Изобретение относится к фармацевтической лекарственной форме, которая содержит опиоид с улучшенной стабильностью при хранении.
Много фармакологически активных соединений обладают потенциалом для злоупотребления и поэтому, преимущественно обеспечиваются в виде защищенных от применения не по назначению фармацевтических лекарственных форм. Известными примерами таких фармакологически активных соединений являются опиоиды.
Известно, что злоумышленники дробят традиционные таблетки, которые содержат опиоиды, для того чтобы разрушить «микрокапсулирование», которое обеспечивает замедленное высвобождение, и затем заглатывают полученный порошок через рот, или втягивают его через нос, ректально, или применяют при помощи инъекции.
Были разработаны различные концепции для предотвращения злоупотребления лекарственными средствами. Одна из концепций основана на механических свойствах фармацевтических лекарственных форм, в частности на увеличении предела прочности на разрыв (сопротивление разрушению). Главным преимуществом таких фармацевтических лекарственных форм состоит в том, что, измельчение, в частности измельчение в порошок, при помощи традиционных средств, таких как размол в ступке или разрушение при помощи молотка, является невозможным, или, по крайней мере, существенно затрудненным.
Такие фармацевтические лекарственные формы являются полезными для предотвращения от злоупотребления лекарственными средствами, в частности фармакологически активным соединением, содержащимся в них, так как они не могут быть измельчены в порошок при помощи традиционных средств и, таким образом, не могут быть применены в виде порошка, например, через нос. Механические свойства, в частности высокий предел прочности на разрыв указанных фармацевтических лекарственных форм обеспечивает им защиту от применения не по назначению. В контексте таких защищенных от применения не по назначению фармацевтических лекарственных форм можно сослаться, например, на WO 2005/016313, WO 2005/016314, WO 2005/063214, WO 2005/102286, WO 2006/002883, WO 2006/002884, WO 2006/002886, WO 2006/082097, WO 2006/082099 и WO 2008/107149.
Проблемой в изготовлении фармацевтических лекарственных форм, которые содержат опиоиды, такие как оксиморфон, гидроморфон, и оксикодон, является их чувствительность как к окислительному разрушению, так и к разложению. Окисление может быть вызвано молекулярным кислородом или радикалами или пероксидами, образованными соединениями, которые контактируют с указанными чувствительными к окислению опиоидами. Фармацевтические наполнители как таковые, например, полиэтиленгликоли, могут вызывать или катализировать окислительное разрушение, например, в ходе процесса изготовления фармацевтических лекарственных форм. Кроме того, молекулярный кислород может образовывать указанные радикалы или пероксиды.
Как правило, разложение отслеживается в стандартных испытаниях на стабильность при хранении, например, при форсированных условиях хранения, таких как 40°С/75% относительной влажности. При этих условиях, разрушение и разложение происходит быстрее, чем при условиях окружающей среды. Государственные органы, которые утверждают лекарственные средства, такие как СНМР (Комитет по медицинским продуктам, предназначенным для человека) и FDA (Управление по контролю за пищевыми продуктами и лекарственными средствами), и международные союзы по гармонизации, такие как ICH (Международная конференция по гармонизации), установили стандартные пределы стабильности при хранении, которые должны быть соблюдены для того, чтобы получить утверждение фармацевтической лекарственной формы.
Определенные проблемы возникают, когда чувствительный к окислению опиоид должен быть подвержен повышенным температурам в ходе процесса производства, такого как экструзия горячего расплава, покрытие пленкой и подобные. При указанных условиях опиоиды становятся еще более чувствительными к окислению. Например, для некоторых известных процессов по изготовлению фармацевтических лекарственных форм, имеющий повышенным пределом прочности на разрыв, является необходимым, чтобы фармацевтическая композиция, содержащая действующий ингредиент, была подвергнута воздействию давления определенной величины при определенной повышенной температуре на протяжении определенного периода времени. В зависимости от составляющих фармацевтической композиции и их количества, температура, давление и время могут быть различными в пределах определенных пределов. Однако, если минимальные требования не удовлетворяются, то предел прочности на разрыв полученной фармацевтической лекарственной формы является слишком низким.
В следствии этого, некоторые традиционные способы изготовления фармацевтических лекарственных форм, в частности фармацевтических лекарственных форм, имеющий повышенным пределом прочности на разрыв, требуют сравнительно жестких условий производства и, таким образом, на сегодняшний день являются не применимыми для изготовления чувствительных к окислению опиоидов. В частности разрыв цепи фармацевтических наполнителей, таких как полиэтиленоксид во время экструзии горячего расплава, приводит к риску образования свободных радикалов и, таким образом, к дальнейшему повышению окислительного стресса.
Более низкие дозировки чувствительных к окислению опиоидов часто демонстрируют более высокий процент окислительного разрушения и разложения, чем более высокие их дозировки. Таким образом, если принимать во внимание стабильность при хранении, то фармацевтические лекарственные формы, содержащие более низкие дозировки чувствительных к окислению опиоидов, требуют особого внимания.
Действие механизмов окисления и химических взаимодействий на стабильность полимерных систем для аморфного Δ9-тетрагидроканнабинола (неопиоид), полученного методом горячего расплава, описан в М.Munjal и др., J.Pharm. Sciences, 95 (11), 2006, 2473-85. Исследование продемонстрировало для этого очень нестабильного лекарственного средства сложную природу взаимодействий, включая совместимость наполнителя и лекарственного средства, применение ингибиторов окисления, образование поперечных связей в полимерных матрицах, уровень рН микросреды и действие влажности.
К.С.WatermaH и др., Pharm. Develop. Tech. 7 (1), 2002, 1-32 делает обзор стабилизации фармацевтических препаратов к действию окислительного разрушения. Рекомендуются различные способы уменьшения окисления. Авторы делают заключение, что в итоге, каждое лекарственное средство представляет собой уникальную ситуацию.
WO 2008/107149 раскрывает лекарственную форму для перорального применения, обладающую повышенным пределом прочности на разрыв, которая может содержать окислительно-восстановительные стабилизаторы, такие как комплексообразующие агенты, например, ЭДТК.
WO 2008/086804 относится к композициям с контролируемым высвобождением, содержащим композицию матрицы, включающую а) полимер или смесь полимеров, b) действующее (активное) вещество и необязательно с) один или более фармацевтически приемлемых наполнителей, которые не содержат спирт, который вызывает быстрое высвобождение дозы и обладает превосходными свойствами в отношении предотвращения злоупотребления лекарственными средствами. Предпочтительно, композиция является устойчивой к выделению и/или выделению в раствор действующего (активного) вещества из композиции при помощи раздробления, расплавления и/или экстракции этиловым спиртом, в результате чего композиция является устойчивой против злоупотребления лекарственными средствами. В качестве ароматического вещества может присутствовать лимонная кислота. Пример 2 относится к композиции, содержащей 7 масс. % лимонной кислоты.
WO 2008/148798 раскрывает слоистую композицию с продленным высвобождением для пролонгированного действия и способ, который обеспечивает пролонгированное действие, например, прием препарата один раз в день должен обеспечить оптимальную абсорбцию активного вещества в желудочно-кишечном тракте, то есть от желудка до прямой кишки.
Не существует общей концепции успешного подавления окислительного разрушения чувствительных к окислению лекарственных средств фармацевтической лекарственной формы. Комплексные индивидуальные механизмы окисления, которые подходят для конкретного лекарственное средства, так же как и множество вероятных факторов, которые оказывают влияние на процессы окисления, требуют широких исследований для каждого конкретного случая, где будут приняты во внимание конкретные обстоятельства.
Кроме того, является известным, что другие компоненты фармацевтических лекарственных форм также могут демонстрировать проблемы со стабильностью, когда их подвергают указанным жестким условиям во время производства. Например, полиэтиленоксид с высокой молекулярной массой имеет тенденцию разлагаться при воздействии экструзии горячего расплава. При этом, разрушение полимера может привести к неконтролируемому профилю высвобождения, в частности когда действующий ингредиент заключен в матрицу полиэтиленоксида, и это может быть другой причиной окислительного разрушения действующего ингредиента радикалами. Когда добавляют подходящие наполнители для того, чтобы стабилизировать полиэтиленоксид с высокой молекулярной массой, такой как DDDa-токоферол, то должно приниматься во внимание то, что указанные наполнители в свою очередь могут иметь отрицательное воздействие на стабильность других компонентов фармацевтической лекарственной формы, например, фармакологически активного соединения.
Задачей настоящего изобретения является обеспечение защищенных от применения не по назначению фармацевтических лекарственных форм, содержащих опиоиды, в частности чувствительные к окислению опиоиды, которые имеют преимущества перед фармацевтическими лекарственными формами предшествующего уровня техники. Фармацевтические лекарственные формы должны обладать улучшенной стабильностью при хранении, так, чтобы они могли содержать чувствительные к окислению опиоиды даже в сравнительно низких дозах. Кроме того, должна существовать возможность приготовления фармацевтических лекарственных форм при помощи традиционных способов традиционных условиях, таких как повышенная температура и давление (например, в случае термоформования посредством экструзии горячего расплава).
Указанная задача была решена с помощью объекта формулы изобретения.
Изобретение относится к термоформованной фармацевтической лекарственной форме, имеющей предел прочности на разрыв, составляющий, по крайней мере, 300 Н и включающей - опиоид (А),
- свободную физиологически приемлемую кислоту (В) в количестве от 0,001 до 5,0 масс. %, из расчета общей массы фармацевтической лекарственной формы, и
- полиалкиленоксид (С), имеющий среднемассовую молекулярную массу Мм, составляющую, по крайней мере, 200 000 г/моль.
Неожиданно было выявлено, что определенные производные морфинана, такие как оксиморфон, окислительно разрушаются до N-оксидов (например, оксиморфон-N-оксид, N-оксиды, как правило, часто считаются токсичными и возможно канцерогенными) после изготовления и хранения соответствующих лекарственных форм и что образование указанных М-оксидов и других продуктов разложения может быть подавлено присутствием подходящего количества кислоты (В) в фармацевтической лекарственной формы в соответствии с изобретением.
Если не привязываться ни к какой теории, стабилизирующее действие кислоты (В) может коррелировать со значением константы ионизации чувствительных к окислению опиоидов. Значение константы ионизации оксиморфона составляет 8.3. Традиционные препаративные формы оксиморфона, которые являются защищенными от применения не по назначению благодаря их повышенному пределу прочности на разрыв, но которые не демонстрируют желательного срока годности, имеют значение рН фактора, которое составляет приблизительно 7.5, когда они растворены в воде. При этих условиях значительное количество оксиморфона присутствует в качестве свободного основания (то есть, не протонированный), который может быть более чувствительным к окислению чем (протонированный) в виде соли. Кроме того, эта концепция поддержана тем фактом, что в отсутствии кислоты (В), лекарственные формы имеют тенденцию иметь желтоватый, бежевый цвет, в то время как присутствие кислоты (В) приводит к более белым, например, бесцветным таблеткам. Таким образом, присутствие кислоты (В) может уменьшить значение рН внутри лекарственной формы, таким образом, улучшая сопротивление лекарственного средства к окислительному разрушению.
Неожиданно было выявлено, что фармацевтические наполнители, которые традиционно используются для того, чтобы улучшить сопротивление лекарственного средства к окислительному разрушению, в частности определенные антиокислители, например, α-токоферол, могут быть противодействующими и скорее ухудшающими, чем улучшающими сопротивление лекарственного средства к окислительному разрушению.
Кроме того, имеются экспериментальные данные того, что неожиданно кислота (В) также является способной к стабилизации полиалкиленоксидов с высокой молекулярной массой к разрушению, таких как полиалкиленоксиды (С), имеющие среднемассовую молекулярную массу Мм, составляющую, по крайней мере, 200000 г/моль.
Фармацевтическая лекарственная форма в соответствии с изобретением термоформируют, предпочтительно с использованием экструзии, хотя другие способы термоформования также могут использоваться для того, чтобы образовать фармацевтическую лекарственную форму в соответствии с изобретением, такие как прессование при повышенной температуре или нагревание таблеток, которые были произведены при помощи традиционного прессования на первом этапе и затем их нагревали выше температуры размягчения полимера в таблетке на втором этапе, для того чтобы сформировать твердые таблетки. В этом отношении, термоформование означает формование, или формовку массы после применения нагрева. В предпочтительном варианте осуществления изобретения фармацевтические лекарственные формы термоформируют при помощи экструзии горячего расплава.
Предпочтительно, фармацевтическая лекарственная форма является монолитной массой. Фармацевтическую лекарственную форму предпочтительно приготавливают при помощи экструзии горячего расплава. Расплавленные экструдированные заготовки предпочтительно нарезают на монолиты, которые затем предпочтительно формуют в таблетки. В этом отношении, термин «таблетки» не должен предпочтительно пониматься как лекарственные формы, которые изготовлены при помощи прессования порошка или гранул (прессовка) а скорее, как сформованные экструдаты.
Фармацевтическая лекарственная форма в соответствии с изобретением содержат, в качестве компонента (А), опиоид (А), предпочтительно чувствительный к окислению опиоид (А), наиболее предпочтительно оксиморфон или оксикодон. Для целей описания термин опиоид (А) также включает свободное основание и их физиологически приемлемые соли.
Согласно индексу АТХ, опиоиды подразделяют на алкалоиды опия природного происхождения, производные фенилпиперидина, производные дифенилпропиламина, производные бензоморфана, производные орипавина, производные морфинана и другие. Примерами алкалоидов опия природного происхождения являются морфий, опиум, гидроморфон, никоморфин, оксикодон, дигидрокодеин, диаморфин, папаверетум и кодеин. Следующие опиоиды (А) представляют собой, например, этилморфин, гидрокодон, оксиморфон и их физиологически приемлемые производные или соединения, предпочтительно их соли и сольваты, предпочтительно их гидрохлориды, физиологически приемлемые энантиомеры, стереоизомеры, диастереомеры и рацематы и их физиологически приемлемые производные, предпочтительно простые простой эфиры, сложные эфиры или амиды.
Кроме того, предпочтительные опиоиды (А) включают N-(1-метил-2-пиперидиноэтил)-N-(2-пиридил)пропионамид, (1R,2R)-3-(3-диметиламино-1-этил-2-метил-пропил)фенол, (1R,2R,4S)-2-(диметиламино)метил-4-(п-фторбензилокси)-1-(м-метоксифенил)циклогексанол, (1R,2R)-3-(2-диметиламинометил-циклогексил)фенол, (1S,2S)-3-(3-диметиламино-1-этил-2-метил-пропил)фенол, (2R,3R)-1-диметиламино-3(3-метоксифенил)-2-метил-пентан-3-ол, (1RS,3RS,6RS)-6-диметиламинометил-1-(3-метоксифенил)-циклогексан-1,3-диол, предпочтительно в качестве рацемата, 3-(2-диметиламинометил-1-гидрокси-циклогексил)фенил 2-(4-изобутил-фенил)пропионат, 3-(2-диметиламинометил-1-гидрокси-циклогексил)фенил 2-(6-метокси-нафтален-2-ил)пропионат, 3-(2-диметиламинометил-циклогекс-1-энил)-фенил 2-(4-изобутил-фенил)пропионат, 3-(2-диметиламинометил-циклогекс-1-энил)-фенил 2-(6-метокси-нафтален-2-ил)пропионат, (RR-SS)-2-ацетокси-4-трифторметил-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, (RR-SS)-2-гидрокси-4-трифторметил-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, (RR-SS)-4-хлор-2-гидрокси-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, (RR-SS)-2-гидрокси-4-метил-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, (RR-SS)-2-гидрокси-4-метокси-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, (RR-SS)-2-гидрокси-5-нитро-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, (RR-SS)-2',4'-дифтор-3-гидрокси-бифенил-4-карбоновой кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фенил сложный эфир, 1,1-(3-диметиламино-3-фенилпентаметилен)-6-фтор-1,3,4,9-тетрагидропирано[3,4-b]индол, в частности его гемицитрат; 1,1-[3-диметиламино-3-(2-тиенил)пентаметилен]-1,3,4,9-тетрагидропирано[3,4-b]индол, в частности его соль лимонной кислоты; и 1,1-[3-диметиламино-3-(2-тиенил)пентаметилен]-1,3,4,9-тетрагидропирано[3,4-b]-6-фтор-индол, в частности его гемицитрат, и соответствующие стереоизомерные соединения, в каждом случае их соответствующие производные, физиологически приемлемые энантиомеры, стереоизомеры, диастереомеры и рацематы и их физиологически приемлемые производные, например, простые эфиры, сложные эфиры или амиды, и в каждом случае их физиологически приемлемые соединения, в частности их соли и сольваты, например гидрохлориды.
Предпочтительные опиоиды (А) имеют общую формулу (I)
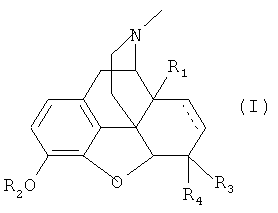
где
R1 представляет собой -Н, -ОН или -OC1-6-алкил;
R2 представляет собой -Н или -C1-6-алкил;
R3 представляет собой -Н или -ОН, и R4 представляет собой -Н; или R3 и
R4 вместе представляют собой =O; и
--- представляет собой необязательную двойную связь;
или их физиологически приемлемые соли.
В частности предпочтительные опиоиды (А) включают оксиморфон, оксикодон, гидроморфон и их физиологически приемлемые соли.
Содержание опиоида (А) в фармацевтической лекарственной формы не ограничено.
Предпочтительно, его содержание находится в пределах от 0,01 до 80 масс.%, более предпочтительно 0,1-50 масс.%, еще более предпочтительно 1-25 масс.%, из расчета общей массы фармацевтической лекарственной формы. В предпочтительном варианте осуществления изобретения содержание опиоида (А) находится в пределах от 7±6 масс.%, более предпочтительно 7±5 масс.%, еще более предпочтительно 5±4 масс.%, 7±4 масс.% или 9±4 масс.%, наиболее предпочтительно 5±3 масс.%, 7±3 масс.% или 9±3 масс.%, и в частности 5±2 масс.%, 7±2 масс.% или 9±2 масс.%, из расчета общей массы фармацевтической лекарственной формы. В другом предпочтительном варианте осуществления изобретения содержание опиоида (А) находится в пределах от 11±10 масс.%, более предпочтительно 11±9 масс.%, еще более предпочтительно 9±6 масс.%, 11±6 масс.%, 13±6 масс.% или 15±6 масс.%, наиболее предпочтительно 11±4 масс.%, 13±4 масс.% или 15±4 масс.%, и в частности 11±2 масс.%, 13±2 масс.% или 15±2 масс.%, из расчета общей массы фармацевтической лекарственной формы. В дополнительном предпочтительном варианте осуществления изобретения содержание опиоида (А) находится в пределах от 20±6 масс.%, более предпочтительно 20±5 масс. %, еще более предпочтительно 20±4 масс.%, наиболее предпочтительно 20±3 масс.%, и в частности 20±2 масс.%, из расчета общей массы фармацевтической лекарственной формы.
Предпочтительно, общее количество опиоида (А), который содержится в фармацевтической лекарственной форме, находится в пределах от 0,01 до 200 мг, более предпочтительно 0,1-190 мг, еще более предпочтительно 1,0-180 мг, и еще более предпочтительно 1,5-160 мг, наиболее предпочтительно 2,0-100 мг и в частности 2,5-80 мг.
В предпочтительном варианте осуществления изобретения опиоид (А) содержится в фармацевтической лекарственной форме в количестве 7,5±5 мг, 10±5 мг, 20±5 мг, 30±5 мг, 40±5 мг, 50±5 мг, 60±5 мг, 70±5 мг, 80±5 мг, 90±5 мг, 100±5 мг, 110±5 мг, 120±5 мг, 130±5, 140±5 мг, 150±5 мг, или 160±5 мг. В другом предпочтительном варианте осуществления изобретения опиоид (А) содержится в фармацевтической лекарственной форме в количестве 5±2.5 мг, 7.5±2.5 мг, 10±2.5 мг, 15±2.5 мг, 20±2.5 мг, 25±2.5 мг, 30±2.5 мг, 35±2.5 мг, 40±2.5 мг, 45±2.5 мг, 50±2.5 мг, 55±2.5 мг, 60±2.5 мг, 65±2.5 мг, 70±2.5 мг, 75±2.5 мг, 80±2.5 мг, 85±2.5 мг, 90±2.5 мг, 95±2.5 мг, 100±2.5 мг, 105±2.5 мг, 110±2.5 мг, 115±2.5 мг, 120±2.5 мг, 125±2.5 мг, 130±2.5 мг, 135±2.5 мг, 140±2.5 мг, 145±2.5 мг, 150±2.5 мг, 155±2.5 мг, или 160±2.5 мг.
В особенно предпочтительном варианте осуществления изобретения опиоид (А) представляет собой оксиморфон, предпочтительно его соль HCI, и фармацевтическая лекарственная форма является подходящей для применения два раза в день. В этом варианте осуществления изобретения опиоид (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 5 до 40 мг. В другом, особенно предпочтительном варианте осуществления изобретения опиоид (А) представляет собой оксиморфон, предпочтительно его соль HCI, и фармацевтическая лекарственная форма является подходящей для применения один раз в день. В этом варианте осуществления изобретения опиоид (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 10 до 80 мг.
В другом в особенно предпочтительном варианте осуществления изобретения опиоид (А) представляет собой оксикодон, предпочтительно его соль HCI, и фармацевтическая лекарственная форма является подходящей для применения два раза в день. В этом варианте осуществления изобретения опиоид (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 5 до 80 мг. В другом особенно предпочтительном варианте осуществления изобретения опиоид (А) представляет собой оксикодон, предпочтительно его соль HCI, и фармацевтическая лекарственная форма является подходящей для применения один раз в день. В этом варианте осуществления изобретения опиоид (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 10 до 320 мг.
В еще другом особенно предпочтительном варианте осуществления изобретения опиоид (А) представляет собой гидроморфон, предпочтительно его соль HCI, и фармацевтическая лекарственная форма является подходящей для применения два раза в день. В этом варианте осуществления изобретения опиоид (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 2 до 52 мг. В другом особенно предпочтительном варианте осуществления изобретения опиоид (А) представляет собой гидроморфон, предпочтительно его соль HCI, и фармацевтическая лекарственная форма является подходящей для применения один раз в день. В этом варианте осуществления изобретения опиоид (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 4 до 104 мг.
Фармацевтическая лекарственная форма в соответствии с изобретением отличается превосходной стабильностью при хранении. Предпочтительно, после хранения в течение 4 недель при температуре 40°С и 75%-й относительной влажности, содержание опиоида (А) составляет, по крайней мере, 98,0 %, более предпочтительно, по крайней мере, 98,5%, еще более предпочтительно, по крайней мере, 99,0%, и еще более предпочтительно, по крайней мере, 99,2%, наиболее предпочтительно, по крайней мере, 99,4% и в частности, по крайней мере, 99,6% от его первоначального содержания перед хранением. Подходящие способы измерения содержания опиоида (А) в фармацевтической лекарственной формы являются известными специалисту в данной области техники. В этом отношении можно сослаться на Европейскую Фармакопею или Фармакопею США, в частности на анализ ВЭЖХ с обратной фазой. Предпочтительно, фармацевтическая лекарственная форма хранится в закрытых, предпочтительно запечатанных контейнерах, предпочтительно как описано в экспериментальном разделе, наиболее предпочтительно снабженных поглотителем кислорода, в частности поглотителем кислорода, который эффективен даже при низкой относительной влажности.
Фармацевтическая лекарственная форма в соответствии с изобретением содержит, в качестве компонента (В), свободную физиологически приемлемую кислоту в количестве от 0,001 до 5,0 масс. %, из расчета общей массы фармацевтической лекарственной формы. Кислота (В) может быть органической или неорганической, жидкой или твердой. Твердые кислоты являются предпочтительными, в частности кристаллические органические или неорганические кислоты.
Кислота (В) является свободной. Это означает, что кислотные функциональные группы кислоты (В) не являются все вместе составляющими соли опиоида (А). Если
опиоид (А) присутствует в качестве соли кислоты, например, в качестве гидрохлорида, то фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно содержит в качестве компонента (В) другую, химически отличную кислоту, которая не присутствует в качестве составляющего соли опиоида (А). Другими словами, монокислоты, которые образуют соль с опиоидом (А), нельзя рассматривать как свободные кислоты (В) в значении настоящего изобретения. Когда кислота (В) имеет больше чем одну кислотную функциональную группу (например, фосфорная кислота), то кислота (В) может присутствовать в качестве составляющего соли опиоида (А), при условии, что, по крайней мере, одна из кислотных функциональных групп кислоты (В) не вовлечена в образование соли, то есть является свободной. При этом является предпочтительным, чтобы все без исключения кислотные функциональные группы кислоты (В) не были вовлечены в образование соли с опиоидом (А). При этом также является возможным, что свободная кислота (В) и кислота, образующая соль с опиоидом (А), являются одинаковыми. В указанных обстоятельствах кислота (В) предпочтительно присутствует в молярном избытке по сравнению с опиоидом (А).
В предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу (например, -CO2H, -SO3H, -РО3Н2, -ОН и подобные), имеющую значение константы ионизации, которое находится в пределах 2,00±1,50, более предпочтительно 2,00±1,25, еще более предпочтительно 2,00±1,00, и еще более предпочтительно 2,00±0,75, наиболее предпочтительно 2,00±0,50 и в частности 2,00±0,25. В другом предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 2,25±1,50, более предпочтительно 2,25±1,25, еще более предпочтительно 2,25±1,00, и еще более предпочтительно 2,25±0,75, наиболее предпочтительно 2,25±0,50 и в частности 2,25±0,25. В другом предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 2,50±1,50, более предпочтительно 2,50±1,25, еще более предпочтительно 2,50±1,00, и еще более предпочтительно 2,50±0,75, наиболее предпочтительно 2,50±0,50 и в частности 2,50±0,25. В другом предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 2,75±1,50, более предпочтительно 2,75±1,25, еще более предпочтительно 2,75±1,00, и еще более предпочтительно 2,75±0,75, наиболее предпочтительно 2,75±0,50 и в частности 2,75±0,25. В другом предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 3,00±1,50, более предпочтительно 3,00±1,25, еще более предпочтительно 3,00±1,00, и еще более предпочтительно 3,00±0,75, наиболее предпочтительно 3,00±0,50 и в частности 3,00±0,25. В еще другом предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 3,25±1,50, более предпочтительно 3,25±1,25, еще более предпочтительно 3,25±1,00, и еще более предпочтительно 3,25±0,75, наиболее предпочтительно 3,25±0,50 и в частности 3,25±0,25.
В еще одном предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 4,50±1,50, более предпочтительно 4,50±1,25, еще более предпочтительно 4,50±1,00, и еще более предпочтительно 4,50±0,75, наиболее предпочтительно 4,50±0,50 и в частности 4,50±0,25. В еще одном предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 4,75±1,50, более предпочтительно 4,75±1,25, еще более предпочтительно 4,75±1,00, и еще более предпочтительно 4,75±0,75, наиболее предпочтительно 4,75±0,50 и в частности 4,75±0,25. В еще одном предпочтительном варианте осуществления изобретения кислота (В) содержит, по крайней мере, одну кислотную функциональную группу, имеющую значение константы ионизации, которое находится в пределах 5,00±1,50, более предпочтительно 5,00±1,25, еще более предпочтительно 5,00±1,00, и еще более предпочтительно 5,00±0,75, наиболее предпочтительно 5,00±0,50 и в частности 5,00±0,25.
Предпочтительно, кислота (В) является органической карбоновой или сульфоновой кислотой, в частности карбоновой кислотой. Мультикарбоновые кислоты и/или гидрокси-карбоновые кислоты в частности являются предпочтительными.
В случае мультикарбоновых кислот их неполные соли должны также рассматриваться как мультикарбоновые кислоты, например, неполные натриевые, калиевые или аммониевые соли. Например, лимонная кислота представляет собой мультикарбоновую кислоту, имеющую три карбоксильные группы. Поскольку остается, по крайней мере, одна протонированная карбоксильная группа (например, натрий дигидроген цитрат или двунатрий гидроген цитрат), то соль должна рассматриваться как мультикарбоновая кислота. Предпочтительно, тем не менее, чтобы все карбоксильные группы мультикарбоновой кислоты были протонированы.
Предпочтительно, кислота (В) имеет низкую молекулярную массу, то есть, не является полимеризованной. Как правило, молекулярная масса кислоты (В) составляет ниже 500 г/моль.
Примеры кислот включают насыщенные и ненасыщенные монокарбоновые кислоты, насыщенные и ненасыщенные бикарбоновой кислоты, трикарбоновой кислоты, α-гидроксикислоты и β-гидроксикислоты монокарбоновых кислот, α-гидроксикислоты и β-гидроксикислоты бикарбоновых кислот, α-гидроксикислоты и β-гидроксикислоты трикарбоновых кислот, кетокислоты, α-кетокислоты, β-кетокислоты, поликарбоновых кислот, полигидроксимонокарбоновых кислот, полигидроксибикарбоновых кислот, полигидрокситрикарбоновых кислот.
Предпочтительно, кислоту (В) выбирают из группы, состоящей из бензилсульфоновой кислоты, лимонной кислоты, α-глюкогептоновая кислота, D-глюконовая кислота, гликолевая кислота, молочная кислота, яблочная кислота, малоновая кислота, миндальная кислота, пропановая кислота, янтарная кислота, винная кислота (d, I, или dI), толуолсульфоновая кислота, валериановая кислота, пальмитиновая кислота, памоевая кислота, себациновая кислота, стеариновая кислота, лауриновая кислота, уксусная кислота, адипиновая кислота, глутаровая кислота, 4-хлорбензилсульфоновая кислота, этандисульфоновой кислота, этилянтарная кислота, фумаровая кислота, галактаровая кислота (муциновая кислота), D-глюкуроновая кислота, 2-оксо-глутаровая кислота, глицерофосфорная кислота, гиппуровая кислота, изотионовая кислота (этанолсульфоноваяя кислота), лактобионовая кислота, малеиновая кислота, малеиновая кислота, 1,5-нафталин-дисульфоновая кислота, 2-нафталин-сульфоновая кислота, пивалевая кислота, терефталевая кислота, тиоциановая кислота, холиновая кислота, н-додецил сульфат, 3-гидрокси-2-нафтойная кислота, 1-гидрокси-2-нафтойная кислота, олеиновая кислота, ундеценовая кислота, аскорбиновая кислота, (+) - камфарная кислота, д-камфарсульфоновая кислота, дихлоруксусная кислота, этансульфоновая кислота, муравьиная кислота, метансульфоновая кислота, никотиновая кислота, оротовая кислота, щавелевая кислота, пикриновая кислота, L-пироглутаминовая кислота, сахарин, салициловая кислота, гентизиновая кислота и/или 4-ацетамидобензойная кислота.
Содержание кислоты (В) находится в пределах от 0,001 до 5,0 масс.%, предпочтительно 0,005-2,5 масс.%, более предпочтительно 0,01-2,0 масс.%, еще более предпочтительно 0,05-1,5 масс.%, наиболее предпочтительно 0,1-1,0 масс.% и в частности 0,2-0,9 масс.%, из расчета общей массы фармацевтической лекарственной формы.
Предпочтительно, кислота (В) является мультикарбоновой кислотой. Более предпочтительно, мультикарбоновую кислоту выбирают из группы, состоящей из лимонной кислоты, малеиновой кислоты и фумаровой кислоты.
Лимонная кислота в частности является предпочтительной.
Мультикарбоновая кислота, предпочтительно лимонная кислота, может присутствовать в своей безводной форме или в качестве сольвата и гидрата, соответственно, например, в качестве моногидрата.
В предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,2±0,18 масс.%, более предпочтительно 0,2±0,15 масс.%, еще более предпочтительно 0,2±0,12 масс.%, и еще более предпочтительно 0,2±0,09 масс.%, наиболее предпочтительно 0,2±0,06 масс.%, и в частности 0,2±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В другом предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,3±0,18 масс.%, более предпочтительно 0,3±0,15 масс.%, еще более предпочтительно 0,3±0,12 масс.%, и еще более предпочтительно 0,3±0,09 масс.%, наиболее предпочтительно 0,3±0,06 масс.%, и в частности 0,3±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще другом предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,4±0,18 масс.%, более предпочтительно 0,4±0,15 масс.%, еще более предпочтительно 0,4±0,12 масс.%, и еще более предпочтительно 0,4±0,09 масс.%, наиболее предпочтительно 0,4±0,06 масс.%, и в частности 0,4±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще одном предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,5±0,18 масс.%, более предпочтительно 0,5±0,15 масс.%, еще более предпочтительно 0,5±0,12 масс.%, и еще более предпочтительно 0,5±0,09 масс.%, наиболее предпочтительно 0,5±0,06 масс.%, и в частности 0,5±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще одном предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,6±0,18 масс.%, более предпочтительно 0,6±0,15 масс.%, еще более предпочтительно 0,6±0,12 масс.%, и еще более предпочтительно 0,6±0,09 масс.%, наиболее предпочтительно 0,6±0,06 масс.%, и в частности 0,6±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще одном предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,7±0,18 масс.%, более предпочтительно 0,7±0,15 масс.%, еще более предпочтительно 0,7±0,12 масс.%, и еще более предпочтительно 0,7±0,09 масс.%, наиболее предпочтительно 0,7±0,06 масс.%, и в частности 0,7±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще одном предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,8±0,18 масс.%, более предпочтительно 0,8±0,15 масс.%, еще более предпочтительно 0,8±0,12 масс.%, и еще более предпочтительно 0,8±0,09 масс.%, наиболее предпочтительно 0,8±0,06 масс.%, и в частности 0,8±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще одном предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,85±0,18 масс.%, более предпочтительно 0,85±0,15 масс.%, еще более предпочтительно 0,85±0,12 масс.%, и еще более предпочтительно 0,85±0,09 масс.%, наиболее предпочтительно 0,85±0,06 масс.%, и в частности 0,85±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В еще другом предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 0,9±0,18 масс.%, более предпочтительно 0,9±0,15 масс.%, еще более предпочтительно 0,9±0,12 масс.%, и еще более предпочтительно 0,9±0,09 масс.%, наиболее предпочтительно 0,9±0,06 масс.%, и в частности 0,9±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В дополнительном предпочтительном варианте осуществления изобретения содержание кислоты (В), предпочтительно лимонной кислоты, находится в пределах 1,0±0,18 масс.%, более предпочтительно 1,0±0,15 масс.%, еще более предпочтительно 1,0±0,12 масс.%, и еще более предпочтительно 1,0±0,09 масс.%, наиболее предпочтительно 1,0±0,06 масс.%, и в частности 1,0±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
Фармацевтическая лекарственная форма в соответствии с изобретением включает, в качестве компонента (С), полиалкиленоксид (С), имеющий среднемассовую молекулярную массу Мм, которая составляет, по крайней мере, 200000 г/моль, предпочтительно, по крайней мере, 500000 г/моль, более предпочтительно, по крайней мере, 750000 г/моль, еще более предпочтительно, по крайней мере, 1000000 г/моль, наиболее предпочтительно, по крайней мере, 2000000 г/моль и в частности находится в пределах от 500000 до 15000000 г/моль.
Предпочтительно, полиалкиленоксид выбирают из группы, состоящей из полиметиленоксида, полиэтиленоксида и полипропиленоксида, их сополимеров и смесей.
Полиалкиленоксид (С) может включать единственный полиалкиленоксид, имеющий определенную среднюю молекулярную массу, или смесь (примесь) различных полимеров, такую как смесь из двух, трех, четырех или пяти полимеров, например, полимеров той же самой химической природы, но различной средней молекулярной массы, полимеров различной химической природы, но той же самой средней молекулярной массы, или полимеров как различной химической природы, так и различной молекулярной массы.
Для целей описания полиалкиленгликоль имеет молекулярная масса до 20000 г/моль, в то время как полиалкиленоксид имеет молекулярную массу больше чем 20000 г/моль. В предпочтительном варианте осуществления изобретения средняя масса как результат всех молекулярных масс всех полиалкиленоксидов, которые содержатся в фармацевтической лекарственной форме, составляет, по крайней мере, 200000 г/моль. Таким образом, полиалкиленгликоли, если таковые имеются, предпочтительно не принимаются во внимание, когда определяют среднемассовую молекулярную массу полиалкиленоксида (С).
Предпочтительно, содержание полиалкиленоксида (С) находится в пределах от 20 до 99 масс.%, более предпочтительно 25-95 масс.%, еще более предпочтительно 30-90 масс.%, и еще более предпочтительно 30-85 масс.%, наиболее предпочтительно 30-80 масс.% и в частности 30-75 масс.%, из расчета общей массы фармацевтической лекарственной формы. В предпочтительном варианте осуществления изобретения содержание полиалкиленоксида составляет, по крайней мере, 20 масс.%, более предпочтительно, по крайней мере, 25 масс.%, еще более предпочтительно, по крайней мере, 30 масс.%, и еще более предпочтительно, по крайней мере, 35 масс.% и в частности, по крайней мере, 40 масс.%.
В предпочтительном варианте осуществления изобретения полное содержание полиалкиленоксида (С) находится в пределах 25±20 масс.%, более предпочтительно 25±15 масс.%, наиболее предпочтительно 25±10 масс.%, и в частности 25±5 масс.%. В другом предпочтительном варианте осуществления изобретения полное содержание полиалкиленоксида (С) находится в пределах 35±20 масс.%, более предпочтительно 35±15 масс.%, наиболее предпочтительно 35±10 масс.%, и в частности 35±5 масс.%. В еще другом предпочтительном варианте осуществления изобретения полное содержание полиалкиленоксида (С) находится в пределах 45±20 масс.%, более предпочтительно 45±15 масс.%, наиболее предпочтительно 45±10 масс.%, и в частности 45±5 масс.%. В еще одном предпочтительном варианте осуществления изобретения полное содержание полиалкиленоксида (С) находится в пределах 55±20 масс.%, более предпочтительно 55±15 масс.%, наиболее предпочтительно 55±10 масс.%, и в частности 55±5 масс.%. В дополнительном предпочтительном варианте осуществления изобретения полное содержание полиалкиленоксида (С) находится в пределах 65±20 масс.%, более предпочтительно 65±15 масс.%, наиболее предпочтительно 65±10 масс.%, и в частности 65±5 масс.%. В еще дополнительном предпочтительном варианте осуществления изобретения, полное содержание полиалкиленоксида (С) находится в пределах 75±20 масс.%, более предпочтительно 75±15 масс.%, наиболее предпочтительно 75±10 масс.%, и в частности 75±5 масс.%. В еще дополнительном предпочтительном варианте осуществления изобретения, полное содержание полиалкиленоксида (С) находится в пределах 80±15 масс.%, более предпочтительно 80±10 масс.%, и наиболее предпочтительно 80±5 масс.%.
В предпочтительном варианте осуществления изобретения полиалкиленоксид (С) гомогенно распределен в фармацевтической лекарственной форме в соответствии с изобретением. Предпочтительно, полиалкиленоксид (С) образует матрицу, в которую заключен опиоид (А). В особенно предпочтительном варианте осуществления изобретения опиоид (А) и полиалкиленоксид (С) непосредственно гомогенно распределены в фармацевтической лекарственной форме так, что фармацевтическая лекарственная форма вообще не содержит сегментов, где либо опиоид (А) присутствует в отсутствии полиалкиленоксида (С), либо полиалкиленоксид (С) присутствует в отсутствии опиоида (А).
Когда фармацевтическая лекарственная форма является покрытой пленочной оболочкой, то полиалкиленоксид (С) предпочтительно гомогенно распределен в сердцевине фармацевтической лекарственной формы, то есть пленочная оболочка предпочтительно не содержит полиалкиленоксида (С). Тем не менее, пленочная оболочка, как таковая, может конечно содержать один или более полимеров, которые, тем не менее, предпочтительно отличаются от полиалкиленоксида (С), который содержится в сердцевине.
Полиалкиленоксид (С) может быть комбинирован с одним или более различными полимерами, выбранными из группы, состоящей из полиалкиленоксида, предпочтительно полиметиленоксида, полиэтиленоксида, полипропиленаоксида; полиэтилена, полипропилена, поливинилхлорида, поликарбоната, полистирола, поливинилпирролидона, поли(алк)акрилата, поли(гидрокси жирных кислот), таких как, например, поли(3-гидроксибутират-со-3-гидроксивалерат) (Biopol®), поли(гидроксивалериановая кислота); поликапролактон, поливиниловый спирт, полиэстерамид, полиэтилен сукцинат, полилактон, полигликолид, полиуретан, полиамид, полилактид, полиацеталь (например, полисахариды необязательно с модифицированными боковыми цепями), полилактид/гликолид, полилактон, полигликолид, полиортосложный эфир, полиангидрид, блоксополимеры полиэтиленгликоля и полибутилентерефталата (Polyactive®), полиангидрид (Polifeprosan), их сополимеры, их блоксополимеры, и смеси, по крайней мере, двух из изложенных полимеров, или других полимеров с вышеупомянутыми характеристиками.
Предпочтительно, молекулярная масса дисперсности Мм/Мч полиалкиленоксида (С) находится в пределах 2,5±2,0, более предпочтительно 2,5±1,5, еще более предпочтительно 2,5±1,0, и еще более предпочтительно 2,5±0,8, наиболее предпочтительно 2,5±0,6, и в частности 2,5±0,4.
Полиалкиленоксид (С) предпочтительно при температуре 25°С обладает вязкостью, которая составляет 30-17 600 сП, более предпочтительно 55-17600 сП, еще более предпочтительно 600-17600 сП и наиболее предпочтительно 4500-17600 сП, при измерении в 5 масс.% водного раствора, используя модель RVF вискозиметр Брукфилда (шпиндель номер 2 / скорость вращения 2 оборота в минуту); которая составляет 400-4000 сП, более предпочтительно 400-800 сП или 2000-4000 сП, при измерении в 2 масс.% сводного раствора, используя упомянутый вискозиметр (шпиндель номер 1 или 3 / скорость вращения 10 оборотов в минуту); или которая составляет 1650-10000 сП, более предпочтительно 1650-5 500 сП, 5500-7500 сП или 7500-10000 сП, при измерении в 1 масс.% водного раствора, используя упомянутый вискозиметр (шпиндель номер 2 / скорость вращения 2 оборота в минуту).
В предпочтительном варианте осуществления изобретения в соответствии с изобретением полиалкиленоксид (С), имеющий среднемассовую молекулярную массу, которая составляет, по крайней мере, 200000 г/моль, комбинируют, по крайней мере, с одним дополнительным полимером, предпочтительно но не обязательно также имеющим среднемассовую молекулярную массу (Мм), которая составляет, по крайней мере, 200000 г/моль, выбранным из группы, состоящей из полиэтилена, полипропилена, поливинилхлорида, поликарбоната, полистирола, полиакрилата, поли(гидрокси жирных кислот), поликапролактона, поливинилового спирта, полиэстерамида, полиэтиленсукцината, полилактона, пол игл и кол ида, полиуретана, поливинилпирролидона, полиамида, полилактида, полилактида/гликолида, полилактона, пол игл и кол ида, полиортосложного эфира, полиангидрида, блоксополимеров полиэтиленгликоля и полибутилентерефталата, полиангидрида, полиацетали, сложных эфиров целлюлозы, простых эфиров целлюлозы и их сополимеров. Сложные эфиры целлюлоза и простые эфиры целлюлозы в частности являются предпочтительными, например, метилцеллюлоза, этилцеллюлоза, гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза и подобные.
В предпочтительном варианте осуществления изобретения, указанный дополнительный полимер не является ни полиалкиленоксидом, ни полиалкиленгликолем. Тем не менее, фармацевтическая лекарственная форма может содержать полиалкиленгликоль, например, в качестве пластификатор, но тогда, фармацевтическая лекарственная форма предпочтительно является ттрехкомпонентной смесью полимеров: полиалкиленоксид (С) + дополнительный полимер + пластификатор.
В особенно предпочтительном варианте осуществления изобретения, указанный дополнительный полимер представляет собой гидрофильный сложный эфир целлюлозы или простой эфир целлюлозы, предпочтительно гидроксипропилметилцеллюлозу (ГПМЦ), гидроксипропилцеллюлозу (ГПЦ) или гидроксиэтилцеллюлозу (ГЭЦ), предпочтительно, имеющий среднюю вязкость (предпочтительно измеренную с помощью капиллярным вискозиметром или вращательным вискозиметром), которая сосотавляет 1000-150000 мПа∙с, более предпочтительно 3000-150000. В предпочтительном варианте осуществления изобретения средняя вязкость находится в пределах 110000±50000 мПа∙с, более предпочтительно 110000±40000 мПа∙с, еще более предпочтительно 110000±30000 мПа∙с, наиболее предпочтительно 110000±20000 мПа∙с, и в частности 100000±10000 мПа∙с.
В предпочтительном варианте осуществления изобретения соотношение удельной массы указанного полиалкиленоксида (С) и указанного дополнительного полимера находится в пределах от 20:1 до 1:20, более предпочтительно 10:1 к 1:10, еще более предпочтительно 7:1-1:5, и еще более предпочтительно 5:1-1:1, наиболее предпочтительно 4:1-1,5:1 и в частности 3:1-2:1. В предпочтительном варианте осуществления изобретения, соотношение удельной массы указанного полиалкиленоксида (С) и указанного дополнительного полимера находится в пределах от 10:1 до 5:1, более предпочтительно 8:1-5:1, наиболее предпочтительно 7:1-5:1.
Предпочтительно, содержание указанного дополнительного полимера составляет 0,5-25 масс.%, более предпочтительно 1,0-20 масс.%, еще более предпочтительно 2,0-22,5 масс.%, и еще более предпочтительно 3,0-20 масс.% и наиболее предпочтительно 4,0-17,5 масс.% и в частности 5,0-15 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления изобретения дополнительный полимер представляет собой сложный эфир целлюлозы или простой эфир целлюлозы, предпочтительно ГПМЦ, содержание которого находится в пределах 10±8 масс.%, более предпочтительно 10±6 масс.%, еще более предпочтительно 10±5 масс.%, и еще более предпочтительно 10±4 масс.%, наиболее предпочтительно 10±3 масс.%, и в частности 10±2 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В другом предпочтительном варианте осуществления изобретения дополнительный полимер представляет собой сложный эфир целлюлозы или простой эфир целлюлозы, предпочтительно ГПМЦ, содержание которого находится в пределах 14±8 масс.%, более предпочтительно 14±6 масс.%, еще более предпочтительно 14±5 масс.%, и еще более предпочтительно 14±4 масс.%, наиболее предпочтительно 14±3 масс.%, и в частности 14±2 масс.%, из расчета общей массы фармацевтической лекарственной формы.
Все полимеры предпочтительно применяются в качестве порошков. Они могут быть растворимыми в воде.
Кроме опиоида (А), кислоты (В) и полиалкиленоксида (С) фармацевтическая лекарственная форма в соответствии с изобретением может содержать дополнительные составляющие, такие как традиционные фармацевтические наполнители.
Предпочтительно, фармацевтическая лекарственная форма включает антиокислитель. Подходящие антиокислители включают аскорбиновую кислоту, α-токоферол (витамин Е), бутилгидроксианизол, бутил гидрокситолуол, соли аскорбиновой кислоты (витамин С), аскорбил пальмитат, монотиоглицерин, конифериловый бензоат, нордигидрогуаяретовая кислота, сложные эфиры галловой кислоты, фосфорная кислота, и их производные, такие как витамин Е-сукцинат или витамин Е-пальмитат и/или бисульфит натрия, более предпочтительно бутилгидрокситолуол (БГТ) или бутилгидроксианизол (БГА) и/или α-токоферол.
Предпочтительно, содержание антиокислителя находится в пределах от 0,001 до 5,0 масс.%, более предпочтительно 0,002-2,5 масс.%, более предпочтительно 0,003-1,5 масс.%, еще более предпочтительно 0,005-1,0 масс.%, и еще более предпочтительно 0,01-0,5 масс.%, наиболее предпочтительно 0,05-0,4 масс.% и в частности 0,1-0,3 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В особенно предпочтительном антиокислитель представляет собой α-токоферол. Неожиданно было выявлено, что α-токоферол стабилизирует полиалкиленоксид и одновременно дестабилизирует определенные опиоиды (А), такие как оксиморфон. Таким образом, в предпочтительном варианте осуществления изобретения, содержание α-токоферола уравновешено между достаточной стабильностью полиалкиленоксида с одной стороны и достаточной стабильностью опиоида (А) с другой стороны.
В предпочтительном варианте осуществления изобретения содержание α-токоферола находится в пределах 0,2±0,18 масс.%, более предпочтительно 0,2±0,15 масс.%, еще более предпочтительно 0,2±0,12 масс.%, и еще более предпочтительно 0,2±0,09 масс.%, наиболее предпочтительно 0,2±0,06 масс.%, и в частности 0,2±0,03 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления изобретения, соотношение удельной массы кислоты (В), предпочтительно лимонной кислоты, и антиокислителя, предпочтительно α-токоферола, находится в пределах от 10:1 до 1:10, более предпочтительно 8:1-1:8, еще более предпочтительно 6:1-1:6, и еще более предпочтительно 5:1-1:4, наиболее предпочтительно 4:1-1:3 и в частности 3:1-1:2.
Фармацевтическая лекарственная форма в соответствии с изобретением также может содержать натуральный, полусинтетический или синтетический воск. Предпочтительными являются воски с, температурой размягчения, которая составляет, по крайней мере, 50°С, более предпочтительно 60°С. В частности предпочтительными являются карнаубский воск и пчелиный воск, в частности карнаубский воск.
Предпочтительно, профиль высвобождения опиоида (А) представляет собой ретардированное высвобождение матричного типа. Предпочтительно, опиоид (А) заключен в матрицу, содержащую полиалкиленоксид, указанная матрица контролирует высвобождение опиоида (А) из фармацевтической лекарственной формы.
В качестве дополнительных материалов матрицы могут применяться физиологически приемлемые материалы, которые являются известными специалисту в данной области техники. В качестве гидрофильных материалов матрицы предпочтительно применяются полимеры, в частности предпочтительно простые эфиры целлюлозы, сложные эфиры целлюлозы и/или акриловые смолы. В качестве материалов матрицы очень часто предпочтительно применяют этилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу, поли(мет)акриловую кислоту и/или их производные, такие как их соли, амиды или сложные эфиры. Материалы матрицы, изготовленные из гидрофобных материалов, таких как гидрофобные полимеры, воски, жиры, длинноцепочечные жирные кислоты, жирный спирты или соответствующие сложные эфиры или простые эфиры или их смеси, также являются предпочтительными. В частности в качестве гидрофобных материалов предпочтительно применяются моно- или дидиглицериды С12-С30 жирных кислот и/или С12-С30 жирных спиртов и/или восков или их смесей. Также в качестве материалов матрицы возможно применять смеси вышеупомянутых гидрофильных и гидрофобных материалов.
Предпочтительно, соотношение удельной массы полиалкиленоксида к опиоиду (А), составляет, по крайней мере, 0.5:1, более предпочтительно, по крайней мере, 1:1, по крайней мере, 2:1, по крайней мере, 3:1, по крайней мере, 4:1, по крайней мере, 5:1, по крайней мере, 6:1, по крайней мере, 7:1, по крайней мере, 8:1 или, по крайней мере, 9:1; еще более предпочтительно, по крайней мере, 10:1 или, по крайней мере, 15:1, и еще более предпочтительно, по крайней мере, 20:1, наиболее предпочтительно, по крайней мере, 30:1 ив частности, по крайней мере, 40:1. В предпочтительном варианте осуществления изобретения соотношение удельной массы полиалкиленоксида к опиоиду (А) находится в пределах от 3:1 до 50:1, более предпочтительно 3:1-40:1 ив частности 3:1-30:1.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно содержит пластификатор. Пластификатор улучшает способность полиалкиленоксида поддаваться обработке. Предпочтительный пластификатор представляет собой полиалкиленгликоль, такой как полиэтиленгликоль, триацетин, жирные кислоты, сложные эфиры жирных кислот, воски и/или микрокристаллические воски. Особенно предпочтительными пластификаторами являются полиэтиленгликоли, такие как ПЭГ6000.
Предпочтительно, содержание пластификатора находится в пределах от 0,1 до 25 масс.%, более предпочтительно 0,5-22,5 масс.%, еще более предпочтительно 1,0-20 масс.%, и еще более предпочтительно 2,5-17,5 масс.%, наиболее предпочтительно 5,0-15 масс.,% и в частности 7,5-12,5 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления изобретения пластификатор представляет собой полиалкиленгликоль, содержание которогно находится в пределах 10±8 масс.%, более предпочтительно 10±6 масс.%, еще более предпочтительно 10±5 масс.%, и еще более предпочтительно 10±4 масс.%, наиболее предпочтительно 10±3 масс.%, и в частности 10±2 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В другом предпочтительном варианте осуществления изобретения пластификатор представляет собой полиалкиленгликоль, содержание которого находится в пределах 15±8 масс.%, более предпочтительно 15±6 масс.%, еще более предпочтительно 15±5 масс.%, и еще более предпочтительно 15±4 масс.%, наиболее предпочтительно 15±3 масс.%, и в частности 15±2 масс.%, из расчета общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления изобретения соотношение удельной массы полиалкиленоксида к полиалкиленгликолю находится в пределах 4,2±2:1, более предпочтительно 4,2±1,5:1, еще более предпочтительно 4,2±1:1, и еще более предпочтительно 4,2±0,5:1, наиболее предпочтительно 4,2±0,2:1, и в частности 4,2±0,1:1. Это соотношение удовлетворяет требованиям относительно высокого содержания полиалкиленоксида и хорошей способности поддаваться экструзии.
Когда лекарственные формы изготавливают из частей, которые получены путем нарезания заготовки экструдат, то масса частей определяет массу получающейся лекарственной формы. Резко выраженное изменение массы указанных частей приводит к соответствующему отклонению массы лекарственной формы от заданной массы. Изменение массы частей сильно зависит от характеристик поверхности заготовки экструдата. Заготовка с полностью гладкой поверхностью позволяет получать части, демонстрирующие небольшое изменение массы. Напротив, волнистая или ребристая поверхность заготовки приводит к тому, что части демонстрируют более значительное изменение массы, таким образом, повышая количество брака.
Было неожиданно выявлено, что характеристики поверхности заготовки экструдата могут зависеть от полиалкиленоксида: соотношения массы полиалкиленгликоля.
Предпочтительные композиции X1-Х32 фармацевтической лекарственной формы в соответствии с изобретением подытожены в таблицах, приведенных ниже:
В предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 100±75 мг, более предпочтительно 100±50 мг, наиболее предпочтительно 100±25 мг. В другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 200±75 мг, более предпочтительно 200±50 мг, наиболее предпочтительно 200±25 мг. В другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 250±75 мг, более предпочтительно 250±50 мг, наиболее предпочтительно 250±25 мг. В еще другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 300±75 мг, более предпочтительно 300±50 мг, наиболее предпочтительно 300±25 мг. В еще одном предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 400±75 мг, более предпочтительно 400±50 мг, наиболее предпочтительно 400±25 мг.
В предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 500±250 мг, более предпочтительно 500±200 мг, наиболее предпочтительно 500±150 мг. В другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 750±250 мг, более предпочтительно 750±200 мг, наиболее предпочтительно 750±150 мг. В другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 1000±250 мг, более предпочтительно 1000±200 мг, наиболее предпочтительно 1000±150 мг. В еще другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма имеет общую массу, которая находится в пределах 1250±250 мг, более предпочтительно 1250±200 мг, наиболее предпочтительно 1250±150 мг.
В предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма в соответствии с изобретением имеет объемную плотность, которая находится в пределах 1,19±0,30 г/см3, более предпочтительно 1,19±0,25 г/см3, еще более предпочтительно 1,19±0,20 г/см3, и еще более предпочтительно 1,19±0,15 г/см3, наиболее предпочтительно 1,19±0,10 г/см3, и в частности 1,19±0,05 г/см3. Предпочтительно, объемная плотность фармацевтической лекарственной формы в соответствии с изобретением составляет 1,1710,02 г/см3, 1,19±0,02 г/см3 или 1,21±0,02 г/см3. Способы измерения плотности лекарственной формы известны специалисту в данной области техники. Объемная плотность лекарственной формы может быть определена, например, при помощи метода ртутной порометрии или метода гелиевой пикнометрии, как описано в Европейской Фармакопеи
Предпочтительно, фармацевтическая лекарственная форма в соответствии с изобретением является подходящей для перорального применения. Тем не менее, также является возможным применять фармацевтическую лекарственную форму посредством различных путей введения и, таким образом, альтернативно фармацевтическая лекарственная форма может быть подходящей для буккального подъязыкового, ректального или влагалищного применения. Имплантаты также возможны.
В предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма в соответствии с изобретением является подходящей для приема один раз в день. В другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма в соответствии с изобретением является подходящей для приема два раза в день. В еще другом предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма в соответствии с изобретением является подходящей для приема три раза в день.
Для целей описания, «два раза в день» означает равные или почти равные промежутки времени, то есть примерно каждые 12 часов, или разные промежутки времени, например, 8 и 16 часов или 10 и 14 часов между отдельными приемами.
Для целей описания, «три раза в день» означает равные или почти равные промежутки времени, то есть, приблизительно каждые 8 часов, или различные промежутки времени, например, 6, 6 и 12 часов; или 7, 7 и 10 часов между отдельными приемами.
Предпочтительно, фармацевтическая лекарственная форма в соответствии с изобретением обуславливает, по крайней мере, частично отстроченное или пролонгированное высвобождение опиоида (А).
Контролируемое или пролонгированное высвобождение понимается в соответствии с изобретением предпочтительно как означающее профиль высвобождения, в котором опиоид (А) высвобождается на протяжении относительно долгого периода с уменьшенной частотой потребления с целью расширенного терапевтического действия. Предпочтительно, значение термина «пролонгированное высвобождение» находится в соответствии с Европейской директивой в отношении номенклатуры профиля высвобождения фармацевтических лекарственных форм (СНМР - Комитет по медицинским продуктам, предназначенным для человека). Оно в частности достигается при помощи перорального приема. Выражение «по крайней мере, частично отсроченное или пролонгированное высвобождение» охватывает в соответствии с изобретением любые фармацевтические лекарственные формы, которые гарантируют модифицированное высвобождение опиоидов (А), которые содержатся в них. Фармацевтические лекарственные формы предпочтительно включают фармацевтические лекарственные формы с нанесенным покрытием или без него, которые изготовлены с использованием определенных вспомогательных веществ, определенных процессов или комбинацией двух возможных вариантов, для того чтобы целенаправленно изменить скорость высвобождения или место высвобождения.
В случае фармацевтических лекарственных форм в соответствии с изобретением, профиль времени высвобождения формы с контролируемым высвобождением может быть модифицирован, например, следующим образом; продленное высвобождение, высвобождение повторного действия, пролонгированное высвобождение и замедленное высвобождение.
Для целей описания «контролируемое высвобождение», предпочтительно означает продукт, в котором высвобождение активного соединения в динамике по времени контролируется с помощью вида и состава препаративной формы. Для целей описания «продленное высвобождение» предпочтительно означает продукт, в котором высвобождение активного соединения отложено на определенное время задержки, после которого высвобождение является свободным. Для целей описания «высвобождение повторного действия» предпочтительно означает продукт, в котором первая порция активного соединения высвобождается с самого начала, за чем следует, по крайней мере, одна следующая порция активного соединения, которая высвобождается впоследствии. Для целей описания «пролонгированное высвобождение» предпочтительно означает продукт, в котором скорость высвобождения активного соединения из препаративной формы после применения была понижена в динамике по времени, для того чтобы поддержать терапевтическое действие, уменьшить токсическое действие, или для некоторых других терапевтических целей. Для целей описания «замедленное высвобождение» предпочтительно означает способ составления лекарственного средства таким образом, чтобы оно высвобождалось в организм непрерывно, на протяжении долгого периода времени, таким образом, уменьшая частоту приема. Для дополнительных деталей может быть сделана ссылка, например, на К.Н.Bauer, Lehrbuch der PharmazeutischeH Technologie, 6-ой издание, WVG Штутгарт, 1999; и Европейскую Фармакопею.
Фармацевтическая лекарственная форма в соответствии с изобретением может включать один или более опиоидов (А), по крайней мере, частично в дополнительной форме с контролируемым высвобождением, где контролируемое высвобождение может быть достигнуто с помощью традиционных материалов и процессов, известных специалисту в данной области техники, например, заключением активного вещества в матрицу с контролируемым высвобождением, или применения одного или более покрытий, которые контролируются высвобождение. Высвобождение активного вещества, тем не менее, нужно контролировать таким образом, чтобы дополнение материалов, задерживающих высвобождение не ослабляло требуемого предела прочности на разрыв. Контролируемое высвобождение из фармацевтической лекарственной формы в соответствии с изобретением предпочтительно достигается заключением вещества в матрицу. Предпочтительно, полиалкиленоксид (С) служит в качестве такой матрицы. Вспомогательные вещества действуют как материалы матрицы, контролируемые высвобождение. Материалы матрицы могут, например, быть гидрофильными, материалами, образующими гель, из которых высвобождение проистекает главным образом путем диффузии, или гидрофобными материалами, из которых высвобождение происходит главным образом путем диффузии из пор в матрице.
Предпочтительно, профиль высвобождения является существенно контролируемым матрицей, предпочтительно путем включения опиоида (А) в матрицу, содержащую полиалкиленоксид (С) и необязательно, дополнительные материалы матрицы. Предпочтительно, профиль высвобождения не является осмотическим. Предпочтительно, кинетика высвобождение не относится к нулевому порядку.
Предпочтительно, при физиологических условиях фармацевтическая лекарственная форма в соответствии с изобретением через 30 минут высвобождает опиоид (А) в количестве 0,1-75 %, через 240 минут в количестве 0,5-95 %, через 480 минут в количестве 1,0-100 % и через 720 минут в количестве 2,5-100%. Следующие предпочтительные профили высвобождения R1-R6 подытожены в таблице, приведенной ниже [все данные в масс. % высвобожденного опиоида (А)]:
Следующие предпочтительные профили высвобождения R1-R6 подытожены в таблице, приведенной ниже [все данные в масс.% высвобожденного опиоида (А)]:
Предпочтительно, профиль высвобождения фармацевтической лекарственной формы в соответствии с настоящим изобретением является стабильным после хранения, предпочтительно после хранения при повышенной температуре, например при 37°С, на протяжении 3 месяцев в запечатанных контейнерах. В этом отношении «стабильный» означает то, что, сравнивая изначальный профиль высвобождения с профилем высвобождения после хранения, в любой заданный момент времени профили высвобождения отличаются друг от друга не более чем на 20 %, более предпочтительно не более чем на 15 %, еще более предпочтительно не более чем на 10 %, и еще более предпочтительно не более чем на 7,5 %, наиболее предпочтительно не более чем на 5,0 % и в частности не более чем на 2,5 %.
Предпочтительно, в условиях in vitro фармацевтическая лекарственная форма высвобождает опиоид (А), изначально содержащийся в фармацевтической лекарственной форме, через 0,5 ч в количестве 1,0-35 масс.%, через 1 ч в количестве 5,0-45 масс.%, через 2 ч в количестве 10-60 масс.%, через 4 ч, по крайней мере, в количестве 15 масс.%, через 6 ч, по крайней мере, в количестве 20 масс.%, через 8 ч, по крайней мере, в количестве 25 масс.% и через 12 ч, по крайней мере, в количестве 30 масс.%.
Подходящие условия in vitro являются известными специалисту в данной области техники. В этом отношении может быть упомянута, например, Европейская Фармакопея. Предпочтительно, профиль высвобождения измеряют при следующих условиях: прибор с лопастью, снабженный грузом, 50 оборотов минуту, 37±5°С, 900 мл воспроизводимой интестинальной жидкости со значением рН, составляющим 6.8 (фосфатный буфер) или со значением рН, составляющим 4.5. В предпочтительном варианте осуществления изобретения, скорость вращения лопасти увеличена до 100 оборотов в минуту.
В предпочтительном варианте осуществления изобретения, после предпочтительно перорального применения фармацевтической лекарственной формы в соответствии с изобретением, in vivo среднее значение пика уровня в плазме (Cmax) в среднем достигается по истечении tmax, которое составляет 4,0±2,5 ч, более предпочтительно по истечении tmax, которое составляет 4,0±2,0 ч, еще более предпочтительно по истечении tmax, которое составляет 4,0±1,5 ч, наиболее предпочтительно по истечении tmax, которое составляет 4,0±1,0 ч и в частности по истечении tmax, которое составляет 4,0±0,5 ч. В другом предпочтительном варианте осуществления изобретения, после предпочтительно перорального применения фармацевтической лекарственной формы в соответствии с изобретением, in vivo среднее значение пика уровня в плазме (Cmax) в среднем достигается по истечении tmax, которое составляет 5,0±2,5 ч, более предпочтительно по истечении tmax, которое составляет 5,0±2,0 ч, еще более предпочтительно по истечении tmax, которое составляет 5,0±1,5 ч, наиболее предпочтительно по истечении tmax, которое составляет 5,0±1,0 ч и в частности по истечении tmax, которое составляет 5,0±0,5 ч. В еще другом предпочтительном варианте осуществления изобретения, после предпочтительно перорального применения фармацевтической лекарственной формы в соответствии с изобретением, in vivo среднее значение пика уровня в плазме (Cmax) в среднем достигается по истечении tmax, которое составляет 6,0±2,5 ч, более предпочтительно по истечении tmax, которое составляет 6,0±2,0 ч, еще более предпочтительно по истечении tmax, которое составляет 6,0±1,5 ч, наиболее предпочтительно по истечении tmax, которое составляет 6,0±1,0 ч и в частности по истечении tmax, которое составляет 6,0±0,5 ч.
В предпочтительном варианте осуществления изобретения среднее значение для t1/2 после предпочтительно перорального применения фармацевтической лекарственной формы в соответствии с изобретением in vivo составляет 4,0±2,5 ч, более предпочтительно 4,0±2,0 ч, еще более предпочтительно 4,0±1,5 ч, наиболее предпочтительно 4,0±1,0 ч, и в частности 4,0±0,5 ч. В другом предпочтительном варианте осуществления изобретения среднее значение для t1/2 после предпочтительно перорального применения фармацевтической лекарственной формы в соответствии с изобретением in vivo составляет предпочтительно 5,0±2,5 ч, более предпочтительно 5,0±2,0 ч, еще более предпочтительно 5,0±1,5 ч, наиболее предпочтительно 5,0±1,0 ч, и в частности 5,0±0,5 ч. В еще другом предпочтительном варианте осуществления изобретения среднее значение для t1/2 после предпочтительно перорального применения фармацевтической лекарственной формы в соответствии с изобретением in vivo составляет предпочтительно 6,0±2,5 ч, более предпочтительно 6,0±2,0 ч, еще более предпочтительно 6,0±1,5 ч, наиболее предпочтительно 6,0±1,0 ч, и в частности 6,0±0,5 ч.
Предпочтительно, фармацевтическая лекарственная форма в соответствии с изобретением содержит покрытие, предпочтительно пленочную оболочку. Подходящие материалы покрытия известны специалисту в данной области техники. Подходящие материалы покрытия являются коммерчески доступными, например, под торговыми марками Opadry® и Eudragit®.
Примеры подходящих материалов включают сложные эфиры целлюлозы и простые эфиры целлюлозы, такие как метилцеллюлоза (МЦ), гидроксипропилметилцеллюлоза (ГПМЦ), гидроксипропилцеллюлоза (ГПЦ), гидроксиэтилцеллюлоза (ГЭЦ), натрий карбоксиметилцеллюлоза (Na-КМЦ), этилцеллюлоза (ЭЦ), ацетатфталат целлюлозы (АФЦ), гидроксипропилметилцеллюлоза фталат (ГПМЦФ); поли(мет)акрилаты, такие как сополимеры аминоалкилметакрилата, сополимеры этилакрилат и метилметакрилата, сополимеры метакриловой кислоты и метилметакрилата, сополимеры метакриловой кислоты и метилметакрилата; винилполимеры, такие как поливинилпирролидон, поливинилацетатфталат, поливиниловый спирт, поливинилацетат; и природные пленкообразующие материалы, такие как шеллак.
В особенно предпочтительном варианте осуществления изобретения покрытие растворимо в воде. В предпочтительном варианте осуществления изобретения покрытие основано на поливиниловом спирте, таком как поливиниловом спирте, частично гидролизируемом, и может дополнительно содержать полиэтиленгликоль, такой как макроголь 3350, и/или красители. В другом предпочтительном варианте осуществления изобретения покрытие основано на гидроксипропилметилцеллюлозе, предпочтительно гипромеллозе типа 2910 имеющей вязкость, составляющую 3-15 мПа∙с.
Покрытие фармацевтической лекарственной формы может увеличить ее стабильность при хранении.
Покрытие может быть устойчивым к желудочному соку и растворяться как функция значения рН среды высвобождения. При помощи этого покрытия возможно обеспечить то, что фармацевтическая лекарственная форма в соответствии с изобретением проходит через желудок нерастворенной, и активное соединение высвобождается только в кишечнике. Покрытие, которое является устойчивым к желудочному соку, предпочтительно растворяется при значении рН, которое лежит в пределах 5 и 7.5. Соответствующие материалы и способы для отсроченного высвобождения активных соединений и для применения покрытий, которые являются устойчивыми к желудочному соку, являются известными специалисту в данной области техники, например, из «Coated Pharmaceutical dosage forms - Fundamentals, Manufacturing Techniques, Biopharmaceutical Aspects, Test Methods and Raw Materials» Kurt H.Bauer, K.Lehmann, HermanH P.Osterwald, Rothgang, Gerhart, 1-ое издание, 1998, Medpharm Scientific Publishers.
В предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма в соответствии с изобретением не содержит веществ, которые раздражают носовые ходы и/или глотку, то есть вещества, которые при применении через носовые ходы и/или глотку вызывают физическую реакцию, которая либо является настолько неприятной для пациента, что он или она не желает или не может продолжать прием, например, жжение, либо физиологически противодействуют применению соответствующего активного соединения, например, из-за увеличенной носовой секреции или чихания. Следующими примерами веществ, которые раздражают носовые ходы и/или глотку, являются те, которые вызывают жжение, зуд, желание чихнуть, увеличенное образование секреции или, по крайней мере, комбинацию двух из этих раздражителей. Соответствующие вещества и их количества, которые традиционно применяются, являются известными специалисту в данной области техники. Некоторые из веществ, которые раздражают носовые ходы и/или глотку, соответственно основаны на одном или более составляющих, или одной или более растительных составляющих свежеприготовленного лекарственное средства. Соответствующие свежеприготовленные активные вещества известны как таковые специалисту в данной области техники, и описаны, например, в «Pharmazeutische Biologie - DrogeH und ihre Inhaltsstoffe» by Prof. Dr.Hildebert Wagner, 2-е, исправленное издание, Gustav Fischer Verlag, Штутгарт-Нью-Йорк, 1982, страницы 82 и след. Соответствующее описание тем самым включено в качестве ссылки и считаются частью данного писания изобретения.
Кроме того, фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно не содержит антагонистов опиоида (А), предпочтительно не содержит антагонистов психотропных веществ, в частности не содержит антагонистов опиоидов (А). Антагонисты, подходящие для данного опиоида (А), являются известными специалисту в данной области техники, и могут присутствовать также или в виде соответствующих производных, в частности сложных эфиров или простых эфиров, или в каждом случае в виде соответствующих физиологически приемлемых соединений, в частности в виде их солей или сольватов. Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно не содержит антагонистов, выбранных из числа группы, включающей налоксон, налтрексон, налмефен, налид, налмексон, налорфин или налуфин, в каждом случае необязательно в виде соответствующих физиологически приемлемого соединения, в частности в виде основания, соли или сольвата; и не содержит нейролептиков, например, соединения, выбраннного из группы, включающей галоперидол, прометацин, флуфеназин, перфеназин, левомепромазин, тиоридазин, перазин, хлорпромазин, хлорпротиксин, зуклопентиксол, флупентиксол, протипендил, зотепин, бенперидол, пипамперон, мелперон и бромперидол.
Фармацевтическая лекарственная форма в соответствии с изобретением, кроме того, предпочтительно не содержит рвотного средства. Рвотные средства являются известными специалисту в данной области техники, и могут присутствовать также или в виде соответствующих производных, в частности сложных эфиров или простых эфиров, или в каждом случае в виде соответствующих физиологически приемлемых соединений, в частности в виде их солей или сольватов. Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно не содержит рвотного средства, основанного на одном или более составляющих корня ипекакуаны (таволга), например, основаного на составляющем эметина, таком как, например, описанный в «Pharmazeutische Biologie - DrogeH und ihre Inhaltsstoffe» профессором by Prof. Dr.Hildebert Wagner, 2-е, исправленное издание, Gustav Fischer Verlag, Штутгарт-Нью-Йорк, 1982. Соответствующее литературное описание тем самым включено в качестве ссылки и как считается частью данного описания изобретения. Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно также не содержит апоморфин в качестве рвотного средства.
Наконец, фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно также не содержит горького вещества. Примеры горьких веществ и их количеств, эффективных для применения, могут быть найдены в US-2003/0064099 A1, соответствующее описание которого должно считаться описанием настоящей заявки и тем самым включено здесь в качестве ссылки. Примерами горьких веществ являются ароматические масла, такие как масло мяты, масло эвкалипта, масло горького миндаля, ментол, фруктовые ароматические вещества, ароматические вещества лимона, апельсина, лайма, грейпфрута или их смесей и/или денатониум бензоат.
Фармацевтическая лекарственная форма в соответствии с изобретением соответственно предпочтительно не содержит ни веществ, которые раздражают носовые ходы и/или глотку, ни антагонистов опиоида (А), ни рвотных средства, ни горьких веществ.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно является подходящей для перорального применения.
Как правило, фармацевтическая лекарственная форма в соответствии с изобретением представлена в виде таблетки. Предпочтительно, фармацевтическая лекарственная форма не представлена ни в виде пленки, и не состоит из множества частиц.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно является защищенной от применения не по назначению. Предпочтительно, защита от применения не по назначению достигается на основании механических характеристик фармацевтической лекарственной формы так, чтобы предотвратить раздробления или, по крайней мере, существенно ему воспрепятствовать. В соответствии с изобретением, термин раздробление означает измельчение в порошок фармацевтической лекарственной формы с применением традиционных средств, обычно доступных для злоумышленника, таких как, например пестик и ступка, молоток, молот или другие традиционные средства, применяемые для того, чтобы измельчить в порошок под воздействием силы. Таким образом, защита от применения не по назначению предпочтительно означает, что измельчение в порошок фармацевтической лекарственной формы с применением традиционных средств предотвращено или, по крайней мере, существенно затруднено.
Предпочтительно, механические характеристики фармацевтической лекарственной формы в соответствии с изобретением, в частности его предел прочности на разрыв, существенно зависят от присутствия и пространственного распределения полиалкиленоксида (С), хотя его простое присутствие обычно не является удовлетворительным, для того чтобы достичь указанных характеристик. Полезные механические характеристики фармацевтической лекарственной формы в соответствии с изобретением не могут быть достигнуты автоматически, посредством простой технологической обработки опиоида (А), кислоты (В), полиалкиленоксида (С), и необязательно дополнительных наполнителей при помощи традиционных методов, подходящих для изготовления фармацевтических лекарственных форм. Фактически, обычно для изготовления необходимо выбрать подходящие аппараты, и необходимо приспособить критические параметры процесса, в частности давление/силу, температуру и время. Таким образом, даже если применяются традиционные аппараты, то протоколы процесса обычно должны приспосабливаться, для того чтобы соответствовать необходимым критериям.
Фармацевтическая лекарственная форма в соответствии с изобретением имеет предел прочности на разрыв, который составляет, по крайней мере, 300 Н, предпочтительно, по крайней мере, 400 Н, более предпочтительно, по крайней мере, 500 Н, еще более предпочтительно, по крайней мере, 750 Н, и еще более предпочтительно, по крайней мере, 1000 Н, наиболее предпочтительно, по крайней мере, 1250 Нив частности, по крайней мере, 1500 Н.
«Предел прочности на разрыв» (сопротивление разрушению) фармацевтической лекарственной формы известно специалисту в данной области техники. В этом отношении может быть упомянуто, например, W.A.Ritschel, Die Tablette, 2. Auflage, Editio Cantor Verlag Aulendorf, 2002; H Liebermann и др., Pharmaceutical dosage forms: Tablets, т. 2, Informa Healthcare; 2 издание, 1990; и Encyclopedia of Pharmaceutical Technology, Informa Healthcare; 1 издание.
Для целей описания предел прочности на разрыв предпочтительно определяют как количество силы, которая необходима, для того чтобы раздробить фармацевтическую лекарственную форму (= разрушающее усилие). По этой причине, для целей описания фармацевтическая лекарственная форма предпочтительно не показывает желательный предел прочности на разрыв, если она разрушается, то есть, разламывается, по крайней мере, на две отдельные части, которые отделены друг от друга. В другом предпочтительном варианте осуществления изобретения, тем не менее, фармацевтическая лекарственная форма считается разрушенной если сила уменьшается на 25 % (предельное значение) самой высокой силы, установленной во время измерения (см. ниже).
Фармацевтические лекарственные формы в соответствии с изобретением отличается от традиционных фармацевтических лекарственных форм тем, что по причине их предела прочности на разрыв, они не могут быть измельчены в порошок применением силы при помощи традиционных средств, таких как, например, пестик и ступка, молоток, молот или другие традиционные средства, используемые для измельчения в порошок, в частности устройства, разработанные для этой цели (дробилки таблеток). В этом отношении «измельчение в порошок» означает дробление на маленькие частицы, которые будут немедленно высвобождать фармакологически активное соединение (А) в подходящую среду. Предотвращение измельчение в порошок фактически исключает пероральное или парентеральное применение, в частности применение не по назначению внутривенно или через нос.
Традиционные таблетки обычно имеют предел прочности на разрыв значительно ниже 200 Н в любом направлении растяжения. Предел прочности на разрыв традиционных круглых таблеток может быть установлен в соответствии со следующей эмпирической формулой: Предел прочности на разрыв [в Н] = 10 × Диаметр Таблетки [в мм]. Таким образом, в соответствии с указанной эмпирической формулой, круглая таблетка, имеющая предел прочности на разрыв, составляющий, по крайней мере, 300 Н, имела бы диаметр, составляющий, по крайней мере, 30 мм). При этом, такую таблетку нельзя проглотить. Вышеупомянутая эмпирическая формула предпочтительно не применяется к фармацевтическим лекарственным формам изобретения, которые не являются традиционными, а скорее особыми.
Кроме того, актуальное значение сила жевания составляет приблизительно 220 Н (см., например, Р.А.Proeschel и др., J Dent Res, 2002, 81 (7), 464-468). Это означает, что традиционные таблетки, имеющие предел прочности на разрыв значительно ниже 200 Н, могут быть раздроблены после непосредственного жевания, в то время как фармацевтическая лекарственная форма в соответствии с изобретением не может.
Более того, когда применяется ускорение свободного падения приблизительно в 9,81 м/с2, 300 Н соответствуют силе тяжести больше чем 30 кг, то есть фармацевтическая лекарственная форма в соответствии с изобретением может предпочтительно противостоять весу больше чем в 30 кг и не быть при этом измельченной в порошок.
Способы для измерения предела прочности на разрыв фармацевтической лекарственной формы известны специалисту в данной области техники. Подходящие устройства являются коммерчески доступными.
Например, предел прочности на разрыв (сопротивление разрушению) может быть устанавливается в соответствии с Европейской Фармакопеи 5.0, 2.9.8 или 6.0, 2.09.08 «Resistance to Crushing of Tablets». Испытание предназначено для того, чтобы определить, при определенных условиях, сопротивление разрушению таблеток, установленное при помощи силы, необходимой для того, чтобы разрушить их путем раздробления. Аппарат состоит из 2 зажимных приспособлений расположенных друг против друга, одно из которых двигается в направлении другого. Плоские поверхности зажимных приспособлений перпендикулярны направлению движения. Дробящие поверхности зажимных приспособлений являются плоскими и являются больше чем зона контакта с таблеткой. Аппарат прокалибрирован, с использованием системы с точностью в 1 Ньютон. Таблетку располагают между зажимными приспособлениями, принимая во внимание, где это применимо, форму, измерительную метку и надпись; для каждого измерения таблетка располагают таким же образом относительно направления применения силы (и направления растяжения, в котором предел прочности на разрыв должна быть установлен). Измерение проводят на 10 таблетках, принимая во внимание, чтобы все фрагменты таблеток были удалены перед каждым новым измерением. Результат выражают как средние, минимальные и максимальные значения установленных сил, все выраженные в Ньютонах.
Подобное описание предела прочности на разрыв (разрушающего усилия) можно найти в Фармакопее США. В качестве альтернативы предел прочности на разрыв может быть устанавливается в соответствии с методом, описанным в ней, где утверждается, что предел прочности на разрыв представляет собой силу, необходимую для того, чтобы привести таблетку в негодность (то есть, разрушить) в определенной плоскости. Таблетки, как правило, помещают между двумя прижимными устройствами, один из которых перемещается, чтобы применить к таблетке силу, достаточную для того, чтобы вызвать разрушение. Для традиционных, круглых (круглое поперечное сечение) таблеток, нагрузка идет через их диаметр (иногда упоминается как диаметральная нагрузка), и разрушение происходит в плоскости. Разрушающее усилие таблеток обычно называют твердостью в фармацевтической литературе; тем не менее, применение этого термина вводит в заблуждение. В материаловедении термин твердость относится к сопротивлению поверхности к проникновению или вдавливанию, применяя небольшие образцы. Термин предел прочности при сжатии также часто используется для того, чтобы описать сопротивление таблеток к применению сжимающей нагрузки. Хотя этот термин описывает истинную природу испытания более точно, чем твердость, подразумевается, что таблетки фактически раздробляются во время испытания, что часто не происходит.
В качестве альтернативы, предел прочности на разрыв (сопротивление разрушению) может быть устанавливается в соответствии с WO 2005/016313, WO 2005/016314, и WO 2006/082099, что может быть расценено как модификация метода, описанного в Европейской Фармакопее. Аппарат, используемый для измерения, предпочтительно является установкой для испытания материалов «Zwick Z 2.5», Fmax = 2.5 кН с максимальным перемещением 1150 мм, которая настраивается при помощи одной опоры и одного шпинделя, с зазором позади в 100 мм и скоростью испытания, которая устанавливается между значениями 0,1 и 800 мм/минут программным обеспечением testControl. Измерение проводят, используя пневматический поршень с завинчивающими вкладками и цилиндр (диаметр 10 мм), датчик силы, Fmax. 1 кН, диаметр = 8 мм, тип 0,5 от 10 Н, тип 1 от 2 Н в соответствии с ISO 7500-1, со свидетельством М об испытании изготовителя в соответствии с DIN (немецкий институт стандартов) 55350-18 (полная сила Zwick Fmax = 1,45 кН) (все аппараты от компании Zwick GmbH & Со. KG, Ульма, Германия) с номером индекса BTC-FR 2.5 ТН. D09 для установки для испытания, номером индекса BTC-LC 0050N. Р01 для датчика силы, номером индекса ВО 70000 S06 для центрирующего устройства.
В предпочтительном варианте осуществления изобретения предел прочности на разрыв устанавливается при помощи установки для испытания предела прочности на разрыв, например, Sotax®, типа НТ100 или типа НТ1 (Альшвиль, Швейцария). Оба типа, Sotax® НТ100 и Sotax® HT1 могут измерять предел прочности на разрыв в соответствии с двум различным принципам измерения: при постоянной скорости (где зажимное приспособление испытательного аппарата двигается при постоянной скорости, устанавливаемой от 5-200 мм/минут) или при постоянной силе (где зажимное приспособление испытательного аппарат увеличивает силу, регулируется последовательно от 5-100 H/с). В любом случае, оба принципа измерения являются подходящими для измерения предела прочности на разрыв фармацевтической лекарственной формы в соответствии с изобретением. Предпочтительно, предел прочности на разрыв устанавливается при постоянной скорости, предпочтительно при постоянной скорости, которая составляет 120 мм/минут.
В предпочтительном варианте осуществления изобретения фармацевтическая лекарственная форма считается разрушенной, если она разломана, по крайней мере, на две отдельные части.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно демонстрирует механическую прочность в широком диапазоне температур, и в дополнение к пределу прочности на разрыв (сопротивлению разрушению) необязательно также достаточную твердость, ударопрочность, ударную упругость, прочность на разрыв и/или модуль продольной упругости, и необязательно также при низких температурах (например, ниже -24°С, ниже -40°С или в жидком азоте), что фактически делает невозможным измельчить ее в порошок непосредственным жеванием, размолом в ступке, раздроблением, и т.д. Таким образом, предпочтительно, сравнительно высокий предел прочности на разрыв фармацевтической лекарственной формы в соответствии с изобретением поддерживается даже при низких или очень низких температурах, например, когда фармацевтическую лекарственную форму сначала охлаждают, чтобы увеличить ее хрупкость, например до температур ниже -25°С, ниже -40°С или даже в жидком азоте.
Фармацевтическая лекарственная форма в соответствии с изобретением отличается определенным уровнем предела прочности на разрыв. Это не означает, что фармацевтическая лекарственная форма должна также демонстрировать определенный уровень твердости. Твердость и предел прочности на разрыв являются различными физическими характеристиками. Поэтому, защита от применения не по назначению фармацевтической лекарственной формы не обязательно зависит от твердости фармацевтической лекарственной формы. Например, по причине ее предела прочности на разрыв, ударопрочности, модуля продольной упругости и прочности на разрыв, соответственно, фармацевтическая лекарственная форма может предпочтительно быть деформирована, например, пластически, будучи подверженной внешней силе, например с использованием молотка, но она не может быть измельченной в порошок, то есть, быть раскрошенной на большое количество фрагментов. Другими словами, фармацевтическая лекарственная форма в соответствии с изобретением отличается определенным уровнем предела прочности на разрыв, но не обязательно также отличается определенным уровнем стабильности формы.
Поэтому, в значении описания, фармацевтическая лекарственная форма, которая деформируется, будучи подверженной силе в определенном направлении растяжения, но при этом не разрушается (пластическая деформация, или текучесть) должна предпочтительно считаться как, имеющая желательный предел прочности на разрыв в указанном направлении растяжения.
В особенно предпочтительном варианте осуществления, изобретение относится к защищенной от применения не по назначению фармацевтической лекарственной форме, имеющий предел прочности на разрыв, составляющий, по крайней мере, 300 Н, и которую при этом термоформируют с помощью экструзии горячего расплава, при указанная фармацевтическая лекарственная форма содержит
- опиоид (А), выбранный из группы, состоящей из оксиморфона, оксикодона, гидроморфона, и их физиологически приемлемых солей;
- свободную физиологически приемлемую мультикарбоновую кислоту (В), предпочтительно лимонную кислоту, где содержание кислоты (В) находится в пределах от 0,001 до 5,0 масс.%, из расчета общей массы фармацевтической лекарственной формы;
- антиокислитель, где содержание антиокислителя, предпочтительно α-токоферола, находится в пределах от 0,001 до 5,0 масс. %, из расчета общей массы фармацевтической лекарственной формы; и
- полиалкиленоксид (С), имеющий среднемассовую молекулярную массу Мм, составляющую, по крайней мере, 200 000 г/моль;
где
- опиоид (А) заключен в матрицу, содержащую полиалкиленоксид (С), где указанная матрица контролирует высвобождение опиоида (А) из фармацевтической лекарственной формы; и
- после хранения в течение 4 недель при температуре 40°С и 75%-й относительной влажности, содержание опиоида (А) составляет, по крайней мере, 98,0% от его изначального содержания перед хранением.
Фармацевтическая лекарственная форма в соответствии с изобретением может быть изготовлена различными способами, особенно предпочтительные из которых объяснены более детально ниже. Некоторые подходящие способы были уже описаны в предшествующем уровне техники. В этом отношении могут быть упомянуты, например, WO 2005/016313, WO 2005/016314, WO 2005/063214, WO 2005/102286, WO 2006/002883, WO 2006/002884, WO 2006/002886, WO 2006/082097 и WO 2006/082099.
Настоящее изобретение также относится к фармацевтическим лекарственным формам, которые могут быть получены любым из способов, описанных ниже.
Как правило, способ изготовления фармацевтической лекарственной формы в соответствии с изобретением предпочтительно включает следующие стадии:
(a) смешивание всех компонентов;
(b) необязательно предварительное формование смеси, полученной на этапе (а), предпочтительно с применением нагрева и/или силы к смеси, полученной на этапе (а), при этом величина примененного нагрева предпочтительно не является достаточной для того, чтобы нагреть полиалкиленоксид (С) до его температуры размягчения;
(c) упрочнение смеси посредством применения нагрева и силы, при этом является возможным применять нагрев во время и/или перед применением силы и величина примененного нагрева является достаточной для того, чтобы нагреть полиалкиленоксид (С), по крайней мере, до его температуры размягчения;
(d) необязательно разделение упрочненной смеси;
(e) необязательно формование фармацевтической лекарственной формы; и
(f) необязательно обеспечение пленочной оболочки.
Нагрев может применяться непосредственно, например, при помощи контактирования или при помощи горячего газа, такого как горячий воздух, или с помощью ультразвука. Сила может быть применена, и/или фармацевтическая лекарственная форма может формоваться, например, при помощи непосредственного таблетирования или с помощью подходящего экструдера, в частности при помощи шнекового экструдера, снабженного двумя шнеками (двухшнековый экструдер) или при помощи экструдера с планетарным механизмом.
Окончательная форма фармацевтической лекарственной формы может либо быть обеспечена во время упрочнения смеси, когда применяют нагрев и силу (стадия (с) или в последующей стадии (стадия (е)). В обоих случаях, смесь всех компонентов находится предпочтительно в пластичном состоянии, то есть предпочтительно, формование проводят при температуре, по крайней мере, выше температуры размягчения полиалкиленоксида (С). Тем не менее, экструзия при более низких температурах, например, при температуре окружающей среды, также является возможной и может быть предпочтительной.
Формование могут проводить, например, при помощи таблеточного пресса, содержащего, пресс-форму и пуансоны соответствующей формы.
Особенно предпочтительный способ изготовления фармацевтической лекарственной формы изобретения включает экструзию горячего расплава. В этом способе фармацевтическую лекарственную форму в соответствии с изобретением изготавливают путем термоформования с помощью экструдера, предпочтительно без какого-либо заметного последующего обесцвечивания экструдата. Неожиданно было выявлено, что кислота (В) способна подавлять обесцвечивания. В отсутствии кислоты (В), экструдат имеет тенденцию окрашиваться от бежевого до желтоватого цвета, в то время как в присутствии кислоты (В) экструдаты являются в основном бесцветными, то есть белыми.
Этот способ отличается тем, что
a) все компоненты смешивают,
b) полученную смесь нагревают в экструдере, по крайней мере, до температуры размягчения полиалкиленоксида (С) и выдавливают через отверстие выхода экструдера с применением силы,
c) все еще пластичный экструдат разделяют на части и формуют в фармацевтическую лекарственную форму или
d) охлажденный и необязательно повторно нагретый разделенный на части экструдат формуют в фармацевтическую лекарственную форму.
Смешивание компонентов в соответствии со стадией способа а) также может происходить в экструдере.
Компоненты могут также быть смешаны в миксере, известном специалисту в данной области техники. Миксер может быть, например, роликовым миксером, миксером со встряхивающим механизмом, миксером со сдвиговыми усилиями или миксером с приложенными усилиями.
До того, как смешать с остальными компонентами, полиалкиленоксид (С) предпочтительно смешивают в соответствии с изобретением с антиокислителем, предпочтительно α-токоферолом. Это может происходить, путем смешивания этих двух компонентов, полиалкиленоксида (С) и антиокислителя, предпочтительно при помощи растворения или суспендирования антиокислителя в высоко неустойчивом растворителе и затем этот раствор или суспензию гомогенно перемешивают с полиалкиленоксидом (С) и затем удаляют растворитель при помощи сушки, предпочтительно в атмосфере инертного газа.
Предпочтительно расплавленную смесь, которая была нагрета в экструдере, по крайней мере, до температуры размягчения полиалкиленоксида (С), выдавливают из экструдера в пресс-формы, по крайней мере, с одним отверстием.
Способ в соответствии с изобретением требует применения подходящих экструдеров, предпочтительно шнековых экструдеров. Шнековые экструдеры, которые оборудованы двумя шнеками (двухшнековые экструдеры), в частности являются предпочтительными.
Экструзию предпочтительно проводят так, чтобы растяжение заготовки в результате экструзии составило не больше чем 30%, то есть когда применяют пресс-форму с отверстием, имеющим диаметр, например 6 мм, то экструдированная заготовка должна иметь диаметр не больше чем 8 мм. Более предпочтительно, растяжение заготовки составляет не больше чем 25%, еще более предпочтительно не больше чем 20%, наиболее предпочтительно не больше чем 15% и в частности не больше чем 10%.
Предпочтительно, экструзию проводят в отсутствии воды, то есть, не добавляют какой-либо воды. Тем не менее, следы воды (например, вызванные атмосферной влажностью) могут присутствовать.
Экструдер предпочтительно включает, по крайней мере, две температурных зоны, с нагреванием смеси, по крайней мере, до температуры размягчения полиалкиленоксида (С), которая происходит в первой зоне, которая расположена внизу от зоны подачи, и необязательно смешивающей зоны. Пропускная способность смеси предпочтительно составляет от 1,0 кг до 15,0 кг/час. В предпочтительном варианте осуществления изобретения пропускная способность составляет от 1,0 до 3,5 кг/час. В другом предпочтительном варианте осуществления изобретения пропускная способность составляет от 4,0 до 15,0 кг/час.
В предпочтительном варианте осуществления изобретения давление в пресс-форме находится в пределах от 25 до 100 бар. Давление в пресс-форме, при этом может регулироваться при помощи размера пресс-формы, температурой и скоростью экструзии.
Размер пресс-формы или размер отверстия выбираются произвольно. Пресс-форма или отверстие могут соответственно быть круглыми, продолговатыми или овальными в поперечном сечении, где круглое поперечное сечение предпочтительно имеет диаметр, составляющий 0,1 мм - 15 мм, и продолговатое поперечное сечение предпочтительно имеет максимальный продольный размер 21 мм и в поперечном направлении размер, который составляет 10 мм. Предпочтительно, пресс-форма или отверстие имеет круглое поперечное сечение. Кожух экструдера, используемого в соответствии с изобретением, может быть нагрет или охлажден. Соответствующий температурный контроль, то есть нагревание или охлаждение, устроен таким образом, чтобы смесь, которая будет экструдировать, показывала, по крайней мере, среднюю температуру (температура продукта), соответствующую температуре размягчения полиалкиленоксида (С) и не повышалась выше температуры, при которой может быть поврежден опиоид (А). Предпочтительно, подходящую температуру смеси, которую предстоит экструдировать, доводят до температуры ниже 180°С, предпочтительно ниже 150°С, но, по крайней мере, до температуры размягчения полиалкиленоксида (С). Типичные температуры экструзии составляют 120°С и 130°С.
В предпочтительном варианте осуществления изобретения крутящий момент экструдера находится в пределах от 30 до 95%. Крутящий момент экструдера при этом может регулироваться при помощи размера пресс-формы, температуры и скорости экструзии.
После экструзии расплавленной смеси и дополнительного охлаждения экструдированной заготовки или экструдированных заготовок, экструдаты предпочтительно разделяют на части. Указанное разделение предпочтительно может быть выполнено путем нарезания экструдатов при помощи автоматических или вращающихся резаков, гидромеханических резаков, проволоки, лезвий или с помощью лазерных резаков.
Предпочтительно, промежуточное или заключительное хранение необязательно разделеннх экструдатов или конечной фармацевтической лекарственной формы в соответствии с изобретением осуществляют в свободной от кислорода атмосфере, что может быть достигнуто, например, при помощи поглотителей кислорода.
Разделенный на части экструдат может быть пресс-формирован в таблетки для придания конечной формы фармацевтической лекарственной форме.
Применение силы в экструдере на смесь которой, по крайней мере, придана пластичность, регулируется при помощи контроля скорости вращения соответствующего устройства в экструдере и его размера, а также при помощи подбора размера выходного отверстия таким образом, чтобы давление, необходимое для экструдирования пластичной смеси, подавалось в экструдер, предпочтительно непосредственно перед экструзией. Параметры экструзии, которые, для каждой конкретной композиции, необходимы для того, чтобы обеспечить фармацевтическую лекарственную форму с желательными механическими характеристиками, могут быть установлены простым предварительным тестированием.
Например, но не ограничиваясь этим, экструзия может быть проведена при помощи двухшнекового экструдера, типа ZSE18 или ZSE27 (компании Leistritz, Нюрнберг, Германия), диаметры шнеков составляют 18 или 27 мм. Могут применяться шнеки, имеющие эксцентричные концы. Может применяться нагреваемая пресс-форма с круглым отверстием, имеющим диаметр 7, 8, или 9 мм. Параметры экструзии могут регулироваться, например, до следующих значений: скорость вращения шнеков: 120 Об/мин; скорость загрузки 2 кг/ч для ZSE 18 или 8 кг/ч для ZSE27; температура продукта: до пресс-формы, 125°С и после пресс-формы 135°С; и температура кожуха: 110°С.
Предпочтительно, экструзию проводят при помощи двухшнековых экструдеров или экструдеров с планетарным механизмов, двухшнековые экструдеры (вращающиеся в одном направлении или вращающиеся в противоположном направлении) являются в частности предпочтительными.
Фармацевтическую лекарственную форму в соответствии с изобретением предпочтительно изготавливают путем термоформования с помощью экструдера без последующего какого-либо заметного обесцвечивания экструдатов.
Способ изготовления фармацевтической лекарственной формы в соответствии с изобретением предпочтительно осуществляется непрерывно. Предпочтительно, способ включает экструзию гомогенной смеси всех компонентов. Является особенно предпочтительным, если полученный таким образом промежуточный продукт, например, заготовка, полученная посредством экструзии, демонстрирует однородные характеристики. В частности желательной является однородная плотность, однородное распределение активного соединения, однородные механические характеристики, однородная пористость, однородная на вид поверхность, и т.д. Только при указанных условиях может быть обеспечена однородность фармакологических характеристик, таких как стабильность профиля высвобождения, и количество отклонений должно поддерживаться низким.
Дополнительный аспект изобретения относится к упаковке, содержащей фармацевтическую лекарственную форму в соответствии с изобретением и поглотитель кислорода. Подходящая упаковочная тара включает блистерные упаковки и флаконы, такие как стеклянные флаконы или флаконы, сделанные из термопластических полимеров.
Подходящие поглотители кислорода известны специалисту в данной области техники. Поглотитель кислорода может быть любым поглотителем, известным в уровне техники, который применяют для того, чтобы очистить кислород. Могут применяться как органические, так и неорганические поглотители кислорода.
В одном варианте осуществления изобретения поглотитель кислорода представляет собой любой металлокомплекс, который демонстрирует кислород-поглощающее действие. Примеры включают комплексы, содержащие один или более сплавов алюминия, алюминия ферросилиция, сурьмы, бериллия, силицида кальция, церия, кобальта, галлия, гафния, железа, магния, никелевого катализатора, селена, кремния, серебра, стронция, титана, цинка, и/или циркония.
В еще одном варианте осуществления изобретения в качестве поглотителей кислорода могут применяться один или более элементов Группы 1А периодической таблицы и их сплавы и соединения. Примеры элементов Группы 1А включают цезий, литий, калий, натрий. Следующие примеры неорганических поглотителей кислорода включают один или более таких соединений, как азид натрия (NaNs), сульфит натрия (Na2SO3), гидразин, и гидроксиламин.
В одном варианте осуществления изобретения поглотитель кислорода представляет собой органическое соединение. Примеры включают один или более таких соединений, как политерпены, аскорбиновая кислота, аминополикарбоновая кислота, циклогександион, тетраметилпиперидон, и гетероциклические соединения с N-замещенными амино группами.
Поглотители кислорода и их применение в упаковке фармацевтического средства являются известными специалисту в данной области техники. В предпочтительном варианте осуществления изобретения поглотитель кислорода выбирают из группы, состоящей из катализируемых металлом окисляемых органических полимеров и антиокислителей. Особенно предпочтительными являются те поглотители кислорода, которые способны проявлять эксплутационные свойства в сухой окружающей среде, которая соответствует относительной влажности, составляющей ниже 60 %, предпочтительно ниже 30 %-ой относительной влажности, и которые комбинируют с поглотителем влаги. Примеры коммерчески доступных поглотителей кислорода, удовлетворяющих этим требованиям, включают Pharmakeep® KD10 и KD20.
Неожиданно было выявлено, что стабильность при хранении фармацевтической лекарственной формы может быть увеличена, если поддерживать низкое содержание кислорода воздушной среды в пределах упаковки. Способы упаковки фармацевтических лекарственных форм и применение подходящих поглотителей кислорода являются известными специалисту в данной области техники. В этом отношении ссылку можно сделать, например, на D.A.Dean, Pharmaceutical Packaging Technology, Taylor&Francis, 1-е изд.; F.A.Paine и др., Packaging Pharmaceutical and Healthcare Products, Springer, 1-е изд.; и O.G.Piringer и др., Plastic Packaging: Interactions with Food and Pharmaceuticals, Wiley-VCH, 2-е изд.
Что касается упаковки, то круглые флаконы, сделанные из полиолефинов, предпочтительно из ПЭНД (полиэтилен низкого давления), являются предпочтительными. Толщина стенки флаконов составляет предпочтительно, по крайней мере, 0,25 мм, более предпочтительно, по крайней мере, 0,5 мм, иначе флакон может быть деформирован.
Что касается крышки упаковки, то упаковка предпочтительно запечатывается под воздействием электромагнитной индукции или термозапечатывается алюминиевой фольгой.
Неожиданно было выявлено, что при помощи выбора соответствующей формы упаковки и вида ее запечатывания, пониженное давление, которое создается под действием поглотителя кислорода (разрежение, составляющее приблизительно 20000 Па = 2 Н/см2) можно регулировать, не вызывая деформации упаковки. Запечатывание под воздействием электромагнитной индукции (например, 3 секунды воздействия энергии) является предпочтительным. Когда запечатывают алюминиевой фольгой 75-и миллилитровый флакон, имеющий отверстие диаметром в 1 дюйм, то разрежение, составляющее 20 000 Па благодаря поглотителю кислорода, приводит к силе приблизительно 10 Н, соответствующей силе, которое оказывает давление веса в 1 кг.
Механическая прочность запечатывания может быть проверена либо при помощи введения соответствующего количества поглотителя кислорода во флакон, его запечатывания и ожидании на протяжении достаточного периода времени, например 2 дней, того, что кислород поглощается и устанавливается разрежение приблизительно в 20000 Па. В качестве альтернативы, флакон может быть запечатана без какого-либо поглотителя кислорода внутри, и веем в 1 кг может быть положен на алюминиевую фольгу, таким образом, моделируя силу.
Дополнительный аспект изобретения относится к применению опиоида (А) для изготовления фармацевтической лекарственной формы, как описано выше, для лечения боли.
Дополнительный аспект изобретения относится к применению фармацевтической лекарственной формы, как описано выше, для предотвращения или препятствования злоупотреблению опиоидом (А), который содержится там.
Дополнительный аспект изобретения относится к применению фармацевтической лекарственной формы, как описано выше, для предотвращения или препятствования случайной передозировке опиоида (А), который содержится там.
В этом отношении, изобретение также относится к применению опиоида (А), как описано выше, и/или полиалкиленоксида (С), как описано выше, для изготовления фармацевтической лекарственной формы в соответствии с изобретением для профилактики и/или лечения заболевания, таким образом, предотвращая передозировку опиоида (А), в частности вследствие раздробления фармацевтической лекарственной формы при помощи механического действия.
Кроме того, изобретение относится к способу профилактики и/или лечения заболевания, который содержит прием фармацевтической лекарственной формы в соответствии с изобретением, таким образом, предотвращая передозировку опиоида (А), в частности вследствие раздробления фармацевтической лекарственной формы при помощи механического действия. Предпочтительно, механическое действие выбираю-то из группы, состоящей из жевания, размола в ступке, раздробления, и применения аппаратов для того, чтобы измельчить в порошок традиционную фармацевтическую продукцию лекарственная форм.
Следующие примеры далее иллюстрируют изобретение, но не должны рассматриваться как ограничение его объема.
Пример 1
Таблетки были изготовлены при помощи экструзии горячего расплава гомогенных смесей различного состава при следующих, одинаковых условиях экструзии:
тип экструдера: экструдер Leistritz ZSE18PH 40D со шнеками с высокими сдвиговыми усилиями и пресс-формой диаметром, составляющим 9 мм
пропускная способность: 1,0 кг/ч
скорость вращения: 100 оборотов минуту
температура рабочего цилиндра: 100°С
температура экструдата: 120°С
экструдат был порезан на части по 325 мг, содержащие приблизительно 5 мг гидрохлорида оксиморфона.
Отдельные составляющие экструдированных смесей, так же как и общее количество продуктов разложения до и после хранения при ускоренных условиях хранения подытожены в таблице, приведенной ниже:
(А): гидрохлорид оксиморфона
ПЭО: полиэтиленоксид Мм 7 млн г/моль
ПЭГ: полиэтиленгликоль 6000
ГПМЦ: гипромеллоза 100000 Па*с
α-ток.: α-токоферол
oNo: оксиморфон-N-оксид (смесь)
S: сумма всех примесей
1: после экструзии, перед хранением
2: после хранения, бутылки из желтого стекла, пластиковая крышка, 4 недели, 40°С, 75%-я относительная влажность
Продукты разложения были проанализированы при помощи ВЭЖХ-УФ. Пик элюирования для оксиморфон-N-оксида не мог быть в достаточной степени по нулевой линии отделен от пика неизвестного продукта разложения (названного «UK 0.83»). Таким образом, оба пика были объединены вместе. В результате сравнения примеров A1 А5 становится очевидным, что содержание оксиморфон-N-оксида перед хранением (oNo1) существенно не меняется, если содержание антиокислителя α-токоферол уменьшают от 1,5 масс. % до 1,0 масс. %, 0,5 масс. %, 0,2 масс. % и даже до 0 масс.%. При этом, после хранения (oNo2) содержание оксиморфон-N-оксида является пропорциональным содержанию α-токоферола. Это является самым неожиданным, потому что оксиморфон-N-оксид является продуктом окисления, и можно было бы ожидать, что антиокислители обычно скорее подавляют, чем поддерживают образование продуктов окисления.
Тем не менее, полное исключение антиокислителя (α-токоферола) имеет свои недостатки. Посредством измерениями вязкости можно продемонстрировать, что полиэтиленоксид с высокой молекулярной массой разлагается после экструзии и/или хранения в отсутствии антиокислителя. Неожиданно было выявлено, что приблизительно 0,2 масс. % α-токоферола является достаточным для того, чтобы стабилизировать полиэтиленоксид; более высокое содержание α-токоферола не приводит к более высоким показателям вязкости полиалкиленоксида и, таким образом, не предотвращает более явно ПЭО от разложения. Таким образом, содержание антиокислителя (α-токоферол) предпочтительно сбалансировано так, чтобы с одной стороны, полиэтиленоксид с высокой молекулярной массой был достаточным образом стабилизирован, и с другой стороны, было сохранено на низком уровне нежелательное образование оксиморфон-N-оксида во время хранения.
Кроме того, при сравнении примеров B1-B4 и примеров C1-C4, становится очевидным, что неполная замена полиэтиленоксида с высокой молекулярной массой или полная замена полиэтиленагликоля альтернативным пластификатором не приводит к существенному уменьшению содержания нежелательного оксиморфон-N-оксида. Это является неожиданным, поскольку можно было бы ожидать, что полиэтиленоксид и полиэтиленгликоль являются потенциальными носителями пероксида, и что их уменьшение привело бы к уменьшению окислительных процессов, таких как окисление оксиморфона до оксиморфон-N-оксида.
Кроме того, при сравнении примеров D1-D5 и E1-Е4, становится очевидным, что добавление физиологически приемлемых кислот, в частности лимонной кислоты, приводит к уменьшению образования оксиморфон-N-оксида. Указанное является более явным, когда количество кислоты увеличено. При концентрации 0,1 масс.%, действие кислоты сравнительно слабо, но при концентрации 0,2 масс.% действие кислоты является более сильным и, кроме того, усиливается, когда концентрация лимонной кислоты увеличивается. Не только количество оксиморфон-N-оксида уменьшается, но также и общее количество продуктов разложения, в частности тех, которые имеют высокое время выдерживания ВЭЖХ.
Пример 2
Таблетки, которые были изготовлены на аналогии с пр. A1, B1, C1, D1 и Е1 выше, были упакованы в различные упаковочные материалы и хранились при температуре 40°С и 75%-й отн. влажности. Продукты разложения до и после хранения при ускоренных условиях хранения подытожены в таблице приведенной ниже:
Неожиданно было выявлено, что включение поглотителя кислорода в упаковку приводит к дополнительной стабилизации лекарственной формы, таким образом, что образование продуктов разложения было ограничено чрезвычайно низкими значениями.
Пример 3:
Таблетки были изготовлены, как описано в примере 1, упакованные во флаконы ПЭНД объемом 75 мл вместе с поглотителем кислорода и влагопоглотителем (Pharmakeep 20 KD), запечатаны с помощью электромагнитной индукции и закрыты пластиковой крышкой.
Отдельные составляющие экструдированных смесей, общее количество продуктов разложения до и после хранения при ускоренных условиях хранения подытожены в таблице, приведенной ниже:
(А): гидрохлорид оксиморфона
ПЭО: полиэтиленоксид Мм 7 млн г/моль
ПЭГ: полиэтиленгликоль 6000
ГПМЦ: гипромеллоза 100000 Па*с
α-ток.: α-токоферол
oNo: оксиморфон-N-оксид (смесь)
S: сумма всех примесей
1: после экструзии, перед хранением
2: после хранения, бутылок ПЭНД, пластиковая крышка запечатаны с помощью электромагнитной индукции, поглотитель кислорода, 4 недели, 40°С, 75%-я отн. Влажность
3: после хранения, флаконы ПЭНД, запечатаны с помощью электромагнитной индукции, пластиковая крышка, поглотитель кислорода, 8 недель, 40°С, 75 %-я отн. влажность
Результаты демонстрируют, что чистота продукта очень высокой после изготовления и что продукт показывает стабильность во время хранения в течение 8 недель при ускоренных условиях деградации, составляющих температуру 40°С/75% отн. влажности.
Пример 4:
Таблетки были изготовлены, как описано в примере 1, но были порезаны на части по 215 мг, которые содержали 5 мг, или 40 мг гидрохлорид оксиморфона, после формования таблетки были покрыты приблизительно по 6.5 мг каждая традиционным пленочным покрытием Opadry II, содержащим поливиниловый спирт, в качестве пленкообразующего наполнителя, упакованы во флаконы ПЭНД объемом 75 мл вместе с поглотителем кислорода и влагопоглотителем (Pharmakeep 20 KD), запечатаны с помощью электромагнитной индукции и закрыты пластиковой крышкой.
Отдельные составляющие экструдированных смесей, общее количество продуктов разложения до и после хранения при ускоренных условиях хранения подытожены в таблице, приведенной ниже:
(А): гидрохлорид оксиморфона
ПЭО: полиэтиленоксид Мм 7 млн г/моль
ПЭГ: полиэтиленгликоль 6000
ГПМЦ: гипромеллоза 100000 Па*с
α-ток.: α-токоферол
oNo: оксиморфон-N-оксид (смесь)
S: сумма всех примесей
1: после экструзии, перед хранением
2: после хранения, флаконы ПЭНД, запечатаны с помощью электромагнитной индукции, пластиковая крышка, поглотитель кислорода, 1 месяц, 40°С, 75%-я отн. влажность
Пример 5:
Наиболее предпочтительная лекарственная форма в соответствии с примером 3 является также подходящей для стабилизации оксикодона. Это может быть продемонстрировано для препаративной формы, содержащей 80 мг гидрохлорид оксикодона, изготовленной по аналогии примера 1, но при этом, экструдат был порезан на части по 400 мг:
(А): оксикодон
ПЭО: полиэтиленоксид Мм 7 млн г/моль
ПЭГ: полиэтиленгликоль 6000
ГПМЦ: гипромеллоза 100000 Па*с
α-ток.: α-токоферол
oNo: оксикодон-N-оксид (примесь D+E)
1: после экструзии, перед хранением
2: после хранения, бутылки из желтого стекла, пластиковая крышка, поглотитель кислорода с влагопоглотителем (Pharmakeep 20KD), 1 месяц, 40°С, 75%-я отн. влажность
Пример 6:
В однократной дозе (40 мг гидрохлорид оксиморфона, таблетки примера 4), рандомизированное, трехстороннее перекрестное исследование с 1 неделей, между приемами пациенты не принимали пищу накануне вечером, и прием пищи осуществлялся через 4 и 10 часов после приема. В пределах ±1 часа после приема препарата воду не пили. Все таблетки запивали 240 мл воды (пример Т).
Образцы для фармакокинетики были взяты перед приемом оксиморфона и 6-ОН-оксиморфона и через 48 часов после приема.
Биоэквивалентность сравнивали с Opana ER® (ссылка R). Результаты подытожены в таблицах, приведенных ниже:
Cl = доверительный интервал
Становится очевидным, что лекарственные формы в соответствии с изобретением, имеющая повышенный предел прочности на разрыв, являются биоэквивалентными в сравнении с традиционными лекарственными формами (Opana ER®).
Пример 7:
Таблетки были изготовлены при одинаковых условиях экструзии горячего расплава из двух гомогенных составляющих смесей h и h:
при следующих, одинаковых условиях экструзии:
тип экструдера: Экструдер Leistritz тип Micro 27GL 40D со шнеками со
средними сдвиговыми усилиями и пресс-формой, составляющей 8 мм в диаметре
пропускная способность: 10 кг/ч
скорость вращения: 120 оборотов минуту
время изготовления: 30 минут
температура самой горячей зоны нагрева: 100°С
температура пресс-формы: 130°С.
Экструдат был порезан на части по 360 мг, содержащие приблизительно 40 мг гидрохлорида оксиморфона.
100 частей были взвешены по отдельности, и было установлено стандартное отклонение от массы. Части с композицией I1 (ПЭО:ЛЭГ = 6,82:1) показали стандартное отклонение 2,3 %, в то время как части с композицией I2 (ПЭО:ПЭГ = 4,21:1) показали стандартное отклонение всего лишь 1,6 %.
Из этих сравнительных исследований, которые показывают неожиданные результаты, становится очевидным, что способность поддаваться обработке эксткдированной массы может быть улучшена с помощью регулирования соотношения ПЭО к ПЭГ.
Пример 8:
Для того чтобы установить, могут ли мультикарбоновые кислоты, кроме лимонной кислоты, также препятствовать образованию оксиморфон-N-оксида, были изготовлены таблетки, как описано в примере 1, содержащие малеиновую кислоту или фумаровую кислоту. Для сравнения также были изготовлены таблетки, содержащие неорганическую соль NaH2PO4. Образцы хранили в открытой посуде при 40°С и 75%-й отн. влажности в течение 4 недель.
Отдельные составляющие экструдированных смесей так же как и общее количество продуктов разложения до и после хранения при ускоренных условиях хранения подытожены в таблице, приведенной ниже:
(А): гидрохлорид оксиморфона
ПЭО: полиэтиленоксид Мм 7 млн г/моль
ПЭГ: полиэтиленгликоль 6000
ГПМЦ: гипромеллоза 100000 Па*с
α-ток.: α-токоферол
*NaH2PO4: примененный в виде 1,3 % дигидрата
оМо: оксиморфон-N-оксид (смесь)
Σ: сумма всех примесей; малеиновая кислота, фумаровая кислота и соответствующие соединения за вычетом из суммы примесей
1: после экструзии, перед хранением
2: после хранения, открытые блюда, 4 недели, 40°С, 75%-я отн. влажность
В случае малеиновой и фумаровой кислоты, указанные соединения, а в случае малеиновая кислоты также и другие соответствующие соединения, были обнаружены во время исследования чистоты в качестве примесей (приблизительно до 40%). Их значения были вычтены из общего числа примесей.
При сравнении примеров J1 и J2-A1 и B1, становится очевидным, что присутствие малеиновой и фумаровой кислоты полностью защищает оксиморфон против окисления до N-оксидов и в большой степени против другого разложения, не смотря на то, что образцы хранились в открытой посуде, а не в закрытых флаконах. Эти результаты сопоставимы с результатами, полученными с лимонной кислотой (пример 1, D4 и Е2-Е4). Образцы, содержащие NaH2PO4(J3) продемонстрировали защиту против образования N-оксидов и другого разложения, когда их сравнивали с препаративными формами, не содержащими какого-либо кислотного соединения (A1 и B1), но в меньшей степени чем мультикарбоновые кислоты, такие как лимонная, малеиновая и фумаровая кислота.
Пример 9:
Для того чтобы установить, может ли присутствие лимонной кислоты также защищать чувствительные к окислению опиоиды, кроме оксиморфона, против N-окисления, таблетки, содержащие гидрохлорид оксикодона, были изготовлены, как описано в примере 1.
Для сравнения также были изготовлены таблетки, содержащие меньшее количество α-токоферола. Образцы хранились в открытой посуде при температуре 40°С и 75 %-й отн. влажности в течение 4 недель.
Отдельные составляющие экструдированных смесей, так же как и общее количество продуктов разложения до и после хранения при ускоренных условиях хранения подытожены в таблице, приведенной ниже:
(А): гидрохлорид оксикодона
ПЭО: полиэтиленоксид Мм 7 млн г/моль
ПЭГ: полиэтиленгликоль 6000
ГПМЦ: гипромеллоза 100000 Па*с
α-ток.: α-токоферол
oNo: оксикодон-N-оксид
Σ: сумма всех примесей
1: после экструзии, перед хранением
2: после хранения, открытая посуда, 4 недели, 40°С, 75%-я отн. влажность
Эти результаты демонстрируют, что лимонная кислота полностью защищает оксикодон против окисления до N-оксидов и в большой степени против другого разложения, не смотря на то, что образцы хранились в открытой посуде, а не в закрытых флаконах. Уменьшение количества α-токоферола приводило к уменьшению разложения в том случае, когда препаративные формы не содержали лимонную кислоту. Эти результаты сопоставимы с результатами, полученными для оксиморфона.
| название | год | авторы | номер документа |
|---|---|---|---|
| СТАБИЛЬНАЯ ПРИ ОКИСЛЕНИИ, ПРОЧНАЯ НА ИЗЛОМ ЛЕКАРСТВЕННАЯ ФОРМА | 2010 |
|
RU2567723C2 |
| ЛЕКАРСТВЕННАЯ ФОРМА | 2009 |
|
RU2493830C2 |
| УСТОЙЧИВАЯ К РАЗРУШЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА, СОДЕРЖАЩАЯ НЕОРГАНИЧЕСКУЮ СОЛЬ | 2011 |
|
RU2604676C2 |
| УСТОЙЧИВАЯ К РАЗРУШЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА, КОТОРАЯ СОДЕРЖИТ АНИОННЫЙ ПОЛИМЕР | 2011 |
|
RU2607499C2 |
| ЗАЩИЩЕННАЯ ОТ ПРИМЕНЕНИЯ НЕ ПО НАЗНАЧЕНИЮ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА | 2005 |
|
RU2396944C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ПРЕПАРАТИВНАЯ ФОРМА | 2004 |
|
RU2362560C2 |
| ЭКСТРУДИРОВАННАЯ ИЗ ГОРЯЧЕГО РАСПЛАВА ФАРМАЦЕВТИЧЕСКАЯ ЛЕКАРСТВЕННАЯ ФОРМА | 2010 |
|
RU2547555C2 |
| ЗАЩИЩЕННАЯ ОТ ПРИМЕНЕНИЯ НЕ ПО НАЗНАЧЕНИЮ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА, СОДЕРЖАЩАЯ (1R, 2R)-3-(3-ДИМЕТИЛАМИНО-1-ЭТИЛ-2-МЕТИЛПРОПИЛ)ФЕНОЛ | 2005 |
|
RU2393847C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОПИОИДНЫЙ АНТАГОНИСТ, ДЛЯ ЛЕЧЕНИЯ ЗАДЕРЖКИ МОЧИ | 2009 |
|
RU2478388C2 |
| ТАПЕНТАДОЛ ДЛЯ ЛЕЧЕНИЯ В СВЯЗИ С ОСТЕОАРТРОЗОМ | 2008 |
|
RU2473337C2 |
Изобретение относится к термоформованной фармацевтической лекарственной форме, имеющий предел прочности на разрыв, составляющий, по крайней мере, 300 Н, при этом указанная лекарственная форма содержит опиоид, свободную физиологически приемлемую мулькарбоновую кислоту в количестве от 0,1 до 5,0 мас.%, из расчета общей массы фармацевтической лекарственной формы, и полиалкиленоксид, имеющий среднемассовую молекулярную массу Мм, составляющую, по крайней мере, 500000 г/моль. Мулькарбоновую кислоту выбирают из группы, состоящей из малеиновой кислоты, фумаровой кислоты, глутаровой кислоты, малоновой кислоты и лимонной кислоты. Опиоид выбирают из группы, состоящей из оксиморфона, оксикодона, гидроморфона и их физиологически приемлемых солей. Фармацевтическая лекарственная форма может быть помещена в упаковку. Фармацевтическая лекарственная форма по изобретению характеризуется улучшенной стабильностью при хранении. 13 з.п. ф-лы,11 табл., 9 пр.
1. Термоформованная фармацевтическая лекарственная форма, имеющая предел прочности на разрыв, составляющий, по крайней мере, 300 Н и содержащая
- опиоид (А),
- свободную физиологически приемлемую мультикарбоновую кислоту (В) в количестве, которое составляет от 0,1 до 5,0 мас.%, из расчета общей массы фармацевтической лекарственной формы, и
- полиалкиленоксид (С), имеющий среднемассовую молекулярную массу Мм, составляющую, по крайней мере, 500000 г/моль.
2. Фармацевтическая лекарственная форма в соответствии с п 1, где мультикарбоновую кислоту выбирают из группы, состоящей из малеиновой кислоты, фумаровой кислоты, глутаровой кислоты, малоновой кислоты и лимонной кислоты.
3. Фармацевтическая лекарственная форма в соответствии с п.1, где содержание кислоты (В) находится в пределах от 0,2 до 2,5 мас.%, из расчета общей массы фармацевтической лекарственной формы.
4. Фармацевтическая лекарственная форма в соответствии с п. 1, которая содержит полиалкиленгликоль, где соотношение удельной массы полиалкиленоксида к полиалкиленгликолю находится в пределах 4.2±2 : 1.
5. Фармацевтическая лекарственная форма в соответствии с п. 1, которая содержит антиокислитель.
6. Фармацевтическая лекарственная форма в соответствии с п. 5, где антиокислитель представляет собой α-токоферол.
7. Фармацевтическая лекарственная форма в соответствии с п. 5 или 6, где содержание антиокислителя находится в пределах от 0,001 до 5,0 мас.% из расчета общей массы фармацевтической лекарственной формы.
8. Фармацевтическая лекарственная форма в соответствии с пунктом 1, где после хранения в течение 4 недель при температуре 40°C и 75%-й относительной влажности, содержание опиоида (А) составляет, по крайней мере, 98,0% его изначального содержания перед хранением.
9. Фармацевтическая лекарственная форма в соответствии с п. 1, где опиоид (А) заключен в матрицу, содержащую полиалкиленоксид (С), при этом указанная матрица контролирует высвобождение опиоида из фармацевтической лекарственной формы.
10. Фармацевтическая лекарственная форма в соответствии с п. 1, где опиоид (А) выбирают из группы, состоящей из оксиморфона, оксикодона, гидроморфона, и их физиологически приемлемых солей.
11. Фармацевтическая лекарственная форма в соответствии с п. 1, где соотношение удельной массы полиалкиленоксида (С) и опиоида (А), составляет, по крайней мере, 1:1.
12. Фармацевтическая лекарственная форма в соответствии с п. 1, которая предназначена для приема один раз в день или два раза в день.
13. Фармацевтическая лекарственная форма в соответствии с п. 1, которая имеет предел прочности на разрыв, составляющий, по крайней мере, 500 Н.
14. Фармацевтическая лекарственная форма в соответствии с любым из пп. 1-13, помещенная в упаковку, содержащую поглотитель кислорода.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 6569506 B1, 27.05.2003 | |||
| RU 2007103712 A, 27.09.2008 | |||
| RU 2005134361 A, 10.05.2006 | |||

