Изобретение относится к способу производства производных 5-метоксиметилпиридин-2,3-дикарбоновой кислоты и дальнейшему преобразованию этих соединений до гербицидных 5-замещенных-2-(2-имидазолин-2-ил) никотиновых кислот, таких как имазамокс.
Производные 2-(2-имидазолин-2-ил) никотиновых кислот, как имазамокс (2-[(RS)-4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил]-5-метоксиметилникотиновая кислота),
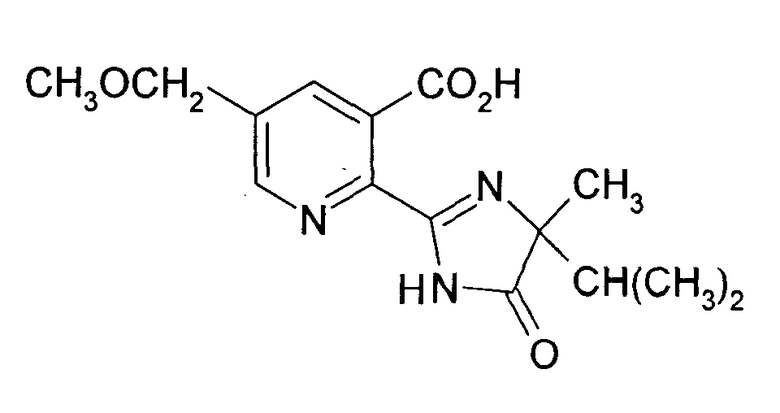
являются пригодными селективными гербицидами, которые действуют как ALS-ингибиторы и могут быть применены в пред- и пост-появление применениях.
Из литературы известны различные способы синтеза этих соединений, смотри, например, ЕР-А 0322616, US 5,378,843, US 5,760,239, EP-A 0933362 или Q. Bi et al, Modern Agrochemicals 6(2)(2007) 10-14.
US 5,378,843 раскрывает синтез 5-(метоксиметил)-2,3-пиридиндикарбоновой кислоты реакцией диметилового сложного эфира [(5,6-дикарбокси-3-пиридил)метил] триметиламмоний бромида с метоксидом натрия/метанолом и NaOH/вода, сопровождаемой ацилированием.
US 5,760,239 раскрывает улучшенный способ преобразования 5,6-дикарбоксил-3-пиридилметил аммоний галогенида до 5-(алкоксиметил)пиридин-2,3-дикарбоксилатной соли в одну стадию закрытой реакции с выделением спирта и основания при температуре около 120-180°С.
Хотя синтез этими способами осуществляется в промышленном масштабе, существует все еще область для его усовершенствования, особенно ввиду экономичных и экологических аспектов, таких как полное усовершенствование выхода или избежание определенных растворителей или реактивов.
Одна задача изобретения состоит в том, чтобы обеспечить улучшенный способ производства 5-метоксиметилпиридин-2,3-дикарбоновых кислот или карбоксилатов. Дальнейшая задача изобретения состоит в том, чтобы обеспечить улучшенный способ преобразования этих соединений до гербицидных 2-(2-имидазолин-2-ил) никотиновых кислот или их производных.
Было найдено, что метоксилирование 5,6-дизамещенных-3-метилпиридинов и дальнейшая реакция с третичными аминами может быть значительно улучшена путем работы в системе метанол/вода и метанолат или смеси метанолата и гидроксида под давлением при температуре около 75-110°С.
Соответственно, в одном объекте изобретения обеспечивается способ производства 2,3-дизамещенного-6-метоксиметилпиридина формулы (I),
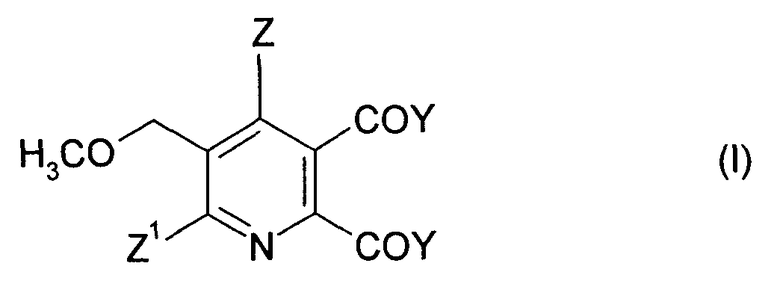
где
Z означает И или галоген;
Z1 означает Н, галоген, CN или NO2;
Y означает ОМ, и
М означает Н, щелочной металл или щелочноземельный металл, включающий стадии:
(i) реакция соединения формулы (II)
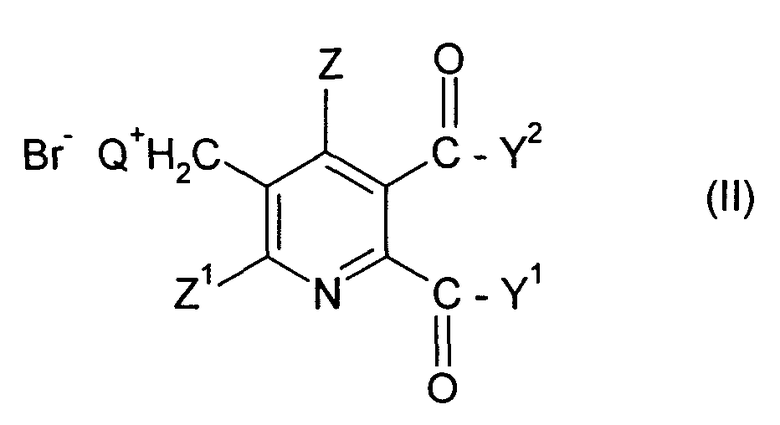
где
Q означает третичный алифатический или циклический, насыщенный, частично ненасыщенный или ароматический амин;
Z означает Н или галоген;
Z1 означает Н, галоген, CN или NO2;
Y1 и Y2 каждый независимо означают OR1, NR1R2, или когда взяты вместе Y1Y2 означает -О-, -S- или -NR3-;
R1 и R2 каждый независимо означают Н,
C1-C4 алкил, необязательно замещенный C1-C4 алкоксигруппой или фенилом, необязательно замещенным от одной до трех C1-C4 алкильными группами, C1-C4 алкоксигруппами или атомами галогена, или
фенил, необязательно замещенный от одной до трех C1-C4 алкильными группами, C1-C4 алкоксигруппами или атомами галогена;
R3 означает Н или C1-C4 алкил,
в смеси метанол/H2O, содержащей по меньшей мере 20 мас.% H2O (в пересчете на суммарное количество воды и бромида (II)), с основанием, содержащим МОСН3 и/или МОН, где М означает щелочной металл или щелочноземельный металл, под давлением в закрытой емкости при температуре от около 75 до 110°С.
В дополнительном объекте изобретения обеспечивается способ получения гербицидного имидазолинонового соединения формулы (III),
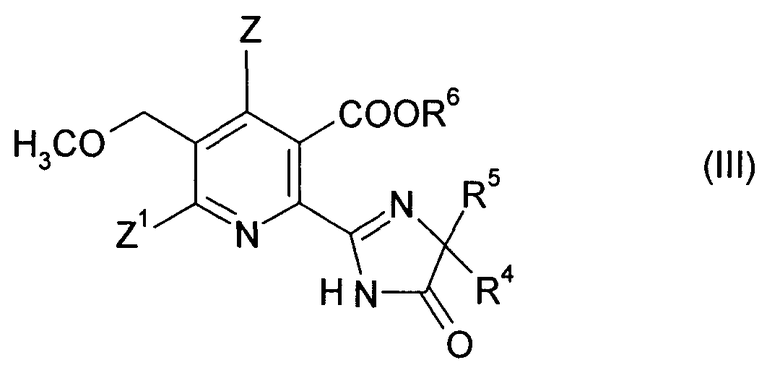
в которой
Z, Z1 являются такими, как определено в формуле (I);
R4 означает C1-C4 алкил;
R5 означает C1-C4 алкил, С3-С6 циклоалкил или R4 и R5, когда взяты вместе с атомом, к которому они присоединены, означают С3-С6 циклоалкильную группу, необязательно замещенную метилом, и
R6 означает водород; группу формулы -N=C (низший алкил)2; C1-C12 алкил, необязательно замещенный одной из следующих групп: C1-С3 алкоксигруппа, галоген, гидроксил, С3-С6-циклоалкил, бензилоксигруппа, фурил, фенил, галофенил, низший алкилфенил, низший алкоксифенил, нитрофенил, карбоксил, низший алкоксикарбонил, цианогруппа или три низший алкиламмоний; С3-C12 алкенил, необязательно замещенный одной из следующих групп: C1-С3 алкоксигруппа, фенил, галоген или низший алкоксикарбонил или двумя C1-С3 алкоксигруппами или двумя группами галогена; С3-С6-циклоалкил, необязательно замещенный одной или двумя C1-С3 алкильными группами; или
катион предпочтительно выбранный из группы, состоящей из щелочных металлов, щелочноземельных металлов, марганца, меди, железа, цинка, кобальта, свинца, серебра, никеля, аммония и органического аммония;
включающий стадии:
(i) получения соединения формулы (I), как описано выше,
(ii) превращения соединения формулы (I) в гербицидное соединение формулы (III).
Способ изобретения приводит к более высоким выходам, более высокой продуктивности (более высокой объемной производительности, более низким фиксированным расходам, более низким затратам сырья (NaOH более дешевый, чем Na-метилат) и улучшенной селективности для соединений формулы (I). Это дает возможность осуществлять способ при более низких температурах, более высоких содержаниях воды и за более короткое время реакции.
Предпочтительными являются соединения формулы (I), где символы имеют следующие значения:
Z предпочтительно означает Н.
Z1 предпочтительно означает Н.
Y предпочтительно означает ОН.
Предпочтительно, все символы в формуле (I) имеют предпочтительные значения.
Особенно предпочтительным соединением формулы (I) является соединение формулы (Ia):
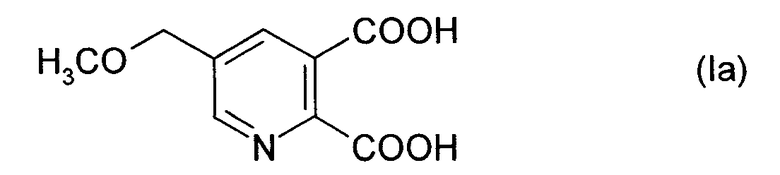
Соединения формулы (II) и их получение известны, например, из US 5,378,843.
Предпочтительными соединениями формулы (II) являются те, которые приводят к предпочтительным соединениям формулы (I). Предпочтительно Y1, Y2 означают OR1 с R1 означающим C1-C4 алкил. Подразумевается, что из-за присутствия воды может иметь место частичный гидролиз диэфира (или других групп).
Q предпочтительно означает
 ;
; 
 ;
; 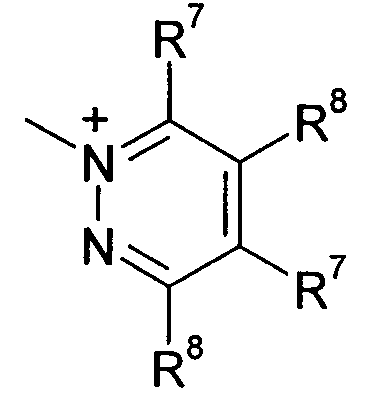 ;
; 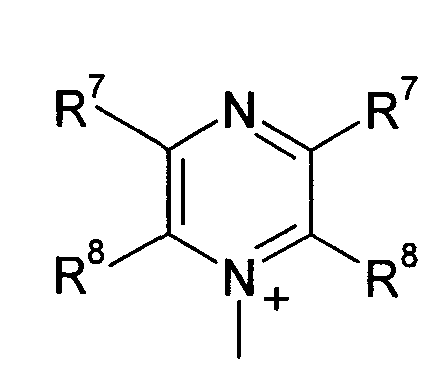 и
и
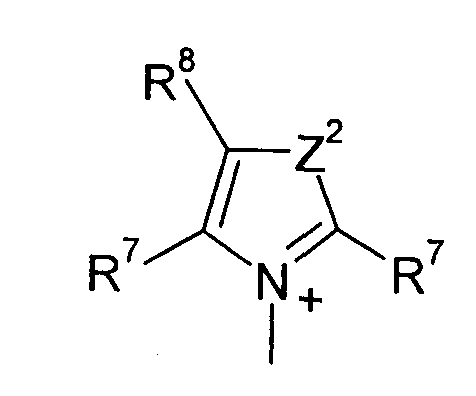 ;
;
Z2 означает О, S или NR12;
R12 означает C1-C4 алкил;
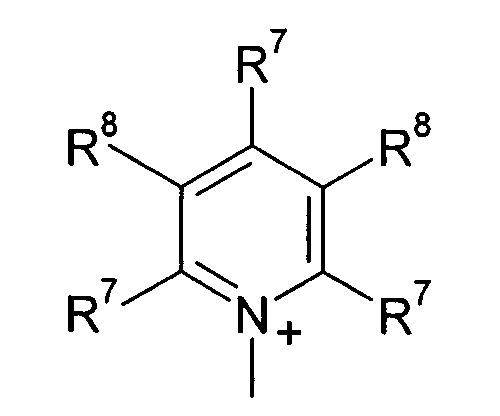
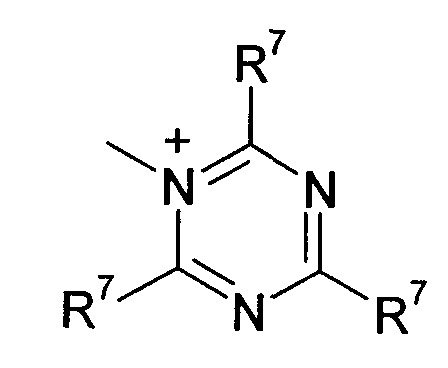
R7 и R8 каждый независимо означают водород, галоген, C1-C4 алкил или C1-C4 алкоксигруппу, или, когда взяты вместе, R7 и R8 образовывают 5- или 6-членное насыщенное или ненасыщенное кольцо, необязательно содержащее О, S, или NR12 и необязательно замещенное от одного до трех атомами галогена, C1-C4 алкильными группами или C1-C4 алкоксигруппами, и
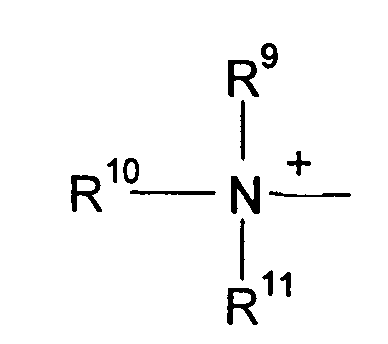
R9, R10 и R11 каждый независимо означают C1-C4 алкил, или R9 и R10, когда взяты вместе, образовывают 5-или 6-членное кольцо, в котором R9 R10 представлены структурой -(CH2)n-, необязательно содержащее О, S или NR9, где n означает целое число 3, 4 или 5, предусмотренный R11 означает C1-C4 алкил.
Q+ более предпочтительно означает ⊕NR9R10R11 или пиридиний, и
R9, R10 и R11 более предпочтительно означают каждый независимо C1-C4 алкил, или R9 и R10, когда взяты вместе, образовывают 5- или 6-членное кольцо, в котором R9R10, представлены структурой: -(СН2)n-, необязательно содержащей О, S или NR12, где n означает целое число или 3, 4 или 5, предусмотренный R11 означает C1-C4 алкил; в особенности предпочтительно R9, R10 и R11 означают C1-C4 алкил.
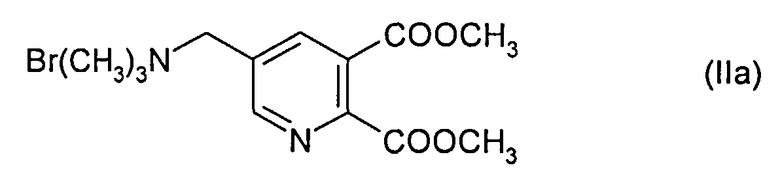
Особенно предпочтительным соединением формулы (II) является соединение формулы (IIa):
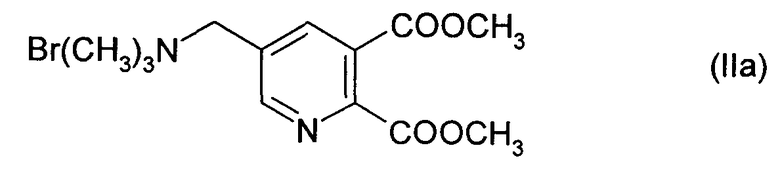
Реакцию проводят в смеси растворителей, содержащей метанол и по меньшей мере 20 мас.% воды (в пересчете на суммарное количество воды и бромида (II)). Предпочтительно количество воды составляет от около 25 до около 75%, более предпочтительно 30-70%, в особенности от около 40 до около 50%. Остаток смеси растворителей представляет собой метанол и до около 50%, предпочтительно до около 20% дополнительных растворителей, предпочтительно выбранных из толуола, хлорбензола и этанола.
Массовое соотношение метанола к бромиду (II) в основном находится в диапазоне от 0.5-25:1, предпочтительно 1-20:1, более предпочтительно 1-10:1, в особенности 2-3:1.
Основание включает МеОМ, МОН или смесь МеОМ и МОН, где М означает щелочной металл или щелочноземельный металл, предпочтительно Na или K, в особенности Na. В случаях смесей МеОМ/МОН, М может быть одинаковым или разным, предпочтительно одинаковым.
Если присутствует МеОМ, молярное соотношение МеОМ, предпочтительно MeONa, к бромиду (II) в основном находится в диапазоне 1-10:1, предпочтительно 1-7.5:1, более предпочтительно 1.25-7:1, в особенности 1.25-2:1.
Если присутствует МОН, молярное соотношение МОН, предпочтительно NaOH, к бромиду (II) в основном находится в диапазоне 0.5-10:1, предпочтительно 1-7:1, более предпочтительно 3-5:1. Если смесь МеОМ и МОН применяется как основание молярное соотношение МОН к бромиду (II) в основном находится в диапазоне 0.5-7.5:1, предпочтительно 1-5:1, более предпочтительно 3-5:1. В одном предпочтительном варианте осуществления не добавляется МОН к реакционной смеси. По экономичным причинам выгодно использовать избыток МОН относительно МеОМ. В другом предпочтительном варианте осуществления применяется только МОН в соотношении 3-10:1, более предпочтительно 4-7:1, более предпочтительно 4.5-6:1.
Молярное соотношение общего количества добавляемого основания/бромид (II) в основном находится от около 2.5-10:1, предпочтительно от около 3-7:1, особенно предпочтительно от около 4.5-6:1. МеОМ обычно добавляют растворенным в метаноле. МОН обычно добавляют растворенным в воде.
Реакцию проводят при температуре в диапазоне от около 75 до 110°С, предпочтительно в диапазоне от около 80 до 105°С, более предпочтительно в диапазоне от около 80 до 100°С.
Реакцию проводят в закрытой емкости, например, в реакторе высокого давления Парра, при повышенном давлении, которое в основном находится в диапазоне от около 1.01 до 5.00 бар, предпочтительно от около 1.02 до 4.00 бар, в особенности от около 1.03 до 3.50 бар. В предпочтительном варианте осуществления внешнее давление не оказано, и реакцию проводят при давлении, сформированным от растворителей при температуре реакции в замкнутой посуде.
Время реакции в основном находится в диапазоне от около 5 до 20 ч, предпочтительно в диапазоне от около 6 до 9 ч, в особенности около 8 ч.
В предпочтительном варианте осуществления бромид (II), содержащий от около 25 до 75 мас.% воды (в пересчете на суммарное количество воды и бромида (II)), взятый в метаноле, водный NaOH медленно добавляют при температуре в диапазоне от около 25 до 40°С, за чем следует NaOCH3 в метаноле, который медленно добавляют при температуре в диапазоне от около 40 до 50°С. Реакционную смесь затем нагревают до температуры реакции (в основном 80-100°С) в реакторе высокого давления, который закрыт, после чего давление растет по мере достижения температуры реакции.
После завершения реакции смесь охлаждают и она может быть обработана согласно известным процедурам, например путем охлаждения, обработкой кислотой, такой как серная кислота, до осаждения соединения (I) и может быть отфильтрована.
В предпочтительном варианте осуществления изобретения обеспечивается способ производства соединения формулы (I), включающий стадии
(i-1) реакция соединения формулы (IV),
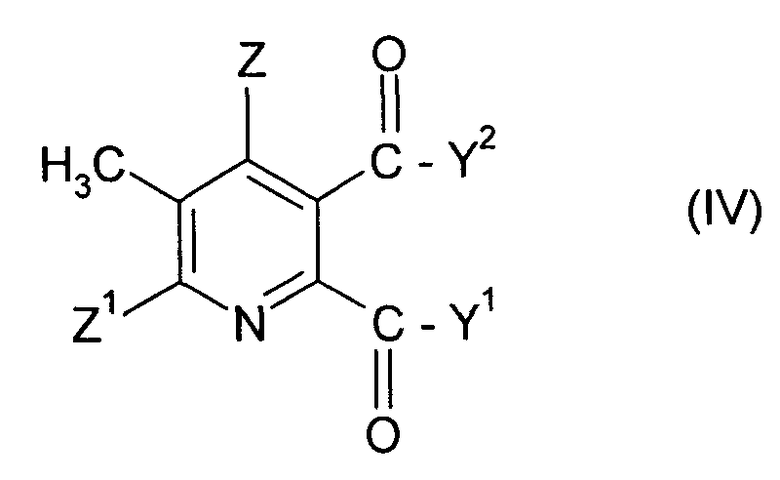
в которой символы имеют значения данные в формуле (II), за исключением того, что Y1, Y2 не являются ОН, с бромом в присутствии радикального инициатора в смеси растворителей, содержащей водную фазу и органическую фазу, где органическая фаза содержит растворитель, выбранный из 1,2-дихлорэтана, хлорбензола, 1,2-дихлорбензола, 1,3-дихлорбензола, 1,4-дихлорбензола и тетрахлорметана, и где величина рН водной фазы составляет от 3 до <8, до получения 3-бромметил-5,6-дизамещенного пиридина соединения (V),
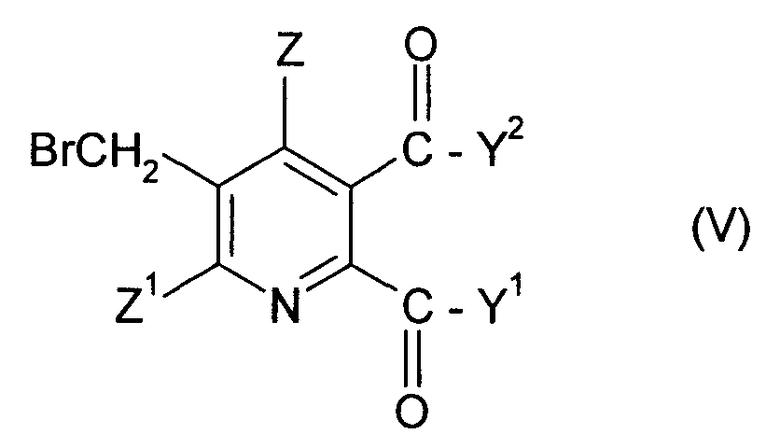
в котором Y1, Y2, Z и Z1 имеют значения данные в формуле (II), за исключением того, что Y1, Y2 не являются ОН, и
(i-2) реакция соединения брома формулы (V) с третичным амином Q в растворителе в интервале температуры около 0-100°С до получения соли аммония (II), и
(i-3) реакция соли аммония (II) в смеси метанол/Н2О, содержащей основание, содержащее МОСН3 и МОН, где М означает щелочной металл, под давлением в закрытой емкости при температуре от около 75 до 110°С.
На стадии (i-1) молярное соотношение пиридинового соединения (IV) к брому в основном находится в диапазоне 1:0.5-1.2, предпочтительно 1:0.6-1.0, более предпочтительно 1:0.7-0.95.
Также возможно работать с половиной эквивалентов брома, и генерировать бром в реакционной смеси из HBr окислителем, таким как H2O2.
Подходящими свободнорадикальными генераторами для инициации реакции являются такие, которые раскладываются при выбранной температуре реакции. Примерами предпочтительных инициаторов являются свободнорадикальные генераторы, такие как азосоединения и пероксиды. Также возможно, несмотря на это, применять редокс-системы, особенно такие, которые основаны на гидропероксидах, таких как гидропероксид кумена.
Радикальные инициаторы, подходящие для применения в способе изобретения, включают 2,2'-азобисизобутиронитрил, 2,2'-азобис(2-метилбутаннитрил), 2,2'-азобис(2,4-диметил-пентаннитрил), 1,1'-азобис(циклогексанкарбонитрил), органические и неорганические пероксиды, такие как пероксид дилауроила, перекись водорода, трет-бутилпероксипивалат, пероксид бензоила и тому подобное, при этом 2,2'-азобисизобутиронитрил, 2,2'-азобис(2-метилбутаннитрил) и пероксид дилауроила являются предпочтительными, и 2,2'-азобисизобутиронитрил и 2,2'-азобис(2-метилбутаннитрил) особенно предпочтительны.
Молярное соотношение инициатора к брому предпочтительно находится в диапазоне 0.04-0.15:1, более предпочтительно 0.06-0.10:1.
Органический растворитель выбран из группы, включающей 1,2-дихлорэтан, хлорбензол, 1,2-дихлорбензол, 1,3-дихлорбензол, 1,4-дихлорбензол и тетрахлорметан, предпочтительно 1,2-Дихлорэтан и хлорбензол. 1,2-дихлорэтан особенно предпочтительный. Также возможны смеси, в особенности дихлорбензолов.
Количество органического растворителя может варьировать в широких пределах. Предпочтительно применяется 900-2000 г, более предпочтительно 1000-1300 г, органического растворителя на моль соединения (II).
Реакционная смесь содержит органическую фазу и водную фазу. Количество водной фазы может варьировать в широких пределах. Предпочтительно применяется 140-500 г, более предпочтительно 140-300 г, особенно 150-200 г воды на моль соединения формулы (II).
Во время реакции величина рН водной фазы сохраняется в диапазоне от 3 до <8, предпочтительно от 3 до 7, более предпочтительно от 4 до 7. Регулирование величины рН может быть достигнуто добавлением подходящего основания, предпочтительно неорганического основания, такого как гидроксид щелочного металла, например, NaOH, или щелочноземельного металла. Водный NaOH является предпочтительным основанием, особенно в разбавленной форме (например, содержащий 5-20 мас.% NaOH).
Чтобы достигнуть желаемого регулирования величины рН, основание может быть добавлено непрерывно в течении реакции, или величина рН непрерывно проверяется, и основание добавляют при помощи связанного автоматизированного устройства дозировки.
В одном предпочтительном варианте осуществления стадию (И) реакции проводят путем растворения соединения (IV) в органической фазе и добавлением воды до образования водной фазы.
Инициатор добавляется в виде чистого соединения или в растворе, при комнатной температуре или при температуре реакции после нагревания. В зависимости от температуры разложения инициатора, часть или даже все количество инициатора должно быть добавлено перед началом дозировки брома. Количество инициатора, которое должно быть добавлено во время добавления брома, также зависит от температуры разложения. Во время реакции бромирования должна быть всегда доступной минимальная концентрация свободных радикалов.
Для 2,2'-азобис(2-метилбутаннитрила) добавляется раствор с инициатором в органическом растворителе. Медленное добавление брома так же, как и основания для регулирования величины рН, может быть начато в одно время или некоторое время спустя. Предпочтено, чтобы начать дозировку брома/основания позже, чтобы иметь достаточное количество свободных радикалов в смеси, когда начинается реакция бромирования. После завершения реакции смесь охлаждают и разделяют фазы. Реакцию в основном проводят при температуре от около 50 до около 120°С, предпочтительно от около 60 до около 90°С.
Реакция может осуществляться при атмосферном давлении или при повышенном давлении до 6 бар. Атмосферное давление является предпочтительным.
Время реакции (для стадии (i-1)) отличается от параметров реакции, но в основном находится между 1 и 24 ч.
Чтобы улучшить полный выход и увеличить селективность реакции, то есть уменьшить формирование нежелательных побочных продуктов диброма и триброма, предпочтительно проводить реакцию только до преобразования 5-60% (в пересчете на количество соединения (IV)), предпочтительно 30-55%. В одном предпочтительном варианте осуществления реакцию проводят до преобразования около 50% (в пересчете на соединение (IV)). Степень преобразования может быть проверена стандартными способами известными специалисту в данной области техники, например, анализом ВЭЖХ.
Когда достигается желаемая степень преобразования, реакция останавливается и фазы разделяют.
Органическая фаза, содержащая продукт стадии (i-1), соединение (V), непрореагировавший исходный материал (IV) и побочные продукты диброма и триброма, могут быть экстрагированы водой для удаления водорастворимых примесей, таких как кислоты и бромид. Продукт (III) может быть отделен известными способами, предпочтительно, несмотря на это, применять органическую фазу без дальнейшей обработки для реакции с третичным амином Q (стадия (i-2)).
Также возможно экстрагировать водную фазу органическим растворителем и соединить с органическими фазами, чтобы увеличить выход соединения (V).
На стадии (i-2) реакции соединение (V) реагирует с третичным амином Q до получения соединения аммония (II).
Предпочтительные третичные амины Q следуют из предпочтительных значений Q в формуле (II) выше, т.е. пиридин и третичные алкиламины NR9R10R11 являются более предпочтительными,
где
R9, R10 и R11 каждый независимо означают C1-C4 алкил, или, R9 и R10, когда взяты вместе, образовывают 5- или 6-членное кольцо, в котором R9R10 представлены структурой: -(СН2)n-, необязательно содержащей О, S или NR12, где n означает целое число 3, 4, или 5, предусмотренный R11 означает C1-C4 алкил, и
R12 означает C1-C4 алкил.
Триметиламин, NMe3, особенно предпочтительны.
В основном применяется избыток третичного амина. В основном применяется 1.1-2, предпочтительно 1.05-1.5 эквивалентов третичного амина на эквивалент соединения (V).
В основном третичный амин, необязательно растворенный в растворителе, медленно добавляют в раствор соединения (V), после чего образуется и осаждается соль (II). В случае предпочтительного амина NMe3, который является газообразным при комнатной температуре, предпочтительно работать в закрытой емкости и насыщать газообразный амин или жидкий амин под давлением в раствор соединения (V).
Стадия (i-2) предпочтительно осуществляется при температуре около 0-70°С, более предпочтительно 5-70°С, особенно предпочтительно 5-40°С. Реакция может быть осуществлена при давлении окружающей среды или при повышенном давлении. В предпочтительном варианте осуществления реакцию проводят в закрытой емкости при давлении растворителя и/или амина, которое растет при повышении температуры реакции.
Обработка реакционной смеси и выделение соединения аммония (II) может быть осуществлена общепринятыми способами, например, соединение (II) может быть отфильтровано.
В предпочтительном варианте осуществления вода добавляется к реакционной смеси, чтобы растворить продукт, соединение (II), и разделяются водная фаза и органическая фаза. Водная фаза может быть дополнительно экстрагирована органическим растворителем, чтобы увеличить чистоту продукта (II), и увеличить выход возвращенного исходного материала (V) в органической фазе. Количество воды должно быть достаточным для образования водной фазы и предпочтительно выбрано так, чтобы образовывать 20-45 мас.% раствора соединения (II) в водной фазе.
Соединение аммония (II) может быть выделено из водной фазы известными способами. В предпочтительном варианте осуществления соединение (II) не выделяют и водная фаза, полученная на стадии (i-2) применяется в последующих реакциях без дополнительной обработки. Такое возможно и выгодно, так как большое количество воды допускается в способе метоксилирования изобретения. Несмотря на это, также возможно смешивать водную фазу с растворителем, который образует азеотроп с водой, например толуол, и вода удаляется азеотропной дистилляцией. Получающаяся суспензия соединения (II) может быть применена для последующих реакций.
В дополнительном предпочтительном варианте осуществления после отделения соединения (V), органическая фаза из стадии (i-2), содержащей до 80% исходного материала (IV) (в пересчете на начальное количество, использованное на стадии (i-1)), возвращается и рециклизуется в способе реакции стадии (i-1). Предпочтительно добавляется дополнительный исходный материал (IV), чтобы компенсировать количество, превращенное в предыдущей стадии (i-1). Таким образом, в принципе, органическая фаза стадии (i-1) может быть рециклизирована любое количество раз, несмотря на это из-за накопления побочных продуктов, главным образом ди- и трибромированных продуктов соединения (IV), в основном выполнимо до 20, предпочтительно до 10 циклов.
В предпочтительном варианте осуществления циклического способа реакции в последнем цикле дополнительный исходный материал (IV) не добавляется, чтобы улучшить полный выход и норму преобразования.
В дополнительном варианте осуществления циклического способа реакции определенное количество органической фазы, предпочтительно около 5-20 мас.%, удаляется, чтобы уменьшить или подавить накопление побочных продуктов в органической фазе. В этом варианте осуществления изобретения фактически отсутствует предел числа циклов, в которых может использоваться органическая фаза.
Соединения формулы (I) представляют собой ценные промежуточные соединения в органическом синтезе. Они особенно пригодны для преобразования до гербицидного имидазолинонового соединения (III).
Дальнейшее преобразование соединения (I) до гербицидных имидазолинонов (III) может быть достигнуто способами, известными в уровне техники.
Способы, которые могут использоваться, чтобы создать имидазолиноновые гербициды, проиллюстрированы в книге "The Imidazolinone Herbicides" под редакцией D. L. Shaner и S. L. O'Connor, изданной 1991 CRC Press, Boca Raton, Florida с особой ссылкой на Главу 2, названную "Synthesis of The Imidazolinone Herbicides", страница 8-14 и ссылки, процитировали там. Следующие патентные литературные ссылки также иллюстрируют способы, которые могут использоваться, чтобы преобразовать пиридиновые дикислоты, сложные эфиры и соли до имидазолиноновых конечных продуктов:
Патенты US №. 5371229; 5250694; 5276157; 5110930; 5122608; 5206368; 4925944; 4921961; 4959476; 5103009; 4816588; 4757146; 4798619; 4766218; 5001254; 5021078; 4723011; 4709036; 4658030; 4608079; 4719303; 4562257; 4518780; 4474962; 4623726; 4750978; 4638068; 4439607; 4459408; 4459409; 4460776; 4125727 и 4758667, и ЕР-А 0041623.
Согласно предпочтительным вариантам осуществления изобретения преобразование соединения (I) до гербицидного имидазолинона (III) осуществляется по аналогии с методами, описанными в ЕР-А 0041623, US 4,518,780 или ЕР-А 0144595.
Согласно этим вариантам осуществления соединение (I) сначала преобразовано в соответствующий ангидрид известными способами, такими как реакция с уксусным ангидридом.
В одном варианте осуществления соединение (III) получают
(i) приготовлением соединения (I), где Y означает ОН, как уже указано выше;
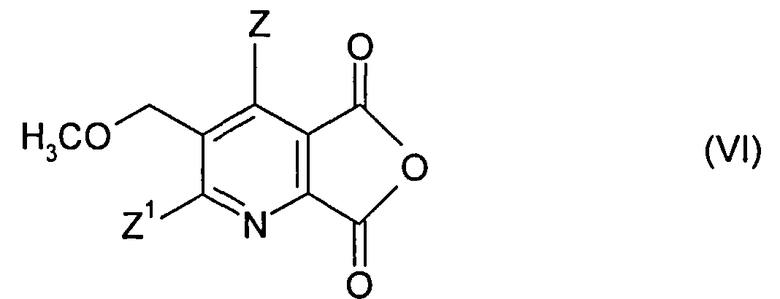
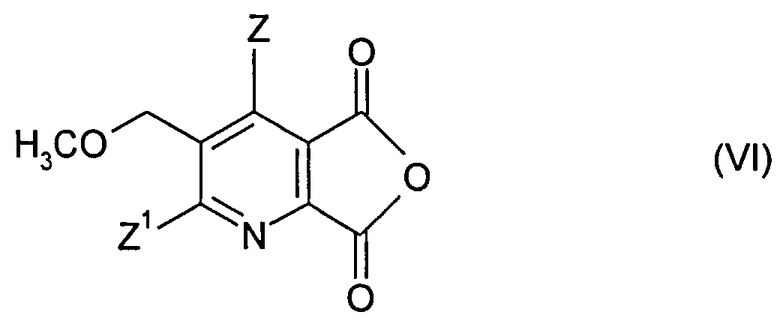
(ii-1) преобразованием соединения (I) до ангидрида (VI),
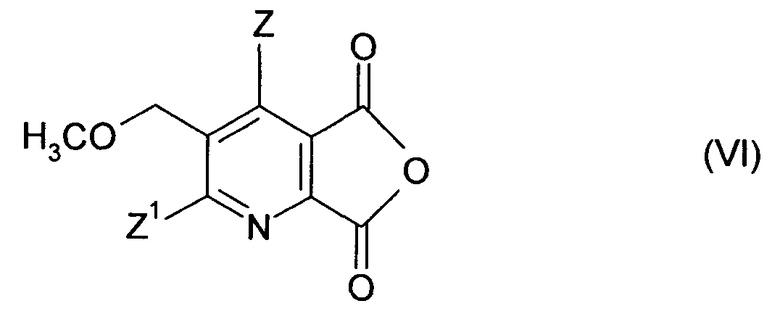
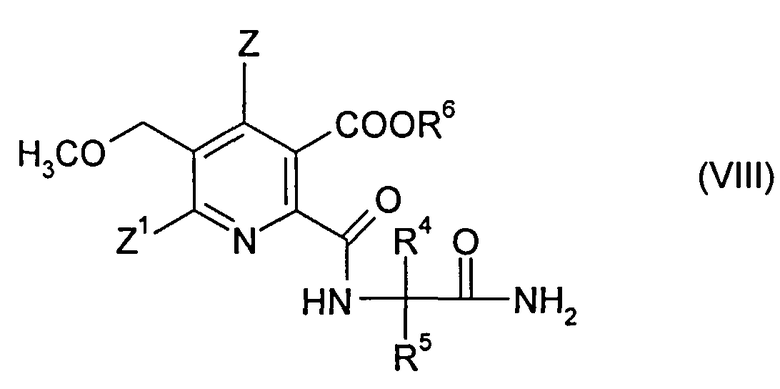
(ii-2) реакцией ангидрида (VI) с 2-аминоалкан карбоксамидом формулы (VII),
 ,
,
где R4 и R5 являются такими, как в формуле (III), до получения амида (VIII),
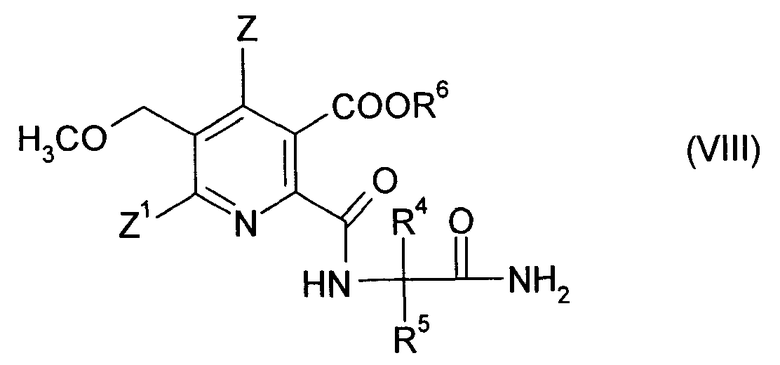
где символы являются такими, как в формуле (III), и
(ii-3) конденсацией амида (VIII) с получением гербицидного имидазолинона (III).
Стадии (ii-2) и (ii-3) могут осуществляться как однореакторная реакция.
В одном варианте осуществления стадия (ii-2) осуществляется по аналогии с способом, раскрытом в примере 10 ЕР-А 0322616. Соединение (I), замещенный 2-аминоалкан карбоксамид (VI) и третичный амин, предпочтительно триэтиламин реагируют в полярном апротонном растворителе, таком как ацетонитрил, с получением соли аммония (VIII), которая может быть окислена до кислоты (VIII).
Альтернативные способы раскрыты в US 4,518,780 и ЕР-А 0144595. В последнем документе раскрыто добавление азотистого основания, выбранного из пиридина, пиколинов, хинолина и лутидина, улучшения региоселективности реакции, т.е. увеличения количества 2-присоединение продукта.
В одном варианте осуществления стадии (ii-3) амидо соединение (VIII), предпочтительно в форме соли аммония (R6 означает HNR3), реагирует с метоксидом щелочного металла, предпочтительно NaOCH3 в метаноле по аналогии с примером 11 ЕР 0322616. Получаемая суспензия выдерживается при кипячении в колбе с обратным холодильником до полного преобразования. После охлаждения смесь окисляют до получения соединения (III), или как соль аммония (ацилирование до рН около 4), или свободная кислота (ацилирование до рН ≤2).
В дополнительном предпочтительном варианте осуществления реакционная смесь из стадии (ii-2) реагирует с метанолом (в основном 2-100 эквивалентов в пересчете на (VIII)) в присутствии водного основания (в основном 3-100 эквивалентов в пересчете на (VIII)), основание предпочтительно выбирают из МОН и МОСН3, где М означает щелочной металл, предпочтительно Na или K, особенно Na.
Реакцию проводят при температуре в диапазоне от 20 до 120°С, предпочтительно 40-90°С. Реакция может быть осуществлена при атмосферном давлении или при повышенном давлении, предпочтительно давление формируется при желаемой температуре реакции. Время реакции в основном составляет от 1 до 8 ч, предпочтительно от 1 до 5 ч.
Выделение продукта (III) может быть достигнуто стандартными способами. В предпочтительном варианте осуществления добавляется вода и отгоняются органические растворители. Остаток может быть перенесен в воду и окислен, после чего осаждается соединение (III). После фильтрования сырой продукт может быть дополнительный очищен, например, путем взбалтывания с водой или перекристаллизацией.
В дополнительном варианте осуществления соединение (III) получают
(i) приготовлением соединения (I), где Y означает ОН, как уже указано выше;
(ii-1) преобразованием соединения (I) до ангидрида (VI),
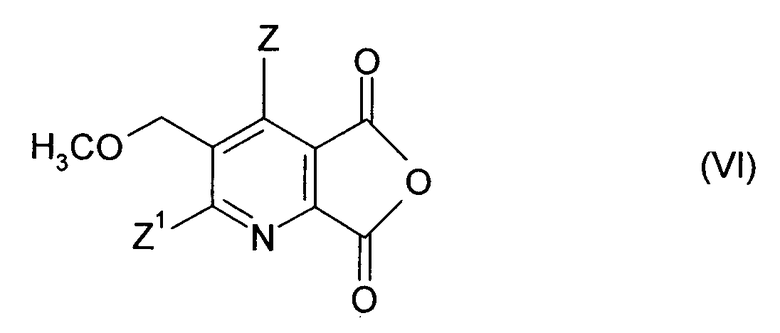
(ii-2) реакцией ангидрида (VI) с аминокарбонитрилом (IX),
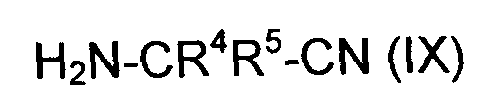
где R4 и R5 являются такими, как в формуле (III),
до получения амидонитрильного соединения (X),
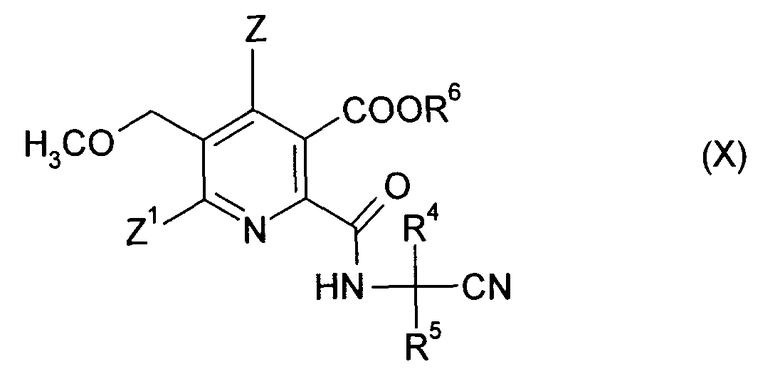
где R4, R5 и R6 являются такими, как в формуле (III),
(ii-3) гидролизом нитрильной группы в соединении (X) до получения амида (VIII),
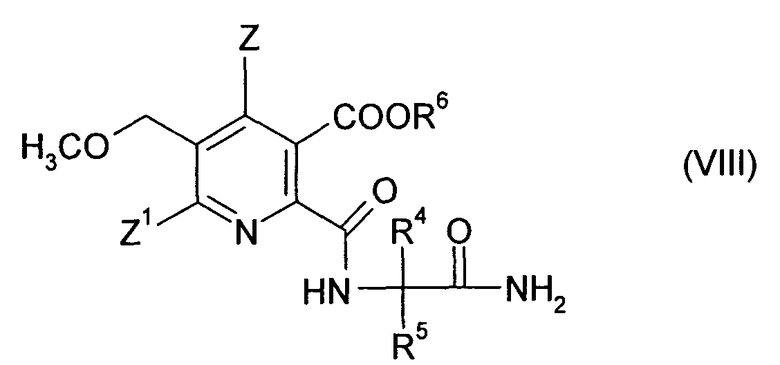
где символы имеют такие же значения, как в формуле (III), и
(ii-4) конденсированием амида (VIII) с получением гербицидного имидазолинона (III).
Получение ангидрида (VI) может быть осуществлено, как описано выше.
Аминокарбонитрилы (IX), которые применяются на стадии (ii-2), коммерчески доступны или могут быть получены способами, известными в уровне техники. В основном применяются 0.8-1.2 эквивалентов аминонитрила (IX) на эквивалент соединения (VI), предпочтительно 0.95-1.1.
Реакцию проводят в растворителе, который предпочтительно выбран из ароматических углеводородов, предпочтительно толуола, мезителенов, хлорированных ароматических углеводородов, таких как хлорбензол, дихлорбензол, хлорированных углеводородов, таких как 1.2-дихлорэтан, дихлорметан, уксусная кислота, и их смесей.
Если уксусная кислота применяется не как основной растворитель, выгодно добавление 0.5-4 эквивалентов, предпочтительно 1-3 эквивалентов (в пересчете на соединение (I)). Дополнительные выгодные добавки, которые улучшают селективность реакции раскрытия цикла (2 в противоположность 3 положению), перечислены в ЕР-А 0144555 и включают пиридин, 4-пиколин, 2-пиколин и хинолин.
Реакцию в основном проводят в интервале температуры от около 40 до около 120°С, предпочтительно от около 60 до около 100°С. Время реакции в основном составляет от около 1 до около 3 ч.
В предпочтительном варианте осуществления ангидрид (VI) растворяют в растворителе и доводят до температуры реакции, и постепенно добавляют аминонитрил (IX). После завершения реакции и охлаждения нитрильное соединение (X) может быть выделено стандартными способами.
В предпочтительном варианте осуществления, несмотря на это, соединение (X) не выделяют, а непосредственно применяют реакционную смесь в следующей стадии гидролизации нитрила (стадия iv-3).
При обычном способе добавляют небольшой избыток (например, 1.1-1.5 эквивалентов в пересчете на (X)) сильной минеральной кислоты, предпочтительно серной кислоты (предпочтительно в концентрации 30-98%) и воды (например, 2-10 эквивалентов) при температуре, которая в основном находится в диапазоне около 30-120°С, предпочтительно 50-90°С. Смесь дополнительно перемешивают до полного преобразования. Время реакции в основном составляет от 1 до 8 ч, предпочтительно 1-5 ч.
Обработка и выделение могут быть достигнуты стандартными способами, такими как водным раствором (например, в виде его соли аммония). В предпочтительном варианте осуществления реакционную смесь сразу применяют в следующей стадии конденсации (ii-4).
В альтернативном варианте осуществления гидролиз нитрильной группы проводится реакцией с водным NaOH/H2O2, как раскрыто, например, в ЕР-А 0144595 и US 4,518,780.
Конденсация амидосоединения (VIII) до гербицидного имидазолинона может осуществляться, как описано выше.
Все из вышеописанных способов особенно предпочтительны для получения соединения формулы (III), где Z и Z1 означают Н, R6 означает Н, R4 означает СН3 и R5 означает СН(СН3)2, т.е. имазамокс.
Изобретение иллюстрируется следующими примерами, не ограничивая его таким образом.
Примеры
Сравнительный Пример 1
Получение 5-(метоксиметил)-2,3-пиридиндикарбоновой кислоты (согласно пр. 3 ЕР-А 0548532)
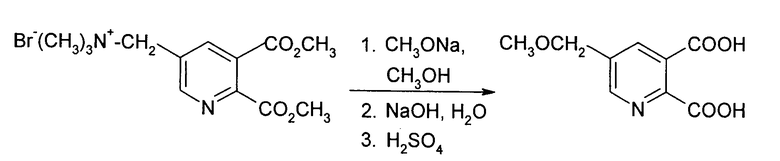
Смесь 25% метоксида натрия в метаноле (270 г, 1.25 моль) и [(5,6-дикарбокси-3-пиридил)-метил]триметиламмоний бромида, диметилового сложного эфира (Ia) (347 г, 1.00 моль) в метаноле (650 мл) нагревали при кипячении в колбе с обратным холодильником на протяжении 1 часа в атмосфере азота. Добавляли воду (1 л) и гидроксид натрия (80.0 г, 2.0 моль) и реакционную смесь дистиллировали до тех пор, пока колба не достигала 100-105°С. Реакционную смесь охлаждали до комнатной температуры, обрабатывали серной кислотой для регулирования рН до величины от 1.5 до 2 и фильтровали с получением твердого осадка. Твердый осадок промывали водой и сушили в вакуум-сушильном шкафу до получения названого продукта в виде белого твердого вещества (т.пл. 161-162°С), которое имеет чистоту больше, чем 99%, определенную анализом ВЭЖХ.
Вода не присутствует на первой стадии реакции (метоксилирование 5-метильной группы), которая требует трудоемкого высушивания или способа выделения твердого вещества для исходного материала. Таким образом способ является менее подходящим для промышленного применения.
Сравнительный Пример 2
Получение 5-(метоксиметил)пиридин-2,3-дикарбоксилата динатрия из [5,6-(дикарбоксилат-3-пиридил)метил]триметиламмоний бромида динатрия (согласно пр. 3 ЕР-А 0747360)
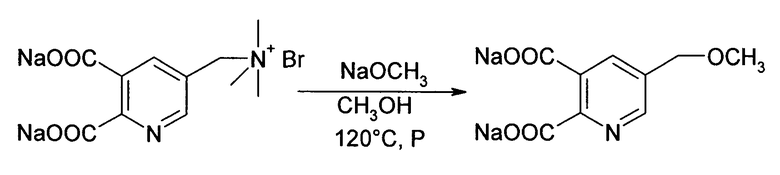
Смесь [(5,6-дикарбоксилат-3-пиридил)метил]триметиламмоний бромида динатрия (5.0 г, 13.8 ммоль) и 25% мас./мас. раствора метоксида натрия в метаноле (4.46 г, 20.7 ммоль NaOCH3) в 75 г метанола нагревали при 120°С на протяжении 21 часа в реакторе высокого давления. Реакцию охлаждали до комнатной температуры, обрабатывали водой и концентрировали до конечной массы 55.03 г. 5.0 г образца анализировали анализом ЖХ (30% CH3CN, 0.77 М H3PO4). Остаток реакционного раствора выпаривали досуха, чтобы получить твердый остаток, определяемый ЯМР анализом.
Несмотря на более высокую температуру время реакции значительно более длительное, чем в способе изобретения.
Пример 1
Получение 5-(метоксиметил)-2,3-пиридиндикарбоновой кислоты
Бромид (IIa), (0.41 моль), содержащий около 40 мас.% воды, растворяли в метаноле. Добавляли водный NaOH на протяжении 30 мин при 35°С, и смесь перемешивали на протяжении еще 15 мин. NaOCH3 в метаноле добавляли на протяжении 20 мин при 45°С.
Реакционную смесь переносили в реактор высокого давления Парра и нагревали до температуры реакции (80-100°С). При около 60°С реактор закрывали, после чего давление растет, достигая около 40-45 Psi (фунт на квадратный дюйм) при температуре реакции. Внешнее давление не оказано.
После 6-8 ч, смесь охлаждали и обработку проводили согласно известным способам.
Например, реакционную смесь охлаждали до комнатной температуры, обрабатывали серной кислотой для регулирования рН до величины от 1.5 до 2 и фильтровали до получения твердого осадка. Твердый осадок промывали водой и сушили в вакуум-сушильном шкафу до получения названого продукта в виде белого твердого вещества (т.пл. 161-162°С), которое имеет чистоту больше, чем 99%, определяемое анализом ВЭЖХ.
Подобным способом были выполнены примеры 2-17, показанные в Таблице 1.
Можно заметить, что более высокие температуры и более длительное время реакции улучшают степень преобразования. Кроме того, преобразование увеличивается добавлением большего количества основания. Способ изобретения может быть выполнен в присутствии большего количества воды по сравнению с Сравнительными Примерами.
Пример 18
Синтез [(5,6-дикарбокси-3-пиридил)метил] триметиламмоний бромида, диметилэфира (IIa)
а) Синтез диметил 5-(бромметил)-2,3-пиридиндикарбоксилата (Va) (50% преобразование)
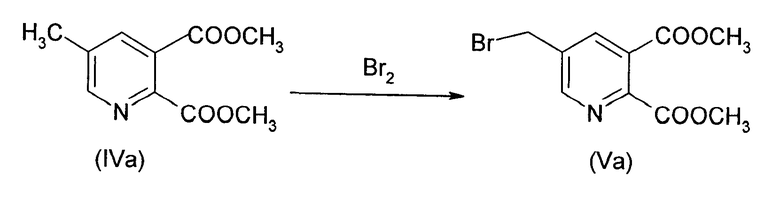
218.4 г (1.0 моль) соединения (Va) растворяли в 1139.0 г 1,2-дихлорэтана (EDC) и добавляли 160.0 г воды и нагревали до 72°С (около 1-2°С ниже дефлегмирования). 14.4 г (0.075 моль) 2,2'-азобис(2-метилбутиронитрила) (Vazo 67) в 160.0 г EDC добавляли на протяжении 2 ч при 72°С. Через 30 минут 143.8 г (0.9 моль) брома добавляли на протяжении 2 ч, при регулировании рН (рН 5-7) при помощи дозирования около 375.0 г водного NaOH (15%). Смесь перемешивали 1 ч для завершения реакции (анализ ВЭЖХ). После охлаждения до 40°С фазы были разделены.
b) Синтез соединения (IIa)
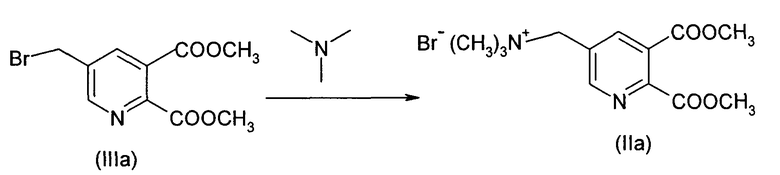
Загружали 288.1 г (1.0 моль) соединения (IIIa) в смеси с ди- и трибромированными побочными продуктами в 3359.0 г EDC (органическая фаза из стадии а, включающая непрореагировавшее соединение (На) и высшие бромированные побочные продукты). Смесь нагревали до 30°С и емкость вакуумировали до 200 мбар.
70.9 г (1.2 моль) триметиламина (ТМА) добавляли к газовой фазе на протяжении 2 ч при 40°С (закрытая система). Смесь перемешивали один дополнительный час (ВЭЖХ контроль преобразования: соединение (IIIa) в растворе <0.1%).
Избыток ТМА отгоняли вместе с EDC (масса: 40% массы EDC), перемещенным на стадию 2 (1344 г) при 50-55°С (370-250 мбар). Величина рН дистиллята была <9. 630.0 г воды разбрызгивали на стенки так, чтобы растворить твердый осадок и смесь перемещали в следующую емкость. Смесь потом перемешивали 0.25 ч, и нижнюю органическую фазу отделяли при 40°С. Добавляли 320.4 г EDC. Смесь перемешивали и нижнюю органическую фазу отделяли при 40°С. Обратную экстракцию повторяли (40°С) с 320.4 г EDC. Две органические фазы обратной экстракции объединяли с первой органической фазой и рециклизировали для следующей партии бромирования (после добавления 50% свежего соединения (IIa) для дальнейшего цикла.
Стадии а) и b) повторяли шесть раз. В последнем цикле соединение (IVa) не добавляли на стадии а) и 0.8 моль ТМА добавляли на стадии b).
Полная степень преобразования соединения (IIa) составляет 96.6%. Выход соединения (IIa) (за 7 циклов) составляет 77.4% с чистотой >95% (как определено при помощи ВЭЖХ).
Изобретение относится к способу получения 2,3-дизамещенного-5-метоксиметилпиридина формулы (I)
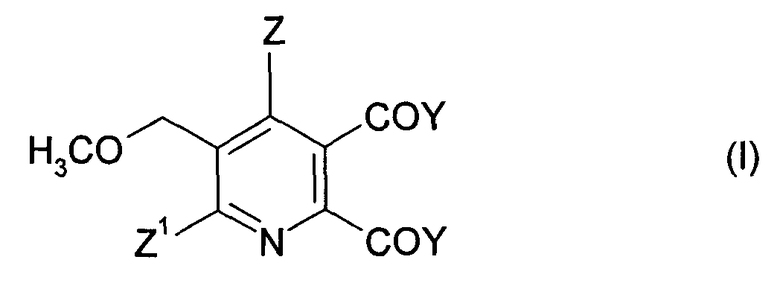
где Z означает Н или галоген; Z1 означает Н, галоген, CN или NO2; Y2 означает ОМ, и М означает щелочной металл или щелочноземельный металл, включающий стадии:
(i) реакция соединения формулы (II)
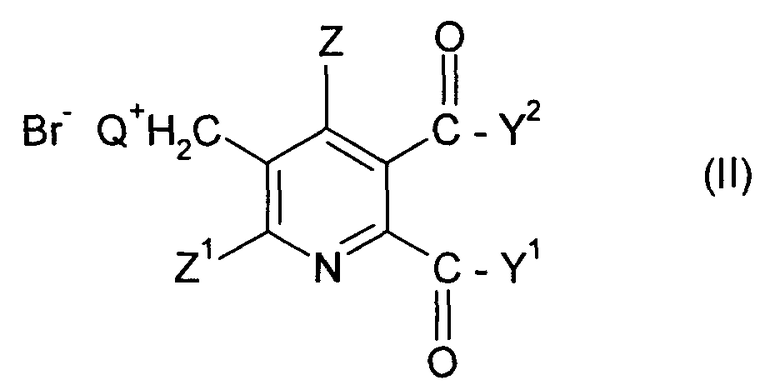
где Q означает третичный алифатический или циклический, насыщенный, частично ненасыщенный или ароматический амин; Z означает Н или галоген; Z1 означает Н, галоген, CN или NO2; Y1 и Y2 каждый независимо означают OR1, NR1R2, или когда взяты вместе Y1Y2 означает -О-, -S- или -NR3-; R1 и R2 каждый независимо означают Н, C1-C4 алкил, необязательно замещенный С1-С4 алкоксигруппой или фенилом, необязательно замещенным от одной до трех C1-C4 алкильными группами, C1-C4 алкоксигруппами или атомами галогена, или фенил, необязательно замещенный от одной до трех C1-C4 алкильными группами, C1-C4 алкоксигруппами или атомами галогена; R3 означает Н или C1-C4 алкил,
в смеси метанол/H2O, содержащей по меньшей мере 20 мас.% H2O (в пересчете на суммарное количество воды и бромида (II)), с основанием, содержащим МОСН3 и/или МОН, где М означает щелочной металл или щелочноземельный металл, под давлением в закрытой емкости при температуре от 75 до 110°С. Соединения формулы (I) являются пригодными промежуточными соединениями в синтезе гербицидных имидазолинонов, таких как имазамокс. 3 н. и 13 з.п. ф-лы, 1 табл., 18 пр.
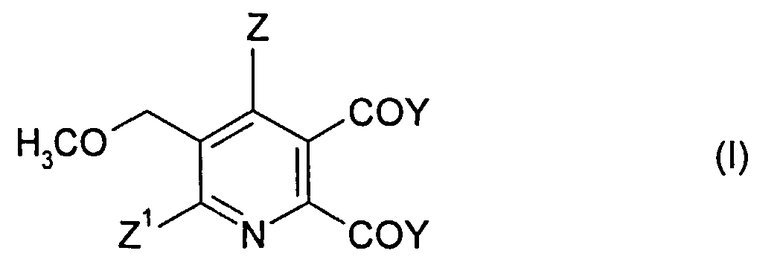
1. Способ получения 2,3-дизамещенного-5-метоксиметилпиридина формулы (I),
где
Z означает Н или галоген;
Z1 означает Н, галоген, CN или NO2;
Y означает ОМ, и
M означает щелочной металл или щелочноземельный металл,
включающий стадии:
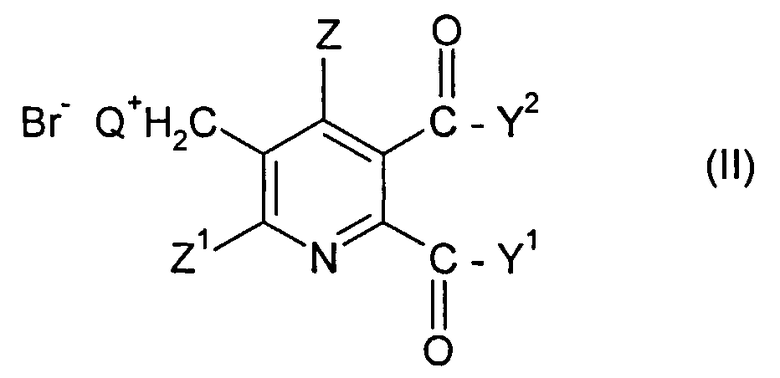
(i) реакция соединения формулы (II)
где
Q означает третичный алифатический или циклический, насыщенный, частично ненасыщенный или ароматический амин;
Z означает Н или галоген;
Z1 означает Н, галоген, CN или NO2;
Y1 и Y2 каждый независимо означают OR1, NR1R2, или когда взяты вместе Y1Y2 означает -O-, -S- или -NR3-;
R1 и R2 каждый независимо означают Н,
C1-C4 алкил, необязательно замещенный С1-С4 алкоксигруппой или фенилом, необязательно замещенным от одной до трех С1-С4 алкильными группами, C1-C4 алкоксигруппами или атомами галогена, или
фенил, необязательно замещенный от одной до трех C1-C4 алкильными группами, C1-C4 алкоксигруппами или атомами галогена;
R3 означает Н или C1-C4 алкил,
в смеси метанол/Н2О, содержащей по меньшей мере 20 мас. % H2O (в пересчете на суммарное количество воды и бромида (II)), с основанием, содержащим МОСН3 и/или МОН, где М означает щелочной металл или щелочноземельный металл, под давлением в закрытой емкости при температуре от 75 до 110°С.
2. Способ по п. 1, включающий стадии:
(i-1) реакция соединения формулы (IV),
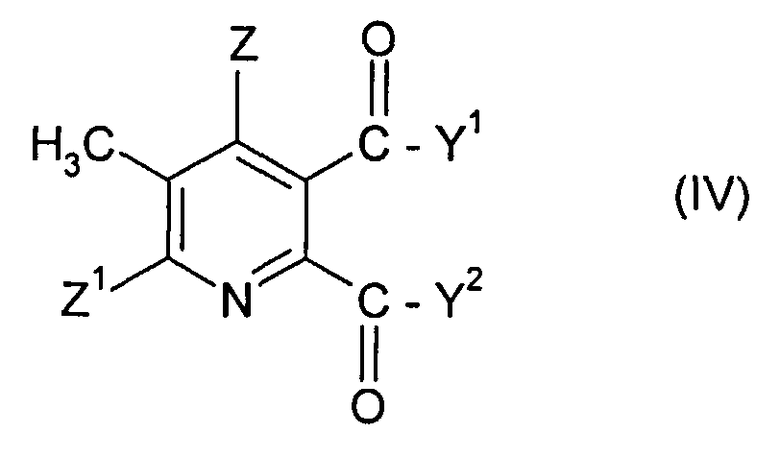
в которой Y1, Y2 не являются ОН и символы в других случаях имеют значения, данные в формуле (II) в п. 1, с бромом в присутствии радикального инициатора в смеси растворителей, содержащей водную фазу и органическую фазу, где органическая фаза включает растворитель, выбранный из 1,2-дихлорэтана, хлорбензола, 1,2-дихлорбензола, 1,3-дихлорбензола, 1,4-дихлорбензола и тетрахлорметана, и где величина рН водной фазы составляет от 3 до <8, до получения 3-бромметил-5,6-дизамещенного пиридинового соединения (V),
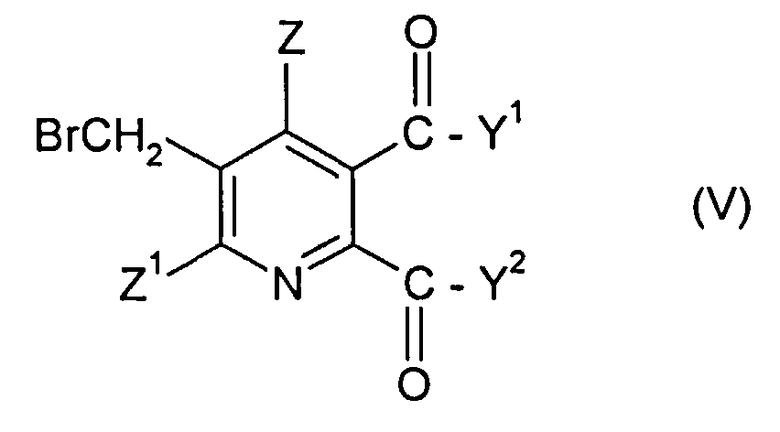
где Y1, Y2 не являются ОН и символы в других случаях имеют значения, данные в формуле (II), и
(i-2) реакция соединения брома формулы (V) с третичным амином Q в растворителе в интервале температуры 0-100°С до получения соли аммония (II), и
(i-3) реакция соли аммония (II) в смеси метанол/Н2О, содержащей основание, содержащее МОСН3 и МОН, где М означает щелочной металл, под давлением в закрытой емкости при температуре от 75 до 110°С.
3. Способ по п. 1 или 2, в котором соединение формулы (II) представляет собой соединение (IIа)
4. Способ по п. 1 или 2, в котором соотношение метанола к соединению (II) находится в диапазоне от 0.5-25:1.
5. Способ по п. 1 или 2, в котором основанием является смесь МеОМ и МОН.
6. Способ по п. 1 или 2, в котором основанием является МеОМ.
7. Способ по п. 5, где молярное соотношение МеОМ к соединению (II) находится в диапазоне от 1-10:1.
8. Способ по п. 6, где молярное соотношение МеОМ к соединению (II) находится в диапазоне от 1-10:1.
9. Способ по п. 1 или 2, в котором основанием является МОН.
10. Способ по п. 5, в котором молярное соотношение МОН к соединению (II) находится в диапазоне от 0.5-10:1.
11. Способ по п. 9, в котором молярное соотношение МОН к соединению (II) находится в диапазоне от 0.5-10:1.
12. Способ по п. 1 или 2, в котором молярное соотношение общего количества основания, добавляемого к бромиду (II) составляет 3-7:1.
13. Способ по п. 1 или 2, в котором температура реакции находится в диапазоне от около 75 до 110°С.
14. Способ по п. 1 или п. 2, который осуществляется при давлении 1.01-5,00 бар.
15. Способ получения гербицидного имидазолинонового соединения формулы (III),
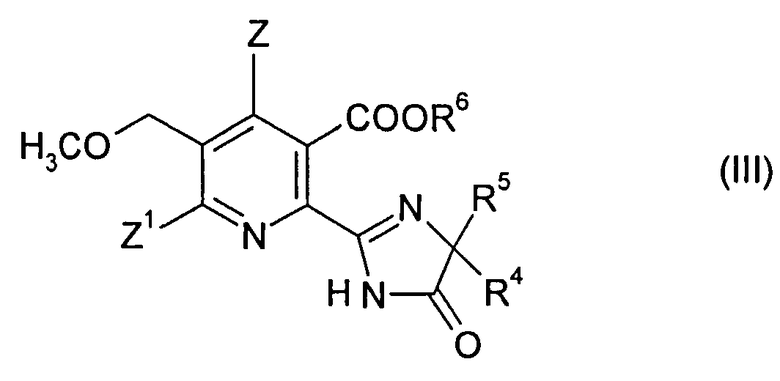
в котором
Z, Z1 являются такими, как определено в формуле (I) в п. 1;
R4 означает C1-C4 алкил;
R5 означает C1-C4 алкил, С3-С6 циклоалкил или R4 и R5, когда взяты вместе с атомом, к которому они присоединены, означают С3-С6 циклоалкильную группу, необязательно замещенную метилом, и
R6 означает водород; группу формулы -N=C (низший алкил)2; C1-C12 алкил, необязательно замещенный одной из следующих групп: С1-С3 алкоксигруппа, галоген, гидроксил, С3-С6-циклоалкил, бензилоксигруппа, фурил, фенил, галофенил, низший алкилфенил, низший алкоксифенил, нитрофенил, карбоксил, низший алкоксикарбонил, цианогруппа или три низший алкиламмоний; С3-С12 алкенил, необязательно замещенный одной из следующих групп: C1-С3 алкоксигруппа, фенил, галоген или низший алкоксикарбонил или двумя C1-С3 алкоксигруппами или двумя группами галогена; С3-С6 циклоалкил, необязательно замещенный одной или двумя C1-С3 алкильными группами; или
катион;
включающий стадии:
(i) получения соединения формулы (I) согласно п. 1, где Y означает ОН,
(ii) превращения соединения формулы (I) в гербицидное соединение формулы (III), путем
(ii-1) преобразования соединения (I) до ангидрида (VI),
где Z, Z1 являются такими, как в формуле (III),
(ii-2) реакции ангидрида (VI) с 2-аминоалкан карбоксамидом формулы (VII),
H2N-CR4R5-CONH2 (VII),
где R4, R5 являются такими, как в формуле (III), до получения амида (VIII),
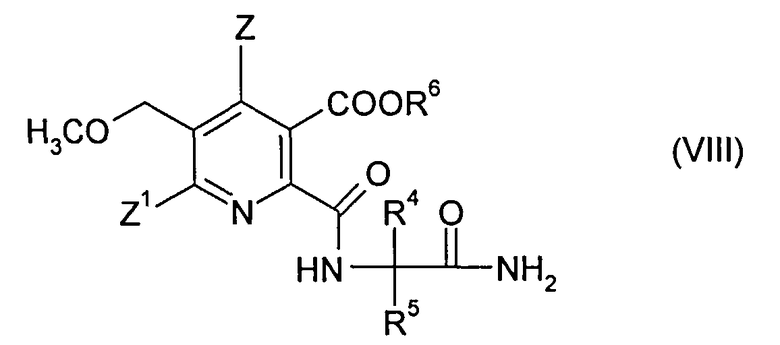
где символы являются такими, как в формуле (III), и
(ii-3) конденсации амида (VIII) с получением гербицидного имидазолинона (III).
16. Способ получения гербицидного имидазолинонового соединения формулы (III)
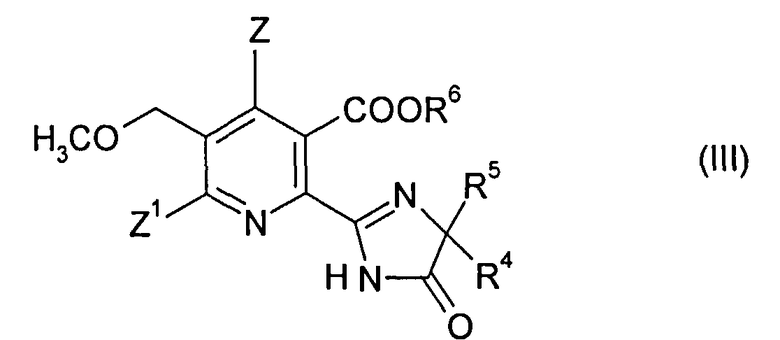
Z, Z1 являются такими, как определено в формуле (I) в п. 1;
R4 означает С1-С4 алкил;
R5 означает C1-C4 алкил, С3-С6 циклоалкил или R4 и R5, когда взяты вместе с атомом, к которому они присоединены, означают С3-С6 циклоалкильную группу, необязательно замещенную метилом, и
R6 означает водород; группу формулы -N=C (низший алкил)2; C1-C12 алкил, необязательно замещенный одной из следующих групп: С1-С3 алкоксигруппа, галоген, гидроксил, С3-С6-циклоалкил, бензилоксигруппа, фурил, фенил, галофенил, низший алкилфенил, низший алкоксифенил, нитрофенил, карбоксил, низший алкоксикарбонил, цианогруппа или три низший алкиламмоний; С3-С12 алкенил, необязательно замещенный одной из следующих групп: C1-С3 алкоксигруппа, фенил, галоген или низший алкоксикарбонил, или двумя C1-С3 алкоксигруппами, или двумя группами галогена; С3-С6 циклоалкил, необязательно замещенный одной или двумя С1-С3 алкильными группами; или
катион;
включающий стадии:
(i) получения соединения формулы (I) согласно п. 1, где Y означает ОН,
(ii) превращения соединения формулы (I) в гербицидное соединение формулы (III), путем
(ii-1) преобразования соединения (I) до ангидрида (VI),
(ii-2) реакции ангидрида (VI) с аминокарбонитрилом (IX), H2N-CR4R5-CN (IX)
где R4 и R5 являются такими, как в формуле (III), до получения амидонитрильного соединения (X),
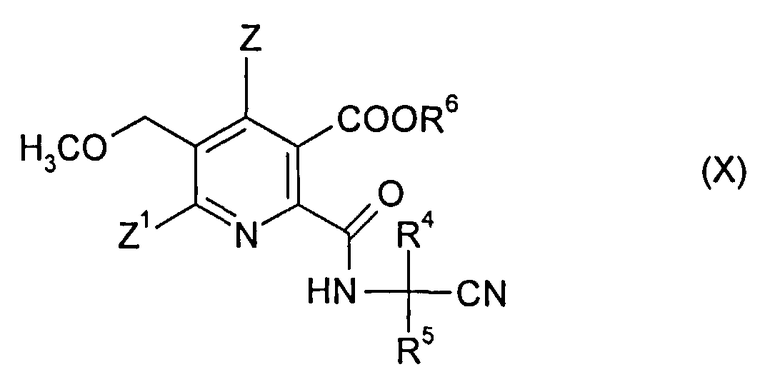
где символы являются такими, как в формуле (III);
(ii-3) гидролиза нитрильной группы в соединении (X) до получения амида (VIII),
где символы имеют такие же значения как в формуле (III) и
(ii-4) конденсирования амида (XI) с получением гербицидного имидазолинона (III).
| Кипятильник для воды | 1921 |
|
SU5A1 |
| 5,6-ДИЗАМЕЩЕННЫЕ 3-ПИРИДИЛМЕТИЛАММОНИЙ ГАЛОГЕНИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 5-(ЗАМЕЩЕННЫЙ МЕТИЛ)-2,3-ПИРИДИНДИКАРБОНОВЫХ КИСЛОТ | 1993 |
|
RU2090558C1 |
| Устройство для одновременного открывания и закрывания дверей кабины и шахты лифта | 1975 |
|
SU548532A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Пуговица | 0 |
|
SU83A1 |
| EP 0 434 965, A2, 19.11.1990 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| EP 0 216 397, A, 22.04.1986 | |||

