Настоящее изобретение относится к катализаторам. В частности, настоящее изобретение относится к способу получения кобальтсодержащего катализатора синтеза углеводородов и способу получения углеводородов, который включает использование указанного катализатора синтеза углеводородов. Настоящее изобретение также относится к катализаторам и продуктам, полученным указанными выше способами.
Сведения о предшествующем уровне техники
Известно, что нанесенные кобальтсодержащие катализаторы синтеза Фишера-Тропша (FTS) могут быть приготовлены путем пропитки носителя катализатора солью кобальта в сочетании с сушкой пропитанного носителя с последующим прокаливанием полученного высушенного пропитанного носителя для получения предшественника катализатора FTS. Предшественник катализатора затем восстанавливают для получения катализатора FTS, содержащего кристаллиты кобальта, диспергированные на поверхности носителя.
При использовании соединения кобальта, такого как нитрат кобальта, для пропитки носителя катализатора лишь относительно низкая нагрузка катализатора на носитель может быть получена за одну стадию пропитки. Исходя из этого, известно об использовании многостадийной пропитки соединением кобальта для увеличения нагрузки кобальта на носитель. В патентах США 6455462, 5733839 и WO 2010/075516 описаны способы многостадийной пропитки для приготовления катализатора FTS. Эти способы включают первую стадию, на которой соль кобальта вводят на носитель катализатора путем пропитки с последующим прокаливанием пропитанного носителя. На второй стадии прокаленный пропитанный носитель первой стадии подвергают второй пропитке солью кобальта с последующим прокаливанием. На третьей стадии прокаленный носитель второй стадии затем активируют, подвергая его воздействию восстановительного газа для обеспечения катализатора FTS.
Также известно, что в приготовлении катализатора FTS стадия прокаливания может быть пропущена, то есть после пропитки носителя катализатора солью кобальта указанный пропитанный носитель напрямую восстанавливают (без предварительного прокаливания) для обеспечения катализатора FTS. Такой способ прямого восстановления описан в WO 2008/090150 и Journal of Catalysis 153 (1995) 108-122 для восстановления носителя катализатора, который был подвергнут одной пропитке расплавленным нитратом кобальта.
Неожиданно было обнаружено, что в случае приготовления катализатора FTS согласно способу по настоящему изобретению, такой катализатор демонстрирует улучшенную активность и/или пониженную селективность в отношении образования метана по сравнению с катализатором, приготовленным в соответствии с описанной выше процедурой двойной пропитки, то есть когда после каждой стадии пропитки следует стадия прокаливания, после чего прокаленный продукт восстанавливают. Кроме того, неожиданно было обнаружено, что способ согласно настоящему изобретению обеспечивает катализатор FTS со сходной FTS-активностью и пониженной селективностью в отношении образования метана по сравнению с катализатором, приготовленным с помощью процедуры двойной пропитки, когда после каждой стадии пропитки следует прямое восстановление пропитанного носителя катализатора. Последний из упомянутых способов также имеет недостаток, который состоит в том, что продукт, образовавшийся после первого цикла пропитки и восстановления, является пирофорным продуктом, который необходимо подвергнуть пассивации перед проведением второй стадии пропитки. Этот недостаток устранен в настоящем изобретении.
Описание изобретения
В соответствии с первым аспектом настоящего изобретения обеспечен способ приготовления кобальтсодержащего катализатора синтеза углеводородов, при этом способ включает:
- прокаливание исходного предшественника катализатора, содержащего носитель катализатора, несущий соединение кобальта, путем термообработки исходного предшественника катализатора в не восстановительных условиях для разложения соединения кобальта и/или для того, чтобы вызвать взаимодействие кобальта с кислородом, для получения, таким образом, прокаленного исходного предшественника катализатора;
- введение соединения кобальта на и/или в прокаленный исходный предшественник катализатора, с тем чтобы прокаленный исходный предшественник катализатора нес данное соединение кобальта, получая, таким образом, последующий предшественник катализатора; и
- непосредственное подвергание последующего предшественника катализатора восстановительным условиям для активации последующего предшественника катализатора для получения, таким образом, кобальтсодержащего катализатора синтеза углеводородов.
Под «не восстановительными условиями» понимаются условия, в которых не происходит восстановление соединения кобальта.
Под «непосредственным подверганием последующего предшественника катализатора восстановительным условиям» понимается, что последующий предшественник катализатора подвергается восстановительным условиям без предварительного подвергания последующего предшественника катализатора термообработке в окислительных условиях, что вызывает окисление нанесенного кобальта или разложение соединения кобальта, то есть без предварительной стадии прокаливания. Предпочтительно, чтобы последующий предшественник катализатора предварительно не подвергался термообработке в не восстановительных условиях, включая окислительные условия, которые вызывают окисление нанесенного кобальта или разложение соединения кобальта.
Исходный предшественник катализатора
Способ может включать приготовление исходного предшественника катализатора путем введения соединения кобальта на и/или в носитель катализатора.
Соединение кобальта, введенное на и/или в носитель катализатора, или соединение кобальта, которое содержит носитель катализатора, может представлять собой любое пригодное органическое или неорганическое соединение кобальта, предпочтительно соль кобальта. Предпочтительно, указанное соединение кобальта представляет собой неорганическое соединение, более предпочтительно, неорганическую соль кобальта. Соединение кобальта может представлять собой нитрат кобальта и, в частности, может представлять собой Со(NO3)2·6Н2О.
Соединение кобальта может быть введено на и/или в носитель катализатора любым подходящим способом, но предпочтительно путем пропитки. Предпочтительно, носитель катализатора пропитывают соединением кобальта путем формирования смеси соединения кобальта; жидкого носителя для соединения кобальта; и носителя катализатора.
Жидкий носитель может содержать растворитель для соединения кобальта и, предпочтительно, соединение кобальта является растворенным в жидком носителе.
Жидкий носитель может представлять собой воду.
Жидкий носитель может представлять собой кислотный жидкий носитель и, предпочтительно, представляет собой кислотную водную композицию. Кислотный жидкий носитель может иметь рН ниже 5, предпочтительно ниже 3, и более предпочтительно ниже 3. Предпочтительно, чтобы значение рН было выше 1, более предпочтительно выше 1.8.
Пропитку можно осуществлять любым подходящим способом пропитки, включая пропитку по влагоемкости или пропитку в суспензии. Пропитка из суспензии является предпочтительной. Предпочтительно, соединение кобальта является растворенным в жидком носителе для того, чтобы объем раствора был больше, чем ху литра, при этом указанный раствор затем смешивают с носителем катализатора, и где х представляет собой объем пор по методу BET носителя катализатора в мл/г носителя, и у представляет собой массу в кг подлежащего пропитке носителя катализатора. Предпочтительно, чтобы объем раствора был больше чем 1.5ху литра и предпочтительно составлял примерно 2ху литра.
Пропитку можно проводить при давлении ниже атмосферного, предпочтительно ниже 85 кПа(а), более предпочтительно при 30 кПа(а) и ниже.
Предпочтительно, пропитку проводят при температуре выше 25°С. Предпочтительно, чтобы температура составляла выше 40°С, более предпочтительно, чтобы температура составляла не ниже 60°С, но предпочтительно не выше 95°С. Таким образом, температура пропитки может быть обозначена Ti, в этом случае, Ti>25°C, предпочтительно >40°С, в частности ≥60°С; однако, предпочтительно Ti≤95°С.
После пропитки может следовать частичная сушка пропитанного носителя, или пропитку и сушку можно проводить в одно и то же время. Предпочтительно, сушку проводят при температуре сушки выше 25°С. Предпочтительно, температура сушки составляет выше 40°С, более предпочтительно, температура сушки составляет не ниже 60°С, но предпочтительно не выше 95°С.Таким образом, температура сушки может быть обозначена Td1, в этом случае, Td1>25°C, предпочтительно >40°С, более предпочтительно ≥60°С; однако, предпочтительно, Td1≤95°С. Предпочтительно, частичную сушку осуществляют в субатмосферных условиях, более предпочтительно ниже 85 кПа(а), наиболее предпочтительно при 30 кПа(а) и ниже.
В одном варианте осуществления изобретения пропитку и частичную сушку можно проводить с использованием процедуры, которая включает первую стадию, на которой носитель катализатора пропитывают (предпочтительно путем пропитки в суспензии) соединением кобальта при температуре выше 25°С и при субатмосферном давлении, и полученный продукт сушат; и, по меньшей мере, одну последующую стадию, на которой полученный частично высушенный продукт первой стадии подвергают обработке при температуре выше 25°С и при субатмосферном давлении таким образом, что температура последующей стадии превышает температуру на первой стадии и/или субатмосферное давление на последующей стадии ниже, чем субатмосферное давление на первой стадии. Эта двухстадийная пропитка может представлять собой способ, описанный в WO 00/20116, который включен в настоящий документ путем отсылки.
Прокаливание исходного предшественника катализатора
Как указано выше, прокаливание осуществляют для разложения соединения кобальта и/или для того, чтобы вызвать взаимодействие соединения кобальта с кислородом. Например, соединение кобальта (такое как нитрат кобальта) может быть превращено в соединение, выбранное из СоО, СоО(ОН), Co3O4, Co2O3, или смесь одного или нескольких из них.
Прокаливание можно проводить любым подходящим способом, например, в ротационной печи для обжига, вертикальной печи или реакторе с псевдоожиженным слоем.
Прокаливание можно проводить в инертной атмосфере, но предпочтительно прокаливание проводится в окислительных условиях. Предпочтительно, окисление проводится в присутствии кислорода, более предпочтительно, на воздухе.
Предпочтительно, прокаливание проводится при температуре выше 95°С, более предпочтительно выше 120°С, еще более предпочтительно выше 130°С, наиболее предпочтительно выше 200°С и предпочтительно не выше 400°С, более предпочтительно не выше 300°С.Таким образом, температура прокаливания может быть обозначена Тс, в этом случае, Tc>95°С, более предпочтительно >120°С, еще более предпочтительно >130°С, наиболее предпочтительно >200°С; однако, предпочтительно, Тс≤400°С, более предпочтительно ≤300°С.
Прокаливание можно проводить с использованием скорости нагрева и объемной скорости воздуха, которые удовлетворяют следующим критериям:
(i) когда скорость нагрева составляет ≤1°С/мин, объемная скорость потока воздуха составляет по меньшей мере 0.76 мn 3/(кг Со(NO3)2·6H2O)/ч; и
(ii) когда скорость нагрева выше чем 1°С/мин, объемная скорость потока воздуха удовлетворяет следующему уравнению:
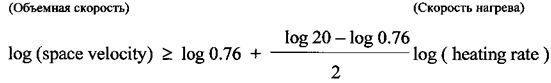
Пропитку, частичную сушку и прокаливание можно повторять для достижения более высоких нагрузок соединения кобальта на носитель катализатора.
Носитель катализатора
Носитель катализатора может представлять собой любой носитель катализатора, пригодный для нанесения кобальта или соединение кобальта на его поверхность.
Носитель катализатора, как правило, представляет собой пористый носитель и предпочтительно является предварительно формованным. Носитель предпочтительно имеет средний диаметр пор между 8 и 50 нанометров, более предпочтительно между 10 и 15 нанометров. Объем пор носителя может составлять между 0.1 и 1 мл/г носителя катализатора, более предпочтительно между 0.3 и 0.9 мл/г носителя катализатора. Предварительно формованный носитель может представлять собой дисперсный носитель, предпочтительно со средним размером частиц между 1 и 500 микрометров, более предпочтительно между 10 и 250 микрометров, еще более предпочтительно между 45 и 200 микрометров.
Носитель катализатора может содержать подложку носителя катализатора и необязательно один или несколько модифицирующих компонентов. Подложка носителя катализатора может быть выбрана из группы, состоящей из окиси алюминия в форме одного или нескольких оксидов алюминия; диоксида кремния (SiO2); диоксида титана (TiO2); оксида магния (MgO) и оксида цинка (ZnO) и их смесей. Предпочтительно, подложка носителя выбрана из группы, состоящей из оксида алюминия в форме одного или нескольких оксидов алюминия; диоксида титана (TiO2) и диоксида кремния (SiO2). Более предпочтительно, подложка носителя представляет собой оксид алюминия в форме одного или нескольких оксидов алюминия. Подложка носителя может представлять собой коммерчески доступный продукт, например, Puralox (торговое название) (доступный от фирмы Sasol Germany GmbH).
Предпочтительно, носитель катализатора включает один или несколько модифицирующих компонентов. Это является тем случаем, когда подложка носителя является растворимой в нейтральном и/или кислотном водном растворе, или когда подложка носителя подвержена гидротермическому воздействию, как описано ниже.
Модифицирующий компонент может содержать компонент, который вызывает одно или более из следующего:
(i) уменьшение растворения носителя катализатора в водной среде;
(ii) подавление подверженности носителя катализатора гидротермическому воздействию (в особенности во время синтеза Фишера-Тропша);
(iii) увеличение объема пор носителя катализатора;
(iv) увеличение прочности и/или износа, и/или стойкости к истиранию носителя катализатора.
В предпочтительном варианте осуществления изобретения модифицирующий компонент уменьшает растворимость носителя катализатора в водной среде и/или подавляет подверженность носителя катализатора гидротермическому воздействию (в особенности во время синтеза Фишера-Тропша). Такая водная среда может включать водный раствор кислоты и/или нейтральный водный раствор, в особенности такую среду, которая присутствует во время пропитки водной фазой на стадии приготовления катализатора. Гидротермическое воздействие рассматривается как спекание носителя катализатора (например, оксида алюминия) во время синтеза углеводородов, в особенности синтеза Фишера-Тропша, из-за воздействия высокой температуры и воды.
Модифицирующий компонент может включать или состоять из Si, Zr, Со, Ti, Cu, Zn, Mn, Ва, Ni, Na, K, Ca, Sn, Cr, Fe, Li, Ti, Sr, Ga, Sb, V, Hf, Th, Ce, Ge, U, Nb, Та, W, La и смесей двух или более из них.
Модифицирующий компонент может быть выбран из группы, состоящей из Si; Zr; Cu; Zn; Mn; Ва; La; W; Ni и смесей одного или нескольких из них.
Предпочтительно, модифицирующий компонент выбран из группы, состоящей из Si и Zr. В предпочтительном варианте осуществления изобретения модифицирующий компонент представляет собой Si.
В случае, когда модифицирующий компонент представляет собой Si, уровень кремния в полученном носителе катализатора может составлять по меньшей мере 0.06 атомов Si на квадратный нанометр носителя катализатора, предпочтительно по меньшей мере 0.13 атомов Si на квадратный нанометр носителя катализатора, и более предпочтительно по меньшей мере 0.26 атомов Si на квадратный нанометр носителя катализатора.
Предпочтительно, верхний уровень составляет 2.8 атомов Si/нм2 носителя катализатора.
В одном предпочтительном варианте осуществления изобретения носитель катализатора содержит подложку носителя катализатора, необязательно включая модифицирующий компонент, выбранный из Si, Zr и W, при этом подложка носителя катализатора выбрана из группы, состоящей из окиси алюминия в форме одного или нескольких оксидов алюминия; диоксида кремния (SiO2) и диоксида титана (TiO2). Предпочтительно, подложка носителя катализатора представляет собой оксид алюминия в форме одного или нескольких оксидов алюминия и предпочтительно включает модифицирующий компонент, который предпочтительно выбран из Si, Zr и W, более предпочтительно Si. В одном предпочтительном варианте осуществления изобретения носитель катализатора может быть выбран из оксида алюминия в форме одного или нескольких оксидов алюминия, диоксида кремния (SiO2), диоксида титана (TiO2), оксида магния (MgO), модифицированного диоксидом кремния оксида алюминия и их смесей. Предпочтительно, носитель представляет собой оксид алюминия, модифицированный диоксидом кремния, например, продукт, доступный под торговой маркой Siralox от фирмы Sasol Germany GmbH. Siralox представляет собой высушенный распылением носитель на основе оксида алюминия, содержащего диоксид кремния. Носитель на основе оксида алюминия, модифицированного диоксидом кремния, может представлять собой продукт, описанный в патенте США 5045519, который включен в настоящий документ путем отсылки.
Один или несколько оксидов алюминия могут быть выбраны из группы, включающей (предпочтительно состоящей) гамма-оксид алюминия, дельта-оксид алюминия, тета-оксид алюминия и смеси двух или более из них. Предпочтительно, группа включает или, предпочтительно, состоит из гамма-оксида алюминия, дельта-оксида алюминия и смеси гамма-оксида алюминия и дельта-оксида алюминия. Носитель катализатора на основе оксида алюминия может представлять собой носитель катализатора, доступный под торговой маркой Puralox, предпочтительно Puralox SCCa 2/150 от фирмы SASOL Germany GmbH. Puralox SCCa 2/150 (торговая марка) представляет собой высушенный распылением носитель на основе оксида алюминия, состоящего из смеси гамма- и дельта-оксида алюминия.
Оксид алюминия предпочтительно представляет собой кристаллическое соединение, которое может быть описано формулой Al2O3·xH2O, где 0<х<1. Термин «оксид алюминия», таким образом, включает Al(ОН)3 и AlO(ОН), но включает соединения, такие как гамма-, дельта- и тета-оксид алюминия.
В предпочтительном варианте осуществления изобретения носитель катализатора или подложка носителя катализатора не представляет собой цеолит.
Последующий предшественник катализатора
Как указано выше, последующий предшественник катализатора готовят путем введения соединения кобальта на и/или в прокаленный исходный предшественник катализатора.
Соединение кобальта может представлять собой любое пригодное органическое или неорганическое соединение кобальта, предпочтительно соль кобальта. Предпочтительно, соединение кобальта представляет собой неорганическое соединение, более предпочтительно, неорганическую соль кобальта. Соединение кобальта может представлять собой нитрат кобальта и, в частности, может представлять собой Со(NO3)2·6H2O.
Предпочтительно, соединение кобальта является таким же, что и соединение кобальта, вводимое на и/или в носитель катализатора для формирования исходного предшественника катализатора.
Соединение кобальта можно вводить на и/или в прокаленный исходный предшественник катализатора любым подходящим способом, но предпочтительно путем пропитки. Предпочтительно, прокаленный исходный предшественник катализатора пропитывают соединением кобальта путем формирования смеси соединения кобальта; жидкого носителя для соединения кобальта и прокаленного исходного предшественника катализатора.
Жидкий носитель может содержать растворитель для соединения кобальта и, предпочтительно, соединение кобальта является растворенным в жидком носителе.
Жидкий носитель может представлять собой воду.
Жидкий носитель может представлять собой кислотный жидкий носитель и, предпочтительно, представляет собой кислотную водную композицию. Кислотный жидкий носитель может иметь рН ниже 5, предпочтительно ниже 3 и более предпочтительно ниже 3. Предпочтительно, чтобы рН было выше 1, более предпочтительно выше 1.8.
Пропитку можно осуществлять любым подходящим способом пропитки, включая пропитку по влагоемкости или пропитку в суспензии. Пропитка в суспензии является предпочтительной. Предпочтительно, соединение кобальта растворяют в жидком носителе для того, чтобы объем раствора был больше, чем ху литра, при этом указанный раствор затем смешивают с прокаленным исходным предшественником катализатора, и где х представляет собой объем пор по методу BET прокаленного исходного предшественника катализатора в мл/г носителя, и у представляет собой массу в кг подлежащего пропитке прокаленного исходного предшественника катализатора. Предпочтительно, объем раствора составляет больше, чем 1.5ху литра и, предпочтительно, составляет примерно 2ху литра.
Пропитку можно проводить при давлении ниже атмосферного, предпочтительно ниже 85 кПа(а), более предпочтительно при 20 кПа(а) и ниже.
Предпочтительно пропитку проводят при температуре выше 25°С. Предпочтительно, температура составляет выше 40°С, более предпочтительно температура составляет по меньшей мере 60°С, но предпочтительно не выше 95°С.
После пропитки может следовать частичная сушка пропитанного носителя, или пропитку и сушку можно проводить в одно и то же время. Предпочтительно, сушку проводят при температуре сушки выше 25°С.Предпочтительно, температура сушки составляет выше 40°С, более предпочтительно температура сушки составляет по меньшей мере 60°С, но предпочтительно не выше 95°С.Таким образом, температура сушки может быть обозначена Td2, в этом случае, Td2>25°C, предпочтительно >40°С, более предпочтительно ≥60°С; однако, предпочтительно, Td2≤95°С.Предпочтительно, частичную сушку осуществляют в субатмосферных условиях, более предпочтительно ниже 85 кПа(а), наиболее предпочтительно при 30 кПа(а) и ниже.
В одном варианте осуществления изобретения пропитку и частичную сушку можно проводить с использованием процедуры, которая включает первую стадию, на которой прокаленный исходный предшественник катализатора пропитывают (предпочтительно пропитка в суспензии) соединением кобальта при температуре выше 25°С и при субатмосферном давлении, и полученный продукт сушат; и по меньшей мере одну последующую стадию, на которой полученный частично высушенный продукт первой стадии подвергают обработке при температуре выше 25°С и субатмосферном давлении таким образом, что температура последующей стадии превышает температуру на первой стадии и/или субатмосферное давление на последующей стадии ниже, чем субатмосферное давление на первой стадии. Эта двухстадийная пропитка может представлять собой способ, описанный в WO 00/20116, который включен в настоящий документ путем отсылки.
В другом варианте осуществления изобретения введение соединения кобальта на и/или в прокаленный исходный предшественник катализатора можно проводить путем пропитки, включающей формирование смеси соединения кобальта, жидкого носителя для соединения кобальта и прокаленного исходного предшественника катализатора, в сочетании с сушкой полученного пропитанного прокаленного исходного предшественника катализатора при температуре выше 25°С для получения последующего предшественника катализатора. Сушку или частичную сушку полученного пропитанного прокаленного исходного предшественника катализатора можно осуществлять одновременно и/или после пропитки прокаленного исходного предшественника катализатора.
Допирующая добавка
Допирующую добавку, повышающую способность к восстановлению активного компонента катализатора, можно также вводить на и/или в носитель катализатора, и/или прокаленный исходный предшественник катализатора. Допирующая добавка может представлять собой металл, выбранный из группы, включающей палладий (Pd), платину (Pt), рутений (Ru), рений (Re) и смесь одного или нескольких их них. Массовое соотношение металла допирующей добавки (в особенности металла палладия или металла платины) к металлу кобальту может составлять от 1:300 до 1:3000.
Допирующую добавку можно вводить во время или после введения соединения кобальта на и/или в носитель катализатора в процессе приготовления исходного предшественника катализатора. Альтернативно или дополнительно добирующую добавку можно вводить во время или после введения соединения кобальта на и/или в прокаленный исходный предшественник катализатора в процессе приготовления последующего предшественника катализатора. Допирующую добавку можно вводить в виде соединения допирующей добавки, которое представляет собой соединение металла, выбранного из группы, включающей палладий (Pd), платину (Pt), рутений (Ru), рений (Re) и смесь одного или нескольких из них. Предпочтительно, соединение допирующей добавки представляет собой неорганическую соль и предпочтительно является растворимым в воде.
Восстановление
Подвергание последующего предшественника катализатора восстановительным условиям может включать приведение в контакт последующего предшественника катализатора с восстановительным газом для активации, таким образом, последующего предшественника катализатора.
Подвергание последующего предшественника катализатора восстановительным условиям может включать приведение в контакт последующего предшественника катализатора с восстановительным газом для активации, таким образом, последующего предшественника катализатора, при этом последующий предшественник катализатора находится при температуре, близкой к температуре сушки при инициировании его контакта с восстановительным газом.
Восстановительный газ может представлять собой СО или может включать СО. Восстановительный газ может содержать или может включать комбинацию СО и Н2. Предпочтительно, тем не менее, чтобы восстановительный газ представлял собой водород или содержащий водород газ. Содержащий водород газ может состоять из водорода и одного или нескольких инертных газов, которые являются инертными в отношении активного катализатора. Содержащий водород газ предпочтительно содержит по меньшей мере 8 объемных % водорода, предпочтительно по меньшей мере 90 объемных % водорода.
Приведение в контакт восстановительного газа с последующим предшественником катализатора можно осуществлять любым подходящим способом. Предпочтительно, последующий предшественник катализатора обеспечен в форме слоя частиц предшественника катализатора с восстановительным газом, пропускаемым через слой частиц. Слой частиц может представлять собой неподвижный слой, но предпочтительно представляет собой псевдоожиженный слой и, предпочтительно, восстановительный газ действует в качестве псевдоожижающей среды для слоя частиц предшественника катализатора.
Восстановление можно проводить при давлении от 0.6 до 1.5 бар(а), предпочтительно от 0.8 до 1.3 бар(а). Альтернативно, давление может составлять от 1.5 бар(а) до 20 бар(а). Более предпочтительно, тем не менее, чтобы давление представляло собой давление, близкое к атмосферному.
Восстановление предпочтительно проводят при температуре, превышающей на 25°С температуру, при которой соединение кобальта и прокаленное соединение кобальта будет восстановлено до активной формы. Предпочтительно, восстановление проводят при температуре выше 150°С и предпочтительно ниже 600°С. Более предпочтительно, восстановление проводят при температуре ниже 500°С, наиболее предпочтительно ниже 450°С.
Во время активации температура может изменяться и предпочтительно, чтобы температура повышалась до максимальной температуры, как указано выше.
Поток восстановительного газа через слой катализатора предпочтительно контролируют для обеспечения поддержания достаточно низкого уровня загрязняющих примесей, образующихся в процессе восстановления. Восстановительный газ может быть рециркулирован и, предпочтительно, рециркулированный восстановительный газ обрабатывают для удаления одной или нескольких загрязняющих примесей, образовавшихся в процессе восстановления. Загрязняющие примеси могут содержать одну или несколько примесей, состоящих из воды и аммиака.
Активацию можно проводить за две или более стадий, во время которых скорость нагревания или объемная скорость восстановительного газа, или и то и другое изменяются.
В процессе восстановления объемная скорость подачи восстановительного газа (GHSV) предпочтительно составляет выше 1 мn 3/(кг Со(NO3)2·H2O)/час, более предпочтительно выше 4 мn 3/(кг Со(NO3)2·H2O)/час. Предпочтительно объемная скорость подачи восстановительного газа (GHSV) составляет ниже 20 мn 3/(кг Со(NO3)2·H2O)/час, более предпочтительно, ниже 15 мn 3/(кг Со(NO3)2·Н2О)/час.
В процессе восстановления скорость нагрева последующего предшественника катализатора предпочтительно составляет выше 0.1°С/мин. Предпочтительно, чтобы скорость нагрева последующего предшественника катализатора не превышала 0.1°С/мин.
Активный кобальтсодержащий катализатор синтеза углеводородов
Активный кобальтсодержащий катализатор синтеза углеводородов может представлять собой катализатор синтеза Фишера-Тропша (FT). Катализатор синтеза FT может быть пригодным для способа, выполняемого в реакторе с неподвижным слоем, реакторе с трехфазным псевдоожиженным слоем или даже реакторе с неподвижным псевдоожиженным слоем. Предпочтительно, способ выполняют в реакторе синтеза FT с трехфазным псевдоожиженным слоем.
Активный кобальтсодержащий катализатор синтеза углеводородов может содержать кобальт при нагрузке от 5 до 70 г Со/100 г носителя катализатора, предпочтительно от 20 до 40 г Со/100 г носителя катализатора, и более предпочтительно от 25 до 35 г Со/100 г носителя катализатора.
В одном предпочтительном варианте осуществления изобретения активный кобальтсодержащий катализатор синтеза углеводородов не подвергается воздействию окислительных условий при температуре, превышающей 100°С, и предпочтительно не подвергается воздействию окислительных условий при температуре, превышающей 50°С перед использованием указанного катализатора в синтезе углеводородов.
В одном варианте осуществления изобретения активный кобальтсодержащий катализатор синтеза углеводородов может быть подвергнут воздействию окислительных условий при температуре, не превышающей 100°С, предпочтительно при температуре, не превышающей 50°С, чтобы таким образом пассивировать катализатор перед использованием в синтезе углеводородов.
В самом предпочтительном варианте осуществления изобретения активный кобальтсодержащий катализатор синтеза углеводородов не подвергают окислению перед использованием указанного катализатора в синтезе углеводородов.
Согласно второму аспекту настоящего изобретения обеспечен кобальтсодержащий катализатор синтеза углеводородов, приготовленный в соответствии со способом по первому аспекту изобретения.
Синтез углеводородов
Согласно третьему аспекту изобретения обеспечен способ получения углеводородов, при этом способ включает приготовление кобальтсодержащего катализатора синтеза углеводородов в соответствии со способом по первому аспекту изобретения; и приведение в контакт водорода с монооксидом углерода при температуре выше 100°С и при давлении по меньшей мере 10 бар в присутствии кобальтсодержащего катализатора синтеза углеводородов, получая, таким образом, углеводороды и, необязательно, оксигенаты углеводородов в процессе синтеза Фишера-Тропша.
В одном предпочтительном варианте осуществления изобретения кобальтсодержащий катализатор синтеза углеводородов не подвергают воздействию окислительных условий при температуре выше 100°С, предпочтительно выше 50°С перед использованием указанного катализатора в синтезе углеводородов.
В наиболее предпочтительном варианте осуществления изобретения кобальтсодержащий катализатор синтеза углеводородов не подвергают окислению перед использованием указанного катализатора в синтезе углеводородов.
Способ получения углеводородов может также включать стадию гидроочистки для превращения углеводородов и необязательно их оксигенатов в жидкие топлива и/или химические продукты.
Согласно четвертому аспекту настоящего изобретения обеспечены продукты, полученные с использованием способа получения углеводородов в соответствии с третьим аспектом изобретения.
Изобретение будет описано далее более подробно с обращением к следующим не ограничивающим примерам.
ПРИМЕРЫ
ПРИМЕР 1 (сравнительный)
НС1825 (30 г Со/0.075 г Pt/100 г Al2O3)
Катализатор готовят посредством двух пропиток в суспензии. После каждой стадии пропитки высушенный промежуточный продукт термически обрабатывают/прокаливают на воздухе.
Первая пропитка: НС1825/1 (16 г Со/0.025 г Pt/100 г Al2O3)
Co(NO3)2·6H2O растворяют в 30 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 5 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют носитель, Si-модифицированный Puralox, для формирования суспензии. Затем следует процедура сушки, представленная ниже, для формирования исходного предшественника катализатора.
Процедура сушки

После сушки в вакууме предшественник (30 г) прокаливают (термически обрабатывают) в вертикальной печи на воздухе (0.06 мn 3/час; GHSV воздуха =4.5 м3 n/(кг Со(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч при 250°С, и после этого охлаждают до комнатной температуры для получения НС1825/1 (16 г Со/0.025 г Pt/100 г Al2O3), то есть прокаленного исходного предшественника катализатора.
Этот материал затем используют для второй пропитки:
Со(NO3)2·6Н2О растворяют в 20 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 4 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют прокаленный промежуточный продукт НС18825/1, и далее следует процедура сушки, описанная выше, для формирования последующего предшественника катализатора.
После сушки в вакууме предшественник (20 г) прокаливают/термически обрабатывают в вертикальной печи на воздухе (0.04 мn 3/час; GHSV воздуха =5.5 м3 n/(кг Co(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч при 250°С, и после этого охлаждают до комнатной температуры для получения требуемого катализатора из ПРИМЕРА 1 (НС1825; 30 г Со/0.075 г Pt/100 г Al2O3).
ПРИМЕР 2 (сравнительный)
С1848С (30 г Со/0.05 г Pt/100 г Al2O3)
Катализатор готовят посредством двух пропиток в суспензии. После каждой стадии пропитки высушенный промежуточный продукт термически обрабатывают в атмосфере водорода.
Первая пропитка: С1848С/1 (16 г Со/0.01 г Pt/100 г Al2O3)
Co(NO3)2·6H2O растворяют в 20 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 4 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют носитель, Si-модифицированный Puralox, для формирования суспензии. Следующую процедуру сушки используют для формирования исходного предшественника катализатора:
Процедура сушки

После сушки в вакууме предшественник (20 г) термически обрабатывают (прямое восстановление) в вертикальной печи в атмосфере водорода (100%) (0.04 мn 3/час; GHSV=4.5 м3 n/(кг Co(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 425°С (6 ч время выдержки при 425°С) и затем охлаждают до комнатной температуры в атмосфере водорода.
Систему продувают аргоном (1 ч) и затем обрабатывают 1% O2 (в аргоне) в течение 1 ч. Температура образца остается на уровне комнатной температуры на протяжении стадии пассивации. Данную процедуру пассивации проводят для обеспечения того, что образец больше не является пирофорным и безопасен в обращении.
Пассивированный материал (С1848С/1) затем используют для второй пропитки:
Co(NO3)2·6H2O растворяют в 10 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 4 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют пассивированный С1848С/1, и далее следует процедура сушки, описанная выше.
После сушки в вакууме предшественник (10 г) термически обрабатывают (прямое восстановление) в вертикальной печи в атмосфере водорода (100%) (0.04 мn 3/час; GHSV=11.1 м3 n/(кг Со(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 0.5°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 6 ч при 425°С, и затем охлаждают до комнатной температуры в атмосфере водорода.
Систему продувают аргоном (1 ч) и затем обрабатывают 1% О2 (в аргоне) в течение 1 ч для получения требуемого катализатора из ПРИМЕРА 2 (С1848С; 30 г Со/0.05 г Pt/100 г Al2O3). Температура образца остается на уровне комнатной температуры на протяжении стадии пассивации. Данную процедуру пассивации проводят для обеспечения того, что образец больше не является пирофорным и безопасен в обращении.
ПРИМЕР 3 (изобретения)
С1846С (30 г Со/0.05 г Pt/100 г Al2O3)
Катализатор готовят посредством двух пропиток в суспензии. После первой стадии пропитки высушенный промежуточный продукт термически обрабатывают (прокаливают) на воздухе, тогда как после второй стадии пропитки высушенный промежуточный продукт термически обрабатывают (прямое восстановление) в атмосфере водорода.
Первая пропитка: С1846С/1 (16 г Со/0.01 г Pt/100 г Al2O3)
Co(NO3)2·6H2O растворяют в 20 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 4 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют носитель, Si-модифицированный Puralox, для формирования суспензии. Далее следует процедура сушки, представленная ниже, для формирования исходного предшественника катализатора: Процедура сушки

После сушки в вакууме предшественник (20 г) прокаливают в вертикальной печи на воздухе (0.04 мn 3/час; GHSV воздуха =4.5 м3 n/(кг Со(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч при 250°С, и после этого охлаждают до комнатной температуры для получения С1846С/1 (16 г Со/0.01 г Pt/100 г Al2O3), то есть прокаленного исходного предшественника катализатора.
Этот материал (С1846С/1) затем используют для второй пропитки:
Co(NO3)2·6H2O растворяют в 10 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 4 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют С1846С/1, и далее следует процедура сушки, описанная выше, для формирования последующего предшественника катализатора.
После сушки в вакууме предшественник (10 г) термически обрабатывают (прямое восстановление) в вертикальной печи в атмосфере водорода (100%) (0.04 мn 3/час; GHSV=11.1 м3 n/(кг Co(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 0.5°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 6 ч при 425°С, и затем охлаждают до комнатной температуры в атмосфере водорода.
Систему продувают аргоном (1 ч) и затем обрабатывают 1% O2 (в аргоне) в течение 1 ч для получения требуемого катализатора из ПРИМЕРА 3 (С1846С; 30 г Со/0.05 г Pt/100 г Al2O3). Температура образца остается на уровне комнатной температуры на протяжении стадии пассивации. Данную процедуру пассивации проводят для обеспечения того, что образец больше не является пирофорным и безопасен в обращении.
ПРИМЕР 4 (изобретения)
С1854А (30 г Со/0.075 г Pt/100 г Al2O3)
Катализатор готовят посредством двух пропиток в суспензии. После первой стадии пропитки высушенный промежуточный продукт термически обрабатывают (прокаливают) на воздухе, тогда как после второй стадии пропитки высушенный промежуточный продукт термически обрабатывают (прямое восстановление) в атмосфере водорода.
Первая пропитка: С1854А/1 (16 г Со/0.025 г Pt/100 г Al2O3)
Co(NO3)2·6H2O растворяют в 40 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 8 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют носитель, Si-модифицированный Puralox, для формирования суспензии. Далее следует процедура сушки, представленная ниже, для формирования исходного предшественника катализатора: Процедура сушки

После сушки в вакууме пропитанный предшественник (45 г) прокаливают в вертикальной печи на воздухе (0.12 мn 3/час; GHSV воздуха =6.0 м3 n/(кг Co(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч при 250°С, и после этого охлаждают до комнатной температуры для получения С1854А/1 (16 г Со/0.025 г Pt/100 г Al2O3), то есть прокаленного исходного предшественника катализатора.
Этот материал (С1854А/1) затем используют для второй пропитки:
Со(NO3)2·6H2O растворяют в 30 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 4 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют С1854А/1, и далее следует процедура сушки, описанная выше, для формирования последующего предшественника катализатора.
После сушки в вакууме предшественник (15 г) термически обрабатывают (прямое восстановление) в вертикальной печи в чистом водороде (100%) (0.06 мn 3/час; GHSV=11.0 м3 n/(кг Co(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 0.5°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 6 ч при 425°С, и после этого охлаждают до комнатной температуры в атмосфере водорода.
Систему продувают аргоном (1 ч) и затем обрабатывают 1% О2 (в аргоне) в течение 1 ч для получения требуемого катализатора из ПРИМЕРА 4 (С1854А; 30 г Со/0.075 г Pt/100 г Al2O3). Температура образца остается на уровне комнатной температуры на протяжении стадии пассивации. Этот процесс пассивации проводят для обеспечения того, что образец больше не является пирофорным и безопасен в обращении.
ПРИМЕР 5
Образцы катализаторов из Примеров 1-4 тестируют на выполнение синтеза Фишера-Тропша (FT). Перед FTS-тестированием образцы повторно восстанавливают в водороде с использованием скорости нагрева 1°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 5 ч при 425°С и охлаждают до 230°С.
FTS-тестирование проводят в лабораторном реакторе с неподвижным слоем с использованием 0.6 г катализатора. FTS выполняют при 230°С с использованием отношения Н2/СО 1.6 и давления 16 бар. GHSV постоянно регулируют для поддержания приблизительно 50% конверсии СО.
Относительная активность и селективность в отношении образования метана для этих Примеров 1-4 представлены в Таблице 1. Относительная FTS-активность выражена относительно характеристики (не раскрытого) референсного катализатора.
Таблица 1: Относительная активность и селективность в отношении образования метана для Примеров 1-4 во время FTS-тестирования в лабораторном реакторе с неподвижным слоем через 140 часов режима работы
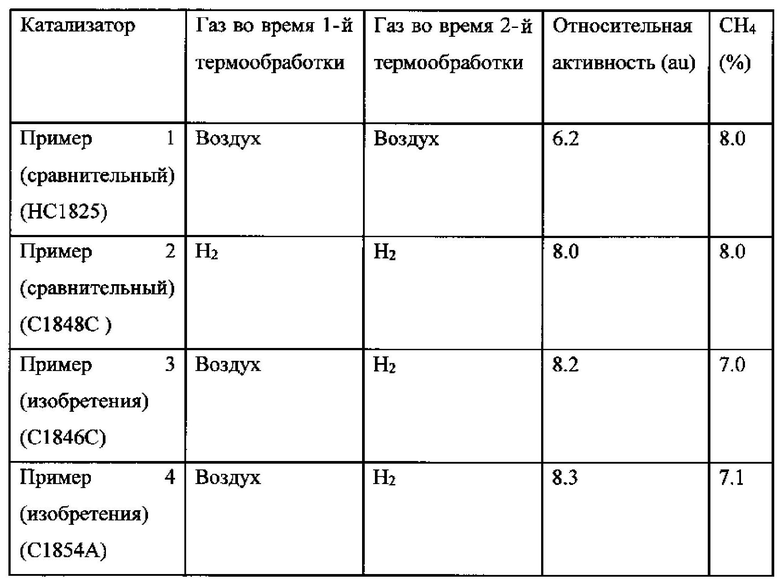
Как можно видеть из Таблицы 1, преимущество Примеров 3 и 4 (оба по изобретению) перед Примером 1 по результатам тестирования в лабораторном реакторе с неподвижным слоем состоит в увеличенной активности (примерно 30%) и пониженной селективности в отношении образования метана.
Кроме того, преимущество Примеров 3 и 4 (оба по изобретению) перед Примером 2 состоит в уменьшенном количестве стадий способа, так как стадию пассивации не проводили после стадий первой пропитки и первой термической обработки, а также пониженной селективности в отношении образования метана.
ПРИМЕР 6 (изобретения)
С1858А (30 г Со/0.075 г Pt/100 г Al2O3)
Данный катализатор готовят способом, аналогичным способу в Примере 4.
Однако, после второй пропитки Co/Pt и сушки в вакууме предшественник (15 г) термически обрабатывают (прямое восстановление) в вертикальной печи в атмосфере смеси водород/азот (10% водорода) (0.06 мn 3/час; GHSV=11.0 м3 n/(кг Со(NO3)2·6H2O)/ч) с использованием скорости изменения температуры 0.5°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 6 ч при 425°С, и затем охлаждают до комнатной температуры в атмосфере водорода.
Систему продувают аргоном (1 ч) и затем обрабатывают 1% O2 (в Аргоне) в течение 1 ч для получения требуемого катализатора из ПРИМЕРА 6 (С1854А; 30 г Со/0.075 г Pt/100 г Al2O3). Температура образца остается на уровне комнатной температуры во время стадии пассивации. Этот процесс пассивации проводят для обеспечения того, что образец больше не является пирофорным и безопасен в обращении.
ПРИМЕР 7
Образец катализатора из Примера 6 тестируют на выполнение синтеза Фишера-Тропша (FTS) способом, аналогичным способу, описанному в Примере 5.
Относительная активность и селективность в отношении образования метана для этого Примера 6 представлены в Таблице 2 и проведено сравнение с Примером 1.
Таблица 2: Относительная активность и селективность в отношении образования метана для Примеров 1 и 6 во время FTS-тестирования в лабораторном реакторе с неподвижным слоем через 140 часов режима работы

Как можно видеть из Таблицы 2, преимущество Примера 6 (изобретения), с использованием в качестве восстановительного газа 10% водорода, перед Примером 1 состоит в увеличенной активности (примерно 25%) и пониженной селективности в отношении образования метана.
ПРИМЕР 8
Образцы катализаторов из Примеров 1 и 4 также тестируют на выполнение синтеза Фишера-Тропша (FTS) в микро CSTR реакторе (т.е. суспензионном реакторе).
Перед FTS-тестированием образцы повторно восстанавливают в водороде с использованием скорости нагрева 1°С/мин до 425°С с последующей выдержкой в течение 5 ч при 425°С, охлаждают до комнатной температуры (примерно 25°С) и выгружают в FTS-воск для предотвращения окисления.
Восстановленный и покрытый воском образец загружают в 1-литровый микро CSTR реактор FTS с использованием 10 г катализатора. FTS выполняют при 230°С с использованием отношения Н2/СО 1.7 и давления 18 бар. GHSV постоянно регулируют для поддержания приблизительно 60% конверсии СО.
Относительная активность и селективность в отношении образования метана для этих Примеров 1 и 4 представлены в Таблице 3. В этом случае относительная FTS активность выражена относительно Примера 1.
Таблица 3: Относительная активность и селективность в отношении образования метана для Примеров 1 и 4 во время FTS-тестирования в микро CSTR реакторе после 200 часов работы
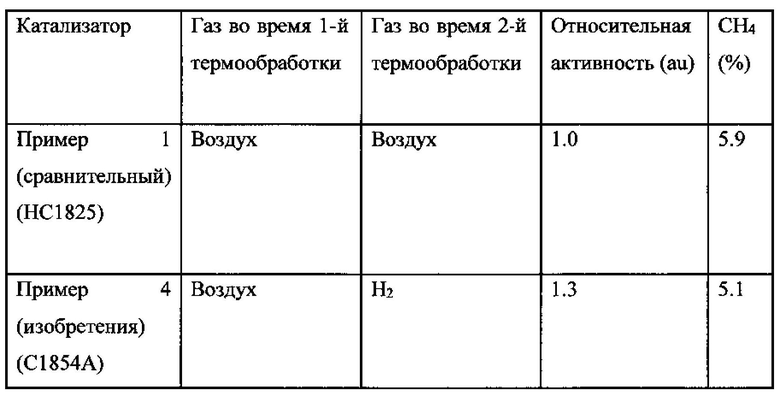
Как можно видеть из Таблицы 3, преимущество Примера 4 (изобретения) перед Примером 1 по результатам тестирования в суспензионном CSTR состоит опять же в увеличенной активности (примерно 30%) и пониженной селективности в отношении образования метана.
ПРИМЕР 9 (сравнительный)
C1855ISAC (30 г Со/0.075 г Pt/100 г Al2O3)
Катализатор готовят посредством двух пропиток в суспензии. После первой пропитки и сушки материал прокаливают на воздухе в условиях псевдоожиженного слоя. После второй пропитки и сушки предшественник катализатора также прокаливают на воздухе в неподвижном слое с последующим восстановлением неподвижного слоя и катализом Фишера-Тропша.
Первая пропитка: С1855/1 (16 г Со/0.025 г Pt/100 г Al2O3)
Co(NO3)2·6H2O растворяют в 15 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 2 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют носитель, Si-модифицированный Puralox, для формирования суспензии. Затем следует процедура сушки, описанная в Примере 1, для формирования исходного предшественника катализатора.
После сушки в вакууме исходный предшественник катализатора (15 г) прокаливают (термически обрабатывают) в вертикальной печи на воздухе (0.03 мn 3/час; GHSV воздуха =4.5 м3 n/(кг Co(NO3)2·6H2O)/4) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч, и охлаждают до комнатной температуры для получения С1855/1 16 г Со/0.025 г Pt/100 г Al2O3, то есть прокаленного исходного предшественника катализатора.
Этот материал затем используют для второй пропитки:
Co(NO3)2·6H2O растворяют в 10 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 2 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют прокаленный промежуточный продукт С1855/1, и такая же процедура сушки следует для формирования последующего предшественника катализатора, С1855РС/2.
После сушки в вакууме предшественник, С1855РС/2 (1.0 г), разбавляют SiC (4 г, зернистость 320), упаковывают в камеру реактора с неподвижным слоем и затем прокаливают/термически обрабатывают на воздухе (60 мл/мин) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч, и охлаждают до комнатной температуры. После прокаливания на воздухе в неподвижном слое образец продувают Ar (30 мин) с последующим восстановлением в водороде с использованием скорости нагрева 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 5 ч. Образец охлаждают в атмосфере Н2 до 230°С с последующим немедленным FTS. FTS выполняют при 230°С с использованием отношения Н2/СО 1.6 и давления 16 бар. GHSV постоянно регулируют для поддержания приблизительно 50% конверсии СО.
ПРИМЕР 10 (изобретения)
C1855ISRC (30 г Со/0.075 г Pt/100 г Al2O3)
Высушенный в вакууме предшественник катализатора из ПРИМЕРА 9, С1855РС/2 (1.0 г), разбавляют SiC (4 г, зернистость 320), упаковывают в камеру реактора с неподвижным слоем и затем термически обрабатывают в атмосфере чистого водорода (100 мл/мин) (прямое восстановление) с использованием скорости изменения температуры 0.5°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 6 ч. Образец охлаждают в атмосфере Н2 до 230°С с последующим немедленным FTS. FTS выполняют при 230°С с использованием отношения Н2/СО 1.6 и давления 16 бар. GHSV постоянно регулируют для поддержания приблизительно 50% конверсии СО.
FT-активность Примера 10 (изобретения) была на 20% лучше соответствующей FT-активности Примера 9 (сравнительный). Селективность в отношении образования метана Примеров 9 и 10 была сходной.
ПРИМЕР 11 (изобретения)
C1855MS (30 г Со/0.075 г Pt/100 г Al2O3)
Катализатор готовят посредством двух пропиток в суспензии. После первой стадии пропитки высушенный промежуточный продукт термически обрабатывают (прокаливают) на воздухе, при этом после стадии второй пропитки высушенный промежуточный продукт термически обрабатывают (прямое восстановление) в атмосфере водорода и загружают в расплавленный воск и тестируют на FTS в микро CSTR суспензионном реакторе при 230°С с использованием отношения Н2/СО 1.7 и давления 18 бар.
Первая пропитка: C1855MS/1 (16 г Со/0.025 г Pt/100 г Al2O3)
Со(NO3)2·6H2O растворяют в 20 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 5 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют носитель, Si-модифицированный Puralox, для формирования раствора суспензии. Затем следует процедура сушки, описанная в Примере 1, для формирования исходного предшественника катализатора.
После сушки в вакууме исходный предшественник катализатора (25 г) прокаливают в вертикальной печи на воздухе (0.07 мn 3/час; GHSV воздуха =6.0 м3 n/(кг Со(NO3)2·6Н2О)/ч) с использованием скорости изменения температуры 1°С/мин от комнатной температуры (примерно 25°С) до 250°С с последующей выдержкой в течение 6 ч, и охлаждают до комнатной температуры для получения C1855MS/1, 16 г Со/0.025 г Pt/100 г Al2O3, то есть прокаленного исходного предшественника катализатора.
Этот материал (C1855MS/1) затем используют для второй пропитки:
Со(NO3)2·6Н2О растворяют в 15 мл дистиллированной воды и (NH3)4Pt(NO3)2 в 3 мл дистиллированной воды. После этого два раствора смешивают. Значение рН доводят до рН 2.0-2.3 с использованием разбавленной HNO3. Раствор перемешивают при 60°С в течение 5-10 минут и затем добавляют C1855MS/1, и далее следует описанная выше процедура сушки для формирования последующего предшественника катализатора.
После сушки в вакууме предшественник (15 г) термически обрабатывают (прямое восстановление) в вертикальной печи в атмосфере чистого водорода (100%) (0.06 мn 3/час; GHSV=11.0 м3 n/(кг Co(NO3)2·6H2O)/4) с использованием скорости изменения температуры 0.5°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 6 ч, и затем охлаждают до комнатной температуры (примерно 25°С) в атмосфере водорода.
Восстановленный катализатор затем загружают в расплавленный воск (25 г) и оставляют для охлаждения и отверждения под слоем аргона. Восстановленный и покрытый воском образец загружают в 1-литровый микро CSTR реактор FTS с использованием 10 г катализатора. FTS проводят при 230°С с использованием отношения H2/CO 1.7 и давления 18 бар. GHSV постоянно регулируют для поддержания приблизительно 60% конверсии СО.
ПРИМЕР 12 (сравнительный)
Катализатор НС1825 (30 г Со/0.075 г Pt/100 г Al2O3) из ПРИМЕРА 1 восстанавливают в вертикальной печи в атмосфере чистого водорода (100%) с использованием скорости изменения температуры 1.0°С/мин от комнатной температуры (примерно 25°С) до 425°С с последующей выдержкой в течение 16 ч, и затем охлаждают до комнатной температуры (примерно 25°С) в атмосфере водорода.
Восстановленный катализатор затем загружают в расплавленный воск (25 г) и оставляют для охлаждения и отверждения под слоем аргона. Восстановленный и покрытый воском образец загружают в 1-литровый микро CSTR реактор FTS с использованием 10 г катализатора. FTS проводят при 230°С с использованием отношения Н2/СО 1.7 и давления 18 бар. GHSV постоянно регулируют для поддержания приблизительно 60% конверсии СО.
FT-активность катализатора из Примера 11 (изобретения) была примерно на 20% лучше соответствующего катализатора, приготовленного посредством двух прокаливаний на воздухе (Пример 12, сравнительный) с последующим восстановлением, покрытием воском и микро суспензионной FTS, тогда как селективность в отношении образования метана была сходной (т.е. 5.9%).
После пропитки и сушки в вакууме исходный или последующий предшественник катализатора, как правило, охлаждают до комнатной температуры (примерно 25°С) перед инициированием стадии прокаливания на воздухе или стадии прямого восстановления в атмосфере водорода. Тем не менее, стадию прокаливания на воздухе или стадию прямого восстановления в атмосфере водорода можно также начинать сразу же после завершения стадии сушки (например, при 90°С) без предварительного охлаждения до комнатной температуры.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2009 |
|
RU2481156C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2592271C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕДШЕСТВЕННИКА КОБАЛЬТСОДЕРЖАЩЕГО КАТАЛИЗАТОРА И СПОСОБ СИНТЕЗА УГЛЕВОДОРОДОВ | 2019 |
|
RU2796695C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2591702C2 |
| КАТАЛИЗАТОРЫ НА ОСНОВЕ КОБАЛЬТА | 2001 |
|
RU2261143C2 |
| КАТАЛИЗАТОРЫ | 2010 |
|
RU2517700C2 |
| КАТАЛИЗАТОРЫ | 2011 |
|
RU2551433C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕДШЕСТВЕННИКА КАТАЛИЗАТОРА И КАТАЛИЗАТОРА ФИШЕРА-ТРОПША НА ОСНОВЕ КОБАЛЬТА | 2001 |
|
RU2298434C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕДШЕСТВЕННИКА КАТАЛИЗАТОРА НА ОСНОВЕ КОБАЛЬТА И СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ФИШЕРА-ТРОПША | 2001 |
|
RU2259234C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ НАНЕСЕННОГО КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША НА ОСНОВЕ КОБАЛЬТА | 2008 |
|
RU2456329C2 |
Изобретение относится к способу получения кобальтсодержащего катализатора синтеза углеводородов, при этом способ включает: прокаливание исходного предшественника катализатора, содержащего носитель катализатора, несущий соединение кобальта, путем термообработки исходного предшественника катализатора в не восстановительных условиях для разложения соединения кобальта и/или для того, чтобы вызвать взаимодействие соединения кобальта с кислородом, получая, таким образом, прокаленный исходный предшественник катализатора; введение соединения кобальта на и/или в прокаленный исходный предшественник катализатора, с тем чтобы прокаленный исходный предшественник катализатора нес данное соединение кобальта, получая, таким образом, последующий предшественник катализатора; и непосредственное подвергание последующего предшественника катализатора восстановительным условиям без предварительного подвергания термообработке в окислительных условиях, которое вызывает окисление соединения кобальта на носителе или разложение соединения кобальта, для активации последующего предшественника катализатора с получением, таким образом, кобальтсодержащего катализатора синтеза углеводородов. Изобретение также относится к способу получения углеводородов в способе синтеза Фишера-Тропша, который заключается в приведении в контакт водорода с монооксидом углерода при температуре выше 100°С и при давлении по меньшей мере 10 бар в присутствии заявленного кобальтсодержащего катализатора. Технический результат заключается в улучшенной активности катализатора и/или пониженной селективности в отношении образования метана. 2 н. и 14 з.п. ф-лы, 3 табл., 12 пр.
1. Способ получения кобальтсодержащего катализатора синтеза углеводородов, при этом способ включает:
- прокаливание исходного предшественника катализатора, содержащего носитель катализатора, несущий соединение кобальта, путем термообработки исходного предшественника катализатора в невосстановительных условиях для разложения соединения кобальта и/или для того, чтобы вызвать взаимодействие соединения кобальта с кислородом, получая, таким образом, прокаленный исходный предшественник катализатора;
- введение соединения кобальта на и/или в прокаленный исходный предшественник катализатора, с тем чтобы прокаленный исходный предшественник катализатора нес данное соединение кобальта, получая, таким образом, последующий предшественник катализатора; и
- непосредственное подвергание последующего предшественника катализатора восстановительным условиям без предварительного подвергания термообработке в окислительных условиях, которое вызывает окисление соединения кобальта на носителе или разложение соединения кобальта, для активации последующего предшественника катализатора с получением, таким образом, кобальтсодержащего катализатора синтеза углеводородов.
2. Способ по п. 1, который включает приготовление исходного предшественника катализатора путем введения соединения кобальта на и/или в носитель катализатора.
3. Способ по п. 1, в котором прокаливание исходного предшественника катализатора проводится в окислительных условиях при температуре выше 95°С, но не выше 400°С.
4. Способ по п. 1, в котором соединение кобальта, которое вводится на и/или в прокаленный исходный предшественник катализатора, является таким же, как и соединение кобальта исходного предшественника катализатора.
5. Способ по п. 4, в котором соединение кобальта представляет собой Co(NO3)2·6H2O.
6. Способ по п. 1, в котором введение соединения кобальта на и/или в прокаленный исходный предшественник катализатора осуществляется путем пропитки в суспензии.
7. Способ по п. 6, в котором пропитка в суспензии проводится при давлении ниже атмосферного ниже 85 кПа и при температуре выше 25°С, но не выше 95°С.
8. Способ по п. 6, в котором после пропитки в суспензии следует частичная сушка пропитанного прокаленного исходного предшественника катализатора при температуре сушки выше 25°С и при давлении ниже атмосферного ниже 85 кПа.
9. Способ по п. 1, в котором подвергание последующего предшественника катализатора восстановительным условиям включает приведение в контакт последующего предшественника катализатора с восстановительным газом, активируя, таким образом, последующий предшественник катализатора.
10. Способ по п. 1, в котором введение соединения кобальта на и/или в прокаленный исходный предшественник катализатора осуществляют путем пропитки, включающей формирование смеси соединения кобальта, жидкого носителя для соединения кобальта и прокаленного исходного предшественника катализатора, и сушку полученного пропитанного прокаленного исходного предшественника катализатора при температуре выше 25°С, для получения последующего предшественника катализатора.
11. Способ по п. 10, в котором подвергание последующего предшественника катализатора восстановительным условиям включает приведение в контакт последующего предшественника катализатора с восстановительным газом, активируя, таким образом, последующий предшественник катализатора, при этом последующий предшественник катализатора находится при температуре, близкой к температуре сушки при приведении его в контакт с восстановительным газом.
12. Способ по п. 9, в котором восстановительный газ представляет собой водород или водородсодержащий газ.
13. Способ по п. 9, в котором приведение в контакт последующего предшественника катализатора с восстановительным газом достигается путем пропускания восстановительного газа через слой частиц последующего предшественника катализатора.
14. Способ по п. 11, в котором восстановительный газ представляет собой водород или содержащий водород газ.
15. Способ по п. 11, в котором приведение в контакт последующего предшественника катализатора с восстановительным газом достигается путем пропускания восстановительного газа через слой частиц последующего предшественника катализатора.
16. Способ получения углеводородов, при этом способ включает получение кобальтсодержащего катализатора синтеза углеводородов согласно способу по любому из пп. 1-15; и приведение в контакт водорода с монооксидом углерода при температуре выше 100°С и при давлении по меньшей мере 10 бар в присутствии кобальтсодержащего катализатора синтеза углеводородов, получая, таким образом, углеводороды в способе синтеза Фишера-Тропша.
| СПОСОБ ПОЛУЧЕНИЯ НОСИТЕЛЯ ДЛЯ КАТАЛИЗАТОРА С ПОВЫШЕННОЙ ГИДРОТЕРМАЛЬНОЙ СТАБИЛЬНОСТЬЮ (ВАРИАНТЫ), КАТАЛИЗАТОР ДЛЯ СИНТЕЗА УГЛЕВОДОРОДОВ И СПОСОБ СИНТЕЗА УГЛЕВОДОРОДОВ ИЗ СИНТЕЗ-ГАЗА | 2003 |
|
RU2340394C2 |
| US 4413064 A, 01.11.1983 | |||
| WO 2010075516 A2, 01.07.2010 | |||
| WO 2010097754 A2, 02.09.2010 | |||
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ УГЛЕВОДОРОДА ИЗ СИНТЕЗ-ГАЗА И СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА | 2004 |
|
RU2311230C2 |

