Изобретение относится к области биотехнологии, а именно к молекулярной биологии, медицинской генетике и оториноларингологии.
Потеря слуха является одним из наиболее распространенных сенсорных расстройств у людей и имеет самый высокий уровень инвалидности в мире [1]. Приблизительно половина случаев потери слуха имеет генетическую этиологию с аутосомно-доминантным, аутосомно-рецессивным, X-сцепленным или митохондриальным типами наследования [2]. Врожденная глухота является одним из частых заболеваний человека, регистрируемая с частотой 1:1000 новорожденных детей. Потеря слуха является как медицинской, так и социальной проблемой. По данным ВОЗ, в мире насчитывается 466 млн. человек с инвалидизирующей потерей слуха, что составляет около 5% от всей популяции земного шара. В России людей с нарушениями слуха насчитывается более 13 млн. человек, из которых более 3 млн. нуждаются в слухопротезировании.
Среди наследственных нарушений слуха наиболее частая форма (около 80%) наследственной потери слуха - несиндромальная аутосомно-рецессивная глухота (DFNB1) [3, 4], относится к сенсоневральным нарушениям слуха, при которых страдает процесс звуковосприятия. Исследования в европейских и азиатских популяциях выявили мутации в генах семейства белков-коннексинов: GJB2 (gap junction β2) и GJB6 (gap junction β6), являющиеся наиболее частой причиной возникновения заболевания. Локализованы эти гены в 13 хромосоме в области 13q11-q13, кодирующих коннексин 26 (Сх26) и коннексин 30 (Сх30) - белки межклеточных щелевых контактов [5].
GJB2, кодирующий белок коннексин-26 ответственен за более чем половину случаев, за которыми следует SLC26A4, MY015A, OTOF, Cdh23 и ТМС1 [6]. Эти мутации происходят с различной частотой в разных популяциях [7], причем c.35delG, c.167delT и c.235delC преобладают в популяциях кавказских, ашкеназских евреев и Восточной Азии соответственно [8-12]. Кроме того, мутации синдрома Пегента в SLC26A4 составляют 10% наследственной потери слуха в большинстве населения мира. В Китае почти 50% пациентов с несиндромальной потерей слуха имеют мутации GJB2 или SLC26A4 [12]. Выявление этих мутаций представляет основной интерес для генетического консультирования. Хотя большое количество случаев вызвано мутациями горячих точек этих генов, как показали молекулярные эпидемиологические исследования, редкие мутации также могут способствовать потере слуха.
Более 300 патогенных вариаций GJB2 были зарегистрированы в базе данных мутаций генов человека [13]. Многие из них имеют высокую этногеографическую специфику распространенности [14].
Мутации GJB2 делают фенотип очень вариабельным. Факторы окружающей среды, модифицирующие гены и другие гены коннексина могут играть роль в потере слуха и его фенотипе [14]. Хотя большинство мутаций GJB2 сосредоточены в кодирующей области экзона 2, несколько мутаций были обнаружены в некодирующей области гена [15-17]. После обследования пациентов с рецессивной мутацией в кодирующей области был выявлен ряд мутаций патогена в некодирующей области GJB2, которые были ответственны за потерю слуха [18, 19]. Реконструкция структуры GJB2, окружающей геномные области, указывает на то, что основатель разветвился во всем мире за последние два или три столетия. Соответственно, мутации GJB2reH могли возникнуть во время миграции человека. Эти выводы имеют важное значение для подхода к политике диагностики и профилактики наследственной глухоты во всем мире [14].
Мутация с.-23 + 1G> А локализована в некодирующих областях экзона 1, что влияет на сплайсинг транскрипта GJB2 и представляет собой однонуклеотидную замену "G" на "А" в позиции 20,192,782 в хромосоме 13 [20]. Мутация с.-23 + 1G> А была впервые идентифицирована Denoyelle et al. [15], затем гетерозиготность и гомозиготность были доказаны у разных пациентов [17, 20-22].
Поскольку мутация с.-23 + 1G> A (chr13: 20766921: C> Т) находится в некодирующей области, она была рассмотрена в меньшем количестве глобальных исследований; однако исследования также изучали некодирующие области в дополнение к области кодирования. Мутация с.-23 + 1G> А представляет собой патогенную мутацию, вызванную аутосомно-рецессивной глухоты, которая находится в некодирующей области экзона 1, которая влияет на сплайсинг транскрипта GJB2 [20].
Эта мутация отмечается с различной частотой среди разных популяций и особенно часто встречается в монгольской, якутской, чешской, венгерской и турецкой популяциях с потерей слуха [23-25]. Некоторые ученые утверждают [14], что мутация в Восточной Азии, Северной и Южной Америке не распространена и имеет наибольшую частоту в Южной Азии и некоторых частях Ближнего Востока и допускает возможность того, что эти районы являются регионом-основателем, из которого они могут распространиться в другие регионы [14].
Эта мутация уменьшалась с севера на юг и с запада на восток, пока не достигла нуля в южных регионах. Можно сказать, что источником этой мутации является северный Иран, который распространился на другие регионы Ирана. Различные исследования показали, что эта мутация является второй наиболее распространенной мутацией после мутации c.35delG в Европе [26] и Иране [27].
Как известно, анамнестические и клинические данные являются недостаточными для постановки правильного диагноза в случае наследственной глухоты. Кроме того, своевременная и точная диагностика этого наследственного дефекта позволит наиболее качественно и в самые ранние сроки начинать лечение и реабилитационные мероприятия для обеспечения полноценной социальной адаптации лиц с наследственными дефектами слуха.
Известно большое количество методов определения генетического полиморфизма, использующих различные способы для анализа уникальной нуклеотидной последовательности ДНК человека. Сюда относят методы, основанные на применении ферментов (полиморфизм длин амплифицированных фрагментов (AFLP); полиморфизм длин рестрикционных фрагментов (RFLP) и пр.), химические методы (химическое расщепление гетеродуплексов, химическое лигирование), методы, основанные на различной электрофоретической подвижности полиморфных участков ДНК, детекция на твердой фазе, хроматографические методы, физические методы.
В частности, полиморфизмы гена GJB2 были исследованы во многих работах [9-11, 28]. Среди способов выявления мутаций в этом гене известен способ обнаружения мутаций 35delG, 167delT и 235delC гена GJB2 с помощью гетеродуплексного анализа [29]. Существенными недостатками метода являются низкая чувствительность, использование дорогостоящих вариантов гелей, а также невозможность определения точной локализации SNP внутри исследуемого фрагмента. Известен также метод SSCP (анализ конформационного полиморфизма одноцепочечной ДНК), предложенный в 1989 году и широко применяемый для детекции однонуклеотидных делеций [30]. Однако этот анализ является достаточно трудоемким и нуждается в дорогостоящих оборудовании и реактивах, а также его длительность составляет около 5 суток. Позже появился способ детекции мутаций методом мультиплексной системы полимеразной цепной реакции (ПЦР) с одной трубкой. Этот способ использовался в том числе и для выявления распространенных мутаций в гене Connexin-26 и был описан в работе Baris I. с соавторами [31]. В данной работе приводится описание проведения эксперимента, включающего выделение ДНК из лимфоцитов периферической крови, проведение мультиплексной полимеразной цепной реакции и амплификацию фрагментов ДНК, содержащих мутации 35delG, 167delT и 235delC гена GJB2. С помощью данного метода можно выявлять гетерозиготное носительство мутаций гена GJB2.
Из уровня техники известны следующие аналоги данного изобретения.
Патент № RU 2317547 С1 «СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ GJB2, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ» [Джемилева Л.У. и др., 2008].
Предметом данного изобретения является проведение мультиплексной полимеразной цепной реакции, в которой амплифицируют одновременно три участка гена GJB2. При наличии определенных аллелей диагностируют гетерозиготное либо гомозиготное носительство мутаций 35delG, 167delT и 235delC.
Патент № RU2555755 C1 «СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ MYO7A, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ И СИНДРОМОМ УШЕРА» [Джемилева Л.У. и др., 2015].
Способ, описанный в данном изобретении включает выделение ДНК из лимфоцитов периферической крови методом фенольно-хлороформной экстракции, проведение ПЦР, амплификацию 18 участков гена MYO7A, детекцию в денатурирующем акриламидном геле и секвенирование. ПЦР проводят с использованием специально подобранных последовательностей олигонуклеотидов, фланкирующих области 18 экзонов гена MYO7A с возможным содержанием различных мутаций.
Патент № RU 2448163 C2 «СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ» [Барашков Н.А. и др., 2012]
Данное изобретение направлено на детекцию 17 связанных с данным заболеванием мутаций в генах GJB2 и GJB6 с помощью ПЦР-амплификации соответствующих областей этих генов, которую проводят в 8 реакционных смесях с использованием специфичных пар праймеров, и последующего анализа полученных ампликонов, проводимого либо без предварительного расщепления эндонуклеазами (при определении мутаций c.312del14, с. 333-334delAA, ΔGJB6-D13S1830, ΔGJB6-D13S1854), либо после гидролиза соответствующими специфичными рестриктазами (при определении мутаций IVS1+1G>A, c.35delG, c.71G>A, c.79G>A, c.167delT, c.235delC, c.224G>A, c.299-300delAT, c.360delGAG, c.341A>G, c.269T>C, с.101Т>С, c.109G>A).
Патент № RU 2505608 C1 «СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИИ с.-53-2A>G В ГЕНЕ ПРЕСТИНА (SLC26A5), ВЫЗЫВАЮЩЕЙ РАЗВИТИЕ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ» [Джемилева Л.У. и др., 2014]
Способ, описанный в данном изобретении включает выделение ДНК из лимфоцитов периферической крови методом фенольно-хлороформной экстракции. Проводят полимеразную цепную реакцию с возможностью проведения анализа флуоресценции по конечной точке. Амплифицируют одновременно два участка гена SLC26A5 в смеси двух пар последовательностей олигонуклеотидов с флуоресцентной меткой: 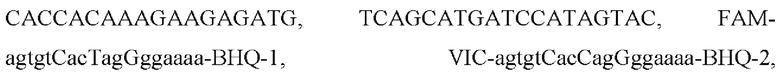 фланкирующих область с возможным содержанием мутации c.-53-2A>G в гене SLC26A5.
фланкирующих область с возможным содержанием мутации c.-53-2A>G в гене SLC26A5.
Патент № RU 2688180 C1 «Способ выявления мутаций гена GJB2, обуславливающих аутосомно-рецессивную глухоту 1А типа» [Пшенникова В.Г. и др., 2019]
В данном изобретении предложен способ, включающий детекцию трех наиболее распространенных в Якутии мутаций c.-23+1G>A, c.35delG и c.109G>A с использованием праймеров и с последующим проведением анализа полиморфизма длин рестрикционных фрагментов с использованием эндонуклеаз AsuHPI, Bsc4I, HindII.
Патент № RU 2627115 C2 «Способ одновременной диагностики наследственных заболеваний» [Саввина М.Т. и др., 2017]
В данном изобретении предложен способ одновременной диагностики наследственных заболеваний на основе использования биочипа с иммобилизованными на его поверхности олигонуклеотидными мишенями, включающий детекцию точковых в генах CUL7, NBAS, DIA1, FAH и, вызывающих 3М синдром, SOPH синдром, наследственную энзимопеническую метгемоглобинемию 1 типа, тирозинемию 1 типа и наследственную несиндромальную глухоту 1А типа, соответственно.
Патент № CN 103352080 В «Набор для определения генов при наследственной потере слуха» [Zheng Weiguo Meng Xiang et al., 2014]
Изобретение раскрывает набор, способный обнаруживать 17 несиндромальных генов предрасположенности к потере слуха. Набор состоит из 17 пар специфичных праймеров для проведения генотипирования генов восприимчивости к потере слуха и позволяет обнаружить одновременно 17 мутаций в четырех наиболее распространенных китайских генах в одной пробирке в течение 3 часов. Набор включает праймерные комбинации 17 полиморфных участков наследственной тугоухости на GJB2 (СХ26) ген, ген SLC26A4 (PDS), ген GJB3 и ген 12SrRNA (MTRNR1), они могут быть использованы для точной оценки дикого типа, типа чистого мутанта или гибридного типа из 17 сайтов, а также для диагностики и скрининга генов потери слуха. Суть предлагаемого набора заключается в обнаружении сайта полиморфизма генетического маркера третьего поколения (однонуклеотидный полиморфизм) и комбинации с техникой обнаружения, применяя флуоресценцию к несиндромальному гену наследственного нарушения слуха.
Прототипом заявляемого изобретения является способ, описанный в заявке RU 2016136727 А, ООО «ТВОЙ ГЕН», Жмырко Е.В., 2018 «Набор олигонуклеотидных праймеров, зондов и способ выявления мутации гена методом аллель-специфической ПЦР амплификации при наследственной несиндромальной глухоте». Выявление мутации гена, с использованием набора праймеров и зондов, методом аллель-специфической ПЦР амплификации при наследственной несиндромальной глухоте проводят по результатам ПЦР в режиме реального времени; при этом наличие/отсутствие гетерозиготного носительства мутации определяют по испускаемому сигналу флуоресценции, при наличии сигнала по каналу FAM генотип интерпретируется как наличие мутации гена в исследуемом образце, при наличии сигнала по каналу HEX - как норма, а при наличии двух сигналов как - гетерозиготное состояние, носительство мутации гена.
Анализ показывает, что в описанных аналогах, как в прототипе, недостатком является сравнительно невысокая достоверность результатов исследования из-за небольшой разницы между получаемыми кривыми флуоресценции для разных аллелей.
Настоящее изобретение направлено на повышение достоверности диагностики на наличие либо отсутствие гетерозиготной / гомозиготной мутации с.-23+1G>А (rs80338940) гена GJB2, определение генотипа человека в биологическом материале (цельная кровь, слюна), а также повышения доступности подобных исследований, поскольку способ может быть осуществлен на стандартном известном оборудовании.
Преимущество состоит в уникальности состава реакционной смеси, в частности в нуклеотидной последовательности праймеров и зондов для диагностики гетерозиготной / гомозиготной мутации с.-23+1G>А (rs80338940) гена GJB2 в биоматериале человека. Определение вариабельных позиций в ДНК осуществляется более надежным методом с использованием стандартного оборудования.
Предложен способ диагностики гетерозиготной и гомозиготной мутаций c.-23+1G>A (rs80338940) гена GJB2 в биоматериале человека методом ПЦР-РВ.
Использование в этих целях метода полимеразной цепной реакции (ПЦР) - является наиболее перспективным. Принцип метода ПЦР основан на использовании процесса амплификации ДНК, заключающегося в повторяющихся циклах температурной денатурации ДНК, отжига праймеров с комплементарными последовательностями и последующей достройки полинуклеотидных цепей с этих праймеров Taq-полимеразой. Основным достоинством метода ПЦР как молекулярно-биологического исследования является его чрезвычайно высокая чувствительность.
В смесь для амплификации введены сигнальные зонды, содержащие флуоресцентные метки Fam и Hex, на каждый вариант определяемого генетического полиморфизма. После окончания ПЦР проводится раунд температурного плавления дуплексов, образованных ампликонами и сигнальными зондами, в результате чего изменяется уровень флуоресценции, который фиксируется и представляется программным обеспечением прибора в виде графика. Если сигнальный зонд частично комплементарен ДНК-мишени, температура плавления такого дуплекса будет ниже температуры плавления дуплекса в случае полной комплементарности зонда. На основании температуры плавления сигнальных зондов проводится интерпретация результатов анализа.
Использование трех флуоресцентных красителей позволяет одновременно определять два аллеля и оценивать количество геномной ДНК в одной пробирке. Исследование с использованием комплектов реагентов для определения генетических полиморфизмов состоит из следующих этапов: выделение ДНК (пробоподготовка) и ПЦР-амплификация в режиме реального времени.
Работа над созданием олигонуклеотидов, которые используются в подобных наборах, строится обычно следующим образом.
1) С помощью открытых и коммерческих баз данных нуклеотидных последовательностей генов человека либо в результате самостоятельного определения нуклеотидной последовательности изучаемого гена выбирается участок гена, в котором возможен генетический полиморфизм (мутация с-23+1G>А).
2) На основании выбранного участка гена с помощью программного обеспечения подбирается последовательность олигонуклеотидов, используемых для проведения ПЦР-реакции.
3) Изготовление праймеров заказывается в сервисном центре.
4) С помощью практических экспериментов доказывается пригодность подобранных последовательностей для конкретных целей (например, для диагностики гетерозиготной / гомозиготной мутации с.-23+1G>А (rs80338940) гена GJB2 в биоматериале человека).
Предлагаемым подходом к детекции генетического полиморфизма является использование двух аллель-специфичных и меченых разными флуоресцентными метками олигонуклеотидов, а также олигонуклеотида, несущего гаситель флуоресценции и гибридизирующегося на матрицу рядом с аллель-специфичным олигонуклеотидом. Гибридизация аллель-специфичного олигонуклеотида на матрицу ведет к переносу энергии с находящегося на нем флуорофора-донора на гаситель флуоресценции расположенного рядом «гасящего» олигонуклеотида. Регистрацию результатов амплификации ведут по окончании ПЦР путем снятия спектра флуоресценции при изменении температуры реакционной смеси в диапазоне от 25 до 80 градусов по Цельсию (так называемые «кривые плавления»). При получении графиков флуоресценции возможен как нагрев, так и охлаждение реакционной смеси в указанном интервале температур. Предлагаемое изобретение делает определение вариабельных позиций в ДНК более надежным и удешевляет подобные исследования благодаря использованию стандартного оборудования.
Техническим результатом, на достижение которого направлено предлагаемое изобретение, является достоверная диагностика на наличие либо отсутствие гетерозиготной / гомозиготной мутации c.-23+1G>A (rs80338940) гена GJB2, определение генотипа человека в биологическом материале (цельная кровь, плазма крови, слюна, образцы тканей), а также повышение доступности подобных исследований, поскольку способ может быть осуществлен на стандартном известном оборудовании. Данный метод вдобавок обеспечивает возможность проводить исследование в одной пробирке, что снижает затраты на исследование.
Указанный результат достигается путем использования при постановке ПЦР набора синтетических олигонуклеотидов для диагностики гетерозиготной / гомозиготной мутации с.-23+1G>А (rs80338940) гена GJB2, вызывающей потерю слуха, включающего в себя одну пару из прямого и обратного праймеров (yakms1 и yakma1 соответственно) и два аллель-специфичных зонда соответствующие генотипу дикого типа и мутантному (yakmpt1 и yakmpt2 соответственно), а также зонд-гаситель (yakmpt3), гибридизирующийся на матрицу рядом с аллель-специфичными зондами:

где FAM - флуоресцентный краситель FAM, присоединенный к нуклеотиду А на 3'-конце пробы, HEX - флуоресцентный краситель HEX, присоединенный к нуклеотиду А на 3'-конце пробы, BHQ1 означает присоединенный к 5'-концевому нуклеотиду темновой гаситель флуоресценции.
Каждый комплект реагентов для определения генетических полиморфизмов методом полимеразной цепной реакции в режиме реального времени включает:
1) Пробирки с реакционной смесью, запаянной парафином. В данную смесь входят указанные праймеры и зонды, специфичные к определенному участку гена GJB2 на каждый вариант определяемого генетического полиморфизма, смесь дезоксирибонуклеотидтрифосфатов четырех типов, реакционный буфер (80 мМ Tris-HCl (рН 8.6 при 25°С), 20 мМ (NH4)2SO4, 3 мМ MgCl2).
И праймеры, и зонды представляют собой синтетические олигонуклеотиды. Праймеры, специфичные к маркерному участку гена, вводятся в реакцию непосредственно для амплификации (наработки) продукта. Зонды (тоже специфичные к данному участку ДНК) имеют в своем составе флуоресцентную метку, интенсивность флуоресценции которой свидетельствует о количестве образующегося продукта, что, в свою очередь, зависит от эффективности работы праймеров, количества стартового материала и других параметров реакции. Праймеры в реакции выступают в качестве компонентов, обеспечивающих прохождение реакции, а зонды - в качестве компонента, обеспечивающего наблюдение за ходом реакции.
2) Раствор фермента Taq-полимеразы.
3) Минеральное масло.
4) Отрицательный контрольный образец - образец, который вводится в эксперимент для контроля возможного загрязнения реактивов продуктами ранее проведенных реакций. Положительный результат в этом образце свидетельствует о необходимости заменить реагенты и переставить эксперимент.
Кроме того, в состав смесей для амплификации, специфичных для каждого генетического полиморфизма, включена система для амплификации фрагмента геномной ДНК человека, которая позволяет контролировать количество ДНК в амплификационной пробирке и исключить ошибки генотипирования. Система для амплификации геномной ДНК человека включает ДНК-зонд, который содержит флуоресцентную метку (Су5) и гаситель флуоресценции.
При этом смесь для амплификации специфична для каждого генетического полиморфизма. ПЦР-буфер, Taq-AT-полимераза, минеральное масло - универсальные реагенты для всех комплектов реагентов для определения генетических полиморфизмов.
Аналогичные наборы, а также реакционные смеси, праймеры для диагностики гетерозиготной / гомозиготной мутации с.-23+1G>А (rs80338940) гена GJB2 в крови и других биологических материалах человека неизвестны.
Осуществление (реализация) изобретения.
1) Биологический материал (цельная кровь, слюна) перед проведением ПЦР с помощью предлагаемого набора реагентов проводится через процедуру пробоподготовки с использованием другого набора (набор реагентов для пробоподготовки не является предметом данного патента); в ходе этой процедуры из биологического материала выделяется ДНК, которую, в свою очередь, используют для ПЦР;
2) Необходимое количество пробирок с подготовленной реакционной смесью, содержащей специфичные синтетические олигонуклеотидные праймеры и зонды, маркируется согласно количеству анализируемых образцов;
3) Во все промаркированные пробирки, не повреждая слой парафина, добавляется раствор Taq-полимеразы;
4) Во все промаркированные пробирки (кроме пробирок К-(отрицательный контрольный образец), вносится выделенная согласно п. 1) ДНК;
5) В пробирку, маркированную К-, вносится отрицательный контрольный образец;
6) Все пробирки устанавливаются в блок термоциклера, амплификация проводится согласно режимам, прописанным в инструкции к набору. Детекция результатов осуществляется детектирующим термоциклером автоматически во время амплификации (устройства: детектирующие термоциклеры ДТ-322, ДТ-96 (ООО «НПО ДНК-Технология»).
Анализ результатов проводится в соответствии с инструкцией к прибору.
При образовании специфичного продукта ДНК-зонд разрушается, действие гасителя на флуоресцентную метку прекращается, что ведет к возрастанию уровня флуоресценции. Количество разрушенных зондов (а, следовательно, и уровень флуоресценции) возрастает пропорционально количеству образовавшихся специфических ампликонов и измеряется на каждом цикле амплификации.
Пробирки (приготовленные как описано выше) устанавливали в детектирующий амплификатор ДТ-прайм (ООО «НПО ДНК-Технология», Москва) и проводили реакцию со следующими параметрами циклирования:
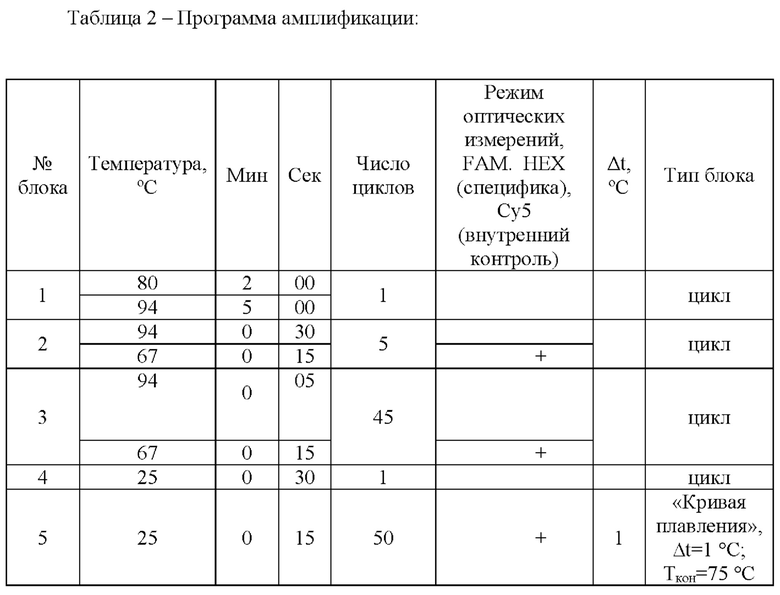

Использованный нами способ генотипирования является вариантом классического метода «примыкающих проб». При определении замен одиночных нуклеотидов вначале проводили ПЦР с праймерами, общими для обоих вариантов последовательности, затем температуру реакционной смеси снижали для гибридизации полученной матрицы с олигонуклеотидными пробами. Для определения варианта последовательности использовали два типа олигонуклеотидов, гибридизирующихся на матрицу рядом. Один из олигонуклеотидов метили флуорофором, другой - гасителем флуоресценции. В реакции использовали один общий олигоиуклеотид с гасителем флуоресценции и пару аллель-специфичных (сиквенс-специфичных) олигонуклеотидов, несущих флуорофор. Олигонуклеотидные пробы, соответствующие тому или иному варианту последовательности, метили различными флуорофорами, что позволило определять оба варианта в одной пробирке. Определение генотипа проводили после ПЦР и гибридизации путем измерения уровня флуоресценции в ходе температурной денатурации дуплексов олигонуклеотидов и полученных матриц (или наоборот -гибридизации). Данное измерение проводили в режиме реального времени, его результатом являлись кривые плавления. Условия реакции подбирали так, чтобы максимизировать разницу в температурах плавления совершенного и несовершенного дуплексов. Таким образом, если анализируемый образец содержал только один вариант последовательности, т.е. был гомозиготен по данному полиморфизму, температура плавления для пробы, образующей совершенный дуплекс, была существенно выше, нежели для пробы, образующей несовершенный дуплекс. Если же анализировали гетерозиготный образец, температуры плавления были практически одинаковы.
На основании экспериментальных данных при сравнении образцов крови методом ПЦР-РВ «в реальном времени» были получены и проанализированы результаты. Детекция и учет результатов осуществляется детектирующим амплификатором автоматически. После окончания программы амплификации на экране появится соответствующее информационное сообщение и будет предложено перейти к анализу результатов.
Для отрицательных контрольных образцов программа фиксирует недостоверный результат. При получении положительного значения (определение генотипа) результаты всей постановочной серии считают недостоверными. В этом случае необходимо проведение мероприятий для устранения возможной контаминации.
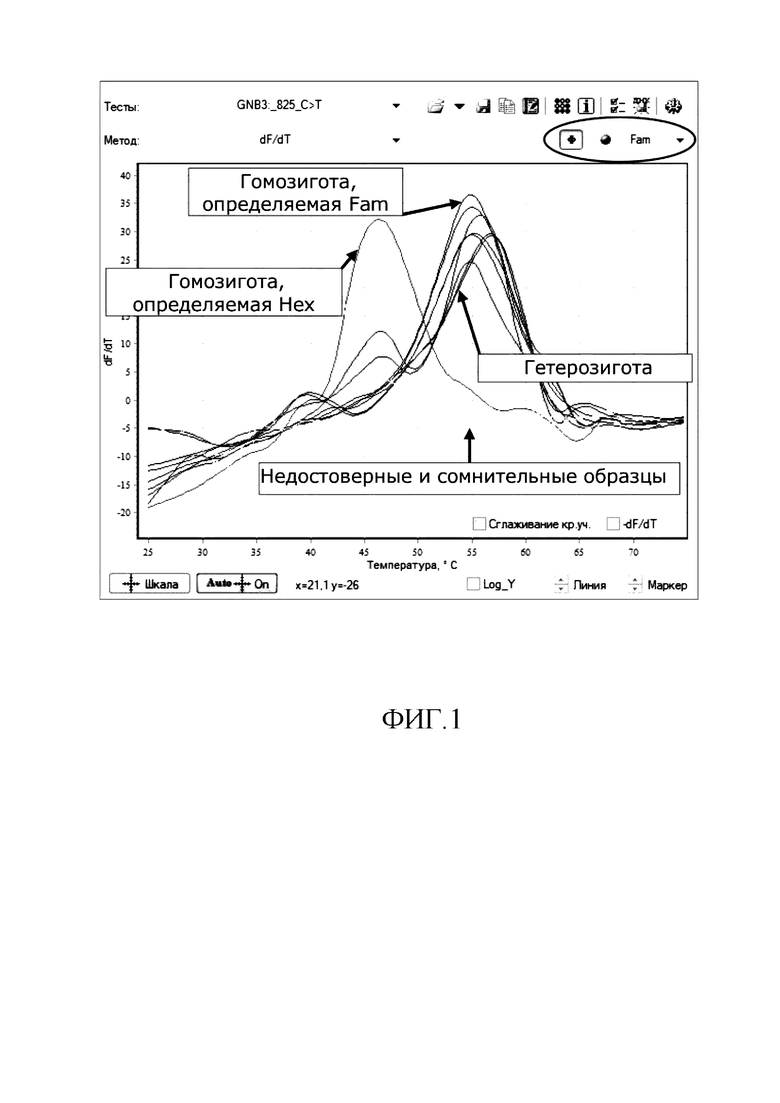
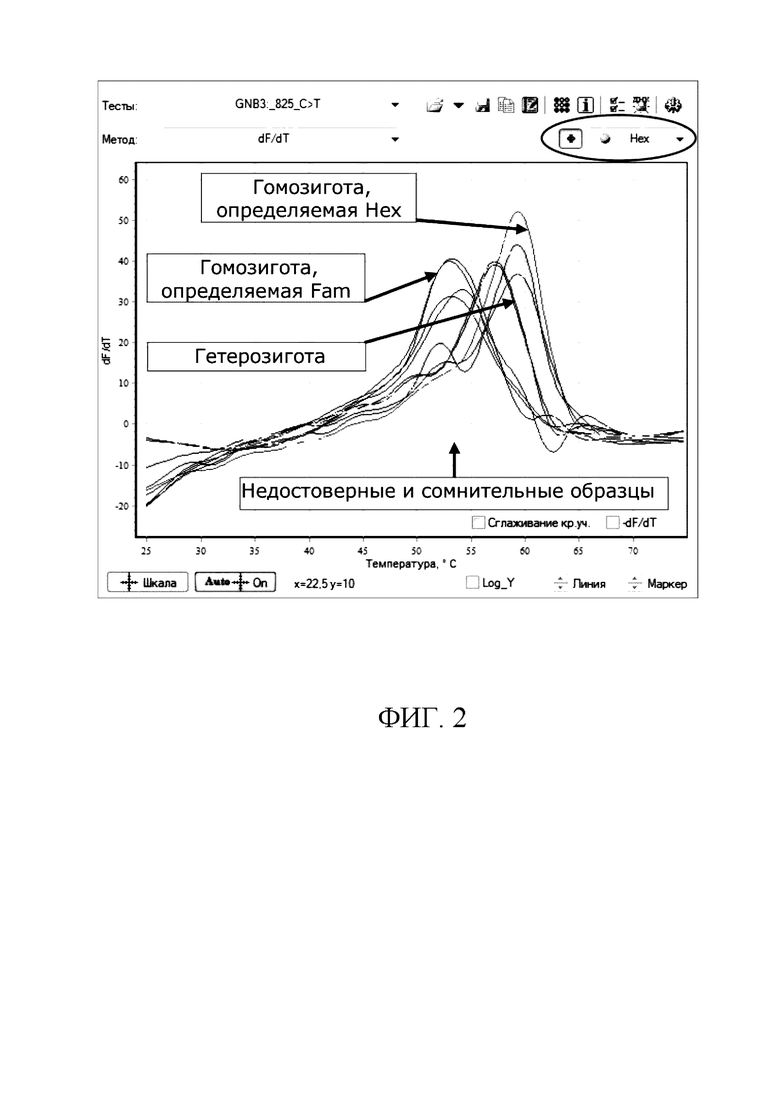
Температуры плавления продуктов амплификации отражаются на графике в виде температурных баров. Образцы, гомозиготные по полиморфизму, определяемому каналом Fam, будут автоматически покрашены в синий цвет; гомозиготные по каналу Hex - в зеленый; гетерозиготные - в красный; сомнительные и недостоверные - в желтый (см. фиг 1-2). При этом отнесение образца к гомозиготе или гетерозиготе по данному аллелю проводят по форме кривых плавления ДНК (по максимуму первой производной графиков флуоресценции); наличие одного пика на графике свидетельствует о гомозиготности образца по аллелю, соответствующему более тугоплавкой флуоресцентно-меченой аллель-специфичной олигонуклеотидной пробе, а наличие двух пиков на графике свидетельствует о гетерозиготности образца.
На фиг. 1 представлен пример интерпретации результатов (канал Fam).
На фиг. 2 представлен пример интерпретации результатов (канал Hex).
Список использованных источников
1. Vos, Т.; Allen, С; Arora, М.; Barber, R.M.; Bhutta, Z.A.; Brown, А.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545-1602
2. Morton CC: Genetics, genomics and gene discovery in the auditory system. Hum Mol Genet. 11:1229-1240. 2002.
3. Wu, X.; Wang, S.; Chen, S.; Wen, Y.; Liu, В.; Xie, W.; Li, D.; Liu, L.; Huang, X.; Sun, Y.; et al. Autosomal Recessive Congenital Sensorineural Hearing Loss due to a Novel Compound Heterozygous PTPRQ Mutation in a Chinese Family. Neural Plast. 2018, 2018, 9425725.
4. Adhikary, В.; Ghosh, S.; Paul, S.; Bankura, В.; Pattanayak, A.K.; Biswas, S.; Maity, В.; Das, M. Spectrum and frequency of GJB2, GJB6 and SLC26A4 gene mutations among nonsyndromic hearing loss patients in eastern part of India. Gene 2015,573,239-245.
5. Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124,E34-E53
6. Hilgert N, Smith RJ and Campa G: Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics. Mutat Res. 681:189-196. 2009.
7. Kenneson A, Van Naarden Braun К and Boyle C: GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. 4:258-274. 2002.
8. Estivill X, Fortina P, Surrey S, et al: Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 351:394-398. 1998.
9. Morell RJ, Kim HJ, Hood LJ, et al: Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 339:1500-1505. 1998.
10. Abe S, Usami S, Shinkawa H, Kelley PM and Kimberling WJ: Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 37:41-43. 2000.
11. Sobe T, Vreugde S, Shahin H, et al: The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum Genet. 106:50-57. 2000.
12. Dai P, Yu F, Han B, et al: GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 7:262009.
13. Stenson PD, Mort M, Ball EV, Evans K, Hay den M, Heywood S, Hussain M, Phillips AD, Cooper DN. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665-77.
14. Azadegan-Dehkordi F., Ahmadi R., Koohiyan M., Hashemzadeh-Chaleshtori M. Update of spectrum c.35delG and c.-23+1G>A mutations on the GJB2 gene in individuals with autosomal recessive nonsyndromic hearing loss. Ann Hum Genet. 2019;83(1):1-10. doi: 10.1111/ahg. 12284.
15. Denoyelle, F., Marlin, S., Weil, D., Moatti, L., Chauvin, P., Garabedian, E. N., & Petit, C. (1999).Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: Implications for genetic counselling. Lancet, 353 (9161): 1298-1303.
16. A1-Qahtani, M. H., Baghlab, I., Chaudhary, A. G., Abuzenadah, A. M., et al (2010). Spectrum of GJB2 mutations in a cohort of nonsyndromic hearing loss cases from the Kingdom of Saudi Arabia. Genetic Testing and Molecular Biomarkers, 14(1), 79-83.
17. Najmabadi, H., & Kahrizi, K. (2014). Genetics of non-syndromic hearing loss in the Middle East. International Journal of Pediatric Otorhinolaryngology, 78 (12), 2026-2036.
18. Matos, T.D., Simoes-Teixeira, H., Caria, H., Cascao, R., et al (2011). Assessing noncoding sequence variants of GJB2 for hearing loss association. Genetics Research International, 2011, 827469.
19. Janecke, A. R., Hirst-Stadlmann, A., Gunther, В., Utermann, В., et al (2002). Progressive hearing loss, and recurrent sudden sensorineural hearing loss associated with GJB2 mutations-Phenotypic spectrum and frequencies of GJB2 mutations in Austria. Human Mutation, 111 (2), 145-153.
20. Angeli, S. I. (2008). Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope, 118 (11), 2014-2023.
21. Riahi, Z., Zainine, R., Mellouli, Y., Hannachi, R., Bouyacoub, et al (2013).Compound heterozygosity for dominant and recessive GJB2 mutations in a Tunisian family and association with successful cochlear implant outcome. International Journal of Pediatric Otorhinolaryngology, 77 (9), 1481-1484.
22. Medica, I., Rudolf, G., Balaban, M., & Peterlm, B. (2005). C.35delG/GJB2 and del(GJB6-D13S1830) mutations in Croatians with prelingual non-syndromic hearing impairment. BMC Ear, Nose and Throat Disorders, 5,11.
23. Barashkov, N. A., Pshennikova, V. G., Posukh, O. L., Teryutin, et al (2016). Spectrum and frequency of the GJB2 gene pathogenic variants in a large cohort of patients with hearing impairment living in a subarctic region of Russia (the Sakha Republic). PLoS One, 11(5), e0156300.
24. Teryutin, F. M., Barashkov, N. A., Kunel'skaya, N. L., Pshennikova, V.G., & Solov'ev, A.V. (2016). The audiological analysis in the patients homozygous for the c.-23+1G>A mutation in the GJB2 gene presenting with the loss of hearing in Yakutiya. Vestn Otorinolaringol, 81 (1), 19-24.
25. Yuan, Y., Yu, F., Wang, G., Huang, S., Yu, R., et al (2010). Prevalence of the GJB2 IVS1+1G>A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. Journal of Translational Medicine, 8, 127.
26. Seeman, P., & Sakmaryova, I. (2006). High prevalence of the IVS 1+1 G to A/GJB2 mutation among Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2. Clinical Genetics, 69(5), 410-413.
27. Zeinali, S., Davoudi-Dehaghani, E., Azadmehr, S., DabbaghBagheri, et al (2015). GJB2 c.-23+1G>A mutation is second most common mutation among Iranian individuals with autosomal recessive hearing loss. European Archives of Otorhinolaryngology, 272 (9), 2255-2259.
28. M.V. Zytsar, N.A. Barashkov, M.S. Bady-Khoo, O.A. Shubina-Olejnik et al. Updated carrier rates for c.35delG (GJB2) associated with hearing loss in Russia and common c.35delG haplotypes in Siberia BMC Medical Genetics volume 19, Article number: 138 (2018)
29. Kelley P.M., Harris D.J., Comer B.C. et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss // American Journal of Human Genetics. - 1998. V. 62. P. 792-799
30. Orita M., Iwahana H., Kanazawa H., Sekya T. Detection of polymorphism of human DNA by gel electrophoresis as single cell conformation polymorphism // Protocols Natl. Acad. Sci. - 1989. - V. 86. - P. 2766-2770
31. Baris I., Koksal V. et al. Myltiplex deletion of common mutations in the connexin-26 gene, Genetic Testing, 2003, v. 7, №1, pp. 63-65
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ диагностики мутации 35delG (rs80338939) гена GJB2 | 2020 |
|
RU2739889C1 |
| Способ диагностики мутации 167delT (rs80338942) гена GJB2 | 2020 |
|
RU2739943C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ GJB2, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2006 |
|
RU2317547C1 |
| СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ | 2010 |
|
RU2448163C2 |
| Способ дифференциальной и подтверждающей молекулярно-генетической диагностики нейросенсорной тугоухости в популяции чувашей | 2021 |
|
RU2768033C1 |
| Способ выявления мутаций гена GJB2, обуславливающих аутосомно-рецессивную глухоту 1А типа | 2017 |
|
RU2688180C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИИ с.-53-2А>G В ГЕНЕ ПРЕСТИНА (SLC26A5), ВЫЗЫВАЮЩЕЙ РАЗВИТИЕ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2012 |
|
RU2505608C1 |
| Способ ДНК-диагностики аутосомно-рецессивной глухоты-103 | 2019 |
|
RU2727684C1 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ MYO7A, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ И СИНДРОМОМ УШЕРА | 2013 |
|
RU2555755C1 |
| Способ редактирования гена GJB2 для исправления патогенного варианта c.del35G в клетках человека, культивируемых in vitro | 2021 |
|
RU2780677C1 |
Изобретение относится к области биотехнологии, а именно к молекулярной биологии, медицинской генетике и оториноларингологии. Раскрыты синтетические олигонуклеотиды для диагностики (генотипирования) мутации с.-23+1G>А (rs80338940) гена GJB2 в биоматериале человека. Праймеры объединены в наборы для выявления ДНК в крови и других биологических материалах методом полимеразной цепной реакции. Изобретение позволяет высокочувствительно проводить диагностику врожденной наследственной глухоты человека в биологическом материале. Использование данного метода обеспечивает также высокочувствительное и объективное описание генотипа человека с определением гомозиготного и гетерозиготного носительства данной мутации. 2 ил., 2 табл.
Способ диагностики мутации с.-23+1G>А (rs80338940) гена GJB2 в биоматериале человека, выбранном из цельной крови, слюны, основанный на полимеразной цепной реакции в режиме реального времени (ПЦР-РВ), отличающийся тем, что при проведении ПЦР-РВ используют праймеры и зонды, соответствующие полиморфному участку rs80338940 гена GJB2:

где FAM - флуоресцентный краситель FAM, присоединенный к нуклеотиду А на 3'-конце пробы, HEX - флуоресцентный краситель HEX, присоединенный к нуклеотиду А на 3'-конце пробы, a BHQ1 означает присоединенный к 5'-концевому нуклеотиду темновой гаситель флуоресценции, на основании полученных графиков кривых плавления ДНК определяют мутацию, при этом при наличии одного пика на графике диагностируют гомозиготное носительство, а при наличии двух пиков - гетерозиготное носительство.
| RU 2016136727 A, 16.03.2018 | |||
| Способ одновременной диагностики наследственных заболеваний | 2015 |
|
RU2627115C2 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В ГЕНЕ GJB2, СОПРОВОЖДАЮЩИХСЯ РАЗВИТИЕМ НЕСИНДРОМАЛЬНОЙ АУТОСОМНО-РЕЦЕССИВНОЙ ГЛУХОТЫ | 2006 |
|
RU2317547C1 |
| MATOS, T.D., et al, Assessing noncoding sequence variants of GJB2 for hearing loss association, Genetics Research International, 2011, 827469. | |||

