Изобретение относится к процессу получения препаратов из плазмы крови, а именно к способу очистки растворов иммуноглобулинов (ИГ) IgG для внутривенного и внутримышечного введения, получаемых методом спиртового фракционирования, от примеси каприловой кислоты (КК), применяемой на стадии инактивации вирусов.
Изобретение позволяет усовершенствовать технологии получения безопасных вирусинактивированных растворов ИГ с низким остаточным содержанием КК при высоком выходе конечного продукта, с сохраненными физико-химическими свойствами и специфической активностью с использованием простого технического приема.
Традиционные схемы спиртового фракционирования и его модификации [1, 2] существенно снижают риск вирусных трансмиссий, но не гарантируют полную инактивацию вирусов [3, 4, 5, 6, 7]. Поэтому для гарантии вирусной безопасности препараты ИГ получают с применением дополнительных стадий обработки [8, 9], направленных на инактивацию и/или удаление вирусов и не оказывающих неблагоприятного воздействия на качество, безопасность продукта и не вызывающих снижения выхода ИГ. Одним из таких способов является каприлатная обработка.
Преимуществами каприлатной обработки [10, 11, 12, 13, 14, 15] относительно других способов инактивации вирусов являются простота исполнения, легкость встраивания в технологию, экологичность, сохранение показателей активности антител и высокая скорость инактивации вирусов. Кроме того, присутствие стабилизаторов иммуноглобулинов в растворе, таких как ПЭГ [16], аминокислоты, моносахара, олигосахара, сахароспирты, не мешают процессу инактивации вирусов [17]. В качестве вируцидного реагента обычно используют КК или каприлат натрия (КН) в конечной концентрации в растворе иммуноглобулина от 6 до 100 мМ [10, 11], при температуре от 15°C до 26°C [12, 13], pH от 4,0 до 6,0 [12, 13, 14], времени воздействия не менее 30 минут [15].
При инактивации вирусов с помощью любых химических реагентов возникает серьезная проблема максимального удаления последних из растворов с сохранением выхода и активности препарата. Превышение допустимого остаточного содержания вирусинактивирующих реагентов в препарате может приводить к серьезным нежелательным последствиям. Присутствие значительных остаточных количеств КК в препаратах может оказывать негативное воздействие на физико-химические свойства и функциональную активность антител, но, что более важно, на безопасность препарата. Так, внутривенное введение КК кроликам может ингибировать реактивность тромбоцитов [18], что может нести опасность при состояниях связанных с кровотечением. Кроме того, ИГ с остаточным содержанием КК около 125 мкг/мл имеет более высокие уровни титров анти-A, анти-B антител [19], что повышает вероятность снижения содержания гемоглобина, эритроцитов [20]. Поэтому получение препаратов ИГ с минимальным остаточным содержанием КК, более низким, чем в существующих препаратах ИГ, является актуальной задачей.
Известны различные методы удаления КК из растворов ИГ: многостадийная последовательная обработка - фосфат кальция, ультрафильтрация, порошковый активированный уголь при нормальных условиях (комнатная температура и нейтральное значение pH), хроматография на ДЭАЭ-сефадексе А50 [21]; комбинированное использование ацетата кальция, диафильтрация против 10 объемов буфера, хроматография [22]; диафильтрация и 1-2-кратная анионообменная хроматография [23]; полиэтиленгликоль, аэросил/диатомит и анионообменная хроматография [24]; осаждение сульфатом аммония и гель-хроматография на S-300 [25]; 1-2-кратная хроматография [26, 27, 28]; осаждение сульфатом аммония [29]; одиночная хроматография [30]; диализ [31, 32].
Однако известные методы удаления КК имеют существенные недостатки. Так, обработка порошковым активированным углем при нормальных условиях pH и температуры приводит к снижению выхода и агрегации ИГ, а также требует проведения дополнительных стадий по удалению угля, включающих центрифугирование и/или фильтрацию [21], при 1-2-кратной хроматографии происходит снижение выхода ИГ как минимум на 10% [26, 27, 28], при осаждении сульфатом аммония выход снижается до 68% [29], диализ является нетехнологичным и неэффективным методом, так как разделение осуществляется только по молекулярным весам и к тому же существует вероятность накопления пирогена [31, 32].
Известен также метод многократной хроматографической очистки, который позволяет снизить остаточное содержание КК до 216 мкг/мл в препаратах Gamunex (Talecris) [33] и Gammaked (Kedrion Biopharma, INC.) [34], но при этом также наблюдается значительное снижение выхода ИГ до 18% [35].
Таким образом, все известные способы, во-первых, не позволяют удалить КК из растворов ИГ ниже 125 мкг/мл, в лучшем случае ниже 40 мкг/мл [36], во-вторых, они обычно многостадийные и сопровождаются значительным снижением выхода ИГ (от 10 до 40%), увеличением длительности технологического процесса и затрат. Поэтому актуальным является поиск альтернативных - более эффективных, экономичных, «быстрых» методов удаления КК из растворов ИГ.
В качестве прототипа настоящего изобретения выбран метод, позволяющий получать раствор ИГ с наиболее низким остаточным содержанием КК от 40 до 50 мкг/мл [36]. Вирусинактивирующую обработку КК иммуноглобулин-содержащей фракции проводят при pH 4,7÷6,0 и температуре 15÷37°C, концентрации КК 6÷100 мМ, ионной силе 0,15 М. Удаление КК осуществляется на трех стадиях при физиологичной концентрации соли и pH не ниже 4,7: а) центрифугирование с получением супернатанта; б) ультрафильтрация на установке 100 КДа и менее; в) осветляющая фильтрация на фильтре BC0025L60SP03A (0,5 мк, картридж фирмы 3М Cuno), иногда с дополнительным центрифугированием. Данный способ позволяет получать растворы ИГ с высокой электрофоретической чистотой более 95% и с низким остаточным содержанием КК от 40 до 50 мкг/мл или от 9,25 до 11,56 мкмоль/г белка с учетом конечной концентрации белка в растворе 30 г/л. Однако удаление КК требует проведения, по крайней мере, двух различных стадий ультрафильтрации и осветляющей фильтрации, при этом стадия ультрафильтрации длительна, так как требует многократных циклов диафильтрации, что несет опасность пирогенизации продукта и сопровождается значительными потерями по белку, поэтому наблюдается довольно низкий выход ИГ 50÷60% с допустимым содержанием агрегатов до 3%.
Задачей заявленного изобретения является усовершенствование технологии получения безопасных вирусинактивированных растворов ИГ с низким остаточным содержанием КК, с сохраненными физико-химическими свойствами, специфической активностью и высоким выходом ИГ.
Технический результат предлагаемого изобретения заключается в разработке способа максимального удаления вирусинактивирующего реагента КК при высоком выходе ИГ, с сохранением физико-химических свойств и специфической активности.
Указанный результат достигается следующим образом: обработку растворов ИГ проводят КК в концентрации 6÷100 мМ, при pH 3,9÷4,6, ионной силе не более 0,03 М, температуре 15÷37°C и перемешивании от 0,5 до 2 ч, а очистку от примеси вирусинактивирующего реагента проводят путем фильтрации через угольный фильтр при температуре 5÷15°C, ионной силе не более 0,03 М и pH 3,9÷4,6. Полученный раствор ИГ содержит КК не более 20 мкг/мл или 2,77 мкмоль/г белка с учетом конечной концентрации белка в растворе 50 г/л, агрегатов не более 1%, выход иммуноглобулина составляет не менее 98%.
В отличие от прототипа вирусинактивирующая обработка КК иммуноглобулин-содержащей фракции проводится при pH 3,9÷4,6 и низкой ионной силе - не более 0,03 М, удаление КК из растворов ИГ осуществляется в одну стадию фильтрации через угольный фильтр при pH 3,9÷4,6, температуре 5÷15°C и ионной силе не более 0,03 М. Выбранное значение pH раствора ИГ 3,9÷4,6 оптимально как с учетом эффективности сорбции КК при максимальном переводе в неионизированную (молекулярную) плохо растворимую в воде форму, которая увеличивается с постепенным снижением pH, в особенности ниже константы диссоциации рКа=4,895 [37], так и увеличения диссоциации связанной с ИГ КК (усиление отталкивания одноименно заряженных ионов с увеличением заряда). Фильтрация вирусинактивированного раствора ИГ при pH ниже 3,9 или выше 4,6 приводит к агрегации и димеризации, снижению эффективности удаления КК и выхода ИГ, а также возрастанию АКА.
Удаление КК из растворов ИГ при температуре 5÷15°C и низкой ионной силе не более 0,03 М создает благоприятные условия для сорбции КК, предупреждения агрегации, димеризации и возрастания АКА, а также повышения выхода ИГ. Для лучшего задерживания КК на угольном фильтре необходимо, чтобы температура раствора иммуноглобулина была более низкой, чем температура плавления КК, равная 16,5°C [38], но выше температуры 0÷4°C, так как при температуре, близкой к температуре замерзания раствора, снижается массообмен, повышается вязкость и, соответственно, снижается эффективность удаления КК.
Отличительные признаки заявленного изобретения позволяют получить раствор ИГ с минимальным содержанием КК - не более 20 мкг/мл, с низким содержанием агрегатов - не более 1%, увеличить выход ИГ - не менее 98%, а также сократить длительность процесса удаления КК.
Согласно настоящему изобретению в качестве исходного материала для обработки используются фракции, содержащие иммуноглобулины классов G, А, М, очищенные от других плазменных белков путем дополнительной стадии спиртового осаждения и/или диа/ультрафильтрации и/или хроматографической очистки. Конкретными возможными примерами таких фракций являются: центрифугат III (Б) или фракция II (паста иммуноглобулина G), комплексный иммуноглобулиновый препарат из фракции III, моноклональные и рекомбинантные антитела, полученные любым известным способом.
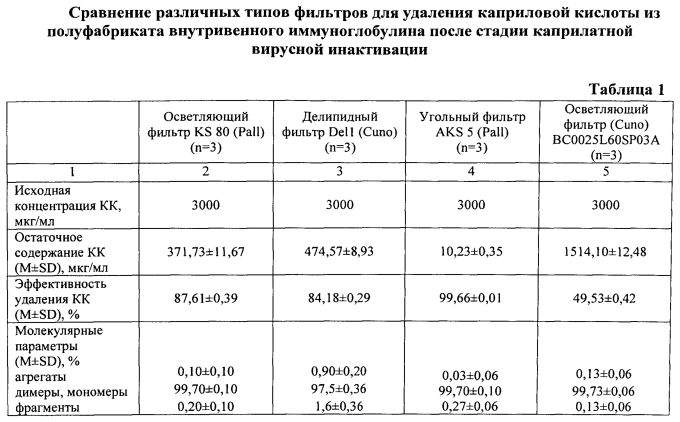
При этом можно использовать как свежевыделенные фракции, так и замороженные при низких температурах, предварительно растворив их в апирогенной воде (воде для инъекций) или подходящем буферном растворе с ионной силой не более 0,03 М. При необходимости раствор ИГ подвергается ультрафильтрации/диафильтрации для удаления этанола, снижения ионной силы до не более 0,03 М, доведения концентрации белка до 1,0÷12,5%. Перед добавлением КК температура раствора ИГ понижается до 0÷4°C. Обработка КК проводится в концентрации, достаточной для создания вирусинактивирующей дозы реагента в растворе 6÷100 мМ, при концентрации белка в растворе 1,0÷12,5%, pH 3,9÷4,6, ионной силе не более 0,03 М и температуре 15÷37°C и перемешивании в течение 0,5÷2 ч, а очистка от примеси вирусинактивирующего реагента проводится путем фильтрации через угольный фильтр марок ZetaCarbon (Cuno) - R53S(LP), R54S(LP), R55S(LP) или AKS (Pall) - AKS 5, AKS 6 при температуре 5÷15°C, ионной силе не более 0,03 М и pH 3,9÷4,6. В таблице 1 представлены результаты сравнения различных типов фильтров для удаления каприловой кислоты из полуфабриката внутривенного иммуноглобулина после стадии каприлатной вирусной инактивации. Из приведенных данных видно, что не угольные фильтры не обеспечивают эффективное удаление КК. Далее в зависимости от готовой формы препарата (жидкая, сухая; внутривенная, внутримышечная) осуществляется доведение значения pH, осмолярности, концентрации стабилизаторов до требуемых значений. При необходимости снижения антикомплементарной активности (АКА) используется любой известный способ.
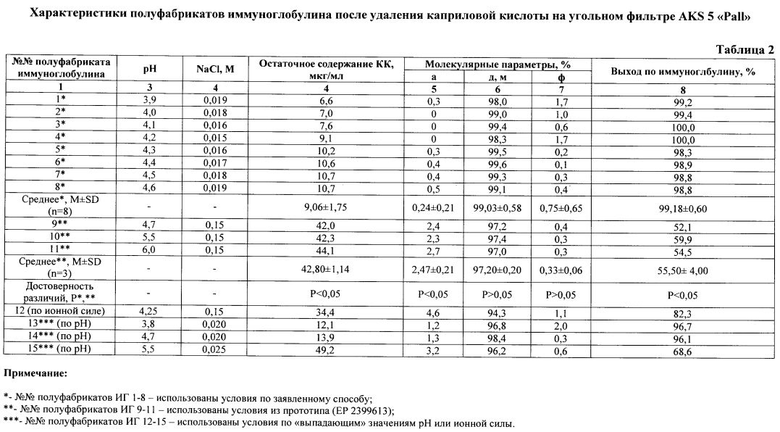
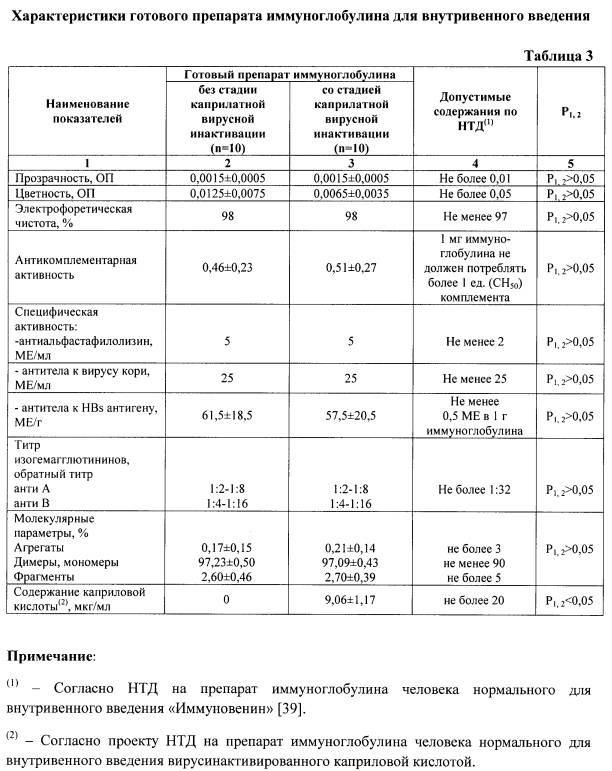
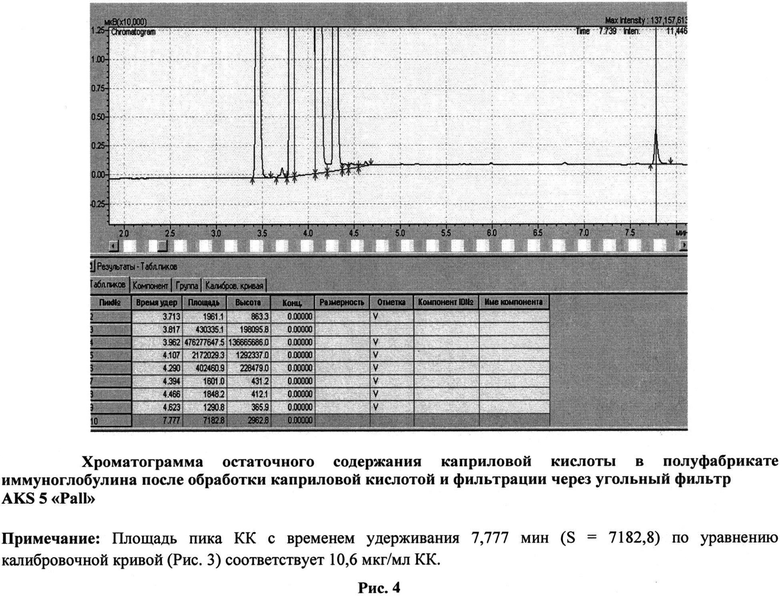
Как видно из представленных данных (табл. 2, рис. 1, 2), использование предлагаемого способа, в отличие от способа-прототипа, позволяет получать препарат без образования агрегатов - не более 1,0%, тогда как в прототипе до 3,0%, и практически без изменения молекулярного состава, а также без снижения выхода - выход не менее 98%. При получении иммуноглобулинов по заявленному способу не выявлено ухудшения физико-химических и биологических свойств препаратов, снижения концентрации антибактериальных и противовирусных антител (табл. 3). Эффективность очистки, рассчитанная по остаточному содержанию КК (не более 20 мкг/мл) (Рис. 3, 4), составляет более 99% (табл. 1).
Контроль полуфабриката и готового препарата иммуноглобулина для внутривенного введения проводится по следующим методикам.
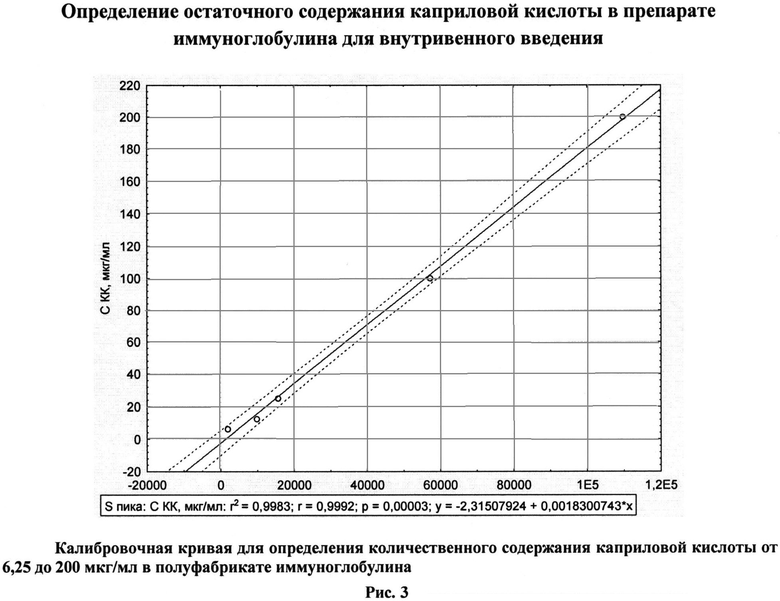
Электрофоретическая однородность определяется методом электрофореза на пленках из ацетата целлюлозы по ФС 42-3874-99, с. 20. Молекулярные параметры - методом гельфильтрации по ФС 42-3874-99, с. 28. Антикомплементарная активность контролируется по МУК 3.3.2.1063-010. Содержание анти-А и анти-В иммунных изогемагглютининов определяется модифицированным методом непрямой гемагглютинации согласно требованиям Европейской Фармакопеи 2010, 7.0, монография 0918. Натрия хлорид - по ФС 42-3874-99, с. 59. Глицин - по МУК 4.1/4.2.588-96, с. 114. Количественное содержание мальтозы и глюкозы определяется специфическими ферментативными наборами: Maltose/Sucrose/D-Glucose («BOEHRINGER MANNHEIM/R-BIOPHARM AG», Германия) или ENZYTEC D-Glucose, Enzytec Maltose («SCIL Diagnostics GmbH, Германия). Определение пирогенности проводится согласно требованиям Европейской Фармакопеи 2010, 7.0, монография 0918. Осмолярность определяется криоскопическим методом на миллиосмометре-криоскопе термоэлектрическом МТ-4. Статистическая обработка результатов проводится с использованием компьютерных программ «Excel 4.0» и «Statistica 5.7». Определение содержания КК проводится методом газовой хроматографии согласно известному методу (Hans J.Т.F. Nelis, Marc F. Lefevere, etc. Chromatographic determination of N-acetyl-DL-tryptophan and octanoic acid in human albumin solutions. Journal of Chromatography, №333, 1985, p.381-387) в нашей модификации. Пробоподготовка состоит из этапов осаждения белка, метилирования и экстракции. К 500 мкл образца с содержанием белка 2,5%, содержащего 20% ацетонитрила, добавляется в два этапа 1 М соляная кислота. Затем проводится экстракция 5 мл гексана, с последующим отделением и высушиванием органической фазы. К сухому остатку добавляется 5% серная кислота в метаноле в объеме 200 мкл и проводится стадия метилирования в течение 10 ч при температуре 50°C. После этого к образцам приливается ледяная вода в объеме 100 мкл и гексан в объеме 400 мкл. Анализируется органическая фаза. Газохроматографический анализ выполняется на газовом хроматографе GC-2010. Используется капиллярная колонка НР-5, 30 м × 0,32 мм с толщиной пленки 0,25 мкм, фирмы Hewlett-Packard (№19091-60312). Предел обнаружения метода составляет 6,25 мкг/мл. Коэффициент корреляции - не менее 0,99.
Пример 1
Выделение иммуноглобулина проводят этанольным методом фракционирования по методу Кона 6. Полученный фильтрат Б (III по Кону) подвергают ультрафильтрации при ионной силе не более 0,015 и значении pH 4,0, с последующей диафильтрацией против воды дистиллированной с величиной pH 3,8. Затем в полуфабрикат иммуноглобулина с концентрацией белка 7,0% и температурой 0÷4°C и pH 4,0 при постоянном перемешивании вводят КК капельно до концентрации 0,3%, постепенно подогревают раствор до 20°C и проводят инкубацию при температуре 20°C и значении pH 3,9 в течение 1 ч при постоянном перемешивании. Затем снижают температуру до 5°C и проводят фильтрацию через угольный фильтр AKS 5. Затем полуфабрикат иммуноглобулина для снижения АКА обрабатывают пепсином в дозе 1:10-6 в присутствии 2% мальтозы. После удаления пепсина фильтрацией через угольный фильтр AKS 5 доводят осмолярность ВВИГ до значения 270÷400 мОсм/кг с использованием вспомогательных веществ: 0,0-0,7% натрия хлорида, и/или мальтозы, и/или пролина, и/или гистидина, и/или глюкозы, и/или глицина. В готовом препарате устанавливают значение pH 5,0÷7,5, содержание белка 4,5÷10,5%. Конечный препарат ВВИГ получают в жидкой или сухой форме.
Пример 2
Выделение иммуноглобулина проводят этанольным методом фракционирования по методу Кона 6. Полученный осадок В (II по Кону) подвергают ультрафильтрации при ионной силе 0 и значении pH 4,5, с последующей диафильтрацией против воды дистиллированной с величиной pH 3,9 и фильтрацией через угольный фильтр AKS 5. Затем в полуфабрикат иммуноглобулина с концентрацией белка 12,0% и температурой 0÷4°C и pH 4,4 вводят КК до концентрации 1,4%, подогревают раствор до 20°C и проводят инкубацию при температуре 20°C и значении pH 4,15 в течение 1 ч при постоянном перемешивании. Затем снижают температуру до 10°C и проводят фильтрацию через угольный фильтр AKS 5. После чего в готовый препарат вводят глицин и натрия хлорид, устанавливают значение pH 6,5-7,5, содержание белка 9,5-10,5% и получают иммуноглобулин для внутримышечного/подкожного введения.
Пример 3
5 кг пасты комплексного иммуноглобулинового препарата, полученной в результате спиртового фракционирования фракции III [40] и состоящей в основном из смеси иммуноглобулинов G, А, М, растворяют в водном растворе глицина (концентрация около 3%), pH 4,25 до концентрации белка 7,5%, ионная сила 0,03. Нерастворившиеся белки отделяют от иммуноглобулина осветляющей и стерилизующей фильтрацией. К раствору добавляют КК до концентрации 0,8%, постепенно подогревают раствор до 20°C и проводят инкубацию при температуре 20°C и значении pH 4,6 в течение 1 ч при постоянном перемешивании. Затем снижают температуру до 15°C и проводят фильтрацию через угольный фильтр AKS 5. После проводят осветляющую и стерилизующую фильтрацию, розлив препарата во флаконы и лиофилизацию.
Пример 4
Раствор моноклональных антител с концентрацией белка 50 г/л, очищенный согласно методу [41], подвергается ультрафильтрации/диафильтрации для снижения ионной силы до 0,025 М и pH до 4,0. Затем в раствор моноклональных антител с температурой 0÷4°C и pH 4,0 при постоянном перемешивании вводят КК капельно до концентрации 0,1%, постепенно подогревают раствор до 20°C и проводят инкубацию при температуре 20°C и значении pH 3,9 в течение 1 ч при постоянном перемешивании. После этого снижают температуру до 5°C и проводят фильтрацию через угольный фильтр AKS 5. Далее вирусинактивированные моноклональные антитела переводятся в конечную лекарственную форму с физиологичными значениями pH и осмолярности.
Таким образом, на основании полученных данных можно сделать вывод, что разработанная технология получения растворов иммуноглобулинов позволяет получать вирусобезопасные препараты с сохраненными физико-химическими и биологическими свойствами, а также специфической активностью. Высокая эффективность очистки достигается простыми технологическими приемами. Очистка проводится без использования дорогостоящих хроматографических сорбентов и ультрафильтрационного оборудования, благодаря чему упрощается технология получения вирусобезопасных препаратов и повышается экономичность и выход.
Литература
1. Cohn E.J., Strong W.L., Mulford D.J., Ashworth J.N., Melin M., Taylor H.L. Preparation and Properties of Serum and Plasma Proteins. IV. A system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids // J. Am Chem Soc. - 1946. - №68. - P.459-475.
2. Oncley J.L., Melin M, Richert D.A., Cameron J.W., Gross P.M. Preparation and properties of serum and plasma proteins. XIX. The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and β1-lipoprotein into subfractions of human plasma // J. Am Chem Soc-1949. - №71. - P.541-550.
3. Ochs H.D., Fischer S.H., Virant F.S., Lee M.L., Kingdon H.S., Wedgwood R.J. Non-A, non-B hepatitis and Intravenous Immunoglobulin // Lancet. - 1985 - №1. - P.404-405.
4. Ochs H.D., Fischer S.H., Virant F.S., Lee M.L., Mankarious S., Kingdon H.S., Wedgwood R.J. Non-A, non-B hepatitis after intravenous gammaglobulin // Lancet. - 1986. №1. - P.322-323.
5. Webster A.D.B., Lever A.M.L. Non-A, non-B hepatitis after intravenous gammaglobilin // Lancet. - 1986 - №1 - P.322.
6. Williams P.E., Yap P.L., Gillon J, Crawford R.J., Galea G., Cuthbertson B. Non-A, non-B hepatitis transmission by intravenous immunoglobulin // Lancet. - 1988. - II - P.501.
7. Pizani G., Cristiano K., Wirz M., Bisso G., Beneduce F., Morace G., Rapicetta M., Gentili G. Prevalence of TT virus in plasma pools and blood products // Br. J. Haemotol. - 1999. - №106 (2) - P. 431-435.
8. Burnouf T, Radosevich M. Reducing the risk of infection from plasma products: specific preventative strategies // Blood Rev. - 2000. - №14 - P.94-110.
9. WHO: Guidelines on viral inactivation and removal procedures intended to assure the viral safety of human blood plasma products. - Geneva - 2003 (www.WHO.intlbloodproducts).
10. Korneyeva M, Hotta J, Lebing W, Rosenthal RS, Franks L, Petteway SR Jr. Enveloped virus inactivation by caprylate: a robust alternative to solvent-detergent treatment in plasma derived intermediates // Biologicals. - 2002, Jun. - №30 (2) - P.153-62.
11. Lundblad J.I. and Seng R.I. Inactivation of Lipid-Enveloped Viruses in Proteins by Caprylate // Vox Sang. - 1991. - №60 (2) - P.75-81.
12. WO 1998024485 (A1). Inactivation of viruses by incubation with caprylate. - Publ. 1998-06-11.
13. Dichtelmuller H, Rudnick D, Kloft M. Inactivation of lipid enveloped viruses by octanoic acid treatment of immunoglobulin solution // Biologicals. - 2002. - №30. - P.135-42.
14. US 4939176 (A). Viral inactivation process. - Publ. 1990-07-03.
15. US 5886154 (A). Chromatographic method for high yield purification and viral inactivation of antibodies. - Publ. 1999-03-23.
16. Parkkinen J, Rahola A, von Bonsdorff L, Tölö H, Törmä E. A modified caprylic acid method for manufacturing immunoglobulin G from human plasma with high yield and efficient virus clearance // Vox Sang. - 2006 Feb. - №90 (2). - P.97-104.
17. US 4446134 (A). Process for heat treatment of aqueous solution containing human blood coagulation factor VIII. - Publ. 1984-05-01.
18. Tangen O, Wallenbeck IA, Bergqvist D. Platelet reactivity ex vivo and in vivo after acute and chronic treatment with sodium caprylate // Scand J Clin Lab Invest. - 1975 Jan. - №35 (1). - P.19-23.
19. Lebing W., Remington К.M., Schreiner C. & Paul H.-I. Properties of a new intravenous immunoglobulin (IGIV-C, 10%) produced by virus inactivation with caprylate and column chromatography // Vox Sanguinis. - 2003. - №84. - P.193-201.
20. Ballow M., Berger M., Bonilla F.A. Pharmacokinetics and tolerability of a new intravenous immunoglobulin preparation, IGIV-C, 10% (Gamunexтм, 10%) // Vox Sanguinis. - 2003. - №84. - P.202-210.
21. US 5075425 (A). Process for the preparation of a pharmaceutical which contains IgG, IgA and IgM and can be administered intravenously. - Publ. 1991-12-24.
22. US 5164487 (A). Manufacturing intravenous tolerable immunoglobulin-G preparation. - Publ. 1992-11-17.
23. WO 2005082937 (A2). A method of providing a purified, virus safe antibody preparation. - Publ. 2005-09-09.
24. WO 2005073252 (A1). Process for the manufacture of virus safe immunoglobulin. - Publ. 2005-08-11.
25. Perosa F, Carbone R, Ferrone S, Dammacco F. Purification of human immunoglobulins by sequential precipitation with caprylic acid and ammonium sulphate // J Immunol Methods. - 1990 Mar 27. - №128 (1) - P.9-16.
26. US 4164495 (A). Method of recovering immunoglobulin using a polyol and an alkanoic acid. - Publ. 1979-08-14.
27. EP 0374625 (A2). Viral inactivation process. - Publ. 1990-06-2.
28. EP 0893450 (A1). Chromatographic method for high yield purification and viral inactivation of IgG. - Publ. 1999-01-27.
29. WO 2006064373 (A2). Methods of purifying immunoglobulins. - Publ. 2006-06-22.
30. US 4164495 (A), Method of recovering immunoglobulin using a polyol and an alkanoic acid. - Publ. 1979-08-14.
31. RU 2108794 (С1). Method of immunoglobulin preparation preparing. - Publ. 1998-04-20.
32. RU 2111001 (C1). Method of immunoglobulin preparation preparing. - Publ. 1998-05-20.
33. Product monograph №133225. Gamunex. - Date of approval. May 13, 2010. - P.1-42.
34. Highlights of prescribing information 08940330. Gammaked, immune globulin injection (human) 10% caprilate/chromatography purified. - 2011, June. - P.1-3.
35. Trejo SR, Hotta JA, Lebing W, Stenland C, Storms RE, Lee DC, Li H, Petteway S, Remington KM. Evaluation of virus and prion reduction in a new intravenous immunoglobulin manufacturing process // Vox Sang. - 2003, Apr. - №84 (3). - P.176-87.
36. EP 2399613 (A1). Fractionation of plasma using caprylic acid. - Publ. 2011-12-28.
37. Dean, J.A. Handbook of Organic Chemistry. - New York, NY: McGraw-Hill Book Co., 1987. - P.8-45.
38. Lide, D.R. CRC Handbook of Chemistry and Physics. - Boca Raton, FL: CRC Press, Taylor & Francis, 2005. - P.3-404.
39. Фармакопейная статья предприятия №42-0493-06. «Иммуновенин» (Иммуноглобулин человека нормальный), лиофилизат для приготовления раствора для внутривенного введения. ФГУП «НПО «Микроген» Минздрава России. - Введ. 07.04.09. - 1-13.
40. RU 2199343 (C1). Method of immunoglobulin preparation preparing. - Publ. 2003-02-27.
41. WO 2013075740 (A1). Antibody purification method. - Publ. 2013-05-30.
Изобретение относится к способу получения препаратов из плазмы крови, а именно к способу очистки растворов иммуноглобулинов, получаемых методом спиртового фракционирования, от примеси каприловой кислоты, применяемой на стадии инактивации вирусов, и может быть использовано в производстве медицинских биологических препаратов. Данный способ включает обработку раствора иммуноглобулина каприловой кислотой в концентрации 6÷100 мМ при температуре 15÷37°C и перемешивании в течение 0,5÷2 часов. Обработку раствора иммуноглобулина каприловой кислотой проводят при ионной силе не более 0,03 М и pH 3,9÷4,6, удаление каприловой кислоты из раствора иммуноглобулина осуществляют фильтрацией через угольный фильтр при температуре 5÷15°C, ионной силе не более 0,03 М и pH 3,9÷4,6; полученный раствор содержит каприловую кислоту не более 20 мкг/мл, количество агрегатов составляет не более 1%, выход иммуноглобулина не менее 98%. Изобретение позволяет усовершенствовать технологию получения безопасных вирусинактивированных растворов иммуноглобулинов с низким остаточным содержанием каприловой кислоты при высоком выходе конечного продукта, с сохраненными физико-химическими свойствами и специфической активностью с использованием простого технического приема. 2 з.п. ф-лы, 4 ил., 3 табл., 4 пр.
1. Способ приготовления вирусинактивированных растворов иммуноглобулинов с низким остаточным содержанием каприловой кислоты, включающий обработку раствора иммуноглобулина каприловой кислотой в концентрации 6÷100 мМ при температуре 15÷37°C и перемешивании в течение 0,5÷2 часов, отличающийся тем, что обработку раствора иммуноглобулина каприловой кислотой проводят при ионной силе не более 0,03 М и pH 3,9÷4,6, удаление каприловой кислоты из раствора иммуноглобулина осуществляют фильтрацией через угольный фильтр при температуре 5÷15°C, ионной силе не более 0,03 М и pH 3,9÷4,6; полученный раствор содержит каприловую кислоту не более 20 мкг/мл, количество агрегатов составляет не более 1%, выход иммуноглобулина не менее 98%.
2. Способ по п. 1, отличающийся тем, что для фильтрации используют угольные фильтры следующих марок ZetaCarbon (Cuno) - R53S(LP), R54S(LP), R55S(LP) или AKS (Pall) - AKS 5, AKS 6.
3. Способ по п. 1, отличающийся тем, что в качестве исходного раствора используют фракции, содержащие иммуноглобулины классов G, А, М: центрифугат III (Б) или фракция II (паста иммуноглобулина G), комплексный иммуноглобулиновый препарат из фракции III, моноклональные и рекомбинантные антитела.
| ИНГИБИТОРЫ ЦИСТЕИНОВОЙ ПРОТЕАЗЫ КАТЕПСИНА | 2005 |
|
RU2399613C2 |
| WO2005073252 A1, 11.08.2005 | |||
| WO2005082937 A2, 09.09.2005 | |||
| H | |||
| DICHTELMULLER et al | |||
| Inactivation of Lipid Enveloped Viruses by Octanoic Acid Treatment of Immunoglobulin Solution | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Способ обделки поверхностей приборов отопления с целью увеличения теплоотдачи | 1919 |
|
SU135A1 |
| W | |||
| LEBING et al | |||
| Properties of a new intravenous immunoglobulin (IGIV-C, 10%) produced by virus inactivation | |||

