Множество расстройств у человека и других млекопитающих вовлекает аномальную резорбцию костей или связано с ней. Такие расстройства включают, без ограничения, остеопороз, остеопороз, вызванный глюкокортикоидами, болезнь Педжета, аномально высокий метаболизм в костной ткани, болезнь периодонта, выпадение зубов, переломы костей, ревматоидный артрит, остеоартрит, остеолиз тканей в зоне протеза, несовершенный остеогенез, гиперкальциемию при злокачественных новообразованиях или множественную миелому. Одним из наиболее частых из указанных расстройств является остеопороз, который чаще всего встречается у женщин в пост-менопаузе. Отеопороз представляет собой системное заболевание скелета, характеризующееся низкой массой кости и нарушением микроархитектуры костной ткани, которое приводит к повышению ломкости костей и чувствительности к переломам. Переломы остеопорозной природы являются основной причиной заболеваемости и смертности у пожилого населения. До 50% женщин и 1/3 мужчин имеют переломы остеопорозной природы. Большой сегмент пожилой популяции уже обладает низкой плотностью кости и высоким риском переломов. В этой связи, имеется выраженная потребность в профилактике и лечении остеопороза и других состояний, связанных с резорбцией кости. Поскольку остеопороз, как и другие расстройства, связанные с потерей костной ткани, представляют собой в основном хронические состояния, считается, что для проведения соответствующей терапии обычно требуется регулярное лечение.
Катепсины принадлежат к папаиновому суперсемейству цистеиновых протеаз. Указанные протеазы функционируют как в нормальных физиологических условиях, так и в случае патологического разложения соединительной ткани. Катепсины выполняют важную роль во внутриклеточной деградации белка, в метаболизме и ремоделировании костной ткани. К настоящему времени было идентифицировано множество катепсинов из различных источников с последующим их секвенированием. Указанные катепсины встречаются в естественном состоянии в большом числе тканей. Некоторые катепсины, такие как катепсины B, C, F, H, L, K, O, S, V, W и Z, были клонированы. Катепсин L вовлекается в нормальный процесс лизосомального протеолиза, а также в развитие некоторых заболеваний, который включает, без ограничения, метастазирование меланом. Катепсин S вовлекается в развитие болезни Альцгеймера, атеросклероза, хронической обструктивной болезни легких и некоторых аутоиммунных расстройств, включающих, без ограничения, ювенильный диабет, рассеянный склероз, пузырчатку обыкновенную, болезнь Грейвса, тяжелую псевдопаралитическую миастению, системную красную волчанку, ревматоидный артрит и тиреоидит Хашимото; аллергических расстройств, включающих без ограничения, астму; и аллогенных иммунных ответов, включающих, без ограничения, реакцию отторжения трансплантатов органов или трансплантатов ткани. Повышенные уровни катепсина B и перераспределение соответствующих ферментов были обнаружены в опухолях, что указывает на его предположительную роль в инвазии и метастазировании опухоли. Кроме того, аберрантная активность катепсина B вовлекается в патологию таких болезненных состояний, как ревматоидный артрит, остеоартрит, инфекция, вызванная Pneumocystisis carinii, острый панкреатит, воспалительные заболевания дыхательных путей и болезни костей и связок.
Катепсины млекопитающих относятся к папаино-подобным цистеиновым протеазам, экспрессируемым болезнетворными паразитами, включающими представителей следующих семейств: простейшие, платигельминты, нематоды и членистоногие. Указанные цистеиновые протеазы играют важную роль в жизненном цикле таких организмов.
Человеческий коллаген типа I, основной коллаген кости, представляет собой хороший субстрат для катепсина К (см. Kafienah, W., et al., 1998, Biochem J 331: 727-732, данная работа включена в настоящее описание полностью в качестве ссылки). В экспериментах in vitro с использованием антисмысловых олигонуклеотидов к катепсину К было продемонстрировано снижение резорбции кости in vitro, что, возможно, связано со снижением трансляции мРНК катепсина К (см. Inui, T., et al., 1997, J Biol Chem 272: 8109-8112, данная работа включена в настоящее описание полностью в качестве ссылки). Кристаллическая структура катепсина К была определена (см. McGrath, M. E., et al., 1997, Nat Struct Biol 4:105-109; Zhao, B., et al., 1997, Nat Struct Biol 4; 109-11, данная работа включена в настоящее описание полностью в качестве ссылки). Кроме того, были разработаны селективные ингибиторы катепсина К пептидной природы (см. Bromme, D., et al., 1996, Biochem J 315: 85-89; Thompson S. K., et al., 1997, Proc Natl Acad Sci USA 94: 14249-14254, данные работы включены в настоящее описание полностью в качестве ссылки). Соответственно, ингибиторы катепсина К могут снижать резорбцию кости. Такие ингибиторы могут использоваться при лечении расстройств, вовлекающих резорбцию кости, таких как остеопороз.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
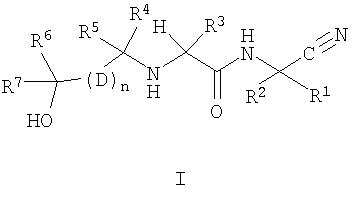
Настоящее изобретение относится к соединениям, которые способны оказывать лечебное и профилактическое воздействие на состояния или заболевания, зависимые от катепсинов, у млекопитающих, при наличии соответствующей потребности. Один вариант настоящего изобретения относится к соединению формулы I и его фармацевтически приемлемым солям, стереоизомерам и N-оксидным производным:
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, имеющим следующую химическую формулу:
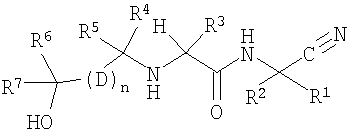
где R1 и R2, взятые вместе с атомом углерода, к которому они присоединяются, образуют С3-4циклоалкил, который может быть необязательно замещен С1-3алкилом;
R3 обозначает С1-6алкил, который замещен одним-четырьмя атомами фтора или одним-четырьмя атомами хлора;
R4 обозначает С1-6алкил, который замещен одним-пятью атомами галогена;
R5 обозначает водород или С1-6алкил, который может быть необязательно замещен одним-пятью атомами галогена;
каждый D обозначает независимо арил или гетероарил;
R6 обозначает водород или С1-6алкил, который может быть необязательно замещен одним-двумя гидроксилами или двумя-шестью атомами галогена;
R7 обозначает водород или С1-6алкил, который может быть необязательно замещен двумя-пятью атомами галогена;
n равно двум;
или к его фармацевтически приемлемым солям, стереоизомерам или N-оксидным производным.
В одном варианте осуществления настоящего изобретения R1 и R2, взятые вместе с атомом углерода, к которому один присоединяются, образуют циклопропил.
В одном варианте осуществления настоящего изобретения D обозначает фенил.
В одном варианте осуществления настоящего изобретения R4 обозначает CF3.
В одном варианте осуществления настоящего изобретения R5 обозначает водород.
В одном варианте осуществления настоящего изобретения R7 обозначает С1-3алкил, замещенный двумя или тремя атомами фтора.
Указанные выше предпочтительные варианты относятся также ко всем сочетаниям конкретных и предпочтительных групп, если особо не указано иное.
Конкретные варианты осуществления настоящего изобретения включают, без ограничения:
N1-(1-цианоциклопропил)-N2-(1-{4'-[2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-(2,2,2-трифтор-1-{4'-[3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-(2,2,2-трифтор-1-{4'-[2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-(2,2,2-трифтор-1-{4'-[1-гидрокси-1-(трифторметил)пропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-(1-{4'-[2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-(1-{4'-[2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1S)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1R)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(R)-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[1-гидрокси-1-(трифторметил)пропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
и их фармацевтически приемлемые соли, стереоизомеры и N-оксидные производные.
Настоящее изобретение охватывает также фармацевтическую композицию, которая включает соединение формулы I, описанное выше, и фармацевтически приемлемый носитель. Настоящее изобретение также охватывает фармацевтическую композицию, которая включает фармацевтически приемлемый носитель и любое из соединений, конкретно указанных в настоящей заявке.
Другой вариант осуществления настоящего изобретения относится к пероральной фармацевтической композиции, включающей соединение формулы I или его фармацевтически приемлемую соль, стереоизомер или N-оксидное производное, для ингибирования резорбции кости при постоянном применении, включающем интервал во введении доз, например, еженедельного введения, введения один раз в две недели, два раза в месяц или ежемесячного введения.
Приведенные и другие аспекты настоящего изобретения будут очевидны из приведенного описания.
Применимость
Соединения согласно настоящему изобретению представляют собой ингибиторы катепсинов и, в этой связи, могут использоваться для лечения или профилактики катепсин-зависимых заболеваний или состояний у млекопитающих, предпочтительно у людей. Конкретно, соединения согласно настоящему изобретению представляют собой ингибиторы катепсина К и, в этой связи, они могут использоваться для лечения или профилактики катепсин К-зависимых заболеваний или состояний у млекопитающих, предпочтительно у людей.
Соединения согласно настоящему изобретению имеют преимущество по сравнению со структурно родственными соединениями, известными в данной области, в том, что они обладают заметно улучшенным фармакокинетическим профилем. А именно, соединения согласно настоящему изобретению обладают прекрасной биологической доступностью, такой как обладает, например, без ограничения, доза 10 мг/кг, вводимая крысам Sprague Dawley, в 0,5-1% метоцеле. Дополнительно, соединения согласно настоящему изобретению обеспечивают более сильное системное воздействие лекарственного агента, чем известные структурно родственные соединения.
Термин «катепсин-зависимые заболевания или состояния» относится к патологическим состояниям, которые зависят от активности одного или нескольких катепсинов. Термин «катепсин К-зависимые заболевания или состояния» относится к патологическим состояниям, которые зависят от активности катепсина К. Заболевания, связанные с активностями катепсина К, включают остеопороз, остеопороз, вызванный глюкокортикоидами, болезнь Педжета, аномально высокий метаболизм в костной ткани, выпадение зубов, переломы костей, ревматоидный артрит, остеоартрит, остеолит околопротезной ткани, несовершенный остеогенез, атеросклероз, ожирение, глаукома, хроническая обструктивная болезнь легких и рак, включающий метастазирование в костях, гиперкальциемию при злокачественных новообразованиях или множественную миелому. При лечении таких состояний соединениями согласно настоящему изобретению требуемое терапевтическое количество данных соединений будет варьироваться в зависимости от конкретного заболевания и может быть легко определено специалистом в данной области. Несмотря на то, что в область настоящего изобретения включены и лечение и профилактика, лечение указанных состояний представляет собой предпочтительный вариант использования.
Один из вариантов осуществления настоящего изобретения относится к способу ингибирования активности катепсина у млекопитающих при наличии такой необходимости, включающему введение указанному млекопитающему терапевтически активного количества любого из соединения или любой из фармацевтических композиций, указанных выше.
Настоящее изобретение в одном из вариантов относится к способу, в соответствии с которым активность катепсина представляет собой активность катепсина К.
Другой вариант настоящего изобретения относится к способу лечения или профилактики катепсин-зависимых состояний у млекопитающих, при наличии такой необходимости, включающему введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше.
Настоящее изобретение в одном из своих вариантов относится к способу, в соответствии с которым активность катепсина представляет собой активность катепсина К.
Другой вариант осуществления настоящего изобретения относится к способу ингибирования разрежения кости у млекопитающего, при наличии такой необходимости, включающему введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Другой вариант осуществления настоящего изобретения относится к способу снижения разрежения кости у млекопитающего, при наличии такой необходимости, включающему введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Еще один вариант осуществления настоящего изобретения относится к способу лечения аномально высокого метаболизма в костной ткани и переломов костей у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Применение ингибиторов катепсина К при ингибировании резорбции кости описано в литературе (см. Stroup, G.B., Lark, M.W., Veber, D.F., Bhattacharrya, A., Blake, S., Dare, L.C., Erhard, K.F., Hoffman, S.J., James, I.E., Marquis, R.W., Ru, Y., Vasko-Moser, J.A., Smith, B.R., Tomaszek, T. and Gowen, M. Potent and selective inhibition of human cathepsin K leads to inhibition of bone resorption in vivo in a nonhuman primate. J. Bone Miner. Res., 16: 1739-1746; 2001; и Votta, B.J., Levy, M. A., Badger, A., Dodds, R.A., James, I.E., Thompson, S., Bossard, M.J., Carr, T., Connor, J.R., Tomaszek T.A., Szewczuk, L., Drake, F.H., Veber, D., and Gowen, M. Peptide aldehyde inhibitors of cathepsin K inhibit bone resorption in vivo and in vitro. J. Bone Miner. Res., 12: 1396-1406; 1997).
Другой вариант осуществления настоящего изобретения относится к способу лечения или профилактики остеопороза у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Применение ингибиторов катепсина К при лечении или профилактике остеопороза, включая остеопороз, вызванный глюкокортикоидами, описано в литературе (см. Saftig, P., Hunziker, E., Wehmeyer, O., Jones, S., Boyde, A., Rommerskirch, W., Moritz, H.D., Schu, P., and Vonfigura, K. Impaired osteoclast bone resorption leads to osteoporosis in cathepsin K-deficient mice. Proc. Natl. Acad. Sci. USA 95: 13453-13458; 1998).
Другой вариант осуществления настоящего изобретения относится к способу лечения или профилактики болезни периодонта или выпадения зубов у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Применение ингибитора катепсина К при лечении или профилактике болезни периодонта или выпадения зубов уже обсуждалось в литературе (см. Sasaki, T., «Differentiation and function of osteoclasts and osontoclasts in mineralized tissue resorption», Microsc. Res. Tech. 2003 Aug 15: 61(6): 483-95).
Другой вариант осуществления настоящего изобретения относится к способу лечения или профилактики ревматоидного артрита или состояния, связанного с ревматоидным артритом, у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что прогрессирующая деструкция околосуставной кости представляет собой основную причину дисфункции суставов и инвалидизации у пациентов с ревматоидным артритом (РА) (см. Goldring SR, «Pathogenesis of bone erosions in rheumatoid arthritis» Curr. Opin. Rheumatol. 2002; 14: 406-10. Analysis of joint tissues from patients with RA have provided evidence that cathepsin K positive osteoclasts are the cell types that mediate the focal bone resorption associated with rheumatoid synovial lesion, см. Hou, W-S, Li, W, Keyszer, G, Weber, E, Levy, R, Klein, MJ, Gravallese, EM, Goldring, SR, Bromme, D, «Comparison of Cathepsin К and S expression within the Rheumatoid and Osteoarthritic Synovium», Arthritis Rheumatism 2002; 46: 663-74). Кроме того, общее разрежение кости представляет собой основную причину заболеваемости, связанную с тяжелыми случаями РА. Частота переломов бедра и позвоночника существенно повышается у пациентов с хроническим РА (см. Gould, A, Sambrook, P, Devlin, J et al., «Osteoclastic activation is the principal mechanism leading to secondary osteoporosis in rheumatoid arthritis», J. Rheumatol. 1998: 25: 1282-9). Применение ингибиторов катепсина К при лечении или профилактике резорбции подсуставной кости и при общем разрежении кости представляет собой рациональную стратегию фармакологического воздействия при прогрессировании ревматоидного артрита.
Другой вариант осуществления настоящего изобретения относится к способу лечения или профилактики прогрессирования остеоартрита у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что остеоартрит (ОА) сопровождается вполне определенными изменениями в суставах, включая эрозию поверхности суставного хряща, оссификацию/остеофитоз околосуставной эндохондральной ткани, и субхондральный костный склероз и образование цист (см. Oettmeier R, Abendroth, K, «Osteoarthtitis and bone: osteologic types of osteoarthritis of the hip», Skeletel Radiol. 1989; 18: 165-74). Недавно появилось сообщение, указывающее на возможность участия субхондрального костного склероза в инициации прогрессирования ОА. Слабоподвижная субхондральная кость, функционирующая как сустав в ответ на повторную импульсную нагрузку, в меньшей степени способна ослаблять и распределять усилия по суставу, и таким образом подвергает его большему механическому стрессу по всей поверхности суставного хряща. Это, в свою очередь, ускоряет изнашивание хряща и фибрилл (см. Radin, EL and Rose RM, «Role of subchondral bone in the initiation and progression of cartilage damage», Clin. Orthop. 1986; 213: 34-40). Ингибирование избыточного разрежения подсуставной кости антирезорбционным агентом, таким как ингибитор катепсина К, приведет, в свою очередь, к ингибированию метаболизма субхондральной кости, что может оказать ингибирующее воздействие на прогрессирование ОА.
В дополнение к указанному выше следует отметить, что недавно была идентифицирована экспрессия белка катепсина К в синовиальных фибробластах, макрофагально-подобных клетках и в хондроцитах из синовия и образцов суставного хряща, взятых от пациентов с ОА (см. Hou, W-S, Li, W, Keyszer, G, Weber, E, Levy, R, Klein, MJ, Gravallese, EM, Goldring, SR, Bromme, D, «Comparison of Cathepsin К and S expression within the Rheumatoid and Osteoarthritic Synovium», Arthritis Rheumatism 2002; 46: 663-74; и Dodds, RA., Connor, JR., Drake, FH., Gowen, M. «Expression of Cathepsin K messenger RNA in giant cells and their precursors in human osteoarthritis synovial tissues». Arthritis Rheumatism 1999; 42: 1588-93; и Konttinen, YT, Mandelin, J, Li, T-F, Salo, J, Lassus, J et al., «Acidic cysteine endoproteinase cathepsin K in the degeneration of the superficial articular hyaline cartilage in osteoarthritis», Arthritis Rheumatism 2002; 46: 953-60). Указанные выше исследования указывают на роль катепсина К в деструкции коллагена типа II в суставном хряще, связанной с прогрессированием остеоартрита. Применение ингибиторов катепсина К при лечении или профилактике остеоартирта в соответствии с настоящим описанием будет, таким образом, основываться на двух разных механизмах, один из которых представляет собой ингибирование остеокласт-зависимого метаболизма субхондральной кости, тогда как второй связан с непосредственным ингибированием дегенерации коллагена типа II в синовии и хряще у пациентов с ОА.
Другой вариант осуществления настоящего изобретения относится к способу лечения остеолиза околопротезной ткани у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Применение ингибиторов катепсина К для лечения остеолиза околопротезной ткани обсуждалось в литературе (см. Mandelin, J., et al., «Interface tissue fibroblasts from loose total hip replacement prosthesis produce receptor activator of nuclear factor-kappaB ligand, osteoprotegerin and cathepsin K», J. Rheumatol. 2005 Apr; 32(4): 713-20).
Другой вариант осуществления настоящего изобретения относится к способу лечения костного заболевания, такого как болезнь Педжета, несовершенный остеогенез и костные поражения при множественной миеломе у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Применение ингибиторов катепсина К для лечения болезни Педжета, несовершенного остеогенеза и костных поражений при множественной миеломе обсуждалось в литературе (см. Lipton, A., «New therapeutic agents for the treatment of bone diseases», Expert Opin Biol Ther. 2005 Jun; 5(6): 817-32).
Другой вариант осуществления настоящего изобретения относится к способу лечения рака у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что катепсин К экспрессируется при карциноме молочной железы человека, раке предстательной железы и хордоме и обладает способностью разрушать костный матрикс (см. Littlewood-Evans AJ, Bilbe G, Bowler WB, Farley D, Wlodarski B, Kokubo T, Inaoka T, Sloane J, Evans DB, Gallagher JA, «The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma», Cancer Res 1997 Dec 1: 57(23): 5386-90; Brubaker KD, Vessella RL, True LD, Thomas R, Corey E, «Cathepsin K mRNA and protein expression in prostate cancer progression», J Bone Miner Res 2003 18, 222-30. Haeckel C, Krueger S, Kuester D, Ostertag H, Samii M, Buehling F, Broemme D, D, Czerniak B, Roessner A, «Expression of cathepsin K in chordoma», Hum Pathol 2000 Jul; 31(7): 834-40).
Другой вариант осуществления настоящего изобретения относится к способу лечения атеросклероза у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что катепсин К экспрессируется в атероме человека и обладает выраженной эластазной активностью (см. Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P, «Expression of the elastolytic cathepsin S and K in himan atheroma and regulation of their production in smooth muscle cells», J Clin Invest 1998 Aug 102, 576-83).
Другой вариант осуществления настоящего изобретения относится к способу лечения ожирения у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что мРНК катепсина К повышается в жировой ткани, по результатам оценки на моделях мышей с ожирением, а также в жировой ткани у мужчин с ожирением (см. Chiellini C, Costa M, Novelli SE, Amri EZ, Benzi L, Bertacca A, Cohen P, Del Prato S, Friedman JM, Maffei M, «Identification of cathepsin K as a novel marker of adiposity in white adipose tissue», J Cell Physiol 2003, 195, 309-21).
Другой вариант осуществления настоящего изобретения относится к способу лечения глаукомы у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Катепсин К экспрессируется на высоком уровне в радужной оболочке, в ресничном теле и пигменте эпителия в сетчатке и в этой связи может использоваться при лечении глаукомы (см. Ortega, J., et al., «Gene Expression of Proteases and Protease Inhibitors in the Human Ciliary Epithelium and ODM-2 cells», Exp. Eye Res (1997) 65, 289-299; Международная публикация WO 2004/058238 (Alcon, Inc.)).
Другой вариант осуществления настоящего изобретения относится к способу лечения хронической обструктивной болезни легкого у млекопитающего, при наличии такой необходимости, включающему введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что катепсин К играет определенную роль в развитии фиброза легкого (см. Buhling, F., et al., «Pivotal role of cathepsin K in lung fibrosis», Am J Pathol. 2004 Jun; 164(6): 2203-16).
Другой вариант осуществления настоящего изобретения относится к способу лечения паразитарных инфекций у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что катепсины млекопитающих близки к цистеиновым протеазам папаинового типа, которые играют важную роль в жизненном цикле таких паразитов. Указанные паразиты вовлекаются в патологию таких заболеваний, как малярия, американский трипаносомоз, африканский трипаносомоз, лейшманиоз, лямблиоз, трихомоноз, амебиаз, шистосомоз, фасциолез, парагонимоз и инвазия кишечными аскаридами (см. Lecaille F, Kaleta J, Bromme D., Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem Rev 2002 102, 4459-88).
Другой вариант осуществления настоящего изобретения относится к способу лечения тяжелого острого респираторного синдрома (SARS) у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше.
Другой вариант осуществления настоящего изобретения относится к способу лечения метастаза кости у млекопитающего, при наличии такой необходимости, включающему введение ему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше. Из литературы известно, что остеокласты отвечают за резорбцию кости и что деструкция кости и гиперкальциемия индуцируются метастазирующими опухолями и осуществляются остеокластами. Соответственно, ингибирование остеокластов может препятствовать деструкции костей и метастазированию опухоли (см. Miyamoto, T. and Suda, T., «Defferentiation and function of osteoclasts», Keio J Med 2003 Mar; 52(1): 1-7).
Другой вариант осуществления настоящего изобретения относится к введению млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, указанных выше, для лечения болезней у млекопитающих, связанных с катепсином S, включающих болезнь Альцгеймера, атеросклероз, хроническую обструктивную болезнь легких, рак и некоторые аутоиммунные расстройства, включающие, без ограничения, ювенильный диабет, рассеянный склероз, пузырчатку обыкновенную, болезнь Грейвса, тяжелую псевдопаралитическую миастению, системную красную волчанку, ревматоидный артрит и тиреоидит Хашимото; аллергические расстройства, включающие, без ограничения, астму; и аллогенные иммунные реакции, включающие, без ограничения, реакцию отторжения трансплантатов органов или трансплантатов ткани. Из литературы известно, что активность катепсина S связана с указанными выше болезненными состояниями (см. Munger JS, Haass C, Lemere CA, Shi GP, Wong WS, Teplow DB, Selkoe DJ, Chapman HA, «Lysosomal processing of amyloid precursor protein to A beta peptides: a distinct role for cathepsin S», Biochem J 1995, 311, 299-305; Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, Sakara Y, Chin MT, Libby P, Shi GP, «Deficiency of cathepsin S reduced atherosclerosis in LDL receptor-deficient mice», J Clin Invest 2003 111, 897-906; Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr, Chapman HA Jr, Shapiro SD, Elias JA, «Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema», J. Clin Invest 2000 106, 1081-93; Shi GP, Sukhova GK, Kuzuya M, Ye Q, Du J, Zhang Y, Pan JH, Lu ML, Cheng XW, Iguchi A, Perrey S, Lee AM, Chapman HA, Libby P, «Deficiency of the cysteine protease cathepsin S impairs microvessel growth», Circ Res 2003 92, 493-500; Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, McNeish JD, Eastman SE, Howard ED, Clarke SR, Rosloniec EF, Elliott EA, Rudensky AY, «Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice», Immunity 1999 10, 207-17).
Примерами осуществления настоящего изобретения является использование любого из указанных выше соединений при получении лекарственного средства для лечения и/или профилактики остеопороза у млекопитающего, при наличии такой необходимости. Еще одним примером осуществления настоящего изобретения является использование любого из указанных выше соединений при получении лекарственного средства для лечения и/или профилактики разрежения кости, резорбции кости, переломов кости, метастаза в кости и/или расстройств, связанных с функционированием катепсина.
Еще одним примером осуществления настоящего изобретения является использование ингибитора катепсина К согласно настоящему изобретению или его фармацевтически приемлемой соли, стереоизомера или N-оксидного производного при производстве лекарственного средства в виде пероральной дозированной формы для лечения расстройства, выбранного из остеопороза, остеопороза, индуцированного глюкокортикоидами, болезни Педжета, аномально высокого метаболизма в костной ткани, болезни периодонта, выпадения зубов, переломов костей, ревматоидного артрита, остеоартрита, остеолита околопротезной ткани, несовершенного остеогенеза, атеросклероза, ожирения, глаукомы, хронической обструктивной болезни легких, метастаза кости, гиперкальциемии при злокачественных новообразованиях или множественной миеломы, у млекопитающего, при необходимости, постоянного лечения с интервалом дозирования, таким как прием дозы один раз в неделю, два раза в неделю, два раза в месяц или один раз в месяц. Примером осуществления настоящего изобретения также является способ лечения расстройства, выбранного из остеопороза, остеопороза, индуцированного глюкокортикоидами, болезни Педжета, аномально высокого метаболизма в костной ткани, болезни периодонта, выпадения зубов, переломов костей, ревматоидного артрита, остеоартрита, остеолита околопротезной ткани, несовершенного остеогенеза, атеросклероза, ожирения, глаукомы, хронической обструктивной болезни легких, метастаза костей, гиперкальциемии при злокачественных новообразованиях или множественной миеломы, путем введения ингибитора катепсина К согласно настоящему изобретению млекопитающему, при наличии такой необходимости, постоянного введения со следующим интервалом во введении доз: введение один раз в неделю, один раз в две недели, два раза месяц или один раз в месяц.
Соединение согласно настоящему изобретению может вводиться млекопитающим, предпочтительно людям, либо само по себе, либо, что предпочтительно, в сочетании с фармацевтически приемлемыми носителями или разбавителями, необязательно вместе с известными вспомогательными компонентами, такими как квасцы, в составе фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Соединения могут вводиться перорально или парентерально, и способы введения включают внутривенное, внутримышечное, внутрибрюшинное, подкожное, ректальное и местное введение.
В случае таблеток для перорального употребления чаще всего добавляют обычно используемые носители, такие как лактоза и кукурузный крахмал, и лубриканты, такие как стеарат магния. Для перорального введения капсул используемый при этом разбавитель включает лактозу и высушенный кукурузный крахмал. Для перорального применения терапевтической композиции согласно настоящему изобретению выбранное соединение может вводиться, например, в виде таблеток или капсул или в виде водного раствора или водной суспензии. Для перорального введения в виде таблетки или капсулы активный лекарственный компонент может быть объединен с принимаемым перорально нетоксичным фармацевтически приемлемым носителем, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния, фосфат дикальция, сульфат кальция, маннит, сорбит и т.п.; для перорального введения в жидкой форме перорально принимаемые лекарственные компоненты могут быть объединены с любым перорально принимаемым нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Кроме того, в случае желательности или необходимости, в смесь могут быть включены подходящие связующие вещества, лубриканты, средства, способствующие разложению, и красители. Подходящие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, натуральные и синтетические камеди, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и т.п. Лубриканты, используемые в указанных дозированных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Средства, способствующие разложению, включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п. В том случае, когда для перорального применения требуются водные суспензии, активный ингредиент объединяют с эмульгатором и средством, способствующим суспендированию. При необходимости могут быть добавлены подсластители и/или вкусовые вещества. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного использования обычно готовят стерильные растворы активного ингредиента и соответствующим образом корректируют pH данных растворов и забуферивают их. Для внутривенного использования общая концентрация растворимых веществ должна контролироваться, с тем, чтобы получаемый препарат был изотоничным.
Соединения согласно настоящему изобретению могут быть также введены в составе систем для липосомальной доставки, такие как небольшие однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы на основе большого числа фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Соединения согласно настоящему изобретению могут также доставляться с использованием моноклональных антител в качестве индивидуальных носителей, с которыми связывают молекулы данного соединения. Соединения согласно настоящему изобретению могут быть также связаны с растворимыми полимерами, используемыми в качестве направляющих носителей для лекарственных компонентов. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламид-фенол, полигидроксиэтиласпартамид-фенол или полиэтиленоксид-полилизин, замещенный пальмитоильными остатками. Кроме того, соединения согласно настоящему изобретению могут быть связаны с полимерами из класса биодеградируемых полимеров, используемых для достижения контролируемого высвобождения лекарственного компонента, например, с полимолочной кислотой, полигликолевой кислотой, сополимерами полимолочной и полигликолевой кислот, полиэпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатическими блок-сополимерами гидрогелей.
Соединения согласно настоящему изобретению также используются в сочетании с известными агентами, которые применяют для лечения или профилактики остеопороза, остеопороза, индуцированного гликокортикоидами, болезни Педжета, аномально высокого метаболизма в костной ткани, болезни периодонта, выпадения зубов, переломов костей, ревматоидного артрита, остеоартрита, остеолита околопротезной ткани, несовершенного остеогенеза, метастаза кости, гипрекальциемии при злокачественных новообразованиях и множественной миеломы. Сочетания соединений, рассматриваемых в настоящем изобретении, с другими агентами, используемыми при лечении или профилактике остеопороза или других костных заболеваний, входят в область настоящего изобретения. Специалист со средним уровнем знаний в данной области в состоянии определить, какие сочетания агентов будут полезными, основываясь на конкретных характеристиках лекарственных препаратов и определенного заболевания. Такие агенты включают следующие: органический бисфосфонат; модулятор рецептора эстрогена; модулятор рецептора андрогена; ингибитор протон-АТФазы остеокластов; ингибитор ГМГ-KoA редуктазы; антагонист рецептора интегрина; витамин D; синтетический аналог витамина D; анаболический агент, такой как паратгормон; нестероидные противовоспалительные средства; селективный ингибитор циклооксигеназы-2; ингибитор интерлейкина-1-бета; ингибитор LOX/COX; ингибитор RANKL; и их фармацевтически приемлемые соли и смеси. Предпочтительное сочетание представляет собой сочетание соединения согласно настоящему изобретению и органического бисфосфоната. Другое предпочтительное сочетание представляет собой сочетание соединения согласно настоящему изобретению и модулятора рецептора эстрогена. Другое предпочтительное сочетание представляет собой сочетание соединения согласно настоящему изобретению и модулятора рецептора андрогена. Другое предпочтительное сочетание представляет собой сочетание соединения согласно настоящему изобретению и анаболического агента остеобластов.
Термин «органический бисфосфонат» включает, без ограничения, соединения общей химической формулы:
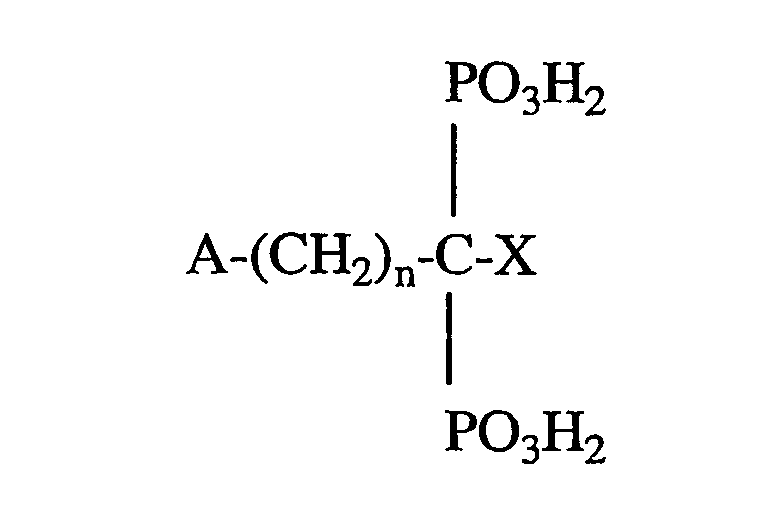
где n равно целому числу от 0 до 7 и A и X независимо выбирают из группы, состоящей из H, OH, галогена, NH2, SH, фенила, C1-С30алкила, C3-C30 разветвленной или циклоалкильной бициклической кольцевой структуры, содержащей два или три N, C1-C30 замещенного алкила, C1-С10алкила, замещенного NH2, C3-C10 разветвленного радикала или циклоалкила, замещенного NH2, C1-C10 диалкила, замещенного NH2, C1-C10 алкокси, C1-C10 алкила, замещенного тио, тиофенилом, галогенфенилтио, C1-C10 алкила, замещенного фенилом, пиридилом, фуранилом, пирролидинилом, имидазолилом, имидазопиридинилом и бензилом, таким образом, что и А, и Х не выбирают из H или OH, когда n равно 0; или A и X, взятые вместе с одним или несколькими атомами углерода, к которым они присоединяются, образуют C3-C10 кольцо.
В указанных выше химических формулах алкильные группы могут быть линейными, разветвленными или циклическими, при условии, что выбрано достаточное количество атомов для химических формул. C1-C30 замещенный алкил может включать большое число заместителей, неограничивающие примеры которых включают заместители, выбранные из группы, состоящей из фенила, пиридила, фуранила, пирролидинила, имидазолила, NH2, C1-С10алкила или диалкила, замещенного NH2, OH, SH и C1-С10алкокси.
Приведенные выше химические формулы также могут включать сложные карбоциклические ароматические и гетероатомные структуры в качестве заместителей А и/или Х, неограничивающие примеры которых включают нафтил, хинолил, изохинолил, адамантил и хлорфенилтио.
Фармацевтически приемлемые соли и производные бисфосфонатов также используются в контексте настоящего изобретения. Неограничивающие примеры солей включают соли, выбранные из группы, состоящей из солей щелочного металла, щелочноземельного металла, аммония и моно-, ди-, три- или тетра-C1-C10 алкил-замещенного аммония. Предпочтительными солями являются соли, выбранные из группы, состоящей из солей натрия, калия, кальция, магния и аммония. Более предпочтительными являются соли натрия. Неограничивающие примеры производных включают производные, выбранные из группы, состоящей из сложных эфиров, гидратов и амидов.
Следует отметить, что термины «бисфосфонат» и «бисфосфонаты», используемые в настоящем описании применительно к терапевтическим агентам, также охватывают дифосфонаты, бифосфоновые кислоты и дифосфоновые кислоты, а также соли и производные указанных материалов. Использование специфической номенклатуры применительно к бисфосфонату или бисфосфонатам никоим образом не ограничивает область настоящего изобретения, если особо не указано иное. В связи с тем, что специалисты в данной области используют в настоящее время сложную номенклатуру, то любая ссылка на удельный вес или процент бисфосфонатного соединения в контексте настоящего изобретения относится к соответствующим показателям применительно к весу активной кислоты, если особо не указано иное. Например, термин «примерно 5 мг ингибирующего резорбцию кости бисфосфоната, выбранного из группы, состоящей из алендроната, его фармацевтически приемлемых солей и смесей, на основе веса активной алендроновой кислоты» означает, что количество выбранного бисфосфонатного соединения рассчитывают на основе 5 мг алендроновой кислоты.
Неограничивающие примеры бисфосфонатов, используемых в контексте настоящего изобретения, включают следующие:
Алендронат, который также известен как алендроновая кислота, 4-амино-1-гидроксибутилиден-1,1-бисфосфоновая кислота, алендронат натрия или тригидрат алендроната мононатрия, тригидрат мононатриевой соли 4-амино-1-гидроксибутилиден-1,1-бисфосфоновой кислоты.
Алендронат описан в патентах США No. 4922007 (Kieczykowski et al.), выданном 1 мая 1990 года; No. 5019651 (Kieczykowski et al.), выданном 28 мая 1991 года; No. 5510517 (Dauer et al.), выданном 23 апреля 1996 года; No. 5648491 (Dauer et al.), выданном 15 июля 1997 года, которые включены в настоящее описание полностью в качестве ссылок.
Циклогептиламинометилен-1,1-бисфосфоновая кислота, YM 175, (Yamanouchi) (инкадронат, ранее известный как цимадронат), описана в патенте США No. 4970335 (Isomura et al.), выданном 13 ноября 1990 года, который включен в настоящее описание полностью в качестве ссылки.
1,1-дихлорметилен-1,1-дифосфоновая кислота (клодроновая кислота) и ее динатриевая соль (клодронат, Procter and Gamble) описаны в патенте Бельгии 672205 (1966) и в J. Org. Chem 32, 4111 (1967), и оба документа включены в настоящее описание полностью в качестве ссылки.
1-гидрокси-3-(1-пирролидинил)-пропилиден-1,1-бисфосфоновая кислота (EB-1053).
1-гидроксиэтан-1,1-дифосфоновая кислота (этидроновая кислота).
1-гидрокси-3-(N-метил-N-пентиламино)пропилиден-1,1-бисфосфоновая кислота, также известная как BM-210955 (Boehringer-Mannheim) (ибандронат), описана в патенте США No. 4927814, выданном 22 мая 1990 года, который включен в настоящее описание полностью в качестве ссылки.
1-гидрокси-2-имидазо-(1,2-а)пиридин-3-этилиден (минодронат).
6-амино-1-гидроксигексилиден-1,1-бисфосфоновая кислота (неридронат).
3-(диметиламино)-1-гидроксипропилиден-1,1-бисфосфоновая кислота (олпадронат).
3-амино-1-гидроксипропилиден-1,1-бисфосфоновая кислота (памидронат).
[2-(2-пиридинил)этилиден]-1,1-бифосфоновая кислота (пиридронат), описана в патенте США No. 4761406, который включен в настоящее описание полностью в качестве ссылки.
1-гидрокси-2-(3-пиридинил)этилиден-1,1-бисфосфоновая кислота (ризедронат).
(4-хлорфенил)тиометан-1,1-бисфосфоновая кислота (тилудронат), описана в патенте США No. 4876248 (Breliere et al.,), выданном 24 октября 1989 года, который включен в настоящее описание полностью в качестве ссылки.
1-гидрокси-2-(1Н-имидазол-1-ил)этилиден-1,1-бисфосфоновая кислота (золедронат).
Неограничивающие примеры бисфосфонатов включают алендронат, цимадронат, клодронат, этидронат, ибандронат, инкадронат, минодронат, неридронат, олпадронат, памидронат, пиридронат, ризедронат, тилудронат и золедронат и их фармацевтически приемлемые соли и сложные эфиры. Особенно предпочтительным бисфосфонатом является алендронат, в особенности натриевая, калиевая, кальциевая, магниевая или аммониевая соль алендроновой кислоты. Примером предпочтительного бисфосфоната является натриевая соль алендроновой кислоты, в особенности гидрат натриевой соли алендроновой кислоты. Соль может быть гидратирована целым числом молей воды или не целым числом молей воды. Другим примером предпочтительного бисфосфоната является гидрат натриевой соли алендроновой кислоты, в особенности когда гидратированная соль представляет собой тригидрат алендроната мононатрия.
Известно, что могут использоваться смеси двух или более бисфософнатных активных агентов.
Точная дозировка органического бисфосфоната будет варьировать в зависимости от режима дозирования, конкретного выбранного бисфосфоната, возраста, размера, пола и состояния здоровья млекопитающего, в частности человека, от природы и тяжести заболевания, подлежащего лечению, и от других соответствующих медицинских и физических факторов. Таким образом, точное фармацевтически эффективное количество не может быть определено заранее и может быть определено специалистом, оказывающим помощь, или врачом. Соответствующие количества могут быть определены на основе данных экспериментальных исследований на моделях животных и результатов клинических испытаний на людях. В основном, подходящее количество бисфосфоната выбирают таким образом, чтобы достичь ингибирующего эффекта в отношении резорбции кости, то есть подбирают количество бисфосфоната, ингибирующего резорбцию кости. В случае людей эффективная пероральная доза бисфосфоната типично составляет от примерно 1,5 до примерно 6000 мкг/кг веса тела и предпочтительно составляет от примерно 10 до примерно 2000 мкг/кг веса тела. В случае тригидрата алендроната мононатрия обычная вводимая доза для людей составляет в основном от примерно 2 мг/день до примерно 40 мг/день, предпочтительно от примерно 5 мг/день до примерно 40 мг/день. В США в настоящее время разрешены дозировки для тригидрата алендроната мононатрия, равные 5 мг/день - для профилактики остеопороза, 10 мг/день - для лечения остеопороза и 40 мг/день для лечения болезни Педжета.
В случае альтернативных вариантов дозирования бисфосфонат может вводиться с интервалами, отличными от ежедневного введения, например, это будет доза, вводимая один раз в неделю, доза, вводимая два раза в неделю, доза, вводимая один раз в две недели, и доза, вводимая два раза в месяц. При использовании еженедельного режима дозирования тригидрат алендроната мононатрия может вводиться в дозировках 35 мг/неделю или 70 мг/неделю.
Термин «селективные модуляторы рецептора эстрогена» относится к соединениям, которые препятствуют связыванию эстрогена с рецептором, независимо от механизма данного эффекта, или ингибируют такое связывание. Примеры модуляторов рецептора эстрогена включают, без ограничения, эстроген, прогестерон, эстрадиол, дролоксифен, ралоксифен, лазофоксифен, TSE-424, тамоксифен, идоксифен, LY353381, LY117081, торемифен, фулвестрант, 4-[7-(2,2-диметил-1-оксопропокси-4-метил-2-[4-[2-(1-пиперидинил)этокси]фенил]-2Н-бензопиран-3-ил]фенил-2,2-диметилпропаноат, 4,4'-дигидроксибензофенон-2,4-динитрофенил-гидразон и SH646.
Термин «модулятор рецептора эстрогена бета» обозначает соединение, которое оказывает селективное агонистское или антагонистское воздействие на рецептор эстрогена бета. ERβ-агонист приводит к повышению транскрипции триптофан-гидроксилазного гена (TPH, ключевой фермент синтеза серотонина) через ERβ-опосредованный механизм. Примеры агонистов рецептора эстрогена бета приведены в международных публикациях PCT WO 01/82923, опубликованной 8 ноября 2001 года, и WO 02/41835, опубликованной 20 мая 2002 года, которые включены в настоящее описание полностью в качестве ссылки.
Термин «модуляторы рецептора андрогена» относится к соединениям, которые препятствуют связыванию андрогенов с рецептором, независимо от механизма данного эффекта, или ингибируют такое связывание. Примеры модуляторов рецептора андрогенов включат финастерид и другие ингибиторы 5α-редуктазы, нилутамид, флутамид, бикалутамид, лиарозол и ацетат абиратерона.
Термин «ингибитор протон-АТФазы остоекласта» относится к ингибитору протон-АТФазы, которая найдена в апикальной мембране остеокласта и которая, как было выявлено, играет важную роль в процессе резорбции костей. Протонный насос представляет собой заманчивую мишень для разработки ингибиторов резорбции кости, и это механизм может быть в принципе использован для лечения и профилактики остеопороза и родственных метаболических заболеваний (см. Farina et al., «Selective inhibitors of the osteoclast vacuolar proton ATPase as novel bone antiresorptive agents», DDT, 4: 163-172 (1999), данные работы включены в настоящее описание в качестве ссылки).
Термин «ингибиторы ГМГ-КоА редуктазы» относится к ингибиторам 3-гидрокси-3-метилглутарил-Коа редуктазы. Соединения, которые обладают ингибирующей активностью в отношении ГМГ-КоА редуктазы, могут быть легко идентифицированы с использованием известных в данной области тестов. Например, см. тесты, описанные или процитированные в патенте США No. 4231938, в колонке 6, и в WO 84/02131, на страницах 30-33. Термины «ГМГ-КоА редуктазы ингибитор» и «ингибитор ГМГ-КоА редуктазы» имеют одно и то же значение при использовании в контексте настоящего описания.
Примеры ингибиторов ГМГ-КоА редуктазы, которые могут использоваться в контексте настоящего изобретения, включают, без ограничения, ловастатин (MEVACOR®; см. патенты США NoNo. 4231938, 4294926 и 4319039), симвастатин (ZOCOR®; см. патенты США NoNo. 4444784, 4820850 и 4916239), правастатин (PRAVACHOL®; см. патенты США NoNo. 4346227, 4537859, 4410629, 5030447 и 5180589), флувастатин (LESCOL®; см. патенты США NoNo. 5354772, 4911165, 4929437, 5189164, 5118853, 5290946 и 5356896), аторвастатин (LIPITOR®; см. патенты США NoNo. 5273995, 4681893, 5489691 и 5342952) и церивастатин (также известный как ривастатин и BAYCHOL®; см. патент США No. 5177080). Структурные формулы указанных и других ингибиторов ГМГ-КоА редуктазы, которые могут использоваться в способах согласно настоящему изобретению, описаны на странице 87 в руководстве M. Yalpani «Cholestrol Lowering Drugs», Chemistry & Industry, pp.85-89 (5 февраля 1996 года) и в патентах США NoNo. 4782084 и 4885314. Термин «ингибитор ГМГ-КоА редуктазы» в контексте настоящего описания включает все фармацевтически приемлемые лактоновые и кислые формы с открытым кольцом (то есть в тех случаях, когда лактоновое кольцо открыто для формирования свободной кислоты), а также соли и сложноэфирные формы соединений, которые обладают активностью по ингибированию ГМГ-КоА редуктазы, и, в этой связи, использование таких солей, сложных эфиров, открытых кислотных и лактоновых форм включено в область настоящего изобретения. Лактоновая часть и соответствующая открытая кислотная форма показаны ниже в виде структур I и II.
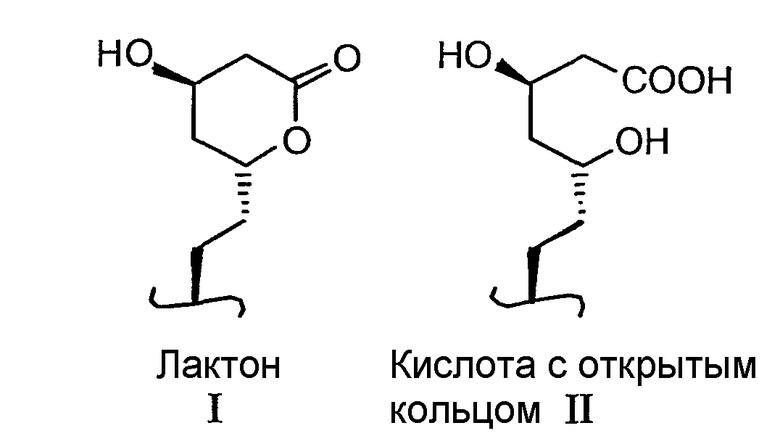
В случае ингибиторов ГМГ-КоА редуктазы, если может существовать открытая кислотная форма, солевая и сложноэфирная форма могут быть предпочтительно образованы из формы кислоты с открытым кольцом, и все такие формы включены в понятие «ингибитор ГМГ-КоА редуктазы», используемое в тексте настоящего описания. Предпочтительно, ингибитор ГМГ-КоА редуктазы выбирают из ловастатина и симвастатина, и наиболее предпочтительно, это симвастатин. В данном описании термин «фармацевтически приемлемые соли» применительно к ингибитору ГМГ-КоА редуктазы обозначает нетоксичные соли соединений, используемых в настоящем изобретении, которые в основном получают путем взаимодействия свободной кислоты с подходящим органическим или неорганическим основанием, в особенности с соединениями, образованными на основе катионов, таких как натрий, калий, алюминий, кальций, литий, магий, цинк и тетраметиламмоний, а также обозначает соли, полученные на основе аминов, таких как аммиак, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлоропрокаин, диэтаноламин, прокаин, N-бензилфенетиламин, 1-п-хлорбензил-2-пирролидин-1'-ил-метилбензимидазол, диэтиламин, пиперазин и трис(гидроксиметил)аминометан. Другие примеры солевых форм ингибиторов ГМГ-КоА редуктазы могут включать, без ограничения, ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдизилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глютамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памоат, пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат.
Сложноэфирные производные описанных соединений-ингибиторов ГМГ-КоА редуктазы могут действовать в качестве пролекарств, которые при абсорбции в кровоток теплокровного животного могут расщепляться таким образом, что будут высвобождать лекарственную форму, позволяя лекарственному компоненту проявлять повышенную терапевтическую эффективность.
Как указывалось выше, термин «антагонисты рецептора интегрина» относится к соединениям, которые оказывают селективное антагонизирующее воздействие, ингибируют связывание или противодействуют связыванию физиологического лиганда с интегрином αvβ3, к соединениям, которые оказывают селективное антагонизирующее воздействие, ингибируют связывание или противодействуют связыванию физиологического лиганда с интегрином αvβ5, к соединениям, которые оказывают антагонизирующее воздействие, ингибируют связывание или противодействуют связыванию физиологического лиганда и с αvβ3 интегрином, и с αvβ5, и к соединениям, которые оказывают антагонизирующее воздействие, ингибируют проявление активности или противодействуют проявлению активности одного или нескольких конкретных интегринов, экспрессированных на эндотелиальных клетках капилляров. Данный термин также относится к антагонистам интегринов αvβ6, αvβ8, α1β1, α2β1, α5β1, α6β1 и α6β4. Данный термин также относится к антагонистам любого сочетания интегринов αvβ3, αvβ5, αvβ6, αvβ8, α1β1, α2β1, α5β1, α6β1 и α6β4. Лоуд с соавт. (H.N.Lode and coworkers, PNAS USA 96: 1591-1596 (1999)) наблюдали синергические эффекты между ангиогенным антагонистом интегрина αv и белком слияния опухолеспецифичного антитела-цитокина (интерлейкин-2) по эрадикации спонтанных опухолевых метастаз. Результаты, полученные указанными авторами, позволяют полагать, что данное сочетание имеет потенциал для лечения рака и метастаза при опухолевом росте. Антагонисты рецептора интегрина αvβ3 ингибируют резорбцию кости через новый механизм, отличный от механизма, посредством которого действуют доступные в настоящее время препараты. Интегрины представляют собой гетеродимерные трансмембранные рецепторы адгезии, которые опосредуют взаимодействие по типу клетка-клетка и клетка-матрица. Субъединицы α и β интегрина взаимодействуют нековалентно и связываются с лигандами внеклеточной матрицы по механизму, зависимому от двухвалентных катионов. Наиболее представленным интегрином на остоекластах является αvβ3 (>107/остекласт), который, по всей видимости, играет роль в ограничении скорости цитоскелетной организации, что важно для миграции и поляризации клеток. Антагонизирующий эффект для αvβ3 выбирают из ингибирования резорбции кости, ингибирования рестеноза, ингибирования дегенерации желтого пятна, подавления артрита и ингибирования рака и метастазирующего роста.
Термин «анаболический агент остеобласта» относится к агентам, которые блокируют построение кости, таким как паратгормон (ПТГ). Было показано, что перемежающееся воздействие паратгормона (ПТГ) или его амино-концевых фрагментов и аналогов противодействует, останавливает, частично реверсирует разрежение кости и стимулирует образование костной ткани у животных и людей. Обсуждение данной темы приводится в работе Демпстера с соавт. (D. W. Dempster et al., «Anabolic actions of parathyroid hormone on bone», Endocr Rev 14: 690-709 (1993)). Проведенные исследования продемонстрировали благоприятные клинические эффекты паратиреоидного гормона по стимуляции образования кости и, в этой связи, повышения массы и прочности кости. Результаты были представлены в работе RM Neer et al., in New Eng J Med 344 1434-1441 (2001).
Дополнительно был показан мощный антикальциурический эффект белковых фрагментов или аналогов паратиреоидного гормона, таких как PTHrP-(1-36) [см. M.A.Syed et al., «Parathyroid hormone-related protein-(1-36) stimulates renal tubular calcium reabsorbtion in normal human volunteers: implications for the pathogenesis of humoral hypercalcemia of malignancy», JCEM 86:L 1525-1531 (2001)], которые могут рассматриваться как потенциальные анаболические агенты для лечении остеопороза.
Термин «витамин D» включает, без ограничения, витамин D3 (холекальциферол) и витамин D2 (эргокальциферол), которые представляют собой естественные биологически неактивные предшественники гидроксилированных биологически активных метаболитов витамина D: 1α-гидроксивитамина D; 25-гидроксивитамина D и 1α,25-гидроксивитамина D. Витамин D2 и витамин D3 имеют одинаковую биологическую эффективность для человека. В случае поступления витамина D2 или витамина D3 в кровоток, они гидроксилируются цитохром-Р450-витамин D-25-гидроксилазой с образованием 25-гидроксивитамина D. Метаболит 25-гидроксивитамин D представляет собой биологически инертный агент, который далее гидроксилируется в почке цитохром Р450-монооксигеназой, 25(OH)-D-1α-гидроксилазой с образованием 1,25-дигидроксивитамина D. При снижении сывороточного уровня кальция наблюдается повышение уровня продукции паратиреоидного гормона (ПТГ), который регулирует кальциевый гомеостаз и повышает плазменный уровень кальция за счет усиления конверсии 25-гидроксивитамина D в 1,25-дигидроксивитамин D.
Считается, что 1,25-дигидроксивитамин D отвечает за эффекты витамина D на кальциевый и костный метаболизм. Метаболит 1,25-дигидрокси является активным гормоном, необходимым для поддержания абсорбции кальция и скелетной целостности. Кальциевый гомеостаз поддерживается 1,25-дигидроксивитамином D за счет индукции моноцитарных стволовых клеток к дифференциации в остеокласты и за счет поддержания уровня кальция в нормальном диапазоне, что приводит к минерализации кости из-за отложения гидроксиапатита кальция на поверхности кости (см. Holick, MF «Vitamin D photobiology, metabolism and clinical application», in Endocrinology, 3rd ed., 990-1013 (1995), edited by DeGroot L, et al.). Однако повышенные уровни 1α,25-дигидроксивитамина D3 могут привести к повышению концентрации кальция в крови и к аномалиям в механизме контроля концентрации кальция в процессе костного метаболизма, что приводит к гиперкальциемии. 1α,25-дигидроксивитамин D3 также опосредованно регулирует активность остеокластов в костном метаболизме и можно ожидать, что его повышенные уровни будут усиливать избыточную резорбцию кости при остеопорозе.
В ряде вариантов осуществления настоящего изобретения выбирают соответствующее количество витамина D, с тем чтобы обеспечить адекватное поступление витамина D в промежутки между дозами, когда отсутствуют помехи для ингибитора катепсина К оказывать ингибирующий эффект на резорбцию кости. В случае пероральных композиций согласно настоящему изобретению, включающих ингибитор катепсина К и витамин D, количество витамина D составляет от примерно 100 МЕ до примерно 60000 МЕ. Неограничивающие примеры перорально принимаемого количества витамина D согласно вариантам осуществления настоящего изобретения включают, без ограничения, дозировки 2800 МЕ, 5600 МЕ, 7000 МЕ, 8400 МЕ, 11200 МЕ, 14000 МЕ, 16800 МЕ или 19600 МЕ. Неограничивающие примеры перорально принимаемого количества витамина D при еженедельном дозировании составляет 2800 МЕ, 5600 МЕ, 7000 МЕ, 8400 МЕ и 11200 МЕ. Неограничивающие примеры перорально принимаемого количества витамина D при ежемесячном введении дозы составляет 11200 МЕ, 14000 МЕ, 15400 МЕ, 16800 МЕ или 19600 МЕ.
Термин «синтетические аналоги витамина D» относится к неприродным соединениям, которые действуют подобно витамину D.
Термин «нестероидные противовоспалительные препараты» или НПВП относится к препаратам, которые ингибируют метаболизм арахидоновой кислоты до провоспалительных простагландинов через циклооксигеназу (COX)-1 и COX-2. Неограничивающие примеры НПВП включают: аспирин, ибупрофен, напроксен, диклофенак, этодолак, фенопрофен, флубипрофен, индометацин, кетопрофен, кеторолак, мелоксикам, набуметон, оксапрозин, пироксикам, сулиндак, толметин, дифлунизал, меклофенамат и фенилбутазон.
Термин «селективный ингибитор циклооксигеназы-2» или ингибитор COX-2 относится к типу нестероидных противовоспалительных препаратов (НПВП), которые ингибируют коэнзим COX-2, что способствует снижению болевых ощущений и воспаления в организме. Неограничивающие примеры ингибиторов COX-2 включают: целекоксиб, эторикоксиб, парекоксиб, рофекоксиб, валдекоксиб и лумиракоксиб.
Термин «ингибитор интелейкина-1 бета» или IL-1β относится к ингибиторам IL-1, который представляет собой растворимый фактор, продуцируемый моноцитами, макрофагами или другими клетками, которые активируют Т-лимфоциты и потенцируют их ответ на митогены или антигены. Неограничивающие примеры ингибиторов IL-1β включают диацереин и реин.
Термин «ингибитор LOX/COX» относится к ингибитору всех трех основных ферментов, вовлекаемых в путь арахидоновой кислоты, а именно: 5-LOX, COX-1 и COX-2. Неограничивающим примером ингибитора LOX/COX является ликофелон.
Термин «ингибитор RANKL» относится к ингибитору рецептора активатора лиганда NF-kB (RANKL), который ранее называли как фактор дифференциации остеокластов (ODF), лиганд остеопротегерина (OPGL) и цитокин, индуцирующий TNF-опосредованную активацию (TRANCE). RANKL представляет собой ключевой стимулятор образования и созревания остеокластов. Неограничивающим примером ингибитора RANKL является AMG-162.
При изготовлении в виде композиций с фиксированной дозой в таких объединенных продуктах используются соединения согласно настоящему изобретению в указанном ниже диапазоне дозировок, а также один или несколько фармацевтически активных агентов в разрешенном интервале дозировок. Альтернативно, соединения согласно настоящему изобретению могут использоваться последовательно вместе с одним или несколькими известными фармацевтически приемлемыми агентами, в том случае, когда использование сочетанных композиций неприемлемо.
Термин «введение» и его варианты (например, «ввод» соединения) применительно к соединению согласно настоящему изобретению обозначает введение соединения или пролекарства данного соединения в систему животного, нуждающегося в лечении. В том случае, когда соединение согласно настоящему изобретению или его пролекарство обеспечивается в сочетании с одним или несколькими активными агентами (например, цитотоксическим агентом и т.п.), термин «введение» и его варианты следует понимать как включающий и одновременное и последовательное введение соединения или его пролекарства и других агентов. Настоящее изобретение охватывают в свою область пролекарства соединений согласно настоящему изобретению. В основном такие пролекарства представляют собой функциональные производные соединения согласно настоящему изобретению, которые могут быть легко превращены in vivo в требуемые соединения. Неограничивающие примеры пролекраств, входящих в область настоящего изобретения, включают сложные эфиры, которые могут гидролизоваться с образованием спиртов согласно настоящему изобретению; кетоны, которые могут восстанавливаться in vivo с образованием спиртов согласно настоящему изобретению. Следует понимать, что в ряде случаев восстановление кетонов может происходить стереоспецифически с образованием преимущественного одного спиртового диастереомера. Другие примеры подходящих пролекарств, а также соответствующие традиционные процедуры отбора и получения таких производных описаны, например, в работе «Design of Prodrugs», ed. H. Bundgaard, Elsevier, 1985, которая включена в настоящее описание полностью в качестве ссылки. Так, применительно к способам лечения согласно настоящему изобретению термин «введение» будет охватывать лечение различных состояний, описанных применительно к данному рассматриваемому соединению или к тем соединениям, которые специфически не описаны, но которые превращаются в указанные соединения in vivo после введения пациенту. Метаболиты указанных соединений включают активные виды, продуцируемые при введении соединений согласно настоящему изобретению в биологическую среду.
В контексте настоящего описания термин «композиция» охватывает продукт, включающий указанные ингредиенты в указанных количествах, а также любой продукт, который образуется, непосредственно или опосредованно, на основе сочетания указанных ингредиентов в указанных количествах.
Термин «терапевтически эффективное количество» в контексте настоящего описания означает, что количество активного соединения или фармацевтического агента, который проявляет биологический или медицинский ответ в ткани, системе, организме животного или человека, а именно, такой, который необходим для исследователя, специалиста в области ветеринарии, врача или другого клинического специалиста.
Термины «лечение» или «терапия» заболевания в контексте настоящего описания включают: предотвращение заболевания, то есть воздействие, с тем чтобы предотвратить развитие клинических симптомов заболевания у млекопитающего, которое может быть подвергнуто воздействию или который предрасположен к данному заболеванию, но у которого отсутствуют или не проявляются симптомы заболевания; подавление заболевания, то есть остановку или снижение темпов развития процесса заболевания или его клинических симптомов; или ослабление заболевания, то есть регрессию заболевания или его клинических симптомов.
Термин «резорбция кости» в контексте настоящего описания относится к процессу, в ходе которого остеокласты разрушают кость.
Термины «один раз в неделю» или «еженедельное дозирование» в контексте настоящего описания означает, что стандартная дозировка, например, стандартная дозировка катепсина К, вводится один раз в неделю, то есть один раз в течение семидневного периода, предпочтительно в один и тот же день каждой недели. При еженедельном приеме стандартную дозировку в основном вводят примерно каждые семь дней. Неограничивающим примером еженедельного приема является введение стандартной дозировки ингибитора катепсина К каждое воскресенье. В основном рекомендуется, чтобы стандартная дозировка для еженедельного введения не вводилась в течение ряда последовательных дней, однако еженедельный прием может включать такой режим, при котором стандартные дозы вводятся в течение двух последовательных дней, попадающих в два разных недельных периода.
Дозировка «один раз в две недели» означает, что стандартная доза ингибитора катепсина К вводится один раз в двухнедельный период, то есть один раз в течение четырнадцатидневного периода, предпочтительно в один и тот же день в течение каждого двухнедельного периода. В формате двухнедельного дозирования каждая стандартная дозировка вводится примерно каждые четырнадцать дней. Неограничивающим примером режима введения один раз в две недели является введение стандартной дозы ингибитора катепсина К через воскресенье. Предпочтительно, чтобы стандартная доза не вводилась в течение ряда последовательных дней, однако режим введения дозы раз в две недели может включать такой режим дозирования, при котором стандартную дозу вводят в течение двух последовательных дней в ходе двух разных двухнедельных периодов.
Введение дозы «два раза в месяц» означает, что стандартную дозу ингибитора катепсина К вводят дважды, то есть два раза в течение календарного месяца. В формате введения два раза в месяц дозу предпочтительно вводят в те же самые два дня каждый месяц. В режиме введения дозы два раза в месяц стандартную дозу предпочтительно вводят примерно каждые четырнадцать-шестнадцать дней. Неограничивающим примером режима дозирования два раза в месяц является введение дозы в начале или примерно в начале месяца и на пятнадцатый или примерно на пятнадцатый день, то есть в середине месяца. Предпочтительно, чтобы стандартные дозировки не вводились в одни и те же или последовательные дни, однако прием дозы дважды в месяц может включать такой режим приема, при котором стандартные дозировки вводятся в течение двух последовательных дней в течение месячного периода или различных месячных периодов. Режим двукратного дозирования в месяц в контексте настоящего описания отличается от режима дозирования один раз в две недели и не включает его, поскольку указанные два режима имеют разную периодичность и приводят к введению различного количества дозировок в течение длительного периода времени. Например, в течение года может быть введено всего примерно двадцать четыре дозы в соответствии с режимом введения два раза в месяц (поскольку в году двенадцать календарных месяцев), тогда как в соответствии с режимом введения дозы один раз в две недели будет введено примерно двадцать шесть доз (поскольку имеется примерно пятьдесят две недели в году).
Термин «один раз в месяц» используется в общепринятом значении, составляющем примерно четыре недели, примерно 30 дней или 1/12 часть календарного года.
Настоящее изобретение также относится к фармацевтической композиции применяемой при лечении остеопороза или других костных заболеваний, включающем введение терапевтически эффективного количества соединения согласно настоящему изобретению, при наличии или в отсутствие фармацевтически приемлемых носителей или разбавителей. Подходящие композиции согласно настоящему изобретению включают водные растворы, содержащие соединения согласно настоящему изобретению и фармацевтически приемлемые носители, например, солевой раствор с заданным уровнем pH, например 7,4. Указанные растворы могут вводиться в кровоток пациента с помощью местной инъекции болюсом.
В том случае, когда соединение согласно настоящему изобретению вводят в организм человека, ежедневная доза может быть определена лечащим врачом и доза в основном будет варьироваться в зависимости от возраста, веса и реакции отдельного пациента, а также от тяжести симптомов заболевания у пациентов.
В одном примерном варианте применения подходящее количество соединения вводят млекопитающему, подвергающемуся лечению состояния, зависимого от катепсина. Пероральные дозировки согласно настоящему изобретению, используемые с целью достижения указанных эффектов, будут варьироваться от примерно 0,01 мг на кг веса тела в день (мг/кг/день) до примерно 100 мг/кг/день, предпочтительно от 0,01 до 10 мг/кг/день и наиболее предпочтительно от 0,1 до 5,0 мг/кг/день. Для перорального введения предпочтительно используют композиции, имеющие форму таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 3,5, 5,0, 10,0, 15,0, 20,0, 25,0, 35,0, 40,0, 50,0, 80,0, 100, 200 и 500 миллиграмм активного ингредиента, для симптоматической корректировки дозы, вводимой пациенту, подлежащему лечению. Указанный лекарственный препарат обычно содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента, предпочтительно от примерно 1 мг до примерно 100 мг активного ингредиента. При внутривенном введении наиболее предпочтительные дозы варьируют от примерно 0,1 мг до примерно 10 мг/кг/минуту при инфузии с постоянной скоростью. Соединения согласно настоящему изобретению могут быть введены с достижением благоприятного эффекта в виде однократной ежедневной дозы или общие ежедневные дозировки могут вводиться в виде раздельных доз два, три или четыре раза в день. Кроме того, соединения согласно настоящему изобретению могут вводиться в непрерывном режиме, включающем интервал дозирования один раз в неделю, один раз в две недели, два раза в месяц и один раз в месяц. Дополнительно, предпочтительные соединения согласно настоящему изобретению могут вводиться в интраназальной форме посредством местного нанесения интраназальных носителей или чрескожным способом, с помощью трансдермальных кожных пластырей, известных специалистам в данной области. В случае введения в составе чрескожной системы доставки режим введения дозы должен быть, разумеется, непрерывным, а не перемежающимся в течение приема.
Соединения согласно настоящему изобретению могут использоваться в сочетании с другими агентами, применяемыми для лечения катепсин-опосредованных состояний. Индивидуальные компоненты таких сочетаний могут вводиться по отдельности в разные временные точки в ходе всего курса лечения или одновременно в составе отдельных стандартных объединенных форм. В этой связи, настоящее изобретение следует рассматривать как охватывающее все такие режимы одновременного или перемежающегося лечения и термин «введение» следует соответствующим образом интерпретировать. Следует понимать, что в контексте настоящего описания сочетание соединений согласно настоящему изобретению с другими агентами, используемыми для лечения катепсин-опосредованных заболеваний, включает в принципе любое сочетание с любой фармацевтической композицией, применяемой для лечения заболеваний, связанных с функционированием эстрогенов.
В этой связи, область настоящего изобретения охватывает использование заявленных в настоящем изобретении соединений в сочетании со вторым агентом, выбранным из: органического бисфосфоната; модулятора рецептора эстрогена; модулятора рецептора андрогена; ингибитора протон-АФТазы остеокластов; ингибитора ГМК-КоА редуктазы; антагониста рецептора интегрина; анаболического агента остеобластов, такого как паратгормон; витамина D; синтетического аналога витамина D; нестероидного противовоспалительного средства; селективного ингибитора циклооксигеназы-2; ингибитора интерлейкина-1-бета; ингибитора LOX/COX; ингибитора RANKL; и их фармацевтически приемлемых солей и смесей.
Указанные и другие аспекты настоящего изобретения станут понятными из описания, приведенного в настоящей заявке.
Определения
Соединения согласно настоящему изобретению могут иметь асимметрические центры, хиральные оси и хиральные планы (как описано в руководстве: E.L.Eliel and S.H.Wilen, Stereochemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, pages 1119-1190) и могут быть представлены в виде рацематов, рацемических смесей и в виде отдельных диастереомеров, со всеми возможными изомерами и их смесями, включая оптические изомеры, которые, все, включены в область настоящего изобретения. Дополнительно, описываемые соединения могут существовать в виде таутомеров, и обе таутомерные формы рассматриваются как входящие в область настоящего изобретения, даже если в описании приведена всего лишь одна таутомерная структура. Например, любую ссылку на приведенное ниже соединение А следует понимать как включающую и таутомерную структуру В, и наоборот, а также их смеси.
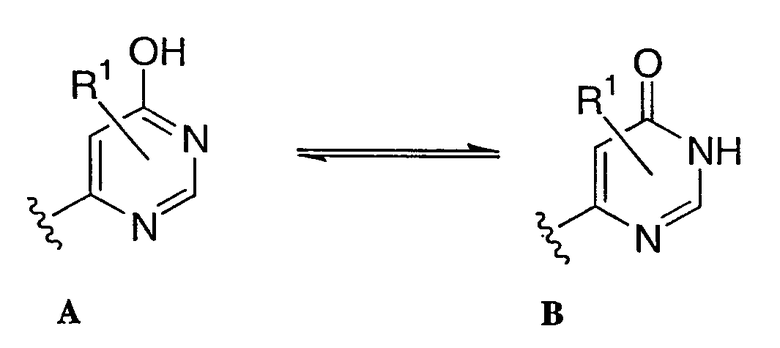
В том случае, когда любая переменная (например, R1, R2, R3 и т.п.) встречается более чем один раз в любом заместителе, ее определение в каждом случае не зависит от любого другого случая встречаемости. Кроме того, сочетание заместителей при переменных возможно только в том случае, если такие сочетания приводят к получению стабильных соединений. Линии, изображенные на кольцевых системах от заместителей, указывают на то, что данная связь может быть присоединена к любому замещаемому атому углерода в кольце. Если кольцевая система является полициклической, то следует понимать, что такая связь присоединяется к любому подходящему атому углерода только на проксимальном кольце.
Следует понимать, что заместители и характер замещения в соединениях согласно настоящему изобретению могут быть выбраны специалистом в данной области таким образом, чтобы получить соединения, которые будут химически стабильными и которые могут быть легко синтезированы по известным методикам, а также по приведенным ниже методикам, из легко доступных исходных материалов. Если заместитель сам замещен более чем одной группой, следует понимать, что указанные множественные группы могут находиться на одном и том же атоме углерода или на разных атомах, при условии, что при этом будет образована стабильная структура. Термин «необязательно замещенный одним или несколькими заместителями» следует понимать как эквивалентный термину «необязательно замещенный по меньшей мере одним заместителем», и в таких случаях предпочтительный вариант будет представлять собой замещение заместителями в количестве от нуля до трех.
В контексте настоящего описания термин «алкил» включает и разветвленные и линейно-цепочечные насыщенные алифатические углеводородные группы, содержащие от одного до десяти атомов углерода, если особо не указано иное. Например, С1-С10 применительно к «С1-С10 алкилу» определяется как включающий группу, содержащую 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода в линейном, разветвленном или циклическом варианте организации. Например, термин «С1-С10 алкил» конкретно включает метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил и т.п.
Термин «циклоалкил» или «карбоцикл» обозначает циклические кольца алканов, включающие в целом от трех до восьми атомов углерода, если особо не оговорено иное, или любое число в пределах данного диапазона (например, это может быть циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил).
В некоторых случаях заместители могут определяться по интервалу атомов углерода, который включает нулевой вариант, такой как (С0-С6) алкилен-арил. Если арил объединяется вместе с фенилом, то указанное определение может включать как сам фенил, так и -CH2Ph, -CH2CH2Ph, -CH(CH3), -CH2CH(CH3)Ph и т.п.
В контексте настоящего описания термин «арил» обозначает любое стабильное моноциклическое или карбоциклическое углерод-содержащее кольцо, включающее до 12 атомов в каждом кольце, где по меньшей мере одно кольцо является ароматическим. Примеры таких арильных элементов включают фенил, нафтил, тетрагидронафтил, инданил, бифенил, фенантрил, антрил или аценафтил. В тех случаях, когда арильный заместитель представляет собой бициклический заместитель и одно кольцо является неароматическим, следует понимать, что присоединение происходит через ароматическое кольцо.
Термин «гетероарил» в контексте настоящего описания обозначает стабильное моноциклическое, бициклическое или трициклическое кольцо, содержащее до 10 атомов в каждом кольце, где по меньшей мере одно кольцо является ароматическим и содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. В свете данного определения гетероарильные группы включают, без ограничения, бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тетразолил, тетразолопиридил, тиадиазолил, тиазолил, тиенил, триазолил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидроиндолил, дигидрохинолинил, метилендиоксибензол, бензотиазолил, бензотиенил, хинолинил, изохинолинил, оксазолил и тетрагидрохинолин. В тех случаях, когда гетероарильный заместитель является бициклическим, одно кольцо является неароматическим или не содержит гетероатомов, следует понимать, что присоединение происходит через ароматическое кольцо или через кольцо, содержащее гетероатом, соответственно. Если гетероарил содержит атом азота, следует понимать, что соответствующие ему N-оксиды также входят в область данного определения.
Как очевидно специалистам в данной области, термины «гало» или «галоген», используемые в настоящем описании, включают хлор, фтор, бром и йод. Термин «кето» обозначает карбонил (С=О).
Настоящее изобретение также включает N-оксидные производные и защищенные соединения формулы I. Например, в том случае, когда соединения формулы I содержат окисляемый атом азота, указанный атом азота может быть превращен в N-оксид по известным методам. Кроме того, когда соединения формулы I содержат группы, такие как гидрокси, карбокси, тио, или любую группу, содержащую один или несколько атомов азота, указанные группы могут быть защищены соответствующими защитными группами. Исчерпывающий перечень подходящих защитных групп приведен в руководстве Грина (T.W.Green, Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 1981), описание которого включено в данную заявку в качестве ссылки. Защищенные производные соединения формулы I могут быть получены с помощью известных методик.
Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают традиционные нетоксичные соли согласно настоящему изобретению, получаемые на основе неорганических или органических кислот. Например, традиционные нетоксичные соли включают соли, полученные на основе неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная кислоты и т.п., а также соли, полученные на основе органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глютаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая, трифторуксусная и т.п. Получение описанных выше фармацевтически приемлемых солей и других типичных фармацевтически приемлемых солей более полно описано в работе Берга с соавт. (Berg et al., «Pharmaceutical Salts», J. Pharm. Sci., 1977: 66:6:1-19), включенной в настоящее описание в качестве ссылки. Фармацевтически приемлемые соли соединений согласно настоящему изобретению могут быть синтезированы из соединений согласно настоящему изобретению, которые содержат основной или кислотный фрагмент, с использованием обычных химических методов. Как правило, соли основных соединений получают либо путем ионообменной хроматографии, либо путем взаимодействия свободного основания со стехиометрическими количествами или с избытком желательной солеобразующей неорганической или органической кислоты в соответствующем растворителе, или с использованием различных сочетаний растворителей. Аналогично, соли кислотных соединений получают при проведении реакции с соответствующим неорганическим или органическим основанием.
Для целей настоящего описания приведенные в нем сокращения имеют следующие значения:
i-BuCOCl = изобутилхлорформиат
t-BuMe2SiCl = трет-бутилдиметилхлоросилан
BuLi = бутиллитий
CH2Cl2 = метиленхлорид
CH3CN = метилцианид
CrO3 = хромат
DAST = диэтиалминосульфуртрифторид
DMF (ДМФ) = N,N-диметилформамид
DMSO (ДМСО) = диметилсульфоксид
DTT (ДТТ) = дитиотрейтол
EDTA (ЭДТА) = этилендиаминтетрауксусная кислота
EtOH = этанол
KOH = гидроксид калия
HATU = гексафторфосфат 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3,-тетраметилурония
HCl = соляная кислота
H5IO6 = надйодная кислота
MeMgBr = бромид метилмагния
MgSO4 = сульфат магния
Na2CO3 = карбонат натрия
NaCl = хлорид натрия
NH4Cl = хлорид аммония
Na2BH4 = боргидрид натрия
PdCl2(dppf) = [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II)
PG = защитная группа
rt (к.т.) = комнатная температура
sat.aq. = насыщенный водный раствор
SiO2 = силикагель
TBAF = фторид тетрабутиламмония
THF (ТГФ)= тетрагидрофуран
tlc (тсх)= тонкослойная хроматография
(Ts)2O = п-толуолсульфоновый ангидрид
Me = метил
Et = этил
Новые соединения согласно настоящему изобретению могут быть получены в соответствии с приведенными общими методами с использованием подходящих материалов, которые иллюстрируются конкретными примерами, приведенными далее. Проиллюстрированные в указанных примерах соединения не следует трактовать как представляющие ограничивающие настоящее изобретение. Приведенные примеры лишь иллюстрируют детали получения соединений по настоящему изобретению. Специалистам в данной области понятно, что для получения данных соединений могут быть использованы различные вариации условий и процессов в приведенных препаративных процедурах. Все указанные значения температур относятся к градусам Цельсия, если особо не указано иное.
Как описано в примере 12 и схеме 1, исходным соединением является коммерчески доступный сложный бета бензиловый эфир N-t-Boc-аспарагиновой кислоты, в рамках данного процесса свободную кислоту активируют по методике, включающей использование смешанного ангидрида (см. Chen, F.M.F; Lee, Y; Steinauer, R., Benoiton, N.L., Can. J. Chem. 1987, 65, 613-618) для восстановления боргидридом натрия. Спирт превращают в удаляемую группу и проводят циклизацию in situ до получения карбамата при нагревании. Сложный эфир обрабатывают избыточным количеством реактива Гриньяра с получением водорастворимого третичного спирта. Указанный спирт обрабатывают DAST с получением фторированного продукта. Гидролиз циклического карбамата и силилирование образовавшегося спирта дают первичный амин. Далее получают имин при азеотропном удалении летучих продуктов. Монолитиирование 1,4-дибромбензола дает нуклеофил, который легко вступает в реакцию с имином с образованием вторичного амина. Полученный спирт подвергают защите и окисляют с образованием кислоты. Стандартная реакция образования амида дает летучий бромсодержащий интермедиат.
Схема 1
Бромсодержащее соединение, полученное по методике схемы 1, может быть превращено в боронатный сложный эфир в условиях реакции с палладием, как показано на схеме 2. В свою очередь, полученный боронатный сложный эфир может быть легко превращен в биарильный продукт при связывании с бромидом в условиях, включающих палладий.
Схема 2
Альтернативно, на схеме 3 показано, что арилбромид может быть превращен в боронатный сложный эфир в реакции, катализируемой палладием. В реакции, включающей палладий, указанный боронатный сложный эфир может быть объединен с бромидом, показанным в схеме 1, с получением биарильного соединения.
Схема 3

ПРИМЕР 1
Синтез N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
Стадия 1:
Получение 1-(4-бромфенил)-2,2-дифторэтанона
К холодному (-78°С) перемешанному раствору 1,4-дибромбензола (86,4 г, 366 ммоль) в тетрагидрофуране (800 мл) добавляют н-бутиллитий (228 мл, 1,6 М в гексанах, 366 ммоль). Полученную смесь перемешивают при температуре -78°С в течение 30 минут и затем к данной взвеси добавляют этилдифторацетат (50 г, 402 ммоль) в течение 2 минут. Указанную смесь перемешивают при температуре -78°С в течение 1 часа. Реакцию гасят добавлением 1N соляной кислоты (250 мл) и оставляют смесь нагреваться до комнатной температуры. Смесь разбавляют метил-трет-бутиловым эфиром (250 мл) и разделяют слои. Органический продукт промывают насыщенным раствором соли (100 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток отгоняют в вакууме с получением дифторкетона в виде блестящего твердого вещества белого цвета.
Стадия 2
Получение (1R)-1-(4-бромфенил)-2,2-дифторэтанола (аналогичен соединению, полученному в работе Ramachandran. P.V. et al., Tet. Asym. 1994, Vol., 5, No.6, pp.1075-86)
Кетон, полученный на стадии 1 примера 1 (2,35 г, 10 ммоль), и коммерчески доступный S-Alpin Borane (3,1 г, 12 ммоль) смешивают при комнатной температуре и перемешивают в течение четырех дней при некотором выделении газа. Через четыре дня результаты 1H ЯМР анализа аликвот демонстрируют полное расходование кетона. Реакционную смесь охлаждают до 0°С для добавления ацетальдегида (168 мкл, 3 ммоль). Баню удаляют и перемешивание продолжают при комнатной температуре в течение 30 минут. Добавляют диэтиловый эфир (20 мл) и затем этаноламин (725 мкл, 12 ммоль). Смесь перемешивают при комнатной температуре в течение часа. Осадок удаляют фильтрованием и промывают пентаном. Фильтрат концентрируют при пониженном давлении и очищают флэш-хроматографией (90% гексаны; 10% этилацетат - 70% гексаны; 30 этилацетат) с образованием желательного материала в виде бесцветного масла. В данной точке оптическую чистоту не проверяют.
Стадия 3:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этил}-L-лейцинамида
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид со стадии 9 в примере 12 (24 г, 53 ммоль), бис(пинаколато)дибор (15,24 г, 60 ммоль) и ацетат калия (16,2 г, 165 ммоль) перемешивают в диоксане (240 мл) и смесь барботируют азотом через пипетку в течение 15 минут. Добавляют комплекс в отношении 1:1 [1,1-бис(бифенилфосфин)ферроцен]дихлорпалладия (II) с дихлорметаном (PdCl2dppf.CH2Cl2, 2,4 г, 3 ммоль) и реакционную смесь погружают в масляную баню при температуре 80°С в атмосфере азота на 105 минут. Результаты оценки по методу 1H ЯМР аликвот демонстрируют полное расходование бромида. Реакционную смесь оставляют охлаждаться до комнатной температуры и большую часть растворителя удаляют при пониженном давлении. Остаток растворяют в минимальном количестве дихлорметана и фильтруют через слой силикагеля. Две фракции собирают и концентрируют при пониженном давлении с получением твердого материала. Наименее полярную фракцию перемешивают при комнатной температуре в течение ночи в 90% гексанах; в смеси с 10% диэтиловым эфиром, а самую полярную фракцию перемешивают при температуре 0°С в течение ночи в гексанах. Обе фракции дают чистый материал в виде твердого вещества не совсем белого цвета.
Стадия 4:
Получение N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (20 г, 40 ммоль) и арилбромид со стадии 2 примера 1 (11,4 г, 48 ммоль) растворяют в диметилформамиде (400 мл) и затем добавляют водный раствор бикарбоната натрия (60 мл, 120 ммоль). Смесь барботируют азотом через пипетку в течение 15 минут. Добавляют комплекс в отношении 1:1 [1,1-бис(бифенилфосфин)ферроцен]дихлорпалладия (II) с дихлорметаном (PdCl2dppf.CH2Cl2, 2,4 г, 3 ммоль) и реакционную смесь погружают в масляную баню при температуре 80°С в атмосфере азота на 16 часов. Большую часть диметилформамида удаляют в роторном испарителе при низком давлении (45°С, примерно 1-5 мм Hg). Остаток растворяют в этилацетате (400 мл) и фильтруют через слой целита. Фильтрат концентрируют при пониженном давлении. Остаток отбирают примерно в 10 мл дихлорметана и разделяют флэш-хроматографией. Наиболее чистые фракции разгоняют в гексанах (100 мл) при температуре 0°С в течение ночи три раза и снова проводят флэш-хроматографию (90% гексаны; 10% этилацетат - 45 гексаны; 55% этилацетат). После выпаривания летучих продуктов при пониженном давлении твердый материал перемешивают в гексанах в течение 2 дней. Продукт все еще содержит 4% пинакола, и партию разделяют на две части. Фракцию А растворяют в минимальном количестве изопропилового спирта при осторожном нагревании. Гексаны добавляют до тех пор, пока раствор помутнеет, и оставляют для охлаждения. Появляются белые кристаллы и реакционную смесь далее охлаждают до температуры 0°С в течение 15 минут. Твердый материал собирают фильтрованием с получением продукта, не загрязненного пинаколом. Фракцию В перемешивают в 20% диэтиловом эфире, 1% этилацетате, 79% гексанах (100 мл) в течение 3 часов при комнатной температуре. Твердый материал, собранный фильтрованием, загрязнен всего лишь 1% пинакола, по данным анализа 1Н ЯМР. Энантиомерный избыток подтверждается хиральным AD-RH с использованием 32% ацетонитрила, 68% воды, 0,1% муравьиной кислоты в изократическом режиме (24 минут для энантиомера и 27 минут для требуемой молекулы). (MH)+ESI=528,0.
ПРИМЕР 2
Синтез N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-лейцинамида
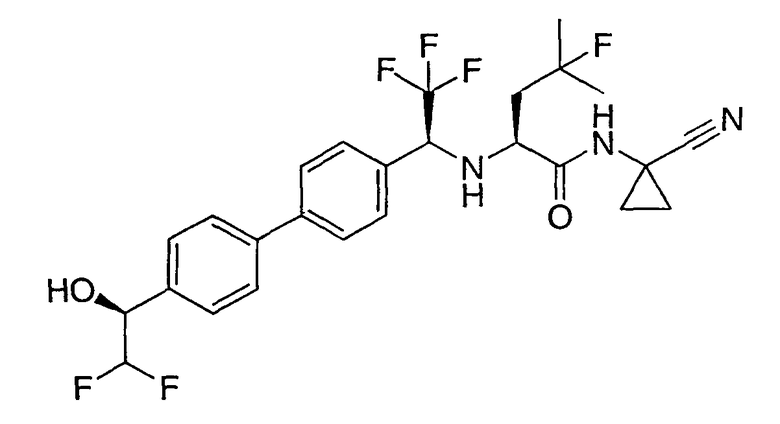
Стадия 1:
Получение (1S)-1-(4-бромфенил)-2,2-дифторэтанола
Кетон, полученный на стадии 1 примера 1 (2,35 г, 10 ммоль), и коммерчески доступный S-Alpin Borane (3,1 г, 12 ммоль) смешивают при комнатной температуре и перемешивают в течение четырех дней при некотором выделении газа. Через четыре дня результаты 1H ЯМР анализа аликвоты демонстрируют наличие исходного материала. Добавляют еще S-Alpin Borane (1 мл) и перемешивание продолжают еще два дня. Результаты 1H ЯМР анализа аликвоты демонстрируют полное расходование кетона. Реакционную смесь охлаждают до 0°С с последующим добавлением ацетальдегида (393 мкл, 7 ммоль). Баню удаляют и перемешивание продолжат при комнатной температуре в течение 30 минут. Добавляют диэтиловый эфир (20 мл) и затем этаноламин (966 мкл, 16 ммоль). Смесь перемешивают при комнатной температуре в течение часа. Осадок удаляют фильтрованием и промывают пентаном. Фильтрат концентрируют при пониженном давлении и очищают флэш-хроматографией (90% гексаны; 10% этилацетат - 70% гексаны; 30% этилацетат) с образованием желательного материала в виде бесцветного масла.
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (200 мг, 0,40 ммоль) и арилбромид со стадии 1 примера 2 (114 мг 0,48 ммоль) обрабатывают по методике стадии 4 примера 1 с получением твердого материала белого цвета (MH)+ ESI=528,0.
ПРИМЕР 3
Синтез N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамида
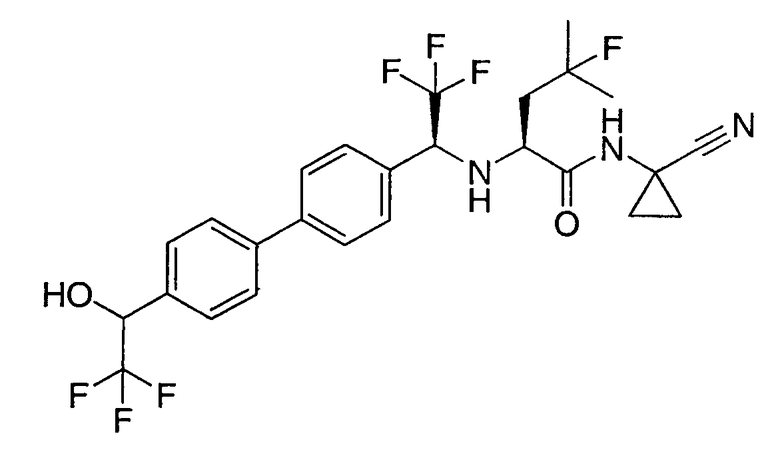
Стадия 1:
Получение 1-(4-бромфенил)-2,2,2-трифторэтанола
К раствору коммерчески доступного 4'-бром-2,2,2-трифторацетофенона (100 мг) при комнатной температуре в 1,9 мл метанола добавляют боргидрид натрия (15 мг). Смесь перемешивают при комнатной температуре в течение ночи. Добавляют воду, проводят экстракцию метил-т-бутиловым эфиром (3×20 мл), промывают водой и насыщенным раствором соли. Далее сушат сульфатом магния и растворитель удаляют при пониженном давлении с получением указанного в заголовке соединения, которое используют в качестве такового на следующей стадии.
1Н ЯМР (CDCl3) δ (ч/млн): 7,55 (2Н, д), 7,35 (2Н, д), 4,92-5,05 (1Н, м), 3,20 (1Н, с).
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамида
Пропускают поток азота через раствор ДМФ (4 мл), N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этил}-L-лейцинамида, описанного на стадии 3 примера 1 (150 мг), 1-(4-бромфенил)-2,2,2-трифторэтанола со стадии 1 примера 3 (92 мг) и 2M Na2CO3 (750 мкл) в течение 15 минут и затем добавляют комплекс (1:1) [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (12 мг). Смесь нагревают до 80°С в течение 3 часов в атмосфере азота. Далее смесь оставляют охлаждаться до комнатной температуры, выливают в лед (20 г) и насыщенный водный раствор бикарбоната натрия (20 мл) и экстрагируют 50% этилацетатом в диэтиловом эфире (3×50 мл). Объединенные экстракты промывают насыщенным раствором соли и сушат сульфатом магния. Удаление растворителя дает остаток, который очищают хроматографией на SiO2 с использованием этилацетат и гексанов (20-50%) в качестве элюента, после чего растирают с использованием диэтилового эфира и гексанов с получением указанного в заголовке соединения.
1Н ЯМР (CD3COCD3) δ (ч/млн): 8,18 (1Н, с), 7,60-7,70 (4Н, м), 7,50-7,55 (1Н, м), 7,33 (1Н, д), 7,28 (1Н, д), 6,40 (1Н, ушир.с), 4,38-4,48 (1Н, м), 3,56 (1Н, т), 2,67-2,69 (1Н, м), 1,92-2,01 (2Н, м), 1,45-1,46 (10Н, м), 1,05-1,11 (3Н, м), 0,92-0,99 (1Н, м), 0,56-0,60 (2Н, м), 0,36-0,38 (2Н, м).
ПРИМЕР 4
Синтез N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[(1S)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамида и N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[(1R)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамида

Стадия 1:
Получение 2-(4-бромфенил)-4,4,4-трифторбутан-2-ола
К раствору 1,4-дибромбензола (2,5 г, 10,6 ммоль) в ТГФ (50 мл) при температуре -78°С добавляют n-BuLi (6,5 мл, 10,4 ммоль; 1,6 М в гексанах) и смесь перемешивают при температуре -78°С в течение 15 минут. Затем добавляют 4,4,4-трифтор-2-бутанон (1,3 г, 10,3 ммоль). После перемешивания еще в течение 15 минут в смесь добавляют водный NH4Cl для гашения реакции и проводят экстракцию этилацетатом. Очистка комби-флэш-хроматографией (колонка 40 г, элюция гексанами-этилацетатом (10%-20%) в течение 20 минут; скорость течения 35 мл/мин, и проводят сбор фракций по 18 мл) дает указанное в заголовке соединение в виде жидкости слегка коричневого цвета.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,5 (4Н, м), 4,64 (1Н, с), 1,64 (3Н, с).
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[(1S)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамида и N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[(1R)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамида
Пропускают поток азота через раствор ДМФ (5 мл), N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этил}-L-лейцинамида, описанного на стадии 3 примера 1 (150 мг), 2-(4-бромфенил)-4,4,4-трифторбутан-2-ола со стадии 1 примера 4 (100 мг) и 2M Na2CO3 (360 мкл) в течение 15 минут и затем добавляют комплекс (1:1) [1,1-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (5 мг). Смесь нагревают до 80°С в течение 3 часов в атмосфере азота. Далее смесь оставляют охлаждаться до комнатной температуры, выливают в лед (20 г) и насыщенный водный раствор бикароната натрия (20 мл) и экстрагируют 50% этилацетатом в диэтиловом эфире (3×50 мл). Объединенные экстракты промывают насыщенным раствором соли и сушат сульфатом магния. Удаление растворителя дает остаток, который очищают хроматографией на силикагеле с использованием автоматизированной системы с насосом для создания градиента CombiFlash (этилацетат/гексан, 20:80-50:50 в течение 25 минут), после чего растирают с использованием диэтилового эфира и гексанов с получением смеси двух стереомеров.
Для разделения инъецируют на Chiralcel OD, 250 X 20 мм (OD00C-СК004) раствор 200 мкл смеси двух диастереомеров (в концентрации 50 мкг/мкл в 33% 2-пропанола и 67% гексанов) N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(3,3,3-трифтор-1-гидрокси-1-метилпропил)бифенил-4-ил]этил}-L-лейцинамида с использованием 33% 2-пропанола в гексанах в качестве растворителя, скорость течения поддерживают на уровне 6 мл/мин и выявление проводят при длине волны 260 нм. После нескольких инъекций N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид выделяют из фракций, элюирующихся в первую очередь, и N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид выделяют из второй группы элюированных фракций.
Первый элюированный диастереомер: 1Н ЯМР (CD3COCD3) δ (ч/млн): 8,19 (1Н, с), 7,74-7,68 (6Н, м), 7,57 (2Н, д), 4,60 (1Н, с), 4,42-4,36 (1Н, м), 3,57-3,53 (1Н, м), 2,78-2,88 (2Н, м), 1,94-2,00 (2Н, м), 1,73 (3Н, с), 1,49-1,31 (8Н, м), 1,07-1,13 (1Н, м), 1,00-0,90 (1Н, м), NH трифторэтиламина не выявляется. (MH)+ APCI=573,9. Данные по стереохимии являются предположительными.
Второй элюированный диастереомер: 1Н ЯМР (CD3COCD3) δ (ч/млн): 8,19 (1Н, с), 7,74-7,68 (6Н, м), 7,57 (2Н, д), 4,60 (1Н, с), 4,42-4,36 (1Н, м), 3,57-3,53 (1Н, м), 2,78-2,88 (2Н, м), 1,94-2,00 (2Н, м), 1,73 (3Н, с), 1,49-1,31 (8Н, м), 1,07-1,13 (1Н, м), 1,00-0,90 (1Н, м), NH трифторэтиламина не выявляется. (MH)+ APCI=574,0. Данные по стереохимии являются предположительными.
ПРИМЕР 5
Синтез N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4'-(R)-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамида
Стадия 1:
Получение (R)-2,2,2-трифтор-1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этанола
Через суспензию (R)-1-(4-бромфенил)-2,2,2-трифторэтанола (2,26 г, 8,86 ммоль), бис(пинаколато)дибора (2,9 г, 11 ммоль) и ацетата калия (3 г, 30 ммоль) в ДМФ (80 мл) пропускают азот для барботирования в течение15 минут. Добавляют комплекс (1:1) [1,1-бис(дифенилфосфин)ферроцен]дихлорпалладия (II) с дихлорметаном (362 мг, 0,44 ммоль) и снова проводят барботирование азотом в течение 10 минут. Далее реакционную смесь перемешивают при температуре 85°С в течение 2 часов, выливают в лед и воду и экстрагируют этилацетатом (2×80 мл). Объединенные экстракты промывают насыщенным раствором NaCl, сушат (MgSO4) и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле (этилацетат/гексан, 5:95-20:80 в течение 25 минут и затем 20:80 в течение 5 минут) с получением указанного в заголовке продукта.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,8 (2Н, д), 7,55 (2Н, д), 5,9 (1Н, ОН), 5,2-5,3 (1Н, м), 1,3 (12Н, с).
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4'-(R)-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамида
Боронатный сложный эфир со стадии 1 примера 5 (250 мг, 0,83 ммоль) и N2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид (374 мг, 0,83 ммоль) со стадии 9 примера 12 объединяют, как было описано на стадии 4 примера 1, с получением указанного в заголовке соединения в виде порошка не совсем белого цвета.
1Н ЯМР (CD3COCD3) δ (ч/млн): 8,1-8,2 (1Н, ушир.с), 7,75-7,8 (4Н, м), 7,7 (2Н, м), 7,6 (2Н, м), 5,9 (1Н, м), 5,25-5,35 (1Н, м), 4,35 (1Н, м), 3,5-3,6 (1Н, м), 1,9-2,1 (2Н, м), 1,2-1,6 (8Н, м), 0,9-1,1 (2Н, м); NH не выявляется. (MH)+ESI=545,8.
ПРИМЕР 6
Синтез N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил]бифенил-4-ил}этил)-L-лейцинамида
Стадия 1:
Получение 2-(4-бромфенил)-1,1,1,3,3,3-гексафторпропан-2-ола
При температуре -78°С к раствору дибромбензола (4,7 г) в тетрагидрофуране (100 мл) добавляют н-бутиллитий (8 мл, 2,5 М в гексанах) и смесь перемешивают в течение 15 минут. Через суспензию в течение примерно 15 минут осторожно пропускают поток гексафторацетона. Затем смесь оставляют для реакции на 1 час. Далее ее выливают в лед, разбавляют хлоридом аммония и экстрагируют этилацетатом. Органический слой сушат сульфатом магния и растворитель удаляют при пониженном давлении при минимальном нагревании. Остаток пропускают через короткую насадку SiO2 с использованием 10% этилацетата/90% гексанов в качестве элюента с получением третичного спирта.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,75-7,8 (4Н, с), 7,65 (1Н, ОН).
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил]бифенил-4-ил}этил)-L-лейцинамида
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этил}-L-лейцинамид, описанный на стадии 3 примера 1 (220 мг, 0,44 ммоль) и бромид со стадии 1 примера 6 (323 мг, 1 ммоль) связывают, как описано на стадии 4 примера 1, с получением указанного в заголовке соединения в виде пены.
1Н ЯМР (CD3COCD3) δ (ч/млн): 8,2 (1Н, ушир.с), 7,85-7,95 (4Н, м), 7,75-7,8 (2Н, д), 7,6 (2Н, д), 7,55 (1Н, ОН), 4,4 (1Н, м), 3,55 (1Н, м), 2,8-2,9 (1Н, м), 1,9-2,1 (2Н, м), 1,4-1,5 (6Н, м), 1,3-1,4 (2Н, м), 1,1 (1Н, м), 0,95 (1Н, м). (MH)+ ESI=614,1.
ПРИМЕР 7
Синтез N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]этил}-L-лейцинамида
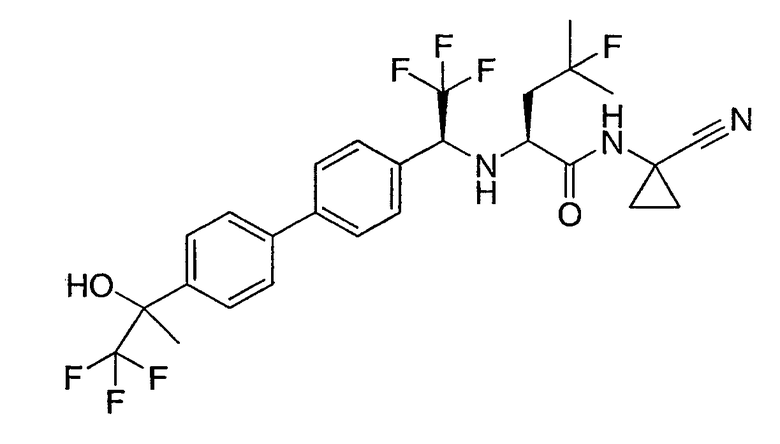
Стадия 1:
Получение 2-(4-бромфенил)-1,1,1-тритфорпропан-2-ола
Коммерческий 1-(4-бромфенил)-2,2,2-трифторэтанон (1 г, 4 ммоль) в диэтиловом эфире (8 мл) охлаждают до температуры -78°С с последующим добавлением коммерчески доступного бромида метилмагния (2,65 мл, 7,9 ммоль, 3М в диэтиловом эфире). Мутную реакционную среду оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь переносят в делительную воронку, содержащую 1,2 М соляной кислоты (20 мл). Указанный водный слой экстрагируют 3 раза этилацетатом (30 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат сульфатом магния и концентрируют при пониженном давлении. Получают прозрачное масло, которое достаточно чистое для использования без дополнительной очистки.
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]этил}-L-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (250 мг) и бромид со стадии 1 примера 7 (150 мг) объединяют по процедуре стадии 4 примера 1 с получением указанного в заголовке соединения в виде порошка белого цвета.
(MH)+ESI=560.
ПРИМЕР 8
Синтез N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[1-гидрокси-1-(трифторметил)пропил]бифенил-4-ил}этил)-L-лейцинамида

Стадия 1:
Получение 2-(4-бромфенил)-1,1,1-трифторбутан-2-ола
К раствору 1,4-дибромбензола (2,5 г, 10,6 ммоль) в ТГФ (50 мл) при температуре -78°С добавляют n-BuLi (6,5 мл, 10 ммоль; 1,6 М в гексанах) и смесь перемешивают при температуре -78°С в течение 15 минут. Затем добавляют 1,1,1-трифтор-2-бутанон (1,3 г, 10 ммоль). После перемешивания еще в течение 15 минут смесь гасят добавлением водного NH4Cl и экстрагируют этилацетатом. Очистка комби-флэш-хроматографией (колонка 40 г; элюция гесанами-этилацетатом (10%-20% в течение 20 минут; скорость течения 35 мл/мин, проводят сбор фракций по 18 мл) приводит к получению указанного в заголовке соединения в виде бесцветной жидкости.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,58 (м, 5Н), 5,52 (с, 1Н), 2,28 (м, 1Н), 2,10 (м, 1Н).
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-4-фтор-N 2 -((1S)-2,2,2-трифтор-1-{4'-[1-гидрокси-1-(трифторметил)пропил]бифенил-4-ил}этил)-L-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (150 мг) и бромид со стадии 1 примера 8 (100 мг) объединяют по процедуре стадии 4 примера 1 с получением указанного в заголовке соединения в виде порошка белого цвета.
(MH)+ESI=574.
ПРИМЕР 9
Синтез N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида и N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
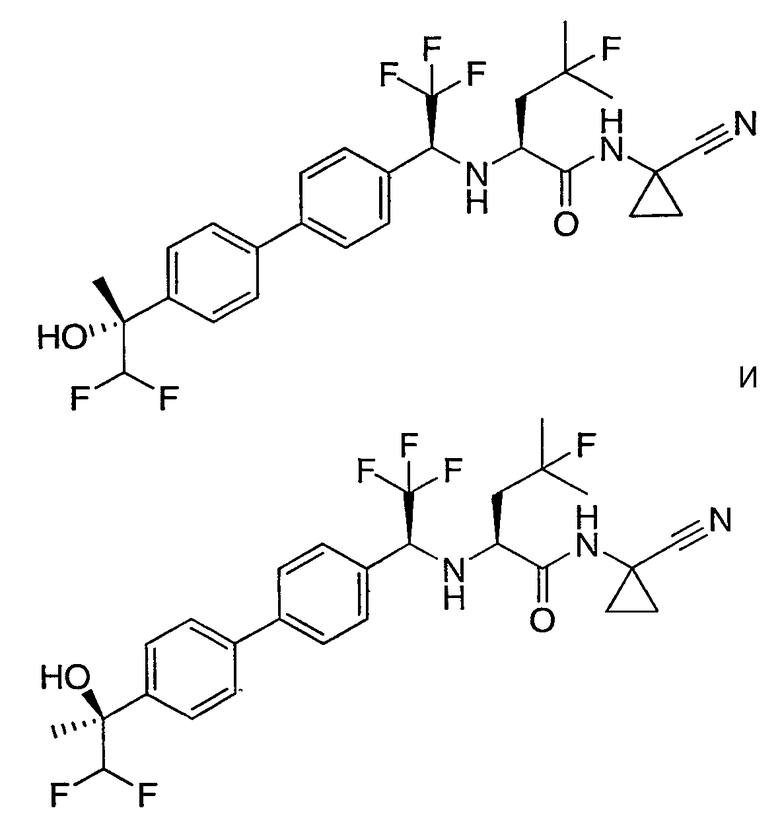
Стадия 1:
Получение 2-(4-бромфенил)-1,1-дифторпропан-2-ола
К раствору 1-(4-бромфенил)-2,2-дифторэтанона (2,5 г, 10,6 ммоль) в ТГФ (60 мл) добавляют хлорид метилмагния (10 мл, 30 ммоль; 3М в ТГФ) при температуре 0°С в течение примерно 10 минут и смесь перемешивают при температуре 0°С в течение 1 часа. Оценка небольшого образца показывает наличие остаточных количеств исходного вещества, в связи с чем добавляют еще хлорид метилмагния (5 мл, 15 ммоль, 3М в ТГФ). После дополнительного перемешивания в течение 15 минут реакцию гасят добавлением в смесь H2O, осторожно подкисляют 1М HCl (100 мл) и экстрагируют этилацетатом. Очистка комби-флэш-хроматографией (колонка 120 г; элюция гесанами/этилацетатом (5%-25%) в течение 20 минут; скорость течения 70 мл/мин, проводят сбор фракций по 25 мл) приводит к получению указанного в заголовке соединения в виде бесцветной жидкости.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,54 (м, 4Н), 5,86 (т, 1Н), 5,10 (с, 1Н), 1,64 (с, 3Н).
Стадия 2:
Получение N 1 -(1-цианоциклопропил)-N 2 -{(1S)-1-[4'-(2,2-дифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (2,5 г) и бромид со стадии 1 примера 9 (1,6 мг) объединяют по процедуре стадии 4 примера 1 с получением смеси диастереомеров в виде порошка белого цвета.
Стадия 3: Разделение диастереомеров
Диастереомерную смесь в отношении 1:1 со стадии 2 примера 9 (80 мг) растворяют в этаноле (2 мл). Смесь соединений разделяют с использованием примерно 10 инъекций (10 × 200 мкл) в полупрепаративную колонку Chiralcel OD (2 см в.д. × 25 см) и далее проводят элюцию 32,5% 2-пропанола в гексанах со скоростью 6 мл/мин. Быстрые фракции, элюируемые на 21-23 минуте, объединяют и концентрируют с получением N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида в виде порошка белого цвета (98% d.e.).
(MH)+ ESI=542
Медленные фракции, элюируемые примерно на 25 минуте, объединяют и концентрируют с получением N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида в виде порошка белого цвета (98% d.e.).
(MH)+ ESI=542
ПРИМЕР 10
Синтез N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
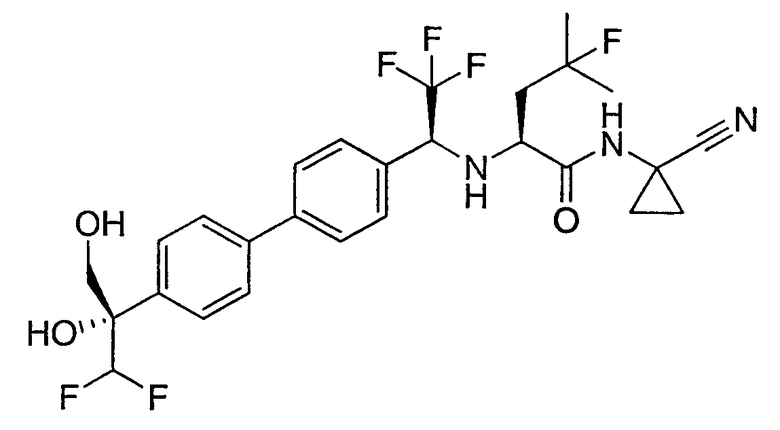
Стадия 1:
Получение 1-бром-4-[1-(дифторметил)винил]бензола
К активированному цинковому порошку (13,3 г, 0,2 моль) в колбе на 1 л добавляют ТГФ (200 мл). Добавляют по каплям в течение примерно 10 минут дийодметан (8,9 мл, 110 ммоль). Смесь перемешивают при комнатной температуре в течение 30 минут. Смесь охлаждают на бане лед-ацетон, добавляют 1М хлорида олова (IV) в CH2Cl2 (22,1 мл, 22,1 ммоль) в течение примерно 15 минут (конец иглы вставляют в смесь).
Смесь перемешивают в течение 15 минут и удаляют охлаждающую баню. Далее смесь перемешивают при комнатной температуре в течение 30 минут. После нового охлаждения на бане лед-ацетон добавляют по каплям в течение примерно 10 минут раствор 1-(4-бромфенил)-2,2-дифторэтанона (5,2 г, 22,12 ммоль) в ТГФ (30 мл). Охлаждающую баню удаляют и смесь перемешивают при комнатной температуре в течение 30 минут. Затем смесь разливают частями на смесь бикарбоната натрия (300 мл, 300 ммоль) и гексанов (300 мл) при температуре 0°С. После перемешивания в течение 15 минут смесь фильтруют через целит. Органический слой отделяют, промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют. Хроматография на силикагеле при проведении элюции гексанами:этилацетатом (20:1) дает указанное в заголовке соединение в виде жидкости бледно-желтого цвета.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,60 (д, 2Н), 7,50 (д, 2Н), 6,70 (т, 1Н), 5,92 (с, 1Н), 5,80 (с, 1Н).
Стадия 2:
Получение (2S)-2-(4-бромфенил)-3,3-дифторпропан-1,2-диола
К колбе на 250 мл, в которую внесен коммерчески доступный препарат AD-mix-alpha (7 г), добавляют трет-бутиловый спирт (25 мл) и H2O (25 мл). Смесь перемешивают при комнатной температуре с получением 2 прозрачных фаз и нижняя фаза имеет желтовато-оранжевый цвет. После охлаждения до 0°С сразу добавляют в виде одной порции 1-бром-4-[1-(дифторметил)винил]бензол (1,1 г, 4,7 ммоль) и смесь перемешивают примерно при температуре 4°С в течение ночи. Получают смесь ярко-желтого цвета. Смесь выдерживают при температуре 0°С и добавляют твердый сульфит натрия (8 г, 64 ммоль). Смесь нагревают до комнатной температуры и перемешивают в течение 30 минут. Добавляют этилацетат (50 мл) и отделяют органический слой. Водный слой экстрагируют этилацетатом (2×10 мл). Объединенные этилацетатные экстракты сушат (Na2SO4) и концентрируют. Очистка комби-флэш-хроматографией (колонка 40 г; элюция гексанами/этилацетатом (20%-60%) в течение 25 минут; скорость течения 35 мл/мин, проводят сбор фракций по 18 мл) с получением указанного в заголовке соединения в виде бесцветного масла.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,56 (с, 4Н), 6,14 (т, 1Н), 5,00 (с, 1Н), 4,40 (т, 1Н), 4,00 (м, 1Н), 3,80 (м, 1Н).
Стадия 3: Получение N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (900 мг) и бромид со стадии 2 примера 10 (450 мг) объединяют по процедуре стадии 4 примера 1 с получением указанного в заголовке соединения в виде порошка белого цвета.
Получают монофосфонатное сложноэфирное производное (Yves Leblanc, et. al., Tetrahedron Asymmetry 2001, 12, 3063-3066) для определения оптической чистоты (94% d.e., Chiralpak AD, 40% 2-пропанола в гексанах, скорость течения 1 мл/мин; время удерживания 7,4 мин).
1Н ЯМР (CD3COCD3) δ (ч/млн): 8,15 (с, 1Н), 7,70 (м, 6Н), 7,55 (д, 2Н), 6,20 (т, 1Н), 4,92 (с, 1Н), 4,35 (м, 2Н), 4,08 (м, 1Н), 3,85 (м, 1Н), 3,52 (м, 1Н), 1,98 (м, 2Н), 1,50-1,28 (м, 8Н), 1,05 (м, 1Н), 0,90 (м, 1Н).
ПРИМЕР 11
Синтез N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
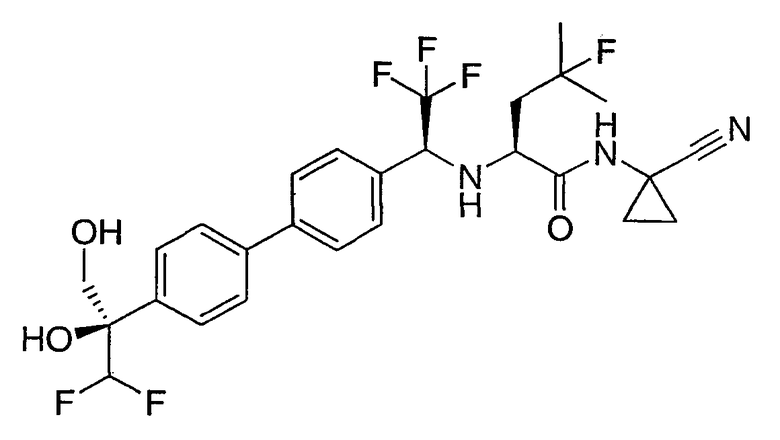
Стадия 1: Получение (2R)-2-(4-бромфенил)-3,3-дифторпропан-1,2-диола
К колбе на 250 мл, в которую внесен коммерческий AD-mix-бета (7 г), добавляют t-BuOH (25 мл) и H2O (25 мл). Смесь перемешивают при комнатной температуре с получением 2 прозрачных фаз и нижняя фаза имеет желтовато-оранжевый цвет. После охлаждения до 0°С добавляют в виде одной порции 1-бром-4-[1-(дифторметил)винил]бензол (1,1 г, 4,7 ммоль) и смесь перемешивают при температуре примерно 4°С в течение ночи. Образуется смесь ярко-желтого цвета. Смесь выдерживают при температуре 0°С и добавляют твердый сульфит натрия (8 г, 64 ммоль). Смесь нагревают до комнатной температуры и перемешивают в течение 30 минут. Добавляют этилацетат (50 мл) и отделяют органический слой. Водный слой экстрагируют этилацетатом (2×10 мл). Объединенные этилацетатные экстракты сушат (Na2SO4) и концентрируют. Очистка комби-флэш-хроматографией (колонка 40 г; элюция гексанами/этилацетатом (20%-60%) в течение 25 минут; скорость течения 35 мл/мин, проводят сбор фракций по 18 мл) приводит к получению указанного в заголовке соединения в виде бесцветного масла.
1Н ЯМР (CD3COCD3) δ (ч/млн): 7,56 (с, 4Н), 6,14 (т, 1Н), 5,00 (с, 1Н), 4,40 (т, 1Н), 4,00 (м, 1Н), 3,80 (м, 1Н).
Стадия 2: Получение N 1 -(1-цианоциклопропил)-N 2 -((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамида
Боронатный сложный эфир со стадии 3 примера 1 (1,5 г) и бромид со стадии 1 примера 11 (740 мг) объединяют по процедуре стадии 4 примера 1 с получением указанного в заголовке соединения в виде порошка белого цвета.
Получают монофосфонатное сложноэфирное производное (Yves Leblanc, et. al., Tetrahedron Asymmetry 2001, 12, 3063-3066) для определения оптической чистоты (94% d.e., Chiralpak AD, 40% 2-пропанол в гексанах, скорость течения 1 мл/мин; время удерживания 11,1 мин.).
1Н ЯМР (CD3COCD3) δ (ч/млн): 8,15 (с, 1Н), 7,70 (м, 6Н), 7,55 (д, 2Н), 6,20 (т, 1Н), 4,92 (с, 1Н), 4,35 (м, 2Н), 4,08 (м, 1Н), 3,85 (м, 1Н), 3,52 (м, 1Н), 1,98 (м, 2Н), 1,50-1,28 (м, 8Н), 1,05 (м, 1Н), 0,90 (м, 1Н).
ПРИМЕР 12
Синтез N 2 -[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N 1 -(1-цианоциклопропил)-4-фтор-L-лейцинамида
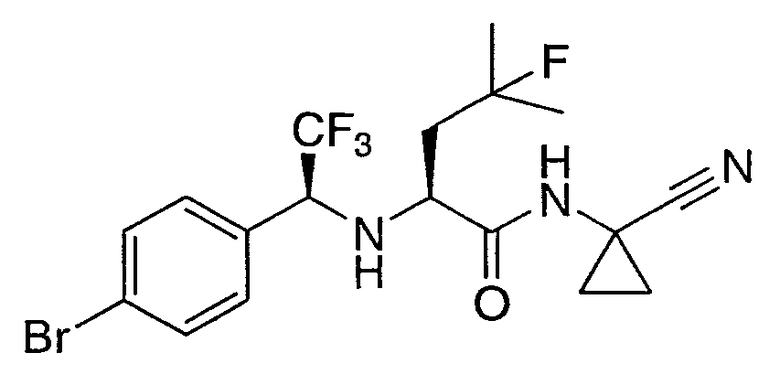
Стадия 1:
Получение бензил-(3S)-3-[(трет-бутоксикарбонил)амино]-4-гидроксибутаноата
4-бензиловый сложный эфир N-(трет-бутоксикарбонил)-L-аспарагиновой кислоты (30 г) растворяют в диметоксиэтане (90 мл) и полученный раствор охлаждают до -5°С. Добавляют N-метилморфолин (10,32 мл) и затем медленно добавляют изобутилхлорформиат (12,66 мл), так чтобы температура реакции поддерживалась ниже -10°С. Смесь оставляют на 0,5 часа. Твердое вещество быстро фильтруют и промывают диметоксиэтаном (90 мл). Фильтрат охлаждают до температуры -50°С и медленно добавляют раствор боргидрида натрия (4,4 г) в воде (45 мл), так чтобы температура реакции поддерживалась в диапазоне от -30°С до -15°С. Затем добавляют воду (500 мл), так чтобы температура реакционной смеси поддерживалась ниже -15°С. Суспензию фильтруют, твердое вещество промывают водой (400 мл) и сушат с получением бензил-(3S)-3-[(трет-бутоксикарбонил)амино]-4-гидроксибутаноата.
1Н ЯМР (CD3COCD3) δ 7,3-7,45 (5Н, м), 5,85-5,95 (1Н, NH), 5,15 (2Н, с), 3,95-4,1 (2Н, м), 3,5-3,7 (2Н, м), 2,55-2,75 (2Н, м), 1,4 (9Н, с).
Стадия 2:
Получение бензил-[(4S)-2-оксо-1,3-оксазолидин-4-ил]ацетата
К раствору спирта (95,7 г) со стадии 1 в дихлорэтане (925 мл) добавляют пиридин (625 мл) и смесь охлаждают до температуры 0-5°С. Добавляют безводный п-толуолсульфоновый ангидрид (105,7 г) и смесь нагревают до комнатной температуры и перемешивают в течение 1 часа, после чего нагревают до 90°С в течение 2 часов. Смесь охлаждают, разбавляют дихлорметаном (1000 мл) и промывают 1N HCl (3×600 мл). Органический слой промывают насыщенным раствором соли, сушат сульфатом натрия и растворители удаляют в вакууме. Остаток очищают хроматографией на SiO2 с использованием смеси (1:1) этилацетата и гексанов и затем этилацетата с получением бензил-[(4S)-2-оксо-1,3-оксазолидин-4-ил]ацетата.
1Н ЯМР (CD3SOCD3) δ 7,8 (1Н, NH), 7,3-7,45 (5Н, м), 5,05-5,15 (2Н, м), 4,4-4,5 (1Н, м), 4,1-4,2 (1Н, м), 4,0-4,05 (1Н, м), 3,6-3,8 (2Н, м).
Стадия 3:
Получение (4S)-4-(2-гидрокси-2-метилпропил)-1,3-оксазолидин-2-она
Бромид метилмагния (227 мл, 3М раствора в диэтиловом эфире) добавляют к смеси толуола (340 мл) и ТГФ (340 мл) при температуре -20°С. Затем добавляют по каплям теплый ТГФ раствор (170 мл) сложного эфира со стадии 2 (40 г), поддерживая температуру на уровне ниже -10°С. Смесь выдерживают в течение 2 часов и затем медленно добавляют к смеси воды (1000 мл) и уксусной кислоты (200 мл), и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Отделяют водный слой и органический слой экстрагируют водой (2×200 мл). Продукт экстрагируют из объединенных водных слоев с использованием дихлорметана и экстрактора, работающего в непрерывном режиме. Дихлорметановый экстракт выпаривают досуха с использованием гептана в качестве сорастворителя для азеотропного удаления уксусной кислоты. Остаток очищают хроматографией на SiO2 с использованием этанола и дихлорметана (1:30) с получением (4S)-4-(2-гидрокси-2-метилпропил)-1,3-оксазолидин-2-она.
1Н ЯМР (CD3COCD3) δ 6,1-6,4 (1Н, NH), 4,45-4,55 (1Н, м), 4,1-4,2 (1Н, м), 3,95-4,05 (1Н, м), 3,7 (1Н, с), 1,65-1,85 (2Н, м), 1,25 (6Н, м).
Стадия 4: Получение (4S)-4-(2-фтор-2-метилпропил)-1,3-оксазолидин-2-она
Добавляют дихлорметановый раствор (100 мл) спирта (47,8 г) со стадии 3 к раствору трифторида (диэтиламино)серы (48,5 г) в дихлорметане (500 мл) при -70°С. Смесь нагревают до комнатной температуры и перемешивают в течение 1 часа. Затем смесь осторожно добавляют к смеси насыщенного водного NaHCO3 (800 мл) при 0°С. Органический слой отделяют и промывают насыщенным водным раствором NaHCO3. Водный слой далее экстрагируют дихлорметаном (100 мл) и объединенные дихлорметановые слои сушат и концентрируют. Остаток очищают хроматографией на SiO2 с использованием смеси этилацетата и гексанов (1:5) и затем этилацетата с получением (4S)-4-(2-фтор-2-метилпропил)-1,3-оксазолидин-2-она.
1Н ЯМР (CD3SOCD3) δ 7,6 (1Н, NH), 4,4-4,5 (1Н, м), 3,95-4,05 (1Н, м), 3,9-3,95 (1Н, м), 1,8-1,95 (2Н, м), 1,25-1,4 (6Н, 2с).
Стадия 5:
Получение (2S)-2-амино-4-фтор-4-метилпентан-1-ола
К раствору фтор-содержащего производного (21,0 г) со стадии 4 в 90% водном этиловом спирте (216 мл) добавляют гидроксид калия (21,9 г). Смесь нагревают при температуре кипения с обратным холодильником в течение 4 часов и охлаждают до комнатной температуры. Затем смесь концентрируют и выпаривают совместно с толуолом (3×300 мл). Остаток растворяют в дихлорметане (500 мл) и перемешивают в течение 0,5 часа. Суспензию фильтруют через целит и целит промывают дихлорметаном (3×100 мл). Фильтрат концентрируют досуха с получением (2S)-2-амино-4-фтор-4-метилпентан-1-ола.
1Н ЯМР (CD3OD) δ 3,4-3,5 (1Н, м), 3,2-3,3 (1Н, м), 3,0-3,1 (1Н, м), 1,5-1,7 (2Н, м), 1,35 (3Н, с), 1,3 (3Н, с).
Стадия 6:
Получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метилпентан-2-амина
Аминоспирт (21,0 г) со стадии 5 растворяют в дихлрметане (300 мл) и раствор охлаждают до 0°С. Добавляют 4-(диметиламино)пиридин (0,051 г) и трет-бутилдиметилсилилхлорид (21 г) и затем триэтиламин (25 мл). Смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь медленно выливают в насыщенный водный раствор хлорида аммония при 0°С и экстрагируют дихлорметаном (3×300 мл). Органические слои промывают насыщенным раствором соли, сушат сульфатом натрия и растворитель удаляют в вакууме с получением (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метилпентан-2-амина.
1Н ЯМР (CD3OD) δ 3,6-3,65 (1Н, м), 3,4-3,5 (1Н, м), 3,1-3,2 (1Н, м), 1,6-1,8 (2Н, м), 1,35-1,45 (6Н, м), 0,93 (9Н, с), 0,1 (6Н, с).
Стадия 7:
Получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метил-N-[(1E)-2,2,2-трифторэтилиден]пентан-2-амина
К раствору амина (31,5 г) из стадии 6 в бензоле (126 г) добавляют метилгемиацеталь трифторацетальдегида (21,6 мл). Раствор нагревают при температуре кипения с обратным холодильником в течение ночи с использованием ловушки Dean-Stark для сбора воды. Реакционную смесь охлаждают до комнатной температуры и концентрируют досуха. Остаток очищают на SiO2 с использованием 4% этилацетата в гексанах с получением (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метилпентан-2-амина.
1Н ЯМР (CD3COCD3) δ 7,9-7,95 (1Н, м), 3,75-3,85 (1Н, м), 3,7-3,75 (1Н, м), 3,53-3,6 (1Н, м), 1,9-2,0 (2Н, м), 1,3-1,4 (6Н, м), 0,9 (9Н, с), 0,1 (3Н, с), 0,05 (3Н, с).
Стадия 8:
Получение (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}-4-фтор-4-метилпентан-1-ола
К раствору 1,4-дибромбензола (0,26 г) в ТГФ (4 мл) при -75°С добавляют n-BuLi (0,42 мл 2,5М раствора гексанов) и смесь выдерживают в течение 20 минут. Добавляют имин (0,329 г) со стадии 7 в ТГФ (2 мл) и смесь выдерживают в течение 2 часов. Затем указанную смесь добавляют к смеси воды (50 мл), NH4Cl (1 г) и дробленого льда. Всю смесь экстрагируют этилацетатом (2×25 мл) и объединенные этилацетатные слои сушат и выпаривают досуха.
Ту же самую процедуру повторяют в увеличенном масштабе с использованием 1,4-дибробензола (1,2 г), n-BuLi (1,84 мл) и имина (1,38 г) и реакционную смесь обрабатывают, как было указано выше. Объединенные остатки из обоих препаратов растворяют в ТГФ (10 мл) и охлаждают до 0°С. Добавляют фторид н-тетрабутиламмония (6 мл из 1М раствора ТГФ) и смесь перемешивают при +5°С в течение 16 часов. Далее смесь выливают в смесь воды (50 мл), хлорида аммония (1 г) и дробленого льда и отделяют органический слой. Далее водный слой экстрагируют этилацетатом (2×15 мл) и объединенные органические слои сушат и концентрируют. Остаток очищают на SiO2 с использованием смеси этилацетата и гексанов (1:5), получая (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}-4-фтор-4-метилпентан-1-ол.
1Н ЯМР (CD3COCD3) δ 7,65 (2Н, м), 7,5 (2Н, м), 4,5-4,6 (1Н, м), 3,8 (1Н, м), 3,6 (1Н, м), 3,3-3,4 (1Н, м), 2,85-2,0 (1Н, м), 2,55 (1Н, м), 1,7-1,9 (2Н, с), 1,3-1,4 (6Н, м).
Стадия 9:
Получение N 2 -[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N 1 -(1-цианоциклопропил)-4-фтор-L-лейцинамида
Суспензию H5IO6/CrO3 (66 мл, 0,44М в CH3CN; см. примечание) охлаждают до 0°С и добавляют по каплям раствор спирта со стадии 8 (1,55 г) в CH3CN (5 мл). Смесь перемешивают при температуре 0-5°С в течение 3,5 часа. Далее ее выливают в Na2HPO4 (200 мл), pH 4 при сильном перемешивании и смесь экстрагируют диэтиловым эфиром (3×50 мл). Объединенные эфирные экстракты промывают водой и насыщенным раствором соли (1:1) и затем разбавленным водным раствором NaHSO3 и насыщенным раствором соли. Смесь сушат сульфатом натрия, фильтруют и растворитель выпаривают досуха с получением N-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-4-фтор-L-лейцина, который используют в качестве такового на следующей стадии.
Примечание. Окислитель (H5IO6/CrO3) получают по методике, описанной в Tetrahedron Letters 39 (1998) 5323-5326, но с использованием CH3CN с чистотой для ВЭЖХ (содержит 0,5% воды); воду не добавляют.
Добавляют диизопропилэтиламин (4,2 мл) к суспензии кислоты (1,5 г) при 0°С, указанной выше, гидрохлориду 1-амино-1-циклопропанкарбонитрила (1,18 г), гексафторфосфату О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (1,94 г) и диметилформамиду (5 мл) и смесь подвергают реакции при комнатной температуре в течение 48 часов. Далее смесь выливают в лед и разбавленный водный хлорид аммония. Смесь экстрагируют этилацетатом и эфиром (1:1) и объединенные органические слои промывают разбавленным Na2HPO4, pH 3 и насыщенным солевым раствором. Растворители выпаривают досуха и остаток очищают хроматографией на SiO2 с использованием этилацетата и гексанов (1:2) с получением N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамида с достаточной степенью чистоты для использования на следующей стадии.
1Н ЯМР (CD3COCD3) δ 8,15 (1Н, NH), 7,6 (2Н, м), 7,45 (2Н, м), 4,35-4,45 (1Н, м), 3,45-3,55 (1Н, м), 1,9-2,1 (2Н, м), 1,75-1,85 (1Н, NH), 1,35-1,55 (8Н, м), 1,1-1,15 (1Н, м), 0,95-1,05 (1Н, м).
Фармацевтическая композиция
В качестве конкретного варианта осуществления настоящего изобретения 100 мг (1R,2R)-N-(цианометил)-5,5-дифтор-2-[4'-(метилтио)-1,1'-бифенил-2-ил]циклогексанкарбоксамида вводят в композицию с достаточно тонко измельченной лактозой с образованием в совокупности 580-590 мг для заполнения твердой желатиновой капсулы с размером 0.
Соединения по настоящему изобретению демонстрируют активность в приведенных ниже тестах. Кроме того, описанные в настоящем изобретении соединения обладают улучшенным фармакологическим профилем по сравнению с ранее известными соединениями.
Тест на катепсин К
Готовят серийные разбавления (1/3) от 500 мкМ до 0,0085 мкМ исследуемых соединений в диметилсульфоксиде (ДМСО). Затем добавляют по 2 мкл каждого разбавления в ДМСО к 50 мкл буфера для тестирования (50 мМ MES, (pH 5,5); 2,5 мМ ЭДТА; 2,5 мМ ДТТ и 10% ДМСО) и 25 мкл человеческого катепсина К (0,4 нМ) в растворе буфера для тестирования. Исследуемые растворы перемешивают в течение 5-10 секунд на встряхивателе для планшетов и инкубируют в течение 15 минут при комнатной температуре. К растворам для тестирования добавляют Z-Leu-Arg-AMC (8 мкМ) в 25 мкл буфера для тестирования. Гидролиз удаляемой группы кумарина (АМС) отслеживают на спектрофлюориметре (Exλ=355 нм; Emλ=460 нм) в течение 10 минут. Процент ингибирования вычисляют по стандартной математической модели на основе кривой доза-ответ применительно к полученным экспериментальным значениям.
Тест на катепсин L
Готовят серийные разбавления (1/3) от 500 мкМ до 0,0085 мкМ исследуемых соединений в диметилсульфоксиде (ДМСО). Затем добавляют по 2 мкл каждого разбавления в ДМСО к 50 мкл буфера для тестирования (50 мМ MES (pH 5,5); 2,5 мМ ЭДТА; 2,5 мМ ДТТ и 10% ДМСО) и 25 мкл человеческого катепсина L (0,5 нМ) в растворе буфера для тестирования. Исследуемые растворы перемешивают в течение 5-10 секунд на встряхивателе для планшетов и инкубируют в течение 15 минут при комнатной температуре. К растворам для тестирования добавляют Z-Leu-Arg-AMC (8 мкМ) в 25 мкл буфера для тестирования. Гидролиз удаляемой группы кумарина (АМС) отслеживают на спектрофлюориметре (Exλ=355 нм; Emλ=460 нм) в течение 10 минут. Процент ингибирования вычисляют по стандартной математической модели на основе кривой доза-ответ применительно к полученным экспериментальным значениям.
Тест на катепсин B
Готовят серийные разбавления (1/3) от 500 мкМ до 0,0085 мкМ исследуемых соединений в диметилсульфоксиде (ДМСО). Затем добавляют по 2 мкл каждого разбавления в ДМСО к 50 мкл буфера для тестирования (50 мМ MES (pH 5,5); 2,5 мМ ЭДТА; 2,5 мМ ДТТ и 10% ДМСО) и 25 мкл человеческого катепсина B (4,0 нМ) в растворе буфера для тестирования. Исследуемые растворы перемешивают в течение 5-10 секунд на встряхивателе для планшетов и инкубируют в течение 15 минут при комнатной температуре. К растворам для тестирования добавляют Z-Leu-Arg-AMC (8 мкМ) в 25 мкл буфера для тестирования. Гидролиз удаляемой группы кумарина (АМС) отслеживают на спектрофлюориметре (Exλ=355 нм; Emλ=460 нм) в течение 10 минут. Процент ингибирования вычисляют по стандартной математической модели на основе кривой доза-ответ применительно к полученным экспериментальным значениям.
Тест на катепсин S
Готовят серийные разбавления (1/3) от 500 мкМ до 0,0085 мкМ исследуемых соединений в диметилсульфоксиде (ДМСО). Затем добавляют по 2 мкл каждого разбавления в ДМСО к 50 мкл буфера для тестирования (50 мМ MES (pH 5,5); 2,5 мМ ЭДТА; 2,5 мМ ДТТ и 10% ДМСО) и 25 мкл человеческого катепсина S (20 нМ) в растворе буфера для тестирования. Исследуемые растворы перемешивают в течение 5-10 секунд на встряхивателе для планшетов и инкубируют в течение 15 минут при комнатной температуре. К растворам для тестирования добавляют Z-Leu-Arg-AMC (8 мкМ) в 25 мкл буфера для тестирования. Гидролиз удаляемой группы кумарина (АМС) отслеживают на спектрофлюориметре (Exλ=355 нм; Emλ=460 нм) в течение 10 минут. Процент ингибирования вычисляют по стандартной математической модели на основе кривой доза-ответ применительно к полученным экспериментальным значениям.
Фармакокинетика у крыс
Фармакокинетика у крыс при пероральном введении (PO)
ПРОЦЕДУРА:
Животных помещают в клетки, кормят и ухаживают за ними в соответствии с требованиями Руководства Канадского Совета по уходу за животными (Guidelines of the Canadian Council on Animal Care).
Самцов крыс Sprague Dawley (вес 250-400 г) выдерживают без пищи в течение ночи перед проведением каждого анализа крови после перорального введения препарата.
Животных помещают один раз в клетку, ограничивающую движение, и клетку плотно закрывают. Нулевой образец крови получают отщипыванием небольшого (1 мм или меньше) кусочка с конца хвоста. Затем хвост встряхивают энергичным, но осторожным движением от конца к основанию для сцеживания крови. Собирают примерно 0,5 мл крови в пробирку с вакуумом, содержащую гепарин.
Соединение получают в объеме, соответствующем стандартной дозе 10 мл/кг, и вводят перорально через зонд с иглой 16 размера 3'' в пищевод.
Далее отбирают кровь для анализа таким же образом, как и проводили отбор образца для анализа крови в нулевой точке, за исключением того, что отпадает необходимость снова отщипывать кончик хвоста. Хвост очищают кусочком марли и проводят встряхивание, как описано выше, для сбора крови в маркированные соответствующим образом пробирки.
Сразу после отбора образца кровь центрифугируют, отделяют плазму и переносят плазму в чистые маркированные флаконы и хранят в морозильнике до анализа.
Типичные временные точки для анализа крови крыс после введения пероральной дозы следующие: 0, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 24 часа.
После отбора крови в точке 4 часа крыс обеспечивают вдоволь пищей. Воду дают в течение всего эксперимента.
Носители
Следующие носители (в соответствующих объемах для введения дозы) могут быть использованы для анализа крови у крыс при пероральном введении препарата:
Соединения для анализа крови после перорального приема могут быть введены инфузией. Для достижения лучшей гомогенности суспензию можно озвучить примерно в течение 5 минут.
Для анализа аликвоты образцов разбавляют с использованием 1,2-1,5 объемов ацетонитрила, необязательно содержащего внутренний стандарт, и центрифугируют для удаления осадка белка. Супернатант инъецируют непосредственно в С-18 колонку для ВЭЖХ со встроенным масс-спектрометром (MS) или устройством для анализа поглощения в ультрафиолетовой области (UV), или для оценки флуоресценции (Fluo). Количественную оценку проводят по стандартной кривой, полученной с использованием чистых образцов крови, в которые внесены известные количества препарата в ацетонитриле, необязательно содержащем внутренний стандарт. Добавляют еще некоторое количество ацетонитрила, необязательно содержащего внутренний стандарт, до достижения 1,2-1,5 объемов от исходного количества крови с целью соответствия объемам в случае образцов. Биологическую доступность (F) оценивают при сравнении площади под кривой (AUC) для внутривенного введения в сравнении с результатами перорального введения.
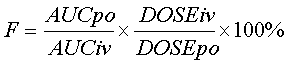
и
AUC=(Cl+C2)*(T2-T1)/2
где С обозначает измеренную концентрацию по данным анализа в MS или UV или Fluo в данной временной точке T.
Фармакокинетика у крыс при внутривенном ведении препарата
ПРОЦЕДУРА:
Крыс помещают в клетки, кормят и ухаживают за ними в соответствии с требованиями руководства Канадского Совета по уходу за животными (Guidelines of the Canadian Council on Animal Care).
Самцов крыс Sprague Dawley (вес 325-375 г) используют в данных исследованиях не натощак. Соединения готовят, как было описано выше, в объеме, соответствующем стандартной дозе 1 мл/кг.
Дозу для внутривенного введения крысам в состоянии сознания вводят через яремную вену с использованием иглы 25 размера. Указанное введение представляет собой нулевую точку эксперимента.
Отбор крови для точки 5 минут проводят путем отщипывания небольшого (1-2 мм) кусочка с кончика хвоста. Затем хвост встряхивают энергичным, но осторожным движением от конца к основанию для сцеживания крови. Собирают примерно 0,5 мл крови в пробирку с вакуумом, содержащую гепарин. Последующий отбор крови проводят таким же образом, за исключением того, что отпадает необходимость отщипывать кончик хвоста. Хвост очищают кусочком марли и отбирают кровь, как описано выше, в пробирки, помеченные соответствующими этикетками.
Типичные временные точки для анализа крови крыс после в/в следующие:
0, 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов
или
0, 5 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 24 часа.
Носители
Для анализа крови у крыс при в/в введении могут быть использованы следующие носители:
В случае декстрозы может быть добавлен бикарбонат натрия, если получился мутный раствор.
Для анализа аликвоты образца разбавляют с использованием 1,2-1,5 объемов ацетонитрила, необязательно содержащего внутренний стандарт, и центрифугируют для удаления остаток белка. Супернатант инъецируют непосредственно в С-18 колонку для ВЭЖХ со встроенным масс-спектрометром (MS), или устройством для выявления поглощения в ультрафиолетовой области (UV) или для оценки флуоресценции (Fluo). Количественную оценку проводят по стандартной кривой, полученной с использованием чистых образцов крови, в которые внесены известные количества лекарственного препарата в ацетонитриле, необязательно содержащем внутренний стандарт. Добавляют еще некоторое количество ацетонитрила, необязательно содержащего внутренний стандарт, до достижения 1,2-1,5 объемов от исходного количества крови с целью соответствия объемам в случае образцов. Биологическую доступность (F) оценивают при сравнении площади под кривой (AUC) для внутривенного введения в сравнении с результатами перорального введения.
и
AUC=(Cl+C2)*(T2-T1)/2
где С обозначает измеренную концентрацию, по данным анализа в MS или UV или Fluo в данной временной точке T.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТАНА | 2010 |
|
RU2572555C2 |
| ПРОИЗВОДНЫЕ ПРОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2010 |
|
RU2535479C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЗЕТИДИНА | 2013 |
|
RU2629114C2 |
| ПИРАЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2012 |
|
RU2617678C2 |
| ИНГИБИТОРЫ БЕТА-СЕКРЕТАЗЫ | 2016 |
|
RU2712272C2 |
| ИНГИБИТОРЫ ЦИСТЕИНПРОТЕАЗ КАТЕПСИНОВ | 2014 |
|
RU2692799C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ МОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2664541C2 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| ПРОНИЦАЕМЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2707292C2 |
Изобретение относится к новым соединениям формулы
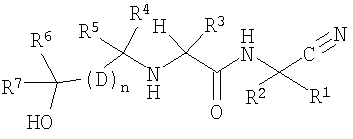 или к их фармацевтически приемлемым солям или стереоизомерам, которые обладают свойствами ингибиторов катепсина К, L, S и В. В формуле R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют С3-4циклоалкил; R3 обозначает C1-6алкил, который замещен одним-четырьмя атомами фтора или хлора; R4 обозначает C1-6алкил, который замещен одним-пятью атомами галогена; R5 обозначает водород; каждый D обозначает независимо фенил; R6 обозначает водород или C1-6алкил, который может быть замещен одним-двумя гидроксилами или двумя-шестью атомами галогена; R7 обозначает C1-6алкил, который может быть замещен двумя-пятью атомами галогена; n равно двум. Изобретение относится также к фармацевтической композиции и к применению указанных соединений для получения лекарственного средства. 3 н. и 5 з. ф-лы.
или к их фармацевтически приемлемым солям или стереоизомерам, которые обладают свойствами ингибиторов катепсина К, L, S и В. В формуле R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют С3-4циклоалкил; R3 обозначает C1-6алкил, который замещен одним-четырьмя атомами фтора или хлора; R4 обозначает C1-6алкил, который замещен одним-пятью атомами галогена; R5 обозначает водород; каждый D обозначает независимо фенил; R6 обозначает водород или C1-6алкил, который может быть замещен одним-двумя гидроксилами или двумя-шестью атомами галогена; R7 обозначает C1-6алкил, который может быть замещен двумя-пятью атомами галогена; n равно двум. Изобретение относится также к фармацевтической композиции и к применению указанных соединений для получения лекарственного средства. 3 н. и 5 з. ф-лы.
1. Соединения формулы:
где R1 и R2, взятые вместе с атомом углерода, к которому они присоединяются, образуют С3-4циклоалкил;
R3 обозначает C1-6алкил, который замещен одним-четырьмя атомами фтора или одним-четырьмя атомами хлора;
R4 обозначает C1-6алкил, который замещен одним-пятью атомами галогена;
R5 обозначает водород;
каждый D обозначает независимо фенил;
R6 обозначает водород или C1-6алкил, который может быть необязательно замещен одним-двумя гидроксилами или двумя-шестью атомами галогена;
R7 обозначает С1-6алкил, который может быть необязательно замещен двумя-пятью атомами галогена;
n равно двум;
или его фармацевтически приемлемая соль или стереоизомер.
2. Соединение по п.1, причем R1 и R2, взятые вместе с атомом углерода, к которому они присоединяются, образуют циклопропил; или его фармацевтически приемлемая соль или стереоизомер.
3. Соединение по п.2, причем R5 обозначает водород и R4 обозначает CF3; или его фармацевтически приемлемая соль или стереоизомер.
4. Соединение по п.3, причем R7 обозначает C1-3алкил, замещенный двумя или тремя атомами фтора; или его фармацевтически приемлемая соль или стереоизомер.
5. Соединение по п.1, которое представляет собой:
N1-(1-цианоциклопропил)-N2-(1-{4'-[2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)-бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-(2,2,2-трифтор-1-{4'-[3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамид;
N1(1-цианоциклопропил)-4-фтор-N2-(2,2,2-трифтор-1-{4'-[2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-(2,2,2-трифтор-1-{4'-[1-гидрокси-1-(трифторметил)пропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-(1-{4'-[2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-(1-{4'-[2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]-бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
или их фармацевтически приемлемую соль.
6. Соединение по п.5, которое представляет:
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидроксиэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамид;
Nl-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1S)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1R)-3,3,3-трифтор-1-гидрокси-1-метилпропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(R)-(2,2,2-трифтор-1-гидроксиэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-[2,2,2-трифтор-1-гидрокси-1-метилэтил)бифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[1-гидрокси-1-(трифторметил)пропил]бифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-метилэтил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1S)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-1-{4'-[(1R)-2,2-дифтор-1-гидрокси-1-(гидроксиметил)этил]бифенил-4-ил}-2,2,2-трифторэтил)-4-фтор-L-лейцинамид;
или их фармацевтически приемлемую соль.
7. Фармацевтическая композиция, обладающая свойствами ингибиторов катепсина К, L, S и В, включающая соединение по п.1 и фармацевтически приемлемый носитель.
8. Применение соединения по п.1 для получения лекарственного средства для лечения: остеопороза, остеопороза, индуцированного гликокортикоидами, болезни Педжета, аномально высокого метаболизма в костной ткани, болезни периодонта, выпадения зубов, переломов костей, ревматоидного артрита, остеоартрита, остеолита околопротезной ткани, несовершенного остеогенеза, атеросклероза, ожирения, глаукомы, хронической обструктивной болезни легких, метастаз в кости, гиперкальциемии при злокачественных новообразованиях или множественной миеломы у животного.
| СПОСОБ СТРОИТЕЛЬСТВА НАЗЕМНОГО МЕТАЛЛИЧЕСКОГО СБОРНО-РАЗБОРНОГО НЕФТЕПРОДУКТОПРОВОДА | 2010 |
|
RU2439415C1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| J | |||
| OBICHAUD et al, Rational design of potent and selective NH-linked aryl/heteroaryl cathepsin К inhibitors, BIOORGANIC & MED | |||
| CHEM | |||
| LETT., 2004, 14, 4291-4295 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

