Область техники, к которой относится изобретение
Настоящее изобретение относится к производным пирролидиния и их применению в качестве фармацевтических средств, в частности к способам получения бромида гликопиррония и его аналогов в промышленных масштабах.
Предпосылки создания изобретения
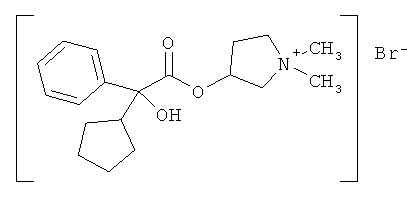
Бромид гликопиррония, также известный как бромид 3-[(циклопентил-гидроксифенилацетил)окси]-1,1-диметилпирролидиния или гликопирролат, представляет собой антимускариновый агент, который в настоящее время применяется в форме инъекции с целью уменьшения секреции в ходе анестезии или принимается перорально для лечения язв желудка.
Он имеет следующую химическую структуру:
В патенте US 2956062 сообщается, что 1-метил-3-пирролидиловый эфир альфа-циклопентилминдальной кислоты может быть получен из метилового эфира альфа-циклопентилминдальной кислоты и что четвертичная соль метилбромида может быть получена путем насыщения раствора 1-метил-3-пирролидилового эфира альфа-циклопентилминдальной кислоты метилбромидом в сухом этилацетате и последующей фильтрации кристаллического твердого вещества, которое образуется при стоянии.
Способ получения 1-метил-3-пирролидилового эфира альфа-циклопентилминдальной кислоты согласно патенту US 2956062 включает транс-этерификацию метилгликолята с аминоспиртом в присутствии металлического натрия, приводящему к гликолятному промежуточному соединению. Металлический натрий является высокореакционноспособным, что представляет риск для здоровья и безопасности, делающий нежелательным его использование в промышленном масштабе для коммерческого производства.
Способ согласно патенту US 2956062 требует получения метилового сложного эфира на предварительной стадии и алкилирования сложных аминоэфиров на последующей стадии с целью образования искомых четвертичных солей аммония.
В способе согласно патенту US 2956062 образуется смесь диастереомеров. Относительные доли диастереомеров могут варьироваться в широких пределах от партии к партии. Этот разброс может вызывать непредсказуемые различия при приготовлении препаратов в виде сухого порошка из бромида гликопиррония, что может вызвать проблемы при приготовлении из подобных сухих порошков препаратов для фармацевтического использования.
В заявке на патент US 2007/0123557 сообщается об антихолинергических 1-(алкоксикарбонилметил)-1-метилпирролидиловых сложных эфирах. Описывается сочетание (R)-циклопентилминдальной кислоты с (R,S)-1-метилпирролидин-3-олом в условиях по Мицунобу с целью получения чистых (R)-стереоизомерных соединений, которые вводят во взаимодействие с бромацетатом с целью получения искомых сложных эфиров. Однако следует отметить, что реагенты, используемые в реакциях по Мицунобу, как правило, диалкилазодикарбоксилаты и трифенилфосфин, представляют риск для здоровья, безопасности и экологии, что делает нежелательным их использование в промышленных масштабах для коммерческого производства. Кроме того, они, как правило, слишком дороги для получения и слишком сложны в использовании в коммерческом производстве.
Таким образом, существует необходимость создания способа получения бромида гликопиррония, который решает вышеуказанные проблемы, присутствующие в известном способе, или представляет собой по меньшей мере полезную альтернативу ему.
Краткое описание изобретения
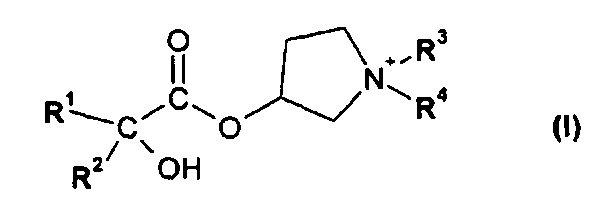
В первом варианте осуществления настоящего изобретения, его объектом является способ получения соединения формулы I
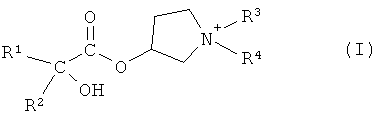
в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил; и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил;
который включает стадии:
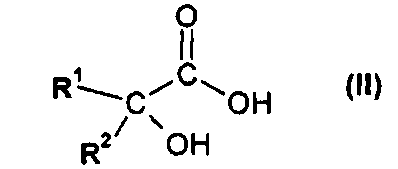
(а) взаимодействия соединения формулы II
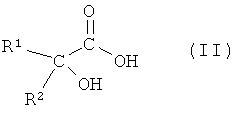
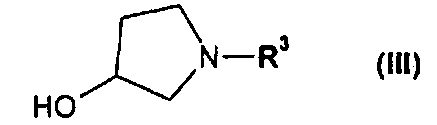
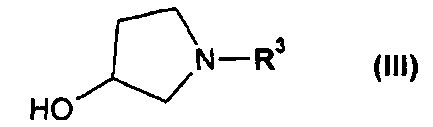
или его соли, где R1 или R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, с соединением формулы III
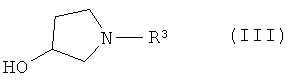
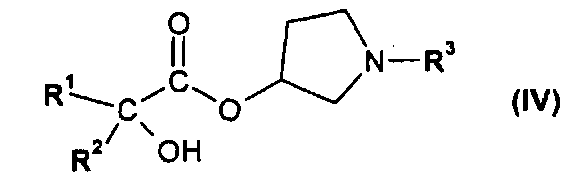
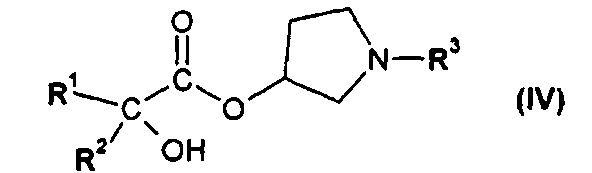
или его производным, способным к образованию сложного эфира, где R3 представляет собой (C1-C8)алкил, с получением соединения формулы IV
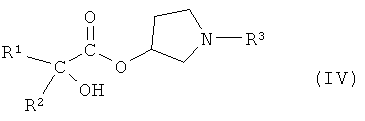 ,
,
где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, и R3 представляет собой (C1-C8)алкил; и
(b) взаимодействия соединения формулы IV, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, и R3 представляет собой (C1-C8)алкил, с соединением формулы V

где R4 представляет собой (C1-C8)алкил, а X представляет собой уходящую группу, с получением соединения формулы I в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил; и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил.
Стадию (а) удобно осуществлять в присутствии агента для сочетания, например карбонилдиимидазола.
Во втором варианте осуществления настоящего изобретения его объектом является способ получения состава в виде сухого порошка для ингаляции, содержащего соединение формулы I
в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил; и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил;
который включает стадии:
(i) взаимодействия соединения формулы II
или его соли, где R1 или R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, с соединением формулы III
или его производным, способным к образованию сложного эфира, где R3 представляет собой (C1-C8)алкил, с получением соединения формулы IV
где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, и R3 представляет собой (C1-C8)алкил;
(ii) взаимодействия соединения формулы IV, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, и R3 представляет собой (C1-C8)алкил, с соединением формулы V
где R4 представляет собой (C1-C8)алкил, а Х представляет собой уходящую группу, с получением соединения формулы I в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил; и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил;
(iii) необязательной очистки лекарственного вещества посредством кристаллизации с получением очищенного лекарственного вещества;
(iv) микронизации лекарственного вещества; и
(v) добавления частиц носителя с получением сухого порошка для ингаляций.
В предпочтительном варианте осуществления настоящего изобретения частицы носителя представляют собой кристаллические сахара, в особенности моногидрат лактозы или безводную лактозу.
В предпочтительном варианте осуществления настоящего изобретения кристаллический гликопирролат микронизируют вместе с агентом, регулирующим силу адгезии. Агент, регулирующий силу адгезии, предпочтительно представляет собой стеарат магния.
Термины
Термины, употребляемые в настоящем изобретении, имеют следующие значения.
Термин «(C1-C8)алкил», как он употребляется в настоящем изобретении, обозначает неразветвленный или разветвленный (C1-C8)алкил, содержащий от 1 до 8 атомов углерода, и может представлять собой, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутл, неразветвленный или разветвленный пентил, неразветвленный или разветвленный гексил, неразветвленный или разветвленный гептил, неразветвленный или разветвленный октил. Подходящим примером «(C1-C8)алкила» является (C1-C4)алкил, в особенности метил.
Термин «(C3-C8)циклоалкил», как он употребляется в настоящем изобретении, обозначает циклоалкил, содержащий от 3 до 8 атомов углерода, и может представлять собой, например циклопропил, циклобутил, циклопентил, метилциклопентил, циклогексил, метилциклогексил, диметилциклогексил, циклогептил, бициклогептил, циклооктил и бициклооктил. Подходящим примером (C3-C8)циклоалкила является (C3-C6)циклоалкил, в особенности циклопропил.
Термин «(C6-C10)арил», как он употребляется в настоящем изобретении, означает ароматическую группу, содержащую от 6 до 10 кольцевых атомов углерода. Примеры (C6-C10)арильных групп включают, но не ограничиваются ими, фенил, инданил, инденил и нафтил. Подходящим примером «(C6-C10)арила» является фенил.
Термин «уходящая группа», как он употребляется в настоящем изобретении, означает химическую группу, которая уходит вместе с электронной парой при гетеролитическом разрыве связи. В данной области техники хорошо известно, что уходящие группы могут принимать много форм, вследствие чего подразумевается, что данный термин включает в свой объем любую химическую группу, которая выполняет вышеуказанную функцию. Уходящие группы могут представлять собой анионы или нейтральные молекулы. Обычными анионными уходящими группами являются галогенид-анионы, такие как Cl-, Br- и I-, и сложные эфиры сульфокислот, такие как пара-толуолсульфонат или «тозилат» (TsO-). Обычные уходящие группы в виде нейтральных молекул представляют собой воду, аммиак и спирты. В способе по настоящему изобретению уходящая группа представляет собой анионную уходящую группу, например Cl-, Br- или I-, в особенности Br-.
Термин «соль», как он употребляется в настоящем изобретении, относится к кислотно-аддитивной соли или соли с основанием соединения по настоящему изобретению. Термин «соли» включает, в частности, «фармацевтически приемлемые соли». Термин «фармацевтически приемлемые соли» относится к солям, которые сохраняют биологическую эффективность и свойства соединений по настоящему изобретению и которые, как правило, не являются нежелательными биологически или иным образом. Во многих случаях, соединения по настоящему изобретению способны к образованию солей с кислотами и/или основаниями, благодаря присутствию амино- и/или карбоксильных групп, или же сходных с ними групп. Фармацевтически приемлемые кислотно-аддитивные соли могут быть образованы с неорганическими кислотами или органическими кислотами, представляя собой, например, ацетат, аспартат, бензоат, безилат, бромид/гидробромид, гидрокарбонат/карбонат, гидросульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофиллинат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидроидодид/иодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат, сукцинат, сульфосалицилат, тартрат, тозилат и трифторацетат.
Неорганические кислоты, производными которых могут быть соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и им подобные. Органические кислоты, из которых могут быть образованы соли, включают например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфокислоту, этансульфокислоту, толуолсульфокислоту, сульфосалициловую кислоту и им подобные. Фармацевтически приемлемые основно-аддитивные соли могут быть образованы неорганическими и органическими основаниями. Неорганические основания, с которыми могут быть образованы соли, включают, например, соли аммония и металлов, находящихся в колонках с I по XII Периодической системы. В некоторых вариантах осуществления настоящего изобретения, соли образованы с натрием, калием, аммонием, кальцием, магнием, железом, серебром, цинком и медью; в частности, подходящие соли включают соли аммония, калия, натрия, кальция и магния. Органические основания, производными от которых могут быть соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая замещенные амины природного происхождения, циклические амины, основные ионообменные смолы и им подобные. Отдельные органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин.
Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, основного или кислотного остатка, с помощью стандартных химических способов. Как правило, такие соли могут быть получены путем введения во взаимодействие этих соединений в форме свободных кислот со стехиометрическим количеством соответствующего основания (такого как гидроксид, карбонат, гидрокарбонат натрия, кальция, магния или калия и им подобные), или путем введения во взаимодействие этих соединений в форме свободных оснований со стехиометрическим количеством соответствующей кислоты. Такие реакции, как правило, осуществляют в воде, органическом растворителе или в их смеси. Как правило, желательно использование неводных сред, таких как диэтиловый эфир, этилацетат, этанол, изопропанол или ацетонитрил, если это осуществимо. Наиболее подходящие примеры соединений формулы I представляют собой бромидные соли.
Термин «цвиттер-ион», как он употребляется в настоящем изобретении, относится к внутренним солям, которые образуются в случаях, когда основная и кислотная группы одновременно присутствуют в одной молекуле. Например, соединения формулы I содержат кислотную карбоксильную группу, которая может существовать в виде цвиттер-иона в сочетании с четвертичным аммонийным атомом азота.
На всем протяжении данного описания, и последующей формулы изобретения, если иное не следует из контекста, слово «включать» или его варианты, такие как «включает» или «включающий», следует понимать как подразумевающее включение указанного объекта в целом или его составной части, или же группы объектов в целом или их частей, но не исключение любого другого объекта в целом или его составной части, или же группы объектов в целом или их частей.
Подробное описание изобретения
Объектом настоящего изобретения является способ получения соединения формулы I:
в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил; и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил.
Наиболее предпочтительным соединением формулы I является бромид гликопиррония или гликопирролат, который имеет следующую химическую структуру:
Бромид гликопиррония имеет два асимметрических центра и, как следствие, существует в виде четырех изомерных форм или стереоизомеров, а именно бромид (3R,2'R)-, (3S,2'R)-, (3R,2'S)- и (3S,2'S)-3-[(циклопентил-гидроксифенилацетил)окси]-1,1-диметилпирролидиния.
Настоящий способ представляет собой двухстадийный способ получения соединений формулы I, в особенности бромида гликопиррония, который может быть осуществлен в одном реакционном сосуде, т.е. является однореакторным способом.
На первой стадии (а) способа по настоящему изобретению соединение формулы II
или его соль, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, вводят во взаимодействие с соединением формулы III
или его производным, способным к образованию сложного эфира, где R3 представляет собой (C1-C8)алкил, с получением соединения формулы IV
,
где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, и R3 представляет собой (C1-C8)алкил.
Реакция может быть осуществлена посредством использования известных способов взаимодействия гидроксильных соединений или их солей (например, натриевых солей) с карбоновыми кислотами или их производными, способными образовывать сложные эфиры, такими как галоидангидриды кислот, или же аналогичных способов, как это описано ниже в примерах. Реакцию обычно осуществляют в органических растворителях, например, в диметилформамиде (ДМФ) или толуоле, в присутствии агента для сочетания, например, 1,1'-карбонилдиимидазола (КДИ), предпочтительно в инертной атмосфере, например, в атмосфере аргона. Подходящие температуры проведения реакции находятся в диапазоне от 0°С до 100°С, предпочтительно от 30°С до 80°С, в особенности около 60°С.
В тех случаях, если агент для сочетания представляет собой 1,1'-карбонилдиимидазол, активное промежуточное соединение представляет собой соединение формулы IIa:
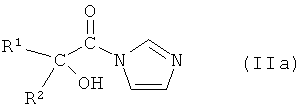
или его соль, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил.
В предпочтительном варианте осуществления настоящего изобретения R1 и R2 представляют собой циклопентил и фенил, соответственно. Таким образом, соединение формулы IIa представляет собой 2-циклопентил-2-гидрокси-1-имидазол-1-ил-2-фенилэтанон.
Соединение формулы I может быть очищено любым подходящим способом, известным в данной области техники, например посредством перекристаллизации, и/или может быть осуществлено удаление каких-либо крупных частиц посредством просеивания.
Нижеследующие подходящие, предпочтительные, более предпочтительные и наиболее предпочтительные варианты осуществления настоящего изобретения могут быть реализованы независимо, совместно или в любом сочетании.
Предпочтительно, R1 и R2 независимо друг от друга представляют собой циклопропил, циклогексил или фенил. Альтернативно, R1 представляет собой, подходящим образом, циклопропил, a R2 предпочтительно представляет собой фенил.
Предпочтительно, R3 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил. Альтернативно, R3 предпочтительно является метилом.
Предпочтительно, R4 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил. Альтернативно, R4 предпочтительно является метилом.
Предпочтительно, Х представляет собой хлор, бром или йод. Альтернативно, Х предпочтительно является бромом.
В предпочтительных вариантах осуществления настоящего изобретения R1 и R2 в соединениях формул II и IV независимо друг от друга представляют собой (C5-C6)циклоалкил или фенил, а R3 в соединениях формул III и IV представляет собой (C1-C4)алкил, в особенности метил.
В другом предпочтительном варианте осуществления настоящего изобретения R1 в соединениях формул II и IV представляет собой (C5-C6)циклоалкил, R2 в соединениях формул II и IV представляет собой фенил, а R3 в соединениях формул III и IV представляет собой (C1-C4)алкил, в особенности метил.
В еще одном предпочтительном варианте осуществления настоящего изобретения в соединениях формул II и IV R1 представляет собой циклопентил, R2 в соединениях формул II и IV представляет собой фенил, а R3 в соединениях формул III и IV представляет собой метил.
На второй стадии (b) способа по настоящему изобретению, соединение формулы IV
где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, и R3 представляет собой (C1-C8)алкил, вводят во взаимодействие с соединением формулы V
где R4 представляет собой (C1-C8)алкил, а Х представляет собой уходящую группу, с получением соединения формулы I в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил; и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил.
Реакция может быть осуществлена с помощью известных способов взаимодействия хинуклидиноловых сложных эфиров с алкилгалогенидами, или же аналогичными способами, как это описано ниже в примерах. Реакция обычно осуществляется в воде или в органическом растворителе, например, в ацетонитриле, диметилформамиде (ДМФ), диметилсульфоксиде (ДМСО), этилацетате или хлороформе. Реакцию осуществляют при температуре от примерно -10°С до примерно 120°С, подходящими являются температуры от примерно -5°С до примерно 80°С, в особенности от примерно 0°С до примерно 60°С.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы V представляет собой метилбромид. Соединение является летучим (температура кипения 4°С), поэтому взаимодействие сначала проводят при температуре от примерно 0°С до примерно 20°С, а затем, перед кристаллизацией, реакционную смесь нагревают до температуры примерно 60°С. Кристаллизация индуцируется охлаждением, т.е. активным или пассивным понижением температуры смеси. В предпочтительном варианте осуществления настоящего изобретения температуру смеси понижают медленно, т.е. в течение нескольких часов, посредством использования коммерчески доступного автоматизированного оборудования. Если то требуется, в реакционную смесь вносятся затравки для облегчения кристаллизации. В предпочтительном варианте осуществления, реакционную смесь охлаждают до температуры примерно 50°С, после чего вносят в нее затравки, а затем медленно охлаждают ее до температуры примерно 15°С.
Выбор растворителя, используемого в реакции алкилирования, может оказывать значительное влияние на выход отдельных стереоизомеров желаемого соединения. Действительно, благоприятным может являться проведение стадии (b) в органическом растворителе, в котором стереоизомеры соединения формулы I обладают различающимися растворимостями. Например, при взаимодействии 1-метилпирролидин-3-илового эфира циклопентилгидроксифенилуксусной кислоты и метилбромида в н-пропаноле с целью получения бромида гликопиррония, продукт, кристаллизующийся из раствора, обогащен бромидами (3S,2'R)- и (3R,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния, тогда как бромиды (3R,2'R)- и (3S,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния, которые гораздо лучше растворимы в пропаноле, имеют тенденцию оставаться в фильтрате.
Нижеследующие подходящие, предпочтительные, более предпочтительные и наиболее предпочтительные варианты осуществления настоящего изобретения могут быть реализованы независимо, совместно или в любом сочетании.
Предпочтительно, R1 и R2 независимо друг от друга представляют собой циклопропил, циклогексил или фенил. Альтернативно, R1 предпочтительно представляет собой циклопропил, а R2 предпочтительно представляет собой фенил.
Предпочтительно, R3 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил. Альтернативно, R3 предпочтительно является метилом.
Предпочтительно, R4 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил. Альтернативно, R4 предпочтительно является метилом.
Предпочтительно, Х представляет собой хлор, бром или йод. Альтернативно, Х предпочтительно является бромом.
Предпочтительно, Х представляет собой остаток сульфо- или фосфокислоты, такой как мезилат, тозилат, бензолсульфонат или метилметанфосфонат.
В предпочтительном варианте осуществления настоящего изобретения R1 и R2 в соединениях формул IV и I независимо друг от друга представляют собой (C5-C6)циклоалкил или фенил, R3 в соединениях формул IV и I представляет собой (C1-C5)алкил, и R4 в соединениях формул V и I представляет собой (C1-C4)алкил, в особенности метил.
В другом предпочтительном варианте осуществления настоящего изобретения R1 в соединениях формул IV и I представляет собой (C5-C6)циклоалкил, R2 в соединениях формул IV и I представляет собой фенил, R3 в соединениях формул IV и I представляет собой (C1-C4)алкил, в особенности метил, и R4 в соединениях формул V и I представляет собой (C1-C4)алкил, в особенности метил.
В еще одном предпочтительном варианте осуществления настоящего изобретения R1 в соединениях формул IV и I представляет собой циклопентил, R2 в соединениях формул IV и I представляет собой фенил, R3 в соединениях формул IV и I представляет собой метил, и R4 в соединениях формул V и I представляет собой метил. Таким образом, соединение формулы I представляет собой гликопирроний в форме соли или цвиттер-иона.
Способ по настоящему изобретению преодолевает различные проблемы, сопутствующие способу получения гликопирролата, который описан в патенте US 2956062. Посредством устранения необходимости получения сложного метилового эфира из кислоты в качестве дополнительной стадии, можно сократить способ, улучшить выход и избежать необходимости использовать трудоемкий метод транс-этерификации, который часто сложно контролировать, сложно оптимизировать и который подразумевает использование опасных реагентов, таких как натрий и гидрид натрия, и опасных условий реакции, таких как образование газообразного водорода. Представляется удобным и эффективным по временным и экономическим затратам то, что исходные вещества представляют собой коммерчески доступные кислоты, а также то, что способ может быть осуществлен в одном реакционном сосуде, т.е. посредством простого однореакторного процесса. Эти преимущества делают способ по настоящему изобретению значительно более подходящим для крупномасштабного промышленного производства, чем способ, описанный в патенте US 2956062.
Предпочтительный вариант осуществления способа по настоящему изобретению в той степени, в которой его конечным продуктом является гликопирролат, представлен и сопоставлен со способом, описанным в патенте US 2956062, на нижеследующей схеме. Согласно этой схеме, гликопирролат получают известным способом посредством стадий 1, 2 и 3, тогда как с помощью способа по настоящему изобретению его получают через стадии 1a и 3:
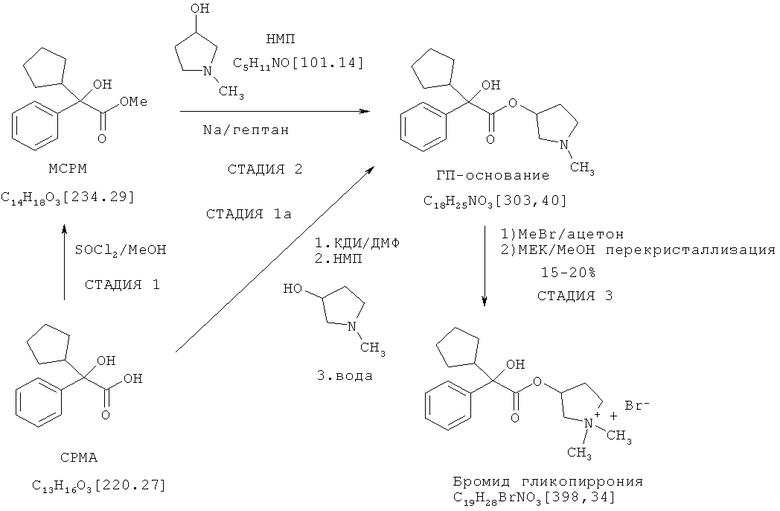
В предпочтительном варианте осуществления настоящего изобретения R1 и R2 в соединении формулы II представляют собой циклопентил и фенил, соответственно, R3 в соединении формулы III представляет собой метил, R4 в соединении формулы V представляет собой метил, и соединение формулы I представляет собой рацемическую смесь бромидов (3S,2'R)- и (3R,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния.
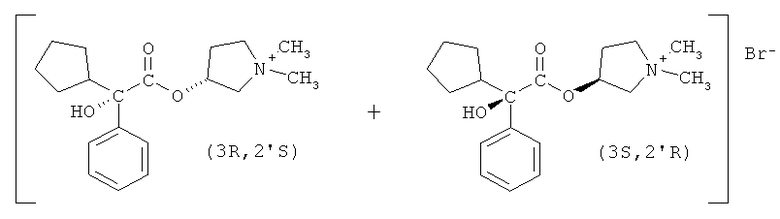
Способ по настоящему изобретению минимизирует изменения относительных количеств этих энантиомеров гликопирролата.
Объектом настоящего изобретения также является способ получения сухих порошкообразных составов для ингаляций, содержащих соединение формулы I
в форме соли или цвиттер-иона, где R1 и R2 независимо друг от друга представляют собой (C3-C8)пиклоалкил или (C6-C10)арил; и R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил. Этот способ включает пять стадий (i)-(v).
Стадии (i) и (ii) идентичны стадиям (а) и (b) вышеупомянутого способа получения лекарственного вещества, которое содержит соединение формулы I в форме соли или цвиттер-иона.
На третьей стадии (iii) способа получения сухого порошкообразного состава для ингаляций, которая является необязательной, лекарственное вещество, содержащее соединение I в форме соли или цвиттер-иона, очищают путем кристаллизации. Эта стадия может быть при необходимости повторена до достижения желаемой чистоты. Лекарственное вещество может быть просеяно для удаления крупных частиц.
На четвертой стадии (iv) способа получения сухого порошкообразного состава для ингаляций, лекарственное вещество, содержащее соединение формулы I в форме соли или цвиттер-иона, необязательно очищенное, согласно стадии (iii), микронизуют. Это уменьшает размер частиц лекарственного вещества, чтобы сделать его подходящим для введения путем ингаляции. Среднемассовый аэродинамический диаметр (MMAD) этих частиц предпочтительно составляет менее 10 микрон (мкм). Частицы, имеющие аэродинамический диаметр более чем 10 мкм, склонны к соударениям со стенками гортани и, в основном, не достигают легких. Частицы, имеющие аэродинамический диаметр в диапазоне от примерно 2 мкм до примерно 5 мкм, в основном распределяются в дыхательных бронхиолах, тогда как меньшие частицы, имеющие аэродинамический диаметр в диапазоне от примерно 0,05 мкм до примерно 3 мкм, скорее всего, оседают в альвеолах и поглощаются кровотоком.
Оборудование для микронизации хорошо известно в данной области техники и включает различные механизмы для дробления и перемалывания, например мельницы компрессионного типа, такие как мельницы для механосинтеза, ударные мельницы, такие как шаровые мельницы, гомогенизаторы и флюидизаторы, а также струйные мельницы. Подходящее оборудование для микронизации включает мешалки с низкой скоростью сдвига, такие как измельчитель порошков Turbula®, и мешалки с высокой скоростью сдвига, такие как измельчитель порошков MiPro®.
В предпочтительном варианте осуществления, кристаллический гликопирролат измельчают с помощью противоточной струйной мельницы Hosokawa Alpine® 100 AFG или спиральной струйной мельницы Hosokawa Alpine® AS100. Другое подходящее оборудование для измельчения струйного типа включает струйные мельницы Hosokawa Alpine® AFG140, AFG200, AFG280 и AFG400.
На пятой стадии (v) процесса получения сухого порошкообразного препарата для ингаляций частицы носителя добавляют к микронизованному кристаллическому лекарственному веществу, что приводит к образованию искомого сухого порошкообразного состава для ингаляций. Частицы носителя делают микронизованное лекарственное вещество менее склонным к когезии и улучшают его сыпучесть. Это делает порошок более удобно применимым в последующих операциях с ним, например при заполнении капсул составом в виде сухого порошка. Микронизованное лекарственное вещество имеет тенденцию прилипать к поверхности частиц носителя, пока оно находится в приборе для ингаляций сухих порошков, но распыляться с поверхности частиц носителя при ингаляции в дыхательные пути, образуя тонкоизмельченную суспензию. Более крупные частицы носителя, в основном, оседают в ротоглоточной полости.
Частицы носителя могут состоять из любого фармакологически инертного вещества или сочетания веществ, приемлемых для ингаляции. Подходящие частицы носителя состоят из одного или более кристаллических сахаров, включающих моносахариды, дисахариды, полисахариды и спирты на основе сахаров, такие как арабиноза, глюкоза, фруктоза, манноза, сахароза, трегалоза, лактоза, мальтоза, крахмалы, декстран, маннит или сорбит. Особенно предпочтительным носителем является лактоза, например моногидрат лактозы или безводная лактоза.
Предпочтительно, практически все частицы носителя (по массе) имеют диаметр от 20 до 1000 мкм, более предпочтительно от 50 до 500 мкм, но в особенности от 0 до 250 мкм. Подходящим является диаметр практически всех частиц носителя (по массе) менее 355 мкм. Это обеспечивает хорошие характеристики потока и поглощения, а также улучшенное высвобождение активных частиц в дыхательных путях, увеличивая оседание активных частиц в нижних отделах легких. Следует понимать, что диаметр частиц, на который ссылаются в настоящем изобретении, представляет собой аэродинамический диаметр частиц.
Если это желательно, в сухой порошкообразный состав для ингаляций включают один или более агентов, регулирующих силу адгезии, таких как стеарат магния. Регулирующий силу адгезии агент приводит к общему увеличению вдыхаемой фракции тонкоизмельченных частиц составов гликопирролата в виде сухого порошка. Он стабилизирует вещества-носители и лекарственное вещество путем подавления или замедления нежелательных морфологических фазовых переходов. Это также увеличивает эффективность дозирования сухих порошкообразных препаратов гликопирролата путем улучшения сыпучести порошка.
Другие подходящие агенты, регулирующие силу адгезии, включают аминокислоты, такие как лейцин, фосфолипиды, такие как лецитин, или производные жирных кислот, такие как стеарат кальция. Однако особенно предпочтительным является стеарат магния. Его предпочтительно добавляют в особо малых количествах, например от 0,1 до 5% стеарата магния по массе, более предпочтительно, от 0,1 до 2% по массе, но особенно предпочтительно, от 0,25 до 1% по массе в отношении всего препарата.
Регулирующий силу адгезии агент предпочтительно находится в форме микрочастиц, но он может быть добавлен в жидкой или твердой форме, причем для некоторых веществ, в особенности тех, которые не могут легко образовывать частицы вещества, и/или в случае, если частицы должны быть особенно мелкими, может быть предпочтительным добавление вещества в жидкой форме, например, в виде суспензии или раствора.
Сухой порошок может содержаться в виде стандартных доз в капсулах, например, из желатина или пластика, или в блистерах (например, алюминиевых или пластиковых), для использования в устройстве для ингаляций сухого порошка, каковое может быть однодозовым или многодозовым устройством. Предпочтительно, общая масса порошка на одну капсулу составляет от 5 мг до 50 мг. Альтернативно, сухой порошок может содержаться в емкости мультидозового ингалятора для сухого порошка (MDDPI), настроенного на введение, например, 3-25 мг сухого порошка за одно срабатывание. Подходящее устройство для введения в сухого порошка в инкапсулированной форме описано в патенте US 3991761 или международной заявке на изобретение WO 05/113042, в то время как подходящие MDDPI-устройства включают приборы, описанные в международных заявках на изобретение WO 97/20589 и WO 97/30743.
Настоящее изобретение проиллюстрировано следующими примерами.
Примеры
Пример 1
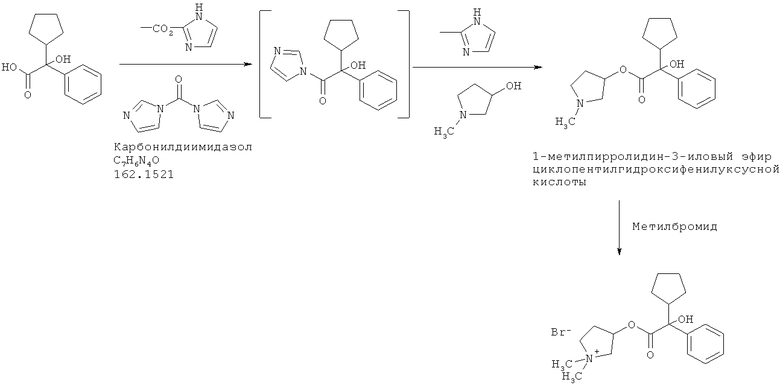
Получение бромидов (3S,2'R)- и (3R,2'S)-3-[(циклопентилгидрокси-фенилацетил)окси]-1,1 -диметилпирролидиния
30 г циклопентилминдальной кислоты, растворенной в 135 г диметилформамида (ДМФ), обрабатывают 27 г карбонилдиимидазола при температуре 18°С (порциями), для образования «активного амида». После добавления 16,9 г 1-метилпирролидин-3-ола, смесь нагревают до температуры 60°С в течение 1 часа и перемешивают в течение 8 часов при этой температуре. После проверки полноты превращения смесь охлаждают и добавляют 200 г воды. Затем смесь экстрагируют 200 г толуола и промывают экстракт водой 3 раза. Концентрируют органическую фазу с целью получения 1-метилпирролидин-3-илового эфира циклопентилгидроксифенилуксусной кислоты в виде примерно 50%-ного раствора в толуоле, готового для использования на следующей стадии.
Раствор разбавляют 120 г н-пропанола и охлаждают до температуры 0°С. В смесь вводят 16,8 г метилбромида, перемешивают смесь в течение 2 часов, и затем постепенно охлаждают до 60°С, испаряя избыток метилбромида в газопромыватель. Затем смесь охлаждают до температуры 50°С и вносят затравку для облегчения кристаллизации. После того медленно понижают температуру до 15°С в течение 18 часов. Затем фильтрацией отделяют твердое вещество, получая после сушки 22,7 г. Вещество состоит в основном из одной пары энантиомеров, рацемической смеси бромидов (3S,2'R)- и (3R,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния, с чистотой более 90% (по данным ВЭЖХ). Другая пара диастереомеров (бромиды (3R,2'R)- и (3S,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния) остается, в основном, в фильтрате, так как эти соединения значительно более растворимы в н-пропаноле, чем другие стереоизомеры.
Получаемое твердое вещество затем перекристаллизовывают из н-пропанола (1:10 по массе) с целью получения чистых бромидов (3S,2'R)- и (3R,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния, то есть чистоты более 99,9%, как определяют с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ).
Этот способ может быть представлен следующей схемой реакций:
Пример 2
Получение 1-метилпирролидин-3-илового эфира циклопентилгидроксифенилуксусной кислоты в толуоле
1 г циклопентилминдальной кислоты суспендируют в 4,7 г толуола и добавляют 1,5 г карбодиимидазола в твердом виде. Через 30 минут добавляют 0,69 г 1-метилпирролидин-3-ола и 20 мг трет-бутилата натрия. Смесь перемешивают при комнатной температуре в течение 18 часов, а затем добавляют воду. После перемешивания фазы разделяют, дважды промывают органическую фазу водой и выпаривают, что приводит к примерно 50%-ному раствору 1-метилпирролидин-3-илового эфира циклопентилгидроксифенилуксусной кислоты в толуоле.
Пример 3
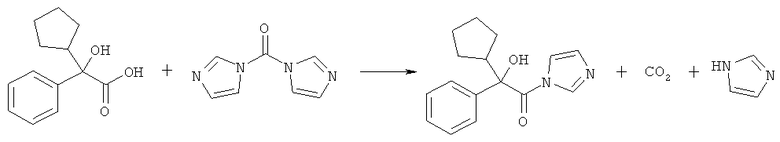
Получение 2-циклопентил-2-гидрокси-1-имидазол-1-ил-2-фенилэтанона, активного промежуточного соединения
Имидазолидиновое производное циклопентилминдальной кислоты получают и выделяют в виде твердого вещества следующим способом:
10 г циклопентилминдальной кислоты суспендируют в 30 мл ацетонитрила и охлаждают смесь до температуры 0°С. Добавляют 10,3 г твердого карбонилдиимидазола и нагревают смесь до комнатной температуры в течение 2 часов. По мере выпадения осадка выделяется диоксид углерода в виде газа. После этого смесь охлаждают до температуры 5°С, фильтруют твердое вещество, промывают его ацетонитрилом и сушат под вакуумом при температуре 40°С, что приводит к 7,3 г чистого 2-циклопентил-2-гидрокси-1-имидазол-1-ил-2-фенилэтанона.
Этот способ представлен следующей схемой реакций:
С помощью масс-спектроскопии (МС) высокого разрешения показано, что молекулярная формула соединения (в виде M+H) представляет собой C16H19O2N2, с точной массой 271,14414 (отклонение от рассчитанной величины составляет 0,14575 мд).
1Н ЯМР (протонный ядерный магнитный резонанс) (600 МГц, ДМСО-(d6): 1,03-1,07 (m, 1H), 1,25-1,30 (m, 1H), 1,35-1,40 (m, 1H), 1,40-1,50 (m, 1H), 1,53-1,56 (m, 2H), 1-60-1,67 (m, 1H), 1,75-1,84 (m, 1H), 1,03-1,85 (8H, 8 вторичных CH2-протонов в циклопентильном кольце, H-C11, H-C12, H-C13, H-C14); 2,7-2,9 (m, 1H, H-C10); 6,76 (1H, H-C5); 6,91 (1H, H-C4); 7,29 (1H, H-C18); 7,39 (2H, H-C17, H-C19); 7,49 (2H, H-C16, H-C20); 7,65 (1H, H-C2).
Соединение было охарактеризовано с помощью ИК-спектроскопии (измерено в тонкой пленке на спектрометре BRUKER TENSOR 27 FT-IR в диапазоне волновых чисел 4000-600 см-1 с разрешением 4 см-1). Отнесения наиболее важных полос даны ниже:
Деформационное CH в монозамещенном бензоле, выход из плоскости
Настоящее изобретение относится к двухстадийному способу получения соединения формулы I
в форме соли или цвиттер-иона, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил. Указанный способ минимизирует отклонения в относительных содержаниях диастереомеров. 2 н. и 9 з.п. ф-лы, 3 пр.
1. Способ получения соединения формулы I
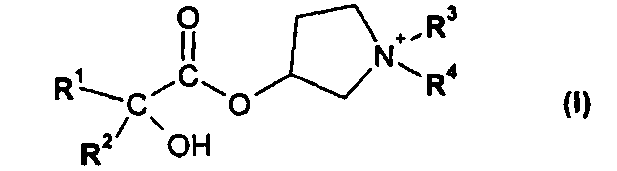
в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил; включающий стадии:
(a) взаимодействия соединения формулы II
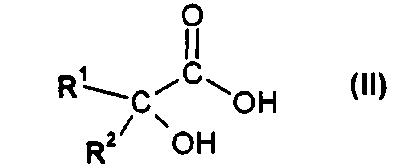
или его соли, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, с соединением формулы III
или его производным, способным образовывать сложный эфир, где R3 представляет собой (C1-C8)алкил, с получением соединения формулы IV
где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и R3 представляет собой (C1-C8)алкил, и
(b) взаимодействия соединения формулы IV, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и R3 представляет собой (C1-C8)алкил, с соединением формулы V

где R4 представляет собой (C1-C8)алкил, а X представляет собой уходящую группу, с получением соединения формулы I в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил.
2. Способ по п.1, в котором
R1 и R2 в соединениях формул II и IV независимо друг от друга представляют собой (C5-C6)циклоалкил или фенил и
R3 в соединениях формул III и IV представляет собой (C1-C4)алкил.
3. Способ по п.1, в котором
R1 и R2 в соединениях формул IV и I независимо друг от друга представляют собой (C5-C6)циклоалкил или фенил;
R3 в соединениях формул IV и I представляет собой (C1-C4)алкил и
R4 в соединениях формул V и I представляет собой (C1-C4)алкил.
4. Способ по любому из предшествующих пунктов, в котором
R1 в соединениях формул II и IV представляет собой циклопропил;
R2 в соединениях формул II и IV представляет собой фенил;
R3 в соединениях формул III и IV представляет собой метил и
R4 в соединении формулы IV представляет собой метил таким образом, что соединение формулы I представляет собой гликопирроний в форме соли или цвиттер-иона.
5. Способ по п.4, в котором соединение формулы I представляет собой рацемическую смесь бромидов (3S,2'R)- и (3R,2'S)-3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния.
6. Способ по п.1, в котором стадию (а) осуществляют в присутствии агента для сочетания.
7. Способ по п.6, в котором агент для сочетания представляет собой карбонилдиимидазол.
8. Способ по п.1, в котором стадию (b) осуществляют в органическом растворителе, в котором стереоизомеры соединения формулы I имеют различающуюся растворимость.
9. Способ получения сухого порошкообразного состава для ингаляций, содержащего соединение формулы I
в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил;
включающий стадии:
(i) взаимодействия соединения формулы II
или его соли, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил, с соединением формулы III
или его производным, способным образовывать сложный эфир, где R3 представляет собой (C1-C8)алкил, с получением соединения формулы IV
где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и R3 представляет собой (C1-C8)алкил;
(ii) взаимодействия соединения формулы IV, где R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и R3 представляет собой (C1-C8)алкил, с соединением формулы V

где R4 представляет собой (C1-C8)алкил, а X представляет собой уходящую группу, с получением соединения формулы I в форме соли или цвиттер-иона, где
R1 и R2 независимо друг от друга представляют собой (C3-C8)циклоалкил или (C6-C10)арил и
R3 и R4 независимо друг от друга представляют собой (C1-C8)алкил;
(iii) необязательной очистки лекарственного вещества посредством кристаллизации с получением очищенного лекарственного вещества;
(iv) микронизации лекарственного вещества; и
(v) добавления частиц носителя с получением сухого порошка для ингаляций.
10. Способ по п.9, в котором на стадии (iv) лекарственное вещество микронизуют совместно с агентом, регулирующим силу адгезии.
11. Способ по п.10, в котором регулирующий силу адгезии агент представляет собой стеарат магния.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНИЯ | 2003 |
|
RU2320657C2 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| GB 1350929, 24.04.1974 | |||
| US 2956062, 11.10.1960 | |||

