Изобретение относится к медицине, а именно к медицинской генетике и оториноларингологии, может быть использовано для диагностики врожденной наследственной глухоты у человека, вызванной мутациями в гене MYO7A.
Врожденная глухота - одно из частых заболеваний человека, регистрируемое с частотой 1:1000 новорожденных детей. К сожалению, существует множество причин ослабления слуха и возникновения глухоты. Потеря слуха является одновременно медицинской и социальной проблемой. Наибольшее значение имеет врожденная глухота как тяжелое и инвалидизирующее заболевание. В вопросах этиологии и патогенеза заболевания остается много неясных аспектов, но считается, что примерно половина всех случаев врожденной глухоты имеет наследственное происхождение. Несмотря на острую социальную значимость проблемы, до сих пор не разработаны способы ранней диагностики и система профилактических мероприятий.
Наследственная глухота относится к тем заболеваниям, в которых анамнестические и клинические данные представляют довольно скудный материал для постановки правильного диагноза с определением этиологии заболевания. Вместе с тем, своевременная и точная диагностика этого наследственного дефекта позволит наиболее качественно и в самые ранние сроки начинать реабилитационные мероприятия для обеспечения полноценной социальной адаптации лиц с наследственными дефектами слуха.
Клинический полиморфизм многих наследственных синдромальных и несиндромальных форм тугоухости/глухоты обусловлен в первую очередь ее значительной генетической гетерогенностью [Morton C.C. et al., 2006; Eisen M.D. et al., 2007]. На данный момент известно более 400 синдромов, сочетающихся с патологией слуха [OMIM].
Одной из наиболее распространенных в мире синдромальных форм глухоты является синдром Ушера (СУ), при различных формах которого наблюдается сочетание нарушения слуха и зрения. СУ - это наследственное заболевание, характеризующееся врожденными нарушениями слуха различной степени, вестибулярной дисфункцией и прогрессирующей пигментной дегенерацией сетчатки (ПДС), приводящей к постепенному сужению полей зрения и слепоте, но характер поражения органов свето- и звуковосприятия может быть различным. СУ встречается у 3-10% больных с врожденной глухотой и у 50% детей со слепоглухотой [Boughman J.A. et al., 1983; Pennings R.J., 2005].
Преимущества предлагаемой разработки перед существующими аналогами заключаются в оптимальности разрабатываемых подходов ДНК-диагностики заболеваний, основанных на выявлении существующего своеобразия генофонда народов Волго-Уральского региона.
Белковый продукт гена MYO7A - миозин VIIA, является структурным компонентом стереоцилий в волосковых клетках внутреннего уха и отвечает за везикулярный транспорт в клетках сетчатки и вестибулярного аппарата и принадлежит суперсемейству необычных (неконвенциональных) миозинов, которые взаимодействуют с актином посредством своего моторного домена и благодаря гидролизу АТФ получают энергию для перемещения вдоль актиновых филамент [Mermall V. et al., 1998].
Как и предполагали многие исследователи, различные мутации в гене MYO7A могут вызывать как несиндромальную глухоту (DFNA11, DFNB2), так и СУ [Ahmed Z.M. et al., 2003; Morton C.C. et al., 2006]. Ген MYO7A состоит из 49 экзонов [Adato A. et al., 1997], занимает примерно 87 kb геномной ДНК и кодирует белок в 2215 аминокислотных остатках [Chen Z.Y. et al., 1996].
На сегодняшний день известно более 400 мутаций, идентифицированных в гене MYO7A у больных с DFNA11, DFNB2 и USH1B (Weston M.D et al., 1996; Ahmed Z.M. et al, 2003; Le Quesne Stabej et al., 2012).
Мутации распределены почти по всем экзонам MYO7A, но большинство из них локализовано в экзонах, кодирующих моторный домен (с 3 по 18 экзон) (Weston M.D et al., 1996; Ahmed Z.M. et al., 2003; Yang et al., 2013).
Известны способы детекции различных мутаций гена MYO7A посредством SSCP-анализа каждого из 49 экзонов (Adato A, Weil D, Kalinski Н, Pel-Or Y, Ayadi H, Petit С, Korostishevsky M, Bonne-Tamir В. 1997. Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am J Hum Genet. 61 (4): 813-821; Weston MD, Kelley PM, Overbeck LD, Wagenaar M, Orten, DJ, Hasson T, Chen Z-Y, et al. 1996) Myosin VIIA mutation screening in 189 Usher syndrome type 1 patients. Am J Hum Genet 59: 1074-1083; Levy G, Levi-Acobas B, Blanchard S, Gerber S, Larget-Piet D, Chenal V, Liu XZ, et al. (1997) Myosin VIIA gene: heterogeneity of the mutations responsible for Usher syndrome type IB. Hum Mol Genet 6: 111-117). Существенными недостатками этих методов являются их трудоемкость, а также большое количество затрачиваемого времени (около 10 суток).
Известен способ детекции мутаций гена MY07A посредством гибридизации на чипах (Sandie Le Guedard-Mereuze, Christel Vache., David Baux, Vale, rie Fauge're, Lise Larrieu, Caroline Abadie, Andreas Janecke, Mireille Claustres, Anne-Francoise Roux, and Sylvie Tuffery-Giraud. 2010, Ex Vivo Splicing Assays of Mutations at Noncanonical Positions of Splice Sites in Usher Genes. Human Mutation, Vol.31, No. 3, 347-355; Prachi Kothiyal, Stephanie Cox, Jonathan Ebert, Ammar Husami, Margaret A Kenna, John H Greinwald, Bruce J Aronow, Heidi L Rehm. High-throughput detection of mutations responsible for childhood hearing loss using resequencing microarrays BMC Biotechnology, 2010, 10:10). Однако гибридизация на чипах - достаточно дорогостоящий и сложный метод, который, к тому же, практически невыполним в условиях диагностических лабораторий лечебно-профилактических учреждений.
Прототипом изобретения является работа Orita М. с соавторами, предложенная в 1989 году (Orita М., Iwahana Н., Kanazawa Н., Sekya Т. Detection of polymorphism of human DNA by gel electrophoresis as single cell conformation polymorphism // Protocols Natl. Acad. Sci. - 1989. - V.86. - P.2766-2770). Разработанный Orita M. с соавторами метод выявления однонуклеотидных замен и делеций позволяет идентифицировать различные мутации в ДНК. Время исследования составляет 4 дня.
Техническим результатом изобретения является упрощение способа и повышение точности определения мутаций гена MYO7A и сокращение времени исследования (до 2-х суток) посредством разработки мультиплексной (триплексной) ПЦР у больных наследственными формами глухоты и у гетерозиготных носителей данных мутаций.
Указанный технический результат достигается тем, что в способе, включающем выделение ДНК из лимфоцитов периферической крови методом фенольно-хлороформной экстракции, проведение мультиплексной полимеразной цепной реакции, амплификацию фрагментов ДНК, предположительно содержащих мутации гена MYO7A, амплифицируют одновременно восемнадцать участков гена MYO7A в шести триплексных системах олигонуклеотидов: триплекс 1 (7 exon) 
 фланкирующих области 18 экзонов с прилегающими сайтами сплайсинга с учетом картины распределения мутаций по экзонам, их локализации, функциональной значимости затрагиваемых доменов белкового продукта гена с последующей детекцией в денатурирующем акриламидном геле.
фланкирующих области 18 экзонов с прилегающими сайтами сплайсинга с учетом картины распределения мутаций по экзонам, их локализации, функциональной значимости затрагиваемых доменов белкового продукта гена с последующей детекцией в денатурирующем акриламидном геле.
Способ осуществляется следующим образом.
ДНК выделяют из лимфоцитов периферической крови. В качестве консерванта используют раствор следующего состава: 0,48% лимонной кислоты, 1,32% цитрата натрия, 1,47% глюкозы. При заборе крови к 1 мл консерванта добавляют 4 мл венозной крови и хорошо перемешивают.
Для получения ДНК необходимой степени чистоты и достаточного молекулярного веса используется метод выделения ДНК из крови фенольно-хлороформной экстракцией, описанный Метью (Mathew С.С. The isolation of high molecular weight eucariotic DNA. // Methods in Molecular Biology / Ed. Walker J.M.Y.L.: Human Press. 1984. - V.2. - P.31-34).
1. Кровь в пробирке с консервантом тщательно перемешивается и переливается в центрифужный стакан объемом 50 мл, туда же добавляется 30 мл охлажденного лизирующего буфера, содержащего 320 мМ сахарозы, 1% раствор тритона Х-100, 5 мМ MgCl2, 10 мМ трис HCl (pH 7,6).
2. Смесь центрифугируется 20 мин при 4000 об/мин.
3. Надосадочную жидкость сливают, к получившемуся осадку приливают 0,4 мл 10% SDS и протеиназу К (концентрация 10 мг/мл). Смесь для лизиса оставляют на 16 часов в термостате при температуре 37°C.
Экстракцию ДНК осуществляют в следующем порядке:
4. К лизату добавляют 0,5 мл фенола, насыщенного 1М трисHCl до pH 7,8.
5. Смесь встряхивают на шейкере и центрифугируют 10 мин при 6000 об/мин.
6. Отбирают водную фазу, содержащую ДНК и неденатурированные белки.
7. Отобранную фазу обрабатывают смесью фенол-хлороформа (1:1), а затем хлороформом.
8. Препараты осаждают двумя объемами охлажденного этанола 96%.
9. Образовавшийся осадок ДНК растворяют в 1,5 мл деионизированной H2O; раствор хранят при -20°C.
В дальнейшем полученную ДНК используют в качестве матрицы для мультиплексной полимеразной цепной реакции (М-ПЦР) для амплификации 18 фрагментов кодирующего региона гена MYO7A. Первичная последовательность (сиквенс) генов была получена из базы генетических последовательностей GenBank® Национального Института Здоровья, США (National Institute of Health) [http://www.ncbi.nlm.nih.gov]. На основе полученной интрон-экзонной структуры соответствующих генов [http://www.dsi.univ-paris5.fr/genatlas/] были подобраны специфические последовательности олигонуклеотидных праймеров с помощью пакета биологических программ: Primer Express® Software v2.0 Applied Biosystems, DNASTAR (Primer select 5.05 - 1993-2002), Primer Premier 5.0.
При подборе синтетических олигонуклеотидов в соответствующих биологических программах были заданы стандартные условия: длина синтетического олигонуклеотида 19-24 п.н., средняя температура отжига 60°C, размер ампликона 250-400 п.н. При отборе подобранных программой пар праймеров в мультиплексной системе учитывались следующие критерии:
1) фланкирование всего кодирующего региона (экзона гена);
2) существенные различия в размерах синтезируемых ПЦР продуктах (30 и более п.н.);
3) минимальное наличие или полное отсутствие гетеро- (димеров) и гомо-комплексов - («шпилек»), образуемых праймерами;
4) незначительные отличия в температуре отжига праймеров;
5) отсутствие гомологии с другими участками человеческого генома.
Для этого, на последнем этапе работы предложенные программой олигонуклеотиды оценивались на степень гомологии с ДНК Homo sapiens посредством сравнения последовательностей праймеров и баз данных на сайте http://www.genebee.msu.su/blast_new/blast.html.
Реакционную смесь для проведения полимеразного синтеза, содержащую 0,8 мкг геномной ДНК, рассчитанное количество каждого олигонуклеотида, 125 мкМ соответствующего дезоксинуклеозидтрифосфата (dNTP) (Promega, USA), помещали в 12,5 мкл однократного буфера для ПЦР следующего состава: 67 mM Tris-HCl, pH 8,8, 6,7 mM MgCl2, 16,6 mM (NH4)2SO4, 0,01% Tween-20. К полученной смеси прибавляли 5 единиц термофильной ДНК-полимеразы, 20-30 мкл стерильного минерального масла. ПЦР проводилась на амплификаторе “Терцик” производства компании «ДНК-технология» с использованием ДНК-полимеразы Thermus aquaticus производства фирмы «Силекс».

Оптимальный температурный режим амплификации для конкретных участков генов подбирался экспериментально и составлял приблизительно 64°C±2°C, с учетом разного G-C контента использованных пар праймеров и первичной последовательности матрицы, а также специфичности самой полимеразной цепной реакции.
Используются шесть триплексных систем олигонуклеотидов: триплекс 1 (7 exon) (F) 5′ 

После проведения ПЦР 10 мкл амплификата смешивали с 3,3 мкл денатурирующей смеси (5M NaOH и 0,5M ЭДТА) и нагревали в течение 15 минут при 42°C. После этого к пробам добавляли 3-4 мкл формамидной краски, смешанной с 0,5% бромфеноловым синим и 0,5% ксиленцианолом, и наносили на 10% полиакриламидный гель с 3% мочевиной (исходное соотношение акриламида/метиленбисакриламида 29:1) и 5% глицерином. Для проведения электрофореза использовали 0,5*TBE буфер.
Электрофорез геля длиной 20 см, толщиной 1 мм проходил при комнатной температуре при напряжении 100 В в течение 20-40 часов при температуре +4°C. Окраска геля проводилась в течение 15 минут 0,09% раствором азотнокислого серебра (AgNO3). Далее гель промывался дистиллированной водой и помещался в раствор, содержащий 40% формалин и NaOH (5М). После проявления гель еще раз промывали дистиллированной водой.
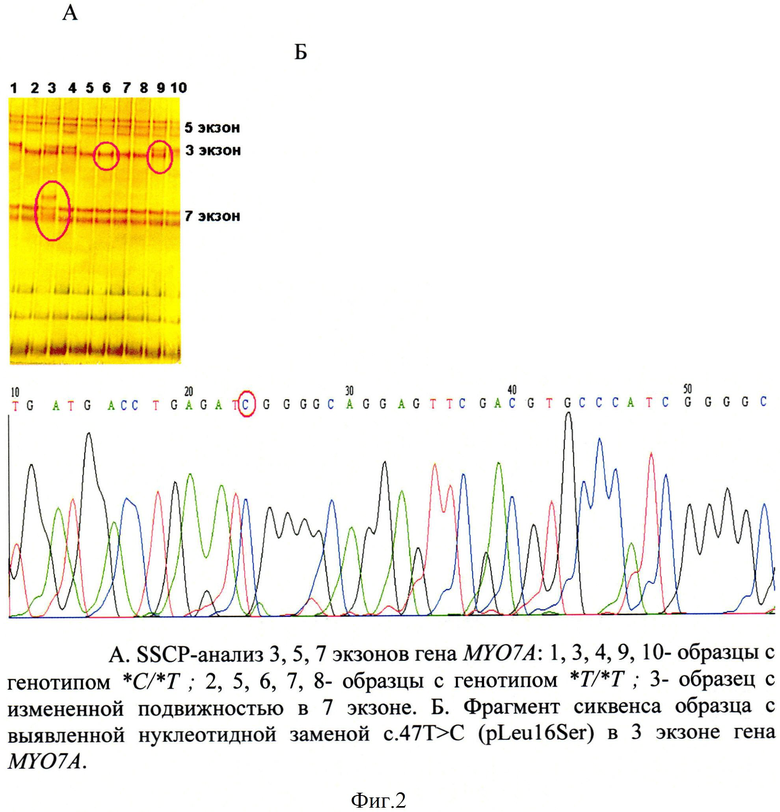
Идентификацию генотипов проводили по критерию присутствия или отсутствия дополнительных полос в сравнении с контрольной ДНК (норма) и панели образцов ДНК с мутациями (фигура 1).
Подготовка матриц для секвенирования включала очистку продуктов ПЦР от избытка праймеров, димеров и дезоксинуклеозидтрифосфатов (dNTP) путем ферментативной обработки экзонуклеазой (ExoI) и последующей очисткой на колонках из сорбента Sepfadex G50 фирмы Fine Chemicals АВ (Упсала, Швеция).
Определение нуклеотидных последовательностей проводили на автоматическом секвенаторе ABI Prism модель 310 (Applied Biosystems) с использованием набора для флюоресцентного мечения DYEnamic ЕТ согласно протоколу фирмы производителя [Amersham Pharmacia Biotech DYEnamic ET Terminator Cycle Sequencing Kit].
Для «прочтения» последовательности нуклеотидов использовали приложение BioEdit v.5.0.9. (1997-2001), а для анализа полученных сиквенсов - MegAlign из пакета программ DNAStar Inc (1993-2002).
На фигуре 1 представлено электрофоретическое разделение продуктов амплификации 6 мультиплексных систем гена MYO7A. Дорожка 1 - молекулярный маркер λpUC19/Pst1; дорожка 2 - триплекс 1 (экзон 7 - 172 п.н., экзон 3 - 220 п.н., экзон 5 - 260 п.н.), дорожка 3 - триплекс 2 (экзон 17 - 275 п.н., экзон 31 - 306 п.н., экзон 13 - 330 п.н.), дорожка 4 - триплекс 3 (экзон 23 - 292 п.н., экзон 35 - 364 п.н., экзон 16 - 383 п.н.), дорожка 5 - триплекс 4 (экзон 41 - 317 п.н., экзон 47 - 344 п.н., экзон 36 - 378 п.н.); дорожка 6 - триплекс 5 (экзон 23 - 295 п.н., экзон 9 - 328 п.н., экзон 16 - 401 п.н.), дорожка 7 - триплекс 6 (экзон 12 - 251 п.н., экзон 10 - 288 п.н., экзон 34 - 359 п.н.).
Предметом исследования послужили образцы ДНК неродственных индивидов. В качестве контроля использовалась ДНК обследованных здоровых доноров.
В качестве конкретных примеров в целом были обследованы 40 неродственных пациентов с диагнозом «синдром Ушера», а также 231 пробанд с диагнозом двухсторонняя нейросенсорная тугоухость предположительно наследственной этиологии, состоящих на учете в Республиканском сурдологическом центре Республиканской детской клинической больницы г.Уфы, члены их семей, а также здоровые доноры, проживающие в Волго-Уральском регионе. Диагноз глухоты или тугоухости устанавливался на основе данных тональной пороговой аудиометрии (аудиометр «GSI-61», Grason Stadler Instruments, USA), акустической импедансометрии (импедансометр «Zodiac 901», Дания) и регистрации отоакустической эмиссии (система «ILO 92», Otodynamics Ltd., Великобритания). Диагноз синдрома Ушера был поставлен на основании аудиологических (отоскопии, чистотональной аудиометрии, вызванной отоакустической эмиссии), офтальмологических (фундоскопии, остроты зрения, полей зрения, электроретинографии) данных, а также анализа вестибулярной функции (электронистагмографии). В ходе исследования уточнялись данные об этнической принадлежности пациентов путем анкетирования и выяснения национальной принадлежности родителей до третьего поколения. При этом особое внимание было уделено выявлению кровнородственных браков в семьях исследованных больных.
Наследственный характер глухоты в семьях устанавливали на основании генеалогических данных и ретроспективного анализа анамнеза больных с целью исключения возможного влияния факторов внешней среды во время пренатального и постнатального развития: учитывали отсутствие инфекций, травм слухового аппарата. В качестве контроля использованы ДНК 50 здоровых индивидов, обследованных в ГУ УфНИИГБ АНРБ и сопоставимых по полу, возрасту и этнической принадлежности с обследуемой группой. Аналогично, для сравнения по этнической принадлежности были выбраны представители трех основных этнических групп - русских, татар и башкир, проживающих на территории РБ. Представители других национальностей не вошли в данную часть исследования из-за их малочисленности в выборке больных.
SSCP - анализ 5, 9, 18, 22, 36 экзонов гена MYO7A не выявил изменений подвижности одноцепочечной ДНК в выборке больных.
В экзоне 3 нами было зафиксировано одинаковое изменение подвижности у 5 неродственных больных. Секвенционный анализ данного экзона позволил обнаружить нуклеотидную замену цитозина на тимин в 47 положении (с.47T>C), приводящую к замене аминокислоты (p.Leu16Ser) в кодирующей части третьего экзона (Фигура 2).
Из всей выборки больных (N=231) из Республики Башкортостан данная нуклеотидная замена в гетерозиготном состоянии была выявлена у 111 больных, что составило частоту 48%, частота аллеля *C при этом была 0,48.
В группе контроля аллель *C не был выявлен. У всех пациентов была установлена наследственная несиндромальная форма потери слуха. В результате проведенного статистического анализа выявлены достоверные различия (χ2=21,068; p>0,000) в распределении частот аллелей гена между выборками здорового контроля и группами пациентов с нарушениями зрения и слуха.
Полученные нами данные согласуются с литературными и подтверждают предположение о возможной патогенетической роли найденной нуклеотидной замены в патогенезе нарушений слуха и зрения.
Идентифицированные нами частоты аллелей T* и C* совпадают с результатами, полученными в центре по изучению полиморфизмов человека в Париже (СЕРН), где мажорным аллелем является C*. Данные, предоставленные другими научно-исследовательскими центрами из Азиатских стран - Японии, Китая, свидетельствуют о том, что в этих популяциях мажорным является другой аллель - C*.
Полученное соотношение частот аллелей, возможно, объясняется этническим своеобразием индивидов, составивших исследуемые выборки пациентов и группу контроля.
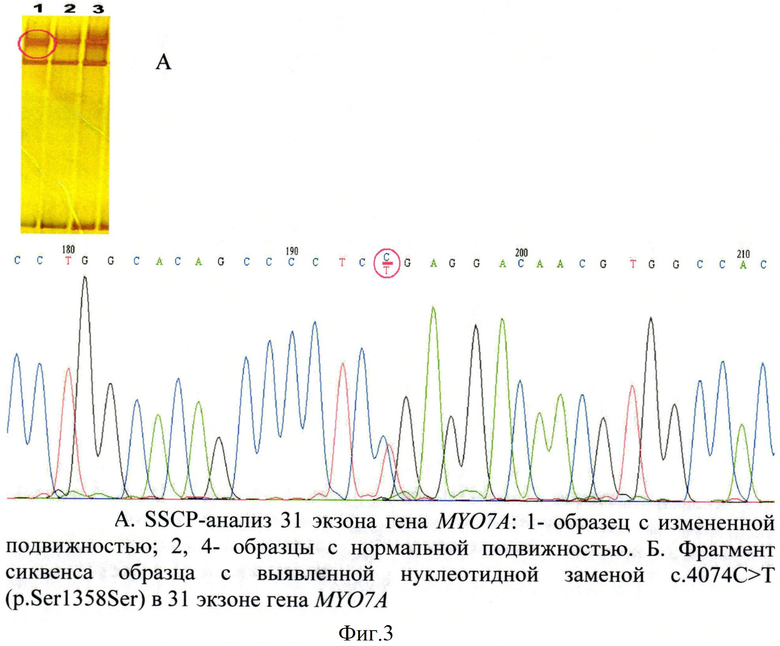
SSCP-анализ ПЦР продуктов, синтезированных с использованием мультиплексной системы (триплекса) на 13, 17, 31 экзоны гена MYO7A обнаружил изменение электрофоретической подвижности в 31 экзоне у двух пациентов с несиндромальной глухотой, последующее секвенирование которого позволило выявить изменение нуклеотидной последовательности в 4074 положении, ранее не описанное в литературе (Фигура 3).
Данный полиморфный вариант был обнаружен у двух пациентов русской этнической принадлежности из Республики Башкортостан с диагнозом врожденной нейросенсорной тугоухости IV степени. В группе контроля С.4074C>T (p.Ser1358Ser) обнаружен не был. Выявленная нуклеотидная замена является синонимичной, на основании чего ее можно считать полиморфизмом по типу транзиции.
Полученные высокие значения частоты p.Leu16Ser среди больных острой нейросенсорной тугоухостью в Башкортостане подтверждают важность определения этого изменения нуклеотидной последовательности в гене MYO7A при медико-генетическом консультировании пациентов с наследственными формами потери слуха.
Пример. Больной Ф.Ш., 1989 год рождения, город Туймазы, Республика Башкортостан. Диагноз синдрома Ушера установлен в 1990 году. В настоящее время пациент использует слуховой аппарат. Сибс пациента также болен синдромом Ушера. При молекулярно-генетическом тестировании у больного, его брата и родителей было взято по 8 мл венозной крови с последующим выделением ДНК и амплификацией трех участков 18 экзонов гена MYO7A в 6 триплексных системах, содержащей 0,8 мкг геномной ДНК, соответствующие кол-ва каждого олигонуклеотида, 125 мкМ каждого дезоксинуклеозидтрифосфата в 12,5 мкл однократного буфера для ПЦР. Затем провели электрофорез амплифицированных ПЦР-продуктов при постоянном напряжении 110 вольт. После окончания электрофореза гель окрасили последовательно раствором азотнокислого серебра и натрия гидрокарбоната в течение 15 минут и проанализировали. Исследование ДНК больного и его сибса выявило мутацию p.Leu16X в компаундгетерозиготном состоянии с мутацией p.Ala26Glu у пациента и его сибса. Мать пациента была носительницей мутации p.Leu16X, а отец имел мутацию p.Ala26Glu в гетерозиготном состоянии. Время исследования составило 4 дня.
В заключение необходимо отметить, что в связи с интенсивным накоплением знаний об основных этиологических, патогенетических механизмах на молекулярно-генетическом уровне, существенно расширяются возможности проведения дополнительных методов исследования, среди которых особенно актуальными становятся методы ДНК-диагностики. Поскольку они позволяют выяснить первопричину заболевания, открытие патогенетических механизмов которого в дальнейшем значительно облегчает терапию заболевания. Полученные в ходе данного исследования данные позволяют оценивать вероятность появления врожденной глухоты и синдрома Ушера у детей из семей с отягощенной наследственностью. Установление с помощью молекулярно-генетических методов необратимого наследственного повреждения слуха с самого рождения позволяет правильно проводить реабилитацию ребенка с повреждениями процесса звуковосприятия и снижать экономические затраты на проведение дорогостоящих диагностических процедур.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ диагностики мутации c.-23+1G>A (rs80338940) гена GJB2 | 2020 |
|
RU2746055C1 |
| Способ диагностики мутации 167delT (rs80338942) гена GJB2 | 2020 |
|
RU2739943C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ GJB2, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2006 |
|
RU2317547C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИИ с.-53-2А>G В ГЕНЕ ПРЕСТИНА (SLC26A5), ВЫЗЫВАЮЩЕЙ РАЗВИТИЕ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2012 |
|
RU2505608C1 |
| Способ диагностики мутации 35delG (rs80338939) гена GJB2 | 2020 |
|
RU2739889C1 |
| СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ | 2010 |
|
RU2448163C2 |
| Способ выявления мутаций гена GJB2, обуславливающих аутосомно-рецессивную глухоту 1А типа | 2017 |
|
RU2688180C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МУТАЦИЙ ГЕНА MTRNR1 ПРИ ОСТРОЙ НЕЙРОСЕНСОРНОЙ ТУГОУХОСТИ, ВЫЗВАННОЙ ПРИМЕНЕНИЕМ АНТИБИОТИКОВ ИЗ ГРУППЫ АМИНОГЛИКОЗИДОВ | 2007 |
|
RU2335541C1 |
| Способ одновременной диагностики наследственных заболеваний | 2015 |
|
RU2627115C2 |
| Способ ДНК-диагностики аутосомно-рецессивной глухоты-103 | 2019 |
|
RU2727684C1 |
Изобретение относится к области биохимии, в частности к способу выявления мутаций в гене MYO7A, сопровождающихся развитием несиндромальной аутосомно-рецессивной глухоты или синдрома Ушера. Способ включает выделение ДНК из лимфоцитов периферической крови методом фенольно-хлороформной экстракции, проведение ПЦР, амплификацию 18 участков гена MYO7A, детекцию в денатурирующем акриламидном геле и секвенирование. ПЦР проводят с использованием специально подобранных последовательностей олигонуклеотидов, фланкирующих области 18 экзонов гена MYO7A с возможным содержанием различных мутаций. Изобретение позволяет упростить способ и повысить точность определения мутаций гена MYO7A, а также сократить время исследования. 3 ил., 1 пр.
Способ выявления мутаций в гене MYO7A, сопровождающихся развитием несиндромальной аутосомно-рецессивной глухоты или синдрома Ушера, включающий выделение ДНК из лимфоцитов периферической крови методом фенольно-хлороформной экстракции, проведение полимеразной цепной реакции, амплификацию фрагментов ДНК, содержащих различные мутации гена MYO7A, отличающийся тем, что амплифицируют одновременно восемнадцать участков гена MYO7A в шести триплексных системах олигонуклеотидов: 
 экзонов с прилегающими сайтами сплайсинга с учетом картины распределения мутаций по экзонам, их локализации, функциональной значимости затрагиваемых доменов белкового продукта гена с последующей детекцией в денатурирующем акриламидном геле и секвенированием.
экзонов с прилегающими сайтами сплайсинга с учетом картины распределения мутаций по экзонам, их локализации, функциональной значимости затрагиваемых доменов белкового продукта гена с последующей детекцией в денатурирующем акриламидном геле и секвенированием.
| WESTON M.D | |||
| et al., Myosin VIIA Mutation Screening in 189 Usher Syndrome Type 1 Patients, Am | |||
| J | |||
| Hum | |||
| Genet., 1996, vol.59, pp.1074-1083 | |||
| ORITA M | |||
| et al., Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms, Proc | |||
| Natl | |||
| Acad | |||
| Sci | |||
| USA, 1989, Vol.86, pp.2766-2770 | |||
| AHMED Z.M | |||
| et al., The |

