3 обозначает водород, галоген, (C1-C3)-алкил, (C1-C3)-алкокси или (C1-C3)-алкилтио, причем каждый из трех остатков, названных последними, не замещен или замещен одно- или многократно галогеном, или одно- или двукратно (C1-C3)-алкокси или (C1-C3)-алкилтио, или остаток формулы NR4R5, (C3-C6)-циклоалкил, (C2-C4)-алкенил, (C2-C4)-алкинил, (C3-C4)-алкенилокси или (C3-C4)-алкинилокси,
R4 и R5 независимо друг от друга обозначают водород, (C1-C4)-алкил, (C3-C4)-алкенил, (C1-C4)-галогеналкил или (C1-C4)-алкокси.
Соли соединений (I) предпочтительно являются соединениями, в которых атом водорода в группе SO2NH-сульфонилмочевины заменен катионом, предпочтительно физиологически совместимым и используемым в защите растений катионом, в особенности катионом щелочного или щелочноземельного металла или, в случае необходимости, замещенным ионом аммония, включая четвертичные ионы аммония. Например, такими катионами являются ионы натрия, калия и аммония.
Соли соединений формулы (I) могут быть образованы посредством присоединения пригодной неорганической или органической кислоты, как, например, HCl, HBr, H2SO4 или HNO3, а также щавелевая кислота или сульфокислота, к основной группе, как, например, аминогруппа или алкиламиногруппа. Пригодные заместители, которые существуют в депротонированной форме, как, например, сульфокислоты или карбоновые кислоты, могут образовывать внутренние соли с протонируемыми с их стороны группами, как аминогруппы.
Также соли могут быть образованы таким образом, что у пригодных функциональных групп, как, например, карбоксильные группы, водород замещен на пригодные для сельского хозяйства катионы. Эти соли являются, например, солями металлов, в особенности солями щелочных или щелочноземельных металлов, в особенности солями натрия и калия, или также солями аммония, солями органических аминов или четвертичными аммониевыми солями.
Особенный интерес представляют соединения формулы (I) или их соли и, соответственно, их получение, в которых группа формулы -CO-Q-R находится в орто-положении к сульфонильной группе сульфонилмочевины (I). Предпочтительными являются соединения (I) или их соли, где Q обозначает атом кислорода, X* обозначает водород или галоген, предпочтительно йод, R обозначает (C1-C4)-алкил, (C2-C4)-алкенил, (C2-C4)-алкинил, (C1-C4)-галогеналкил, или (C1-C4)-алкокси(C1-C4)-алкил, предпочтительно метил или этил, в особенности метил. Далее предпочтительными являются соединения (I) и их соли, в которых группа формулы -CO-Q-R находится в орто-положении к сульфонильной группе сульфонилмочевины, X* обозначает галоген, предпочтительно йод, и X* находится в пара-положении к группе формулы -CO-Q-R. При этом далее предпочтительными являются и сами соединения (I), и их соли, где Z обозначает атом азота и Y обозначает атом азота или группу формулы CH, предпочтительно Y обозначает атом азота.
Из заявок WO-A-92/13845 и WO 01/23368 и цитируемой там литературы известно, что фенилсульфогалогениды, замещенные в фенильном кольце остатками производных карбоновых кислот и дополнительно другими остатками как галоген, посредством аммонолиза подвергают превращению до сульфоамидов, которые после фосгенирования подвергают превращению до изоцианатов и последующими реакциями с гетероциклическими аминосоединениями до сульфонилмочевин формулы (I) или их солей.
Исходное сырье (упомянутый фенилсульфохлорид) согласно WO 92/13845 получают из замещенных аминобензойных кислот после этерификации, диазотирования, взаимодействия с SO2 в присутствии катализаторов Cu (реакция Меервейна) и окислительного расщепления полученного дисульфида с газообразным хлором в соляной кислоте.
Однако известный синтез из-за выхода и числа стадий является неудовлетворительным. В особенности получение многофункционального фенилсульфохлорида является дорогостоящим и не оптимальным по выходу. Кроме того, для последующего фосгенирования нужна особенная, дорогостоящая техника для проведения и контроля способа. Поэтому задачей является разработка альтернативного способа, обладающего преимуществами по сравнению с известными способами в отношении одного аспекта, предпочтительно нескольких аспектов.
Объектом изобретения является способ получения вышеупомянутых фенилсульфонилмочевин формулы (I) и их солей, отличающийся тем, что
a) соединение формулы (II)
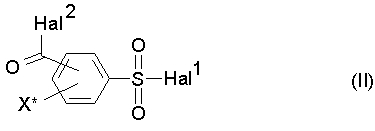
где
Hal1 обозначает атом галогена, предпочтительно хлор или бром, в особенности хлор,
Hal2 обозначает атом галогена, предпочтительно хлор или бром, в особенности хлор, и
X* определен как в формуле (I),
подвергают взаимодействию с соединением формулы R-Q-H или его солью с образованием соединения формулы (III),
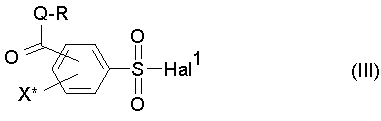
причем R, Q и X определены как в формуле (I), и Hal1 определен как в формуле (II), и
(b) или с промежуточным выделением или без промежуточного выделения
(b1) полученное соединение (III) подвергают аммонолизу с получением сульфоамида формулы (IV),
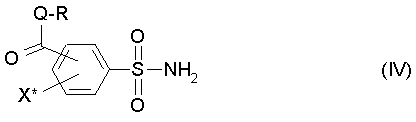
где R, Q и X* определены как в формуле (III),
и соединение (IV), с промежуточным выделением или без промежуточного выделения, подвергают взаимодействию с фосгеном с получением фенилсульфонилизоцианата формулы (V),
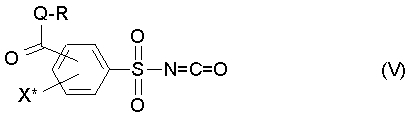
где R, Q и X* определены как в формуле (III),
или
(b2) полученное соединение (III) подвергают взаимодействию с цианатом, например, цианатом щелочного металла, с получением изоцианата формулы (V) или его сольватированного (стабилизированного) производного,
и
(c) с промежуточным выделением или, предпочтительно, без промежуточного выделения изоцианат формулы (V) или его стабилизированное производное подвергают взаимодействию с гетероциклическим амином формулы (VI)
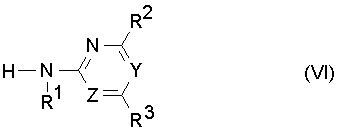
где R1, R2, R3, Y и Z определены как в формуле (I),
с получением сульфонилмочевины формулы (I) или ее соли.
Некоторые соединения формулы (II) (определенные дигалогениды) являются новыми и также являются объектом изобретения. Получение соединений формулы (II) (дигалогенидов) также является объектом изобретения.
Следующим объектом изобретения также является способ согласно стадии (a) (селективная этерификация) с использованием соединений (II).
Следующим объектом изобретения также является способ согласно стадии (b2) (превращение с использованием цианата), предпочтительно в комбинации со стадией (с).
В определениях формул (I)-(VI) и всех последующих формул остатки алкил, алкокси, галогеналкил, галогеналкокси, алкиламино и алкилтио, а также соответствующие незамещенные и/или замещенные остатки в углеводородном фрагменте могут быть, смотря по обстоятельствам, неразветвленными или разветвленными.
Если специально не указано, для этих остатков является предпочтительным небольшой углеводородный фрагмент, например, от 1 до 6 С-атомов, в особенности, от 1 до 4 С-атомов, или для ненасыщенных групп от 2 до 6 С-атомов, в особенности от 2 до 4 С-атомов. Алкильные остатки, также и в составе остатков, таких как алкокси, галогеналкил и т.д. обозначают, например, метил, этил, н- или изопропил, н-, изо-, трет.- или 2-бутил, пентил, гексил, как н-гексил, изогексил и 1,3-диметилбутил, гептил, как н-гептил, 1-метилгексил и 1,4-диметилпентил, алкенильные и алкинильные остатки имеют значение возможных ненасыщенных остатков, соответствующих алкильным остаткам, причем имеется, по крайней мере, одна двойная связь или тройная связь, предпочтительно одна двойная связь или тройная связь. Алкенил обозначает, например, аллил, 1-метилпроп-2-ен-1-ил, 2-метил-проп-2-ен-1-ил, бут-2-ен-1-ил, бут-3-ен-1-ил, 1-метил-бут-3-ен-1-ил и 1-метил-бут-2-ен-1-ил; алкинил обозначает, например, пропаргил, бут-2-ин-1-ил, бут-3-ин-1-ил, 1-метил-бут-3-ин-1-ил.
Циклоалкил обозначает карбоциклическую, насыщенную кольцевую систему с предпочтительно 3-8 С-атомами, предпочтительно 3-6 С-атомами, например, циклопропил, циклобутил, циклопентил или циклогексил.
Галоген обозначает, например, фтор, хлор, бром или йод. В определении остатков термином «галоген» обозначают галогеновый остаток, то есть атом галогена. Галогеналкил, -алкенил и -алкинил обозначают частично или полностью (посредством одинаковых или разных атомов галогена) замещенный галогеном, предпочтительно фтором, хлором и/или бромом, в особенности фтором или хлором, алкил, алкенил или алкинил, например, моногалогеналкил (=моногалогеналкил), пергалогеналкил, CF3, CHF2, CH2F, CF3CF2, CH2FCHCl, CCl3, CHCl2, CH2CH2Cl; галогеналкокси обозначает, например, OCF3, OCHF2, OCH2F, CF3CF2O, OCH2CF3 и OCH2CH2Cl; соответственно это распространяется на галогеналкенилы и другие остатки, замещенные галогенами.
Арил обозначает карбоциклическую ароматическую систему, например, моно-, би- или полициклическую ароматическую систему, например, фенил, нафтил, тетрагидронафтил, инденил, инданил, пенталенил, флуоренил и похожие, предпочтительно фенил.
Углеводородный остаток содержит исключительно С-атомы и Н-атомы и может быть неразветвленным, разветвленным или циклическим, насыщенным, ненасыщенным или ароматическим, или может содержать комбинацию одинаковых или разных других вышеназванных углеводородных остатков. Например, термин «углеводородный остаток» включает остатки: алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкил-алкил, циклоалкенил-алкил, арил как фенил или нафтил, бензил, фенэтил и т.д. Углеводородный остаток содержит предпочтительно 1-30 С-атомов, в особенности 1-24 С-атомов, если не указано иное.
Если основной фрагмент замещен «посредством одного или нескольких остатков» из перечня остатков (=групп) или одной группой остатков, определенной как родовая, то это включает, соответственно, одновременное замещение несколькими одинаковыми и/или структурно различающимися остатками группы или группой, определенной как родовая.
Замещенные остатки, как замещенный углеводородный остаток, например, замещенный алкильный, алкенильный, алкинильный, арильный, фенильный, бензильный остаток обозначают, например, замещенный остаток, образованный от незамещенного основного фрагмента, причем заместителями являются, например, один или несколько, предпочтительно, 1,2 или 3 остатка из группы: галоген, алкокси, алкилтио, гидрокси, амино, нитро, карбокси, циано, ацидо, алкоксикарбонил, алкилкарбонил, формил, карбамоил, моно- и диалкиламинокарбонил, замещенный амино, как ациламино, моно- и диалкиламино, алкилсульфинил и алкилсульфонил и, в случае циклических остатков, также алкил, галогеналкил, алкилтиоалкил, алкоксиалкил, в случае необходимости, замещенные моно- и диалкиламиноалкил и гидроксиалкил.
Предпочтительно заместители выбраны из группы: галоген, алкокси, алкилтио, гидрокси, амино, нитро, циано, моно- и диалкиламино, в случае циклических остатков также алкил и галогеналкил.
В понятие «замещенные остатки», например, замещенные углеводородные остатки, как замещенный алкил и т.д., в качестве заместителей дополнительно к названным насыщенным углеводородным остаткам включены соответствующие ненасыщенные алифатические и ароматические остатки, например, в случае необходимости, замещенный алкенил, алкинил, алкенилокси, алкинилокси, фенил, фенокси и т.д. В случае замещенных циклических остатков с алифатическими частями в кольце также включены циклические системы с такими заместителями, которые соединены с кольцом двойной связью, например, замещены алкилиденовой группой как метилиден или этилиден.
Заместители, названные в качестве примеров («первый ряд заместителей»), если они содержат углеводородную часть, могут быть там замещены далее («второй ряд заместителей»), например, одним из заместителей, определенным для первого ряда заместителей. Соответственно, возможны другие ряды заместителей. Предпочтительно понятие «замещенный остаток» включает только один или два ряда заместителей.
Для названных заместителей, смотря по обстоятельствам, является предпочтительным число С-атомов, ранее названное предпочтительным для остатков с углеводородной частью.
Соединения формулы (II) отчасти известны. Так, в заявке US-A-4110373 описана реакция, в случае необходимости, замещенных бензотрихлоридов посредством олеума. Среди прочих там упомянуто получение 4-хлор-3-хлорсульфонил-бензоилхлорида и 3-хлор-5-хлорсульфонил-бензоилхлорида.
Далее, из заявки NL-A-7603612 известно получение 2-хлорсульфонил-бензоилхлорида из 2-сульфобензойной кислоты посредством реакции с фосгеном в качестве галогенирующего средства в полярных апротонных растворителях как ДМФ. В качестве побочного продукта при этом образуется в значительных количествах дихлортолилсульфон (3,3-дихлор-1,1-диоксо-бензо-1-тиа-2-оксолан). В целом способ описан также для производных сульфобензойных кислот, дополнительно галогенированных или нитрированных в бензойном кольце.
Соединения (I) и (III) до (VI) описаны в общем и частично конкретно в публикациях EP-A-007687, WO-A-92/13845 и WO-A-01/23368 и цитируемой там литературе. В особенности, частью данного описания и изобретения должно быть содержание заявок WO-A-92/13845 и WO-A-01/23368, в отношении стадий способа и названных там предпочтительных соединений формулы (I) и их исходных продуктов.
Особенно предпочтительными для способа получения являются соединения формулы (II), где Q обозначает атом кислорода, X* обозначает водород или галоген, предпочтительно йод, R обозначает (C1-C4)-алкил, (C2-C4)-алкенил, (C2-C4)-алкинил, (C1-C4)-галогеналкил, или (C1-C4)-алкокси(C1-C4)-алкил, предпочтительно метил или этил, в особенности метил. Предпочтительно обозначают Hal1 атом хлора и Hal2 атом хлора.
Также предпочтительным является способ получения с использованием соединений формулы (II), где галогенангидридная группа карбоновой кислоты находится в орто-положении к сульфогалогенидной группе. Далее предпочтительным является способ получения с помощью соединений формулы (II), где галогенангидридная группа карбоновой кислоты находится в орто-положении к сульфогалогенидной группе и X*, обозначающий атом галогена, предпочтительно йод, находится в пара-положении к галогеналкильной группе карбоновой кислоты.
Поэтому также предпочтителен способ, при котором на стадии a) соединения формулы (IIa) используют в качестве соединений формулы (II):
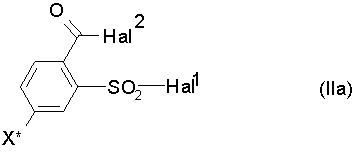
где Hal1 обозначает атом галогена, предпочтительно атом хлора или брома, в особенности атом хлора, Hal2 обозначает атом галогена, предпочтительно атом хлора или брома, в особенности атом хлора, и X* определен как в формуле (I), предпочтительно обозначает галоген, в особенности йод.
В целом предпочтительным является способ с промежуточными продуктами и соединениями формулы (I), которые имеют в фенильном кольце заместитель, соответствующий соединениям (IIa).
Особенно предпочтительным также является способ согласно изобретению, характеризующийся комбинацией признаков, названных предпочтительными.
Согласно изобретению, реакция соединения формулы (II) с соединением формулы R-Q-H или его солью с образованием соединения формулы (III) представляет собой селективную реакцию дигалогенида с нуклеофильным соединением R-Q-H.
Взаимодействие проводят с помощью спирта или тиоспирта (Q обозначает атом кислорода или атом серы) и/или их солью, причем соль используют непосредственно или могут получать в реакционной смеси из спирта в присутствии других оснований. Условия реакции целесообразно выбирать так, чтобы по возможности избежать побочных реакций по сульфогалогенидной группе. В качестве побочных реакций имеют в виду, например, этерификацию сульфогалогенида до сложного эфира сульфокислоты с последующей внутримолекулярной переэтерификацией с образованием сульфокислотной группы и дальнейших последовательных реакций.
Относительно хорошие выходы желаемого сложного моноэфира (III) замещенной галогенсульфонилбензойной кислоты получают, например, если взаимодействие проводят в инертном органическом растворителе и/или разбавителе (в дальнейшем коротко обозначенном как «растворитель»), контролируя температуру. В качестве инертного органического растворителя имеют в виду, например, относительно неполярные апротонные органические растворители как
- алифатические и ароматические углеводороды, например, минеральные масла, эфиры нефти, н-пентан, н-гексан, циклопентан, циклогексан или толуол, ксилол, мезитилен, производные нафталина, ®Solvesso 200 (высококипящую смесь ароматических углеводородов);
- галогенированные алифатические и ароматические углеводороды, например, метиленхлорид, дихлорэтан или хлорбензол, хлортолуол или дихлорбензол или
- смеси названных растворителей.
При больших загрузках, как правило, используют высококипящие органические растворители как толуол, ксилол, мезитилен, хлорбензол, хлортолуол или дихлорбензол, или их смеси.
В качестве спиртов или тиоспиртов используют соединения R-Q-H, соответствующие остатку R-Q в формуле (I). При этом предпочтительными являются (C1-C4)-спирты как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, в особенности метанол и этанол. Как правило, используют 1 мольный эквивалент или избыток, например, 1-20 мольных эквивалентов, предпочтительно 1-8 мольных эквивалентов, в особенности 1-6 мольных эквивалентов соединения R-Q-H на моль соединения формулы (II) (дигалогенид, например, дихлорид). В случае метанола предпочтительно используют 1-8 мольных эквивалентов, в особенности 1-6 мольных эквивалентов, очень предпочтительно 3-6 мольных эквивалентов.
В качестве солей спиртов или тиоспиртов имеют в виду, например, соли щелочных металлов, предпочтительно соли натрия или калия, или соли щелочноземельных металлов. Как правило, используют 1 мольный эквивалент или избыток, например, 1-10 мольных эквивалентов, предпочтительно 1-3 мольных эквивалента, в особенности 1-2 мольных эквивалента соли соединения R-Q-H на моль соединения формулы (II) (дигалогенида). В случае соли метанола, например, метилата натрия, предпочтительно используют 1-10 мольных эквивалентов, в особенности 1-3 мольных эквивалента, очень предпочтительно 1-2 мольных эквивалента метилата.
Температура реакции при этерификации может быть целенаправленно оптимизирована в зависимости от компонентов и их долей в реакционной смеси с помощью предварительных опытов, и чаще всего находится в области от -20°C до 100°C. В случае, если для этерификации используют спирт или тиоспирт, предпочтительно (C1-C4)-алканол, в особенности метанол, пригодная температура реакции, как правило, находится в области от -10°C до 70°C, предпочтительно 20 до 40°C. В случае использования солей соединений R-Q-H оптимальная температура реакции чаще всего имеет сравнительно более низкое значение. Как правило, пригодная температура реакции при этерификации с солью, предпочтительно солью щелочного или щелочноземельного металла (C1-C4)-алканола или также тиоспирта, в особенности метилата натрия или метилата калия, находится в области от -20°C до 50°C, предпочтительно -10 до 35°C, в особенности 0 до 25°C.
Для обработки реакционной исходной смеси можно действовать согласно общепринятым методикам. После окончания реакции с избытком спирта, избыточный спирт может быть отогнан, например, под вакуумом, и затем реакционную смесь для извлечения образующихся солей подают в воду и затем продукт экстрагируют органическим растворителем. Альтернативно исходную смесь можно подавать непосредственно в воду и экстрагировать растворителем.
Следующее превращение полученного сложного эфира галогенсульфонилбензойной кислоты (III) (например, сложного эфира хлорсульфонилбензойной кислоты) можно проводить согласно или аналогично известным способам, например, описанным в WO-A-92/13845 и WO 01/23368 и цитируемой там литературе. Известные способы включают аммонолиз соединения формулы (III) с образованием сульфоамида формулы (IV), фосгенирование соединения (IV) с образованием фенилсульфонилизоцианата формулы (V) и последующей реакции присоединения (соединения) с гетероциклическим амином формулы (VI) с образованием сульфонилмочевины формулы (I) или ее соли. На способы проведения аммонолиза, фосгенирования и присоединения в этом отношении ссылаются на WO 01/23368.
Альтернативно вышеописанным способам, соединение формулы (III) может быть подвергнуто превращению с цианатом, например, металлической солью цианата, особенно цианатом щелочного металла, с образованием изоцианата формулы (V) или его сольватированного (стабилизированного) производного.
В качестве цианатов пригодны цианаты с катионами из группы катионов металлов или органических катионов, как стерически затрудненные органические ионы аммония. Предпочтительными являются, например, цианаты щелочных металлов, предпочтительно цианат натрия и цианат калия, или также цианаты щелочноземельных металлов. Используемый цианат или смесь цианатов вводят в количестве, достаточном для превращения до соединения (V). Как правило, для этого достаточно эквимолярного количества цианата или небольшого избытка, предпочтительно 1-2 мольных эквивалента, в особенности 1-1,5 мольных эквивалента цианата в расчете на соединение (III).
Взаимодействие с цианатом проводят, как правило, в апротонном полярном растворителе. Пригодными растворителями и/или разбавителями являются апротонные органические растворители, которые инертны в условиях реакции, например, такие как
- простые эфиры, как диэтиловый эфир, ди-н-пропиловый эфир, диизопропиловый эфир, метил-трет.бутиловый эфир, тетрагидрофуран (ТГФ), диоксан, моноалкиловый и диалкиловый эфир алкиленгликоля, как, например, монометиловый эфир пропиленгликоля, моноэтиловый эфир пропиленгликоля, монометиловый или моноэтиловый эфир этиленгликоля, диметоксиэтан, диглим, триглим и тетраглим;
- амиды, как диметилформамид (ДМФ), диметилацетамид и N-метилпирролидон;
- кетоны, как ацетон, циклогексанон, метилизобутилкетон (МИБК) (MIBK);
- нитрилы, как ацетонитрил, пропионитрил, бутиронитрил и бензонитрил;
- сульфоксиды и сульфоны, как диметилсульфоксид (ДМСО) (DMSO) и сульфолан,
а также смеси из двух или нескольких вышеназванных растворителей или разбавителей.
Технологически особенный интерес вызывают, как правило, такие растворители, которые могут легко удаляться из продукта посредством дистилляции.
Время реакции можно варьировать в зависимости от размера частиц цианата, поскольку он присутствует в реакционной смеси в твердом виде.
Предпочтительно апротонные растворители выбирают из группы: простые эфиры, например, диизопропиловый эфир и метил-трет.-бутиловый эфир, кетоны, например, метилизобутилкетон (МИБК) и нитрилы, как ацетонитрил.
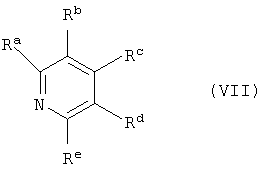
В большинстве случаев предпочтительно проводят реакцию с образованием изоцианата формулы (V) при добавлении в реакционную смесь N-гетероароматических соединений (азотсодержащих гетероциклов) в качестве катализатора или стабилизатора, например, пиридина и производных пиридина формулы (VII),
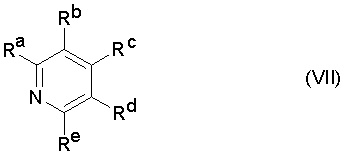
где
Ra, Rb, Rc, Rd и Re обозначают, смотря по обстоятельствам, независимо друг от друга водород, (C1-C6)алкил, (C2-C6)алкенил, (C2-C6)алкинил или (C1-C6)алкокси, или два соседних остатка совместно с С-атомами первого кольца, с которыми они соединены, образуют конденсированное карбоциклическое кольцо с 4 до 8 С-атомами или гетероциклическое кольцо с 4 до 8 атомами в кольце и 1,2 или 3 гетероатомами в кольце из группы N, O и S.
Предпочтительно Ra, Rb, Rc, Rd и Re обозначают, смотря по обстоятельствам, независимо друг от друга водород или один, два или три остатка (C1-C4)алкил или (C1-C4)алкокси, в особенности метил или этил, и остальные обозначают, смотря по обстоятельствам, водород.
Примерами пригодных N-гетероароматических соединений являются пиридин или замещенные пиридины как алкилпиридины, например, пиколины (например, 2-метилпиридин, 3-метилпиридин или 4-метилпиридин) или лутидины (например, 2,4-диметилпиридин, 2,6-диметилпиридин, 2,3-диметилпиридин, 2,5-диметилпиридин, 3,4-диметилпиридин или 3,5-диметилпиридин), или их смеси. Количество N-гетероароматических соединений может варьироваться в широкой области. Целесообразно использовать 0,8 до 2 мольных эквивалентов, предпочтительно 0,9 до 1,5 мольных эквивалентов, в особенности 1 до 1,3 мольных эквивалентов N-гетероароматических соединений на моль сложного эфира галогенсульфонилбензойной кислоты (III), предпочтительно сложного эфира хлорсульфонилбензойной кислоты, в расчете на моль образующегося изоцианата (V).
В случае Y=CH (пиримидин-2-ил) добавление каталитических количеств пиридина или производного пиридина может благоприятно сказываться на получении изоцианата в присутствии цианата.
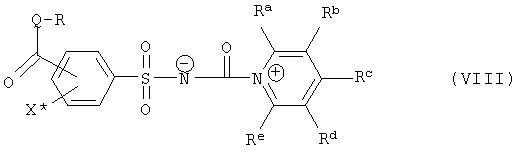
При добавлении стабилизатора из группы N-гетероароматических соединений продукт полностью или частично находится не в форме изоцианата, а в форме стабильного промежуточного продукта в растворе, который в случае использования предпочтительно названных (производных) пиридина формулы (VII) обозначает соединение формулы (VIII),
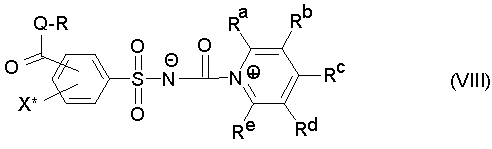
где R, Ra, Rb, Rc, Rd, Re, Q и X* определены как в формуле (III) или формуле (VII).
В случае использования пиридина в качестве стабилизатора образуется соответствующий промежуточный продукт формулы (VIIIa),
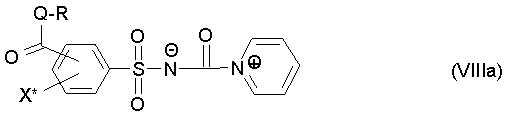
где
R, Q и X* определены как в формуле (III).
Промежуточные продукты формул (VIII) или (VIIIa) являются новыми, и поэтому также относятся к объектам изобретения. Особенно предпочтительны промежуточные продукты формулы (VIIIb)
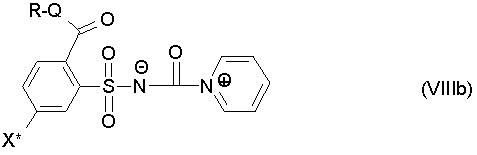
где
R, Q и X* определены как в формуле (III).
Соединения формул (VIII), (VIIIa) и (VIIIb) подтверждены, например, спектроскопическим путем. Они характеризуются характерными сдвигами полос колебаний карбонила в инфракрасном спектре по сравнению с полосой карбонила соответствующего изоцианата формулы (V).
Температура реакции при взаимодействии соединения (III) с цианатом может варьироваться в широких пределах и оптимизироваться в предварительных испытаниях. Она имеет предпочтительно умеренные значения в области от -30°C до 70°C, в особенности от -10°C до 30°C.
Изоцианат (V) затем может быть подвергнут взаимодействию, аналогично изоцианату из варианта (b1), с гетероциклическим амином (VI) до сульфонилмочевины (I). В случае добавления катализатора или стабилизатора целесообразно сначала нейтрализовать кислотой, предпочтительно безводной кислотой, например, хлористым водородом или органической кислотой.
В одном варианте способа реакционную смесь из реакции получения изоцианата после получения цианата непосредственно используют для реакции присоединения изоцианата или стабилизированного изоцианата с образованием сульфонилмочевины формулы (I). Для этого, в случае необходимости, имеющееся N-гетероароматическое соединение нейтрализуют посредством добавления кислоты (как упомянуто выше) и реакционную смесь смешивают с гетероциклическим амином (VI). Альтернативно может быть добавлен гетероциклический амин (VI) и лишь затем добавлена кислота для нейтрализации катализатора/стабилизатора.
Взаимодействие соединений (V) и (VI) проводят, как правило, в органическом растворителе. В качестве растворителя при этом могут использоваться полярные или относительно неполярные апротонные растворители. Предпочтительно при превращениях используют те же растворители, что и при получении изоцианата (V).
Реакцию предпочтительно проводят при температуре в области от 0°С до 100°С, в особенности от 20 до 80°С, очень предпочтительно от 20 до 40°С.
В расчете на один моль сульфонилизоцианата формулы (V) предпочтительно используют 1-1,2 моля, в особенности 1-1,1 моля, особенно предпочтительно 1-1,05 моля амина формулы (VI).
Обработка реакционной смеси после присоединения может происходить по обычным методикам, причем сульфонилмочевины формулы (I) могут быть выделены, например, в несолевой форме или после взаимодействия с основаниями или, в случае необходимости, также с кислотами - в виде солей.
Способ согласно изобретению обеспечивает получение сульфонилмочевин (I) или их солей простым путем, исходя из дихлорида формулы (II) с относительно хорошими, вплоть до превосходных, выходами за три или четыре стадии. В варианте трехстадийного способа согласно изобретению фосгенирование исключают.
Соединения формулы (II) могут быть получены способом, в котором соединение формулы (IX) или его соль,

где X* определен как в формуле (II),
подвергают взаимодействию с одним или несколькими галогенирующими средствами из группы галогенангидридов кислот серы или фосфора на одной или нескольких стадиях реакции с образованием соединения формулы (II), предпочтительно с дихлоридом.
Вышеупомянутый способ является новым и также является объектом изобретения. Соединения формулы (IX) известны, например, WO 95/26952 или могут быть получены аналогично известным способам.
Соединения формулы (II) отчасти известны. Так, в заявке US-A-4110373 описана реакция, в случае необходимости, замещенных бензотрихлоридов посредством олеума. Среди прочих там упомянуто получение 4-хлор-3-хлорсульфонил-бензоилхлорида и 3-хлор-5-хлорсульфонил-бензоилхлорида.
Далее, из заявки NL-A-7603612 известно получение 2-хлорсульфонил-бензоилхлорида из 2-сульфобензойной кислоты посредством реакции с фосгеном в качестве галогенирующего средства в полярных апротонных растворителях как ДМФ. В качестве побочного продукта при этом образуется в значительных количествах дихлортолилсульфон (3,3-дихлор-1,1-диоксо-бензо-1-тиа-2-оксолан). В целом способ описан также для производных сульфобензойных кислот, дополнительно галогенированных или нитрированных в бензольном кольце.
Соединения формулы (II) или формулы (IIa), где галогенсульфонильная группа находится в орто-положении к галогенангидридной группе карбоновой кислоты и которые в пара-положении к галогенангидридной группе карбоновой кислоты дополнительно галогенированы, предпочтительно йодированы, также являются новыми и относятся к объектам изобретения (соединения (IIa)).
Вследствие того что известные методы галогенирования фосгеном приводят к побочным реакциям, была потребность в разработке альтернативного способа, позволяющего получать соединения (II) или (IIa).
В качестве галогенирующих средств имеют в виду неорганические галогенангидриды кислот серы и фосфора, например, тионилгалогениды, как тионилфторид или тионилхлорид, или сульфурилгалогениды, как сульфурилхлорид, или фосфоргалогениды, как трихлорид фосфора, фосфорилхлорид, пентахлорид фосфора, трибромид фосфора. Предпочтительно получение дихлоридов (формула (II) или (IIa), где Hal1=Cl и Hal2=Cl), предпочтительно с тиохлоридом, трихлоридом фосфора, фосфорилхлоридом или пентахлоридом фосфора, в особенности с тионилхлоридом.
Галогенирующее средство используют, как правило, в количестве одного реакционного эквивалента на реакционную группу или в избытке. При этом следует принимать во внимание общепринятые реакционные эквиваленты, например, для хлорирующего средства тионилхлорида 1 реакционный эквивалент, для PCl3 и POCl3 три реакционных эквивалента, для PCl5 в зависимости от условий реакции один или четыре реакционных эквивалента.
На моль соединения формулы (IX) по стехиометрии требуется по меньшей мере 2 эквивалента галогенирующего средства. Как правило, в зависимости от галогенирующего средства достаточно 2-10 эквивалентов галогенирующего средства на моль дикислоты. В случае тионилхлорида используют предпочтительно 4-8 молей на моль соединения формулы (IX). Избыток галогенирующего средства и, в случае необходимости, образующийся галогенводород удаляют, предпочтительно, непрерывно во время или после реакции из продукта реакции или химически связывают. В случае тионилхлорида чаще всего возможно избыток отгонять из продукта.
Галогенирование соединения (IX) проводят, как правило, в присутствии инертного (относительно) неполярного органического растворителя, но в отдельных случаях его также можно проводить в массе. В качестве растворителей имеют в виду многочисленные инертные растворители, предпочтительно, широко используемые неполярные растворители, которые в условиях галогенирования не реагируют или значительно не реагируют с галогенирующим средством или продуктом реакции. Например, пригодными растворителями являются:
- алифатические и ароматические углеводороды, например, минеральные масла, простые эфиры нефти, н-пентан, н-гексан, циклопентан, циклогексан или толуол, ксилол, мезитилен, производные нафталина, ®Solvesso 200 (высококипящая смесь ароматических углеводородов);
- галогенированные алифатические и ароматические углеводороды, например метиленхлорид, дихлорэтан или хлорбензол, хлортолуол или дихлорбензол,
а также смеси двух или нескольких вышеназванных растворителей или разбавителей. Предпочтительно пригодны такие инертные растворители, которые также могут быть использованы на предварительной стадии или последующей стадии всего способа. Например, для этого пригодны, в случае необходимости, галогенированные ароматические углеводороды, предпочтительно высококипящие органические растворители как ксилол, толуол, мезитилен, хлорбензол или дихлорбензол, или их смеси.
Реакция галогенирования может катализироваться путем добавления полярных слабо нуклеофильных основных соединений. Например, для этого могут быть использованы стерически затрудненные аминные основания, например, в виде трехзамещенных аминов или азотсодержащих гетероциклов. В принципе пригодны, например, триэтиламин, пиридин, алкилпиридины (например, пиколины, лутидины), DBU (1,8-диазабицикло[5.4.0]ундек-7-ен), DABCO (1,4-диазабицикло[2.2.2]октан) или амиды, как диалкилформамиды: DMF (диметилформамид), диэтилформамид, ди-н-пропилформамид, ди-изопропилформамид, ди-н-бутилформамид или ди-н-пентилформамид, или диметилацетамид. С практической точки зрения, учитывая облегчение обработки, очистку или эффективность, доля катализатора должна быть выбрана возможно более малой. Часто для ускорения реакции достаточно количество от 0,01 до 2 мольных эквивалентов, предпочтительно 0,02 до 1 мольного эквивалента катализатора в расчете на соединение формулы (IX). Пригодная температура реакции может варьироваться в зависимости от галогенирующего средства и катализатора и просто определяется в предварительных опытах. Как правило, она находится в области от -10°C до температуры кипения соответствующего растворителя, предпочтительно 20°C до 150°C, в особенности 40°C до 120°C, причем нижнюю границу температуры реакции определяют путем установленного превращения.
В принципе можно проводить реакцию, в которой эддукт формулы (IX), чаще всего в массе, или растворенный или суспендированный в растворителе, подают с катализатором при повышенной температуре (выше температуры, при которой проводили бы реакцию) и затем добавляют галогенирующий агент. Несмотря на повышенную температуру, может случиться, что реакция начинается замедленно, проходит относительно медленно и/или проходит не полностью.
Альтернативно этому галогенирующий агент, предпочтительно хлорирующий агент может быть подан с катализатором при повышенной температуре, предпочтительно от 60 до 80°C, и затем добавлен эдукт, или в виде твердого вещества в массе, или в суспензии в растворителе. Тем самым можно избежать большого избытка эдукта в реакторе.
После полного окончания реакции избыток галогенирующего агента предпочтительно отгоняют, что, в случае необходимости, может происходить при пониженном давлении. Вследствие реакционной способности дигалогенида формулы (II) его после получения, предпочтительно без промежуточного выделения, непосредственно перерабатывают далее, то есть, например, путем описанной ранее моноэтерификации до соединения формулы (III) или его соли.
В следующих примерах количественные данные приведены в расчете на вес, если нет других указаний.
Пример 1. Получение дигалогенида
Получение 2-хлорсульфонил-4-йод-бензоилхлорида путем добавки твердого 3-йод-6-карбоксибензолсульфоната калия
650 г (5,3 моль) тионилхлорида (технический, >97%-ный) загружают в токе азота, нагревают до 70°C и медленно добавляют 8 г (0,11 моль) диметилформамида. После окончания добавления перемешивают при заданной температуре 15 минут, до того, как с помощью шнека для твердых веществ так быстро добавляют 242 г (0,661 моль)монокальциевой соли3-йод-6-карбоксибензолсульфокислоты, чтобы возник хорошо контролируемый поток отходящего газа. После окончания добавления температуру внутри реактора медленно повышают до 85°C и в течение 2 часов поддерживают эту температуру. Затем избыточный тионилхлорид отгоняют при температуре 90-100°C. Во время отгонки давление снижают до минимально 180 мбар. Получают расплав продукта с суспендированным тонкокристаллическим хлоридом калия.
Для удаления остаточного тионилхлорида добавляют 100 мл ксилола и отгоняют в вакууме. Полученная суспензия после добавки ксилола может быть использована в последующей этерификации без дальнейшей обработки. Альтернативно может быть проведена обработка, в которой смесь разбавляют 400 мл ксилола и после охлаждения до приблизительно30°C фильтруют в инертной атмосфере. Осадок дважды промывают ксилолом, и очищенный фильтрат освобождают от растворителя под вакуумом. Получают 238 г (0,652 моль=98,6% d. Th.) слегка желтоватого твердого вещества (чистота>99%, ВЭЖХ).
Пример 2.Получение дигалогенида
Получение 2-хлорсульфонил-4-йод-бензоилхлорида при добавлении ксилольной суспензии 3-йод-6-карбоксибензолсульфоната калия
650 г (5,3 моль) тионилхлорида (технический, >97%-ный) загружают в токе азота, нагревают до 70°C и медленно добавляют 8 г (52 ммоль) ди-н-бутилформамида. После окончания добавления перемешивают при заданной температуре 15 минут. Температуру повышают до 85°C, перед тем как быстро добавляют хорошо перемешанную суспензию тонко размельченной монокальциевой соли3-йод-6-карбоксибензолсульфокислоты в количестве 242 г (0,74 моль) в 400 мл толуола, чтобы возник хорошо контролируемый поток отходящего газа. После окончания добавления смывают небольшим количеством ксилола и выдерживают в течение 2 часов при этой температуре. Затем избыточный тионилхлорид отгоняют при температуре 90-100°C на ректификационной колонне. Во время отгонки давление снижают до минимально 180 мбар. Отгоняют до тех пор, пока в кубе колонны температура кипения ксилола достигнет стабильного значения при соответствующем давлении. В случае необходимости отогнанный ксилол пополняют путем добавки свежего ксилола. Полученная реакционная смесь может быть обработана как в примере 1 или непосредственно использована для последующей этерификации.
Пример 3.Получение дигалогенида
Получение 2-хлорсульфонил-4-йод-бензоилхлорида путем превращения 4-йод-2-сульфобензойной кислоты
20,9 г 4-йод-2-сульфобензойной кислоты суспендируют в 80 мл оксихлорида фосфора (POCl3). При перемешивании добавляют 26,7 г пентахлорида фосфора (PCl5). Медленно нагревают до температуры 100-110°C, причем от температуры внутри реактора приблизительно 40°C начинается сильное газообразование. По истечении часа после проведения реакции реакционную смесь охлаждают до комнатной температуры, и растворитель отгоняют на роторном испарителе. Получают 28,3 г сырого 2-хлорсульфонил-4-йод-бензоилхлорида.
Пример 4. Получение соединения (III)
Получение метилового эфира 2-хлорсульфонил-4-йод-бензойной кислоты.
Реакционную смесь, полученную как в примере 1 или 2, охлаждают до 20-25°C и прикапывают 140 мл (110,6 г) метанола таким образом, чтобы температура внутри не превышала 28°C. После окончательного добавления перемешивают вплоть до полного превращения и обрабатывают по варианту А или В.
Вариант A: Избыточный метанол полностью отгоняют в вакууме при температуре менее 30°C совместно с некоторым количеством ксилола. Разбавляют ксилолом до общего объема 1320 мл и в три порции промывают водой. Отделяют органическую фазу и сушат посредством азеотропной перегонки в вакууме. Продукт для последующей реакции может быть использован в форме ксилольного раствора.
Альтернативно растворитель отгоняют под вакуумом и получают 237 г (=99,5% тель-кель* (без гарантии качества)) основного соединения в виде застывшего расплава с чистотой 98% (ВЭЖХ), (97,4% d.Th.*).
* = в расчете на используемую монокалиевую соль 3-йод-6-карбоксибензолсульфокислоты.
Вариант B: Разбавляют ксилолом до общего объема 1320 мл и в три порции промывают водой. Отделяют органическую фазу и сушат посредством азеотропной перегонки в вакууме. Продукт для последующей реакции может быть использован в виде ксилольного раствора.
Альтернативно растворитель отгоняют под вакуумом и получают 235 г (=98,6% тель-кель* (без гарантии качества)) основного соединения в виде застывшего расплава с чистотой 97% (ВЭЖХ), (95,6% d.Th.*).
* = в расчете на используемую монокалиевую соль 3-йод-6-карбоксибензолсульфокислоты.
Пример 5:Получение соединения (III) из дихлорангидрида кислоты
Получение проп-2-инилового эфира 2-хлорсульфонил-4-йод-бензойной кислоты
28,3 г сырого дихлорангидрида кислоты, например, из примера 4, растворяют в 150 мл хлороформа и смешивают с 7,7 мл пропаргилового спирта. Кипятят в течение 3 часов, охлаждают до комнатной температуры и выливают в 200 мл ледяной воды. Водную фазу нейтрализуют NaHCO3, разделяют фазы и еще два раза экстрагируют водную фазу дихлорметаном. Объединенные органические фазы сушат над Na2SO4, и растворитель отгоняют на роторном испарителе. Получают 23,5 г сырого проп-2-инилового эфира 2-хлорсульфонил-4-йод-бензойной кислоты.
Пример 6: Получение соединения (III) из галоген-сульфобензойной кислоты
Получение проп-2-инилового эфира 2-хлорсульфонил-4-йод-бензойной кислоты
98,4 г 4-йод-2-сульфобензойной кислоты растворяют в растворе 33,7 г гидроксида калия (лепешки) в 150 мл воды и 500 мл метанола. Затем растворитель отгоняют на роторном испарителе и сушат осадок под высоким вакуумом. Получают 133,9 г сырой дикалиевой соли исходного соединения. К 66 г этой соли прикапывают 150 мл тионилхлорида и добавляют еще 4,7 мл диметилформамида (ДМФ). Кипятят до прекращения газовыделения, и затем отгоняют растворитель при исключении влаги на роторном испарителе. Затем прикапывают при охлаждении 150 мл пропаргилового спирта и перемешивают 6 часов при комнатной температуре. Реакционный раствор упаривают, добавляют 150 мл воды, отсасывают и фильтровальную массу хорошо промывают водой. Осадок на фильтре суспендируют в дихлорметане, фильтруют и отделяют органическую фазу. Получают 35,2 г проп-2-инилового эфира 2-хлорсульфонил-4-йод-бензойной кислоты с температурой плавления 69-73°C. Дополнительно фаза содержит 14,3 г 4-йод-2-сульфобензойной кислоты в виде продукта гидролиза.
Пример 7: Получение метилового эфира 4-йод-[3-(4-метокси-6-метил-1,3,5-триазин-2-ил)-уреидосульфонил]-бензойной кислоты
106 г метилового эфира 2-хлорсульфонил-4-йод-бензойной кислоты (97%-ного) растворяют в 500 г ацетонитрила и смешивают с 23 г технического цианата натрия (95%). Смесь охлаждают до 6 -10°C и добавляют 25 г пиридина в 100 мл ацетонитрила в течение 2-4 часов и перемешивают вплоть до полного превращения (получение стабилизированного изоцианата).
Затем добавляют 43 г технического 2-амино-4-метокси-6-метил-1,3,5-триазина и охлаждают до 0°C. Затем подают 12 г сухого хлористого водорода (в виде газа) под поверхность и после окончательного введения при 40°C перемешивают вплоть до полного превращения. После отгонки ацетонитрила при пониженном давлении отфильтровывают осадок и промывают ацетонитрилом. Осадок, суспендированный со смесью из ацетона и разбавленной соляной кислоты, вновь фильтруют, промывают водой и ацетоном и сушат под вакуумом. Получают 114 г (77,3% d. Th.) основного соединения в виде белого порошка (чистота: более 98%, ВЭЖХ).
Изобретение касается способа получения соединения формулы (I)
или его соли, где Q, X*, Y, Z, R, R1, R2 и R3 определены как в пункте 1, (Ia), (II): (R*=Hal, R**=Hal), (III): (R*=-Q-R, R**=Hal), (V): R*=-Q-R, R**=-NCO), отличающегося тем, что а) соединение (II), где Hal=атом галогена, подвергают моноэтерификации до соединения(III),
b) или b1) подвергают аммонолизу и фосгенированию до соединения (V), или b2) подвергают взаимодействию с цианатом до соединения (V), и
с) полученное соединение (V) подвергают взаимодействию с аминогетероциклом H2N-Het (Het=гетероцикл как в (I)) до соединения (I) или его соли. Некоторые соединения (II) и стабилизированные N-гетероароматическими соединениями промежуточные продукты (VIII) соединений (V) являются новыми. Соединения (II) могут быть получены из соответствующих сульфобензойных кислот путем взаимодействия с галоидангидридами кислот серы и фосфора в неполярных органических растворителях. 6 н. и 13 з.п. ф-лы.
где Q обозначает кислород или серу,
X* обозначает водород, галоген, циано, нитро, (С1-С3)-алкил или метокси,
Y, Z независимо друг от друга обозначают СН или N, причем Y и Z одновременно не являются СН,
R обозначает водород, (С1-С12)-алкил, (С2-С10)-алкенил, (С2-С10)-алкинил, (С1-С6)-алкил, одно-четырехкратно замещенный остатком из группы: галоген, (С1-С4)-алкокси, (С1-С4)-алкилтио, CN, [(С1-С4)-алкокси]-карбонил и (С2-С6)-алкенил; или (С3-С8)-циклоалкил, незамещенный или замещенный остатком из группы: (С1-С4)-алкил, (С1-С4)-алкокси, (C1-C4)-алкилтио и галоген; (С5-С8)-циклоалкенил, фенил-(С1-С4)-алкил, в фенильном остатке незамещенный или замещенный одним или несколькими остатками из группы: галоген, (С1-С4)-алкил, (С1-С4)-алкокси, (С1-С4)-галогеналкил, (С1-С4)-алкилтио, [(С1-С4)-алкокси]-карбонил, [(С1-С4)-алкил]-карбонилокси, карбамоил, [(С1-С4)-алкил]-карбониламино, [(С1--C4)-алкил]-аминокарбонил, ди-[(C1-C4)-алкил]-аминокарбонил и нитро; или остаток формул A-1 - A-10
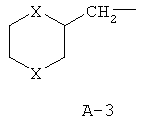
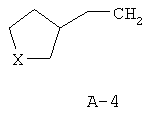
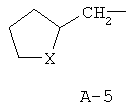
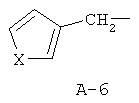
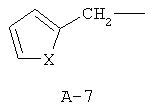
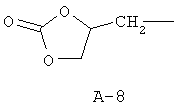
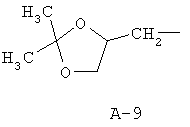
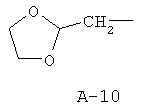
причем в формулах A-1 - A-10
X или оба X независимо друг от друга обозначают O, S, S(O) или SO2,
R1 обозначает водород или (C1-C3)-алкил,
R2 обозначает водород, галоген, (C1-C3)-алкил или (C1-C3)-алкокси, причем каждый из обоих остатков, названных последними, не замещен или одно- или многократно замещен галогеном или (C1-C3)-алкокси,
R3 обозначает водород, галоген, (C1-C3)-алкил, (C1-C3)-алкокси или (C1-C3)-алкилтио, причем каждый из трех остатков, названных последними, незамещен или одно- или многократно замещен галогеном, или одно- или двукратно (C1-C3)-алкокси или (C1-C3)-алкилтио; или остаток формулы NR4R5, (C3-C6)-циклоалкил, (C2-C4)-алкенил, (C2-C4)-алкинил, (C3-C4)-алкенилокси или (C3-C4)-алкинилокси,
R4 и R5 независимо друг от друга обозначают водород, (C1-C4)-алкил, (C3-C4)-алкенил, (C1-C4)-галогеналкил или (C1-C4)-алкокси,
отличающийся тем, что
a) соединение формулы (II)
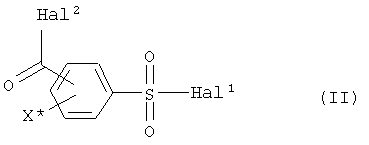
где Hal1 обозначает атом галогена,
Hal2 обозначает атом галогена,
X* определен, как в формуле (I),
посредством реакции с соединением формулы R-Q-H или его солью подвергают взаимодействию с образованием соединения формулы (III)
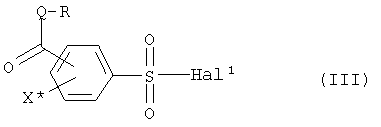
причем R, Q и X определены, как в формуле (I), и Hal1 определен, как в формуле (II), и
(b) или с промежуточным выделением или без него
(b1) полученное соединение (III) подвергают аммонолизу с получением сульфоамида формулы (IV)
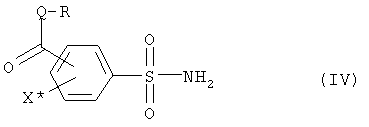
где R, Q и X* определены, как в формуле (III),
и соединение (IV) с промежуточным выделением или без промежуточного выделения подвергают взаимодействию с фосгеном с получением фенилсульфонилизоцианата формулы (V)
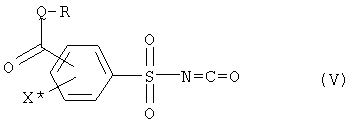
где R, Q и X* определены, как в формуле (III),
или
(b2) полученное соединение (III) подвергают взаимодействию с цианатом с получением изоцианата формулы (V) или его сольватированного (стабилизированного) производного,
и
(с) с промежуточным выделением или, предпочтительно, без промежуточного выделения изоцианата формулы (V) или его стабилизированное производное подвергают взаимодействию с гетероциклическим амином формулы (VI)
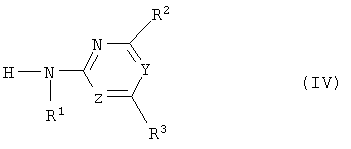
где R1, R2, R3, Y и Z определены, как в формуле (I),
с получением сульфонилмочевины формулы (I) или ее соли.
Q обозначает атом кислорода,
X* обозначает атом водорода или атом галогена,
R обозначает (С1-С4)-алкил, (С2-С4)-алкенил, (С2-С4)-алкинил, (С1-С4)-галогеналкил или (С1-С4)-алкокси(С1-С4)-алкил,
R1 обозначает атом водорода,
R2 обозначает (С1-С4)-алкил или (С1-С4)-алкокси,
R3 обозначает (С1-С4)-алкил или (С1-С4)-алкокси,
Y обозначает атом азота или группу формулы СН и
Z обозначает атом азота.
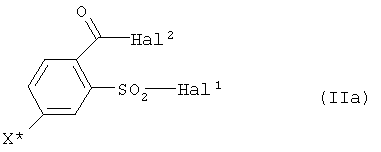
где Hal1 обозначает галоген,
Hal обозначает галоген и
X* определен, как в формуле (I).
X* обозначает атом йода,
R обозначает метил или этил,
R1 обозначает атом водорода,
R2 обозначает метокси,
R3 обозначает метил, и
Y обозначает атом азота,
Z обозначает атом азота.
где R, Q и X* определены, как в формуле (I) по п.1, и Ra, Rb, Rc, Rd и Re, смотря по обстоятельствам, независимо друг от друга обозначают водород, (С1-С6)алкил, (С2-С6)алкенил, (С2-С6)алкинил или (С1-С6)алкокси, или два соседних остатка вместе с соединенными с ними С-атомами первого кольца образуют конденсированное карбоциклическое кольцо с 4 до 8 С-атомами или гетероциклическое кольцо с 4 до 8 атомами в кольце и 1, 2 или 3 гетероатомами в кольце из группы N, О и S.
причем Hal1 обозначает атом галогена,
подвергают взаимодействию с цианатом в присутствии соединения формулы (VII)
причем в формулах (III) и (VII) остатки R, Ra, Rb, Rc, Rd, Re, Q и X* определены, как в формуле (VIII).
где Hal1 и Hal2 независимо друг от друга, смотря по обстоятельствам, обозначают атом галогена, и X* обозначает водород, галоген, циано, нитро, (С1-С3)-алкил или метокси, отличающийся тем, что соединение формулы (IX) или его соль

где X* определен, как в формуле (II),
подвергают взаимодействию с одним или несколькими галогенирующими средствами из группы неорганических галогенангидридов кислот серы или фосфора на одной или нескольких стадиях реакции с образованием соединения формулы (II).
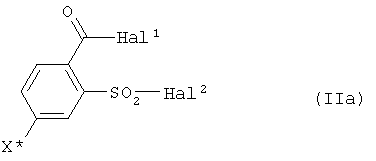
где Hal1, Hal2 независимо друг от друга, смотря по обстоятельствам, обозначают атом галогена и X* обозначает атом йода.
где R и Q определены, как в формуле (I), и X* определен, как в формуле (I), подвергают взаимодействию с цианатом с получением изоцианата формулы (V)
где R, Q и X* определены, как в формуле (III),
или его сольватированного (стабилизированного) производного,
и
полученное соединение (V) или его стабилизированное производное подвергают взаимодействию с гетероциклическим амином формулы (VI) 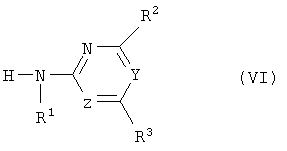
где R1, R2, R3, Y и Z определены, как в формуле (I),
с получением сульфонилмочевины формулы (I) или ее соли.
| WO 9213845 А1, 20.08.1992 | |||
| Устройство для прессования творога | 1959 |
|
SU123368A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ N-(1,3,5-ТРИАЗИН-2-ИЛ)АМИНОКАРБОНИЛАРИЛСУЛЬФОНАМИДОВ | 1995 |
|
RU2103263C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ N-[(1,3,5-ТРИАЗИН-2-ИЛ)АМИНОКАРБОНИЛ]БЕНЗОЛСУЛЬФОНАМИДОВ | 1984 |
|
SU1233456A1 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ NAMPT | 2011 |
|
RU2616612C2 |

