ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики. В частности, настоящее изобретение относится к производному галогеналлиламина или его фармацевтически приемлемой соли, сложному эфиру, стереоизомеру или таутомеру, фармацевтическому составу и фармацевтической композиции, содержащей это соединение, и к применению его для профилактики и/или лечения заболеваний, связанных c белком SSAO/VAP-1 или опосредованных им.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Как класс аминоксидаз, особенно чувствительных к семикарбазиду, семикарбазид-чувствительная аминоксидаза (SSAO) широко распространена in vivo, а также на клеточных мембранах и в плазме. В эндотелиальных клетках SSAO существует в виде белка-1 сосудистой адгезии (VAP-1). В настоящее время считается, что основная физиологическая функция SSAO in vivo состоит в том, чтобы участвовать в метаболизме аминов, катализировать окислительное дезаминирование короткоцепочечных первичных аминов (таких как метиламин, аминоацетон и тому подобное) и генерировать соответствующие альдегиды, перекись водорода и аммиак. Структура SSAO содержит ион двухвалентной меди с хинонильной группой в качестве кофермента. SSAO не имеет определенного субстрата, и ее основными субстратами являются алифатические и ароматические первичные амины.
В WO 2013163675A1 описано производное 3-галогеналлиламина в качестве ингибитора SSAO/VAP-1 (показано как формула I), обладающее ингибирующей активностью в отношении фермента SSAO/VAP-1, и, в частности, описано соединение 23, также называемое PXS-4728, структура которого выглядит следующим образом:
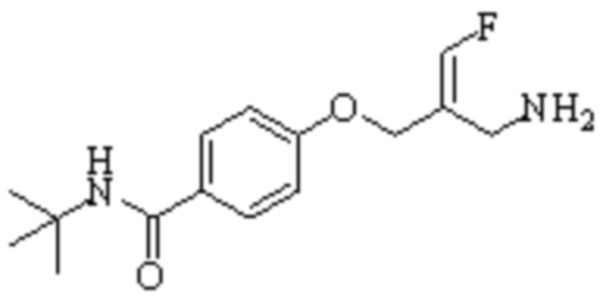 .
.
В настоящее время ингибитор SSAO/VAP-1 еще не разработан. Ингибитор SSAO/VAP-1 по настоящему изобретению может быть использован для эффективного облегчения симптомов и поражений в условиях дисбаланса различных болезненных состояний, которые связаны со сверхэкспрессией SSAO/VAP-1 и т. д., что имеет большие перспективы его применения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
С учетом вышеуказанной проблемы в данной области авторы изобретения провели углубленное исследование и в результате разработали новое производное галогеналлиламина (в дальнейшем иногда также именуемое как «соединение по настоящему изобретению) или его фармацевтически приемлемую соль, сложный эфир, стереоизомер или таутомер в качестве ингибитора SSAO/VAP-1. Такое соединение-ингибитор проявляет превосходную ингибирующую активность в отношении белка SSAO/VAP-1, поэтому его можно использовать для предотвращения и/или лечения заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им.
Более того, соединение-ингибитор SSAO/VAP-1 по настоящему изобретению демонстрирует превосходное ингибирование белка SSAO/VAP-1 и демонстрирует отличную селективность в отношении белка rhAOC1 и белка MAO, предотвращая, таким образом, другие ненужные побочные эффекты при профилактике и/или лечении заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им.
Кроме того, по сравнению с существующими лекарственными средствами соединение-ингибитор SSAO/VAP-1 по настоящему изобретению с трудом проникает через гематоэнцефалический барьер, поэтому соединение по настоящему изобретению имеет очень низкий токсический риск для нервной системы, демонстрируя отличную лекарственную безопасность.
В частности, в настоящем изобретении предложены следующие технические решения.
Техническое решение 1. Соединение, представленное формулой I ниже, или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер:
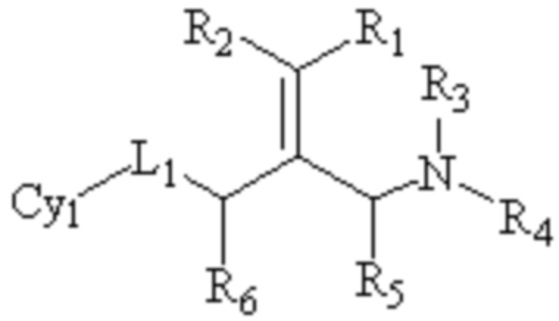
I
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила, необязательно замещенного заместителем A, или вместе с атомом N, связанным с ним, образуют 5-10 членный азотсодержащий гетероциклил, необязательно замещенный заместителем A;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила, необязательно замещенного заместителем A;
L1 представляет собой связь или -CRʼRʺ-, -NRʼ-, -S-, -SO2-, -S(O)-, -SONRʼ-, -SO2NRʼ- или -NRʼCONRʼ-, и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила, необязательно замещенного заместителем A;
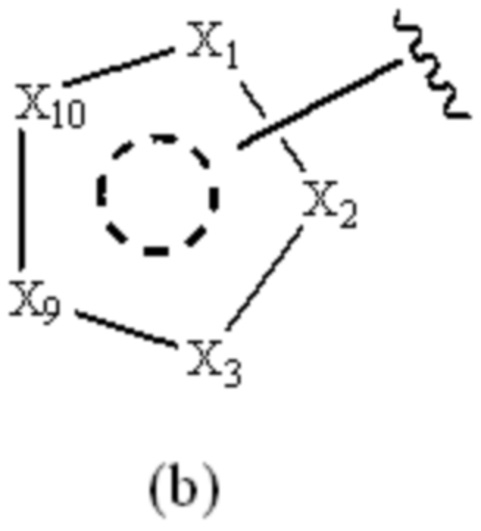
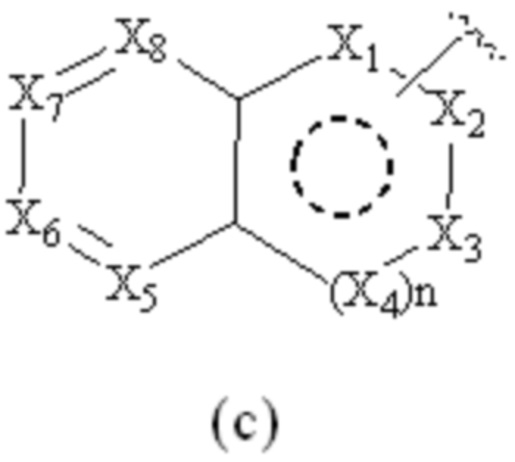
Cy1 представляет собой группу, представленную общей формулой (A), (a), (b) или (c) ниже, которая является незамещенной или замещена одним или несколькими Ra:
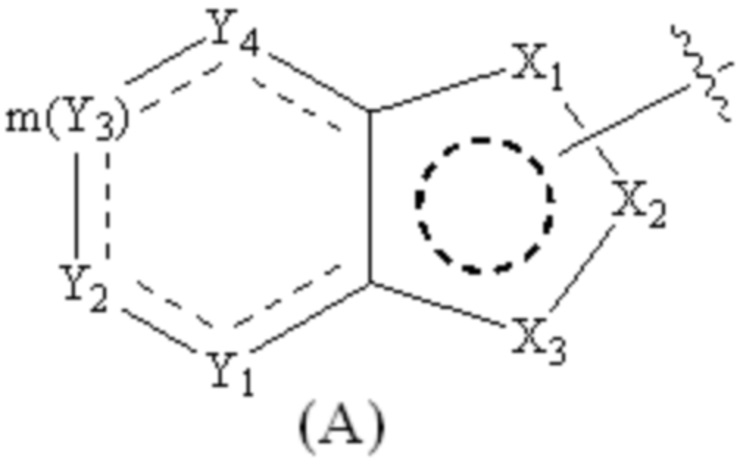
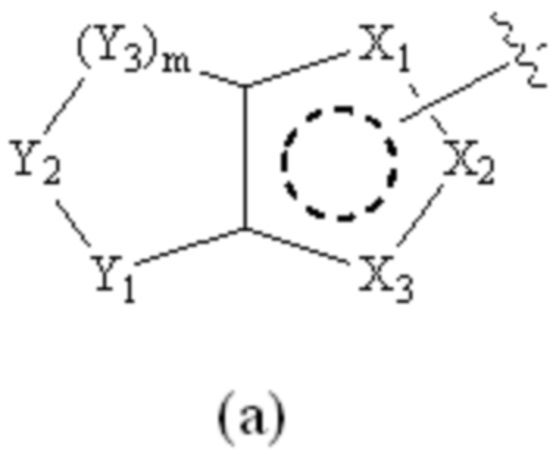
 ;
;
m обозначает целое число 0 до 3, и n обозначает целое число 0 до 2;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CRcRc, NRd, O и S;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CRcRc, NRd, O и S, X5, X6, X7 и X8, каждый, независимо, выбраны из CRcRc и NRd, и по меньшей мере один из X1, X2 и X3 представляет собой NRd;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбоксила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C2-6 алкенила, необязательно замещенного одним или несколькими Rb, C2-6 алкинила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламиносульфонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминосульфонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламиносульфонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминосульфонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb, и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb, и
Cy2, каждый, независимо, выбран из 3-12 членного циклоалкила, 3-12 членного циклоалкенила, 3-12 членного гетероциклила, 6-10 членного арила и 5-14 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкиламиносульфонила, (C1-6 алкил)2 аминосульфонила, C1-6 алкилсульфониламино и C1-6 алкилсульфонила;
заместители A, каждый, независимо, выбраны из гидроксила, амино, карбоксила, циано, нитро, галоген, аминокарбонила, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, амино C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкилсульфониламино, C1-6 алкиламиносульфонила, (C1-6 алкил)2 аминосульфонила, C1-6 галогеналкила, C1-6 галогеналкокси, C1-6 алкилсульфонила, C1-6 алкилтио, 3-12 членного циклоалкила, 6-10 членного арила, 3-12 членного гетероциклила, 5-14 членного гетероарила и оксо;
Rc отсутствует или, когда он присутствует, каждый, независимо, выбран из атома водорода; или два Rc вместе образуют оксо группу;
Rd отсутствует или, когда он присутствует, каждый, независимо, выбран из атома водорода;
 обозначает одинарную связь или двойную связь;
обозначает одинарную связь или двойную связь;
 обозначает двойную связь, необязательно имеющуюся в кольцевой структуре;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре;
при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra; и
при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O.
Техническое решение 2: Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с техническим решением 1,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 представляет собой связь или -CRʼRʺ-, -NRʼ- или -S-, и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила;
m обозначает целое число 0 до 3, и n обозначает целое число 0 до 2;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CRcRc и NRd;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CRcRc и NRd, и по меньшей мере один из X1, X2 и X3 представляет собой NRd;
X5, X6, X7 и X8, каждый, независимо, выбраны из CRcRc и NRd;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
заместители A, каждый, независимо, выбраны из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 галогеналкила, C1-6 галогеналкокси, C1-6 алкилтио, 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила, 5-10 членного гетероарила и оксо;
предпочтительно, каждый Ra независимо выбран из гидроксила, амино, циано, галогена, аминокарбонила, C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкокси, необязательно замещенного одним или несколькими Rb, C1-4 алкокси C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкокси C1-4 алкокси, необязательно замещенного одним или несколькими Rb, C1-4 алкилтио, необязательно замещенного одним или несколькими Rb, C1-4 алкилтио C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-4 алкиламино C1-4 алкила, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 амино C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-4 алкиламинокарбонил C1-4 алкила, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 аминокарбонил C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбониламино C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбонил C1-4 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-4 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-4 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb;
предпочтительно, каждый Rb независимо выбран из гидроксила, амино, циано, галогена, аминокарбонила, C1-4 алкила, C1-4 алкокси, амино C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси C1-4 алкила, C1-4 алкокси C1-4 алкокси, амино C1-4 алкокси, C1-4 галогеналкокси, C1-4 алкилтио, C1-4 алкиламино, (C1-4 алкил)2 амино, C1-4 алкиламинокарбонила, (C1-4 алкил)2 аминокарбонила, C1-4 алкилкарбониламино и C1-4 алкилкарбонила;
предпочтительно, заместители A, каждый, независимо, выбраны из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-4 алкила, C1-4 алкокси, амино C1-4 алкила, C1-4 алкокси C1-4 алкила, C1-4 алкокси C1-4 алкокси, амино C1-4 алкокси, C1-4 алкиламино, (C1-4 алкил)2 амино, C1-4 алкиламинокарбонила, (C1-4 алкил)2 аминокарбонила, C1-4 алкилкарбониламино, C1-4 алкилкарбонила, C1-4 галогеналкила, C1-4 галогеналкокси, C1-4 алкилтио, 3-6 членный циклоалкила, 5-10 членного гетероциклила, фенила, нафтила, 5-10 членного гетероарила и оксо;
предпочтительно, по меньшей мере один из Y1, Y2, Y3 и Y4 представляет собой C=O;
предпочтительно, в формуле (A), когда Ra присутствует, по меньшей мере один Ra присоединен к одному из Y1, Y2, Y3 и Y4;
предпочтительно, в формуле (a), когда Ra присутствует, по меньшей мере один Ra присоединен к одному из Y1, Y2 и Y3;
предпочтительно, в формуле (c) по меньшей мере один Ra присутствует, и по меньшей мере один Ra присоединен к одному из X5, X6, X7 и X8;
предпочтительно, в формуле (A) группа L1 присоединена к X1, X2 или X3 в формуле (A);
предпочтительно, в формуле (A) группа L1 присоединена к X1, X2 или X3 в формуле (A);
предпочтительно, в формуле (c) группа L1 присоединена к X1, X2, X3 или X4 в формуле (c);
предпочтительно, группа L1 присоединена к атому N.
Техническое решение 3. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с техническим решением 1 или техническим решением 2:
где Cy1 представляет собой группу, представленную общей формулой (A-1), (A-2), (A-3), (a), (b) или (c) ниже, которая является незамещенной или замещена одним или несколькими Ra:
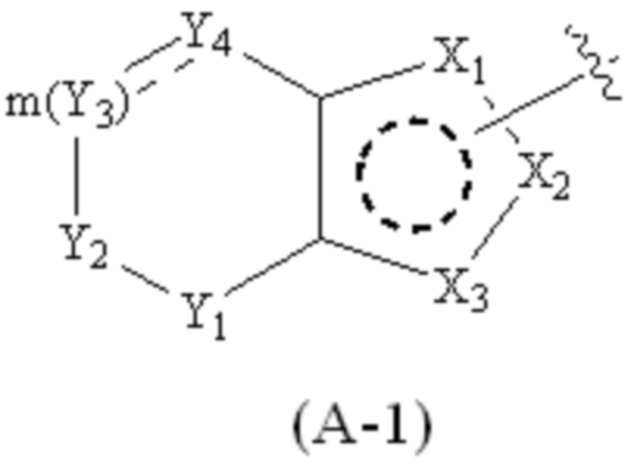
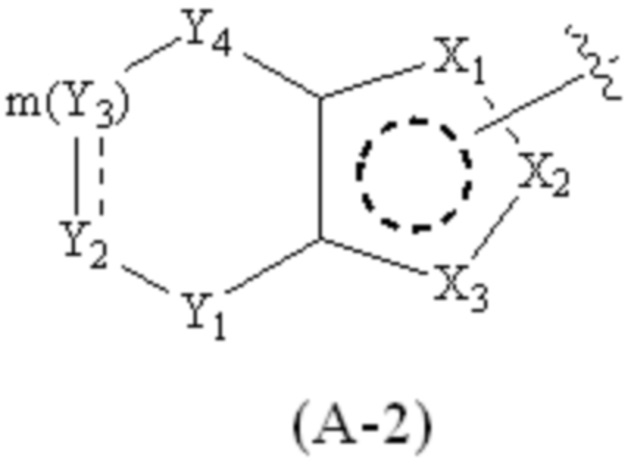
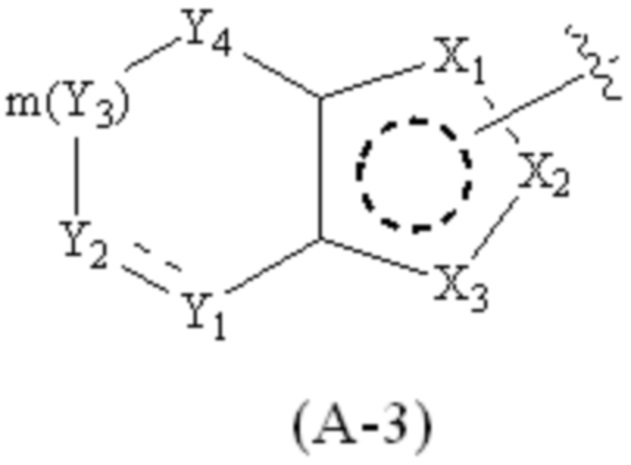
.
Техническое решение 4. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с любым из технических решений 1-3,
где Cy1 представляет собой группу, представленную общей формулой (A-11), (a-1), (a-2), (b-1), (c-1) или (c-2) ниже, которая является незамещенной или замещена одним или несколькими Ra:
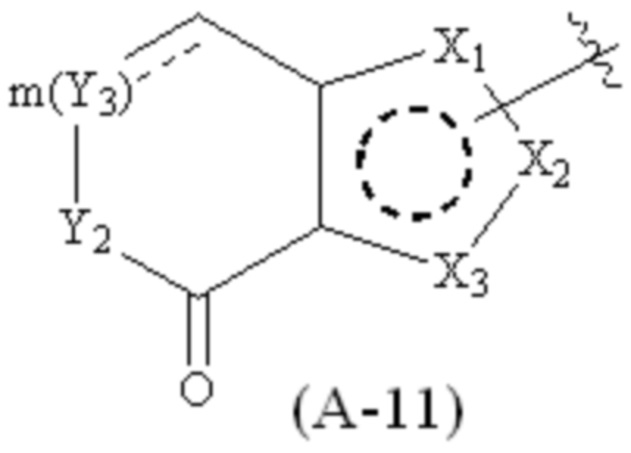
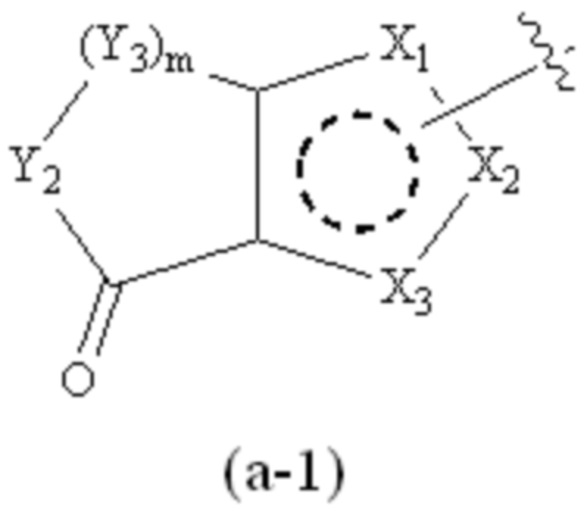
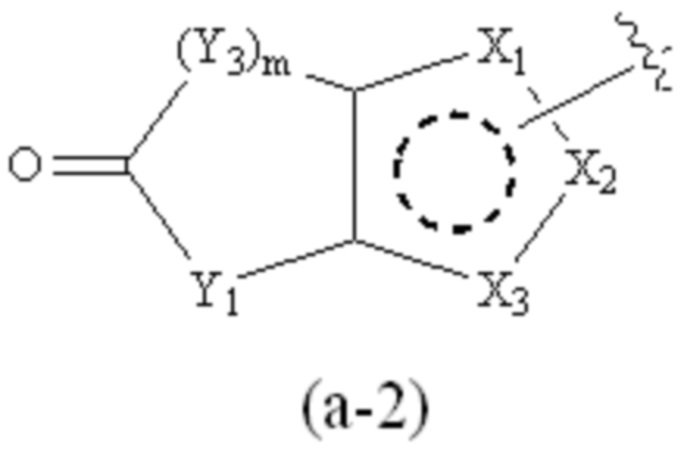
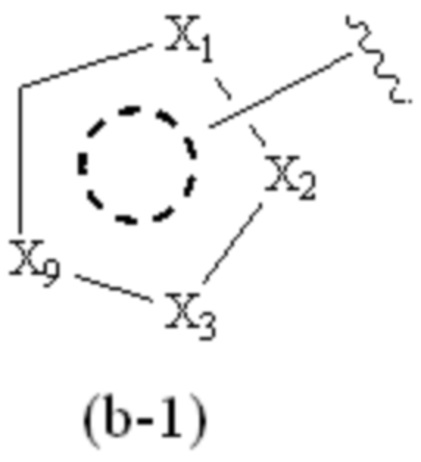
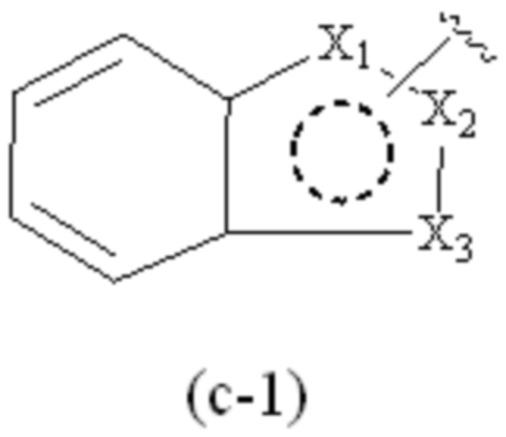
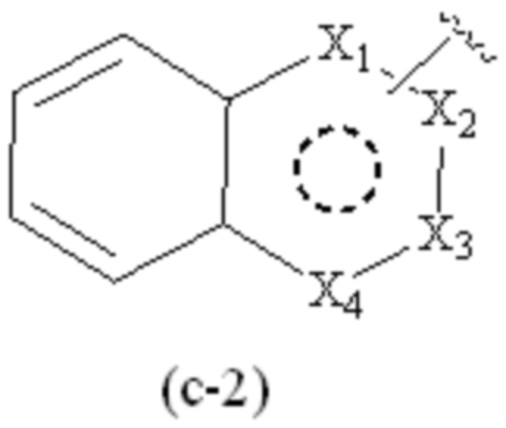 ;
;
где m обозначает целое число, равное 1 или 2;
Y1, Y2 и Y3, каждый, независимо, выбраны из CH2, NH, CH и N;
X1, X2, X3, X4 и X9, каждый, независимо, выбраны из CH2, CH, N, NH и C=O, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
при условии, что, когда Cy1 соответствует группе формулы (c-1) или (c-2), группа формулы (c-1) или (c-2) является замещенной одним или несколькими Ra, и
при условии, что, когда Cy1 соответствует формуле (b-1), X1, X2, X3 и X9 не представляют собой C=O;
предпочтительно, в формуле (A-11), когда Ra присутствует, по меньшей мере один Ra присоединен к Y2;
предпочтительно, в формуле (a-1), когда Ra присутствует, по меньшей мере один Ra присоединен к Y2;
предпочтительно, в формуле (a-2), когда Ra присутствует, по меньшей мере один Ra присоединен к Y1;
предпочтительно, в формуле (b-1), когда Ra присутствует, по меньшей мере один Ra присоединен к циклическому атому углерода;
предпочтительно, в формулах (c-1) и (c-2), по меньшей мере один Ra присутствует, и по меньшей мере один Ra присоединен к группе бензольного кольца.
Техническое решение 5. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с любым из технических решений 1-4,
где Cy1 представляет собой группу, представленную общей формулой (A-11) или (a-1) ниже, которая является незамещенной или замещена одним или несколькими Ra:
;
m обозначает целое число, равное 1 или 2;
Y2 и Y3, каждый, независимо, выбраны из CH2, NH, CH и N;
X1, X2 и X3, каждый, независимо, выбраны из CH2, CH, N, NH и C=O, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, и Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
предпочтительно, Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила, нафтила и 5-6 членного гетероарила;
предпочтительно, Ra, каждый, независимо, выбран из галогена, циано, C1-6 алкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-6 членного циклоалкила, 3-8 членного циклоалкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фенила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, и 5-6 членного гетероарила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
более предпочтительно, Ra, каждый, независимо, выбран из галогена, циано, C1-4 алкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-5 членного циклоалкила, 3-5 членного циклоалкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фенила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, и 5-6 членного содержащего азот гетероарила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
предпочтительно, в формуле (A-11) X1 и X2, каждый, независимо, выбраны из CH2, CH, N и NH;
предпочтительно, в формуле (a-1) X1 и X2, каждый, независимо, выбраны из CH2, CH, N и NH;
предпочтительно, в формулах (A-11) и (a-1) Y2 представляет собой NH;
предпочтительно, в формулах (A-11) и (a-1) Y3 представляет собой CH2 или CH;
предпочтительно, в формулах (A-11) и (a-1), когда Ra присутствует, по меньшей мере один Ra присоединен к положению Y2;
предпочтительно, в формулах (A-11) и (a-1) группа L1 присоединена к X1, X2 или X3;
предпочтительно, в формулах (A-11) и (a-1), группа L1 присоединена к атому N.
Техническое решение 6. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с техническим решением 5,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 представляет собой связь;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
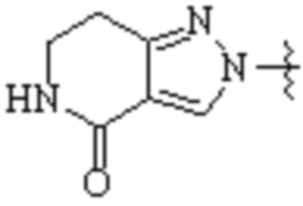 ,
, 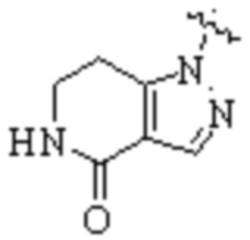 ,
, 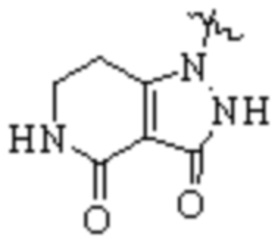 ,
, 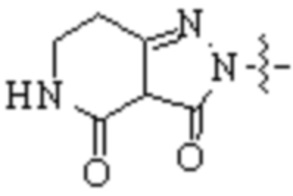 ,
, 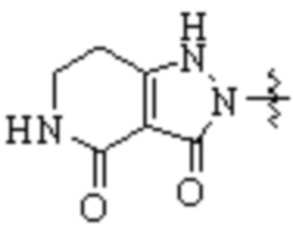 ,
,  ,
, 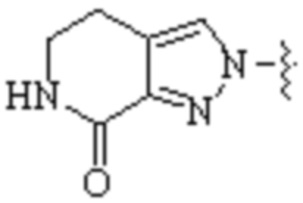 ,
, 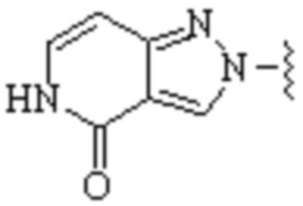 ,
, 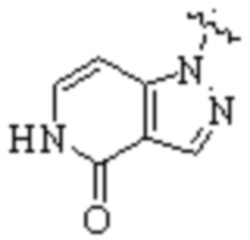 ,
, 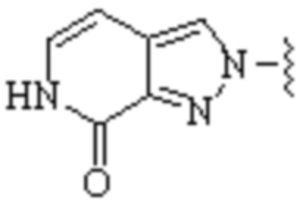 ,
, 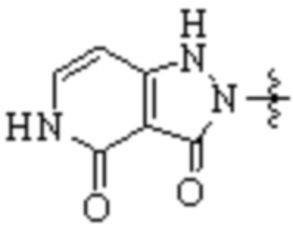 ,
, 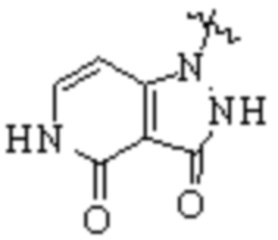 ,
, 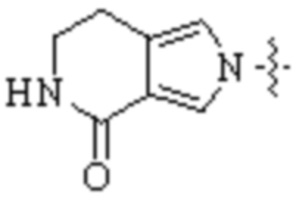 ,
, 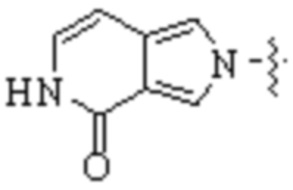 ,
, 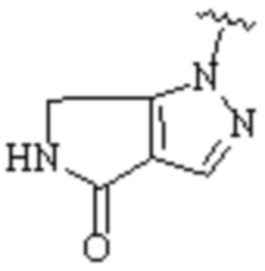 ,
, 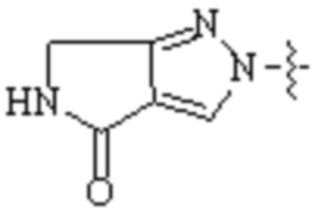 ,
, 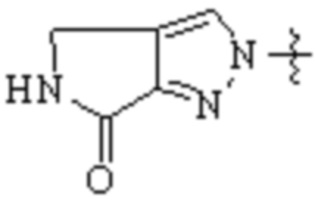 ,
,  ;
;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb и Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
предпочтительно, Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила и 5-6 членного гетероарила;
предпочтительно, Ra, каждый, независимо, выбран из галогена, циано, C1-6 алкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-6 членного циклоалкила, 3-8 членного циклоалкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фенила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, и 5-6 членного гетероарила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
Более предпочтительно, Ra, каждый, независимо, выбран из фтора; хлора; брома; циано; метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила и трет-бутила, необязательно замещенных по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-5 членного циклоалкила; циклопропила, циклобутила и циклопентила, необязательно замещенных по меньшей мере одним из галогена и C1-6 алкила; фенила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила; и пирролила, имидазолила, пиразолила, оксазолила и тиазолила, необязательно замещенных по меньшей мере одним из галогена и C1-6 алкила.
Техническое решение 7. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с любым из технических решений 1-4,
где Cy1 представляет собой группу, представленную общей формулой (b-1) ниже, которая является незамещенной или замещена одним или несколькими Ra:
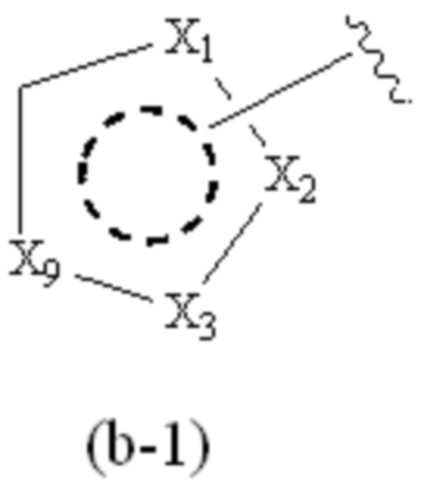 ;
;
X1, X2, X3 и X9, каждый, независимо, выбраны из CH2, CH, N и NH, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb, и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
предпочтительно, Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила и 5-6 членного гетероарила;
предпочтительно, Ra, каждый, независимо, выбран из C1-6 алкила, необязательно замещенного галогеном; галогена; аминокарбонила; C1-6 алкиламинокарбонила, необязательно замещенного галогеном; (C1-6 алкил)2 аминокарбонила, необязательно замещенного галогеном; 3-8 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; 5-6 членного гетероариламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; и 5-10 членного гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
более предпочтительно, Ra, каждый, независимо, выбран из аминокарбонила, C1-4 алкиламинокарбонила, необязательно замещенного галогеном, (C1-4 алкил)2 аминокарбонила, необязательно замещенного галогеном, 3-5 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси и 5-6 членного азотсодержащего гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
Предпочтительно, в формуле (b-1) группа L1 присоединена к атому N в формуле (b-1); и
предпочтительно, в формуле (b-1), когда Ra присутствует, по меньшей мере один Ra присоединен к атому углерода формулы (b-1).
Техническое решение 8. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с техническим решением 7,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 представляет собой связь;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
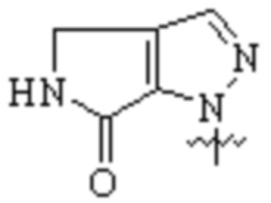
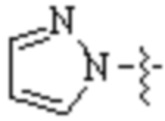 ,
, 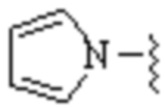 или
или  ;
;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb, и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
предпочтительно, Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила и 5-6 членного гетероарила;
предпочтительно, Ra, каждый, независимо, выбран из C1-6 алкила, необязательно замещенного галогеном; галогена; аминокарбонила; C1-6 алкиламинокарбонила, необязательно замещенного галогеном; (C1-6 алкил)2 аминокарбонила, необязательно замещенного галогеном; 3-8 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; 5-6 членного гетероариламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; и 5-10 членного гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
более предпочтительно, Ra, каждый, независимо, выбран из аминокарбонила, метиламинокарбонила, необязательно замещенного галогеном, этиламинокарбонила, необязательно замещенного галогеном, пропиламинокарбонила, необязательно замещенного галогеном, изопропиламинокарбонила, необязательно замещенного галогеном, н-аминокарбонила, необязательно замещенного галогеном, изобутиламинокарбонила, необязательно замещенного галогеном, втор-бутиламинокарбонила, необязательно замещенного галогеном, трет-бутиламинокарбонила, необязательно замещенного галогеном, циклопропиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, циклобутиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, циклопентиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, пирролидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, пиперидилкарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила и пиперазинилкарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила.
Техническое решение 9. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с любым из технических решений 1-4,
где Cy1 представляет собой группу, представленную общей формулой (c-1) ниже, которая замещена одним или несколькими Ra:
,
X1, X2 и X3, каждый, независимо, выбраны из CH2, CH, N, NH и C=O, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb, и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
предпочтительно, Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила и 5-6 членного гетероарила;
предпочтительно, Ra, каждый, независимо, выбран из C1-6 алкила, необязательно замещенного галогеном; галогена; аминокарбонила; C1-6 алкиламинокарбонила, необязательно замещенного галогеном; (C1-6 алкил)2 аминокарбонила, необязательно замещенного галогеном; 3-8 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; 5-6 членного гетероариламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; и 5-10 членного гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
более предпочтительно, Ra, каждый, независимо, выбран из аминокарбонила, C1-4 алкиламинокарбонила, необязательно замещенного галогеном, (C1-4 алкил)2 аминокарбонила, необязательно замещенного галогеном, 3-5 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси и 5-6 членного азотсодержащего гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
предпочтительно, в формуле (c-1) по меньшей мере один Ra присоединен к группе бензольного кольца в формуле (c-1);
предпочтительно, в формуле (c-1) группа L1 присоединена к X1, X2 или X3;
предпочтительно, в формуле (c-1) группа L1 присоединена к атому N в формуле (c-1).
Техническое решение 10. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с техническим решением 9,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, выбраны из водорода и C1-6 алкила;
L1 представляет собой связь;
Cy1 представляет собой следующую группу, замещенную одним или несколькими заместителями Ra:
 ;
;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb, и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb;
Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила и 5-10 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
предпочтительно, Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила и 5-6 членного гетероарила;
предпочтительно, Ra, каждый, независимо, выбран из C1-6 алкила, необязательно замещенного галогеном; галогена; аминокарбонила; C1-6 алкиламинокарбонила, необязательно замещенного галогеном; (C1-6 алкил)2 аминокарбонила, необязательно замещенного галогеном; 3-8 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; 5-6 членного гетероариламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; и 5-10 членного гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси;
предпочтительно, Ra, каждый, независимо, выбран из аминокарбонила, метиламинокарбонила, необязательно замещенного галогеном, этиламинокарбонила, необязательно замещенного галогеном, пропиламинокарбонила, необязательно замещенного галогеном, изопропиламинокарбонила, необязательно замещенного галогеном, н-аминокарбонила, необязательно замещенного галогеном, изобутиламинокарбонила, необязательно замещенного галогеном, втор-бутиламинокарбонила, необязательно замещенного галогеном, трет-бутиламинокарбонила, необязательно замещенного галогеном, циклопропиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, циклобутиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, циклопентиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; азетидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пирролидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пиперидилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пиперазинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, и морфолинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
Техническое решение 11. Соединение или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер в соответствии с техническим решением 1. Соединение выбрано из следующих:
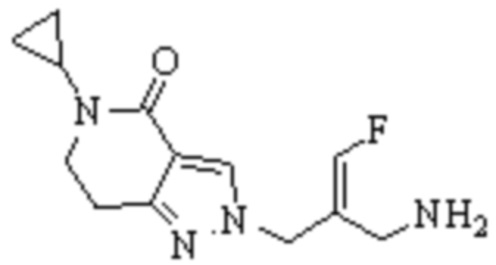
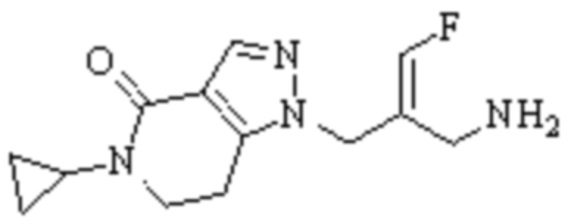
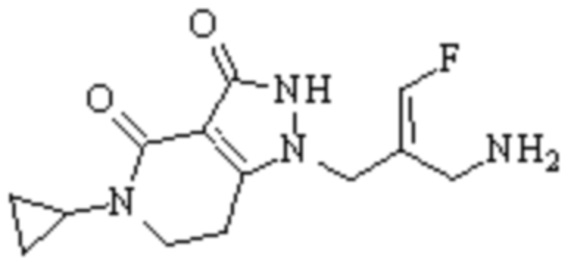
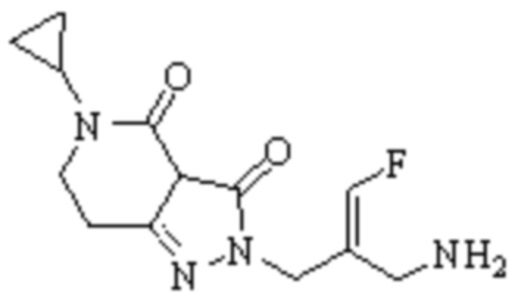
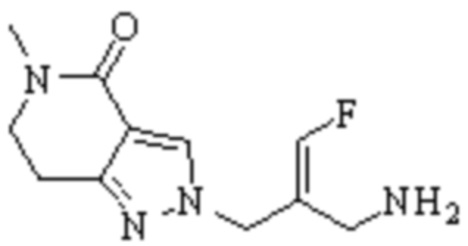
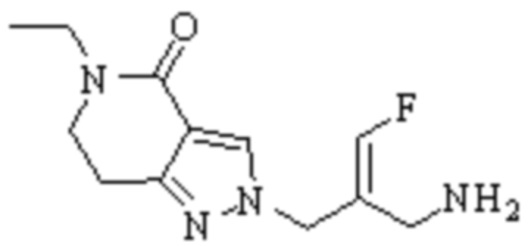
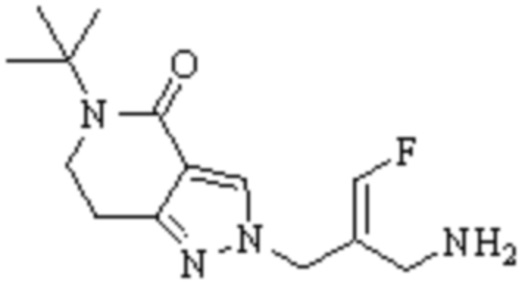
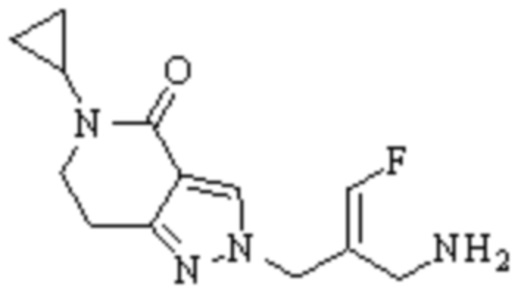
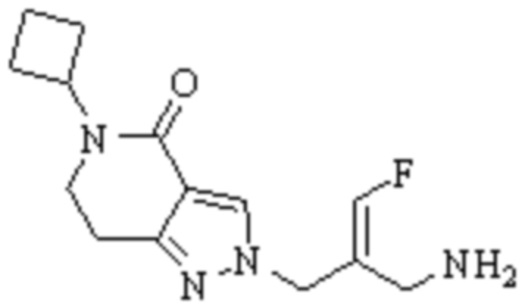
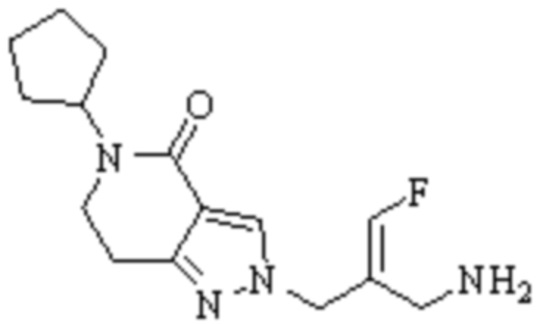
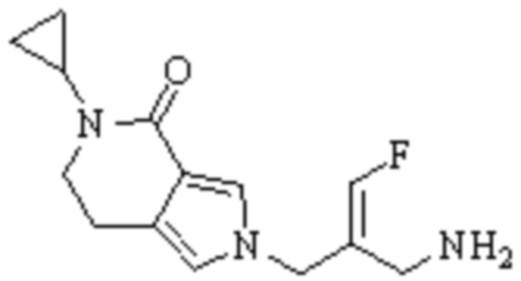
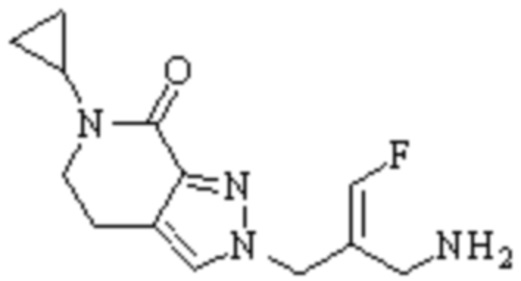
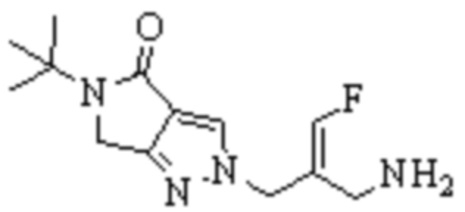
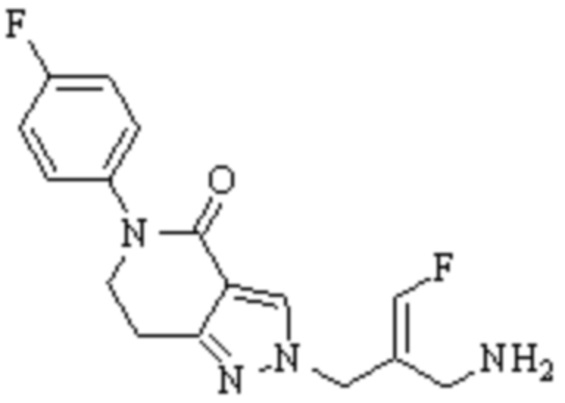
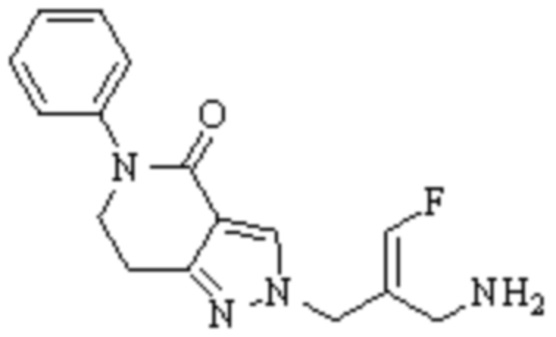
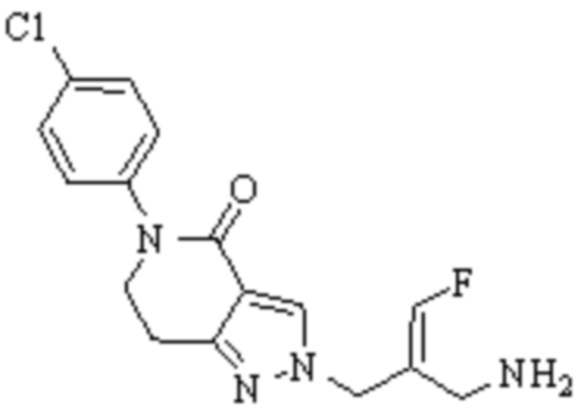
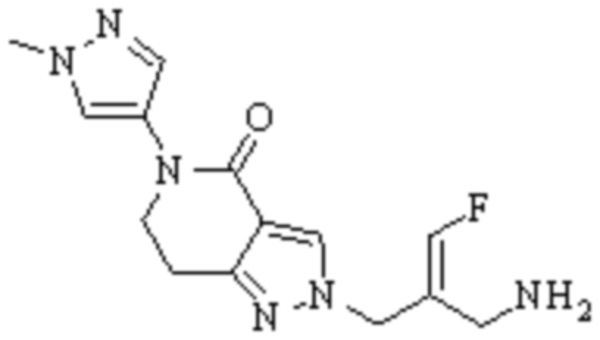
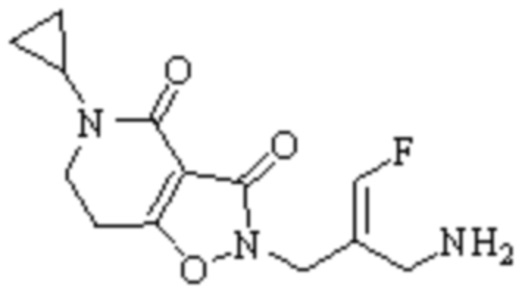
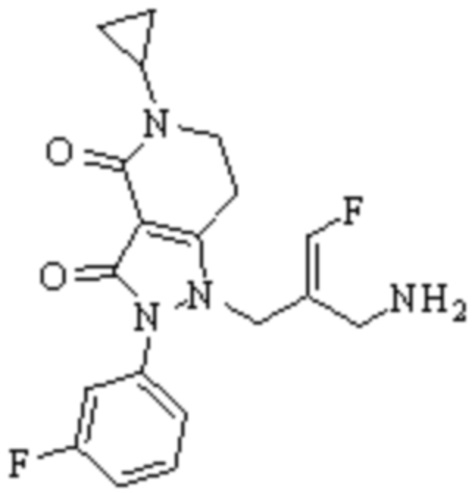
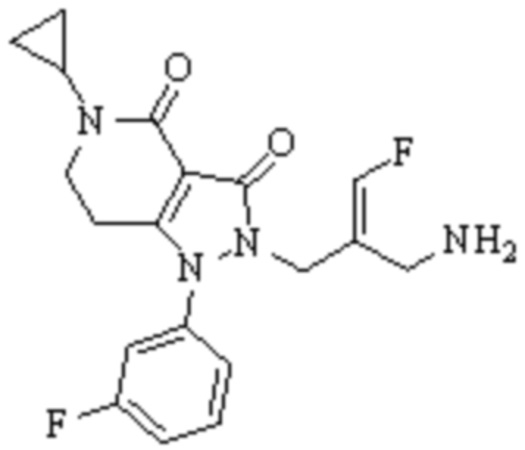
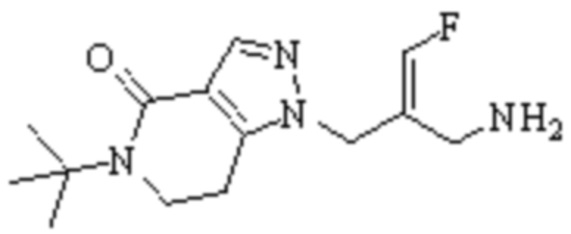

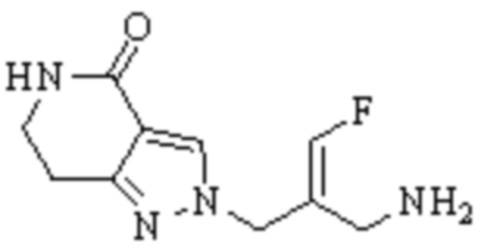
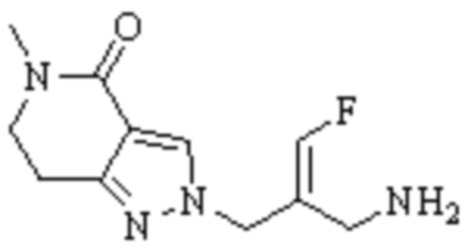
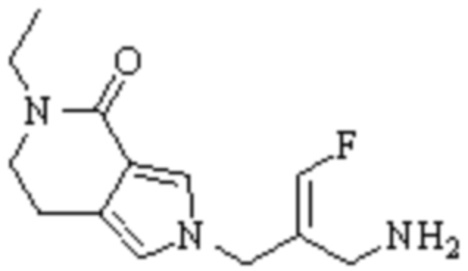
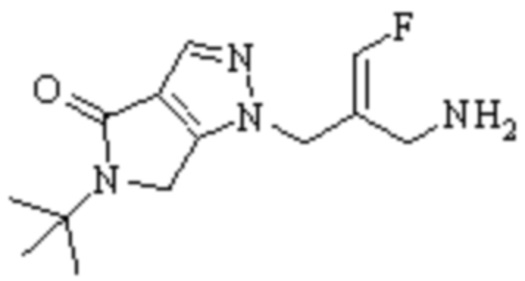
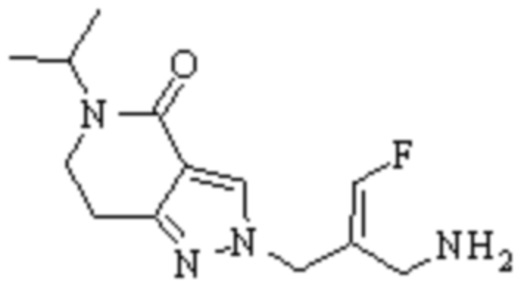
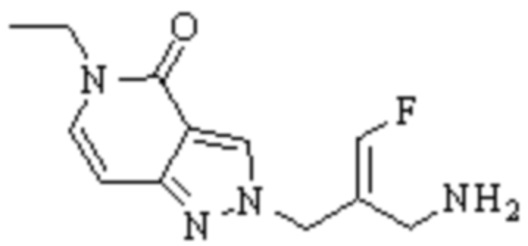
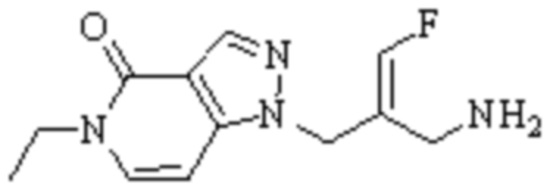
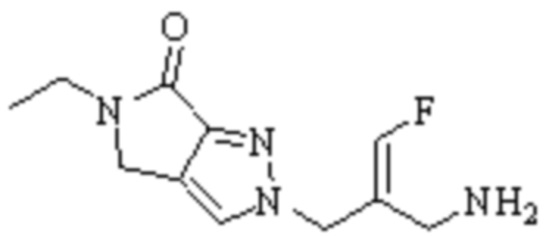
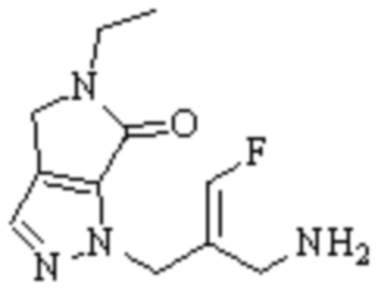
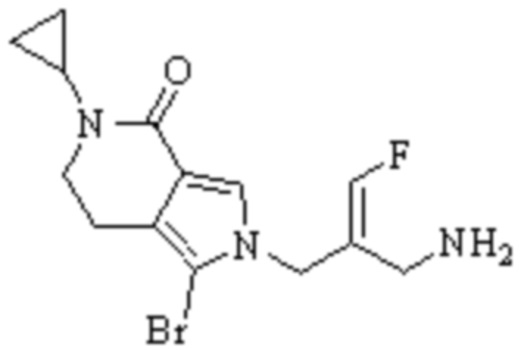
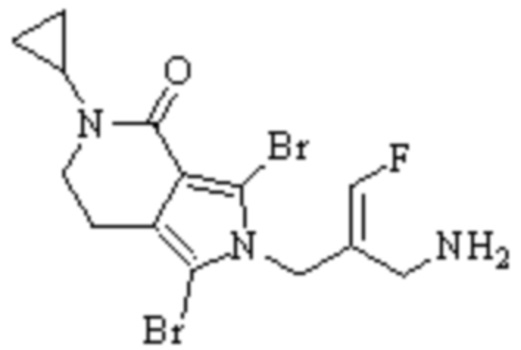
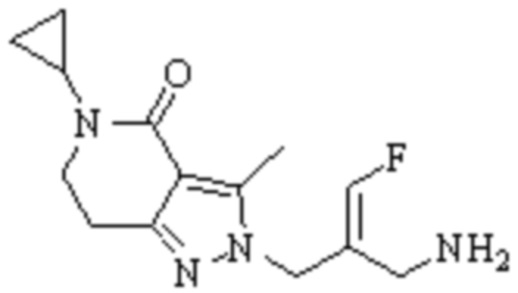
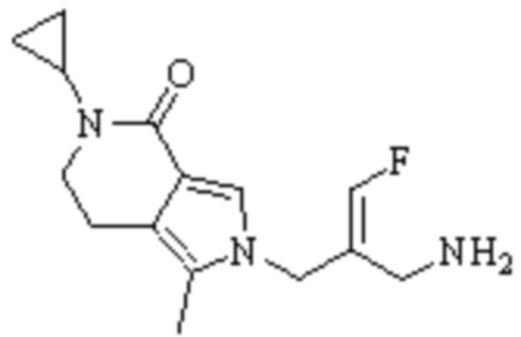
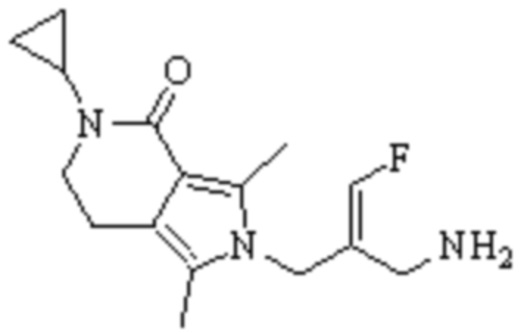
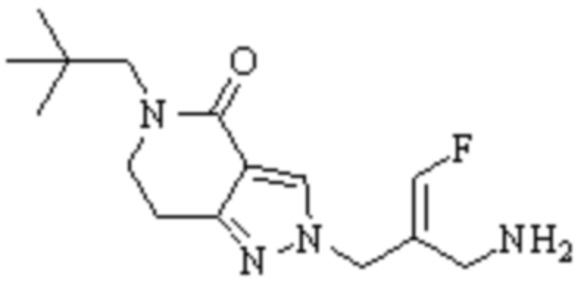
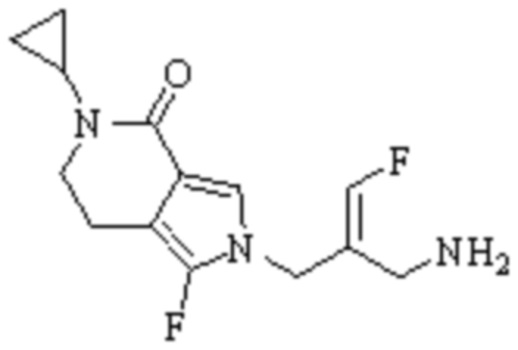
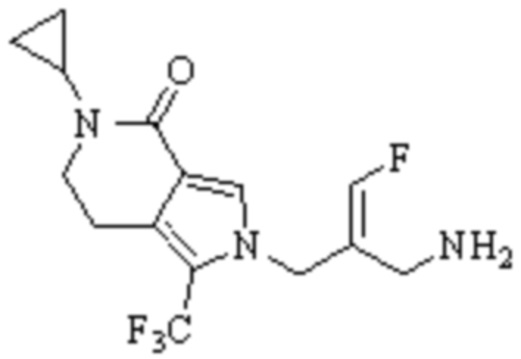
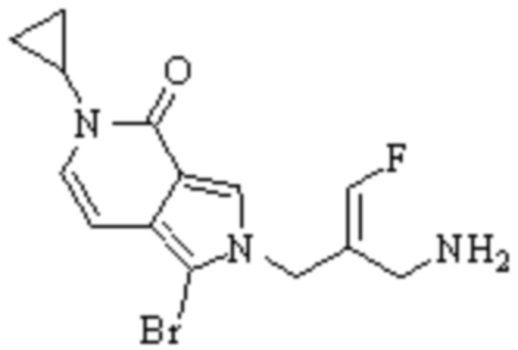
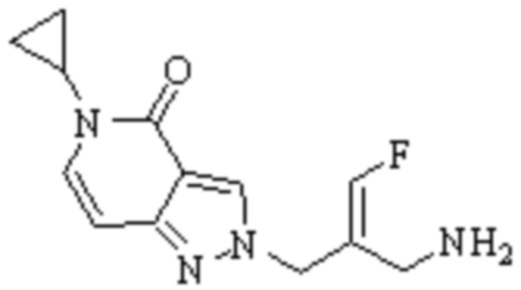
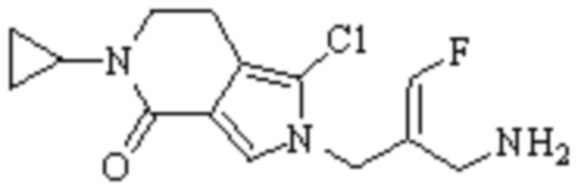
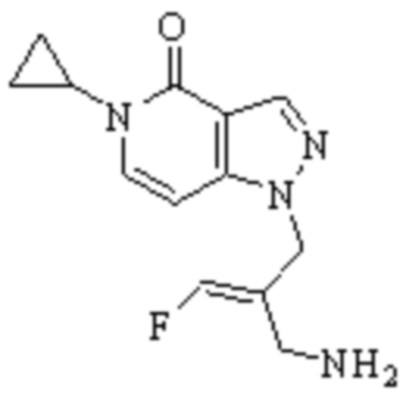
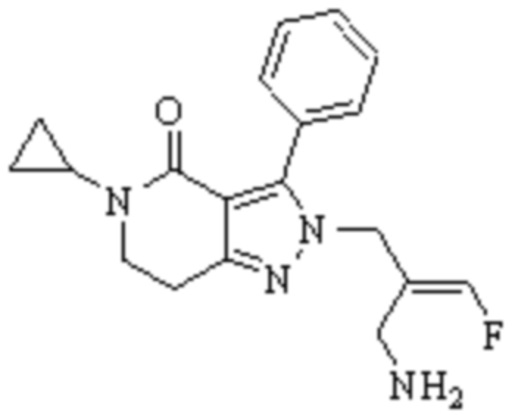

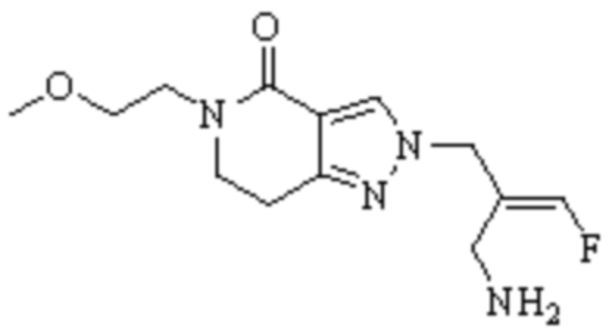
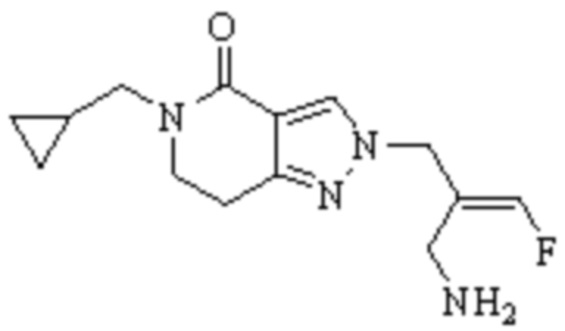
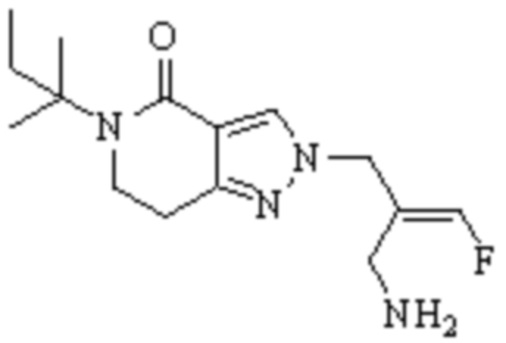
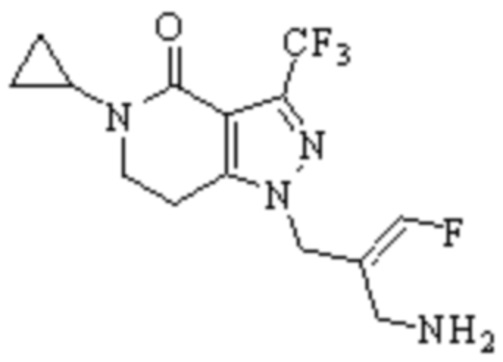
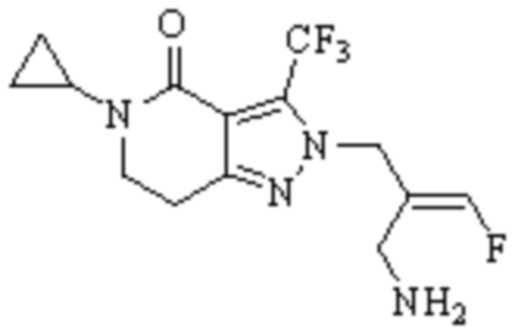
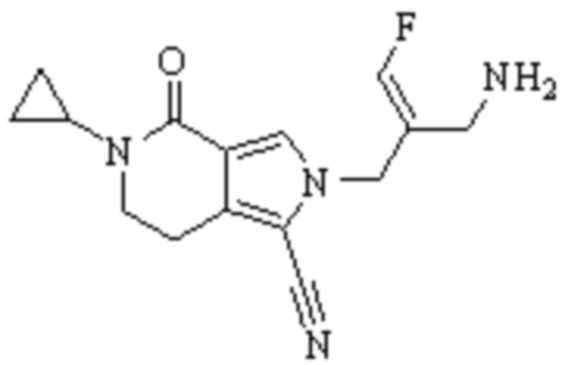
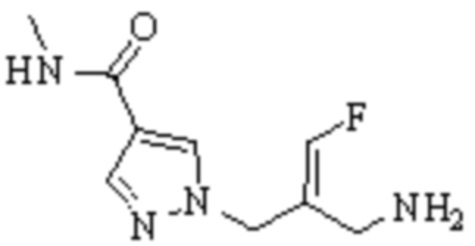
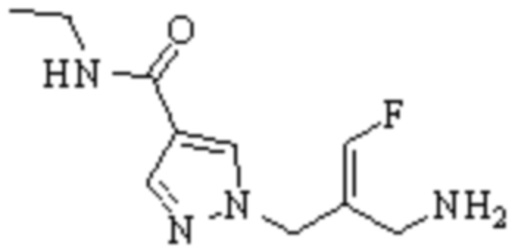
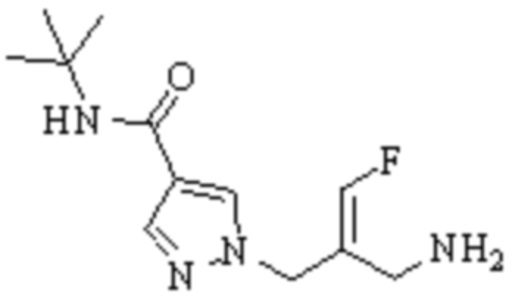
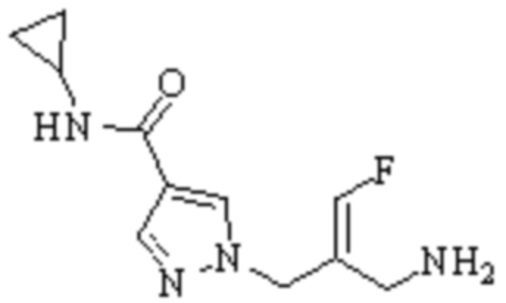
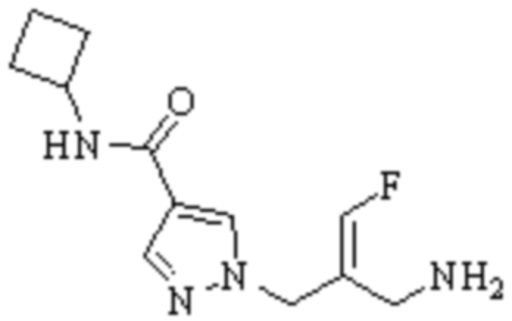
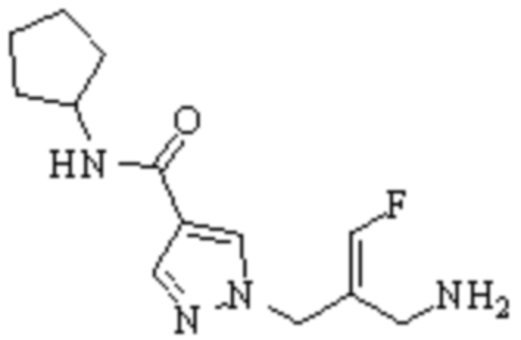
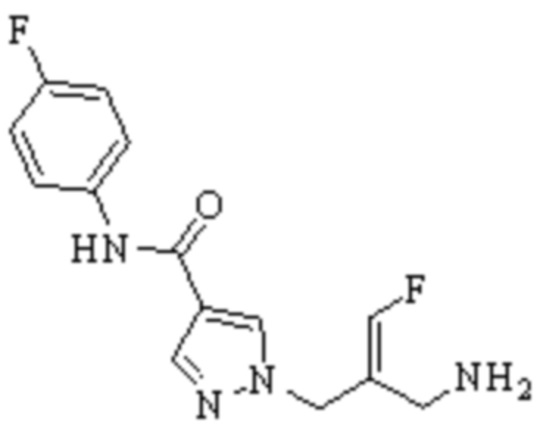
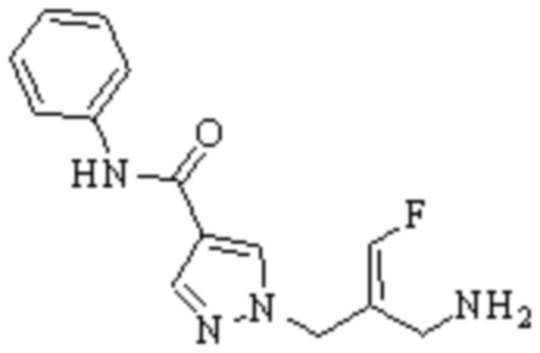
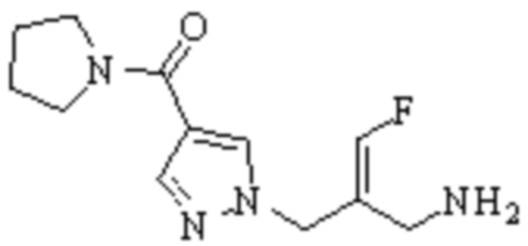
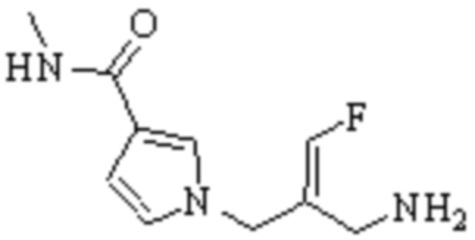
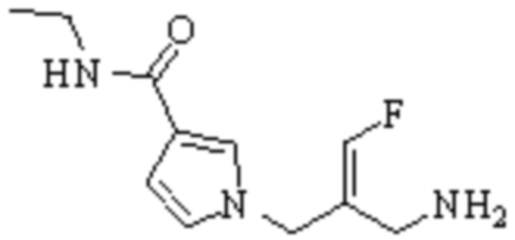
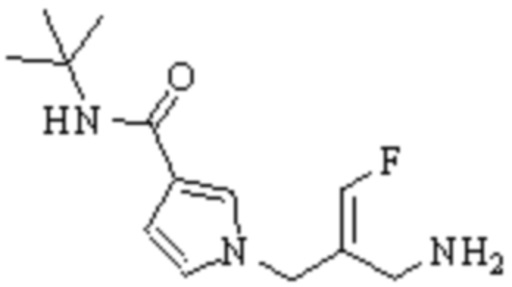
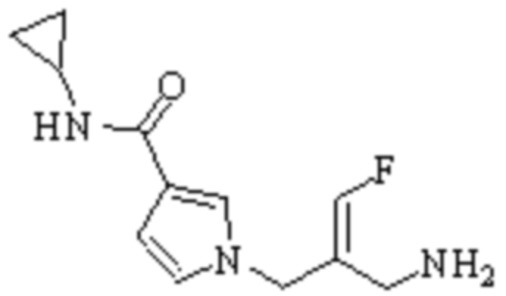
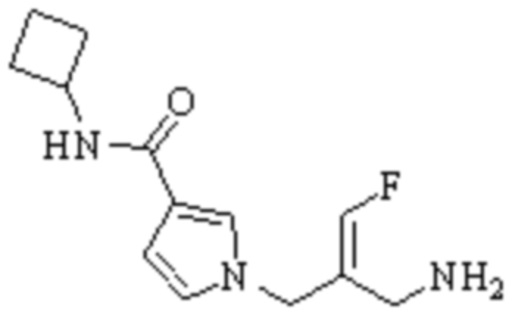
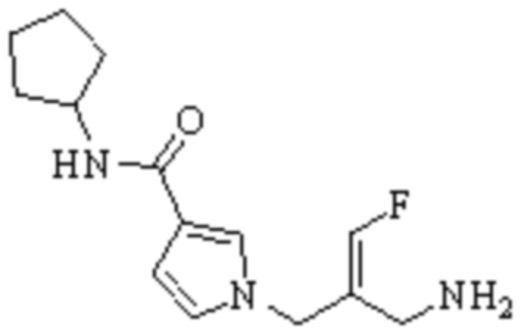
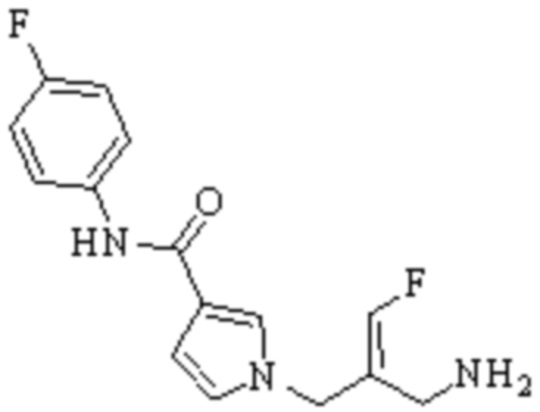
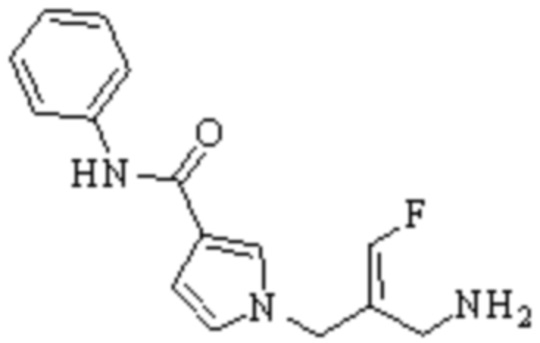
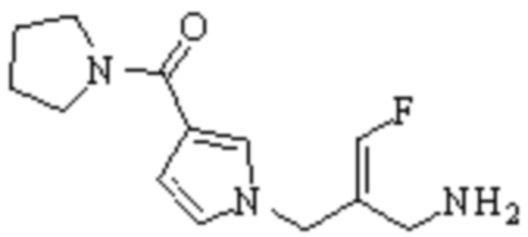
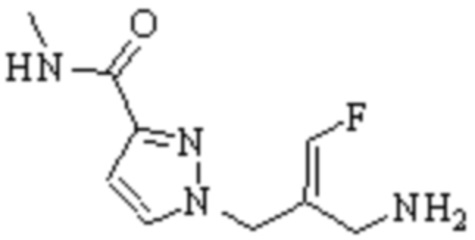
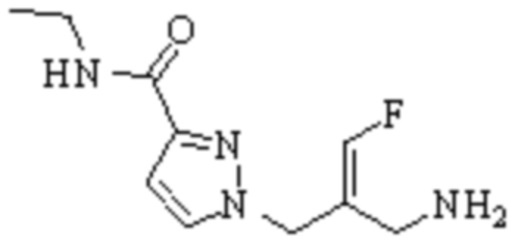
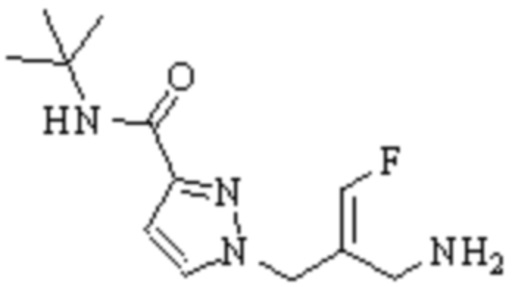
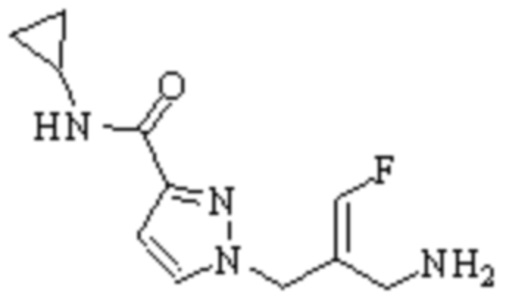

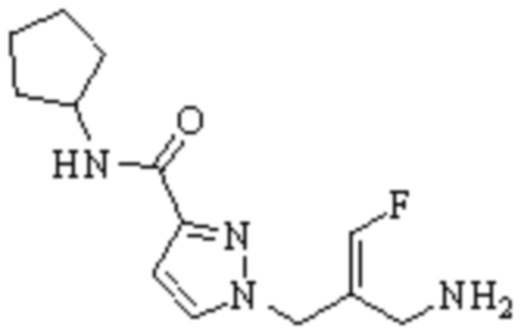
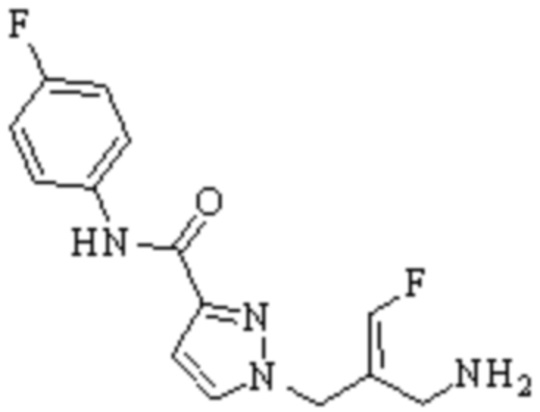
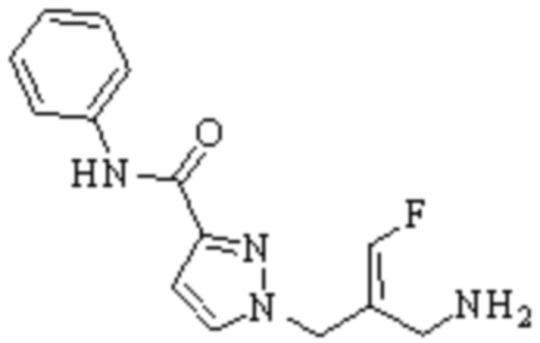
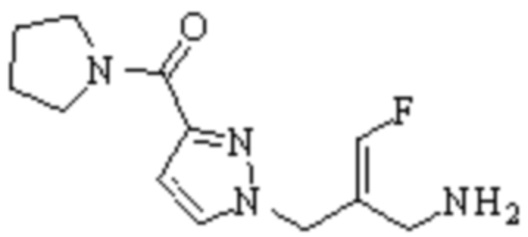
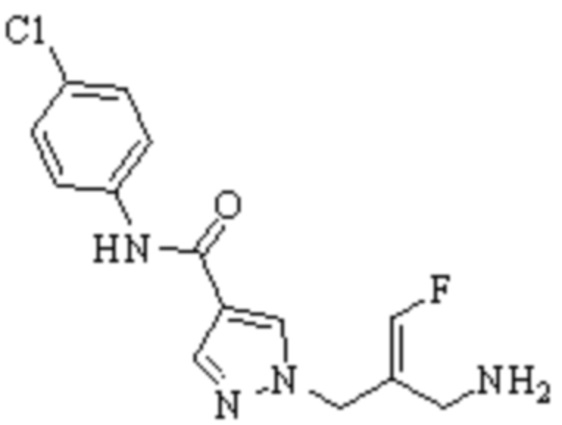
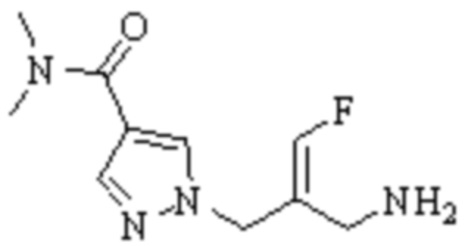
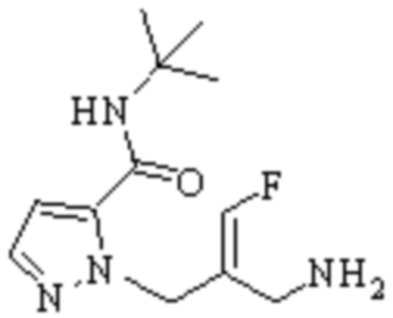
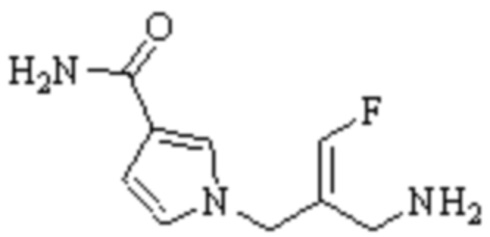
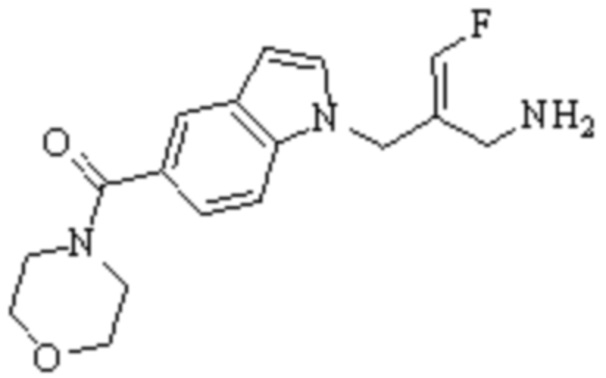
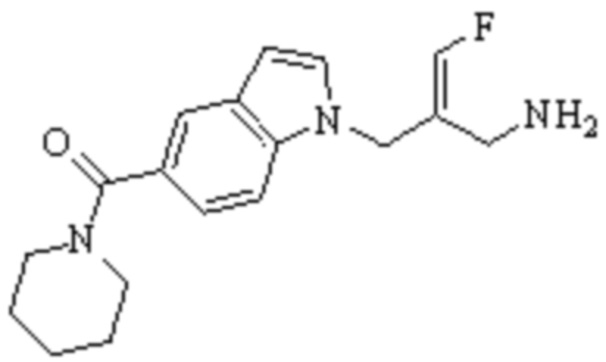
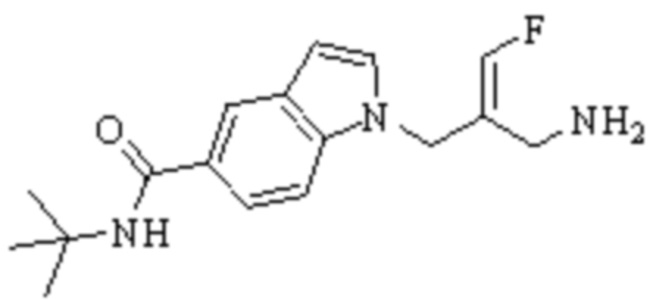
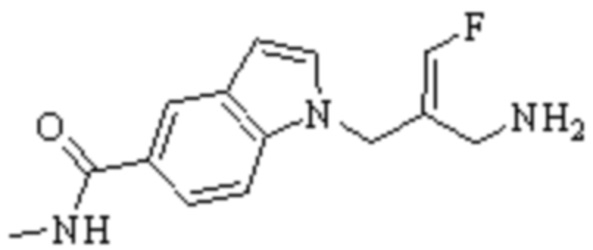
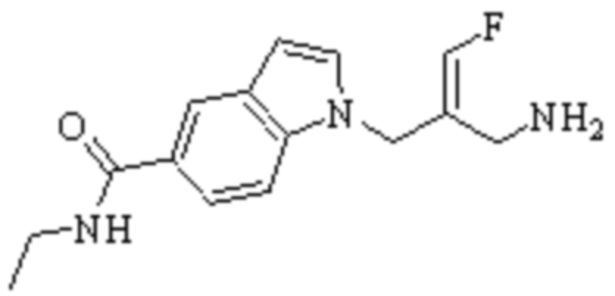
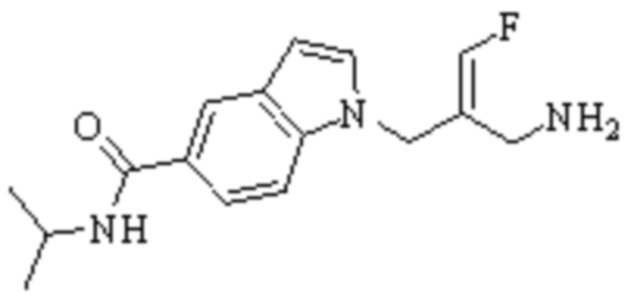
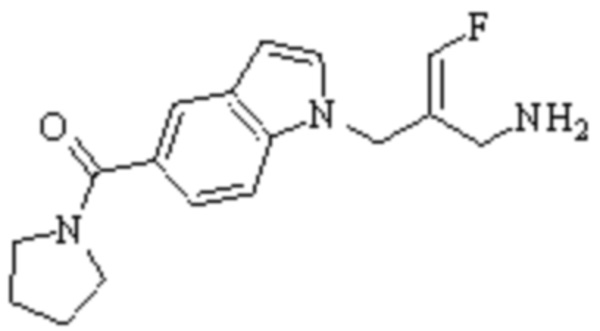
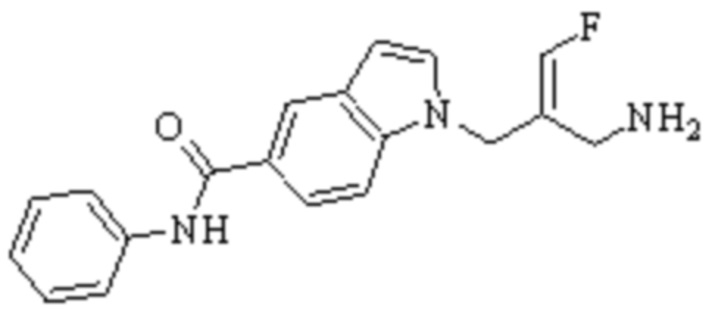
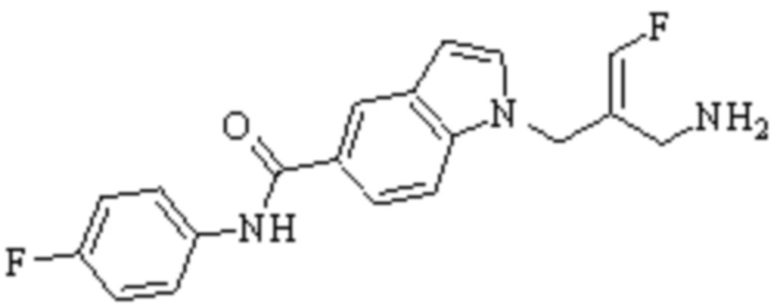
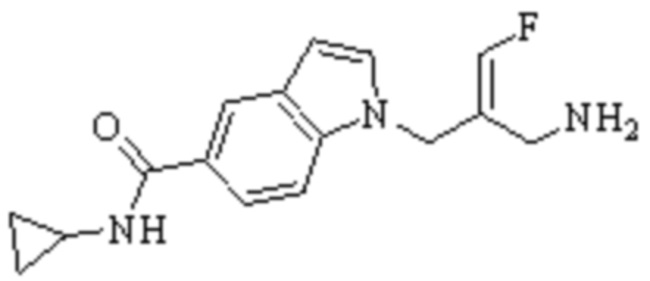

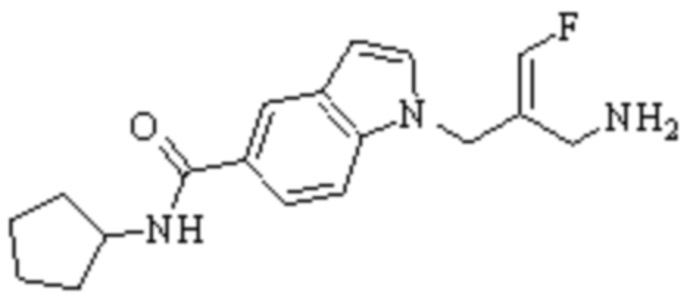
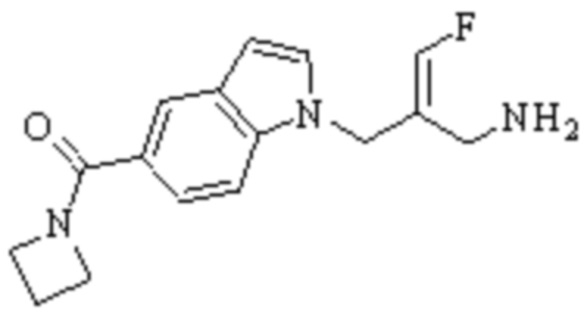

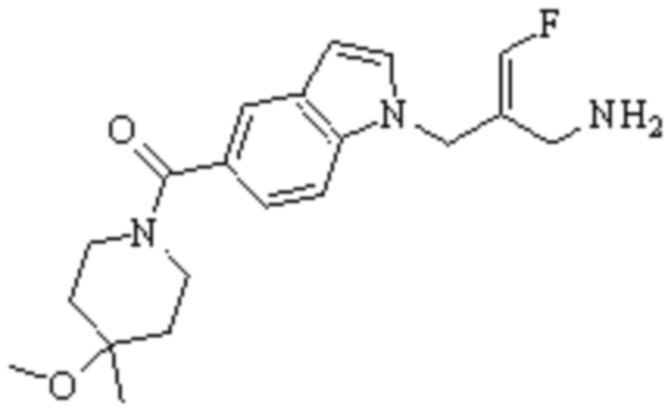
Техническое решение 12. Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль, сложный эфир, стереоизомер или таутомер в соответствии с любым из технических решений 1-11, где фармацевтическая композиция необязательно содержит один или несколько фармацевтически приемлемых носителей.
Техническое решение 13. Применение соединения или его фармацевтически приемлемой соли, сложного эфира, стереоизомера или таутомера в соответствии с любым из технических решений 1-11 или фармацевтическая композиция в соответствии с техническим решением 12 при производстве лекарственного препарата для профилактики и/или лечения заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им.
ЭФФЕКТ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено новое производное галогеналлиламина, которое является эффективным при профилактике и/или лечении заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им. В частности, соединение, представленное формулой I, и его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер по настоящему изобретению проявляют превосходную ингибирующую активность в отношении белка SSAO/VAP-1 и, таким образом, могут использоваться для профилактики и/или лечения заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им.
Более того, соединение по настоящему изобретению демонстрирует превосходное ингибирование белка SSAO/VAP-1 и демонстрирует превосходную селективность в отношении белка rhAOC1 и белка MAO. Следовательно, соединение по настоящему изобретению позволяет избежать других нежелательных побочных эффектов при предотвращении и/или лечении заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им.
Кроме того, по сравнению с существующими лекарственными средствами соединение по настоящему изобретению с трудом проникает через гематоэнцефалический барьер. Следовательно, соединение по настоящему изобретению имеет очень низкий риск токсичности для нервной системы, демонстрируя превосходную безопасность лекарственного средства.
Следовательно, настоящее изобретение может предоставить высокобезопасное соединение или его фармацевтически приемлемую соль, сложный эфир, стереоизомер или таутомер, которые могут предотвращать и/или лечить заболевания, связанные с белком SSAO/VAP-1 или опосредованные им.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Варианты осуществления настоящего изобретения описаны далее более подробно в связи с конкретными вариантами осуществления, но специалистам в данной области техники понятно, что конкретные варианты осуществления, описанные ниже, используются только для иллюстрации настоящего изобретения и не должны рассматриваться как ограничение объема настоящего изобретения. Напротив, настоящее изобретение предназначено для охвата всех альтернатив, модификаций и эквивалентов, которые могут быть включены в объем изобретения, определенный формулой изобретения. Если не указано иное, различные варианты осуществления настоящего изобретения можно комбинировать любым способом, и преобразования, модификации и изменения технических решений, полученные таким образом, также входят в объем настоящего изобретения
Определения
В настоящем изобретении выражение «Ca-b группа» (a и b обозначают целое число ≥1, и a<b) означает, что «группа» содержит от a до b атомов углерода, например, C1-6 алкил представляет собой алкил с 1-6 атомами углерода, C1-6 алкокси представляет собой алкокси с 1-6 атомами углерода, C3-8 циклоалкил представляет собой циклоалкил с 3-8 атомами углерода и C1-6 алкокси C1-6 алкил представляет собой группу, образованную связыванием алкокси, имеющего 1-6 атомов углерода, с алкилом, имеющим 1-6 атомов углерода. Кроме того, В настоящем изобретении выражение «группа» может также относиться к группе, содержащей две или более подгрупп, и в этом случае выражение «Ca-b» определяет количество атомов углерода во всей группе, содержащей две или более подгрупп. Например, выражение «C7-12 алкиларил» означает, общее количество атомов углерода в алкиларильной группе, содержащей алкильный фрагмент и арильный фрагмент, составляет 7-10, то есть оно может быть отнесено, но не ограничивается ими, к C1-6 алкилфенилу или к C1-2 алкилнафтилу.
В настоящем изобретении «группа» представляет собой одновалентную группу или двух- или более валентную группу, отвечающую требуемой валентности. Например, «циклоалкил» (обозначаемый также как циклоалкильная группа) включает одновалентную группу, полученную удалением атома водорода из циклоалкана, а также двух- или более валентную группу, полученную удалением двух или более атомов водорода от одного и того же атома углерода или двух или более разных атомов углерода циклоалкана. Например, когда «циклоалкил» служит концевой группой, он связан с другими частями структуры соединения в виде одновалентной группы, когда не несет заместителей; и когда он несет заместители, циклоалкил имеет соответствующее число валентностей (число заместителей +1) в соответствии с числом содержащихся заместителей. Специалисты в данной области могут однозначно определить валентность «группы». Кроме того, в настоящем изобретении, если «группа» представляет собой двух- или более валентную группу, предпочтительно соединять эти связи в группе с разными атомами (такими как, но не ограничиваясь этим, атомы углерода, атомы азота и т.д.).
«Галоген» или «атом галогена», описанные в настоящем изобретении, относится к фтору, хлору, брому и йоду, предпочтительно, фтору и хлору.
«C1-6 алкил», описанный в настоящем изобретении, относится к линейной или разветвленной алкильной группе, полученной удалением атома водорода у алкановой группы, содержащей от 1 до 6 атомов углерода, и включает линейный C1-6 алкил и разветвленный C1-6 алкил. Фактически, специалистам в данной области хорошо известно, что C1-6 алкил имеет по крайней мере три атома углерода, когда он имеет разветвленную цепь (разветвленный C1-6 алкил). Примеры «C1-6 алкила» могут включать, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутила н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1-метил-2-метилпропил и тому подобное. Термин «C1-4 алкил» относится к вышеуказанным примерам, содержащим от 1 до 4 атомов углерода.
«C2-6 алкенил», описанный в настоящем изобретении, относится к линейной или разветвленной алкенильной группе, полученной удалением атома водорода у алкена, содержащего от 2 до 6 атомов углерода и по меньшей мере одну двойную углерод-углеродную связь, например, винил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 1,3-бутадиен-1-ил, 1-пентен-3-ил, 2-пентен-1-ил, 3-пентен-1-ил, 3-пентен-2-ил, 1,3-пентадиен-1-ил, 1,4-пентадиен-3-ил, 1-гексен-3-ил, 1,4-гексадиен-1-ил и тому подобное. Предпочтительно, «C2-6 алкенил» содержит двойную углерод-углеродную связь.
«C2-6 алкинил», описанный в настоящем изобретении, относится к линейной или разветвленной алкенильной группе, полученной удалением атома водорода у алкина, содержащего от 2 до 6 атомов углерода и по меньшей мере одну тройную углерод-углеродную связь, например, этинил, пропинил, 2-бутин-1-ил, 2-пентин-1-ил, 3-пентин-1-ил, 4-метил-2-пентин-1-ил, 2-гексин-1-ил, 3-гексин-2-ил, 3-гексин-1-ил, 3-гексин-2-ил и тому подобное. Предпочтительно, «C2-6 алкинил» содержит тройную углерод-углеродную связь.
«C1-6 алкокси», описанный в настоящем изобретении, относится к группе, полученной путем присоединения через атомы кислорода «C1-6 алкила», определенного выше, к другим частям химической структурной формулы, то есть относится к группе «C1-6 алкил-O-», такой как группы, полученные связыванием с -O- групп, перечисленных в отношении вышеуказанного «C1-6 алкила», включая, но этим не ограничиваясь, метокси, этокси, пропокси, изопропокси, н-бутокси, трет-бутокси, н-пентилокси, неопентилокси, н-гексилокси и тому подобное. «C1-4 алкокси» относится к вышеуказанным примерам, содержащим от 1 до 4 атомов углерода, то есть к группе «C1-4 алкил-O-».
«C1-6 алкокси C1-6 алкокси» относится к группе, образованной замещением одного или нескольких атомов водорода в C1-6 алкокси на C1-6 алкокси.
«C1-6 алкиламино (C1-6 алкиламино)», «(C1-6 алкил)2 амино», «C1-6 алкилкарбониламино», «C1-6 алкиламинокарбонил», «C1-6 алкилкарбонил», «C1-6 алкиламиносульфонил», «C1-6 алкилсульфониламино», «C1-6 алкилсульфонил», «C1-6 алкилтио (C1-6 алкилтио)» и тому подобные, описанные в настоящем документе, относятся к группам, образованным путем присоединения соответствующего C1-6 алкила к соответствующим группам, таким как -NH2, -CO-NH2-, -NH2-CO-, -CO-, -NH2SO2-, -SO2NH2-, -SO2- и -S-.
«C1-6 алкокси C1-6 алкил», «C1-6 алкилтио C1-6 алкил», «C1-6 алкиламино C1-6 алкил», «C1-6 алкиламинокарбонил C1-6 алкил», «C1-6 алкилкарбониламино C1-6 алкил», «C1-6 алкилкарбонил C1-6 алкил», «C1-6 алкиламиносульфонил C1-6 алкил», «C1-6 алкилсульфониламино C1-6 алкил», «C1-6 алкилсульфонил C1-6 алкил» и тому подобные, описанные в настоящем документе, относятся к группам, образованным путем замещения одного или нескольких атомов водорода в C1-6 алкиле у C1-6 алкокси, C1-6 алкилтио, C1-6 алкиламино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкиламиносульфонила, C1-6 алкилсульфониламино и C1-6 алкилсульфонила.
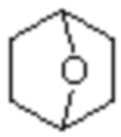
«Полициклическое кольцо» в настоящем изобретении относится к структуре многокольцевой системы, образованной двумя или более кольцевыми структурами, соединенными орто-конденсированной, спиро- или мостиковой связью. Орто-конденсированное кольцо относится к полициклической структуре, образованной двумя или более кольцевыми структурами, разделяющими два соседних кольцевых атома (то есть разделяющими связь) друг с другом. Мостиковое кольцо относится к полициклической структуре, образованной двумя или более кольцевыми структурами, разделяющими два несмежных кольцевых атома друг с другом. Спиро-кольцо относится к полициклической структуре, образованной двумя или более кольцевыми структурами, разделяющими один кольцевой атом друг с другом.
«Циклоалкил», описанный в настоящем изобретении, относится к одновалентной группе или двухвалентной группе (если требуется), полученной из циклоалкана, и циклоалкан включает моноциклический циклоалкан или полициклический циклоалкан и может иметь 3, 4, 5, 6, 7, 8 , 9, 10, 11 или 12 атомов углерода. Если не указано иное, несколько членный циклоалкил включает все возможные образованные моноциклические и полициклические (включая конденсированные в виде орто-, спиро- или мостиковые) случаи. Циклоалкил может представлять собой 3-12 членную одновалентную, двух- или более (при необходимости) валентную группу, 3-10 членную одновалентную, двух- или более (при необходимости) валентную группу, 3-8 членную одновалентную, двух- или более (при необходимости) валентную группу, 3-6 членную одновалентную, двух- или более (при необходимости) валентную группу, 4-6 членную одновалентную, двух- или более (при необходимости) валентную группу или 5-7 членную одновалентную, двух- или более (при необходимости) валентную группу.
(Одновалентный, двух- или более валентный) моноциклический циклоалкил может представлять собой 3-12 членный циклоалкил, 3-10 членный циклоалкил, 3-8 членный циклоалкил, 3-6 членный циклоалкил, 4-6 членный циклоалкил или 5-7 членный циклоалкил, примеры которого включают, но этим не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентил-1,3-диил, циклогексил-1,4-диил, циклогептил-1,4-диил и тому подобное.
(Одновалентный, двух- или более валентный) полициклический циклоалкил включает орто-конденсированный циклоалкил, мостиковый циклоалкил и спиро-циклоалкил.
(Одновалентный, двух- или более валентный) орто-конденсированный циклоалкил может представлять собой 6-12 членный орто-конденсированный циклоалкил или 7-10 членный орто-конденсированный циклоалкил, примеры которого включают, но этим не ограничиваются, бицикло[3.1.1]гептил, бицикло[2.2.1]гептил, бицикло[2.2.2]октанил, бицикло[3.2.2]нонил, бицикло[3.3.1] нонил и бицикло[4.2.1]нонил.
«Циклоалкенил», описанный в настоящем изобретении, относится к группе, в которой имеется по меньшей мере одна двойная углерод-углеродная связь (предпочтительно, имеется двойная углерод-углеродная связь) в вышеуказанной циклоалкильной группе.
«Циклоалкил» и «циклоалкенил» также могут представлять собой одновалентные группы, полученные удалением атома водорода у 6-12-членного спирокольца или 7-11-членного спирокольца, или двухвалентные группы (если требуется), полученные удалением атома водорода у каждого из двух различных атомов углерода. Примеры спиро-кольца включают, но этим не ограничиваются:  ,
,  ,
,  ,
, 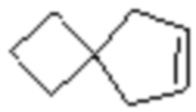 ,
, 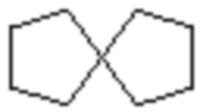 ,
, 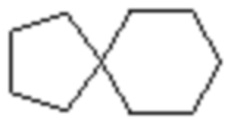 ,
, 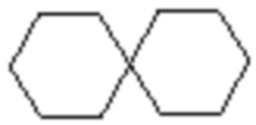 и
и 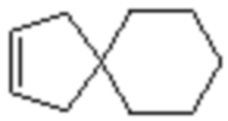 .
.
«Циклоалкил» и «циклоалкенил» также могут представлять собой одновалентные группы, полученные удалением атома водорода у 6-12 членного мостикового кольца или 7-11 членного мостикового кольца, или двухвалентные группы (если требуется), полученные удалением атома водорода у каждого из двух различных атомов углерода. Примеры мостикового кольца включают, но этим не ограничиваются: 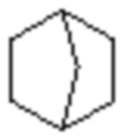 ,
,  ,
, 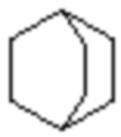 ,
,  ,
,  ,
,  и
и  .
.
Следовательно, если не указано иное, «3-12-членный циклоалкенил», описанный в настоящем изобретении, включает все возможные образованные моноциклические и полициклические (включая конденсированные в орто-, спиро- или мостиковой форме) случаи. Это группа, которая имеет по меньшей мере одну двойную углерод-углеродную связь в 3-12-членной одновалентной, двух- или более (при необходимости) валентной циклоалкильной группе, перечисленной выше. Например, она может представлять собой одновалентную или двухвалентную группу, производную 3-8-членного циклоалкенила, 7-11-членного спироциклоалкенила, 7-11-членного орто-конденсированного циклоалкенила, 6-11-членного мостикового циклоалкенила или тому подобное. Примеры включают циклобутенил, циклопентенил, циклогексенил, 1,4-циклогексадиенил, циклогептенил, 1,4-циклогептадиенил, циклооктенил и 1,5-циклооктадиенил.
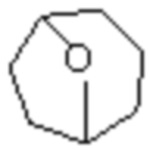
«Гетероциклил», описанный в настоящем изобретении, относится к неароматической одновалентной или двухвалентной циклической группе, образованной замещением по меньшей мере одного циклического атома углерода в вышеуказанном циклоалкиле на гетероатом, выбранный из O, S и N, предпочтительно, и не имеющей или имеющей двойную углерод-углеродную связь. Предпочтительно, это гетероциклил, полученный замещением кольцевых атомов углерода вышеуказанного циклического алкила от 1 до 3 гетероатомами, выбранными из O, S и N. Кроме того, гетероциклил, описанный в настоящем изобретении, дополнительно включает случай, когда атомы углерода или атомы серы в качестве кольцевых атомов замещены кислородом или азотом, например, замещены кольцевые атомы углерода, C(=O), S(=O), S(=O)2 и S(=O)(=NH).
В частности, «гетероциклил» по настоящему изобретению может представлять собой группу с 3, 4, 5, 6, 7, 8, 9, 10, 11 и 12 кольцевыми атомами. Он может представлять собой 3-14 членный гетероциклил, 3-12 членный гетероциклил, 3-10 членный гетероциклил, 4-10 членный гетероциклил, 3-8 членный гетероциклил, 4-12 членный гетероциклил, 4-8 членный гетероциклил, 4-6 членный гетероциклил или 5-10 членный гетероциклил.
Кроме того, «гетероциклил» дополнительно включает одновалентную, двух- или более (при необходимости) валентную моноциклическую гетероциклильную систему или одновалентную, двух- или более (при необходимости) валентную полициклическую гетероциклильную систему (также называемую полициклической системой) и включает насыщенную или ненасыщенную гетероциклильную группу, и он не является целиком ароматическим. Если не указано иное, он включает все возможно образованные моноциклические, полициклические (включая конденсированные в орто-, спиро- или мостиковой форме), насыщенные и ненасыщенные случаи и не является целиком ароматическим.
Одновалентный, двух- или более (при необходимости) валентный моноциклический гетероциклил может представлять собой 3-14 членный гетероциклил, 3-12 членный гетероциклил, 3-10 членный гетероциклил, 4-10 членный гетероциклил, 3-8 членный гетероциклил, 4-12 членный гетероциклил, 4-8 членный гетероциклил, 4-6 членный гетероциклил, 5-10 членный гетероциклил, 3-8 членный насыщенный гетероциклил, 3-6 членный гетероциклил, 4-12 членный гетероциклил, 4-7 членный гетероциклил, 4-6 членный гетероциклил, 5-10 членный гетероциклил, 5-7 членный гетероциклил, 5-6 членный гетероциклил, 5-6 членный кислородсодержащий гетероциклил, 5-6 членный азотсодержащий гетероциклил, 5-6 членный насыщенный гетероциклил, 5-7 членный насыщенный гетероциклил или тому подобное, который может представлять собой насыщенный, частично насыщенный или ненасыщенный, но не ароматический. Его примеры включают, но этим не ограничиваются: азациклопропил, 2H-азациклопропил, диазациклопропил, 3H-диазациклопропил, азетидинил, 1,4-диоксациклогексил, 1,3-диоксациклогексил, 1,3-диоксациклопентил, 1,4-диоксациклогексадиенил, тетрагидрофурил, дигидропирролил, пирролидинил, имидазолидинил, 4,5-дигидроимидазолил, пиразолидинил, 4,5-дигидропиразолил, 2,5-дигидротиенил, тетрагидротиенил, 4,5-дигидротиазолил, пиперидил, пиперазинил, морфолинил, гексагидропиримидинил, гексагидропиридазинил, 4,5-дигидроксазолил, 4,5-дигидроизоксазолил, 2,3-дигидроизоксазолил, 2H-1,2-оксазинил, 6H-1,3-оксазинил, 4H-1,3-тиазинил, 6H-1,3-тиазинил, 2H-пиранил, 2H-пиран-2-он, 3,4-дигидро-2H-пиранил, 1,1-диоксотетрагидротиапиранил, 1,1-диоксотетрагидротиенил, 1-имино-1-оксо-тетрагидротиобутилциклил, 1-имино-1-оксо-тетрагидротиенил, 1-имино-1-оксо-гексагидротиапиранила и тому подобное.
Одновалентный, двух- или более валентный (при необходимости) полициклический гетероциклил включает орто-конденсированный гетероциклил, спиро-гетероциклил и мостиковый гетероциклил, которые могут представлять собой насыщенный, частично насыщенный или ненасыщенный, но не ароматический.
Орто-конденсированный гетероциклил может представлять собой 6-12 членный орто-конденсированный гетероциклил, 7-10 членный орто-конденсированный гетероциклил, 6-10 членный орто-конденсированный циклил, 6-12 членный насыщенный орто-конденсированный гетероциклил, 7-8 членный насыщенный орто-конденсированный гетероциклил или 8 членный насыщенный орто-конденсированный гетероциклил, и его примеры включают, но этим не ограничиваются: 3-азабицикло[3.1.0]гексил, 3,6-диазабицикло[3.2.0]гептил, 3,8-диазабицикло[4.2.0]октил, 3,7-диазабицикло[4.2.0]октил, октагидропирроло[3,4-c]пирролил, октагидропирроло[3,4-b]пирролил, октагидропирроло[3,4-b][1,4]оксазинил, октагидро-1H-пирроло[3,4-c]пиридил, 2,3-дигидробензофуран-2-ил, 2,3-дигидробензофурил-3-ил, индолин-1-ил, индолин-2-ил, индолин-3-ил, 2,3-дигидробензотиофен-2-ил, октагидро-1H-индолил, октагидробензофурил, октагидроциклопента[c]пирролил, гексагидроциклопента[c]фурил, 2,2-диоксогексагидроциклопента[c]тиенил и 2-имино-2-оксо-октагидроциклопента[c]тиенил.
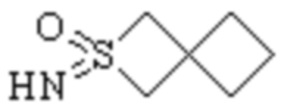
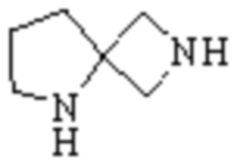
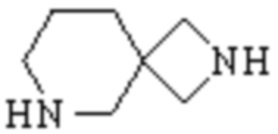
Спиро-гетероциклил может представлять собой одновалентную группу, полученную удалением атома водорода у 6-12 членного спиро гетероциклического кольца, 7-11 членного спиро гетероциклического кольца, 6-12 членного насыщенного спиро гетероциклического кольца или 7 членного насыщенного спиро гетероциклического кольца, или двухвалентную группу (при необходимости), полученную удалением атома водорода у каждого из двух различных атомов углерода, и примеры спиро гетероциклила включают но этим не ограничиваются:  ,
,  ,
,  ,
,  ,
, 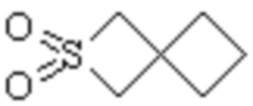 ,
,  ,
,  ,
,  ,
, 
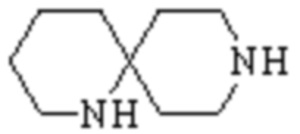 ,
, 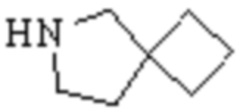 ,
,  ,
,  ,
, 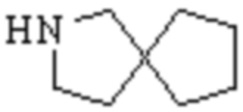 и
и 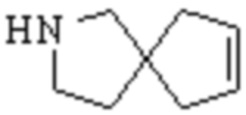 .
.
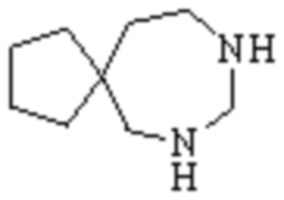
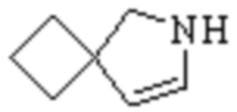
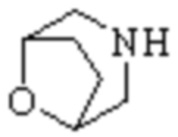
Мостиковый гетероциклил может представлять собой одновалентную группу, полученную удалением атома водорода у 6-12-ленного мостикового гетероциклического кольца, 7-11-членного мостикового гетероциклического кольца, 6-12-членного насыщенного мостикового гетероциклического кольца или 7-8-членного насыщенного мостикового гетероциклического кольца, или двухвалентную группу (при необходимости), полученную удалением атома водорода каждого из двух различных атомов углерода, и примеры мостикового гетероциклила включают, но не ограничиваются ими 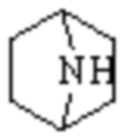 ,
, 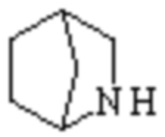 ,
, 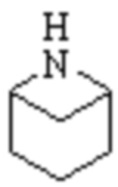 ,
, 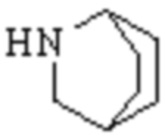 ,
, 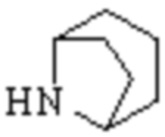 ,
,  ,
, 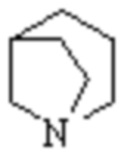 ,
,  ,
,  ,
,  ,
,  ,
, 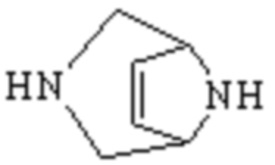 ,
, 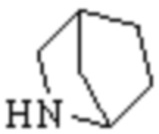 ,
, 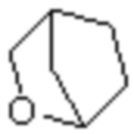 ,
,  и
и 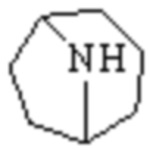 .
.
«Арил», описанный в настоящем изобретении, относится к одновалентной группе или двух- или более (при необходимости) валентной группе, производной ароматического карбоциклического углеводорода, и ароматический карбоциклический углеводород включает 6-8-членный моноциклический ароматический углеводород и 8-14-членный полициклический ароматический углеводород. 6-8-членный моноциклический арил представляет собой, например, фенил. 8-14-членный полициклический арил представляет собой, например, нафтил, фенантрил, антрил и тому подобное. Двухвалентный арил может включать, например, фенилен, нафтилен и тому подобное.
«Гетероарил», описанный в настоящем изобретении, может представлять собой 5-14 членный гетероарил, 5-10 членный гетероарил или 5-6 членный гетероарил и относится к ароматической одновалентной или двухвалентной циклической группе с 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 кольцевыми атомами, которые имеют по меньшей мере один гетероатом, выбранный из O, S и N. Предпочтительно, он содержит от 1 до 3 кольцевых гетероатомов. Кроме того, гетероарил дополнительно включает случай, когда атомы углерода или атомы серы в качестве кольцевых атомов замещены кислородом или азотом, например, когда замещены атомы углерода C(=O), S(=O), S(=O)2 и S(=O)(=NH). Гетероароматическое кольцо, описанное в настоящем изобретении, может представлять собой моноциклическую систему или полициклическую систему (конденсированную в орто-, спиро- или мостиковой форме). Гетероарил включает моноциклический гетероарил и полициклический гетероарил. Если не указано иное, несколько членный гетероарил включает все возможные моноциклические, полициклические, полностью ароматические и частично ароматические группы. Моноциклический гетероарил может представлять собой, например, 5-7 членный гетероарил или 5-6 членный гетероарил, примеры которого включают, но этим не ограничиваются, фурил, имидазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, оксазолил, изоксазолил, пиридил, пиридонил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, тетразолил, тиадиазолил, тиазолил, тиенил, триазолил и триазинил. Полициклический гетероарил может представлять собой 8-12 членный орто-конденсированный гетероарил или 9-10 членный орто-конденсированный гетероарил, примеры которого включают, но этим не ограничиваются, бензимидазолил, бензофурил, изобензофурил, бензотиенил, бензотиенил, бензооксадиазолил, бензотиазолил, циннолинил, индазолил, индолил, изоиндолил, изохинолинил, нафтиридинил, пуринил, хинолинил, хиноксалинил и хиназолинил. Гетероарил также может представлять собой двухвалентные группы, производные вышеуказанных групп.
В настоящем изобретении «гетероатом» означает атом, выбранный из S, O и N. Кроме того, в некоторых случаях также включены случаи окисления или азотирования S или O.
«3-6-членное кольцо», «3-8-членное кольцо», «4-6-членное кольцо» и «4-7-членное кольцо», описанные в настоящем изобретении, относятся к химически возможным кольцевым структурам с 3-6 кольцевыми атомами, 3-8 кольцевыми атомами, 4-6 кольцевыми атомами и 4-7 кольцевыми атомами; кольцевые атомы, необязательно, могут быть выбраны из C, N, O, S, C(=O), S(=O), S(=O)2 и S(=O)(=NH), и образованные кольцевые структуры могут быть моноциклическими, конденсированными полициклическими, насыщенными, частично насыщенными или ароматическими. В частности, примеры может представлять собой вышеуказанные группы, перечисленные как циклоалкил, циклоалкенил, гетероциклил, арил и гетероарил.
В «амино» или группе, содержащей «амино», описанной в настоящем изобретении, группа/подгруппа с амино в качестве конца может быть представлена как -N(Re)2, где Re, каждый, независимо, выбран из водорода, вышеуказанного C1-6 алкила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного C2-6 алкенила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного C2-6 алкинила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного циклоалкила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного циклоалкенила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного арила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного гетероарила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного гетероциклила, необязательно замещенного заместителем A по настоящему изобретению, вышеуказанного Cy2 по настоящему изобретению, необязательно замещенного заместителем A по настоящему изобретению, и других групп (включая, но этим не ограничиваясь, карбонил, сульфонил и тому подобное) соединенного с амино в группе по настоящему изобретению, содержащей «амино». В настоящем изобретении группа/подгруппа с амино в качестве конца в группе, содержащей «амино», означает, что амино связан с двумя Re и затем связан с другими группами. Например, в случае «(C1-6 алкил)2 амино» это соответствует случаю группы с амино в качестве конца в группе, содержащей «амино»; в случае «C1-6 алкиламино» это соответствует случаю группы с амино в качестве конца в группе, содержащей «амино», в которой один Re уже представляет собой «C1-6 алкил», а другой Re представляет собой группу, перечисленную выше; в случае «(C1-6 алкил)2 амино C1-6 алкила», содержащийся в группе «(C1-6 алкил)2 амино» соответствует случаю подгруппы с амино в качестве конца в группе, содержащей «амино», то есть является амином, связанным сначала с двумя C1-6 алкилами (соответствующими Re) и затем с C1-6 алкилом; в случае «(C1-6 алкил)2 аминокарбонила», содержащийся в группе «(C1-6 алкил)2 амино» соответствует случаю подгруппы с амино в качестве конца в группе, содержащей «амино», то есть является амином, связанным сначала с двумя C1-6 алкилами (соответствующими Re) и затем с карбонилом; в случае «C1-6 алкиламинокарбонила», содержащийся в группе «C1-6 алкиламино» соответствует случаю подгруппы с амино в качестве конца в группе, содержащей «амино», то есть амино сначала связан с C1-6 алкилом (соответствующим Re) и затем с карбонилом, и один Re в группе уже представляет собой «C1-6 алкил», а другой Re представляет собой группу, перечисленную выше; в случае «амино C1-6 алкила», содержащийся в группе «амино» соответствует случаю подгруппы с амино в качестве конца в группе, содержащей «амино», и амино в группе может быть представлен как -N(Re)2, то есть группа может быть представлена как «N(Re)2-C1-6 алкил». Таким образом, N в амино по настоящему изобретению является трехвалентным (  ), две связи в котором могут быть связаны с Re. Кроме того, в настоящем изобретении два Re в «-N(Re)2» вместе с атомом N могут образовывать азотсодержащий гетероциклил, имеющий определение, описанное выше в настоящем изобретении.
), две связи в котором могут быть связаны с Re. Кроме того, в настоящем изобретении два Re в «-N(Re)2» вместе с атомом N могут образовывать азотсодержащий гетероциклил, имеющий определение, описанное выше в настоящем изобретении.
В настоящем изобретении термин «необязательно замещенный» или «необязательно им замещенный» означает, что любая часть фрагмента, известная специалистам в данной области техники как доступная для замещения, может быть незамещенной или замещенной заместителем, описанным в настоящем изобретении, где, если присутствуют один или несколько заместителей, каждый заместитель может быть выбран независимо. Что касается замещения, количество заместителей определяется в соответствии с количеством положений, которые могут быть замещены в замещаемой группе, и может представлять собой 1 заместитель, 2 заместителя, 3 заместителя, 4 заместителя, 5 заместителей, 6 заместителей, 7 заместителей, 8 заместителей или более, при условии, что количество положений, которые могут быть замещены в замещаемой группе, не превышено. При наличии заместителей «один или несколько» заместителей указывают на то, что присутствует не менее одного заместителя, и конкретное количество заместителей варьируется в зависимости от замещаемой группы, и может представлять собой 1 заместитель, 2 заместителя, 3 заместителя, 4 заместителя, 5 заместителей, 6 заместителей, 7 заместителей, 8 заместителей или более, при условии, что количество положений, которые могут быть замещены в замещаемой группе, не превышено.
В настоящем изобретении выражение «необязательно замещенный», добавленное перед названием группы, или «необязательно замещенный», добавленное после названия группы, указывает на то, что все подгруппы в группе могут быть необязательно замещены. Например, в случае «C7-12 алкиларил, необязательно замещенного галогеном», алкильный фрагмент может быть замещен галогеном, арильный фрагмент может быть замещен галогеном, или и алкильный фрагмент, и арильный фрагмент могут быть замещены галогеном.
В настоящем изобретении в кольцевой структуре обозначает двойную связь, необязательно имеющуюся в кольце, и могут быть 1, 2 или 3 двойные связи, ограниченные максимальным количеством двойных связей, которые могут существовать в кольце. Например, в 5-членном кольце могут существовать одна или две двойные связи; и в 6-членном кольце могут существовать одна, две или три двойные связи.
В настоящем изобретении обозначает одинарную связь или двойную связь.
В настоящем изобретении «отсутствие» определенной группы может означать, что сама группа отсутствует; например, в случае выражения «Rd отсутствует», это означает, что, когда образующий кольцо атом N соединен с соседними атомами одинарными связями и двойными связями в кольце, Rd отсутствует в определении «NRd». Кроме того, это также может означать, что группа представляет собой связь; например, в случае выражения «L1 отсутствует», в настоящем изобретении это означает, что L1 представляет собой связь, позволяющую группе Cy1 напрямую связываться с атомом углерода, связанным с группой R6.
В настоящем изобретении заместитель в выражении «необязательно замещенном заместителями» может быть «заместителем А», описанным в настоящем изобретении. Количество заместителей определяется в соответствии с количеством положений, которые могут быть замещены в замещенной группе, и может представлять собой 1 заместитель, 2 заместителя, 3 заместителя, 4 заместителя, 5 заместителей, 6 заместителей, 7 заместителей, 8 заместителей или более.
В настоящем изобретении валентности всех групп, заместителей, участков химической связи, атомов и т. д. не нарушают общеизвестных правил в области химии. Например, атомы углерода четырехвалентны, атомы азота трехвалентны, атомы кислорода двухвалентны, а атомы водорода одновалентны.
Конкретно, в настоящем изобретении предложено соединение, представленное формулой (I) ниже, или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер:
I
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила, необязательно замещенного заместителем A, или образуют 5-10 членный азотсодержащий гетероциклил, необязательно замещенный заместителем A вместе с атомом N, связанным с ним;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила, необязательно замещенного заместителем A;
L1 представляет собой связь или -CRʼRʺ-, -NRʼ-, -S-, -SO2-, -S(O)-, -SONRʼ-, -SO2NRʼ- или -NRʼCONRʼ- и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила, необязательно замещенного заместителем A;
Cy1 представляет собой группу, представленную общей формулой (A), (a), (b) или (c) ниже, которая является незамещенной или замещена одним или несколькими Ra:
;
m обозначает целое число 0 до 3, и n обозначает целое число 0 до 2;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CRcRc, NRd, O и S;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CRcRc, NRd, O и S, X5, X6, X7 и X8, каждый, независимо, выбраны из CRcRc и NRd, и по меньшей мере один из X1, X2 и X3 представляет собой NRd;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбоксила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C2-6 алкенила, необязательно замещенного одним или несколькими Rb, C2-6 алкинила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламиносульфонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминосульфонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламиносульфонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминосульфонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилсульфонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb, и
Cy2, каждый, независимо, выбран из 3-12 членного циклоалкила, 3-12 членного циклоалкенила, 3-12 членного гетероциклила, 6-10 членного арила и 5-14 членного гетероарила;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкиламиносульфонила, (C1-6 алкил)2 аминосульфонила, C1-6 алкилсульфониламино и C1-6 алкилсульфонила;
заместители A, каждый, независимо, выбраны из гидроксила, амино, карбоксила, циано, нитро, галоген, аминокарбонила, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, амино C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкилсульфониламино, C1-6 алкиламиносульфонила, (C1-6 алкил)2 аминосульфонила, C1-6 галогеналкила, C1-6 галогеналкокси, C1-6 алкилсульфонила, C1-6 алкилтио, 3-12 членного циклоалкила, 6-10 членный арила, 3-12 членного гетероциклила, 5-14 членного гетероарила и оксо;
Rc отсутствует или, когда он присутствует, каждый, независимо, выбран из атома водорода; или два Rc вместе образуют оксо группу;
Rd отсутствует или, когда он присутствует, каждый, независимо, выбран из атома водорода;
 обозначает одинарную связь или двойную связь;
обозначает одинарную связь или двойную связь;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре;
при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra; и
при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O.
В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород. В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода, фтора, хлора, брома и йода и оба R1 и R2 не представляют собой водород. В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода, фтора и хлора и оба R1 и R2 не представляют собой водород. В одном варианте осуществления настоящего изобретения R1 представляет собой водород и R2 представляет собой фтор. В одном варианте осуществления настоящего изобретения R1 и R2 вместе со связанными с ними атомами N образуют 5-8 азотсодержащий гетероциклил.
В одном варианте осуществления настоящего изобретения R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила. В одном варианте осуществления настоящего изобретения R3 и R4, каждый, независимо, выбраны из водорода и C1-4 алкила. В одном варианте осуществления настоящего изобретения R3 и R4, каждый, независимо, выбраны из водорода, метила, этила, пропила, изопропила, бутила, втор-бутила и трет-бутила. В одном варианте осуществления настоящего изобретения R3 и R4 представляют собой водород. В одном варианте осуществления настоящего изобретения R3 и R4 вместе со связанными с ними атомами N образуют 5-6 членный азотсодержащий гетероциклил. В одном варианте осуществления настоящего изобретения R3 и R4 вместе со связанными с ними атомами N образуют пирролинил, пирролидинил, пиперидил или морфолинил.
В одном варианте осуществления настоящего изобретения R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила. В одном варианте осуществления настоящего изобретения R5 и R6, каждый, независимо, выбраны из водорода и C1-4 алкила. В одном варианте осуществления настоящего изобретения R5 и R6, каждый, независимо, выбраны из водорода, метила, этила, пропила, изопропила, бутила, втор-бутила и трет-бутила. В одном варианте осуществления настоящего изобретения R5 и R6 представляют собой водород.
В одном варианте осуществления настоящего изобретения L1 представляет собой связь или -CRʼRʺ-, -NRʼ- или -S-, и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила. В одном варианте осуществления настоящего изобретения L1 представляет собой связь, -NRʼ- или -S- и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила. В одном варианте осуществления настоящего изобретения L1 представляет собой связь или -NRʼ- и Rʼ выбран из водорода и C1-6 алкила. В одном варианте осуществления настоящего изобретения L1 представляет собой связь.
В одном варианте осуществления настоящего изобретения в формуле (A), когда Ra присутствует, по меньшей мере один Ra присоединен к любому из Y1, Y2, Y3 и Y4. В одном варианте осуществления настоящего изобретения в формуле (a), когда Ra присутствует, по меньшей мере один Ra присоединен к любому из Y1, Y2 и Y3. В одном варианте осуществления настоящего изобретения в формуле (c) по меньшей мере один Ra присутствует и по меньшей мере один Ra присоединен к любому из X5, X6, X7 и X8.
В одном варианте осуществления настоящего изобретения в формуле (A) группа L1 присоединена к X1, X2 или X3 в формуле (A). В одном варианте осуществления настоящего изобретения в формуле (a) группа L1 присоединена к X1, X2 или X3 в формуле (a). В одном варианте осуществления настоящего изобретения в формуле (c) группа L1 присоединена к X1, X2, X3 или X4 в формуле (c).
В одном варианте осуществления настоящего изобретения группа L1 присоединена к атому N.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, представленную общей формулой (A-1), (A-2), (A-3), (a), (b) или (c) ниже, которая является незамещенной или замещена одним или несколькими Ra:
.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, представленную общей формулой (A-11), (a-1), (a-2), (b-1), (c-1) или (c-2) ниже, которая является незамещенной или замещена одним или несколькими Ra:
;
В одном варианте осуществления настоящего изобретения, когда Cy1 соответствует группе формулы (c-1) или (c-2), группа формулы (c-1) или (c-2) является замещенной одним или несколькими Ra.
В одном варианте осуществления настоящего изобретения, когда Cy1 соответствует формуле (b-1), X1, X2, X3 и X9 не представляют собой C=O.
В одном варианте осуществления настоящего изобретения в формуле (A-11), когда Ra присутствует, по меньшей мере один Ra присоединен к Y2. В одном варианте осуществления настоящего изобретения в формуле (a-1), когда Ra присутствует, по меньшей мере один Ra присоединен к Y2. В одном варианте осуществления настоящего изобретения в формуле (a-2), когда Ra присутствует, по меньшей мере один Ra присоединен к Y1. В одном варианте осуществления настоящего изобретения в формуле (b-1), когда Ra присутствует, по меньшей мере один Ra присоединен к образующему кольцу атому углерода. В одном варианте осуществления настоящего изобретения в формулах (c-1) и (c-2) по меньшей мере один Ra присутствует и по меньшей мере один Ra присоединен к группе бензольного кольца.
В одном варианте осуществления настоящего изобретения m обозначает целое число от 0 до 3. В одном варианте осуществления настоящего изобретения m обозначает 1 или 2. В одном варианте осуществления настоящего изобретения n обозначает целое число от 0 до 2. В одном варианте осуществления настоящего изобретения n обозначает 1 или 2.
В одном варианте осуществления настоящего изобретения Rc отсутствует или, когда он присутствует, каждый, независимо, выбран из атома водорода; или два Rc вместе образуют оксо группу.
В одном варианте осуществления настоящего изобретения Rd отсутствует или, каждый, независимо, выбран из атома водорода.
В одном варианте осуществления настоящего изобретения Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CRcRc и NRd. В одном варианте осуществления настоящего изобретения по меньшей мере один из Y1, Y2, Y3 и Y4 представляет собой C=O. В одном варианте осуществления настоящего изобретения Y1, Y2 и Y3, каждый, независимо, выбраны из CH2, NH, CH и N.
В одном варианте осуществления настоящего изобретения X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CRcRc и NRd и по меньшей мере один из X1, X2 и X3 представляет собой N или NRd. В одном варианте осуществления настоящего изобретения X1, X2, X3, X4 и X9, каждый, независимо, выбраны из CH2, CH, N, NH и C=O и по меньшей мере один из X1, X2 и X3 представляет собой N или NH.
В одном варианте осуществления настоящего изобретения X5, X6, X7 и X8, каждый, независимо, выбраны из CRcRc и NRd.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, представленную общей формулой (A-11) или (a-1) ниже, которая является незамещенной или замещена одним или несколькими Ra:
;
В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1), Y2 и Y3, каждый, независимо, выбраны из CH2, NH, CH и N. В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1) X1, X2 и X3, каждый, независимо, выбраны из CH2, CH, N, NH и C=O и по меньшей мере один из X1, X2 и X3 представляет собой N или NH.
В одном варианте осуществления настоящего изобретения в формуле (A-11) X1 и X2, каждый, независимо, выбраны из CH2, CH, N и NH. В одном варианте осуществления настоящего изобретения в формуле (a-1) X1 и X2, каждый, независимо, выбраны из CH2, CH, N и NH. В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1) Y2 представляет собой NH. В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1) Y3 представляет собой CH2 или CH. В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1), когда Ra присутствует, по меньшей мере один Ra присоединен к положении Y2. В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1) группа L1 присоединена к X1, X2 или X3. В одном варианте осуществления настоящего изобретения в формулах (A-11) и (a-1) группа L1 присоединена к атому N.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой одну из следующих групп, которая является незамещенной или замещена одним или несколькими Ra:
, , , , , , , , , , , , , , , , или ;
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, представленную общей формулой (b-1) ниже, которая является незамещенной или замещена одним или несколькими Ra:
.
В одном варианте осуществления настоящего изобретения в формуле (b-1), X1, X2, X3 и X9, каждый, независимо, выбраны из CH2, CH, N и NH и по меньшей мере один из X1, X2 и X3 представляет собой N или NH.
В одном варианте осуществления настоящего изобретения в формуле (b-1) группа L1 присоединена к атому N в формуле (b-1). В одном варианте осуществления настоящего изобретения в формуле (b-1), когда Ra присутствует, по меньшей мере один Ra присоединен к атому углерода формулы (b-1).
В одном варианте осуществления настоящего изобретения Cy1 представляет собой одну из следующих групп, которая является незамещенной или замещена одним или несколькими Ra:
, или ;
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, представленную общей формулой (c-1) ниже, которая замещена одним или несколькими Ra:
.
В одном варианте осуществления настоящего изобретения в формуле (c-1) X1, X2 и X3, каждый, независимо, выбраны из CH2, CH, N, NH и C=O и по меньшей мере один из X1, X2 и X3 представляет собой N или NH.
В одном варианте осуществления настоящего изобретения в формуле (c-1) по меньшей мере один Ra присоединен к группе бензольного кольца в формуле (c-1). В одном варианте осуществления настоящего изобретения в формуле (c-1) группа L1 присоединена к X1, X2 или X3. В одном варианте осуществления настоящего изобретения в формуле (c-1) группа L1 присоединена к атому N в формуле (c-1).
В одном варианте осуществления настоящего изобретения Cy1 представляет собой следующую группу, которая замещена одним или несколькими заместителями Ra:
.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио, необязательно замещенного одним или несколькими Rb, C1-6 алкилтио C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb, и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 амино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, (C1-6 алкил)2 аминокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, и Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкокси C1-6 алкокси, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино, необязательно замещенного одним или несколькими Rb, C1-6 алкиламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкиламинокарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбониламино C1-6 алкила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-6 алкилкарбонил C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-6 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb и Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из галогена, циано, C1-6 алкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-6 членного циклоалкила, 3-8 членного циклоалкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фенила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, и 5-6 членного гетероарила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
В одном варианте осуществления настоящего изобретения Ra, каждый, независимо, выбран из C1-6 алкила, необязательно замещенного галогеном; галогена; аминокарбонила; C1-6 алкиламинокарбонила, необязательно замещенного галогеном; (C1-6 алкил)2 аминокарбонила, необязательно замещенного галогеном; 3-8 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; 5-6 членного гетероариламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; и 5-10 членного гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, циано, галогена, аминокарбонила, C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкокси, необязательно замещенного одним или несколькими Rb, C1-4 алкокси C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкокси C1-4 алкокси, необязательно замещенного одним или несколькими Rb, C1-4 алкилтио, необязательно замещенного одним или несколькими Rb, C1-4 алкилтио C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкиламино, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 амино, необязательно замещенного одним или несколькими Rb, C1-4 алкиламино C1-4 алкила, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 амино C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкиламинокарбонила, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 аминокарбонила, необязательно замещенного одним или несколькими Rb, C1-4 алкиламинокарбонил C1-4 алкила, необязательно замещенного одним или несколькими Rb, (C1-4 алкил)2 аминокарбонил C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбониламино, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбониламино C1-4 алкила, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбонила, необязательно замещенного одним или несколькими Rb, C1-4 алкилкарбонил C1-4 алкила, необязательно замещенного одним или несколькими Rb, Cy2-, необязательно замещенного одним или несколькими Rb, Cy2-C1-4 алкила, необязательно замещенного одним или несколькими Rb, Cy2-C1-4 алкокси, необязательно замещенного одним или несколькими Rb, Cy2-карбонила, необязательно замещенного одним или несколькими Rb, Cy2-аминокарбонила, необязательно замещенного одним или несколькими Rb и Cy2-карбониламино, необязательно замещенного одним или несколькими Rb.
В одном варианте осуществления настоящего изобретения Ra, каждый, независимо, выбран из галогена, циано,C1-4 алкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-5 членного циклоалкила, 3-5 членного циклоалкила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фенила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, и 5-6 членного азотсодержащего гетероарила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси. В одном варианте осуществления настоящего изобретения Ra, каждый, независимо, выбран из фтора; хлора; брома; циано; метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила и трет-бутила, необязательно замещенных по меньшей мере одним из галогена, C1-6 алкила, C1-6 алкокси и 3-5 членного циклоалкила; циклопропила, циклобутила и циклопентила, необязательно замещенных по меньшей мере одним из галогена и C1-6 алкила; фенила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила; и пирролила, имидазолила, пиразолила, оксазолила и тиазолила, необязательно замещенных по меньшей мере одним из галогена и C1-6 алкила.
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из аминокарбонила, C1-4 алкиламинокарбонила, необязательно замещенного галогеном, (C1-4 алкил)2 аминокарбонила, необязательно замещенного галогеном, 3-5 членного циклоалкиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси и 5-6 членного азотсодержащего гетероциклилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
В одном варианте осуществления настоящего изобретения Ra, каждый, независимо, выбран из аминокарбонила, метиламинокарбонила, необязательно замещенного галогеном, этиламинокарбонила, необязательно замещенного галогеном, пропиламинокарбонила, необязательно замещенного галогеном, изопропиламинокарбонила, необязательно замещенного галогеном, н-аминокарбонила, необязательно замещенного галогеном, изобутиламинокарбонила, необязательно замещенного галогеном, втор-бутиламинокарбонила, необязательно замещенного галогеном, трет-бутиламинокарбонила, необязательно замещенного галогеном, циклопропиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, циклобутиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, циклопентиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, пирролидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила, пиперидилкарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила и пиперазинилкарбонила, необязательно замещенного по меньшей мере одним из галогена и C1-6 алкила.
В одном варианте осуществления настоящего изобретения Ra, каждый, независимо, выбран из аминокарбонила, метиламинокарбонила, необязательно замещенного галогеном, этиламинокарбонила, необязательно замещенного галогеном, пропиламинокарбонила, необязательно замещенного галогеном, изопропиламинокарбонила, необязательно замещенного галогеном, н-аминокарбонила, необязательно замещенного галогеном, изобутиламинокарбонила, необязательно замещенного галогеном, втор-бутиламинокарбонила, необязательно замещенного галогеном, трет-бутиламинокарбонила, необязательно замещенного галогеном, циклопропиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, циклобутиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, циклопентиламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; фениламинокарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси; азетидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пирролидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пиперидилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пиперазинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси и морфолинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси.
В одном варианте осуществления настоящего изобретения, когда Ra, каждый, независимо, выбран из азетидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пирролидинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пиперидилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, пиперазинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси и морфолинилкарбонила, необязательно замещенного по меньшей мере одним из галогена, C1-6 алкила и C1-6 алкокси, кольцевой атом азот, расположенный на кольце, связан с карбонилом (C=O).
В одном варианте осуществления настоящего изобретения Cy2, каждый, независимо, выбран из 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила и 5-10 членного гетероарила. В одном варианте осуществления настоящего изобретения Cy2, каждый, независимо, выбран из 3-6 членного циклоалкила, 5-6 членного гетероциклила, фенила, нафтила и 5-6 членного гетероарила.
В одном варианте осуществления настоящего изобретения каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонил. В одном варианте осуществления настоящего изобретения каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, аминокарбонила, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 галогеналкокси, C1-6 алкилтио, C1-6 алкиламино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила.
В одном варианте осуществления настоящего изобретения каждый Rb независимо выбран из гидроксила, амино, циано, галогена, аминокарбонила, C1-4 алкила, C1-4 алкокси, амино C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси C1-4 алкила, C1-4 алкокси C1-4 алкокси, амино C1-4 алкокси, C1-4 галогеналкокси, C1-4 алкилтио, C1-4 алкиламино, (C1-4 алкил)2 амино, C1-4 алкиламинокарбонила, (C1-4 алкил)2 аминокарбонила, C1-4 алкилкарбониламино и C1-4 алкилкарбонила.
В одном варианте осуществления настоящего изобретения заместители A, каждый, независимо, выбраны из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, амино C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, амино C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 галогеналкила, C1-6 галогеналкокси, C1-6 алкилтио, 3-8 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила, 5-10 членного гетероарила и оксо.
В одном варианте осуществления настоящего изобретения заместители A, каждый, независимо, выбраны из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-4 алкила, C1-4 алкокси, амино C1-4 алкила, C1-4 алкокси C1-4 алкила, C1-4 алкокси C1-4 алкокси, амино C1-4 алкокси, C1-4 алкиламино, (C1-4 алкил)2 амино, C1-4 алкиламинокарбонила, (C1-4 алкил)2 аминокарбонила, C1-4 алкилкарбониламино, C1-4 алкилкарбонила, C1-4 галогеналкила, C1-4 галогеналкокси, C1-4 алкилтио, 3-6 членного циклоалкила, 5-10 членного гетероциклила, фенила, нафтила, 5-10 членного гетероарила и оксо.
В одном варианте осуществления настоящего изобретения в формулах (b) и (b-1) аминокарбонильная группа ( 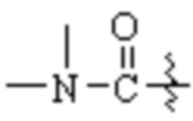 , где атом N связан с вышеуказанной группой, указанной в настоящем изобретении, или атом N содержится в азотсодержащем гетероциклическом кольце) связана с кольцевой структурой. В одном варианте осуществления настоящего изобретения в формулах (b) и (b-1) аминокарбонильная группа ( , где атом N связан с вышеуказанной группой, указанной в настоящем изобретении, или атом N содержится в азотсодержащем гетероциклическом кольце) присоединена к кольцевому атому углерода.
, где атом N связан с вышеуказанной группой, указанной в настоящем изобретении, или атом N содержится в азотсодержащем гетероциклическом кольце) связана с кольцевой структурой. В одном варианте осуществления настоящего изобретения в формулах (b) и (b-1) аминокарбонильная группа ( , где атом N связан с вышеуказанной группой, указанной в настоящем изобретении, или атом N содержится в азотсодержащем гетероциклическом кольце) присоединена к кольцевому атому углерода.
В одном варианте осуществления настоящего изобретения в формуле (c) X5, X6, X7 или X8 связан с аминокарбонильной группой ( , где атом N связан с вышеуказанной группой, указанной в настоящем изобретении, или атом N содержится в азотсодержащем гетероциклическом кольце). В одном варианте осуществления настоящего изобретения в формулах (c-1) и (c-2) аминокарбонильная группа ( , где атом N связан с вышеуказанной группой, указанной в настоящем изобретении, или атом N содержится в азотсодержащем гетероциклическом кольце) присоединена к бензольной кольцевой структуре. В одном варианте осуществления настоящего изобретения в формуле (c) X5, X6, X7 и X8, каждый, независимо, представляют собой CH.
В одном варианте осуществления настоящего изобретения в формуле (c) X5, X6, X7 и X8 не представляют собой C=O.
В одном варианте осуществления настоящего изобретения в формулах (A-1) и (A-2) Y1 представляет собой C=O и Y2 представляет собой NRd.
В одном варианте осуществления настоящего изобретения кольцевой атом азот, расположенный на кольце, связан с карбонилом (C=O) с образованием аминокарбонила.
В одном варианте осуществления настоящего изобретения предложено соединение по настоящему изобретению или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер, и соединение выбрано из следующих:
В одном варианте осуществления настоящего изобретения предложена фармацевтическая композиция, которая содержит соединение по настоящему изобретению или его фармацевтически приемлемую соль, сложный эфир, стереоизомер или таутомер, описанные выше, и также необязательно содержит один или несколько фармацевтически приемлемых носителей.
В одном варианте осуществления настоящего изобретения предложено применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, стереоизомера или таутомера, описанных выше, или фармацевтической композиции по настоящему изобретению, описанной выше, при производстве лекарственного препарата для профилактики и/или лечения заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им.
В одном варианте осуществления настоящего изобретения предложен способ профилактики и/или лечения заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им, где фармацевтическую композицию соединения по настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, стереоизомера или таутомера, описанных выше, или фармацевтическую композицию, описанную выше, вводят субъекту, нуждающемуся в лечении, в эффективной дозе.
В одном варианте осуществления настоящего изобретения предложено соединение формулы I или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер:
I
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила, или вместе с присоединенным к нему атомом N образуют 5-10 членный азотсодержащий гетероцикл, необязательно замещенный заместителем;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 отсутствует или представляет собой -CRʼRʺ-, -N-, -O-, -S-, -SO2-, S(O), -SONRʼ-, -SO2NRʼ- или -NRʼCONRʼ-, и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила;
Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными общими формулами (A), (a), (b) или (c) ниже:
;
m обозначает целое число 0 до 3, и n обозначает целое число 0 до 2;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CH2, CH, NH, N, O, S и C=O;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CH2, CH, N, O, S, NH и C=O, X5, X6, X7 и X8, каждый, независимо, выбраны из CH и N, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкилтио C1-6 алкила, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламино C1-6 алкила, C1-6 алкиламинокарбонила, C1-6 алкиламинокарбонил C1-6 алкила, C1-6 алкилкарбониламино, C1-6 алкилкарбониламино C1-6 алкила, (C1-6 алкил)2 амино C1-6 алкила, C1-6 алкилкарбонила, C1-6 алкилкарбонил C1-6 алкила, C1-6 алкиламиносульфонила, C1-6 алкиламиносульфонил C1-6 алкила, C1-6 алкилсульфониламино, C1-6 алкилсульфониламино C1-6 алкила, C1-6 алкилсульфонила, C1-6 алкилсульфонил C1-6 алкила, Cy2-, Cy2-C1-6 алкила, Cy2-C1-6 алкокси, Cy2- карбонила и Cy2- аминокарбонила, незамещенного или замещенного одним или несколькими Rb,
Cy2 представляет собой 3-12 членный циклоалкил, 3-12 членный циклоалкенил, 3-12 членный гетероциклил, арил или 5-14 членный гетероарил;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкиламиносульфонила, C1-6 алкилсульфониламино и C1-6 алкилсульфонила;
при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra;
при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O;
обозначает одинарную связь или двойную связь; и
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
В одном варианте осуществления настоящего изобретения предложено соединение формулы I, представленной ниже, или его фармацевтически приемлемая соль, сложный эфир, стереоизомер или таутомер:
I
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила, или вместе с присоединенным к нему атомом N образуют 5-10 членный азотсодержащий гетероцикл, необязательно замещенный заместителем;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 отсутствует или представляет собой -CRʼRʺ-, -N-, -O-, -S-, -SO2-, S(O), -SONRʼ-, -SO2NRʼ- или -NRʼCONRʼ-, и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила;
Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными общей формулой (A-1), (A-2), (A-3), (a), (b) или (c) ниже:
;
m обозначает целое число 0 до 3, и n обозначает целое число 0 до 2;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CH2, CH, NH, N, O, S и C=O;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CH2, CH, N, O, S, NH и C=O, X5, X6, X7 и X8, каждый, независимо, выбраны из CH и N, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкилтио C1-6 алкила, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламино C1-6 алкила, C1-6 алкиламинокарбонила, C1-6 алкиламинокарбонил C1-6 алкила, C1-6 алкилкарбониламино, C1-6 алкилкарбониламино C1-6 алкила, (C1-6 алкил)2 амино C1-6 алкила, C1-6 алкилкарбонила, C1-6 алкилкарбонил C1-6 алкила, C1-6 алкиламиносульфонила, C1-6 алкиламиносульфонил C1-6 алкила, C1-6 алкилсульфониламино, C1-6 алкилсульфониламино C1-6 алкила, C1-6 алкилсульфонила, C1-6 алкилсульфонил C1-6 алкила, Cy2-, Cy2-C1-6 алкила, Cy2-C1-6 алкокси, Cy2- карбонила и Cy2- аминокарбонила, незамещенного или замещенного одним или несколькими Rb, и
Cy2 представляет собой 3-12 членный циклоалкил, 3-12 членный циклоалкенил, 3-12 членный гетероциклил, арил или 5-14 членный гетероарил;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино, C1-6 алкилкарбонила, C1-6 алкиламиносульфонила, C1-6 алкилсульфониламино и C1-6 алкилсульфонила;
при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra;
при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O;
обозначает одинарную связь или двойную связь;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 отсутствует или представляет собой -CRʼRʺ-, -N-, -O- или -S- и Rʼ и Rʺ, каждый, независимо, выбраны из водорода и C1-6 алкила;
Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными общей формулой (A-1), (A-2), (A-3), (a), (b) или (c) ниже:
;
m обозначает целое число 0 до 3, и n обозначает целое число 0 до 2;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CH2, CH, NH, N и C=O;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CH2, CH, N, NH и C=O, X5, X6, X7 и X8, каждый, независимо, выбраны из CH и N, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкилтио C1-6 алкила, C1-6 алкиламино, C1-6 алкиламино C1-6 алкила, C1-6 алкиламинокарбонила, C1-6 алкиламинокарбонил C1-6 алкила, C1-6 алкилкарбониламино, C1-6 алкилкарбониламино C1-6 алкила, C1-6 алкилкарбонила, C1-6 алкилкарбонил C1-6 алкила, Cy2, Cy2-C1-6 алкила, Cy2-C1-6 алкокси, Cy2-карбонил и Cy2-аминокарбонила, незамещенного или замещенного одним или несколькими заместителями Rb, и
Cy2 представляет собой 3-8 членный циклоалкил, 5-10 членный гетероциклил, фенил или 5-10 членный гетероарил;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкиламино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила;
при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra;
при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O; и
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными общей формулой (A-11), (a-1), (a-2), (b-1), (c-1) или (c-2) ниже:
;
m обозначает целое число, равное 1 или 2;
Y1, Y2 и Y3, каждый, независимо, выбраны из CH2, CH, NH и N;
X1, X2, X3, X4 и X9, каждый, независимо, выбраны из CH2, CH, N, NH и C=O, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
при условии, что, когда Cy1 соответствует группе формулы (c-1) или (c-2), группа формулы (c-1) или (c-2) является замещенной одним или несколькими Ra;
при условии, что, когда Cy1 соответствует формуле (b-1), X1, X2, X3 и X9 не представляют собой C=O; и
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными общей формулой (A-11) или (a-1) ниже:
;
m обозначает целое число, равное 1 или 2;
Y2 и Y3, каждый, независимо, выбраны из CH2, CH, NH и N;
X1, X2 и X3, каждый, независимо, выбраны из CH2, CH, N и NH, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 отсутствует;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
, , , , ,
, , , , ,
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, C1-6 алкиламино C1-6 алкила, C1-6 алкиламинокарбонила, C1-6 алкиламинокарбонил C1-6 алкила, C1-6 алкилкарбониламино, C1-6 алкилкарбониламино C1-6 алкила, C1-6 алкилкарбонила, C1-6 алкилкарбонил C1-6 алкила, Cy2, Cy2-C1-6 алкила, Cy2-C1-6 алкокси, Cy2-карбонил и Cy2-аминокарбонила, незамещенного или замещенного одним или несколькими заместителями Rb, и
Cy2 представляет собой 3-8 членный циклоалкил, 5-10 членный гетероциклил, фенил или 5-10 членный гетероарил;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкиламино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонил;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре; и
предпочтительно, Cy2 представляет собой 3-6 членный циклоалкил, 5-6 членный гетероциклил, фенил или 5-6 членный гетероарил.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными общей формулой (b-1) ниже:
,
X1, X2, X3 и X9, каждый, независимо, выбраны из CH2, CH, N и NH, и по меньшей мере один из X1, X2 и X3 представляет собой N или NH;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре;
при условии, что в общей формуле (b-1), X1, X2, X3 и X9 не представляют собой C=O.
В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, выбраны из водорода и C1-6 алкила;
R5 и R6, каждый, независимо, выбраны из водорода и C1-6 алкила;
L1 отсутствует;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
и ;
каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, C1-6 алкиламино C1-6 алкила, C1-6 алкиламинокарбонила, C1-6 алкиламинокарбонил C1-6 алкила, C1-6 алкилкарбониламино, C1-6 алкилкарбониламино C1-6 алкила, C1-6 алкилкарбонила, C1-6 алкилкарбонил C1-6 алкила, Cy2, Cy2-C1-6 алкила, Cy2-C1-6 алкокси, Cy2-карбонил и Cy2-аминокарбонила, незамещенного или замещенного одним или несколькими заместителями Rb, и
Cy2 представляет собой 3-8 членный циклоалкил, 5-10 членный гетероциклил, фенил или 5-10 членный гетероарил;
каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкила, C1-6 алкокси C1-6 алкокси, C1-6 алкилтио, C1-6 алкиламино, C1-6 алкиламинокарбонила, C1-6 алкилкарбониламино и C1-6 алкилкарбонила.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
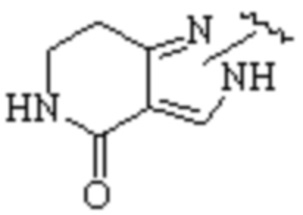 ,
, 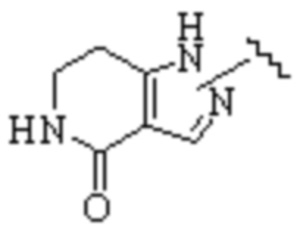 ,
, 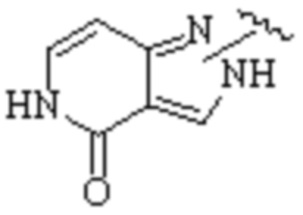 , ,
, , 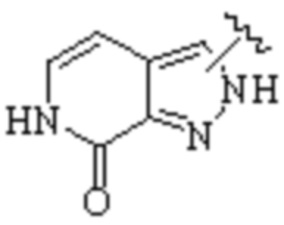 ,
,
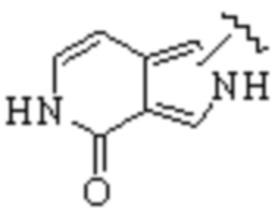 ,
, 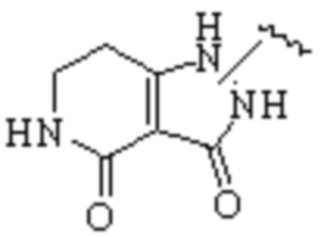 ,
, 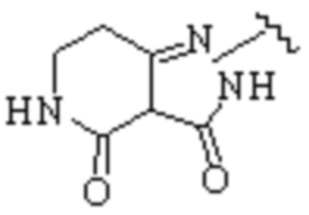 ,
, 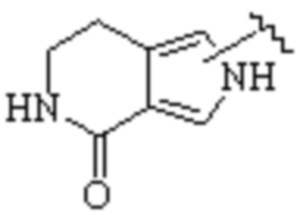 ,
, 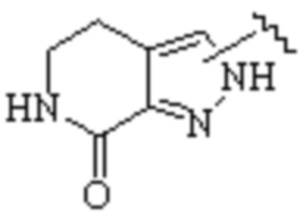 ,
, 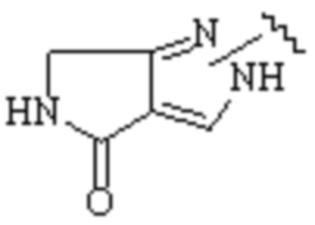 ,
, 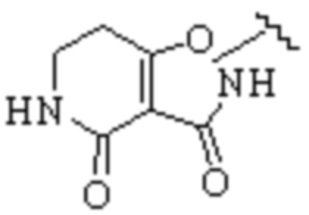 ,
, 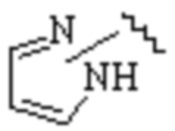 ,
,  ,
,  ,
,  .
.
В одном варианте осуществления настоящего изобретения R1 и R2, каждый, независимо, выбраны из водорода и фтора и оба R1 и R2 не представляют собой водород.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой одну из следующих групп, замещенную одним или несколькими Ra:
, , , , ,
, , , , ,
, , , , .
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила, C1-6 алкокси, C1-6 алкиламинокарбонила, Cy2, Cy2-карбонила и Cy2-аминокарбонила, незамещенного или замещенного одним или несколькими заместителями Rb, и
Cy2 представляет собой 3-6 членный циклоалкил, 5-6 членный гетероциклил, фенил или 5-6 членный гетероарил.
В одном варианте осуществления настоящего изобретения каждый Rb независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена, C1-6 алкила и C1-6 алкокси.
В одном варианте осуществления настоящего изобретения Cy1 представляет собой одну из следующих групп, замещенную одним или несколькими Ra:
 , , , ,
, , , ,
, , , ,
В одном варианте осуществления настоящего изобретения каждый Ra независимо выбран из гидроксила, амино, карбоксила, циано, нитро, галогена и C1-6 алкила и 3-6 членного циклоалкила, незамещенного или замещенного одним или несколькими заместителями Ra.
В одном варианте осуществления настоящего изобретения каждый Rb независимо выбран из гидроксила, амино, циано, нитро и галогена.
«Фармацевтически приемлемая соль», описанная в настоящем изобретении, относится к фармацевтически приемлемой аддитивной соли кислоты или основания соединения формулы I или его к аддитивной соли его сольвата. Когда в соединении присутствует кислотная функциональная группа (такая как -COOH, -OH, -SO3H и тому подобное), кислотная функциональная группа может образовывать соль с подходящим неорганическим или органическим катионом (основанием), включая соль со щелочным металлом, щелочноземельным металлом и тому подобное, соль аммония и соль, образованную с азотистым органическим основанием. Когда в соединении присутствует основная функциональная группа (такая как -NH2 и тому подобное), основная функциональная группа может образовывать соль с подходящим неорганическим или органическим анионом (кислотой), включая соль, образованную неорганической кислотой или органической кислотой. Такая «фармацевтически приемлемая соль» включает, но этим не ограничивается, соли кислот, такие как гидрохлорид, гидробромид, гидройодат, сульфат, фосфат, нитрат, бензолсульфонат, бензоат, п-толуолсульфонат, 2,3-дигидроксисукцинат, камфорсульфонат, цитрат, метансульфонат, этансульфонат, пропансульфонат, фумарат, глюконат, глутамат, гидроксиэтилсульфонат, лактат, малеат, малат, манделат, муконат, памоат, пантотенат, сукцинат, тартрат и тому подобное, предпочтительно, бензоат, бензолсульфонат, п-толуолсульфонат, метансульфонат, цитрат, малеат, фумарат, тартрат, соли алкановой кислоты (HOOC-(CH2)n-COOH (где n равно от 0 до 4)) (такие как формиат, ацетат и пропионат). Кроме того, «фармацевтически приемлемая соль» также включает, но этим не ограничивается, соли, образованные следующими основаниями: аргинин, бетаин, кофеин, холин, N, Nʼ-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этил-морфолин, N-этилпиперидин, меглумин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, прокаин, пурин, теобромин, триэтиламин, триметиламин, трипропиламин и трометамин; и кроме того, «фармацевтически приемлемая соль» также может представлять собой литиевую соль, натриевую соль, калиевую соль, кальциевую соль, магниевую соль, цинковую соль, бариевую соль, алюминиевую соль, соль трехвалентного железа, соль меди, соль двухвалентного железа, соль марганца, соль двухвалентного марганца или тому подобное.
«Фармацевтически приемлемый сложный эфир» соединения по настоящему изобретению относится к сложному эфиру соединения по настоящему изобретению, который гидролизуется in vivo, и включает сложный эфир, который может легко разлагаться в организме человека, предоставляя исходное соединение или его соль. Подходящие сложноэфирные группы включают, например, сложноэфирные группы, полученные из фармацевтически приемлемых алифатических карбоновых кислот (в частности, алкановой кислоты, алкеновой кислоты, циклической алкановой кислоты и алкановой двухосновной кислоты), где каждый алкил или алкенил, предпочтительно, имеет шесть или меньше атомов углерода. Типичные примеры конкретного сложного эфира включают, но этим не ограничиваются, формиат, ацетат, пропионат, бутират, акрилат и этилсукцинат.
В настоящем изобретении в процессе реакции атом N аминогруппы может быть необязательно защищен аминозащитной группой. «Аминозащитная группа» относится к химической группе, которая связана с аминогруппой и может быть легко удалена в определенных условиях, и включает, но не ограничивается ими, алкоксикарбонильные группы, ацильные группы и алкильные группы, такие как трет-бутоксикарбонил, бензилоксикарбонил, флуоренилметоксикарбонил, аллилоксикарбонил, фталоил, бензил, п-метоксибензил, трифенилметил и тому подобное. Специалисты в данной области могут осуществить соответствующий выбор и работу в соответствии с общепринятым руководством в данной области техники Greeneʼs Protective Groups in Organic Synthesis (4th edition).
Фраза «фармацевтически приемлемый» означает, что вещество или композиция должны быть фармацевтически и/или токсикологически совместимыми с другими ингредиентами, содержащимися в препарате и/или фармацевтической композиции.
«Изомеры», описанные в настоящем изобретении, включают стереоизомер и таутомер.
Стереоизомер означает, что энантиомер будет образовываться, когда в соединении присутствует асимметричный атом углерода, или что цис-транс-изомер будет образовываться, когда в соединении присутствует двойная связь углерод-углерод или кольцевая структура.
«Таутомер» означает изомер функциональной группы, который образуется из-за быстрого перемещения определенного атома между двумя положениями в молекуле, и таутомер представляет собой особый изомер функциональной группы. Например, таутомер карбонильного соединения, содержащего α-H, может представлять собой:  или
или 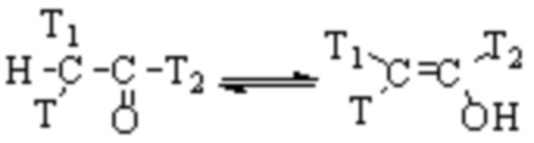 , где T, T1 и T2, каждый, независимо, выбраны из любой группы, которая соответствует правилу связывания соединения.
, где T, T1 и T2, каждый, независимо, выбраны из любой группы, которая соответствует правилу связывания соединения.
«Таутомер» также может представлять собой, например, другие прототропные таутомеры, в частности, такие как фенол-кето таутомер, нитрозо-оксимино таутомер и имин-енаминный таутомер. Однако этим не ограничивается, и специалисты в данной области могут легко судить о существовании у соединения таутомера и его конкретной формы.
Таким образом, все энантиомеры, диастереомеры, рацематы, цис-транс-изомеры, геометрические изомеры, эпимеры, таутомеры и их смеси соединения формулы I включены в объем настоящего изобретения.
Фармацевтическая композиция по настоящему изобретению содержит по меньшей мере одно соединение формулы I и его фармацевтически приемлемую соль, сложный эфир, стереоизомер и таутомер, и, необязательно, один или несколько фармацевтически приемлемых носителей.
Фармацевтическая композиция по настоящему изобретению может вводиться пациенту или субъекту, нуждающемуся в профилактике и/или лечении, любым подходящим способом, известным в данной области, например, пероральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, внутрикожный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, транспульмональный, местный (включая буккальный и сублингвальный), вагинальный, внутрибрюшинный, внутрилегочный, интраназальный и другие способы введения.
Фармацевтическая композиция по настоящему изобретению может быть изготовлена в виде обычного твердого препарата, такого как таблетка, капсула, пилюля, гранула и тому подобное, и также может быть изготовлена в виде жидкого препарата для перорального применения, такого как пероральный раствор, пероральная суспензия, сироп и тому подобное. При изготовлении перорального препарата могут быть добавлены один или несколько подходящих эксципиентов, разбавителей, подсластителей, солюбилизаторов, лубрикантов, связующих, разрыхлителей таблеток, стабилизаторов, консервантов и инкапсулирующих материалов. Для парентерального введения фармацевтическая композиция может быть изготовлена в виде инъекции, включая раствор для инъекций, стерильный порошок для инъекций и концентрированный раствор для инъекций. Инъекцию можно проводить обычным способом, существующим в фармацевтической области, и во время процесса приготовления нельзя добавлять никакую добавку или можно добавлять подходящую добавку в соответствии со свойствами лекарственного средства. Для ректального введения фармацевтическая композиция может быть изготовлена в виде суппозитория и тому подобное. Для транспульмонального введения фармацевтическая композиция может быть изготовлена в виде ингаляционного средства, спрея или тому подобного. В настоящем изобретении подходящие твердые носители включают, но этим не ограничиваются, целлюлозу, глюкозу, лактозу, маннит, стеарат магния, карбонат магния, карбонат натрия, сахарин натрия, сахарозу, декстрин, тальк, крахмал, пектин, желатин, трагакант, аравийскую камедь, альгинат натрия, п-гидроксилбензоат, метилцеллюлозу, карбоксиметилцеллюлозу натрия, низкоплавкий воск, масло какао и тому подобное Подходящие жидкие носители включают, но этим не ограничиваются, воду, этанол, полиол (такой как глицерин, пропиленгликоль, жидкий полиэтиленгликоль и т.д.), растительное масло, глицерид и их смеси).
Способы изготовления фармацевтической композиции по настоящему изобретению являются общеизвестными. Фармацевтическую композицию по настоящему изобретению получают известными способами, включая обычное смешивание, гранулирование, таблетирование, нанесение покрытия, растворение или лиофилизацию.
Фармацевтический состав, предпочтительно, находится в виде стандартной лекарственной формы. В этой форме состав подразделяется на стандартные дозировки, содержащие соответствующее количество активных компонентов. Стандартная лекарственная форма может быть расфасована в упаковки, содержащие дискретное количество препарата, такие как упакованные таблетки, капсулы или порошки во флаконе или ампуле.
Дозировка лекарственного средства зависит от различных факторов, включая возраст, вес и состояние пациента, а также способ введения. Точная вводимая доза определяется на основании заключения лечащего врача. Обычная доза для введения активного соединения может составлять, например, от около 0,01 до около 100 мг/день, от около 0,05 до около 75 мг/день, от около 0,1 до около 50 мг/день или от около 5 до около 10 мг/день. Желаемая дозировка также зависит от конкретного применяемого соединения, тяжести заболевания, пути введения, веса и состояния здоровья пациента и заключения лечащего врача.
Соединение по настоящему изобретению также включает соединение, в котором один или несколько атомов водорода, атомов фтора, атомов углерода, атомов азота, атомов кислорода и атомов серы заменены радиоизотопами или стабильными изотопами. Эти меченые соединения можно использовать для метаболических или фармакокинетических исследований, биологического анализа в качестве лигандов для рецепторов и тому подобное.
Соединение по настоящему изобретению может быть использовано для лечения и/или профилактики заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им, которое включает введение субъекту соединения по настоящему изобретению.
Фармацевтическая композиция, содержащая соединение по настоящему изобретению может быть использована для лечения и/или профилактики заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им, которое включает введение субъекту соединения по настоящему изобретению.
Способ получения соединения формулы (I) по настоящему изобретению
Соединение по настоящему изобретению можно получить рядом способов, включая стандартные химические методы. Если не указано иное, любая переменная, определенная выше, будет иметь значение, определенное выше. Примеры общих способов синтеза представлены на следующих схемах, и их можно легко модифицировать для получения других соединений по настоящему изобретению. Специалисты в данной области могут проводить следующие реакции в соответствии с обычными методами (такими как Organic Synthesis (2nd edition), Michael B. Smith etc.), изученными в данной области. Конкретные соединения настоящего изобретения были специально получены в примерах.
В одном варианте осуществления настоящего изобретения соединение общей формулы (I) получали посредством реакции соединения формулы (SM1) и соединения формулы (SM2),
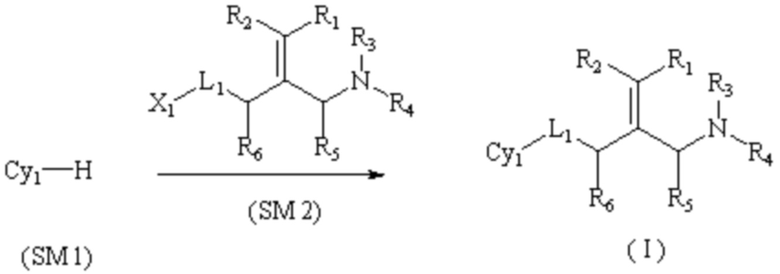 ,
,
где Cy1, R1, R2, R3, R4, R5, R6 и L1 имеют значения, описанные выше; и X1 представляет собой удаляемую группу, включая но этим не ограничиваясь, галоген или сульфонат.
Кроме того, когда R3 и R4 представляют собой водород, в процессе получения в группе  необходимо защищать водород, имеющийся на N, таким образом образуя
необходимо защищать водород, имеющийся на N, таким образом образуя  , где G1 и G2 представляют собой аминозащитные группы.
, где G1 и G2 представляют собой аминозащитные группы.
«Аминозащитные группы» представляют собой защитные группы, обычно используемые специалистами в данной области, такие как трет-бутоксикарбонил, бензилоксикарбонил, трет-бутил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трифторацетил, хлорацетил, трифенилметил, тетрагидропиранил, 4-метоксибензил, 2,4-диметоксибензил, o-нитробензолсульфонил и фталоил. Более того, способы введения защитных групп и удаления защитных групп у аминогрупп также могут быть осуществлены методами, известными специалистам в данной области. Например, можно сослаться на стадии, описанные в Protective Groups in Organic Synthesis (3rd edition).
В одном варианте осуществления настоящего изобретения соединение общей формулы (Iʼ) получали посредством проведения следующих стадий:
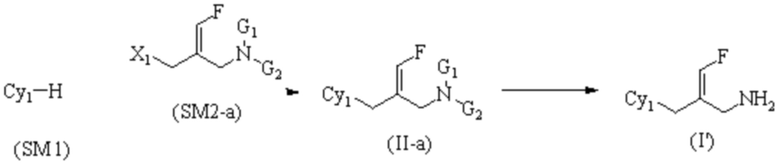
где определения Cy1, G1, G2 и X1 имеют значения, описанные выше.
(1) Соединение формулы (SM1) растворяли в органическом растворителе1 и к раствору добавляли подходящее основание для осуществления взаимодействия с соединением формулы (SM2-a) с получением соединения формулы (II-a);
(2) Соединение формулы (II-a) растворяли в органическом растворителе2 и к раствору добавляли подходящий реагент для удаления защитной группы, получая таким образом соединение формулы (I).
В одном варианте осуществления настоящего изобретения соединение общей формулы (Iʼ) получали посредством проведения следующих стадий:
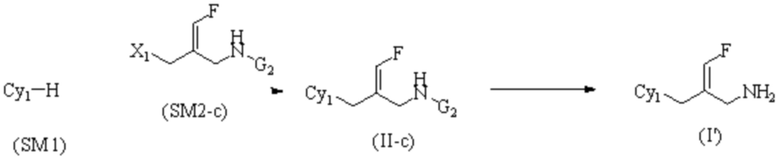
где определения Cy1 и X1 имеют значения, описанные выше; и
G2 выбран из трет-бутоксикарбонила, бензилоксикарбонила, трет-бутила, 9-флуоренилметоксикарбонила, аллилоксикарбонила, трифторацетила, хлорацетила, трифенилметила, тетрагидропиранила, 4-метоксибензила, 2,4-диметоксибензила и o-нитробензолсульфонила.
(1) Соединение формулы (SM1) растворяли в органическом растворителе1 и к раствору добавляли соединение формулы (SM2-c) и основание с получением соединения формулы (II-c);
(2) Соединение формулы (II-c) растворяли в органическом растворителе2 и к раствору добавляли подходящий реагент для удаления защитной группы, получая таким образом соединение формулы (Ic).
В одном варианте осуществления настоящего изобретения соединение общей формулы (Iʼ) получали посредством проведения следующих стадий:
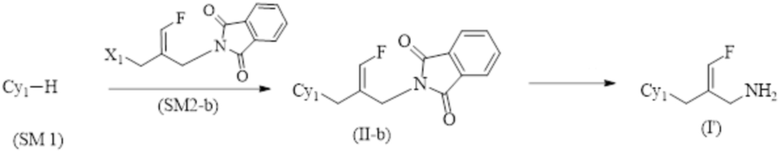
где определения Cy1 и X1 имеют значения, описанные выше.
(1) Соединение формулы (SM1) растворяли в органическом растворителе1 и к раствору добавляли подходящее основание для осуществления взаимодействия с соединением формулы (SM2-b) с получением соединения формулы (II-b);
(2) Соединение формулы (II-b) растворяли в органическом растворителе2 и к раствору добавляли гидразингидрат для осуществления гидразинолиза, таким образом получали соединение формулы (Ib).
В одном варианте осуществления настоящего изобретения органическим растворителем1 является ДМФ, DMA, ACN, метанол, этанол, изопропанол или ТГФ.
В одном варианте осуществления настоящего изобретения органическим растворителем2 является метанол, этанол или изопропанол.
В одном варианте осуществления настоящего изобретения основание представляет собой гидрид натрия, карбонат цезия или карбонат калия.
В одном варианте осуществления настоящего изобретения агентом для удаления защитной группы является хлористоводородная кислота, трифторуксусная кислота, бромистоводородная кислота, триметилйодсилан или тому подобное.
В одном варианте осуществления настоящего изобретения в процессе реакции для получения целевого соединения может быть добавлен катализатор межфазного переноса, и катализатор межфазного переноса может представлять собой катализатор, обычно используемый в данной области техники, включая, но этим не ограничиваясь, например, ацетат меди, хлорид меди, палладий на углероде, хлорид железа, ацетат палладия, дихлорид [1,1-бис(дифенилфосфино)ферроцен]палладия, бромид тетрабутиламмония, хлорид бензилтриэтиламмония, хлорид тетрабутиламмония и тому подобное.
«Подходящий агент для удаления защитных групп» относится к реагенту, который специалисты в данной области используют для проведения реакции удаления защитных групп путем выбора соответствующей кислоты, основания или окислителя в соответствии с различными типами аминозащитных групп G1 и G2 в химической структуре. Могут быть приняты те, которые внесены в руководство Protective Groups in Organic Synthesis (3rd edition).
В настоящем изобретении «кислота» может представлять собой кислоту, обычно используемую в данной области, включая органические кислоты и неорганические кислоты. Органические кислоты могут включать, например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, трифторуксусную кислоту, лимонную кислоту, молочную кислоту, винную кислоту, щавелевую кислоту, малеиновую кислоту, фумаровую кислоту, миндальную кислоту, глутаровую кислоту, яблочную кислоту, бензойную кислоту, фталевая кислота, аскорбиновую кислоту, бензолсульфоновую кислота, п-толуолсульфоновую кислоту, метансульфоновую кислоту и этилсульфоновую кислоту; и неорганические кислоты могут включать, например, хлористоводородную кислоту, серную кислоту, азотную кислоту, угольную кислоту, бромистоводородную кислоту, фосфорную кислоту, йодистоводородную кислоту и тому подобное. Предпочтительной является хлористоводородная кислота.
В настоящем изобретении «основание» может представлять собой основание, обычно используемое в данной области, включая органические основания и неорганические основания. Органические основания могут включать, например, метиламин, этиламин, пропиламин, N, N-диизопропилэтиламин, триметиламин, триэтиламин, N-метилморфолин, дициклогексиламин, этаноламин, диэтаноламин, триэтаноламин, меглумин, диэтаноламин, этилендиамин, пиридин, метилпиридин, хинолин и тому подобное; и неорганические основания могут включать, например, гидроксиды, карбонаты и бикарбонаты щелочных металлов (таких как литий, натрий, калий и цезий); гидроксиды, карбонаты и бикарбонаты щелочноземельных металлов (магний, кальций, стронций и барий); трет-бутоксид натрия, трет-бутоксид калия, этоксид натрия и тому подобное.
В настоящем изобретении «окислитель» может быть окислителем, обычно используемым в данной области, включая, помимо прочего, нитрат церия-аммония, 2,3-дихлор-5,6-дициано-п-бензохинон, хлорид меди, диоксид марганца, перманганат, дихромат, пероксиуксусную кислоту, пероксибензойную кислоту и тому подобное.
В настоящем изобретении «органический растворитель1» относится к отдельному или смешанному органическому растворителю, обычно используемому в данной области, включая, но не ограничиваясь ими, простые эфиры, алканы, галогеналканы, ароматические углеводороды, спирты и тому подобное. В частности, органический растворитель может представлять собой N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ароматические углеводороды (например, толуол, бензол, диметилбензол, триметилбензол и тому подобное), насыщенные углеводороды (такие как циклогексан, гексан и тому подобное), галогенуглеводороды (такие как дихлорметан, хлороформ, 1,2-дихлорэтан и тому подобное), простые эфиры (такие как тетрагидрофуран, диэтиловый эфир, диоксан, 1,2-диметоксиэтан и тому подобное), сложные эфиры (такие как метилацетат, этилацетат и тому подобное), кетоны (такие как ацетон, метилэтилкетон и тому подобное), нитрилы (такие как ацетонитрил и тому подобное), спирты (такие как метанол, этанол, изопропанол, трет-бутанол и тому подобное), воду, смешанные водные растворители или тому подобное.
В настоящем изобретении «органическом растворителе2» относится к отдельному или смешанному органическому растворителю, обычно используемому в данной области, включая, но не ограничиваясь ими, простые эфиры, алканы, галогеналканы, ароматические углеводороды, спирты и тому подобное. В частности, органический растворитель может представлять собой N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ароматические углеводороды (например, толуол, бензол, диметилбензол, триметилбензол и тому подобное), насыщенные углеводороды (такие как циклогексан, гексан и тому подобное), галогенуглеводороды (такие как дихлорметан, хлороформ, 1,2-дихлорэтан и тому подобное), простые эфиры (такие как тетрагидрофуран, диэтиловый эфир, диоксан, 1,2-диметоксиэтан и тому подобное), сложные эфиры (такие как метилацетат, этилацетат и тому подобное), кетоны (такие как ацетон, метилэтилкетон и тому подобное), нитрилы (такие как ацетонитрил и тому подобное), спирты (такие как метанол, этанол, изопропанол, трет-бутанол и тому подобное), воду, смешанные водные растворители или тому подобное.
В настоящем изобретении в вышеуказанном процессе реакции температура реакции может быть отрегулирована по мере необходимости, например, высокая температура, комнатная температура, низкая температура и т. д. Высокая температура обычно относится к температуре выше 30°C, и при необходимости может быть выполняться нагревание. Комнатная температура обычно составляет от 15°C до 30°C. Под низкой температурой обычно понимается температура ниже 15°C, и при необходимости может быть выполнено охлаждение.
Примеры
Если в примерах не указаны конкретные условия проведения реакции, должны быть приняты обычные условия или условия, рекомендованные производителями. Все используемые реагенты или инструменты без производителей являются коммерчески доступными традиционными продуктами.
В настоящем изобретении, если не указано иное: (i) температура выражается в градусах Цельсия (°C), и процесс осуществляют при комнатной температуре; (ii) процесс реакции отслеживают с помощью тонкослойной хроматографии (ТСХ) или ЖХ-МС; (iii) конечный продукт имеет четкие данные спектроскопии протонного ядерного магнитного резонанса (1H-ЯМР) и данные масс-спектрометрии (МС).
Сокращения и английские выражения, используемые в настоящем изобретении, имеют следующие значения:
ДХМ: дихлорметан
DIPEA: N, N-диизопропилэтиламин
Boc: трет-бутоксикарбонил
(Boc)2O: ди-трет-бутил дикарбонат
TEA: триэтиламин
ДМСО: диметилсульфоксид
DMA или DMAc: диметилацетамид
DMAP: 4-диметиламинопиридин
ДМФ: N, N-диметилформамид
EDCI: 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид
EtOH: этанол
EtONa: этоксид натрия
водн. HCl: разбавленная соляная кислота
NaH: гидрид натрия
N2H4: гидразин
AcONa: ацетат натрия
Ac2O: уксусный ангидрид
ACN: ацетонитрил
ТГФ: тетрагидрофуран
CuI: йодид меди
Cs2Co3: карбонат цезия
K2CO3: карбонат калия
EA: этилацетат
MeOH: метанол
MTBE: метиловый трет-бутиловый эфир
PE: петролейный эфир
HATU: 2-(7-азабензотриазол-1-ил)-N, N,Nʼ,Nʼ-тетраметилуроний гексафторфосфат
TBAB: тетрабутиламмоний бромид
(E)-2-(2-(бромметил)-3-фтораллил)изоиндол-1,3-дион получали согласно ссылке на способ синтеза промежуточного соединения (Ian A. McDonald, Philipe Bey. A general preparation of fluoroallylamine enzyme inhibitors incorporating a β -substituted heteroatom. Tetrahedron Letters, Vo1.26, No.32, pp 3807-3810, 1985), о котором сообщили Ian A. McDonald, et al.
Пример 1: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A1)
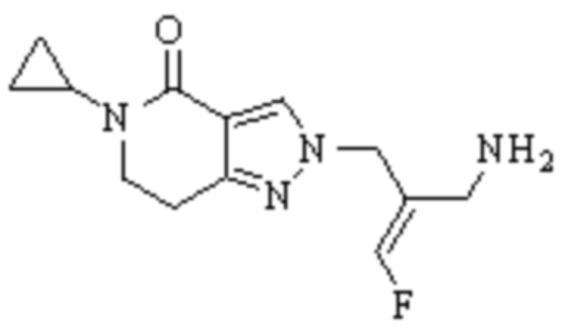
Стадия 1: Синтез (E)-1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-диона

Продукт циклопропилпиперидин-2,4-дион (5 г, 32,6 ммоль, 1,0 экв.) медленно добавляли к ДМФ-DMA (5 г, 41,8 ммоль, 1,28 экв.) и подвергали взаимодействию при температуре 25°C в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционную систему концентрировали с получением сырого продукта (теоретический выход: 6,789 г), который напрямую использовали на следующей стадии.
Стадия 2: Синтез 5-циклопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c] пиридин-4-она
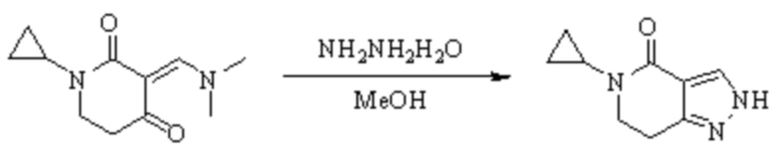
Сырой продукт (E)-1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-дион (6,789 г, 32,6 ммоль), полученный на предшествующей стадии, растворяли в MeOH (50 мл). Затем к раствору добавляли 85%-ный гидразингидрат (2,1 г, 35,9 ммоль, 1,1 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 0,5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали и оставляли до тех пор, пока не образовалось большое количество твердого осадка белого цвета. Затем в полученную смесь добавляли небольшое количество MTBE и фильтровали в вакууме. Слой на фильтре перекристаллизовывали с 2-кратным объемом 95%-ного EtOH, фильтровали в вакууме, и сушили при температуре 50°C с получением продукта (3,7 г, выход за две стадии: 64%).
Стадия 3: Синтез (E)-2-(2-((5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндол-1,3-диона
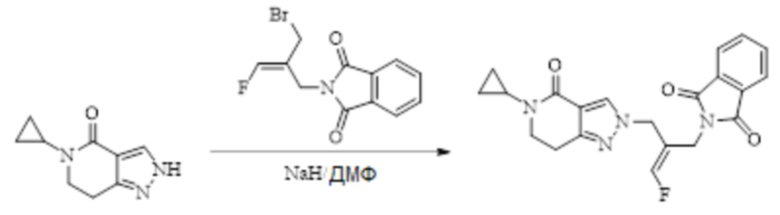
Промежуточный 5-циклопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c] пиридин-4-он (1,0 г, 5,643 ммоль, 1 экв.) растворяли в ДМФ (2,5 мл). После охлаждения до температуры 0°C к раствору в атмосфере N2 добавляли 60%-ный NaH (248 мг, 6,207 ммоль, 1,1 экв.) и перемешивали в течение 30 мин в атмосфере N2. Затем добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндол-1,3-диона (2,019 г, 6,722 ммоль, 1,2 экв.) в ДМФ (2,5 мл) и подвергали взаимодействию при комнатной температуре 18°C в течение 1 ч После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (10 мл) и экстрагировали смесью ДХМ:MeOH=10:1 (15 мл × 3). Органическую фазу промывали водой, сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (элюент ДХМ:MeOH=200:1) с получением продукта (583 мг, выход: 26,2%).
Стадия 4: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
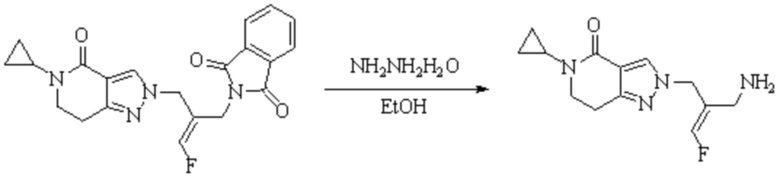
Промежуточный (E)-2-(2-((5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндол-1,3-дион (583 мг, 1,478 ммоль, 1 экв.) растворяли в EtOH (15 мл). Затем к раствору добавляли 85%-ный гидразингидрат (305 мг, 5,174 ммоль, 3,5 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали в вакууме и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (70 мг, выход: 17,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,12 (с, 1H), 6,79-7,00 (д, 1H), 4,70-4,71 (д, 2H), 3,48-3,51 (т, 2H), 3,04-3,05 (д, 2H), 2,75-2,78 (т, 2H), 2,59-2,65 (м, 1H), 0,71-0,76 (м, 2H), 0,59-0,61 (м, 2H).
Молекулярная формула: C13H17FN4O, молекулярная масса: 264,30, ЖХ-МС (полож., m/z)=265,25 [M+H]+.
Пример 2: Синтез гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A2)

Стадия 1: Синтез (E)-1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-диона
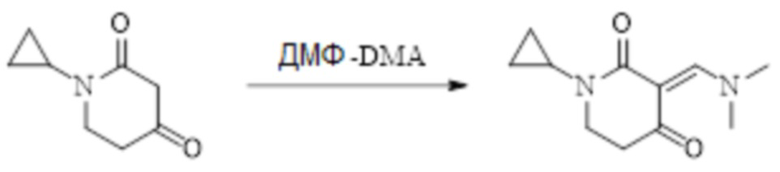
Продукт циклопропилпиперидин-2,4-дион (20 г, 0,130 моль, 1,0 экв.) медленно добавляли к смеси ДМФ-DMA (19,9 г, 0,167 моль, 1,28 экв.) и смесь перемешивали при температуре 25°C в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционную систему концентрировали при температуре 50°C с получением сырого продукта, который напрямую использовали на следующей стадии.
Стадия 2: Синтез 5-циклопропил-1,5,6,7-тетрагидро-4H-пиразоло[4,3-c] пиридин-4-она
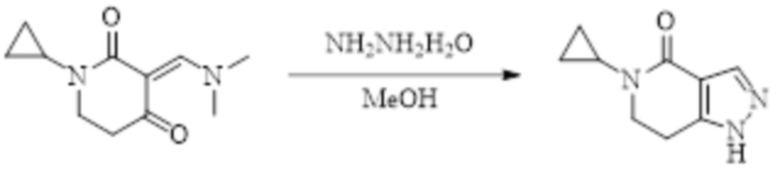
Промежуточный (E)-1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-дион (27,073 г, 0,130 моль) растворяли в MeOH (100 мл). Затем к раствору добавляли 85%-ный гидразингидрат (8,4 г, 0,143 моль, 1,1 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали с получением сырого продукта. Сырой продукт перекристаллизовывали из 95%-ного EtOH и фильтровали в вакууме, и слой на фильтре сушили при температуре 50°C с получением продукта (16 г, выход: 69,6%).
Стадия 3: Синтез (E)-2-(2-((5-циклопропил-4-оксо-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндол-1,3-диона
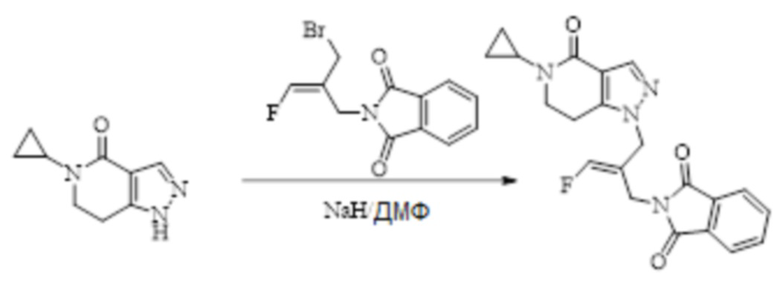
Промежуточный 5-циклопропил-1,5,6,7-тетрагидро-4H-пиразоло[4,3-H]пиридин-4-он (2000 мг, 11,286 ммоль, 1 экв.) растворяли в ДМФ (5 мл). После охлаждения до температуры 0°C к раствору добавляли 60% NaH (496 мг, 12,414 ммоль, 1,1 экв.) в атмосфере N2 и перемешивали в течение 30 мин в атмосфере N2. Затем добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндол-1,3-диона (4037 мг, 13,543 ммоль, 1,2 экв.) в ДМФ (5 мл) и подвергали взаимодействию при комнатной температуре в 19°C в течение ночи. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (20 мл) и экстрагировали ДХМ:MeOH=10:1 (30 мл × 3). Органическую фазу промывали водой, сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (элюент ДХМ:MeOH=200:1) с получением продукта (100 мг, выход: 2,2%).
Стадия 4: Синтез гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
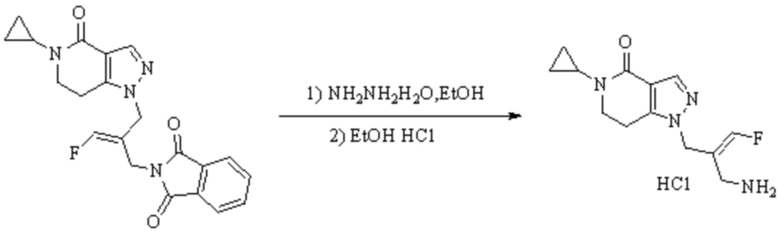
Промежуточный (E)-2-(2-((5-циклопропил-4-оксо-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндол-1,3-дион (70 мг, 0,177 ммоль, 1 экв.) растворяли в EtOH (1,75 мл). Затем к раствору добавляли 85%-ный гидразингидрат (36 мг, 0,621 ммоль, 3,5 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали в вакууме и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением сырого продукта. Сырой продукт растворяли в небольшом количестве этанола. К раствору добавляли хлорид водорода в этаноле (0,1 мл) и перемешивали в течение 1 ч, при этом образовывался осадок. Полученную смесь фильтровали в вакууме и слой на фильтре сушили с получением продукта (40 мг, выход: 74,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,46 (с, 3H), 7,72 (с, 1H), 7,06-7,26 (д, 1H), 4,89 (д, 2H), 3,54-3,57 (т, 2H), 3,43-3,44 (д, 2H), 2,98-3,02 (т, 2H), 2,57-2,62 (м, 1H), 0,71-0,76 (т, 2H), 0,56-0,60 (м, 2H).
Молекулярная формула: C13H18ClFN4O, молекулярная масса: 300,76, ЖХ-МС (полож., m/z)=264,8 [M+H]+.
Пример 3: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2, 5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A6)
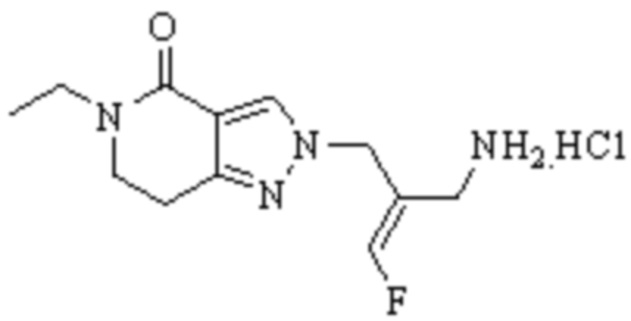
Стадия 1: Синтез промежуточного (E)-(3-фтор-2-((5-этил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамата
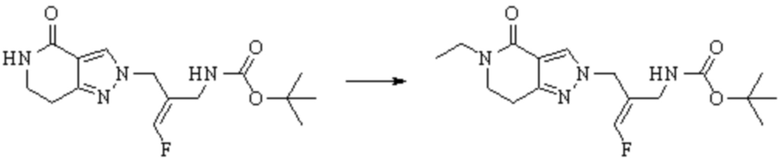
Промежуточный (E)-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (1,0 г, 3,08 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10 мл). Затем к раствору добавляли гидрид натрия (160 мг, 4,0 ммоль, 1,3 экв., 60%) и подвергали взаимодействию при комнатной температуре в течение 30 мин. К реакционному раствору добавляли йодэтан (576 мг, 3,7 ммоль, 1,2 экв.) и подвергали взаимодействию при нагревании при температуре 60°C в течение 4 ч. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционный сосуд добавляли воду (10 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили и концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (MeOH:ДХМ=1:20) с получением продукта (180 мг, выход: 16%).
Стадия 2: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
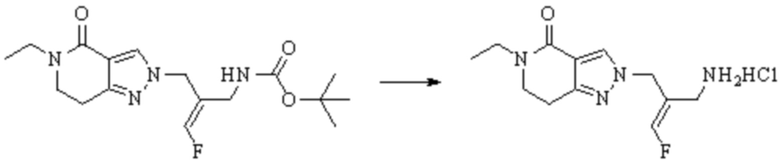
Промежуточный (E)-(3-фтор-2-((5-этил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (180 мг, 0,51 ммоль, 1,0 экв.) растворяли в этаноле (2 мл). Затем к раствору добавляли раствор хлорида водорода в этаноле (2 мл) подвергали взаимодействию в течение 12 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении, растворяли в воде и лиофилизовали с получением продукта (120 мг, выход: 83%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,47 (с, 3H), 8,27 (с, 1H), 7,37 (с, 0,5H), 7,16 (с, 0,5H), 4,93 (с, 2H), 3,52-3,56 (м, 2H), 3,39-3,42 (м, 2H), 3,32-3,33 (д, 2H), 2,81-2,84 (м, 2H), 1,04-1,07 (м, 3H).
Молекулярная формула: C12H17FN4O, молекулярная масса: 252,29, ЖХ-МС (полож., m/z)=253,22 [M+H]+.
Пример 4: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-трет-бутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A7)

Стадия 1: Синтез этил 3-(трет-бутиламино)пропионата
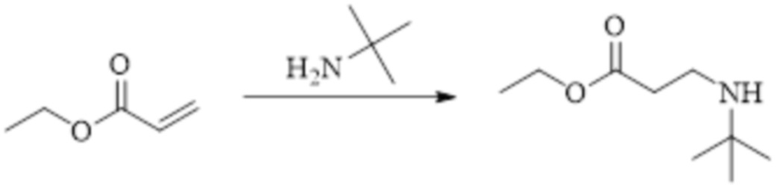
трет-Бутиламин (87,66 г, 1,20 моль, 1,2 экв.) растворяли в этаноле (400 мл). Затем на ледяной бане в раствор медленно по каплям добавляли этилакрилат (100,00 г, 1,00 моль, 1,0 экв.) и подвергали взаимодействию при комнатной температуре в течение ночи. После того, как не оставалось исходных продуктов, как обнаруживалось по данным ГХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (173 г, выход: 99%).
Стадия 2: Синтез этил 3-(трет-бутил(3-этокси-3-оксопропил)амино)-3-оксопропионата
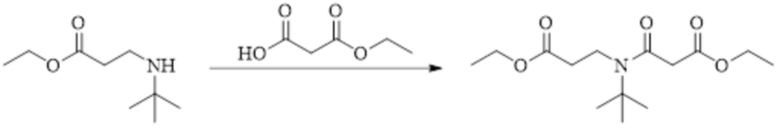
Промежуточный этил 3-(трет-бутиламино)пропионат (70,00 г, 0,404 моль, 1,0 экв.), моноэтил малонат (53,37 г, 0,404 моль, 1,0 экв.), 4-диметиламинопиридин (9,88 г, 0,0808 ммоль, 0,2 экв.) и триэтиламин (102,30 г, 1,010 моль, 2,5 экв.) растворяли в дихлорметане (490 мл) и раствор перемешивали на ледяной бане в течение 15 мин. Затем партиями добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (93,02 г, 0,485 моль, 1,2 экв.), и раствору давали взаимодействовать при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, на ледяной бане в реакционный сосуд добавляли воду и концентрированную соляную кислоту (3:1, 360 мл) и смесь перемешивали в течение 15 мин, затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (300 мл). Органические фазы объединяли, промывали последовательно насыщенным водным раствором бикарбоната натрия (800 мл) и насыщенным водным раствором хлорида натрия (800 мл × 2), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (114 г, выход: 98,2%).
Стадия 3: Синтез 1-трет-бутилпиперидин-2,4-диона
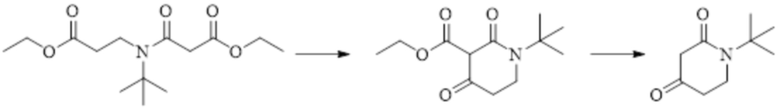
Этил 3-(трет-бутил(3-этокси-3-оксопропил)амино)-3-оксопропионат (114,00 г, 0,397 моль, 1,0 экв.) и этоксид натрия (53,99 г, 0,793 моль, 2,0 экв.) растворяли в этаноле (456 мл) и подвергали взаимодействию при температуре 80°C в течение 4 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением натриевой соли промежуточного этил 1-трет-бутил-2,4-диоксопиперидин-3-карбоксилата. Затем добавляли воду и pH смеси доводили до 2 с помощью концентрированной соляной кислоты. Реакцию проводили при температуре 85°C в течение 5 ч и затем при температуре 95°C в течение 1,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и добавляли хлорид натрия до насыщения реакционного раствора. Экстрагировали дихлорметаном (400 мл × 3). Органическую фазу сушили и концентрировали с получением маслянистого сырого продукта. После охлаждения в осадок выпадали твердые продукты. Затем добавляли MTBE и смесь перемешивали, так что в осадок выпадало большое количество твердых продуктов. Полученную смесь фильтровали в вакууме и слой на фильтре промывали небольшим количеством MTBE и сушили с получением продукта (45,3 г, выход: 67,48%).
Стадия 4: Синтез 5-трет-бутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c] пиридин-4-она
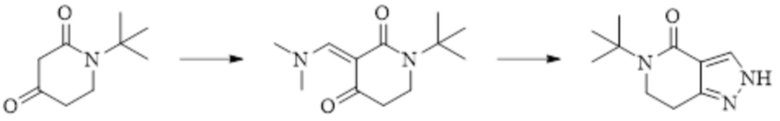
Промежуточный 1-трет-бутилпиперидин-2,4-дион (40 г, 0,207 моль, 1,0 экв.) растворяли в N, N-диметилформамид диметилацетале (35,04 г, 0,265 моль, 1,28 экв.) и подвергали взаимодействию при комнатной температуре в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением промежуточного (E)-1-трет-бутил-3-((диметиламино) метилен)пиперидин-2,4-диона. К промежуточному продукту добавляли метанол (200 мл) и гидразингидрат (15,3 г, 0,228 моль, 1,1 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении, добавляли MTBE (300 мл), и перемешивали в течение ночи, и в осадок выпадало большое количество твердых продуктов. Полученную смесь фильтровали в вакууме и слой на фильтре промывали MTBE и сушили с получением продукта (40,00 г, выход: 87,7%).
Стадия 5: Синтез (E)-2-(2-((5-трет-бутил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
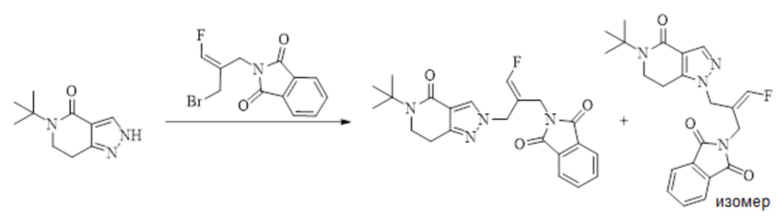
5-трет-Бутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (1,08 г, 5,59 ммоль, 1,0 экв.) растворяли в ДМФ (10 мл). После охлаждения до температуры 0°C к раствору добавляли NaH (массовая доля: 60%, 246 мг, 6,147 ммоль, 1,1 экв.), перемешивали в течение 30 мин, добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-диона (2,00 г, 6,706 ммоль, 1,2 экв.) в ДМФ (5 мл) и подвергали взаимодействию при комнатной температуре в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционную смесь добавляли воду (12 мл) и экстрагировали смесью дихлорметана и метанола (10:1, 25 мл × 3), затем жидкости разделяли. Органические фазы объединяли, опять промывали водой, сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=3:1-2:1, с 0,5% триэтиламина) с получением продукта (E)-2-(2-((5-трет-бутил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона (1,3 г, выход: 56,7%) и изомера положения (E)-2-(2-((5-(трет-бутил)-4-оксо-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-диона (350 мг, выход: 15,3%).
Стадия 6: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-трет-бутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
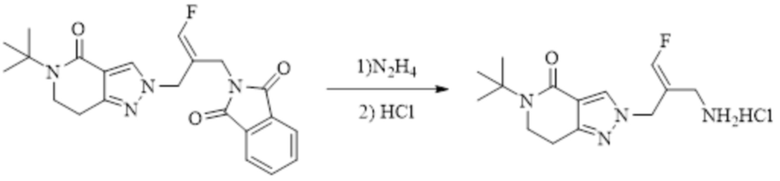
(E)-2-(2-((5-трет-Бутил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (1000 мг, 2,438 ммоль, 1,0 экв.) растворяли в EtOH (30 мл). Затем к раствору добавляли гидразингидрат (502,4 мг, 8,531 ммоль, 3,5 экв., 85%) и кипятили с обратным холодильником в течение 2 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор фильтровали в вакууме и фильтрат концентрировали при пониженном давлении. К полученному раствору добавляли EA (30 мл), кипятили с обратным холодильником и фильтровали горячим. Фильтрат концентрировали при пониженном давлении, разбавляли небольшим количеством этанола, добавляли раствор хлорида водорода в этаноле, и перемешивали при комнатной температуре в течение 30 мин. В полученную смесь добавляли ацетонитрил и концентрировали при пониженном давлении с получением продукта гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-трет-бутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (747,9 мг, выход: 98%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,51 (шир. с, 3H), 8,22 (с, 1H), 7,16-7,36 (д, J=82 Гц, 1H), 4,93 (с, 2H), 3,52-3,56 (т, 2H), 3,31-3,32 (д,2H), 2,73-2,76 (т, 2H), 2,08 (с, 1H), 1,42 (с, 9H).
Молекулярная формула: C14H21FN4O, молекулярная масса: 280,35, ЖХ-МС (полож., m/z)=281,23 [M+H]+.
Пример 5: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклобутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A9)
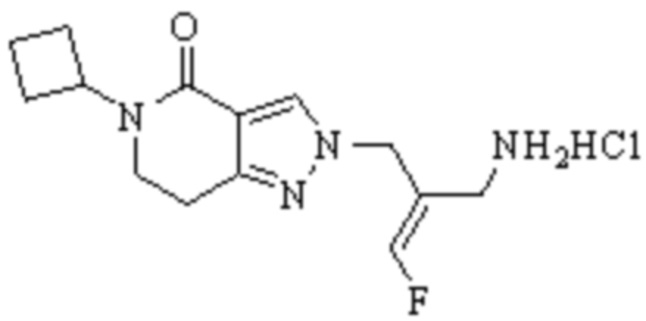
Стадия 1: Синтез этил 3-(циклобутиламино)пропионата
Продукт циклобутиламин (4,26 г, 59,93 ммоль, 1,2 экв.) растворяли в этаноле (50 мл). Затем на ледяной бане в раствор медленно по каплям добавляли этилакрилат (5,0 г, 49,94 ммоль, 1,0 экв.) и подвергали взаимодействию в течение 12 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении при температуре 80°C с получением продукта (8,55 г, выход: 100%).
Стадия 2: Синтез этил 3-(циклобутил(3-этокси-3-оксопропил)амино)-3-оксопропионата
Промежуточный этил 3-(циклобутиламино)пропионат (8,55 г, 49,94 ммоль, 1,0 экв.) растворяли в дихлорметане (100 мл). Затем раствор охлаждали до температуры 0°C на водяной бане со льдом, добавляли по каплям моноэтил малонат (6,6 г, 49,94 ммоль, 1,0 экв.), и после добавления перемешивали в течение 10 мин. Затем в смесь последовательно добавляли триэтиламин (12,6 г, 124,85 ммоль, 2,5 экв.), 4-диметиламинопиридин (609,6 мг, 4,99 ммоль, 0,1 экв.) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (11,5 г, 59,93 ммоль, 1,2 экв.) и подвергали взаимодействию в течение 12 ч. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли 2,5 моль/л соляной кислоты (100 мл) и перемешивали в течение 10 мин, затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (50 мл). Органические фазы объединяли, промывали последовательно насыщенным водным раствором карбоната натрия (50 мл) и водой(50 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (10,1 г, выход: 71,1%).
Стадия 3: Синтез этил 1-циклобутил-2,4-диоксопиперидин-3-карбоксилата
Промежуточный этил 3-(циклобутил(3-этокси-3-оксопропил)амино)-3-оксопропионат (10,1 г, 35,40 ммоль, 1,0 экв.) растворяли в этаноле (50 мл). К раствору добавляли этоксид натрия (6,0 г, 88,49 ммоль, 2,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (8,47 г, выход: 100%).
Стадия 4: Синтез 1-циклобутилпиперидин-2,4-диона
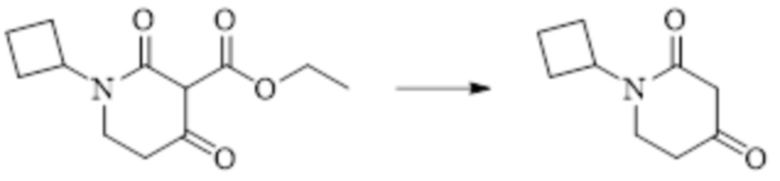
Промежуточный этил 1-циклобутил-2,4-диоксопиперидин-3-карбоксилат (8,47 г, 35,40 ммоль, 1,0 экв.) растворяли в воде (20 мл) и концентрированной соляной кислоте (30 мл) и подвергали взаимодействию при температуре 120°C в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры, и экстрагировали дихлорметаном (50 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали с получением продукта (3 г, выход: 50,8%).
Стадия 5: Синтез 1-циклобутил-3-((диметиламино)метилен)пиперидин-2,4-диона
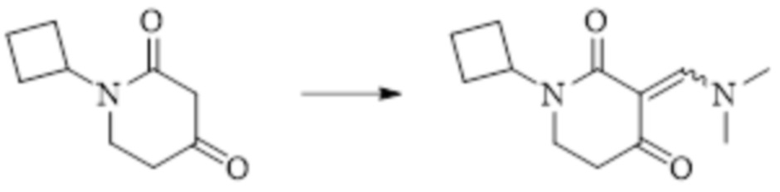
Промежуточный 1-циклобутилпиперидин-2,4-дион (3 г, 17,94 ммоль, 1,0 экв.) растворяли в дихлорметане (2 мл). К раствору добавляли N, N-диметилформамид диметилацеталь (2,35 г, 19,74 ммоль, 1,1 экв.) и подвергали взаимодействию при комнатной температуре в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (3,98 г, выход: 100%).
Стадия 6: Синтез 5-циклобутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

Промежуточный (E)-1-циклобутил-3-((диметиламино)метилен)пиперидин-2,4-дион (3,98 г, 17,94 ммоль, 1,0 экв.) и гидразингидрат (1,16 г, 19,73 ммоль, 1,1 экв.) растворяли в метаноле (4 мл). Затем раствор нагревали до температуры кипения с обратным холодильником в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (ДХМ:MeOH=100:1-50:1) с получением продукта (2,1 г, выход: 61,2%).
Стадия 7: Синтез (E)-2-(2-((5-циклобутил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
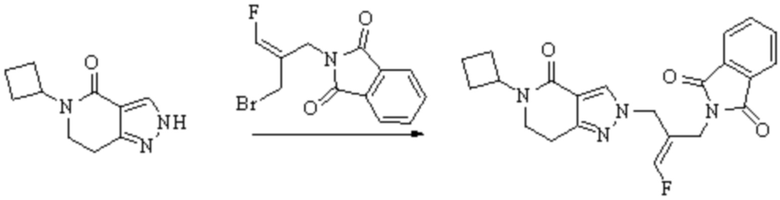
Промежуточный 5-циклобутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (1,0 г, 5,23 ммоль, 1,0 экв.) растворяли в ДМФ (5 мл). Раствор охлаждали на водяной бане со льдом, добавляли NaH (массовая доля: 60%, 230 мг, 5,75 ммоль, 1,1 экв.), перемешивали при комнатной температуре в течение 30 мин и затем добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (2,0 г, 6,29 ммоль, 1,2 экв.) в ДМФ (5 мл). После добавления реакция протекала в течение 1 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органическую фазу промывали водой (50 мл × 2), сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (PE:EA=1:2) с получением продукта (572 мг, выход: 27,2%).
Стадия 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклобутил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
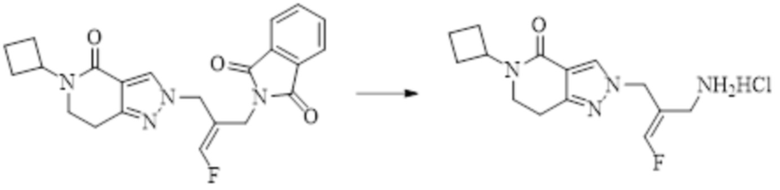
Промежуточный (E)-2-(2-((5-циклобутил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (572 мг, 1,40 ммоль, 1,0 экв.) растворяли в EtOH (10 мл). Затем к раствору добавляли гидразингидрат (245 мг, 4,90 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 3 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме, и фильтрат концентрировали. Сырой продукт суспендировали в дихлорметане (10 мл) и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением маслянистой жидкости (296 мг). Полученный продукт растворяли в дихлорметане (2 мл), добавляли по каплям раствор хлорида водорода в этаноле (129 мг), перемешивали в течение 10 мин, и концентрировали при пониженном давлении с получением продукта (256 мг, выход: 58%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,43 (с, 3H), 8,26 (с, 1H), 7,36 (с, 0,5H), 7,15 (с, 0,5H), 4,93-4,92 (м, 3H), 3,57-3,54 (м, 2H), 3,34-3,33 (м, 2H), 2,83-2,80 (м, 2H), 2,20-2,13 (м, 2H), 2,06-1,99 (м, 2H), 1,67-1,66 (м, 2H).
Молекулярная формула: C14H19FN4O, молекулярная масса: 278,33, ЖХ-МС (полож., m/z)=279,19 [M+H]+.
Пример 6: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопентил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A10)
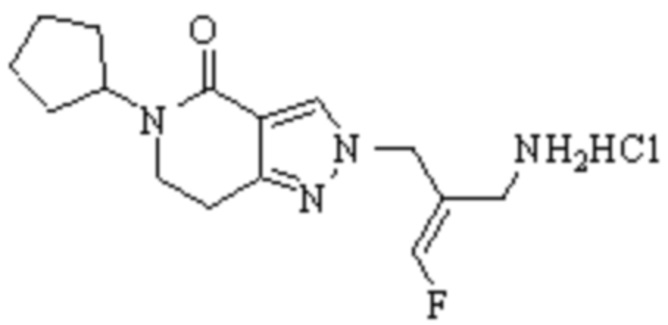
Стадия 1: Синтез промежуточного этил 3-(циклопентиламино)пропионата
Продукт циклопентиламин (5,1 г, 60 ммоль, 1,0 экв.) растворяли в этаноле (10 мл). Затем на ледяной бане в раствор медленно по каплям добавляли этилакрилат (5,0 г, 50 ммоль, 1,0 экв.) и подвергали взаимодействию в течение 12 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (9,2 г, выход: 99%).
Стадия 2: Синтез промежуточного этил 3-(циклопентил(3-этокси-3-оксопропил)амино)-3-оксопропионата
Промежуточный этил 3-(циклопентиламино)пропионат (8,2 г, 44,26 ммоль, 1,0 экв.), моноэтил малонат (5,85 г, 44,26 ммоль, 1,0 экв.), 4-диметиламинопиридин (1,08 г, 8,85 ммоль, 0,2 экв.) и триэтиламин (10,3 г, 101,8 ммоль, 2,3 экв.) растворяли в дихлорметане (100 мл). После перемешивания в течение 5 мин к раствору партиями добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (8,49 г, 53,11 ммоль, 1,2 экв.) подвергали взаимодействию в течение 12 ч. После завершения реакции, как определялось данными ТСХ, в реакционный сосуд добавляли воду и концентрированную соляную кислоту (3:1, 100 мл) и смесь перемешивали в течение 10 мин, затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (100 мл). Органические фазы объединяли, промывали последовательно насыщенным водным раствором карбоната натрия (100 мл) и водой (100 мл × 2), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (11,5 г, выход: 87%).
Стадия 3: Синтез промежуточного этил 1-циклопентил-2,4-диоксопиперидин-3-карбоксилата
Промежуточный этил 3-(циклопентил(3-этокси-3-оксопропил)амино)-3-оксопропионат (11,5 г, 38,4 ммоль, 1,0 экв.) и этоксид натрия (5,23 г, 76,8 ммоль, 2,0 экв.) растворяли в этаноле (100 мл) и подвергали взаимодействию при температуре 80°C в течение 2 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении с получением продукта (9,73 г, выход: 100%).
Стадия 4: Синтез промежуточного 1-циклопентилпиперидин-2,4-диона
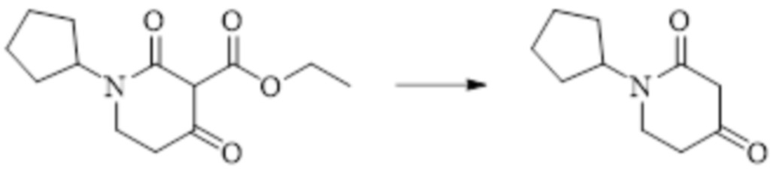
Промежуточный этил 1-циклопентил-2,4-диоксопиперидин-3-карбоксилат (9,72 г, 38,4 ммоль, 1,0 экв.) растворяли в воде (50 мл) и концентрированной соляной кислоте (20 мл) и подвергали взаимодействию при температуре 120°C в течение 1,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, раствор охлаждали до комнатной температуры и добавляли твердый хлорид натрия до насыщения. Экстрагировали дихлорметаном (100 мл × 3). Органическую фазу сушили и концентрировали с получением продукта (6,95 г, выход: 100%).
Стадия 5: Синтез промежуточного 1-циклопентил-3-((диметиламино)метилен)пиперидин-2,4-диона
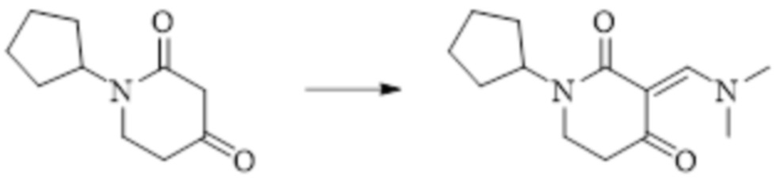
Промежуточный 1-циклопентилпиперидин-2,4-дион (6,95 г, 38,4 ммоль, 1,0 экв.) растворяли в N, N-диметилформамид диметилацетале (5,04 г, 42,24 ммоль, 1,1 экв.) и подвергали взаимодействию в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении с получением продукта (9,07 г, выход: 100%).
Стадия 6: Синтез промежуточного 5-циклопентил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

Промежуточный 1-циклопентил-3-((диметиламино)метилен)пиперидин-2,4-дион (9,07 г, 38,4 ммоль, 1,0 экв.) и гидразингидрат (2,114 г, 42,24 ммоль, 1,1 экв.) растворяли в метаноле (50 мл) и подвергали взаимодействию при температуре 60°C в течение 40 мин. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Сырой продукт сначала очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=50:1), и затем суспендировали в метил трет-бутиловом эфире (50 мл) и фильтровали в вакууме с получением продукта (3,1 г, выход: 39%).
Стадия 7: Синтез промежуточного (E)-2-(2-((5-циклопентил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
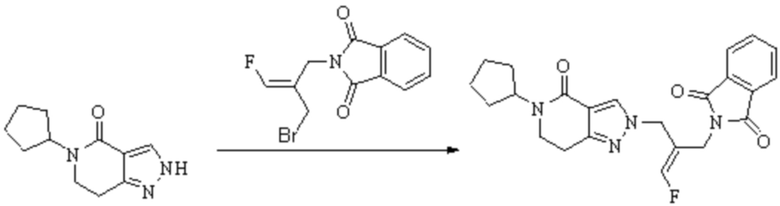
Промежуточный 5-циклопентил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (1,0 г, 4,87 ммоль, 1,0 экв.) растворяли в ДМФ (5 мл). Затем к раствору добавляли гидрид натрия (214 мг, 5,36 ммоль, 1,12 экв., 60%), перемешивали в течение 30 мин и затем по каплям добавляли раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-диона (1,74 г, 5,84 ммоль, 1,2 экв.) в ДМФ (5 мл) и подвергали взаимодействию в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционную смесь добавляли воду (10 мл) и экстрагировали этилацетатом (50 мл × 2), затем жидкости разделяли. Органическую фазу промывали водой (50 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1) с получением продукта (700 мг, выход: 34%).
Стадия 8: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопентил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
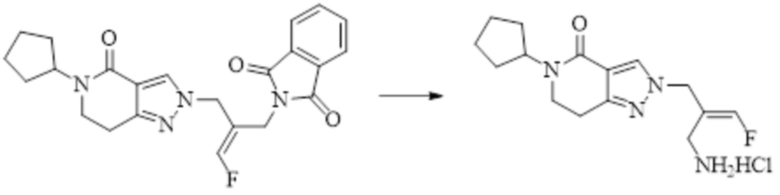
Промежуточный (E)-2-(2-((5-циклопентил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (700 мг, 1,65 ммоль, 1,0 экв.) растворяли в EtOH (10 мл). Затем к раствору добавляли гидразингидрат (290 мг, 5,77 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 30 мин. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор фильтровали в вакууме и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1). Полученное масло растворяли в метаноле (2 мл). К полученному раствору добавляли по каплям раствор хлорида водорода в этаноле (0,25 мл), перемешивали в течение 30 мин, и концентрировали при пониженном давлении с получением продукта (230 мг, выход: 42%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,37 (с, 3H), 8,24 (с, 1H), 7,36 (с, 0,5H), 7,16 (с, 0,5H), 4,91 (с, 3H), 3,42-3,45 (м, 2H), 3,34-3,35 (д, 2H), 2,78-2,81 (м, 2H), 1,69 (с, 4H), 1,54 (с, 4H).
Молекулярная формула: C15H21FN4O, молекулярная масса: 292,36, ЖХ-МС (полож., m/z)=293,20 [M+H]+.
Пример 7: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(2H)-она (соединение A13)

Стадия 1: Синтез промежуточного этил трет-бутил глицината
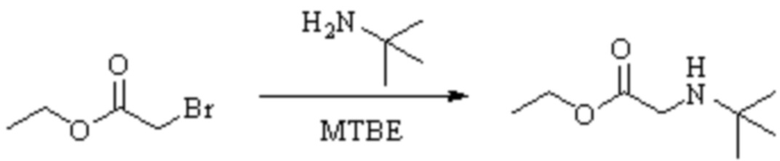
Этил бромацетат (10,0 г, 59,88 ммоль, 1,0 экв.) добавляли по каплям к раствору трет-бутил амина (21,9 г, 299,40 ммоль, 5,0 экв.) в метил трет-бутиловом эфире (200 мл). После добавления смесь перемешивали при комнатной температуре в течение 40 ч. Реакционный раствор фильтровали и фильтрат концентрировали. В концентрированный раствор добавляли метил трет-бутиловый эфир (100 мл), промывали последовательно водой (30 мл) и насыщенным солевым раствором (50 мл) и фильтровали и фильтрат концентрировали с получением продукта (8,5 г, выход: 89,2%).
Стадия 2: Синтез промежуточного этил 3-(трет-бутил(2-этокси-2-оксоэтил)амино)-3-оксопропионата

Этил трет-бутил глицинат (6,0 г, 37,68 ммоль, 1,0 экв.), моноэтил малонат (5,48 г, 41,15 ммоль, 1,1 экв.), триэтиламин (8,77 г, 86,67 ммоль, 2,3 экв.) и DMAP (921 мг, 7,54 ммоль, 0,2 экв.) последовательно добавляли к ДХМ (150 мл), затем к ДХМ партиями добавляли EDCI (8,62 г, 45,22 ммоль, 1,2 экв.). Смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, реакционный раствор выливали в 1 моль/л соляную кислоту (300 мл), затем жидкости разделяли. Органическую фазу промывали последовательно водой (50 мл) и насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением продукта (10,0 г, выход: 97,1%).
Стадия 3: Синтез промежуточного этил 1-(трет-бутил)-2,4-диоксопирролидин-3-карбоксилата
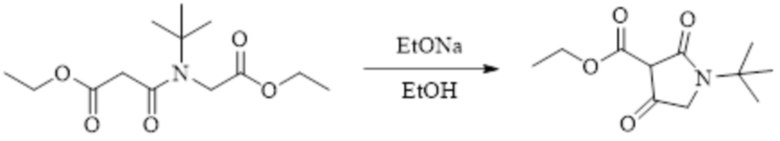
Этоксид натрия (4,98 г, 73,17 ммоль, 2,0 экв.) добавляли в этанол (100 мл), затем при перемешивании при комнатной температуре в раствор этоксида натрия в этаноле добавляли этил 3-(трет-бутил(2-этокси-2-оксоэтил)амино)-3-оксопропионат (10,0 г, 36,59 ммоль, 1,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 4: Синтез промежуточного 1-(трет-бутил)пирролидин-2,4-диона
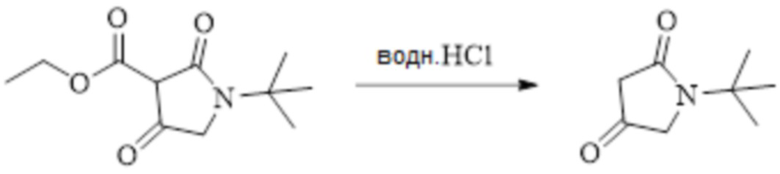
Этил 1-(трет-бутил)-2,4-диоксопирролидин-3-карбоксилат (8,31 г, 36,57 ммоль, 1,0 экв.) партиями добавляли к 1 моль/л соляной кислоте (150 мл). После добавления смесь перемешивали при температуре 85°C в течение 4 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и добавляли ДХМ (200 мл), затем жидкости разделяли. Органическую фазу промывали водой (50 мл) и насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением продукта (4,2 г, выход за две стадии: 74,0%).
Стадия 5: Синтез промежуточного 1-(трет-бутил)-3-((диметиламино)метилен)пирролидин-2,4-диона
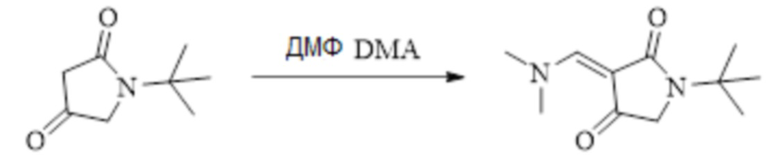
1-(трет-Бутил)пирролидин-2,4-дион (2,20 г, 14,18 ммоль, 1,0 экв.) добавляли к 1,1-диметокси-N, N-диметилметиламину (1,69 г, 14,18 ммоль, 1,0 экв.). Затем смесь перемешивали при комнатной температуре в течение 0,5 ч. После завершения реакции, как определялось данными ТСХ, метанол упаривали при пониженном давлении с получением продукта (сырой продукт, рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 6: Синтез промежуточного 1-бензил-5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(1H)-она
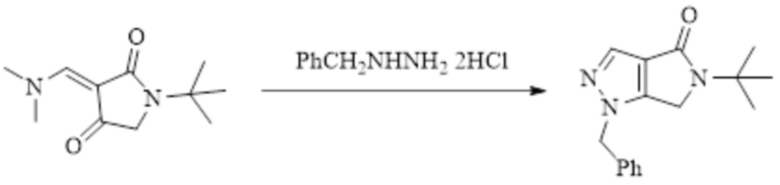
Бензилгидразин дигидрохлорид (2,76 г, 14,17 ммоль, 1,0 экв.) и 1-(трет-бутил)-3-((диметиламино)метилен)пирролидин-2,4-дион (2,98 г, 14,17 ммоль, 1,0 экв.) последовательно добавляли к этанолу (30 мл) и смесь перемешивали при температуре 20°C в течение 1 ч. Затем смесь нагревали до температуры 90°C и подвергали взаимодействию в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры, выливали в насыщенный водный раствор бикарбоната натрия (50 мл) и экстрагировали ДХМ (25 мл × 3). Органическую фазу промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 1:1) с получением продукта (3,0 г, выход за две стадии: 78,6%).
Стадия 7: Синтез промежуточного 5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(2H)-она
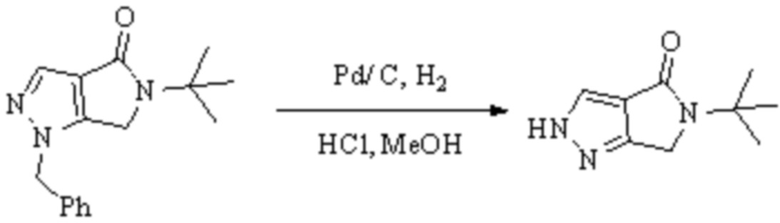
Промежуточный 1-бензил-5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(1H)-он (3,0 г, 11,14 ммоль, 1,0 экв.) растворяли в метаноле (45 мл). Затем к раствору добавляли концентрированную соляную кислоту (1,0 мл) и влажный палладий на угле (1,0 г) и подвергали взаимодействию в атмосфере водорода в течение 40 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали через целит и фильтрат концентрировали с получением продукта (1,8 г, выход: 90,2%).
Стадия 8: Синтез промежуточного (E)-2-(2-((5-(трет-бутил)-4-оксо-5,6-дигидропирроло[3,4-c]пиразол-2(4H)-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
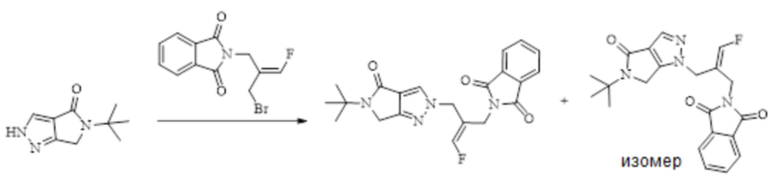
5-(трет-Бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(2H)-он (359 мг, 2,01 ммоль, 1,2 экв.), карбонат цезия (1,20 г, 3,69 ммоль, 2,2 экв.) и (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (500 мг, 1,68 ммоль, 1,0 экв.) добавляли к DMAc (10 мл). Затем смесь перемешивали при температуре 55°C в течение 16 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали и слой на фильтре промывали этилацетатом (30 мл). Органическую фазу промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (этилацетат) с получением продукта (320 мг, 0,81 ммоль, выход: 48,0%) и изомера положения (E)-2-(2-((5-(трет-бутил)-4-оксо-5,6-дигидропирроло[3,4-c]пиразол-1(4H)-ил)метил)-3-фтораллил)изоиндол-1,3-диона (100 мг, выход: 15,0%).
Стадия 9: Синтез соединения (E)-2-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(2H)-она
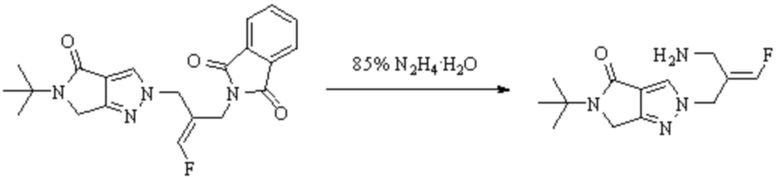
Промежуточный (E)-2-(2-((5-(трет-бутил)-4-оксо-5,6-дигидропирроло[3,4-c]пиразол-2(4H)-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (320 мг, 0,81 ммоль, 1,0 экв.) и 85%-ный гидразингидрат (190 мг, 3,23 ммоль, 4,0 экв.) последовательно добавляли в этанол (8,0 мл). Затем смесь перемешивали при температуре 40°C в течение 15 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали, и слой на фильтре промывали небольшим количеством этанола. Фильтрат концентрировали, добавляли абсолютный этанол (4 мл) и фильтровали, и полученный фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (дихлорметан : метанол, 5:1, об./об.) с получением продукта (85 мг, выход: 39,54%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,05 (с, 1H), 6,87-7,08 (д, 1H), 4,83 (д, 2H), 4,43 (с, 2H), 3,16 (с, 2H), 3,10 (д, 2H), 1,43 (с, 9H).
Молекулярная формула: C13H19FN4O, молекулярная масса: 266,32, ЖХ-МС (m/z)=267,24 [M+H]+.
Пример 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(4-фторфенил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридина (соединение A14)
Стадия 1: Синтез промежуточного (E)-(3-фтор-2-((5-(4-фторфенил)-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамата
Промежуточный (E)-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (200 мг, 0,62 моль, 1,0 экв.) растворяли в N,N-диметилацетамиде (2 мл). Затем к раствору добавляли 1-фтор-4-йодбензол (412,9 мг, 1,86 ммоль, 3,0 экв.), безводный карбонат калия (171,1 мг, 1,24 ммоль, 2,0 экв.) и йодид меди (11,8 мг, 0,06 ммоль, 0,1 экв.) и нагревали до температуры 130°C в атмосфере азота и подвергали взаимодействию в течение 16 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры, добавляли воду (20 мл) и экстрагировали этилацетатом (20 мл × 3), затем жидкости разделяли. Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (MeOH:ДХМ=1:20) с получением продукта (97 мг, выход: 40,5%).
Стадия 2: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(4-фторфенил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридина
Промежуточный (E)-(3-фтор-2-((5-(4-фторфенил)-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (97 мг, 0,23 ммоль, 1,0 экв.) растворяли в 30%-ном растворе хлорида водорода в этаноле (2 мл). Затем раствор перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали обращенно-фазовой колоночной хроматографией (элюировали 0,5%-ным водным раствором трифторуксусной кислоты) и лиофилизовали с получением продукта (25 мг, выход: 30,6%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,37-8,35 (с, 4H), 7,39-7,34 (м, 2,5H), 7,25-7,19 (м, 2,5H), 4,94 (м, 2H), 3,96-3,93 (м, 2H), 3,39-3,38 (м, 2H), 3,00-2,97 (м, 2H).
Молекулярная формула: C16H16F2N4O, молекулярная масса: 318,33, ЖХ-МС (полож., m/z)=319,13 [M+H]+.
Пример 9: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-2-(3-фторфенил)-1,2,6,7-тетрагидро-3H-пиразоло[4,3-c]пиридин-3,4(5H)-диона (соединение A19)
Стадия 1: Синтез 5-циклопропил-2-(3-фторфенил)-1,2,6,7-тетрагидро-3H-пиразоло[4,3-c]пиридин-3,4(5H)-диона
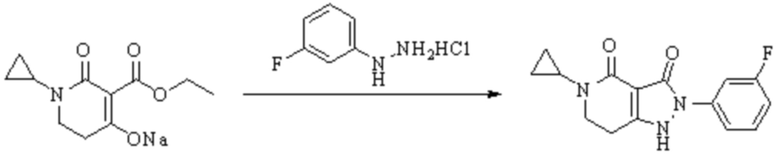
Алкоксид 1-циклопропил-5-(этоксикарбонил)-6-оксо-1,2,3,6-тетрагидропиридин-4-натрия (1,0 г, 4,05 ммоль) и м-фторфенилгидразин гидрохлорид (656,3 мг, 4,05 ммоль) добавляли в этанол (10 мл). Затем смесь нагревали при температуре 80°C и подвергали взаимодействию в течение ночи. После завершения реакции, как определялось данными ТСХ, в реакционный раствор добавляли водный раствор лимонной кислоты до установления pH равным 6-7. Водную фазу экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=40:1-20:1) с получением продукта (440,0 мг, выход: 37,8%).
Стадия 2: Синтез (E)-5-циклопропил-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-2-(3-фторфенил)-1,2,6,7-тетрагидро-3H-пиразоло[4,3-c]пиридин-3,4(5H)-диона

К DMA (8 мл) добавляли 5-циклопропил-2-(3-фторфенил)-1,2,6,7-тетрагидро-3H-пиразоло[4,3-c]пиридин-3,4(5H)-дион (440,0 мг, 1,54 ммоль), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (551,0 мг, 1,85 ммоль) и карбонат калия (425,6 мг, 3,08 ммоль). Затем смесь перемешивали при комнатной температуре в течение 12 ч и фильтровали в вакууме, и фильтрат концентрировали при пониженном давлении, добавляли этилацетат (50 мл), промывали водой (10 мл × 4) и насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=200:1-40:1) с получением продукта (450,0 мг, выход: 58,0%).
Стадия 3: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-2-(3-фторфенил)-1,2,6,7-тетрагидро-3H-пиразоло[4,3-c]пиридин-3,4(5H)-диона
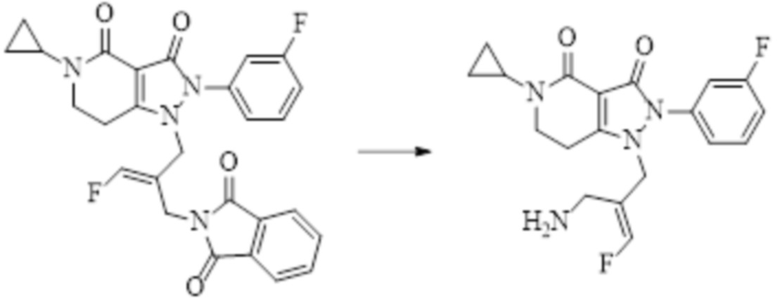
(E)-5-Циклопропил-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-2-(3-фторфенил)-1,2,6,7-тетрагидро-3H-пиразоло[4,3-c]пиридин-3,4(5H)-дион (450,0 мг, 0,893 ммоль) добавляли к этанолу (5 мл) и гидразингидрату (49% мас., 446,8 мг, 8,93 ммоль). Затем смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (100,0 мг, выход: 29,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 7,95 (с, 2H), 7,57-7,47 (м, 3H), 7,38 (с, 0,5H), 7,29-7,24 (м, 1H), 7,18 (с, 0,5H), 5,19-5,18 (д, 2H), 3,61-3,58 (т, 2H), 3,57-3,51 (д, 2H), 2,85-2,81 (т, 2H), 2,73-2,69 (м, 1H), 0,79-0,76 (м, 2H), 0,69-0,66 (м, 2H).
Молекулярная формула: C19H20F2N4O2, молекулярная масса: 375,24, ЖХ-МС (полож., m/z)=375,22 [M+H]+.
Пример 10: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-1,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A21)
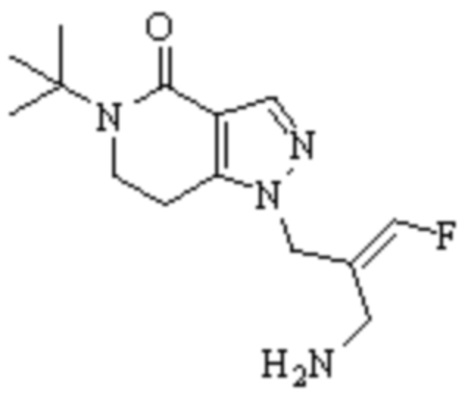
Стадия 1: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-1,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

(E)-2-(2-((5-(трет-Бутил)-4-оксо-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (350 мг, 0,853 ммоль, 1,0 экв.), полученный в примере 4, растворяли в EtOH (15 мл). Затем добавляли гидразингидрат (175,8 мг, 2,984 ммоль, 3,5 экв.) и реакционную смесь кипятили с обратным холодильником в течение 2 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор фильтровали в вакууме и фильтрат концентрировали при пониженном давлении. К полученному раствору добавляли EA (20 мл), кипятили с обратным холодильником и фильтровали горячим. После охлаждения фильтрат фильтровали в вакууме, и полученный фильтрат концентрировали при пониженном давлении с получением продукта (E)-1-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-1,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (192,2 мг, выход: 80,0%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 7,63 (с, 1H), 6,78-6,99 (д, J=84 Гц, 1H), 4,70 (д, 2H), 3,56-3,59 (т, 2H), 3,04-3,05 (д, 2H), 2,90-2,94 (т, 2H), 1,43 (с, 9H).
Молекулярная формула: C14H21FN4O, молекулярная масса: 280,35, ЖХ-МС (полож., m/z)=281,22 [M+H]+.
Пример 11: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A22)
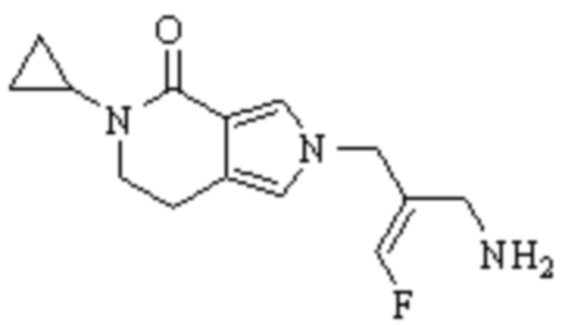
Стадия 1: Синтез промежуточного 1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-диона
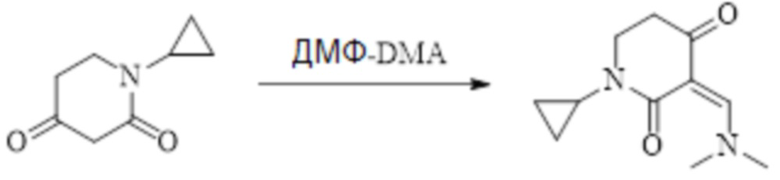
1-Циклопропилпиперидин-2,4-дион (2,50 г, 16,33 ммоль, 1,0 экв.) добавляли к 1,1-диметокси-N, N-диметилметиламину (2,14 г, 17,95 ммоль, 1,1 экв.). Затем смесь перемешивали при комнатной температуре в течение 0,5 ч. После завершения реакции, как определялось данными ТСХ, избыток 1,1-диметокси-N,N-диметилметиламина удаляли упариванием при пониженном давлении с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 2: Синтез промежуточного ((1-циклопропил-2,4-диоксопиперидин-3-илиден)метил)глицина
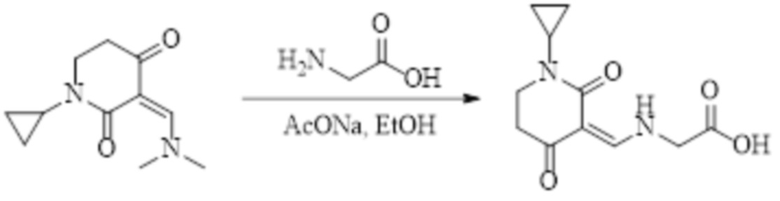
Сырой продукт 1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-дион (3,40 г, 16,33 ммоль, 1,0 экв.), полученный на предыдущей стадии, глицин (1,23 г, 16,33 ммоль, 1,0 экв.) и ацетат натрия (1,61 г, 19,59 ммоль, 1,2 экв.) добавляли к этанолу (50 мл). Затем смесь перемешивали при температуре 50°C до завершения реакции. Реакционный раствор концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 3: Синтез промежуточного 2-ацетил-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
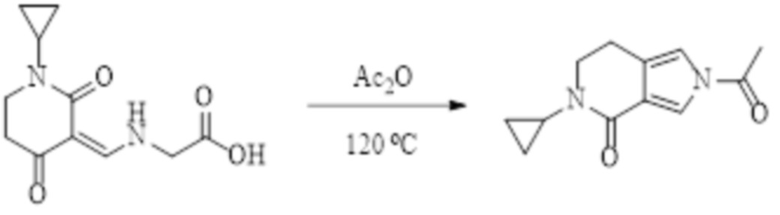
((1-Циклопропил-2,4-диоксопиперидин-3-илиден)метил)глицин (3,89 г, 16,33 ммоль, 1,0 экв.) добавляли к уксусному ангидриду (40 мл). Затем смесь перемешивали при температуре 120°C в течение 5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали и выливали в насыщенный водный раствор бикарбоната натрия (100 мл). Экстрагировали этилацетатом (30 мл × 2). Органическую фазу промывали насыщенным солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 4: Синтез промежуточного 5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
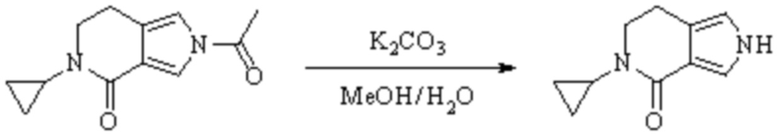
2-Ацетил-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (3,55 г, 16,33 ммоль, 1,0 экв.) и карбонат калия (4,51 г, 32,66 ммоль, 2,0 экв.) последовательно добавляли в смешанный растворитель из метанола (30 мл) и воды (30 мл). Затем смесь перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали, добавляли этилацетат (100 мл), промывали последовательно водой (30 мл) и насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (петролейный эфир: этилацетат=1:1-0:1, об./об.) с получением продукта (950 мг, выход за четыре стадии: 33,1%).
Стадия 5: Синтез промежуточного (Z)-2-(2-((5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
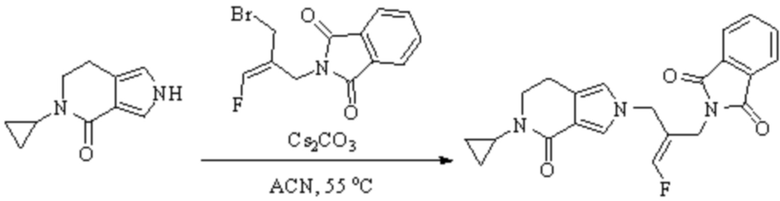
5-Циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (900 мг, 5,11 ммоль, 1,0 экв.), карбонат цезия (2,50 г, 7,66 ммоль, 1,5 экв.) и (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (1,67 г, 5,62 ммоль, 1,1 экв.) добавляли к ацетонитрилу (30 мл). Затем смесь перемешивали при температуре 55°C в течение 16 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали, и слой на фильтре один раз промывали этилацетатом (15 мл) и концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 1:1-0:1, об./об.) с получением продукта (1,05 г, 2,67 ммоль, выход: 52,3%).
Стадия 6: Синтез соединения (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
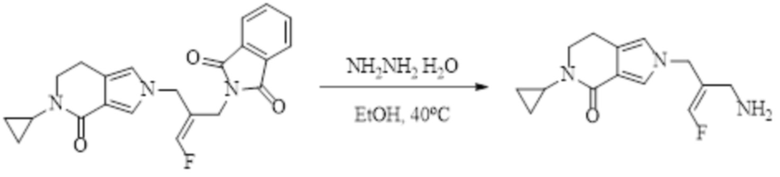
(Z)-2-(2-((5-Циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (600 мг, 1,53 ммоль, 1,0 экв.) и 85%-ный гидразингидрат (898 мг, 15,25 ммоль, 10 экв.) последовательно добавляли к этанол (15 мл). Затем смесь перемешивали при температуре 40°C в течение 5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали, и слой на фильтре промывали небольшим количеством этанола. Фильтрат концентрировали, добавляли абсолютный этанол (10 мл) и фильтровали, и полученный фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (дихлорметан : метанол, 5:1, об./об.) с получением продукта (105 мг, выход: 26,2%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,09 (шир. с, 2H), 7,30 (с, 1H), 7,31-7,10 (д, 1H), 6,66 (с, 1H), 4,70 (с, 2H), 3,37-3,41 (м, 2H), 3,20 (с, 2H), 2,60-2,63 (м, 3H), 0,69-0,74 (м, 2H), 0,55-0,56 (м, 2H).
Молекулярная формула: C14H18FN3O, молекулярная масса: 263,32, ЖХ-МС (m/z)=264,20 [M+H]+.
Пример 12: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A23)
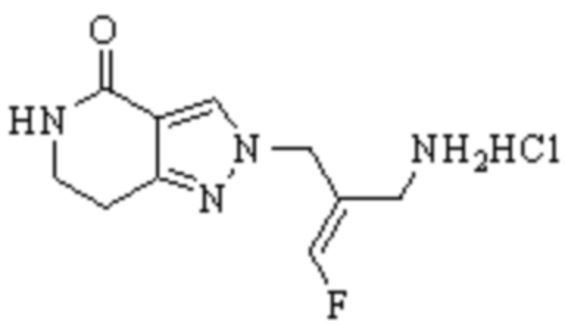
Стадия 1: Синтез промежуточного (E)-2-(2-((5-(трет-бутил)-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
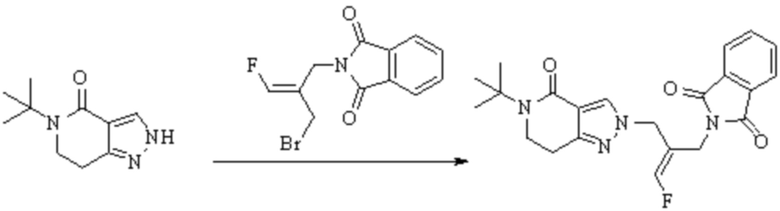
Промежуточный 5-(трет-бутил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (5,0 г, 25,87 ммоль, 1,0 экв.) растворяли в ДМФ (20 мл). В атмосфере азота раствор охлаждали до температуры -10°C и добавляли гидрид натрия (1,14 г, 28,46 ммоль, 1,1 экв., 60%), и подвергали взаимодействию в течение 30 мин. Затем в реакционный раствор медленно добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-диона (9,3 г, 31,04 ммоль, 1,2 экв.) в ДМФ (10 мл) и подвергали взаимодействию в течение 2 ч. После завершения реакции, как определялось с помощью ЖХ-МС, к реакционному раствору добавляли насыщенный водный хлорида аммония раствор (100 мл) и экстрагировали этилацетатом (100 мл × 3). Органическую фазу промывали водой (100 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1) с получением продукта (5,9 г, выход: 59%).
Стадия 2: Синтез промежуточного (E)-2-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-диона
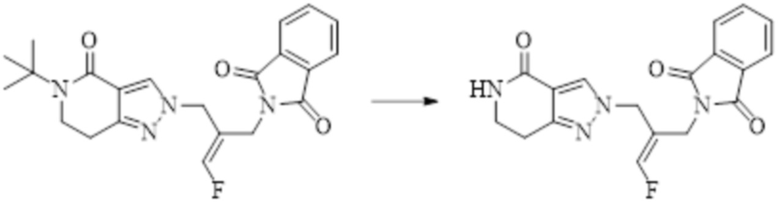
Промежуточный (E)-2-(2-((5-(трет-бутил)-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (6,3 г, 15,34 ммоль, 1,0 экв.) растворяли в концентрированной соляной кислоте (30 мл) и этаноле (30 мл) и подвергали взаимодействию при температуре 60°C в течение 12 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении. Сырой продукт суспендировали в этилацетате (50 мл) и фильтровали в вакууме с получением продукта (5,4 г, выход: 99%).
Стадия 3: Синтез промежуточного (E)-2-(2-(аминометил)-3-фтораллил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

Промежуточный (E)-2-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-дион (5,4 г, 15,24 ммоль, 1,0 экв.) и гидразингидрат (3,05 г, 60,96 ммоль, 4,0 экв.) растворяли в этаноле (100 мл) и подвергали взаимодействию при температуре 80°C в течение 1,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и фильтровали. Маточную жидкость концентрировали при пониженном давлении с получением продукта (3,4 г, выход: 100%).
Стадия 4: Синтез промежуточного (E)-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамата
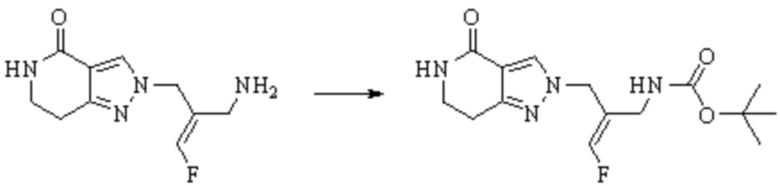
Промежуточный (E)-2-(2-(аминометил)-3-фтораллил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (3,4 г, 15,24 ммоль, 1,0 экв.), ди-трет-бутил дикарбонат (6,65 г, 30,48 ммоль, 2,0 экв.), 4-диметиламинопиридин (171 мг, 1,52 ммоль, 0,1 экв.) и триэтиламин (2,31 г, 22,86 ммоль, 1,5 экв.) растворяли в дихлорметане (50 мл) и подвергали взаимодействию в течение 2,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, к реакционному раствору добавляли насыщенный водный раствор карбоната натрия (50 мл) и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу сушили и концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:60) с получением продукта (2,57 г, выход: 51%).
Стадия 5: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-2, 5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
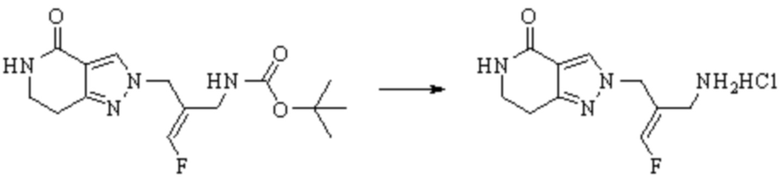
Промежуточный (E)-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (200 мг, 0,61 ммоль, 1,0 экв.) растворяли в этаноле (2 мл). Затем к раствору добавляли раствор хлорида водорода в этаноле (2 мл) и подвергали взаимодействию в течение 2,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении. Сырой продукт суспендировали в этилацетате (5 мл) и фильтровали в вакууме, и слой на фильтре сушили с получением продукта (150 мг, выход: 93%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 8,55 (с, 3H), 8,30 (с, 1H), 7,44 (с, 1H), 7,37 (с, 0,5H), 7,17 (с, 0,5H), 4,97 (с, 2H), 3,35-3,38 (м, 2H), 3,31-3,33 (д, 2H), 2,72-2,76 (м, 2H).
Молекулярная формула: C10H13FN4O, молекулярная масса: 224,24, ЖХ-МС (полож., m/z)=225,16 [M+H]+.
Пример 13: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-метил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A24)
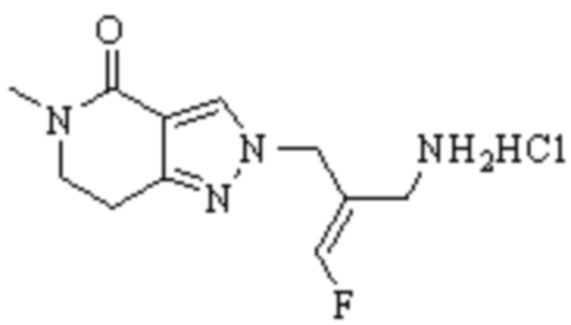
Стадия 1: Синтез промежуточного (E)-(3-фтор-2-((5-метил-4-оксо-4,5,6, 7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамата
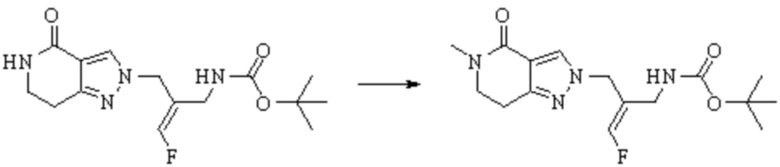
Промежуточный (E)-(3-фтор-2-((4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (500 мг, 1,54 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (5 мл). Затем к раствору добавляли гидрид натрия (80 мг, 2,0 ммоль, 1,3 экв., 60%) и подвергали взаимодействию при комнатной температуре в течение 30 мин. К реакционному раствору добавляли йодметан (263 мг, 1,85 ммоль, 1,2 экв.) и подвергали взаимодействию в течение 2 ч. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционный сосуд добавляли воду (5 мл) и этилацетат (10 мл × 3) экстрагировали. Органическую фазу сушили и концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:80) с получением продукта (100 мг, выход: 19%).
Стадия 2: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-метил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
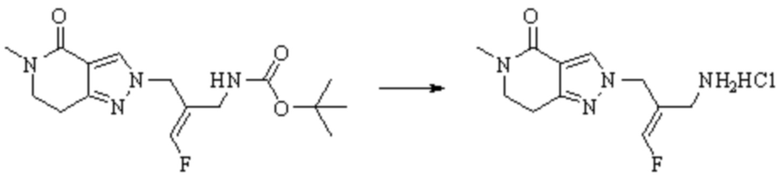
Промежуточный (E)-(3-фтор-2-((5-метил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)трет-бутил карбамат (100 мг, 0,29 ммоль, 1,0 экв.) растворяли в этаноле (1 мл). Затем к раствору добавляли раствор хлорида водорода в этаноле (1 мл) и подвергали взаимодействию в течение 12 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении с получением продукта (55 мг, выход: 69%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,41 (с, 3H), 8,25 (с, 1H), 7,37 (с, 0,5H), 7,16 (с, 0,5H), 4,91-4,92 (д, 2H), 3,52-3,56 (м, 2H), 3,33-3,35 (д, 2H), 2,92 (с, 3H), 2,83-2,85 (м, 2H).
Молекулярная формула: C11H15FN4O, молекулярная масса: 238,27, ЖХ-МС (полож., m/z)=239,19 [M+H]+.
Пример 14: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A25)
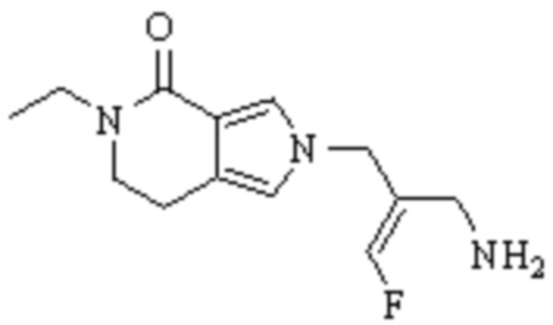
Стадия 1: Синтез промежуточного этил 3-(этиламино)пропионата
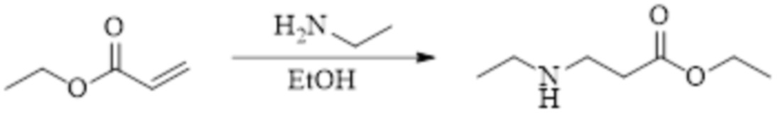
65%-ный водный раствор этиламина (29,1 г, 419,51 ммоль, 1,4 экв., 65%) растворяли в этаноле (100 мл). Затем к раствору по каплям добавляли раствор этил акрилата (30,00 г, 299,65 ммоль, 1,0 экв.) в этаноле (20 мл) (1,5 ч). После добавления смесь перемешивали при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали с получением продукта (42,0 г, выход: 96,5%).
Стадия 2: Синтез промежуточного этил 3-((3-этокси-3-оксопропил)(этил)амино)-3-оксопропионата

Промежуточный этил 3-(этиламино)пропионат (20,0 г, 137,74 ммоль, 1,0 экв.), моноэтил малонат (18,20 г, 137,74 ммоль, 1,0 экв.), триэтиламин (32,06 г, 316,80 ммоль, 2,3 экв.) и DMAP (3,37 г, 27,55 ммоль, 0,2 экв.) последовательно добавляли к ДХМ (500 мл), затем партиями добавляли EDCI (31,69 г, 165,29 ммоль, 1,2 экв.). Смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, реакционный раствор выливали в воду (300 мл), и pH доводили до 3-4 с помощью 3 моль/л соляной кислоты, затем жидкости разделяли. Органическую фазу промывали последовательно водой (150 мл × 2) и насыщенным солевым раствором (300 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением продукта (29,0 г, выход: 81,2%).
Стадия 3: Синтез промежуточного этил 1-этил-2,4-диоксопиперидин-3-карбоксилата
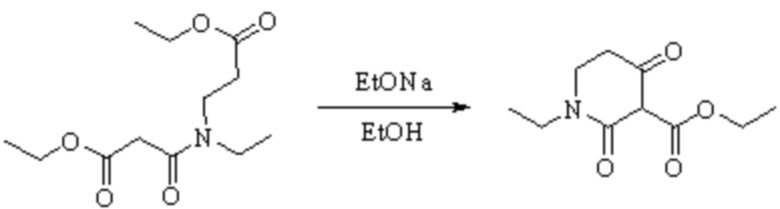
Натрий (5,14 г, 223,68 ммоль, 2,0 экв.) добавляли к этанолу (150 мл) и смесь перемешивали до завершения реакции. Затем в раствор этоксида натрия в этаноле добавляли этил 3-((3-этокси-3-оксопропил)(этил)амино)-3-оксопропионат (29,0 г, 118,84 ммоль, 1,0 экв.). После добавления смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 4: Синтез промежуточного 1-этилпиперидин-2,4-диона
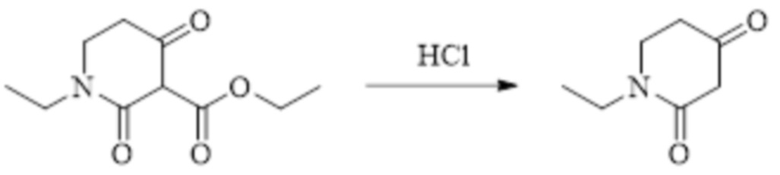
Этил 1-этил-2,4-диоксопиперидин-3-карбоксилат (23,85 г, 111,84 ммоль, 1,0 экв.) партиями добавляли к 2 моль/л соляной кислоты (300 мл). Затем смесь нагревали до температуры 100°C и подвергали взаимодействию в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и экстрагировали ДХМ (150 мл × 3). Органическую фазу промывали последовательно водой (100 мл) и насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением продукта (12,0 г, выход за две стадии: 76,0%).
Стадия 5: Синтез промежуточного 1-этил-3-((диметиламино)метилен)пиперидин-2,4-диона
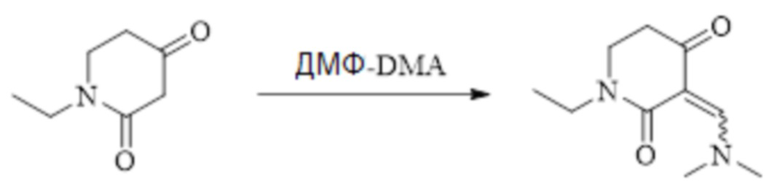
Промежуточный 1-этилпиперидин-2,4-дион (6,00 г, 42,50 ммоль, 1,0 экв.) добавляли к 1,1-диметокси-N,N-диметилметиламину (5,32 г, 44,63 ммоль, 1,05 экв.). Затем смесь перемешивали при комнатной температуре в течение 0,5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 6: Синтез промежуточного ((1-этил-2,4-диоксопиперидин-3-илиден)метил)глицина
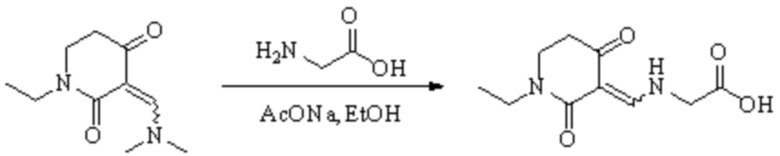
Промежуточный 1-этил-3-((диметиламино)метилен)пиперидин-2,4-дион (8,85 г, 42,49 ммоль, 1,0 экв.), глицин (3,19 г, 42,49 ммоль, 1,0 экв.) и ацетат натрия (4,18 г, 50,99 ммоль, 1,2 экв.) добавляли к этанолу (100 мл). Затем смесь перемешивали при температуре 50°C до завершения реакции. Реакционный раствор концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 7: Синтез промежуточного 2-ацетил-5-этил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
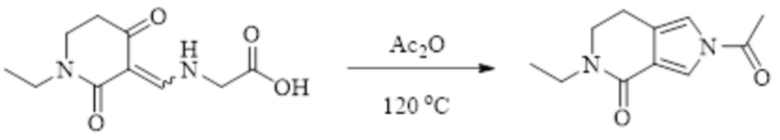
Промежуточный ((1-этил-2,4-диоксопиперидин-3-илиден)метил)глицин (10,12 г, 42,49 ммоль, 1,0 экв.) добавляли к уксусному ангидриду (80 мл). Затем смесь перемешивали при температуре 120°C в течение 5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали, и остаток выливали в насыщенный водный бикарбоната натрия раствор (200 мл). Экстрагировали этилацетатом (50 мл × 3). Органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением сырого продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 8: Синтез промежуточного 5-этил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
Промежуточный 2-ацетил-5-этил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (8,76 г, 42,49 ммоль, 1,0 экв.) и карбонат калия (11,74 г, 84,98 ммоль, 2,0 экв.) последовательно добавляли в смешанный растворитель из метанола (50 мл) и воды (50 мл). Затем смесь перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали, добавляли этилацетат (200 мл), промывали последовательно водой (50 мл) и насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (этилацетат: петролейный эфир, 1:1-1:0, об./об.) с получением продукта (2,2 г, выход за четыре стадии: 31,53%).
Стадия 9: Синтез промежуточного (Z)-2-(2-((5-этил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
5-Этил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (1,5 г, 9,13 ммоль, 1,0 экв.), карбонат цезия (4,46 г, 13,70 ммоль, 1,5 экв.) и (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (3,00 г, 10,05 ммоль, 1,1 экв.) добавляли к DMA (20 мл). Затем смесь перемешивали при температуре 55°C в течение 16 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали, и слой на фильтре промывали этилацетатом (40 мл). Органическую фазу промывали последовательно водой (20 мл × 2) и насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (этилацетат) с получением продукта (1,45 г, выход: 41,6%).
Стадия 10: Синтез соединения (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она

(Z)-2-(2-((5-Этил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (1,45 г, 3,80 ммоль, 1,0 экв.) и 85%-ный гидразингидрат (1,12 г, 19,01 ммоль, 5 экв.) последовательно добавляли к этанолу (20 мл). Затем смесь перемешивали при температуре 40°C в течение 15 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали, и слой на фильтре промывали небольшим количеством этанол. Фильтрат концентрировали, добавляли абсолютный этанол (10 мл) и фильтровали, и полученный фильтрат концентрировали с получением сырого продукта (750 мг), и 375 мг сырого продукта очищали с помощью препаративной тонкослойной хроматографии (дихлорметан : метанол, 5:1, об./об.) с получением продукта (85 мг, выход: 17,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 7,22 (с, 1H), 6,96-7,17 (д, 1H), 6,61 (с, 1H), 4,58 (д, 2H), 3,37-3,44 (м, 4H), 3,13 (д, 2H), 2,65-2,68 (т, 2H), 1,02-1,06 (т, 3H).
Молекулярная формула: C13H18FN3O, молекулярная масса: 251,31, ЖХ-МС (m/z)=252,28 [M+H]+.
Пример 15: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(1H)-она (соединение A26)
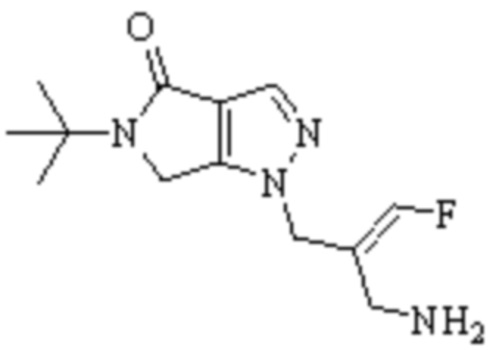
Стадия 1: Синтез соединения (E)-1-(2-(аминометил)-3-фтораллил)-5-(трет-бутил)-5,6-дигидропирроло[3,4-c]пиразол-4(1H)-она
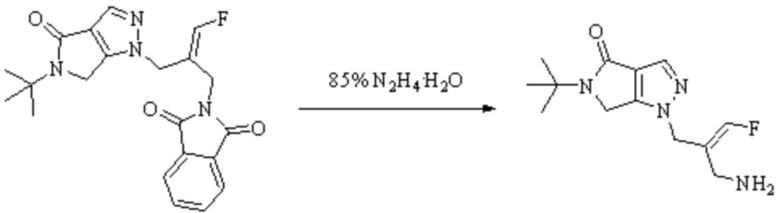
(E)-2-(2-((5-(трет-бутил)-4-оксо-5,6-дигидропирроло[3,4-c]пиразол-1(4H)-ил)метил)-3-фтораллил)изоиндол-1,3-дион (100 мг, 0,25 ммоль, 1,0 экв.), полученный в примере 7, и 85%-ный гидразингидрат (59 мг, 1,01 ммоль, 4,0 экв.) последовательно добавляли к этанолу (5,0 мл). Затем смесь перемешивали при температуре 40°C в течение 5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали, и слой на фильтре промывали небольшим количеством этанол. Фильтрат концентрировали, добавляли абсолютный этанол (3,0 мл) и фильтровали, и полученный фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (дихлорметан : метанол, 5:1, об./об.) с получением продукта (35 мг, выход: 52,1%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 7,58 (с, 1H), 7,02-7,23 (д, 1H), 4,85 (д, 2H), 4,54 (с, 2H), 3,26 (д, 2H), 1,43 (м, 9H).
Молекулярная формула: C13H19FN4O, молекулярная масса: 266,32, ЖХ-МС (m/z)=267,22 [M+H+].
Пример 16: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-изопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A27)
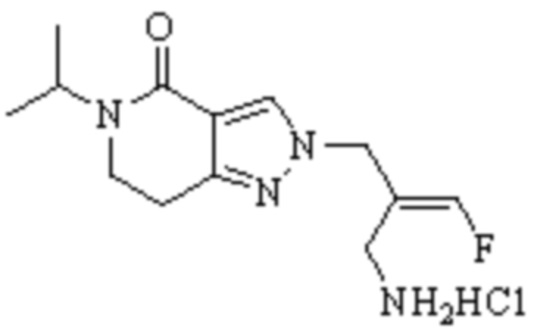
Стадия 1: Синтез этил 3-(изопропиламино)пропионата
Продукт изопропиламин (7,09 г, 0,12 моль, 1,2 экв.) растворяли в этаноле (20 мл). Затем на ледяной бане в раствор медленно добавляли по каплям этилакрилат (10 г, 0,1 моль, 1,0 экв.) и подвергали взаимодействию в течение 12 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (15 г, выход: 94%).
Стадия 2: Синтез этил 3-(изопропил(3-этокси-3-оксопропил)амино)-3-оксопропионата
Промежуточный этил 3-(изопропиламино)пропионат (15 г, 94,2 ммоль, 1,0 экв.), моноэтил малонат (12,44 г, 94,2 ммоль, 1,0 экв.), 4-диметиламинопиридин (2,3 г, 18,84 ммоль, 0,2 экв.) и триэтиламин (21,9 г, 220 ммоль, 2,3 экв.) растворяли в дихлорметане (150 мл) и раствор перемешивали в течение 10 мин. Затем на ледяной бане партиями добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (21,66 г, 113 ммоль, 1,2 экв.). После добавления раствор подвергали взаимодействию при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду и концентрированную соляную кислоту (4:1, 150 мл), и смесь перемешивали в течение 10 мин, затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (100 мл × 2). Органические фазы объединяли, промывали последовательно насыщенным водным раствором карбоната натрия (150 мл) и водой (150 мл × 2), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (17,25 г, выход: 67%).
Стадия 3: Синтез этил 1-изопропил-2,4-диоксопиперидин-3-карбоксилата
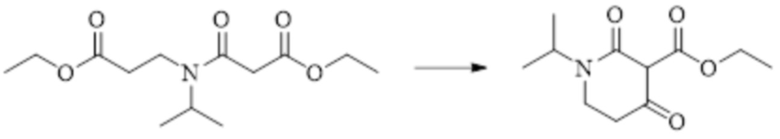
Промежуточный этил 3-(изопропил(3-этокси-3-оксопропил)амино)-3-оксопропионат (17,25 г, 63,11 ммоль, 1,0 экв.) и этоксид натрия (8,59 г, 126 ммоль, 2,0 экв.) растворяли в этаноле (100 мл) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (14,34 г, выход: 100%).
Стадия 4: Синтез 1-изопропилпиперидин-2,4-диона
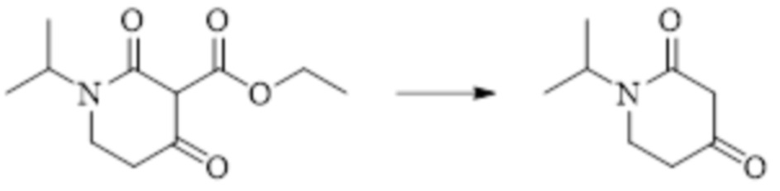
Промежуточный этил 1-изопропил-2,4-диоксопиперидин-3-карбоксилат (14,23 г, 63,11 ммоль, 1,0 экв.) растворяли в воде (80 мл) и концентрированной соляной кислоте (20 мл) и подвергали взаимодействию при температуре 110°C в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и экстрагировали дихлорметаном (100 мл × 3). Органическую фазу сушили и концентрировали с получением продукта (7,14 г, выход: 72%).
Стадия 5: Синтез 1-изопропил-3-((диметиламино)метилен)пиперидин-2,4-диона
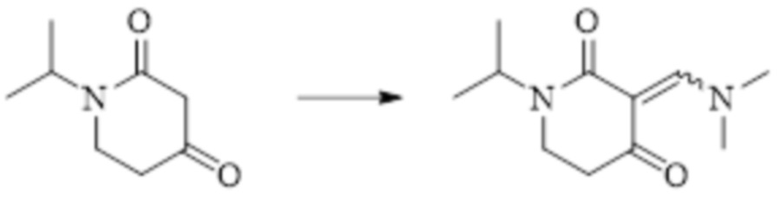
Промежуточный 1-изопропилпиперидин-2,4-дион (7,14 г, 46 ммоль, 1,0 экв.) растворяли в N, N-диметилформамид диметилацетале (6,03 г, 50,6 ммоль, 1,1 экв.) и подвергали взаимодействию в течение 0,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении с получением продукта (9,67 г, выход: 100%).
Стадия 6: Синтез 5-изопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

Промежуточный 1-изопропил-3-((диметиламино)метилен)пиперидин-2,4-дион (9,67 г, 46 ммоль, 1,0 экв.) и гидразингидрат (2,506 г, 50,06 ммоль, 1,1 экв.) растворяли в метаноле (50 мл) и подвергали взаимодействию при температуре 60°C в течение 30 мин. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Сырой продукт перекристаллизовывали этилацетатом (80 мл) и фильтровали в вакууме, и слой на фильтре сушили с получением продукта (4,5 г, выход: 54%).
Стадия 7: Синтез (E)-2-(2-((5-изопропил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
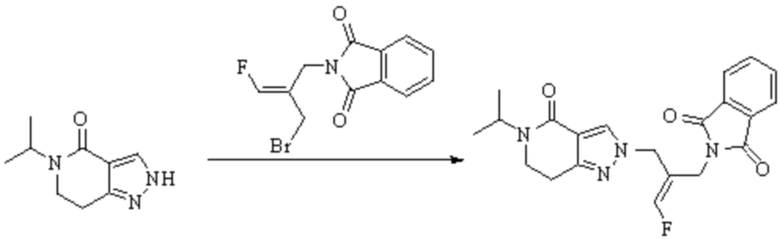
Промежуточный 5-изопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (300 мг, 1,67 ммоль, 1,0 экв.) растворяли в ДМФ (1,5 мл). Затем к раствору добавляли NaH (87 мг, 2,21 ммоль, 1,3 экв.), перемешивали в течение 30 мин, и затем по каплям добавляли раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-диона (599 мг, 2,0 ммоль, 1,2 экв.) в ДМФ (1,5 мл) и подвергали взаимодействию в течение 30 мин. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционную смесь добавляли воду (10 мл) и экстрагировали этилацетатом (20 мл × 2). Органическую фазу промывали водой (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:2) с получением продукта (200 мг, выход: 30%).
Стадия 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-изопропил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
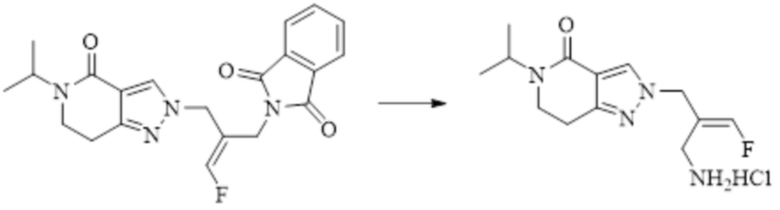
Промежуточный (E)-2-(2-((5-изопропил-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (200 мг, 0,5 ммоль, 1,0 экв.) растворяли в EtOH (2 мл). Затем к раствору добавляли гидразингидрат (88 мг, 1,75 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 30 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением маслянистой жидкости. В маслянистую жидкость добавляли метанол (2 мл), затем раствор хлорида водорода в этаноле (0,15 мл), перемешивали в течение 5 мин и концентрировали при пониженном давлении с получением продукта (60 мг, выход: 39%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,24 (с, 3H), 8,20 (с, 1H), 7,36 (с, 0,5H), 7,16 (с, 0,5H), 4,86-4,87 (д, 2H), 4,73-4,80 (м, 1H), 3,40-3,44 (м, 2H), 3,34 (с, 2H), 2,77-2,79 (м, 2H), 1,11 (с, 3H), 1,09 (с, 3H).
Молекулярная формула: C13H20ClFN4O, молекулярная масса: 302,78, ЖХ-МС (полож., m/z)=267,28 [M+H]+.
Пример 17: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-этил-1H-пиразол-4-карбоксамида (соединение B1)
Стадия 1: Синтез метил 1H-пиразол-4-карбоксилата

1H-Пиразол-4-карбоновую кислоту (10,0 г, 0,0892 моль, 1,0 экв.) растворяли в метаноле (36,0 мл). Затем при температуре 0°C к раствору добавляли по каплям серную кислоту (10,0 мл) и нагревали до температуры 75°C и подвергали взаимодействию в течение ночи. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор выливали в холодную воду (50,0 мл). Добавляли по каплям насыщенный водный раствор карбоната натрия до установления pH равным 8-9 и экстрагировали этилацетатом (20,0 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением продукта (10,37 г, выход: 92,2%).
Стадия 2: Синтез метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-4-карбоксилата
Метил 1H-пиразол-4-карбоксилат (2,0 г, 15,86 ммоль, 1,0 экв.), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (5,2 г, 17,44 ммоль, 1,1 экв.) и карбонат калия (3,29 г, 23,79 ммоль, 1,5 экв.) растворяли в ацетонитриле (20,0 мл) и подвергали взаимодействию при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (30,0 мл) и экстрагировали этилацетатом (20,0 мл × 2). Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением продукта (6,0 г, сырой продукт).
Стадия 3: Синтез метил (E)-1-(2-(аминометил)-3-фтораллил)-1H-пиразол-4-карбоксилата
Метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-4-карбоксилат (6,0 г, сырой продукт) растворяли в этаноле (60,0 мл). Затем к раствору добавляли гидразингидрат (10,27 г, 174,44 ммоль, 85%) и подвергали взаимодействию в течение ночи при комнатной температуре. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении, и затем добавляли толуолом. Полученную смесь концентрировали при пониженном давлении с получением продукта (5,0 г, сырой продукт).
Стадия 4: Синтез метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоксилата
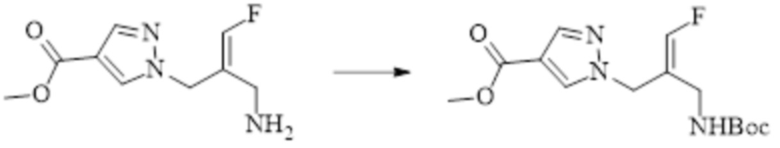
Метил (E)-1-(2-(аминометил)-3-фтораллил)-1H-пиразол-4-карбоксилат (5,0 г, сырой продукт) и карбонат натрия (1,85 г, 17,44 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (20,0 мл) и воде (10,0 мл) и подвергали взаимодействию при температуре 40°C в течение 2,5 ч. После завершения взаимодействия продуктов, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, экстрагировали этилацетатом (40,0 мл × 2), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (3,2 г, выход за три стадии: 64,4%).
Стадия 5: Синтез (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоновой кислоты

Метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоксилат (1,98 г, 6,32 ммоль, 1,0 экв.) и NaOH (1,517 г, 37,915 ммоль) растворяли в метаноле (9,0 мл) и воде (9,0 мл) и подвергали взаимодействию при температуре 60°C в течение 4 ч. После завершения взаимодействия продуктов, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли воду (20,0 мл). Добавляли лимонную кислоту до установления pH равным 5-6 и экстрагировали дихлорметаном (20,0 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт суспендировали в небольшом количестве этилацетата (2,5 мл) и фильтровали в вакууме с получением продукта как слой на фильтре (690,0 мг, выход: 36,5%).
Стадия 6: Синтез (E)-(3-фтор-2-((4-(метилкарбамоил)-1H-пиразол-1-ил)метил)аллил)трет-бутил карбамата
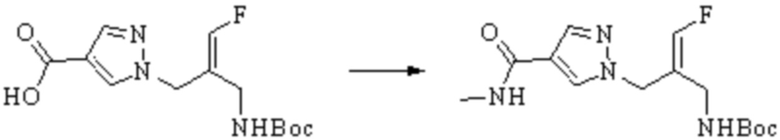
(E)-1-(2-((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоновую кислоту (150,0 мг, 0,501 ммоль, 1,0 экв.) и DIPEA (272,0 мг, 2,11 ммоль, 4,2 экв.) растворяли в ДМФ (2,0 мл). В атмосфере азота при температуре 0°C к раствору добавляли HATU (286,0 мг, 0,752 ммоль, 1,5 экв.), перемешивали в течение 0,5 ч и затем добавляли гидрохлорид метилмамина (41,0 мг, 0,601 ммоль, 1,2 экв.). Смесь медленно нагревали до комнатной температуры в течение ночи. После завершения реакции, как определялось данными ТСХ, в реакционный раствор добавляли этилацетат (40,0 мл) и смесь промывали последовательно насыщенным водным раствором карбоната натрия (4,0 мл), насыщенным водным раствором хлорида аммония (4,0 мл) и насыщенным водным раствором хлорида натрия (4,0 мл). Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=150:1-30:1) с получением продукта (133,0 мг, выход: 85%).
Стадия 7: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-метил-1H-пиразол-4-карбоксамида

(E)-(3-Фтор-2-((4-(метилкарбамоил)-1H-пиразол-1-ил)метил)аллил)трет-бутил карбамат (133,0 мг, 0,426 ммоль, 1,0 экв.) растворяли в этаноле (1,0 мл). Затем по каплям к раствору добавляли раствор хлорида водорода в этаноле (1,0 мл) и подвергали взаимодействию при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=8:1) с получением продукта (80,0 мг, выход: 88,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,16 (с, 1H), 8,03-8,01 (д, 1H), 7,82 (с, 1H), 7,03-6,82 (д, 1H), 4,76-4,75 (д, 2H), 3,06-3,05 (д, 2H), 2,71-2,69 (д, 3H), 1,71 (с, 2H).
Молекулярная формула: C9H13FN4O, молекулярная масса: 212,23, ЖХ-МС (полож., m/z)=213,19 [M+H]+
Пример 18: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-этил-1H-пиразол-4-карбоксамида (соединение B2)
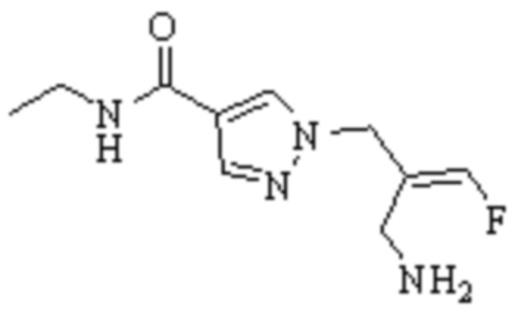
Стадия 1: Синтез трет-бутил (E)-(2-((4-(этилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
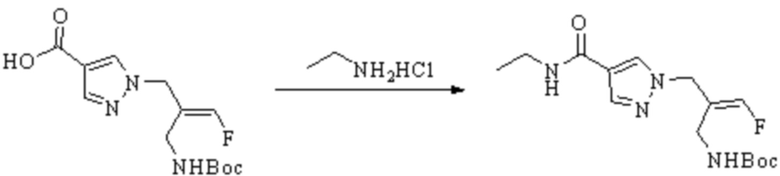
(E)-1-(2-((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоновую кислоту (500,0 мг, 1,67 ммоль, 1,0 экв.) добавляли к ДМФ (4,0 мл) и на ледяной бане добавляли DIPEA (1,29 г, 10,02 ммоль, 6,0 экв.) и HATU (952,5 г, 2,50 ммоль, 1,5 экв.). Смесь перемешивали в течение 2 ч, и затем добавляли гидрохлорид этиламина (340,4 мг, 4,17 ммоль, 2,5 экв.). Полученную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 12 ч. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли EA (70 мл) и водный карбонат натрия раствор (40 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (426,0 мг, выход: 78,1%).
Стадия 2: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-этил-1H-пиразол-4-карбоксамида
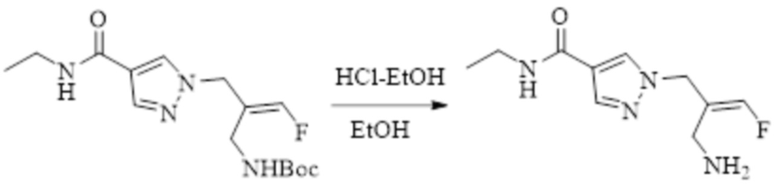
(E)-(2-((4-(Этилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)трет-бутил карбамат (426,0 мг, 1,30 ммоль, 1,0 экв.) добавляли к EtOH (4 мл) и на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (4 мл). Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и pH доводили до 7-8 насыщенным водным раствором карбоната натрия. Добавляли ДХМ (50 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (157,0 мг, выход: 53,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,16 (с, 1H), 8,04-8,06 (м, 1H), 7,83 (с, 1H), 7,03 (с, 0,5H), 6,82 (с, 0,5H), 4,75 (м, 2H), 3,18-3,22 (м, 2H), 3,04-3,05 (м, 2H), 1,55 (с, 2H), 1,06-1,10 (м, 3H).
Молекулярная формула: C10H15FN4O, молекулярная масса: 226,26, ЖХ-МС (полож., m/z)=227,21 [M+H]+.
Пример 19: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-(трет-бутил)-1H-пиразол-4-карбоксамида (соединение B3)
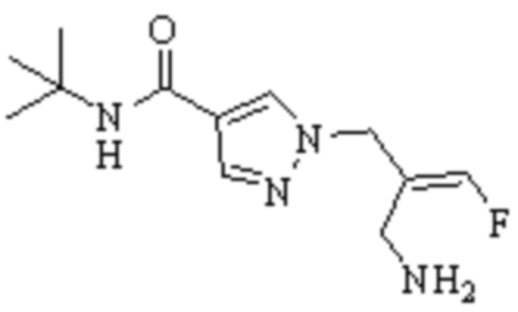
Стадия 1: Синтез трет-бутил (E)-(2-((4-(трет-бутилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата

(E)-1-(2-((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоновую кислоту (440,0 мг, 1,47 ммоль, 1,0 экв.) добавляли к ДМФ (4,5 мл) и на ледяной бане добавляли DIPEA (285,0 мг, 2,20 ммоль, 1,5 экв.) и HATU (1,67 г, 4,41 ммоль, 3,0 экв.). Смесь перемешивали на ледяной бане в течение 2 ч и добавляли трет-бутиламин (150,5 мг, 2,05 ммоль, 1,4 экв.). Полученную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 12 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, и добавляли EA (100 мл) и водный раствор карбоната натрия (60 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (400,0 мг, выход: 76,7%).
Стадия 2: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-(трет-бутил)-1H-пиразол-4-карбоксамида
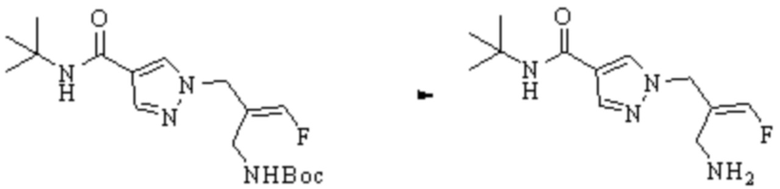
трет-Бутил (E)-(2-((4-(трет-бутилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамат (300,0 мг, 0,84 ммоль, 1,0 экв.) добавляли к EtOH (4 мл) и на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (4 мл). Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и pH доводили до 7-8 насыщенным водным раствором карбоната натрия. Добавляли ДХМ (30 мл), затем жидкости разделяли. Водную фазу экстрагировали н-бутанолом (10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (92,0 мг, выход: 43%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,19 (с, 1H), 7,83 (с, 1H), 7,36 (с, 1H), 7,03 (с, 0,5H), 6,82 (с, 0,5H), 4,73-4,74 (д, 2H), 3,05 (д, 2H), 1,53-1,70 (с, 2H), 1,34 (с, 9H).
Молекулярная формула: C12H19FN4O, молекулярная масса: 254,31, ЖХ-МС (полож., m/z)=255,24 [M+H]+.
Пример 20: Синтез гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-N-циклопропил-1H-пиразол-4-карбоксамида (соединение B4)
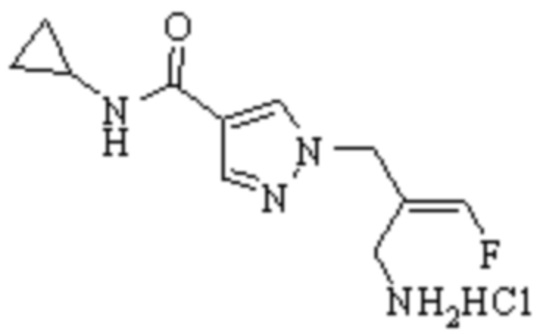
Стадия 1: Синтез трет-бутил (E)-(2-((4-(циклопропилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
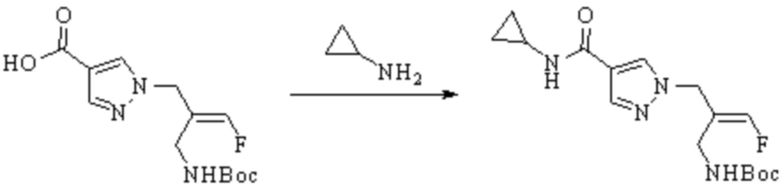
(E)-1-(2-((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоновую кислоту (133,0 мг, 0,44 ммоль, 1,0 экв.) добавляли к ДМФ (3,0 мл), на ледяной бане добавляли DIPEA (170,4 мг, 1,32 ммоль, 3,0 экв.) и HATU (253,4 мг, 0,667 ммоль, 1,5 экв.), перемешивали в течение 2 ч и затем добавляли циклопропиламин (32,6 мг, 0,57 ммоль, 1,3 экв.). Смесь выдерживали при комнатной температуре и перемешивали в течение 12 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли EA (50 мл), насыщенный водный раствор хлорида аммония (30 мл) и насыщенный водный раствор хлорида натрия (30 мл), и жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле с получением продукта (97,6 мг, выход: 65,5%).
Стадия 2: Синтез гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-N-циклопропил-1H-пиразол-4-карбоксамида
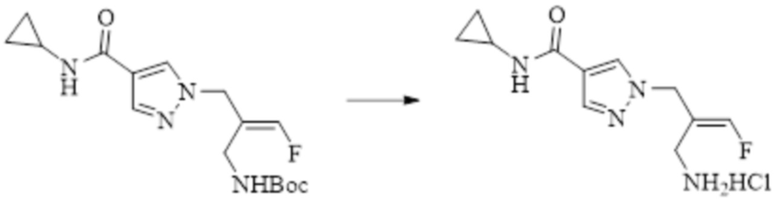
трет-Бутил (E)-2-((4-(циклопропилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамат (97,6 мг, 0,28 ммоль, 1,0 экв.) добавляли в EtOH (3 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (3 мл) и смесь выдерживали при комнатной температуре и перемешивали в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (35,7 мг, выход: 46,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,27 (с, 2H), 8,24 (с, 1H), 8,13-8,14 (д, 1H), 7,89 (с, 1H), 7,35 (с, 0,5H), 7,15 (с, 0,5H), 4,89-4,90 (д, 2H), 2,71-2,75 (м, 1H), 0,64-0,67 (м, 2H), 0,49-0,51 (м, 2H).
Молекулярная формула: C11H15FN4O, молекулярная масса: 238,27, ЖХ-МС (полож., m/z)=239,21 [M+H]+.
Пример 21: Синтез гидрохлорида (E)-1-(2-аминометил-3-фтораллил)-N-этил-1H-пиррол-3-карбоксамида (соединение B11)
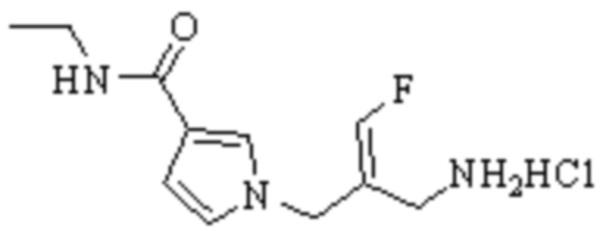
Стадия 1: Синтез метил (Z)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиррол-3-карбоксилата
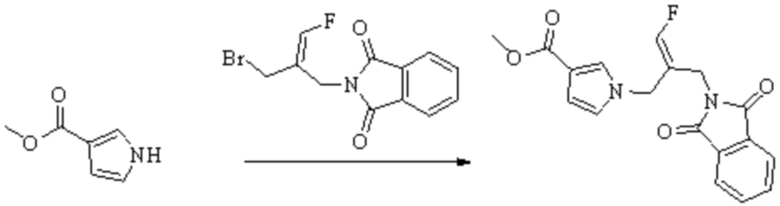
Метил 1H-пиррол-3-карбоксилат (1,77 г, 14,19 ммоль, 1,0 экв.), (E)-2-(2-бромметил-3-фтораллил)изоиндолин-1,3-дион (5,50 г, 18,45 ммоль, 1,3 экв.) и карбонат калия (5,88 г, 42,57 ммоль, 3,0 экв.) добавляли в ДМФ (100 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, добавляли этилацетат (100 мл) и промывали водой (50 мл × 4). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали в вакууме, и фильтрат концентрировали при пониженном давлении с получением продукта (5,89 г сырого продукта).
Стадия 2: Синтез метил (E)-1-(2-аминометил-3-фтораллил)-1H-пиррол-3-карбоксилата
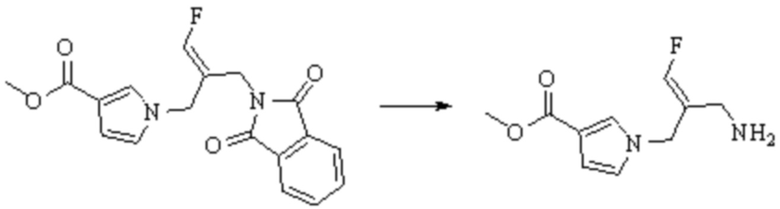
Метил (Z)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиррол-3-карбоксилат (5,89 г сырого продукта) и гидразингидрат (85%, 10,16 г, 172,6 ммоль, 10,0 экв.) добавляли в этанол (60 мл) и смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли этилацетат (60 мл) и промывали насыщенным водным раствором NH4Cl (30 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и фильтровали в вакууме, и фильтрат концентрировали при пониженном давлении с получением продукта (1,96 г сырого продукта).
Стадия 3: Синтез метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил-3-фтораллил)-1H-пиррол-3-карбоксилата
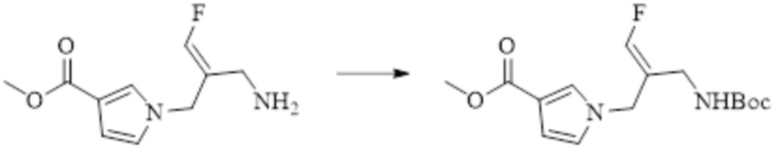
Метил (E)-1-(2-аминометил-3-фтораллил)-1H-пиррол-3-карбоксилат (1,96 г сырого продукта) и триэтиламин (1,40 г, 13,85 ммоль, 1,5 экв.) добавляли в тетрагидрофуран (20 мл), добавляли по каплям (Boc)2O (2,22 г, 10,16 ммоль, 1,1 экв.) и смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли этилацетат (80 мл), и для промывки добавляли насыщенный водный раствор NH4Cl (40 мл × 2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали в вакууме и концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле с получением продукта (2,87 г, выход за три стадии: 65,0%).
Стадия 4: Синтез (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиррол-3-карбоновой кислоты

Метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил-3-фтораллил)-1H-пиррол-3-карбоксилат (2,87 г, 9,19 ммоль, 1,0 экв.) растворяли в метаноле (15 мл). К раствору добавляли водный раствор (15 мл) NaOH (2,21 г, 55,13 ммоль, 6,0 экв.) и перемешивали при температуре 60°C в течение 4 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, добавляли 5% водный раствор лимонной кислоты до установления pH равным 4 и экстрагировали ДХМ (50 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (2,00 г, выход: 73,0%).
Стадия 5: Синтез трет-бутил (E)-(2-((3-(этилкарбамоил)-1H-пиррол-1-ил)метил)-3-фтораллил)карбамата
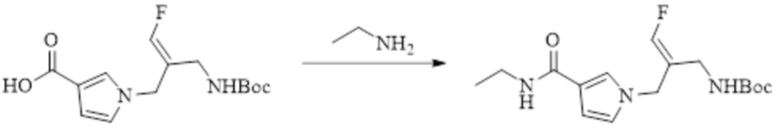
(E)-1-(2-(((трет-Бутоксикарбонил)амино)метил-3-фтораллил)-1H-пиррол-3-карбоновую кислоту (200,0 мг, 0,67 ммоль, 1,0 экв.) добавляли в ДМФ (2 мл), на ледяной бане добавляли DIPEA (519,8 мг, 4,02 ммоль, 6,0 экв.) и HATU (382,4 мг, 1,01 ммоль, 1,5 экв.) и смесь перемешивали в течение 1 ч. В смесь добавляли гидрохлорид этиламина (164,0 мг, 2,01 ммоль, 3,0 экв.) и выдерживали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Концентрат растворяли в EA (60 мл), и для промывки последовательно использовали насыщенный водный раствор NaHCO3 (50 мл), насыщенный водный раствор NH4Cl (50 мл) и насыщенный солевой раствор (50 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=40:1) с получением продукта (133,0 мг, выход: 61,0%).
Стадия 6: Синтез гидрохлорида (E)-1-(2-аминометил-3-фтораллил)-N-этил-1H-пиррол-3-карбоксамида

трет-Бутил (E)-(2-((3-(этилкарбамоил)-1H-пиррол-1-ил)метил)-3-фтораллил)карбамат (133,0 мг, 0,41 ммоль) добавляли в EtOH (5,0 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (5,0 мл) и смесь выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, сырой продукт суспендировали в метил трет-бутиловом эфире в течение 2 ч и фильтровали с получением продукта (93,0 мг, выход: 86,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,55 (с, 3H), 7,84 (с, 1H), 7,37-7,36 (т, 1H), 7,32 (с, 0,5H), 7,11 (с, 0,5H), 6,88-6,87 (т, 1H), 6,49-6,47 (кв, 1H), 4,74-4,73 (д, 2H), 3,24-3,16 (м, 4H), 1,06 (т, 3H).
Молекулярная формула: C11H17ClN3FO, молекулярная масса: 261,73, ЖХ-МС (полож., m/z)=226,18 [M+H]+.
Пример 22: Синтез (E)-1-(2-аминометил-3-фтораллил)-N-трет-бутил-1H-пиррол-3-карбоксамида (соединение B12)
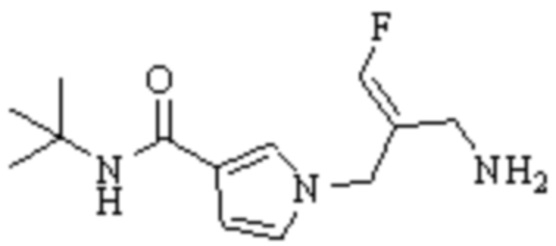
Стадия 1: Синтез трет-бутил (E)-(2-((3-(трет-бутилкарбамоил)-1H-пиррол-1-ил)метил)-3-фтораллил)карбамата
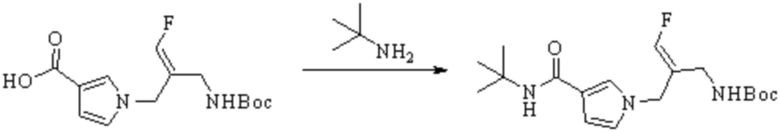
(E)-1-(2-(((трет-Бутоксикарбонил)амино)метил-3-фтораллил)-1H-пиррол-3-карбоновую кислоту (200,0 мг, 0,67 ммоль, 1,05 экв.) добавляли в ДМФ (2 мл), на ледяной бане добавляли DIPEA (248,3 мг, 1,92 ммоль, 3,0 экв.) и HATU (365,2 мг, 0,96 ммоль, 1,5 экв.) и смесь перемешивали на ледяной бане в течение 1 ч. В смесь добавляли трет-бутиламин (51,5 мг, 0,70 ммоль, 1,1 экв.) и выдерживали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Концентрат растворяли в EA (60 мл) и для промывки последовательно использовали насыщенный водный раствор NaHCO3 (50 мл), насыщенный водный раствор NH4Cl (50 мл) и насыщенный солевой раствор (50 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (165,0 мг, выход: 69,6%).
Стадия 2: Синтез (E)-1-(2-аминометил-3-фтораллил)-N-трет-бутил-1H-пиррол-3-карбоксамида
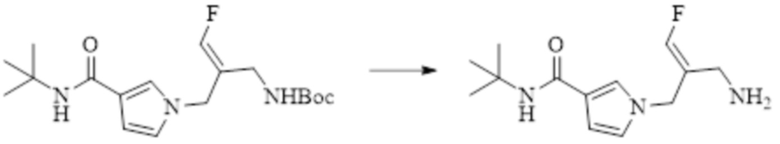
трет-Бутил (E)-(2-((3-(трет-бутилкарбамоил)-1H-пиррол-1-ил)метил)-3-фтораллил)карбамат (165,0 мг, 0,47 ммоль) добавляли в EtOH (5,0 мл), на ледяной бане добавляли по каплям раствор хлористоводородной кислоты в этаноле (5,0 мл) и смесь выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли ДХМ (40 мл) и 15% водный раствор NaOH (40 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении. После выделения и очистки путем препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) получали (E)-1-(2-аминометил-3-фтораллил)-N-трет-бутил-1H-пиррол-3-карбоксамид (77,0 мг, выход: 65,1%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 7,31 (с, 1H), 7,01 (с, 0,5H), 6,96 (с, 1H), 6,79 (с, 0,5H), 6,72-6,71 (т, 1H), 6,44-6,43 (д, 1H), 4,50-4,49 (д, 2H), 3,03-3,02 (д, 2H), 1,71 (с, 2H), 1,33 (с, 9H).
Молекулярная формула: C13H20FN3O, молекулярная масса: 253,32, ЖХ-МС (полож., m/z)=254,24 [M+H]+.
Пример 23: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-этил-1H-пиразол-3-карбоксамида (соединение B20)
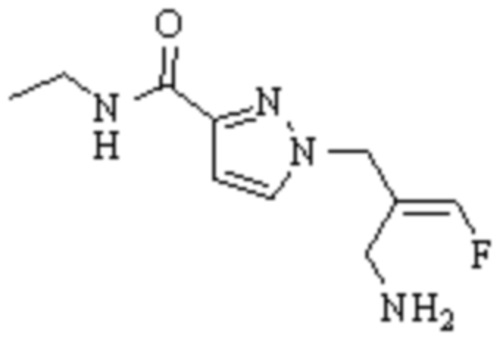
Стадия 1: Синтез трет-бутил (E)-(2-((3-(этилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
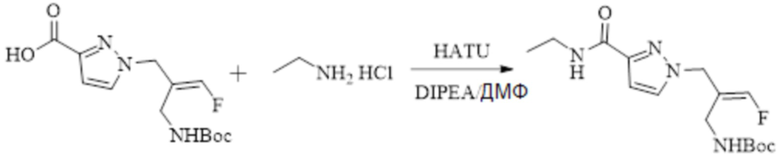
(E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-3-карбоновую кислоту (233,5 мг, 0,78 ммоль, 1,0 экв.) добавляли в ДМФ (3 мл), на ледяной бане добавляли DIPEA (604,5 мг, 4,68 ммоль, 6,0 экв.) и HATU (444,8 мг, 1,17 ммоль, 1,5 экв.), перемешивали в течение 2 ч и затем добавляли гидрохлорид этиламина (159,0 мг, 1,95 ммоль, 2,5 экв.). Смесь выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли EA (40 мл) и насыщенный водный раствор карбоната натрия (50 мл), затем жидкости разделяли. Органическую фазу промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (153,0 мг, выход: 60,2%).
Стадия 2: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-этил-1H-пиразол-3-карбоксамида
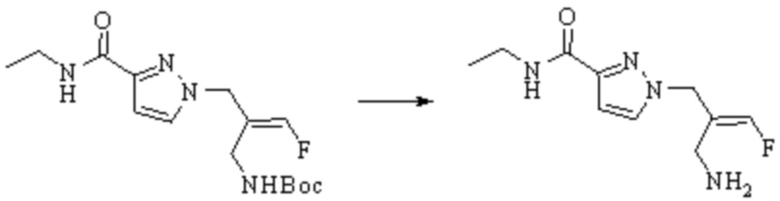
трет-Бутил (E)-(2-((3-(этилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил) карбамат (153,0 мг, 0,46 ммоль, 1,0 экв.) добавляли в EtOH (3,0 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (3,0 мл) и смесь выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли ДХМ (25 мл) и насыщенный водный раствор карбоната натрия (25 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (30,1 мг, выход: 28,6%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,01-8,03 (м, 1H), 7,81 (м, 1H), 7,00 (с, 0,5H), 6,79 (с, 0,5H), 6,61-6,62 (м, 1H), 4,79 (м, 2H), 3,20-3,26 (м, 2H), 3,06-3,07 (м, 2H), 1,69 (с, 2H), 1,06-1,10 (м, 3H).
Молекулярная формула: C10H15FN4O, молекулярная масса: 226,26, ЖХ-МС (полож., m/z)=227,21 [M+H]+.
Пример 24: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-(трет-бутил)-1H-пиразол-3-карбоксамида (соединение B21)
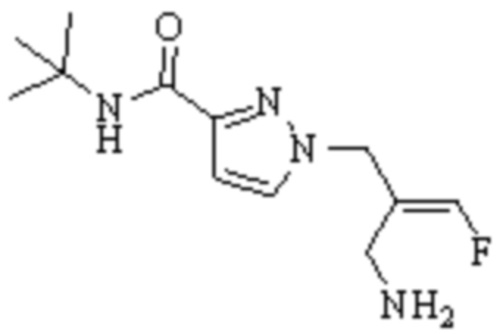
Стадия 1: Синтез метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-3-карбоксилата
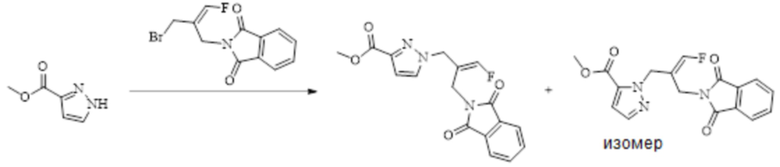
(E)-2-(2-(Бромметил)-3-фтораллил)изоиндолин-1,3-дион (3,0 г, 10,0 ммоль, 1,0 экв.), карбонат калия (2,0 г, 15,0 ммоль, 1,5 экв.) и метил 1H-пиразол-3-карбоксилат (1,5 г) добавляли в ДМФ (10 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, ДМФ удаляли концентрированием при пониженном давлении, добавляли EA (100 мл) и воду (200 мл) и жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали при пониженном давлении и сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=6:1-1:1) с получением продукта метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-3-карбоксилата (2,25 г, выход: 66,1%) и изомера положения метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-5-карбоксилата (772,5 мг, выход: 22,7%).
Стадия 2: Синтез метил (E)-1-(2-(аминометил)-3-фтораллил)-1H-пиразол-3-карбоксилата
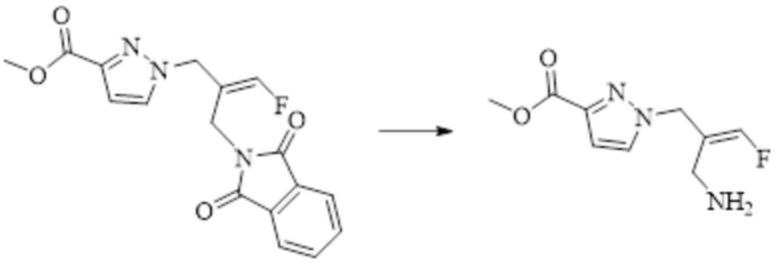
Метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-3-карбоксилат (2,25 г, 6,55 ммоль, 1,0 экв.) и гидразингидрат (984,2 мг, 19,6 ммоль, 3,0 экв.) добавляли в этанол (20 мл). Полученный раствор перемешивали при комнатной температуре в течение 12 ч и фильтровали в вакууме. Фильтрат концентрировали, суспендировали в этаноле (20 мл) и фильтровали, и полученный таким образом фильтрат концентрировали при пониженном давлении с получением продукта (1,79 г сырого продукта).
Стадия 3: Синтез метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-3-карбоксилата
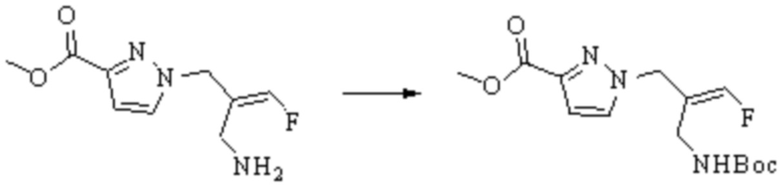
Метил (E)-1-(2-(аминометил)-3-хлораллил)-1H-пиразол-3-карбоксилат (1,79 г сырого продукта), триэтиламин (993,6 мг, 9,82 ммоль, 1,5 экв.) и ди-трет-бутил дикарбонат (1,85 г, 8,51 ммоль, 1,3 экв.) добавляли в ТГФ (40 мл) и полученный раствор перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли EA (100 мл) и насыщенный водный раствор хлорида аммония (150 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=8:1-5:1) с получением продукта (545,1 мг, выход за две стадии: 27,2%).
Стадия 4: Синтез (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-3-карбоновой кислоты

Метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-3-карбоксилат (545,0 мг, 1,73 ммоль, 1,0 экв.) растворяли в метаноле (5 мл). К раствору добавляли водный раствор NaOH (6 моль/л, 2,5 мл) и перемешивали при температуре 40°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли 5% водный раствор лимонной кислоты для достижения pH системы равным 4 и экстрагировали ДХМ (50 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (490,7 мг сырого продукта).
Стадия 5: Синтез трет-бутил (E)-(2-((3-(трет-бутилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
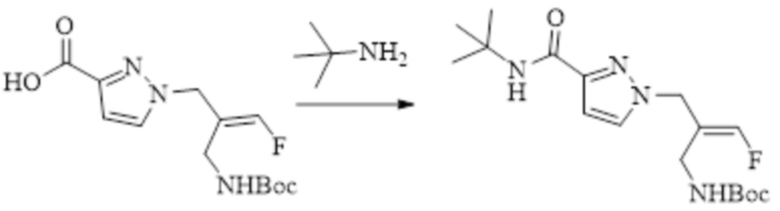
(E)-1-(2-(((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-3-карбоновую кислоту (296,0 мг, 0,98 ммоль, 1,0 экв.) добавляли в ДМФ (5 мл), на ледяной бане добавляли DIPEA (382,8 мг, 2,96 ммоль, 3,0 экв.) и HATU (563,5 мг, 1,48 ммоль, 1,5 экв.), перемешивали в течение 2 ч и добавляли трет-бутиламин (86,8 мг, 1,18 ммоль, 1,2 экв.). Смесь выдерживали при комнатной температуре и перемешивали в течение 12 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли EA (60 мл) и насыщенный водный раствор карбоната натрия (50 мл), затем жидкости разделяли. Органическую фазу промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (200,0 мг, выход за две стадии: 32,6%).
Стадия 6: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-(трет-бутил)-1H-пиразол-3-карбоксамида
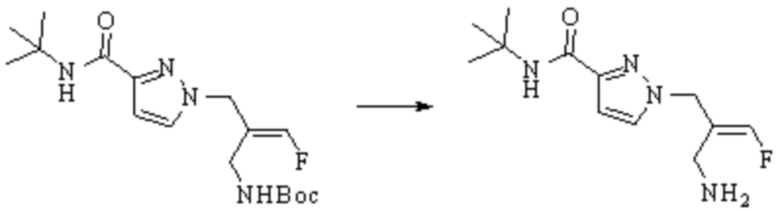
трет-Бутил (E)-(2-((3-(трет-бутилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамат (200,0 мг, 0,56 ммоль, 1,0 экв.) добавляли в EtOH (3,0 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (3,0 мл) и смесь выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли ДХМ (40 мл) и насыщенный водный раствор карбоната натрия (40 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (118,0 мг, выход: 82,5%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 7,81-7,82 (д, 1H), 7,04 (с, 1H), 7,02 (с, 0,5H), 6,81 (с, 0,5H), 6,60-6,61 (д, 1H), 4,78-4,79 (д, 2H), 3,06-3,07 (д, 2H), 1,36 (с, 9H).
Молекулярная формула: C12H19FN4O, молекулярная масса: 254,31, ЖХ-МС (полож., m/z)=255,25 [M+H]+.
Пример 25: Синтез (E)-1-(2-аминометил-3-фтораллил)-N-4-хлорфенил-1H-пиразол-4-карбоксамида (соединение B28)
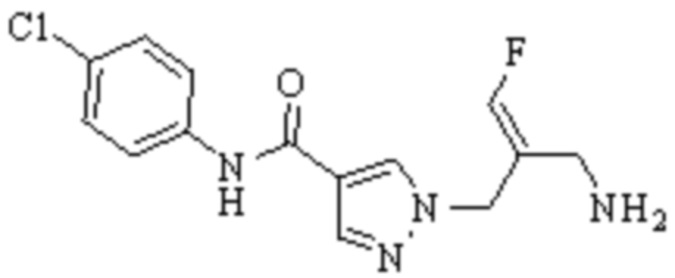
Стадия 1: Синтез трет-бутил (E)-(2-((4-((4-хлорфенил)карбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
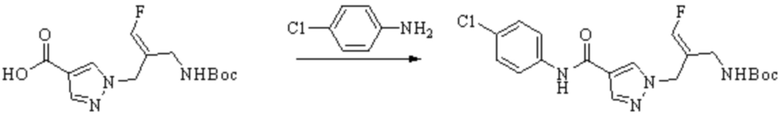
(E)-1-(2-(((трет-Бутоксикарбонил)амино)метил-3-фтораллил)-1H-пиразол-4-карбоновую кислоту (190,0 мг, 0,64 ммоль, 1,0 экв.) добавляли в ДМФ (2 мл), на ледяной бане добавляли DIPEA (246,1 мг, 1,90 ммоль, 3,0 экв.) и HATU (362,1 мг, 0,95 ммоль, 1,5 экв.) и смесь перемешивали на ледяной бане в течение 1 ч. В смесь добавляли 4-хлоранилин (89,1 мг, 0,70 ммоль, 1,1 экв.) и выдерживали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Концентрат растворяли в EA (60 мл) и для промывки последовательно использовали насыщенный водный раствор NaHCO3 (50 мл), насыщенный водный раствор NH4Cl (50 мл) и насыщенный солевой раствор (50 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (90,0 мг, выход: 34,7%).
Стадия 2: Синтез (E)-1-(2-аминометил-3-фтораллил)-N-4-хлорфенил-1H-пиразол-4-карбоксамида
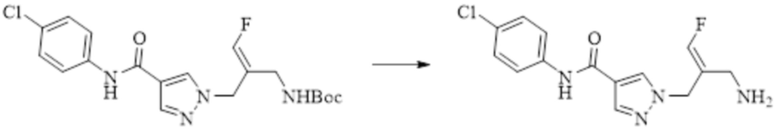
трет-Бутил (E)-(2-((4-((4-хлорфенил)карбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамат (90,0 мг, 0,22 ммоль) добавляли в EtOH (5,0 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (5,0 мл) и смесь выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли ДХМ (40 мл) и 15% водный раствор NaOH (40 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали при пониженном давлении и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (48,0 мг, выход: 70,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 9,96 (с, 1H), 8,40 (с, 1H), 8,04 (с, 1H), 7,75-7,73 (д, 2H), 7,41-7,38 (д, 2H), 7,09 (с, 0,5H), 6,87 (с, 0,5H), 4,82 (с, 2H), 3,09 (с, 2H), 1,81 (с, 2H).
Молекулярная формула: C14H14N4ClFO, молекулярная масса: 308,74, ЖХ-МС (полож., m/z)=309,09 [M+H]+.
Пример 26: (E)-1-(2-(Аминометил)-3-фтораллил)-N,N-диметил-1H-пиразол-4-карбоксамид (соединение B29)
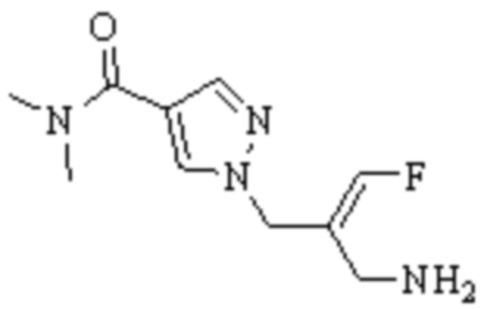
Стадия 1: Синтез трет-бутил (E)-(2-((4-(диметилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
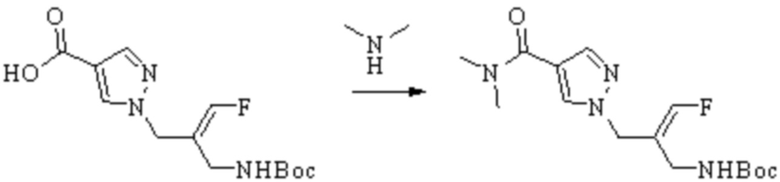
(E)-1-(2-((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-4-карбоновую кислоту (150,0 мг, 0,501 ммоль, 1,0 экв.) и DIPEA (272,0 мг, 2,105 ммоль, 4,2 экв.) растворяли в ДМФ (2,0 мл). В атмосфере азота при температуре 0°C к раствору добавляли HATU (286,0 мг, 0,752 ммоль, 1,5 экв.), перемешивали в течение 0,5 ч, добавляли раствор диэтиламина в метаноле (90,4 мг, 0,601 ммоль, 1,2 экв., 30%) и выдерживали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, в реакционный раствор добавляли этилацетат (40,0 мл) и смесь промывали последовательно насыщенным водным раствором карбоната натрия (4,0 мл), насыщенным водным раствором хлорида аммония (4,0 мл) и насыщенным водным раствором хлорида натрия (4,0 мл). Органическую фазу сушили над безводным сульфатом натрия и концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=120:1-30:1) с получением продукта (132,0 мг, выход: 80,7%).
Стадия 2: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N, N-диметил-1H-пиразол-4-карбоксамида
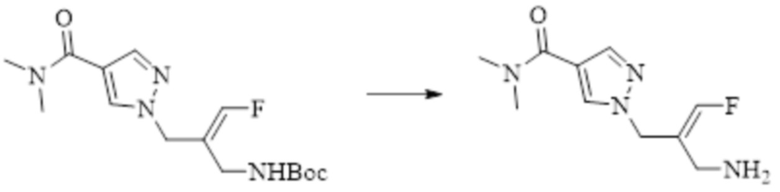
трет-Бутил(E)-(2-((4-(диметилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамат (132,0 мг, 0,404 ммоль, 1,0 экв.) растворяли в этаноле (1,0 мл). К раствору добавляли по каплям раствор хлорида водорода в этаноле (1,5 мл) и подвергали взаимодействию при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (37,0 мг, выход: 40,4%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,16 (с, 1H), 7,74 (с, 1H), 7,03-6,81 (д, 1H), 4,77 (с, 2H), 3,35-2,95 (м, 8H), 1,75 (с, 2H).
Молекулярная формула: C10H15FN4O, молекулярная масса: 226,26, ЖХ-МС (полож., m/z)=227,19 [M+H]+
Пример 27: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-(трет-бутил)-1H-пиразол-5-карбоксамида (соединение B30)
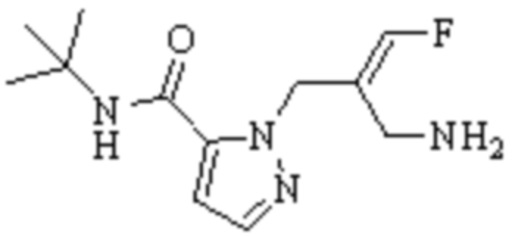
Стадия 1: Синтез метил (E)-1-(2-(аминометил)-3-фтораллил)-1H-пиразол-5-карбоксилата
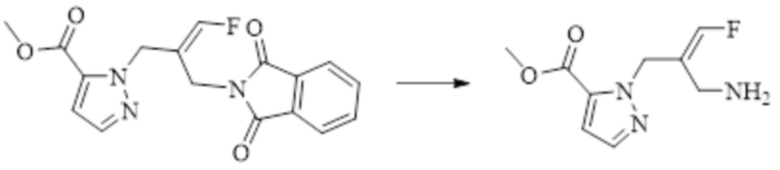
Метил (E)-1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-1H-пиразол-5-карбоксилат (772,5 мг, 2,25 ммоль, 1,0 экв.) добавляли в этанол (20 мл), добавляли гидразингидрат (337,5 мг, 6,75 ммоль, 3,0 экв.) и смесь перемешивали при комнатной температуре в течение 5 ч. После завершения реакции, как определялось данными ТСХ, добавляли ДХМ (150 мл) и воду (80 мл) и жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (441,6 мг сырого продукта).
Стадия 2: Синтез метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-5-карбоксилата
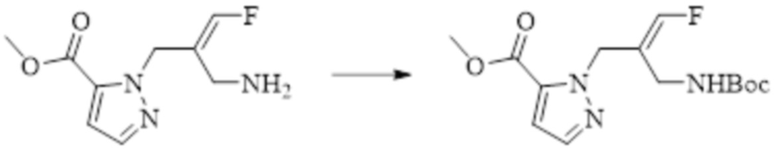
Метил (E)-1-(2-(аминометил)-3-хлораллил)-1H-пиразол-5-карбоксилат (441,6 мг сырого продукта) растворяли в ТГФ (7 мл). К раствору добавляли триэтиламин (290,0 мг, 2,86 ммоль) и ди-трет-бутил дикарбонат (541,9 мг, 2,48 ммоль) и перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как определялось данными ТСХ, к реакционному раствору добавляли EA (50 мл) и насыщенный водный раствор хлорида аммония (60 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=6:1-1:1) с получением продукта (176,6 мг, выход за две стадии: 27,2%).
Стадия 3: Синтез (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-5-карбоновой кислоты
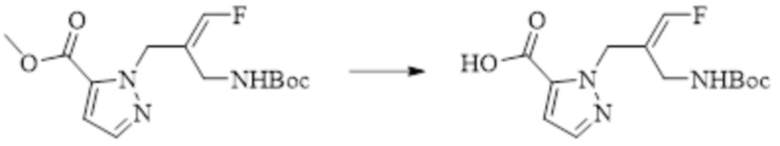
Метил (E)-1-(2-(((трет-бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-5-карбоксилат (176,6 мг, 0,56 ммоль, 1,0 экв.) растворяли в метаноле (2 мл). К раствору добавляли водный раствор NaOH (6 моль/л, 1 мл) и перемешивали при температуре 40°C в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли 5% водный раствор лимонной кислоты для достижения pH системы равным 4 и экстрагировали ДХМ (20 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением продукта (124,3 мг, выход: 74,1%).
Стадия 4: Синтез трет-бутил (E)-(2-((5-(трет-бутилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамата
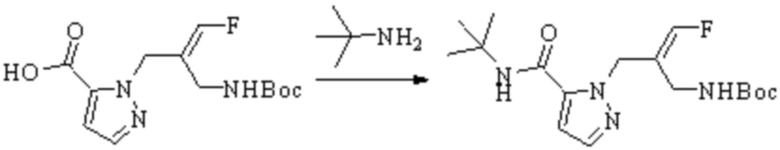
(E)-1-(2-(((трет-Бутоксикарбонил)амино)метил)-3-фтораллил)-1H-пиразол-5-карбоновую кислоту (124,3 мг, 0,41 ммоль, 1,0 экв.) добавляли в ДМФ (3 мл), на ледяной бане добавляли DIPEA (160,7 мг, 1,24 ммоль, 3,0 экв.) и HATU (236,6 мг, 0,62 ммоль, 1,5 экв.), перемешивали в течение 2 ч и затем добавляли трет-бутиламин (36,4 мг, 0,49 ммоль, 1,2 экв.). Выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли EA (50 мл) и насыщенный водный раствор карбоната натрия (50 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (100,0 мг, выход: 68,0%).
Стадия 5: Синтез (E)-1-(2-(аминометил)-3-фтораллил)-N-(трет-бутил)-1H-пиразол-5-карбоксамида
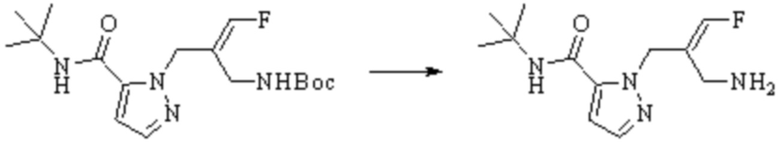
трет-Бутил (E)-(2-((5-(трет-бутилкарбамоил)-1H-пиразол-1-ил)метил)-3-фтораллил)карбамат (100,0 мг, 0,28 ммоль, 1,0 экв.) добавляли в EtOH (2,5 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (2,5 мл) и выдерживали при комнатной температуре и перемешивали в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и добавляли ДХМ (30 мл) и насыщенный водный раствор карбоната натрия (40 мл), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (23,0 мг, выход: 32,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,0 (с, 1H), 7,48 (д, 1H), 6,92 (с, 0,5H), 6,80-6,81 (д, 1H), 6,70 (с, 0,5H), 5,07 (д, 2H), 3,05-3,06 (д, 2H), 1,36 (с, 9H).
Молекулярная формула: C12H19FN4O, молекулярная масса: 254,31, ЖХ-МС (полож., m/z)=255,22 [M+H]+.
Пример 28: Синтез гидрохлорида (E)-1-(2-аминометил-3-фтораллил)-1H-пиррол-3-карбоксамида (соединение B31)
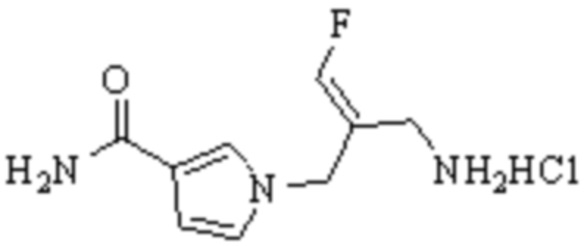
Стадия 1: Синтез трет-бутил (E)-(2-((3-карбамоил-1H-пиррол-1-ил)метил)-3-фтораллил)карбамата
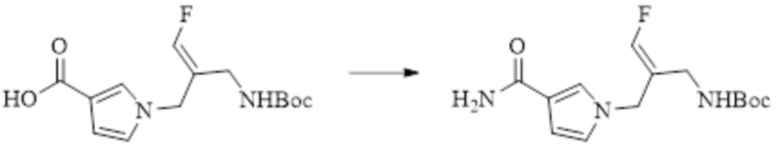
(E)-1-(2-(((трет-Бутоксикарбонил)амино)метил-3-фтораллил)-1H-пиррол-3-карбоновую кислоту (200,0 мг, 0,67 ммоль, 1,0 экв.) добавляли в ДМФ (2 мл), на ледяной бане добавляли DIPEA (259,9 мг, 2,01 ммоль, 3,0 экв.) и HATU (382,4 мг, 1,01 ммоль, 1,5 экв.) и смесь перемешивали в течение 1 ч. В смесь добавляли раствор аммиака в метаноле (5%, 685,1 мг, 2,01 ммоль, 3,0 экв.), выдерживали при комнатной температуре и перемешивали в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Концентрат растворяли в EA (60 мл) и для промывки последовательно использовали насыщенный водный раствор NaHCO3 (50 мл), насыщенный водный раствор NH4Cl (50 мл) и насыщенный солевой раствор (50 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (155,0 мг, выход: 77,8%).
Стадия 2: Синтез гидрохлорида (E)-1-(2-аминометил-3-фтораллил)-1H-пиррол-3-карбоксамида
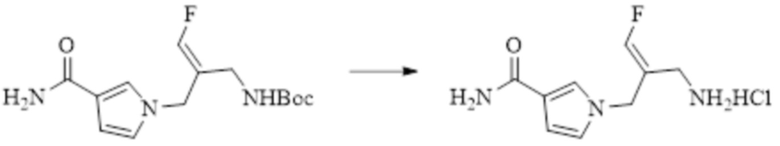
трет-Бутил (E)-(2-((3-карбамоил-1H-пиррол-1-ил)метил)-3-фтораллил) карбамат (155,0 мг, 0,52 ммоль) добавляли в EtOH (5,0 мл), на ледяной бане добавляли по каплям раствор хлорида водорода в этаноле (5,0 мл) и выдерживали при комнатной температуре и перемешивали в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, суспендировали в метил трет-бутиловом эфире в течение 2 ч и фильтровали. Слой на фильтре собирали с получением продукта (94,0 мг, выход: 77,2%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,54 (с, 3H), 7,64 (с, 2H), 7,41-7,40 (т, 1H), 7,31 (с, 0,5H), 7,11 (с, 0,5H), 6,88-6,87 (т, 1H), 6,50-6,49 (кв, 1H), 4,74-4,73 (д, 2H), 3,25-3,23 (д, 2H).
Молекулярная формула: C9H12N3FO, молекулярная масса: 197,21, ЖХ-МС (полож., m/z)=198,21 [M+H]+.
Пример 29: Получение гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A28) и гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-этил-1,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A29)
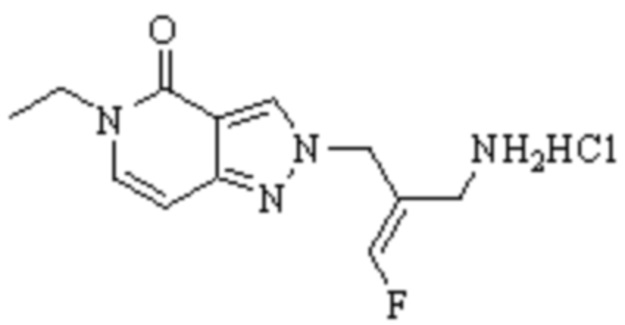 A28
A28 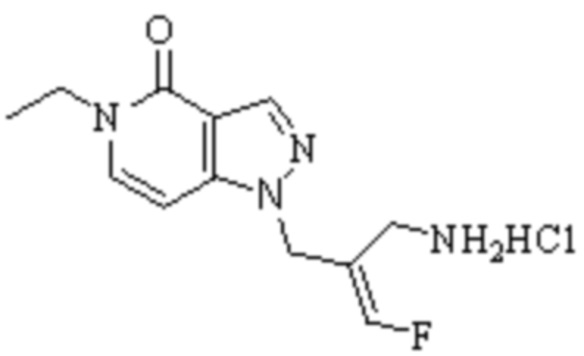 A29
A29
Стадия 1: Синтез 4-хлор-2-(4-метоксибензил)-2H-пиразоло[4,3-c]пиридина
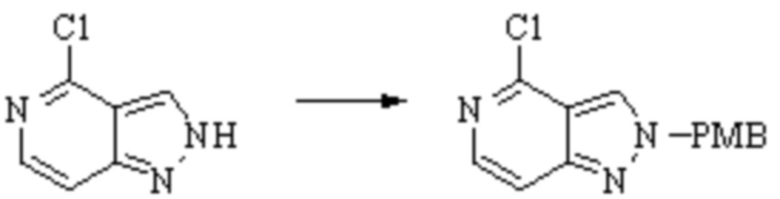
Соединения 4-хлор-2H-пиразоло[4,3-c]пиридин (5,0 г, 32,55 ммоль, 1,0 экв.), 4-метоксибензилхлорид (5,58 г, 35,81 ммоль, 1,1 экв.) и карбонат калия (8,98 г, 65,1 ммоль, 2,0 экв.) растворяли в ДМФ (30 мл) и подвергали взаимодействию при комнатной температуре в течение 1,5 ч. После того как не оставалось исходных веществ, как определялось данными ЖХ-МС, реакционный раствор выливали в воду (50 мл) и экстрагировали этилацетатом (80 мл × 2) и органическую фазу промывали водой (100 мл × 2), сушили и концентрировали с получением продукта (8,9 г, выход: 100%).
Стадия 2: Синтез промежуточного 2-(4-метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
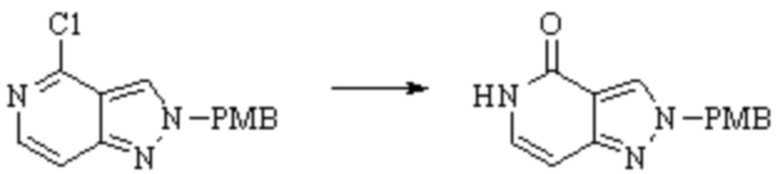
Промежуточный 4-хлор-2-(4-метоксибензил)-2H-пиразоло[4,3-c]пиридин (8,9 г, 32,51 ммоль, 1,0 экв.) растворяли в уксусной кислоте (80 мл) и воде (20 мл) и подвергали взаимодействию при температуре 100°C в течение 12 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении, сырой продукт суспендировали в метил трет-бутиловом эфире (100 мл) и фильтровали в вакууме с получением продукта (8,3 г, выход: 100%).
Стадия 3: Синтез 5-этил-2-(4-метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
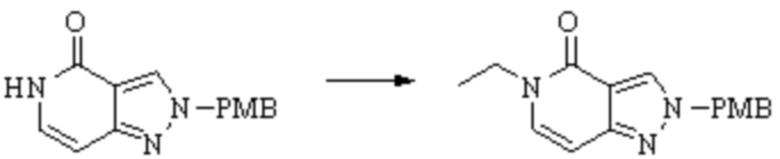
Промежуточный 2-(4-метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он (8,3 г, 32,51 ммоль, 1,0 экв.) и гидрид натрия (1,95 г, 48,76 ммоль, 1,5 экв.) растворяли в ДМФ (80 мл). Смесь перемешивали в течение 30 мин, добавляли по каплям йодэтан (7,6 г, 48,76 ммоль, 1,5 экв.) и подвергали взаимодействию при температуре 60°C в течение 30 мин. После завершения реакции, как определялось с помощью ЖХ-МС, в сосуд медленно добавляли воду (80 мл) и экстрагировали этилацетатом (100 мл × 4). Органическую фазу промывали водой (200 мл × 4), сушили и концентрировали при пониженном давлении и сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1) с получением продукта (5,0 г, выход: 54%).
Стадия 4: Синтез промежуточного 5-этил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
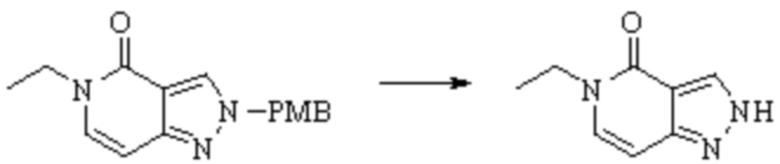
Промежуточный 5-этил-2-(4-метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он (5,0 г, 17,64 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (50 мл) и подвергали взаимодействию при температуре 75°C в течение 12 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении. Сырой продукт сначала очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1), затем суспендировали в метил трет-бутиловом эфире (10 мл) и фильтровали в вакууме с получением продукта (1,1 г, выход: 38%).
Стадия 5: Синтез (E)-2-(2-((5-этил-4-оксо-4,5-дигидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона и (E)-2-(2-((5-этил-4-оксо-4,5-дигидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
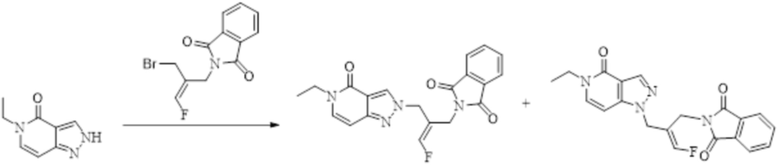
Промежуточный 5-этил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он (500 мг, 3,06 ммоль, 1,0 экв.) растворяли в ДМФ (3 мл). К раствору добавляли NaH (159 мг, 3,98 ммоль, 1,3 экв.), перемешивали в течение 30 мин и затем добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил) изоиндолин-1,3-дион (1,004 г, 3,37 ммоль, 1,1 экв.) в ДМФ (2 мл) и подвергали взаимодействию в течение 30 мин. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционную смесь добавляли воду (10 мл) и экстрагировали этилацетатом (20 мл × 2), затем жидкости разделяли. Органическую фазу промывали водой (20 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт суспендировали в метил трет-бутиловом эфире (10 мл) и фильтровали в вакууме с получением смеси (E)-2-(2-((5-этил-4-оксо-4,5-дигидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона и (E)-2-(2-((5-этил-4-оксо-4,5-дигидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-диона (390 мг).
Стадия 6: Гидрохлорид (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она и гидрохлорид (E)-1-(2-(аминометил)-3-фтораллил)-5-этил-1,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
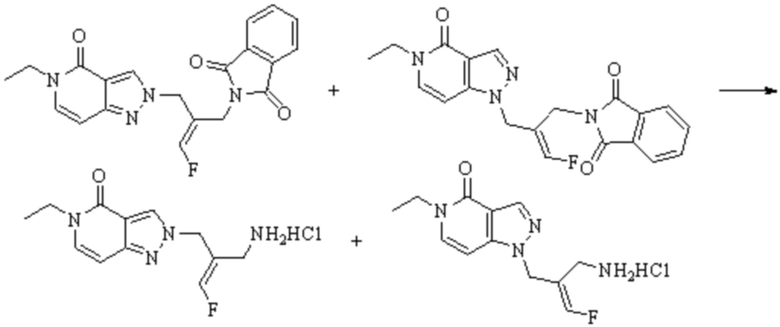
(E)-2-(2-((5-Этил-4-оксо-4,5-дигидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион и (E)-2-(2-((5-этил-4-оксо-4,5-дигидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (390 мг, 1,02 ммоль, 1,0 экв.) растворяли в EtOH (8 мл). К раствору добавляли гидразингидрат (178 мг, 3,57 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 30 мин. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме и фильтрат концентрировали при пониженном давлении. Сырой продукт сначала очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) и в полученный продукт добавляли метанол (1 мл) для растворения. В раствор затем по каплям добавляли раствор хлорида водорода в этаноле (0,015 мл), перемешивали в течение 5 мин и концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (дихлорметан : изопропанол : аммиак = 10:1:0,5) с получением гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-этил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она (18 мг, выход: 6,1%) с низким значением Rf, соответствующее гидрохлориду соединения A28.
1H ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,65 (с, 1H), 8,27 (с, 3H), 7,44 (с, 0,5H), 7,35-7,37 (д, 1H), 7,24 (с, 0,5H), 6,48-6,50 (м, 1H), 5,07-5,08 (д, 2H), 3,88-3,90 (м, 2H), 3,38-3,39 (м, 2H), 1,17-1,21 (м, 3H).
Молекулярная формула: C12H16ClFN4O, молекулярная масса: 286,74, ЖХ-МС (полож., m/z)=251,21 [M+H]+.
И (E)-1-(2-(аминометил)-3-фтораллил)-5-этил-1,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он гидрохлорид (13 мг, выход: 4,4%) с высоким значением Rf, соответствующее гидрохлориду соединения A29.
1H ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,38 (с, 3H), 8,10 (с, 1H), 7,62-7,64 (д, 1H), 7,36 (с, 0,5 H), 7,16 (с, 0,5H), 6,90-6,91 (д, 1H), 5,06-5,10 (д, 2H), 3,95-3,97 (м, 2H), 3,38-3,39 (м, 2H), 1,19-1,23 (м, 3H).
Молекулярная формула: C12H16ClFN4O, молекулярная масса: 286,74, ЖХ-МС (полож., m/z)=251,19 [M+H]+.
Пример 30: Синтез гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-этил-4,5-дигидропирроло[3,4-c]пиразол-6(1H)-она (соединение A31)
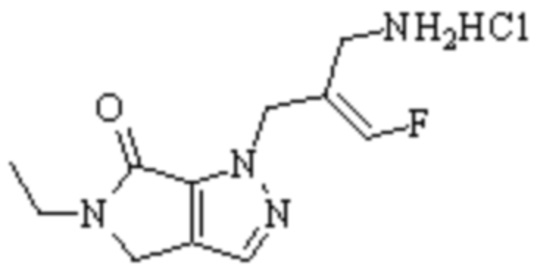
Стадия 1: Синтез этил (E)-2-(2-карбамоилгидразоно)пропионата
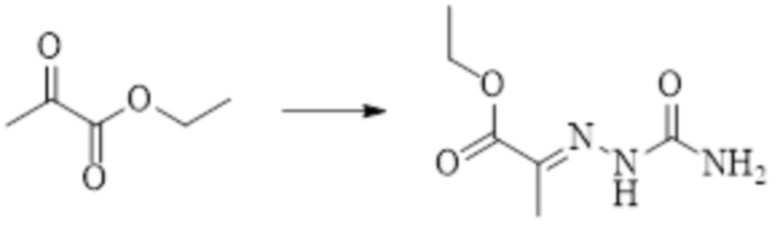
Соединения этил 2-оксопропионат (21 г, 0,18 моль, 1,0 экв.) и гидрохлорид семикарбазида (20 г, 0,18 моль, 1,0 экв.) растворяли в воде (150 мл). К раствору добавляли ацетат натрия (29 г, 0,35 моль, 1,9 экв.) и перемешивали при комнатной температуре в течение 12 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали в вакууме и слой на фильтре промывали небольшим количеством воды и сушили с получением продукта (29 г, выход: 94%), который напрямую использовали на следующей стадии.
Стадия 2: Синтез этил 4-формил-1H-пиразол-5-карбоксилата
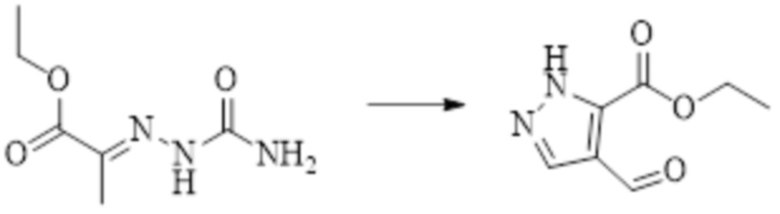
На ледяной бане в ДМФ (71,3 мл) добавляли по каплям оксихлорид фосфора (32,8 мл). После добавления ледяную баню удаляли и выдерживали при комнатной температуре и перемешивали в течение 30 мин. Реакционный раствор нагревали до температуры 40°C и добавляли этил (E)-2-(2-карбамоилгидразоно)пропионат (26,6 г, 0,154 моль, 1,0 экв.). После добавления смесь нагревали при температуре 80°C и подвергали взаимодействию в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор выливали в ледяную воду и при перемешивании pH раствора доводили до 10 с помощью водного раствора гидроксида натрия (массовая доля: 50%). Водный раствор нагревали до температуры 50°C до полного растворения, на ледяной бане pH раствора доводили до 7 с помощью концентрированной соляной кислоты и экстрагировали этилацетатом (2 × 200 мл). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали и сырой продукт суспендировали в ДХМ с получением продукта (14 г, выход: 66%).
Стадия 3: Синтез этил 4-формил-1-(4-метоксибензил)-1H-пиразол-5-карбоксилата
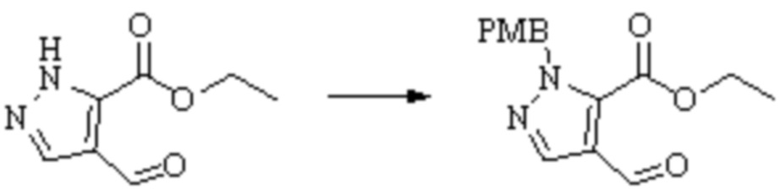
Промежуточный этил 4-формил-1H-пиразол-5-карбоксилат (10,5 г, 0,06 моль, 1,0 экв.) растворяли в ДМФ (60 мл). К раствору добавляли карбонат калия (25,9 г, 0,18 моль, 3,0 экв.) и п-метоксибензилхлорид (12,2 г, 0,078 моль, 1,3 экв.) и перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (200 мл) и экстрагировали этилацетатом (2 × 200 мл). Органическую фазу промывали насыщенным солевым раствором (2 × 200 мл), сушили над безводным сульфатом натрия, фильтровали в вакууме и концентрировали и сырой продукт очищали путем колоночной хроматографии на силикагеле (EA:PE=0-1:2) с получением продукта (12,5 г, выход: 69%).
Стадия 4: Синтез этил 4-((этиламино)метил)-1-(4-метоксибензил)-1H-пиразол-5-карбоксилата
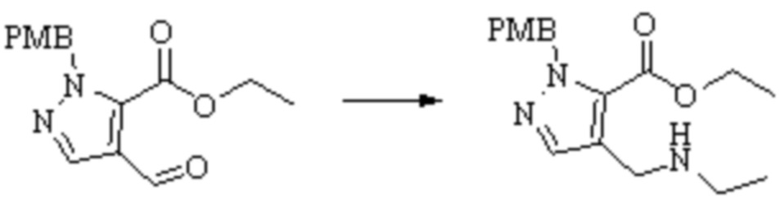
Гидрохлорид этиламина (4,0 г, 48,4 ммоль, 4,0 экв.) растворяли в MeOH (20 мл). К раствору добавляли триэтиламин (4,9 г, 48,4 ммоль, 4,0 экв.) и перемешивали в течение 10 мин. К реакционному раствору добавляли этил 4-формил-1-(4-метоксибензил)-1H-пиразол-5-карбоксилат (3,5 г, 12,1 ммоль, 1,0 экв.) и уксусную кислоту (0,5 мл) и перемешивали при комнатной температуре в течение 30 мин. К реакционному раствору добавляли цианоборгидрид натрия (2,3 г, 36,3 ммоль, 3,0 экв.) и подвергали взаимодействию при комнатной температуре в течение 12 ч. После завершения реакции, как определялось с помощью ЖХ-МС, pH раствора доводили до 10 насыщенным водным раствором бикарбоната натрия и экстрагировали ДХМ (2 × 30 мл). Органическую фазу промывали насыщенным солевым раствором (2 × 20 мл), сушили над безводным сульфатом натрия, фильтровали в вакууме и концентрировали с получением продукта (4,3 г, выход: 100%).
Стадия 5: Синтез 4-((этиламино)метил)-1-(4-метоксибензил)-1H-пиразол-5-карбоновой кислоты

Промежуточный этил 4-((этиламино)метил)-1-(4-метоксибензил)-1H-пиразол-5-карбоксилат (4,3 г сырого продукта, 13,5 ммоль, 1,0 экв.) растворяли в MeOH (20 мл) и воде (20 мл). К раствору добавляли моногидрат гидроксида лития (1,7 г, 40,6 ммоль, 3,0 экв.) и перемешивали при температуре 50°C в течение 3 ч. После завершения реакции, как определялось с помощью ЖХ-МС, pH раствора доводили до 2 с помощью 2 моль/л водного раствора соляной кислоты и лиофилизовали с получением продукта (5 г сырого продукта, выход: 100%).
Стадия 6: Синтез 5-этил-1-(4-метоксибензил)-4,5-дигидропирроло[3,4-c]пиразол-6(1H)-она
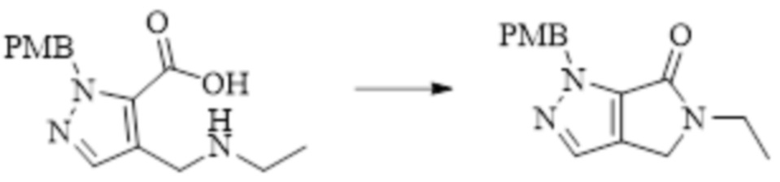
Промежуточную 4-((этиламино)метил)-1-(4-метоксибензил)-1H-пиразол-5-карбоновую кислоту (5,0 г сырого продукта, 17,28ммоль, 1,0 экв.) растворяли в ДМФ (30 мл). К раствору добавляли HATU (8,5 г, 22,46 ммоль, 1,9 экв.) и DIPEA (6,7 г, 51,84 ммоль, 3,0 экв.) и перемешивали в течение 1,5 ч. После завершения реакции, как определялось с помощью ЖХ-МС, в реакционную смесь добавляли воду (50 мл), экстрагировали этилацетатом (2 × 50 мл) и концентрировали и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (1,4 г, выход за три стадии: 30%).
Стадия 7: Синтез 5-этил-4,5-дигидропирроло[3,4-c]пиразол-6(1H)-она

Промежуточный 5-этил-1-(4-метоксибензил)-4,5-дигидропирроло[3,4-c]пиразол-6(1H)-он (1,4 г, 5,16 ммоль, 1,0 экв.) растворяли в ТФУ (8 мл). К раствору добавляли анизол (0,15 мл) и перемешивали при температуре 80°C в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали, и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (560 мг, выход: 72%).
Стадия 8: Синтез (E)-2-(2-((5-этил-6-оксо-5,6-дигидропирроло[3,4-c]пиразол-1(4H)-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
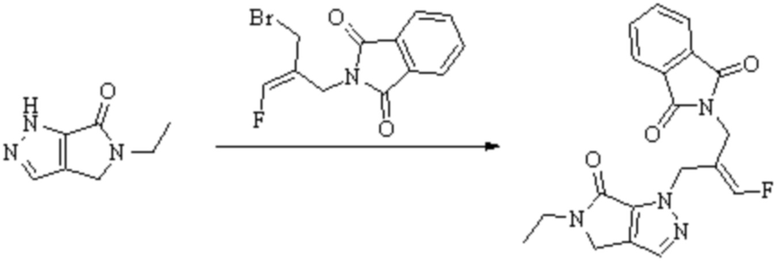
Промежуточный 5-этил-4,5-дигидропирроло[3,4-c]пиразол-6(1H)-он (560 мг, 3,7 ммоль, 1,0 экв.) растворяли в ДМФ (10 мл). К раствору добавляли карбонат калия (1,5 г, 11,1 ммоль, 3,0 экв.) и (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (1,3 г, 4,4 ммоль, 1,2 экв.) и перемешивали при комнатной температуре в течение 12 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (30 мл) и экстрагировали этилацетатом (2×30 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали в вакууме и концентрировали и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (300 мг, выход: 22%).
Стадия 9: Синтез гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-этил-4, 5-дигидропирроло[3,4-c]пиразол-6(1H)-она
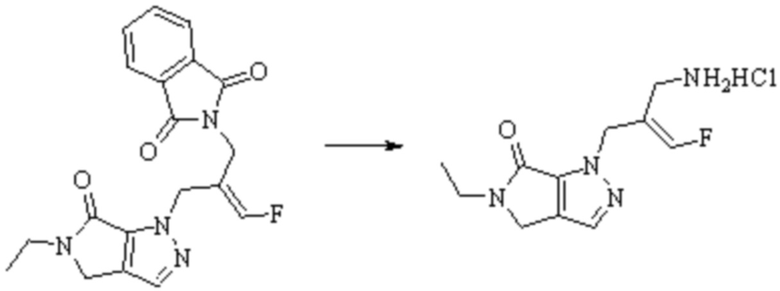
Промежуточный (E)-2-(2-((5-этил-6-оксо-5,6-дигидропирроло[3,4-c]пиразол-1(4H)-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (300 мг, 0,814 ммоль, 1,0 экв.) растворяли в EtOH (5 мл). К раствору добавляли гидразингидрат (203,8 мг, 4,072 ммоль, 5,0 экв.) и перемешивали при температуре 45°C в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры. Твердые продукты удаляли фильтрованием и фильтрат концентрировали. Твердые продукты опять удаляли фильтрованием и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=5:1). Полученное масло растворяли в ДХМ (3 мл) и к раствору по каплям добавляли раствор хлорида водорода в этаноле (1 мл). После завершения реакции, как определялось данными ТСХ, раствор концентрировали с получением продукта (90 мг, выход: 40%).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,35 (с, 3H), 7,54 (с, 1H), 7,31 (с, 0,5H), 7,11 (с, 0,5H), 4,99 (д, 2H), 4,23-4,29 (д, 2H), 3,46-3,47 (д, 4H), 1,16 (м, 3H).
Молекулярная формула: C11H16ClFN4O, молекулярная масса: 238,27, ЖХ-МС (m/z)=239,21 [M+H]+.
Пример 31: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-1-бром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A32)

Стадия 1: Синтез промежуточного 1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-диона
1-Циклопропилпиперазин-2,4-дион (50 г, 326,6 ммоль, 1 экв.) добавляли в 1,1-диметокси-N,N-диметилметиламин (42,8 г, 359 ммоль, 1,1 экв.) и смесь перемешивали при комнатной температуре в течение 0,5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор напрямую использовали на следующей стадии.
Стадия 2: Синтез промежуточного ((1-циклопропил-2,4-диоксопиперидин-3-илиден)метил)глицина
Раствор 1-циклопропил-3-((диметиламино)метилен)пиперидин-2,4-диона, полученного на предшествующей стадии, растворяли в этаноле (1000 мл). К раствору добавляли глицин (24,5 г, 326,6 ммоль, 1 экв.) и ацетат натрия (32,2 г, 391,8 ммоль, 1,2 экв.) и нагревали до температуры 50°C и подвергали взаимодействию в течение 6 ч. Когда реакция завершалась, осуществляли концентрирование с получением продукта, который напрямую использовали на следующей стадии.
Стадия 3: Синтез промежуточного 2-ацетил-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
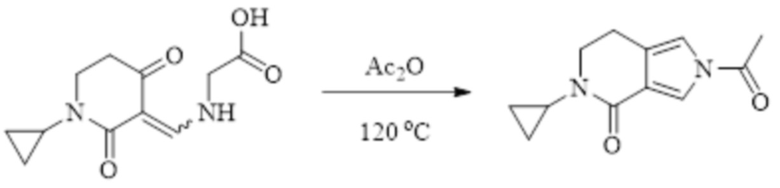
Сырой продукт ((1-циклопропил-2,4-диоксопиперидин-3-илиден)метил) глицин, полученный на предшествующей стадии, добавляли к уксусному ангидриду (1000 мл), смесь нагревали до температуры 120°C и подвергали взаимодействию в течение 5 ч. После завершения реакции уксусный ангидрид удаляли упариванием, концентрат выливали в насыщенный водный раствор бикарбоната натрия и pH доводили до нейтрального. Экстрагировали этилацетатом (300 мл × 4). Органические фазы объединяли, сушили и концентрировали с получением продукта (рассчитано в соответствии с теоретическим выходом), который напрямую использовали на следующей стадии.
Стадия 4: Синтез промежуточного 5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
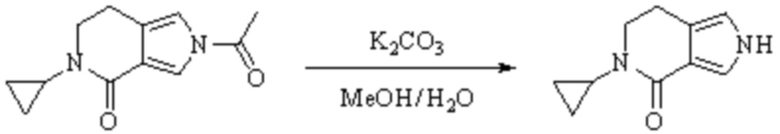
Сырой продукт 2-ацетил-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он растворяли в метаноле (300 мл). К раствору добавляли раствор карбонат калия (33,9 г, 464,3 ммоль, 1,5 экв.) в воде (300 мл) и перемешивали при комнатной температуре в течение 20 мин. После завершения реакции, как определялось данными ТСХ, осуществляли концентрирование для удаления метанола, pH доводили до слабокислотного и экстрагировали этилацетатом. Органические фазы объединяли, сушили и концентрировали с получением сырого продукта (37,8 г). Сырой продукт подвергали колоночной хроматографии на силикагеле (этилацетат:петролейный эфир, 1:1-1:0, об./об.) с получением продукта (13,05 г, выход за четыре стадии: 21,4%).
Стадия 5: Синтез промежуточного 1-бром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
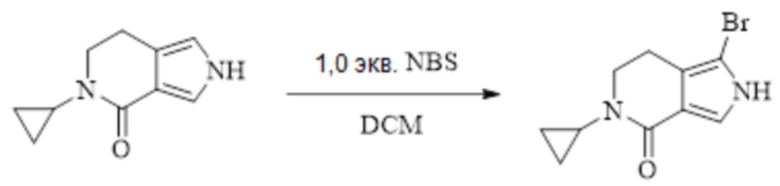
5-Циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (3 г, 17 ммоль, 1 экв.) растворяли в ДХМ (50 мл). К раствору партиями добавляли NBS (3 г, 17 ммоль, 1 экв.) на водяной бане со льдом подвергали взаимодействию при температуре 0°C в течение 30 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор последовательно промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органическую фазу сушили и концентрировали с получением сырого продукта (4,47 г), который подвергали колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 3:1, об./об.) с получением продукта (2,82 г, выход: 65,6%).
Стадия 6: Синтез промежуточного трет-бутил (E)-(2-((1-бром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамата
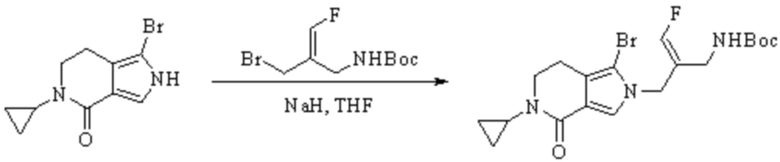
1-Бром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (2,14 г, 8,37 ммоль, 1 экв.) растворяли в безводном ТГФ (40 мл). На водяной бане со льдом медленно добавляли партиями NaH (670 мг, 16,74 ммоль). После добавления смесь перемешивали при температуре 0°C в течение 30 мин и добавляли трет-бутил (E)-(2-(бромметил)-3-фтораллил)карбамат (2,34 г, 8,74 ммоль, 1,05 экв.) и подвергали взаимодействию при комнатной температуре в течение 40 ч. После завершения реакции, как определялось данными ТСХ, насыщенный водный раствор хлорида аммония добавляли для гашения реакции. Органическую фазу промывали водой и насыщенным солевым раствором последовательно, сушили и концентрировали с получением сырого продукта (3,89 г). Сырой продукт подвергали колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 4:1-3:1, об./об.) с получением продукта (1,77 г, выход: 47,8%).
Стадия 7: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-1-бром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
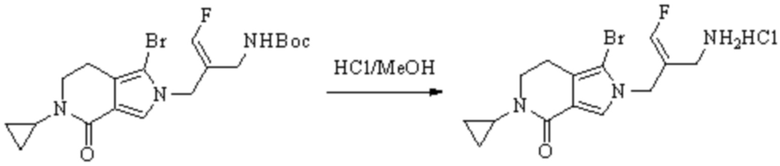
трет-Бутил (E)-(2-((1-бром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (350 мг, 0,791 ммоль, 1 экв.) растворяли в растворе хлорида водорода в этаноле (12 мл) и смесь перемешивали до завершения реакции. Реакционный раствор концентрировали и лиофилизовали с получением продукта (134 мг, выход: 49,6%).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,41 (шир. с, 3H), 7,55 (с, 1H), 7,30-7,02 (д, 1H), 4,76 (д, 2H), 3,42-3,47 (м, 2H), 3,29-3,30 (м, 2H), 2,56-2,65 (м, 3H), 0,71-0,77 (м, 2H), 0,56-0,60 (м, 2H).
Молекулярная формула: C14H17BrFN3O, молекулярная масса: 342,21, ЖХ-МС (m/z)=343,92 [M+H]+.
Пример 32: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-1,3-дибром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A33)
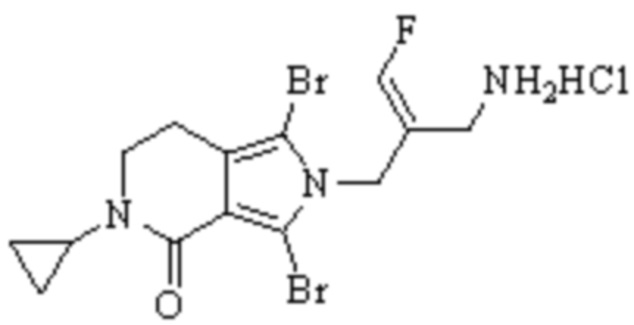
Стадия 1: Синтез 1,3-дибром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
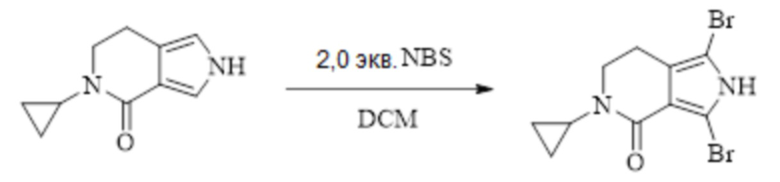
5-Циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (3 г, 17 ммоль, 1 экв.) растворяли в ДХМ (50 мл). К раствору на водяной бане со льдом партиями добавляли NBS (6 г, 34 ммоль, 2 экв.) и подвергали взаимодействию при комнатной температуре в течение 20 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор последовательно промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органическую фазу сушили и концентрировали с получением сырого продукта (6,03 г), который подвергали колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 5:1-4:1, об./об.) с получением продукта (3,33 г, выход: 58,4%).
Стадия 2: Синтез трет-бутил (E)-(2-((1,3-дибром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамата
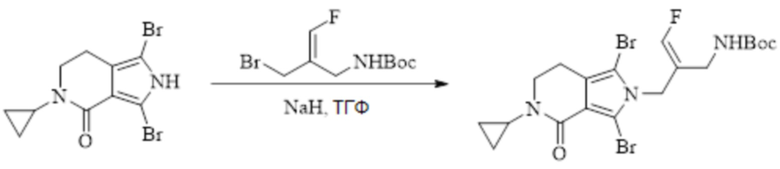
1,3-Дибром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (3,83 г, 11,4 ммоль, 1,0 экв.) растворяли в безводном ТГФ (60 мл). На водяной бане со льдом медленно добавляли NaH (912 мг, 22,8 ммоль, 2,0 экв.). После добавления смесь перемешивали при температуре 0°C в течение 30 мин и добавляли трет-бутил (E)-(2-(бромметил)-3-фтораллил)карбамат (3,06 г, 11,4 ммоль, 1,0 экв.) и подвергали взаимодействию при комнатной температуре в течение 40 ч. После завершения реакции, как определялось данными ТСХ, для гашения реакции добавляли насыщенный водный раствор хлорида аммония. Органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили и концентрировали с получением сырого продукта (6,17 г). Сырой продукт подвергали колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 4:1-2:1, об./об.) с получением продукта (3,98 г, выход: 67,5%).
Стадия 3: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-1,3-дибром-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
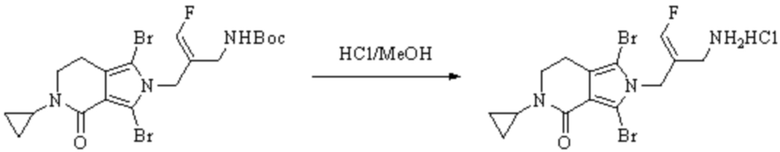
трет-Бутил (E)-(2-((1,3-дибром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (400 мг, 0,767 ммоль, 1 экв.) растворяли в растворе хлорида водорода в этаноле (12 мл) и смесь перемешивали до завершения реакции. Реакционный раствор концентрировали и лиофилизовали с получением продукта (103 мг, выход: 31,9%).
1H-ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,53 (шир. с, 3H), 6,58-6,85 (д, 1H), 4,81-4,82 (д, 2H), 3,43-3,45 (м, 4H), 2,60-2,62 (м, 3H), 0,71-0,78 (м, 2H), 0,56-0,61 (м, 2H).
Молекулярная формула: C14H16Br2FN3O, молекулярная масса: 421,11, ЖХ-МС (m/z)=421,94 [M+H]+.
Пример 33: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1-метил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A35)

Стадия 1: Синтез промежуточного трет-бутил (E)-(2-((5-циклопропил-1-метил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамата
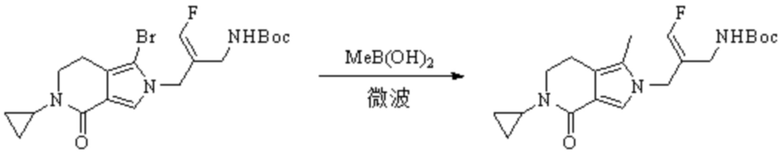
трет-Бутил (E)-(2-((1-бром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (500 мг, 1,13 ммоль, 1 экв.), фосфат калия (720 мг, 3,39 ммоль, 3 экв.), метилбороновую кислоту (271 мг, 4,52 ммоль, 4 экв.) и трициклогексилфосфин (31,7 мг, 0,113 ммоль, 0,1 экв.) помещали в пробирку для микроволнового воздействия и добавляли толуол (30 мл). После барботирования N2 в течение 5 мин добавляли Pd2(dba)3 (52 мг, 0,0565 ммоль, 0,05 экв.). Реакцию проводили при воздействии микроволнового воздействия при температуре 120°C в течение 1 ч. После завершения реакции реакционный раствор концентрировали, сырой продукт очищали обращенно фазовой колоночной хроматографией (CH3CN:H2O=1:4) и лиофилизовали с получением продукта (230 мг, выход: 53,9%).
Стадия 2: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1-метил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
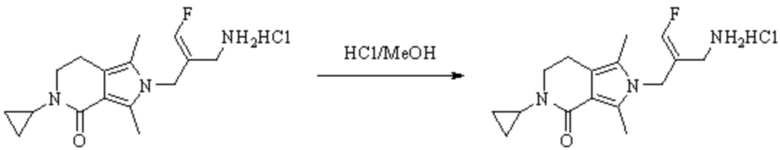
трет-Бутил (E)-(2-((5-циклопропил-1-метил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (230 мг, 0,609 ммоль, 1 экв.) растворяли в растворе хлорида водорода в этаноле (10 мл) и смесь перемешивали до завершения реакции. Реакционный раствор концентрировали и лиофилизовали с получением продукта (134,7 мг, выход: 79,7%).
1H-ЯМР (300 МГц, ДМСО-d6) δ (м.д.): 8,62 (шир. с, 3H), 7,33 (с, 1H), 6,87-7,14 (д, 1H), 4,74 (с, 2H), 3,37-3,41 (м, 2H), 3,24 (м, 2H), 2,52-2,63 (м, 3H), 2,07 (с, 3H), 0,68-0,72 (м, 2H), 0,55-0,58 (м, 2H).
Молекулярная формула: C15H21ClFN3O, молекулярная масса: 277,34, ЖХ-МС (m/z)=278,09 [M+H]+.
Пример 34: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,3-диметил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A36)
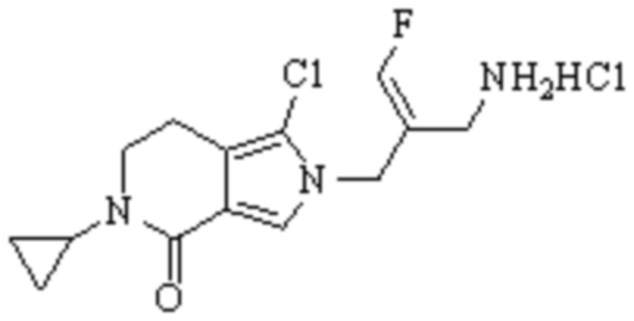
Стадия 1: Синтез трет-бутил (E)-(2-((5-циклопропил-1-метил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамата

трет-Бутил (E)-(2-((1-бром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (500 мг, 0,96 ммоль, 1 экв.), метилбороновую кислоту (230 мг, 3,84 ммоль, 4 экв.) и фосфат калия (1,63 г, 7,68 ммоль, 8 экв.) помещали в колбу, добавляли 1,4-диоксан (12 мл), заполняли азотом после вакуумирования и добавляли Pd(PPh3)4 (60 мг, 0,05 ммоль, 0,05 экв.). Смесь перемешивали в атмосфере азота в течение 5 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали и добавляли этилацетат. Для промывки последовательно использовали воду и насыщенным солевым раствором, и после сушки и концентрирования получали сырой продукт (570 мг). Сырой продукт очищали обращенно-фазовой колоночной хроматографией (CH3CN:H2O=1:3) с получением продукта (170 мг, выход: 45,2%).
Стадия 2: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,3-диметил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
Гидрохлорид (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,3-диметил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (170 мг, 0,434 ммоль, 1 экв.) растворяли в растворе хлорида водорода в этаноле (10 мл) и раствор перемешивали до завершения реакции. Реакционный раствор концентрировали и лиофилизовали с получением продукта (102,8 мг, выход: 81,6%).
1H-ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,62 (шир. с, 3H), 5,85-6,12 (д, 1H), 4,66 (с, 2H), 3,46 (с, 2H), 3,33-3,46 (м, 2H), 2,51-2,58 (м, 3H), 2,42 (с, 3H), 2,05 (с, 3H), 0,69-0,71 (м, 2H), 0,54 (м, 2H).
Молекулярная формула: C16H23ClFN3O, молекулярная масса: 291,37, ЖХ-МС (m/z)=292,20 [M+H]+
Пример 35: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-1-хлор-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она (соединение A42)
Стадия 1: Синтез 1-хлор-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
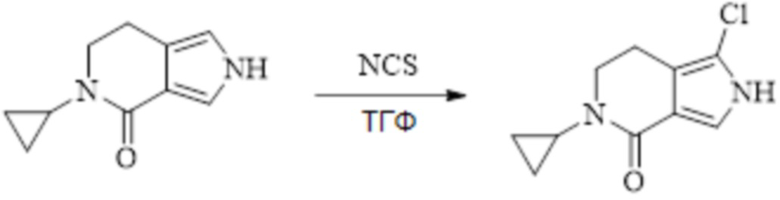
5-Циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (600 г, 3,4 ммоль, 1 экв.) растворяли в ТГФ (20 мл). На водяной бане со льдом к раствору партиями добавляли NCS (455 мг, 3,4 ммоль, 1 экв.) и подвергали взаимодействию при комнатной температуре в течение 30 мин. После завершения реакции, как определялось данными ТСХ, ТГФ удаляли упариванием при пониженном давлении, добавляли ДХМ (30 мл) и для осуществления промывки последовательно использовали воду (30 мл) и насыщенный солевой раствор (30 мл). Органическую фазу сушили и концентрировали с получением сырого продукта (820 мг). Сырой продукт очищали путем колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=3:1, 1:1, об./об.) с получением продукта (540 мг, выход: 75,4%).
Стадия 2: Синтез трет-бутил (E)-(2-((1-хлор-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамата
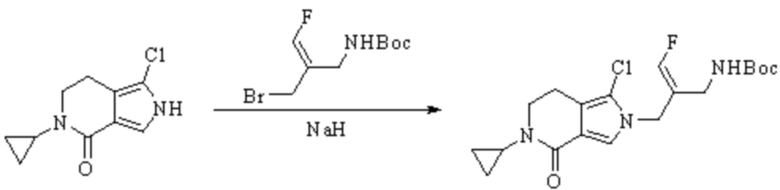
1-Хлор-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-он (500 мг, 2,37 ммоль, 1 экв.) растворяли в безводном ТГФ (20 мл). На водяной бане со льдом медленно партиями добавляли NaH (190 мг, 4,74 ммоль). После добавления смесь перемешивали при температуре 0°C в течение 30 мин и добавляли трет-бутил (E)-(2-(бромметил)-3-фтораллил)карбамат (668 мг, 2,49 ммоль, 1,05 экв.) и подвергали взаимодействию при комнатной температуре в течение 23 ч. После завершения реакции, как определялось данными ТСХ, для гашения реакции добавляли насыщенный водный раствор хлорида аммония. Органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили и концентрировали с получением сырого продукта (1,03 г) и сырой продукт очищали путем колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 2,5:1-2:1, об./об.) с получением продукта (320 мг, выход: 34%).
Стадия 3: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-1-хлор-5-циклопропил-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
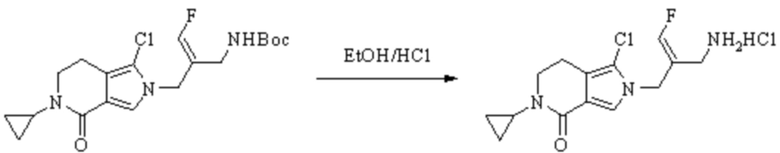
трет-Бутил (E)-(2-((1-хлор-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (320 мг, 0,804 ммоль, 1 экв.) растворяли в растворе хлорида водорода в этаноле (10 мл) и смесь перемешивали при комнатной температуре в течение 4 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали с получением сырого продукта (200 мг). Сырой продукт очищали хроматографией на колонке C-18 (CH3CN:H2O=1:4) и лиофилизовали с получением продукта (34 мг, выход:14,2%).
1H-ЯМР (300 МГц, ДМСО-d6) δ (м.д.): 8,41 (шир. с, 3H), 7,55 (с, 1H), 7,02-7,30 (д, 1H), 4,76 (с, 2H), 3,42-3,47 (м, 2H), 3,29-3,30 (м, 2H), 2,61-2,66 (м, 1H), 2,55-2,59 (м, 2H), 0,70-0,77 (м, 2H), 0,52-0,59 (м, 2H).
Молекулярная формула: C14H17ClFN3O, молекулярная масса: 297,10, ЖХ-МС (m/z)=297,74 [M+H]+.
Пример 36: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A41) и гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A44)
Стадия 1: Синтез 5-циклопропил-2-(4-метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
2-(4-Метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он (6,4 г, 32,51 ммоль, 1,0 экв.), циклопропилбороновую кислоту (10,77 г, 125,35 ммоль, 5,0 экв.), ацетат меди (13,66 г, 75,21 ммоль, 3,0 экв.) и пиридин (9,92 г, 125,35 ммоль, 5,0 экв.) растворяли в толуоле (150 мл) и подвергали взаимодействию при температуре 100°C на воздухе в течение 47 ч. Реакционный раствор концентрировали при пониженном давлении и сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1) с получением продукта (2,0 г, выход: 27%).
Стадия 2: Синтез 5-циклопропил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
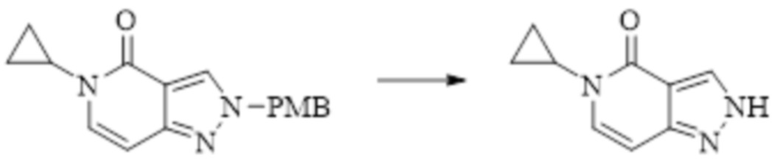
5-Циклопропил-2-(4-метоксибензил)-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он (2,0 г, 6,77 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (20 мл) и подвергали взаимодействию при температуре 75°C в течение 14 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:50) с получением продукта (1,0 г, выход: 84%).
Стадия 3: Синтез (E)-2-(2-((5-циклопропил-4-оксо-4,5-дигидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона и (E)-2-(2-((5-циклопропил-4-оксо-4,5-дигидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-диона

5-Циклопропил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-он (1,0 г, 5,71 ммоль, 1,0 экв.), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (1,87 г, 6,28 ммоль, 1,1 экв.), карбонат калия (867 мг, 6,28 ммоль, 1,1 экв.) и TBAB (184 мг, 0,57 ммоль, 0,1 экв.) растворяли в абсолютном этаноле (8 мл) подвергали взаимодействию в течение 19 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1) с получением смеси (E)-2-(2-((5-циклопропил-4-оксо-4,5-дигидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона и (E)-2-(2-((5-циклопропил-4-оксо-4,5-дигидро-1H-пиразоло[4,3-c]пиридин-1-ил)метил)-3-фтораллил)изоиндолин-1,3-диона (1,8 г, выход: 80%).
Стадия 4: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она и гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она
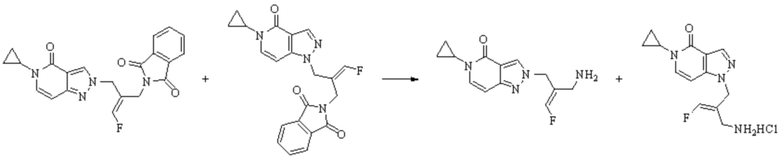
Смесь (1,4 г, 3,57 ммоль, 1,0 экв.), полученную на предшествующей стадии, и гидразингидрат (335 мг, 5,35 ммоль, 1,5 экв.) растворяли в EtOH (20 мл) подвергали взаимодействию при температуре 80°C в течение 20 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме и фильтрат концентрировали при пониженном давлении. Сырой продукт растворяли в дихлорметане, раствор фильтровали в вакууме и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (дихлорметан : изопропанол : аммиака = 10:1:0,5) с получением (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-2,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она с низким значением Rf (350 мг, выход: 37%), представляющее собой соединение A41.
1H ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,55 (с, 1H), 6,87-7,02 (д, 1H), 4,91 (с, 2H), 3,18 (с, 1H), 3,05 (с, 2H), 1,60 (с, 2H), 0,96-0,97 (д, 2H), 0,79 (с, 2H).
Молекулярная формула: C13H15FN4O, молекулярная масса: 262,29, ЖХ-МС (полож., m/z)=263,13 [M+H]+.
Продукт с высоким значением Rf растворяли в этаноле. К раствору добавляли раствор хлорида водорода в этаноле, концентрировали при пониженном давлении и лиофилизовали с получением гидрохлорида (E)-1-(2-(аминометил)-3-фтораллил)-5-циклопропил-1,5-дигидро-4H-пиразоло[4,3-c]пиридин-4-она (220 мг, выход: 20%), представляющее собой соединение A44.
1H ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,48 (с, 3H), 8,08 (с, 1H), 7,44-7,46 (д, 1H), 7,35 (с, 0,5H), 7,08 (с, 0,5H), 6,86-6,88 (д, 1H), 5,10-5,11 (д, 2H), 3,36 (с, 2H), 3,22-3,26 (м, 1H), 0,98-1,00 (д, 2H), 0,81-0,84 (м, 2H).
Молекулярная формула: C13H15FN4O, молекулярная масса: 262,29, ЖХ-МС (полож., m/z)=263,10 [M+H]+.
Пример 37: Синтез (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(морфолино)метанона (соединение C1)
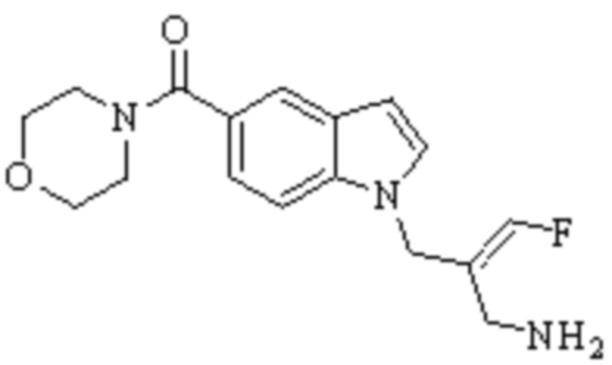
Стадия 1: Синтез (Z)-2-(3-фтор-2-((5-(морфолин-4-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-диона
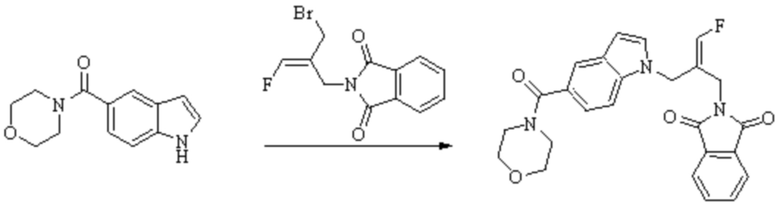
(1H-Индол-5-ил)(морфолино)метанон (1,00 г, 4,34 ммоль, 1 экв.) растворяли в ДМФ (10 мл) и раствор охлаждали до температуры 0°C. К раствору в атмосфере азота добавляли NaH (массовая доля: 60%, 0,19 г, 4,77 ммоль, 1,1 экв.) и перемешивали в течение 30 мин в атмосфере N2. К раствору по каплям добавляли раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-диона (1,55 г, 5,21 ммоль, 1,2 экв.) в ДМФ (10 мл), и выдерживали при комнатной температуре в течение ночи. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (60 мл) и экстрагировали EA (80 мл × 3). Органические фазы объединяли, опять промывали водой, сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=150:1) с получением продукта (1,42 г, выход: 73,2%).
Стадия 2: Синтез (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(морфолино)метанона
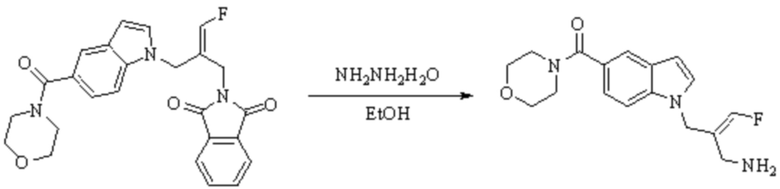
Промежуточный (Z)-2-(3-фтор-2-((5-(морфолин-4-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-дион (1,42 г, 3,17 ммоль, 1 экв.) растворяли в EtOH (35 мл). К раствору добавляли 85% гидразингидрата (0,65 г, 11,08 ммоль, 3,5 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 2 ч. После завершения реакции, как определялось данными ТСХ, фильтровали в вакууме и концентрировали. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (0,13 г, выход: 13,1%).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 7,64-7,65 (т, 2H), 7,59-7,60 (д, 1H), 7,20-7,22 (м, 1H), 7,04-7,25 (д, J=84 Гц, 1H), 6,55-6,56 (д, 1H), 4,96 (д, 2H), 3,60 (с, 4H), 3,52 (с, 4H), 3,17 (с, 2H), 3,12 (д, 2H).
Молекулярная формула: C17H20FN3O2, молекулярная масса: 317,36, ЖХ-МС (полож., m/z)=318,19 [M+H]+.
Пример 38: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(азетидин-1-ил)метанона (соединение C13)
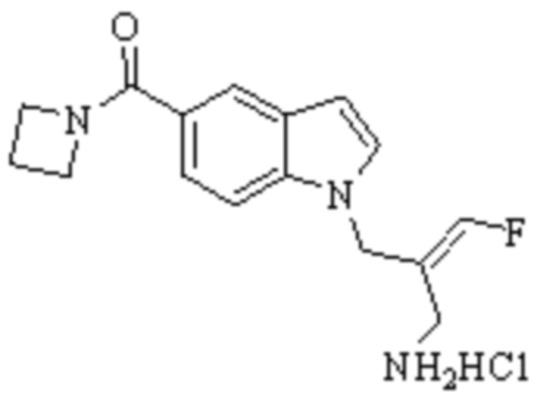
Стадия 1: Синтез азетидин-1-ил(1H-индол-5-ил)метанона
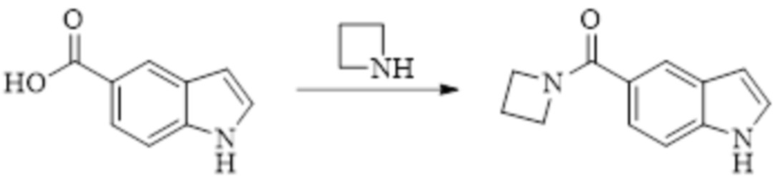
Соединения 1H-индол-5-карбоновую кислоту (1 г, 6,2 ммоль, 1,0 экв.) и DIPEA (2,4 г, 18,6 ммоль, 3,0 экв.) растворяли в N, N-диметилацетамиде (5 мл), и после вакуумирования заполняли азотом. Раствор охлаждали до температуры 0°C, добавляли HATU (3,5 г, 9,3 ммоль, 1,5 экв.) и подвергали взаимодействию в течение 0,5 ч. К реакционному раствору добавляли азетидин (708,6 мг, 12,41 ммоль, 2,0 экв.) и подвергали взаимодействию в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, в реакционную смесь добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:100-1:50). К полученному твердому продукту добавляли небольшое количество этилацетат и фильтровали, и слой на фильтре сушили с получением продукта (1,1 г, выход: 88,7%).
Стадия 2: Синтез (Z)-2-(2-((5-(азетидин-1-карбонил)-1H-индол-1-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
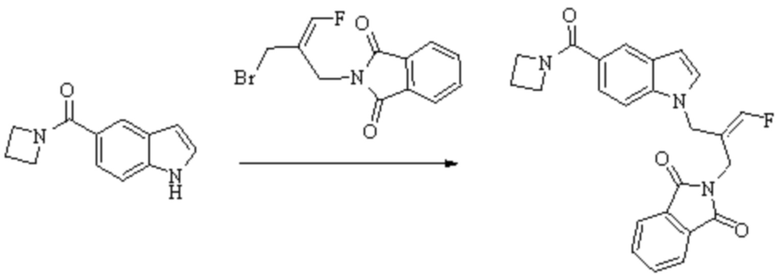
Промежуточный азетидин-1-ил(1H-индол-5-ил)метанон (500 мг, 2,5 ммоль, 1,0 экв.) растворяли в N, N-диметилацетамиде (5 мл). После вакуумирования заполняли азотом и раствор охлаждали до температуры 0°C, добавляли гидрид натрия (массовая доля: 60%, 109,8 мг, 2,75 ммоль, 1,1 экв.) и перемешивали в течение 0,5 ч. В смесь добавляли (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (894,3 мг, 3 ммоль, 1,2 экв.) и подвергали взаимодействию в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, к реакционному раствору добавляли насыщенный водный раствор хлорида аммония (50 мл) и воду (50 мл), перемешивали в течение 10 мин и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали водой (50 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:100-1:60) с получением продукта (643 мг, выход: 64,3%).
Стадия 3: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(азетидин-1-ил)метанона
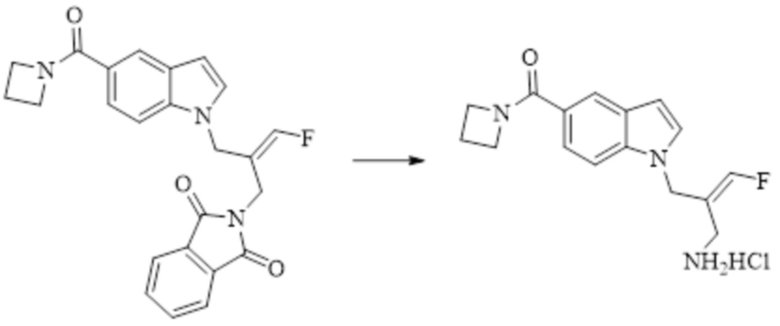
Промежуточный (Z)-2-(2-((5-(азетидин-1-карбонил)-1H-индол-1-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (643 мг, 1,59 ммоль, 1,0 экв.) растворяли в EtOH (10 мл). К раствору добавляли 80%-ный гидразингидрат (348 мг, 5,56 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Фильтрат концентрировали, суспендировали в дихлорметане (10 мл) и фильтровали, и полученный таким образом фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением маслянистой жидкости (260 мг). В маслянистую жидкость добавляли дихлорметан (5 мл). В смесь добавляли 20%-ный раствор хлорида водорода в этаноле (104 мг) по каплям, перемешивали в течение 10 мин и концентрировали при пониженном давлении с получением продукта (238 мг, выход: 46,2%).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 7,88 (д, 1H), 7,67-7,64 (д, 2H), 7,47-7,43 (д, 1H), 7,38 (с, 0,5H), 7,11 (с, 0,5H), 6,60-6,59 (с, 1H), 5,04-5,03 (с, 2H), 4,34 (м, 2H), 4,06 (м, 2H), 3,23-3,17 (м, 4H), 2,31-2,21 (м, 2H).
Молекулярная формула: C16H19ClFN3O, молекулярная масса: 323,8, ЖХ-МС (полож., m/z)=287,65 [M+H]+.
Пример 39: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(4-метоксипиперидин-1-ил)метанона (соединение C14)
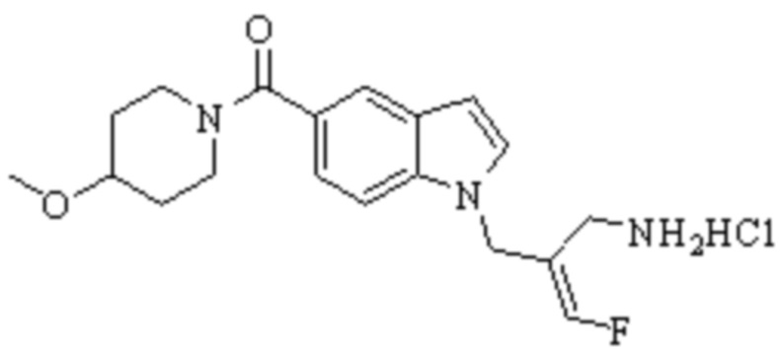
Стадия 1: Синтез (1H-индол-5-ил)(4-метоксипиперидин-1-ил)метанона
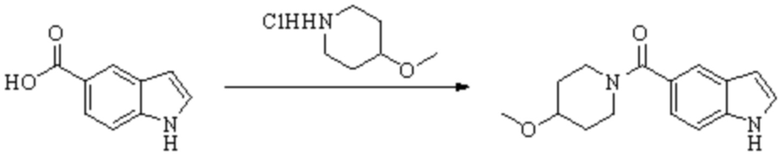
Соединения 1H-индол-5-карбоновую кислоту (1 г, 6,2 ммоль, 1,0 экв.) и DIPEA (2,4 г, 18,6 ммоль, 3,0 экв.) растворяли в N,N-диметилацетамиде (5 мл) и после вакуумирования заполняли азотом. Раствор охлаждали до температуры 0°C, добавляли HATU (3,5 г, 9,3 ммоль, 1,5 экв.) и подвергали взаимодействию в течение 0,5 ч. К реакционному раствору добавляли гидрохлорид 4-метоксипиперидина (1,88 г, 12,41 ммоль, 2,0 экв.) и подвергали взаимодействию в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, к реакционному раствору добавляли насыщенный хлорида аммония (50 мл) и воду (50 мл), перемешивали в течение 10 мин и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали водой (50 мл), сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (1,6 г, выход: 100%).
Стадия 2: Синтез (Z)-2-(3-фтор-2-((5-(4-метоксипиперидин-1-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-диона
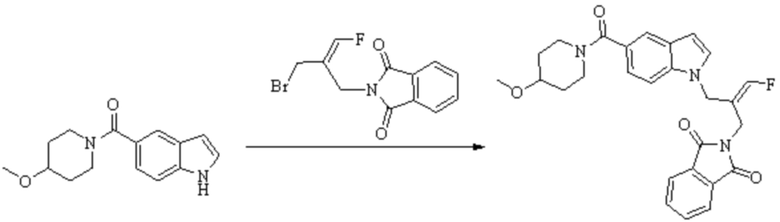
Промежуточный (1H-индол-5-ил)(4-метоксипиперидин-1-ил)метанон (500 мг, 1,94 ммоль, 1,0 экв.) растворяли в N, N-диметилацетамиде (5 мл). После вакуумирования заполняли азотом и раствор охлаждали до температуры 0°C, добавляли гидрид натрия (массовая доля: 60%, 85,2 мг, 2,13 ммоль, 1,1 экв.) и перемешивали в течение 0,5 ч. В смесь добавляли (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (694 мг, 2,33 ммоль, 1,2 экв.) и подвергали взаимодействию в течение 18 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, к реакционному раствору добавляли насыщенный водный раствор хлорида аммония (50 мл) и воду (20 мл), перемешивали в течение 10 мин и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали водой (50 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (MeOH:ДХМ=1:20) с получением продукта (431 мг, выход: 48,1%).
Стадия 3: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(4-метоксипиперидин-1-ил)метанона
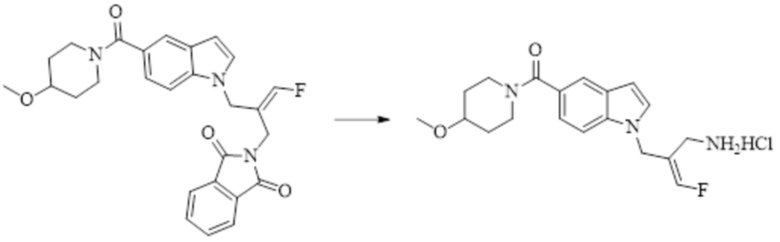
Промежуточный (Z)-2-(3-фтор-2-((5-(4-метоксипиперидин-1-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-дион (431 мг, 0,93 ммоль, 1,0 экв.) растворяли в EtOH (10 мл). К раствору добавляли 80%-ный гидразингидрат (204,5 мг, 3,26 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Фильтрат концентрировали, суспендировали в дихлорметане (10 мл) и фильтровали и полученный таким образом фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением маслянистой жидкости (86 мг). В маслянистую жидкость добавляли дихлорметан (5 мл). В смесь по каплям добавляли 20% раствор хлорида водорода в этаноле (45 мг), перемешивали в течение 10 мин и концентрировали при пониженном давлении с получением продукта (84 мг, выход: 23,6%).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,44 (с, 3H), 7,67-7,63 (д, 3H), 7,40 (с, 0,5H), 7,27-7,18 (д, 1H), 7,13 (с, 0,5H), 6,57-6,56 (д, 1H), 5,04 (м, 2H), 3,76 (м, 2H), 3,21-3,21 (м, 7H), 1,84 (м, 2H), 1,44-1,40 (м, 2H).
Молекулярная формула: C19H25ClFN3O2, молекулярная масса: 381,88, ЖХ-МС (полож., m/z)=346,17 [M+H]+.
Пример 40: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(4-метокси-4-метилпиперидин-1-ил)метанона (соединение C15)
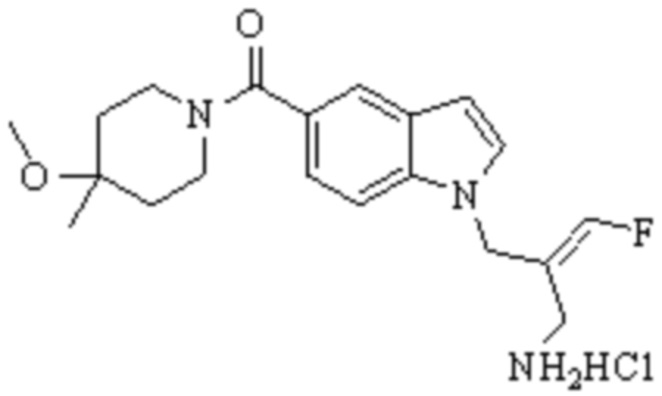
Стадия 1: Синтез (1H-индол-5-ил)(4-метокси-4-метилпиперидин-1-ил)метанона
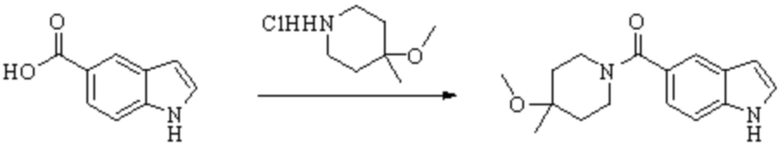
Соединения 1H-индол-5-карбоновую кислоту (1 г, 6,2 ммоль, 1,0 экв.) и DIPEA (2,4 г, 18,6 ммоль, 3,0 экв.) растворяли в N, N-диметилацетамиде (5 мл) и после вакуумирования заполняли азотом. Раствор охлаждали до температуры 0°C и добавляли HATU (3,5 г, 9,3 ммоль, 1,5 экв.) и подвергали взаимодействию в течение 0,5 ч. К реакционному раствору добавляли гидрохлорид 4-метокси-4-метилпиперидина (1,88 г, 12,41 ммоль, 2,0 экв.) и подвергали взаимодействию в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, в реакционную смесь добавляли воду (50 мл), перемешивали в течение 10 мин и экстрагировали этилацетатом (50 мл × 3), затем жидкости разделяли. Органические фазы объединяли, промывали водой (50 мл), сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (1,68 г, выход: 100%).
Стадия 2: Синтез (Z)-2-(3-фтор-2-((5-(4-метокси-4-метилпиперидин-1-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-диона
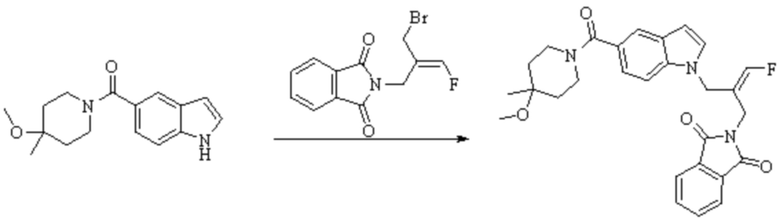
Промежуточный (1H-индол-5-ил)(4-метокси-4-метилпиперидин-1-ил)метанон (500 мг, 1,84 ммоль, 1,0 экв.) растворяли в N, N-диметилацетамиде (5 мл). После вакуумирования заполняли азотом и раствор охлаждали до температуры 0°C, добавляли гидрид натрия (массовая доля: 60%, 80,9 мг, 2,13 ммоль, 1,1 экв.) и перемешивали в течение 0,5 ч. В смесь добавляли (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (658,2 мг, 2,20 ммоль, 1,2 экв.) и подвергали взаимодействию в течение 1 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, к реакционному раствору добавляли насыщенный водный раствор хлорида аммония (50 мл) и воду (50 мл), перемешивали в течение ночи и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали водой (50 мл), сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении с получением маслянистой жидкости. Маслянистую жидкость очищали с помощью препаративной тонкослойной хроматографии (MeOH:ДХМ=1:20) с получением продукта (421 мг, выход: 48,1%).
Стадия 3: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(4-метокси-4-метилпиперидин-1-ил)метанона
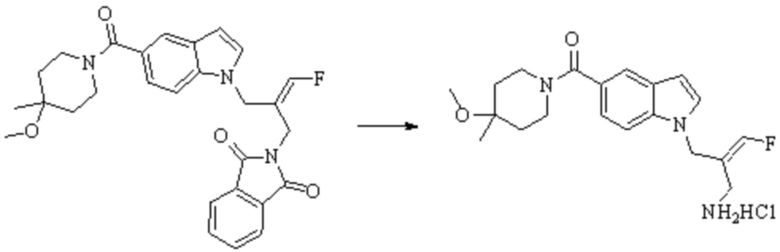
Промежуточный (Z)-2-(3-фтор-2-((5-(4-метокси-4-метилпиперидин-1-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-дион (421 мг, 0,88 ммоль, 1,0 экв.) растворяли в EtOH (15 мл). К раствору добавляли 80%-ный гидразингидрат (193,9 мг, 3,10 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Фильтрат концентрировали с получением белого твердого вещества. Дихлорметан (10 мл) добавляли для суспендирования, затем фильтровали. Маточную жидкость концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10-15:1) с получением продукта (200 мг). Добавляли дихлорметан (5 мл), добавляли по каплям 20% раствор хлорида водорода в этаноле (102 мг) и перемешивали в течение 10 мин. Продукт (200 мг, выход: 57%) получали упариванием при пониженном давлении.
1H-ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,46 (с, 3H), 7,66-7,62 (д, 3H), 7,41 (с, 0,5H), 7,21-7,18 (д, 1H), 7,13 (с, 0,5 H), 6,56-6,55 (с, 1H), 5,04-5,04 (м, 2H), 3,43 (м, 2H), 3,26-3,20 (м, 4H), 3,12 (с, 3H), 1,67 (м, 2H), 1,49-1,42 (м, 2H), 1,14 (с, 3H).
Молекулярная формула: C20H27ClFN3O2, молекулярная масса: 395,9, LC+MS(полож., m/z)=360,14 [M+H]+.
Пример 41: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-неоцикло-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A37) гидрохлорид
Стадия 1: Синтез этил 3-(неоциклоамино)пропионата
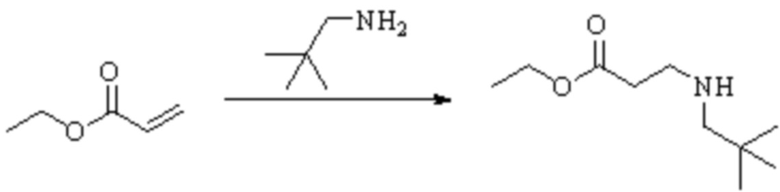
Неоциклоамин (31,38 г, 0,36 моль, 1,2 экв.) растворяли в этаноле (100 мл). При температуре 10°C медленно по каплям добавляли этилакрилат (30 г, 0,3 моль, 1,0 экв.) и после окончания добавления реакцию проводили при температуре 20°C в течение 3 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (56,18 г, выход: 100%).
Стадия 2: Синтез этил 3-((3-этокси-3-оксопропил)(неоцикло)амино)-3-оксопропаноата

Промежуточный этил 3-(неоциклоамино)пропионат (56,18 г, 0,3 моль, 1,0 экв.), этил малонат калия (61,27 г, 0,36 моль, 1,2 экв.), 4-диметиламинопиридин (7,33 г, 0,06 моль, 0,2 экв.) и триэтиламин (39,46 г, 0,39 моль, 1,3 экв.) растворяли в дихлорметане (300 мл) и раствор перемешивали в течение 5 мин. На ледяной бане партиями к раствору добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (69,01 г, 0,36 моль, 1,2 экв.) и подвергали взаимодействию в течение 15 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (200 мл), затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (200 мл). Органические фазы объединяли и концентрировали при пониженном давлении. Сырой продукт растворяли в этилацетате (200 мл), pH доводили до 5 с помощью соляной кислоты и жидкости разделяли. Органическую фазу промывали насыщенным водным раствором бикарбоната натрия (200 мл), сушили над безводным сульфатом натрия и фильтровали, и концентрировали при пониженном давлении с получением продукта (85 г, выход: 94%).
Стадия 3: Синтез этил 1-неоцикло-2,4-диоксопиперидин-3-карбоксилата
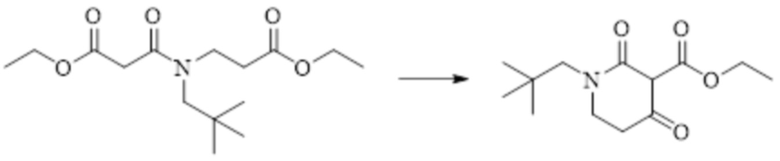
Промежуточный этил 3-((3-этокси-3-оксопропил)неоцикло)амино)-3-оксопропионат (85 г, 0,282 ммоль, 1,0 экв.) и трет-бутоксид натрия (32,48 г, 0,338 моль, 1,2 экв.) растворяли в этаноле (400 мл) и подвергали взаимодействию при температуре 60°C в течение 30 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта, который напрямую использовали на следующей стадии.
Стадия 4: Синтез 1-неоциклопиперидин-2,4-диона
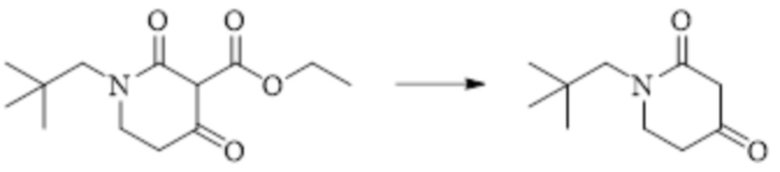
Сырой продукт, полученный на предшествующей стадии, растворяли в воде. pH доводили до 3 и раствор нагревали до температуры 90°C подвергали взаимодействию в течение 3 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и добавляли твердый хлорид натрия до насыщения. Экстрагировали этилацетатом (300 мл × 3) и органическую фазу сушили и концентрировали. Сырой продукт суспендировали в MTBE:PE (1:1) и фильтровали в вакууме. Слой на фильтре сушили с получением продукта (42 г, выход за две стадии: 81%).
Стадия 5: Синтез 3-((диметиламино)метилен)-1-неоциклопиперидин-2,4-диона
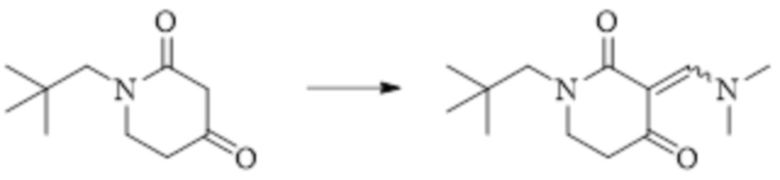
1-Неоциклопиперидин-2,4-дион (5 г, 27,28 ммоль, 1,0 экв.) партиями добавляли в N, N-диметилформамид диметилацеталь (3,58 г, 30 ммоль, 1,1 экв.) и подвергали взаимодействию в течение 30 мин. После завершения реакции, как определялось данными ТСХ, концентрировали при пониженном давлении и сырой продукт обрабатывали изопропанолом с получением продукта (6,5 г, выход: 100%).
Стадия 6: Синтез 5-неоцикло-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

Промежуточный 3-((диметиламино)метилен)-1-неоциклопиперидин-2,4-дион (6,5 г, 27,28 ммоль, 1,0 экв.) и гидразингидрат (1,77 г, 30 ммоль, 1,1 экв.) растворяли в изопропаноле (30 мл) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении и сырой продукт очищали путем колоночной хроматографии на силикагеле (этилацетат) с получением продукта (4,4 г, выход: 77%).
Стадия 7: Синтез (E)-2-(3-фтор-2-((5-неоцикло-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-диона
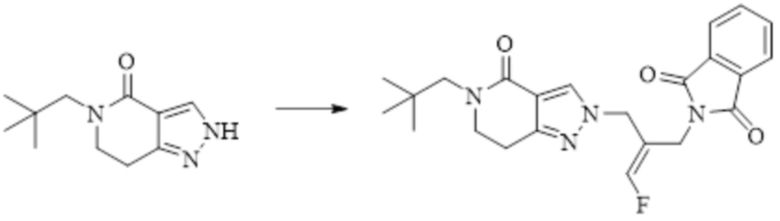
5-Неоцикло-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (4,4 г, 21,22 ммоль, 1,0 экв.), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1, 3-дион (6,96 г, 23,34 ммоль, 1,1 экв.), карбонат калия (3,22 г, 23,34 ммоль, 1,1 экв.) и TBAB (684 мг, 2,12 ммоль, 0,1 экв.) растворяли в абсолютном этаноле (50 мл) и подвергали взаимодействию в течение 17 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (PE:EA=1:1) с получением продукта (4,0 г, выход: 44%).
Стадия 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-неоцикло-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
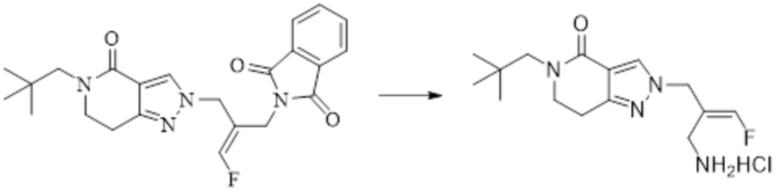
Промежуточный (E)-2-(3-фтор-2-((5-неоцикло-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-дион (4,0 мг, 9,42 ммоль, 1,0 экв.) и гидразингидрат (884 мг, 14,13 ммоль, 1,5 экв.) растворяли в EtOH (40 мл) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Фильтрат концентрировали при пониженном давлении. Сырой продукт суспендировали в изопропилацетате (20 мл) и фильтровали в вакууме. Затем на ледяной бане к фильтрату добавляли 20% раствор хлорида водорода в этаноле (2,06 г, 1,2 экв.) и смесь перемешивали в течение 10 мин. Осаждалось большое количество твердого вещества белого цвета. Фильтровали в вакууме, и слой на фильтре сушили при температуре 40°C с получением продукта (2,32 г, выход: 74%).
1Н ЯМР (300 МГц, ДМСО-d6) δ (м.д.):8,49 (с, 3H), 8,27 (с, 1H), 7,41 (с, 0,5H),7,13 (с, 0,5H), 4,94(д, 2H), 3,55-3,59 (т, 2H), 3,34-3,35(д, 2H),3,20 (с, 2H) 2,81-2,85 (т, 2H), 0,91 (с, 9H).
Молекулярная формула: C15H24ClFN4O, молекулярная масса: 330,83, ЖХ-МС (полож., m/z)=295,12 [M+H]+.
Пример 42: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(пирролидин-1-ил)метанона (соединение C7)
Стадия 1: Синтез (Z)-2-(3-фтор-2-((5-(пирролидин-1-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-диона
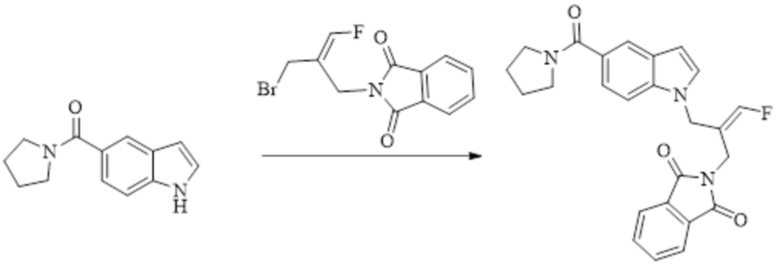
Промежуточный (1H-индол-5-ил)(пирролидин-1-ил)метанон (500 мг, 2,33 ммоль, 1,0 экв.) растворяли в N, N-диметилацетамиде (3 мл). После вакуумирования заполняли азотом и раствор охлаждали до температуры 0°C, добавляли гидрид натрия (массовая доля: 60%, 102,6 мг, 2,56 ммоль, 1,1 экв.) и перемешивали в течение 0,5 ч. В смесь добавляли по каплям раствор (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-диона (833,5 мг, 2,80 ммоль, 1,2 экв.) в N, N-диметилацетамиде (2 мл) и подвергали взаимодействию в течение 1 ч. Когда оставалось менее 10% исходных соединений, как определялось данными ЖХ-МС, к реакционному раствору добавляли насыщенный водный раствор хлорида аммония (50 мл), перемешивали в течение 10 мин и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли и промывали водой (50 мл), жидкости разделяли и сушили с помощью безводного сульфата магния, затем фильтровали. Фильтрат концентрировали при пониженном давлении и сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=20:1) с получением продукта (755 мг, выход: 77,8%).
Стадия 2: Синтез гидрохлорида (E)-(1-(2-(аминометил)-3-фтораллил)-1H-индол-5-ил)(пирролидин-1-ил)метанона
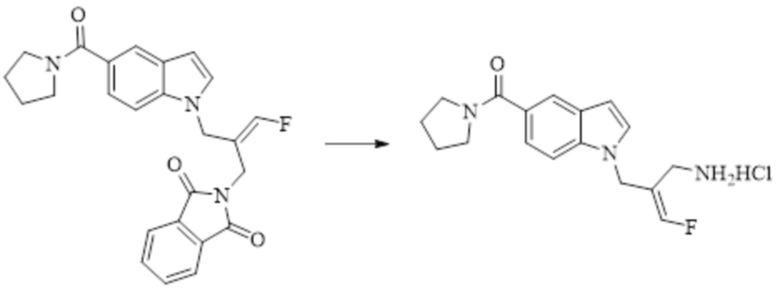
Промежуточный (Z)-2-(3-фтор-2-((5-(пирролидин-1-карбонил)-1H-индол-1-ил)метил)аллил)изоиндолин-1,3-дион (755 мг, 1,81 ммоль, 1,0 экв.) растворяли в EtOH (10 мл), к раствору добавляли гидразингидрат (массовая доля: 80%, 397 мг, 6,34 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 2 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры, фильтровали в вакууме и промывали этанолом. Фильтрат концентрировали с получением белого твердого вещества, которое суспендировали в дихлорметане (10 мл) в течение 5 мин и фильтровали. Слой на фильтре промывали ДХМ. Фильтраты объединяли и концентрировали при пониженном давлении с получением маслянистой жидкости (487 мг), которую очищали тонкослойной хроматографией (ДХМ:MeOH=10:1) с получением маслянистой жидкости (357 мг). Для растворения в маслянистую жидкость добавляли дихлорметан (5 мл). К раствору добавляли 20% раствор хлорида водорода в этаноле (104,2 мг), перемешивали в течение 10 мин и концентрировали при пониженном давлении с получением твердого вещества светло-желтого цвета (325 мг). Твердый продукт очищали препаративной ВЭЖХ (0,05% соляная кислота:вода:ацетонитрил) с получением продукта (120 мг, выход: 19,6%).
1H-ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,58 (с, 3H), 7,78-7,77 (с, 1H), 7,70-7,64 (д, 2H), 7,41 (с, 0,5H), 7,35-7,32 (д, 1H), 7,13 (с, 0,5H), 6,56-6,55 (д, 1H), 5,08-5,07 (д, 2H), 3,47 (м, 4H), 3,24-3,23 (д, 2H), 1,85-1,82 (м, 4H).
Молекулярная формула: C17H21ClFN3O, молекулярная масса: 337,82, ЖХ-МС (полож., m/z)=302,10 [M+H+].
Пример 43: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-6,7-дигидроизоксазоло[4,5-c]пиридин-3,4(2H,5H)-диона (соединение A18)
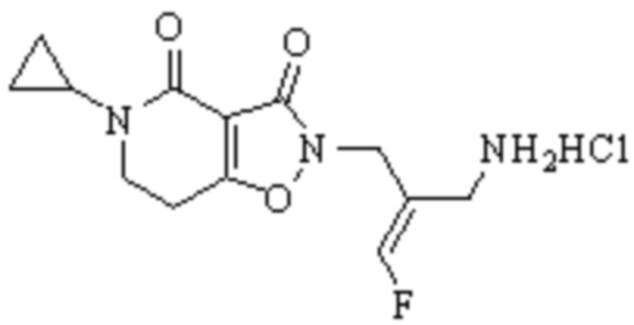
Стадия 1: Синтез 5-циклопропил-6,7-дигидроизоксазоло[4,5-c]пиридин-3,4(2H,5H)-диона
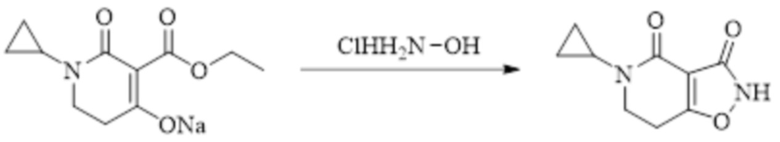
Натрия 1-циклопропил-5-(этоксикарбонил)-6-оксо-1,2,3,6-тетрагидропиридин-4-олат (10,0 г, 0,040 моль, 1,0 экв.), гидроксид натрия (19,4 г, 0,485 моль, 12 экв.), гидрохлорид гидроксиламина (30,9 г, 0,444 моль, 11 экв.) добавляли в этанол (100 мл) и воду (10 мл), при этом при комнатной температуре pH доводили до 7-8 и реакцию проводили при температуре 50°C в течение 17 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, добавляли этанол (100 мл), нагревали до температуры 60°C для растворения и фильтровали горячим. Фильтрат концентрировали при пониженном давлении с получением продукта (5,1 г, выход: 64,97%).
Стадия 2: Синтез (E)-5-циклопропил-2-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-6,7-дигидроизоксазоло[4,5-c]пиридин-3,4(2H,5H)-диона
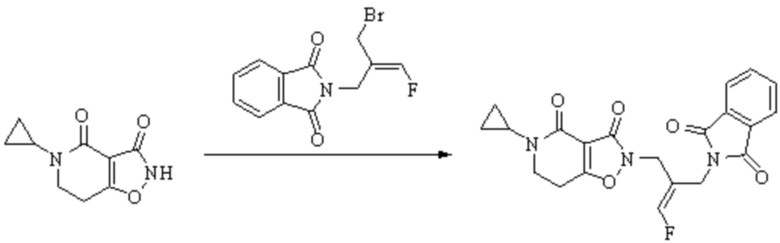
Промежуточный 5-циклопропил-6,7-дигидроизоксазоло[4,5-c]пиридин-3,4 (2H,5H)-дион (1,00 г, 5,15 ммоль, 1 экв.) растворяли в ДМФ (15 мл). К раствору при температуре 0°C добавляли гидрид натрия (массовая доля: 60%, 0,25 г, 6,18 ммоль, 1,2 экв.) и поддерживая эту температуру перемешивали в течение 15 мин. В смесь добавляли (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1, 3-дион (1,84 г, 6,18 ммоль, 1,2 экв.) и подвергали взаимодействию при температуре 60°C в течение 17 ч. Реакционный раствор охлаждали, добавляли воду (15 мл) и экстрагировали дихлорметаном (30 мл × 3), затем жидкости разделяли. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении и сырой продукт разделяли путем колоночной хроматографии на силикагеле (ДХМ:MeOH=200:1) с получением продукта (688 мг, выход: 32,4%).
Стадия 3: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-6,7-дигидроизоксазоло[4,5-c]пиридин-3,4(2H,5H)-диона
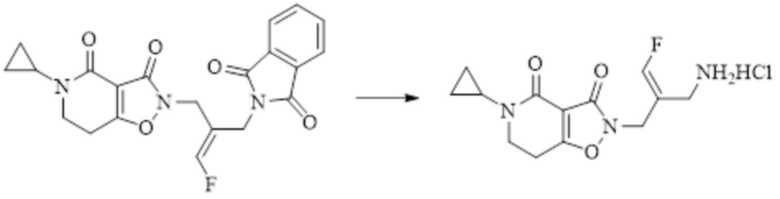
Промежуточный (E)-5-циклопропил-2-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фтораллил)-6,7-дигидроизоксазоло[4,5-c]пиридин-3,4(2H,5H)-дион (545 мг, 1,32 ммоль, 1,0 экв.) растворяли в абсолютном этаноле (20 мл). К раствору добавляли гидразингидрат (85%, 273 мг, 4,64 ммоль, 3,5 экв.) и кипятили с обратным холодильником в течение 2 ч до завершения реакции. Реакционный раствор фильтровали горячим. Фильтрат охлаждали и затем фильтровали в вакууме. Фильтрат концентрировали при пониженном давлении, добавляли EA (20 мл), кипятили с обратным холодильником при температуре 85°C и фильтровали горячим. Фильтрат охлаждали и затем опять фильтровали и полученный таким образом фильтрат концентрировали при пониженном давлении. Сырой продукт разделяли путем препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением маслянистого продукта. В маслянистый продукт добавляли небольшое количество этанола для растворения, раствор хлорида водорода в этаноле добавляли по каплям и осаждалось твердое вещество белого цвета. Фильтровали в вакууме с получением продукта (62,92 мг, выход: 16,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,40 (с, 3H), 7,22-7,42 (д, J=80 Гц, 1H), 4,74 (с, 2H), 3,53-3,56 (т, 2H), 3,49 (с, 2H), 3,05-3,08 (т, 2H), 2,53-2,57 (м, 1H), 0,70-0,74 (м, 2H), 0,54-0,58 (м, 2H).
Молекулярная формула: C13H17ClFN3O3, молекулярная масса: 317,75, ЖХ-МС (полож., m/z)=282,15 [M+H]+.
Пример 44: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-3-фенил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A45)
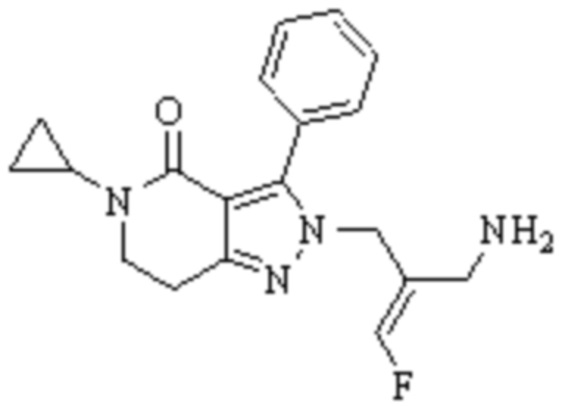
Стадия 1: Синтез 3-бензоил-1-циклопропилпиперидин-2,4-диона
В реакционный сосуд помещали 1-циклопропилпиперидин-2,4-дион (50,00 г, 0,33 моль, 1,0 экв.) и добавляли ТГФ (500 мл). Продукты охлаждали до температуры 0°C, добавляли трет-бутоксид натрия (66,60 г, 0,69 моль, 2,1 экв.) и перемешивали в течение 30 мин. При температуре 0°C медленно по каплям добавляли бензоилхлорид (55,06 г, 0,39 моль, 1,2 экв.). После добавления выдерживали при комнатной температуре в течение 5 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду для разбавления, pH доводили до 3 и экстрагировали EA (100 мл × 3). Сушили с помощью безводного сульфата натрия, затем концентрировали при пониженном давлении. Сырой продукт разделяли путем колоночной хроматографии на силикагеле (ДХМ:MeOH=100:1-50:1) с получением продукта (28,3 г, выход: 33,7%).
Стадия 2: Синтез 5-циклопропил-3-фенил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
Гидразингидрат (85%, 9,72 г, 0,16 моль, 1,5 экв.) растворяли в этаноле (100 мл) и добавляли соляную кислоту до установления pH равным 3. Добавляли 3-бензоил-1-циклопропилпиперидин-2,4-дион (28,30 г, 0,11 моль, 1,0 экв.) и pH доводили до 6. Смесь нагревали до температуры кипения с обратным холодильником в течение 2 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт сначала разделяли путем колоночной хроматографии на силикагеле (ДХМ:MeOH=50:1) и затем перекристаллизовывали в абсолютном этаноле (50 мл) с получением продукта (8,0 г, выход: 28,7%).
Стадия 3: Синтез (E)-2-(2-((5-циклопропил-4-оксо-3-фенил-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона

Промежуточный 5-циклопропил-3-фенил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (3,00 г, 0,012 моль, 1 экв.), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (3,89 г, 0,013 моль, 1,1 экв.), карбонат калия (1,80 г, 0,013 моль, 1,1 экв.) и тетрабутиламмоний бромид (0,39 г, 0,001 моль, 0,1 экв.) добавляли в абсолютный этанол (30 мл) и смесь перемешивали при комнатной температуре в течение 72 ч. Когда оставалось небольшое количество пиразола, как определялось данными ТСХ, реакционный раствор фильтровали в вакууме. Слой на фильтре промывали этанол. Фильтраты объединяли, добавляли MTBE (5 мл), концентрировали до половины объема при пониженном давлении и охлаждали до комнатной температуры, осаждалось большое количество твердого вещества белого цвета. Фильтровали в вакууме, затем сушили. Сырой продукт перекристаллизовывали из этанола (15 мл) с получением продукта (1,8 г, выход: 32,1%).
Стадия 4: Синтез (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-3-фенил-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
(E)-2-(2-((5-Циклопропил-4-оксо-3-фенил-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (1,80 г, 3,83 ммоль, 1,0 экв.) растворяли в EtOH (18 мл), к раствору добавляли гидразингидрат (85%, 0,34 г, 5,74 ммоль, 1,5 экв.) и подвергали взаимодействию при кипячении с обратным холодильником в течение 1 ч. Когда реакция завершалась, фильтровали в вакууме. Фильтрат концентрировали при пониженном давлении, добавляли изопропилацетат (30 мл), нагревали при кипении с обратным холодильником в течение 30 мин и охлаждали до комнатной температуры и осаждалось небольшое количество твердого вещества. Затем фильтровали в вакууме, фильтрат концентрировали при пониженном давлении и осаждалось большое количество твердого продукта. Фильтровали в вакууме и слой на фильтре промывали небольшим количеством изопропилацетата, концентрировали и сушили с получением продукта (850 мг, выход: 65,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м. д.): 8,06-8,08 (м, 2H), 7,32-7,40 (м, 3H), 6,82-7,04 (д, J=84 Гц, 1H), 4,76-4,77 (д, 2H), 3,57-3,60 (т, 2H), 3,32 (шир. с, 2H), 3,12-3,13 (д, 2H), 2,98-3,02 (т, 2H), 2,60-2,67 (м, 1H), 0,74-0,79 (м, 2H), 0,66-0,72 (м, 2H).
Молекулярная формула: C19H21FN4O, молекулярная масса: 340,40, ЖХ-МС (полож., m/z)=341,12 [M+H]+.
Пример 45: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1-(3-фторфенил)-2,5,6,7-тетрагидро-4H-пирроло[3,4c]пиридин-4-она (соединение A46)
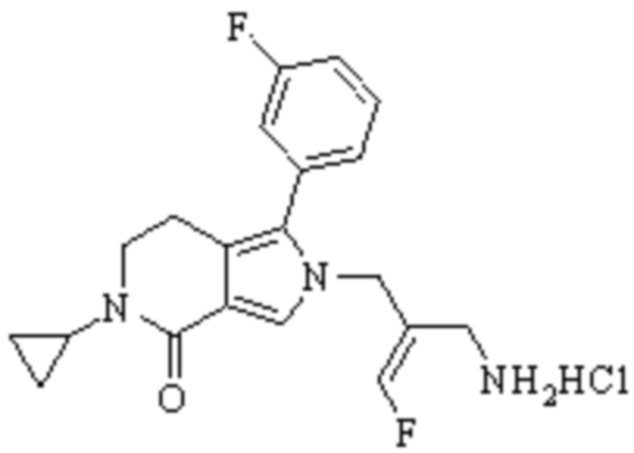
Синтез промежуточного трет-бутил (E)-(2-((5-циклопропил-1-(3-фторфенил)-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамата
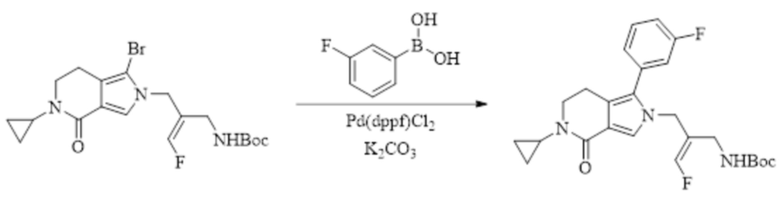
трет-Бутил (E)-(2-((1-бром-5-циклопропил-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (150 мг, 0,339 ммоль, 1 экв.) и м-фторфенилбороновую кислоту (72 мг, 0,509 ммоль, 1,5 экв.) растворяли в 1,4-диоксане (5 мл). К раствору добавляли водный раствор (1 мл) карбоната калия (118 мг, 0,848 ммоль, 2,5 экв.) и в атмосфере N2 добавляли Pd(dppf)Cl2 (28 мг, 0,0339 ммоль, 0,1 экв.). После добавления реакцию проводили при температуре 90°C в течение 17 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор фильтровали через целит. К фильтрату последовательно добавляли этилацетат (20 мл), промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали с получением сырого продукта (190 мг), который очищали с помощью препаративной тонкослойной хроматографии (PE:EA=1:1) с получением продукта (70 мг, выход: 45%).
Стадия 2: Синтез соединения гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-циклопропил-1-(3-фторфенил)-2,5,6,7-тетрагидро-4H-пирроло[3,4-c]пиридин-4-она
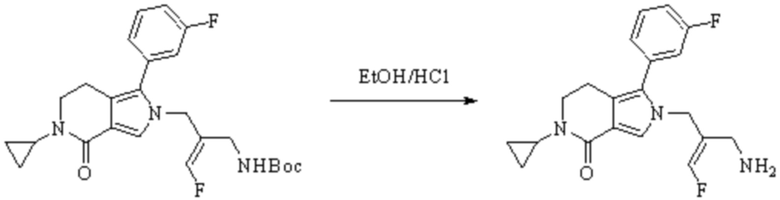
трет-Бутил (E)-(2-((5-циклопропил-1-(3-фторфенил)-4-оксо-4,5,6,7-тетрагидро-2H-пирроло[3,4-c]пиридин-2-ил)метил)-3-фтораллил)карбамат (70 мг, 0,153 ммоль, 1 экв.) растворяли в растворе хлорида водорода в этаноле (5 мл) и раствор перемешивали в течение 3 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор концентрировали и лиофилизовали с получением продукта (28 мг, выход: 51%).
1H-ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,32 (шир. с, 3H), 7,46-7,52 (м, 2H), 7,19-7,22 (м, 3H), 6,36-6,63 (д, 1H), 4,81 (с, 2H), 3,40-3,44 (т, 2H), 3,12-3,13 (м, 2H), 2,59-2,67 (м, 3H), 0,72-0,78 (м, 2H), 0,58-0,60 (м, 2H).
Молекулярная формула: C20H21F2N3O, молекулярная масса: 357,17, ЖХ-МС (m/z)=358,12 [M+H]+.
Пример 46: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(2-метоксиэтил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A47)
Стадия 1: Синтез этил 3-((2-метоксиэтил)амино)пропионата
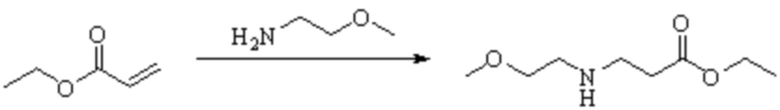
Продукт 2-метоксиэтан-1-амин (10 г, 133,14 ммоль, 1,0 экв.) растворяли в абсолютном этаноле (100 мл). На ледяной бане медленно по каплям добавляли этилакрилат (11,32 г, 113,06 ммоль, 0,85 экв.). После добавления реакцию проводили в течение 2 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении при температуре 80°C с получением сырого продукта, который использовали на следующей стадии в соответствии с теоретическим количеством.
Стадия 2: Синтез этил 3-((3-этокси-3-оксопропил)(2-метоксиэтил)амино)-3-оксопропионата
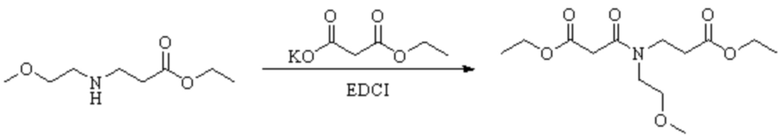
Промежуточный этил 3-((2-метоксиэтил)амино)пропионат (133,14 ммоль, 1,0 экв.) растворяли в дихлорметане (100 мл). Раствор охлаждали до температуры 0°C ледяной водой и последовательно добавляли этил малонат калия (19,2 г, 133,14 ммоль, 1,0 экв.), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (26 г, 135,63 ммоль, 1,2 экв.), 4-диметиламинопиридин (1,38 г, 11,32 ммоль, 0,1 экв.) и триэтиламин (17,2 г, 169,98 ммоль, 1,5 экв.). После добавления продукты выдерживали при комнатной температуре в течение 16 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, добавляли воду (200 мл), добавляли по каплям концентрированную соляную кислота (20 мл), pH доводили до примерно 5 и жидкости разделяли. Водную фазу экстрагировали дихлорметаном (100 мл). Органические фазы объединяли, последовательно промывали 5% водным раствором бикарбоната натрия (100 мл) и водой (100 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением маслянистой жидкости желтого цвета (26,2 г, выход за две стадии: 80,3%).
Стадия 3: Синтез этил 1-(2-метоксиэтил)-2,4-диоксопиперидин-3-карбоксилата

Промежуточный этил 3-((3-этокси-3-оксопропил)(2-метоксиэтил)амино)-3-оксопропионат (26,2 г, 90,55 ммоль, 1,0 экв.) растворяли в абсолютном этаноле (100 мл). К раствору добавляли этоксид натрия (15,4 г, 226,38 ммоль, 2,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта.
Стадия 4: Синтез 1-(2-метоксиэтил)пиперидин-2,4-диона
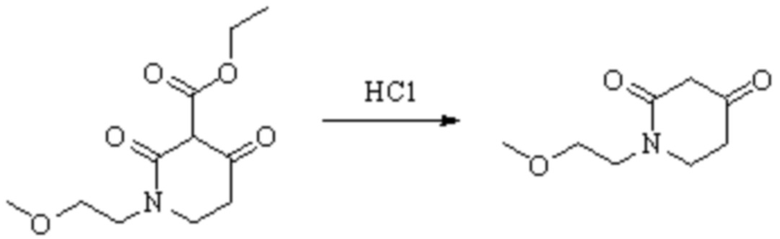
Промежуточный этил 1-(2-метоксиэтил)-2,4-диоксопиперидин-3-карбоксилат (90,55 ммоль, 1,0 экв.) растворяли в воде (50 мл). К раствору по каплям добавляли концентрированную соляную кислоту (20 мл) и нагревали при температуре 80°C для взаимодействия в течение 3 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и экстрагировали дихлорметаном (100 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали с получением продукта, который использовали на следующей стадии в соответствии с теоретическим количеством.
Стадия 5: Синтез 3-((диметиламино)метилен)-1-(2-метоксиэтил)пиперидин-2,4-диона
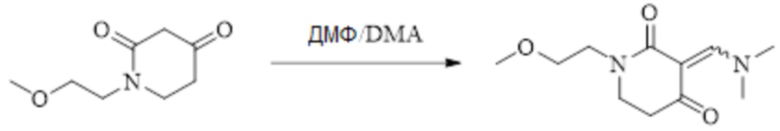
К промежуточному 1-(2-метоксиэтил)пиперидин-2,4-диону (90,55 ммоль, 1,0 экв.) добавляли по каплям N, N-диметилформамид диметилацеталь (11,8 г, 99,60 ммоль, 1,1 экв.) с интенсивным выделением тепла. После добавления реакцию проводили при комнатной температуре в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта, который использовали на следующей стадии в соответствии с теоретическим количеством.
Стадия 6: Синтез 5-(2-метоксиэтил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
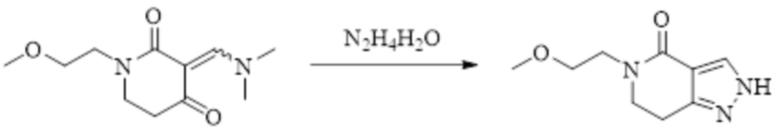
Промежуточный 3-((диметиламино)метилен)-1-(2-метоксиэтил)пиперидин-2,4-дион (20,4 г, 90,55 ммоль, 1,0 экв.) и гидразингидрат (массовая доля: 80%, 6,23 г, 99,60 ммоль, 1,1 экв.) растворяли в метаноле (20 мл). Раствор нагревали до температуры 70°C подвергали взаимодействию в течение 0,5 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=100:1-60:1) с получением продукта (10,6 г, выход за четыре стадии: 60,2%).
Стадия 7: Синтез (E)-2-(3-фтор-2-((5-(2-метоксиэтил)-4-оксо-4,5,6, 7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-диона
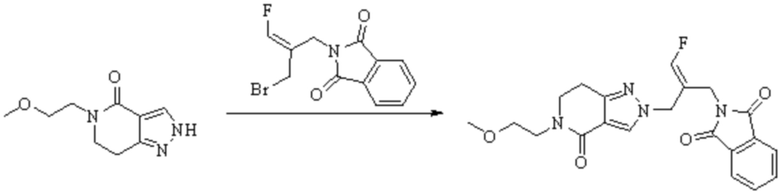
Промежуточный 5-(2-метоксиэтил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (5,0 г, 25,61 ммоль, 1,0 экв.), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (8,4 г, 28,17 ммоль, 1,1 экв.), безводный карбонат калия (3,9 г, 28,17 ммоль, 1,1 экв.) и тетрабутиламмоний бромид (825,6 мг, 2,56 ммоль, 0,1 экв.) добавляли в абсолютный этанол (50 мл) и раствор перемешивали при комнатной температуре в течение 20 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, фильтровали. Слой на фильтре промывали этилацетатом. Фильтрат концентрировали при пониженном давлении, добавляли этилацетат (50 мл) и фильтровали. Полученный таким образом фильтрат концентрировали при пониженном давлении с получением сырого продукта (11 г). Часть сырого продукта (2 г) очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=60:1) с получением продукта (907 мг).
Стадия 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(2-метоксиэтил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
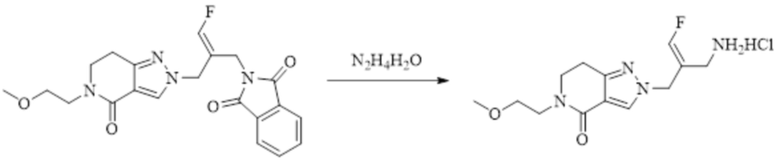
Промежуточный (E)-2-(3-фтор-2-((5-(2-метоксиэтил)-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-дион (900 мг, 2,18 ммоль, 1,0 экв.) растворяли в этаноле (10 мл). Смесь нагревали для растворения и добавляли гидразингидрат (массовая доля: 85%, 128,3 мг, 3,27 ммоль, 1,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 3 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Слой на фильтре промывали дихлорметаном. Фильтрат концентрировали, суспендировали в дихлорметане (10 мл) и фильтровали, и полученный таким образом фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (338 мг). Добавляли дихлорметан (5 мл). В смесь добавляли по каплям раствор хлорида водорода в этаноле (массовая доля: 20%, 218 мг), перемешивали в течение 10 мин и концентрировали при пониженном давлении с получением продукта (317 мг, выход: 45,6%).
1Н ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,34 (с, 3H), 8,10 (с, 1H), 7,40 (с, 0,5H), 7,13 (с, 0,5H), 4,90-4,89 (д, 2H), 3,62-3,54 (м, 4H), 3,48-3,45 (м, 2H), 3,39 (м, 2H), 3,25 (с, 3H), 2,84-2,79 (м, 2H).
Молекулярная формула: C13H20ClFN4O2, молекулярная масса: 318,78, ЖХ-МС (полож., m/z)=283,07 [M+H]+.
Пример 47: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(циклопропилметил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A48)
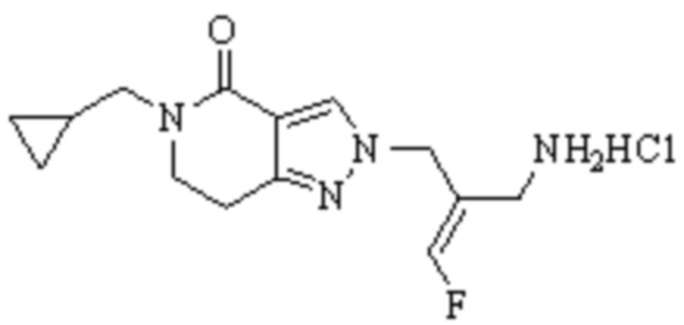
Стадия 1: Синтез этил 3-((циклопропилметил)амино)пропионата
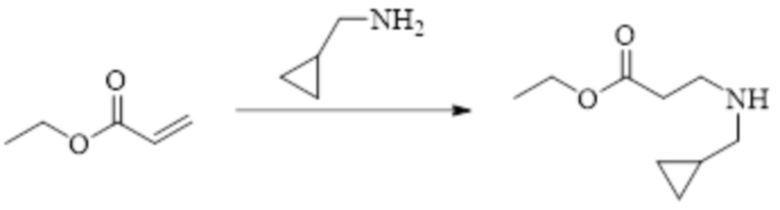
Продукт циклопропанметанамин (10 г, 99,88 ммоль, 1,0 экв.) растворяли в этаноле (30 мл). На ледяной бане к раствору медленно по каплям добавляли этилакрилат (12,67 г, 89,89 ммоль, 0,9 экв.) и после добавления реакцию проводили при комнатной температуре в течение 16 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (15,39 г, выход: 100%).
Стадия 2: Синтез этил 3-((циклопропилметил)(3-этокси-3-оксопропил)амино)-3-оксопропионат

Промежуточный этил 3-((циклопропилметил)амино)пропионат (15,39 г, 89,89 ммоль, 1,0 экв.), этил малонат калия (18,36 г, 107,87 ммоль, 1,2 экв.), 4-диметиламинопиридин (2,2 г, 17,98 ммоль, 0,2 экв.) и триэтиламин (11,83 г, 116,83 ммоль, 1,3 экв.) растворяли в дихлорметане (150 мл). К раствору партиями добавляли гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (20,68 г, 107,87 ммоль, 1,2 экв.) и подвергали взаимодействию в течение 23 ч. После завершения реакции, как определялось данными ТСХ, в реакционную смесь добавляли воду (100 мл) и соляную кислоту (50 мл), затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (200 мл). Органические фазы объединяли и концентрировали при пониженном давлении с получением продукта (21 г, выход: 81,8%).
Стадия 3: Синтез этил 1-(циклопропилметил)-2,4-диоксопиперидин-3-карбоксилата

Промежуточный этил 3-((циклопропилметил)(3-этокси-3-оксопропил)амино)-3-оксопропионат (21 г, 73,59 ммоль, 1,0 экв.) и трет-бутоксид натрия (8,49 г, 88,3 ммоль, 1,2 экв.) растворяли в этаноле (100 мл) и подвергали взаимодействию при температуре 80°C в течение 20 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (17,6 г, выход: 100%).
Стадия 4: Синтез 1-(циклопропилметил)пиперидин-2,4-диона
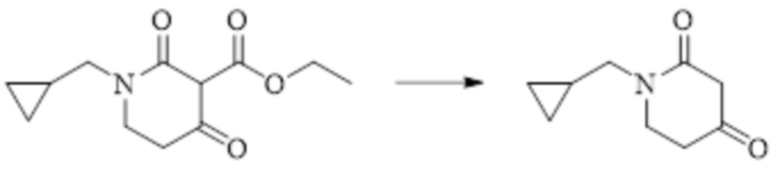
Промежуточный этил 1-(циклопропилметил)-2,4-диоксопиперидин-3-карбоксилат (17,6 г, 73,59 ммоль, 1,0 экв.) растворяли в соляной кислоте (10 мл) и воде (100 мл) и подвергали взаимодействию при температуре 80°C в течение 3 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и добавляли твердый хлорид натрия до насыщения. Экстрагировали дихлорметан (100 мл × 3) и органическую фазу сушили и концентрировали с получением продукта (11 г, выход: 89%).
Стадия 5: Синтез 1-(циклопропилметил)-3-((диметиламино)метилен)пиперидин-2,4-диона
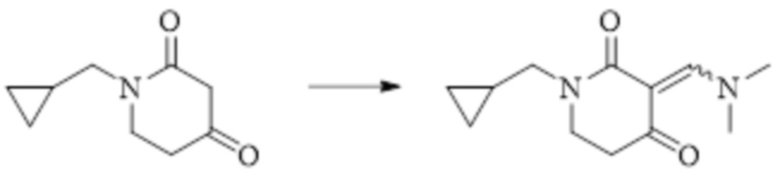
Промежуточный 1-(циклопропилметил)пиперидин-2,4-дион (11 г, 65,78 ммоль, 1,0 экв.) растворяли в N,N-диметилформамид диметилацетале (8,62 г, 72,36 ммоль, 1,1 экв.) и подвергали взаимодействию при комнатной температуре в течение 40 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта (15,08 г, выход: 100%).
Стадия 6: Синтез 5-(циклопропилметил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
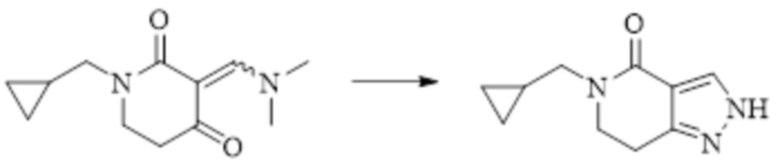
Промежуточный 1-(циклопропилметил)-3-((диметиламино)метилен)пиперидин-2,4-дион (15,08 г, 65,78 ммоль, 1,0 экв.) и гидразингидрат (4,26 г, 72,36 ммоль, 1,1 экв.) растворяли в изопропаноле (100 мл) и подвергали взаимодействию при температуре 80°C в течение 30 мин. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:40) с получением продукта (8,2 г, выход: 65%).
Стадия 7: Синтез (E)-2-(2-((5-(циклопропилметил)-4-оксо-4,5,6, 7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-диона
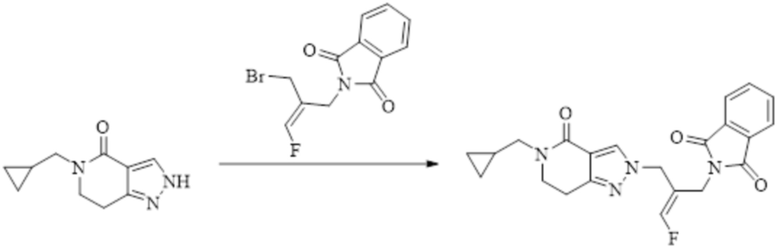
Промежуточный 5-(циклопропилметил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (4,2 г, 21,96 ммоль, 1,0 экв.), (E)-2-(2-(бромметил)-3-фтораллил)изоиндолин-1,3-дион (7,18 г, 24,16 ммоль, 1,1 экв.), карбонат калия (3,34 г, 24,16 ммоль, 1,1 экв.) и тетрабутиламмоний бромид (709 мг, 2,2 ммоль, 0,1 экв.) добавляли в абсолютный этанол (30 мл) и подвергали взаимодействию в течение 15 ч. После завершения реакции, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении, и этанол удаляли. Добавляли ДХМ (50 мл), перемешивали в течение 30 мин и фильтровали в вакууме. Маточную жидкость концентрировали при пониженном давлении с получением 8,0 г неочищенных масляных капель желтого цвета. 2,0 г неочищенных масляных капель желтого цвета очищали с помощью препаративной тонкослойной хроматографии (MeOH:ДХМ=1:20) с получением продукта (1,3 г, выход: 56%).
Стадия 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(циклопропилметил)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
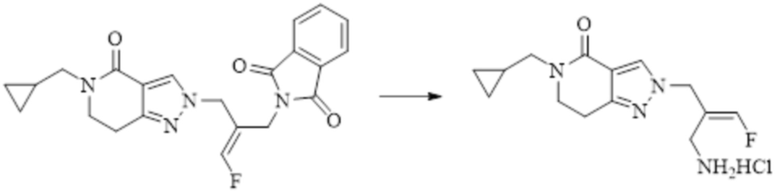
Промежуточный (E)-2-(2-((5-(циклопропилметил)-4-оксо-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)-3-фтораллил)изоиндолин-1,3-дион (1,3 г, 3,18 ммоль, 1,0 экв.) и гидразингидрат (281 мг, 4,77 ммоль, 1,5 экв.) растворяли в EtOH (20 мл) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После завершения реакции, как определялось с помощью ЖХ-МС, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Фильтрат концентрировали при пониженном давлении. Сырой продукт растворяли в изопропилацетате, раствор перемешивали в течение 30 мин и фильтровали в вакууме. В фильтрат на ледяной бане добавляли раствор хлорида водорода в этаноле и концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (MeOH:ДХМ=1:15) с получением продукта (800 мг, выход: 80%).
1Н ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,47 (с, 3H), 8,28 (с, 1H), 7,40 (с, 0,5H), 7,13 (с, 0,5H), 4,93-4,94 (д, 2H), 3,61-3,65 (м, 2H), 3,33-3,34 (д, 2H), 3,27-3,29 (д, 2H), 2,82-2,86 (м, 2H), 0,96-1,00 (м, 1H), 0,40-0,50 (м, 2H), 0,20-0,30 (м, 2H).
Молекулярная формула: C14H20ClFN4O, молекулярная масса: 314,79, ЖХ-МС (полож., m/z)=279,11 [M+H]+.
Пример 48: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(трет-цикло)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она (соединение A49)
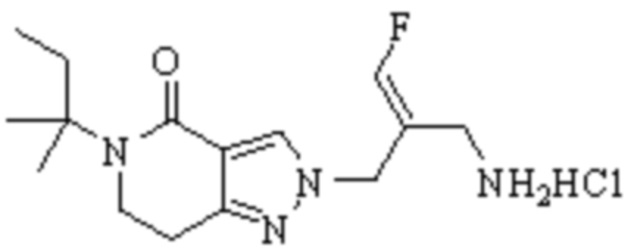
Стадия 1: Синтез этил 3-(трет-циклоамино)пропаноата
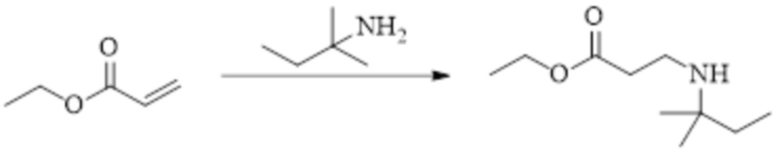
Продукт 2-метилбутил-2-амин (7,44 г, 85,35 ммоль, 1,0 экв.) растворяли в абсолютном этаноле (75 мл). На ледяной бане медленно добавляли по каплям этилакрилат (7,26 г, 72,54 ммоль, 0,85 экв.). После добавления выдерживали при комнатной температуре в течение 9 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении при температуре 80°C с получением продукта, который использовали на следующей стадии в соответствии с теоретическим количеством.
Стадия 2: Синтез этил 3-((3-этокси-3-оксопропил)(трет-цикло)амино)-3-оксопропаноата
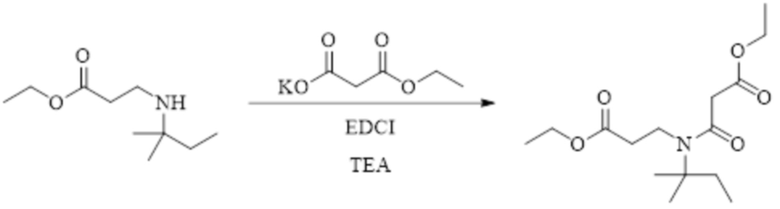
Промежуточный этил 3-(трет-циклоамино)пропаноат (85,35 ммоль, 1,0 экв.) растворяли в дихлорметане (150 мл). К раствору последовательно добавляли этил малонат калия (14,5 г, 85,35 ммоль, 1,0 экв.), триэтиламин (12,9 г, 128 ммоль, 1,5 экв.) и 4-диметиламинопиридин (1 г, 8,53 ммоль, 0,1 экв.), охлаждали до температуры 0°C ледяной водой и добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимид (19,6 г, 102,42 ммоль, 1,2 экв.). После добавления выдерживали при комнатной температуре в течение 15 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, в реакционную смесь добавляли воду (100 мл), охлаждали ледяной водой и добавляли по каплям концентрированную соляную кислоту (20 мл, в массовая доле) до установления pH равным примерно 4-5, затем жидкости разделяли. Водную фазу экстрагировали дихлорметаном (100 мл). Органические фазы объединяли и добавляли воду (100 мл), добавляли карбонат натрия до установления pH равным примерно 8-9, и жидкости разделяли. Органическую фазу промывали водой (100 мл), сушили над безводным сульфатом магния и фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (14,5 г, выход за две стадии: 56,4%).
Стадия 3: Синтез этил 2,4-диоксо-1-(трет-цикло)пиперидин-3-карбоксилата
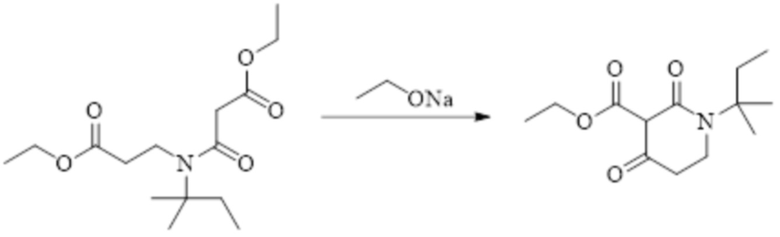
Промежуточный этил 3-((3-этокси-3-оксопропил)(трет-цикло)амино)-3-оксопропаноат (14,5 г, 48,11 ммоль, 1,0 экв.) растворяли в абсолютном этаноле (100 мл). К раствору добавляли этоксид натрия (8,2 г, 120,2 ммоль, 2,5 экв.) подвергали взаимодействию при температуре 80°C в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта, который использовали на следующей стадии в соответствии с теоретическим количеством.
Стадия 4: Синтез 1-(трет-цикло)пиперидин-2,4-диона
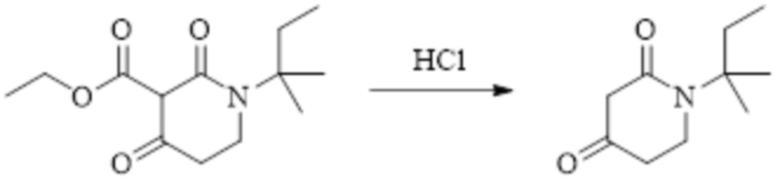
Промежуточный этил 2,4-диоксо-1-(трет-цикло)пиперидин-3-карбоксилат (48,11 ммоль, 1,0 экв.) растворяли в воде (100 мл). К раствору добавляли по каплям концентрированную соляную кислоту (10 мл, в массовая доле) и подвергали взаимодействию при температуре 80°C в течение 3 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до температуры 0°C и экстрагировали дихлорметаном (100 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали с получением продукта (8,4 г, выход за две стадии: 95,5%).
Стадия 5: Синтез 3-((диметиламино)метилен)-1-(трет-цикло)пиперидин-2,4-диона
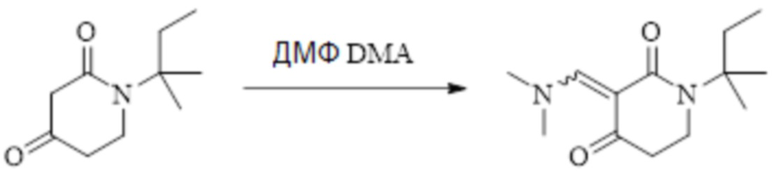
К промежуточному 1-(трет-цикло)пиперидин-2,4-диону (8,4 г, 45,84 ммоль, 1,0 экв.) добавляли по каплям N, N-диметилформамид диметилацеталь (6,0 г, 50,42 ммоль, 1,1 экв.) с интенсивным выделением тепла. После добавления реакцию проводили при комнатной температуре в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении с получением продукта, который использовали на следующей стадии в соответствии с теоретическим количеством.
Стадия 6: Синтез 5-(трет-цикло)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она

Промежуточный 3-((диметиламино)метилен)-1-(трет-цикло)пиперидин-2,4-дион (45,84 ммоль, 1,0 экв.) и гидразингидрат (массовая доля: 80%, 3,15 г, 50,42 ммоль, 1,1 экв.) растворяли в изопропаноле (100 мл). Раствор нагревали до температуры кипения с обратным холодильником в течение 0,5 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор концентрировали при пониженном давлении. Сырой продукт очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH=100:1-50:1) с получением продукта (4,29 г, выход за две стадии: 45,2%).
Стадия 7: Синтез (E)-2-(3-фтор-2-((4-оксо-5-(трет-цикло)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-диона
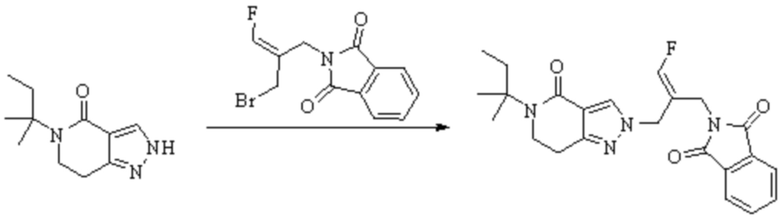
Промежуточный 5-(трет-цикло)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-он (2,0 г, 9,65 ммоль, 1,0 экв.), (E)-2-(2-(бромметил)-3-фтораллил) изоиндолин-1,3-дион (3,16 г, 10,61 ммоль, 1,1 экв.), безводный карбонат калия (1,46 г, 10,61 ммоль, 1,1 экв.) и тетрабутиламмоний бромид (341,7 мг, 1,06 ммоль, 0,1 экв.) добавляли в абсолютный этанол (20 мл) и смесь перемешивали при комнатной температуре в течение 48 ч. Когда оставалось небольшое количество исходных веществ, как определялось данными ТСХ, фильтровали. Слой на фильтре промывали этилацетатом. Фильтрат концентрировали при пониженном давлении с получением сырого продукта (4,9 г). Сырой продукт (2 г) очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=60:1) с получением маслянистой жидкости (821 мг).
Стадия 8: Синтез гидрохлорида (E)-2-(2-(аминометил)-3-фтораллил)-5-(трет-цикло)-2,5,6,7-тетрагидро-4H-пиразоло[4,3-c]пиридин-4-она
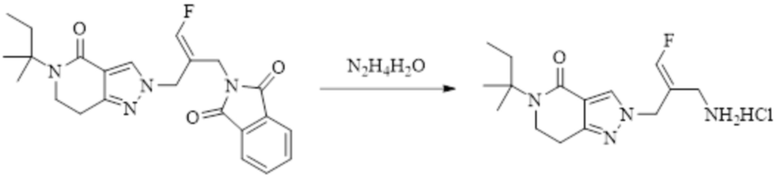
Промежуточный (E)-2-(3-фтор-2-((4-оксо-5-(трет-цикло)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-2-ил)метил)аллил)изоиндолин-1,3-дион (821 мг, 1,93 ммоль, 1,0 экв.) растворяли в этаноле (10 мл). К раствору добавляли гидразингидрат (массовая доля: 85%, 398,6 мг, 6,77 ммоль, 3,5 экв.) и подвергали взаимодействию при температуре 80°C в течение 1 ч. После того как не оставалось исходных веществ, как определялось данными ТСХ, реакционный раствор охлаждали до комнатной температуры и фильтровали в вакууме. Слой на фильтре промывали этанолом. Фильтрат концентрировали, суспендировали в дихлорметане (10 мл) и фильтровали, и полученный таким образом фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной тонкослойной хроматографии (ДХМ:MeOH=10:1) с получением продукта (329 мг). Продукт растворяли в дихлорметане (10 мл). К раствору по каплям добавляли раствор хлорида водорода в этаноле (массовая доля: 20%, 200 мг) и перемешивали в течение 5 мин. Концентрировали при пониженном давлении с получением продукта (352 мг, выход: 61,9%).
1Н ЯМР (300 МГц, ДМСО-d6) δ (м. д.): 8,38 (с, 3H), 8,17 (с, 1H), 7,40 (с, 0,5H), 7,13 (с, 0,5H), 4,89-4,88 (д, 2H), 3,54-3,50 (м, 2H), 3,36-3,35 (м, 2H), 2,76-2,71 (м, 2H), 1,94-1,87 (м, 2H), 1,38 (с, 6H), 0,79-0,74 (м, 3H).
Молекулярная формула: C15H24ClFN4O, молекулярная масса: 330,83, ЖХ-МС (полож., m/z)=295,11 [M+H]+.
В следующих биологических примерах настоящего изобретения все дозировки выражены для соединений в свободном виде.
Биологический пример 1: Анализ ферментативной активности
Образцы: Соединения по настоящему изобретению, показанные в таблице 1, которые были получены в соответствии с примерами способов
1. Ингибирующая активность соединений в отношении фермента rhVAP-1
(1) Лабораторные приборы, расходные материалы и реагенты
Многофункциональный считыватель микропланшетов (MD, FlexStation3), 96-луночный планшет с черным непрозрачным дном (Corning) и RhVAP-1 (PeproTech)
(2) Приготовление растворов соединений с градиентом концентрации
Брали соответствующее количество исследуемого соединения, растворяли в ДМСО до 10 мМ и затем хранили. Перед исследованием соответствующее количество маточного раствора 10 мМ исследуемого соединения разбавляли ДМСО, так что получали 1 мМ раствор. Затем раствор градиентно разбавляли в 3 раза ДМСО, всего 10 градиентов концентрации. Затем использовали PBS для 100-кратного разбавления, так что были приготовлены растворы соединений с 10-кратной серией концентраций.
(3) Приготовление растворов ферментов
К порошку rhVAP-1 добавляли соответствующее количество белкового разбавителя, получая 1 мг/мл маточного раствора для хранения. Перед исследованием для получения растворов ферментов с 4-кратной концентрацией для разведения использовали PBS.
(4) Приготовление смесей субстратов 2-х кратных концентраций
Отвешивали соответствующее количество бензиламина и растворяли в PBS с получением 200 мМ раствора бензиламина. К раствору бензиламина добавляли 2 мМ маточного раствора Amplex Red и 500 Ед/мл маточного раствора HRP и разбавляли PBS с получением смесей субстратов с 2-кратными концентрациями.
(5) Метод исследования
10 мкл растворов соединений различных концентраций, 25 мкл растворов фермента rhVAP-1 с 4-кратной концентрацией и 15 мкл PBS сначала добавляли в 96-луночный планшет, встряхивали для равномерного перемешивания и затем инкубировали при 37°C в течение 30 мин. Затем в каждую лунку добавляли 50 мкл смеси субстратов с 2-кратными концентрациями, и сразу использовали устройство для чтения микропланшетов для определения интенсивности флуоресценции каждой лунки с возбуждающим светом 565 нм и эмиссионным светом 590 нм в течение 5 минут каждый раз, всего 25 минут. Степень ингибирования рассчитывалась по следующей формуле:
V (RFU/мин)=(Ft(RFU)-F0(RFU))/(время (мин))
Степень ингибирования (%)=100%-Vсоед.(RFU/мин)/Vмакс.(RFU/мин)×100%
V: скорость изменения флуоресценции; Ft: показание флуоресценции в момент времени t; F0: начальное считывание флуоресценции; Время: продолжительность t; Vсоед.: скорость изменения флуоресценции исследуемого соединения; Vмакс.: максимальная скорость изменения флуоресценции отверстий.
(6) Подгонка кривой доза-ответ
С логарифмическим значением концентрации в качестве оси X и процентом ингибирования в качестве оси Y, логарифм (ингибитор) в зависимости от наклона переменная-отклик, для построения кривой доза-ответ использовали аналитическое программное обеспечение GraphPad Prism 5, таким образом получая значение IC50 активности каждого соединения в отношении фермента.
2. Селективность соединений в отношении фермента rhAOC1
(1) Лабораторные приборы, расходные материалы и реагенты
Считыватель микропланшетов (Perkin Elmer, Nivo 5S), 96-луночный планшет с черным непрозрачным дном (Corning) и rhAOC1(R&D)
(2) Приготовление растворов соединений с градиентом концентрации
Брали соответствующее количество исследуемого соединения, растворяли в ДМСО до 10 мМ и затем хранили. Перед исследованием отбирали соответствующее количество маточного раствора с 10 мМ исследуемого соединения и градиентно разбавляли в 3 раза ДМСО, всего 10 градиентов концентрации. Затем каждый градиент концентрации разбавляли в 10 раз 0,1 М PBS.
(3) Приготовление растворов ферментов
Брали соответствующее количество маточного раствора rhAOC1 с концентрацией 0,441 мг/мл и разбавляли соответствующим количеством 50 мМ буферного раствора HEPES для получения растворов ферментов с 4-кратной концентрацией.
(4) Приготовление смесей субстратов 2-х кратных концентраций
Отвешивали соответствующее количество гистамина и растворяли в 50 мМ буферного раствора HEPES с получением 20 мМ раствора гистамина. К раствору гистамина добавляли 2 мМ маточного раствора Amplex Red и 500 Ед/мл маточного раствора HRP и разбавляли 50 мМ буферного раствора HEPES с получением смесей субстратов с 2-кратными концентрациями.
(5) Метод исследования
10 мкл растворов соединений различных концентраций, 25 мкл растворов фермента rhAOC1 с 4-кратными концентрациями и 15 мкл 50 мМ буферного раствора HEPES сначала добавляли в 96-луночный планшет, встряхивали для равномерного перемешивания и затем инкубировали при температуре 37°C в течение 30 мин. Затем в каждую лунку добавляли 50 мкл смеси субстратов с 2-кратными концентрациями и сразу использовали считыватель микропланшетов для определения интенсивности флуоресценции каждой лунки с возбуждающим светом 580 нм (20 нм) и испускающим светом 620 нм (10 нм) в течение 5 минут каждый раз, всего 30 минут. Степень ингибирования рассчитывалась по следующей формуле:
V (RFU/мин)=(Ft(RFU)-F0(RFU))/(время (мин))
Степень ингибирования (%)=100%-Vсоед.(RFU/мин)/Vмакс.(RFU/мин)×100%
V: скорость изменения флуоресценции; Ft: показание флуоресценции в момент времени t; F0: начальное считывание флуоресценции; Время: продолжительность t; Vсоед.: скорость изменения флуоресценции исследуемого соединения; Vмакс.: максимальная скорость изменения флуоресценции отверстий.
(6) Подгонка кривой доза-ответ
С логарифмическим значением концентрации в качестве оси X и процентом ингибирования в качестве оси Y, логарифм (ингибитор) в зависимости от наклона переменная-отклик, для построения кривой доза-ответ использовали аналитическое программное обеспечение GraphPad Prism 5, таким образом получая значение IC50 активности каждого соединения в отношении фермента.
3. Результаты исследования представлены в виде таблицы 1
IC50 (нМ)
IC50 (нМ)
Из приведенной выше таблицы видно, что соединения по настоящему изобретению демонстрируют превосходное ингибирование фермента rhVAP-1 и демонстрируют превосходную селективность в отношении фермента rhAOC1. Это указывает на то, что соединения по настоящему изобретению могут быть использованы для профилактики и/или лечения заболеваний, связанных с увеличением экспрессии или повышением активности фермента VAP-1. Более того, поскольку соединения по настоящему изобретению превосходно ингибируют фермент VAP-1 и демонстрируют превосходную селективность в отношении фермента rhAOC1, побочные эффекты, вызванные ингибированием фермента rhAOC1, возникать не будут.
Биологический пример 2: Анализ ферментативной активности
Образцы: Соединения по настоящему изобретению, показанные в таблице 2, которые были получены в соответствии с примерами способов
1. Ингибирующая активность соединений в отношении rhVAP-1 Enzyme
(1) Лабораторные приборы, расходные материалы и реагенты
Считыватель микропланшетов (Perkin Elmer, Nivo 5S), 96-луночный планшет с черным непрозрачным дном (Corning) и rhVAP-1(PeproTech)
(2) Приготовление растворов соединений с градиентом концентрации
Брали соответствующее количество исследуемого соединения, растворяли в ДМСО до 10 мМ и затем хранили. Перед исследованием отбирали соответствующее количество маточного раствора с 10 мМ исследуемого соединения и градиентно разбавляли в 3 раза ДМСО, всего 10 градиентов концентрации. Затем каждый градиент концентрации разбавляли в 100 раз 0,1 М PBS.
(3) Приготовление растворов ферментов
К порошку rhVAP-1 добавляли соответствующее количество белкового разбавителя, получая 1 мг/мл маточного раствора для хранения. Перед исследованием для получения растворов ферментов с 4-кратной концентрацией для разведения использовали PBS.
(4) Приготовление смесей субстратов 2-х кратных концентраций
Отвешивали соответствующее количество бензиламина и растворяли в PBS с получением 200 мМ раствора бензиламина. К раствору бензиламина добавляли 2 мМ маточного раствора Amplex Red и 500 Ед/мл маточного раствора HRP и разбавляли PBS с получением смесей субстратов с 2-кратными концентрациями.
(5) Метод исследования
10 мкл растворов соединений различных концентраций, 25 мкл растворов фермента rhVAP-1 с 4-кратными концентрациями и 15 мкл 50 мМ буферного раствора HEPES сначала добавляли в 96-луночный планшет, встряхивали для равномерного перемешивания и затем инкубировали при температуре 37°C в течение 30 мин. Затем в каждую лунку добавляли 50 мкл смеси субстратов с 2-кратными концентрациями и сразу использовали считыватель микропланшетов для определения с возбуждающим светом 580 нм (20 нм) и испускающим светом 620 нм (10 нм) в течение 5 минут каждый раз, всего 30 минут. Степень ингибирования рассчитывалась по следующей формуле:
V (RFU/мин)=(Ft(RFU)-F0(RFU))/(время (мин))
Степень ингибирования (%)=100%-Vсоед.(RFU/мин)/Vмакс.(RFU/мин)×100%
V: скорость изменения флуоресценции; Ft: показание флуоресценции в момент времени t; F0: начальное считывание флуоресценции; Время: продолжительность t; Vсоед.: скорость изменения флуоресценции исследуемого соединения; Vмакс.: максимальная скорость изменения флуоресценции отверстий.
(6) Подгонка кривой доза-ответ
С логарифмическим значением концентрации в качестве оси X и процентом ингибирования в качестве оси Y, логарифм (ингибитор) в зависимости от наклона переменная-отклик, для построения кривой доза-ответ использовали аналитическое программное обеспечение GraphPad Prism 5, таким образом получая значение IC50 активности каждого соединения в отношении фермента.
2. Селективность соединений в отношении фермента rhAOC1
(1) Лабораторные приборы, расходные материалы и реагенты
Считыватель микропланшетов (Perkin Elmer, Nivo 5S), 96-луночный планшет с черным непрозрачным дном (Corning) и rhAOC1(R&D)
(2) Приготовление растворов соединений с градиентом концентрации
Брали соответствующее количество исследуемого соединения, растворяли в ДМСО до 10 мМ и затем хранили. Перед исследованием отбирали соответствующее количество маточного раствора с 10 мМ исследуемого соединения и градиентно разбавляли в 3 раза ДМСО, всего 10 градиентов концентрации. Затем каждый градиент концентрации разбавляли в 10 раз 0,1 М PBS.
(3) Приготовление растворов ферментов
Брали соответствующее количество маточного раствора rhAOC1 с концентрацией 0,441 мг/мл и разбавляли соответствующим количеством 50 мМ буферного раствора HEPES для получения растворов ферментов с 4-кратной концентрацией.
(4) Приготовление смесей субстратов 2-х кратных концентраций
Отвешивали соответствующее количество гистамина и растворяли в 50 мМ буферного раствора HEPES с получением 20 мМ раствора гистамина. К раствору гистамина добавляли 2 мМ маточного раствора Amplex Red и 500 Ед/мл маточного раствора HRP и разбавляли 50 мМ буферного раствора HEPES с получением смесей субстратов с 2-кратными концентрациями.
(5) Метод исследования
10 мкл растворов соединений различных концентраций, 25 мкл растворов фермента rhAOC1 с 4-кратными концентрациями и 15 мкл 50 мМ буферного раствора HEPES сначала добавляли в 96-луночный планшет, встряхивали для равномерного перемешивания и затем инкубировали при температуре 37°C в течение 30 мин. Затем в каждую лунку добавляли 50 мкл смеси субстратов с 2х концентрациями,, и сразу использовали считыватель микропланшетов для определения интенсивности флуоресценции каждой лунки с возбуждающим светом 580 нм (20 нм) и испускающим светом 620 нм (10 нм) в течение 5 минут каждый раз, всего 30 минут. Степень ингибирования рассчитывалась по следующей формуле:
V (RFU/мин)=(Ft(RFU)-F0(RFU))/(время (мин))
Степень ингибирования (%)=100%-Vсоед.(RFU/мин)/Vмакс.(RFU/мин)×100%
V: скорость изменения флуоресценции; Ft: показание флуоресценции в момент времени t; F0: начальное считывание флуоресценции; Время: продолжительность t; Vсоед.: скорость изменения флуоресценции исследуемого соединения; Vмакс.: максимальная скорость изменения флуоресценции отверстий.
(6) Подгонка кривой доза-ответ
С логарифмическим значением концентрации в качестве оси X и процентом ингибирования в качестве оси Y, логарифм (ингибитор) в зависимости от наклона переменная-отклик, для построения кривой доза-ответ использовали аналитическое программное обеспечение GraphPad Prism 5, таким образом получая значение IC50 активности каждого соединения в отношении фермента.
3. Результаты исследования представлены в таблице 2
IC50 (нМ)
IC50 (нМ)
NA - не исследовали.
Из приведенной выше таблицы видно, что соединения по настоящему изобретению демонстрируют превосходное ингибирование фермента rhVAP-1 и демонстрируют превосходную селективность в отношении фермента rhAOC1. Это указывает на то, что соединения по настоящему изобретению могут быть использованы для профилактики и/или лечения заболеваний, связанных с увеличением экспрессии или повышением активности фермента VAP-1. Более того, поскольку соединения по настоящему изобретению превосходно ингибируют фермент VAP-1 и демонстрируют превосходную селективность в отношении фермента rhAOC1, побочные эффекты, вызванные ингибированием фермента rhAOC1, возникать не будут.
Биологический пример 3: Селективность соединений по настоящему изобретению в отношении фермента MAO-A/B
(1) Лабораторные приборы, расходные материалы и реагенты
Считыватель микропланшетов (Perkin Elmer, EnVision), 384-луночный планшет (Perkin Elmer), центрифуга (Eppendorf), MAO-Glo™ (Promega), MAO-A (активный мотив) и MAO-B (активный мотив).
(2) Приготовление растворов соединений с градиентом концентрации
Брали соответствующее количество исследуемого соединения, растворяли в ДМСО до 10 мМ и затем хранили. Раствор затем градиентно разбавляли в 4 раза ДМСО, всего 6 градиентов концентрации.
(3) Приготовление растворов ферментов
Маточный раствор MAO-A/B разбавляли экспериментальным буфером для MAO-A/B для получения растворов ферментов с 2-кратными концентрациями.
(4) Приготовление смесей субстратов 2-х кратных концентраций
Маточный раствор смеси субстратов MAO-A/B разбавляли экспериментальным буфером для MAO-A/B с получением смесей субстратов с 2-кратными концентрациями.
(5) Метод исследования
200 нл растворов соединений или растворителей различных концентраций и 10 мкл растворов ферментов MAO-A/B с 2-кратной концентрацией добавляли в 384-луночный планшет, центрифугировали при 1000 об/мин в течение 60 с, встряхивали для равномерного перемешивания и затем инкубировали при комнатной температуре в течение 15 мин. Затем в каждую лунку добавляли 10 мкл смеси субстратов с 2-кратными концентрациями и инициировали реакцию. 384-луночный планшет центрифугировали при 1000 об/мин в течение 60 с, встряхивали для равномерного перемешивания и затем инкубировали при комнатной температуре в течение 60 мин. Для остановки реакции добавляли 20 мкл тест-стоп-раствора. Центрифугирование проводили при 1000 об/мин в течение 60 с и встряхивали для равномерного перемешивания. После 30 мин стояния считывали с помощью устройства для чтения микропланшетов.
Степень ингибирования рассчитывалась по следующей формуле:
Степень ингибирования (%)=(сигнал_макс.-сигнал_образец)/(сигнал_макс.-сигнал_мин)×100
(6) Подгонка кривой доза-ответ
С логарифмическим значением концентрации в качестве оси X и процентом ингибирования в качестве оси Y, логарифм (ингибитор) в зависимости от наклона переменная-отклик, для построения кривой доза-ответ использовали аналитическое программное обеспечение GraphPad Prism 5, таким образом получая значение IC50 активности каждого соединения в отношении фермента.
3. Результаты исследования представлены в таблице 3
IC50 (нМ)
IC50 (нМ)
Из таблицы 3 выше видно, что соединения по настоящему изобретению обладают хорошей ингибирующей активностью в отношении фермента rhVAP-1 и демонстрируют превосходную селективность в отношении моноаминоксидазы (MAO). Подводя итог, можно увидеть из таблицы 1, таблицы 2 и таблицы 3 выше, что при лечении и/или профилактике заболеваний, связанных с ферментом SSAO/VAP-1, соединения по настоящему изобретению не имеют побочных эффектов, вызванных ингибированием фермента rhAOC1 и фермента МАО.
Биологический пример 4: Оценка гематоэнцефалического барьера крысы для соединений по настоящему изобретению
(1) Введение животным и метод отбора образцов:
Исследуемое соединение A1, соединение A41 и PXS-4728 растворяли в физиологическом растворе для приготовления растворов. Приготовленные растворы вводили отдельно внутрижелудочно крысам SD в дозе 10 мг/кг. Время взятия проб крови и ткани головного мозга составляло: 0,0167 ч, 1 ч и 6 ч. В каждый момент времени образцы собирали у трех крыс SD.
(2) Сбор образцов:
В день исследования животных фиксировали для сбора 0,15 мл крови из яремной вены в каждый установленный момент времени, и образцы цельной крови помещали в пробирки для антикоагуляции, содержащие EDTA-K2. После завершения забора крови у животных сразу выполняли перфузию сердца, а после перфузии сердца собирали образцы ткани головного мозга.
(3) Обработка образцов:
После центрифугирования образцов цельной крови в условиях 1524 g в течение 10 минут верхние образцы плазмы собирали в пробирки для образцов. Образцы ткани взвешивали, добавляли 20% водный раствор метанола в соотношении массы к объему 1:5 (ткань : гомогенат) и гомогенизировали. Биологические образцы хранили при температуре от -40°C до -20°C для анализа.
(4) Способ анализа образцов:
Образец плазмы. Образцы для исследования доставали из холодильника, размораживали естественным путем при комнатной температуре, а затем перемешивали в течение 5 минут, и 20 мкл образца плазмы аккуратно вносили пипеткой в центрифужные пробирки объемом 1,5 мл. Добавляли 100 мкл рабочего раствора внутреннего стандарта (раствор 5 нг/мл верапамила и 50 нг/мл глибенкламида в ацетонитриле) и равномерно перемешивали. После 1 мин взбалтывания центрифугировали при 13000 об/мин в течение 8 мин. 40 мкл супернатанта аккуратно пипеткой вносили в 96-луночный планшет, предварительно добавляя 160 мкл воды на лунку. Равномерное перемешивание осуществляли путем взбалтывания в течение 10 мин, затем анализировали ЖХ-МС/МС.
Образец ткани головного мозга: Образцы для исследования доставали из холодильника, размораживали естественным путем при комнатной температуре, а затем перемешивали в течение 5 минут, и 50 мкл образца плазмы аккуратно вносили пипеткой в центрифужные пробирки на 1,5 мл. Добавляли 250 мкл рабочего раствора внутреннего стандарта (раствор 5 нг/мл верапамила и 50 нг/мл глибенкламида в ацетонитриле) и равномерно перемешивали. После 1 мин взбалтывания центрифугировали при 13000 об/мин в течение 8 мин. 30 мкл супернатанта аккуратно пипеткой вносили в 96-луночный планшет, предварительно добавляя 150 мкл воды на лунку. Равномерное перемешивание осуществляли путем взбалтывания в течение 10 мин, затем анализировали ЖХ-МС/МС.
(5) Метод обработки данных:
Для получения результатов по концентрации исследуемых соединений использовали Analyst 1.6.2 от компании AB Sciex. Для расчета таких параметров, как среднее значение, стандартное отклонение и коэффициент вариации использовали Microsoft Excel (за исключением значений, выводимых непосредственно Analyst 1.6.2).
(6) Результаты представлены в таблице 4
BLOQ означает, что концентрация ниже предела количественного определения; и NA представляет собой неисчислимое
Из приведенной выше таблицы 4 видно, что соединения A1 и A41 по настоящему изобретению даже с течением времени имеют такие низкие концентрации в тканях мозга, что эти соединения находятся ниже предела обнаружения. Это указывает на то, что соединения настоящего изобретения с трудом могут проходить через гематоэнцефалический барьер, поэтому токсический риск соединений настоящего изобретения для нервной системы очень низок.
Биологический пример 5: Оценка PK соединений на крысах
Введение животным и отбора образцов:
Исследуемые соединения A1, A6, A32, A41 и A42, каждое, растворяли в физиологическом растворе для приготовления растворов. Растворы соединений вводили отдельно внутрижелудочно крысам SD в дозе 5,0 мг/кг. Время для сбора крови составляло 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа.
Каждое из исследуемых соединений A1, A6, A32, A41 и A42 растворяли в физиологическом растворе для приготовления растворов. Растворы соединений отдельно вводили крысам SD внутривенно в дозе 1,0 мг/кг. Точки времени для сбора крови составляли 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа.
Катетеризацию вен проводили накануне введения. После введения из яремной вены собрали около 300 мкл крови и помещали в пробирки для антикоагуляции, содержащие EDTA-K2. Подготовку проводили в течение 30 мин после забора крови. Образцы плазмы получали центрифугированием при 8000 об/мин в течение 6 минут при температуре 4°C и перед исследованием плазмы хранили в холодильнике при температуре -80°C.
Способ анализа образцов:
(1) образцы для исследования доставали из холодильника с температурой -80°C, размораживали естественным путем при комнатной температуре, а затем перемешивали в течение 5 минут;
(2) 20 мкл образца плазмы аккуратно вносили пипеткой в центрифужные пробирки объемом 1,5 мл;
(3) добавляли 200 мкл рабочего раствора внутреннего стандарта (раствор толбутамида в метаноле) с концентрацией 100 нг/мл и равномерно перемешивали;
(4) через 5 мин взбалтывания центрифугировали при 12000 об/мин в течение 5 мин;
(5) 50 мкл супернатанта аккуратно пипеткой вносили в 96-луночный планшет, предварительно добавляя 150 мкл воды на лунку;
(6) равномерное перемешивание осуществляли путем взбалтывания в течение 5 мин, затем анализировали ЖХ-МС/МС.
Метод обработки данных:
Для получения результатов по концентрации исследуемых соединений использовали Analyst 1.6.3 от компании AB Sciex. Для расчета таких параметров, как среднее значение, стандартное отклонение и коэффициент вариации использовали Microsoft Excel (за исключением значений, выводимых непосредственно Analyst 1.6.3).
Программное обеспечение Pharsight Phoenix 6.1 NCA было адаптировано для расчета фармакокинетических параметров (Tmax было медианным).
Полученные результаты:
(л/кг)
(л/ч/кг)
(ч)
Примечание: Tz1/2: конечный период полураспада; Cl_obs: скорость очистки; Vz_obs: кажущийся объем распределения; Tmax: время максимальной наблюдаемой концентрации в плазме; AUCinf: площадь под кривой зависимости концентрации в плазме от времени 0-∞; F%: абсолютная биодоступность
Из результатов в таблице 5 видно, что соединения по настоящему изобретению имеют низкие скорости клиренса и превосходную абсолютную биодоступность при пероральном введении, что указывает на то, что соединения по настоящему изобретению обладают превосходными фармакокинетическими свойствами.
Промышленная применимость
Производное галогеналлиламина по настоящему изобретению может быть использовано для профилактики и/или лечения заболеваний, связанных с белком SSAO/VAP-1 или опосредованных им. Более того, соединение по настоящему изобретению демонстрирует превосходную селективную ингибирующую активность в отношении фермента VAP-1 по сравнению с ферментом rhAOC1 и ферментом МАО и вряд ли может вызывать другие побочные эффекты. Кроме того, по сравнению с существующим лекарственным средством соединение по настоящему изобретению с трудом проникает через гематоэнцефалический барьер, поэтому соединение по настоящему изобретению имеет очень низкий токсический риск для нервной системы, то есть безопасность соединения настоящее изобретение является превосходной.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ MAT2A И СПОСОБЫ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2809987C2 |
| ПРОТИВОГЕЛЬМИНТНЫЕ ДЕПСИПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2707298C2 |
| ПРОТИВОГЕЛЬМИНТНЫЕ ДЕПСИПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2715556C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ IRAK1/4 ИНГИБИТОРОВ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2730016C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| СПОСОБЫ И МАТЕРИАЛЫ ДЛЯ ПОВЫШЕНИЯ УРОВНЕЙ ПОЛИПЕПТИДА ФАКТОРА ТРАНСКРИПЦИИ EB | 2020 |
|
RU2838857C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
Изобретение относится к соединению формулы I или к его фармацевтически приемлемой соли, где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород; R3 и R4, каждый, независимо, представляют собой водород; R5 и R6, каждый, независимо, представляют собой водород; L1 отсутствует; Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными в общей формуле (A-1), (А-2), (А-3), (a), (b) или (c) ниже:
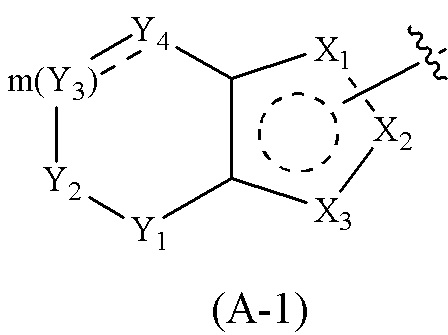
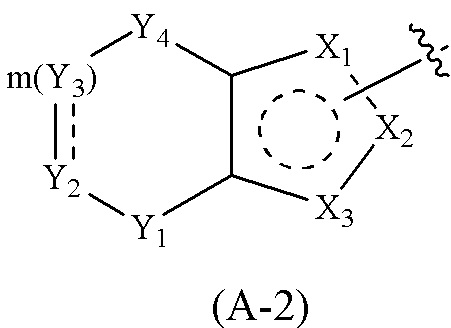
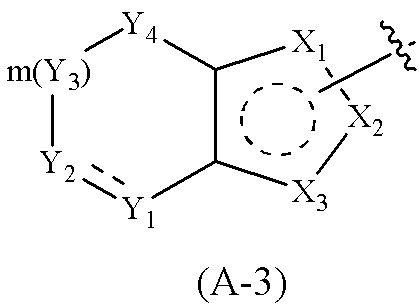
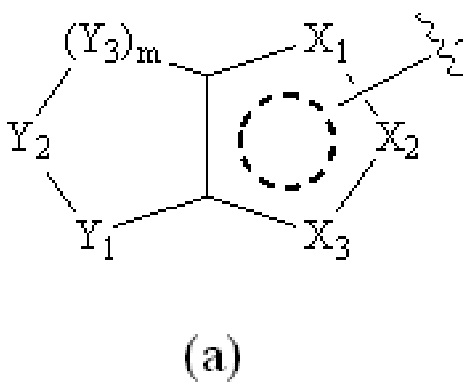
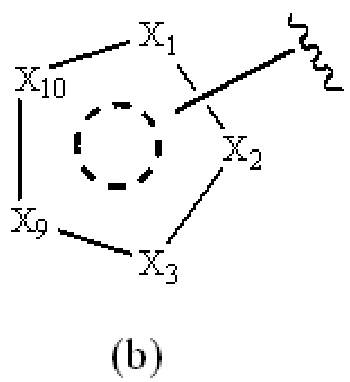
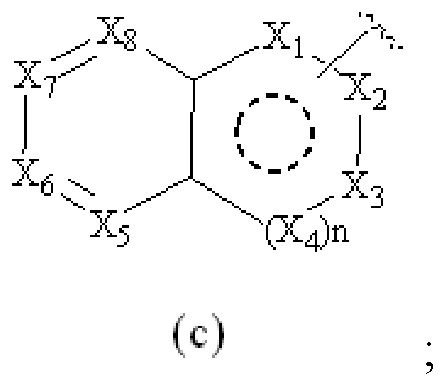
m обозначает целое число, которое обозначает 1 или 2 и n обозначает 0; Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CH2, СН, NH и C=O; X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CH, N, O и C=O; X5, X6, X7 и X8, каждый, независимо, выбраны из CH и N и по меньшей мере один из X1, X2 и X3 представляет собой N; каждый Ra независимо выбран из циано, галогена, C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкиламинокарбонила, аминокарбонила, ди(С1-6 алкил)аминокарбонила, Cy2-, Cy2-C1-6 алкила, Cy2-карбонила и Cy2-аминокарбонила, незамещенного или замещенного одним-тремя Rb, Cy2 представляет собой 3-6-членный циклоалкил, 4-6-членный гетероциклил, который относится к неароматической одновалентной циклической группе, образованной замещением циклических атомов углерода от 1 до 2 гетероатомов, выбранных из O и N, или фенил; каждый Rb независимо выбран из галогена, C1-6 алкила и C1-6 алкокси; при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra; при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O;  обозначает одинарную связь или двойную связь; и
обозначает одинарную связь или двойную связь; и  обозначает двойную связь, необязательно имеющуюся в кольцевой структуре. Изобретение также относится к фармацевтической композиции на основе указанного соединения. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при производстве лекарственного средства для профилактики и/или лечения заболевания, связанного с белком SSAO/VAP-1. 3 н. и 8 з.п. ф-лы, 5 табл., 48 пр.
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре. Изобретение также относится к фармацевтической композиции на основе указанного соединения. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при производстве лекарственного средства для профилактики и/или лечения заболевания, связанного с белком SSAO/VAP-1. 3 н. и 8 з.п. ф-лы, 5 табл., 48 пр.
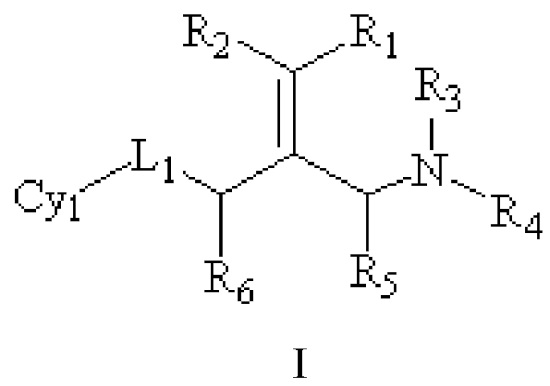
1. Соединение формулы I или его фармацевтически приемлемая соль
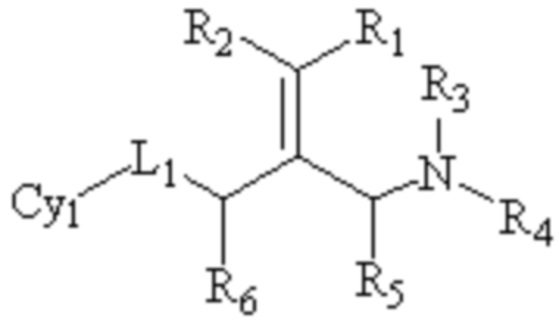
I,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, представляют собой водород;
R5 и R6, каждый, независимо, представляют собой водород;
L1 отсутствует;
Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными в общей формуле (A-1), (А-2), (А-3), (a), (b) или (c) ниже:

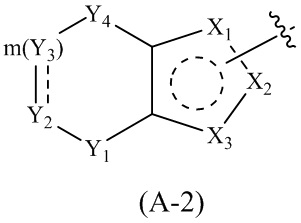
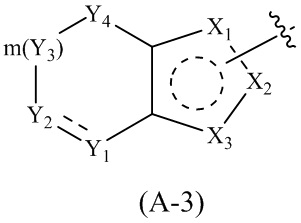
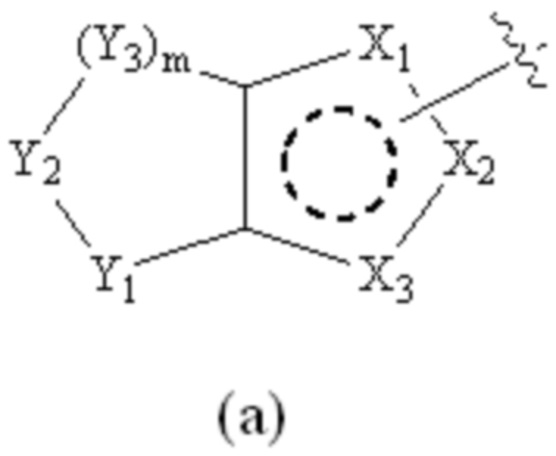
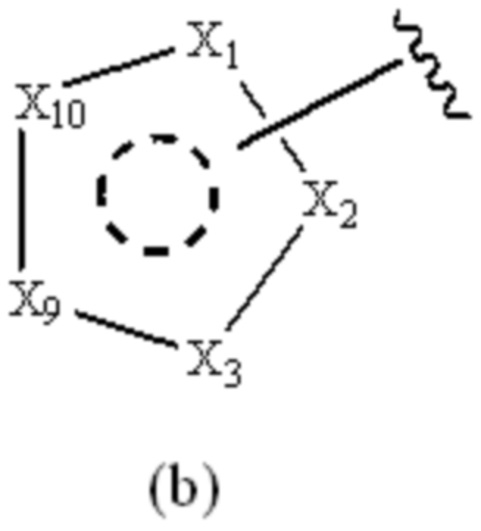
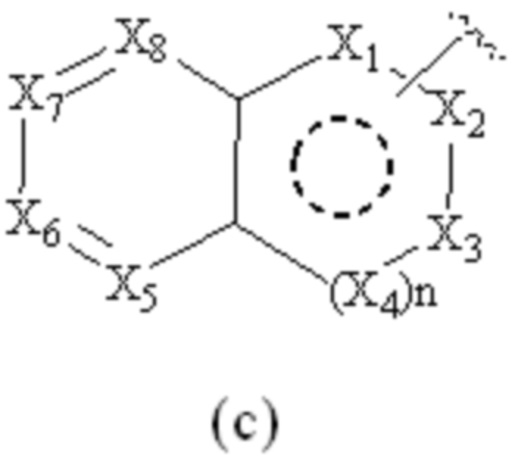 ;
;
m обозначает целое число, которое обозначает 1 или 2, и n обозначает 0;
Y1, Y2, Y3 и Y4, каждый, независимо, выбраны из CH2, СН, NH, и C=O;
X1, X2, X3, X4, X9 и X10, каждый, независимо, выбраны из CH, N, O и C=O, X5, X6, X7 и X8, каждый, независимо, выбраны из CH и N и по меньшей мере один из X1, X2 и X3 представляет собой N;
каждый Ra независимо выбран из циано, галогена, C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкиламинокарбонила, аминокарбонила, ди(С1-6 алкил)аминокарбонила, Cy2-, Cy2-C1-6 алкила, Cy2-карбонила и Cy2-аминокарбонила, незамещенного или замещенного одним-тремя Rb,
Cy2 представляет собой 3-6-членный циклоалкил, 4-6-членный гетероциклил, который относится к неароматической одновалентной циклической группе, образованной замещением циклических атомов углерода от 1 до 2 гетероатомов, выбранных из O и N, или фенил;
каждый Rb независимо выбран из галогена, C1-6 алкила и C1-6 алкокси;
при условии, что, когда Cy1 соответствует группе формулы (c), группа формулы (c) является замещенной одним или несколькими Ra;
при условии, что, когда Cy1 соответствует формуле (b), X1, X2, X3, X9 и X10 не представляют собой C=O;
 обозначает одинарную связь или двойную связь; и
обозначает одинарную связь или двойную связь; и
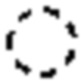 обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
2. Соединение или его фармацевтически приемлемая соль по п.1,
где Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными в общей формуле (A-11), (a-1), (a-2), (b-1) или (c-1) ниже:
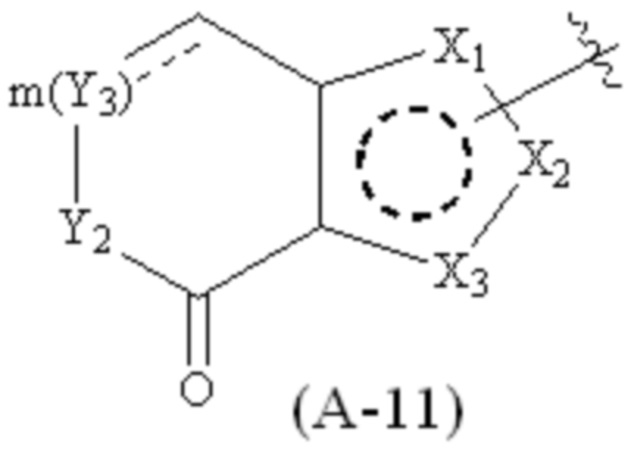
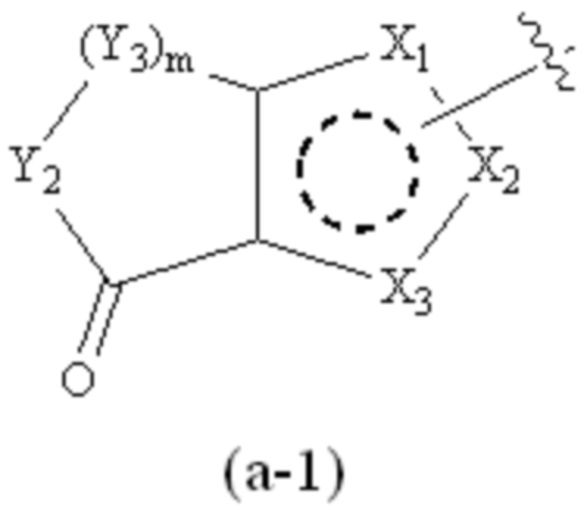
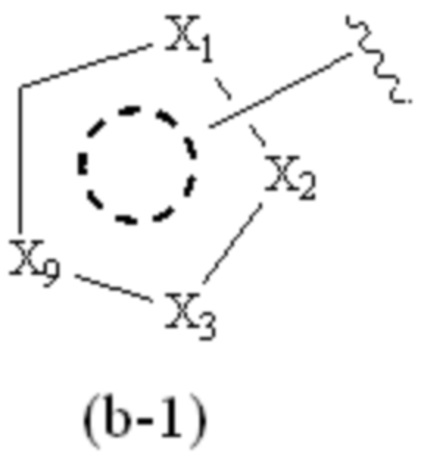
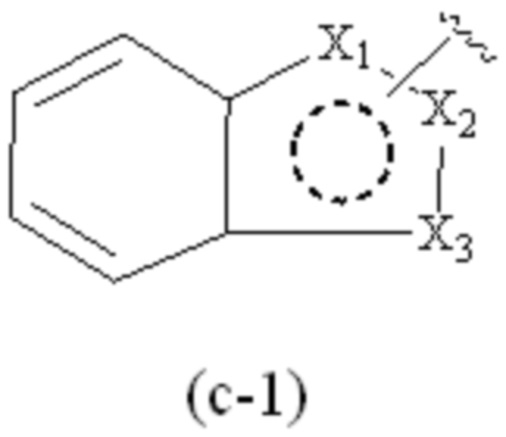 ;
;
m обозначает целое число, равное 1 или 2;
Y1, Y2 и Y3, каждый, независимо, выбраны из CH2, СН и NH;
X1, X2, X3 и X9, каждый, независимо, выбраны из CH, N и C=O и по меньшей мере один из X1, X2 и X3 представляет собой N;
при условии, что, когда Cy1 соответствует группе формулы (c-1), группа формулы (c-1) является замещенной одним или несколькими Ra;
при условии, что, когда Cy1 соответствует формуле (b-1), X1, X2, X3 и X9 не представляют собой C=O; и
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
3. Соединение или его фармацевтически приемлемая соль по п.2,
где Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными в общей формуле (A-11) или (a-1) ниже:
;
m обозначает целое число, равное 1 или 2;
Y2 и Y3, каждый, независимо, выбраны из CH2, СН и NH;
X1, X2 и X3, каждый, независимо, выбраны из CH и N и по меньшей мере один из X1, X2 и X3 представляет собой N; и
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре.
4. Соединение или его фармацевтически приемлемая соль по п.3,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, представляют собой водород;
R5 и R6, каждый, независимо, представляют собой водород;
L1 отсутствует;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
 ,
,  ,
,  ,
,  ,
, 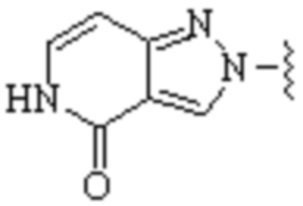 ,
,
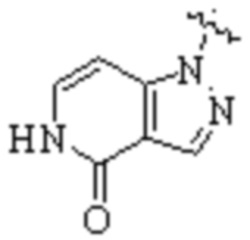 ,
, 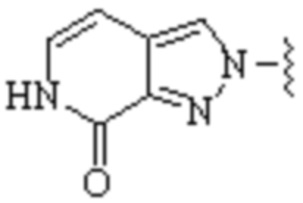 ,
, 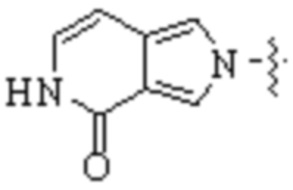 , и
, и 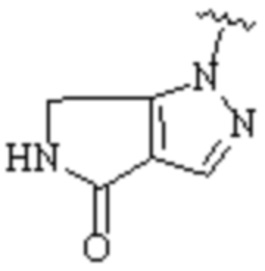 ;
;
каждый Ra независимо выбран из циано, галогена, C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкиламинокарбонила, аминокарбонила, ди(С1-6 алкил)аминокарбонила, Cy2, Cy2-C1-6 алкила, Cy2-карбонила и Cy2-аминокарбонила, незамещенного или замещенного одним-тремя заместителями Rb,
Cy2 представляет собой 3-6-членный циклоалкил, 4-6-членный гетероциклил, который относится к неароматической одновалентной циклической группе, образованной замещением циклических атомов углерода от 1 до 2 гетероатомов, выбранных из O и N, или фенил;
каждый Rb независимо выбран из галогена, C1-6 алкила и C1-6 алкокси;
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре; и
предпочтительно Cy2 представляет собой 3-6-членный циклоалкил, 5-6-членный гетероциклил или фенил.
5. Соединение или его фармацевтически приемлемая соль по п.4,
где Cy1 представляет собой группу, которая является незамещенной или замещена одним или несколькими Ra, показанными в общей формуле (b-1) ниже:
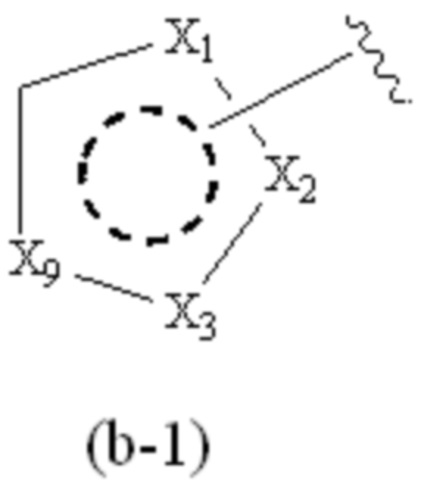 ;
;
X1, X2, X3 и X9, каждый, независимо, выбраны из CH и N и по меньшей мере один из X1, X2 и X3 представляет собой N; и
обозначает двойную связь, необязательно имеющуюся в кольцевой структуре;
при условии, что в общей формуле (b-1) X1, X2, X3 и X9 не представляют собой C=O.
6. Соединение или его фармацевтически приемлемая соль по п.5,
где R1 и R2, каждый, независимо, выбраны из водорода и галогена и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, представляют собой водород;
R5 и R6, каждый, независимо, представляют собой водород;
L1 отсутствует;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
 и
и  ;
;
каждый Ra независимо выбран из циано, галогена, C1-6 алкила, C1-6 алкокси C1-6 алкила, C1-6 алкиламинокарбонила, аминокарбонила, ди(С1-6 алкил)аминокарбонила, Cy2, Cy2-C1-6 алкила, Cy2-карбонила и Cy2-аминокарбонила, незамещенного или замещенного одним-тремя заместителями Rb,
Cy2 представляет собой 3-6-членный циклоалкил, 4-6-членный гетероциклил, который относится к неароматической одновалентной циклической группе, образованной замещением циклических атомов углерода от 1 до 2 гетероатомов, выбранных из O и N, или фенил; и
каждый Rb независимо выбран из галогена, C1-6 алкила и C1-6 алкокси.
7. Соединение или его фармацевтически приемлемая соль по п.1,
где Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими Ra:
 ,
,  ,
,  , ,
, ,  ,
,
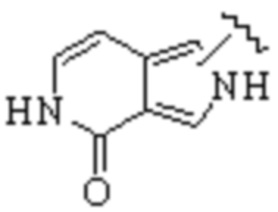 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 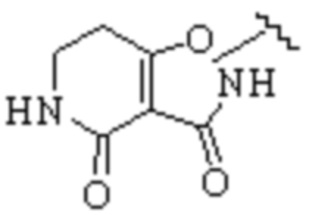 ,
, 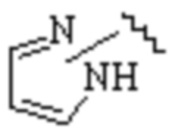 ,
, 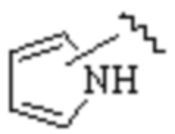 ,
, 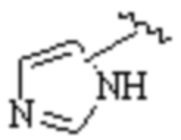 и
и  .
.
8. Соединение или его фармацевтически приемлемая соль по п.7,
где R1 и R2, каждый, независимо, выбраны из водорода и фтора и оба R1 и R2 не представляют собой водород;
R3 и R4, каждый, независимо, представляют собой водород;
R5 и R6 представляют собой водород;
L1 отсутствует;
Cy1 представляет собой одну из следующих групп, незамещенную или замещенную одним или несколькими заместителями Ra:
, , , , ,
, , 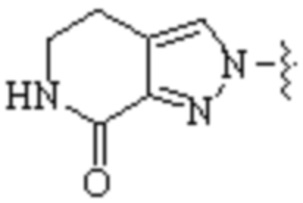 , , , , ,
, , , , , 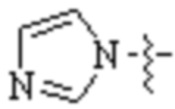 и
и  .
.
каждый Ra независимо выбран из циано, галогена, C1-6 алкила, C1-6 алкиламинокарбонила, Cy2, Cy2-карбонила и Cy2-аминокарбонила, незамещенного или замещенного одним-тремя заместителями Rb,
Cy2 представляет собой 3-6-членный циклоалкил, 5-6-членный гетероциклил или фенил;
каждый Rb независимо выбран из галогена, C1-6 алкила и C1-6 алкокси;
предпочтительно Cy1 представляет собой одну из следующих групп, замещенную одним или несколькими заместителями Ra:
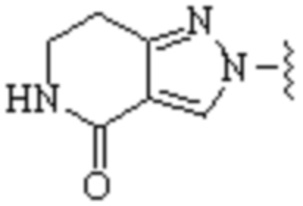 , , , ,
, , , ,
, , и ;
предпочтительно каждый Ra независимо выбран из циано, галогена, C1-6 алкила и 3-6-членного циклоалкила, незамещенного или замещенного одним-тремя заместителями Rb; и
предпочтительно каждый Rb представляет собой галоген.
9. Соединение или его фармацевтически приемлемая соль по п.1, где соединение выбрано из следующих:
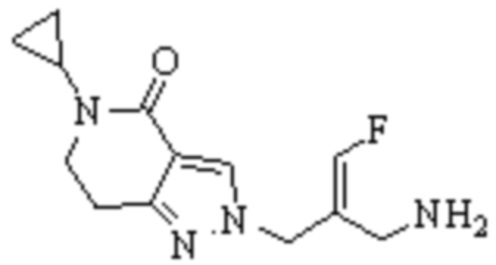
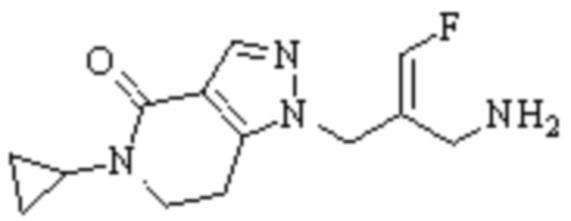

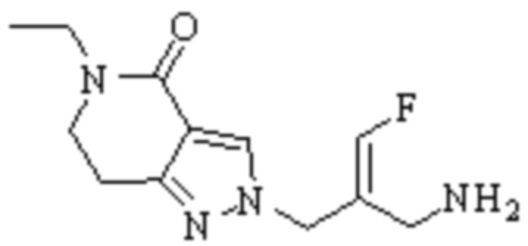
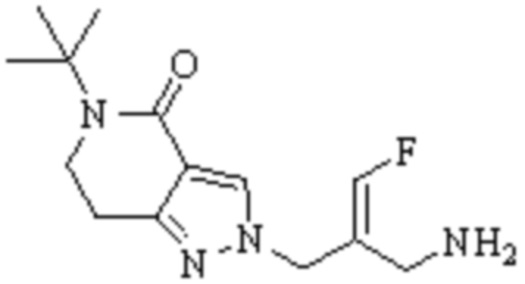
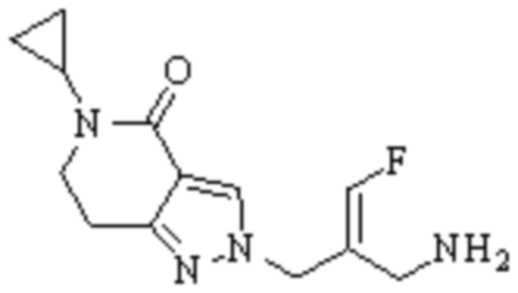
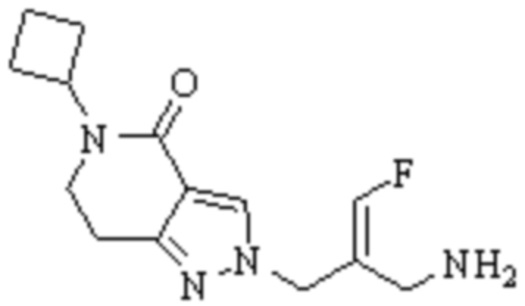

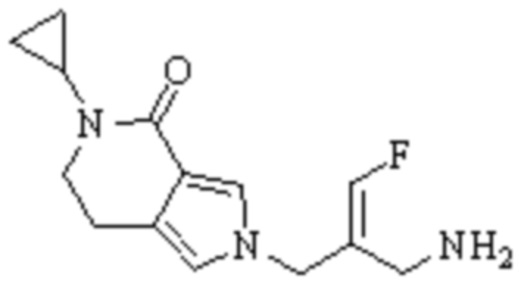
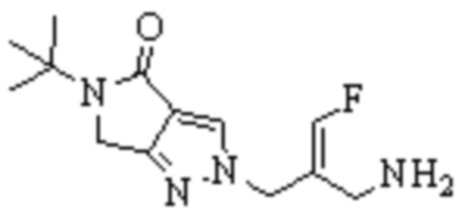
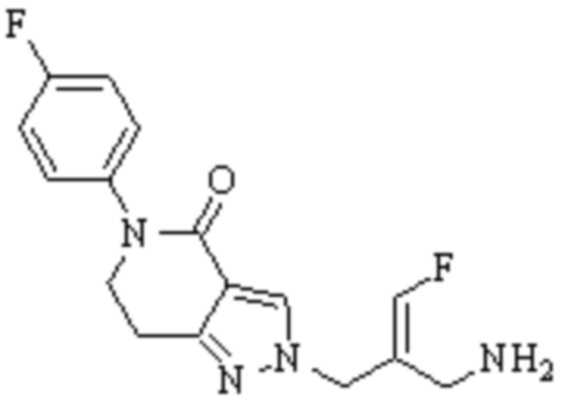

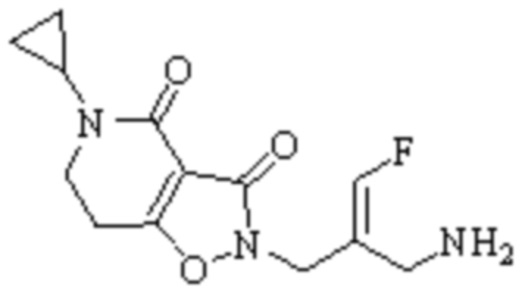
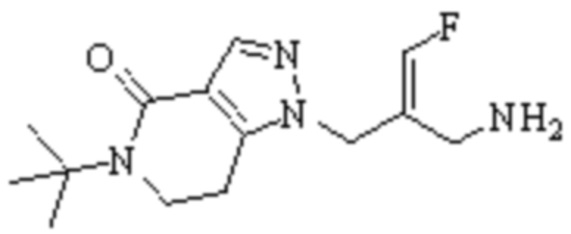

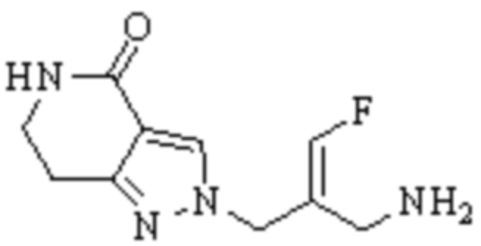
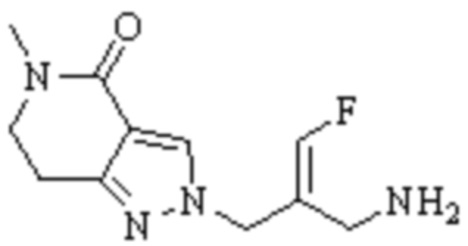
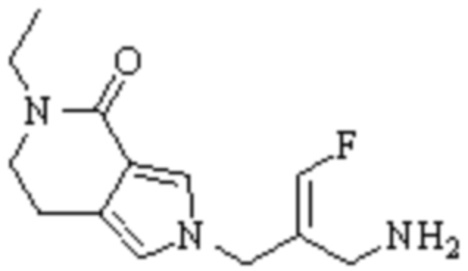
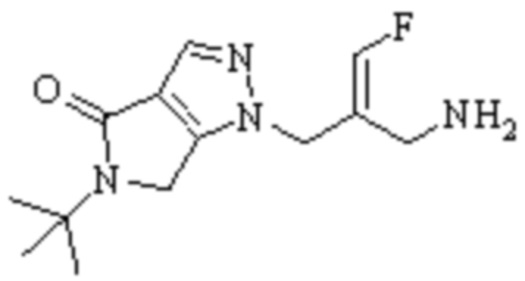
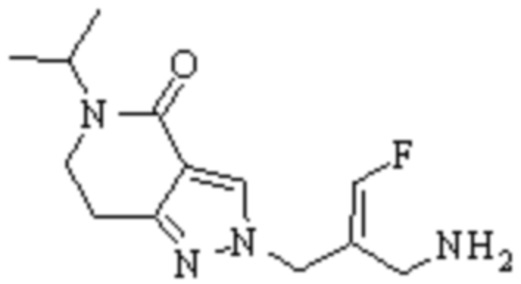
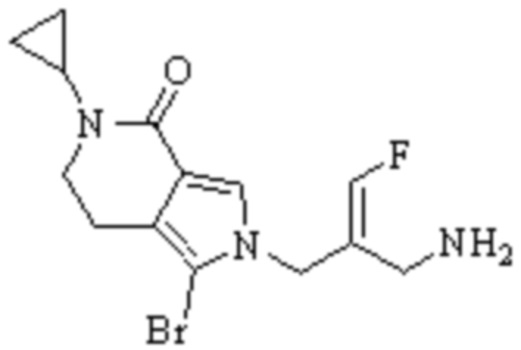
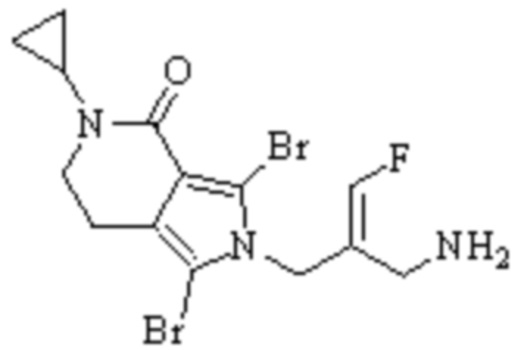
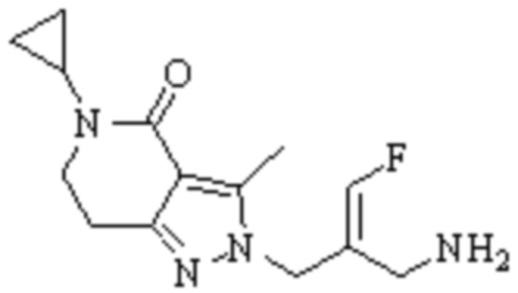
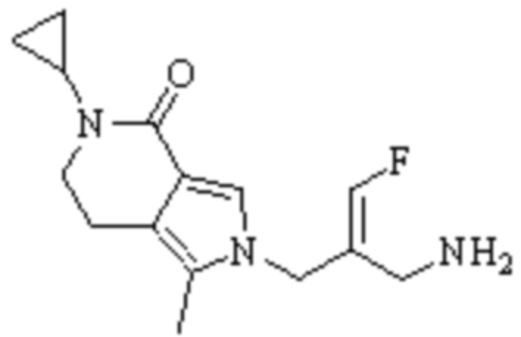
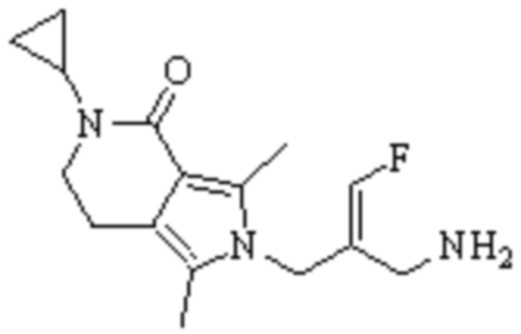
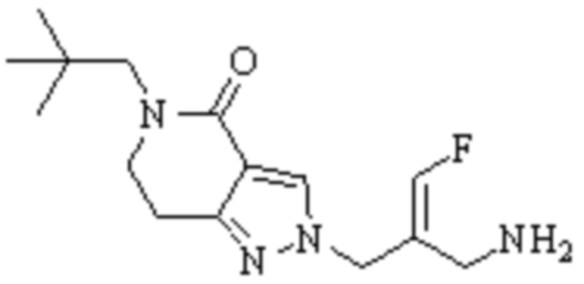
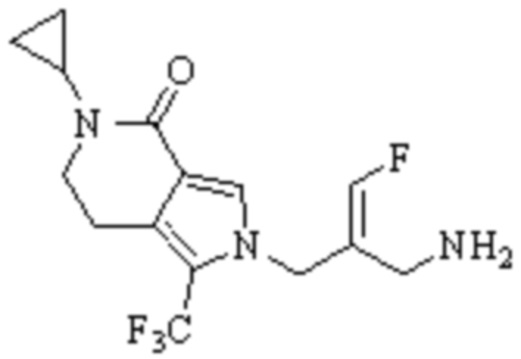
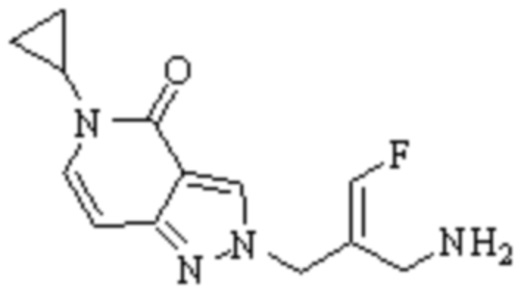
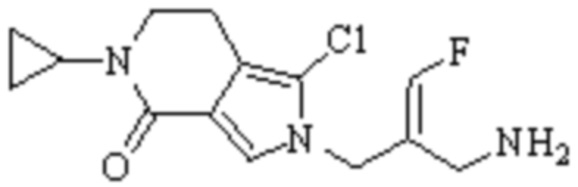
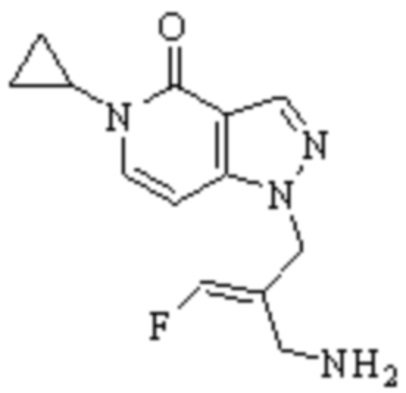
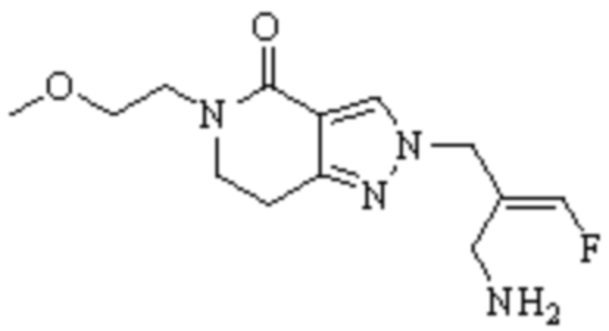
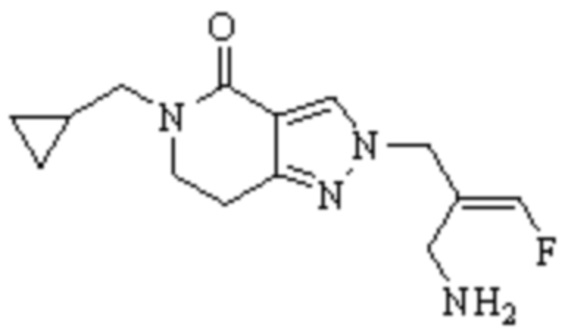
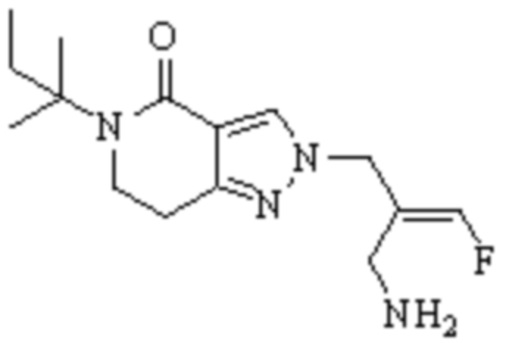
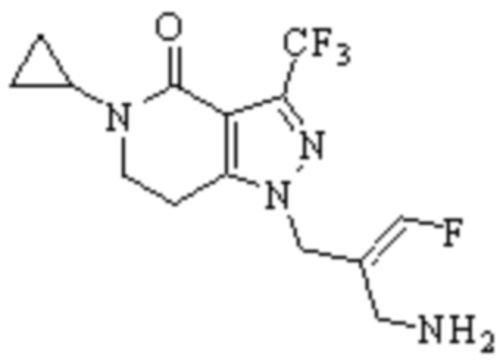
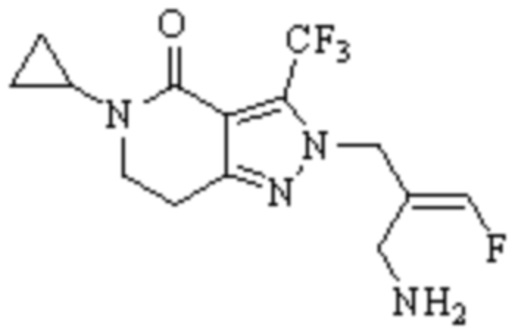
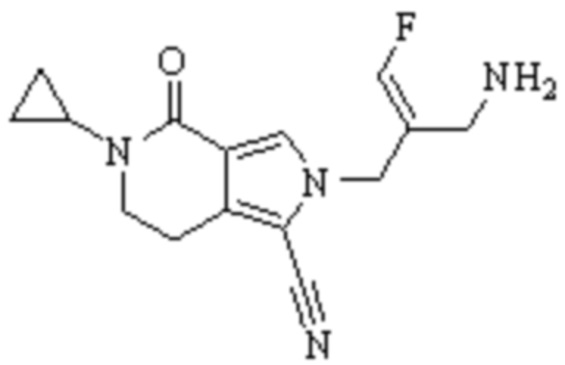
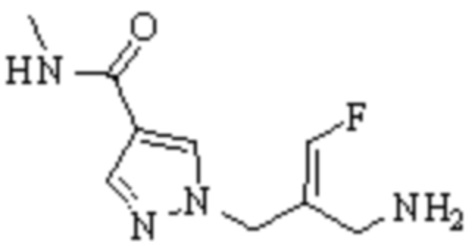

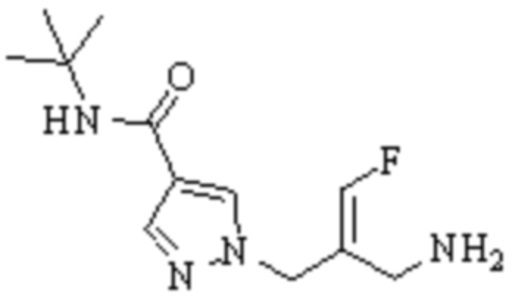
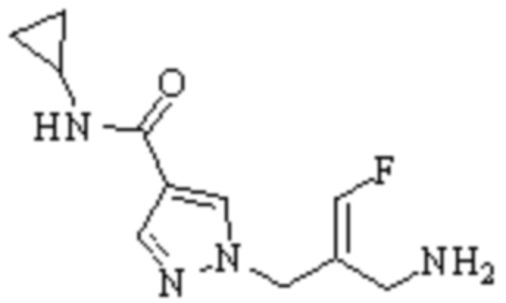


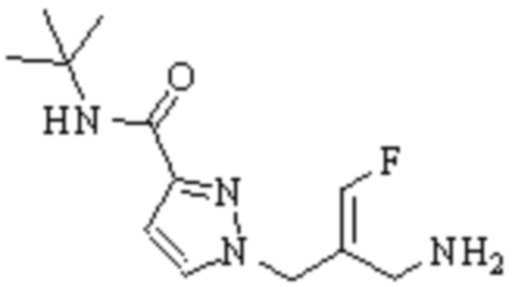
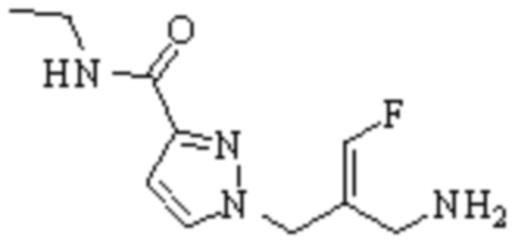
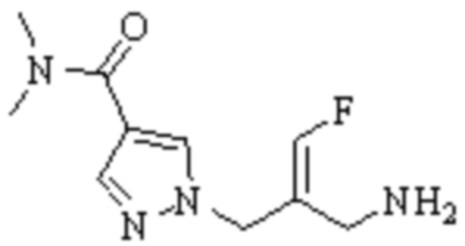
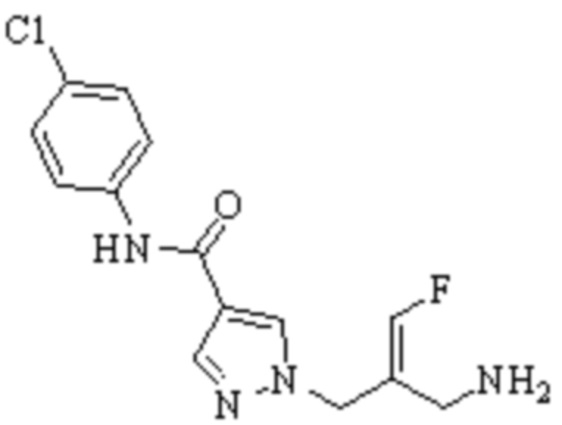
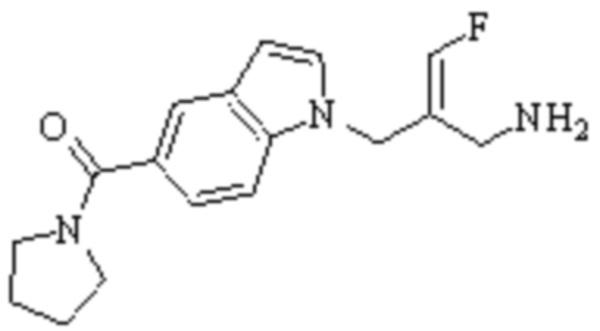
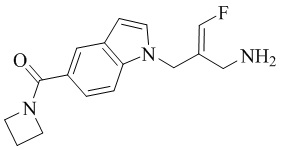
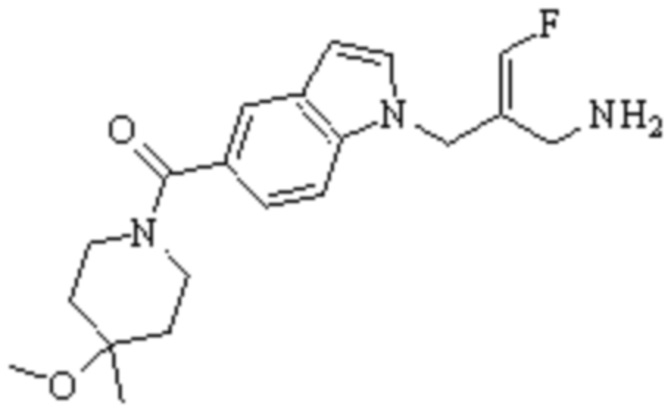

10. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении фермента SSAO/VAP-1, содержащая соединение или его фармацевтически приемлемую соль по любому из пп.1-9, где фармацевтическая композиция необязательно содержит один или несколько фармацевтически приемлемых носителей.
11. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-9 или фармацевтической композиции по п.10 при производстве лекарственного средства для предотвращения и/или лечения заболевания, связанного с белком SSAO/VAP-1 или опосредованного им.
| WO 2018151985 A1, 23.08.2018 | |||
| WO 2018196677 A1, 01.11.2018 | |||
| WO 2013163675 A1, 07.11.2013 | |||
| WO 2018028517 A1, 15.02.2018 | |||
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ VAP-1 ДЛЯ ЛЕЧЕНИЯ ФИБРОЗНЫХ БОЛЕЗНЕЙ | 2010 |
|
RU2667963C1 |

