Область техники, к которой относится изобретение
Предложены способы получения соединений 2-(1-фенилэтил)изоиндолин-1-она, полезных для снижения уровней или активности фактора некроза опухолей α у млекопитающих. Такие соединения 2-(1-фенилэтил)изоиндолин-1-она включают 7-нитро-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он, 7-амино-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он и циклопропил-N-{2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]-3-оксоиндолин-4-ил}карбоксамид.
Предшествующий уровень техники
Фактором некроза опухолей α, или TNF-α, является цитокин, который выделяется, прежде всего, одноядерными фагоцитами в ответ на многие иммуностимуляторы. При введении животным или людям он может вызывать воспаление, лихорадку, действие на сердечно-сосудистую систему, кровоизлияние, коагуляцию и острофазные ответы, подобно наблюдаемым во время острых инфекций и шоковых состояний. Чрезмерное или нерегулируемое продуцирование TNF-α таким образом вовлечено в ряд болезненных состояний. Эти болезненные состояния включают эндотоксикоз и/или синдром токсического шока (Tracey et al., Nature 330, 662-664 (1987) и Hinshaw et al., Circ. Shock 30, 279-292 (1990)); ревматический артрит, болезнь Крона, кахесию (Dezube et al., Lancet, 335 (8690), 662 (1990)) и синдром расстройства дыхания у взрослых (ARDS), при которых TNF-α в концентрации, превышающей 12000 пг/мл был обнаружен в легочной жидкости, полученной путем аспирации у ARDS пациентов (Millar etal., Lancet 2 (8665), 712-714 (1989)). Системное вливание рекомбинантного TNF-α также приводит к изменениям, обычно наблюдаемым при ARDS (Ferrai-Baliviera et al., Arch. Surg. 124 (12), 1400-1405 (1989)). Некоторые соединения 2-(1-фенилэтил)изоиндолин-1-она показаны в литературе как снижающие уровни TNF-α, такой как патент США 6667316 и 6020358 и публикации патентных заявок США № 2004/0254214 и 2004/0204448, все из которых включены в настоящее описание во всей свой полноте посредством ссылки.
Существующие способы синтеза соединений 2-(1-фенилэтил)изоиндолин-1-она описаны в патентах США 6667316 и 6020358 и публикациях патентных заявок США № 2004/0254214 и 2004/0204448. В то время как эти способы полезны при получении соединений 2-(1-фенилэтил)изоиндолин-1-она, желательны альтернативные способы получения соединений 2-(1-фенилэтил)изоиндолин-1-она, в частности, для получения в промышленном масштабе.
Цитирование любой ссылки в настоящем описании не должно рассматриваться в качестве допущения, что такая ссылка является прототипом по отношению к настоящему изобретению.
Сущность изобретения
Предложены эффективные способы получения соединений 2-(1-фенилэтил)изоиндолин-1-она, таких как 7-нитро-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он, 7-амино-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он и циклопропил-N-{2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]-3-оксоиндолин-4-ил}карбоксамид.
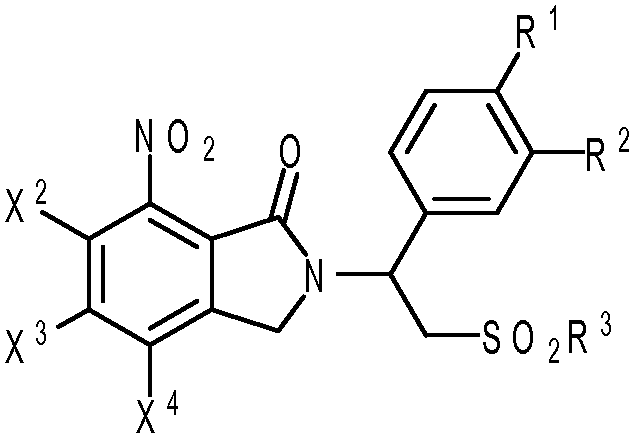
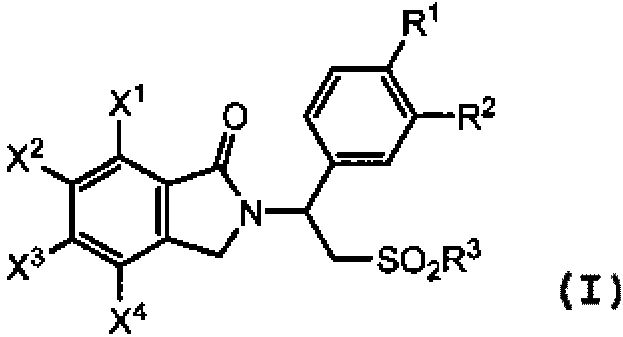
В одном аспекте предложен способ получения соединения изоиндолин-1-она формулы (I):
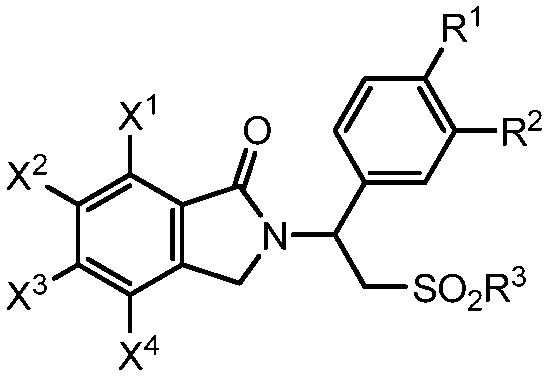 (I)
(I)
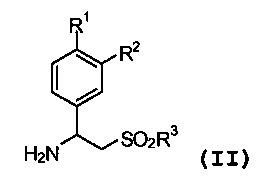
или его фармацевтически приемлемой соли, или его сольвата или полиморфа, включающий стадию взаимодействия первичного амина формулы (II):
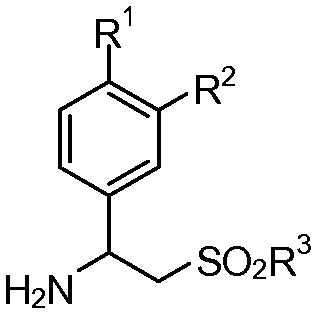 (II)
(II)
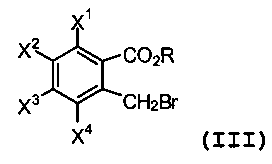
или его соли с эфиром 2-(бромметил)бензойной кислоты формулы (III):
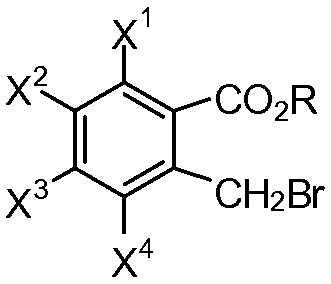 (III)
(III)
в присутствии неорганического основания, где
R представляет собой алкил или арил;
каждый из R1 и R2 независимо представляет собой водород, алкил из 1-4 атомов углерода, алкоксигруппу из 1-4 атомов углерода, цианогруппу или циклоалкоксигруппу из 3-18 атомов углерода;
R3 представляют собой гидроксигруппу, алкил из 1-8 атомов углерода, фенил, бензил или NR4R5;
каждый из X1, X2, X3 и X4 независимо представляет собой водород, галоген, алкил из 1-4 атомов углерода, алкоксигруппу из 1-4 атомов углерода, нитрогруппу, цианогруппу, гидроксигруппу или -NR4'R5'; или любые два из X1, X2, X3 и X4 при смежных атомах углерода вместе с изображенным фениленовым кольцом представляют собой нафтилиден;
каждый из R4 и R5 независимо представляет собой водород, алкил из 1-8 атомов углерода, фенил или бензил; или один из R4 и R5 представляет собой водород, а другой представляют собой -COR6 или -SO2R6; или R4 и R5, взятые вместе, представляют собой тетраметилен, пентаметилен, гексаметилен или -CH2CH2X5CH2CH2-, где X5 представляет собой -O-, -S- или -NH-;
каждый из R4' и R5' независимо представляет собой водород, алкил из 1-8 атомов углерода, фенил или бензил; или один из R4' и R5' представляет собой водород, а другой представляет собой -COR6' или -SO2R6'; или R4' и R5', взятые вместе, представляют собой тетраметилен, пентаметилен, гексаметилен или -CH2CH2X5'CH2CH2-, где X5' представляет собой -О-, -S- или -NH-; и
каждый из R6 и R6' независимо представляет собой водород, алкил из 1-8 атомов углерода, циклоалкил из 3-8 атомов углерода или фенил.
В некоторых вариантах осуществления каждый из R1 и R2 в формуле (I) независимо представляет собой алкоксигруппу из 1-4 атомов углерода. В частном варианте осуществления R1 представляет собой метоксигруппу, и R2 представляет собой этоксигруппу. В других вариантах осуществления каждый из X2 X3 и X4 в формуле (I) представляет собой водород; и X1 представляет собой нитрогруппу, -NH2 или -NHCOR6', где R6' представляет собой водород, алкил из 1-8 атомов углерода, циклоалкил из 3-8 атомов углерода или фенил. В одном варианте осуществления X1 представляет собой -NHCOR6', и R6' представляет собой циклопропил. В некоторых вариантах осуществления R3 представляет собой метил. В другом варианте осуществления R представляет собой метил.
В частном варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой 7-нитро-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он, где X1 представляет собой нитрогруппу; каждый из X2, X3 и X4 представляет собой водород; R3 представляет собой метил; R1 метоксигруппу; и R2 представляют собой этоксигруппу.
В другом варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой 7-амино-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он, где X1 представляет собой -NH2; каждый из X2, X3 и X4 представляет собой водород; R3 представляет собой метил; R1 представляет собой метоксигруппу; и R2 представляет собой этоксигруппу.
В другом варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой циклопропил-N-{2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]-3-оксоиндолин-4-ил}карбоксамид, где X1 представляет собой NHCO-циклопропил; каждый из X2, X3 и X4 представляет собой водород; R3 представляет собой метил; R1 представляет собой метоксигруппу; и R2 представляет собой этоксигруппу.
Подробное описание изобретения
Определения
Как использовано в настоящем описании и если не указано иное, термины "гало", "галоген" или подобные означают -F, -Cl, -Br или -I.
Как использовано в настоящем описании и если не указано иное, термины "алкил" или "алкильная группа" означают одновалентную насыщенную разветвленную или неразветвленную углеводородную цепь, содержащую от 1 до 8 атомов углерода. Неограничивающими примерами таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, гексил, гептил и октил. Алкильная группа может быть незамещенной или замещена одним или более подходящими заместителями.
Как использовано в настоящем описании и если не указано иное, термины "алкокси" или "алкоксигруппа" означают алкильную группу, связанную с остатком молекулы через эфирный атом кислорода. Неограничивающими примерами таких алкоксигрупп являются метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси и трет-бутоксигруппа. Алкоксигруппа может быть незамещенной или замещена одним или более подходящими заместителями.
Как использовано в настоящем описании и если не указано иное, термины "циклоалкил" или "циклоалкильная группа" означают одновалентную циклическую углеводородную цепь, которая может насыщенной или ненасыщенной. Если не указано иное, такие цепи могут содержать от 3 до 18 атомов углерода и включают моноциклоалкильные, полициклоалкильные и бензоциклоалкильные структуры. Моноциклоалкильная структура относится к группам, содержащим одну кольцевую группу. Полициклоалкильная структура означает углеводородные системы, содержащие две или более кольцевых систем с одним или более общими кольцевыми атомами углерода; т.е. спиро, конденсированные или мостиковые структуры. Бензоциклоалкил означает моноциклоалкильную группу, конденсированную с бензогруппой. Неограничивающими примерами моноциклоалкильных групп являются циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил, циклотридецил, циклотетрадецил, циклопентадецил, циклогексадецил, циклогептадецил и циклооктадецил. Неограничивающие примеры полициклоалкила включают декагидронафталин, спиро[4.5]децил, бицикло[2.2.1]гептил, бицикло[3.2.1]октил, пинанил, норбомил и бицикло[2.2.2]октил. Неограничивающие примеры бензоциклоалкила включают тетрагидронафтил, инданил и 1,2-бензоциклогептанил. Циклоалкильная группа может быть незамещенной или замещена одним или более подходящими заместителями.
Как использовано в настоящем описании и если не указано иное, термины "циклоалкокси" или "циклоалкоксигруппа" означают циклоалкильную группу, как описано выше, которая представляет собой моноциклоалкильную, полициклоалкильную или бензоциклоалкильную структуру, связанную с остатком молекулы через эфирный атом кислорода. Циклоалкоксигруппа может быть незамещенной или замещена одним или более подходящими заместителями.
Как использовано в настоящем описании и если не указано иное, термин "замещенный", используемый для описания соединения или химического фрагмента, означает, что по меньшей мере один водородный атом такого соединения или химического фрагмента заменен вторым химическим фрагментом. Вторым химическим фрагментом может быть любой подходящий заместитель, который не аннулирует синтетическую или фармацевтическую пригодность предложенных в настоящем изобретении соединений или промежуточных соединений, полезных для их получения. Примеры подходящих заместителей включают, но не ограничиваются ими: алкил; алкенил; алкинил; арил; циклоалкил; алкоксигруппу; CN; OH; галоген, C(O)OH; COгалоген; O(СО)галоген; CF3, N3; NO2, NH2; NH(алкил); N(алкил)2; NH(арил); N(арил)2; (СО)NH2; (СО)NH(алкил); (СО)N(алкил)2; (СО)NH(арил) и (СО)N(арил)2. Специалист в данной области сможет легко выбрать подходящий заместитель на основе стабильности и фармакологической и синтетической активности соединений, предложенных в настоящем изобретении.
Как использовано в настоящем описании и если не указано иное, композиция, которая "по существу не содержит" соединения, означает, что композиция содержит приблизительно менее чем 20% масс., более предпочтительно, приблизительно менее чем 10% масс., еще более предпочтительно, приблизительно менее чем 5% масс., и наиболее предпочтительно, приблизительно менее чем 3% масс. данного соединения.
Как использовано в данном описании и если не указано иное, термин "стереохимически чистый" означает композицию, которая содержит один стереоизомер соединения и по существу не содержит других стереоизомеров этого соединения. Например, стереомерно чистая композиция соединения, имеющего один хиральный центр, не будет по существу содержать противоположного энантиомера этого соединения. Стереомерно чистая композиция соединения, имеющего два хиральных центра, не будет по существу содержать другого диастереомера этого соединения. Конкретное стереомерно чистое соединение содержит приблизительно более 80% масс. одного стереоизомера соединения и приблизительно менее 20% масс. других стереоизомеров данного соединения, более предпочтительно, приблизительно более 90% масс. одного стереоизомера соединения и приблизительно менее 10% масс. других стереоизомеров данного соединения, еще более предпочтительно, приблизительно более 95% масс. одного стереоизомера соединения и приблизительно менее 5% масс. других стереоизомеров данного соединения, и наиболее предпочтительно, приблизительно более 97% масс. одного стереоизомера соединения и приблизительно менее 3% масс. других стереоизомеров данного соединения.
Как использовано в данном описании и если не указано иное, термин "энантиомерно чистый" означает стереомерно чистую композицию соединения, имеющего один хиральный центр.
Как использовано в настоящем описании и если не указано иное, термины "рацемический" или "рацемат" означают приблизительно 50% одного энантиомера и приблизительно 50% соответствующего энантиомера относительно всех хиральных центров в молекуле. Соединения, предложенные в настоящем изобретении, охватывают все энантиомерно чистые, энантиомерно обогащенные, диастереомерно чистые, диастереомерно обогащенные и рацемические смеси данных соединений.
Как использовано в настоящем описании и если не указано иное, термины "способ(ы) получения" или "способ(ы) для получения" относятся к способам, раскрытым в настоящем изобретении, которые полезны при получении соединения, предложенного в настоящем изобретении. Модификации к способам (например, исходные вещества, реагенты, защитные группы, растворители, температуры, время реакций, очистка), раскрытым в данном описании, также охвачены настоящим изобретением.
Как использовано в настоящем описании и если не указано иное, термины "добавление", "взаимодействие" или подобные означают приведение в контакт одного реактанта, реагента, растворителя, катализатора, реакционноспособной группы или подобного с другим реактантом, реагентом, растворителем, катализатором, реакционноспособной группой или подобным. Реактанты, реагенты, растворители, катализаторы, реакционноспособная группа или подобное могут быть добавлены индивидуально, одновременно или по отдельности и могут быть добавлены в любом порядке. Они могут быть добавлены в присутствии или отсутствии нагревания и необязательно могут быть добавлены в инертной атмосфере. "Взаимодействие" может относиться к образованию in situ или внутримолекулярной реакции, где реакционноспособные группы находятся в одной и той же молекуле.
Как использовано в данном описании и если не указано иное, реакция, которая "по существу завершена" или подходит "по существу к завершению" означает, что реакция содержит приблизительно более 80% по процентному выходу, более предпочтительно, приблизительно более 90% по процентному выходу, еще более предпочтительно, приблизительно более 95% по процентному выходу, и наиболее предпочтительно, приблизительно более 97% по процентному выходу желаемого продукта реакции.
Как использовано в настоящем описании и если не указано иное, термин "фармацевтически приемлемая соль" включает, но, не ограничиваясь ими, соли кислых или основных групп, которые могут присутствовать в соединениях, предложенных в настоящем изобретении. Соединения, предложенные в настоящем изобретении, которые являются основными по природе, способны образовывать большое разнообразие солей с различными неорганическими и органическими кислотами. Кислоты, которые можно использовать при получении фармацевтически приемлемых солей таких основных соединений, являются такими, которые образуют соли, включающие фармакологически приемлемые анионы, включая, но, не ограничиваясь ими, ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, эдетат кальция, камсилат, карбонат, хлорид, бромид, йодид, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эсилат, фумарат, глюцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидроксинафтоат, изэтионат, лактат, лактобионат, малат, малеат, манделат, мезилат, метилсульфат, мускат, напсилат, нитрат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сукцинат, сульфат, таннат, тартрат, теоклат, триэтиодид и памоат. Соединения, предложенные в настоящем изобретении, которые включают аминогруппу, также могут образовывать фармацевтически приемлемые соли с различными аминокислотами, в дополнение к указанным выше кислотам. Соединения, предложенные в настоящем изобретении, которые являются кислыми по природе, способны образовывать основные соли с различными фармакологически приемлемыми катионами. Неограничивающие примеры таких солей включают соли щелочных металлов или щелочно-земельных металлов и, в частности, соли кальция, магния, натрия, лития, цинка, калия и железа.
Как использовано в настоящем описании и если не указано иное, термин "гидрат" означает соединение, предложенное в настоящем изобретении, или его соль, которые дополнительно включают стехиометрическое или нестехиометрическое количество воды, связанной нековалентными силами межмолекулярного взаимодействия.
Как использовано в настоящем описании и если не указано иное, термин "сольват" означает сольват, образованный ассоциацией одной или более молекул растворителя с соединением, предложенным в настоящем изобретении. Термин "сольват" включает гидраты (например, моногидрат, дигидрат, тригидрат, тетрагидрат и т.п.).
Как использовано в настоящем описании и если не указано иное, термин "полиморф" означает твердые кристаллические формы соединения, предложенного в настоящем изобретении, или его комплекс. Различные полиморфы одного и того же соединения могут проявлять различные физические, химические и/или спектроскопические свойства.
Как использовано в настоящем описании и если не указано иное, фраза "заболевания или состояния, связанные с аномально высоким уровнем или активностью TNF-α" означает заболевания или состояния, которые не будут возникать, переносить или вызвать симптомы, если уровень или активность TNF-α будет ниже, или заболевания или состояния, которые могут быть предотвращены или вылечены путем понижения уровня или активности TNF-α.
Как использовано в настоящем описании и если не указано иное, термины "лечить" и "лечение" предусматривают действие, необходимое тогда, когда пациент страдает указанным заболеванием или нарушением, которое уменьшает тяжесть заболевания или симптомы заболевания или нарушения или замедляет прогрессирование симптомов заболевания или нарушения.
Акронимы или символы для групп или реагентов имеют следующие определения: ВЭЖХ = высокоэффективная жидкостная хроматография, CH3CN = ацетонитрил; ДМФА = диметилформамид, ДМСО = диметилсульфоксид, ТГФ = тетрагидрофуран, CH3OAc = метилацетат, EtOAc = этилацетат, AIBN = 2,2'-азобисизобутиронитрил, DBH = 1,3-дибром-5,5-диметилгидантоин и DIPEA = N,N-диизопропилэтиламин.
Если есть несоответствие между изображенной структурой и названием, данным этой структуре, изображенная структура должна иметь большее значение. Кроме того, если стереохимия структуры или ее части не обозначена, например, жирными или пунктирными линиями, структура или ее часть должны быть интерпретированы, как охватывающие любые ее стереоизомеры.
Способы, предложенные в настоящем изобретении, могут быть поняты более полно путем следующего подробного описания и иллюстративных примеров, которые предназначены для иллюстрации неограничивающих вариантов осуществления настоящего изобретения.
Способы
В настоящем изобретении предложены способы получения соединений 2-(1-фенилэтил)изоиндолин-1-она. В конкретных вариантах осуществления способы, предложенные в настоящем изобретении, охватывают эффективные средства для крупномасштабного или коммерческого получения соединений 2-(1-фенилэтил)изоиндолин-1-она.
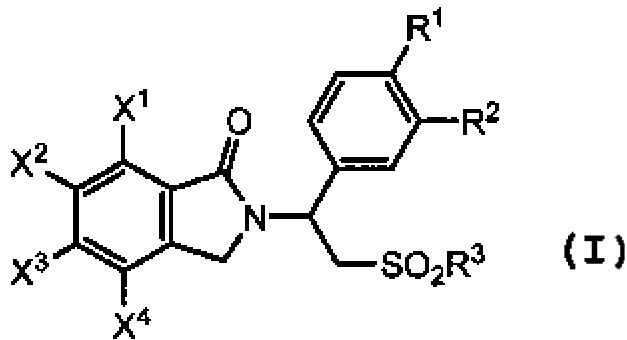
В некоторых вариантах осуществления настоящего изобретения предложены способы получения соединений изоиндолин-1-она формулы (I):
(I)
или их фармацевтически приемлемых солей, сольватов, включая гидраты, или их полиморфы, включающие стадию взаимодействия первичного амина формулы (II):
(II)
или его соли с эфиром 2-(бромметил)бензойной кислоты формулы (III):
(III)
в присутствии неорганического основания,
где R представляет собой алкил или арил;
каждый из R1 и R2 независимо представляет собой водород, алкил из 1-4 атомов углерода, алкоксигруппу из 1-4 атомов углерода, цианогруппу или циклоалкоксигруппу из 3-18 атомов углерода;
R3 представляет собой гидроксигруппу, алкил из 1-8 атомов углерода, фенил, бензил или NR4R5;
каждый из X1, X2, X3 и X4 независимо представляет собой водород, галоген, алкил из 1-4 атомов углерода, алкоксигруппу из 1-4 атомов углерода, нитрогруппу, цианогруппу, гидроксигруппу или -NR4'R5'; или любые два из X1, X2, X3 и X4 при смежных атомах углерода вместе с изображенным фениленовым кольцом образуют нафтилиден;
каждый из R4 и R5 независимо представляет собой водород, алкил из 1-8 атомов углерода, фенил или бензил; или один из R4 и R5 представляет собой водород, а другой представляет собой -COR6, или -SO2R6; или R4 и R5, взятые вместе, представляют собой тетраметилен, пентаметилен, гексаметилен или -CH2CH2X5CH2CH2-, где X5 представляет собой -O-, -S- или -NH-;
каждый из R4' и R5' независимо представляет собой водород, алкил из 1-8 атомов углерода, фенил или бензил; или один из R4' и R5' представляет собой водород, а другой представляет собой -COR6' или -SO2R6'; или R4' и R5', взятые вместе, представляют собой тетраметилен, пентаметилен, гексаметилен или -CH2CH2X5'CH2CH2-, где X5' представляет собой -O-, -S- или -NH-; и
каждый из R6 и R6' независимо представляет собой водород, алкил из 1-8 атомов углерода, циклоалкил из 3-8 атомов углерода или фенил.
Может быть использовано любое неорганическое основание, которое может катализировать или поддерживать реакцию нуклеофильного замещения между бензилбромидом и первичным амином, такую как начальная реакция между соединением формулы (II) и соединением формулы (III). Неограничивающие примеры подходящих неорганических оснований включают гидроксиды металлов, такие как гидроксид калия и гидроксид натрия, карбонаты металлов, такие как карбонат калия и карбонат натрия, гидрокарбонаты металлов, такие как гидрокарбонат натрия и гидрокарбонат калия, гидриды металлов и их комбинации. В одном варианте осуществления неорганическое основание представляет собой гидрокарбонат натрия. В другом варианте осуществления неорганическое основание представляет собой карбонат калия. Мольное отношение неорганического основания к соединению формулы (I) может составлять диапазон приблизительно от 1:1 до приблизительно 3:1. В некоторых вариантах осуществления мольное отношение неорганического основания к соединению формулы (I) составляет приблизительно от 1,5:1 до приблизительно 2,5:1. В других вариантах осуществления мольное отношение неорганического основания к соединению формулы (I) составляет приблизительно от 2,0:1 до приблизительно 2,2:1.
Взаимодействие между соединением формулы (II) и соединением формулы (III) может протекать в растворителе, таком как ацетонитрил, этилацетат, кетоны, такие как ацетон и метилэтилкетон, эфиры, такие как диэтиловый эфир и тетрагидрофуран, дихлорметан, хлороформ, N-метилпирролидинон, диметилформамид, диметилсульфоксид и их комбинации. Как правило, выбор подходящего растворителя может быть основан на многих факторах, таких как растворимость неорганического основания в растворителе, основность или кислотность растворителя и действие растворителя на основность неорганического основания. В одном варианте осуществления растворитель представляет собой ацетонитрил, и неорганическое основание представляет собой карбонат калия. В другом варианте осуществления растворитель представляет собой диметилформамид, и неорганическое основание представляет собой гидрокарбонат натрия.
Температура реакции может быть любой температурой, подходящей для реакции между соединением формулы (II) и соединением формулы (III), в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления температура реакции составляет приблизительно от 20°C до приблизительно 120°C. В некоторых представляющих интерес вариантах осуществления температура реакции составляет приблизительно от 50°C до приблизительно 100°C. В других представляющих интерес вариантах осуществления температура реакции составляет приблизительно от 70°C до приблизительно 100°C. В частном варианте осуществления растворитель представляет собой ацетонитрил, и температурой реакции является точка кипения ацетонитрила, т.е. 81-82°C.
Время протекания реакции может быть любым подходящим временем для реакции между соединением формулы (II) и соединением формулы (III), в зависимости от специалиста в данной области. Как правило, чем выше температура реакции, тем меньше время протекания реакции. Например, в конкретных вариантах осуществления время протекания реакции составляет приблизительно от 1 до приблизительно 24 часов. В некоторых представляющих интерес вариантах осуществления время протекания реакции составляет приблизительно от 1 до приблизительно 5 часов. В частном представляющем интерес варианте осуществления время протекания реакции составляет приблизительно 2-4 часа при 81-82°C.
Отношение эфира 2-(бромметил)бензойной кислоты формулы (III) к первичному амину формулы (II) может быть любым мольным отношением, подходящим для реакции между соединением формулы (II) и соединением формулы (III), в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления мольное отношение соединения формулы (III) к соединению формулы (II) может составлять приблизительно от 1:0,8 до приблизительно 1:1,3. В других вариантах осуществления мольное отношение соединения формулы (III) к соединению формулы (II) составляет приблизительно от 1:0,9 до приблизительно 1:1,2. В дополнительных вариантах осуществления мольное отношение соединения формулы (III) к соединению формулы (II) составляет приблизительно от 1:1 до приблизительно 1:1,1.
Если желательна рацемическая смесь соединения формулы (I), можно использовать рацемическую смесь соединения формулы (II). Наоборот, если желательно энантиомерно чистое соединение формулы (I), можно использовать энантиомерно чистое соединение формулы (II). Некоторые неограничивающие примеры соединения формулы (II) включают (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламин и (1R)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламин. Альтернативно, если желательно энантиомерно чистое соединение формулы (I), может быть получена рацемическая смесь соединения формулы (I) и затем разделена на энантиомеры обычными способами разделения, такими как биологическое разделение и химическое разделение. Как правило, биологическое разделение проводят при использовании микроорганизмов, которые метаболизируют один конкретный энантиомер, оставляя другой энантиомер нетронутым. При химическом разделении рацемическую смесь переводят в смесь двух диастереоизомеров, которые могут быть разделены обычными способами, такими как фракционированная кристаллизация и хроматография. После разделения, диастереоизомерные формы могут быть переведены обратно в энантиомеры. В одном варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой рацемическую смесь. В другом варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой (+)-энантиомер. В дополнительном варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой (-)-энантиомер.
В некоторых вариантах осуществления соединения изоиндолин-1-она формулы (I) каждый из R1 и R2 независимо представляет собой алкоксигруппу из 1-4 углеродных атомов. В частном варианте осуществления R1 представляет собой метоксигруппу, и R2 представляет собой этоксигруппу. В других вариантах осуществления каждый из X2, X3 и X4 представляет собой водород; и X1 представляет собой нитрогруппу, -NH2 или -NHCOR6', где R6' представляет собой водород, алкил из 1-8 атомов углерода, циклоалкил 3-8 атомов углерода или фенил. В одном варианте осуществления X1 представляет собой -NHCOR6' , и R6' представляет собой циклопропил. В некоторых вариантах осуществления R3 представляет собой метил. В другом варианте осуществления R представляет собой метил.
В частном варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой соединение (1), т.е. (1S)-7-нитро-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-он, где X1 представляет собой нитрогруппу; каждый из X2, X3 и X4 представляет собой водород; R3 представляет собой метил; R1 представляет собой метоксигруппу; и R2 представляет собой этоксигруппу. Относительно схемы А ниже, соединение (1) может быть получено путем взаимодействия между соединением (2) (т.е. (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламином) и соединением (3) (т.е. метил-2-бромметил-6-нитробензоатом) в присутствии неорганического катализатора, такого как карбонат калия и гидрокарбонат натрия. В другом варианте осуществления (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламин) заменен на (1R)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламин) для получения (1N)-7-нитро-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-она.
Схема А

Взаимодействие между соединением (2) и соединением (3) может протекать в растворителе. В некоторых вариантах осуществления растворитель представляет собой ацетонитрил, время протекания реакции составляет приблизительно 1-24 часа, и неорганический катализатор представляет собой карбонат калия. В других вариантах осуществления время протекания реакции составляет приблизительно 2-4 часа, и реакция протекает в кипящем с обратным холодильником ацетонитриле. В дополнительных вариантах осуществления реакция протекает в ДМФА в течение приблизительно 15-18 часов при температуре приблизительно 70-100°C в присутствии гидрокарбоната натрия.
Необязательно соединение изоиндолин-1-она формулы (I) может быть переведено в кислую соль путем взаимодействия соединения формулы (I) с кислотой в мольном отношении, например, приблизительно 1:1. Неограничивающие примеры подходящих кислот включают метансульфоновую кислоту, трифторуксусную кислоту, 4-(трифторметил)бензойную кислоту, п-толуолсульфоновую кислоту, хлористоводородную кислоту, азотную кислоту, серную и фосфорную кислоту. В одном варианте осуществления соединение изоиндолин-1-она формулы (I) переводят в гидрохлорид при помощи 12 н. хлористоводородной кислоты при температуре приблизительно от 0°C до приблизительно 22°C.
Первичный амин формулы (II) может быть получен путем взаимодействия между сульфоном и производным бензальдегида и другими способами, известными в данной области. Взаимодействие между сульфонами и производными бензальдегида описано в патенте США 6020358 и публикации патентной заявки США № 2004/0204448, оба из которых включены в настоящее описание посредством ссылки.
Эфир 2-(бромметил)бензойной кислоты формулы (III) может быть получен любым способом, известным в данной области. В конкретных вариантах осуществления эфир 2-(бромметил)бензойной кислоты формулы (III) может быть получен путем взаимодействия эфира 2-метилбензойной кислоты формулы (IV):
 (IV)
(IV)
с бромирующим агентом, где R, X1, X2, X3 и X4 являются такими, как определено выше.
Бромирующий агент может быть любым известным бромирующим агентом, который может заместить бензольный водород группой брома. Неограничивающие примеры подходящих бромирующих агентов включают 1,3-дибром-5,5-диметилгидантоин, N-бромсуккцинимид, бромтрихлорметан, комплексное соединение брома и сополимера стирол-винилпиридина, бром, бромид меди (II), смесь бромата натрия и бромтриметилсилана, и их комбинации. В некоторых вариантах осуществления бромирующий агент представляет собой 1,3-дибром-5,5-диметилгидантоин. Некоторые полезные бромирующие агенты описаны, например, в Baldwin et al., Synthetic Commun., 1976, 6 (2), 109; Lee et al., Bull. Korean. Chem. Soc., 1995, 16, 371; Stephenson, Org. Synth., 1963, Collective Vol 4, 984; Pizey, Synthetic Reagent, Halsted Press, New-York, 1974, Vol 2, 1-63; Sket et al., J. Org. Chem., 1986, 51, 929; и Chaintreau et al., Synth. Comm, 1981, 11, 669, все из которых включены в настоящее описание посредством ссылки.
Необязательно реакция бромирования бензольного кольца между соединением формулы (IV) и бромирующим агентом может протекать в присутствии инициатора свободно-радикальной реакции. Свободный радикал представляет собой, как правило, атом или группу атомов, которые имеют, по меньшей мере, один неспаренный электрон. Инициатор свободно-радикальной реакции представляет собой, как правило, вещество, которое способно инициировать образование свободных радикалов. Любой инициатор свободно-радикальной реакции, известный в данной области, может быть использован в реакции бромирования бензольного кольца между соединением формулы (IV) и бромирующим агентом. Неограничивающие примеры подходящих инициаторов свободно-радикальной реакции включают азосоединения, диалкилпероксиды, гидропероксиды, органические полиоксиды, диацилпероксиды, сложные пероксиэфиры, многоатомные пероксиды, металлорганические пероксиды и их комбинации. Некоторые инициаторы свободно-радикальной реакции описаны в публикации Denisov et al., Handbook of free radical initiators, 2003, John Wiley & Sons, Inc., Hoboken, New Jersey, которая включена в настоящее описание посредством ссылки. В некоторых вариантах осуществления инициатором свободно-радикальной реакции является 2,2'-азобисизобутиронитрил, 2,2'-азобис(2,4-диметилвалеронитрил), 2,2'-азобис(метоксидиметилвалеронитрил), 2,2'-азобис(2-метилбутиронитрил), 1,1'-азобис(цианоциклогексан), 4,4'-азобис(4-циановалерьяновая кислота) или бензоилпероксид, все из которых могут быть куплены у поставщика, такого как Dupont и Aldrich Chemicals; или могут быть получены согласно известным способам синтеза. В частном варианте осуществления инициатор свободно-радикальной реакции представляет собой 2,2'-азобисизобутиронитрил.
Отношение инициатора свободно-радикальной реакции к бромирующему агенту может быть любым мольным отношением, подходящим для реакции бромирования бензольного кольца между бромирующим агентом и соединением формулы (IV), в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления мольное отношение инициатора свободно-радикальной реакции к бромирующему агенту может составлять приблизительно от 0,01:1 до приблизительно 0,5:1. В других вариантах осуществления мольное отношение инициатора свободно-радикальной реакции к бромирующему агенту составляет приблизительно от 0,05:1 до приблизительно 0,25:1. В дополнительных вариантах осуществления мольное отношение инициатора свободно-радикальной реакции к бромирующему агенту составляет приблизительно от 0,07:1 до приблизительно 0,15:1.
Температура реакции может быть любой температурой, подходящей для реакции между бромирующим агентом и соединением формулы (IV), в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления температура реакции составляет приблизительно от 20°C до приблизительно 120°C. В некоторых представляющих интерес вариантах осуществления температура реакции составляет приблизительно от 40°C до приблизительно 90°C. В других представляющих интерес вариантах осуществления температура реакции составляет приблизительно от 50°C до приблизительно 70°C. В частном варианте осуществления растворитель представляет собой метилацетат, и температурой реакции является температура кипения метилацетата с обратным холодильником, т.е. приблизительно от 55 и 60°C.
Время протекания реакции может быть любым временем, подходящим для реакции между бромирующим агентом и соединением формулы (IV), в зависимости от специалиста в данной области. Как правило, чем выше температура реакции, тем меньше время протекания реакции. Например, в конкретных вариантах осуществления время протекания реакции составляет приблизительно от 1 до приблизительно 24 часа. В некоторых представляющих интерес вариантах осуществления время протекания реакции составляет приблизительно от 1 до приблизительно 10 часов. В частном, представляющем интерес варианте осуществления время протекания реакции составляет приблизительно 6-8 часов при температуре приблизительно от 55 до 60°C.
Отношение бромирующего агента к соединению формулы (IV) может быть любым мольным отношением, подходящим для реакции между бромирующим агентом и соединением формулы (IV), в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления мольное отношение бромирующего агента к соединению формулы (IV) составляет приблизительно от 0,5:1 до приблизительно 1,5:1. В других вариантах осуществления мольное отношение составляет от 0,75:1 до приблизительно 1:1. В дополнительных вариантах осуществления мольное отношение составляет от 0,55:1 до приблизительно 0,75:1.
Реакция бромирования бензольного кольца может протекать в растворителе. Может быть использован любой растворитель, который не реагирует с бромирующим агентом. Неограничивающие примеры подходящих растворителей включают метилацетат, ацетонитрил, этилацетат, эфиры, такие как диэтиловый эфир и тетрагидрофуран, дихлорметан, хлороформ, N-метилпирролидинон, диметилформамид, диметилсульфоксид и их комбинации.
В некоторых вариантах осуществления эфира 2-метилбензойной кислоты формулы (IV) каждый из X2, X3 и X4 представляет собой водород; и X1 представляет собой нитрогруппу, -NH2 или -NHCOR6', где R6' представляет собой водород, алкил из 1-8 атомов углерода, циклоалкил из 3-8 атомов углерода или фенил. В других вариантах осуществления X1 представляет собой -NHCOR6', и R6' представляет собой циклопропил. В дополнительных вариантах осуществления R представляет собой метил.
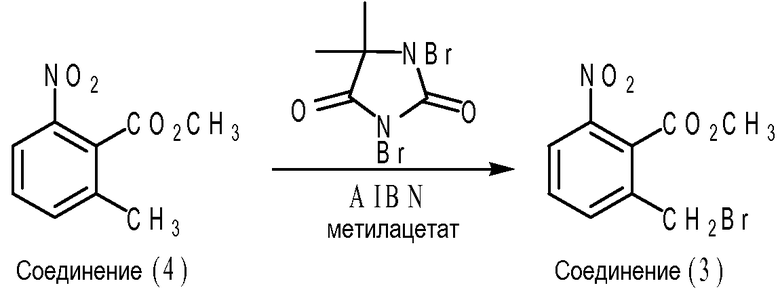
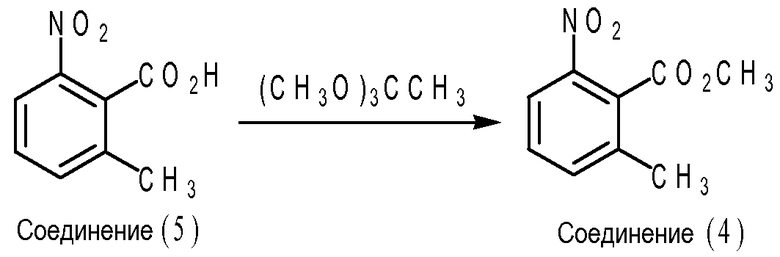
Частным вариантом осуществления эфира 2-метилбнзойной кислоты формулы (IV) является соединение (3), т.е. метил 2-бромметил-6-нитробензоат, где X1 представляет собой нитрогруппу; каждый из X2, X3 и X4 представляет собой водород; R представляет собой метил. Что касается схемы B ниже, соединение (3) может быть получено путем взаимодействия между соединением (4) (т.е. метил-2-метил-6-нитробензоата) и 1,3-дибром-5,5-диметилгидантоином (DBH) в присутствии инициатора свободно-радикальной реакции, такого как 2,2'-азобисизобутиронитрил (AIBN) в метилацетате. В частном варианте осуществления мольное отношение соединения (4) к DBH к AIBN составляет приблизительно 1,02 к приблизительно 0,57 к приблизительно 0,05.
Схема В
Эфир 2-метилбензойной кислоты формулы (IV) может быть куплен у коммерческого поставщика или получен путем взаимодействия этерифицирующего агента с 2-метилбензойной кислотой формулы (V):
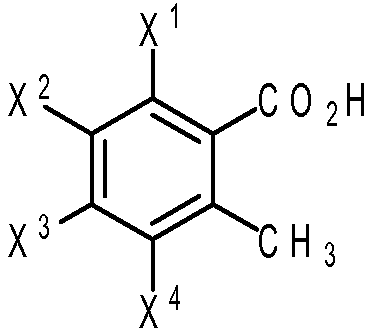 (V)
(V)
где X1, X2, X3 и X4 являются такими, как определено выше.
Для реакции этерификации может быть использован любой этерифицирующий агент, который может преобразовывать группу -CO2H в соединении формулы (V) в группу -CO2R. В некоторых вариантах осуществления реакция этерификации может быть катализирована или промотирована кислотой Брэнстеда, кислотой Льюиса, ионообменными смолами, цеолитами и т.п. В других вариантах осуществления реакция этерификации может быть катализирована или промотирована основными катализаторами, такими как амины, карбонаты металлов, гидрокарбонаты металлов, гидроксиды металлов и т.п. Неограничивающие примеры подходящего агента этерификации включают спирты, алкоголяты металлов, сложные эфиры, алкилгалиды, диазометан и сложные орто-эфиры. Этерификация кислот для образования сложных эфиров описана в публикации Junzo Otera "Esterification: Meethods, Reactions, and Applications” Wiley-VCH Verlag GmbH & Co. KGaA, Wcinheim, p. 3-174 (2003), которая включена в настоящее описание посредством ссылки. Этерификация карбоновых кислот со сложными орто-эфирами описана в литературе, такой как Yoshino et al., Synlett, 2004, 9, 1604; и Trujillo et al., Tetrahedron Lett., 1993, 34, 7355, обе из которых включены в настоящее описание посредством ссылки. Неограничивающие примеры подходящих орто-эфиров включают триметилортоацетат, триметилортоформиат, триэтилортоформиат, триэтилортоацетат, триэтилортопропионат и т.п. В некоторых вариантах осуществления этерифицирующий агент представляет собой сложный орто-эфир. В дополнительных вариантах осуществления орто-эфиром является триметилортоацетат.
Температура реакции этерификации может быть любой температурой, подходящей для реакции между этерифицирующим агентом и соединением формулы (V), в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления температура реакции этерификации составляет приблизительно от 0°C до приблизительно 120°C. В некоторых представляющих интерес вариантах осуществления температура этерификации составляет приблизительно от 20°C до приблизительно 100°C. В других представляющих интерес вариантах осуществления температура этерификации составляет приблизительно от 80°C до приблизительно 120°C. В частном варианте осуществления этерифицирующий агент представляет собой триметилортоацетат, и температура реакции составляет приблизительно от 95 до 100°C.
Время протекания реакции может быть любым временем, подходящим, подходящей для реакции между этерифицирующим агентом и соединением формулы (V), в зависимости от специалиста в данной области. Как правило, чем выше температура реакции, тем меньше время протекания реакции. Например, в конкретных вариантах осуществления время протекания реакции составляет приблизительно от 1 до приблизительно 24 часов. В некоторых представляющих интерес вариантах осуществления время протекания реакции составляет приблизительно от 1 до приблизительно 10 часов. В частном, представляющем интерес варианте осуществления этерифицирующий агент представляет собой триметилортоацетат, и время протекания реакции составляет приблизительно 1-2 часа приблизительно при температуре от 95 до 100°C.
Отношение этерифицирующего агента к соединению формулы (V) может быть любым мольным отношением, подходящим для реакции этерификации, в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления мольное отношение этерифицирующего агента к соединению формулы (V) составляет приблизительно от 2:1 до приблизительно 0,5:1. В других вариантах осуществления мольное отношение составляет от 1,75:1 до приблизительно 0,75:1. В дополнительных вариантах осуществления настоящего изобретения мольное отношение составляет от 1,5:1 до приблизительно 1:1.
Реакция этерификации может протекать в отсутствии или присутствии растворителя. В некоторых вариантах осуществления реакция этерификации протекает в отсутствии растворителя. В других вариантах осуществления настоящего изобретения реакция этерификации протекает в присутствии растворителя. Может быть использован любой растворитель, который не реагирует со этерифицирующим агентом. Неограничивающие примеры подходящих растворителей включают метилацетат, ацетонитрил, этилацетат, простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, дихлорметан, хлороформ, N-метилпирролидинон, диметилформамид, диметилсульфоксид, ионные жидкости, и их комбинации. Как правило, ионная жидкость может быть любой органической солью с низкой температурой плавления, предпочтительно, ниже 100°C, и более предпочтительно, ниже комнатной температуры. Известно, что использование ионной жидкости в качестве растворителя может улучшить выход реакций этерификации. Неограничивающие примеры подходящих ионных жидкостей включают ионные жидкости без галогенов (например, 1-этил-3-метилимидазолийтозилат, 1-бутил-3-метилимидазолийоктилсульфат и 1-бутил-3-метилимидазолий-2-(2-метоксиэтокси)этилсульфат), соединения имидазолия (например, 1-этил-3-метилимидазолийбромид, гексафторфосфат 1-этил-3-метилимидазолия и гексафторфосфат 1-бутил-3-метилимидазолия) и соединения пиридина (например, 1-бутил-4-метилпиридинийхлорид и гексафторфосфат 1-бутил-4-метилпиридиния), соединения фосфония, соединения тетраалкиламмония и их комбинации. Некоторые ионные жидкости описаны Wasserscheid et al., в Angew. Chem. Int. Ed. 2000, 39, 3772; Welton, Chem. Rev. 1999, 99, 2071; Sheldon, Chem. Commun. 2001, 2399; и Dupont et al., Chem. Rev. 2002 102, 3667, все из которых включены в настоящее описание посредством ссылки.
В некоторых вариантах осуществления эфира 2-метилбензойной кислоты формулы (V) каждый из X2, X3 и X4 представляет собой водород; и X1 представляет собой нитрогруппу, -NH2 или -NHCOR6', где R6' представляет собой водород, алкил из 1-8 атомов углерода, циклоалкил из 3-8 атомов углерода или фенил. В других вариантах осуществления X1 представляет собой NHCOR6', и R6' представляет собой циклопропил.
В частном варианте осуществления эфир 2-метилбензойной кислоты формулы (IV) представляет собой соединение (4), т.е. метил 2-метил-6-нитробензоат, где X1 представляет собой нитрогруппу; каждый из X2, X3 и X4 является водородом; R представляет собой метил. Что касается схемы С ниже, соединение (4) может быть получено путем взаимодействия между соединением (5) (т.е. 2-метил-6-нитробензойной кислотой) и триметилортоацетатом в отсутствии растворителя или катализатора. Температура реакции между триметилортоацетатом и 2-метилбензойной кислотой формулы (IV) может составлять приблизительно от 80 до приблизительно 120°C. Мольное отношение триметилортоацетата к соединению формулы (V) может составлять приблизительно от 1:1 до приблизительно 2:1. В частном варианте осуществления температура реакции составляет приблизительно от 95 до приблизительно 100°C, и мольное отношение триметилортоацетата к соединению формулы (V) составляет приблизительно 1,5:1.
Схема С
В одном конкретном варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой соединение (4), т.е. 7-нитроизоиндолин-1-он, имеющий следующую формулу:
 (VI)
(VI)
где X2, X3, X4, R1, R2 и R3 являются такими, как определено выше. В некоторых вариантах осуществления каждый из X2, X3 и X4 представляет собой водород; каждый из R1 и R2 независимо представляет собой алкоксигруппу из 1-4 атомов углерода; и R3 представляет собой алкил из 1-8 атомов углерода. В дополнительном варианте осуществления R1 представляет собой метоксигруппу; R2 представляет собой этоксигруппу; и R3 представляет собой метил.
В другом варианте осуществления соединение изоиндолин-1-она формулы (I) представляет собой соединение 7-аминоизоиндолин-1-она, имеющее следующую формулу:
 (VII)
(VII)
где X2, X3, X4, R1, R2 и R3 являются такими, как определено выше. Соединение 7-аминоизоиндолин-1-она формулы (VII) может быть получено путем взаимодействия первичного амина формулы (II) с эфиром 2-(бромметил)бензойной кислоты формулы (III), где X1 представляет собой -NH2. Как правило, группа первичного амина формулы (II) является более реакционноспособной, чем ароматическая аминогруппа формулы (VII) в реакциях нуклеофильного замещения. Однако при необходимости, ароматическая аминогруппа формулы (VII) может быть защищена посредством защитной группы перед взаимодействием и удалена после взаимодействия. Может быть использована любая аминозащитная группа, известная специалисту в данной области. Неограничивающие примеры подходящих аминозащитных групп включают ацильные группы (например, формил, ацетил и бензоил), производные мочевины и уретана, и алкильные и арильные производные. Некоторые аминозащитные группы описаны в публикации Jif MacOmie "Protective groups in organic chemistry" Plenum Pub.Corp., Глава 2 (1973), которая включена в настоящее описание посредством ссылки. Взаимодействие между соединением формулы (II) и соединением формулы (III) рассмотрено выше.
Альтернативно, соединение 7-аминоизоиндолин-1-она формулы (VII), может быть получено путем восстановления соединения 7-нитроизоиндолин-1-она формулы (VI) восстановителем. Восстановитель может быть любым известным восстановителем в данной области, который может восстанавливать нитрогруппу до первичного амина. Неограничивающие примеры таких восстановителей включают водород плюс катализатор (каталитическое гидрирование), восстанавливающие металлы в кислоте, такой как хлористоводородная и уксусная кислота, сульфид натрия в растворе гидроксида аммония, цинк в растворе формиата аммония, магний в растворе моноформиата гидразиния и дихлорид олова в разбавленной хлористоводородной кислоте. Неограничивающие примеры подходящего катализатора гидрирования включают палладий (Pd) и платину (Pt). Неограничивающие примеры подходящих восстанавливающих металлов включают железо, амальгаму цинка, цинк и олово. В частном варианте осуществления восстановитель представляет собой водород плюс катализатор. В дополнительном варианте осуществления настоящего изобретения катализатор представляет собой Pd катализатор. В другом варианте осуществления катализатор представляет собой 5% Pd/C. В другом варианте осуществления катализатор представляет собой 10% Pd/C.
Каталитическое гидрирование, как правило, проводят при давлении водорода, которое ведет реакцию по существу к завершению. В частном варианте осуществления каталитическое гидрирование проводят при давлении водорода приблизительно от 2,7 и 3,5 бар (приблизительно 40 и 50 фунт/кв.дюйм или приблизительно 5332 и 6666 Па).
В одном варианте осуществления каталитическое гидрирование осуществляют при температуре окружающей среды. Каталитическое гидрирование, как правило, проводят до тех пор, пока реакция не становится по существу завершенной. В частном варианте осуществления каталитическое гидрирование проводят в течение приблизительно 1-24 часов при температуре приблизительно от 15°C до приблизительно 50°C. В дополнительном варианте осуществления каталитическое гидрирование проводят в течение приблизительно 4-6 часов при температуре приблизительно 35-45°C.
Каталитическое гидрирование может протекать в растворителе. В одном варианте осуществления каталитическое гидрирование проводят в протонных растворителях, таких как спирты, вода и их комбинации. В дополнительном варианте осуществления спиртовой растворитель выбирают из группы, состоящей из метанола, этанола, пропанола, изопропанола, бутанола, изобутанола, трет-бутанола и их комбинаций. В другом варианте осуществления каталитическое гидрирование проводят в неполярном, апротонном растворителе, таком как 1,4-диоксан. В еще одном варианте осуществления каталитическое гидрирование проводят в полярном, апротонном растворителе, таком как этилацетат, ацетонитрил, ацетон, ДМСО, ДМФА и ТГФ. В одном представляющем интерес варианте осуществления растворитель представляет собой протонный растворитель. В дополнительном представляющем интерес варианте осуществления растворителем для каталитического гидрирования является этилацетат.
В некоторых вариантах осуществления соединения 7-аминоизоиндолин-1-она формулы (VII) каждый из X2, X3 и X4 представляет собой водород. В других вариантах осуществления каждый из R1 и R2 независимо представляет собой алкоксигруппу из 1-4 атомов углерода; и R3 представляет собой алкил из 1-8 атомов углерода. В других вариантах осуществления R1 представляет собой метоксигруппу, и R2 представляет собой этоксигруппу. В дополнительных вариантах осуществления R3 представляет собой метил.
В частном варианте осуществления соединение 7-аминоизоиндолин-1-она формулы (VII) представляет собой соединение (6), где X1 представляет собой -NH2; каждый из X2, X3 и X4 представляет собой водород; R1 представляет собой метоксигруппу; R2 представляет собой этоксигруппу; и R3 представляет собой метил. Что касается схемы D ниже, соединение (6) может быть получено путем восстановления соединения (1) водородом в присутствии 10% катализатора Pd/C. Каталитическое гидрирование может протекать приблизительно при 40°C в течение приблизительно 4-6 часов в этилацетате в присутствии 10% Pd/C. В дополнительном варианте осуществления каталитическое гидрирование протекает при давлении водорода приблизительно от 40 и 45 фунт/кв.дюйм или 2,7 до 3,1 бар.
Схема D
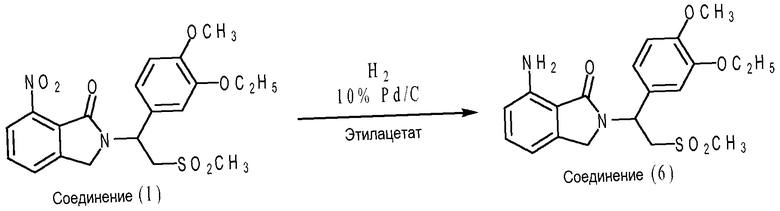
Если желательна рацемическая смесь соединения (6), можно использовать рацемическую смесь соединения (1). Наоборот, если желательно энантиомерно чистое соединение (6), можно использовать энантиомерно чистое соединение (1). Альтернативно, если желательно энантиомерно чистое соединение (6), можно получить рацемическую смесь соединения (6) и затем разделить на энантиомеры обычными способами разделения, такими как биологическое разделение и химическое разделение. В одном варианте осуществления соединение (6) является рацемической смесью. В другом варианте осуществления соединение (6) представляет собой (+)-энантиомер. В дополнительном варианте осуществления соединение (6) представляет собой (-)-энантиомер.
Необязательно, соединение 7-аминоизоиндолин-1-она формулы (VII) может быть переведено в кислую соль путем взаимодействия соединения формулы (VII) с кислотой в мольном отношении 1:1. Неограничивающие примеры подходящей кислоты включают метансульфоновую кислоту, трифторуксусную кислоту, 4-(трифторметил)бензойную кислоту, п-толуолсульфоновую кислоту, хлористоводородную кислоту, азотную кислоту, серную и фосфорную кислоту. В одном варианте осуществления соединение формулы (VII) переводят в гидрохлоридную соль при помощи 12н. хлористоводородной кислоты при температуре от 0°C до 22°C.
В другом варианте осуществления соединение формулы (I) является амидным соединением, имеющим следующую формулу:
 (VIII)
(VIII)
где X2, X3, X4, R1, R2 и R3 являются такими, как определено выше; и R6' представляет собой алкил из 1-8 атомов углерода, циклоалкил из 3-8 атомов углерода или фенил. Амидное соединение формулы (VIII) может быть получено путем взаимодействия первичного амина формулы (II) с эфиром 2-(бромметил)бензойной кислоты формулы (III), где X1 представляет собой -NHCOR6'. Взаимодействие между соединением формулы (II) и соединением формулы (III) рассмотрено выше.
Альтернативно, амидное соединение формулы (VIII) может быть получено путем взаимодействия 7-аминоизоиндолин-1-она формулы (VII) или его кислой соли с ацилгалогенидом, имеющим формулу R6'-C(O)-На, где R6' является таким, как определено выше, и На является F, Cl, Br или I. Взаимодействие между соединением формулы (VII) или его кислой солью и ацилгалогенидом может протекать в растворителе, таком как этилацетат, ацетон, метилэтилкетон, диэтиловый эфир, тетрагидрофуран, ацетонитрил, дихлорметан, хлороформ, N-метилпирролидинон, диметилформамид, диметилсульфоксид и их смеси. В одном варианте осуществления растворителем является этилацетат.
Температура реакции ацилирования может быть любой температурой, подходящей для реакции ацилирования, в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления настоящего изобретения температура реакции ацилирования между ацилгалогенидом и соединением формулы (VII) или его кислой солью может составлять приблизительно от 0°C до приблизительно 50°C. В одном представляющем интерес варианте осуществления настоящего изобретения, температура реакции составляет приблизительно от 15°C до приблизительно 25°C.
Необязательно, реакция ацилирования может протекать в присутствии основного катализатора, такого как органические амины. Неограничивающие примеры органических аминов включают триэтиламин, N,N-диизопропилэтиламин, пиридин и DBU, имидазол и их комбинации. В одном представляющем интерес варианте осуществления катализатор представляет собой триэтиламин. В другом представляющем интерес варианте осуществления катализатор представляет собой имидазол. В дополнительном представляющем интерес варианте осуществления настоящего изобретения катализатор представляет собой N,N-диизопропилэтиламин.
Время протекания реакции ацилирования может быть любым временем, подходящим для реакции ацилирования, в зависимости от специалиста в данной области. Как правило, чем выше температура реакции, тем меньше время протекания реакции. Например, в конкретных вариантах осуществления время протекания реакции ацилирования изменяется от 1 до 24 часов. В одном представляющем интерес варианте осуществления настоящего изобретения время протекания реакции составляет приблизительно от 4 до приблизительно 6 часов при температуре реакции от 20°C до 25°C.
В одном варианте осуществления ацилгалогенид добавляют к раствору соединения формулы (VII) с последующим добавлением основного катализатора. В другом варианте осуществления основный катализатор добавляют к раствору соединения формулы (VII) с последующим добавлением ацилгалогенида. В другом варианте осуществления мольное отношение основного катализатора к соединению формулы (VII) составляет приблизительно от 2:1 до приблизительно 1:2. В дополнительном варианте осуществления мольное отношение составляет приблизительно от 1,4:1 до приблизительно 1:1.
Как правило, любой ацилгалогенид, который может взаимодействовать с первичным амином или вторичным амином, может быть использован для данного варианта осуществления. Неограничивающие примеры подходящих ацилгалоидных соединений включают циклопропанкарбонилхлорид, циклобутанкарбонилхлорид, циклопентанкарбонилхлорид, циклогексанкарбонилхлорид, циклопентилацетилхлорид, 1-метилциклогексанкарбонилхлорид, 3-циклопентилпропаноилхлорид и циклогептанкарбонилхлорид, все из которых могут быть получены у коммерческих поставщиков, таких как Aldrich Chemicals, Milwaukee, WI, или получены путем галогенирования соответствующих карбоновых кислот (R6'COOH, где R6' является таким, как определено выше) галогенирующим агентом. Галогенирующим агентом может быть PY3, PY5 или SOY2, где Y представляет собой F, Cl, Br или I. Например, ацилхлорид (такой как циклогептанкарбонилхлорид) может быть получен путем взаимодействия соответствующей карбоновой кислоты (циклогептанкарбоновой кислоты) с SOCl2 или PCl5. Подобным образом, ацилбромид может быть получен путем взаимодействия соответствующей карбоновой кислоты с PBr5.
Отношение ацилгалогенида к соединению формулы (VII) может быть любым мольным отношением, подходящим для реакции ацилирования, в зависимости от специалиста в данной области. Например, в конкретных вариантах осуществления мольное отношение ацилгалогенида к соединению формулы (VII) составляет приблизительно от 2:1 до приблизительно 0,5:1. В других вариантах осуществления настоящего изобретения мольное отношение составляет приблизительно от 1,75:1 до приблизительно 0,75:1. В дополнительных вариантах осуществления мольное отношение составляет приблизительно от 1,2:1 до приблизительно 1:1.
Ацилированное соединение формулы (VIII) может быть очищено путем перекристаллизации из растворителя. В одном варианте осуществления растворитель представляет собой тетрагидрофуран, этанол, N-метилпирролидинон, метанол, этилацетат, изопропанол, уксусную кислоту, воду или их комбинацию. В дополнительном варианте осуществления растворитель представляет собой смесь тетрагидрофурана и этилового спирта в объемном отношении от 3:1 до 1:3.
В некоторых вариантах осуществления амидного соединения формулы (VIII), R6' представляет собой циклоалкил из 3-8 атомов углерода. В дополнительных вариантах осуществления R6' представляет собой циклопропил. В других вариантах осуществления каждый из R1 и R2 независимо представляет собой алкоксигруппу из 1-4 углеродных атомов; и R3 представляет собой алкил из 1-8 атомов углерода. В дополнительных вариантах осуществления R1 представляет собой метоксигруппу, и R2 представляет собой этоксигруппу. В других вариантах осуществления каждый из X2, X3 и X4 представляет собой водород. В других вариантах осуществления R3 представляет собой метил.
В частном варианте осуществления амидное соединение формулы (VIII) представляет собой соединение (7), где X1 представляет собой -NHCO-циклопропил; каждый из X2, X3 и X4 представляет собой водород; R1 представляет собой метоксигруппу; R2 представляет собой этоксигруппу; и R3 представляет собой метил. Что касается схемы E ниже, соединение (7) может быть получено, например, путем взаимодействия соединения (6) с циклопропилкарбонилхлоридом в присутствии N,N-диизопропилэтиламина. Реакция ацилирования может протекать, например, при температуре реакции от 20°C до 25°C в течение приблизительно от 4 до приблизительно 6 часов в этилацетате. Мольное отношение соединения (6) к циклопропилкарбонилхлориду к N,N-диизопропилэтиламину составляет приблизительно 1:1,05:1,2.
Схема Е

Если желательна рацемическая смесь соединения (7), то можно использовать рацемическую смесь соединения (6). Наоборот, если желательно энантиомерно чистое соединение (7), то можно использовать энантиомерно чистое соединение (6). Альтернативно, если желательно энантиомерно чистое соединение (7), можно использовать рацемическую смесь соединения (7) и затем разделить на энантиомеры обычными способами разделения, такими как биологическое разделение и химическое разделение.
Соединения изоиндолин-1-она формулы (I) могут быть использованы при получении фармацевтических композиций для лечения широкого спектра заболеваний и состояний, включая, но не ограничиваясь ими, воспалительные заболевания, аутоиммунные заболевания, различные типы рака, заболевания сердца, генетические заболевания, аллергические заболевания, остеопороз и волчанка.
Как правило, фармацевтические композиции могут содержать, по меньшей мере, соединение изоиндолин-1-она формулы (I) или его фармацевтически приемлемую соль, сольват или его стереоизомер и могут быть введены пациентам для лечения широкого спектра заболеваний и состояний.
Необязательно, фармацевтические композиции и дозированные формы могут дополнительно содержать один или более носителей, эксципиентов, разбавителей или активных агентов. В некоторых вариантах осуществления фармацевтические композиции можно применять при получении индивидуальных, отдельных единичных дозированных форм. Отдельная единичная дозированная форма являются подходящей для перорального, мукозального (например, подъязычного, назального, вагинального, кистозного, ректального, препуциального, глазного, буккального или ушного), парентерального (например, подкожного, внутривенного, болюсного вливания, внутримышечного или внутриартериального), местного (например, глазные капли или другие глазные формы), трансдермального или чрескожного введения пациентам. Неограничивающие примеры дозированных форм включают таблетки; каплеты; капсулы, такие как мягкие эластичные желатиновые капсулы; крахмальные капсулы; пастилки; ромбовидные пастилки; дисперсии; суппозитории; порошки; аэрозоли (например, назальные спреи или ингаляторы); гели; жидкие дозированные формы, подходящие для перорального или мукозального введения пациентам, включая суспензии (например, водные или неводные жидкие суспензии, эмульсии масло в воде или жидкие эмульсии вода в масле), растворы и эликсиры; жидкие дозированные формы, подходящие для парентерального введения пациенту; глазные капли или офтальмологические формы, подходящие для местного введения; и стерильные твердые вещества (например, кристаллические или аморфные твердые вещества), которые могут быть ресуспендированы для получения жидких дозированных форм, подходящих для парентерального введения пациентам.
Частные варианты осуществления, предложенные в настоящем описании, проиллюстрированы синтезом 7-нитро-2-[1-(3-этокси-4-метоксифенило)-2-(метилсульфонил)этил]изоиндолин-1-она и циклопропил-N-{2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]-3-оксоизоиндолин-4-ил}карбоксамида. Модификации переменных, включая, но не ограничиваясь ими, растворители для реакции, время протекания реакции, температура реакции, реагенты, исходные вещества и функциональные группы в частных вариантах осуществления синтеза 7-нитро-2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]изоиндолин-1-она и циклопропил-N-{2-[1-(3-этокси-4-метоксифенил)-2-(метилсульфонил)этил]-3-оксоизоиндолин-4-ил}карбоксамида будут очевидны специалистам в данной области.
Примеры
Пример 1 - Получение метил-2-метил-6-нитробензоата
Смесь 2-метил-6-нитробензойной кислоты (300,0 г, 1,66 моль, от Acros Organics, Моррис Plains, NJ) и триметилортоацетата (298,3 г, 2,48 моль, от Aldrich Chemicals, Milwauke, WI) загружали в 3 л 3-горлую колбу приблизительно при 20-25°C в атмосфере азота. Реакционную смесь постепенно нагревали до внутренней температуры 95-100°C, и низкокипящие компоненты, образовавшиеся во время реакции, отгоняли. Через 2 часа реакционную смесь охлаждали до 20-25°C в течение 1-2 часов. После того, как гептан (1,50 л, от Aldrich Chemicals) загружали в реакционную смесь в течение 1,0-1,5 часов, реакционную смесь затравливали кристаллами метил 2-метил-6-нитробензоата (0,5 г), когда она становилась мутной. Суспензию охлаждали до 0-5°C в течение 0,5-1 часа и выдерживали при 0-5°C в течение еще 1,5-2 часов. Твердое вещество отделяли фильтрованием в вакууме, промывали гептаном (3×300 мл) и сушили до постоянной массы на лотке при 30-35°C в вакууме при давлении 100-120 торр. Выход метил 2-метил-6-нитробензоата составил 292,0 г (91%) из расчета на 300,0 г 2-метил-6-нитробензойной кислоты. Было обнаружено, что продукт имеет чистоту >99%, измеренную ВЭЖХ на основе процента площади, и содержание воды составляет <0,1%, измеренное путем титрования по Карлу Фишеру.
Пример 2 - Получение метил 2-бромметил-6-нитробензоата
Смесь метил 2-метил-6-нитробензоата (200,0 г, 1,02 моль, ранее полученного), 1,3-дибром-5,5-диметилгидантоина (DBH, 162,0 г, 0,57 моль, от Aldrich Chemicals) и метилацетата (1,20 л от Aldrich Chemicals) загружали в 3 л 3-горлую колбу приблизительно при 20-25°C в атмосфере азота. После того, как реакционную смесь кипятили с обратным холодильником в течение 0,5-1 часа, раствор 2,2'-азобисизобутиронитрила (AIBN, 8,6 г, 52 ммоль от Aldrich Chemicals) в 100 мл метилацетата добавляли в течение 15-30 минут. Реакционную смесь кипятили с обратным холодильником в течение 6,5-8 часов до тех пор, пока количество непрореагировавшего 2-метил-6-нитробензоата не составило менее 5-10%. Реакционную смесь охлаждали до 15-18°C и выдерживали при 15-18°C в течение 50-60 минут. Твердое вещество отфильтровывали, промывали охлажденным (т.е. 5-10°C) метилацетатом (2×100 мл) до тех пор, пока содержание метил 2-бромметил-6-нитробензоата не составило менее 3% в твердом веществе. Затем, после того, как гептан (1,00 л) добавляли к фильтрату, верхний слой, представлявший собой органическую фазу, промывали 2% насыщенным раствором соли (2×500 мл) и деминерализованной водой (1-2×500 мл) до тех пор, пока содержание непрореагировавшего 5,5-диметилгидантоина, согласно измерению ВЭЖХ (процент площади при 210 нм), не составило менее 0,5%. После того, как раствор концентрировали при пониженном давлении для удаления приблизительно 1,80-1,90 л метилацетата, добавляли метил-трет-бутиловый эфир (MTBE, 300 мл). После того, как реакционную смесь кипятили с обратным холодильником при 65-70°C в течение 10-15 минут, раствор охлаждали до 50-55°C в течение 0,5-1 часа и затравливали 500 мг кристаллов метил 2-бромметил-6-нитробензоата при 45-50°C. Суспензию охлаждали до 20-25°C и выдерживали при 20-25°C в течение 2-3 часов. Твердое вещество отделяли фильтрованием, промывали при 5-10°C охлажденной смесью гептана и MTBE в объемном отношении 1:2 (2×100 мл) и сушили до постоянной массы при 20-25°C в вакууме при давлении 100-120 торр. Выход метил 2-бромметил-6-нитробензоата составил 185,2 г (66%) из расчета на 200,0 метил 2-метил-6-нитробензоата. Было обнаружено, что продукт имеет чистоту >98%, измеренную ВЭЖХ на основе процента площади, и содержание воды составляет <0,1%, измеренное путем титрования по Карлу Фишеру.
Пример 3 - Получение (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламина
После того, как смесь (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламиной соли N-ацетил-L-лейцина (1,10 кг, 2,46 моль), деминерализованной воды (4,40 л) и дихлорметана (DCM, 5,50 л) загружали в реактор, раствор гидроксида натрия (196,0 г, 4,90 моль) в 1,00 л деминерализованной воды загружали в реактор приблизительно в течение 5 минут при 15-25°C. Полученную смесь перемешивали в течение по меньшей мере 10 минут при 15-25°C и затем водной и органической фазе давали возможность разделиться. pH верхней водной фазы поддерживали или доводили до pH 13-14. Фазы разделяли и верхнюю водную фазу экстрагировали DCM (2×4,4 л). pH водной фазы поддерживали равной 13-14 при всех экстракциях. Экстракты DCM объединяли и промывали деминерализованной водой (3,3 л) до тех пор, пока pH водной фазы не достигал 11 или меньше. DCM удаляли в вакууме при температуре ниже 35°C. Содержание воды в твердом остатке должно составлять <0,1% масс./масс., измеренное путем титрования по Карлу Фишеру. Твердый остаток сушили азеотропно с большим количеством DCM. Твердый остаток сушили до постоянной массы в вакууме при 30-35°C с получением (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламина в виде белого порошка (639,0-672,0 г, выход 95-100%).
Пример 4A - Получение соединения (1)
Соединение (1) получали следующим способом. Смесь метил 2-бромметил-6-нитробензоата (100,0 г, 365 ммоль, полученного ранее в примере 2), (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламина (104,7 г, 383 ммоль, полученного ранее в примере 3), гидрокарбоната натрия (67,5 г, 8,03 моль от Aldrich Chemicals) и диметилформамида (500 мл) загружали в 1 л 3-горлую колбу при комнатной температуре в атмосфере азота. Реакционную смесь постепенно нагревали до внутренней температуры 70-75°C в течение двух часов, пока содержание непрореагировавшего метил 2-бромметил-6-нитробензоата не составило менее <2%. Реакционную смесь постепенно нагревали до внутренней температуры 95-100°C в течение 18 часов. Реакционную смесь охлаждали до 20-25°C и переносили в 1 л капельную воронку. После того, как очищенную воду (1500 мл) загружали в 5 л 3-горлую колбу, реакционную смесь из капельной воронки добавляли в воду в 5 л 3-горлой колбе при комнатной температуре в течение 1-2 часов, поддерживая внутреннюю температуру ниже 30°C. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Твердое вещество отфильтровывали в вакууме, промывали водой (3×300 мл) и метанолом (2×400 мл) и затем загружали в 2 л 3-горлую колбу с последующим добавлением метанола (1000 мл). Смесь кипятили с обратным холодильником в течение 1 часа. Смесь охлаждали до комнатной температуры. Твердое вещество отделяли фильтрованием в вакууме, промывали 200 мл метанола (2 объема) и сушили до постоянной массы при 40-45°C в вакууме при давлении 100-120 торр. Выход соединения (1) составлял 123,0 г (78%) из расчета на 100,0 г метил 2-бромметил-6-нитробензоата. Было обнаружено, что продукт имеет чистоту >99%, измеренную ВЭЖХ на основе процента площади, и содержание воды составляет <0,1%, измеренное путем титрования по Карлу Фишеру.
Пример 4B - Альтернативное получение соединения (1)
Соединение (1) получали также следующим способом. Смесь метил 2-бромметил-6-нитробензоата (100,0 г, 365 ммоль, полученного ранее в примере 2), (1S)-1-(3-этокси-4-метоксифенил)-2-метансульфонилэтиламина (104,7 г, 383 ммоль, полученного ранее в примере 3) и порошка карбоната калия (100,8 г, 730 ммоль от Aldrich Chemicals) суспендировали в ацетонитриле (500 мл) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником при 81-83°C в течение приблизительно двух часов, пока содержание непрореагировавшего метил 2-бромметил-6-нитробензоата не составило менее 2%. После того, как реакционную смесь охлаждали до 45-50°C, добавляли метанол (200 мл) в течение 5-10 минут. После смеси давали возможность охладиться до 20-25°C и перемешивали в течение 2 часов, добавляли деминерализованную воду (1,40 л) в течение 0,5-1 час и перемешивали при 20-25°C в течение 30 минут и при 0-5°C в течение 1-2 часов. Твердое вещество отфильтровывали, промывали деминерализованной водой (3×300 мл) и сушили до тех пор, пока содержание воды не составило <10%, измеренное путем титрования по Карлу Фишеру. Твердое вещество суспендировали в метаноле (750 мл) и кипятили с обратным холодильником в течение 1-1,5 часов. Суспензию охлаждали до 0-5°C в течение 1,5-2 часов и выдерживали при 0-5°C в течение 1-1,5 часов. Твердое вещество отфильтровывали, промывали при 0-5°C метанолом (2×200 мл) и гептаном (200 мл), и затем сушили при 40-45°C в вакууме до постоянной массы. Выход соединения (1) составлял 148,0 г (93%) из расчета на 100,0 г метил 2-бромметил-6-нитробензоата. Было обнаружено, что продукт имеет чистоту >99%, измеренную ВЭЖХ на основе процента площади, и содержание воды составляет <1,0%, измеренное путем титрования по Карлу Фишеру.
Пример 5 - Получение соединения (7)
Смесь соединения (1) (60 г, 138 ммоль, полученного выше в примере 4), 10% Pd/C (50% влажность, 2,4 г, 4% масс. от Johnson Matthey, Лондон, Великобритания), этилацетата (780 мл) загружали в сосуд Парра при комнатной температуре в атмосфере азота. После того, как смесь продували три раза азотом и три раза водородом, реакционную смесь нагревали до 40°C и затем прекращали нагревание. Реакционную смесь перемешивали в атмосфере водорода при давлении от 40 до 45 фунт/кв.дюйм в течение более 4-6 часов, пока содержание промежуточного продукта гидроксиламина не составило <3%. Реакционную смесь охлаждали до 20-25°C. Реакционную смесь фильтровали через слой целита (толщиной 1 дюйм) и затем промывали слой этилацетатом (120 мл). Фильтрат переносили в 3 л 3-горлую колбу, снабженную 50 мл капельной воронкой. После того, как N,N-диизопропилэтиламин (29 мл, 165 ммоль) загружали в колбу, в капельную воронку загружали циклопропилкарбонилхлорид (13,0 мл, 145 ммоль от Aldrich Chemicals). Циклопропилкарбонилхлорид добавляли при комнатной температуре в течение 1-2 часов при внутренней температуре ниже 30°C. Реакционную смесь перемешивали в течение 2-4 часов при комнатной температуре. После того, как добавили гептан (300 мл), реакционную смесь перемешивали в течение 4-6 часов. Твердое вещество отделяли фильтрованием в вакууме, промывали 2н. HCl (2×300 мл), водой (2×300 мл) и затем гептаном (2×300 мл). Неочищенный продукт сушили при 40-45°C в вакууме при давлении 100-120 торр до постоянной массы. Выход неочищенного продукта соединения (7) составил 58 г (88%) из расчета на 60,0 г Соединения (1).
Пример 6 - Перекристаллизация соединения (7)
Смесь неочищенного соединения (7) (95,2 г, полученного ранее в примере 5) и тетрагидрофурана (ТГФ, 1,43 л) загружали в 3 л колбу при 20-25°C в атмосфере азота. Суспензию нагревали до 60-65°C до тех пор, пока не достигали растворения. Суспензию фильтровали при 45-50°C, и твердое вещество промывали 95 мл ТГФ, предварительно нагретого до 45-55°C. После того, как приблизительно 950-1150 мл ТГФ было отогнано при нормальном давлении в течение более 30-60 минут, добавляли абсолютный этиловый спирт (950 мл) при 55-60°C в течение более 5-10 минут. Приблизительно 350-400 мл растворителей удаляли при нормальном давлении до тех пор, пока внутренняя температура не достигала 72-74°C. Полученную суспензию кипятили с обратным холодильником при 72-75°C в течение 30-60 минут, охлаждали до 20-25°C в течение более 1-2 часов и выдерживали при 20-25°C в течение еще 1-2 часов. Твердое вещество отделяли фильтрованием в вакууме, промывали абсолютным этиловым спиртом (240-280 мл) и гептаном (240-280 мл) и затем сушили на лотке при 50-55°C в вакууме при давлении 130-140 торр до постоянной массы. Выход не совсем белого кристаллического продукта составил (88,0-91,0 г, 92-96%).
Настоящее изобретение не должно быть ограничено частными вариантами осуществления настоящего изобретения, раскрытыми в примерах, которые предназначены для иллюстрации, и любые функционально эквивалентные варианты осуществления настоящего изобретения входят в объем предложенного описания. В действительности, различные модификации в дополнение к показанным и описанным в настоящем описании будут очевидны специалистам в данной области и подразумевается, что они входят в объем приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГАЛОГЕНАЛЛИЛАМИНА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2793850C2 |
| СРЕДСТВО ДЛЯ БОРЬБЫ С БОЛЕЗНЯМИ РАСТЕНИЙ | 2018 |
|
RU2762833C2 |
| НОВОЕ ИЗОИНДОЛИНОВОЕ ПРОИЗВОДНОЕ, ВКЛЮЧАЮЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2728829C1 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА | 2005 |
|
RU2372989C2 |
| ПУТИ ПРИМЕНЕНИЯ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2020 |
|
RU2795503C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА КОЛОНИЕСТИМУЛИРУЮЩЕГО ФАКТОРА-1 (CSF-1R) | 2016 |
|
RU2748884C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2012 |
|
RU2614976C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2019 |
|
RU2794895C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 3-(4-(2,4-ДИФТОРБЕНЗИЛОКСИ)-3-БРОМ-6-МЕТИЛ-2-ОКСОПИРИДИН-1(2Н)-ИЛ)-N,4-ДИМЕТИЛБЕНЗАМИДА | 2007 |
|
RU2411236C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОБУТАНА | 2019 |
|
RU2807184C2 |
Изобретение относится к способу получения соединения, имеющего приведенную ниже формулу (I), или его фармацевтически приемлемой соли. Предложенный способ включает стадию взаимодействия первичного амина формулы (II) или его соли с эфиром 2-(бромметил)бензойной кислоты формулы (III) в присутствии неорганического основания. В приведенных формулах R представляет собой C1-8алкил; каждый из R1 и R2 независимо представляет собой алкил из 1-4 атомов углерода или алкоксигруппу из 1-4 атомов углерода; R3 представляет собой алкил из 1-8 атомов углерода; и один из X1, X2, X3 и X4 независимо представляет собой NHC(O)R6′, NO2 или NH2, и другие представляют собой водород; и R6′ представляет собой С3-С8циклоалкил. Технический результат - разработка альтернативного способа получения соединений 2-(1-фенил)изоиндолин-1-она для получения их в промышленном масштабе с высоким выходом. 16 з.п. ф-лы, 5 пр.
1. Способ получения соединения формулы:
или его фармацевтически приемлемой соли, включающий стадию взаимодействия первичного амина формулы (II):
или его соли,
с эфиром 2-(бромметил)бензойной кислоты формулы (III):
в присутствии неорганического основания, где R представляет собой C1-8алкил;
каждый из R1 и R2 независимо представляет собой алкил из 1-4 атомов углерода или алкоксигруппу из 1-4 атомов углерода;
R3 представляет собой алкил из 1-8 атомов углерода; и
один из X1, X2, X3 и X4 независимо представляет собой NHC(O)R6′, NO2 или NH2, и другие представляют собой водород; и
R6′ представляет собой С3-С8циклоалкил.
2. Способ по п. 1, где неорганическое основание представляет собой гидроксид металла, карбонат металла, гидрокарбонат металла, гидрид металла или их комбинацию.
3. Способ по п. 2, где гидроксид металла представляет собой гидроксид калия или гидроксид натрия.
4. Способ по п. 2, где карбонат металла представляет собой карбонат калия или карбонат натрия.
5. Способ по п. 2, где гидрокарбонат металла представляет собой гидрокарбонат калия или гидрокарбонат натрия.
6. Способ по п. 1, где растворителем, в котором протекает реакция между соединением (II) и соединением (III), является ацетонитрил.
7. Способ по п. 6, где температура реакции представляет собой точку кипения ацетонитрила.
8. Способ по п. 1, где мольное отношение соединения формулы (III) к соединению формулы (II) составляет приблизительно от 1:0,8 до приблизительно 1:1,3.
9. Способ по п. 1, где применяют энантиомерно чистый (S)-изомер формулы (II).
10. Способ по п. 1, где применяют энантиомерно чистый (R)-изомер формулы (II).
11. Способ по п. 1, где X1 представляет собой нитрогруппу.
12. Способ по п. 11, дополнительно включающий стадию восстановления нитрогруппы с использованием восстановителя для получения 7-аминоизоиндолин-1-она формулы (VII):

13. Способ по п. 12, где восстановитель представляет собой Pd/C и водород.
14. Способ по п. 12, дополнительно включающий стадию взаимодействия соединения формулы (VII) с ацилгалогенидом (R6′-С(О)-галоген) для получения амидного соединения формулы (VIII):
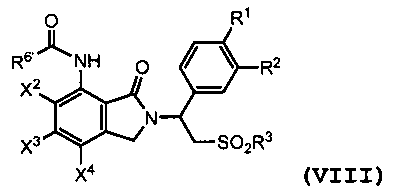
15. Способ по п. 14, где применяют энантиомерно чистый (S)-изомер формулы (VII).
16. Способ по п. 14, где применяют энантиомерно чистый (R)-изомер, соответствующий (VII).
17. Способ по любому из пп. 11, 12 и 14-16, где Х2-Х4, все представляют собой водород, R1 представляет собой метоксигруппу, R2 представляет собой этоксигруппу, и R3 представляет собой метил.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 2004060313 A2, 22.07.2004 | |||
| ИММУНОТЕРАПЕВТИЧЕСКИЕ ИМИДЫ/АМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ФДЭ IV И ФНО | 1997 |
|
RU2177471C2 |

