ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По этой заявке испрашивается приоритет временной заявки на патент США 61/139479, поданной 19 декабря 2008, содержание которой полностью включено в настоящее описание путем ссылки.
УРОВЕНЬ ТЕХНИКИ
Апоптоз теперь считается существенным биологическим процессом, ответственным за гомеостаз тканей всех живых организмов. В частности, было показано, что у млекопитающих он регулирует раннее эмбриональное развитие. Позднее в течение жизни гибель клеток представляет собой механизм по умолчанию, с помощью которого удаляются потенциально опасные клетки (например, клетки, несущие канцерогенные дефекты). Были раскрыты несколько путей апоптоза, и один из самых важных включает семейство белков Bcl-2, которые являются ключевыми регуляторами митохондриального (также называемого "внутренним") пути апоптоза. См., Danial, N.N. and Korsmeyer, SJ. Cell (2004) 116, 205-219. Для этого семейства белков характерны области структурной гомологии BH1, BH2, BH3 и BH4. Семейство белков Bcl-2 может быть далее классифицировано на три подсемейства в зависимости от того, сколько из областей гомологии содержит каждый белок и от его биологической активности (то есть имеет ли он про- или антиапоптотическую функцию).
Первая подгруппа включает белки, имеющие все 4 домена гомологии, то есть BH1, BH2, BH3 и BH4. Их общий эффект является антиапоптотическим, который призван защитить клетку от начала процесса гибели клетки. Такие белки как, например, Bcl-2, Bcl-w, BCl-XL, MCl-1 и Bfl-1/A1 являются членами этой первой подгруппы. Белки, принадлежащие ко второй подгруппе, содержат три домена гомологии BH1, BH2 и BH3 и имеют проапоптотический эффект. Двумя главными представителями белков этой второй подгруппы являются Bax и Bak. Наконец, третья подгруппа состоит из белков, содержащих только домен BH3, и члены этой подгруппы обычно упоминаются как "белки BH3-only." Их биологическое действие на клетку является про-апоптотическим. Bim, Bid, Bad, Bik, Noxa, Hrk, Bmf и Puma являются примерами этого третьего подсемейства белков. Точный механизм, которым семейство белков Bcl-2 регулирует гибель клеток, все еще полностью не известен, и понимание этого механизма является активной областью исследований в научном сообществе. В одной гипотезе регулирования гибели клеток семейством белков Bcl-2, белки BH3-only далее категоризируются либо как белки "активаторы" (например, Bim и Bid), либо как "сенсибилизаторы" (например, Bad, Bik, Noxa, Hrk, Bmf и Puma) в зависимости от их регулирующей функции.
Ключ к гомеостазу ткани достигает тонкого баланса во взаимодействиях среди всех трех подгрупп белков в клетках. В недавних исследованиях была сделана попытка объяснить механизмы, которыми проапоптотические и антиапоптотические подгруппы семейства белков Bcl-2 взаимодействуют так, чтобы позволить клетке перенести программированную клеточную гибель. После получения внутри- или внеклеточных сигналов в клетках происходит посттрансляционная или транскрипционная активация белков BH3-only. Белки BH3-only являются первичными индукторами апоптотического каскада, который включает, в качестве одной стадии, активацию про-апоптотических белков Bax и Bak на митохондриальной мембране в клетках. После активации Bax и/или Bak, которые либо уже заякорены на митохондриальной мембране или мигрируют к этой мембране, Bax и/или Bak олигомеризуются, приводя к повышению проницаемости наружной мембраны митохондрий (MOMP), высвобождению цитохрома C и даунстрим-активации эффекторных каспаз, в конечном счете приводя к апоптозу клетки. Некоторые исследователи выдвигают гипотезу, что некоторые белки BH3-only (например, Puma, Bim, Bid) являются "активаторами", поскольку эти белки непосредственно рекрутируют проапоптотические белки Bax и Bak, чтобы инициировать MOMP, в то время как другие белки BH3-only (например, Bad, Bik и Noxa) являются "сенсибилизаторами" и вызывают олигомеризацию Bax и Bak косвенно, связывая антиапоптотические белки (например, Bcl-2, BCl-XL, Bcl-w, Mcl-1) и вытесняя и "освобождая" белки "активаторы" BH3-only, которые впоследствии связываются с проапоптотическими белками и активизируют их (например, Bax, Bak), вызывая гибель клеток. Другие исследователи предполагают, что антиапоптотические белки рекрутируют и секвестрируют Bax и Bak непосредственно и все белки BH3-only регулируют это взаимодействие, связываясь с антиапоптотическими белками (например, Bcl-2, BCl-XL, Bcl-w, Mcl-1), что приводит к высвобождению Bax и Bak. См., Adams, J.M. and Cory S. Oncogene (2007) 26, 1324-1337; Willis, S.N. et al. Science (2007) 315, 856-859. Хотя точные взаимодействия, через которые анти- и проапоптотические белки семейства Bcl-2 регулируют апоптоз, остаются предметом обсуждений, существует значительное число научных доказательств, показывающих, что соединения, которые ингибируют связывание белков BH3-only с антиапоптотическими белками семейства Bcl-2, промотируют апоптоз в клетках.
Подвергшиеся дисрегуляции пути апопотоза участвуют в патологии многих существенных заболеваний, таких как нейродегенеративные состояния (апрегулируемый апоптоз), такие как, например, болезнь Альцгеймера; и пролиферативные заболевания (даунрегулируемый апоптоз), такие как, например, рак, аутоиммунные заболевания и предтромбозные состояния.
В одном аспекте, участие даунрегулируемого апоптоза (и более конкретно семейства белков Bcl-2) в начале злокачественного процесса открыло новый путь нацеливания этого все еще неуловимого заболевания. Исследование показало, например, что антиапоптотические белки, Bcl-2 и BCl-XL, суперэкспрессируются во многих типах раковых клеток. См., Zhang J.Y., Nature Reviews/Drug Discovery, (2002) 1, 101; Kirkin, V. et al. Biochimica et Biophysica Acta (2004) 1644, 229-249; and Amundson, S.A. et al. Cancer Research (2000) 60, 6101-6110. Эффектом этой дерегуляции является выживание измененных клеток, которые в противном случае подверглись бы апоптозу в нормальных условиях. Копирование этих дефектов, связанных с нерегулируемой пролиферацией, как считается, является исходной точкой злокачественного процесса. Дополнительно, исследование показало, что белки BH3-only могут действовать как супрессоры опухоли, когда они экспрессируются в организме больных животных.
Эти открытия, так же как многочисленные другие, сделали возможным появление новых стратегий в разработке лекарственного средства для нацеливания рака. Если бы малая молекула, которая могла бы миметировать эффект белков BH3-only, была в состоянии поступить в клетку и преодолеть суперэкспрессию антиапоптотического белка, было бы возможно снова запустить апоптотический процесс. Эта стратегия может иметь преимущество в том, что она может облегчить проблему лекарственной резистентности, которая является обычным последствием дерегуляции апоптоза (аномальное выживание).
Исследователи также продемонстрировали, что тромбоциты также содержат необходимые апоптотические механизмы {например, Bax, Bak, BCl-XL, Bcl-2, цитохром c, каспаза-9, каспаза-3 и APAF-1) для осуществления запрограммированной гибели клеток через внутренний путь апоптоза. Хотя продукция тромбоцитов крови представляет собой нормальный физиологический процесс, множество заболеваний вызывается либо усиливается избытком или нежелательной активацией тромбоцитов. Вышесказанное позволяет предположить, что терапевтические средства, способные к ингибированию антиапоптотических белков в тромбоцитах и уменьшению числа тромбоцитов у млекопитающих, могут быть использованы в лечении предтромбозных состояний и заболеваний, которые характеризуются избытком или нежелательной активацией тромбоцитов.
Abbott Laboratories Inc. разработал класс малых молекул, являющихся миметиками белка BH3-only, то есть АВТ-737 и АВТ-263, которые в значительной степени связываются с субпопуляциями антиапоптотических белков Bcl-2, включая Bcl-2, Bcl-w и BCl-XL, но только слабо связываются с Mck-1 и А1 и показывают механизм-основанную цитотоксичность. Эти соединения были проверены в экспериментальном исследовании и продемонстрировали цитотоксическую активность в некоторых моделях ксенотрансплантата в качестве единственных средств, а также усиливали эффекты множества химиотерапевтических средств на других моделях ксенотрансплантата при использовании в комбинации. См., Tse, C. et a Cancer Res (2008) 68, 3421-3428; и van Delft, M.F. et a Cancer Cell (2006) 10, 389-399. Эти исследования in vivo позволяют предположить потенциальную полезность ингибиторов антиапоптотических белков семейства Bcl-2 для лечения заболеваний, которые включают дисрегуляцию пути апоптоза.
Естественные уровни экспрессии членов семейства антиапоптотических белков Bcl-2 варьируют в различных типах клеток. Например, в молодых тромбоцитах, белок BCl-XL высоко экспрессируется и играет важную роль в регуляции клеточной гибели (продолжительность жизни) тромбоцитов. Кроме того, в некоторых типах раковых клеток выживание раковых клеток приписывается дисрегуляции пути апоптоза, вызванному суперэкспрессией одного или более членов семейства антиапоптотических белков Bcl-2. Ввиду важной роли семейства белков Bcl-2 в регуляции апоптоза как в раковых, так и в нормальных (то есть нераковых) клетках, и признанной межклеточной вариабельности типа экспрессии белков семейства Bcl-2, предпочтительно располагать малой молекулой-ингибитором, которая бы селективно нацеливала и предпочтительно связывалась с одним типом или субпопуляцией антиапоптотического белка (белков) Bcl-2, например, антиапоптотическому члену семейства Bcl-2, который суперэкспрессируется при определенном типе рака. Такое селективное соединение также может иметь определенные преимущества в клинических условиях, обеспечивая, например, среди прочего гибкость в выборе режима введения, сниженный токсический эффект нацеливания в нормальных клетках (например, лимфопения наблюдалась у мышей, дефицитных в отношении Bcl-2). См., Nakayama, K. et al. PNAS (1994) 91, 3700-3704.
Ввиду вышеизложенного, в данной области существует потребность в терапевтических средствах на основе малой молекулы, которая может селективно ингибировать активность одного типа или субпопуляции антиапоптотических белков Bcl-2, например, антиапоптотического белка BCl-XL. Настоящее изобретение отвечает по меньшей мере этой потребности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте настоящее изобретение относится к соединению Формулы I
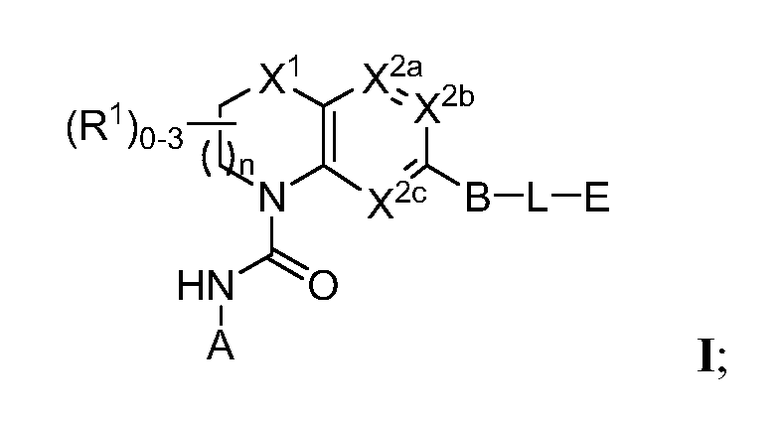
или к его фармацевтически приемлемой соли, в которой R1 является независимо членом, выбранным из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила и галогена. В Формуле I индекс n означает целое число от 0 до 2, причем, когда n=0, тогда, X1 обозначает -CH2-, -C(H)(Ra)- или -C(Ra)2. X1 обозначает член, выбранный из группы, состоящей из -CH2-,-C(H)(Ra)-, -C(Ra)2, -O-, -N(H)-, -N(Ra)-, -N(C(O)Ra)-, -N(C(O)ORa)-, -N(S(O)2Ra)-, -N(S(O)Ra)-, -S-, -S(O)-, -S(O)2-, в котором Ra выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила, C3-6 циклоалкила и галогена. X2a, X2b и X2c, каждый независимо, выбраны из группы, состоящей из C(H), C(R2) и N, причем по меньшей мере один из X2a и X2b обозначает C(H) или C(R2); и причем R2 независимо выбран из группы, состоящей из -ORb, -NRbRc, -SRb, -C(O)ORC, -C(O)NRbRc, -NRbC(O)Rd, -S(O)2Rd, -S(O)Rd, -S(O)2NRbRc, -Rd, галогена, -CN и -NO2. Для заместителя R2, Rb и RC, каждый независимо, выбраны из группы, состоящей из водорода, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галогеналкила, или в случае необходимости Rb и RC, вместе с атомом, к которому каждый из них присоединен, образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла; и Rd выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила и C1-6 галогеналкила. В Формуле I A обозначает член, выбранный из группы, состоящей из:

в которых R3 независимо выбран из группы, состоящей из -NReRf, -ORe, -CN, -NO2, галогена, -C(O)OR6, -C(O)NReRf, -NReC(O)Rf, -NReS(O)2Rg, -NR6S(O) Rg, -S(O)2Rg, S(O)Rg и -Rg. Для группы R3, Re и Rf в каждом случае каждый независимо выбраны из группы, состоящей из водорода, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галогеналкила и -(CH2)1-4 фенила, или Re и Rf, или Re и Rg вместе с атомом, к которому каждый из них присоединен, в случае необходимости образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла; и Rg выбран из группы, состоящей из C1-4 алкила, C2-4 алкенила, C2-4 алкинила и C1-4 галогеналкила. B обозначает член, выбранный из группы, состоящей из:
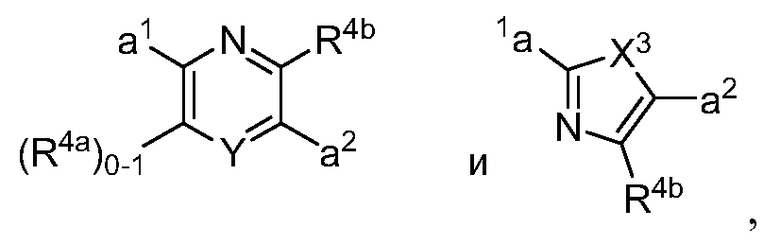
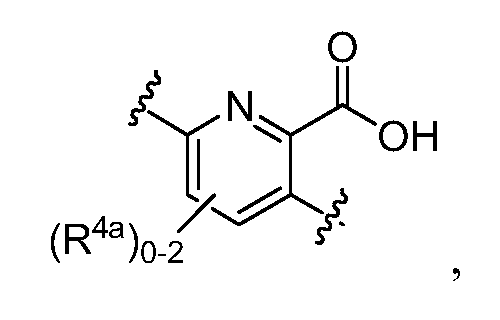
в которых Y обозначает N, C(H) или C(R4a); X3 обозначает -N(H), -N(C1-3 алкил), O или S; R4a, в случае его присутствия, независимо выбран из C1-4 алкила, C1-4 галогеналкила, C2-4 алкенила, C2-4 алкинила, галогена и -CN. R4b, в каждом случае, независимо выбран из группы, состоящей из -C(O)ORj, -C(O)NRhRi, -C(O)Ri, -NRhC(O)Ri, -NRhC(O)NRhRi, -OC(O)NRhRi, -NRhC(O)ORj, -C(=NORh)NRhRi -NRhC(=NCN)NRhRi, -NRhS(O)2NRhRi, -S(O)2RJ, -S(O)2NRhRi, -N(Rh)S(O)2Ri, -NRhC(=NRi)NRhRi, -C(=S)NRhRi, -C(=NRh)NRhRi, галогена, -NO2 и -CN, в которых Rh и Ri в каждом случае, каждый независимо, выбраны из группы, состоящей из водорода, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C1-6 галогеналкила, фенила и -(CH2)1-4 фенила, или Rh и Ri, или Rh и Rj, вместе с атомом, к которому каждый из них присоединен, в случае необходимости образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. Rj выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила, C3-7 циклоалкила, фенила и -(CH2)1-4 фенила; или, альтернативно, R4 выбран из группы, состоящей из

в которых Rk выбран из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила и C1-6 галогеналкила. В Формуле I L отсутствует или обозначает член, выбранный из группы, состоящей из C6-10 арилен-C1-6 гетероалкилена, C5-9 гетероарилен-C1-6 гетероалкилена, C1-6 гетероалкилена, C1-6 алкилена, C1-6 галогеналкилена, C2-6 алкенилена, C2-6 алкинилена, -NH-, -S- и -O-, причем алкиленовая, алкениленовая, алкиниленовая или гетероалкиленовая части группы L замещены от 0 до 4 раз заместителями R5a, выбранными из группы, состоящей из галогена, -Rm и =О, и ароматические части группы L замещены от 0 до 4 раз заместителями R5b, выбранными из группы, состоящей из галогена, -ORn, -NRnRo, -Rn, -NO2 и CN; причем Rm выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 гетероалкила и C1-6 галогеналкила. В случае необходимости любые два заместителя R5a, присоединенные к одному и тому же или к разным атомам L, могут вместе образовывать 5-7-членное карбоциклическое кольцо или 5-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла; и причем Rn и Ro, в каждом случае, выбраны из группы, состоящей из водорода, C1-6 алкила, C2-6 алкенила, C2-6 алкинила и C1-6 галогеналкила, и причем в случае необходимости Rn и Ro, вместе с атомами, к которым каждый из них присоединен, вместе образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. В Формуле I E обозначает водород или галоген; или, альтернативно, E выбран из группы, состоящей из фенила, C5-6 гетероарила, C3-7 гетероциклоалкила и C3-7 циклоалкила, и в случае необходимости с E могут быть сконденсированы 1 или 2 кольца, независимо выбранных из группы, состоящей из 3-7-членного карбоциклического кольца, 3-7-членного гетероциклического кольца, бензольного кольца и 5-6-членного гетероароматического кольца, причем E и каждое кольцо, в случае необходимости конденсированное с E, независимо замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из галогена, -NRpRq, -SRP, -ORP, -C(O)ORP, -C(О)NRpRq, -C(O)RP, -NRpC(О)Rq, -OC(O)Rr, -NRpC(О)NRpRq, -OC(О)NRpRq, -NRpC(О)ORr, -C(=NORp)NRpRq, -NRpC(=N-CN)NRpRq, -NRpS(О)2NRpRq, -S(O)2Rr, -S(O)2NRpRq, -Rr, -Rs, -NO2, -N3, =О, -CN, -Z1-NRpRq, -Z1SRp, -Z1-OR13, -Z1-C(O)ORp, -Z1-C(O)NRpRq, -Z1-C(O)Rp, -Z1-NRpC(O)Rq, -Z1-OC(O)Rr, -Z1-NRpC(O)NRpRq, -Z1-OC(O)NRpRq, -Z1-NRpC(O)ORr, -Z1- C(=NORp)NRpRq, -Z1-NRpC(=N-CN)NRpRq, -Z1-NRpS(O)2NRpRq, -Z1S(O)2Rr, -Z1-S(O)2NRpRq, -Z1-NO2, -Z1-N3, -Z1-Rs и -Z1-CN. В Формуле I Z1 выбран из группы, состоящей из C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена, C1-6 гетероалкилена, C3-7 циклоалкилена и C3-7 гетероциклоалкилена; Rp и Rq, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила, фенила и -(CH2)1-4-фенила; Rr выбран из группы, состоящей из C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила, фенила и -(CH2)1-4-фенила. В случае необходимости в составе каждого заместителя R6 Rp и Rq или Rp и Rr, вместе с атомом, к которому каждый из них присоединен, в случае необходимости вместе образуют 3-7-членное гетероциклическое кольцо, в случае необходимости включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. Rs выбран из группы, состоящей из фенила, C5-6 гетероарила, C3-7 гетероциклоалкила, C3-7 циклоалкила и с Rs может быть конденсировано 1 или 2 кольца, которые, каждый независимо, выбраны из группы, состоящей из 5-7-членного карбоциклического кольца, 5-7-членного гетероциклического кольца, бензольного кольца и 5-6-членного гетероароматического кольца, и Rs и каждое кольцо, в случае необходимости конденсированное с Rs, каждый независимо, замещены от 0 до 5 раз заместителями R7, выбранными из группы, состоящей из галогена, -NRtRu, -SRt, -ORt, -C(O)ORt, -C(O)NRtRu, -C(O)Rt, -NRtC(О)RV, -OC(О)RV, -NRtC(O)NRtRu, -OC(O)NRtRr, -NRtC(O)ORv, -C(=NORt)NRtRu, NRtC(=N-CN)NRtRu, -NRtS(O)2NRtRu, -S(O)2RV, -S(O)2NRtRu, -Rv, -NO2, -N3, =О, -CN, -Z2- NRtRu, -Z2-SRt, -Z2-ORt, -Z2-C(O)ORt, -Z2-C(O)NRtRu, -Z2-C(O)RV, -Z2-NRtC(O)Ru, -Z2-OC(О)Rv, -Z2-NRtC(O)NRtRu, -Z2-OC(O)NRtRu, -Z2-NRtC(О)ORv, -Z2-C(=NORt)NRtRu, -Z2-NRtC(=N-CN)NRtRu, -Z2-NRtS(O)2NRtRu, -Z2-S(O)2RV, -Z2-S(O)2NRtRu, -Z2-NO2, -Z2-N3 и -Z2-CN. Z2 выбран из группы, состоящей из C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена, C1-6 гетероалкилена, Rt и Ru, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила и -(CH2)1-4-фенила, C3-7 циклоалкила и C3-7 гетероциклоалкила; Rv выбран из C1-4 алкила, C1-4 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила и -(CH2)1-4-фенила. В каждом заместителе R7, Rt и Ru или Rt и Rv, вместе с атомом, к которому каждый из них присоединен, в случае необходимости вместе образуют 3-7-членное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранные из N, O и S в качестве вершин цикла.
В другом аспекте, настоящее изобретение относится к фармацевтическим композициям, включающим соединения Формулы I, а также к способам применения соединений Формулы I для лечения заболеваний и состояний (например, рака, тромбоцитемии и т.д.), характеризующихся экспрессией или суперэкспрессией антиапоптотических белков Bcl-2, например, антиапоптотических белков Bcl-XL.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 показывает некоторые подформулы соединений по изобретению, то есть подформулы I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-k, I-m, I-n, I-o и I-p.
Фиг. 2A, Фиг. 2B, Фиг. 2C, Фиг. 2d, Фиг. 2E и Фиг. 2F показывают некоторые варианты групп E для соединений Формулы I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
1. Определения
В рамках изобретения термин "алкил", отдельно или как часть другого заместителя, означает, если не указано иное, углеводородный радикал с прямой или разветвленной цепью, имеющий указанное число атомов углерода (то есть C1-6 означает от одного до восьми атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" относится к ненасыщенному алкильному радикалу, имеющему одну или более двойных связей и включает моно- и полигалогензамещенные варианты. Точно так же термин "алкинил" относится к ненасыщенному алкильному радикалу, имеющему одну или более тройных связей и включает моно- и полигалогензамещенные варианты. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил, и высшие гомологи и изомеры. Термины "циклоалкил", "карбоциклический" и "карбоцикл" используются взаимозаменяемо и, когда они используются отдельно или как часть другого заместителя, относятся к углеводородным кольцам, имеющим обозначенное число кольцевых атомов (например, C3-6 циклоалкил) и являющимся полностью насыщенными или имеющим не более одной двойной связи между вершинами цикла. В рамках изобретения "циклоалкил", "карбоциклический" или "карбоцикл" также относятся к бициклическим, полициклическим и спироциклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, пинан, бицикло[2.2.2]октан, адамантан, норборен, спироциклический C5-12 алкан и т.д. "Циклоалкил", "карбоциклическое" кольцо или "карбоцикл" может быть присоединен к остатку молекулы через кольцевой атом углерода или, если это также указано, альтернативно, "циклоалкил", "карбоциклическое" кольцо или "карбоцикл" может быть конденсирован с остатком молекулы. Неограничивающие примеры "циклоалкила", "карбоциклического" кольца или "карбоцикла", конденсированного, например, с бензольным кольцом, включают 1,2,3,4-тетрагидронафталин, 2,3-дигидро-1H-инден, (Z)-6,9-дигидро-5H-бензо[7]аннулен и т.п.
Термин "гетероалкил", отдельно или в комбинации с другим термином, означает, если не указано иное, стабильный углеводородный радикал с прямой или разветвленной цепью, состоящий из указанного числа атомов углерода и от одного до трех гетероатомов, выбранных из группы, состоящей из O, N, Si и S, и причем атомы азота и серы могут быть окислены, и гетероатом азота может быть кватернизован. Гетероатом(ы) O, N и S может быть помещен в любое внутреннее положение гетероалкильной группы. Гетероатом Si может быть помещен в любое положение гетероалкильной группы, включая положение, в котором алкильная группа связана с остатком молекулы. "Гетероалкил" может содержать до трех единиц ненасыщенности (например, двойные и тройные связи), и также включает моно- и поли-галогензамещенные варианты или их комбинации. Примеры "гетероалкила" включают -CH2-CH2-O-CH3, -CH2-CH2-O-CF3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CHs)-CH3, -CH2-S-CH2-CH3, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -Si(CH3)3, -CH2-CH=N-OCH3 и -CH=CH=N(CH3)-CH3. Кроме того, в случае "гетероалкила" до двух гетероатомов могут быть последовательными, как, например, в -CH2-NH-OCH3 и -CH2-O-Si(CH3)3.
Термины "гетероциклоалкил", "гетероциклический" и "гетероцикл" используются взаимозаменяемо, и когда они используются отдельно или как часть другого заместителя, они относятся к циклоалкильной группе, которая содержит от одного до пяти гетероатомов, выбранных из N, O и S, причем атомы азота и серы могут быть окислены, и атом(ы) азота может быть кватернизован. Специалисту понятно, относительно "гетероциклоалкила", "гетероциклического" и "гетероцикла", имеющего указанное число атомов углерода (например, "C3-7 гетероциклоалкил"), что по меньшей мере один, и возможно до пяти, если это осуществимо, атомов углерода заменены гетероатомом. Например, "C3 гетероциклоалкил" включает, среди других возможностей, оксиранил, который имеет два атома углерода плюс один атом кислорода в качестве членов кольца. Если не указано иное, "гетероциклоалкил", "гетероциклическое" кольцо и "гетероцикл" может быть моноциклической, бициклической, спироциклической или полициклической кольцевой системой. Неограничивающие примеры "гетероциклоалкила", "гетероциклической" группы и "гетероцикла" включают пирролидин, пиперидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, диоксолан, фталимид, пиперидин, пиримидин-2,4(1Н,3H)-дион, пиримидин-4-он, пиримидин-2-он, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин, тропан и т.п. "Гетероциклоалкил", "гетероциклическая" группа или "гетероцикл" может быть присоединен(а) к остатку молекулы через кольцевой углерод, гетероатом, или альтернативно, если это также указано, "гетероциклоалкил", "гетероциклическая" группа или "гетероцикл" может быть конденсирован(а) с остатком молекулы. Неограничивающие примеры "гетероциклоалкила", "гетероциклического" кольца или "гетероцикла", конденсированного с, например, бензольным кольцом, включают изохроман, 2,3-дигидробензофуран, (Z)-4,5-дигидро-1H-бензо[b]азепин и т.п. Если не указано иное, "гетероциклоалкил", "гетероциклическое" кольцо и "гетероцикл" включают моно- и полигалогензамещенные варианты.
Термин "алкилен", отдельно или как часть другого заместителя, означает двухвалентный радикал, полученный из алкана или галогеналкана, как иллюстрируется -CH2CH2CH2CH2- и -CF2CF2-. Как правило, алкильная (или алкиленовая) группа имеет от 1 до 24 атомов углерода, причем группы, имеющие 10 или менее атомов углерода, являются в настоящем изобретении предпочтительными. "Алкенилен" и "алкинилен" относятся к ненасыщенным формам "алкилена", имеющим двойные или тройные связи, соответственно, включая моно и полигалогензамещенные варианты.
Термин "гетероалкилен", отдельно или как часть другого заместителя, означает двухвалентный радикал, полученный из гетероалкила, как иллюстрируется -O- CH2-CH2-CH2-CH2-O-, -О-CH2, -CH2-O-, -CH2-CH2-S-CH2CH2- и -CH2-S-CH2-CH2-NH-CH2-, -О-CH2-CH=CH-, -CH2-CH=C(H)CH2-O-CH2-, -О-CH2-CH=CH-, -S-CH2-OC-, -CF2-O-. В случае гетероалкиленовых групп гетероатом может также занимать любое или оба из концов цепи (например, алкиленокси, алкилендиокси, алкиленамино, алкилендиамино и т.п.). В рамках изобретения термин "гетероалкилен" также относится к моно- и полигалогензамещенным вариантам.
Термины "алкокси", "алкиламино" и "алкилтио" (или тиоалкокси) используются в их обычном смысле и относятся к алкильным группам, присоединенным к остатку молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Дополнительно, в случае диалкиламиногрупп, алкильные части могут быть одинаковыми или разными и могут также вместе образовывать 3-7-членное кольцо с атомом азота, к которому каждый из них присоединен. Соответственно, группа, представленная как -NRiRii включает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин "галоген", сам по себе или как часть другого заместителя, означает, если не указано иное, атом фтора, хлора, брома или йода. Дополнительно, такие термины как "галогеналкил" включают моногалогеналкил и полигалогеналкил. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "арил" означает, если не указано иное, полиненасыщенную, обычно ароматическую, углеводородную группу, которая может представлять собой единственное кольцо или множество колец (до трех колец), которые конденсированы вместе. Термин "гетероарил" относится к арильным группам (или кольцам), которые содержат от одного до пяти гетероатомов, выбранных из N, O и S, причем атомы азота и серы могут быть окислены, и атом(ы) азота могут быть окислены. Гетероарильная группа может быть связана с остатком молекулы через гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и дифенил, в то время как неограничивающие примеры гетероарильных групп включает пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолинил, фталазинил, бензотриазинил, пуринил, бензимидазолил, бензопиразолил, бензотриазолил, бензизоксазолил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридинил, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Дополнительные заместители для каждой вышеуказанной арильной и гетероарильной кольцевой системы могут быть выбраны из, но не ограничены ими, группы приемлемых заместителей, описанных далее.
В рамках изобретения термин "арилен" в общем относится к любому арилу, который является двухвалентным радикалом. В качестве более конкретного примера, "фенилен" относится к двухвалентному фенильному кольцевому радикалу. Термины, "1,2-арилен", "1,3-арилен" или "1,4-арилен" относятся к геометрическим изомерам особого арилена, в которых две группы, присоединенные к арилу, как изображено в формуле, расположены в орто-, мета- или пара-положении в геометрическом отношении к арилу, соответственно.
В рамках изобретения термин "гетероарилен" в общем относится к любому гетероарилу, который является двухвалентным радикалом. В качестве более конкретного примера, "пиридилен" относится к двухвалентному пиридильному кольцевому радикалу.
Специалисту понятно, относительно терминов "гетероарил" и "гетероарилен", имеющие определяемое число атомов углерода (например, "C5-6 гетероарил" или "C5-9 гетероарилен"), что по меньшей мере один и, если это осуществимо, до пяти из указанных атомов углерода заменены гетероатомом. C5 гетероарил, например, может представлять собой пирролил или, как другой пример, представлять собой тиазолил, среди других возможностей.
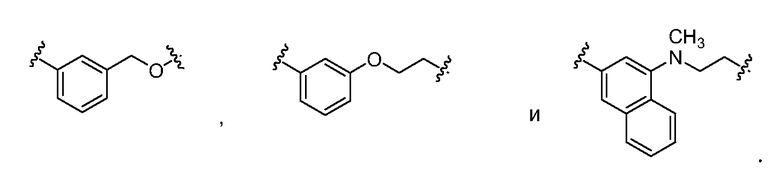
В рамках изобретения, комбинированный термин "арилен-гетероалкилен" в общем относится к двухвалентному радикалу, состоящему из арильной группы и гетероалкильной группы, которые являются ковалентно присоединенными друг к другу, и причем арильная и алкильная группа, каждая, включает дополнительный радикальный центр, к которому может быть присоединена другая группа. Примеры арилен-гетероалкилена включают, но не ограничены:
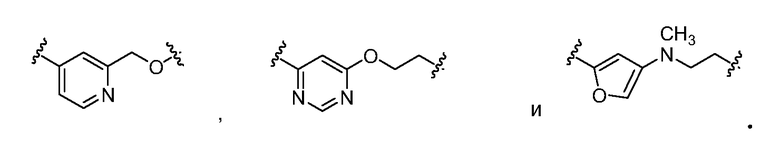
Точно так же термин "гетероарилен-гетероалкилен" относится к двухвалентному радикалу, состоящему из гетероарильной группы и гетероалкильной группы, которые являются ковалентно присоединенными друг к другу, и причем гетероарильная и гетероалкильная группа, каждая, включает дополнительный радикальный центр, к которому присоединена другая группа. Примеры гетероарилен-гетероалкилена включают, но не ограничены
Вышеупомянутые термины (например, "алкил," "арил" и "гетероарил"), в некоторых вариантах осуществления, включают как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикала приведены ниже.
Заместители для алкильных радикалов (включая группы, часто называемые алкиленом, алкенилом, алкинилом, гетероалкилом, гетероциклоалкилом и циклоалкилом) могут представлять собой различные группы, включая, но не ограничиваясь ими, -галоген, -OR', -NR'R'', -SR, -SiR'R''R''', -OC(O)R, -C(O)R'', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'''C(O)NR'R'', -NR11C(O)2R'', -NHC(NH2)=NH, -NRC(NH2)=NH, -NHC(NH2)=NR, -NR'''C(NR'R'')=N-CN, -NR'''C(NR'R'')=NOR', -NHC(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR'S(O)2R'', -NR'''S(O)2NR'R'', -CN, =О, =S, =N-OH и -NO2 числом от нуля до (2m'+l), где m' является общим числом атомов углерода в таком радикале. R', R'' и R''' каждый независимо относится к группам, включая, среди прочего, например, водород, незамещенный C1-6 алкил, незамещенный гетероалкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный C1-6 алкил, C1-6 алкокси или C1-6 тиоалкокси, или незамещенный арил-C1-4 алкил, незамещенный гетероарил, замещенный гетероарил. Когда R' и R'' присоединены к одному и тому же атому азота, они могут вместе с атомом азота образовывать 3-, 4-, 5-, 6- или 7-членное кольцо. Например, -NR'R'' включает 1-пирролидинил и 4-морфолинил. Другие заместители для алкильных радикалов, включая гетероалкил, алкилен, включают например, =О, =NR', =N-OR', =N-CN, =NH, причем R' включают заместители, как описано выше.
Точно так же заместители для арильной и гетероарильной групп варьируют и в целом могут быть выбраны из группы, включающей, но не ограниченной ими, -галоген, -OR', -OC(O)R', -NR'R'', -SR', -R', -CN, -NO2, -CO2R', -CONR'R'', -C(O)R', -OC(O)NR'R'', -NR''C(O)R', -NR''C(O)2R', -NR'C(O)NR''R''', -NHC(NH2)=NH, -NR'C(NH2)=NH, -NHC(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR'S(O)2R'', -N3, перфтор-C1-4 алкокси, и перфтор-C1-4 алкил, числом от нуля до общего числа открытых валентностей на ароматической кольцевой системе; и где R', R'' и R''' могут быть независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C3-6 циклоалкила, C2-6 алкенила, C2-6 алкинила, незамещенного арила и гетероарила, (незамещенный арил)-C1-4 алкила и незамещенного арилокси-C1-4 алкила. Другие подходящие заместители включают каждый из вышеупомянутых заместителей арила, присоединенных к кольцевому атому алкиленовой связкой из 1-4 атомов углерода.
В рамках изобретения, волнистая линия, "~", которая пересекает простую, двойную или тройную связь в любой химической структуре, изображенной здесь, обозначает место присоединения простой, двойной или тройной связи к остатку молекулы.
В рамках изобретения, "соединение по изобретению" относится к соединению Формулы I или любому его конкретному варианту; или к любому стереоизомеру, геометрическому изомеру, таутомеру, сольвату, метаболитам или фармацевтически приемлемой соли или пролекарству соединения Формулы I или его вариантов.
Для описания числа раз, которые заместитель (например, R10) может быть присоединен к химической структуре, показанной в этой заявке, заместитель (например, R10) указывают в круглых скобках и возможное число раз отмечают как нижний диапазон. Например, "-(R10)0-4" означает, что группа R10 может отсутствовать или может присутствовать вплоть до четырех раз.
В рамках изобретения термин "гетероатом" включает кислород (O), азот (N), серу (S) и кремний (Si).
II. Соединения
В одном аспекте настоящее изобретение относится к соединению Формулы I

или к его фармацевтически приемлемой соли, в которой R1 является независимо членом, выбранным из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила и галогена. В Формуле I индекс n означает целое число от 0 до 2, причем, когда n=0, тогда, X1 обозначает -CH2-, -C(H)(Ra)- или -C(Ra)2. X1 обозначает член, выбранный из группы, состоящей из -CH2-,-C(H)(Ra)-, -C(Ra)2, -O-, -N(H)-, -N(Ra)-, -N(C(O)Ra)-, -N(C(O)ORa)-, -N(S(O)2Ra)-, -N(S(O)Ra)-, -S-, -S(O)-, -S(O)2-, в котором Ra выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила, C3-6 циклоалкила и галогена. X2a, X2b и X2c, каждый независимо, выбраны из группы, состоящей из C(H), C(R2) и N, причем по меньшей мере один из X2a и X2b обозначает C(H) или C(R2); и причем R2 независимо выбран из группы, состоящей из -ORb, -NRbRc, -SRb, -C(O)ORC, -C(O)NRbRc, -NRbC(O)Rd, -S(O)2Rd, -S(O)Rd, -S(O)2NRbRc, -Rd, галогена, -CN и -NO2. Для заместителя R2, Rb и RC, каждый независимо, выбраны из группы, состоящей из водорода, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галогеналкила, или в случае необходимости Rb и RC, вместе с атомом, к которому каждый из них присоединен, образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла; и Rd выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила и C1-6 галогеналкила. В Формуле I A обозначает член, выбранный из группы, состоящей из:

в которых R3 независимо выбран из группы, состоящей из -NReRf, -ORe, -CN, -NO2, галогена, -C(O)OR6, -C(O)NReRf, -NReC(O)Rf, -NReS(O)2Rg, -NR6S(O) Rg, -S(O)2Rg, S(O)Rg и -Rg. Для группы R3, Re и Rf в каждом случае каждый независимо выбраны из группы, состоящей из водорода, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галогеналкила и -(CH2)1-4 фенила, или Re и Rf, или Re и Rg вместе с атомом, к которому каждый из них присоединен, в случае необходимости образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла; и Rg выбран из группы, состоящей из C1-4 алкила, C2-4 алкенила, C2-4 алкинила и C1-4 галогеналкила. B обозначает член, выбранный из группы, состоящей из:

в которых Y обозначает N, C(H) или C(R4a); X3 обозначает -N(H), -N(C1-3 алкил), O или S; R4a, в случае его присутствия, независимо выбран из C1-4 алкила, C1-4 галогеналкила, C2-4 алкенила, C2-4 алкинила, галогена и -CN. R4b, в каждом случае, независимо выбран из группы, состоящей из -C(O)ORj, -C(O)NRhRi, -C(O)Ri, -NRhC(O)Ri, -NRhC(O)NRhRi, -OC(O)NRhRi, -NRhC(O)ORj, -C(=NORh)NRhRi -NRhC(=NCN)NRhRi, -NRhS(O)2NRhRi, -S(O)2RJ, -S(O)2NRhRi, -N(Rh)S(O)2Ri, -NRhC(=NRi)NRhRi, -C(=S)NRhRi, -C(=NRh)NRhRi, галогена, -NO2 и -CN, в которых Rh и Ri в каждом случае, каждый независимо, выбраны из группы, состоящей из водорода, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C1-6 галогеналкила, фенила и -(CH2)1-4 фенила, или Rh и Ri, или Rh и Rj, вместе с атомом, к которому каждый из них присоединен, в случае необходимости образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. Rj выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила, C3-7 циклоалкила, фенила и -(CH2)1-4 фенила; или, альтернативно, R4 выбран из группы, состоящей из

в которых Rk выбран из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила и C1-6 галогеналкила. В Формуле I L отсутствует или обозначает член, выбранный из группы, состоящей из C6-10 арилен-C1-6 гетероалкилена, C5-9 гетероарилен-C1-6 гетероалкилена, C1-6 гетероалкилена, C1-6 алкилена, C1-6 галогеналкилена, C2-6 алкенилена, C2-6 алкинилена, -NH-, -S- и -O-, причем алкиленовая, алкениленовая, алкиниленовая или гетероалкиленовая части группы L замещены от 0 до 4 раз заместителями R5a, выбранными из группы, состоящей из галогена, -Rm и =О, и ароматические части группы L замещены от 0 до 4 раз заместителями R5b, выбранными из группы, состоящей из галогена, -ORn, -NRnRo, -Rn, -NO2 и CN; причем Rm выбран из группы, состоящей из C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 гетероалкила и C1-6 галогеналкила. В случае необходимости любые два заместителя R5a, присоединенные к одному и тому же или к разным атомам L, могут вместе образовывать 5-7-членное карбоциклическое кольцо или 5-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла; и причем Rn и Ro, в каждом случае, выбраны из группы, состоящей из водорода, C1-6 алкила, C2-6 алкенила, C2-6 алкинила и C1-6 галогеналкила, и причем в случае необходимости Rn и Ro, вместе с атомами, к которым каждый из них присоединен, вместе образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. В Формуле I E обозначает водород или галоген; или, альтернативно, E выбран из группы, состоящей из фенила, C5-6 гетероарила, C3-7 гетероциклоалкила и C3-7 циклоалкила, и в случае необходимости с E могут быть сконденсированы 1 или 2 кольца, независимо выбранных из группы, состоящей из 3-7-членного карбоциклического кольца, 3-7-членного гетероциклического кольца, бензольного кольца и 5-6-членного гетероароматического кольца, причем E и каждое кольцо, в случае необходимости конденсированное с E, независимо замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из галогена, -NRpRq, -SRP, -ORP, -C(O)ORP, -C(О)NRpRq, -C(O)RP, -NRpC(О)Rq, -OC(O)Rr, -NRpC(О)NRpRq, -OC(О)NRpRq, -NRpC(О)ORr, -C(=NORp)NRpRq, -NRpC(=N-CN)NRpRq, -NRpS(О)2NRpRq, -S(O)2Rr, -S(O)2NRpRq, -Rr, -Rs, -NO2, -N3, =О, -CN, -Z1-NRpRq, -Z1SRp, -Z1-OR13, -Z1-C(O)ORp, -Z1-C(O)NRpRq, -Z1-C(O)Rp, -Z1-NRpC(O)Rq, -Z1-OC(O)Rr, -Z1-NRpC(O)NRpRq, -Z1-OC(O)NRpRq, -Z1-NRpC(O)ORr, -Z1- C(=NORp)NRpRq, -Z1-NRpC(=N-CN)NRpRq, -Z1-NRpS(O)2NRpRq, -Z1S(O)2Rr, -Z1-S(O)2NRpRq, -Z1-NO2, -Z1-N3, -Z1-Rs и -Z1-CN. В Формуле I Z1 выбран из группы, состоящей из C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена, C1-6 гетероалкилена, C3-7 циклоалкилена и C3-7 гетероциклоалкилена; Rp и Rq, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила, фенила и -(CH2)1-4-фенила; Rr выбран из группы, состоящей из C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила, фенила и -(CH2)1-4-фенила. В случае необходимости в составе каждого заместителя R6 Rp и Rq или Rp и Rr, вместе с атомом, к которому каждый из них присоединен, в случае необходимости вместе образуют 3-7-членное гетероциклическое кольцо, в случае необходимости включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. Rs выбран из группы, состоящей из фенила, C5-6 гетероарила, C3-7 гетероциклоалкила, C3-7 циклоалкила и с Rs может быть конденсировано 1 или 2 кольца, которые, каждый независимо, выбраны из группы, состоящей из 5-7-членного карбоциклического кольца, 5-7-членного гетероциклического кольца, бензольного кольца и 5-6-членного гетероароматического кольца, и Rs и каждое кольцо, в случае необходимости конденсированное с Rs, каждый независимо, замещены от 0 до 5 раз заместителями R7, выбранными из группы, состоящей из галогена, -NRtRu, -SRt, -ORt, -C(O)ORt, -C(O)NRtRu, -C(O)Rt, -NRtC(О)RV, -OC(О)RV, -NRtC(O)NRtRu, -OC(O)NRtRr, -NRtC(O)ORv, -C(=NORt)NRtRu, -NRtC(=N-CN)NRtRu, -NRtS(O)2NRtRu, -S(O)2RV, -S(O)2NRtRu, -Rv, -NO2, -N3, =О, -CN, -Z2- NRtRu, -Z2-SRt, -Z2-ORt, -Z2-C(O)ORt, -Z2-C(O)NRtRu, -Z2-C(O)RV, -Z2-NRtC(O)Ru, -Z2-OC(О)Rv, -Z2-NRtC(O)NRtRu, -Z2-OC(O)NRtRu, -Z2-NRtC(О)ORv, -Z2-C(=NORt)NRtRu, -Z2-NRtC(=N-CN)NRtRu, -Z2-NRtS(O)2NRtRu, -Z2-S(O)2RV, -Z2-S(O)2NRtRu, -Z2-NO2, -Z2-N3 и -Z2-CN. Z2 выбран из группы, состоящей из C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена, C1-6 гетероалкилена, Rt и Ru, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила и -(CH2)1-4-фенила, C3-7 циклоалкила и C3-7 гетероциклоалкила; Rv выбран из C1-4 алкила, C1-4 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила и -(CH2)1-4-фенила. В каждом заместителе R7, Rt и Ru или Rt и Rv, вместе с атомом, к которому каждый из них присоединен, в случае необходимости вместе образуют 3-7-членное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранные из N, O и S в качестве вершин цикла.
В первом варианте осуществления соединение Формулы I имеет формулу, выбранную из группы, состоящей из Формулы I-a, Формулы I-b, Формулы I-c, Формулы I-d, Формулы I-e, Формулы I-f, Формулы I-g, Формулы I-h, Формулы I-i, Формулы I-j, Формулы I-k, Формулы I-m, Формулы I-n, Формулы I-o и Формулы I-p, как показано на Фиг. 1.
Во втором варианте осуществления и в некоторых аспектах первого варианта осуществления, соединение Формулы I имеет Формулу I-a,

в которой R1 отсутствует. X1 выбран из группы, состоящей из -CH2-, -C(H)(Ra)-, -C(Ra)2, -O-, -N(H)-, -N(Ra)-, -N(C(O)Ra)-, -N(C(O)ORa)-, -N(S(O)2Ra)-, -N(S(O)Ra)-, -S-, -S(O)- и -S(O)2-. A обозначает

B обозначает член, выбранный из группы, состоящей из:

в которых R4b выбран из группы, состоящей из

В третьем варианте осуществления в некоторых аспектах второго варианта соединений по изобретению, в которых X1 обозначает -CH2- и -O-.
В четвертом варианте осуществления в некоторых аспектах первого варианта соединения по изобретению соединение имеет Формулу I-a

в которой R1 отсутствует. X1 обозначает -CH2-, -C(H)(Ra)-, -C(Ra)2, -O-, -N(H)-, -N(Ra)-, -N(C(O)Ra)-, -N(C(O)ORa)-, -N(S(O)2Ra)-, -N(S(O)Ra)-, -S-, -S(O)- или -S(O)2-. A обозначает

B обозначает член, выбранный из группы, состоящей из:
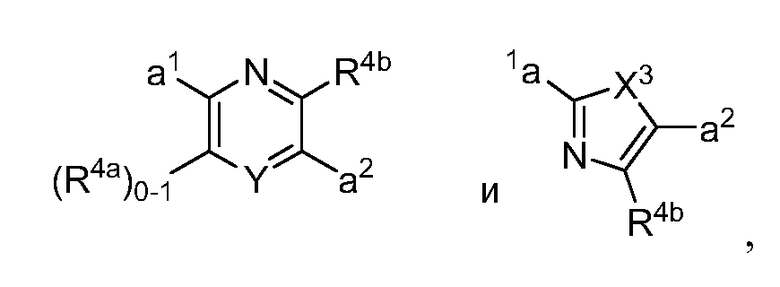
в которых R4b выбран из группы, состоящей из -C(O)ORj, -C(O)NRhRi, -C(O)Ri, -NRhC(O)Ri, -NRhC(O)NRhRi, -OC(O)NRhRi, -NRhC(О)ORj, -C(=NORh)NRhRi, -NRhC(=NCN)NRhRi, -NRhS(O)2NRhRi, -S(O)2Rj, -S(O)2NRhRi, -N(Rh)S(O)2Ri, -NRhC(=NRi)NRhRi, -C(=S)NRhRi, -C(=NRh)NRhRi, галогена, -NO2 и -CN.
В пятом варианте осуществления и в некоторых аспектах четвертого варианта соединений по изобретению X1 обозначает -CH2- или -О-.
В шестом варианте осуществления для соединений Формулы I или первого, второго или четвертого варианта осуществления B обозначает
в которой R4a, в случае его присутствия, выбран из группы, состоящей из галогена и C1-4 алкила.
В седьмом варианте осуществления для соединений Формулы I или первого, второго или четвертого варианта осуществления B обозначает

В восьмом варианте осуществления для соединений Формулы I или первого, второго, четвертого, шестого или седьмого варианта осуществления L отсутствует или обозначает в случае необходимости замещенную группу, выбранную из группы, состоящей из C6-10 арилен-C1-6 гетероалкилена, C5-9 гетероарилен-C1-6 гетероалкилена, C1-6 гетероалкилена, C2-6 гетероалкенилена, C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена и -О-. E обозначает кольцо, выбранное из группы, состоящей из фенила, C5-6 гетероарила, C3-7 гетероциклоалкила, C3-7 циклоалкила, и причем с Е в случае необходимости конденсировано 5-7-членное гетероциклическое кольцо, бензольное кольцо или 5-6-членное гетероароматическое кольцо, и где E и указанное кольцо, в случае необходимости конденсированное с E, замещено от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из фтора, хлора, брома, -NRpRq, -SRP, -ORP, -C(O)ORP, -C(О)NRpRq, -C(O)RP, -NRpC(О)Rq, -OC(O)Rr, -NRpC(О)NRpRq, -OC(О)NRpRq, -NRpC(O)ORr, -S(O)2Rr, -S(O)2NRpRq, -Rr, -Rs, -NO2, -N3, -CN, -Z1-NRpRq, -Z1-SRp, -Z1-ORP, -Z1-OC(O)NRpRq, -Z1-NRpC(O)ORr, -Z1-S(O)2Rr, -Z1-Rs и -Z1-S(O)2NRpRq. Z1 выбран из группы, состоящей из C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена и C1-6 гетероалкилена. Rp и Rq, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила, фенила и -(CH2)1-4-фенила. Rr выбран из C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила и -(CH2)1-4-фенила. В случае необходимости в каждом заместителе R6 Rp и Rq или Rp и Rr, вместе с атомом, к которому каждый из них присоединен, вместе образуют 3-7-членное гетероциклическое кольцо, включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла. Rs обозначает фенил, 5-6-членное гетероароматическое кольцо или 5-7-членное гетероциклическое кольцо, причем с Rs может быть конденсировано бензольное кольцо, 5-6-членное гетероароматическое кольцо или 5-7-членное гетероциклическое кольцо. Rs и указанное кольцо, в случае необходимости конденсированное с Rs, замещено от 0 до 3 раз заместителями R7, выбранными из группы, состоящей из галогена, -NRtRu, -SRt, -ORt, -OC(O)NRtRu, -NRtC(O)ORv, -Rv, -Z2-NRtRu, -Z2-OC(О)NRtRu, -Z2-NRtC(О)ORv и -CN. Z выбран из группы, состоящей из C1-6 алкилена, C2-6 алкенилена, C2-6 алкинилена и C1-6 гетероалкилена, причем Rt и Ru, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила и C3-7 гетероциклоалкила. Rv выбран из C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила и -(CH2)1-4-фенила. В случае необходимости в каждом заместителе R7 Rt и Ru или Rt и Rv, вместе с атомами, к которым каждый из них присоединен, вместе образуют 3-7-членное гетероциклическое кольцо, в случае необходимости включающее 1-2 гетероатома, выбранных из N, O и S в качестве вершин цикла.
В девятом варианте осуществления, в некоторых аспектах шестого варианта соединений по изобретению, L отсутствует, и E выбран из группы, состоящей из фенила и пиридила, и причем с E может быть конденсировано кольцо, выбранное из группы, состоящей из пиримидин-4-она, пиримидин-2-она, бензольного кольца, пиридина, пиррольного кольца, пиразольного кольца, имидазольного кольца, фурана и тиофена, причем E и кольцо, в случае необходимости конденсированное с E, каждое, может быть в случае необходимости независимо замещено.
В десятом варианте осуществления для соединений Формулы I или первого, второго, четвертого, шестого, седьмого или восьмого варианта осуществления L обозначает C1-4 алкилен или C1-4 гетероалкилен.
В одиннадцатом варианте осуществления для соединений Формулы I или первого, второго, четвертого, шестого, седьмого или восьмого варианта осуществления L выбран из группы, состоящей из:

В двенадцатом варианте осуществления, в некоторых аспектах десятого или одиннадцатого вариантов соединений по изобретению, L присутствует, и E выбран из группы, состоящей из фенила, пиридила, пиримидинила, пиперидинила, пиперазинила, морфолино, пирролидинила, пирролидонила и циклобутила. С E могут быть конденсированы пиридин, бензольное кольцо, пиримидин-4-он, пиримидин-2-он или диоксолановое кольцо, и причем E и указанное кольцо, в случае необходимости конденсированное с E, замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из фтора, хлора, -NRpRq, -SRP, -S(O)2Rr, -ORP, -NRpC(O)ORr, -Rr, -Z1-NRPRq, -Z1-NRpC(O)ORr и -Rs. Rp и Rq, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C2-6 алкенилена, C2-6 алкинилена и C1-6 гетероалкилена. Rr выбран из C1-6 алкила, C1-6 галогеналкила, C2-6 алкенила, C2-6 алкинила, C3-7 циклоалкила, C3-7 гетероциклоалкила и -(CH2)1-4-фенила. В случае необходимости в каждом заместителе R6 Rp и Rq или Rp и Rr вместе образуют 3-6-членное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранные из N, O и S в качестве вершин цикла.
В тринадцатом варианте осуществления для соединений Формулы I или первого, второго, четвертого, шестого, седьмого или восьмого варианта осуществления E обозначает фенил и является замещенным в мета- или пара-положении в случае необходимости замещенной группой Rs, имеющей формулу, выбранную из группы, состоящей из:

В четырнадцатом варианте осуществления для соединений Формулы I или первого, второго, четвертого, шестого, седьмого или восьмого варианта осуществления E выбран из группы, показанной на Фиг. 2-A и Фиг. 2-B.
В пятнадцатом варианте осуществления для соединений Формулы I или первого, второго, четвертого, шестого, седьмого или восьмого варианта осуществления E выбран из группы, показанной на Фиг. 2-C, Фиг. 2-D, Фиг. 2-E и Фиг. 2-F.
В шестнадцатом варианте осуществления для соединений Формулы I или первого, второго или четвертого варианта осуществления L отсутствует, и E обозначает водород или галоген.
В семнадцатом варианте осуществления для соединений Формулы I или первого, второго или четвертого варианта осуществления L выбран из группы, состоящей из C1-6 гетероалкилена, C1-6 алкилена, C2-6 алкенилена и C2-6 алкинилена, каждый из которых может быть независимо замещен; и E обозначает водород.
В восемнадцатом варианте осуществления, в некоторых аспектах семнадцатого варианта осуществления соединений по изобретению, L обозначает в случае необходимости замещенный C1-4 гетероалкилен.
В девятнадцатом варианте осуществления соединения Формулы I выбраны из группы, представленной в Таблице 1.

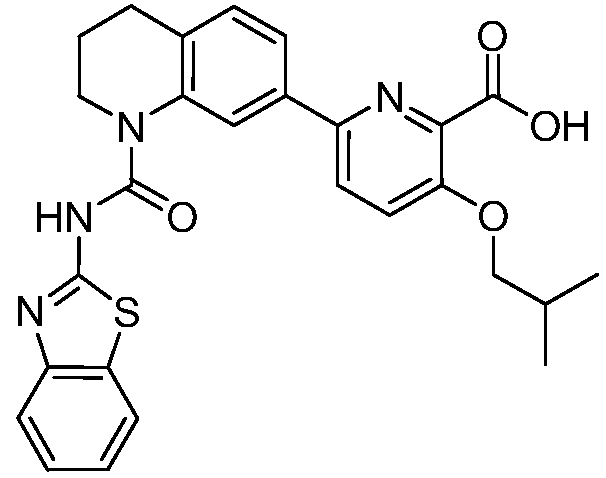



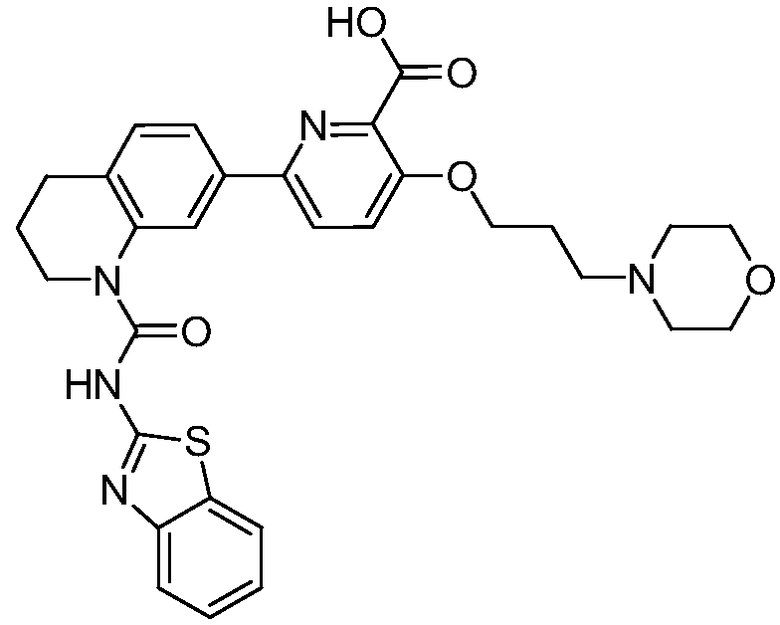
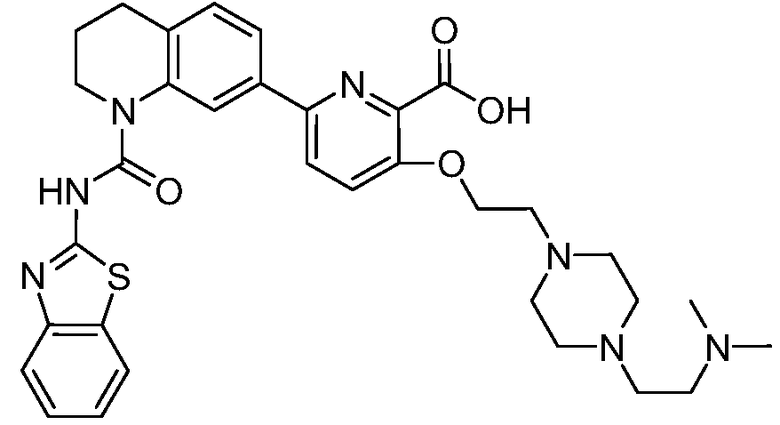
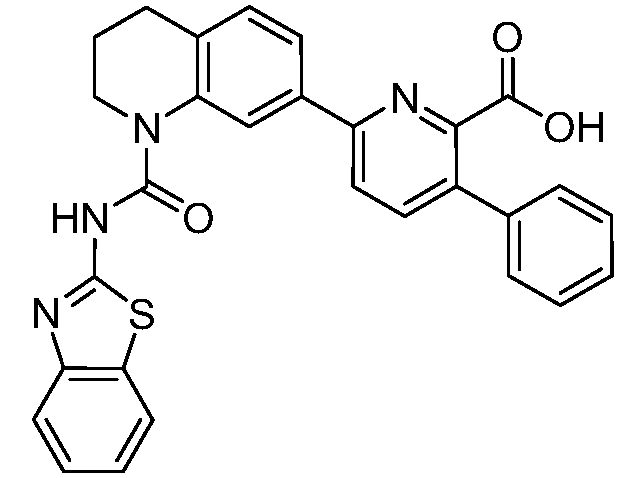
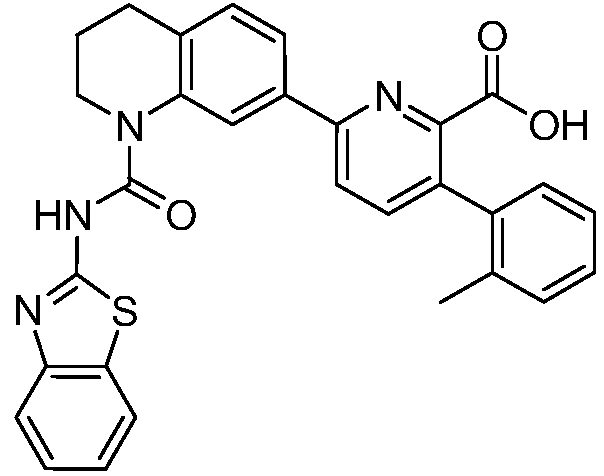



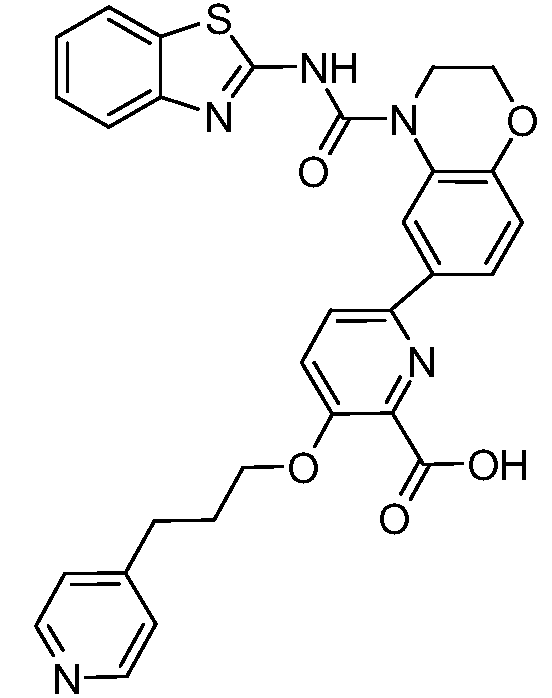


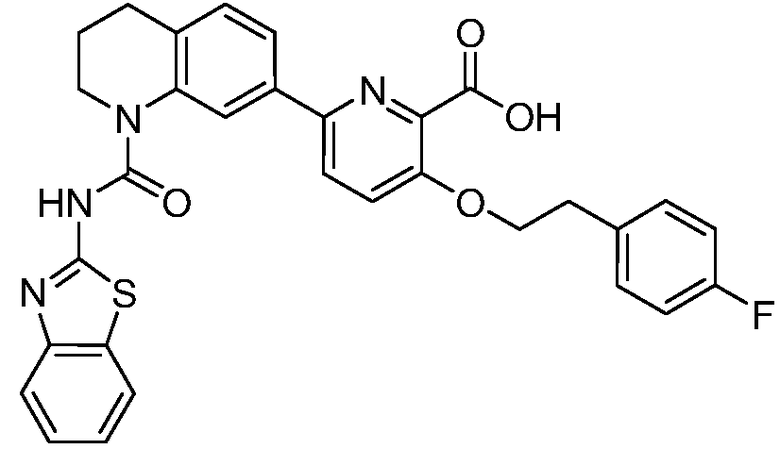
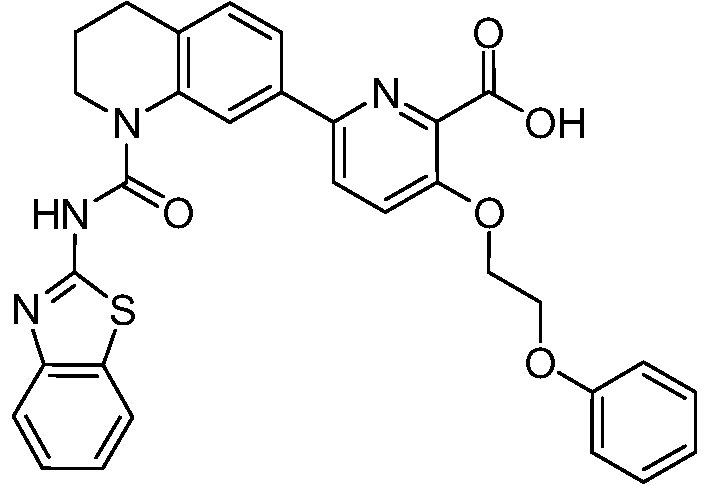
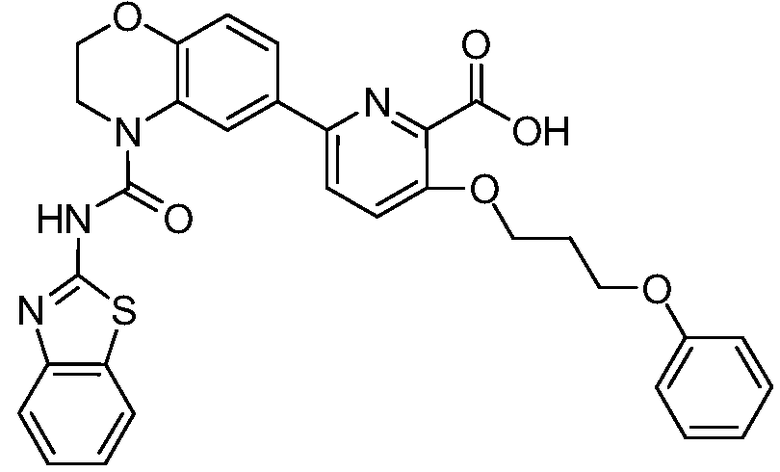


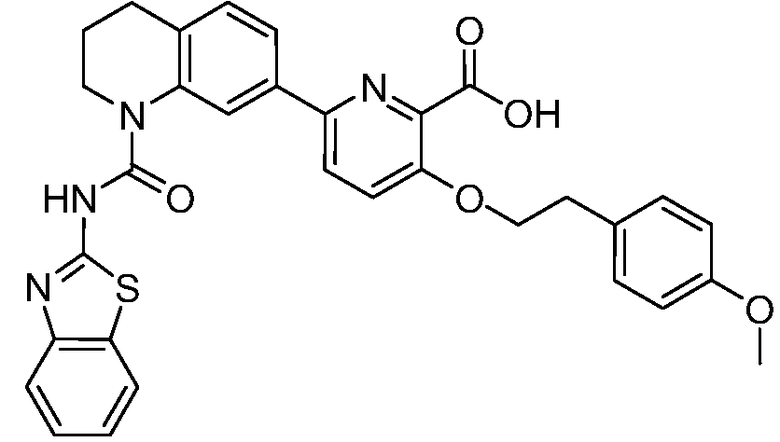


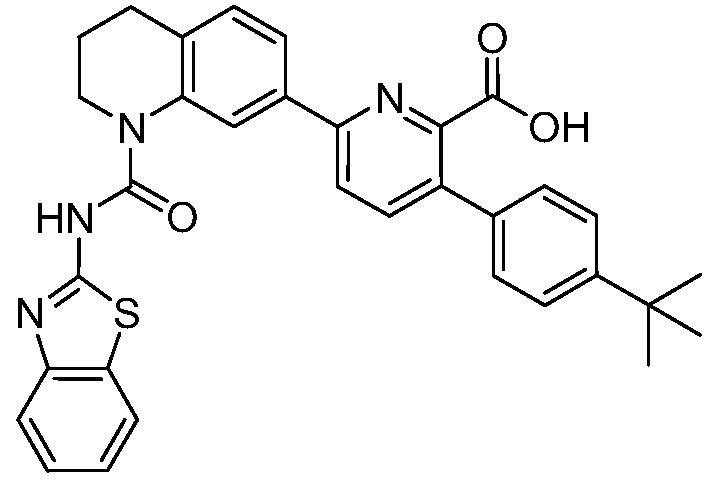


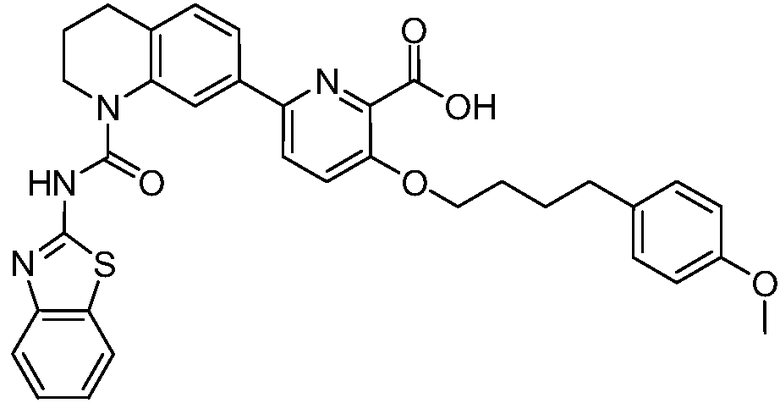

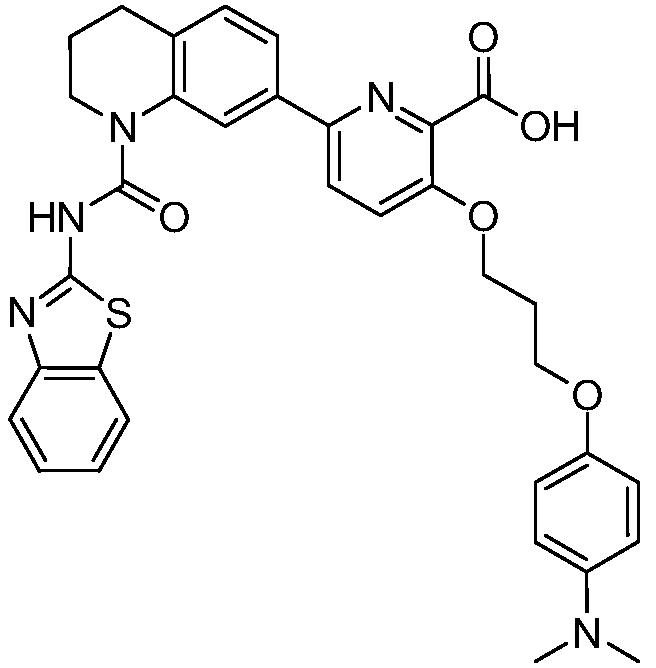

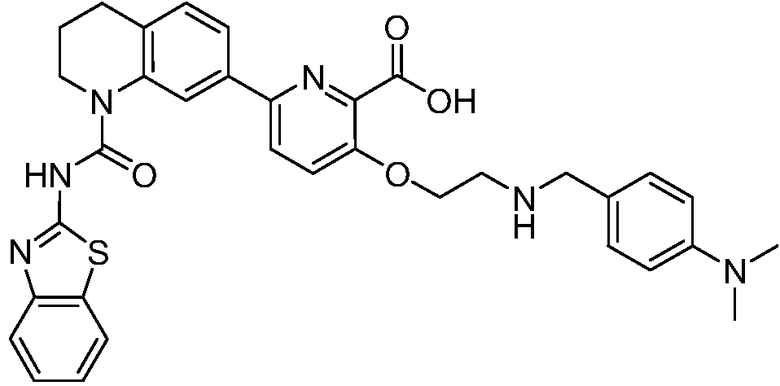

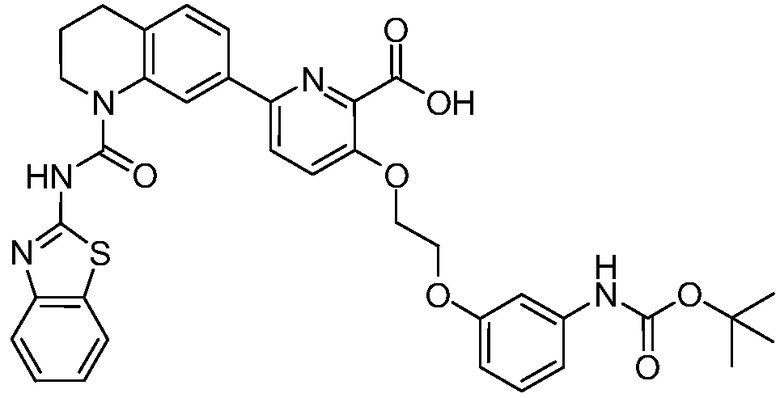
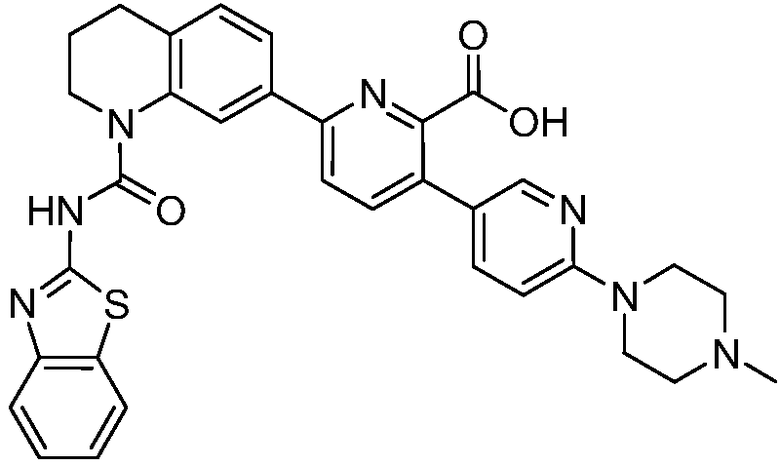
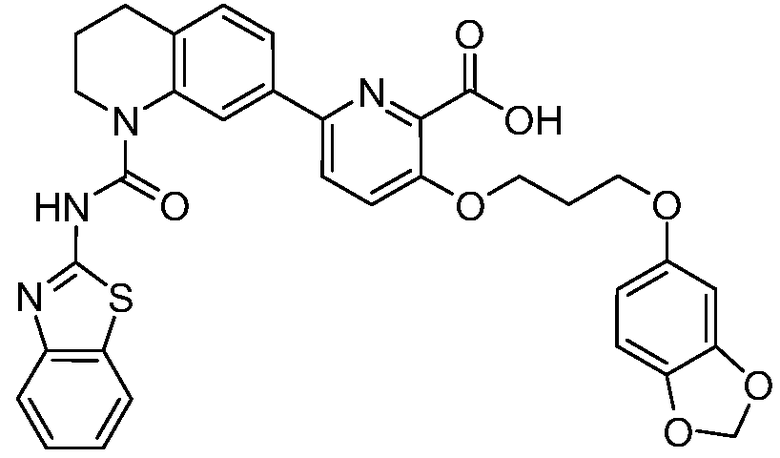
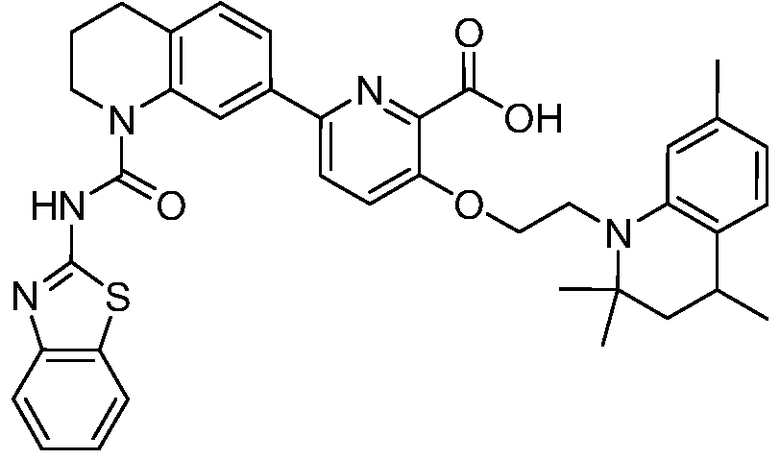


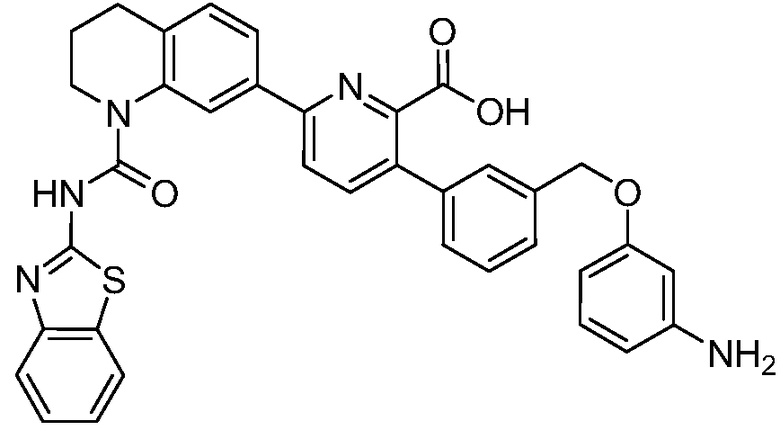


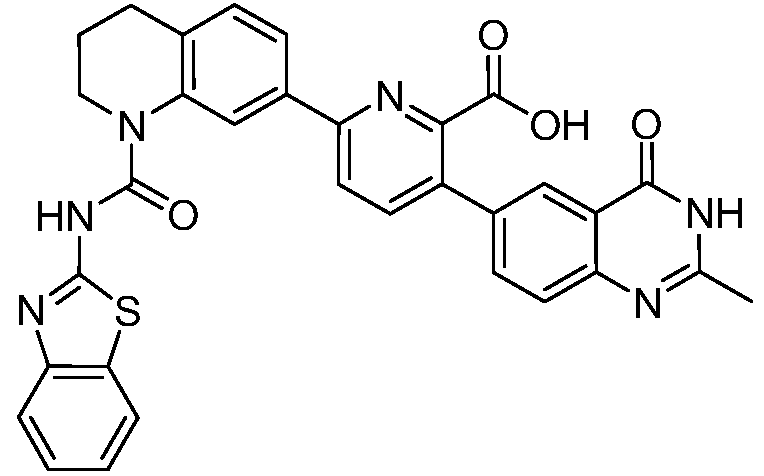
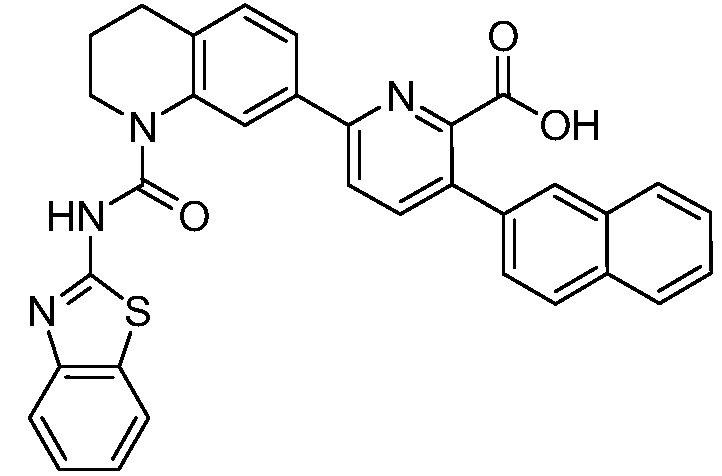



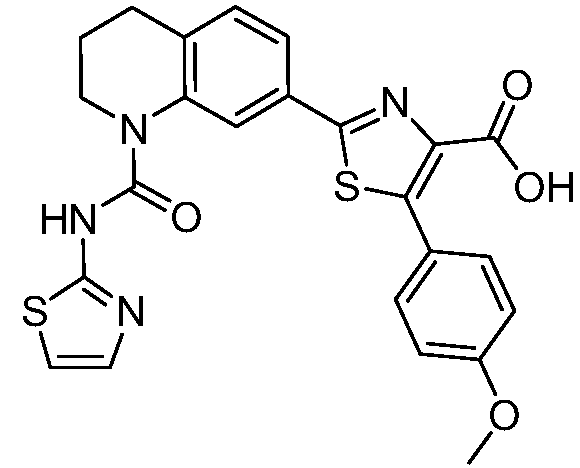
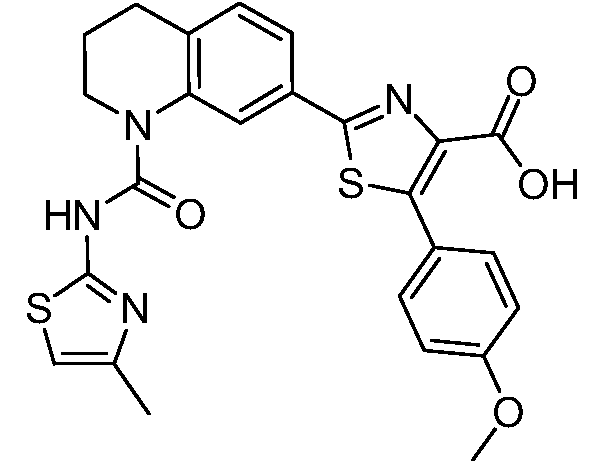
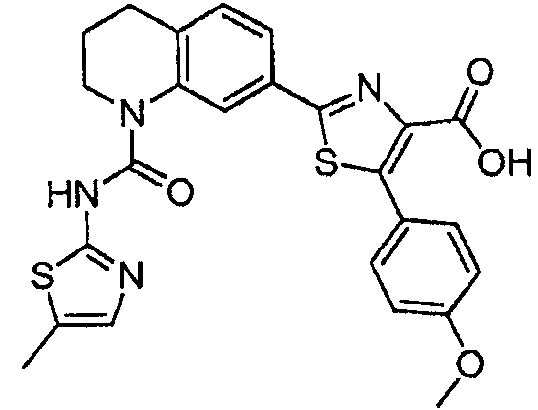

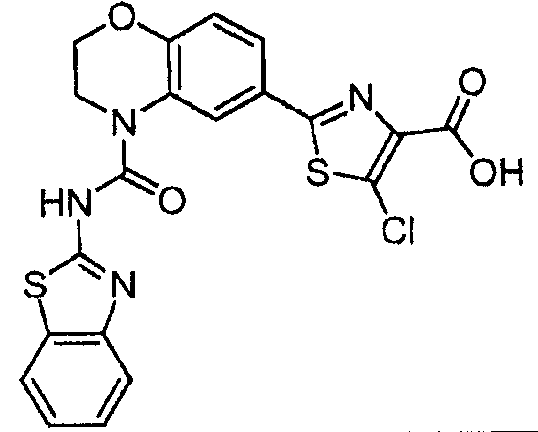

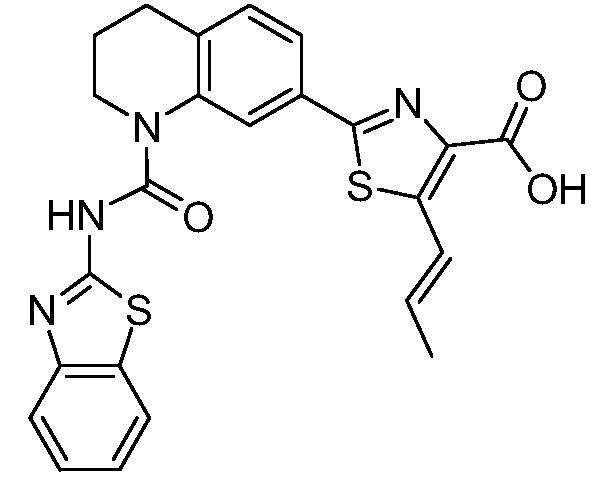
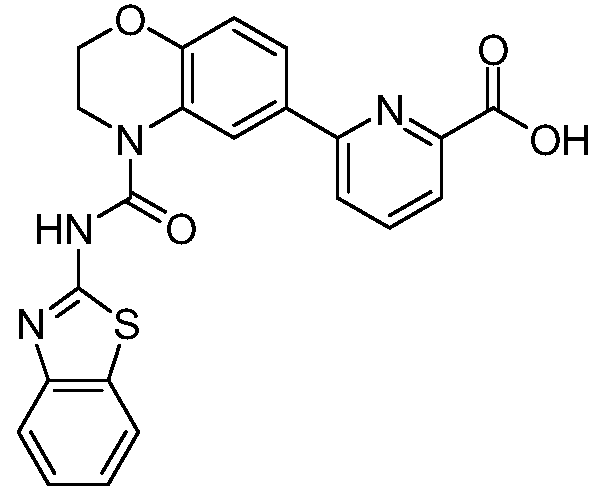
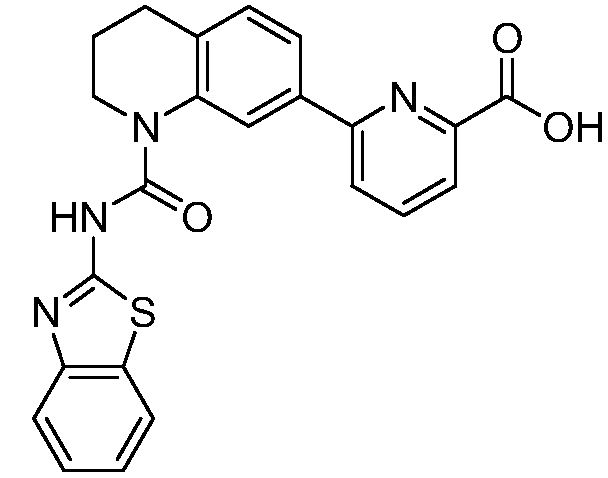
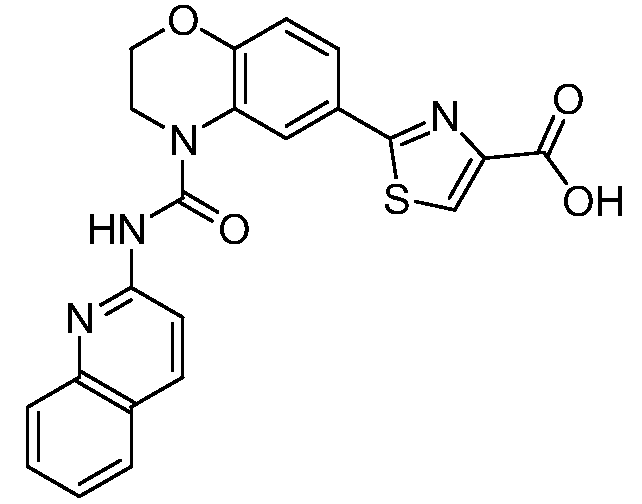
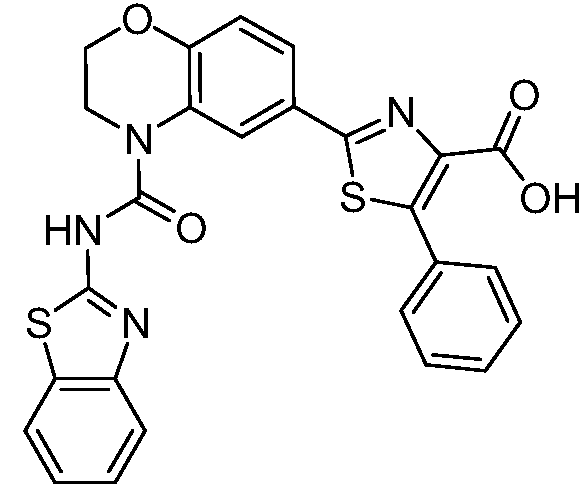
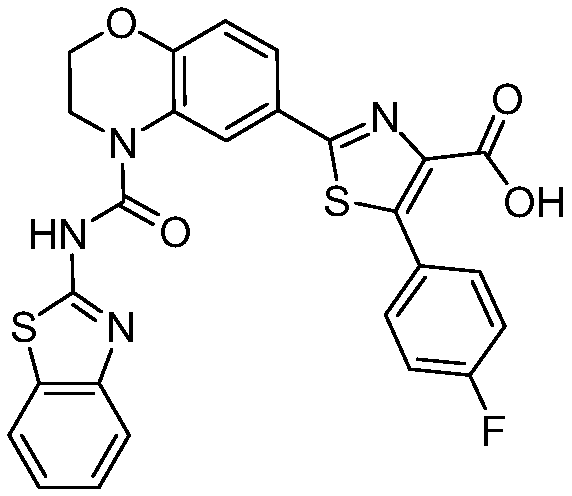
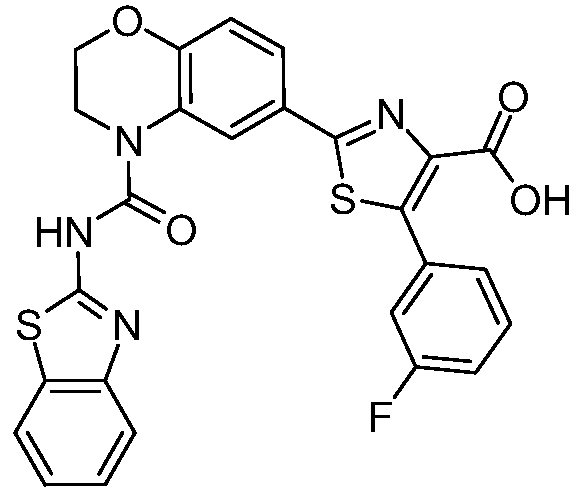

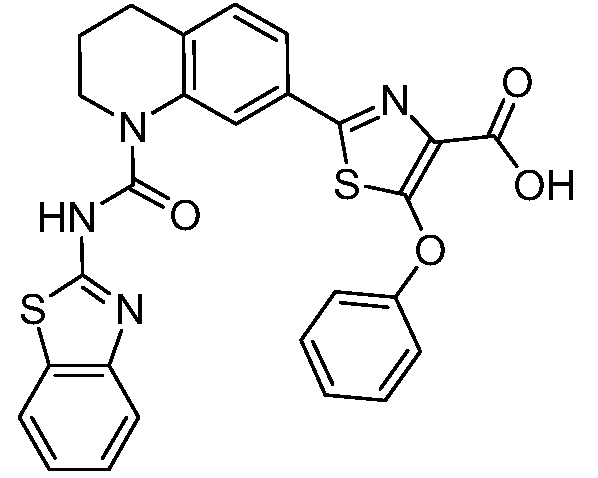
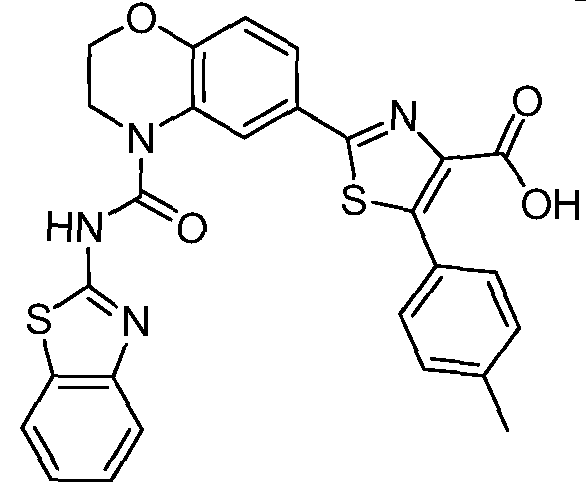
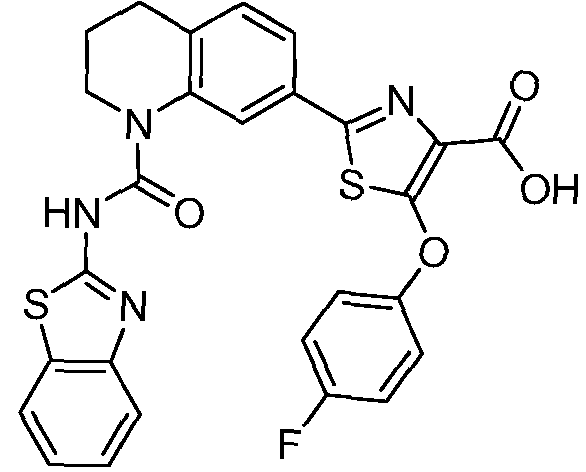



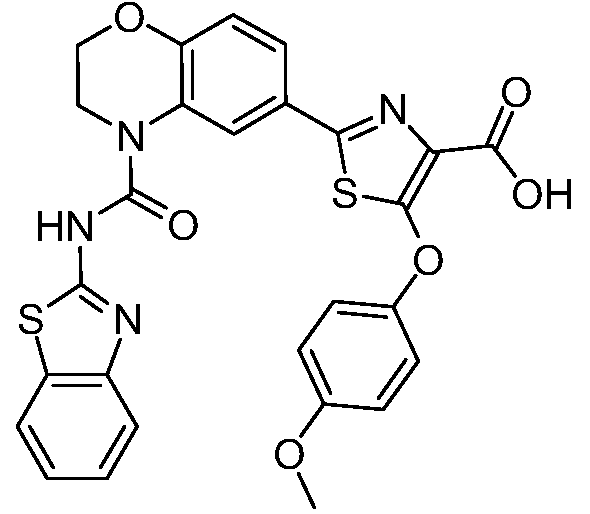


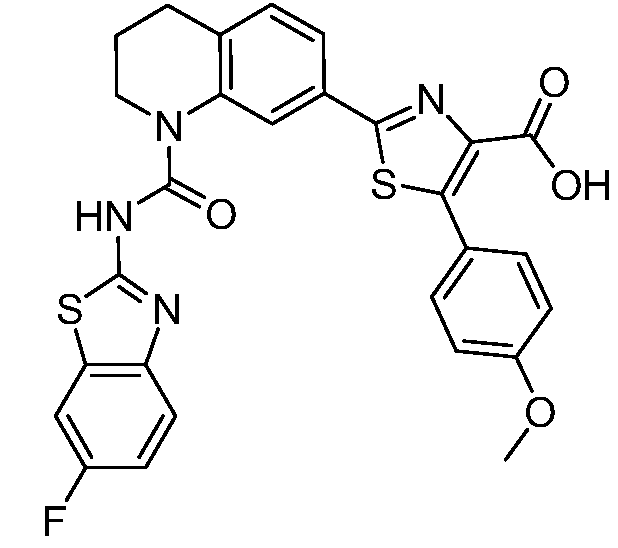
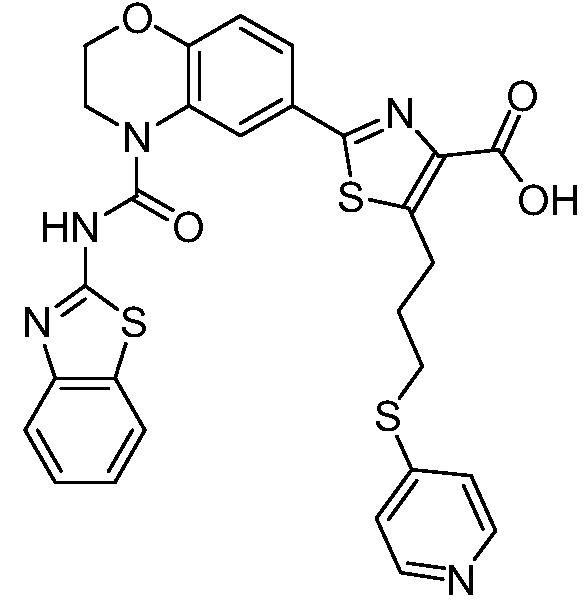

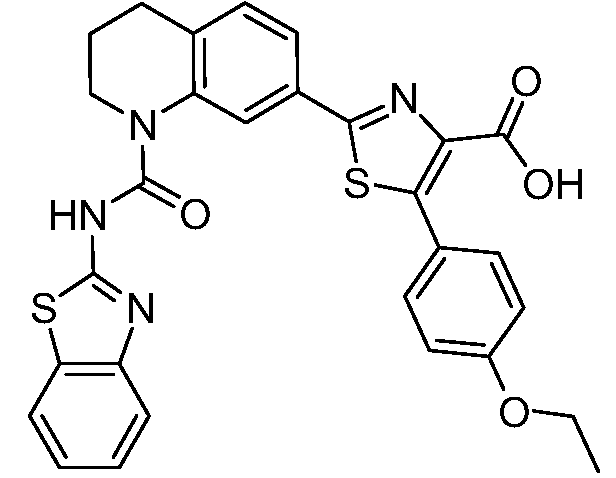





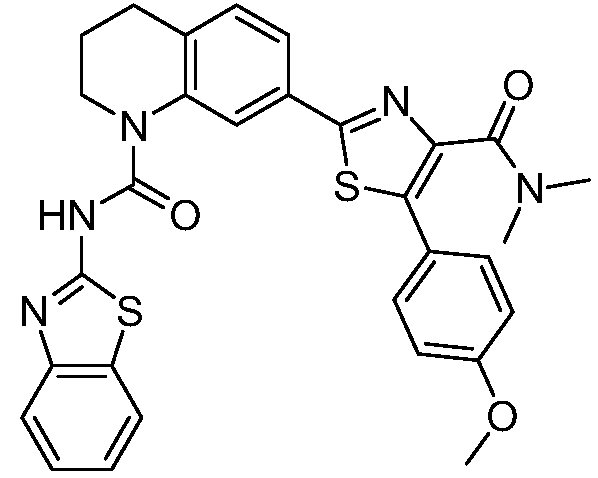
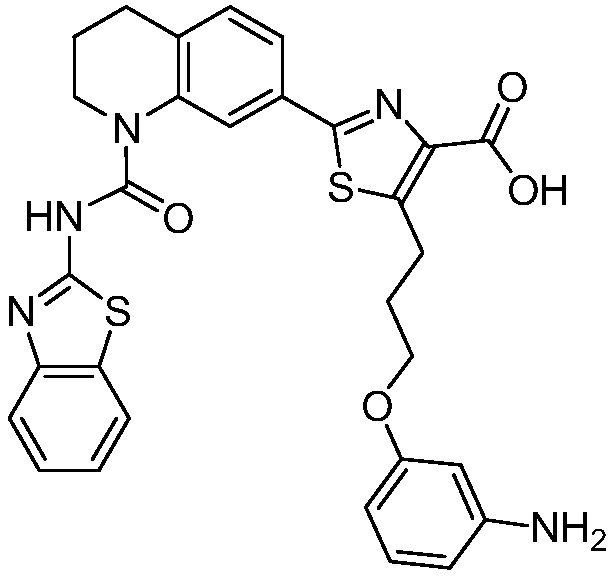
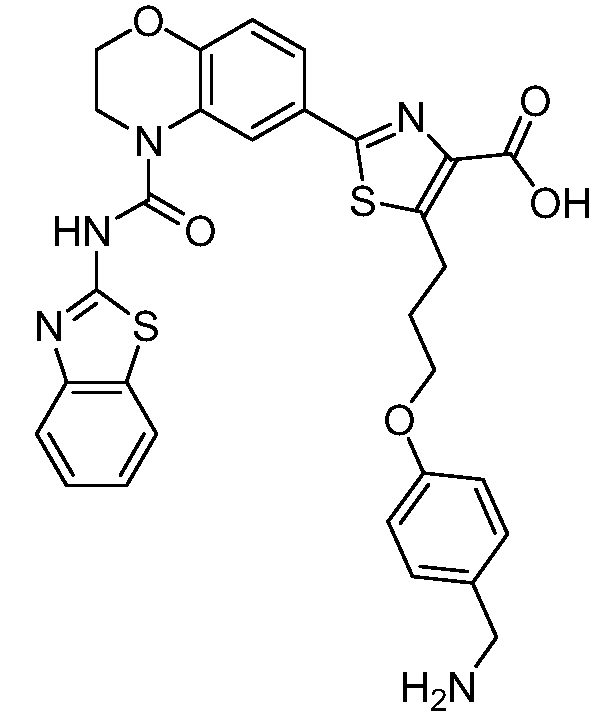

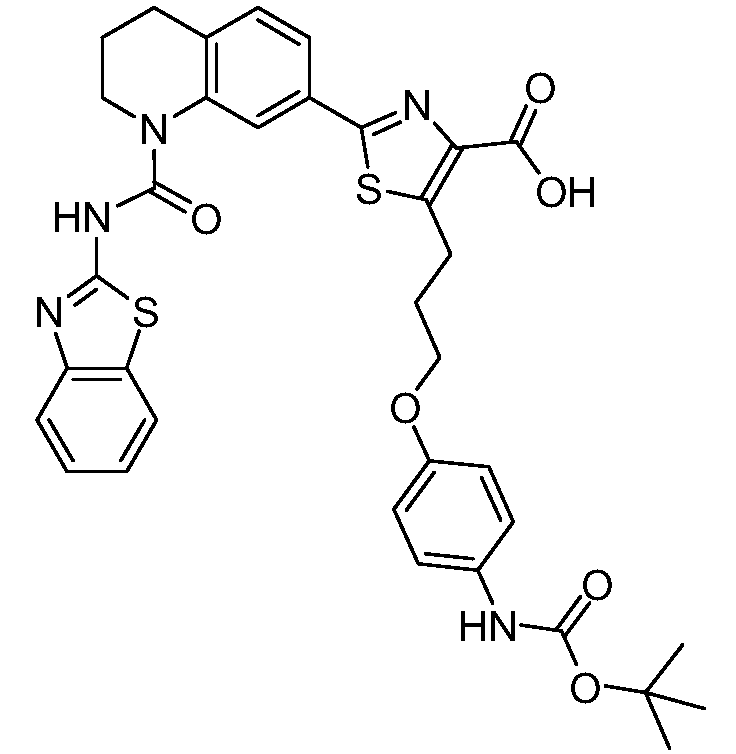
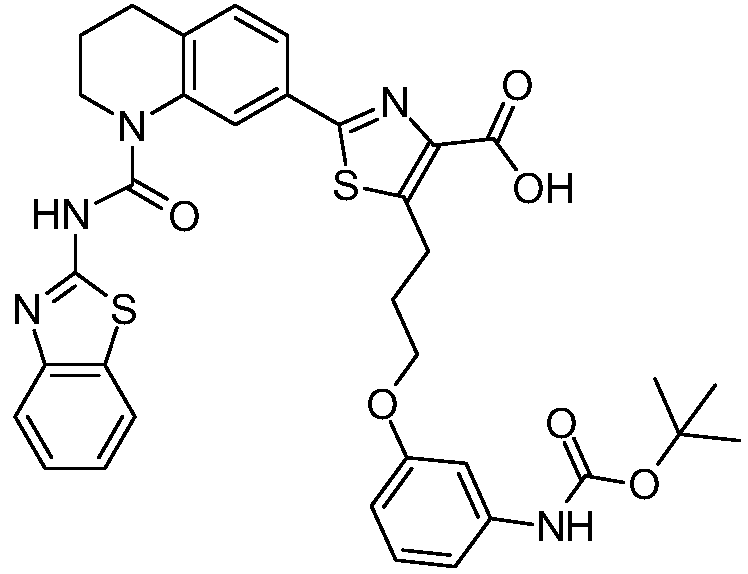

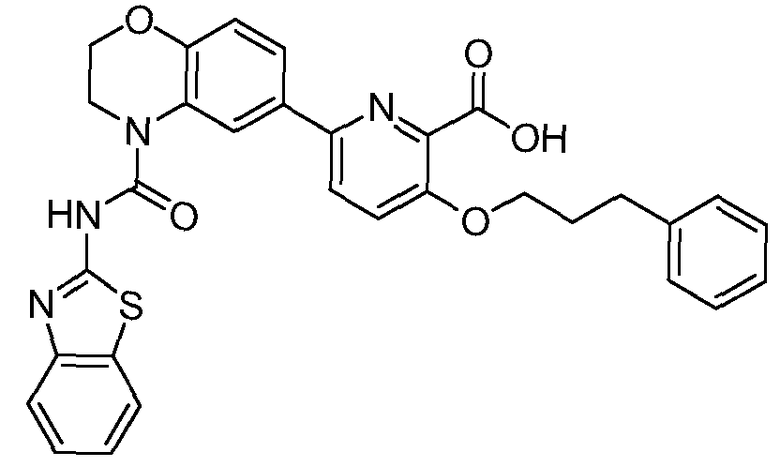

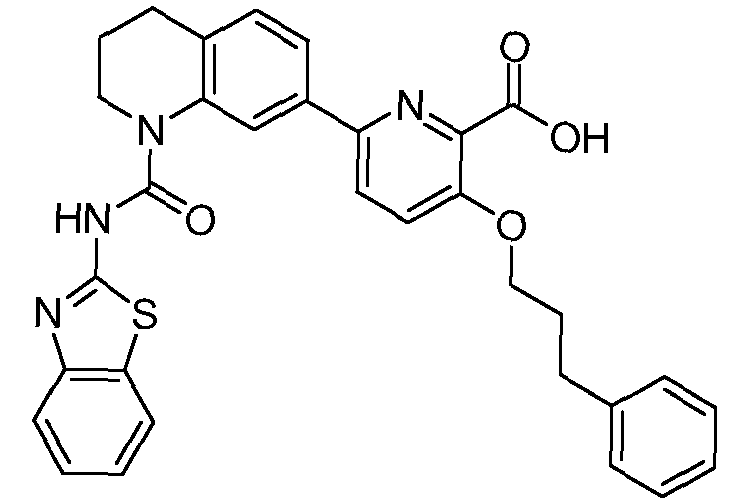

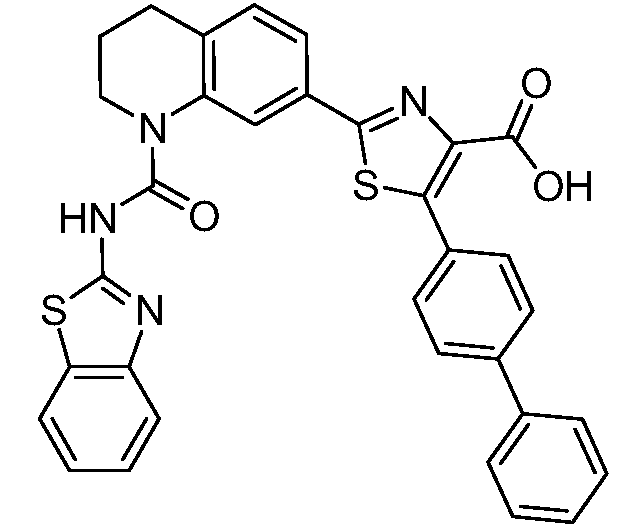


Общие процедуры синтеза
Соединения по изобретению могут быть получены способами синтеза, известными в данной области техники, некоторые из которых описаны ниже в иллюстративных целях.
Соединения Формулы I, имеющие тетрагидрохинолиновое ядро могут быть получены, как показано на Схеме 1 ниже.
Схема 1

Как показано на Схеме 1, внутримолекулярное ацилирование Фриделя-Крафта (например, с использованием AlCl3) замещенной галогеном фенилалкановой кислоты (i) может дать кетон (ii). Превращение ii в O-силилированное производное оксима (iii) может быть осуществлено объединением ii с гидрохлоридом оксима в основных условиях (например, карбонат калия) с последующим введением силильной группы полученного оксима с использованием, например, триалкилсилил хлорида (R3SiCl). Промотируемая кислотой Льюиса перегруппировка Бекмана соединения iv с последующим восстановлением промежуточного имминиевого соединения может дать тетрагидрохинолин v. На Схеме 1, индекс n представляет собой целое число от 1 до 3.
Получение соединений Формулы I, имеющих бензо-конденсированное оксазиновое ядро, може быть осуществлено в соответствии с процедурой синтеза, показанной на Схеме 2 ниже.
Схема 2
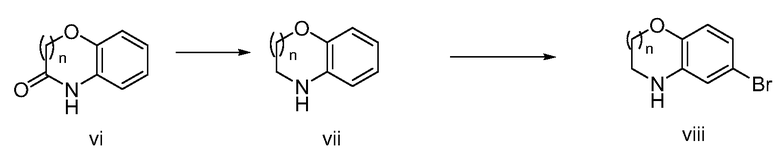
Как показано на Схеме 2, гидридное восстановление амида vi, например, с использованием LiAlH4, может дать бензо-конденсированный оксазин vii. Бромирование vii с использованием, например, N-бромсукцинимида может дать бромидный продукт viii. На Схеме 2 индекс означает целое число от 1 до 3
Получение некоторых аза-производных соединений Формулы I может быть осуществлено, как показано на Схеме 3 ниже.
Схема 3

Как показано на Схеме 3, алкилирование гидроксинитропиридина ix дигалогеналканом, например, дибромэтаном, может дать алкилированный продукт x. Восстановление нитрогруппы в x с использованием, например, железного порошка в уксусной кислоте может дать соответствующий аминный продукт xi, который после нагревания в основных условиях может дать циклизованный оксазиновый продукт xii.
Бромидные промежуточные соединения v, viii и xii могут быть дополнительно модифицированы, как показано на Схеме 4 ниже для соединения v.
Схема 4
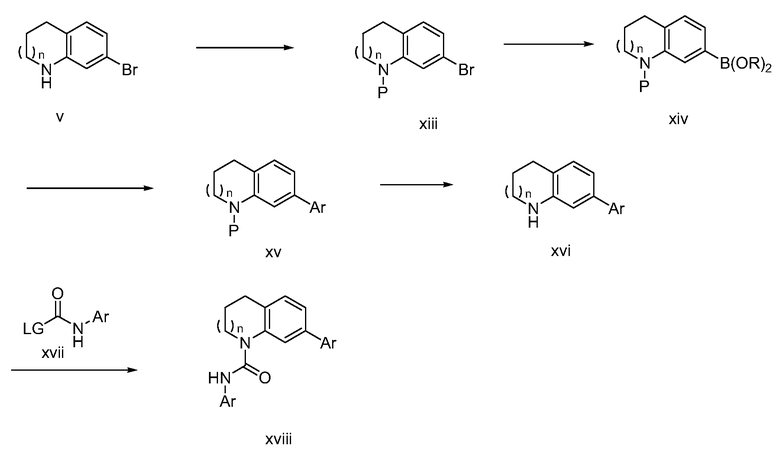
Например, вторичная аминогруппа в v может быть замещена защитной группой (P), (например, ВОС, ацил) с получением xiii. Дальнейшее превращение соединения xiii в его соответствующий боронатный сложный эфир может быть осуществлено опосредуемым палладием сочетанием с пинаколдиборановым реактивом с получением боронатного сложного эфира xiv. Сложный боронатный эфир xiv может использоваться в опосредуемой палладием реакции сочетания Miyaura-Suzuki с арил- или гетероарилгалогенидом (ArX) с получением арильного соединения xv. Последующее удаление защитной группы P на xv с последующим сочетанием xvi с производным карбонила xvii (в котором LG обозначает уходящую группу (например, хлорид, имидазол, бромид) может дать уреидо-соединение xviii.
Дополнительные синтетические превращения, которые могут использоваться для превращения промежуточного соединения xviii (а также родственных соединений viii и xii) в соединения Формулы I описаны подробно в разделе Примеров.
III. Композиции
В дополнение к одному или более соединений, приведенных выше (или их стереоизомерам, геометрическим изомерам, таутомерам, сольватам, метаболитам или фармацевтически приемлемым солям, или пролекарствам), композиции для модуляции активности семейства белков Bcl-2 у человека и животных обычно содержат фармацевтический носитель или разбавитель. В одном варианте осуществления изобретение относится к фармацевтической композиции, включающей соединение Формулы I и по меньшей мере один фармацевтически приемлемый разбавитель, носитель или эксципиент.
Термин "композиция", в рамках изобретения, охватывает продукт, включающий указанные ингредиенты в указанных количествах, а также любой продукт, являющийся, прямо или косвенно, результатом комбинации указанных ингредиентов в указанных количествах. "Фармацевтически приемлемый" означает, что носитель, разбавитель или эксципиент должен быть совместим с другими ингредиентами состава и не быть вредным для реципиента.
Чтобы использовать соединение по изобретению для терапевтического лечения (включая профилактическое лечение) пациента, его обычно составляют в соответствии с обычной фармацевтической практикой в форме фармацевтической композиции. Типичную фармацевтическую композицию получают, смешивая соединение согласно настоящему изобретению и носитель, разбавитель или эксципиент. Подходящие носители, разбавители и эксципиенты известны специалисту и включают такие материалы, как углеводы, воски, растворимые и/или набухающие в воде полимеры, гидрофильные или гидрофобные материалы, желатин, масла, растворители, воду и т.п. Конкретный используемый носитель, разбавитель или эксципиент будут зависеть от средств и цели, с которой применяется соединение согласно настоящему изобретению. Растворители обычно выбирают из растворителей, известных специалистам как безопасные (GRAS) для введения млекопитающему. В целом, безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые являются растворимыми в воде или смешивающимися с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ 400, ПЭГ 300 и т.д.) и их смеси. Составы могут также включать один или более буферов, стабилизаторов, поверхностно-активных веществ, смачивающих веществ, лубрикантов, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, замутнителей, глидантов, веществ для улучшения технологических свойств, красителей, подслащивающих веществ, отдушек, вкусовых агентов и других известных добавок для обеспечения изящного вида лекарственного средства (то есть соединения согласно настоящему изобретению или его фармацевтической композиции) или вспомогательных веществ для производства фармацевтического продукта (то есть лекарственного средства).
Составы могут быть получены с использованием обычных процедур растворения и смешивания. Например, лекарственное вещество (то есть соединение согласно настоящему изобретению или стабилизированную форму этого соединения (например, комплекс с производным циклодекстрина или другим известным комплексообразующим средством)) растворяют в подходящем растворителе в присутствии одного или более эксципиентов, описанных выше. Соединение согласно настоящему изобретению обычно составляют в фармацевтические формы, чтобы обеспечить легко контролируемую дозировку лекарственного средства и обеспечить согласие пациента с предписанным режимом.
Фармацевтическая композиция (или состав) для применения может быть упакована различным образом в зависимости от способа, используемого для введения лекарственного средства. В целом, изделие для распределения включает емкость, содержащую фармацевтический состав в подходящей форме. Подходящие емкости известны специалисту и включают такие материалы, как баллоны (пластмассовые и стеклянные), пакетики, ампулы, полиэтиленовые пакеты, металлические цилиндры и т.п. Емкость может также включать индикатор вскрытия для предотвращения несанкционированного доступа к содержимому упаковки. Кроме того, емкость имеет этикетку, которая описывает содержимое емкости. Этикетка может также включать соответствующие предупреждения.
Фармацевтические композиции соединения согласно настоящему изобретению могут быть получены для различных путей и типов введения. Например, соединение по изобретению (например, соединение Формулы I), имеющее желаемую степень чистоты, может в случае необходимости быть смешано с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (см., Remington: The Science and Practice of Pharmacy: Remington the Science and Practice of Pharmacy (2005) 21st Edition, Lippincott Williams & Wilkins, Philidelphia, PA), в форме лиофилизированного состава, молотого порошка или водного раствора. Композиции могут быть получены путем смешивания при температуре окружающей среды при подходящем рН и при желаемой степени чистоты с физиологически приемлемыми носителями, то есть носителями, которые являются нетоксичными для реципиентов в используемых дозах и концентрациях. Величина рН состава зависит главным образом от конкретного использования и концентрации соединения, но может составлять от приблизительно 3 до приблизительно 8. Состав в ацетатном буфере при рН 5 представляет собой подходящий вариант осуществления.
Соединение по изобретению (например, соединение Формулы I) для применения согласно изобретению предпочтительно является стерильным. В частности, композиции или составы, которые используются для введения in vivo, должны быть стерильными. Такая стерилизация легко может быть осуществлена фильтрацией через мембраны для стерильной фильтрации.
Соединение по изобретению обычно может быть сохранено в форме твердой композиции, лиофилизированного состава или водного раствора.
Фармацевтическую композицию по изобретению составляют, дозируют и вводят таким образом, то есть в таких количествах, концентрациях, согласно таким схемам, курсу, в таких носителях и такими путями введения, который совместим с хорошей медицинской практикой. Факторы для рассмотрения в этом контексте включают конкретное подвергаемое лечению нарушение, конкретное получающее лечение млекопитающее, клиническое состояние индивидуального пациента, причину нарушения, участок доставки средства, способ введения, схему введения и другие факторы, известные врачам. "Терапевтически эффективное количество" вводимого соединения зависит от таких соображений и представляет собой минимальное количество, необходимое для предотвращения, облегчения или лечения заболеваний, которые могут быть охарактеризованы экспрессией или суперэкспрессией белков Bcl-XL. Такое количество предпочтительно ниже количества, которое является токсичным для хозяина.
Как общее предложение, начальное фармацевтически эффективное количество соединения-ингибитора по изобретению, вводимого парентерально, на дозу будет находиться в диапазоне приблизительно 0,01-100 мг/кг, а именно, от приблизительно 0,1 до 20 мг/кг массы тела пациента в сутки, причем обычный начальный диапазон используемого соединения составляет от 0,3 до 15 мг/кг/сутки.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмоний хлорид; гексаметоний хлорид; бензалконий хлорид, бензетоний хлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцин; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее чем приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; агенты для образованию хелатных соединений, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, комплекты Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Активный фармацевтический ингредиент по изобретению (например, соединение Формулы I) может также быть заключен в микрокапсулы, полученные, например, методами коацервации или межфазной полимеризации, например, микрокапсулы из гидроксиметилцеллюлозы или желатина и поли-(метилметакрилатные) микрокапсулы, соответственно; в коллоидных системах для доставки лекарственного средства, например, липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах; или в макроэмульсиях. Такие методики раскрыты в Remington: The Science and Practice of Pharmacy: Remington the Science and Practice of Pharmacy (2005) 21st Edition, Lippincott Williams & Wilkins, Philidelphia, PA.
Могут быть получены композиции замедленного высвобождения соединения по изобретению (например, соединения Формулы I). Подходящие примеры композиций замедленного высвобождения включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие соединение Формулы I, причем эти матрицы находятся в форме формованных изделий, например, пленок или микрокапсул. Примеры матриц замедленного высвобождения включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поливиниловый спирт)), полилактиды (патент США 3773919), сополимеры L-глутаминовой кислоты и гамма-этил-1-глутамата, неразлагаемый этилен-винилацетат, разлагаемые сополимеры молочная кислота-гликолевая кислота, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочная кислота-гликолевая кислота и лейпролид ацетата) и поли-D-(-)-3-гидроксимасляная кислота.
Фармацевтические композиции включают подходящие для путей введения, подробно описанных здесь. Композиции могут быть представлены в стандартной лекарственной форме и могут быть получены любым из способов, известных в области фармации. Методики и составы можно найти в Remington: The Science and Practice of Pharmacy: Remington the Science and Practice of Pharmacy (2005) 21st Edition, Lippincott Williams & Wilkins, Philidelphia, PA. Такие способы включают стадию объединения активного ингредиента с носителем, который составляет один или более вспомогательных ингредиентов. В целом составы получают однородным и тесным объединением активного ингредиента с жидкими носителями или тонко раздробленными твердыми носителями или с обоими, и затем, в случае необходимости, формированием продукта.
Фармацевтические композиции соединения по изобретению (например, соединения Формулы I), подходящие для перорального введения, могут быть получены в форме отдельных единиц, таких как пилюли, капсулы, облатки или таблетки, каждая из которых содержит предопределенное количество соединения по изобретению.
Прессованные таблетки могут быть получены прессованием в подходящем механизме активного ингредиента в сыпучей форме, такой как порошок или гранулы, в случае необходимости смешанного со связующим, лубрикантом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Отлитые под давлением таблетки могут быть получены отливкой в подходящем механизме смеси порошкового активного ингредиента, увлажненного инертным жидким разбавителем. Таблетки могут быть покрыты или насечены и в случае необходимости составлены так, чтобы обеспечить медленное или контролируемое высвобождение активного ингредиента.
Таблетки, пастилки, таблетки для рассасывания, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, например, желатиновые капсулы, сиропы или эликсиры могут быть получены для перорального использования. Составы соединения по изобретению (например, соединения Формулы I), предназначенные для перорального использования, могут быть получены согласно любому способу, известному в области производства фармацевтических композиций, и такие композиции могут содержать одно или более средств, включая подсластители, вкусовые агенты, красители и консерванты, чтобы обеспечить приемлемый препарат. Таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым эксципиентом, которые являются подходящими для производства таблеток, являются приемлемыми. Эти эксципиенты могут быть, например, инертными разбавителями, такими как карбонат кальция или натрия, лактоза, фосфат кальция или натрия; гранулирующими и дезинтегрирующими средствами, такими как кукурузный крахмал или альгиновая кислота; связующими, такими как крахмал, желатин или гуммиарабик; и лубрикантами, такими как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут быть покрыты известными методиками, включая микроинкапсулирование, для задержки разложения и адсорбции в желудочно-кишечном тракте и таким образом обеспечения пролонгированного действия в течение более длительного периода. Например, может использоваться пролонгирующий материал, такой как глицерил моностеарат или глицерил дистеарат, отдельно или с воском.
Для лечения глаз или других внешних тканей, например, рта и кожи, составы предпочтительно наносят в форме топической мази или крема, содержащих активный ингредиент(ы) в количестве, например, от 0,075 до 20% вес./вес. Когда они составлены в форме мази, активный ингредиент может использоваться либо с парафиновой, либо с водорастворимой основой мази. Альтернативно, активные ингредиенты могут быть составлены в форме крема с основой для крема масло-в-воде.
Если желательно, водная фаза основы для крема может включать многоатомный спирт, то есть спирт, имеющий две или более гидроксильных групп, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ 400) и их смеси. Топические составы могут по желанию включать соединение, которое усиливает абсорбцию или проникновение активного ингредиента через кожу или другие затрагиваемые области. Примеры таких кожных усилителей проникновения включают диметилсульфоксид и родственные аналоги.
Масляная фаза эмульсий по изобретению может быть составлена из известных ингредиентов известным образом. Хотя фаза может включать просто эмульгатор, она по желанию включает смесь по меньшей мере одного эмульгатора с жиром или маслом, или и с жиром, и с маслом. Предпочтительно, гидрофильный эмульгатор включен вместе с липофильным эмульгатором, который действует как стабилизатор. Она также предпочтительно включает и масло, и жир. Вместе эмульгатор(ы) со стабилизатором(ами) или без стабилизатора(ов) составляет так называемый эмульгирующий воск, и воск вместе с маслом и жиром составляет так называемую эмульгирующую основу мази, которая формирует масляную дисперсную фазу составов крема. Эмульгаторы и стабилизаторы эмульсий, подходящие для использования в составе по изобретению, включают TWEEN® 60, SPAN® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерил моностеарат и лаурилсульфат натрия.
Водные суспензии соединения по изобретению (например, соединения Формулы I) содержат активные вещества в смеси с эксципиентами, подходящими для получения водных суспензий. Такие эксципиенты включают суспендирующий агент, такой как карбоксиметилцеллюлоза натрия, кроскармеллоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакант и гуммиарабик, и диспергирующие или смачивающие вещества, такие как натуральный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтилен стеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида с неполным эфиром многоатомного спирта, полученным из жирной кислоты и ангидрида гексита (например, полиэтиленоксид сорбитан моноолеат). Водная суспензия может также содержать один или более консервантов, таких как этил- или н-пропил-п-гидроксибензоат, один или более красителей, один или более вкусовых агентов и один или более подсластителей, таких как сахароза или сахарин.
Фармацевтическая композиция соединения по изобретению (например, соединения Формулы I) может быть в форме стерильного инъецируемого препарата, такого как стерильная инъецируемая водная или масляная суспензия. Эта суспензия может быть составлена согласно известному уровню техники с использованием подходящих диспергирующих или смачивающих веществ и суспендирующих агентов, которые были упомянуты выше. Стерильная инъецируемая композиция может также быть стерильным инъецируемым раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле, или может быть получена в форме лиофилизированного порошка. Среди приемлемых носителей и растворителей, которые могут использоваться, можно назвать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла могут традиционно использоваться в качестве растворителя или суспендирующей среды. С этой целью может использоваться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, могут аналогично использоваться в получении препаратов для инъекций.
Количество активного ингредиента, который может быть объединен с материалом-носителем с получением стандартной лекарственной формы, варьирует в зависимости от получающего лечение хозяина и конкретного способа введения. Например, композиция для замедленного высвобождения, предназначенная для перорального введения человеку, может содержать от приблизительно 1 до 1000 мг активного вещества, составленного с подходящим и удобным количеством материала-носителя, которое может варьировать от приблизительно 5 до приблизительно 95% (вес:вес) от общей массы композиции. Фармацевтическая композиция может быть получена таким образом, чтобы обеспечить легко измеримые количества для введения. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать от приблизительно 3 до 500 мкг активного ингредиента на миллилитр раствора, чтобы обеспечить инфузию подходящего объема со скоростью приблизительно 30 мл/ч.
Композиции, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекции, которые могут содержать антиоксиданты, буферы, противомикробные добавки и растворенные вещества, которые делают состав изотоническим по отношению к крови намеченного реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители.
Композиции, подходящие для топического введения в глаз, также включают глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, особенно водном растворителе для активного ингредиента. Активный ингредиент предпочтительно присутствует в таких составах в концентрации от приблизительно 0,5 до 20% вес./вес., например, от приблизительно 0,5 до 10% вес./вес., например, приблизительно 1,5% вес./вес.
Композиции, подходящие для топического введения в рот, включают таблетки, включающие активный ингредиент в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, включающие активный ингредиент в инертном основании, таком как желатин и глицерин, или сахароза и гуммиарабик; и жидкости для полоскания рта, включающие активный ингредиент в подходящем жидком носителе.
Композиции для ректального введения могут быть представлены как суппозиторий с подходящей основой, включающей, например, масло какао или салицилат.
Композиции, подходящие для интрапульмонарного или носового введения, имеют размер частиц, например, в диапазоне от 0,1 до 500 микрон (включая размер частиц в диапазоне от 0,1 до 500 микрон с микронным инкрементом, таким как 0,5, 1, 30 микрон, 35 микрон и т.д.), и их вводят быстрой ингаляцией через носовой проход или ингаляцией через рот для достижения альвеолярных мешков. Подходящие составы включают водные или масляные растворы активного ингредиента. Составы, подходящие для аэрозольного или сухого порошкового введения, могут быть получены согласно обычным способам и могут быть доставлены с другими терапевтическими средствами, такими как соединения, ранее используемые в лечении или профилактике нарушений, как описано ниже.
Композиции, подходящие для влагалищного введения, могут быть представлены как маточные кольца, тампоны, кремы, гели, пасты, пены или составы спрея, содержащие, в дополнение к активному ингредиенту, такие носители, которые считаются подходящими в данной области.
Фармацевтические композиции могут быть упакованы в однодозные или мультидозные емкости, например, закрытые ампулы и пузырьки, и могут быть сохранены в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды, для инъекции непосредственно перед использованием. Экстемпоральные растворы и суспензии для инъекции получают из стерильных порошков, гранул и таблеток предварительно описанного вида. Предпочтительными лекарственными составами являются такие, которые содержат суточную дозу или единицу суточной суб-дозы, как описано здесь выше, или их подходящую часть, активного ингредиента.
Изобретение далее относится к ветеринарным композициям, включающим по меньшей мере один активный ингредиент (например, соединение Формулы I), как определено выше, вместе с ветеринарным носителем. Ветеринарные носители представляют собой материалы, пригодные для введения композиции, и могут быть твердыми, жидкими или газообразными материалами, которые являются в других отношениях инертными или приемлемыми в области ветеринарии и совместимы с активным ингредиентом. Эти ветеринарные композиции могут вводиться парентерально, перорально или любым другим желаемым путем.
IV. Способы применения
Соединения по изобретению (то есть соединения Формулы I) (или их стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты или фармацевтически приемлемые соли, или пролекарства) связываются с семейством антиапоптотических белков Bcl-2 и, в некоторых аспектах, конкретно антиапоптотических белков Bcl-xL и ингибируют их активность; и поэтому могут быть использованы в лечении заболеваний, состояний и/или нарушений, включая, но не ограничиваясь ими, заболевания, характеризующиеся экспрессией или суперэкспрессией членов семейства антиапоптотических белков Bcl-2, и, в некоторых вариантах осуществления, заболевания, характеризующиеся экспрессией или суперэкспрессией белков Bcl-xL. Соответственно, частный аспект этого изобретения включает способ лечения у пациента заболеваний или состояний, которые могут быть охарактеризованы экспрессией или суперэкспрессией членов семейства антиапоптотических белков Bcl-2. В рамках этого аспекта, в некоторых вариантах осуществления, заболевание или состояние представляет собой рак. Соединения по изобретению могут селективно связываться с подгруппой антиапоптотических белков Bcl-2, например, Bcl-xL, по сравнению с белками Bcl-2, Bcl-w или Mcl-1. В некоторых вариантах осуществления соединения по изобретению показывают по меньшей мере 2-кратную, 50-кратную, 100-кратную, 1000-кратную, 10000-кратную, 20000-кратную или 30000-кратную селективность связывания с белком Bcl-xL по сравнению с белком Bcl-2. В некоторых вариантах осуществления соединения по изобретению показывают по меньшей мере 2-кратную, 50-кратную, 100-кратную, 1000-кратную, 10000-кратную, 20000-кратную или 30000-кратную селективность связывания с белком Bcl-xL по сравнению с белком Mcl-1. В некоторых вариантах осуществления соединения по изобретению показывают по меньшей мере 2-кратную, 50-кратную, 100-кратную, 1000-кратную, 10000-кратную, 20000-кратную или 30000-кратную селективность связывания с белком Bcl-xL по сравнению с белком Bcl-w. В одном варианте осуществления способ включает введение пациенту терапевтически эффективного количества соединения по изобретению, например, соединения Формулы I, (или его стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли, или пролекарства). В другом варианте осуществления настоящее изобретение относится к способам лечения у пациента заболеваний и состояний, которые характеризуются экспрессией или суперэкспрессией антиапоптотического белка Bcl-xL, включающим введение пациенту терапевтически эффективного количества соединения Формулы I или его фармацевтической композиции. В одном аспекте указанные композиции для лечения заболеваний и состояний, в ходе которых экспрессируется или суперэкспрессируется антиапоптотический белок Bcl-xL, включают эксципиент и терапевтически эффективное количество соединения Формулы I.
Также изобретение относится к применению соединения по изобретению, например, Формулы I, (или его стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли, или пролекарства), в получении лекарственного средства для лечения заболеваний и состояний, описанных здесь, у пациента, страдающего таким нарушением.
Соединения по изобретению могут вводиться любым путем, подходящим для подвергаемого лечению состояния. Подходящие пути включают пероральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, кожный, внутриоболочковый и эпидуральный), чрескожный, ректальный, носовой, топический (включая щечный и подъязычный), влагалищный, внутрибрюшинный, внутрилегочный и внутриносовой. Для местного иммуносупрессивного лечения соединения могут вводиться внутрь поврежденного участка, включая перфузию или другой контакт трансплантата с ингибитором перед трансплантацией. Следует понимать, что предпочтительный путь может варьировать в зависимости от, например, состояния реципиента. Если соединение вводят перорально, оно может быть составлено в форме пилюли, капсулы, таблетки и т.д. с фармацевтически приемлемым носителем или эксципиентом. Если соединение вводят парентерально, оно может быть составлено с фармацевтически приемлемым парентеральным носителем и в стандартной лекарственной инъецируемой форме, как подробно описано ниже.
Доза для лечения человека может составлять от приблизительно 10 мг до приблизительно 1000 мг соединения Формулы I. Типичная доза может составлять от приблизительно 100 мг до приблизительно 300 мг соединения. Доза может вводиться один раз в сутки (QID), два раза в сутки (BID) или более часто, в зависимости от фармакокинетических и фармакодинамических свойств, включая абсорбцию, распределение, метаболизм и экскрецию конкретного соединения. Кроме того, на дозировку и режим введения могут влиять факторы токсичности. При пероральном введении пилюля, капсула или таблетка могут приниматься один раз в сутки или реже в течение указанного промежутка времени. Режим может быть повторен в виде множества циклов терапии.
В другом варианте осуществления настоящее изобретение относится к композициям, включающим фармацевтически приемлемый эксципиент и терапевтически эффективное количество соединения Формулы I, для лечения заболеваний или состояний аномального роста клеток и/или дерегуляции апоптоза, таких как рак, мезотелиома, рак мочевого пузыря, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак яичника, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, карцинома шейки матки, рак влагалища, рак вульвы, рак кости, рак яичника, рак шейки матки, рак толстой кишки, рак прямой кишки, рак анальной области, рак желудка, рак желудочно-кишечного тракта (желудка, колоректальный и двенадцатиперстной кишки), хронический лимфоцитарный лейкоз, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечника, саркома мягких тканей, рак уретры, рак полового члена, рак яичек, гепатоцеллюлярный рак (печени и желчного протока), первичная или вторичная опухоль центральной нервной системы, первичная или вторичная опухоль головного мозга, болезнь Ходжкина, хронический или острый лейкоза, хронический миелоидный лейкоз, лимфоцитарные лимфомы, лимфобластный лейкоз, фолликулярная лимфома, лимфоидные злокачественные процессы Т-клеточного или В-клеточного происхождения, меланомы, множественная миелома, рак полости рта, рак яичника, немелкоклеточный рак легкого, рак предстательной железы, мелкоклеточный рак легкого, рак почки и мочеточника, почечно-клеточный рак, рак почечной лоханки, опухоли центральной нервной системы, первичная лимфома центральной нервной системы, неходжкинская лимфома, опухоли позвоночника, глиома ствола головного мозга, аденома гипофиза, адренокортикальный рак, рак желчного пузыря, рак селезенки, холангиокарцинома, фибросаркома, нейробластома, ретинобластома или их комбинация.
В другом варианте осуществления настоящее изобретение относится к способу лечения мезотелиомы, рака мочевого пузыря, рака поджелудочной железы, рака кожи, рака головы или шеи, кожной или внутриглазной меланомы, рака яичника, рака молочной железы, рака матки, рака маточных труб, рака эндометрия, карциномы шейки матки, рака влагалища, рака вульвы, рака кости, рака яичника, рака шейки матки, рака толстой кишки, рака прямой кишки, рака анальной области, рака желудка, рака желудочно-кишечного тракта (желудка, колоректальный и двенадцатиперстной кишки), хронического лимфоцитарного лейкоза, рака пищевода, рака тонкой кишки, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечника, саркомы мягких тканей, рака уретры, рака полового члена, рака яичек, гепатоцеллюлярного рака (печени и желчного протока), первичной или вторичной опухоли центральной нервной системы, первичной или вторичной опухоли головного мозга, болезни Ходжкина, хронического или острого лейкоза, хронического миелоидного лейкоза, лимфоцитарных лимфом, лимфобластного лейкоза, фолликулярной лимфомы, лимфоидных злокачественных процессов Т-клеточного или В-клеточного происхождения, меланом, множественной миеломы, рака полости рта, рака яичника, немелкоклеточного рака легкого, рака предстательной железы, мелкоклеточного рака легкого, рака почки и мочеточника, почечно-клеточного рака, рака почечной лоханки, опухолей центральной нервной системы, первичной лимфомы центральной нервной системы, неходжкинской лимфомы, опухолей позвоночника, глиомы ствола головного мозга, аденомы гипофиза, адренокортикального рака, рака желчного пузыря, рака селезенки, холангиокарциномы, фибросаркомы, нейробластомы, ретинобластомы, или комбинации одного или более вышеуказанных видов рака у пациента, причем указанный способ включает введение пациенту терапевтически эффективного количества соединения Формулы I.
В еще одном варианте осуществления настоящее изобретение относится к способам лечения аутоиммунного заболевания у млекопитающего, включающим введение млекопитающему терапевтически приемлемого количества соединения, имеющего Формулу I. Участие белков Bcl-2 в иммунных и аутоиммунных заболеваниях описано в Puck, J.M. et al, Current Allergy and Asthma Reports 2003, 3, 378-384; Shimazaki, K. et al., British Journal ofHaematology 2000, 110(3), 584-90; Rengan, R. et al., Blood 2000, 95(4), 1283-92; and Holzelova, E. et al., New England Journal of Medicine 2004, 351(14), 1409-1418.
Аутоиммунные нарушения включают синдром приобретенного иммуннодефицита (СПИД), аутоиммунный лимфопролиферативный синдром, гемолитическую анемию, воспалительные заболевания и тромбоцитопению, острое или хроническое иммунопатологическое заболевание, связанное с трансплантацией органа, болезнь Аддисона, аллергические заболевания, алопецию, алопецию ареата, атероматозное заболевание/артериосклероз, атеросклероз, артрит (включая остеоартрит, ювенильный хронический артрит, септический артрит, артрит Лайма, псориатический артрит и реактивный артрит), аутоиммунное буллезное заболевание, абеталипопротемию, заболевания, связанные с приобретенным иммуннодефицитом, острое иммунопатологическое заболевание, связанное с трансплантацией органа, приобретенный акроцианоз, острые и хронические паразитарные или инфекционные процессы, острый панкреатит, острый отказ почек, острый ревматизм, острый поперечный миелит, аденокарциномы, атриальные эктопические систолы, (острый) респираторный дистресс-синдром взрослых, комплекс СПИД-деменция, алкогольный цирроз, вызванное алкоголем повреждение печени, вызванный алкоголем гепатит, аллергический конъюнктивит, аллергический контактный дерматит, аллергический ринит, аллергию и астму, отторжение аллотрансплантата, дефицит альфа-1-антитрипсина, болезнь Альцгеймера, боковой амиотрофический склероз, анемию, стенокардию, заболевание легкого, связанное с анкилозирующим спондилитом, дегенерацию клеток переднего рога, опосредуемую антителом цитотоксичность, антифосфолипидный синдром, антирецепторные гиперсензитивные реакции, аортальные и периферические аневризмы, расслоение аорты, артериальную гипертензию, артериосклероз, артериовенозный свищ, артропатию, астению, астму, атаксию, атопическую аллергию, фибрилляцию предсердий (постоянную или пароксизмальную), мерцание предсердий, атриовентрикулярную блокаду, атрофический аутоиммунный гипотиреоз, аутоиммунную гемолитическую анемию, аутоиммунный гепатит, аутоиммунный гепатит типа 1 (классический аутоиммунный или лупоидный гепатит), аутоиммунно опосредованную гипогликемию, аутоиммунную нейтропению, аутоиммунную тромбоцитопению, аутоиммунное заболевание щитовидной железы, В-клеточную лимфому, отторжение костного трансплантата, отторжение трансплантата костного мозга (ВМТ), облитерирующий бронхиолит, блокада ножки пучка Гиса, ожоги, кахексию, аритмии сердца, синдром кардиостимулятора, кардиальные опухоли, кардиомиопатию, воспалительную реакцию при экстрапульмональном кровообращении, отторжение трансплантата хряща, дегенерации коры мозжечка, мозжечковые расстройства, хаотическую или мультифокальную предсердную тахикардию, сопутствующие заболевания при химиотерапии, хламидиоз, холеостаз, хронический алкоголизм, хронический активный гепатит, синдром хронической усталости, хроническое иммунопатологическое заболевание, связанное с трансплантацией органа, хроническую эозинофильную пневмонию, хронические воспалительные патологии, хронический кожно-слизистый кандидоз, хроническое обструктивное заболевание легких (COPD), хроническую интоксикацию салициловой кислотой, колоректальные общие вариабельные иммуннодефициты (общая вариабельная гипогаммаглобулинемия), конъюнктивит, интерстициальное заболевание легкого, связанное с коллагенозом, контактный дерматит, Кумбс-положительную гемолитическую анемию, легочное сердце, болезнь Крейцфельда-Якоба, криптогенный аутоиммунный гепатит, криптогенный фиброзирующий альвеолит, культуронегативный сепсис, муковисцидоз, сопутствующие заболевания при терапии цитокинами, болезнь Крона, деменцию боксеров, демиелинизирующие заболевания, геморрагическую лихорадку, дерматит, склеродермию при дерматите, дерматологические состояния, дерматомиозит/полимиозит-связанное заболевание легкого, диабет, диабетическое артериосклеротическое заболевание, сахарный диабет, болезнь диффузных телец Леви, расширенную кардиомиопатию, расширенную застойную кардиомиопатию, дисковидную красную волчанку, нарушения, связанные с базальными ганглиями, диссеминированную внутрисосудистую коагуляцию, синдром Дауна в среднем возрасте, лекарственное интерстициальное заболевание легкого, лекарственный гепатит, лекарственные нарушения движения, вызванные лекарственными средствами, которые блокируют допамин ЦНС, рецепторы, лекарственную сензитивность, экзему, энцефаломиелит, эндокардит, эндокринопатию, энтеропатический синовит, эпиглоттит, инфекцию вирусом Эпштейн-Барра, эритромелалгию, экстрапирамидальные и мозжечковые нарушения, семейный гематофагоцитарный лимфогистиоцитоз, отторжение имплантата эмбрионального тимуса, атаксию Фридриха, функциональные периферические артериальные нарушения, женское бесплодие, фиброз, фиброзное заболевание легкого, грибковый сепсис, газовую гангрену, язву желудка, гигантоклеточный артериит, гломерулярный нефрит, гломерулонефриты, синдром Гудпасчера, зобный аутоиммунный гипотиреоз (болезнь Хасимото), подагрический артрит, отторжение трансплантата любого органа или ткани, болезнь трансплантат против хозяина, грамотрицательный сепсиса, грамположительный сепсис, гранулемы вследствие внутриклеточных организмов, инфекция стрептококками группы В (GBS), болезнь Грейвса, заболевание легкого, связанное с гемосидерозом, волосатоклеточный лейкоз, волосатоклеточный лейкоз, болезнь Галлервордена-Шпатца, тиреоидит Хасимото, поллиноз, отторжение сердечного трансплантата, гемахроматоз, гематопоэтические злокачественные процессы (лейкоз и лимфома), гемолитическую анемию, гемолитический уремический синдром/тромболитическую тромбоцитопеническую пурпуру, кровотечение, пурпуру Хеноха-Шенлейна, гепатит A, гепатит B, гепатит C, ВИЧ инфекцию/ВИЧ невропатию, болезнь Ходжкина, гипопаратиреоз, хорею Гентингтона, гиперкинетические нарушения движения, гиперсензитивные реакции, гиперсензитивный пневмонит, гипертиреоз, гипокинетические нарушения движения, оценку гипоталамо-гипофизарно-надпочечниковой оси, идиопатическую болезнь Аддисона, идиопатическую лейкопению, идиопатический легочный фиброз, идиопатическую тромбоцитопению, идиосинкратическое заболевание печени, инфантильную спинальную мышечную атрофию, инфекционные заболевания, воспаление аорты, воспалительное заболевание кишечника, инсулинзависимый сахарный диабет, промежуточный пневмонит, иридоциклит/увеит/оптический неврит, повреждение при ишемии-реперфузии, ишемический инсульт, ювенильную пернициозную анемию, ювенильный ревматоидный артрит, ювенильную спинальную мышечную атрофию, саркому Капоши, болезнь Кавасаки, отторжение почечного трансплантата, легионеллез, лейшманиаз, лепру, поражения кортикоспинальной системы, болезнь линейного IgA, липидемию, отторжения трансплантата печени, болезнь Лайма, лимфодермию, лимфоцитарное инфильтративное заболевание легкого, малярию, мужское идиопатическое бесплодие или NOS, злокачественный гистиоцитоз, злокачественную меланому, менингит, менингококкемию, микроскопический васкулит почек, головную боль при мигрени, митохондриальное мультисистемное нарушение, смешанный коллагеноз, заболевание легкого, связанное со смешанным коллагенозом, моноклональную гаммопатию, множественную миелому, мультисистемные дегенерации (Mencel Dejerine-Thomas Shi-Drager и Machado-Joseph), миалгический энцефалит/синдром хронической усталости, миастению гравис, микроскопический васкулит почек, внутриклеточную mycobacterium avium, mycobacterium tuberculosis, миелодипластический синдром, инфаркт миокарда, миокардиальные ишемические нарушения, носоглоточный рак, хроническое заболевание легкого новорожденных, нефрит, нефроз, нефротический синдром, нейродегенеративные заболевания, нейрогенные мышечные атрофии, нейтропеническую лихорадку, неалкогольный стеатогепатит, окклюзию брюшной аорты и ее ответвлений, обтурирующие артериальные нарушения, отторжения трансплантата органа, орхит/эпидидимит, процедуры реверсирования орхита/вазэктомии, органомегалию, остеоартроз, остеопороз, нарушения овуляции, отторжения трансплантата поджелудочной железы, паразитарные заболевания, отторжения трансплантата паращитовидной железы, болезнь Паркинсона, тазовое воспалительное заболевание, пузырчатку обыкновенную, пузырчатка листовидную, пемфигоид, многолетний ринит, перикардиальное заболевание, периферическое атеросклеротическое заболевание, периферические сосудистые нарушения, перитонит, пернициозную анемию, факогенный увеит, пневмонию Pneumocystis carinii, пневмонию, синдром POEMS (полиневропатия, органомегалия, эндокринопатия, моноклональная гаммопатия и поражение кожи), постперфузионный синдром, синдром системного воспалительного ответа, синдром кардиотомии пост-MI, постинфекционное промежуточное заболевание легкого, преждевременное нарушение овуляции, первичный желчный цирроз, первичный склерозирующий гепатит, первичную миксоэдему, первичную легочную артериальную гипертензию, первичный склерозирующий холангит, первичный васкулит, прогрессивный надъядерный паралич, псориаз, псориаз типа 1, псориаз типа 2, псориатичесую артропатию, вторичную легочную гипертензию вследствие коллагеноза, легочное проявление узелкового полиартериита, поствоспалительное интерстициальное заболевание легкого, лучевой фиброз, лучевую терапию, феномен и болезнь Рейно, болезнь Рефсума, тахикардию с постоянным сближенным QRS, болезнь Рейтера, почечное заболевание NOS, вазоренальную гипертензию, реперфузионное повреждение, рестриктивную кардиомиопатию, связанное с ревматоидным артритом интерстициальное заболевание легкого, ревматоидный спондилит, саркоидоз, синдром Шмидта, склеродермию, сенильную хорею, сенильную деменцию типа телец Леви, септический синдром, септический шок, серонегативные артропатии, шок, серповидноклеточную анемию, связанное с болезнью Шегрена заболевание легкого, синдром Шегрена, отторжение аллотрансплантата кожи, синдром поражения кожи, отторжение трансплантата тонкого кишечника, аутоиммунитет спермы, рассеянный склероз (все подтипы), спинальную атаксию, дегенерации спинного мозга, спондилоартропатию, спорадическая плюригландулярная недостаточность типа I, спорадическая плюригландулярная недостаточность типа II, болезнь Стилла, стрептококковый миозит, инсульт, структурные поражения мозжечка, подострый склерозирующий панэнцефалит, симпатическую офтальмию, обморок, сифилис сердечно-сосудистой системы, системную анафилаксию, синдром системного воспалительного ответа, системное начало ювенильного ревматоидного артрита, системную красную волчанку, связанное с системной красной волчанкой заболевание легкого, системный склероз, связанное с системным склерозом интерстициальное заболевание легкого, Т-клеточные или FAB ALL, болезнь Такаясу/артериит, телеангиэктазию, Тип Th2- и Тип Th1-опосредованные заболевания, облитерирующий тромбангиит, тромбоцитопению, тиреоидит, токсичность, синдром токсического шока, трансплантаты, травма/кровотечение, аутоиммунный гепатит типа 2 (гепатит антител анти-LKM), инсулинорезистентность типа B с акантокератодермией, гиперсензитивные реакции типа III, гиперсензитивные реакции типа IV, артропатию с язвенным колитом, неспецифический язвенный колит, нестабильную стенокардию, уремию, уросепсис, крапивницу, увеит, болезни клапанов сердца, варикозные заболевания вен, васкулит, васкулитическое диффузное заболевание легкого, венозные заболевания, венозный тромбоз, фибрилляцию желудочков, витилиго, острое заболевание печени, вирусные и грибковые инфекции, витальный энцефалит/асептический менингит, витально-ассоциированный гемафагоцитарный синдром, гранулематоз Вегенера, синдром Вернике-Корскова, гепатолентикулярную дегенерацию, отторжение ксенотрансплантата любого органа или ткани, иерсиниозную и связанную с сальмонеллой артропатии и т.п.
В одном варианте осуществления соединение по изобретению (например, соединение Формулы I), или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемая соль, пролекарство, используется в качестве противоракового средства или как дополнительное средство для лечения рака в комбинированной терапии. Специалист легко может определить, действительно ли соединение-кандидат лечит злокачественное состояние в любом конкретном типе клеток, индивидуально или в комбинации. В некоторых аспектах этого варианта осуществления соединения по изобретению используются в сочетании с другими видами лечения, включая обычную хирургию, лучевую терапию и химиотерапию, для лечения рака.
Такие виды лечения могут включать один или больше следующих категорий противораковых средств: алкилирующие агенты, ингибиторы ангиогенеза, антитела, антиметаболиты, антимитотические средства, антипролиферативные средства, ингибиторы Аврора-киназы, промооторы апоптоза (например, ингибиторы Bcl-xL, Bcl-w и Bfl-1), активаторы пути рецептора смерти, ингибиторы Bcr-Abl киназы, BiTE (Bi-Specific T cell Engager) антитела, модификаторы биологического ответа, ингибиторы циклинзависимой киназы, ингибиторы клеточного цикла, ингибиторы циклооксигеназы-2, двойные связывающие вариабельные домены белки (DVD), ингибиторы рецептора гомолога онкогена вируса лейкемии (ErbB2), ингибиторы фактора роста, ингибиторы белка теплового шока (HSP)-90, ингибиторы гистон деацетилазы (HDAC), гормональные терапевтические средства, иммунологические средства, ингибиторы встраивающихся в апоптотические белки (IAPs) антибиотиков, ингибиторы киназы, ингибиторы мишеней рапамицина млекопитающих, ингибиторы микроРНК митоген-активируемой внеклеточной регулируемой сигналом киназы, поливалентные связывающие белки, нестероидные противовоспалительные лекарственные средства (NSAID), ингибиторы поли АДФ(аденозин дифосфат)-рибоза полимеразы (PARP), платиновые химиотерапевтические средства, ингибиторы поло-подобной киназы (PIk), ингибиторы протеосом, аналоги пурина, аналоги пиримидина, ингибиторы рецептора тирозин-киназы, ретиноидные/дельтоидные алкалоиды растений, малые ингибирующие рибонуклеиновые кислоты (siРНК), ингибиторы топоизомеразы, их комбинации и т.п.
BiTE антитела представляют собой би-специфические антитела, которые направляют Т-клетки к атаке на раковые клетки, одновременно связывая эти две клетки. Т-клетка затем атакует целевую раковую клетку. Примеры BiTE антител включают адекатумумаб (Micromet MT201), блинатумомаб (Micromet МТ103) и т.п. Не будучи ограниченным теорией, один из механизмов, которыми Т-клетки инициируют апоптоз целевой раковой клетки, представляет собой экзоцитоз компонентов цитолитических гранул, которые включают перфорин и гранзим B. В этом отношении было показано, что Bcl-2 аттенуируют индукцию апоптоза как перфорином, так и гранзимом B. Эти данные позволяют предположить, что ингибирование Bcl-2 может усиливать цитотоксические эффекты, проявляемые Т-клетками при нацеливании на раковые клетки (V.R. Sutton, D.L. Vaux and J.A. Trapani (1997) J. of Immunology. 158 (12): 5783).
SiРНК представляют собой молекулы, имеющие эндогенные основания РНК или химически модифицированные нуклеотиды. Эти модификации не противодействуют клеточной активности, а только придают увеличенную стабильность и/или увеличенный клеточный потенциал. Примеры химических модификаций включают фосфоротиоатные группы, 2'-дезоксинуклеотид, 2'-OCH3-содержащие рибонуклеотиды, 2'-F-рибонуклеотиды, 2'-метоксиэтил рибонуклеотиды, их комбинации и т.п. siРНК могут иметь варьирующую длину (например, 10-200 пар оснований) и структуру (например, шпильки, одинарная/двойная нить, петли, разрывы/пропуски, несовпадения) и процессируются в клетках, обеспечивая заглушение активного гена. Двунитевая siРНК (dsРНК) может иметь одно и то же число нуклеотидов на каждой нити (тупоконечные концы) или асимметричные концы (выступы). Выступ 1-2 нуклеотидов может находиться на смысловой и/или антисмысловой цепи, а также на 5'- и/или 3'-концах данной цепи. Например, было показано, что нацеливающие siРНК Mcl-1 усиливают активность АВТ-263, (то есть N-(4-(4-((2-(4-хлорфенил)-5,5-диметил-1-циклогекс-1-ен-1-ил)метил)пиперазин-1-ил)бензоил)-4-(((1R)-3-(морфолин-4-ил)-1-((фенилсульфанил)метил)пропил)амино)-3-((трифторметил)сульфонил)бензолсульфонамида) или АВТ-737 (то есть N-(4-(4-((4'-хлор(1,1'-дифенил)-2-ил)метил)пиперазин-1-ил)бензоил)-4-(((lR)-3-(диметиламино)-1-((фенилсульфанил)метил)пропил)амино)-3-нитробензолсульфонамида) на множественных линиях опухолевых клеток (Tse et. al (2008) Cancer Research. 68(9): 3421 и цитируемые там ссылки).
Поливалентные связывающие белки представляют собой связывающие белки, включающие два или более антигенсвязывающих сайтов. Поливалентные связывающие белки составляют так, чтобы они имели три или более антигенсвязывающих сайтов, и обычно они представляют собой искусственные антитела. Термин "мультиспецифический связывающий белок" означает связывающий белок, способный к связыванию двух или более родственных или неродственных мишеней. Связывающие белки с двойным вариабельным доменом (DVD) представляют собой четырехвалентные или поливалентные связывающие белки, включающие два или более антигенсвязывающих сайтов. Такие DVD могут быть моноспецифическими (то есть способными к связыванию одного антигена) или мультиспецифическими (то есть способными к связыванию двух или более антигенов). DVD связывающие белки, включающие два полипептида DVD тяжелой цепи и два полипептида DVD легкой цепи, называют DVD Ig. Каждая половина DVD Ig включает полипептид DVD тяжелой цепи, полипептид DVD легкой цепи и два антигенсвязывающих сайта. Каждый связывающий сайт включает вариабельный домен тяжелой цепи и вариабельный домен легкой цепи с в общей сложности 6 CDR, участвующими в связывании антигена, на антигенсвязывающий сайт.
Алкилирующие агенты включают алтретамин, AMD-473, AP-5280, апазихон, бендамустин, бросталлицин, бусульфан, карбохон, кармустин (BCNU), хлорамбуцил, CLORETAZINE® (ларомустин, VNP 40101M), циклофосфамид, декарбазин, эстрамустин, фотемустин, глуфосфамид, ифосфамид, KW-2170, ломустин (CCNU), мафосфамид, мелфалан, митобронитол, митолактол, нимустин, N-оксид азотной горчицы, ранимустин, темозоломид, тиотепа, TREANDA® (бендамустин), треосульфан, рофосфамид и т.п.
Ингибиторы ангиогенеза включают ингибиторы эндотелий-специфического рецептора тирозин киназы (Tie-2), ингибиторы рецептора эпидермального фактора роста (EGFR), ингибиторы рецептора фактора роста инсулина-2 (IGFR-2), ингибиторы матрикс металлопротеиназы-2 (MMP-2), ингибиторы матрикс металлопротеиназы-9 (MMP-9), ингибиторы рецептора тромбоцитарного фактора роста (PDGFR), аналоги тромбоспондина, ингибиторы тирозинкиназы рецептора сосудистого эндотелиального фактора роста (VEGFR) и т.п.
Антиметаболиты включают ALIMTA® (пеметрексед двунатриевый, LY231514, MTA), 5-азацитидин, XELODA® (капецитабин), кармофур, LEUSTAT® (кладрибин), клофарабин, цитарабин, цитарабин окфосфат, 2-окси-6-аминопиримидин арабинозид, децитабин, дефероксамин, доксифлуридин, эфлорнитин, EICAR (5-этинил-1-β-D-рибофуранозилимидазол-4-карбоксамид), эноцитабин, этинилцитидин, флударабин, 5-фтороурацил, индивидуально или в комбинации с лейковорином, GEMZAR® (гемцитабин), (гидроксикарбамид, ALKERAN® (мельфалан), меркаптопурин, 6-меркаптопурин рибозид, метотрексат, микофеноловая кислота, неларабин, нолатрексед, окфосфат, пелитрексол, пентостатин, ралтитрексед, рибавирин, триапин, триметрексат, S-1, тиазофурин, тегафур, TS-1, видарабин, UFT и т.п.
Ингибиторы Аврора-киназы включают AZD-1152, MLN-8054, VX-680 и т.п.
Белки-ингибиторы семейства Bcl-2 включают АТ-101 ((-)госсипол), GENASENSE® (G3139 или облимерсен (Bcl-2-нацеливающий антисмысловой олигонуклеотид)), IPI-194, IPI-565, N-(4-(4-((4'-хлор(1,1'-дифенил)-2-ил)метил)пиперазин-1-ил)бензоил)-4-(((1R)-3-(диметиламино)-1-((фенилсульфанил)метил)пропил)амино)-3-нитробензолсульфонамид) (АВТ-737), N-(4-(4-((2-(4-хлорфенил)-5,5-диметил-1-циклогекс-1-ен-1-ил)метил)пиперазин-1-ил)бензоил)-4-(((1R)-3-(морфолин-4-ил)-1-((фенилсульфанил)метил)пропил)амино)-3-((трифторметил)сульфонил)бензолсульфонамид (АВТ-263), GX-070 (обатоклакс) и т.п.
Ингибиторы Bcr-Abl киназы включают DASATINIB® (BMS-354825), GLEEVEC® (иматиниб) и т.п.
Ингибиторы CDK включают AZD-5438, BMI-1040, BMS-032, BMS-387, CVT-2584, флавопиридол, GPC-286199, MCS-5A, PD0332991, PHA-690509, селициклиб (CYC-202, R-росковитин), ZK-304709 и т.п.
Ингибиторы СОХ-2 включают АВТ-963, ARCOXIA® (эторикоксиб), BEXTRA® (валдекоксиб), BMS347070, CELEBREX® (целекоксиб), СОХ-189 (лумиракоксиб), СТ-3, DERAMAXX® (деракоксиб), JTE-522, 4-метил-2-(3,4-диметилфенил)-1-(4-сульфамоилфенил-1H-пиррол), МК-663 (эторикоксиб), NS-398, парекоксиб, RS-57067, SC-58125, SD-8381, SVT-2016, S-2474, T-614, VIOXX® (rofecoxib) и т.п.
Ингибиторы EGFR включают ABX-EGF, анти-EGFR иммунолипосомы, EGF-вакцину, EMD-7200, ERBITUX® (цетуксимаб), HR3, антитела IgA, IRESSA® (гефитиниб), TARCEVA® (эрлотиниб или OSI-774), TP-38, слитый белок EGFR, TYKERB® (лапатиниб) и т.п.
Ингибиторы рецептора ErbB2 включают CP-724-714, CI-1033 (канертиниб), HERCEPTIN® (трастузумаб), TYKERB® (лапатиниб), OMNITARG® (2C4, петузумаб), TAK-165, GW-572016 (ионафарниб), GW-282974, EKB-569, PI-166, dHER2 (вакцина HER2), APC-8024 (вакцина HER-2), анти-HER/2neu биспецифическое антитело, B7.her2IgG3, AS HER2 трифункциональные биспецифические антитела, mAB AR-209, mAB 2B-1 и т.п.
Ингибиторы гистон деацетилазы включают депсипептид, LAQ-824, MS-275, трапоксин, субероиланилид гидроксамовую кислоту (SAHA), TSA, вальпроевую кислоту и т.п.
Ингибиторы HSP-90 включают 17-AAG-nab, 17-AAG, CNF-101, CNF-1010, CNF-2024, 17-DMAG, гелданамицин, IPI-504, KOS-953, MYCOGRAB® (человеческое рекомбинантное антитело к HSP-90), NCS-683664, PU24FC1, PU-3, радицикол, SNX-2112, STA-9090 VER49009 и т.п.
Активаторы пути смертельного рецептора включают TRAIL, антитела или другие агенты, которые нацеливают смертельные рецепторы (например, DR4 и DR5), такие как апомаб, конатумумаб, ETR2-ST01, GDC0145, лексатумумаб, HGS-1029, LBY-135, PRO-1762 и трастузумаб.
Ингибиторы MEK включают ARRY-142886, ARRY-438162 PD-325901, PD-98059 и т.п.
Ингибиторы mTOR включают AP-23573, CCI-779, эверолимус, RAD-001, рапамицин, темсиролимус и т.п.
Нестероидные противовоспалительные лекарственные средства включают AMIGESIC® (салсалат), DOLOBID® (дифлунисал), MOTRIN® (ибупрофен), ORUDIS® (кетопрофен), RELAFEN® (набуметон), FELDENE® (пироксикам), крем ибупрофена, ALEVE® (напроксен) и NAPROSYN® (напроксен), VOLTAREN® (диклофенак), INDOCIN® (индометацин), CLINORIL® (сулиндак), TOLECTIN® (толметин), LODINE® (этодолак), TORADOL® (кеторолак), DAYPRO® (оксапрозин) и т.п.
Ингибиторы PDGFR включают C-451, СР-673, СР-868596 и т.п.
Платиновые химиотерапевтические средства включают цисплатин, ELOXATIN® (оксалиплатин), эптаплатин, лобаплатин, недаплатин, PARAPLATIN® (карбоплатин), сатраплатин, пикоплатин и т.п.
Ингибиторы поло-подобных киназ включают BI-2536 и т.п.
Аналоги тромбоспондина включают АВТ-510, АВТ-567, АВТ-898, TSP-1 и т.п.
Ингибиторы VEGFR включают AVASTIN® (бевацизумаб), АВТ-869, AEE-788, ANGIOZYME™ (рибозим, который ингибирует ангиогенез (Ribozyme Pharmaceuticals (Boulder, CO.) and Chiron, (Emeryville, CA)), акситиниб (AG-13736), AZD-2171, СР-547 632, IM-862, MACUGEN (пегаптамиб), NEXAVAR® (сорафениб, BAY43-9006), пазопаниб (GW-786034), ваталаниб (PTK-787, ZK-222584), SUTENT® (сунитиниб, SU-11248), ловушка VEGF, ZACTIMA™ (вандетаниб, ZD-6474) и т.п.
Антибиотики включают встраивающиеся антибиотики акларубицин, актиномицин D, амрубицин, аннамицин, адриамицин, BLENOXANE® (блеомицин), даунорубицин, CAELYX® или MYOCET® (липосомальный доксорубицин), элсамитруцин, эпирбицин, гларбуицин, ZAVEDOS® (идарубицин), митомицин C, неморубицин, неокарциностатин, пепломицин, пирарубицин, ребеккамицин, стималамер, стрептозоцин, VALSTAR® (валрубицин), зиностатин и т.п.
Ингибиторы топоизомеразы включают акларубицин, 9-аминокамптотецин, амонафид, амсакрин, бекатекарин, белотекан, BN-80915, CAMPTOSAR® (иринотекан гидрохлорид), камптотецин, CARDIOXANE® (дексразоксин), дифломотекан, эдотекарин, ELLENCE® или PHARMORUBICIN® (эпирубицин), этопозид, экзатекан, 10-гидроксикамптотецин, гиматекан, луртотекан, митоксантрон, оратецин, пирарбуцин, пиксантрон, рубитекан, собузоксан, SN-38, тафлупозид, топотекан и т.п.
Антитела включают AVASTIN® (бевацизумаб), CD40-специфические антитела, chTNT-1/B, денозумаб, ERBITUX® (цетуксимаб), HUMAX-CD4® (занолимумаб), IGF1R-специфические антитела, линтузумаб, PANOREX® (эдреколомаб), RENCAREX® (WX G250), RITUXAN® (ритуксимаб), тицилимумаб, трастузумаб и т.п.
Гормональные терапевтические средства включают ARIMIDEX® (анастрозол), AROMASIN® (эксеместан), арзоксифен, CASODEX® (бикалутамид), CETROTIDE® (цетрореликс), дегареликс, деслорелин, DESOPAN® (трилостан), дексаметазон, DROGENIL® (флутамид), EVISTA® (ралоксифен), AFEMA™ (фадрозол), FARESTON® (торемифен), FASLODEX® (фулвестрант), FEMARA® (летрозол), форместан, глюкокортикоиды, HECTOROL® (доксеркальциферол), RENAGEL® (севеламер карбонат), лазофоксифен, лейпролид ацетат, MEGACE® (мегестерол), MIFEPREX® (мифепристон), NILANDRON™ (нилутамид), NOLVADEX® (тамоксифен цитрат), PLENAXIS™ (абареликс), преднизон, PROPECIA® (финастерид), рилостан, SUPREFACT® (бусерелин), TRELSTAR® (гормон, высвобождающий лютеинизирующий гормон (LHRH)), VANTAS® (гистрелин имплант), VETORYL® (трилостан или модрастан), ZOLADEX® (фосрелин, госерелин) и т.п.
Дельтоиды и ретиноиды включают сеокальцитол (ЕВ1089, СВ1093), лексакальцитрол (KH1060), фенретинид, PANRETIN® (алиретиноин), ATRAGEN® (липосомальный третиноин), TARGRETIN® (бексаротен), LGD-1550 и т.п.
Ингибиторы PARP включают АВТ-888, олапариб, KU-59436, AZD-2281, AG-014699, DSI-201, BGP-15, INO-1001, ONO-2231 и т.п.
Алкалоиды растения включают, но не ограничены ими, винкристин, винбластин, виндезин, винорелбин и т.п.
Ингибиторы протеасомы включают VELCADE® (бортезомиб), MG132, NPI-0052, PR-171 и т.п.
Примеры иммунологических средств включают интерфероны и другие иммуноусиливающие средства. Интерфероны включают альфа-интерферон, альфа-2a-интерферон, альфа-2b-интерферон, бета-интерферон, гамма-1а-интерферон, ACTIMMUNE® (гамма-1b-интерферон) или гамма-n1-интерферон, их комбинации и т.п. Другие средства включают ALFAFERONE®, (IFN-α), ВАМ-002 (окисленный глутатион), BEROMUN® (тазонермин), BEXXAR® (тозитумомаб), CAMPATH® (алемтузумаб), CTLA4 (цитотоксический лимфоцитарный антиген 4), декарбазин, денилейкин, эпратузумаб, GRANOCYTE® (ленограстим), лентинан, лейкоцитарный альфа-интерферон, имихимод, MDX-010 (анти-CTLA-4), вакцину меланомы, митумомаб, молграмостим, MYLOTARG™ (гемтузумаб озогамицин), NEUPOGEN® (филграстим), OncoVAC-CL, OVAREX® (ореговомаб), пемтумомаб (Y-muHMFGl), PROVENGE® (сипулейцел-T), саргарамостим, сизофилан, тецелейкин, THERACYS® (Бацилла Кальметт-Герена), убенимекс, VIRULIZIN® (иммунотерапевтический, Lorus Pharmaceuticals), Z-100 (Специфическое вещество Maruyama (SSM)), WF-10 (тетрахлордекаоксид (TCDO)), PROLEUKIN® (алдеслейкин), ZADAXIN® (тималфазин), ZENAPAX® (даклизумаб), ZEVALIN® (90Y-ибритумомаб тиуксетан) и т.п.
Модификаторы биологического ответа представляют собой средства, которые модифицируют защитные механизмы живых организмов или биологические ответы, такие как выживание, рост или дифференцировка клеток ткани, направляя их таким образом, чтобы они имели противоопухолевую активность, и включают крестин, лентинан, сизофиран, пицибанил PF-3512676 (CpG-8954), убенимекс и т.п.
Аналоги пиримидина включают цитарабин (ara C или Арабинозид C), 2-окси-6-аминопиримидин арабинозид, доксифлуридин, FLUDARA® (флударабин), 5-FU (5-фтороурацил), флоксуридин, GEMZAR® (гемцитабин), TOMUDEX® (ратитрексед), TROXATYL™ (триацетилуридин троксацитабин) и т.п.
Аналоги пурина включают LANVIS® (тиогуанин) и PURI-NETHOL® (меркаптопурин).
Антимитотические средства включают батабулин, эпотилон D (KOS-862), N-(2-((4-гидроксифенил)амино)пиридин-3-ил)-4-метоксибензолсульфонамид, иксабепилон (BMS-247550), паклитаксел, TAXOTERE® (доцетаксел), PNU100940 (109881), патупилон, XRP-9881 (ларотаксел), винфлунин, ZK-EPO (синтетический эпотилон) и т.п.
Соединения по изобретению могут также использоваться как радиосенсибилизаторы, которые усиливают эффективность лучевой терапии. Примеры лучевой терапии включают внешнюю лучевую терапию, телетерапию, брахитерапию и лучевую терапию с закрытым, открытым источником и т.п.
Дополнительно, соединения, имеющие Формулу I, могут быть скомбинированы с другими химиотерапевтическими средствами, такими как ABRAXANE™ (ABI-007), АВТ-100 (ингибитор фарнезилтрансферазы), ADVEXIN® (вакцина Ad5CMV-p53), ALTOCOR® или MEVACOR® (ловастатин), AMPLIGEN® (поли I:поли C12U, синтетическая РНК), APTOSYN® (экзисулинд), AREDIA® (памидроновая кислота), арглабин, L-аспарагиназа, атаместан (1-метил-3,17-дион-андроста-1,4-диен), AVAGE® (тазаротен), AVE-8062 (производное комбреастатина) BEC2 (митумомаб), кахектин или кахексин (фактор некроза опухоли), канваксин (вакцина), CEAVAC® (раковая вакцина), CELEUK® (целмолейкин), CEPLENE® (гистамин дигидрохлорид), CERVARIX® (вакцина человеческого папилломавируса), CHOP® (C:CYTOXAN® (циклофосфамид); Н:ADRIAMYCIN® (гидроксидоксорубицин); O:Винкристин (ONCOVIN®); P:преднизон), CYPAT™ (ципротерон ацетат), комбрестатин A4P, DAB(389)EGF (каталитический и транслокационный домены токсина дифтерии, слитые через линкер His-Ala с человеческим эпидермальным фактором роста) или TransMID-107R™ (токсины дифтерии), дакарбазин, дактиномицин, 5,6-диметилксантенон-4-уксусная кислота (DMXAA), энилурацил, EVIZON™ (скваламин лактат), DIMERICINE® (лосьон липосомы T4N5), дискодермолид, DX-8951f (экзатекан мезилат), энзастаурин, EPO906 (эпитилон B), GARDASIL® (четырехвалентная рекомбинантная вакцина папилломавируса человека (Типы 6, 11, 16, 18)), GASTRIMMUNE®, GENASENSE®, GMK (вакцина ганглиозид конъюгата), GVAX® (вакцина рака предстательной железы), галофугинон, гистерелин, гидроксикарбамид, ибандроновая кислота, IGN-101, IL-13-PE38, IL-13-PE38QQR (цинтредекин безудотокс), IL-13-псевдомонадный экзотоксин, интерферон-α, интерферон-γ, JUNOVAN™ или MEPACT™ (мифамуртид), лонафарниб, 5,10-метилентетрагидрофолат, милтефозин (гексадецилфосфохолин), NEOVASTAT® (АЕ-941), NEUTREXIN® (триметрексат глюкуронат), NIPENT® (пентостатин), ONCONASE® (фермент рибонуклеаза), ONCOPHAGE® (лечение вакциной меланомы), ONCOVAX® (Вакцина IL-2), ORATHECIN™ (рубитекан), OSIDEM® (клеточное лекарственное средство на основе антител), OVAREX® Mab (мышиное моноклональное антитело), паклитаксел, PANDIMEX™ (агликон сапонины из женьшеня, включающие 20(S)протопанаксадиол (aPPD) и 20(S)протопанаксатриол (аРРТ)), панитумумаб, PANVAC®-VF (вакцина для исследования рака), пегаспаргаза, ПЭГ Интерферон A, феноксодиол, прокарбазин, ребимастат, REMOVAB® (катумаксомаб), REVLIMID® (леналидомид), RSR13 (эфапроксирал), SOMATULINE® LA (ланреотид), SORIATANE® (ацитретин), стауроспорин (Streptomyces staurospores), талабостат (PT100), TARGRETIN® (бексаротен), TAXOPREXIN® (DHA-паклитаксел), TELCYTA® (канфосфамид, TLK286), темилифен, TEMODAR® (темозоломид), тесмилифен, талидомид, THERATOPE® (STn-KLH), тимитак (2-амино-3,4-дигидро-6-метил-4-оксо-5-(4-пиридилтио)хиназолин дигидрохлорид), TNFERADE™ (аденовектор: носитель ДНК, содержащий ген фактора некроза опухоли-α), TRACLEER® или ZAVESCA® (босентан), третиноин (Ретин-А), тетрандрин, TRISENOX® (триоксид мышьяка), VIRULIZIN®, украин (производное алкалоидов растения чистотела большого), витаксин (анти-альфабета3 антитело), XCYTRIN® (мотексафин гадолиний), XINLAY™ (атрасентан), XYOTAX™ (паклитаксел полиглумекс), YONDELIS® (трабектедин), ZD-6126, ZINECARD® (дексразоксан), ZOMETA® (золендроновая кислота), зорубицин и т.п.
Средства комбинированной терапии могут вводиться в одновременном или последовательном режиме. При последовательном введении комбинация может вводиться в двух или более введений. Комбинированное введение включает совместное введение с использованием отдельных составов или единственного фармацевтического состава и последовательное введение в любом порядке, причем предпочтительно существует интервал времени, в течение которого оба (или все) активные средства одновременно проявляют свою биологическую активность.
Соответственно, в другом варианте осуществления, настоящее изобретение относится к композициям для лечения заболеваний у пациента, в ходе которого экспрессируется или суперэкспрессируется антиапоптотический белок Bcl-xL, причем указанные композиции включают эксципиент и терапевтически эффективное количество соединения Формулы I и терапевтически эффективное количество одного дополнительного терапевтического средства или более одного дополнительного терапевтического средства.
В другом варианте осуществления настоящее изобретение относится к способам лечения заболеваний у пациента, в ходе которого экспрессируется или суперэкспрессируется антиапоптотический белок Bcl-xL, причем указанные способы включают введение пациенту терапевтически эффективного количества соединения Формулы I и терапевтически эффективного количества одного дополнительного терапевтического средства или более одного дополнительного терапевтического средства.
В другом варианте осуществления настоящее изобретение относится к способам лечения мезотелиомы, рака мочевого пузыря, рака поджелудочной железы, рака кожи, рака головы или шеи, кожной или внутриглазной меланомы, рака яичника, рака молочной железы, рака матки, рака маточных труб, рака эндометрия, рака шейки матки, рака влагалища, рака вульвы, рака кости, рака яичника, рака шейки матки, рака толстой кишки, рака прямой кишки, рак анальной области, рака желудка, рака желудочно-кишечного тракта (желудка, колоректального, и двенадцатиперстной кишки), хронического лимфоцитарного лейкоза, рака желудка, рака тонкой кишки, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечника, саркомы мягких тканей, рака уретры, рака полового члена, рака яичек, гепатоцеллюлярного рака (печени и желчного протока), первичной или вторичной опухоли центральной нервной системы, первичной или вторичной опухоли головного мозга, болезни Ходжкина, хронического или острого лейкоза, хронического миелоидного лейкоза, лимфоцитарных лимфом, лимфобластного лейкоза, фолликулярной лимфомы, лимфоидных злокачественных новообразований Т-клеточного или В-клеточного происхождения, меланомы, множественной миеломы, рака полости рта, рака яичника, немелкоклеточного рака легких, рака предстательной железы, мелкоклеточного рака легких, рака почек и мочеточников, почечно-клеточного рака, рака почечной лоханки, опухолей центральной нервной системы, первичной лимфомы центральной нервной системы, неходжкинской лимфомы, опухолей оси позвоночника, глиомы ствола головного мозга, аденомы гипофиза, адренокортикального рака, рака желчного пузыря, рака селезенки, холангиокарциномы, фибросаркомы, нейробластомы, ретинобластомы или комбинации одного или более вышеуказанных видов рака у пациента, причем указанные способы включают введение пациенту терапевтически эффективных количеств соединения Формулы I или его фармацевтической композиции и одного или более одного из средств из числа этопозида винкристина СНОР, ритуксимаба, рапамицина, R-СНОР или бортезомиба.
Подходящие дозировки для любого из вышеуказанных совместно вводимых средств представляют собой дозировки, используемые в настоящее время, и могут быть снижены вследствие совместного действия (синергизм) недавно идентифицированного средства и других химиотерапевтических агентов или средств для лечения.
Комбинированная терапия может обеспечить "синергизм" и оказаться "синергической", то есть достигнутый эффект, когда активные ингредиенты используются вместе, больше чем сумма эффектов использования соединений по-отдельности. Синергический эффект может быть достигнут, когда активные ингредиенты: (1) совместно составлены и вводятся или доставляются одновременно в комбинированном составе лекарственной формы; (2) доставляются поочередно или параллельно как отдельные составы; или (3) в каком-то другом режиме. При доставке в рамках поочередной терапии синергический эффект может быть достигнут, когда соединения вводят или доставляют последовательно, например, различными инъекциями в отдельных шприцах, отдельных пилюлях или капсулах, или в отдельных инфузиях. В общем, в ходе поочередной терапии эффективную дозу каждого активного ингредиента вводят последовательно, то есть серийно, тогда как в комбинированной терапии эффективные дозы двух или более активных ингредиентов вводят вместе.
В еще одном варианте осуществления настоящее изобретение относится к способам лечения заболеваний или состояний, вызванных, усиленных или являющихся следствием избытком или нежелательной активации тромбоцитов у пациента, включающим введение пациенту соединения Формулы I или его фармацевтической композиции.
Заболевания, вызванные или усиленные избытком или нежелательной активацией тромбоцитов включают, но не ограничены ими, эссенциальную тромбоцитемию, истинную полицитемию, M7 острый миелогенный лейкоз, рестеноз, сердечно-сосудистое заболевание, послеоперационную антитромбоцитарную терапию, связанные с введением устройств тромбы и осложнения, связанные с ними и т.п. Об участии тромбоцитов в эссенциальной тромбоцитемии сообщалось в Seminars in Hematology (2005), 42(4), 230-238 and also in New Eng. J. Med., 2005, 353:1, 33-45. Об участии тромбоцитов в истинной полицитемии сообщалось в Seminars in Thrombosis and Hemostatis (2006), 32(3), 267-275. Об участии тромбоцитов в рестенозе сообщалось в Journal of Clinical Pathology (2006), 59(3), 232-239. Об участии тромбоцитов в сердечно-сосудистом заболевании сообщалось в International Journal of Clinical Practice (2003), 57(10), 898-905. Об участии тромбоцитов в послеоперационной антитромбоцитарной терапии сообщалось в Journal of Thrombosis Thrombolysis. Заболевания или состояния, развивающиеся в результате повышенных уровней тромбоцитов, кровотечение, тромбоз или другое тромбоэмболическое осложнение, инициацию или ухудшение других заболеваний или нарушений крови, таких как синдром "липких тромбоцитов".
В одном варианте осуществления настоящее изобретение относится к способам уменьшения числа тромбоцитов у пациента и лечения предтромбозных состояний и заболеваний, которые характеризуются избытком или нежелательной активацией тромбоцитов, путем введения пациенту соединения Формулы I.
В другом варианте осуществления настоящее изобретение относится к способам лечения эссенциальной тромбоцитемии у пациента, включающим введение пациенту соединения Формулы I. В некоторых аспектах этого варианта осуществления, в одном варианте осуществления настоящее изобретение относится к способу уменьшения числа тромбоцитов у пациента и лечения эссенциальной тромбоцитемии.
В другом варианте осуществления настоящее изобретение относится к способам лечения истинной полицитемии у пациента, включающим введение пациенту соединения Формулы I, которое ингибирует активность члена семейства белков Bcl-xL. В некоторых аспектах этого варианта осуществления, в одном варианте осуществления настоящее изобретение относится к способам уменьшения числа тромбоцитов у пациента и лечения истинной полицитемии.
В некоторых аспектах соединение или фармацевтическая композиция, используемые в каждом из вышеописанных способов по изобретению, представляют собой соединение, выбранное из Таблицы 1, или фармацевтическую композицию, включающую соединение, выбранное из Таблицы 1.
В некоторых аспектах настоящая заявка относится к применению соединения, как описано здесь (например, соединения Формулы I), или его стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли, пролекарства, для лечения заболевания или состояния. Примеры заболеваний или состояний включают, например, любое из заболеваний или состояний, вызванных, усиленных или являющихся следствием избытка или нежелательной активации тромбоцитов, обсужденных выше, любой вид рака, обсужденный выше, или любое из аутоиммунных заболеваний, обсужденных выше. В некоторых вариантах применения, соединение (например, соединение Формулы I) или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемая соль, пролекарство, используются индивидуально. В другом варианте осуществления соединение (например, соединение Формулы I) или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемая соль, пролекарство, используются как часть комбинированной терапии, как обсуждено выше.
Следующие примеры приведены просто для иллюстрации изобретения. Представленные примеры не должны быть рассмотрены, как в какой бы то ни было степени ограничивающие объем изобретения.
V. ПРИМЕРЫ
В Примерах, описанных ниже, если не указано иное, все температуры приведены в градусах Цельсия. Реактивы были приобретены у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, TCI или Maybridge, и использовались без дальнейшей очистки, если не указано иное. Реакции, описанные ниже, осуществляли обычно под давлением выше положительного давления азота или аргона или в трубке для сушки (если не указано иное) в безводных растворителях, и реакционные колбы были обычно оснащены резиновыми перегородками для введения субстратов и реактивов через шприц. Стеклянную посуду высушивали в сушильном шкафу и/или нагреванием. Хроматографию на колонках проводили на системе Biotage (Изготовитель: Dyax Corporation), включающей колонку с силикагелем, или на картридже с силикагелем SEP PAK® (Waters) или на системе для хроматографии ISCO (Изготовитель: Teledyne ISCO), включающей колонку с силикагелем, или вручную с использованием стеклянной колонки, в целом согласно методикам, описанным W.C. Still (см. Still, W. C; Kahn, M.; Mitra, A. J Org. Chem. 1978, 43(14), 2923-2925). Спектры 1Н ЯМР регистрировали на Varian или подобном приборе, функционирующем при 300 или 400 МГц. Спектры 1Н ЯМР получали в растворах дейтеризованного CDCl3, d6-ДМСО, CH3OD или d6-ацетона (выражены в ppm (ч./млн)). Когда указаны мультиплетные пики, используются следующие сокращения: с (синглет), д (дублет), т (триплет), м (мультиплет), ушир. (уширенный), дд (дублет дублетов), дт (дублет триплетов). Константы связывания, когда они приведены, выражены в Герц (Гц). Если возможно, формирование продукта в реакционных смесях может быть проверено LC/MS, осуществляемой либо на Agilent 1200 серии LC, присоединенной к четырехполюсному масс-спектрометру 6140, с использованием колонки Supelco Ascentis Express C18 с линейным градиентом 5-95% ацетонитрил/вода (с 0,1% трифторуксусной кислоты в каждой мобильной фазе) в течение 1,4 минуты и с поддержанием при 95% в течение 0,3 минуты, либо на PE Sciex API 150 EX с использованием колонки Phenomenex DNYC monolithic C18 с линейным градиентом 5%-95% ацетонитрил/вода (с 0,1% трифторуксусной кислоты в каждой мобильной фазе) в течение 5 минут и с поддержанием при 95% в течение 1 минуты, или на подобной системе.
Все сокращения, используемые для описанных реактивов, условий реакции или используемому оборудованию, находятся в соответствии с определениями, сформулированными в "List of standard abbreviations and acronyms", ежегодно публикуемом в Journal of Organic Chemistry (журнал Американского Химического Общества). Следующие сокращения, используемые здесь, имеют следующие значения: rt = температура окружающей среды, DMAP = 2,6-диметиламинопиридин; DCM = дихлорметан, THF = тетрагидрофуран, TEA = триэтиламин, EtOAc = этилацетат, TFA = трифторуксусная кислота, LC/MS = жидкостная хроматография/масс-спектрометрия, APCI = химическая ионизация при атмосферном давлении, DCI = десорбционная химическая ионизация, MS = масс-спектрометрия, MeOH = метанол, DMA = N,N-диметилацетамид, EtOH = этанол, Hex = гексан(ы), ESI = ионизация с электрораспылением, PMB = параметоксибензил, РЕ = петролейный эфир, DIAD = диизопропил азодикарбоксилат, DMF = N,N-диметилформамид, DCE = дихлорэтан и Et3N = триэтиламин.
Пример 1
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновой кислоты (1):

Стадия 1: Получение 3-(4-бромфенил)пропионовой кислоты (1A)

2,2-диметил-[1,3]диоксан-4,6-дион (120 г, 0,84 моль) и 4-бром-бензальдегид (153,4 г, 0,84 моль) растворяли в 300 мл ТЕА и уксусной кислоты. Реакционную смесь нагревали с обратным холодильником в течение ночи. В этот момент реакционную смесь разбавляли водой (4 л) и подкисляли 6M HCl (1 л) до рН=2. Осажденное твердое вещество отфильтровывали и промывали разбавленной HCl, затем растворяли в 5%-ном растворе NaHCO3. Водный солевой раствор промывали простым эфиром (4x500 мл), фильтровали и подкисляли разбавленной HCl. Затем фильтровали и высушивали над вакуумом, получая 100 г (67%) желаемого продукта 3-(4-бромфенил)пропионовой кислоты (1A): 1Н ЯМР (DMSO-d6, 400 МГц), δ 2,5 (м, 2H), 2,8 (м, 2H), 7,2 (м, 2H), 7,5 (м, 2H), 12,2 (с, 1H).
Стадия 2: Получение 6-бром-2,3-дигидро-1Н-инден-1-она (1B)

3-(4-бромфенил)пропионовую кислоту (1A) (34,5 г, 0,15 ммоль) растворяли в 40 мл SOCl2, и смесь нагревали с обратным холодильником в течение 90 минут. В этот момент растворитель удаляли при пониженном давлении, чтобы количественно получить 3-(4-бромфенил)пропионил хлорид, который использовали немедленно на следующей стадии.
3-(4-бромфенил)пропионил хлорид (36,9 г, 0,15 моль) растворяли в 300 мл DCM, и немедленно добавляли AlCl3 (23,76 г, 0,18 моль). При завершении реакции смесь нагревали при 40°C и нагревали с обратным холодильником в течение 90 минут и затем выливали на лед. Добавляли разбавленный водный раствор HCl, и смесь экстрагировали простым диэтиловым эфиром. Органический слой промывали 2M водным раствором HCl, водой и солевым раствором. Продукт очищали хроматографией на колонках на силикагеле с элюированием Смесью РЕ:ЕА (20:1), получая твердое вещество светло-коричневого цвета. После удаления растворителя под вакуумом получали 23 г (75%) желаемого продукта 6-бром-2,3-дигидро-1Н-инден-1-она (1B): 1Н ЯМР (DMSO-d6, 400 МГц) δ 2,6 (м, 2H), 3,1 (м, 2H), 7,55 (м, 1H), 7,1 (м, lH), 7,85 (м, 1H).
Стадия 3:
Получение (Z)-6-бром-2,3-дигидро-1Н-инден-1-он оксима (1C)
К суспензии гидрохлорида гидроксиламина (12,15 г, 142,95 ммоль, 1,5 экв.) в 200 мл абсолютного этанола добавляли ацетат калия (14 г, 142,95 ммоль, 1,5 экв.), затем 6-бром-2,3-дигидро-1Н-инден-1-он (1B) (20 г, 95,3 ммоль). Реакционную смесь нагревали с обратным холодильником в течение часа (TLC 6:1 гексаны/EtOAc показала, что желаемое соединение (1C) соответствует исходному кетону TLC). Реакционную смесь затем концентрировали. К остатку добавляли воду, и осадок собирали фильтрацией. Твердые частицы промывали водой и высушивали, получая 11,5 г (89%) желаемого продукта (Z)-6-бром-2,3-дигидро-1Н-инден-1-он оксима (1C): 1Н ЯМР (DMSO-d6, 400 МГц): δ 2,7 (м, 2H), 2,9 (м, 2H), 7,25 (м, 1H), 7,45 (м, 1H), 7,6 (с, 1H), 11,05 (с, 1Н).
Стадия 4: Получение (Z)-6-бром-2,3-дигидро-1Н-инден-1-он O-триизопропилсилил оксима (1D)
Раствор имидазола (11 г, 162 ммоль) в 100 мл сухого DCM добавляли по каплям к раствору (Z)-6-бром-2,3-дигидро-1Н-инден-1-он оксима (1C) (18,2 г, 81 ммоль) и триизопропилсилил хлорида (23,3 г, 121,5 ммоль) в растворе в 40 мл сухого DCM. Реакционную смесь перемешивали в течение 16 часов при температуре окружающей среды. Затем ее фильтровали, чтобы удалить осадок, и фильтрат концентрировали под вакуумом. Сырой материал очищали хроматографией на колонках на силикагеле, получая (Z)-6-бром-2,3-дигидро-1Н-инден-1-он оксим O-триизопропилсилил (1D). Сырой продукт немедленно использовали на следующей стадии.
Стадия 5: Получение 7-бром-1,2,3,4-тетрагидрохинолина (1Е)
BF3.Et2O (49,2 г, 56,7 мл, 346,2 ммоль) добавляли к раствору (Z)-6-бром-2,3-дигидро-1Н-инден-1-он оксим O-триизопропилсилила (1D) (66 г, 173,1 ммоль) в 400 мл сухого Et2O. Затем добавляли по каплям Me2S-BH3 (31,5 г, 39,48 мл 415,5 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 48 часов (с последующей TLC 100%-ым петролейным эфиром до полного исчезновения исходного материала). Смесь охлаждали до 0°C и осторожно по каплям к смеси добавляли лед/вода, затем добавляли 60 мл раствора 1:1 вода/концентрированная HCl. Эту реакционную смесь нагревали до 70°C в течение 80 минут и затем давали охладиться до температуры окружающей среды и перемешивали в течение 3,5 часов, промывали Et2O. Оставшуюся водную фазу подщелачивали до >10 с 5M NaOH и экстрагировали три раза Et2O. Объединенные органические фазы промывали солевым раствором и высушивали над K2CO3. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом РЕ:EtOAc (100:1), получая 11,2 г (31%) желаемого продукта 7-бром-1,2,3,4-тетрагидрохинолина (1Е): 1Н ЯМР (DMSO-d6, 400 МГц): δ 1,7 (м, 2H), 2,6 (м, 2H), 3,15 (м, 2H), 5,9 (с, 1H), 6,45 (м, 1H), 6,55 (м, 1H), 6,75 (м, 1H). LC/MS (APCI): м/z 211,7 (M+H).
Стадия 6: Получение 1-(7-бром-3,4-дигидрохинолин-1(2H)-ил)этанона (1F)
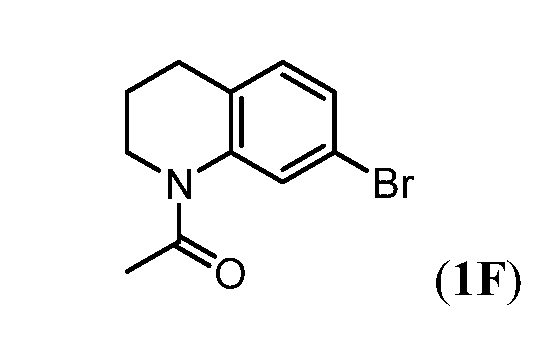
К 7-бром-1,2,3,4-тетрагидрохинолину (1Е) (1,0 г, 4,7 ммоль) в DCM (30 мл) добавляли ТЕА (1,6 мл, 12 ммоль), уксусный ангидрид (1,1 мл, 12 ммоль) и пиридин (1,0 мл, 10,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 24 часов, контролируя LCMS. К реакционной смеси добавляли ТЕА (1,6 мл, 12 ммоль), уксусный ангидрид (1,1 мл, 12,0 ммоль) и пиридин (1,0 мл, 10 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 24 часов. Реакционную смесь концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 100% EtOAc в гексанах, получая 1,09 г (91%) желаемого продукта 1-(7-бром-3,4-дигидрохинолин-1(2H)-ил)этанона (1F): LC/MS (APCI): m/z 255,7 (M+H).
Стадия 7:
Получение 1-(7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолин-1(2Н)-ил)этанона (1G)

К 1-(7-бром-3,4-дигидрохинолин-1(2H)-ил)этанону (1F) (6,0 г, 24 ммоль), ацетату калия (4,63 г, 47 ммоль), бис(трифенилфосфин)палладий (II) хлориду (1,66 г, 4,7 ммоль) и биспинаколборонату (7,79 г, 31 ммоль) добавляли толуол (100 мл). Азот барботировали через реакционную смесь в течение 5 минут. Реакционную смесь нагревали до 100°C и перемешивали в течение ночи. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли EtOAc, фильтровали через Целит и концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 80% EtOAc в гексанах, получая 7,1 г (100%) желаемого продукта 1-(7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолин-1(2H)-ил)этанона (1G): LC/MS (APCI): m/z 301,0 (M+H).
Стадия 8:
Получение 2-амино-5-хлортиазол-4-этилкарбоксилата (1H)
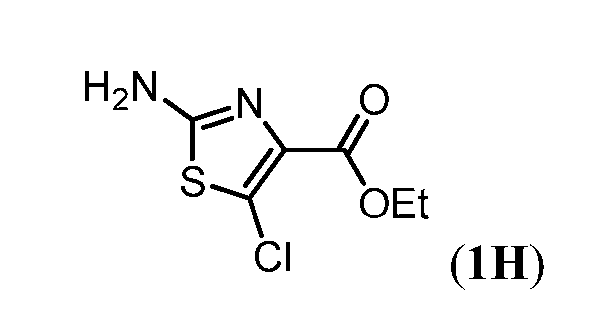
2-аминотиазол-4-этилкарбоксилат (4,72 г, 27,4 ммоль) объединяли с N-хлорсукцинимидом (4,0 г, 30 ммоль) и растворяли в ацетонитриле (50 мл). Смесь нагревали с обратным холодильником при умеренной температуре (>81,5°C) в течение 5 часов. Затем колбу охлаждали до температуры окружающей среды, и к смеси добавляли обесцвечивающий углерод, затем смесь разбавляли EtOAc. Полученную суспензию фильтровали через слой кремнезема и концентрировали при пониженном давлении, получая 5,57 г (98%) желаемого 2-амино-5-хлортиазол-4-этилкарбоксилата (1H). Аналитические данные относительно этого соединения соответствовали ранее описанным (см., South, M.S.J. Heterocyclic Chem. 1991, 28, 1003).
Стадия 9:
Получение 2-бром-5-хлортиазол-4-этилкарбоксилата (1I)
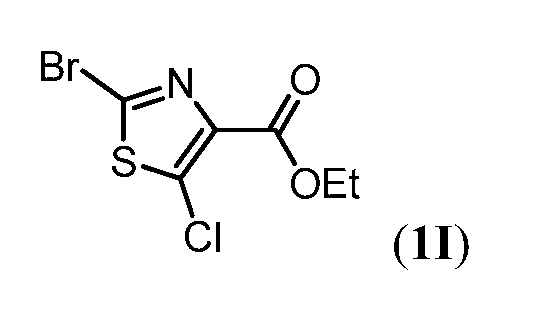
2-амино-5-хлортиазол-4-этилкарбоксилат (1H) (14,08 г, 68,1 ммоль) добавляли в частях к раствору нитрита трет-бутила (12,0 мл, 102 ммоль) и бромида меди (II) (22,8 г, 102 ммоль) в ацетонитриле (351 мл) при температуре окружающей среды. Полученную смесь нагревали при 80°C в течение 2 часов. Затем смесь охлаждали до температуры окружающей среды и разделяли между DCM (120 мл), водой (75 мл) и концентрированной HCl (6 мл). Водный слой экстрагировали DCM (2x150 мл), и объединенные органические экстракты промывали последовательно водой и солевым раствором и высушивали над твердым MgSO4. Твердые частицы фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 30% EtOAc в гексанах, получая 8,83 г (47,9%) желаемого 2-бром-5-хлортиазол-4-этилкарбоксилата (II). Аналитический отчет для этого соединения соответствовал описанному ранее (см., Hodgetts, K. J.; Kershaw, M. T. Org. Lett. 2002, 4, 1363).
Стадия 10: Получение 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-хлортиазол-4-этилкарбоксилата (1J)
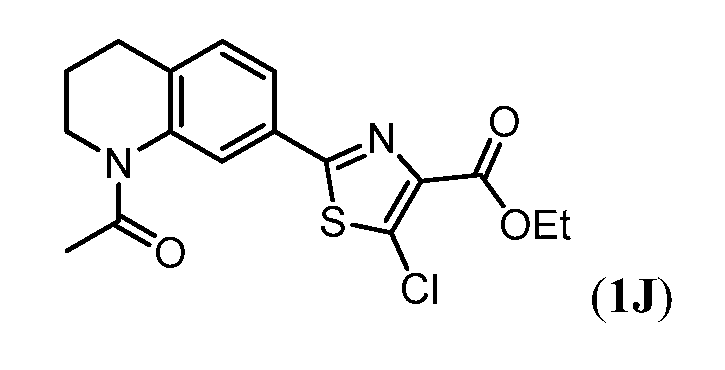
К 1-(7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолин-1(2H)-ил)этанону (IG) (1,24 г, 4,12 ммоль), 2-бром-5-хлортиазол-4-этилкарбоксилату (II) (1,12 г, 4,12 ммоль), хлориду лития (0,52 г, 12,4 ммоль), тетракис(трифенилфосфин)палладию (0,476 г, 0,412 ммоль) и карбонату цезия (4,03 г, 12,4 ммоль) добавляли 1,4-диоксан (30 мл) и воду (10 мл). Азот барботировали через реакционную смесь в течение 5 минут. Реакционную смесь нагревали до 100°C и перемешивали в течение ночи. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли EtOAc, фильтровали через Целит и концентрировали при пониженном давлении. К сырому остатку добавляли EtOAc и воду. Слои разделяли, и водный слой экстрагировали EtOAc, и объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 80% EtOAc в гексанах, получая 0,505 г (34%) желаемого 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-хлортиазол-4-этилкарбоксилата (1J): LC/MS (APCI): m/z 365,3 (M+H).
Стадия 11: Получение 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-этилкарбоксилата (1К)
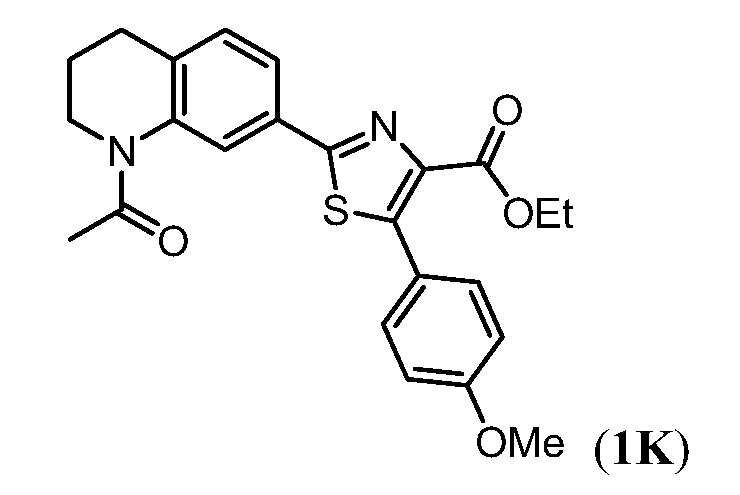
К 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-хлортиазол-4-этилкарбоксилату (1J) (0,450 г, 1,23 ммоль), 4-метоксифенилбороновой кислоте (0,281 г, 1,85 ммоль), хлориду лития (0,157 г, 3,70 ммоль), тетракис(трифенилфосфин) палладию (0) (0,142 г, 0,12 ммоль) и карбонату цезия (1,2 г, 3,70 ммоль) добавляли 1,4-диоксан (20 мл) и воду (10 мл). Азот барботировали через реакционную смесь в течение 5 минут. Реакционную смесь нагревали до 100°C и перемешивали в течение ночи. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли EtOAc, фильтровали через Целит и концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 80% EtOAc в гексанах, получая 0,483 г (90%) желаемого 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-этилкарбоксилата (1К): LC/MS (APCI): m/z 436,8 (M+H).
Стадия 12:
Получение 5-(4-метоксифенил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (1L)

К 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-этилкарбоксилату (1К) (0,483 г, 1,11 ммоль) и KOH (1,24 г, 22,1 ммоль) добавляли MeOH (10 мл) и воду (45 мл). Реакционной смеси давали нагреться до 55°C и перемешивали 48 часов. Реакцию проверяли LCMS. К реакционной смеси добавляли (0,5 г, 8,9 ммоль). Реакционной смеси давали нагреться до 55°C и перемешивали в течение 24 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. К сырому остатку добавляли EtOAc и воду. Слои разделяли, и водный экстракт подкисляли до ~pH6 с 1 н. HCl и экстрагировали EtOAc. Объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 0,350 г (86%) желаемого продукта 5-(4-метоксифенил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (1L): LC/MS (APCI): m/z 367,0 (M+H).
Стадия 13: Получение N-(бензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамида (1M)

К CDI (1,6 г, 10 ммоль) в ацетонитриле (30 мл) медленно добавляли 2-аминобензотиазол (1,0 г, 6,6 ммоль) и перемешивали при температуре окружающей среды в течение 48 часов. Реакционную смесь фильтровали, промывали ацетонитрилом и высушивали, получая 1,44 г (88%) желаемого продукта N-(бензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамида (1M): LC/MS (APCI): m/z 245,1 (M+H).
Стадия 14:
Получение целевого соединения 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновой кислоты (1)

Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновую кислоту (1) получали в соответствии со следующей процедурой: К 5-(4-метоксифенил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоте (1L) (0,070 г, 0,19 ммоль) и N-(бензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамиду (1M) (0,051 г, 0,21 ммоль) добавляли DMF (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. К реакционной смеси добавляли EtOAc и воду. Слои разделяли, и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой остаток очищали ВЭЖХ с обратной фазой, получая 0,028 г (27%) желаемого продукта 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновой кислоты (1): LC/MS (APCI): m/z 543,1 (M+H).
Пример 2
Синтез N-(тиазол-2-ил)-1Н-имидазол-1-карбоксамида (2)
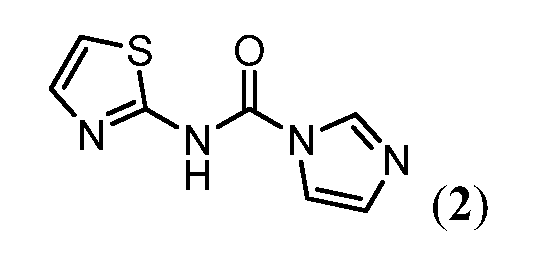
Целевое соединение N-(тиазол-2-ил)-1Н-имидазол-1-карбоксамид (2) получали в соответствии со следующей процедурой: N-(тиазол-2-ил)-1Н-имидазол-1-карбоксамид (2) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-аминотиазол использовался вместо 2-аминобензотиазола на стадии 13 примера 1. LC/MS (APCI): m/z 195,0 (M+H).
Пример 3
Синтез N-(5-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамида (3)

Целевое соединение N-(5-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамид (3) получали в соответствии со следующей процедурой: N-(5-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамид (3) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-амино-5-метилтиазол использовался вместо 2-аминобензотиазола на стадии 13 примера 1. LC/MS (APCI): m/z 209,2 (M+H).
Пример 4
Синтез N-(4-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамида (4)
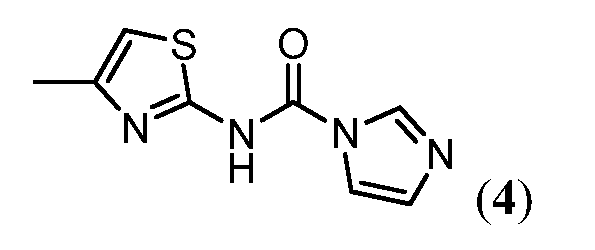
Целевое соединение N-(4-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамид (4) получали в соответствии со следующей процедурой: N-(4-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамид (4) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-амино-4-метилтиазол использовался вместо 2-аминобензотиазола на стадии 13 примера 1. LC/MS (APCI): m/z 208,9 (М+Η).
Пример 5
Синтез N-(6-фторбензо[d]тиазол-2-ил)-1H-имидазол-1-карбоксамида (5)

Целевое соединение N-(6-фторбензо[d]тиазол-2-ил)-1H-имидазол-1-карбоксамид (5) получали в соответствии со следующей процедурой: N-(6-фторбензо[d]тиазол-2-ил)-1H-имидазол-1-карбоксамид (5) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-амино-6-фторбензотиазол использовался вместо 2-аминобензотиазола на стадии 13 примера 1.
Пример 6
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (6)
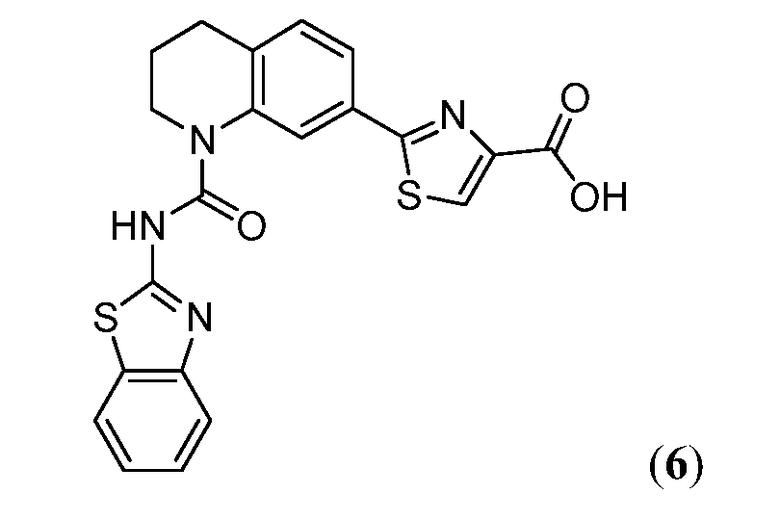
Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (6) получали в соответствии со следующей процедурой: 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (6) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-бромтиазол-4-метилкарбоксилат использовался вместо 2-бром-5-хлортиазол-4-этилкарбоксилата (1I) на стадии 10 примера 1. LC/MS (APCI): m/z 437,51 (M+H).
Пример 7
Синтез 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновой кислоты (7):
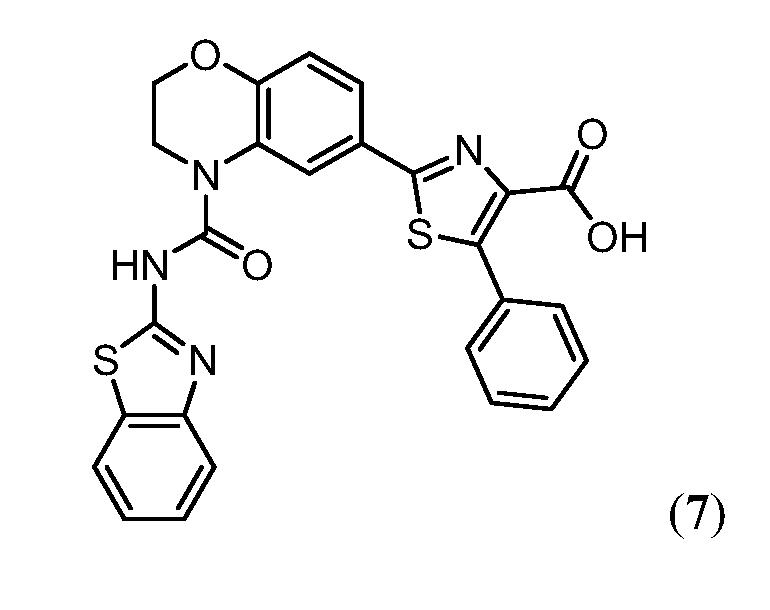
Стадия 1:
Получение 6-бром-3,4-дигидро-2Н-бензо[b][1,4]оксазина (7A)

2H-бензо[b][1,4]оксазин-3(4H)-он (10,0 г, 43,8 ммоль) растворяли в тетрагидрофуране (180 мл). Литий тетрагидроалюминат (2,5 г, 66 ммоль) добавляли к раствору при температуре окружающей среды. Смесь затем перемешивали под атмосферой азота, пока TLC не показала завершение реакции (приблизительно 12 ч). Реакционную смесь лили в насыщенную смесь соли Рочелла (Калий натрий тартрат) и простого диэтилового эфира и энергично перемешивали до разделения фаз. Экстрагировали EtOAc (70 мл) и концентрировали при пониженном давлении, получая 8,3 г (88%) желаемого продукта 6-бром-3,4-дигидро-2Н-бензо[b][1,4]оксазина (7A), который использовали без дальнейшей очистки.
Стадия 2: Получение 1-(6-бром-2H-бензо[b][1,4]оксазин-4(3H)-ил)этанона (7B)
6-бром-2H-бензо[b][1,4]оксазин-3(4H)-он (7A) (2,5 г, 11,7 ммоль) забирали DCM (100 мл) и обрабатывали ТЕА (4,07 мл, 29,2 ммоль), затем Ac2O (2,75 мл, 29,2 ммоль). Добавляли 4-DMAP (0,285 г, 2,34 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 50% EtOAc в гексанах, получая 2,62 г (87%) желаемого соединения 1-(6-бром-2Н-бензо[b][1,4]оксазин-4(3H)-ил)этанона (7B).
Стадия 3:
Получение 1-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-бензо[b][1,4]оксазин-4(3H)-ил)этанона (7C)

1-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-бензо[6][1,4]оксазин-4(3H)-ил)этанон (7C) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 1-(6-бром-2H-бензо[6][1,4]оксазин-4(3H)-ил)этанон (7B) использовался вместо 1-(7-бром-3,4-дигидрохинолин-1(2H)-ил)этанона (1F) на стадии 7 примера 1. LC/MS (APCI): m/z 303,1 (M+H).
Стадия 4: Получение 2-(4-ацетил-3,4-дигидро-2H-бензо[b][1,4] оксазин-6-ил)-5-хлортиазол-4-этилкарбоксилата (7D)
2-(4-ацетил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-хлортиазол-4-этилкарбоксилат (7D) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 1-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-бензо[b][1,4]оксазин-4(3H)-ил)этанон (7C) использовался вместо 1-(7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолин-1(2Н)-ил)этанона (1G) на стадии 10 примера 1. LC/MS (APCI): m/e 366,1 (M+H).
Стадия 5: Получение 2-(4-ацетил-3,4-дигидро-2H-бензо[b][1,4] оксазин-6-ил)-5-фенилтиазол-4-этилкарбоксилата (7E)

2-(4-ацетил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-этилкарбоксилат (7E) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-(4-ацетил-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-хлортиазол-4-этилкарбоксилат (7D) использовался вместо 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-хлортиазол-4-этилкарбоксилата (1J) и фенилбороновая кислота использовалась вместо 4-метоксифенилбороновой кислоты на стадии 11 примера 1. LC/MS (APCI): m/z 409,1 (M+H).
Стадия 6: Получение 2-(3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновой кислоты (7F)

2-(3,4-Дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновую кислоту (7F) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-(4-ацетил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-этилкарбоксилат (7E) использовали вместо 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-этилкарбоксилата (1К) на стадии 12 примера 1. LC/MS (APCI): m/z 339,0 (M+H).
Стадия 7:
Получение целевого соединения 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновой кислоты (7)
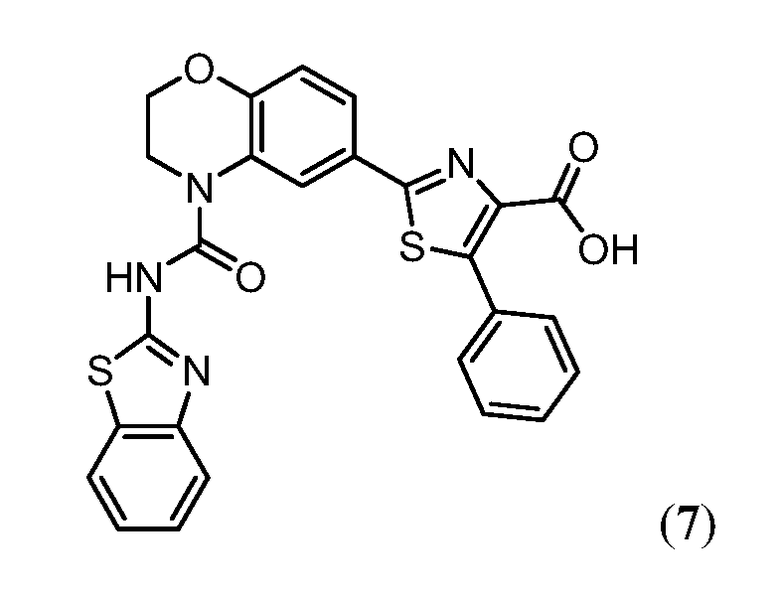
Целевое соединение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновую кислоту (7) получали в соответствии со следующей процедурой: 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4] оксазин-6-ил)-5-фенилтиазол-4-карбоновую кислоту (7) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 2-(3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновая кислота (7F) использовался вместо 5-(4-метоксифенил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (1L) на стадии 14 примера 1. LC/MS (APCI): m/z 515,1 (M+H).
Пример 8
Синтез 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(4-метоксифенил)тиазол-4-карбоновой кислоты (8):
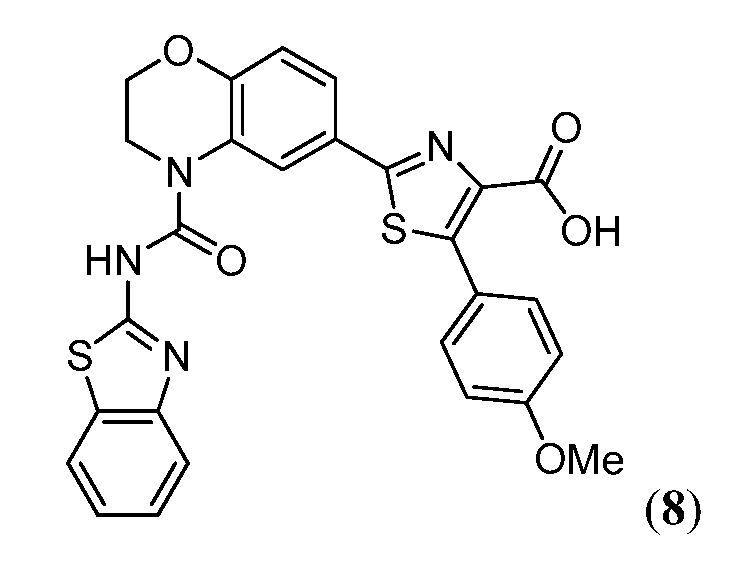
Целевое соединение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-метоксифенил)тиазол-4-карбоновую кислоту (8) получали в соответствии со следующей процедурой: 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-метоксифенил)тиазол-4-карбоновую кислоту (8) получали согласно процедуре, подобной описанной в примере 7, кроме того, что 4-метоксифенилбороновая кислота использовался вместо фенилбороновой кислоты на стадии 5 примера 7. LC/MS (APCI): m/z 545,1 (M+H).
Пример 9
Синтез 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(3-метоксифенил)тиазол-4-карбоновой кислоты (9):

Целевое соединение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(3-метоксифенил)тиазол-4-карбоновую кислоту (9) получали в соответствии со следующей процедурой: 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(3-метоксифенил)тиазол-4-карбоновую кислоту (9) получали согласно процедуре, подобной описанной в примере 7, кроме того, что 3-метоксифенилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 5 примера 7. LC/MS (APCI): m/z 545,1 (M+H).
Пример 10
Синтез 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(4-фторфенил)тиазол-4-карбоновой кислоты (10):

Целевое соединение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-фторфенил)тиазол-4-карбоновую кислоту (10) получали в соответствии со следующей процедурой: 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-фторфенил)тиазол-4-карбоновую кислоту (10) получали согласно процедуре, подобной описанной в примере 7, кроме того, что 4-фторфенилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 5 примера 7. LC/MS (APCI): m/z 533,1 (M+H).
Пример 11
Синтез 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(3-фторфенил)тиазол-4-карбоновой кислоты (11):

Целевое соединение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(3-фторфенил)тиазол-4-карбоновую кислоту (11) получали в соответствии со следующей процедурой: 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(3-фторфенил)тиазол-4-карбоновую кислоту (11) получали согласно процедуре, подобной описанной в примере 7, кроме того, что 3-фторфенилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 5 примера 7. LC/MS (APCI): m/z 533,1 (M+H).
Пример 12
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-изопропоксифенил)тиазол-4-карбоновой кислоты (12):
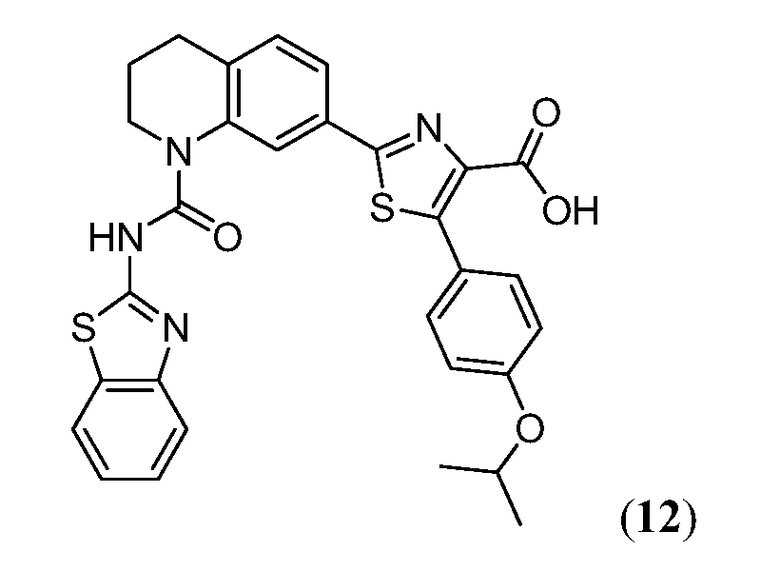
Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-изопропоксифенил)тиазол-4-карбоновую кислоту (12) получали в соответствии со следующей процедурой: 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-изопропоксифенил)тиазол-4-карбоновую кислоту (12) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 4-изопропоксифенилбороновая кислота использовалась вместо 4-метоксифенилбороновой кислоты на стадии 11 примера 1. LC/MS (APCI): m/z 571,2 (M+H).
Пример 13
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(бифенил-4-ил)тиазол-4-карбоновой кислоты (13):

Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(бифенил-4-ил)тиазол-4-карбоновую кислоту (13) получали в соответствии со следующей процедурой: 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(бифенил-4-ил)тиазол-4-карбоновую кислоту (13) получали согласно процедуре, подобной описанной в примере 1, кроме того, что 4-бифенилбороновая кислота использовалась вместо 4-метоксифенилбороновой кислоты на стадии 11 примера 1. LC/MS (APCI): m/z 589,1 (M+H).
Пример 14
Синтез (E)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-стирилтиазол-4-карбоновой кислоты (14):
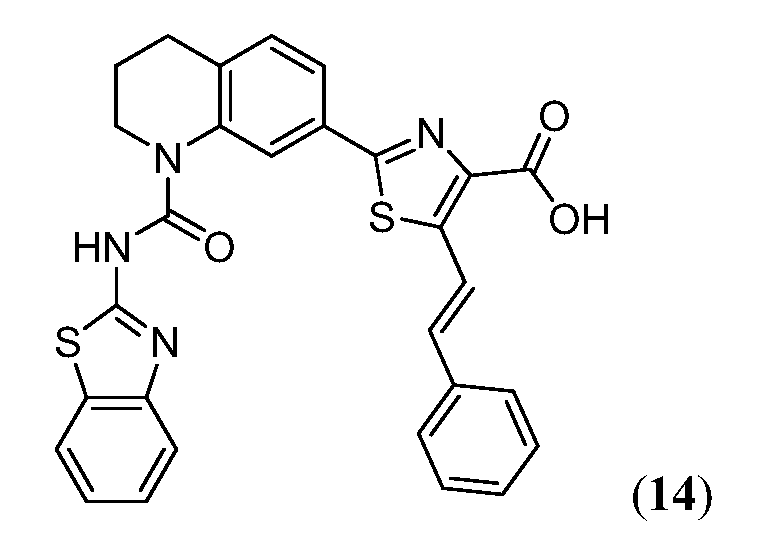
Целевое соединение (E)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-стирилтиазол-4-карбоновую кислоту (14) получали в соответствии со следующей процедурой: (E)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-стирилтиазол-4-карбоновую кислоту (14) получали согласно процедуре, подобной описанной в примере 1, кроме того, что E-фенетилбороновая кислота использовалась вместо 4-метоксифенилбороновой кислоты на стадии 11 примера 1. LC/MS (APCI): m/z 539,1 (M+H).
Пример 15
Синтез 5-(4-метоксифенил)-2-(1-(тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (15):

Целевое соединение 5-(4-метоксифенил)-2-(1-(тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (15) получали в соответствии со следующей процедурой: 5-(4-метоксифенил)-2-(1-(тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (15) получали согласно процедуре, подобной описанной в примере 1, кроме того, что N-(тиазол-2-ил)-1Н-имидазол-1-карбоксамид (2) использовался вместо N-(бензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамида (1M) на стадии 14 примера 1. LC/MS (APCI): m/z 493,1 (M+H).
Пример 16
Синтез 5-(4-метоксифенил)-2-(1-(5-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (16):
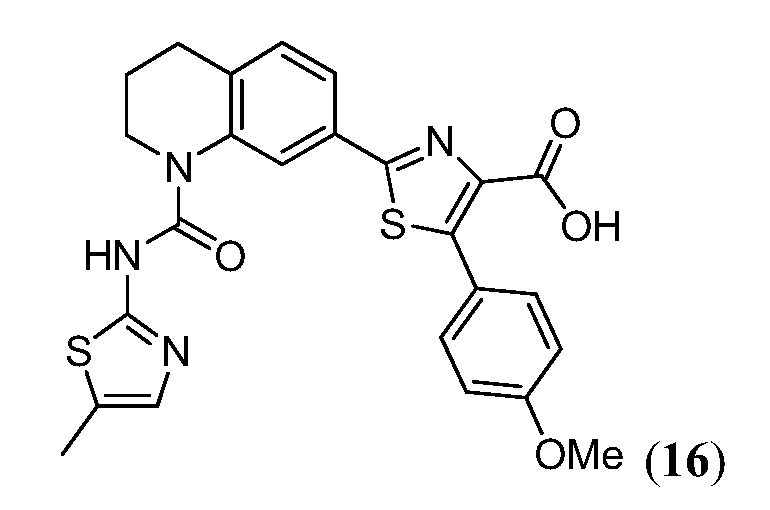
Целевое соединение 5-(4-метоксифенил)-2-(1-(5-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (16) получали в соответствии со следующей процедурой: 5-(4-метоксифенил)-2-(1-(5-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (16) получали согласно процедуре, подобной описанной в примере 1, кроме того, что N-(5-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамид (3) использовался вместо N-(бензо[d]тиазол-2-ил)-1H-имидазол-1-карбоксамида (1M) на стадии 14 примера 1. LC/MS (APCI): m/z 507,1 (M+H).
Пример 17
Синтез 5-(4-метоксифенил)-2-(1-(4-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (17):

Целевое соединение 5-(4-метоксифенил)-2-(1-(4-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (17) получали в соответствии со следующей процедурой: 5-(4-метоксифенил)-2-(1-(4-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (17) получали согласно процедуре, подобной описанной в примере 1, кроме того, что N-(4-метилтиазол-2-ил)-1Н-имидазол-1-карбоксамид (4) использовался вместо N-(бензо[d]тиазол-2-ил)-1H-имидазол-1-карбоксамида (1M) на стадии 14 примера 1. LC/MS (APCI): m/z 507,1 (M+H).
Пример 18
Синтез 2-(1-(6-фторбензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновой кислоты (18):
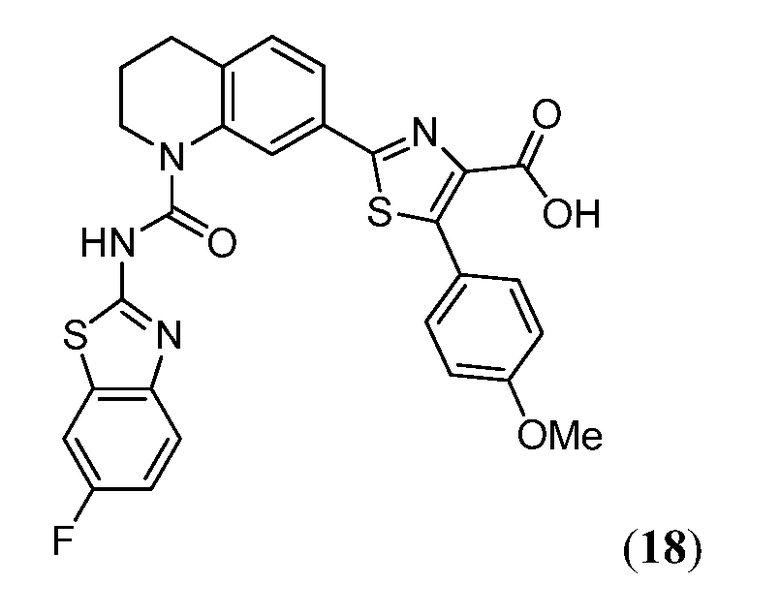
Целевое соединение 2-(1-(6-фторбензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновую кислоту (18) получали в соответствии со следующей процедурой: 2-(1-(6-Фторбензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновую кислоту (18) получали согласно процедуре, подобной описанной в примере 1, кроме того, что N-(6-фторбензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамид (5) использовался вместо N-(бензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамида (1M) на стадии 14 примера 1. LC/MS (APCI): m/z 561,1 (M+H).
Пример 19
Синтез 4-нитрофенил бензо[d]тиазол-2-илкарбамата (19):

Целевое соединение 4-нитрофенил бензо[d]тиазол-2-илкарбамат (19) получали в соответствии со следующей процедурой: К 2-аминобензотиазолу (5,0 г, 30 ммоль) добавляли DCM (36 мл) и реакционную смесь перемешивали при 0°C в течение 15 минут. Затем добавляли п-нитрофенил хлорформиат (6,64 г, 33 ммоль) в DCM (60 мл), затем пиридин (2,66 мл, 33 ммоль) в DCM (36 мл). Реакционную смесь перемешивали и давали нагреться до температуры окружающей среды в течение ночи. Реакционную смесь фильтровали, промывали DCM и высушивали, получая 9,22 г (90%) желаемого продукта 4-нитрофенил бензо[d]тиазол-2-илкарбамата (19): LC/MS (APCI): m/z 316,0 (M+H).
Пример 20
Синтез 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-карбоновой кислоты (20):
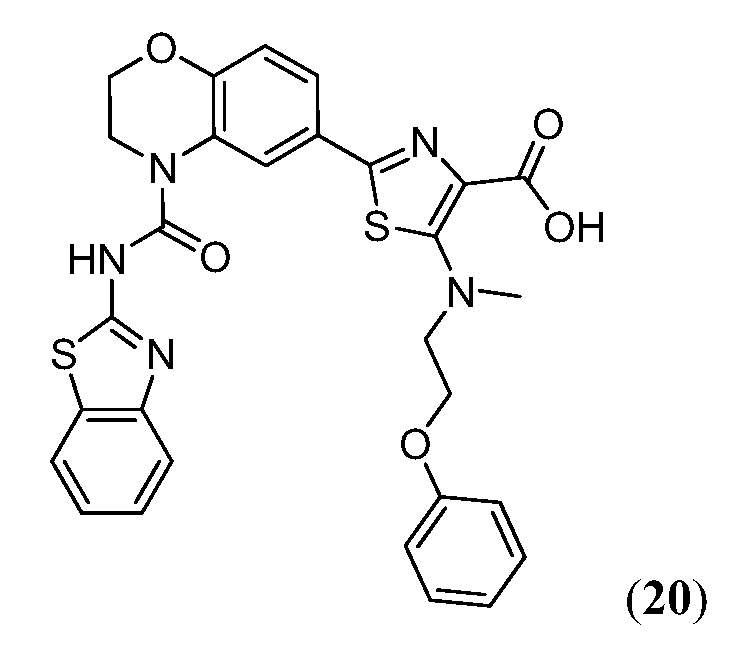
Стадия 1: Получение 2-(4-ацетил-3,4-дигидро-2H-бензо[b][1,4] оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилата (20A)
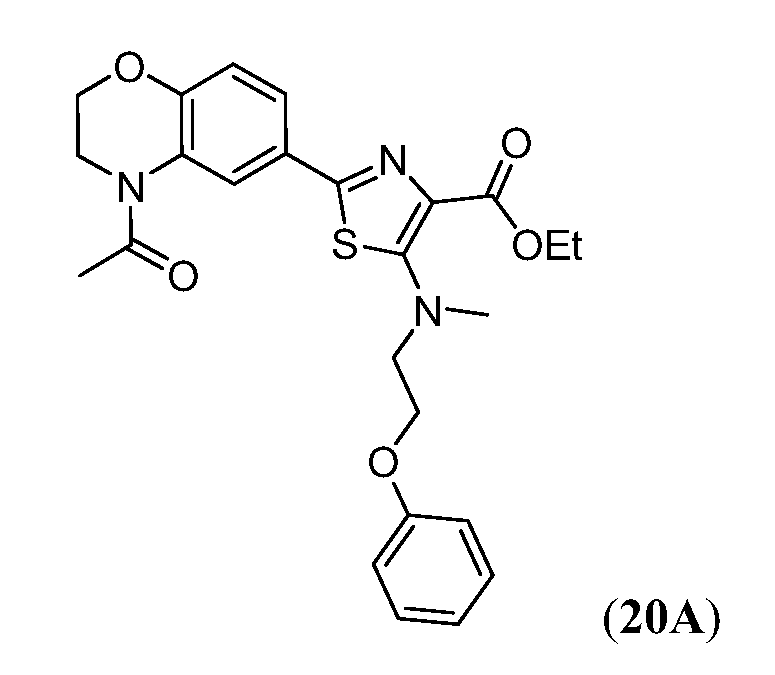
К 2-(4-ацетил-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-хлортиазол-4-этилкарбоксилату (7D) (0,15 г, 0,41 ммоль) и метил-(2-феноксиэтил)-амину (1,24 г, 8,1 ммоль) добавляли DMF (10 мл). Реакционной смеси давали нагреться до 70°C в течение 48 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 80% EtOAc в гексанах, получая 0,102 г (52%) желаемого продукта 2-(4-ацетил-3,4-дигидро-2Н-бензо[b][1,4] оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилата (20A): LC/MS (APCI): m/z 482,1 (M+H).
Стадия 2: Получение 2-(3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилата (20B)

К 2-(4-ацетил-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилату (20A) (0,102 г, 0,212 ммоль) добавляли этанол (4 мл) и 6н. HCl (6 мл). Реакционной смеси давали нагреться до 60°C и перемешивали в течение 8 часов. Реакционной смеси давали охладиться до температуры окружающей среды. К реакционной смеси добавляли EtOAc и насыщенный раствор бикарбоната натрия. Слои разделяли и экстрагировали EtOAc, и объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 0,079 г (85%) желаемого продукта 2-(3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилата (20B): LC/MS (APCI): m/z 440,1 (M+H).
Стадия 3: Получение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилата (20C)

К 2-(3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилату (20B) (0,150 г, 0,34 ммоль) и 4-нитрофенилбензо[d]тиазол-2-илкарбамату (19) (0,349 г, 0,11 ммоль) добавляли ацетонитрил (10 мл). Реакционной смеси давали нагреться до 85°C и перемешивали в течение 8 часов. Затем реакционной смеси давали нагреться при 70°C и перемешивали в течение ночи. Реакционной смеси позволяли охладиться до температуры окружающей среды и концентрировали при пониженном давлении. К концентрату добавляли EtOAc и воду и фильтровали. Слои фильтрата разделяли и экстрагировали EtOAc. Объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 0,21 г (количественный выход) 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилата (20C): LC/MS (APCI): m/z 616,3 (M+H).
Стадия 4: Получение целевого соединения 2-(4-(бензо[d] тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-карбоновой кислоты (20)
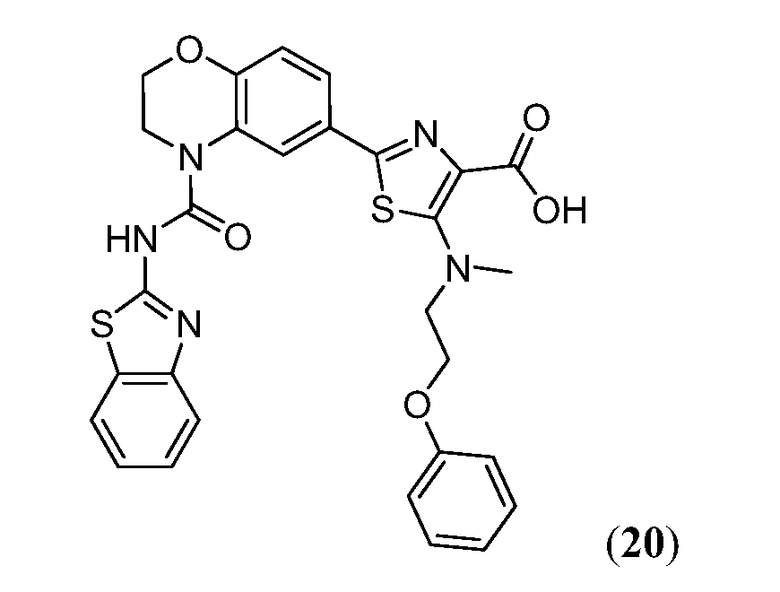
Целевое соединение 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил) амино)тиазол-4-карбоновую кислоту (20) получали в соответствии со следующей процедурой: К 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-этилкарбоксилату (20C) (0,210 г, 0,34 ммоль) и KOH (0,1 г, 1,8 ммоль) добавляли MeOH (2 мл) и воду (5 мл). Реакционной смеси давали нагреться до 55°C и перемешивали 24 часа. Реакцию проверяли LCMS. К реакционной смеси добавляли (0,1 г, 1,8 ммоль). Реакционной смеси давали нагреться до 55°C и перемешивали в течение 24 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. К сырому остатку добавляли EtOAc и воду. Слои разделяли и экстрагировали EtOAc, и объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой остаток очищали ВЭЖХ с обратной фазой, получая 0,015 г (8%) желаемого продукта 2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4] оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-карбоновой кислоты (20): LC/MS (APCI): m/z 588,1 (M+H).
Пример 21
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-карбоновой кислоты (21):
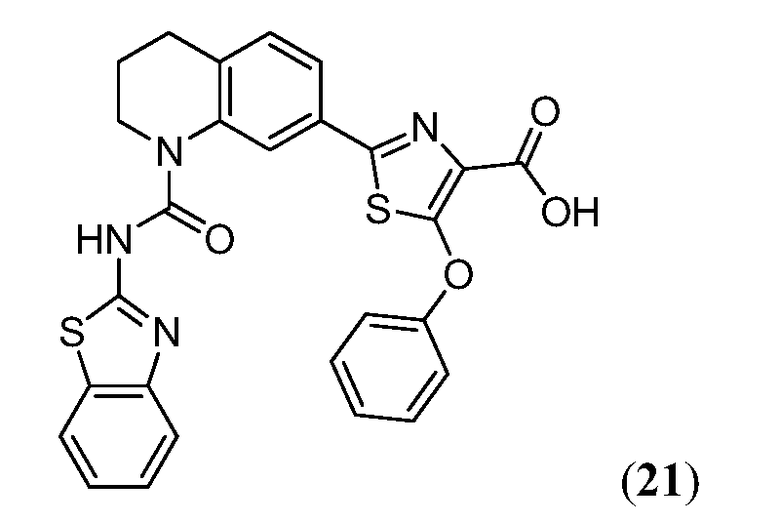
Стадия 1: Получение 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-этилкарбоксилата (21A)

К 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-хлортиазол-4-этилкарбоксилату (1J) (0,250 г, 0,69 ммоль), фенолу (0,129 г, 1,4 ммоль) и KOH (0,284 г, 2,1 ммоль) добавляли DMF (10 мл). Реакционной смеси давали нагреться до 70°C в течение 72 часов. Реакционную смесь охлаждали до температуры окружающей среды и разбавляли EtOAc и водой. Слои разделяли, и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 80% EtOAc в гексанах, получая 0,208 г (72%) этил 2-(1-ацетил-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-карбоксилата (21A): LC/MS (APCI): m/z 424,0 (M+H).
Стадия 2: Получение целевого соединения 2-(1-(бензо[d] тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-карбоновой кислоты (21)
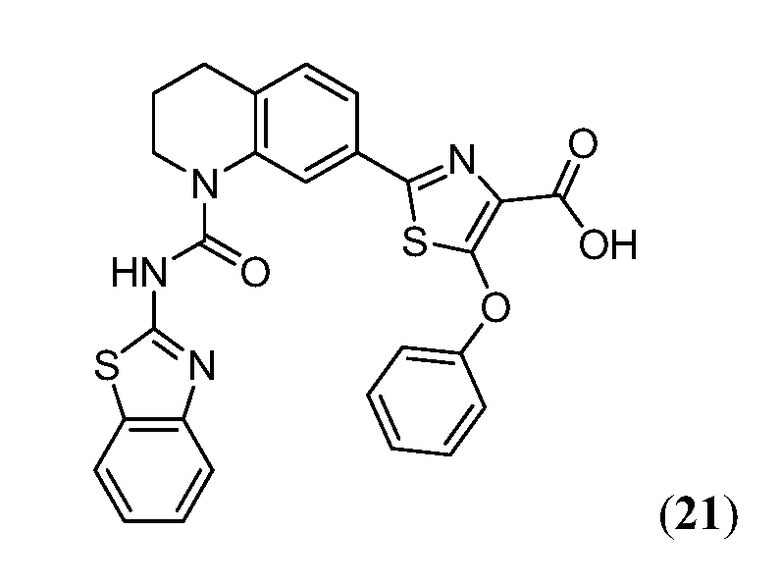
Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-карбоновую кислоту (21) получали в соответствии со следующей процедурой: 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-карбоновую кислоту (21) получали согласно процедуре, подобной описанной на стадии 12 и стадии 14 примера 1. LC/MS (APCI): m/z 529,1 (M+H).
Пример 22
Синтез 4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенола (22):
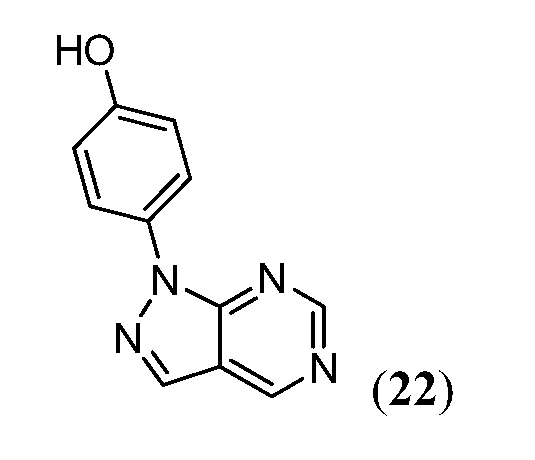
Стадия 1: Получение 5-амино-1-(4-бромфенил)-1Н-пиразол-4-карбоновой кислоты (22A)
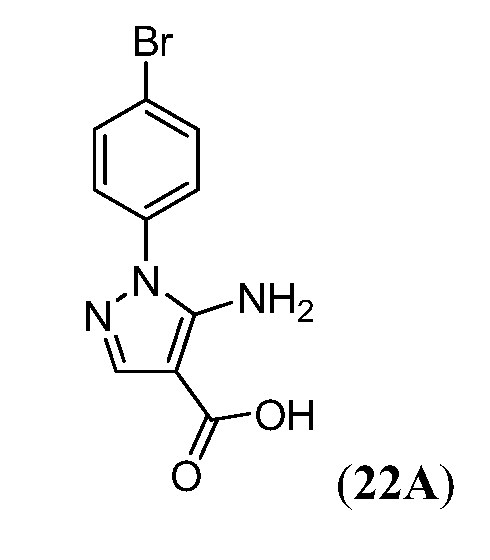
Смесь (Z)-этил-2-циано-3-этоксиакрилата (2,099 г, 12,41 ммоль), 4-бромфенилгидразин гидрохлорида (2,76 г, 12,41 ммоль) и Na2CO3 (0,789 г, 7,44 ммоль) в этаноле (30 мл) нагревали с обратным холодильником в течение 5 часов и немного концентрировали при пониженном давлении. Осадок собирали фильтрацией, промывали простым эфиром, и высушивали, получая сложный эфир. Сложный эфир растворяли в THF (5 мл) и MeOH (25 мл). К реакционной смеси добавляли 10%-ый NaOH (154 мл). Полученную смесь перемешивали при 50°C в течение ночи и концентрировали. Добавляли небольшое количество воды, и полученный раствор нейтрализовали с помощью HCl до рН 6. Белый осадок собирали, промывали водой и высушивали, получая желаемое соединение 5-амино-1-(4-бромфенил)-1Н-пиразол-4-карбоновую кислоту (22A): 1Н ЯМР (400 МГц, DMSO-D6) ppm (ч./млн) 12,09 (1H, с), 7,72 (2H, д), 7,52 (2H, д), 6,35 (2H, с).
Стадия 2: Получение 1-(4-бромфенил)-1Н-пиразоло[3,4-d]пиримидина (22B)

К раствору 1,3,5-триазина (0,575 г, 7,09 ммоль) и 5-амино-1-(4-бромфенил)-1Н-пиразол-4-карбоновой кислоты (22A) (2 г, 7,09 ммоль) в ДМСО (30 мл) добавляли эфират трифторида бора (1,08 мл, 8,51 ммоль). Полученную смесь нагревали при 120°C в течение 20 часов, охлаждали, разбавляли EtOAc и промывали 1%-ым NaOH и солевым раствором. Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении. Остаток растирали с небольшим количеством EtOAc, и осадок собирали, получая желаемый продукт 1-(4-бромфенил)-1Н-пиразоло[3,4-d]пиримидин (22B): 1Н ЯМР (400 МГц, DMSO-D6) δ ppm 9,47 (1H, с), 9,16 (1H, с), 8,68 (1H, с), 8,22 (2H, д), 7,80 (2H, д).
Стадия 3: Получение 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1Н-пиразоло[3,4-d]пиримидина (22C)

Смесь 1-(4-бромфенил)-1Н-пиразоло[3,4-d]пиримидина (22B) (500 мг, 1,817 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (508 мг, 1,999 ммоль), аддукта PdCl2(dppf)-CH2Cl2 (74,2 мг, 0,091 ммоль) и ацетата калия (535 мг, 5,45 ммоль) в ДМСО (15 мл) очищали азотом и затем нагревали при 80°C в течение ночи. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 20% EtOAc в DCM, получая желаемое соединение 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1Н-пиразоло[3,4-d]пиримидин (22C). 1Н ЯМР (400 МГц, DMSO-D6) δ ч./млн 9,49 (1H, с), 9,20 (1H, с), 8,70 (1H, с), 8,33 (2H, д), 7,90 (2H, д), 1,33 (12H, с).
Стадия 4: Получение целевого соединения 4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенола (22)

Целевое соединение 4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенол (22) получали в соответствии со следующей процедурой: К раствору 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1Н-пиразоло[3,4-d]пиримидина (22C) (100 мг, 0,31 ммоль) в THF (5 мл) добавляли NaOH (0,248 мл, 0,621 ммоль) и пероксид водорода (0,048 мл, 0,466 ммоль). Смесь перемешивали при 0°C в течение 30 минут, реакционную смесь концентрировали, и остаток растворяли в воде (8 мл). Водный раствор подкисляли разбавленной HCl. Белый осадок собирали, промывали водой и высушивали, получая целевое соединение 4-(1H-пиразоло[3,4-d]пиримидин-1-ил)фенол (22): 1Н ЯМР (400 МГц, DMSO-D6) δ ч./млн 9,75 (1H, с), 9,44 (1H, с), 9,09 (1H, с), 8,59 (1H, с), 7,82-7,99 (2H, м), 6,83 -7,05 (2H, м).
Пример 23
Синтез 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси) пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (23):

Стадия 1: Получение 3-бром-6-хлор-2-оксоэтилгексаноата (23A)
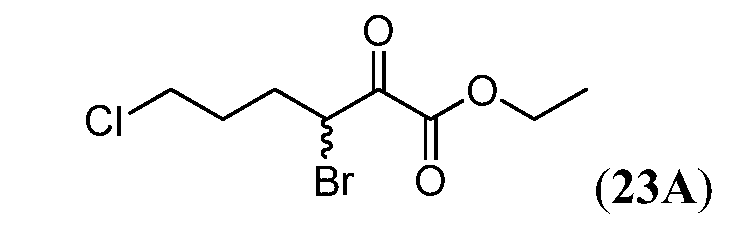
6-хлор-2-оксоэтилгексаноат (2,9 г, 15 ммоль) в тетрахлорметане (30 мл) обрабатывали бромом (0,85 мл, 16,5 ммоль) и перемешивали при температуре окружающей среды в течение 1 часа. Реакционную смесь разбавляли EtOAc, промывали раствором Na2S2O3, водой и солевым раствором, высушивали (MgSO4), фильтровали и концентрировали при пониженном давлении. Концентрат очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 10% EtOAc в гексанах, получая (95%) желаемого продукта 3-бром-6-хлор-2-оксоэтилгексаноата (23A) в форме светло-желтого масла: 1Н ЯМР (300 МГц, DMSO-d6) δ ч./млн 5,25 (1H, дд), 4,29 (2H, кв), 3,71 (2H, т), 2,16 (1H, м), 1,91 (1H, м), 1,29 (3H, т).
Стадия 2: Получение 2-амино-5-(3-хлорпропил)тиазол-4-этилкарбоксилата (23B)
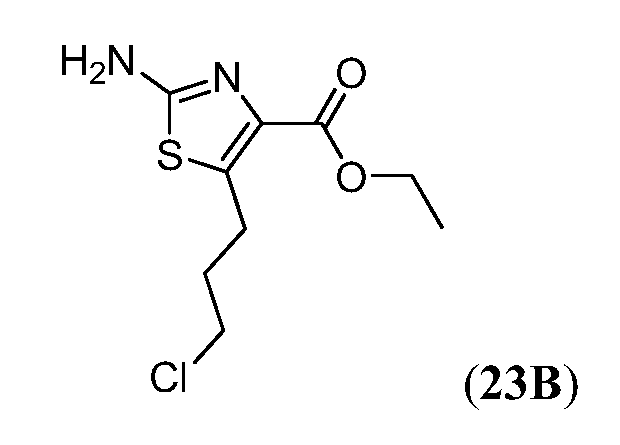
К раствору 3-бром-6-хлор-2-оксоэтилгексаноата (23A) (0,845 г, 3,11 ммоль) в ацетоне (6 мл) добавляли тиомочевину (0,284 г, 3,73 ммоль) и нагревали с обратным холодильником в течение ночи. Реакционную смесь концентрировали при пониженном давлении. Добавляли EtOAc и гексаны в отношении 1:9 и перемешивали в течение 1 часа. Добавляли ацетон, чтобы растворить материал, и концентрировали при пониженном давлении, получая 0,718 г (93%) желаемого продукта 2-амино-5-(3-хлорпропил)тиазол-4-этилкарбоксилата (23B): LC/MS (APCI): m/z 249,5 (M+H).
Стадия 3: Получение 2-бром-5-(3-хлорпропил)тиазол-4-этилкарбоксилата (23C)
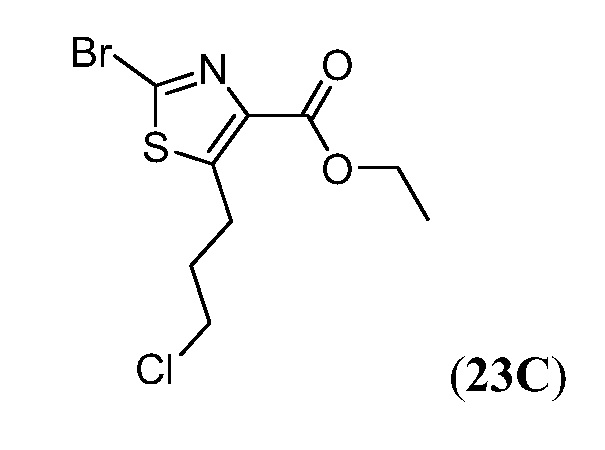
2-амино-5-(3-хлорпропил)тиазол-4-этилкарбоксилат (23B) (3,3 г, 13 ммоль) в ацетонитриле (15 мл) слегка нагревали, чтобы растворить исходный материал. Добавляли трет-бутил нитрит (2,05 г, 19,9 ммоль), и реакционную смесь перемешивали в течение 10 минут. Затем к реакционной смеси добавляли бромид меди (II) (5,04 г, 22,6 ммоль). Реакционную смесь нагревали при 80°C и перемешивали в течение 2 часов. Добавляли DCM и 1 н. HCl, слои разделяли, и водный слой экстрагировали 3x с DCM. Органические экстракты экстрагировали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой остаток очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 60% EtOAc в гексане, получая 1,45 г (35%) желаемого продукта 2-бром-5-(3-хлорпропил)тиазол-4-этилкарбоксилата (23C): LC/MS (APCI): m/z 313,8 (M+H).
Стадия 4: Получение 7-бром-3,4 дигидрохинолина-1(2H)-трет-бутилкарбоксилата (23D)
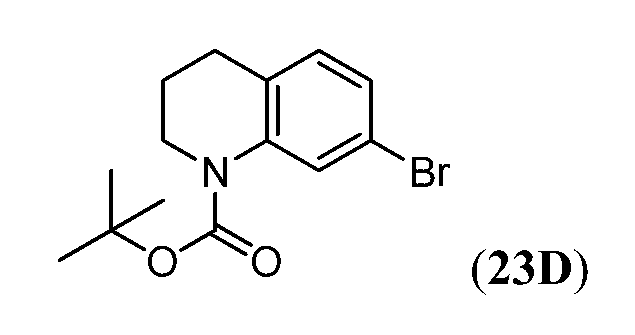
7-бром-1,2,3,4-тетрагидрохинолин (1Е) (1,0 г, 4,7 ммоль), ди-трет-бутилдикарбонат (3,1 г, 14 ммоль) объединяли и нагревали до 100°C в течение 1 часа. Охлаждали до температуры окружающей среды и очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 25% EtOAc в гексанах, получая 1,46 г (99%) желаемого соединения 7-бром-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (23D).
Стадия 5:
Получение 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (23E)
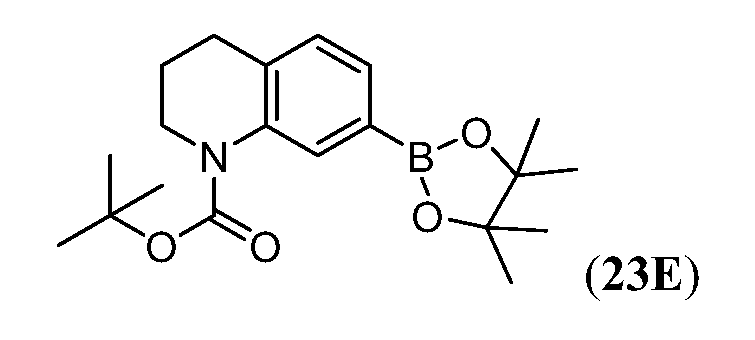
7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолинов-1(2Н)-трет-бутилкарбоксилат (23E), получали согласно процедуре, подобной описанной в примере 1, кроме того, что 7-бром-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилат (23D), использовался вместо 1-(7-бром-3,4-дигидрохинолин-1(2Н)-ил)этанона (1F) на стадии 7 примера 1.
Стадия 6: Получение 2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-хлорпропил)тиазол-4-этилкарбоксилата (23F)
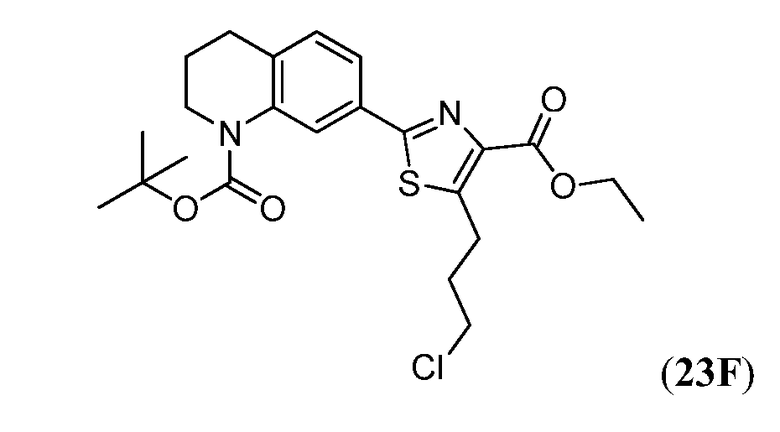
2-бром-5-(3-хлорпропил)тиазол-4-этилкарбоксилат (23C) (0,790 г, 2,5 ммоль) растворяли в диоксане, добавляли 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилат (23E) (0,910 г, 2,5 ммоль), затем хлорид лития (0,320 г, 7,6 ммоль), тетракис(трифенилфосфин)палладий (0) (0,290 г, 0,25 ммоль), карбонат цезия (2,5 г, 7,6 ммоль) и воду. Эту смесь помещали в закрытую трубку, реакционную смесь эвакуировали, смывали азотом и нагревали при 100°C в течение ночи. Реакционную смесь фильтровали через Целит, элюировали EtOAc, концентрировали при пониженном давлении и в сухом виде загружали на силикагель. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 50% EtOAc в гексанах, получая 0,943 г (80%) желаемого продукта 2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-хлорпропил)тиазол-4-этилкарбоксилата (23F): LC/MS (APCI): m/z 466,1 (M+H).
Стадия 7: Получение 2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-йодпропил)тиазол-4-этилкарбоксилата (23G)
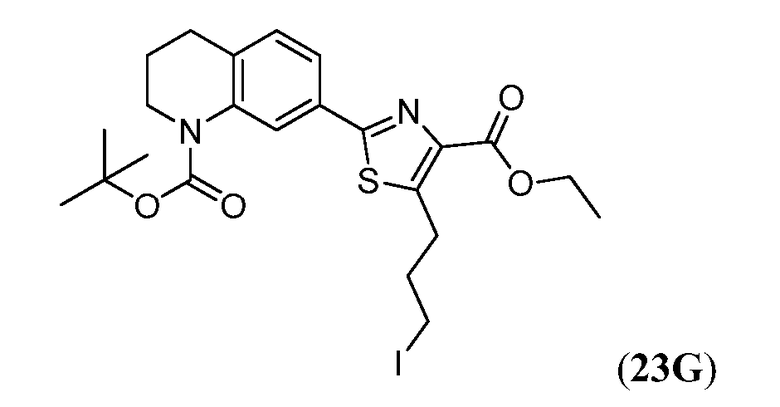
2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-хлорпропил)тиазол-4-этилкарбоксилат (23F) (0,943 г, 2,02 ммоль) растворяли в ацетонитриле (10 мл). К этой реакционной смеси добавляли NaI (0,309 г, 2,06 ммоль) и нагревали до 80°C. Реакционную смесь перемешивали в течение 48 часов. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Сырой материал растворяли в EtOAc, промывали водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 0,370 г (97%) желаемого продукта 2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-йодпропил)тиазол-4-этилкарбоксилата (23G): LC/MS (APCI): m/z 557,2 (M+H).
Стадия 8: Получение 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилата (23Н)

К NaH (10 мг, 0,43 ммоль) и 4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенолу (22) (100 мг, 0,47 ммоль) добавляли DMF (1 мл) и перемешивали в атмосфере азота в течение 10 минут. К реакционной смеси добавляли 2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-йодпропил)тиазол-4-этилкарбоксилат (23G) (240 мг, 0,43 ммоль) в DMF (2 мл) и затем перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли EtOAc, промывали последовательно NaHCO3, водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. LC/MS (APCI): m/z 641,3 (M+H).
Этот материал растворяли в смеси 1:1 DCM:TFA и перемешивали в течение 90 минут. Реакционную смесь концентрировали при пониженном давлении, растворяли в EtOAc, промывали насыщенным раствором NaHCO3, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал загружали в сухом виде на силикагель и очищали хроматографией на колонках с силикагелем с элюированием градиентом от 0 до 70% EtOAc в гексанах, получая 93 мг (40%) желаемого продукта 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилата (23Н): LC/MS (APCI): m/z 541,2 (M+H).
Стадия 9: Получение 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилата (23I)
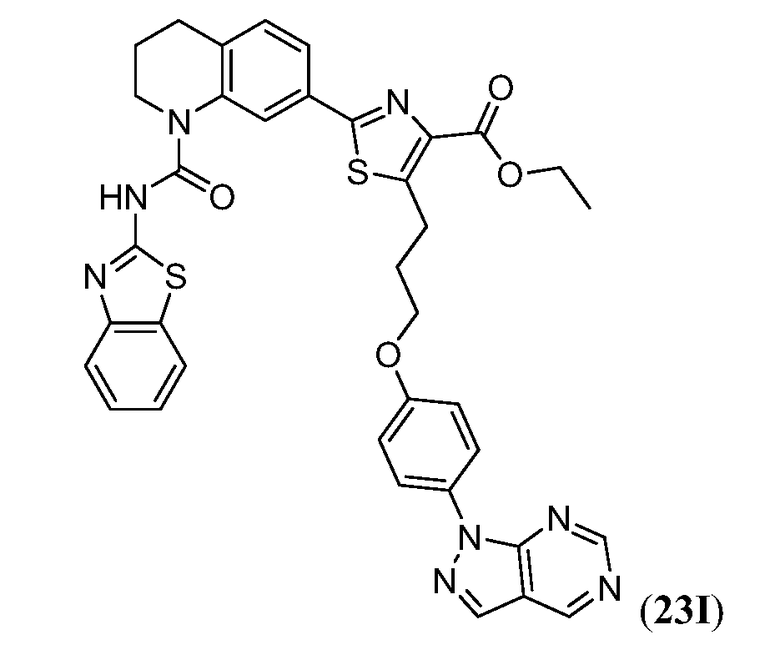
5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилат (23I), получали согласно процедуре, подобной описанной в примере 1, кроме того, что 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилат (23Н) использовался вместо 5-(4-метоксифенил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (1L) на стадии 14 примера 1. Реакционную смесь фильтровали и промывали водой и MeOH, получая желаемый продукт 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилат (23I), который использовали без дальнейшей очистки. LC/MS (APCI): m/z 717,6 (M+H).
Стадия 10:
Получение целевого соединения 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (23)

Целевое соединение 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновую кислоту (23) получали в соответствии со следующей процедурой: К 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилату (23I) (120 мг, 0,17 ммоль) добавляли MeOH и THF. LiOH (20 мг, 0,85 ммоль) растворяли в воде и добавляли к реакционной смеси. Реакционную смесь перемешивали и нагревали при 60°C в течение двух часов. Реакционную смесь охлаждали и помещали на ночь в холодильник. Осадок переносили в пробирку для центрифугирования и центрифугировали при 17000 об/мин. Осадок промывали водой. затем осадок добавляли к ацетонитрилу и воде и лиофилизировали, получая 69 мг (59%) желаемого продукта 5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновой кислоты (23): 1Н ЯМР (400 МГц, DMSO) δ 9,44 (с, 1H), 9,10 (с, 1H), 8,61 (с, 1H), 8,04 (д, J=8,7, 2H), 7,66 (с, 1H), 7,45 (д, J=34,2, 2H), 7,20 (т, J=18,3, 5H), 4,12 (с, 3H), 3,95 (с, 3H), 2,79 (с, 2H), 2,16 (с, 3H), 1,89 (с, 2H), -0,00 (с, 3H).
Пример 24
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-карбоновой кислоты (24):

Стадия 1: Получение 5-(3-феноксипропил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилата (24A)
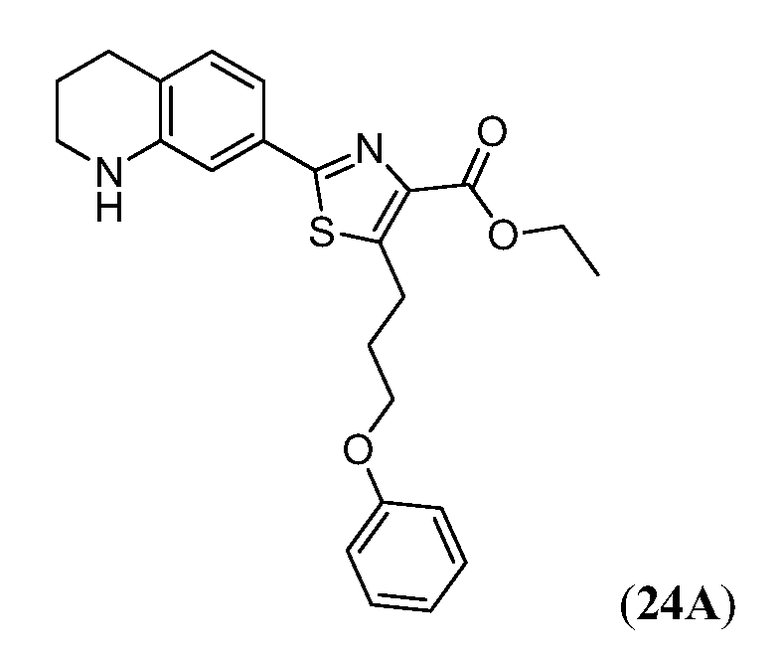
К K2CO3 (32 мг, 0,23 ммоль), фенолу (28 мг, 0,30 ммоль) и 18-crown-6 (180 мг, 0,70 ммоль) добавляли DMF (1 мл). Через 5 минут к реакционной смеси добавляли 2-(1-(трет-бутоксикарбонил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-йодпропил)тиазол-4-этилкарбоксилат (23G) (129 мг, 0,23 ммоль) в DMF (1 мл) и перемешивали при температуре окружающей среды в течение 72 часов. Реакцию проверяли LCMS, и в смеси оставался исходный материал. К реакционной смеси добавляли K2CO3 (32 мг, 0,23 ммоль) и фенол (28 мг, 0,30 ммоль) и перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли EtOAc, промывали насыщенным NaHCO3, водой, солевым раствором, высушивали над MgSO4 и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках на силикагеле, получая 50 мг как смесь исходного материала и продукта. К DMF (1 мл) добавляли NaH (4 мг) и фенол (13 мг) и перемешивали в течение 10 минут. К реакционной смеси добавляли выделенный материал (50 мг) как смесь исходного материала и продукта и перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли EtOAc, промывали насыщенным NaHCO3, водой, солевым раствором, высушивали с помощью MgSO4 и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках на силикагеле, получая 22 мг желаемого продукта.
Этот материал растворяли в смеси 1:1 DCM:TFA и перемешивали в течение 90 минут. Реакционную смесь концентрировали при пониженном давлении, получая желательный продукт 5-(3-феноксипропил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилат (24A): LC/MS (APCI): m/z 423,2 (M+H).
Стадия 2: Получение целевого соединения 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-карбоновой кислоты (24)
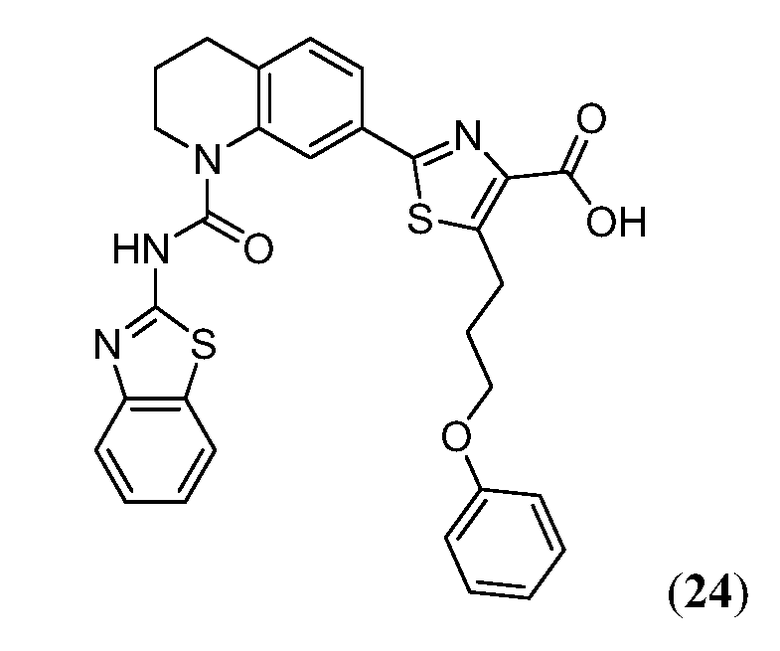
Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-карбоновую кислоту (24) получали в соответствии со следующей процедурой: К 5-(3-феноксипропил)-2-(1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-этилкарбоксилату (24A) (18 мг, 0,0,42 ммоль) и N-(бензо[d]тиазол-2-ил)-1Н-имидазол-1-карбоксамиду (1M) (10 мг, 0,042 ммоль) добавляли DMF (2 мл) и перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли EtOAc, промывали последовательно NaHCO3, водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-этилкарбоксилат. LC/MS (APCI): m/z 599,5 (M+H).
К 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-этилкарбоксилату в смеси 1:1 MeOH и THF (2 мл) добавляли LiOH (5 мг, 0,21 ммоль), растворенный в воде (2 мл). Реакционную смесь перемешивали при 60°C в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. К этому сырому материалу добавляли воду и MeOH и оставляли на ночь. Полученный осадок отфильтровывали и промывали водой. К этому твердому веществу добавляли воду и ацетонитрил и лиофилизировали, получая 12 мг желаемого продукта 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-карбоновой кислоты (24). 1Н ЯМР (400 МГц, DMSO) δ 8,62 (с, 1H), 7,60 (с, 2H), 7,51-7,33 (м, 3H), 7,27 (т, J=8,0, 3H), 7,17 (т, J=7,7, 2H), 6,92 (дд, J=7,5, 14,2, 5H), 4,03 (т, J=6,4, 3H), 3,95 (с, 2H), 3,42 (д, J=7,8, 4H), 2,78 (с, 2H), 2,10 (с, 3H), 1,88 (д, J=9,5, 3H).
Пример 25
Синтез 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)пиколиновой кислоты (25):

Стадия 1: Получение 6-бром-трет-бутилпиколината (25A)

П-толуолсульфонил хлорид (9,0 г, 47,2 ммоль) добавляли частями к смеси 2-бром-пиколиновой кислоты (4,02 г, 19,9 ммоль) в трет-бутаноле (36 мл) и пиридине (10,8 мл, 134 ммоль) при 0°C, и смесь перемешивали при температуре окружающей среды в течение 14 часов. Медленно добавляли водный раствор бикарбоната натрия. Осадок отфильтровывали, промывали водой и высушивали в вакууме, получая 4,04 г (79%) желаемого продукта 6-бром-трет-бутилпиколината (25A): 1Н ЯМР (300 МГц, CDC13) δ 7,97 (1H, дд), 7,64 (2H, м), 1,62 (9H, с).
Стадия 2: Получение 2-((4-бром-2-нитрофенил)амино(метил)) этанола (25B)
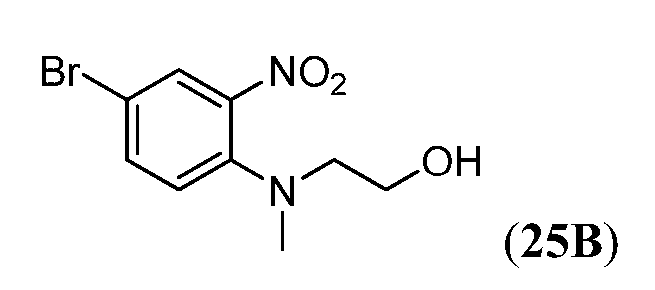
5-бром-2-фторнитробензол (5,0 г, 22,7 ммоль), 2-(метиламино)этанол (4,26 г, 56,8 ммоль) и K2CO3 (9,4 г, 68,1 ммоль) в DMF (20 мл) нагревали до 60°C в течение 1,5 часов, охлаждали до температуры окружающей среды, разбавляли водой и экстрагировали EtOAc. Органические фазы промывали водой, солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 6,98 г (100%) желаемого продукта 2-((4-бром-2-нитрофенил)амино(метил))этанола (25B): 1Н ЯМР (300 МГц, CDC13) δ 7,87 (1H, д), 7,51 (1H, дд), 7,10 (1H, д), 3,77 (2H, т), 3,39 (2H, т), 2,83 (3H, с).
Стадия 3: Получение 2-((2-амино-4-бромфенил)амино(метил)) этанола (25C)
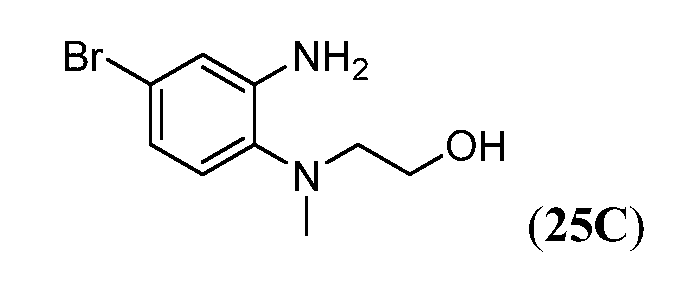
Железо (1,5 г, 26,2 ммоль) и хлорид аммония (0,39 г, 1,0 экв.) в воде (8 мл) нагревали с обратным холодильником в течение 0,5 часа. Медленно добавляли 2-((4-бром-2-нитрофенил)амино(метил))этанол (25B) (2,0 г, 7,27 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 14 часов, охлаждали до температуры окружающей среды, разбавляли водой и EtOAc и фильтровали через Целит. Водную фазу экстрагировали EtOAc. Объединенные органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 60:40-40:60, получая 1,36 г (76%) желаемого продукта 2-((2-амино-4-бромфенил)амино(метил))этанола (25C): 1Н ЯМР (300 МГц, CDC13) δ 6,92-6,82 (3H, м), 3,68 (2H, т), 3,04 (2H, т), 2,70 (3H, с).
Стадия 4: Получение 4-бром-N1-(2-хлорэтил)-N1-метилбензол-1,2 диамина (25D)
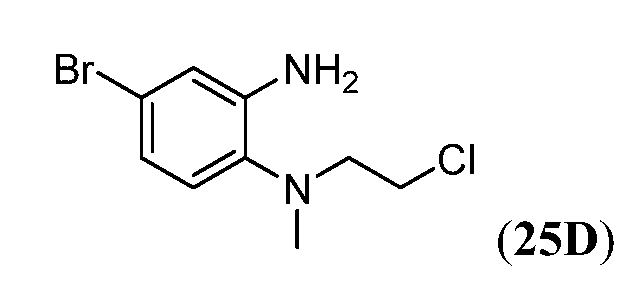
Тионилхлорид (0,78 г, 6,74 ммоль) добавляли по каплям к раствору 2-((2-амино-4-бромфенил)амино(метил))этанола (25C) (1,376 г, 5,62 ммоль) в DCM (50 мл) и DMF (10 капель) при 0°C. Смеси давали нагреться до температуры окружающей среды и перемешивали в течение 1 часа. Затем смесь нагревали до 35°C в течение 2,5 часов, концентрировали, разбавляли 1 н. NaOH и экстрагировали DCM. Органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE:EtOAc 100:0-85:15, получая 0,611 г (41%) желаемого продукта 4-бром-N1-(2-хлорэтил)-N1-метилбензол-1,2 диамина (25D): 1Н ЯМР (300 МГц, CDCl3) δ 6,88-6,79 (3H, м), 4,20 (2H, ушир.с), 3,56 (2H, т), 3,17 (2H, т), 2,67 (3H, с).
Стадия 5:
Получение 6-бром-1-метил-1,2,3,4-тетрагидрохиноксалина (25E)
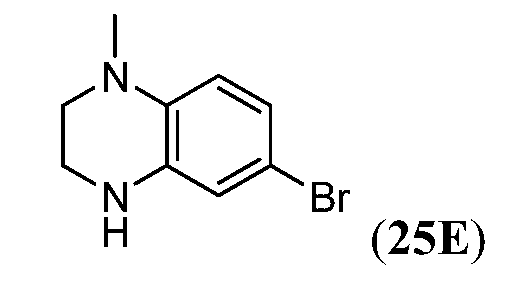
Раствор 4-бром-N1-(2-хлорэтил)-N1-метилбензол-1,2-диамина (25D) (0,611 г, 2,3 ммоль) и K2CO3 (0,634 г, 4,6 ммоль) в DMF (7 мл) нагревали до 80°C в течение 1 часа и затем при 100°C в течение 1,5 часов, охлаждали до температуры окружающей среды, разбавляли водой и экстрагировали EtOAc. Органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 0,528 г (100%) желаемого продукта 6-бром-1-метил-1,2,3,4-тетрагидрохиноксалина (25E): 1Н ЯМР (300 МГц, CDC13) δ 6,72 (1H, дд), 6,56 (1H, д), 6,38 (1H, д), 3,74 (1H, ушир.с), 3,47 (2H, т), 3,22 (2H, т), 2,82 (3H, с).
Стадия 6: Получение 1-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохиноксалина (25F)
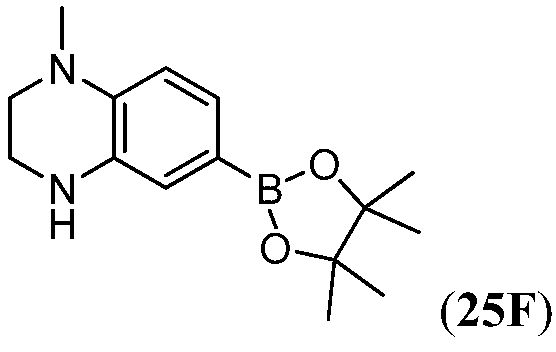
Раствор 6-бром-1-метил-1,2,3,4-тетрагидрохиноксалина (25E) (200 мг, 0,88 ммоль), бис(пинаколято)дибора (304 мг, 1,20 ммоль), ацетата калия (204 мг, 2,08 ммоль), трис(дибензилиденацетон)-дипалладия (0) (20 мг, 0,022 ммоль), и 2-дициклогексилфосфино-2',4',6'-три-изо-пропил-1,1'-дифенила (42 мг, 0,09 ммоль) в безводном диоксане (3,5 мл) нагревали при 100°C в течение 45 минут, охлаждали до температуры окружающей среды, разбавляли водой и экстрагировали EtOAc. Органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE:EtOAc 100:0-85:15, получая 247 мг (100%) желаемого продукта 1-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохиноксалина (25F): 1Н ЯМР (300 МГц, DMSO-d6) δ 6,81 (1H, дд), 6,71 (1H, д), 6,38 (1H, д), 5,38 (1H, ушир.с), 3,29 (2H, т), 3,18 (2H, т), 2,78 (3H, с), 1,22 (12H,с).
Стадия 7: Получение 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколината (25G)
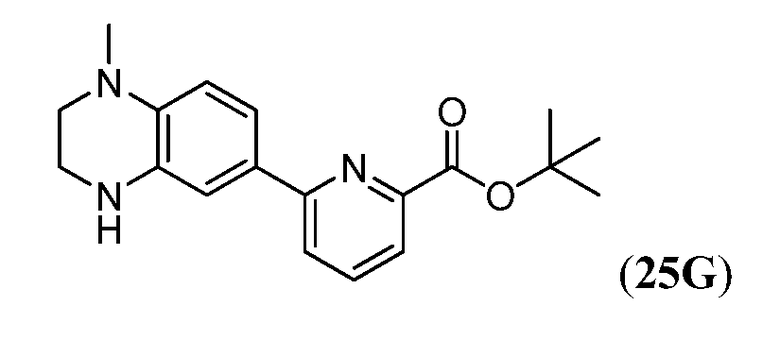
6-бром-трет-бутилпиколинат (25A) (157 мг, 0,61 ммоль) в 1,4-диоксане (1,0 мл) при температуре окружающей среды добавляли по каплям к 1-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохиноксалину (25F) (167 мг, 0,61 ммоль), K2CO3 (207 мг, 1,52 ммоль), тетрабутиламмоний бромиду (19,5 мг, 0,061 ммоль), и дихлорбис(трифенилфосфин)палладию (II) (17,9 мг, 0,024 ммоль) в 1,4-диоксане (2,0 мл) и воде (1,0 мл). Смесь нагревали в микроволновом реакторе (150 Вт) до 100°C в течение 50 минут, охлаждали до 20°C, разбавляли водой и экстрагировали EtOAc. Органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE:EtOAc 100:0-70:30, получая 76 мг (38%) желаемого продукта 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколината (25G): 1Н ЯМР (300 МГц, MeOD) δ 7,84-7,78 (3H, м), 7,35 (1H, д), 7,30 (1H, д), 6,62 (1H, д), 3,41 (2H, т), 3,31 (2H, т), 2,92 (3Н, ушир.с), 1,64 (9H, с).
Стадия 8: Получение 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколината (25Н)
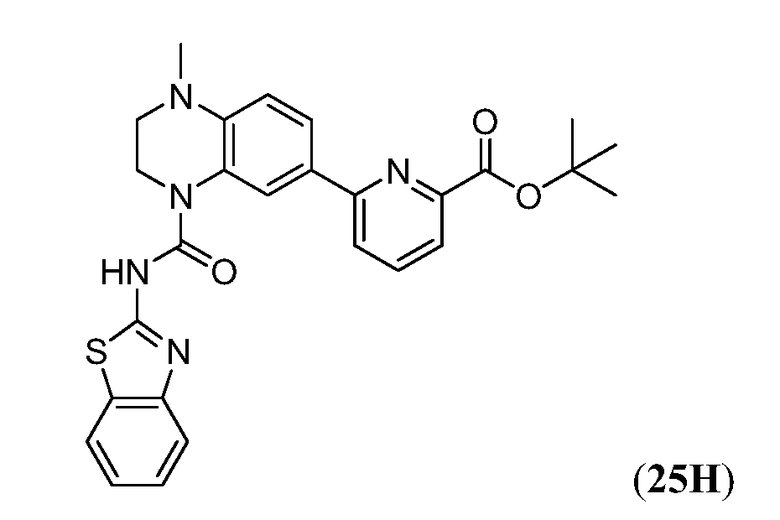
Смесь 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколината (25G) (36 мг, 0,11 ммоль) и 4-нитрофенил бензо[d]тиазол-2-илкарбамата (19) (42 мг, 0,13 ммоль) в безводном ацетонитриле (1,0 мл) нагревали с обратным холодильником в течение 3 часов, охлаждали до температуры окружающей среды, концентрировали при пониженном давлении, и сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE:EtOAc 100:0-70:30, получая 47 мг (85%) желаемого продукта 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколината (25Н): 1Н ЯМР (300 МГц, CDCl3) δ 8,14 (1H, с), 7,88-7,77 (2H, м), 7,74 (2H, д), 7,54 (1H, д), 7,34 (1H, т), 7,22 (1H, т), 6,71 (1H, д), 3,97 (2H, т), 3,49 (2H, т), 3,02 (3H, с), 1,56 (9H, с).
Стадия 9:
Получение целевого соединения 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)пиколиновой кислоты (25)

Целевое соединение 6-(4-(бензо[d]тиазол-2-илкарбамоил)-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)пиколиновую кислоту (25) получали следующей процедурой: Трифторуксусную кислоту (2,0 мл) добавляли по каплям к 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколинату (25Н) (79 мг, 0,16 ммоль) в DCM (2,0 мл) при 0°C. Смеси давали нагреться до температуры окружающей среды и перемешивали в течение 72 часов. Смесь концентрировали, растирали с простым диэтиловым эфиром, фильтровали и высушивали в вакууме, получая 62 мг (70%) желаемого продукта 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)пиколиновой кислоты (25): 1Н ЯМР (300 МГц, DMSO-d6) δ 8,28 (1H, с), 8,01-7,72 (5H, м), 7,45-7,30 (2H, м), 7,21 (1H, т), 6,82 (1H, д), 4,03 (2H, т), 3,45 (2H, т), 3,00 (3H, с), MS (ESI(+)): м/z 446,0 (M+H).
Пример 26
Синтез 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)пиколиновой кислоты (26):

Стадия 1: Получение 3-гидроксиметилпиколината (26A)
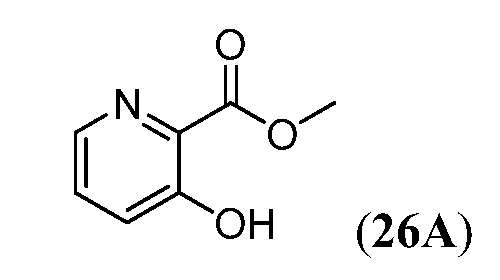
Концентрированную серную кислоту (19 мл) добавляли по каплям при температуре окружающей среды к смеси 3-гидроксипиколиновой кислоты (23,0 г, 165 ммоль) в метаноле (700 мл). Смесь нагревали с обратным холодильником в течение 72 часов, охлаждали и концентрировали. Смесь нейтрализовали бикарбонатом и экстрагировали EtOAc. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали при пониженном давлении, получая 21,7 г (85%) желаемого продукта, 3-гидроксиметилпиколината (26A): 1Н ЯМР (300 МГц, CDCl3) δ 10,63 (1H, с), 8,28 (1H, дд), 7,39 (2H, м), 4,06 (3H, с).
Стадия 2: Получение 6-бром-3-гидроксиметилпиколината (26B)
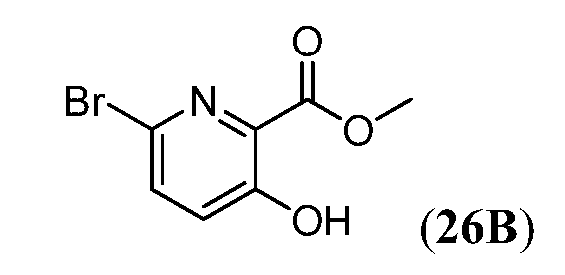
Бром (5,02 г, 31,4 ммоль) добавляли по каплям при температуре окружающей среды к смеси 3-гидроксиметилпиколината (26A) (3,67 г, 26,4 ммоль) в воде (75 мл), и смесь перемешивали в течение 2,5 часов. Осадок отфильтровывали, промывали водой и высушивали в вакууме, получая 4,18 г (75%) желаемого продукта 6-бром-3-гидроксиметилпиколината (26B): 1Н ЯМР (300 МГц, DMSO-d6) δ 10,67 (1H, с), 7,69 (1H, д), 7,42 (1H, д), 3,88 (3H, с).
Стадия 3:
Получение 6-бром-3-(3-феноксипропокси)метилпиколината (26C)
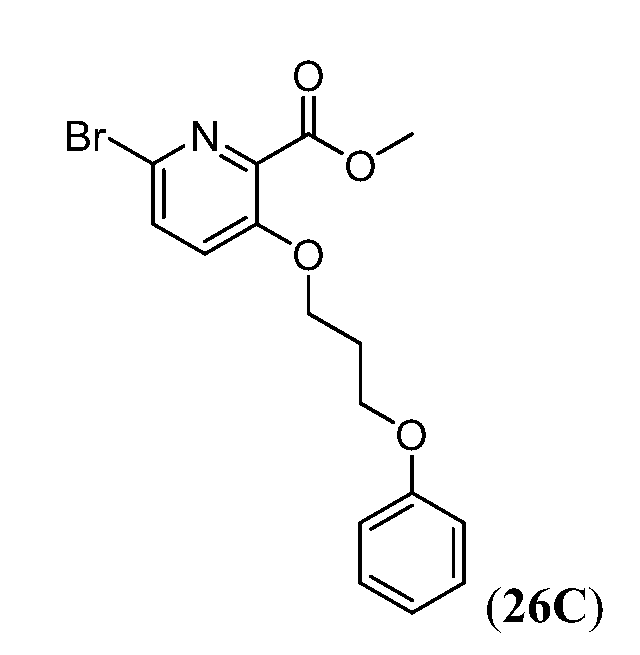
Гидрид натрия (60% в минеральном масле, 172 мг, 4,53 ммоль) добавляли при температуре окружающей среды к смеси 6-бром-3-гидроксиметилпиколината (26B) (1,0 г, 4,31 ммоль) и 3-феноксипропил бромидов (927 мг, 4,31 ммоль) в DMA (20 мл). Смесь нагревали до 100°C в течение 1,75 часов, охлаждали, разбавляли 10%-ной лимонной кислотой и экстрагировали EtOAc. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали при пониженном давлении. Сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE:EtOAc 100:0-75:25, получая 1,35 г (86%) желаемого продукта 6-бром-3-(3-феноксипропокси)метилпиколината (26C): 1Н ЯМР (300 МГц, CDC13) δ 7,52 (1H, д), 7,27 (2H, м), 6,92 (2H, м), 4,25 (2H, т), 4,19 (2H, т), 3,92 (3H, с), 2,30 (2H, т).
Стадия 4: Получение 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)метилпиколината (26D)
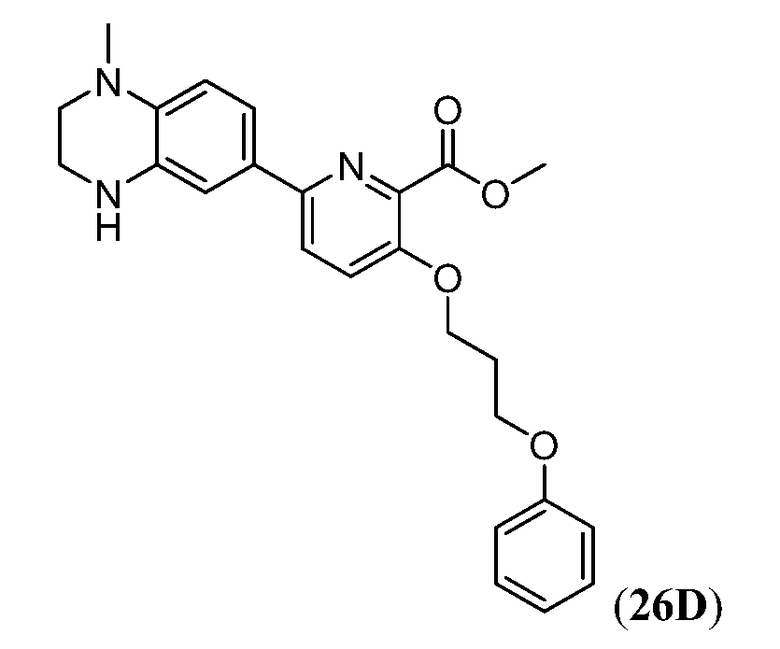
6-бром-3-(3-феноксипропокси)метилпиколинат (26C) (141 мг, 0,38 ммоль) в 1,4-диоксане (1,0 мл) добавляли по каплям при температуре окружающей среды к смеси 1-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохиноксалина (25F) (105 мг, 0,38 ммоль), K2CO3 (207 мг, 1,52 ммоль), тетрабутиламмоний бромида (12,1 мг, 0,038 ммоль) и дихлорбис(трифенилфосфин)палладия (II) (11,3 мг, 0,015 ммоль) в 1,4-диоксане (2,5 мл) и воде (1,3 мл). Смесь нагревали в микроволновом реакторе (150 Вт) до 100°C в течение 5 минут, охлаждали, разбавляли водой и экстрагировали EtOAc. Органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE:EtOAc 100:0-70:30, получая 27 мг (16%) желаемого продукта 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)метилпиколината (26D): 1Н ЯМР (300 МГц, CDCl3) δ 7,71-7,23 (6H, м), 6,92 (3H, м), 6,58 (1H, д), 4,31-4,13 (6H, м), 3,98 (2H, т), 3,92 (3H, с), 2,30 (2H, т).
Стадия 5: Получение 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)метилпиколината (26E)
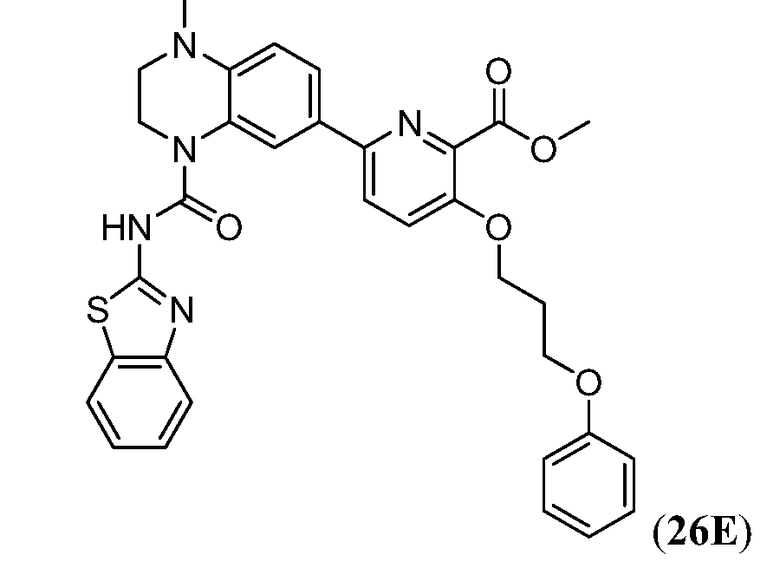
6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)метилпиколинат (26E) (24 мг, 64%) получали согласно процедуре, подобной описанной в примере 25, кроме того, что 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)метилпиколинат (26D) использовали вместо 6-(1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)трет-бутилпиколината (25G) на стадии 8 примера 25. 1Н ЯМР (300 МГц, CDC13) δ 7,98 (1H, с), 7,78 (1H, д), 7,72 (1H, д), 7,48 (1H, д), 7,39 (2H, д), 7,30-7,17 (4H, м), 6,97-6,90 (3H, м), 6,50 (1H, д), 4,29 (2H, т), 4,22 (2H, т), 3,90 (2H, т), 3,79 (3H, с), 3,38 (2H, т), 2,94 (3H, с), 2,32 (2H, т).
Стадия 6: Получение целевого соединения 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)пиколиновой кислоты (26)
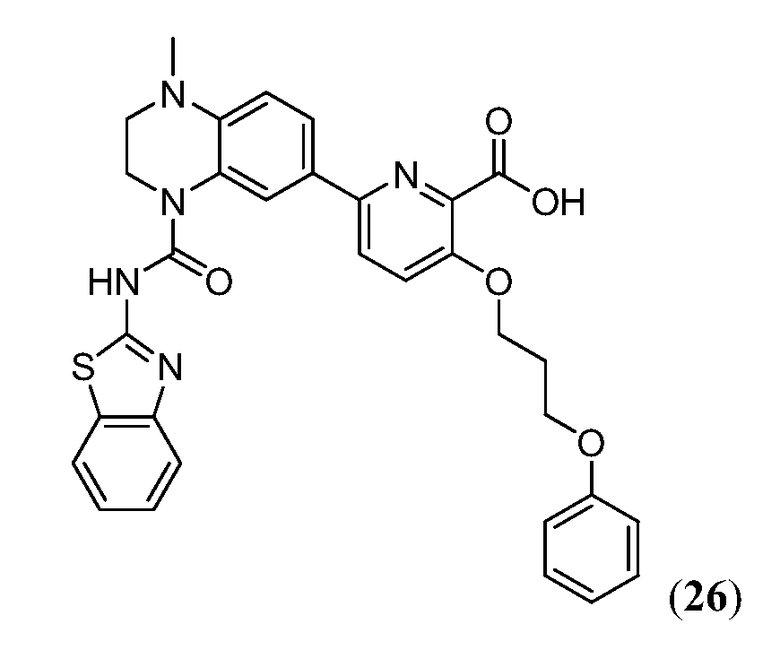
Целевое соединение 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)пиколиновую кислоту (26) получали в соответствии со следующей процедурой: LiOH (12 мг, 10 экв.) добавляли при температуре окружающей среды к смеси 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)метилпиколината (26E) (25 мг, 0,041 ммоль) в MeOH (0,5 мл) и воде (0,25 мл). Смесь нагревали до 55°C в течение 14 часов, охлаждали до 0°C и подкисляли 1 н. HCl до pH~4. Полученный осадок отфильтровывали, промывали водой и высушивали в вакууме, получая 15 мг (61%) желаемого продукта 6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)пиколиновой кислоты (26): 1Н ЯМР (300 МГц, DMSO-d6) δ 8,13 (1H, ушир.с), 7,83 (1H, д), 7,80 (1H, д), 7,65 (2H, м), 7,37 (1H, т), 7,32-7,18 (4H, м), 6,94 (3H, м), 6,79 (1H, д), 4,26 (2H, т), 4,15 (2H, т), 4,02 (2H, т), 3,41 (2H, т), 2,97 (3Н, с), 2,17 (2H, т), MS (ESI(+)): м/z 596,1 (M+H).
Пример 27
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)пиколиновой кислоты (27):

Стадия 1:
Получение 6-бром-3-(2-феноксиэтокси)метилпиколината (27A)

Смесь 6-бром-3-гидроксиметилпиколината (26B) (308 мг, 1,33 ммоль) и 2-феноксиэтанола (183 мг, 1,33 ммоль) перемешивали в сухом DCM (10 мл). Добавляли PPh3-полистирол (665 мг, 2,0 ммоль), затем по каплям добавляли DIAD (0,387 мл, 2,0 ммоль). Смесь перемешивали в течение 48 часов, затем фильтровали через Целит и концентрировали при пониженном давлении. Сырой материал очищали флэш-хроматографией на колонке с силикагелем с элюированием градиентом PE/EtOAc 9:1-7:3, получая 334 мг (72%) желаемого соединения 6-бром-3-(2-феноксиэтокси)метилпиколината (27A): 1Н ЯМР (300 МГц, CDC13) δ 7,58-6,90 (7H, м), 4,43 (2H, м), 4,34 (2H, м), 3.21 (3H, c).
Стадия 2:
Получение 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолина (27B)
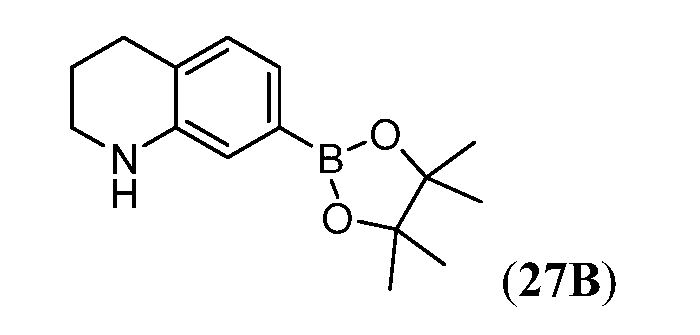
7-бром-1,2,3,4-тетрагидрохинолин (1Е) (375 мг, 1,77 ммоль) в безводном 1,4-диоксане (3 мл) добавляли по каплям при 95°C к смеси бис(пинаколято)дибора (628 мг, 2,48 ммоль), ацетата калия (416 мг, 4,24 ммоль), трис(дибензилиденацетон)дипалладия(0) (40 мг, 0,044 ммоль) и 2-дициклогексилфосфино-2',4',6'-три-изо-пропил-1,1'-дифенил (84 мг, 0,18 ммоль) в безводном 1,4-диоксане (4 мл). Реакционную смесь перемешивали при 95°C в течение 3,5 часов, охлаждали до температуры окружающей среды, разбавляли EtOAc, промывали водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал осаждали из смеси EtOAc/PE, получая 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолин (27B): 1Н ЯМР (300 МГц, CDC13) δ 7,08 (1H, дд), 6,97-6,95 (2H, м), 3,31 (2H, т), 2,78 (2H, т), 1,93 (2H, м), 1,32 (12H, с).
Стадия 3:
Получение 3-(2-феноксиэтокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (27C)
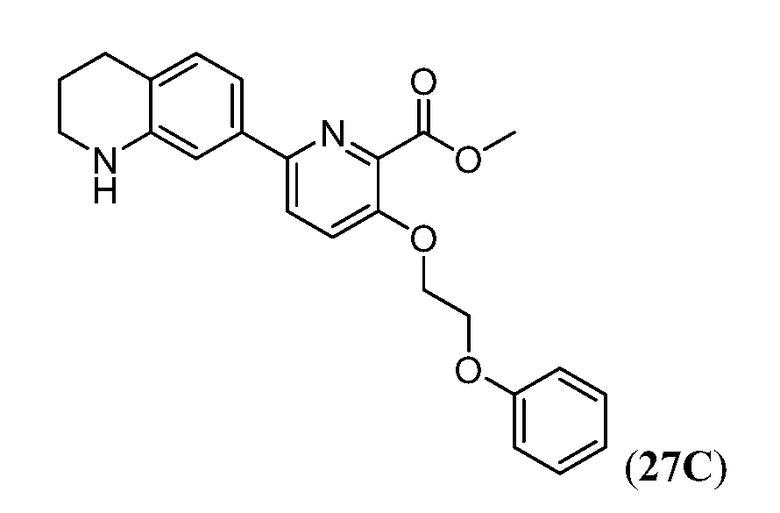
6-бром-3-(2-феноксиэтокси)метилпиколинат (27A) (327 мг, 0,93 ммоль), 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолин (27B) (181 мг, 0,7 ммоль), тетрабутиламмоний бромид (30 мг, 0,09 ммоль), дихлорбис(трифенилфосфин)палладий (II) (26 мг, 0,04 ммоль) и K2CO3 (322 мг, 2,33 ммоль) перемешивали в 1,4-диоксане (2 мл) и воде (1 мл) при 90°C в течение 45 минут. Реакционной смеси давали охладиться до температуры окружающей среды и разбавляли EtOAc (5 мл), промывали H2O, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE/EtOAc 9:1-7:3, получая 205 мг (72%) желаемого продукта 3-(2-феноксиэтокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (27C): 1Н ЯМР (300 МГц, CDC13) δ 7,69 (1H, м), 7,44 (1H, м), 7,27 (2H, м), 7,13 (2H, с), 6,94 (4H, м), 4,40 (2H, м), 4,34 (2H, м), 3,90 (3H, с), 3,27 (2H, т), 2,76 (2H, т), 1,92 (2H, м).
Стадия 4: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)метилпиколината (27D)

3-(2-феноксиэтокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (27C) (81 мг, 0,2 ммоль) и 4-нитрофенилбензо[d]тиазол-2-илкарбамат (19) (127 мг, 0,4 ммоль) нагревали с обратным холодильником в ацетонитриле (5 мл) в течение 24 часов. После охлаждения смесь концентрировали при пониженном давлении и очищали хроматографией на колонках с силикагелем с элюированием градиентом от РЕ (100%) до PE/EtOAc (4:6), получая 114 мг (97%) желаемого продукта 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)метилпиколината (27D). 1Н ЯМР (300 МГц, CDC13) δ 8,12 (1H, с), 7,85 (1H, д), 7,73 (1H, м), 7,44 (3H, м), 7,29 (4H, м), 7,09 (1H, м), 6,94 (3H, м), 4,44 (2H, м), 4,37 (2H, м), 3,88 (2H, м), 3,78 (3H, м), 2,74 (2H, м), 2,00 (2H, м).
Стадия 5: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)пиколиновой кислоты (27)
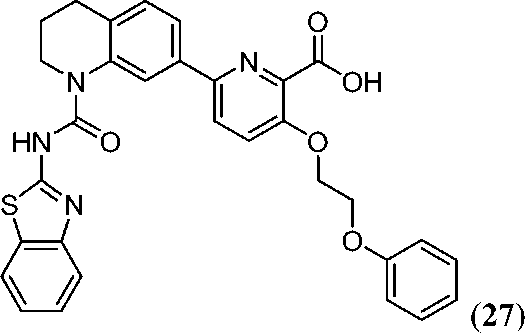
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)пиколиновую кислоту (27) получали в соответствии со следующей процедурой: смесь 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)метилпиколината (27D) (114 мг, 0,20 ммоль) и LiOH (18 мг, 0,75 ммоль) перемешивали в MeOH (3 мл) и воде (1 мл) в течение 20 часов. Смесь разбавляли простым диэтиловым эфиром (2 мл), и водный слой отделяли и подкисляли 1M HCl. Осадок отфильтровывали, промывали EtOAc и высушивали, получая 83 мг (75%) желаемого продукта 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)пиколиновой кислоты (27): 1Н ЯМР (300 МГц, DMSO-d6) δ 7,98 (1H, д), 7,77 (2H, д), 7,65 (1H, м), 7,38-7,18 (6H, м), 6,99-6,91 (4H, м), 4,46 (2H, м), 4,31 (2H, м), 3,93 (2H, м), 2,79 (2H, м), 1,90 (2H, м), LC/MS (APCI): м/z 567,2 (M+H).
Пример 28
Синтез 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-карбоновой кислоты (28):
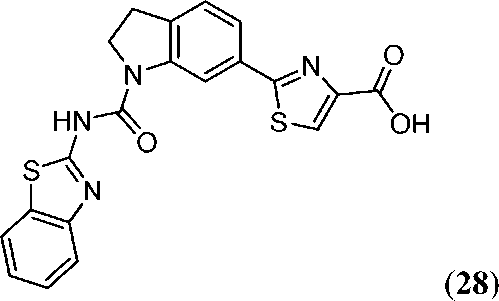
Стадия 1:
Получение 6-броминдолин-1-трет-бутилкарбоксилата (28A)
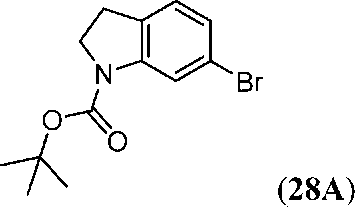
6-бром-индолин гидрохлорид (118 мг, 0,5 ммоль), (Вос)2O (137 мг, 0,63 ммоль) и K2CO3 (76 мг, 0,55 ммоль) перемешивали в MeOH (5 мл) в течение 72 часов. Смесь разбавляли DCM (10 мл), промывали водой, органический слой высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 156 мг (96%) желаемого продукта 6-броминдолин-1-трет-бутилкарбоксилата (28A): 1Н ЯМР (300 МГц, CDC13) δ 7,02 (3H, м), 3,97 (2H, т), 3,02 (2H, т), 1,53 (9H, с).
Стадия 2: Получение 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолин-1-трет-бутилкарбоксилата (28B)
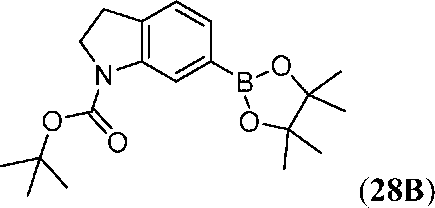
Смесь 6-броминдолин-1-трет-бутилкарбоксилата (28A) (156 мг, 0,52 ммоль), бис(пинаколято)дибора (173 мг, 0,68 ммоль), Pd(dppf)2Cl2 (9 мг, 0,012 ммоль 2,2 мол.%) и ацетата калия (154 мг, 1,57 ммоль) в толуоле (3 мл) нагревали до 150°C в микроволновом реакторе в течение 40 мин. После охлаждения смесь разбавляли DCM (5 мл), промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE/EtOAc (9:1), получая 69 мг (38%) желаемого продукта 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолин-1-трет-бутилкарбоксилата (28B): 1Н ЯМР (300 МГц, CDC13) δ 7,41 (2H, м), 7,12 (1H, м), 3,96 (2H, т), 3,09 (2H, т), 1,57 (9H, с), 1,30 (12H, с).
Стадия 3: Получение 2-(1-(трет-бутоксикарбонил)индолин-6-ил)тиазол-4-этилкарбоксилата (28C)
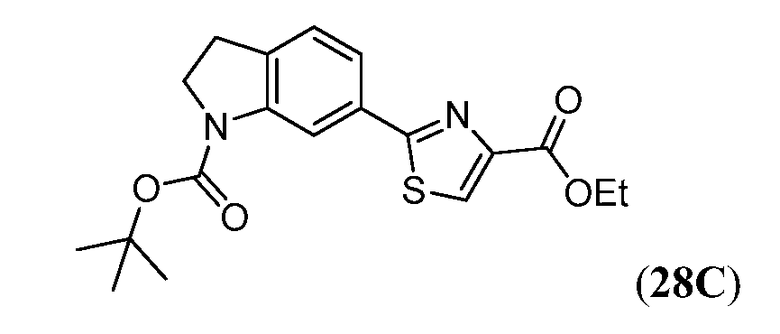
Смесь 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолин-1-трет-бутилкарбоксилата (28B) (69 мг, 0,2 ммоль), 2-бромтиазол-4-этилкарбоксилата (43 мг, 0,2 ммоль), K2CO3 (69 мг, 0,5 ммоль), тетрабутиламмоний бромида (6,0 мг, 0,02 ммоль) и дихлорбис(трифенилфосфин)палладия (II) (6,0 мг, 0,003 ммоль) в 1,4-диоксане (6 мл) и воде (1 мл) нагревали до 90°C в течение 24 часов. После охлаждения смесь экстрагировали DCM, промывали H2O, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE/EtOAc (9:1), получая 46 мг (61%) желаемого продукта 2-(1-(трет-бутоксикарбонил)индолин-6-ил)тиазол-4-этилкарбоксилата (28C): 1Н ЯМР (300 МГц, CDCl3) δ 8,13 (1H, с), 7,18 (1H, дд), 6,95 (1H, дд), 6,43 (1H, дд), 4,44 (2H, м), 3,99 (2H, м), 3,11 (1H, т), 2,97 (1H, т), 1,54 (9H, с), 1,45 (3H, т)/ LC/MS (APCI): м/z 375,0 (M+H).
Стадия 4:
Получение 2-(индолин-6-ил)тиазол-4-этилкарбоксилата (28D)
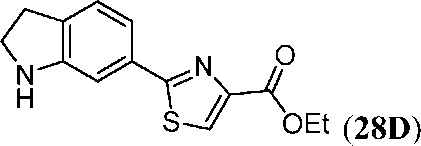
К 2-(1-(трет-бутоксикарбонил)индолин-6-ил)тиазол-4-этилкарбоксилату (28C) (46 мг, 0,12 ммоль) добавляли DCM (5 мл) и TFA (1 мл) и перемешивали в течение 2,5 часов. Смесь концентрировали при пониженном давлении, получая 56 мг желаемого продукта 2-(индолин-6-ил)тиазол-4-этилкарбоксилата (28D): LC/MS (APCI): m/z 275,0 (M+H).
Стадия 5:
Получение 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-этилкарбоксилата (28E)

Смесь 2-(индолин-6-ил)тиазол-4-этилкарбоксилата (28D) (56 мг, 0,14 ммоль), K2CO3 (22 мг, 0,16 ммоль) и 4-нитрофенилбензо[d]тиазол-2-илкарбамата (19) (48 мг, 0,15 ммоль) нагревали с обратным холодильником в ацетонитриле (3 мл) в течение 5,5 часов. Реакционную смесь охлаждали до температуры окружающей среды и разбавляли EtOAc (5 мл), промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE/EtOAc (1:1), получая 22 мг (34%) желаемого продукта 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-этилкарбоксилата (28E): 1Н ЯМР (300 МГц, CDC13) δ 8,67 (1H, с), 8,12 (1H, с), 7,68 (2H, м), 7,52 (1H, м), 7,38 (1H, м), 7,25 (2H, м), 4,43 (2H, кв), 4,22 (2H, м), 3,18 (2H, м) 1,45 (3H, т)/ LC/MS (APCI): м/z 451,1 (M+H).
Стадия 6: Получение целевого соединения 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-карбоновой кислоты (28)

Целевое соединение 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-карбоновую кислоту (28) получали в соответствии со следующей процедурой: смесь 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-этилкарбоксилата (28E) (22 мг, 0,05 ммоль) и LiOH (12 мг, 0,49 ммоль) перемешивали в MeOH (2 мл) и воде (1 мл) в течение 20 часов. Смесь разбавляли EtOAc (5 мл) и подкисляли 1M HCl. Органический слой отделяли, промывали H2O, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали ВЭЖХ, получая 3,0 мг (14%) желаемого продукта 2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-карбоновой кислоты (28). LC/MS (APCI): m/z 423,1 (M+H), 844,8 (2M+1).
Пример 29
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилпиколиновой кислоты (29):

Стадия 1:
Получение 3-(бензилокси)-6-бромметилпиколината (29A)

Бензил бромид (3,87 г, 22,6 ммоль) добавляли к суспензии 6-бром-3-гидроксиметилпиколината (5,00 г, 21,6 ммоль) и K2CO3 (5,96 г, 43,1 ммоль) в безводном DMF (40 мл). Реакционную смесь нагревали при 60°C в течение 18 часов, охлаждали до температуры окружающей среды и вливали в смесь лед/вода. После перемешивания в течение 2,5 часов осадок собирали фильтрацией, промывали водой и высушивали, получая желаемый продукт 3-(бензилокси)-6-бромметилпиколинат (29A).
Стадия 2:
Получение 3-(бензилокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29B)

К 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолину (27B) (1,16 г, 4,48 ммоль), 3-(бензилокси)-6-бромметилпиколинату (29A) (1,44 г, 4,48 ммоль), K2CO3 (1,546 г, 11,2 ммоль), тетрабутиламмоний бромиду (0,144 г, 0,45 ммоль) и дихлорбис(трифенилфосфин)палладию (II) (126 мг, 0,18 ммоль) добавляли 1,4-диоксан (30 мл) и воду (15 мл). Реакционную смесь нагревали до 90°C в течение 1,5 часов, охлаждали до температуры окружающей среды, концентрировали, разбавляли EtOAc, промывали последовательно водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал осаждали из смеси EtOAc/PE, получая продукт 3-(бензилокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (29B). Дополнительный материал отделяли от фильтрата хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 73:27-63:37, получая желаемый продукт (29B): 1Н ЯМР (300 МГц, CDC13) δ 7,67 (1H, д), 7,48-7,44 (2H, м), 7,43-7,31 (4H, м), 7,21 (1H, д), 7,16 (1H, дд), 7,01 (1H, д) 5,21 (2H, с), 3,98 (3H, с), 3,34 (2H, м), 2,79 (2H, т), 1,96 (2H, м).
Стадия 3:
Получение 7-(5-(бензилокси)-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29C)

Смесь 3-(бензилокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29B) (3,41 г, 9,10 ммоль) и ди-трет-бутил бикарбоната (5,96 г, 27,3 ммоль) нагревали до 100°C в течение 17 часов, охлаждали до температуры окружающей среды, разбавляли DCM, промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 83:17-65:35, получая желаемый продукт 7-(5-(бензилокси)-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилат (29C): 1Н ЯМР (300 МГц, CDC13) δ 8,27 (1H, д), 7,75 (1H, д), 7,58 (1H, дд), 7,49-7,30 (6H, м), 7,13 (1H, д), 5,23 (2H, с), 3,99 (3H, с), 3,73 (2H, м), 2,79 (2H, т), 1,94 (2H, м), 1,55 (9H, с).
Стадия 4:
Получение 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29D)

К раствору 7-(5-(бензилокси)-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29C) (2,72 г, 5,74 ммоль) в EtOAc (80 мл) и EtOH (80 мл) и ледяной уксусной кислоте (~30 капель) добавляли 10%-ый Pd-C (420 мг). Реакционную смесь перемешивали под атмосферой водорода в течение 24 часов, фильтровали через слой Целита и концентрировали, получая желаемый продукт 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилат (29D): 1Н ЯМР (300 МГц, CDC13) δ 10,67 (1H, с), 8,30 (1H, д), 7,86 (1H, д), 7,56 (1H, дд), 7,42 (1H, д), 7,15 (1H, д), 4,05 (3H, с), 3,74 (2H, м), 2,80 (2H, т), 1,95 (2H, м), 1,56 (9H, с).
Стадия 5: Получение 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29E)
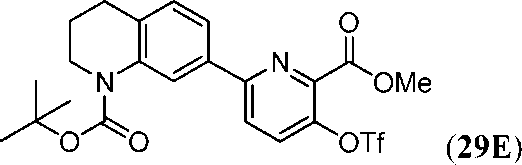
Ангидрид трифторметансульфоновой кислоты (1,93 г, 6,86 ммоль) добавляли по каплям при 0°C к 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилату (29D) (2,40 г, 6,23 ммоль) и ТЕА (0,95 г, 9,35 ммоль) в безводном DCM (25 мл). Реакционную смесь перемешивали при 0°C в течение 1,5 часов, разбавляли DCM, промывали 10%-ной лимонной кислотой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 88:12-82:18, получая 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилат (29E): 1Н ЯМР (300 МГц, CDC13) δ 8,40 (1H? д), 7,95 (1H, д), 7,71 (1H, д), 7,66 (1H, дд), 7,19 (1H, д), 4,04 (3H, с), 3,75 (2H, м), 2,82 (2H, т), 1,96 (2H, м), 1,56 (9H, с).
Стадия 6: Получение 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколината (29F).

Трифторуксусную кислоту (18 мл) добавляли к 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилату (29E) (2,00 г, 3,9 ммоль) в безводном DCM (25 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1,5 часов, концентрировали при пониженном давлении, разбавляли DCM и промывали насыщенным NaHCO3. Водную фазу экстрагировали DCM. Объединенные органические фазы высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая желаемый продукт 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколинат (29F) с достаточной чистотой для последующего использования. 1Н ЯМР (300 МГц, CDC13) δ 7,88 (1H д), 7,66 (1H, д), 7,39 (1H, д), 7,29 (1H, дд), 7,08 (1H, д), 4,03 (3H, с), 3,39 (2H, м), 2,83 (2H, т), 2,02 (2H, м).
Стадия 7: Получение 7-(6-(метоксикарбонил)-5-фенилпиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29G)

7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилат (29E) (40 мг, 0,077 ммоль), фенилбороновую кислоту (11 мг, 0,093 ммоль, 1,2 экв.), K2CO3 (27 мг, 0,19 ммоль), тетрабутиламмоний бромид (2,5 мг, 0,0077 ммоль) и дихлорбис(трифенилфосфин)палладий (II) (2,2 мг, 0,003 ммоль) в 1,4-диоксане (0,5 мл) и воде (0,25 мл) нагревали до 90°C в течение 75 минут, охлаждали до температуры окружающей среды, разбавляли EtOAc, промывали водой, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 88:12-82:18, получая желаемый продукт 7-(6-(метоксикарбонил)-5-фенилпиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилат (29G): 1Н ЯМР (300 МГц, CDC13) δ 8,41 (1H, д), 7,88 (1H, д), 7,80 (1H, д), 7,71 (1H, дд), 7,48-7,37 (5H, м), 7,19 (1H, д), 3,80-3,70 (5 H, м), 2,82 (2H, т), 1,96 (2H, м), 1,57 (9H, с).
Стадия 8: Получение 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н)

3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (29Н) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 7-(6-(метоксикарбонил)-5-фенилпиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилат (29G) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29E) на стадии 6 примера 29.
Стадия 9: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколината (29I).

Смесь 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н) (17 мг, 0,049 ммоль) и 4-нитрофенилбензо[d]тиазол-2-илкарбамата (19) (15,5 мг, 0,049 ммоль) в безводном CH3CN (0,8 мл) нагревали с обратным холодильником в течение 6 часов, охлаждали до температуры окружающей среды, концентрировали при пониженном давлении, и сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 72:28-55:45, получая желаемый продукт 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколинат (29I): 1Н ЯМР (300 МГц, CDCl3) δ 8,23 (1H, д), 8,13 (1H, д), 7,85-7,76 (3H, м), 7,66 (1H, д), 7,48-7,35 (4H, м), 7,34-7,28 (2H, м), 6,89 (1H, д), 3,98 (2H, т), 3,71 (3H, с), 2,85 (2H, т), 2,09 (2H, м).
Стадия 10: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилпиколиновой кислоты (29)
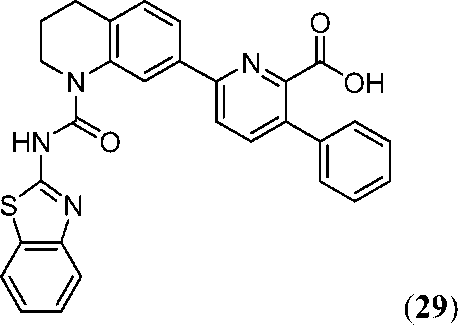
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилпиколиновую кислоту (29) получали в соответствии со следующей процедурой: К 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколинату (29I) (23 мг, 0,044 ммоль) в MeOH (0,55 мл) и воде (0,11 мл) добавляли водный раствор LiOH (0,066 мл 2,0 м., 0,13 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 15 часов, концентрировали при пониженном давлении, чтобы удалить MeOH, и разбавляли водой. Устанавливали рН~4 с помощью 2М HCl, и полученный осадок отделяли фильтрацией и промывали водой. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом DCM:MeOH 98:2-92:8, получая желаемый продукт 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилпиколиновую кислоту (29): MS (ESI (+)): m/z 507,1 (M+H).
Пример 30
Синтез: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-трет-бутилфенил)пиколиновой кислоты (30):
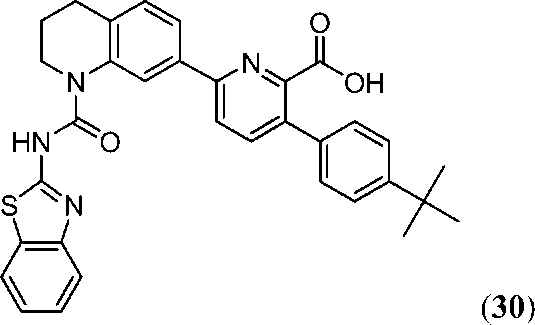
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-трет-бутилфенил)пиколиновую кислоту (30) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-трет-бутилфенил)пиколиновую кислоту (30) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 4-трет-бутилфенилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,43 (1H, с), 7,94-7,85 (2H, м), 7,81 (1H, д), 7,76 (1H, дд), 7,55-7,42 (5H, м), 7,37 (1H, дт), 7,28 (1H, д), 7,19 (1H, дт), 3,96 (2H, т), 2,83 (2H, т), 1,94 (2H, м), 1,32 (9H, с). MS (ESI(+)): м/z 563,2 (M+H).
Пример 31
Синтез: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(нафталин-2-ил)пиколиновой кислоты (31):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(нафталин-2-ил)пиколиновую кислоту (31) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(нафталин-2-ил)пиколиновую кислоту (31) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 2-нафталинебороновая кислота использовалась вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,47 (1H, с), 8,15-7,95 (5 H, м), 7,82 (2 H, м), 7,65-7,55 (3H, м), 7,48 (1H, д), 7,40-7,27 (2H, м), 7,26-7,17 (2H, м), 3,98 (2H, т), 2,85 (2H, т), 1,96 (2H, м). MS (ESI(+)) м/z 557,0 (M+H).
Пример 32
Синтез: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-6'-(4-метилпиперазин-1-ил)-3,3'-бипиридин-2-карбоновой кислоты (32):
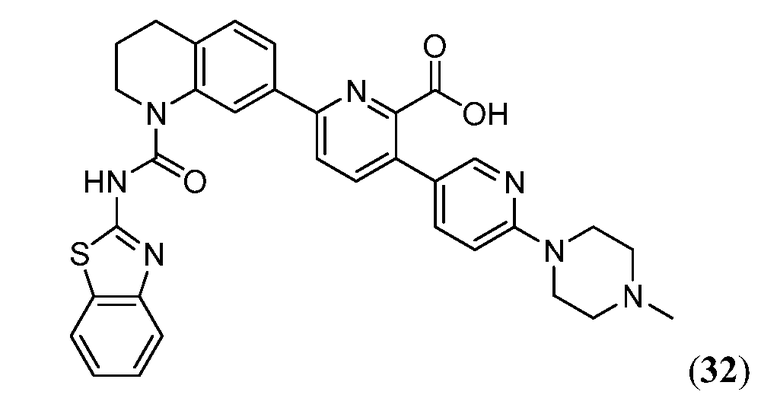
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-6'-(4-метилпиперазин-1-ил)-3,3'-бипиридин-2-карбоновую кислоту (32) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-6'-(4-метилпиперазин-1-ил)-3,3'-бипиридин-2-карбоновую кислоту (32) получали согласно процедуре, подобной описанной в примере 29, кроме того, что пинаколовый эфир 2-(4-метилпиперазин-1-ил)пиридин-5-бороновой кислоты использовался вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,52 (1H, с), 8,37 (1H, д), 7,86 (1H, дд), 7,76-7,62 (4H, м), 7,46 (1H, д), 7,30 (1H, дт), 7,21 (1H, д), 7,12 (1H, дт), 6,82 (1H, д), 3,96 (2H, т), 3,60-3,40 (8H, м), 2,81 (2H, т), 2,22 (3H, с), 1,92 (2H, м). MS (ESI(+)): м/z 606,0 (M+H).
Пример 33
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1Н-индол-5-ил)пиколиновой кислоты (33):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1Н-индол-5-ил)пиколиновую кислоту (33) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1Н-индол-5-ил)пиколиновую кислоту (33) получали согласно процедуре, подобной описанной в примере 29, кроме того, что пинаколовый эфир 1-трет-бутоксикарбонилиндол-5-бороновой кислоты использовался вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 11,15 (1H, с), 8,45 (1H, с), 7,90-7,71 (5H, м), 7,50-7,16 (7H, м), 6,46 (1H, с), 3,96 (2H, м), 2,83 (2H, т), 1,95 (2H, м). MS (ESI(+)): м/z 546,0 (M+H).
Пример 34
Синтез 3-(3-((3-аминофенокси)метил)фенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (34):
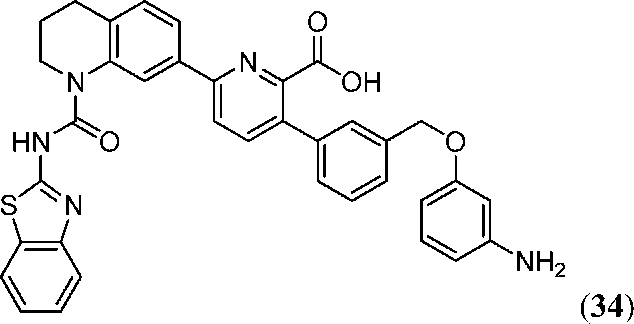
Стадия 1:
Получение 3-(3-бромбензилокси)фенилтрет-бутилкарбамата (34A)

К 3-гидроксифенил-трет-бутилкарбамату (0,245 г, 1,17 ммоль), K2CO3 (404 мг, 2,93 ммоль) и 3-бромбензил бромиду (322 мг, 1,29 ммоль) добавляли безводный DMF (5 мл). Реакционную смесь нагревали до 60°C в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды, вливали в воду, и полученный осадок собирали фильтрацией и промывали водой, получая желаемый продукт 3-(3-бромбензилокси)фенил-трет-бутилкарбамат (34A) с достаточной чистотой для последующего использования.
Стадия 2:
Получение 3-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилокси)фенил-трет-бутилкарбамата (34B)
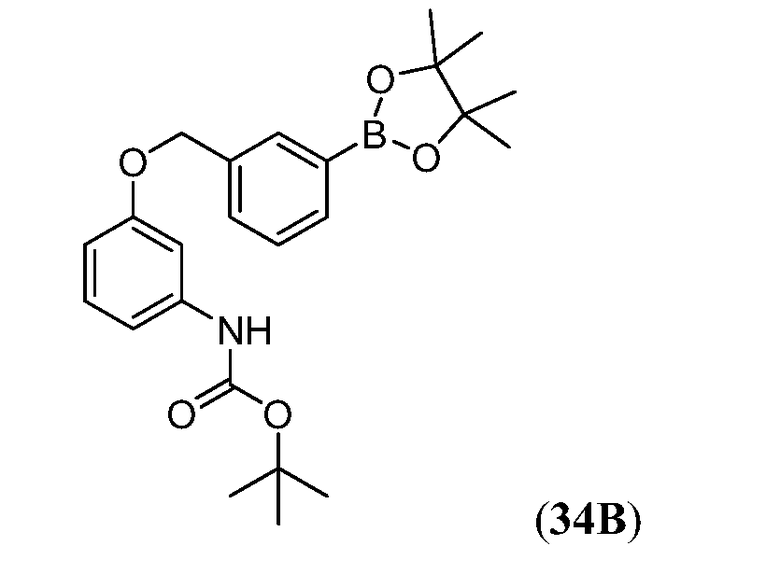
3-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилокси)фенил-трет-бутилкарбамат (34B) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(3-бромбензилокси)фенил-трет-бутилкарбамат (34A) использовался вместо 7-бром-1,2,3,4-тетрагидрохинолина (1Е) на стадии 2 примера 27.
Стадия 3:
Получение 3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил) фенил)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (34C)
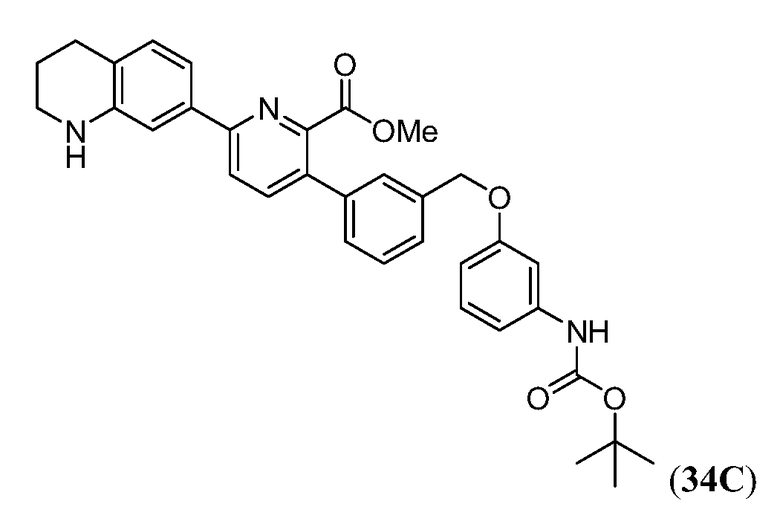
3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)-6-(1,2,3,4-тетрагидрохинолин-7-ил)пиколинат (34C) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколинат (29F) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилата (29E) и 3-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилокси)фенил-трет-бутилкарбамат (34B) использовался вместо фенилбороновой кислоты на стадии 7 примера 29.
Стадия 4: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)метилпиколината (34D)

6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)метилпиколинат (34D) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (34C) использовался вместо 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н) на стадии 9 примера 29.
Стадия 5:
Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино) фенокси)метил)фенил)пиколиновой кислоты (34E)
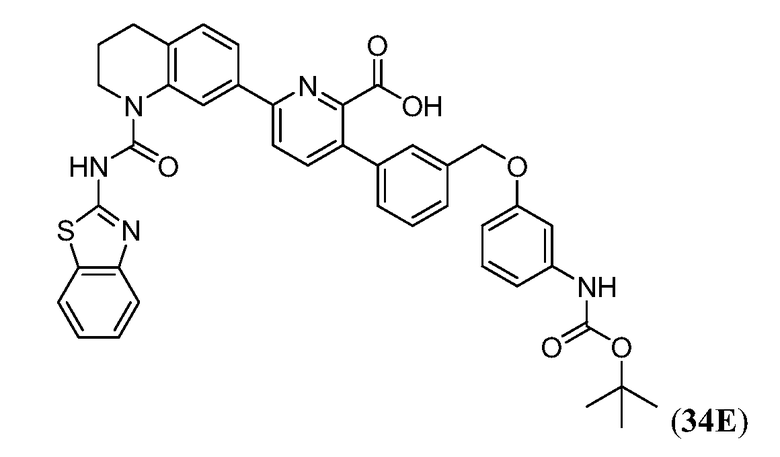
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)пиколиновую кислоту (34E) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)метилпиколинат (34D) использовался вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколината (29I) на стадии 10 примера 29.
Стадия 6: Получение целевого соединения 3-(3-((3-аминофенокси)метил)фенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (34)
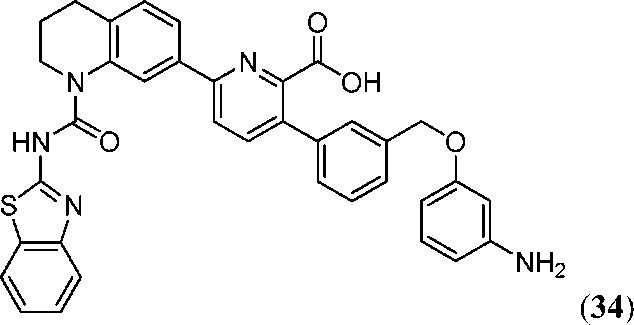
Целевое соединение 3-(3-((3-аминофенокси)метил)фенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (34) получали в соответствии со следующей процедурой: Трифторуксусную кислоту (0,5 мл) добавляли к раствору 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)пиколиновой кислоты (34E) (0,016 г, 0,022 ммоль) в безводном DCM (0,75 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1,5 часов, концентрировали при пониженном давлении, и сырой материал растирали в простом диэтиловом эфире, получая желаемый продукт 3-(3-((3-аминофенокси)метил)фенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (34): 1Н ЯМР (300 МГц, DMSO) δ 8,43 (1H, с, шир.), 8,08 (1H, д), 7,99 (1H, д), 7,85-7,77 (2H, м), 7,67 (1H, с), 7,55-7,41 (4H, м), 7,37 (1H, т), 7,31 (1H, д), 7,22 (1H, т), 7,12 (1H, т), 6,59-7,50 (2H, м), 6,47 (1H, д), 5,11 (2H, с), 3,97 (2H, т), 2,84 (2H, т), 1,96 (2H, м). MS (ESI(+)) м/z 628,1 (М+Н).
Пример 35
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1-бензил-1Н-пиразол-4-ил)пиколиновой кислоты (35):
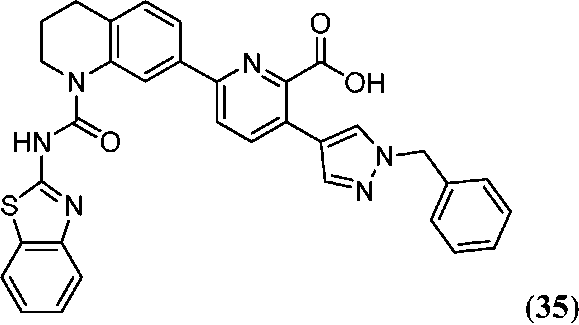
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1-бензил-1H-пиразол-4-ил)пиколиновую кислоту (35) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1-бензил-1Н-пиразол-4-ил)пиколиновую кислоту (35) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколинат (29F) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси) пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилата (29E) и пинаколовый эфир 1-бензил-1Н-пиразол-4-бороновой кислоты использовался вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,38 (1H, с, шир.), 8,19 (1H, с), 8,04 (1H, д), 7,95 (1H, д), 7,85-7,78 (2H, м), 7,75 (1H, дд), 7,47 (1H, д, шир.), 7,40-7,18 (8H, м), 5,38 (2H, с), 3,95 (2H, т), 2,82 (2H, т), 1,94 (2H, м), MS(ESI(+)): м/z 587,1 (M+H).
Пример 36
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-o-толилпиколиновая кислота (36):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-o-толилпиколиновую кислоту (36) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-o-толилпиколиновую кислоту (36) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколинат (29F) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилата (29E), и 2-метилфенилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,45 (1H, с, шир.), 8,05 (1H, д), 7,86-7,78 (3H, м), 7,47 (1H, д, щир.), 7,37 (1H, дт), 7,33-7,18 (5H, м), 7,14 (1H, д), 3,98 (2H, т), 2,85 (2H, т), 2,13 (3H, с), 1,96 (2H, м). MS(ESI(+)): м/z 521,1 (M+H).
Пример 37
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-метил-4-оксо-3,4-дигидрохиназолин-6-ил)пиколиновой кислоты (37):

Стадия 1:
Получение N-ацетил-N-(4-бром-2-цианофенил)ацетамида (37A)
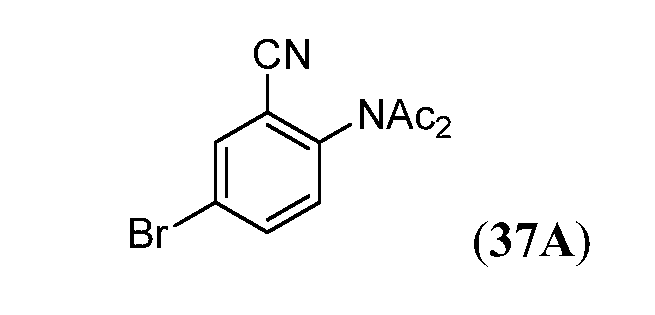
4-Диметиламинопиридин (6,2 мг, 0,05 ммоль) добавляли к раствору 2-амино-5-бромбензонитрила (50 мг, 0,25 ммоль) в пиридине (1 мл) и уксусном ангидриде (0,26 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов, концентрировали, разбавляли EtOAc, промывали 1М HCl, насыщенным NaHCO3, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием смесью PE:EtOAc 3:1, получая желаемый продукт N-ацетил-N-(4-бром-2-цианофенил)ацетамида (37A).
Стадия 2: Получение N-(2-циано-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетамида (37B)

N-(2-циано-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетамид (37B) получали согласно процедуре, подобной описанной в примере 29, кроме того, что N-ацетил-N-(4-бром-2-цианофенил)ацетамид (37A) использовался вместо 7-бром-1,2,3,4-тетрагидрохинолина (1Е) на стадии 2 примера 27.
Стадия 3: Получение 3-(4-ацетамидо-3-цианофенил)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (37C)

3-(4-ацетамидо-3-цианофенил)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (37C) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколинат (29F) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолина-1(2Н)-трет-бутилкарбоксилата (29E), и N-(2-циано-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетамид (37B) использовался вместо фенилбороновой кислоты на стадии 7 примера 29.
Стадия 4: Получение 3-(4-ацетамидо-3-цианофенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (37D)

3-(4-ацетамидо-3-цианофенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (37D) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(4-ацетамидо-3-цианофенил)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (37C) использовался вместо 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н) на стадии 9 примера 29.
Стадия 5: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-метил-4-оксо-3,4-дигидрохиназолин-6-ил)пиколиновой кислоты (37)
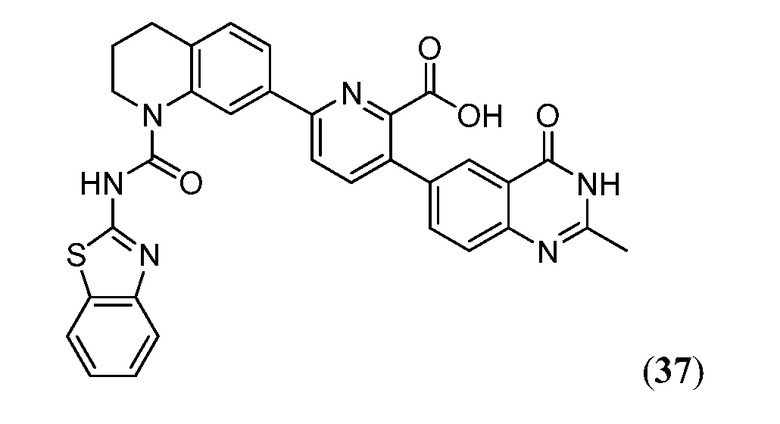
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-метил-4-оксо-3,4-дигидрохиназолин-6-ил)пиколиновую кислоту (37) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-метил-4-оксо-3,4-дигидрохиназолин-6-ил)пиколиновую кислоту (37) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(4-ацетамидо-3-цианофенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (37d) использовался вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколината (29I) на стадии 10 примера 29, и реакционную смесь нагревали до 50°C в течение 4 дней. MS (ESI (+)): m/z 589,0 (M+H).
Пример 38
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(метилсульфонил)фенил)пиколиновой кислоты (38):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(метилсульфонил)фенил)пиколиновую кислоту (38) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(метилсульфонил)фенил)пиколиновую кислоту (38) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1,2,3,4-тетрагидрохинолин-7-ил)-3-(трифторметилсульфонилокси)метилпиколинат (29F) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилата (29E), и 3-(метилсульфонил)фенилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,47 (1H, с), 8,07-8,00 (3H, м), 7,95 (1H, дт), 7,89 (1H, д), 7,84-7,79 (2H, м), 7,74 (1H, т), 7,48 (1H, д), 7,37 (1H, дт), 7,30 (1H, д), 7,22 (1H, дт), 3,97 (2H, т), 3,26 (3H, с), 2,84 (2H, т), 1,95 (2H, м), MS(ESI(+)): м/z 585,0 (M+H).
Пример 39
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)пиколиновой кислоты (39):

Стадия 1:
Получение 2-(3-(диметиламино)фенокси)этилацетата (39A)
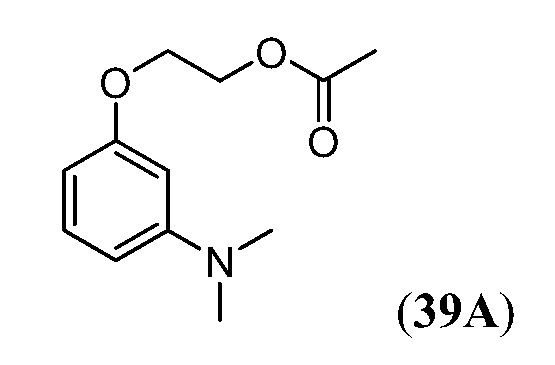
2-бромэтил ацетат (1,34 г, 4,01 ммоль) добавляли к суспензии 3-(диметиламино)фенола (0,50 г, 3,64 ммоль) и K2CO3 (1,5 г, 10,8 ммоль) в безводном DMF (3 мл). Реакционную смесь нагревали до 80°C в течение 18 часов, охлаждали до температуры окружающей среды, разбавляли водой, экстрагировали DCM. Органические фазы промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 90:10-80:20, получая желаемое соединение 2-(3-(диметиламино)фенокси)этилацетат (39A).
Стадия 2:
Получение 2-(3-(диметиламино)фенокси)этанола (39B)

Каталитическое количество метилата натрия (достаточное для получения основного раствора) добавляли к раствору 2-(3-(диметиламино)фенокси)этилацетата (39A) (0,545 г, 2,44 ммоль) в безводном MeOH (7 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 2,5 часов, после чего добавляли небольшое количество кислой смолы (Dowex 50WX8-200). Реакционную смесь фильтровали, концентрировали при пониженном давлении, и сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 66:34-50:50, получая желаемое соединение 2-(3-(диметиламино)фенокси)этанол (39B).
Стадия 3:
Получение 6-бром-3-(2-(3-(диметиламино)фенокси)этокси) метилпиколината (39C)

Связанный с полистиролом PPh3 (0,77 г в 1 ммоль/г, 0,77 ммоль, 1,5 экв.) добавляли к раствору 6-бром-3-гидроксиметилпиколината (0,119 г, 0,51 ммоль) и 2-(3-(диметиламино)фенокси)этанола (39B) (0,093 г, 0,51 ммоль) в сухом DCM (5 мл). К этой смеси по каплям добавляли диэтил азодикарбоксилат (0,134 мг, 0,77 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 4 часов, фильтровали, чтобы удалить смолу, и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 75:25-60:40, получая желаемый продукт 6-бром-3-(2-(3-(диметиламино)фенокси)этокси)метилпиколинат (39C).
Стадия 4: Получение 3-(2-(3-(диметиламино)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (39D)

3-(2-(3-(диметиламино)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (39D) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-бром-3-(2-(3-(диметиламино)фенокси)этокси)метилпиколинат (39C) использовался вместо 3-(бензилокси)-6-бромпиколината (29A) на стадии 2 примера 29.
Стадия 5: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)метилпиколината (39E)

6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)метилпиколинат (39E) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(2-(3-(диметиламино)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (39D) использовался вместо 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н) на стадии 9 примера 29.
Стадия 6: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)пиколиновая кислота (39)

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)пиколиновую кислоту (39) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)пиколиновую кислоту (39) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)метилпиколинат (39E) использовался вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколината (29I) на стадии 10 примера 29. 1Н ЯМР (300 МГц, CDC13) δ 8,20 (1H, с), 7,84 (1H, д), 7,69 (1H, д), 7,40-7,10 (5H, м), 6,92 (1H, с, шир.), 6,40-6,28 (3H, м), 4,53 (2H, т), 4,42 (2H, т), 3,82 (2H, с, шир.), 2,92 (6H, с), 2,69 (2H, т), 1,96 (2H, м), MS (ESI(+)) м/z 610,0 (M+H).
Пример 40
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(3-(диметиламино)фенокси)пропокси)пиколиновой кислоты (40):
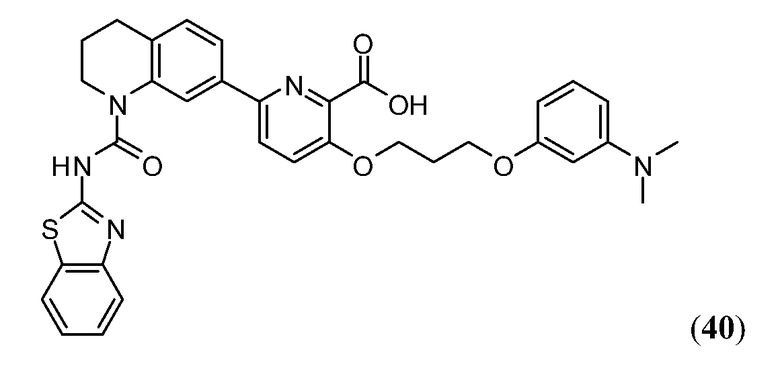
Стадия 1:
Получение 6-бром-3-(4-метоксибензилокси)метилпиколината (40A)

4-метоксибензил бромид (0,095 г, 0,47 ммоль) добавляли по каплям к суспензии 6-бром-3-гидроксиметилпиколината (26B) (0,100 г, 0,43 ммоль) и K2CO3 (0,089 г, 0,65 ммоль) в безводном ацетоне (2,5 мл). Реакционную смесь нагревали с обратным холодильником в течение 3,5 часов, охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Добавляли воду, и смесь экстрагировали DCM. Органические фазы промывали 1М NaOH, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Концентрат очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc от 84:16 до 72:28, получая желаемый продукт 6-бром-3-(4-метоксибензилокси)метилпиколинат (40A).
Стадия 2: Получение 3-(4-метоксибензилокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (40B)
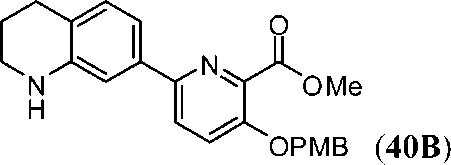
3-(4-метоксибензилокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)пиколинат (40B) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-бром-3-(4-метоксибензилокси)метилпиколинат (40A) использовался вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29.
Стадия 3: Получение 3-гидрокси-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (40C)
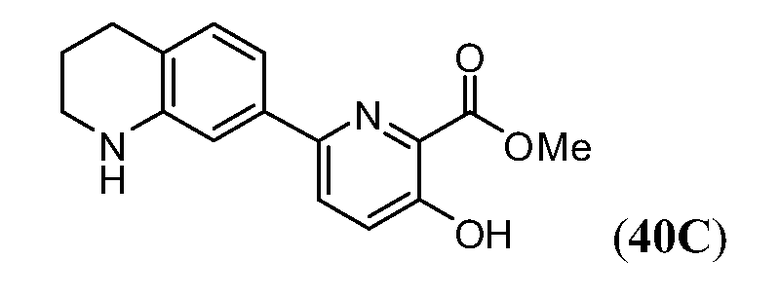
3-гидрокси-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (40C) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(4-метоксибензилокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (40B) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29E) на стадии 6 примера 29.
Стадия 4:
Получение 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29D)

Раствор ди-трет-бутил бикарбоната (0,057 г, 0,26 ммоль) и 3-гидрокси-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (40C) (0,067 г, 0,24 ммоль) в сухом THF (3 мл) нагревали с обратным холодильником в течение 17 часов, охлаждали до температуры окружающей среды, разбавляли с EtOAc и промывали насыщенным NaHCO3. Водную фазу экстрагировали EtOAc, и объединенные органические фазы высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc от 88:12 до 75:25, получая желаемый продукт 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4 дигидрохинолина-1(2H)-трет-бутилкарбоксилат (29D).
Стадия 5:
Получение 3-(3-(диметиламино)фенокси)пропилацетата (40D)
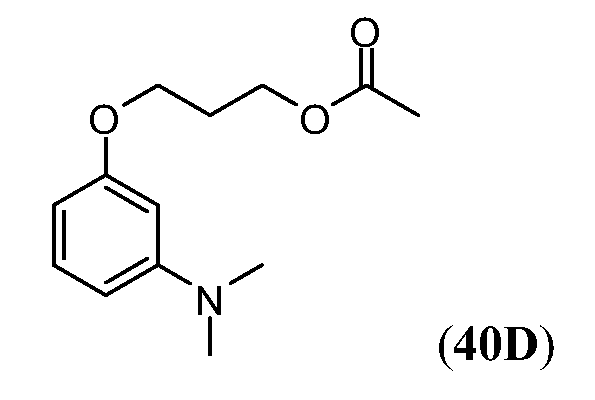
3-(3-(Диметиламино)фенокси)пропилацетат (40D) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 2-бромпропил ацетат использовался вместо 2-бромэтил ацетата на стадии 1 примера 39.
Стадия 6:
Получение 3-(3-(диметиламино)фенокси)пропан-1-ола (40E)

3-(3-(диметиламино)фенокси)пропан-1-ол (40E) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(3-(диметиламино)фенокси)пропилацетат (40D) использовался вместо 2-(3-(диметиламино)фенокси)этилацетат (39A) на стадии 2 примера 39.
Стадия 7: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(3-(диметиламино)фенокси)пропокси)пиколиновой кислоты (40)

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(3-(диметиламино)фенокси)пропокси)пиколиновую кислоту (40) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(3-(диметиламино)фенокси)пропокси)пиколиновую кислоту (40) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 7-(5-гидрокси-6-(метоксикарбонил) пиридин-2-ил)-3,4 дигидрохинолина-1(2H)-трет-бутилкарбоксилат (29D) использовался вместо 6-бром-3-гидроксиметилпиколината (26B), и 3-(3-(диметиламино)фенокси)пропан-1-ол (40E) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39. 1Н ЯМР (300 МГц, CDC13) δ 8,12 (1H, с), 7,82 (1H, д), 7,71 (1H, д), 7,50 (1H, д), 7,40-7,30 (2H, м), 7,26-7,09 (3H, м), 6,97 (1H, с, шир.), 6,38-6,29 (3H, м), 4,35 (2H, т), 4,27 (2H, т), 3,79 (2H, с, шир.), 2,92 (6H, с), 2,68 (2H, т), 2,36 (2H, м), 1,92 (2H, м). MS (ESI(+)): м/z 624,1 (M+H).
Пример 41
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2Н)-ил)этокси)пиколиновой кислоты (41):
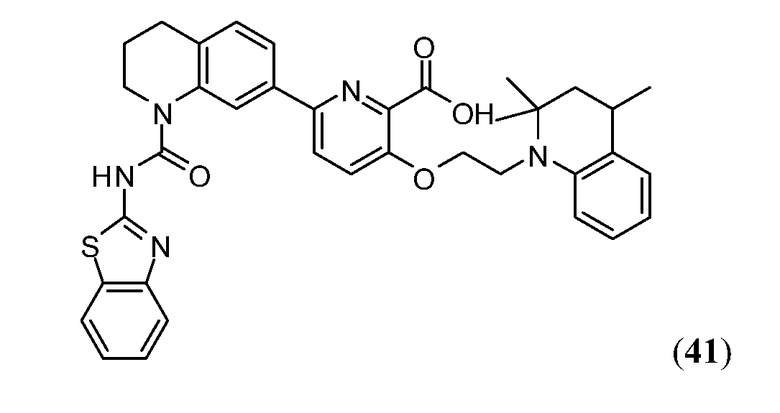
Стадия 1: Получение 6-бром-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2Н)-ил)этокси)пиколиновой кислоты (41A)
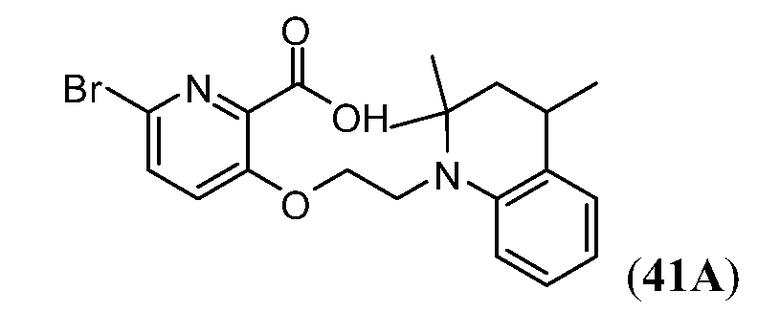
6-бром-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2H)-ил)этокси)пиколиновую кислоту (41A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 1-(2-гидроксиэтил)-1,2,3,4-тетрагидро-2,2,4,7-тетраметилхинолин использовался вместо 2-(3-(диметиламино фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 2: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2H)-ил)этокси)пиколиновой кислоты (41)

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2H)-ил)этокси)пиколиновую кислоту (41) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2H)-ил)этокси)пиколиновую кислоту (41) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 6-бром-3-(2-(2,2,4-триметил-3,4-дигидрохинолин-1(2H)-ил)этокси)пиколиновая кислота (41A) использовалась вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39. 1Н ЯМР (300 МГц, DMSO) δ 8,31 (1H, с), 7,79 (1H, д), 7,74 (1H, д), 7,62 (1H, дд), 7,55-7,45 (2H, м), 7,35 (1H, дт), 7,22-7,16 (2H, м), 6,96 (1H, д), 6,42-6,38 (2H, м), 4,12 (2H, м), 3,93 (2H, т), 3,74 (2H, м), 3,41 (2H, м), 2,79 (2H, т), 2,18 (3H, с), 1,90 (2H, м), 1,76 (1H, м), 1,30 (3H, с), 1,22 (3H, д), 1,12 (3H, с).
Пример 42
Синтез 3-(2-(3-аминофенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (42):
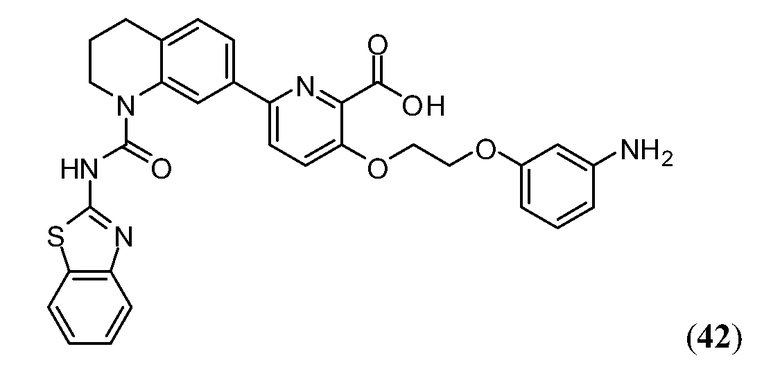
Стадия 1:
Получение 3-гидроксифенил-трет-бутилкарбамата (42A)
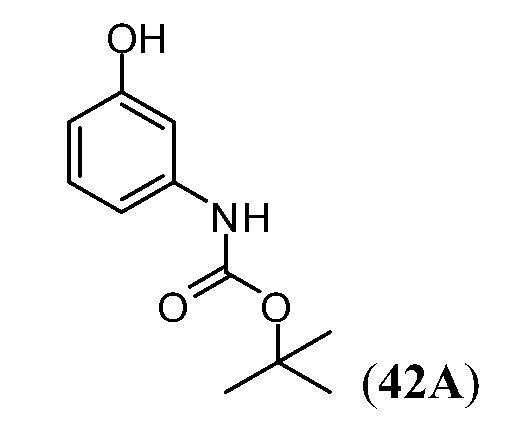
3-гидроксифенил-трет-бутилкарбамат (42A) получали согласно процедуре, подобной описанной в примере 40, кроме того, что 3-аминофенол использовался вместо 3-гидрокси-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (40C) на стадии 4 примера 40.
Стадия 2:
Получение 2-(3-(трет-бутоксикарбониламино)фенокси) этилбензоата (42B)

2-(3-(трет-бутоксикарбониламино)фенокси)этилбензоат (42B) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-гидроксифенил-трет-бутилкарбамат (42A) использовался вместо 3-(диметиламино)фенола и 2-бромэтилбензоат использовался вместо 2-бромэтилацетата на стадии 1 примера 39.
Стадия 3: Получение 3-(2-гидроксиэтокси)фенил-трет-бутилкарбамата (42C)
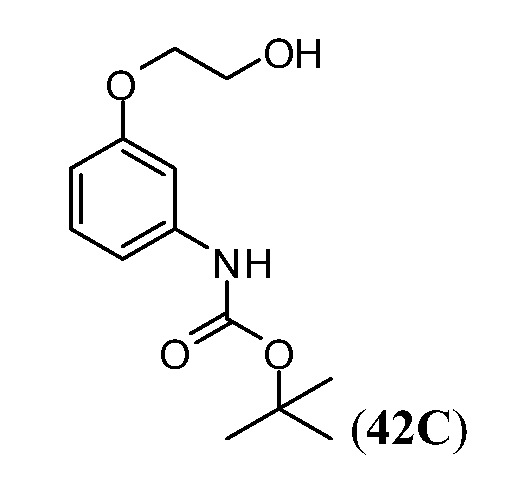
3-(2-гидроксиэтокси)фенил-трет-бутилкарбамат (42C) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 2-(3-(трет-бутоксикарбониламино)фенокси)этилбензоат (42B) использовался вместо 2-(3-(диметиламино)фенокси)этилацетата (39A) на стадии 2 примера 39.
Стадия 4:
Получение 6-бром-3-(2-(3-(трет-бутоксикарбониламино)фенокси) этокси)метилпиколината (42D)

6-бром-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)пиколинат (42D) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(2-гидроксиэтокси)фенил-трет-бутилкарбамат (42C) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 5:
Получение 3-(2-(3-(трет-бутоксикарбониламино)фенокси) этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (42E)
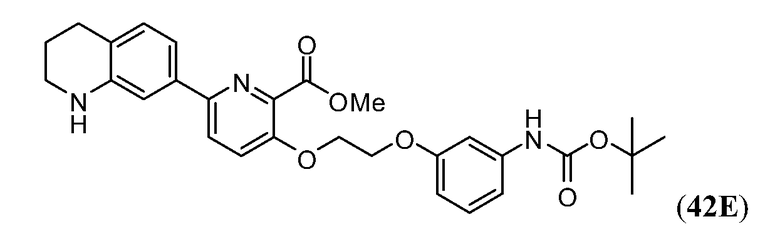
3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (42E) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-бром-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси) пиколинат (42D) использовался вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29.
Стадия 6: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)метилпиколината (42F)

6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)метилпиколинат (42F) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(2-{3-{трет-бутоксикарбониламино)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (42E) использовался вместо 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н) на стадии 9 примера 29.
Стадия 7: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновой кислоты (42G).
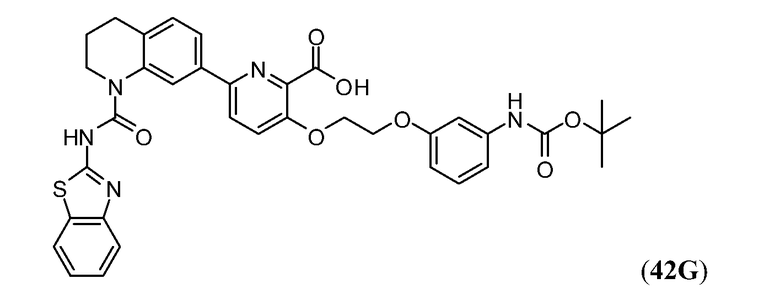
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновую кислоту (42G) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)метилпиколинат (42F) использовался вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколината (29I) на стадии 10 примера 29. 1Н ЯМР (300 МГц, CDCl3) δ 8,19 (1H, с), 7,85 (1H, д), 7,70 (1H, дд), 7,60 (1H, д), 7,35-7,13 (5H, м), 6,97-6,85 (3H, м), 6,62 (1H, дд), 4,52 (2H, т), 4,44 (2H, т), 3,81 (2H, с, шир.), 2,68 (2H, т), 1,96 (2H, м), 1,48 (9H, с).
Стадия 8: Получение целевого соединения 3-(2-(3-аминофенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (42)

Целевое соединение 3-(2-(3-аминофенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (42) получали в соответствии со следующей процедурой: соединение 3-(2-(3-аминофенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (42) получали согласно процедуре, подобной описанной в примере 34, кроме того, что 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновая кислота (42G) использовалась вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)пиколиновой кислоты (34E) на стадии 6 примера 34. 1Н ЯМР (300 МГц, DMSO) δ 8,29 (1H, с), 7,97 (1H, д), 7,81 (1H, д), 7,77 (1H, д), 7,68 (1H, дд), 7,46 (1H, м, шир.), 7,37 (1H, дт), 7,29-7,12 (3H, м), 6,61-6,50 (3H, м), 4,47 (2H, т), 4,28 (2H, т), 3,94 (2H, т), 2,81 (2H, т), 1,94 (2H, м). MS (ESI(+)): м/z 582,1 (M+H).
Пример 43
Синтез 3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (43):

Стадия 1:
Получение 3-(бензо[d][1,3]диоксол-5-илокси)пропилацетата (43A)

3-(бензо[d][1,3]диоксол-5-илокси)пропилацетат (43A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что сезамол использовался вместо 3-(диметиламино)фенола, и 2-бромпропил ацетат использовался вместо 2-бромэтил ацетата на стадии 1 примера 39.
Стадия 2: Получение 3-(бензо[d][1,3]диоксол-5-илокси)пропан-1-ола (43B)

3-(бензо[d][1,3]диоксол-5-илокси)пропан-1-ол (43B) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(бензо[d][1,3]диоксол-5-илокси)пропилацетат (43A) использовался вместо 2-(3-(диметиламино)фенокси)этилацетата (39A) на стадии 2 примера 39.
Стадия 3: Получение 3-(3-(бензо[d][1,3]диоксол-5-илокси) пропокси)-6-бромметилпиколината (43C)

3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-бромметилпиколинат (43C) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(бензо[d][1,3]диоксол-5-илокси)пропан-1-ол (43B) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 4: Получение целевого соединения 3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (43)
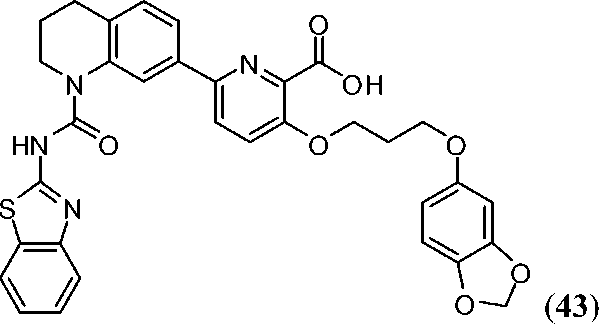
Целевое соединение 3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (43) получали в соответствии со следующей процедурой: 3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (43) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-бромометиликолинат (43C) использовали вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29.
Пример 44
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(хинолин-8-илокси)пропокси)пиколиновой кислоты (44):

Стадия 1: Получение 3-(хинолин-8-илокси)пропилацетата (44A)

3-(хинолин-8-илокси)пропилацетат (44A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 2-бромпропил ацетат использовали вместо 2-бромэтил ацетата, и 8-гидроксихинолин использовали вместо 3-(диметиламино)фенола на стадии 1 примера 39.
Стадия 2: Получение 3-(хинолин-8-илокси)пропан-1-ол (44B)
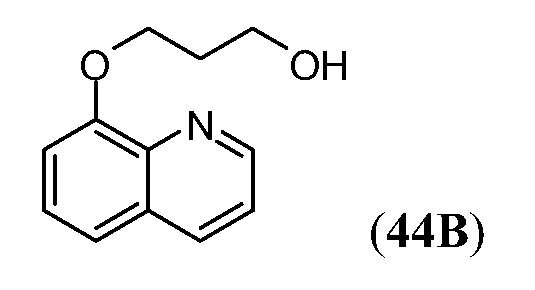
3-(хинолин-8-илокси)пропан-1-ол (44B) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(хинолин-8-илокси)пропил ацетат (44A) использовался вместо 2-(3-(диметиламино)фенокси)этилацетата (39A) на стадии 2 примера 39.
Стадия 3: Получение 6-бром-3-(3-(хинолин-8-илокси)пропокси)метилпиколината (44C)

6-бром-3-(3-(хинолин-8-илокси)пропокси)метилпиколинат (44C) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(хинолин-8-илокси)пропан-1-ол (44B) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 4: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(хинолин-8-илокси)пропокси)пиколиновой кислоты (44)
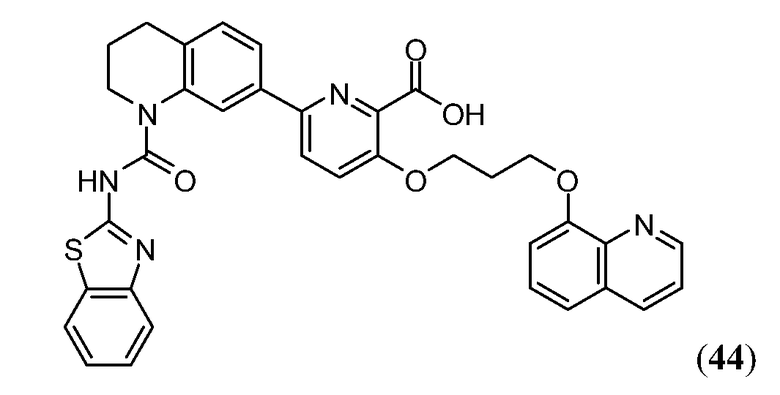
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(хинолин-8-илокси)пропокси)пиколиновую кислоту (44) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(хинолин-8-илокси)пропокси)пиколиновую кислоту (44) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-бром-3-(3-(хинолин-8-илокси)пропокси)метилпиколинат (44C) использовался вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29.
Пример 45
Синтез 3-(2-(4-(аминометил)фенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (45):
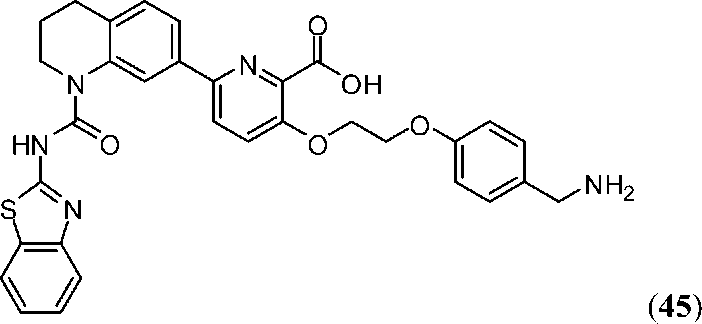
Стадия 1: Получение 4-(бензилокси)бензил-трет-бутилкарбамат (45A)

Ди-трет-бутил бикарбонат (0,934 г, 4,3 ммоль) добавляли к раствору 4-бензилоксибензиламина (0,83 г, 3,89 ммоль) и NaOH (0,171 г, 4,3 ммоль) в воде (15 мл) и THF (5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 4 часов, концентрировали при пониженном давлении, чтобы удалить THF, и экстрагировали DCM. Органические фазы высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc от 92:8 до 87:13, получая желаемый продукт 4-(бензилокси)бензил-трет-бутилкарбамат (45A).
Стадия 2:
Получение 4-гидроксибензил-трет-бутилкарбамат (45B)
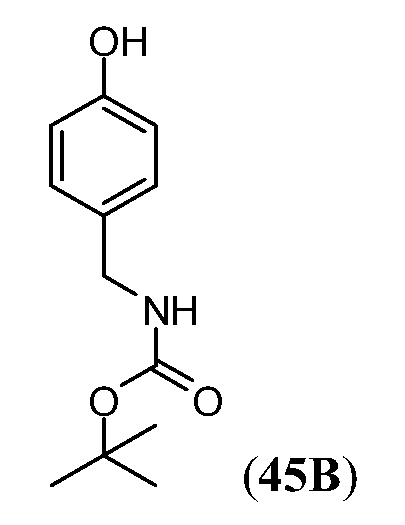
4-гидроксибензил-трет-бутилкарбамат (45B) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 4-(бензилокси)бензил-трет-бутилкарбамат (45A) использовался вместо 7-(5-(бензилокси)-6-(метоксикарбонил)пиридин-2-ил)-3,4 дигидрохинолина-1(2H)-трет-бутилкарбоксилата (29C) на стадии 4 примера 29.
Стадия 3: Получение 2-(4-((трет-бутоксикарбониламино)метил)фенокси)этилацетата (45C)
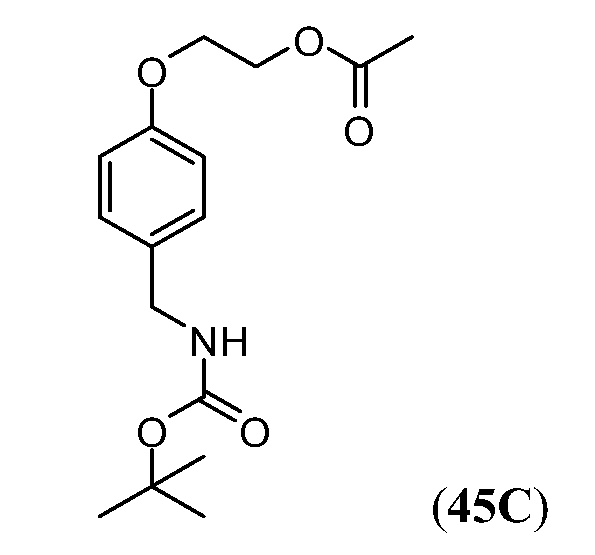
2-(4-((трет-бутоксикарбониламино)метил)фенокси)этилацетат (45C) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 4-гидроксибензил-трет-бутилкарбамат (45B) использовался вместо 3-(диметиламино)фенола на стадии 1 примера 39.
Стадия 4: Получение 4-(2-гидроксиэтокси)бензил-трет-бутилкарбамата (45D)
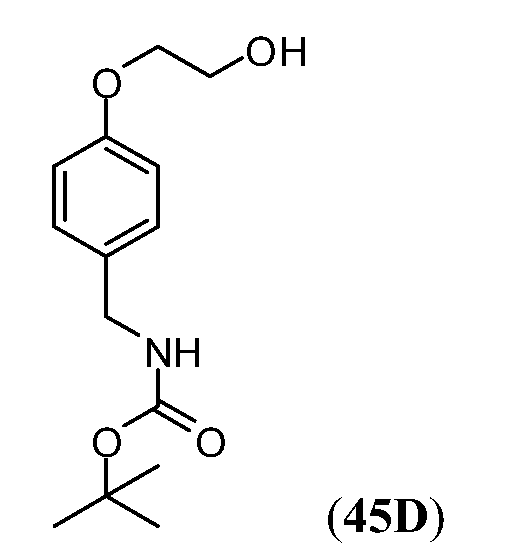
4-(2-гидроксиэтокси)бензил-трет-бутилкарбамат (45D) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 2-(4-((трет-бутоксикарбониламино)метил)фенокси)этилацетат (45C) использовался вместо 2-(3-(диметиламино)фенокси)этилацетата (39A) на стадии 2 примера 39.
Стадия 5: Получение 6-бром-3-(2-{4-{{трет-бутоксикарбониламино)метил)фенокси)этокси)метилпиколината (45E)
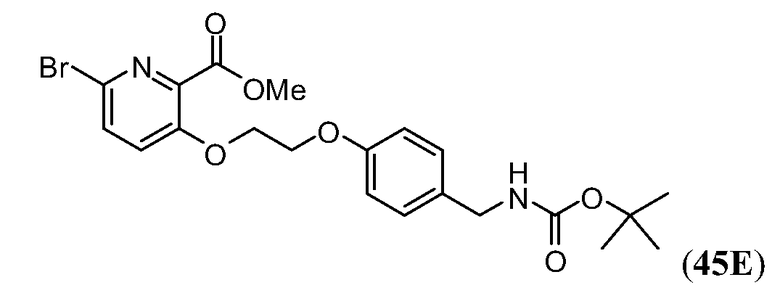
6-бром-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)метилпиколинат (45E) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 4-(2-гидроксиэтокси)бензил-трет-бутилкарбамат (45D) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 6: Получение 3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (45F)
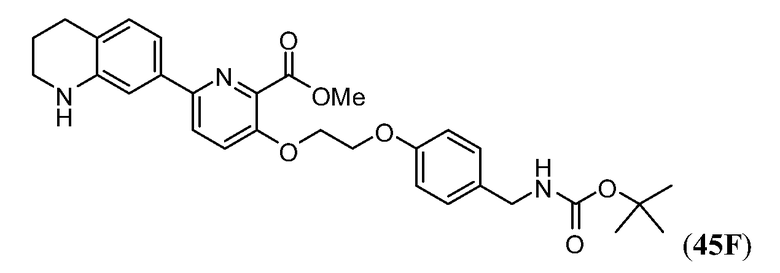
3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (45F) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-бром-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)метилпиколинат (45E) использовался вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29.
Стадия 7: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси) этокси)метилпиколината (45G)
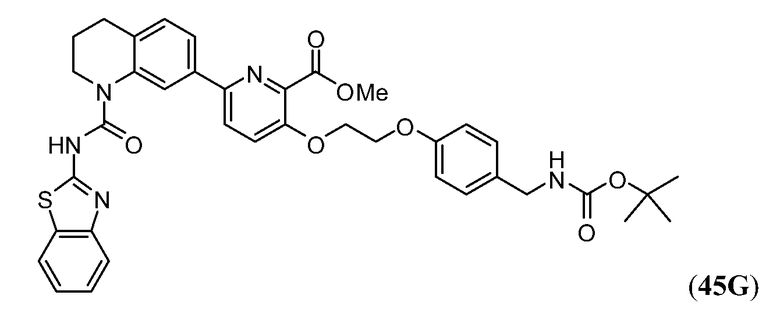
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)метилпиколинат (45G) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколинат (45F) использовался вместо 3-фенил-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (29Н) на стадии 9 примера 29.
Стадия 8: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)пиколиновой кислоты (45Н)
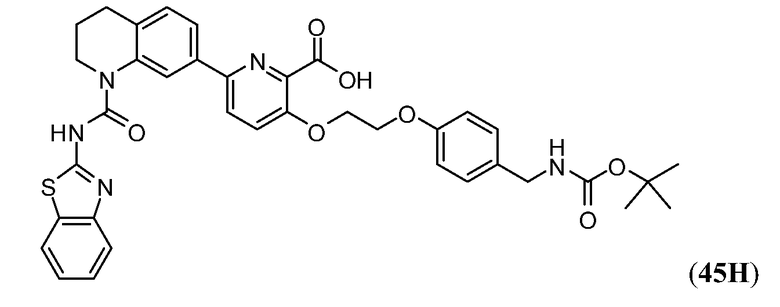
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)пиколиновую кислоту (45Н) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)метилпиколинат (45G) использовался вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилметилпиколината (29I) на стадии 10 примера 29.
Стадия 9: Получение целевого соединения 3-(2-(4-(аминометил)фенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновой кислоты (45)
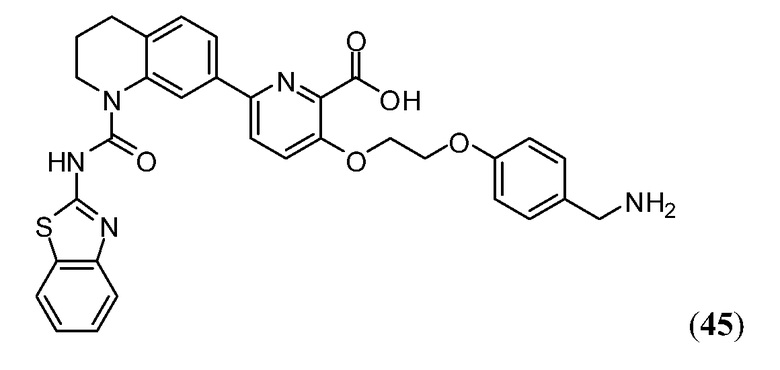
Целевое соединение 3-(2-(4-(аминометил)фенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (45) получали в соответствии со следующей процедурой: соединение 3-(2-(4-(аминометил)фенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновую кислоту (45) получали согласно процедуре, подобной описанной в примере 34, кроме того, что 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-((трет-бутоксикарбониламино)метил)фенокси)этокси)пиколиновая кислота (45Н) использовалась вместо 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)пиколиновой кислоты (34E) на стадии 6 примера 34. 1Н ЯМР (300 МГц, DMSO) δ 8,29 (1H, с, шир.), 8,04 (1H, с, v шир.), 7,98 (1H, д), 7,81 (1H, д), 7,77 (1H, д), 7,68 (1H, дд), 7,45 (1H, с, шир.), 7,41-7,35 (3H, м), 7,28-7,19 (2H, м), 7,04 (2H, д), 4,48 (2H, м), 4,35 (2H, м), 3,96 (4H, м), 2,81 (2H, т), 1,92 (2H, м). MS (ESI(+)): м/z 596,1 (M+H).
Пример 46
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(бифенил-4-илметокси)пиколиновой кислоты (46):
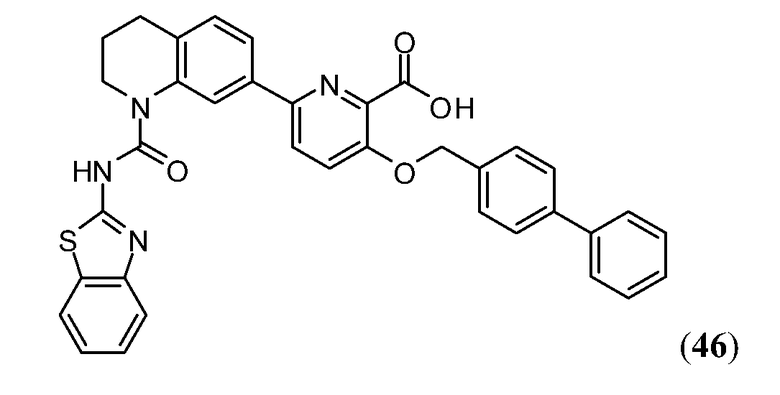
Стадия 1: Получение 3-(бифенил-4-илметокси)-6-бромметилпиколината (46A)

3-(бифенил-4-илметокси)-6-бромметилпиколинат (46A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 4-бифенилметанол использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 2: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(бифенил-4-илметокси)пиколиновой кислоты (46)
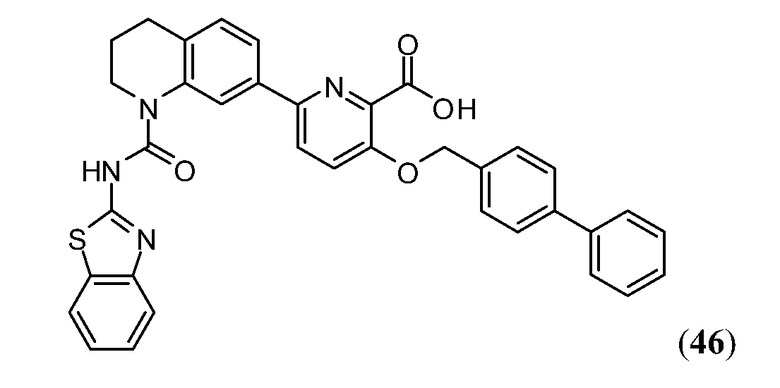
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(бифенил-4-илметокси)пиколиновую кислоту (46) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(бифенил-4-илметокси)пиколиновую кислоту (46) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 3-(бифенил-4-илметокси)-6-бромметилпиколинат (46A) использовался вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29.
Пример 47
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенокси)пропокси)пиколиновой кислоты (47):
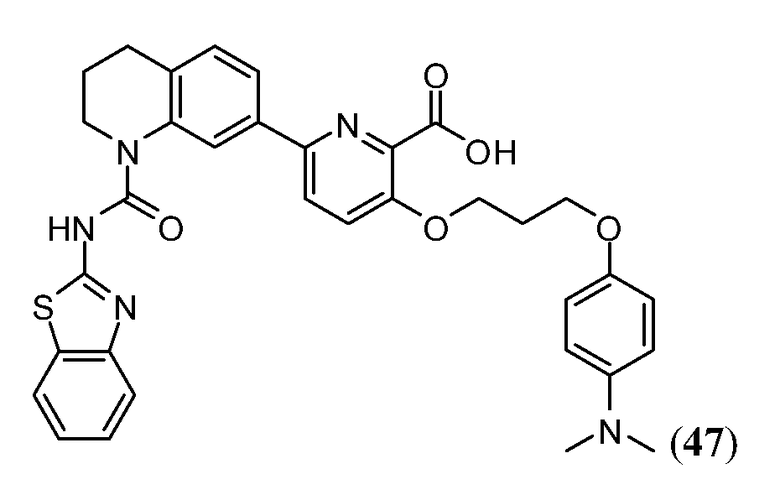
Стадия 1: Получение 3-(4-(диметиламино)фенокси)пропилацетата (47A)

3-(4-(диметиламино)фенокси)пропилацетат (47A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 2-бромпропил ацетат использовался вместо 2-бромэтил ацетата, и 4-(диметиламино)фенол использовался вместо 3-(диметиламино)фенола на стадии 1 примера 39.
Стадия 2:
Получение 3-(4-(диметиламино)фенокси)пропан-1-ола (47B)

3-(4-(диметиламино)фенокси)пропан-1-ол (47B) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(4-(диметиламино)фенокси)пропилацетат (47A) использовался вместо 2-(3-(диметиламино)фенокси)этилацетат (39A) на стадии 2 примера 39.
Стадия 3: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенокси)пропокси)пиколиновой кислоты (47)
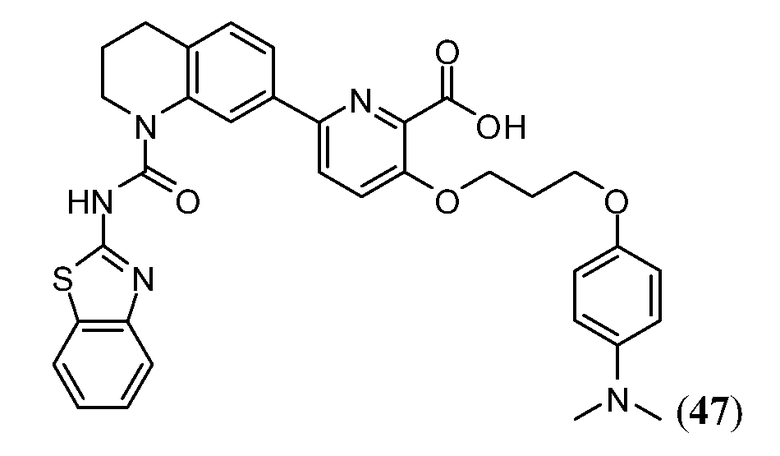
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенокси)пропокси)пиколиновую кислоту (47) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенокси)пропокси)пиколиновую кислоту (47) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолин-1(2H)-трет-бутилкарбоксилат (29D) использовался вместо 6-бром-3-гидроксиметилпиколината (26B), и 3-(4-(диметиламино) фенокси)пропан-1-ол (47B) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39. 1Н ЯМР (300 МГц, DMSO) δ 8,28 (1H, с), 7,95 (1H, д), 7,81 (1H, д), 7,71 (1H, д), 7,67 (1H, дд), 7,47 (1H, д, щир.), 7,37 (1H, дт), 7,28-7,19 (2H, м), 6,90-6,75 (4H, м), 4,26 (2H, т), 4,08 (2H, т), 3,94 (2H, т), 2,84-2,77 (8H, м), 2,14 (2H, м), 1,93 (2H, м). MS (ESI(+)): м/z 624,1 (M+H).
Пример 48
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенил)пропокси)пиколиновой кислоты (48):
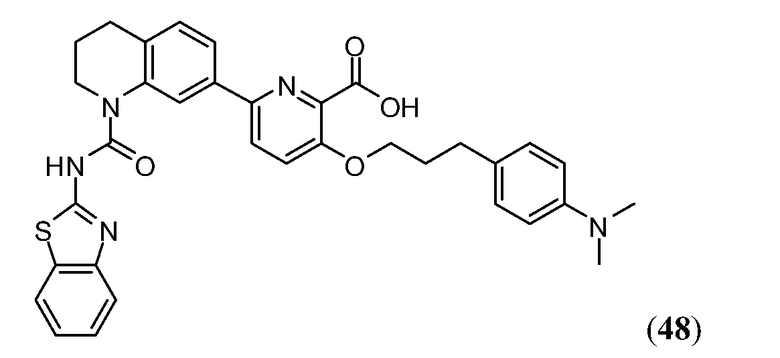
Стадия 1: Получение 3-(4-(диметиламино)фенил)пропан-1-ола (48A)
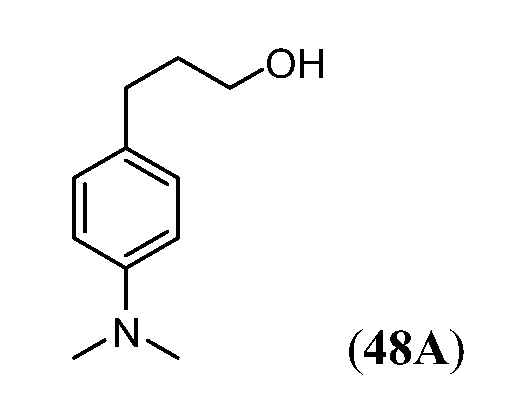
LiAlH4 (0,066 г, 1,71 ммоль) добавляли к раствору 4-(диметиламино)циннамальдегида (0,10 г, 0,57 ммоль) в безводном THF (2 мл). Реакционную смесь нагревали с обратным холодильником в течение 2 часов и охлаждали до температуры окружающей среды. К реакционной смеси добавляли по каплям 1M HCl. Реакционную смесь перемешивали в течение 5 минут, после чего добавляли насыщенный NaHCO3, и смесь экстрагировали DCM. Органические фазы высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом PE:EtOAc 2:1, получая желаемый продукт 3-(4-(диметиламино)фенил)пропан-1-ол (48A).
Стадия 2: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенил)пропокси)пиколиновой кислоты (48)

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенил)пропокси)пиколиновую кислоту (48) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенил)пропокси)пиколиновую кислоту (48) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 7-(5-гидрокси-6-(метоксикарбонил)пиридин-2-ил)-3,4-дигидрохинолина-1(2H)-трет-бутилкарбоксилат (29D) использовался вместо 6-бром-3-гидроксиметилпиколината (26B), и 3-(4-(диметиламино)фенил)пропан-1-ол (48A) использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39. 1Н ЯМР (300 МГц, DMSO) δ 8,28 (1H, с), 7,93 (1H, д), 7,81 (1H, д), 7,70-7,62 (2H, м), 7,48 (1H, д, шир.), 7,37 (1H, дт), 7,27-7,12 (6H, м), 4,08 (2H, т), 3,94 (2H, т), 2,93 (6H, с), 2,81 (2H, т), 2,71 (2H, т), 2,02-1,90 (4H, м). MS (ESI(+)): м/z 608,1 (M+H).
Пример 49
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновой кислоты (49):
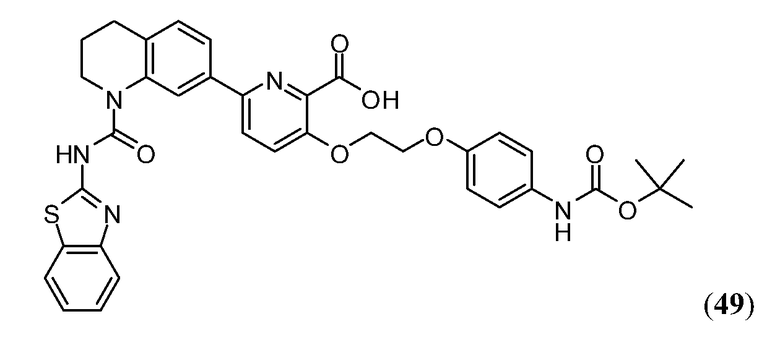
Стадия 1: Получение 6-бром-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)метилпиколината (49A)

6-бром-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)метилпиколинат (49A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 4-(2-гидроксиэтокси)фенил-трет-бутилкарбамат использовался вместо 2-(3-(диметиламино)фенокси)этанола (39B) на стадии 3 примера 39.
Стадия 4: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновой кислоты (49)

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновую кислоту (49) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновую кислоту (49) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-бром-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)метилпиколинат (49A) использовался вместо 3-(бензилокси)-6-бромметилпиколината (29A) на стадии 2 примера 29. 1Н ЯМР (300 МГц, CDCl3): δ 8,18 (1H, с), 7,97 (1H, д), 7,80 (1H, д), 7,68-7,60 (2H, м), 7,48 (1H, т), 7,39 (1H, т), 7,26 (1H, м), 7,20-7,15 (2H, м), 6,89 (2H, д), 6,37 (1H, с), 4,53 (2H, м), 4,01 (2Н, м), 4,02 (2H, т), 2,83 (2H, т), 2,08 (2H, м), 1,50 (9H, с).
Пример 50
Синтез (Е)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторстирил)пиколиновой кислоты (50):

Целевое соединение (Е)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторстирил)пиколиновую кислоту (50) получали в соответствии со следующей процедурой: (Е)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторстирил)пиколиновую кислоту (50) получали согласно процедуре, подобной описанной в примере 29, кроме того, что транс-2-(4-фторфенил)винилбороновая кислота использовалась вместо фенилбороновой кислоты на стадии 7 примера 29. 1Н ЯМР (300 МГц, DMSO) δ 8,64 (1H, с), 8,14 (1H, д), 7,78 (1H, д), 7,69 (1H, д), 7,63-7,52 (4H, м), 7,41 (1H, д), 7,27-7,13 (5H, м), 6,97 (1H, т), 3,96 (1H, т), 2,80 (2H, т), 1,88 (2H, м). MS (ESI(+)): м/z 551,0 (M+H).
Пример 51
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(диметиламино)бензиламино)этокси)пиколиновой кислоты (51):

Стадия 1:
Получение 2-(4-(диметиламино)бензиламино)этанола (51A)

Раствор 2-(диметиламино)бензальдегида (0,50 г, 3,35 ммоль) и этаноламина (0,409 г, 6,7 ммоль) в безводном EtOH (13 мл) перемешивали при температуре окружающей среды в течение 17 часов, после чего добавляли натрий триацетоксиборгидрид (1,42 г, 6,7 ммоль). Реакционную смесь перемешивали в течение 4,5 часов, добавляли воду и подщелачивали до рН 12 с помощью 5М NaOH. Смесь экстрагировали EtOAc, и органические фазы высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием градиентом DCM:MeOH от 9:1 до 1:1, получая желаемый продукт 2-(4-(диметиламино)бензиламино)этанол (51A).
Стадия 2: Получение 4-(диметиламино)бензил(2-гидроксиэтил)трет-бутилкарбамата (51B)
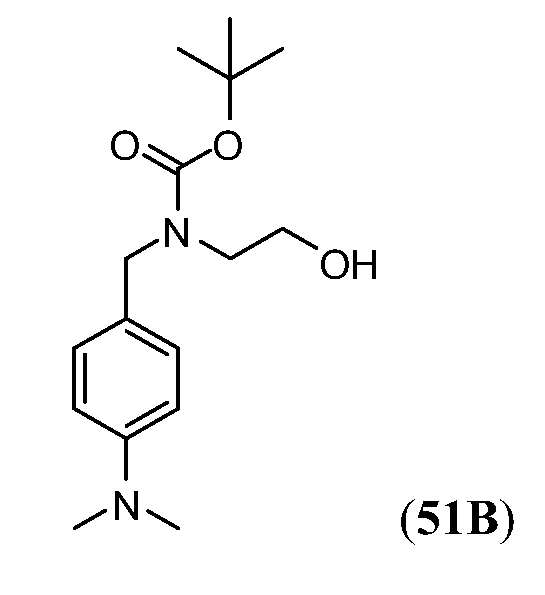
4-(диметиламино)бензил(2-гидроксиэтил)трет-бутилкарбамат (51B) получали согласно процедуре, подобной описанной в примере 40, кроме того, что 2-(4-(диметиламино)бензиламино)этанол (51A) использовался вместо 3-гидрокси-6-(1,2,3,4-тетрагидрохинолин-7-ил)метилпиколината (40C) на стадии 4 примера 40.
Стадия 3: Получение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(диметиламино)бензиламино)этокси)пиколиновой кислоты (51)
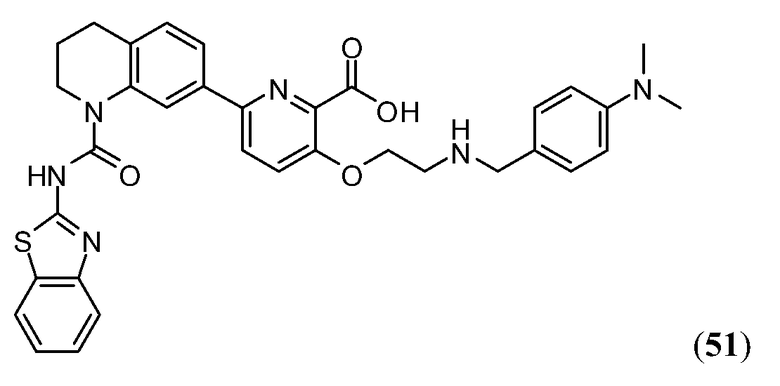
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(диметиламино)бензиламино)этокси)пиколиновую кислоту (51) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(диметиламино)бензиламино)этокси)пиколиновую кислоту (51) получали согласно процедуре, подобной описанной в примере 45, кроме того, что 4-(диметиламино)бензил(2-гидроксиэтил)трет-бутилкарбамат (51B) использовался вместо 4-(2-гидроксиэтокси)бензил-трет-бутилкарбамата (45D) на стадии 5 примера 45. 1Н ЯМР (300 МГц, DMSO) δ 8,82 (2H, с, шир.), 8,31 (1H, с, шир.), 8,04 (1H, д), 7,81 (1H, д), 7,77 (1H, д), 7,71 (1H, дд), 7,47 (1H, с, шир.), 7,41-7,20 (4H, м), 6,75 (2H, д), 4,43 (2H, т), 4,19 (1H, с, шир.), 3,95 (2H, т, шир.), 3,37 (2H, с, шир.), 2,91 (6H, с), 2,82 (2H, т), 1,94 (2H, м). MS(ESI(+)): м/z 623,0 (M+H).
Пример 52
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этокси)пиколиновой кислоты (52):

Стадия 1: Получение 2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этилацетата (52A)
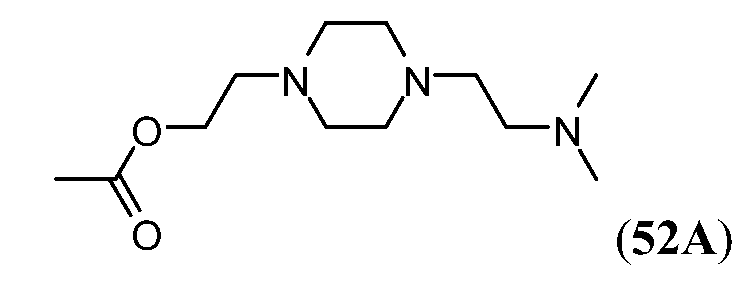
2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этилацетат (52A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 1-(2-диметиламиноэтил)-пиперазин использовался вместо 3-(диметиламино)фенола на стадии 1 примера 39.
Стадия 2: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этокси)пиколиновой кислоты (52)
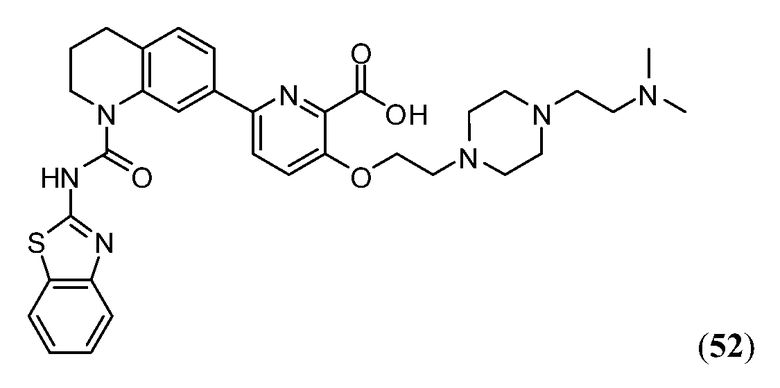
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этокси)пиколиновую кислоту (52) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этокси)пиколиновую кислоту (52) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этилацетат (52A) использовался вместо 2-(3-(диметиламино)фенокси)этилацетата (39A) на стадии 2 примера 39. 1Н ЯМР (300 МГц, DMSO) δ 8,31 (1H, с), 8,23 (1H, с), 7,83-7,74 (2H, м), 7,63 (1H, дд), 7,58 (1H, д), 7,48 (1H, д), 7,36 (1H, т), 7,24-7,16 (2H, м), 4,24 (4H, м), 3,93 (4H, м), 2,80 (4H, м), 2,64 (4H, с), 2,47 (4H, с), 2,25 (6H, с), 1,94 (2H, м). MS (ESI(+)): м/z 630,1 (M+H).
Пример 53
Синтез 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-феноксипропокси)пиколиновой кислоты (53):

Стадия 1: Получение 6-бром-2H-бензо[b][1,4]оксазин-4(3H)-трет-бутилкарбоксилата (53A)
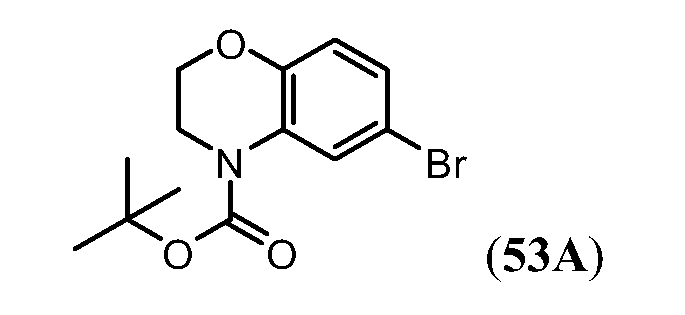
6-бром-2H-бензо[b][1,4]оксазин-4(3H)-трет-бутилкарбоксилат (53A), получали согласно процедуре, подобной описанной в примере 23, кроме того, что 6-бром-3,4-дигидро-2Н-бензо[b][1,4]оксазин (7A) использовался вместо 7-бром-1,2,3,4-тетрагидрохинолина (1Е) на стадии 4 примера 23.
Стадия 2: Получение 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Η)-трет-бутилкарбоксилата (53B)

6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Η)-трет-бутилкарбоксилат (53B) получали согласно процедуре, подобной описанной в примере 23, кроме того, что 6-бром-2H-бензо[b][1,4]оксазин-4(3Н)-трет-бутилкарбоксилат (53A) использовался вместо 7-бром-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (23D) на стадии 5 примера 23.
Стадия 3: Получение 6-(6-(метоксикарбонил)-5-(3-феноксипропокси)пиридин-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Н)-трет-бутилкарбоксилата (53C)

Смесь 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-бензо[b][1,4]оксазин-4(3H)-трет-бутилкарбоксилат (53B) (72 мг, 0,20 ммоль), 6-бром-3-(3-феноксипропокси)метилпиколинат (26C) (72 мг, 0,20 ммоль), K2CO3 (30 мг, 0,22 ммоль в 0,5 мл воды), тетрабутил аммоний бромид (64 мг, 0,20 ммоль) и дихлорбис(трифенилфосфин)палладий (II) (10 мг, каталитическое количество) в 1,4-диоксане (5 мл) нагревали до 90°C в течение 6 часов. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли EtOAc, фильтровали через Целит и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на колонках с силикагелем с элюированием 20%-ым EtOAc в гексанах, получая желаемый продукт 6-(6-(метоксикарбонил)-5-(3-феноксипропокси)пиридин-2-ил)-2H-бензо[b][1,4]оксазин-4(3H)-трет-бутилкарбоксилат (53C). 1Н ЯМР (300 МГц, CDC13): 8,49 (1H, с), 7,74 (1H, д), 7,60 (1H, д), 7,38 (1H, д), 7,24 (2H, м), 6,90 (4H, м), 4,31 (2H, т), 4,18 (2H, т), 4,25 (2H, т), 3,95 (3H, с), 3,85 (2H, т), 2,3 (2H, м), 1,58 (9H, с). LCMS (APCI): м/z 521,1 (M+H).
Стадия 4: Получение целевого соединения 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-феноксипропокси)пиколиновой кислоты (53).

Целевое соединение 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-феноксипропокси)пиколиновую кислоту (53) получали в соответствии со следующей процедурой: 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-феноксипропокси)пиколиновую кислоту (53) получали согласно процедуре, подобной описанной в примере 29, кроме того, что 6-(6-(метоксикарбонил)-5-(3-феноксипропокси)пиридин-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Н)-трет-бутилкарбоксилат (53C) использовался вместо 7-(6-(метоксикарбонил)-5-(трифторметилсульфонилокси)пиридин-2-ил)-3,4-дигидрохинолин-1(2Н)-трет-бутилкарбоксилата (29E) на стадии 6 примера 29. LCMS (APCI): m/z 583,1 (М+H).
Пример 54
Синтез 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-(пиридин-4-ил)пропокси)пиколиновой кислоты (54):
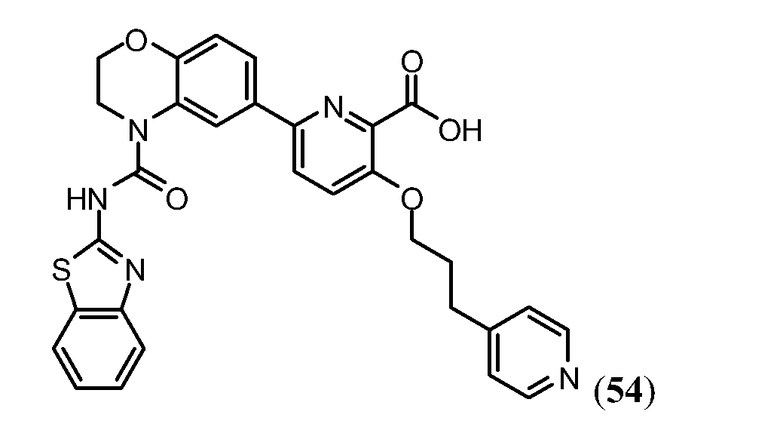
Стадия 1: Получение 6-бром-3-(3-(пиридин-4-ил)пропокси) метилпиколината (54A)
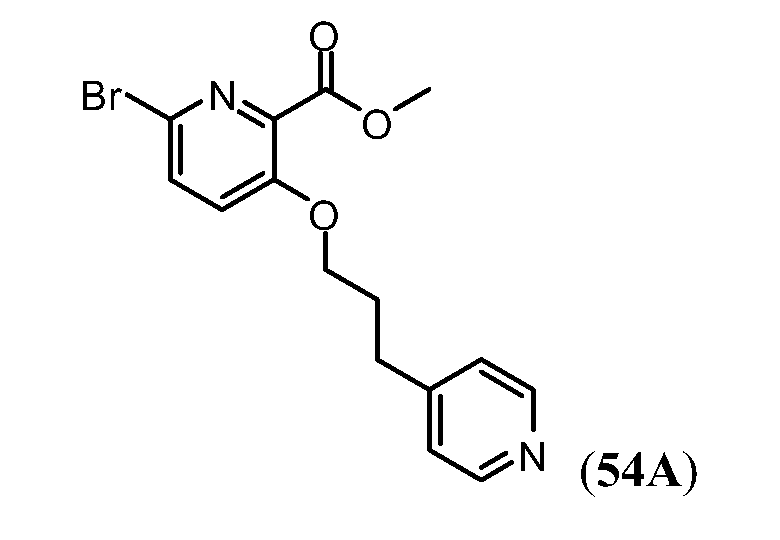
6-бром-3-(3-(пиридин-4-ил)пропокси)метилпиколинат (54A) получали согласно процедуре, подобной описанной в примере 39, кроме того, что 3-(пиридин-4-ил)пропан-1-ол использовался вместо 2-(3-(диметиламино)фенокси)этанола на стадии 3 примера 39.
Стадия 2: Получение целевого соединения 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-(пиридин-4-ил)пропокси)пиколиновой кислоты (54)

Целевое соединение 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-3-(3-(пиридин-4-ил)пропокси)пиколиновую кислоту (54) получали в соответствии со следующей процедурой: 6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-3-(3-(пиридин-4-ил)пропокси)пиколиновую кислоту (54) получали согласно процедуре, подобной описанной в примере 53, кроме того, что 6-бром-3-(3-(пиридин-4-ил)пропокси)метилпиколинат (54A) использовался вместо 6-бром-3-(3-феноксипропокси)метилпиколината (26C) на стадии 3 примера 53. 1Н ЯМР (300 МГц, MeOD): 8,42 (3H, м), 7,7 (2H, м), 7,6 (1H, д)), 7,3-7,4 (5H, м), 7,2 (1H, т), 6,9 (1H, м), 4,3 (2H, т), 4,1-4,2 (4H, м), 2,9 (2H, т), 2,2 (2H, т). LCMS (APCI): м/z 568 (M+H).
Пример 55
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-морфолинопропокси)пиколиновой кислоты (55):
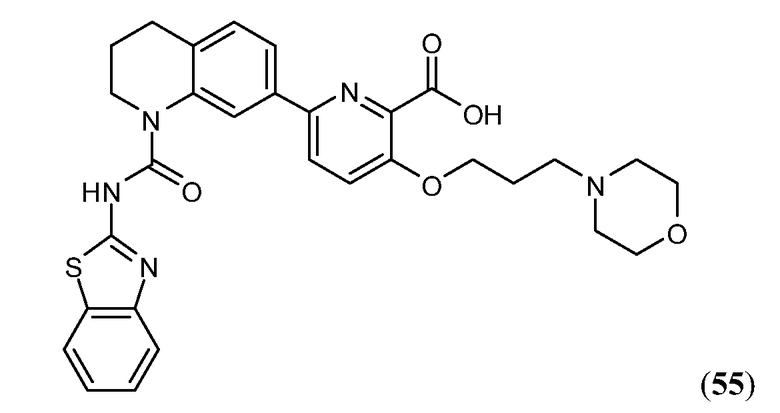
Стадия 1:
Получение 6-бром-3-гидроксипиколиновой кислоты (55A)

6-бром-3-гидроксиметилпиколинат (26B) (464 мг, 2 ммоль) в MeOH (0?5 мл) добавляли к раствору LiOH (200 мг в 2 мл воды). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. Продукт осаждали из раствора и фильтровали, промывали водой и высушивали при пониженном давлении, получая желаемый продукт 6-бром-3-гидроксипиколиновую кислоту (55A). 1Н ЯМР (300 МГц, CDC13): 7,6 (1H, д), 7,32 (1H, д).
Стадия 2: Получение смолы 55B

Смолу Ванга (1 г, Novabiochem, 1.1 ммоль/г) аккуратно смешивали на вибросите со смесью 6-бром-3-гидроксипиколиновой кислоты (55A) (436 мг, 2 ммоль), 1,3-диизопропилкарбодиимида (252 мг, 2 ммоль) и DMAP (2 ммоль) в смеси DCM/DMF (1:1, 10 мл) при 60°C в течение 16 часов. Смолу тщательно промывали смесью DCM/DMF (1:1, 3x5 мл) и DCM (3x5 мл) и высушивали при пониженном давлении в течение 2-3 часов, получая смолу 55B.
Стадия 3: Получение смолы 55C
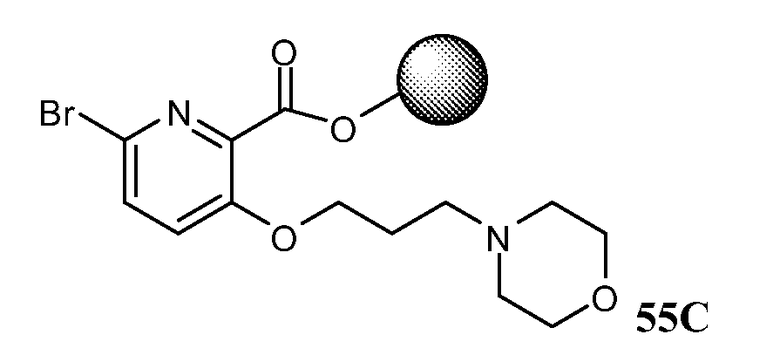
Смолу 55B (0,2 г, 0,22 ммоль) инкубировали со смесью 4-(3-гидроксипропил)морфолина (145 мг, 1 ммоль), диэтилазодикарбоксилата (174 мг, 1 ммоль) и трифенилфосфина (262 мг, 1 ммоль) в DCM (10 мл) при температуре окружающей среды в течение 4 часов. Полученную смолу фильтровали и тщательно промывали DMF (3x5 мл), MeOH (5 мл) и DCM (3x5 мл) и высушивали при пониженном давлении в течение 30 минут, получая смолу 55C.
Стадия 4: Получение смолы 55D

Смолу 55C (0,2 г, 0,11 ммоль) инкубировали с 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидрохинолином (27B) (103,6 мг, 0,4 ммоль), бис-(трифенилфосфин)палладий хлоридом (10 мг, каталитическое количество), тетрабутил аммоний фторидом (0,5 мл 1M раствора в THF) и K2CO3 (25 мг, растворенным в 3 каплях воды) в 5 мл смеси DCM:THF (1:1) при 60°C в течение 16 часов. Полученную смолу фильтровали и тщательно промывали DMF (3x5 мл), MeOH (3x5 мл) и DCM (3x5 мл) и высушивали при пониженном давлении в течение 30 минут, получая смолу 55D.
Стадия 5: Получение смолы 55E

Смолу 55D (0,05 г, 023 ммоль) инкубировали со смесью 4-нитрофенилбензо[d]тиазол-2-илкарбамата (19) (1 ммоль) в ацетонитриле (2 мл) при 65°C в течение 16 часов. Полученную смолу фильтровали и тщательно промывали DMF (3x5 мл), MeOH (5 мл) и DCM (3x5 мл) и высушивали при пониженном давлении в течение 30 минут, получая смолу 55E.
Стадия 6: Получение целевого соединения 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-морфолинопропокси)пиколиновой кислоты (55)

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-морфолинопропокси)пиколиновую кислоту (55) получали в соответствии со следующей процедурой: Смолу 55E инкубировали с 20% тетрафторуксусной кислоты в DCM (2 мл) в течение 1 часа. Смолу тщательно фильтровали, и супернатант высушивали в токе азота. Остаток повторно суспендировали в MeOH и очищали хроматографией на колонках с силикагелем с элюированием 10%-ым EtOAc в гексанах, получая желаемый продукт 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-морфолинопропокси)пиколиновую кислоту (55): LCMS (APCI): m/z 574,0 (М+H).
Пример 56
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-4-ил)этокси)пиколиновой кислоты (56):
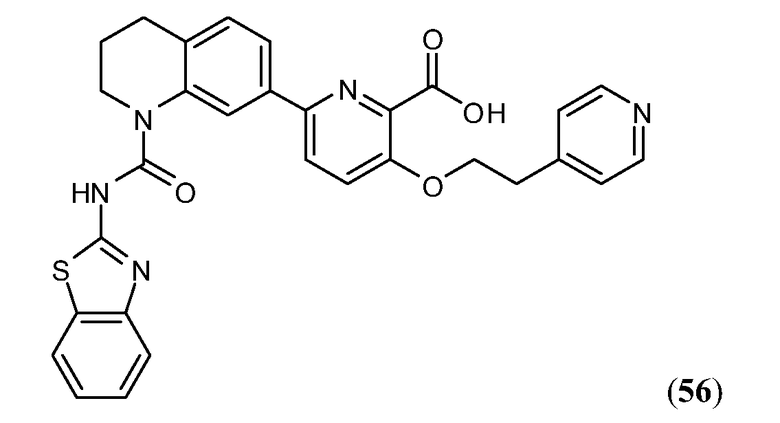
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-4-ил)этокси)пиколиновую кислоту (56) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-4-ил)этокси)пиколиновую кислоту (56) получали согласно процедуре, подобной описанной в примере 55, кроме того, что 4-пиридинэтанол использовался вместо 4-(3-гидроксипропил)морфолина на стадии 3 примера 55. 1Н ЯМР (300 МГц, MeOD): 8,45 (2H, д), 8,2 (1H, с), 7,7-7,6 (3H, м), 7,5-7,3 (5H, м), 7,2 (2H, д), 4,30 (2H, т), 3,95 (2H, т), 3,20 (2H, т), 2,85 (2H, т), 2,1 (2H, м). LCMS(APCI): м/z 552,0 (M+H).
Пример 57
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-2-ил)этокси)пиколиновой кислоты (57):
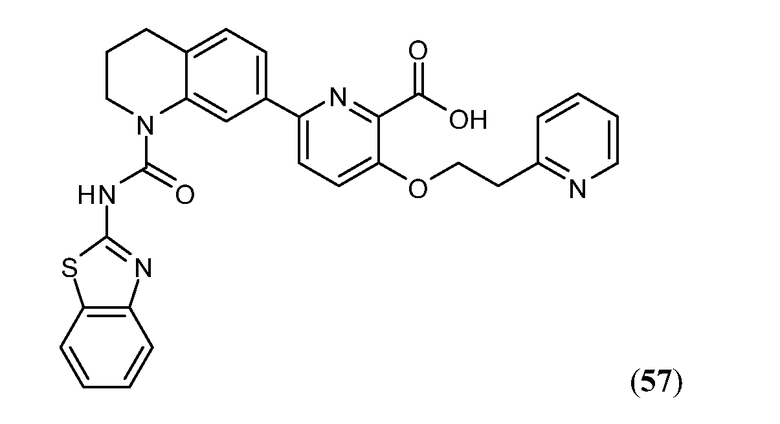
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-2-ил)этокси)пиколиновую кислоту (57) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-2-ил)этокси)пиколиновую кислоту (57) получали согласно процедуре, подобной описанной в примере 56, кроме того, что 2-пиридинэтанол использовался вместо 4-(3-гидроксипропил)морфолина на стадии 3 примера 55. 1Н ЯМР (300 МГц, MeOD): 8,2-8,45 (3H, м), 7,8-7,1 (10H, компл. м), 4,60 (2H, т), 4,0 (2H, т), 3,1 (2H, м), 2,85 (2H, т), 2,0 (2H, м). LCMS (APCI): м/z 552,0 (M+H).
Пример 58
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(циклобутилметокси)пиколиновой кислоты (58):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(циклобутилметокси)пиколиновую кислоту (58) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(циклобутилметокси)пиколиновую кислоту (58) получали согласно процедуре, подобной описанной в примере 55, кроме того, что циклобутанметанол использовался вместо 4-(3-4-гидроксипропил)морфолина на стадии 3 примера 55. 1Н ЯМР (300 МГц, MeOD): 8,15 (1H, с), 7,7-7,2 (8H, компл. м), 4,10 (2H, д), 3,9 (2H, м), 2,80 (2H, т), 2,2-1,8 (9H, м). LCMS (APCI): м/z 515,1 (M+H).
Пример 59
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)- 3-гидроксипиколиновой кислоты (59):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-гидроксипиколиновую кислоту (59) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-гидроксипиколиновую кислоту (59) получали согласно процедуре, подобной описанной в примере 55, кроме того, что смолу 55B использовали вместо смолы 55C стадия 4 примера 55. 1Н ЯМР (300 МГц, MeOD): 8,2 (1H, с), 8,0 (1H, д), 7,75 (1H, д), 7,70 (1H, д), 7,5 (1H, д), 7,4-7,2 (3H, м), 6,6 (1H, д), 3,95 (2H, т), 2,85 (2H, т), 2,0 (2H, м). LCMS (APCI): м/z 447,1 (M+H).
Пример 60
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторфенэтокси)пиколиновой кислоты (60):

Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторфенэтокси)пиколиновую кислоту (60) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторфенэтокси)пиколиновую кислоту (60) получали согласно процедуре, подобной описанной в примере 56, кроме того, что 4-фторфенетиловый спирт использовался вместо 4-(3-4гидроксипропил)морфолина на стадии 3 примера 55. 1Н ЯМР (300 МГц, MeOD): 8,15 (1H, с), 7,7 (2H, т), 7,60-6,90 (10H, компл. м), 4,2 (2H, т), 3,9 (2H, т), 3,1 (2H, т), 2,8 (2H, т), 2,05 (2H, м). LCMS (APCI): м/z 569,2 (M+H).
Пример 61
Синтез 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-(диметиламино) фенэтокси)пиколиновой кислоты (61):
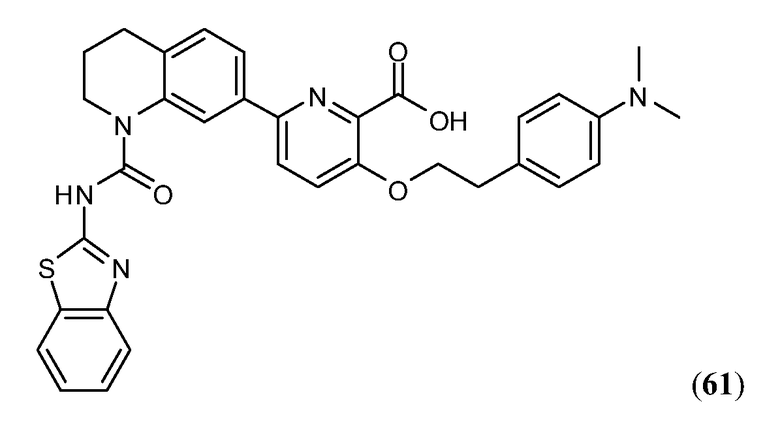
Целевое соединение 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-(диметиламино)фенэтокси)пиколиновую кислоту (61) получали в соответствии со следующей процедурой: 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-(диметиламино)фенэтокси)пиколиновую кислоту (61) получали согласно процедуре, подобной описанной в примере 55, кроме того, что 4-(диметиламино)фенетиловый спирт использовался вместо 4-(3-4гидроксипропил)морфолина на стадии 3 примера 55. 1Н ЯМР (300 МГц, MeOD): 8,15 (1H, с), 7,7 (3H, м), 7,5 (1H, д), 7,4-7,3 (2H, м), 7,3-7,1 (4H, м), 6,75 (2H, д), 4,15 (2H, т), 3,9 (2H, т), 3,0 (2H, т), 2,85 (8H, м), 2,0 (2H, м). LCMS (APCI): м/z 594,1 (M+H).
Пример 62
Синтез 3-амино-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиразин-2-карбоновой кислоты (62):
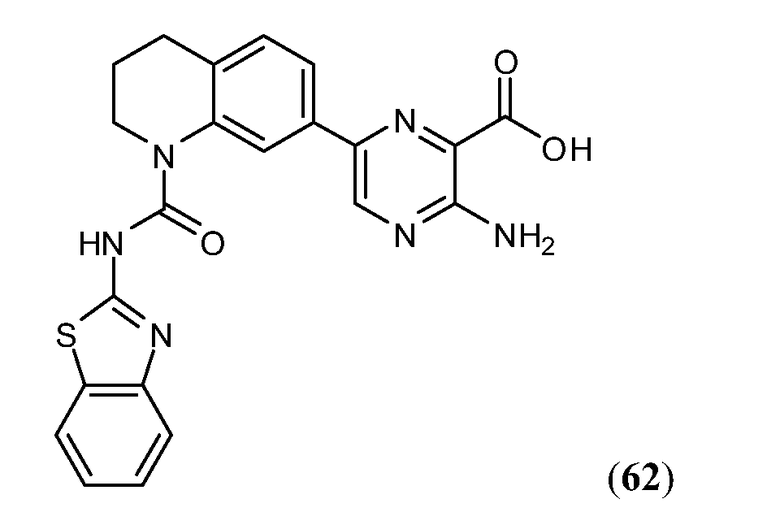
Целевое соединение 3-амино-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиразин-2-карбоновую кислоту (62) получали в соответствии со следующей процедурой: 3-амино-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиразин-2-карбоновую кислоту (62) получали согласно процедуре, подобной описанной в примере 55. LC/MS (APCI) m/z 447 (M+H).
Пример 64
Измерение конкуренции соединений по изобретению с F-Bak за сайт связывания белков семейства Bcl-2 (Bcl-XL) с использованием теста связывания Time Resolved Fluorescence Resonance Energy Transfer (TR-FRET):
Тестируемые соединения последовательно разбавляли в ДМСО, начиная с 50 мкМ (2x стартовая концентрация; 10% ДМСО), и 10 мкл переносили на планшет с 384 лунками. Затем 10 мкл смеси белок/зонд/антитело добавляли к каждой лунке в конечных концентрациях, перечисленных в Таблице 2.
Образцы затем смешивали на вибросите в течение 1 минуты, затем инкубировали в течение дополнительных 2 часов при температуре окружающей среды. Для каждого тестового планшета смесь зонд/антитело и белок/антитело/зонд включали в качестве отрицательного и положительного контроля, соответственно. Флюоресценцию измеряли на Envision (Perkin Elmer), используя фильтр возбуждения 340/35 нм и фильтры эмиссии 520/525 (F-Bak) и 495/510 нм (Tb-меченное анти-his антитело). Константы диссоциации (Ki) определяли, используя уравнение Ванга (см. Wang, Z.X. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995 360: 111-114). Тест TR-FRET может быть проведен в присутствии варьирующих концентраций человеческой сыворотки (HS) или эмбриональной телячьей сыворотки (FBS).
Для сравнения, измерение конкуренции соединений по изобретению в отношении сайтов связывания других белков семейства Bcl-2 (например, Bcl-2, Mcl-1) с использованием теста связывания TR-FRET осуществляли, заменяя GST-Bcl-XL в TR-FRET другим GST-меченным белком, например, GST-Bcl-2, GST-Mcl-1, полученным в лаборатории.
Результаты теста TR-FRET (Ki в микромоль) для соединений 1, 28, 38, 63, 68, 71, 75, 80, 81, 92 и 94 в Таблице 1 составляют 0,197, 0,0009, 0,0002, 0,012, 0,47, 0,007, 0,001, 0,0007, 0,66, 0,0005 и 0,000003, соответственно.
В одном варианте осуществления соединения по изобретению селективно ингибируют белок семейства Bcl-2, Bcl-XL, по сравнению с другими белками семейства Bcl-2, такими как Bcl-2 и Mcl-1. Для сравнения, данные (Ki в микромоль) измерения конкуренции некоторых соединений по изобретению (то есть соединений 1, 28, 38, 63, 71, 75, 80, 81, 92 и 94 в Таблице 1) с F-Bak за сайт связывания Bcl-2 с использованием теста связывания TR-FRET составляют 1,2, 0,15, 0,01, 0,018, 0,50, 0,272, 0,167, 0,66, 0,23 и 0,000003, соответственно.
Пример 65
Измерение конкуренции соединений по изобретению с Bim26-мером за сайт связывания белков семейства Bcl-2 с использованием теста связывания Bcl-XL Alpha Screen:
Тест AlphaScreen™ для белков BH3 использовали, чтобы идентифицировать активные малые молекулы скрининга белка семейства Bcl-2, например, скрининга Bcl-xL, hmMcl-1. Чтобы определить точную оценку IC50, соединения обычно проверяли в стартовых концентрациях, 100 мкМ и/или 1 мкМ, и последовательно титровали 3-кратно в 11 разведениях.
В этом тесте используют технологию Alphascreen™, которая основана на покрытых гидрогелем акцепторных и донорских гранулах, которые имеют функциональные группы для конъюгации с белком (например, GST-hmMcl-1, GST-Bcl-xL или GST-биотином) и пептидом (Биотин-Bak, Биотин-Bim), соответственно. Гранулы вводят в момент, когда белок и пептиды взаимодействуют. Донорские гранулы содержат фотосенсибилизатор, который преобразует кислород в возбужденную форму O2 при возбуждении 680 нм. Энергия трансформируется из синглетного кислорода и вступает в реакцию с хемилюминесцентами на акцепторной грануле, приводя к эмиссии света с длиной волны 520-620 нм. Соединения по изобретению, при их добавлении в реакцию, могут уменьшить интенсивность люминесценции, в зависимости от ингибирования близости акцепторных и донорских гранул. На основании этой информации определяли IC50 каждого соединения.
Материалы
Белки GST-Bcl-xL, GST-hmMcl-1 и биотинилированный GST получали в лаборатории и сохраняли в форме сток-растворов при -80°C. Пептиды биотинилированный-Bak и биотинилированный-Bim покупали у Auspep и сохраняли как 500 мкМ сток-растворы в 100%-ном ДМСО при -80°C. Набор для детекции GST (Глутатион-S-Трансферазы) ALPHASCREEN™ получали от Perkin Elmer Lifesciences (Cat #6760603R). Proxiplates, белые 384-луночные планшеты с плоским дном приобретали у Interpath Services, Мельбурн (Cat #784075). Крышки для планшетов приобретали у Prescience, Мельбурн (Cat#784075). ДМСО покупали у AnalaR. Планшеты с 384 глубокими лунками и планшеты из полипропилена на 50 мкл с V-образным дном покупали у Matrical.
Получение соединений
Соединения по изобретению получали в форме 10 мМ стоков с 100%-ым ДМСО в день перед проведением теста. 12 мкл 100% ДМСО и 6 мкл 10 мМ соединения (то есть 3,333 мМ, конечное количество 100 мкМ) добавляли в ряды 1 и 12 в планшеты из полипропилена на 50 мкл с V-образным дном. Для получения конечной концентрации соединения 1 мкМ, в отдельном планшете Matrical, 28 мкл 100% ДМСО и 2 мкл 10 мМ соединения добавляли в лунку, хорошо смешивали, отбирали 2 мкл этого раствора и добавляли к 38 мкл 100% ДМСО. 20 мкл этого раствора добавляли к тестовому планшету Matrical. Несколько контрольных соединений включали в тестируемые планшеты. Для контрольных лунок в соответствующие лунки каждого планшета добавляли только 15 мкл 100% ДМСО. Планшеты с соединениями затем последовательно разбавляли в 2 раза, используя MiniTrak. Как только титрования были закончены, планшет с соединениями немедленно покрывали крышками из фольги, чтобы предотвратить испарение.
Получение буфера
Готовили свежие тестовый буфер и буфер для гранул. Каждый планшет с титруемым соединением тестировали в двух повторах. Следующие объемы были достаточными для 12 Proxiplates (4 тестовых планшета в двойном экземпляре в каждом из тестов Bclxl, hmMcl и подсчета).
Получение белка и пептида; и проведение теста
1. Тестовый буфер и буфер для гранул использовались для получения акцепторных и донорских растворов. Гранулы ALPHASCREEN™ являются светочувствительными, и поэтому их получают в затемненном помещении. 2,5 мкл гранул добавляли в 1 мл буфера.
2. Объем белка или пептида для добавления вычисляли, используя следующую формулу:
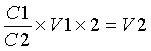
C1 = Конечная концентрация белка/пептида
C2 = Стоковая концентрация белка/пептида
V1 = Полный объем акцепторного/донорского раствора
V2 = Объем стока белка/пептида для добавления к акцепторному/донорскому раствору
3. Компоненты теста получали как отдельные акцепторные и донорские растворы. Акцепторный раствор содержал акцепторные гранулы и целевой белок, в то время как донорский раствор содержал донорские гранулы и биотинилированный пептид.
10 мл
10 мл
0,32 мкл
[4 нМ]
4. После получения растворы оставляли для инкубации в течение 30 минут при комнатной температуре, чтобы позволить гранулам связаться с белком и пептидом.
5. 50 мкл раствора Bcl-xL, 50 мкл раствора hmMcl-1 и 50 мкл биотинилированного-GST добавляли в отдельные глубокие лунки на планшете для проведения теста. Контрольные 50 мкл тестового буфера/буфера для гранул добавляли в отдельные лунки планшета (без белка).
6. 50 мкл раствора Bim и 50 мкл раствора Bak добавляли в отдельные глубокие лунки планшета.
7. 0,3 мкл образца из планшета с соединением переносили в каждый планшет для проведения теста.
8. Инкубировали в течение 30 минут при температуре окружающей среды, затем добавляли 5 мкл донорского раствора. После добавления донорского раствора, планшеты мягко встряхивали постукиванием и герметизировали индивидуально адгезивной пленкой.
9. Планшеты затем загружали на спектрофотометр для прочтения планшетов Envision 2103 для анализа.
Анализ данных
Процент ингибирования вычисляли, используя следующее уравнение:

x=ОЕФ, полученные после обработки соединением
μ-=ОЕФ, полученные для отрицательных контролей (контроли без белка)
μ+=ОЕФ, полученные для положительных контролей (контроли с носителем ДМСО)
Значения IC50 получали построением нелинейных наименьших площадей вышеполученных данных, например, для уравнения XLfit3 205: y=A+((B-A)/(1+((C/x)^D))).
Качество результатов теста было проверено путем определения фактора Z Prime для каждого планшета для проведения теста, где Z Prime => 0,5 для результатов рассматривали как достоверный (Zhang et al, J Biomol Screening, 4:67-73, 1999).
Результаты Alphascreen (IC50 в микромоль) для некоторых соединений по изобретению, т.е., соединений, представленных в Таблице 1, приведены ниже в порядке, в котором они представлены в Таблице 1: 0,36, 0,84, 5,39, 1,44, 0,53, 0,76, 1,79, 0,36, 0,53, 0,035, 0,009, 0,005, 15,7, 0,11, 0,2, 10,09, 0,08, 0,045, 0,002, 0,53, 0,003, 100,0, 0,23, 0,095, 0,025, 0,035, 0,045, 0,007, 24,0, 0,045, 0,04, 0,01, 0,37, 0,002, 0,005, 0,007, 0,01, 0,003, 0,0009, 0,14, 0,005, 0,01, 0,008, 0,015, 0,01, 0,01, 0,69, 100, 0,295, 7,35, 12,83, 1,7, 0,66, NA, 0,01, 5,12, 0,72, 56,0, 0,40, 0,47, 0,15, 0,015, 0,05, 0,31, 0,23, 0,11, 0,20, 0,54, 0,68, 10,0, 0,04, 10,1, 0,008, 0,003, 0,007, 0,39, 0,12, 0,0006, 0,006, 0,006, 0,38, 0,0007, 0,005, 0,027, 0,0005, NA, NA, 0,065, 0,235, 0,01, 0,14, 0,002, 0,04 и 0,0005. В рамках изобретения, сокращение "NA" означает, что данные для соединения не доступны.
Пример 66
Тест на жизнеспособность клеток:
Общее:
Эффективность соединений согласно настоящему изобретению может также быть определена в клеточных тестах на уничтожение с использованием различных линий клеток и моделей опухолей на мышах. Например, их активность в отношении жизнеспособности клеток может быть оценена на группе культивируемых онкогенных и неонкогенных линий клеток, а также первичной популяции мышиных или человеческих клеток. В одном примере набора условий 5000-20000 клеток культивируют при 37°C и 10%-ном CO2 в подходящих средах для выращивания (например, 100 мкл модифицированной по Дульбекко среды Игла, дополненной 10%-ной эмбриональной телячьей сывороткой, аспарагиназой и 2-меркаптоэтанолом в случае pre-B Eμ-Myc опухолей мышей) в 96-луночных планшетах. Жизнеспособность клеток и полное число клеток могут быть проверены после инкубации в течение от нескольких часов до нескольких дней с 1 нМ - 100 мкМ соединений для идентификации тех, которые уничтожают клетки при EC50<10 мкМ. Жизнеспособность клеток может быть определена на основании способности клеток исключать пропидий йодид (10 мкг/мл путем анализа иммунофлюоресценции при длине волны эмиссии 660-675 нм на поточном цитометре (BD FAC Scan) или люминесцентной детекцией после инкубации с CELL TITER-GLO®. Альтернативно может использоваться высокопроизводительный колориметрический тест, такой как водный нерадиоактивный тест пролиферации клеток CELLTITER® 96 (Promega). Гибель клеток путем апоптоза подтверждают предварительной инкубацией клеток с 50 мкМ ингибитора каспазы, такого как zVAD-fmk.
a. Тест жизнеспособности клеток для Эмбриональных Фибробластов Мыши Mcl-1-/- (MEF):
Нейтрализация антиапоптотических белков Bcl-xL и Mcl-1 в нормальных клетках требуется перед тем, как клетка подвергнется апоптозу, через даунстрим-путь Bax/Bak см. Chen, L. et al. MoI. Cell (2005) 17, 393-403; Willis, S.N. et al. Genes Dev. (2005) 19, 1294-1305. Соединение, которое нацеливает Bcl-xL, не должно воздействовать на нормальные клетки, но может уничтожать некоторые раковые клетки, если они основаны больше на Bcl-xL и меньше на других антиапоптотических белках, например, Mcl-1, для выживания. Для того, чтобы отразить это, соединения по изобретению были проверены в отношении эффекта на выживание эмбриональных фибробластов мыши (MEFs) дикого типа (wt), MEFs с двойным нокаутом Bax/Bak (BB DKO), MEFs, которые экспрессируют Noxa, и MEFs, которые экспрессируют Bad. Noxa специфично нейтрализует Mcl-1. Следовательно, MEFs, которые экспрессируют Noxa, отражают типы раковых клеток, которые для выживания основываются на Bcl-xL и должны быть намного более чувствительными к уничтожению соединением, нацеливающим Bcl-xL, чем MEFs, где как Bcl-xL, так и Mcl-1 являются защитными.
В этом тесте клетки Mcl-1(-/-) использовали, чтобы подтвердить, что апоптоз клеток в присутствии малых молекул-миметиков BH3 был следствием преимущественно инактивации Bcl-xL. Эта инактивация оставляет Bax/Bak естественными и приводит к апоптозу. Люминесцентный тест жизнеспособности клеток CELLTITER-GLO представляет собой гомогенный способ определения числа жизнеспособных клеток в культуре, основанный на количественной оценке АТФ. Количество АТФ коррелирует с наличием метаболически активных клеток таким образом, что после лизиса клеток количество АТФ является пропорциональным сумме измеренной люминесценции.
Материалы
Эмбриональные фибробласты мыши Mcl-1(-/-) (MEFs) представляют собой адгерентную линию клеток, полученную в лаборатории. MEFs были выращены в колбах для культуры тканей Iwaki 75 см2 (cat # 3123-075) со средой FMA, которая состоит из:
• 89% DME Kelso
• 10% инактивированной нагреванием эмбриональной телячьей сыворотки (FCS) (Hyclone cat # SH30396.03)
• 1% 10 мМ аспарагина (Fluka cat # 11149)
• 275 мкл 1:2000 разведения 2-меркаптоэтанола добавляли к конечному объему 500 мл FMA (Sigma cat #M7522; разбавленного в MT-PBS)
FMA сохраняли при 4°C и использовали при 37°C. MEFs культивировали в среде FMA и собирали в MT-PBS и трипсин. Для тестирования жизнеспособности клеток MEF, клетки высевали отдельно в планшетах с 10% FCS-FMA и 1% FCS-FMA.
1% FCS-FMA состоит из:
• 98% DME Kelso
• 1% инактивированной нагреванием эмбриональной телячьей сыворотки (FCS) (Hyclone cat # SH30396.03)
• 1% 10 мМ аспарагина (Fluka cat # 11149)
• 275 мкл 1:2000 разведения 2-меркаптоэтанола добавляли к конечному объему 500 мл FMA (Sigma cat #M7522; разбавленного в MT-PBS)
Тесты проводили, используя 384-луночные планшеты Greiner для культуры тканей с плоским прозрачным дном (Interpath #781098). Соединения составляли в 384-луночных планшетах на 25 мкл с V-образным дном Matrical (cat #MP101-2-PP), герметизировали алюминиевой фольгой от Beckman Coulter (cat #538619) и сохраняли при 12°C в течение ночи. Получение соединения и титрование осуществляли в ДМСО уровня AnalaR (Merck cat #1.02952.2500). Используемым тестом детекции жизнеспособности клеток был CELLTITRE-GLO™, который является коммерчески доступным от Promega (cat # G7572), который сохраняли при -20°C и использовали при 37°C.
Автоматизированные системы, которые могут использоваться в этом тесте, включают: 1) диспенсер Multidrop - The Multidrop 384 (ThermoLabsystems) использовали, чтобы распределить клетки асептически в планшеты для проведения теста; 2) MiniTrak - система MiniTrak от Perkin Elmer использовалась для титрования планшетов с соединением; 3) Zymark - систему Zymark Sciclone ALH3000 с 100 нл наконечником pintool использовали для добавления соединения к клеткам; 4) En Vision - спектрофотометр для прочтения планшетов En Vision использовали, чтобы измерить жизнеспособность через детекцию люминесценции.
Соединения по изобретению получали как 10 мМ раствор в 100%-ном ДМСО и сохраняли при -20°C. Соединения размораживали при комнатной температуре и распределяли в 384-луночные планшеты Matrical. Стандартные соединения контроля, например 32,3 мМ этопозида, добавляли к планшетам в качестве контроля.
Планшеты могут быть герметизированы крышками из фольги и сохранены при 12°C в течение ночи. Планшеты с соединением оставляли для оттаивания при комнатной температуре, и соединения титровали 1:3 в 100% ДМСО на MiniTrak (см. раздел способов ниже - день 3).
Способ
1. День один - Разделение клеток
Среду аспирировали и клетки Mcl-1(-/-) промывали 10 мл нагретого MT-PBS. MT-PBS аспирировали и добавляли 1 мл трипсина. Колбы T75 инкубировали при 37°C до отслоения клеток. 4 мл 10% среды FCS FMA добавляли к обработанным трипсином клеткам, и весь объем помещали в 50 мл пробирку для центрифугирования и центрифугировали в течение 3 минут при 250g. Супернатант аспирировали, и осадок повторно суспендировали в 10 мл 10% FCS FMA. 3 мл этой суспензии клеток добавляли в чистую колбу на 75 см2, содержащую 17 мл 10% среды FCS FMA, таким образом выполняя разделение 3:10. Оставшуюся суспензию клеток использовали, чтобы осуществить разделение 1:50 в другой колбе на 75 см2 для дальнейшего культивирования.
2. День два - Засевание планшета для проведения теста и получение планшета с соединениями
Клетки собирали согласно стадии 1 способа, и осадок после центрифугирования повторно суспендиовали в 3 мл 10% FCS FMA. Число клеток определяли, рассчитывая в гемоцитометре Neubauer, и вычисляли разведение, чтобы достигнуть плотности 1x104 мл клеток мл-1 (500 клеток на лунку в 50 мкл среды). Отдельное разведение получали в 50 мл раствора 10% FCS FMA и 50 мл раствора 1% FCS FMA, соответственно.
Получали четыре планшета для проведения теста на планшет с соединениями. Два 384-луночных планшета содержали клетки Mcl-1(-/-) в 10% FCS FMA, и другие два планшета содержали клетки Mcl-1(-/-)в 1% FCS FMA.
Используя Multidrop, 25 мкл клеток распределяли асептически во все 384 лунки планшета для проведения теста. Планшеты оставляли для отстаивания в один слой при комнатной температуре в течение приблизительно 30 минут (минимизируя краевые эффекты) и затем помещали в один слой в термостат при 37°C. Планшеты оставляли для инкубации в течение ночи.
3. День три - Титрование планшетов с соединениями и обработка клеток
Планшеты с соединениями титровали, выполняя 3-кратные 11-точечные разведения, используя 100% ДМСО, на MiniTrak. После титрования соединений, 100 нл соединений добавляли к планшетам с клетками, используя Zymark Sciclone Pintool. Это было 1:250 разведение соединения, таким образом самая высокая конечная концентрация соединения была 40 мкМ. Планшеты затем возвращали в термостат при 37°C и инкубировали в течение ночи.
4. День четыре - Анализ жизнеспособности
Раствор CELLTITRE-GLO™ получали согласно инструкциям изготовителя восстановлением Субстрата CELLTITRE-GLO™ с использованием Буфера CELLTITRE-GLO™ и сохраняли после использования при -80°C. Планшеты удаляли из термостата и оставляли, чтобы уравновесить с комнатной температурой в течение 15 минут. 25 мкл разбавленного CELLTITRE-GLO™ добавляли в каждую лунку планшетов для проведения теста, используя Multidrop. Планшеты взбалтывали на вибросите для планшетов в течение 15 минут, после чего считывали на Envision с использованием протокола люминесценции.
Анализ данных
Процент ингибирования вычисляли, используя следующее уравнение:
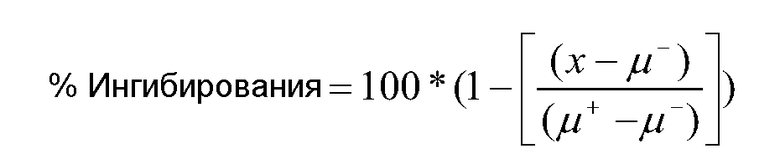
x=CPS, полученный после обработки образца соединения
μ=CPS, полученный для отрицательного контроля
μ+=CPS, полученный для положительного контроля
Значения IC50 получали построением нелинейных наименьших площадей вышеполученных данных с использованием, например, логарифмического соответствия с 4 параметрами (XLFit 4 eqn #205; y=A+((B-A)/(l+((C/x)ΛD))).
Качество результатов теста было проверено определением Z' фактора для каждого планшета для проведения теста, где Z'≥0,5 для результатов рассматривали как значимый (Zhang et al, J Biomol Screening, 4:67-73, 1999).
Результаты жизнеспособности клеток MEF Mcl-1-/- КО (то есть EC50 в микромоль, и тест, выполненный в присутствии 1%-ной Эмбриональной бычьей сыворотки) для некоторых соединений по изобретению, то есть соединений 74, 79, 90 и 92 в Таблице 1, составляют 0,06, 0,21, 0,59, 0,15, соответственно.
b. Тест жизнеспособности клеток для тромбоцитов
Обогащенную тромбоцитами плазму (PRP) инкубировали с соединением по изобретению в течение приблизительно 4 часов при 37°C. После инкубации тромбоциты уравновешивали с комнатной температурой в течение 20 минут и затем добавляли равный объем реактива CELL TITER-GLO™ (Promega Corporation). Образцы смешивали в течение двух минут и затем давали уравновеситься в течение дополнительных 10 минут при комнатной температуре. Люминесценцию, продуцированную образцами, количественно определяли, используя спектрофотометр для прочтения планшетов LJL Analyst.
c. Жизнеспособность клеток человеческой линии опухолевых клеток NCI-H146
Человеческие клетки мелкоклеточного рака легкого NCI-H146 (ATCC, Manassas, VA) высевали в плотности 50000 клеток на лунку в планшеты для культуры тканей с 96 лунками в полном объеме 100 мкл среды для культуры ткани, дополненной 10%-ной человеческой сывороткой (Invitrogen, Карлсбад, Калифорния) и обрабатывали 2-кратным серийным разведением представляющих интерес соединений от 10 мкМ до 0,020 мкл. Каждая концентрация была проверена в двойном экземпляре по меньшей мере 3 раза. Число жизнеспособных клеток после 48 часов обработки соединением определяли, используя Водный нерадиоактивный тест пролиферации клеток MTS CELLTITER96® согласно рекомендациям изготовителя (Promega Corp., Madison, WI).
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2525116C2 |
| АНТИТЕЛА К В7-Н3 И КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА | 2017 |
|
RU2764651C2 |
| ПРОИЗВОДНЫЕ 8-КАРБАМОИЛ-2-(2,3-ДИЗАМЕЩЕННОГО ПИРИД-6-ИЛ)-1,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ ИНДУЦИРУЮЩИХ АПОПТОЗ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2625315C2 |
| СРЕДСТВА, ВЫЗЫВАЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ И ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2594282C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКОГО АМИНОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОХИНОЛИНА ИЛИ РАЦЕМИЧЕСКОГО АМИНОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОИЗОХИНОЛИНА, СПОСОБЫ ИХ РАЗДЕЛЕНИЯ И РАЦЕМИЗАЦИИ, СПОСОБ ПОЛУЧЕНИЯ КЕТОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОХИНОЛИНА ИЛИ КЕТОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОИЗОХИНОЛИНА, СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРА КОНДЕНСИРОВАННОГО БИЦИКЛИЧЕСКОГО КОЛЬЦА, ЗАМЕЩЕННОГО ПЕРВИЧНЫМ АМИНОМ, ПРОИЗВОДНЫЕ 5,6,7,8-ТЕТРАГИДРОХИНОЛИНА | 2002 |
|
RU2308451C2 |
| АМИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ СНИЖЕНИЯ СЕКРЕЦИИ АПОЛИПОПРОТЕИНА В, СОЕДИНЕНИЕ | 1996 |
|
RU2141478C1 |
| АЗЕТИДИНИЛДИАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИН-ЛИПАЗЫ | 2010 |
|
RU2549547C2 |
| СТИМУЛЯТОРЫ СЕКРЕЦИИ ГОРМОНА РОСТА | 1996 |
|
RU2172742C2 |
| СРЕДСТВА, ИНДУЦИРУЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА, ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2568611C2 |
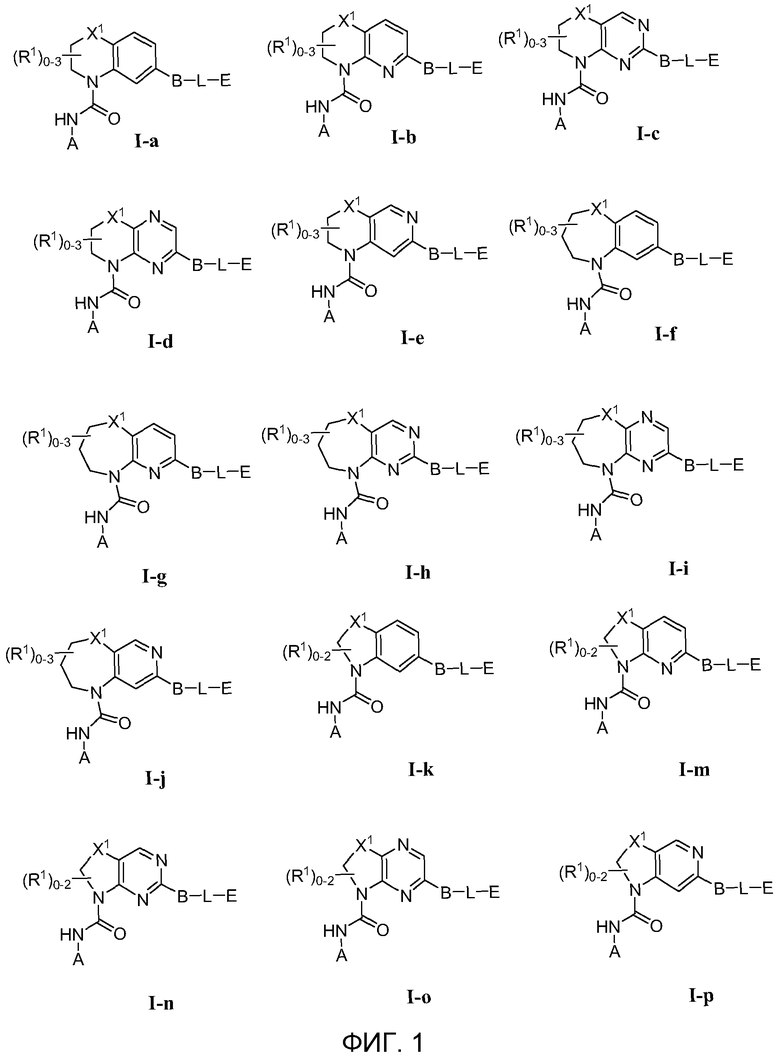
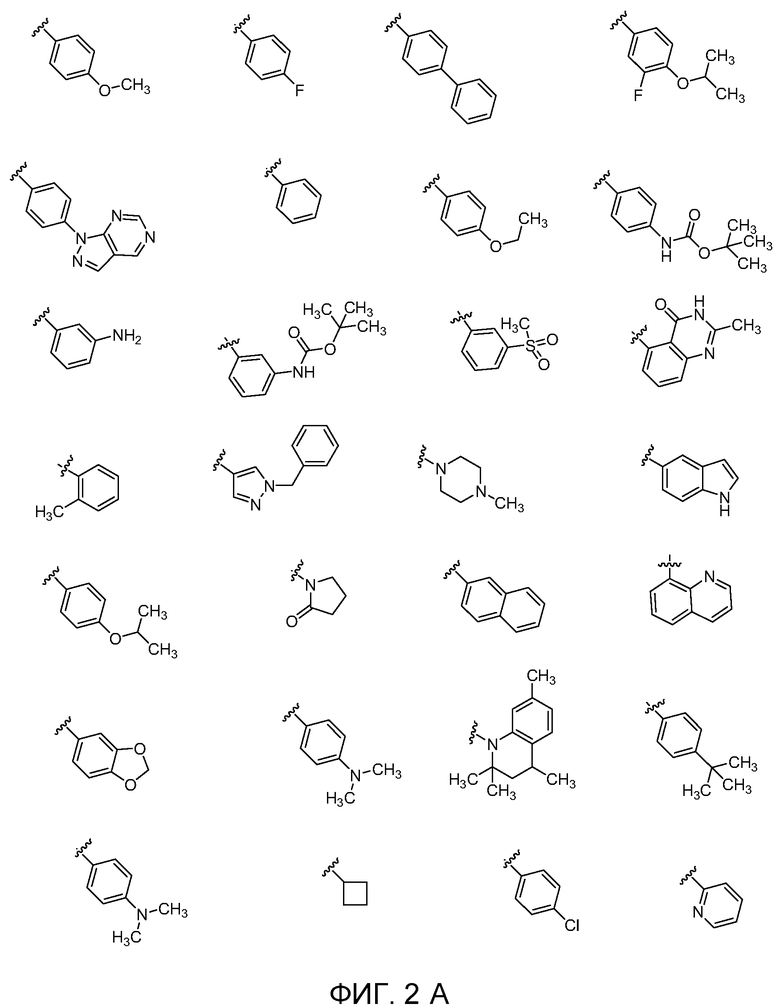
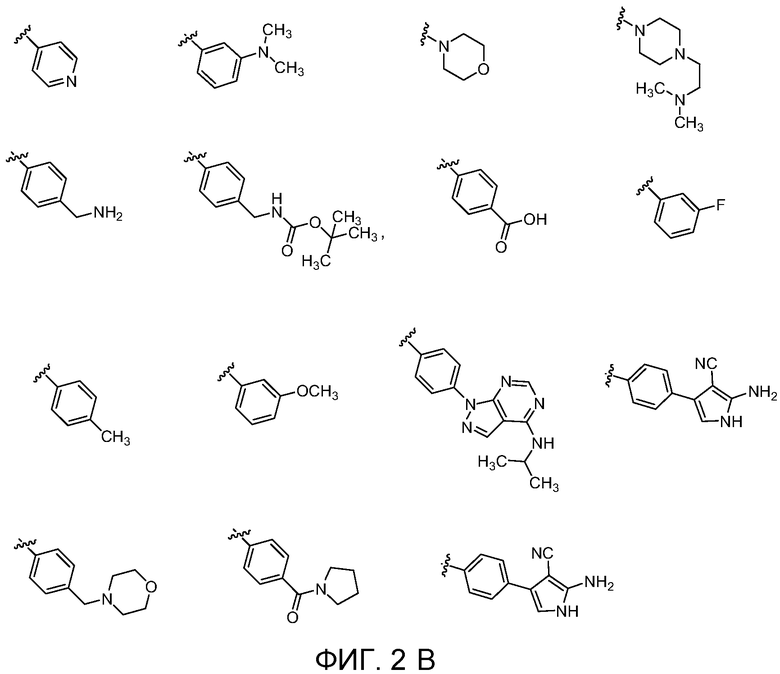


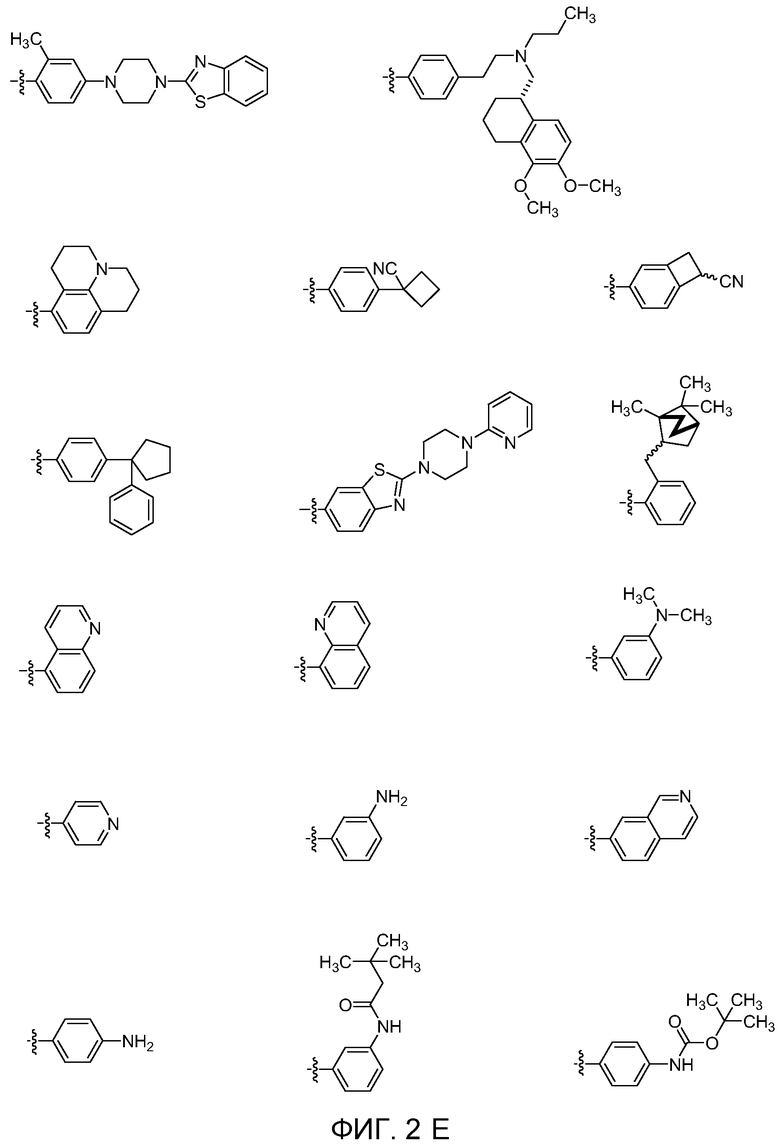

Изобретение относится к соединению или его фармацевтически приемлемой соли, причем соединение а) имеет Формулу I, в которой R1 отсутствует; индекс n означает целое число от 0 до 2, причем, когда n = 0, тогда X1 обозначает -СН2-; X1 обозначает член, выбранный из группы, состоящей из -СН2-, -O-, -N(H)- и -N(Ra)-, где Ra выбран из группы, состоящей из C1-6алкила; Х2а, X2b и Х2с, каждый независимо, выбраны из группы, состоящей из С(Н); А обозначает член, выбранный из группы (аа), где R3 независимо выбран из группы, состоящей из галогена и -Rg, где Rg выбран из группы, состоящей из C1-4алкила; В обозначает член, выбранный из группы соединений (ааа), где R4a отсутствует; L отсутствует или обозначает член, выбранный из группы, состоящей из С6арилен-С1-6гетероалкилена, C1-6гетероалкилена, С1-6алкилена, С2-6алкенилена, С2-6алкинилена и -O-, причем гетероалкилен включает 1 или 2 гетероатома, выбранных из группы, состоящей из N, О или S; Е обозначает водород или галоген; или, альтернативно, Е выбран из группы, состоящей из фенила, С5-6гетероарила, причем С5-6гетероарил выбран из группы, состоящей из пиразола, пиридина и пиразина, С3-7гетероциклоалкила, причем С3-7гетероциклоалкил выбран из группы, состоящей из морфолина, пирролидина, пиперидина и пиперазина, и С3-7циклоалкила, и в случае необходимости с Е может быть сконденсировано 1 кольцо, независимо выбранное из группы, состоящей из 5-членного гетероциклического кольца, включающего 2 гетероатома О, бензольного кольца и 5-6-членного гетероароматического кольца, включающего 1 или 2 гетероатома N, причем Е и 1 кольцо, в случае необходимости конденсированное с Е, независимо замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из галогена, -NRpRq, -ORр, -C(O)ORр, -NRpC(O)ORr, -S(O)2Rr, -Rr, -Rs, =O, -Z1-NRpRq и -Z1-Rs; причем Z1 обозначает C1-6алкилен; Rp и Rq, каждый независимо, выбраны из водорода и C1-6алкила; Rr выбран из группы, состоящей из C1-6алкила и фенила; Rs выбран из группы, состоящей из фенила и пиразола, и с Rs может быть конденсировано 1 пиримидиновое кольцо. Или соединение b) выбрано из группы, состоящей из 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)-N,N-диметилтиазол-4-карбоксамида, 2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-фторфенокси)тиазол-4-карбоксамида и N-(бензо[d]тиазол-2-ил)-6-(6-(метилсульфонилкарбамоил)-5-(3-фенилпропокси)пиридин-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Н)-карбоксамида. Изобретение также относится к конкретным соединениям. Соединения по изобретению предназначены для фармацевтической композиции, имеющей ингибирующую активность в отношении антиапоптотического белка BCl-XL. Фармацевтическая композиция включает терапевтически эффективное количество соединения формулы а) или b), или его фармацевтически приемлемой соли, и по меньшей мере один фармацевтически приемлемый разбавитель, носитель или эксципиент. Технический результат изобретения - новые соединения, ингибирующие антиапоптотический белок BCl-XL. 5 н. и 16 з.п. ф-лы, 2 табл., 7 ил., 66 пр.
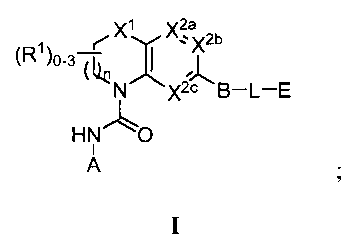
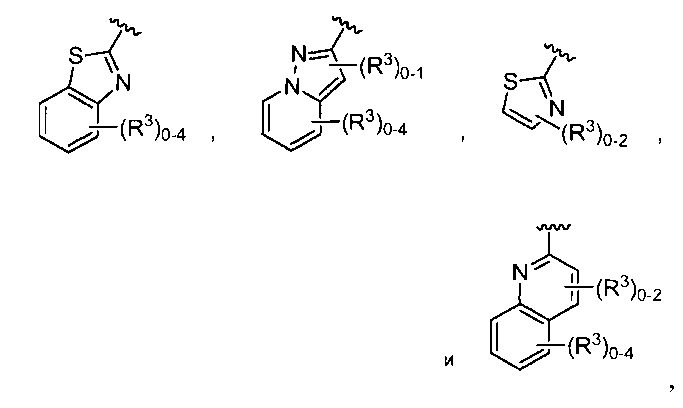
(аа)
 и
и 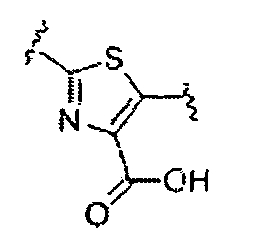
(ааа)
1. Соединение, или его фармацевтически приемлемая соль, причем соединение а) имеет Формулу I

в которой R1 отсутствует;
индекс n означает целое число от 0 до 2, причем, когда n=0, тогда X1 обозначает -СН2-;
X1 обозначает член, выбранный из группы, состоящей из -СН2-, -O-, -N(H)- и -N(Ra)-, где Ra выбран из группы, состоящей из C1-6алкила;
Х2а, X2b и Х2с, каждый независимо, выбраны из группы, состоящей из С(Н);
А обозначает член, выбранный из группы, состоящей из:

в которых R3 независимо выбран из группы, состоящей из галогена и -Rg, где Rg выбран из группы, состоящей из C1-4алкила;
В обозначает член, выбранный из группы, состоящей из:
и ,
в которых R4a отсутствует;
L отсутствует или обозначает член, выбранный из группы, состоящей из С6арилен-С1-6гетероалкилена, C1-6гетероалкилена, С1-6алкилена, С2-6алкенилена, С2-6алкинилена и -O-, причем гетероалкилен включает 1 или 2 гетероатома, выбранных из группы, состоящей из N, О или S;
Е обозначает водород или галоген; или, альтернативно, Е выбран из группы, состоящей из фенила, С5-6гетероарила, причем С5-6гетероарил выбран из группы, состоящей из пиразола, пиридина и пиразина, С3-7гетероциклоалкила, причем С3-7гетероциклоалкил выбран из группы, состоящей из морфолина, пирролидина, пиперидина и пиперазина, и С3-7циклоалкила, и в случае необходимости с Е может быть сконденсировано 1 кольцо, независимо выбранное из группы, состоящей из 5-членного гетероциклического кольца, включающего 2 гетероатома О, бензольного кольца и 5-6-членного гетероароматического кольца, включающего 1 или 2 гетероатома N, причем Е и 1 кольцо, в случае необходимости конденсированное с Е, независимо замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из галогена, -NRpRq, -ORр, -C(O)ORр, -NRpC(O)ORr, -S(O)2Rr, -Rr, -Rs, =O, -Z1-NRpRq и -Z1-Rs; причем Z1 обозначает C1-6алкилен; Rp и Rq, каждый независимо, выбраны из водорода и C1-6алкила; Rr выбран из группы, состоящей из C1-6алкила и фенила; Rs выбран из группы, состоящей из фенила и пиразола, и с Rs может быть конденсировано 1 пиримидиновое кольцо
или b) выбрано из группы, состоящей из
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)-N,N-диметилтиазол-4-карбоксамида,
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-фторфенокси)тиазол-4-карбоксамида
и
N-(бензо[d]тиазол-2-ил)-6-(6-(метилсульфонилкарбамоил)-5-(3-фенилпропокси)пиридин-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Н)-карбоксамида.
2. Соединение по п.1, или его фармацевтически приемлемая соль, причем соединение имеет формулу, выбранную из группы, состоящей из
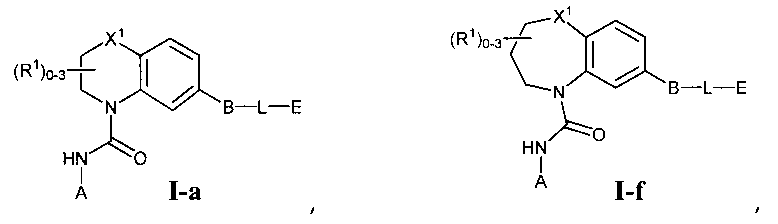
и
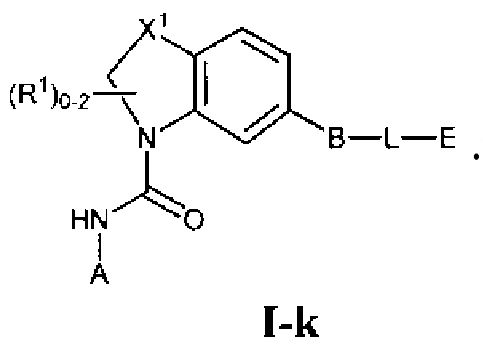
3. Соединение по п.1, или его фармацевтически приемлемая соль, причем соединение Формулы I имеет Формулу I-а,
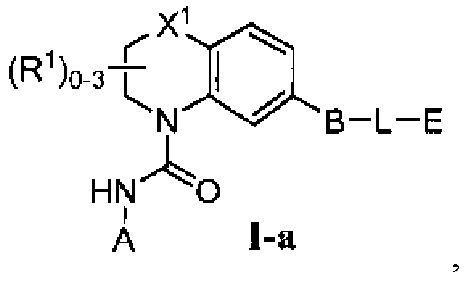
в которой R1 отсутствует;
X1 обозначает -СН2-, -O-, -N(Н)-, -N(Ra) -; и
А обозначает

4. Соединение по п.3, или его фармацевтически приемлемая соль, в котором X1 обозначает -СН2- или -О-.
5. Соединение по п.1, или его фармацевтически приемлемая соль, в котором в Формуле I В обозначает
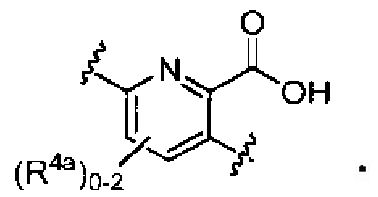
6. Соединение по п.1, или его фармацевтически приемлемая соль, в котором В обозначает
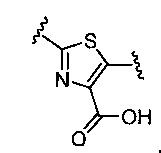
7. Соединение по одному из пп.1-5, или его фармацевтически приемлемая соль, в котором L отсутствует или выбран из группы, состоящей из С6арилен-С1-6гетероалкилена, причем гетероалкилен включает 1 или 2 гетероатома, выбранных из N, О или S, C1-6гетероалкилена, С1-6алкилена, С2-6алкенилена, С2-6алкинилена и -О-; и
Е обозначает кольцо, выбранное из группы, состоящей из фенила, С5-6гетероарила, причем С5-6гетероарил выбран из группы, состоящей из пиразола, пиридина и пиразина, С3-7гетероциклоалкила, причем гетероциклоалкил выбран из группы, состоящей из морфолина, пирролидина, пиперидина и пиперазина, и С3-7циклоалкила, и причем с Е в случае необходимости конденсировано 5-6-членное гетероароматическое кольцо, причем 5-6-членное гетероароматическое кольцо включает 1 или 2 гетероатома N, и где Е и указанное кольцо, в случае необходимости конденсированное с Е, замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из фтора, хлора, брома, -NRpRq, -ORр, -C(O)ORр, -NRpC(O)ORr, -Rr, -Rs, -Z1-NRpRq и -Z1-Rs; причем Z1 обозначает C1-6алкилен; Rp и Rq, каждый независимо, выбраны из водорода и C1-6алкила; Rr обозначает С1-6алкил; Rs обозначает фенил или пиразол, причем с Rs может быть конденсировано 1 пиримидиновое кольцо.
8. Соединение по п.5, или его фармацевтически приемлемая соль, в котором L отсутствует, и Е выбран из группы, состоящей из фенила и пиридила, и причем с Е может быть конденсировано кольцо, выбранное из группы, состоящей из пиримидин-4-она, пиримидин-2-она, бензольного кольца, пиридинового кольца, пиррольного кольца, пиразольного кольца и имидазольного кольца, причем Е и кольцо, в случае необходимости конденсированное с Е, каждое, может быть, в случае необходимости, независимо замещено.
9. Соединение по п.1, или его фармацевтически приемлемая соль, в котором L обозначает C1-4алкилен или C1-4гетероалкилен.
10. Соединение по п.1, или его фармацевтически приемлемая соль, в котором L выбрано из группы, состоящей из:
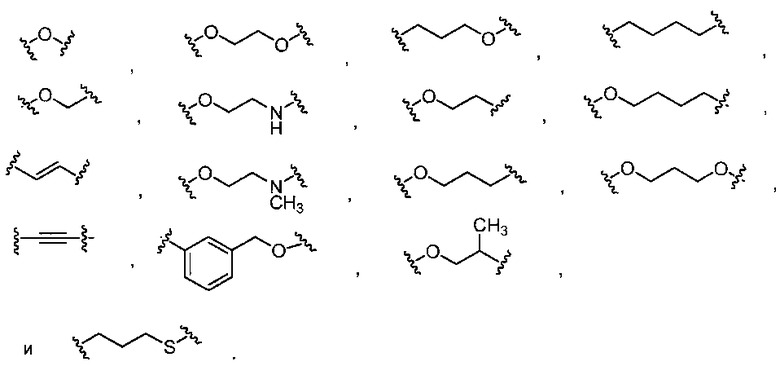
11. Соединение по п.9 или 10, или его фармацевтически приемлемая соль, в котором L присутствует, и Е выбран из группы, состоящей из фенила, пиридила, пиперидинила, пиперазинила, морфолино, пирролидинила, пирролидонила и циклобутила, и причем с Е могут быть конденсированы пиридин, бензольное кольцо, пиримидин-4-он или пиримидин-2-он, и причем Е и указанное кольцо, в случае необходимости конденсированное с Е, замещены от 0 до 5 раз заместителями R6, выбранными из группы, состоящей из фтора, хлора, - NRpRq, -ORp, -NRpC(O)ORr, -Rr, -Z1NRpRq и -Rs, причем Rp и Rq, каждый независимо, выбраны из водорода и С1-6алкила.
12. Соединение по одному из пп.1-6, или его фармацевтически приемлемая соль, в котором Е обозначает фенил и является замещенным в мета- или пара-положении в случае необходимости замещенной группой Rs, имеющей формулу, выбранную из группы, состоящей из:
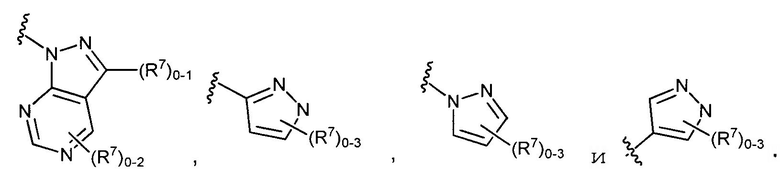
13. Соединение по одному из пп.1-6, или его фармацевтически приемлемая соль, в котором Е выбран из группы, состоящей из:

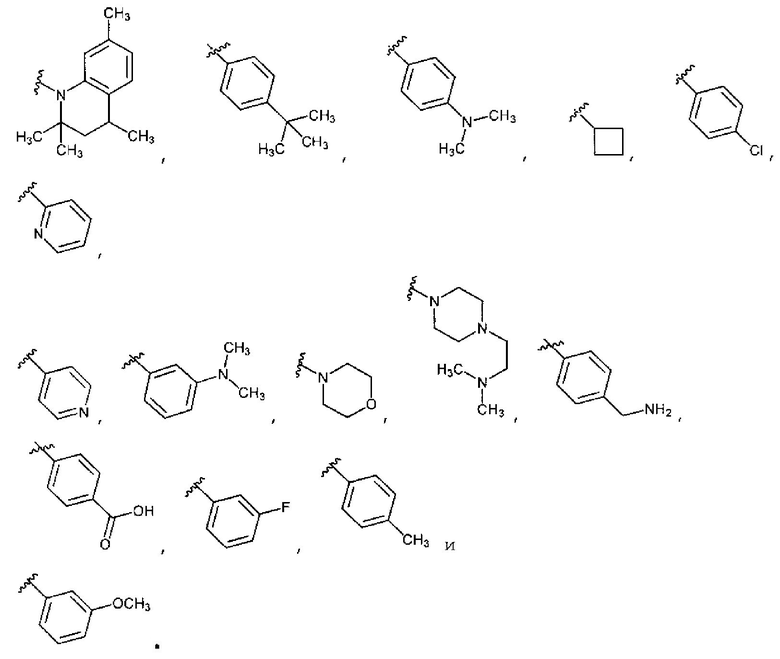
14. Соединение по одному из пп.1-6, или его фармацевтически приемлемая соль, в котором Е выбрано из группы, состоящей из:

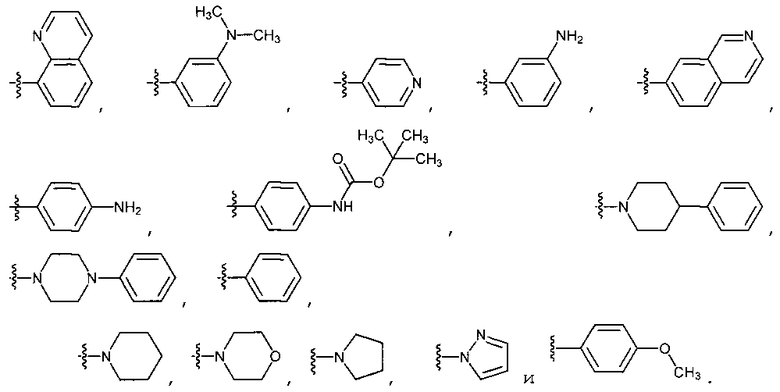
15. Соединение по п.1 или 3, или его фармацевтически приемлемая соль, в котором L отсутствует и Е обозначает водород или галоген.
16. Соединение по п.1 или 3, или его фармацевтически приемлемая соль, в котором L выбран из группы, состоящей из C1-6гетероалкилена, C1-6алкилена, С2-6алкенилена и С2-6алкинилена, и Е обозначает водород.
17. Соединение по п.16, или его фармацевтически приемлемая соль, в котором L обозначает C1-4гетероалкилен.
18. Соединение или его фармацевтически приемлемая соль, причем указанное соединение выбрано из группы, состоящей из следующих соединений:
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота,
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-гидроксипиколиновая кислота;
6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-изобутоксипиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-бутоксипиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(циклобутилметокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(2-оксопирролидин-1-ил)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-морфолинопропокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(2-(диметиламино)этил)пиперазин-1-ил)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-фенилпиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-о-толилпиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(метилсульфонил)фенил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-2-ил)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(пиридин-4-ил)этокси)пиколиновая кислота;
6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-3-(3-(пиридин-4-ил)пропокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(фенилэтинил)пиколиновая кислота;
(Е)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторстирил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-фторфенэтокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-феноксиэтокси)пиколиновая кислота;
6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-3-(3-феноксипропокси)пиколиновая кислота;
3-(2-(3-аминофенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-хлорофенил)пропокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-метоксифенэтокси)пиколиновая кислота;
6-(4-(бензо[d]тиазол-2-илкарбамоил)-1-метил-1,2,3,4-тетрагидрохиноксалин-6-ил)-3-(3-феноксипропокси)пиколиновая кислота;
3-(2-(4-(аминометил)фенокси)этокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-трет-бутилфенил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-(диметиламино)фенэтокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(диметиламино)фенокси)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(4-(4-метоксифенил)бутокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенил)пропокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенокси)пропокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(3-(диметиламино)фенокси)пропокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(диметиламино)бензиламино)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(4-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(3-(трет-бутоксикарбониламино)фенокси)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-6′-(4-метилпиперазин-1-ил)-3,3′-бипиридин-2-карбоновая кислота;
3-(3-(бензо[d][1,3]диоксол-5-илокси)пропокси)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-(2,2,4,7-тетраметил-3,4-дигидрохинолин-1(2Н)-ил)этокси)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1-бензил-1Н-пиразол-4-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(бифенил-4-илметокси)пиколиновая кислота;
3-(3-((3-аминофенокси)метил)фенил)-6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-((3-(трет-бутоксикарбониламино)фенокси)метил)фенил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(1Н-индол-5-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(2-метил-4-оксо-3,4-дигидрохиназолин-6-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(нафталин-2-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(хинолин-8-илокси)пропокси)пиколиновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)индолин-6-ил)тиазол-4-карбоновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-2,3,4,5-тетрагидро-1Н-бензо[b]азепин-8-ил)-3-(3-фенилпропокси)пиколиновая кислота;
5-(4-метоксифенил)-2-(1-(тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота;
5-(4-метоксифенил)-2-(1-(4-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота;
5-(4-метоксифенил)-2-(1-(5-метилтиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-хлоротиазол-4-карбоновая кислота;
(Е)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(проп-1-енил)тиазол-4-карбоновая кислота;
6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)пиколиновая кислота;
2-(4-(хинолин-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-фенилтиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-фторфенил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(3-фторфенил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенилтиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-фенокситиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-п-толилтиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-фторфенокси)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-метоксифенил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(3-метоксифенил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-карбоксифенил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-метоксифенокси)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-фторфенокси)тиазол-4-карбоксамид;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновая кислота;
2-(1-(6-фторбензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(3-(пиридин-4-илтио)пропил)тиазол-4-карбоновая кислота;
(Е)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-стирилтиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-этоксифенил)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(метил(2-феноксиэтил)амино)тиазол-4-карбоновая кислота;
2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-5-(4-фенилбутил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-феноксипропил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-изопропоксифенил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-фтор-4-изопропоксифенил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(4-метоксифенил)-N,N-диметилтиазол-4-карбоксамид;
5-(3-(3-аминофенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота;
5-(3-(4-(аминометил)фенокси)пропил)-2-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-(4-трет-бутилфенокси)пропил)тиазол-4-карбоновая кислота;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(3-(4-(трет-бутоксикарбониламино)фенокси)пропил)тиазол-4-карбоновая кислота;
6-(4-(бензо[d]тиазол-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)-3-(3-фенилпропокси)пиколиновая кислота;
3-(3-фенилпропокси)-6-(4-(пиразоло[1,5-а]пиридин-2-илкарбамоил)-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)пиколиновая кислота;
6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-фенилпропокси)пиколиновая кислота;
N-(бензо[d]тиазол-2-ил)-6-(6-(метилсульфонилкарбамоил)-5-(3-фенилпропокси)пиридин-2-ил)-2Н-бензо[b][1,4]оксазин-4(3Н)-карбоксамид;
2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-5-(бифенил-4-ил)тиазол-4-карбоновая кислота;
5-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота; и
5-(3-(4-(1Н-пиразоло[3,4-d]пиримидин-1-ил)фенокси)пропил)-2-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)тиазол-4-карбоновая кислота.
19. Фармацевтическая композиция, имеющая ингибирующую активность в отношении антиапоптотического белка BCl-XL, включающая терапевтически эффективное количество соединения по п.1, или его фармацевтически приемлемой соли, и по меньшей мере один фармацевтически приемлемый разбавитель, носитель или эксципиент.
20. 6-(1-(бензо[d]тиазол-2-илкарбамоил)-1,2,3,4-тетрагидрохинолин-7-ил)-3-(3-(4-(диметиламино)фенокси)пропокси)пиколиновая кислота или ее фармацевтически приемлемая соль.
21. Фармацевтическая композиция, имеющая ингибирующую активность в отношении антиапоптотического белка BCl-XL, включающая терапевтически эффективное количество соединения по п.20, или его фармацевтически приемлемой соли, и по меньшей мере один фармацевтически приемлемый разбавитель, носитель или эксципиент.
| US 6699857 B1, 02.03.2004 | |||
| US 4107167 A, 15.08.1978 | |||
| US 7091227 В2, 15.08.2006 | |||
| ШАХТНАЯ ПОДЪЕМНАЯ УСТАНОВКА | 0 |
|
SU233198A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

