Изобретение относится к области органического синтеза, в частности, к способу получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов посредством шестистадийного синтеза из 1,1-диалкилпроп-2-ин-1-олов и замещенных бензофенонов. Данные соединения, относящиеся к классу коксибов [1], проявляют свойства селективных ингибиторов фермента циклооксигеназы-2 и могут быть использованы в качестве нестероидных противовоспалительных лекарственных препаратов для длительной терапии заболеваний суставов [1].
Основная терапия ревматоидных заболеваний и болей воспалительного генеза связана с применением нестероидных противовоспалительных препаратов (НПВС, NSAIDs). Однако, длительное применение классических НПВС таких как аспирин, ибупрофен, диклофенак, индометацин, напроксен и т.д., приводит к развитию желудочно-кишечных язв, кровотечений и других осложнений [2, 3]. Причина этих побочных эффектов заключается в неселективном ингибировании ферментов циклооксигеназы (COX) 1-го и 2-го типов. Из двух основных ферментов циклооксигеназы COX-1 и COX-2 за развитие воспалительного процесса отвечает только COX-2, тогда как COX-1 участвует в жизненно важной регуляции секреции в желудочно-кишечном тракте (ЖКТ). Структурно эти ферменты чрезвычайно схожи, и их избирательное ингибирование затруднительно [4].
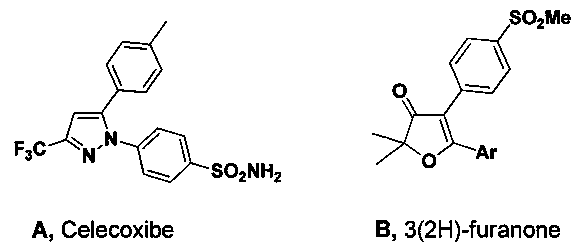
В начале 2000-х годов на фармацевтический рынок были выведены НПВС второго поколения, относящихся к классу селективных ингибиторов фермента COX-2. Типичные примеры таких НПВС: рофекоксиб (″Vioxx″, Merck), целекоксиб (″Celebrex″, Pfizer), вальдекоксиб (″Bextra″, G.D. Searle & Company), эторикоксиб (″Arcoxia″, Merck). Данный класс препаратов объединяет строение молекулы действующего вещества - все это 1,2-диарилзамещенные гетероциклические соединения, содержащие в своем составе группы SO2NH2 или SO2Me, отвечающие за селективность ингибирования COX-2 [5].
Результаты первичных доклинических испытаний противовоспалительной активности некоторых новых соединений класса коксибов, представляющих из себя 2,2-диалкил-4,5-диарилфуран-3(2H)-оны, проведенные южнокорейскими учеными [6], показали, что сходные по структуре с целекоксибом (A) производные 2,2-диметил-5-[4′-(метилсульфонил)фенил]-4-арил-3(2H)-фураноны (B) также являются эффективными селективными ингибиторами COX-2. В экспериментах in vivo по ингибированию воспалительного отека лап крыс, данные соединения проявляют противовоспалительную активность IC50 на уровне 0.05-0.01 мкг/мл и селективность COX-2/COX-1 от 100 до 1000 в зависимости от заместителей в арильном кольце (Ar).
Таким образом, разработка новых способов синтеза перспективных противовоспалительных препаратов класса 2,2-диалкил-4,5-диарилфуран-3(2H)-онов представляет существенный практический интерес.
Известен способ получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов посредством 3-х стадийного one-pot синтеза, включающего реакции конденсации 2-бромо-2-метилпропаноил цианида и замещенных ацетофенонов (a) и внутримолекулярной циклизации (b), который изображен на схеме 1 [7].
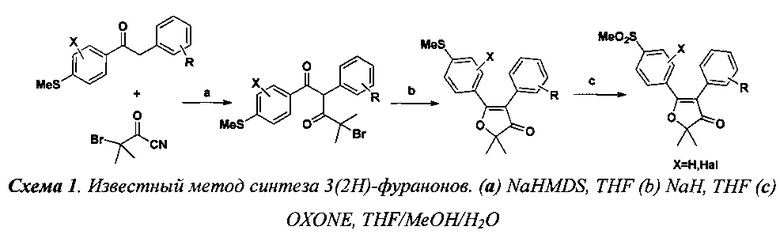
Недостатком данного способа является низкая коммерческая доступность исходных соединений: 2-бром-2,2-диалкилацетил цианидов и 1,2-диарилэтанонов. Стоимость 1 грамма 2,2-диметил-5-[4′-(метилсульфонил)фенил]-4-фенил-3(2H)-фуранона, синтезируемого по этой схеме, исходя из стоимости исходных соединений (по базе ) составляет от 100 до 250 USD, что делает малорентабельным производство данного препарата.
Наиболее близким аналогом заявляемого изобретения является способ получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов посредством шестистадийного синтеза, основанного на реакции 1,1-диалкилпроп-2-ин-1-олов с замещенными бензальдегидами и бутиллитием в ТГФ при -78°C и включающего в качестве ключевой реакцию Сузуки с использованием тетракис-(трифенилфосфин)палладия(0) на заключительных стадиях процесса [8]. Недостатком этого способа является применение ацетангидрида и жидкого брома на стадии бромирования, а также необходимость использования дорогого и цитотоксичного [9] палладиевого катализатора на заключительной стадии синтеза, что требует проведения тщательной хроматографической очистки конечного препарата.
Техническая задача, решаемая заявленным изобретением, состоит в разработке метода синтеза 2,2-диалкил-4,5-диарилфуран-3(2H)-онов общей формулы (1),
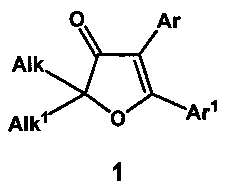
где Alk и Alk1=CH3, CH3CH2 или другие алкильные или циклоалкильные группы, а Ar и Ar1=Ph, p-X-C6H4 (X=H, MeO, MeS, MeSO, MeSO2, Hal) и другие замещенные или незамещенные арильные группы, исходящего из дешевых продуктов многотоннажного органического синтеза, не нуждающегося в использовании палладиевых катализаторов на заключительных стадиях и использующего малотоксичные реагенты на остальных стадиях процесса.
Технический результат, получаемый при реализации заявленного изобретения, заключается в повышении суммарного выхода целевого продукта, снижении его себестоимости и исключении металл-катализируемых реакций, а также реакций, требующих применения опасных или высокотоксичных реагентов.
Указанный технический результат достигается тем, что:
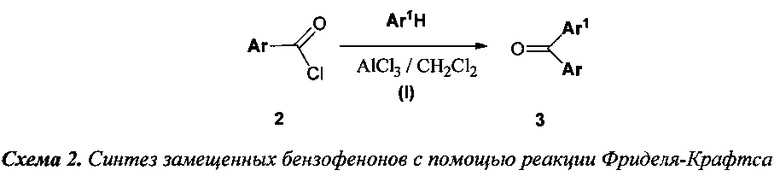
1) для получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов используется шестистадийная схема синтеза, в которой исходными веществами служат 1,1-диалкилпроп-2-ин-1-олы и замещенные бензофеноны (3), получаемые реакцией Фриделя-Крафтса (Стадия I) из хлороангидридов бензойных кислот и производных бензола по схеме 2;
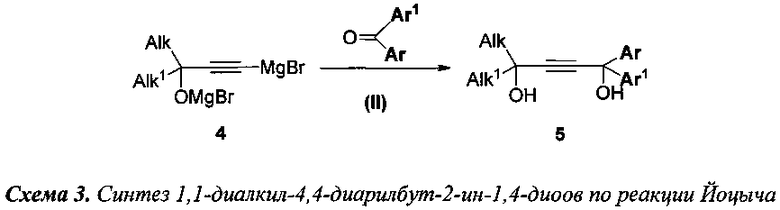
2) 1,1-диалкил-4,4-диарилбут-2-ин-1,4-диолы (5) получают взаимодействием магниевых производных 1,1-диалкилпроп-2-ин-1-олов (4) с замещенными бензофенонами (3) в условиях реакции Йоцыча (Стадия II) так, как изображено на схеме 3;
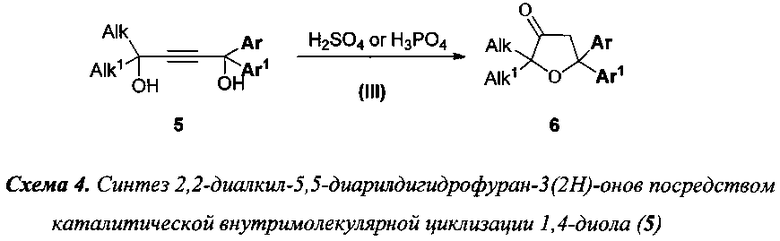
3) 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-оны (6) получают внутримолекулярной циклизацией (Стадия III) 1,1-диалкил-4,4-диарилбут-2-ин-1,4-диолов (5) под действием 5÷15% раствора серной или орто-фосфорной кислоты в метаноле или этаноле согласно схеме 4;
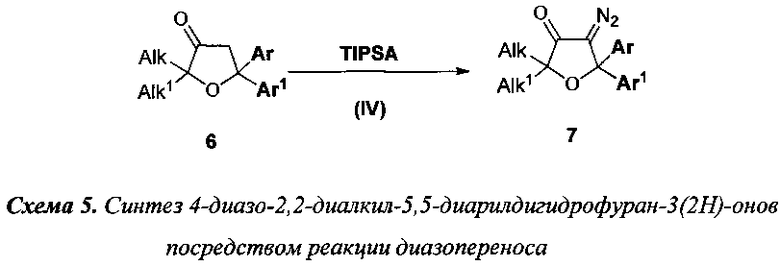
4) для синтеза 4-диазо-2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов (7) используют взаимодействие 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов (6) с 2,4,6-трет-изопропилсульфонилазидом (TIPSA) или n-толуолсульфанилазидом в бензоле или толуоле в присутствии катализаторов межфазного переноса (для гетерофазной реакции) и основания, например 50% водного раствора KOH или 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU). Данная реакция (Стадия IV) отображена на схеме 5;
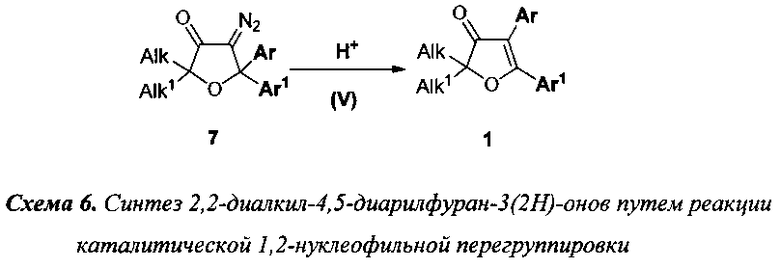
5) превращение 4-диазо-2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов (7) в целевые 2,2-диалкил-4,5-диарилфуран-3(2H)-оны (1) осуществляется посредством каталитической 1,2-нуклеофильной перегруппировки (Стадия V), протекающей под действием сильных кислот, таких как H2SO4, CF3COOH, H3PO4 и др., в неполярном или полярном растворителе, например в н-гексане, толуоле, хлороформе и муравьиной кислоте (схема 6);
6) в соединениях (1) превращение тиометильной группы в метилсульфинильную, а также метилсульфинильной в метилсульфоновую группу осуществляют окислением 35% водным раствором пероксида водорода в среде уксусной кислоты в присутствии каталитических количеств серной кислоты (Окисление, стадия VI), при мольном соотношении 1:1 пероксида водорода к 2,2-диалкил-4,5-диарилфуран-3(2H)-ону. Аналогично, превращение как тиометильной, так и метилсульфинильной групп в метилсульфоновую группу проводится с количественным выходом тем же окислителем в тех же условиях под действием мольного избытка пероксида водорода (схема 7);
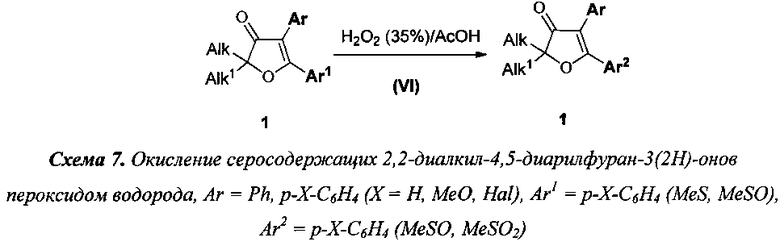
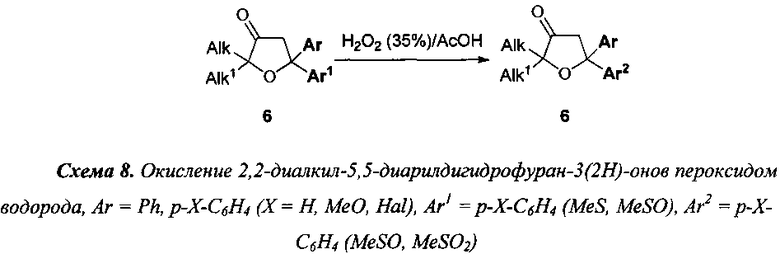
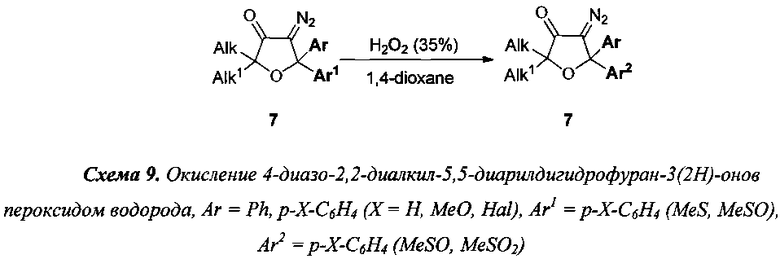
7) указанное в предыдущем пункте окисление серосодержащих групп, может быть осуществлено непосредственно после стадии III в тех же условиях, что указаны в описании стадии VI (схема 8). Кроме того, аналогичное окисление может быть осуществлено непосредственно после стадии IV, если реакцию проводить в 1,4-диоксане в соответствии с условиями стадии VI (схема 9);
Заявляемое изобретение имеет следующие отличительные признаки и преимущества:
1. Использование в качестве исходных соединений коммерчески доступных 1,1-диалкилпроп-2-ин-1-олов, замещенных бензофенонов, аренов и хлороангидридов замещенных бензойных кислот.
2. Использование на ключевой стадии процесса реакцию 1,2-нуклеофильной перегруппировки диазокетонов в условиях катализа сильными кислотами, что позволяет вводить в одну стадию арильную группу в 4-ое положение фуранового кольца и генерировать кратную связь между 4 и 5 положениями с выходами 91-99%.
3. Повышение выхода до практически количественного на заключительных стадиях синтеза (V и VI) при отсутствии необходимости тщательной очистки целевых продуктов.
4. Возможность окисления серосодержащих групп (стадия 6) непосредственно после стадий III, IV и V, что позволяет оптимизировать процесс и увеличить выходы целевых соединений (1).
5. Отсутствие необходимости использования металл-каталитизируемых реакций, в частности, реакции Сузуки, и палладиевых катализаторов в процессе.
6. Отсутствие необходимости использования литийорганических реагентов, таких как бутиллитий, и проведения реакций при низких температурах (ниже 0°C).
7. Отсутствие стадий галогенирования в заявленном процессе.
Апробация заявленного изобретения была выполнена в Санкт-Петербургском государственном университете с использованием оборудования ресурсных центров СПбГУ «Магнито-резонансные методы исследования» и «Методы анализа состава вещества». Результаты исследования представлены в виде конкретных примеров реализации.
Примеры выполнения способа
Изобретение иллюстрируется приведенными ниже примерами, которые обеспечивают только иллюстрацию и не могут рассматриваться как ограничивающие возможности настоящего изобретения.
Спектры ЯМР 1H (400.13 МГц) и 13С (100.61 МГц) регистрировали на приборе «Bruker 400 MHz Avance» в дейтерохлороформе, внутренний стандарт - CHCl3 (7.26 ррт). ИК спектры получены на спектрофотометре «Perkin-Elmer ΒΧΙΙ» в диапазоне 4000-400 см-1 в растворителе тетрахлоруглероде. Для снятия масс-спектров использовали хромато-масс спектрометр «Bruker microTOF». Температуру плавления измеряли на приборе «Buchi В-540».
Реакции проводили в безводных растворителях, очищенных по стандартным методикам. Для препаративного разделения смесей использовали колоночную хроматографию на силикагеле (Silicagel L, 40-100 µm) в градиентном режиме. Аналитическую ТСХ проводили на пластинах Silicagel Merck 60 F254 (Германия). Элюэнты: петролейный эфир-дихлорметан, толуол-этанол.
Соединения, приведенные в описании без ссылок, приготовлены по стандартным лабораторным методикам либо являются коммерчески доступными веществами.
Пример 1.
Получение 2,2-диметил-5-[4′-(метилсульфонил)фенил]-4-фенилфуран-3(2Н)-она (Схема 10, 1а)
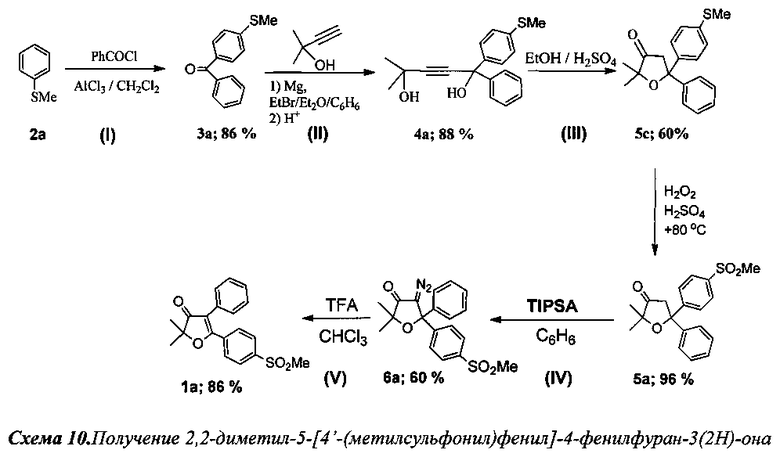
Стадия I. Получение 4′-(метилтио)фенил(фенил)метанона (3а).
К охлажденному до 0°C раствору 50 мл (51 г, 0.4 моль) тиоанизола (2а) в 200 мл дихлорметана при перемешивании добавили порциями 60 г безводного AlCl3. Затем в течение 1 ч при интенсивном охлаждении до 0°C по каплям добавили 46 мл (56 г, 0.4 моль) хлористого бензоила, при этом смесь приобрела оранжевый цвет и большую вязкость. После добавления всего количества хлористого бензоила смесь кипятили с обратным холодильником в течение еще 3 ч. По завершению реакции оранжевый осадок комплекса отфильтровали на фильтре Шотта, промыли дихлорметаном, гидролизовали, в начале, ледяной водной 3÷5% H2SO4, а затем проводили гидролиз до полного обесцвечивания продукта при кипячении. Затвердевший при охлаждении продукт был растворен в дихлорметане, промыт H2O и высушен Na2SO4. После отгонки растворителя получили 66.5 г 4′-(метилтио)фенил(фенил)метанона (3а). Темно-оранжевый маточный раствор комплекса в дихлорметане гидролизовали кипячением с 3÷5% водным раствором H2SO4 до полного обесцвечивания, затем органический слой отделили от водного, водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические фракции были промыты водой и высушены Na2SO4. После отгонки растворителя продукт был дополнительно перекристаллизован из н-гексана и высушен на воздухе. Получили 11.9 г соединения 3а. Общий выход 78.4 г (86%).
4′-(Метилтио)фенил(фенил)метанон (3а), бесцветные кристаллы, т.пл. 79°C. Спектр ЯМР 1H (400 МГц, 42 мг в 0.8 мл CDCl3), δ, м.д.: 2.53 с (3Н, SCH3), 7.29 д (2Н), 7.47 т (2Н), 7.53-7.61 м (1H), 7.74 д (2Н), 7.76 д (2Н). Спектр ЯМР 13С (100 МГц, 42 мг в 0.8 мл CDCl3), δ, м.д.: 14.8 (SCH3), 124.8, 128.2, 129.8, 130.6, 132.1, 133.6, 137.8, 145.2 (все CArAr′), 195.8 (C=O). Масс-спектр HRMS, m/z: 229.0689 [МН]+.
Стадия II. Получение 2-метил-5-фенил-5-(4′-метилтиофенил)пент-3-ин-2,5-диола (4а).
К раствору EtMgBr, приготовленному из 10 г (0.417 моль) магния и 45.4 г (0.420 моль) этилбромида, в 120 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 15.7 мл (0.162 моль) 2-метилбут-3-ин-2-ола в 100 мл абсолютного бензола. Смесь кипятили в течение 2 ч, затем при охлаждении добавили по каплям раствор 26 г (0.115 моль) соединения 3а в 200 мл бензола и кипятили в течение 4 ч. Затем смесь охладили в водяной бане и осторожно гидролизовали при помощи 150 мл 10% раствора соляной кислоты. Органический слой отделили от водного. Водный слой экстрагировали бензолом (3×50 мл). Объединенные органические фракции промыли раствором NaHCO3, водой и высушили Na2SO4. После отгонки растворителя полученное желтое маслообразное вещество высушили в вакууме при давлении 0.02÷0.1 торр в течение 2 ч. Выход 32.4 г (88%). Без дополнительной очистки полученное вещество использовали на следующей стадии.
2-Метил-5-фенил-5-(4′-(метилтио)фенил)пент-3-ин-2,5-диол (4а), бесцветное маслообразное вещество. Спектр ЯМР 1H (400 МГц, 21 мг в 0.8 мл CDCl3), δ, м.д.: 1.57 с (6Н, 2CH3), 2.53 с (3Н, SCH3), 7.18 д (2Н), 7.22-7.28 м (1Н), 7.31 т (2Н), 7.47 д (2Н), 7.53-7.57 м (2Н). Спектр ЯМР 13С (100 МГц, 21 мг в 0.8 мл CDCl3), δ, м.д.: 15.6 (SCH3), 31.3 (2CH3), 65.3 (C(CH3)2), 73.9, 84.7 (C3≡C 4), 91.8 (C 3≡C4), 125.8, 126.2, 126.4, 127.7, 128.4, 138.0, 141.8 (все CArAr′), 144.8. ИК спектр (в CCl4), см-1: 700.9 сл., 908.5 сл., 951.7 сл., 999.6 сл., 1167.6 сл., 1325.6 сл., 1449.0 сл., 1490.0 сл., 2854.7 ср., 2926.6 с, 3606.5 сл. Масс-спектр HRMS, m/z: 335.1087 [MNa]+.
Стадия III. Получение 2,2-диметил-5-[4′-(метилтио)фенил]-5-фенил-дигидрофуран-3(2Н)-она (5с).
31.6 г (0.101 моль) соединения 4а растворили в 100 мл кипящего абсолютного этанола и при перемешивании по каплям добавили 5 мл (0.09 моль) 98% серной кислоты, растворенной в 15 мл этанола. Смесь кипятили в течение 2 ч до окончания реакции (контроль по ТСХ). Затем смесь охладили до 0°C для кристаллизации. Образовавшийся маслообразный коричневый слой отделили от маточника и высушили от этанола в вакууме при давлении 0.02÷0.1 торр. в течение 5 ч. Получившийся при этом кристаллический продукт промыли на фильтре Шотта водой, ледяным н-гексаном и высушили в вакууме. Получили 18.2 г светло-желтого порошка. Выпавший через сутки из маточного раствора светло-желтый осадок отфильтровали на фильтре Шотта, промыли водой, ледяным н-гексаном и высушили в вакууме. Получено еще 5.1 г чистого вещества. Общий выход 23.3 г (60%) соединения 5с.
2,2-Диметил-5-[4′-(метилтио)фенил]-5-фенил-дигидрофуран-3-он (5с), бесцветные кристаллы, т.пл. 65°C. Спектр ЯМР 1H (400 МГц, 31 мг в 0.8 мл CDCl3), δ, м.д.: 1.21 д (6Н, 2CH3), 2.45 с (3Н, SCH3), 3.30 с (2Н, CH2), 7.18 д (2Н), 7.20-7.26 м (1Н), 7.27-7.35 м (4Н), 7.39 д (2Н). Спектр ЯМР 13С (100 МГц, 31 мг в 0.8 мл CDCl3), δ, м.д.: 15.6 (SCH3), 25.3 (2CH3), 47.9 (CH2), 81.4 (C(CH3)2), 82.4, 125.9, 126.2, 126.6, 127.4, 128.4, 137.7, 142.9, 146.0, 216.9 (C=O). ИК спектр (в CCl4), см-1: 502.3 сл., 528.6 сл., 961.0 сл., 994.0 сл., 1094.9 сл., 1133.1 сл., 1173.2 сл., 1281.4 сл., 1376.1 сл., 1446.8 сл., 1493.2 сл., 1599.5 сл., 1760.1 с, 2924.6 сл., 2983.7 сл., 3028.3 сл., 3064.4 сл. Масс-спектр HRMS, m/z: 313.1259 [МН]+.
Стадия окисления. Получение 2,2-диметил-5-[4′-(метилсулъфонил)фенил]-5-фенилдигидрофурано-3(2Н)-она (5а).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл уксусной кислоты и добавили при перемешивании 300 мг (0.96 ммоль) соединения 5с, затем по каплям добавили смесь 2 мл 35% водного раствора (22 ммоль) пероксида водорода с 4 мл уксусной кислоты и 0.1 мл 98% H2SO4. Смесь перемешивали в течение 2 ч при температуре +70-80°C. Затем смесь вылили в 50 мл воды и экстрагировали CH2Cl2 (4×15 мл), промыли раствором NaHCO3 и водой (3×10 мл) для удаления остатков пероксида водорода и кислот, а затем высушили K2CO3. После отгонки растворителя, получили 318 мг бесцветного маслообразного вещества. Выход 318 мг (96%) соединения 5а.
2,2-Диметил-5-[4′-(метилсульфонил)фенил]-5-фенил-дигидрофуран-3(2H)-он (5а), бесцветное масло. ЯМР 1H (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CH2Cl2=5.30 м.д.) δ, м.д.: 1.22 с (3Н, CH3), 1.24 с (3Н, CH3), 3.04 с (3Н, SO2CH3), 3.28 д (1Н, CH2), 3.50 д (1Н, CH2), 7.26-7.29 м (1Н), 7.34 т (2Н), 7.43 д (2Н), 7.78 дд (4Н). ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 24.9 (CH3), 25.4 (CH3), 44.39 (SO2CH3), 47.36 (CH2), 81.68 (C5), 82.16 (C2), 125.8, 126.6, 127.5, 127.9, 128.6, 139.4, 144.7, 152.6 (все CArAr′), 215.6 (C=O). ИК спектр (в CCl4), см-1: 954.5 ср., 996.8 сл., 1157.1 ср., 1328.6 ср., 1597.3 сл., 1762.5 ср. (C=O), 2932.3 сл., 2983 сл. Масс-спектр HRMS: m/z=345.1159 [ΜΗ+], 362.1420 [MNH4 +], 367.0970 [MNa]+.
Стадия IV. Получение 4-диазо-2,2-диметил-5-[4′-(метилсульфонил)фенил]-5-фенил-дигидрофуран-3(2Н)-она (6а).
К смеси 385 мг (1.12 ммоль) соединения 5с, 140 мг (0.52 ммоль) (n-Bu)4NBr, 400 мг (1.30 ммоль) 2,4,6-трис-(изопропил)бензолсульфонилазида и 30 мг (0.112 ммоль) 18-краун-6 в 30 мл абсолютного бензола при охлаждении по каплям добавили 30 мл холодного 50% раствора КОН. При этом смесь сразу же окрашивалась в красный цвет. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч до завершения реакции (контроль по ТСХ). Органический слой отделили, водный слой подвергли экстракции бензолом (3×15 мл). Объединенные органические фракции промыли водой (4×15 мл) и высушили MgSO4. После отгонки растворителя продукт очистили на короткой колонке с силикагелем. Выход: 250 мг (60%) соединения 6а.
4-Диазо-2,2-диметил-5-[4′-(метилсульфонил)фенил]-5-фенилдигидрофуран-3(2H)-он (6а), желтое маслообразное вещество. Спектр ЯМР 1H (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.36 д (6Н, 2CH3), 3.08 с (3Н, SO2CH3), 7.28-7.30 м (2Н), 7.37-7.42 м (3Н), 7.76 дд (4Н). Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.): 25.2 (CH3), 44.4 (SO2CH3), 63.7 (C=N2), 83.6 (C5), 85.9 (C2), 126.2, 127.3, 127.8, 128.8, 128.9, 140.5, 142.6, 149.9 (все CArAr′), 196.2 (C=O). ИК спектр (в CCl4), см-1: 955.4 сл., 1026.1 сл., 1160.3 ср., 1226.9 сл., 1330.3 ср., 1344.6 ср., 1699.4 ср. (C=O), 2092.2 с. (C=N2), 2982.7 сл. Масс-спектр HRMS: m/z=371.1070 [МН+].
Стадия V. Получение 2,2-диметил-5-[4′-(метилсульфонил)фенил]-4-фенилфуран-3(2Н)-она (1а).
К раствору 200 мг (0.54 ммоль) соединения 6а в 5 мл хлороформа по каплям добавили 0.25 мл (3.25 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты. Смесь кипятили 3 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3 и водой (2×3 мл), высушили K2CO3. После отгонки растворителя получили 185 мг (99%) смеси двух региоизомерных 2,2-диалкил-4,5-диарилфуран-3(2H)-онов в виде бесцветного маслообразного вещества. Анализ с помощью 1H ЯМР с внутренним стандартом (1,2-тетрахлорэтан) показал образование двух основных продуктов в соотношении 5.67 (86%) :1 (14%). Реакционная смесь (185 мг) была разделена на колонке с силикагелем. Выход 155 мг (84%) соединения 1а.
2,2-Диметил-5-[4′-(метилсульфонил)фенил]-4-фенилфуран-3(2H)-он (1а), бесцветные кристаллы, т.пл. 201-202°C. Спектр ЯМР 1H (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.58 с (6Н, 2CH3), 3.06 с (3Н, SO2CH3), 7.26-7.40 м (5Н), 7.88 дд (4Н). Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 23.3 (2CH3), 43.3 (SO2 CH3), 87.5 (C2), 115.6 (C4), 127.4, 128.2, 128.9, 129.2, 129.4, 135.1, 142.8 (все CArAr′), 175.2 (C5), 205.2 (C=O). ИК спектр (в CCl4), см-1: 668.5 ср., 954.8 ср., 1152.5 с, 1331.7 ср., 1703.6 ср (C=O). Масс-спектр HRMS: m/z=343.0996 [МН+]
Пример 2.
Получение 2,2-диметил-5-[4-(метилсульфинил)фенил]-4-фенилфуран-3(2Н)-она (Схема 11, 1b).
Проведение реакций на стадиях I-II аналогично примеру 1.
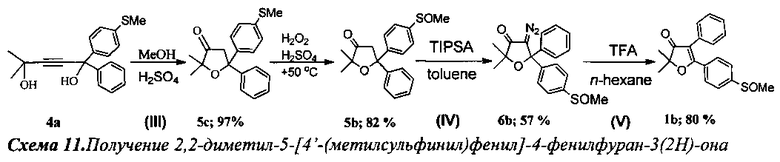
Стадия III. Получение 2,2-диметил-5-[4′-(метилтио)фенил]-5-фенил-дигидрофуран-3(2Н)-она (5с).
3 г (9.6 ммоль) соединения 4а растворили в 50 мл кипящего метанола и при перемешивании по каплям добавили 15 мл 10% (объемн.) раствора серной кислоты в метаноле (30 ммоль H2SO4). Смесь кипятили в течение 1 ч до окончания реакции (контроль по ТСХ). По окончании реакции смесь вылили в 400 мл воды и экстрагировали хлористым метиленом (4×40 мл). Органический слой отделили, промыли раствором NaHCO3 и водой (2×10 мл), высушили Na2SO4. После удаления растворителя под вакуумом получили 2.93 г желтого маслообразного вещества. Выход 2.93 г (97%) соединения 5с. Продукт чист по спектру ЯМР 1H и использовался на последующих стадиях без дополнительной очистки.
Стадия окисления. Получение 2,2-диметил-5-[4′-(метилсульфинил)фенил]-5-фенил-дигидрофуран-3(2Н)-она (5b).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл уксусной кислоты и растворили в ней при +50°C 300 мг (0.96 ммоль) соединения 5с. Затем в течение 1 ч по каплям добавили смесь из 100 мг 35% водного раствора пероксида водорода (0.96 ммоль) в 3 мл уксусной кислоты и 0.05 мл 98% H2SO4. Реакционную смесь перемешивали в течение 3 ч при +50-55°C (контроль реакции по ТСХ). После завершения реакции смесь вылили в воду, экстрагировали CH2Cl2 (4×15 мл), органическую фракцию промыли раствором NaHCO3 и водой (3×5 мл), а затем высушили K2CO3. После отгонки растворителя получено 260 мг маслообразного вещества. Выход: 260 мг (82%) соединения 5b. При правильном исполнении продукт не требует дополнительной очистки.
2,2-Диметил-5-(4′-(метилсульфинил)фенил)-5-фенил-дигидрофуран-3(2H)-он (5b), бесцветное маслообразное вещество. Спектр ЯМР 1H (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CH2Cl2 5.30 м.д.), δ, м.д.: 1.20 с (3Н, CH3), 1.28 с (3Н, CH3), 2.72 с (3Н, SOCH3), 3.28 д (1Н, СН2), 3.44 д (1H, CH2), 7.26-7.28 м (1Н), 7.34 т (2Н), 7.43 д (2Н), 7.61 с (4Н). Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3 77.00 м.д.): 25.0 (CH3), 25.4 (CH3), 43.7 (SOCH3), 47.6 (CH2), 81.5 (C5), 82.2 (C2), 123.6, 125.8, 126.8, 127.67, 128.45, 144.67, 145.17, 149.69 (все CArAr′), 216.0 (C=O). Спектр ИК (в CCl4), см-1: 955.7 сл., 1061.24 сл., 1171.9 сл., 1717.9 сл., 1761.4 с. (C=O), 2859.1 сл., 2927.1 сл., 2959.4 сл. Масс-спектр HRMS: m/z=329.1204 [ΜΗ+], 367.0759 [MK+].
Стадия IV. Получение 4-диазо-2,2-диметил-5-[4′-(метилсулъфинил)фенил]-5-фенил-дигидрофуран-3(2Н)-она (6b).
К смеси 520 мг (1.58 ммоль) соединения 5b, 208 мг (0.78 ммоль) ButNBr, 561 мг (1.82 ммоль) 2,4,6-трис-(изопропил)бензолсульфонилазида и 42 мг (0.157 ммоль) 18-краун-6 в 50 мл абсолютного толуола при 0°C по каплям добавили 30 мл охлажденного 50% водного раствора КОН. Смесь сразу же окрасилась в ярко-красный цвет. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч до завершения реакции (контроль по ТСХ). Затем отделили органический слой, водный слой подвергли экстракции толуолом (3×15 мл). Объединенные органические вытяжки промыли водой (4×10 мл) и высушили MgSO4. После азеотропной отгонки растворителя (с этанолом) продукт очистили на короткой колонке с силикагелем. Получили 320 мг (57%) соединения 6b.
4-Диазо-2,2-диметил-5-[4′-(метилсульфинил)фенил]-5-фенил-дигидрофуран-3(2H)-он (6b), желтое маслообразное вещество. Спектр ЯМР 1Н (400 МГц, 21 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.32 д (6Н, 2CH3), 2.72 с (3Н, SOCH3), 7.27-7.38 м (5Н), 7.56 дд (4Н). Спектр ЯМР 13С (100 МГц, 21 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 25.1 (2CH3), 47.7 (SOCH3), 63.9 (C=N2), 83.6 (C5), 85.6 (C2), 123.8, 126.1, 127.3, 128.6, 128.6, 142.9, 145.9, 147.0 (все CArAr′), 196.4 (C=O). Спектр ИК (в CCl4), см-1: 1025. 5 сл., 1061.9 сл., 1090.3 сл., 1162.9 сл., 1226.8 сл., 1345.16 ср., 1698.26 ср. (C=O), 2091.85 с. (C=N2), 2925.7 сл., 2959 сл., 2983.5 сл. Масс-спектр HRMS: m/z=355.1119 [MH+], 377.0939 [MNa+].
Стадия V. Получение 2,2-диметил-5-[4′-(метилсульфинил)фенил]-4-фенилфуран-3(2Н)-она (1b).
К раствору 186 мг (0.53 ммоль) соединения 6b в 25 мл н-гексана по каплям было добавлено 0.26 мл (3.40 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты. Смесь кипятили 30 мин до полного исчезновения исходного диазокетона (контроль по ТСХ). По окончании реакции смесь промыли раствором NaHCO3 и водой (3×5 мл), а затем высушили K2CO3. После отгонки растворителя получили 173 мг светло-желтого масла, содержащего два региоизомерных 2,2-диалкил-4,5-диарилфуран-3(2H)-она в соотношении 4 (80%): 1 (20%). Общий выход реакции по двум продуктам: 173 мг (99%). Колоночная хроматография реакционной смеси приводит к выделению чистого 1b с выходом 138 мг (80%).
2,2-Диметил-5-[4′-(метилсульфинил)фенил]-4-фенилфуран-3(2H)-он (1b), бесцветные кристаллы, т.пл. 178-179°C. Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.57 с (6Н, 2CH3), 2.74 с (3Н, SOCH3), 7.28-7.39 м (5Н), 7.71 дд (4Н). Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.4 (2CH3), 43.8 (SOCH3), 87.3 (C2), 114.7 (C4), 123.6, 127.9, 128.8, 129.2, 129.4, 132.6, 149.5 (все CArAr′), 176.2 (C5), 205.3 (C=O). Спектр ИК (в CCl4), см-1: 696.4 сл., 951.3 сл., 1090.6 ср., 1238.0 сл., 1385.4 ср., 1400.9 ср., 1618.5 ср., 1701.7 с. (CO), 2857.4 сл., 2931.9 сл., 2981.3 сл., 3059.2 сл. Масс-спектр HRMS: m/z=349.0874 [MNa]+.
Пример 3.
Получение 2,2-диметил-5-[4′-(метилтио)фенил]-4-фенилфуран-3(2Н)-она (Схема 12, 1с).
Проведение реакций на стадиях I-II аналогично примеру 1.
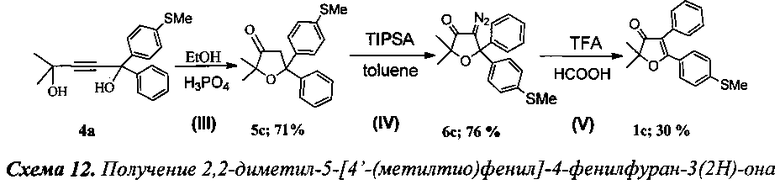
Стадия III. Получение 2,2-диметил-5-[4′-(метилтио)фенил]-5-фенил-дигидрофуран-3(2Н)-она (5с).
3 г (9.6 ммоль) соединения 4а растворили в 30 мл кипящего этанола и при перемешивании по каплям добавили 15 мл 20% (объемн.) раствора орто-фосфорной кислоты в этаноле (52 ммоль H3PO4). Смесь кипятили в течение 4 ч до окончания реакции (контроль по ТСХ). По окончании реакции смесь вылили в 250 мл воды и экстрагировали хлористым метиленом (4×40 мл). Органический слой отделили, промыли раствором NaHCO3 и водой (3×10 мл), высушили Na2SO4. После удаления растворителя под вакуумом получено 2.73 г желтого маслообразного продукта. Полученное вещество дополнительно очистили на колонке с силикагелем. Выход 2.13 г (71%) соединения 5с.
Стадия IV. Получение 4-диазо-2,2-диметил-5-[4′-(метилтио)фенил]-5-фенил-дигидрофуран-3(2Н)-она (6с).
К смеси 250 мг (0.8 ммоль) соединения 5с и 310 мг (0.99 ммоль) 2,4,6-трис(изопропил)-бензолсульфонилазида в 15 мл абсолютного толуола при охлаждении по каплям добавили 152 мг (1 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU). При этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 45°С в течение 6 ч до завершения реакции (контроль по ТСХ). Затем реакционную смесь вылили на колонку с силикагелем и хроматографировали в градиентном режиме смесью н-гексан - хлористый метилен. После азеотропной отгонки растворителя (с этанолом) кристаллический продукт дополнительно высушили под вакуумом. Выход 208 мг (76%) соединения 6с.
4-Диазо-2,2-диметил-5-(4′-(метилтио)фенил)-5-фенилдигидрофуран-3(2Н)-он (6с), желтые кристаллы, т.пл. 134-135°C. Спектр ЯМР 1Н (400 МГц, 40 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.35 с (6Н, 2CH3), 2.48 с (3Н, SCH3), 7.23 с (4Н), 7.29-7.41 м (5Н). Спектр ЯМР 13С (100 МГц, 40 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.4 (SCH3), 25.2 с (2CH3), 83.9 (C5), 85.5 (C(CH3)2), 126.1, 126.3, 126.9, 128.4, 128.5, 139.1, 140.4, 143.5 (все CArAr′), 196.9 (C=O). ИК спектр (в CCl4), см-1: 540.9 сл., 698.0 сл., 1024.8 сл., 1094.2 сл., 1164.7 сл., 1225.9 сл., 1345.6 ср., 1377.6 сл., 1493.0 сл., 1696.3 ср. (C=O), 2091.2 с. (C=N2), 2926.1 сл., 2982.4 сл. Масс-спектр, m/z: 361.0987 [MNa]+.
Стадия V. Получение 2,2-диметил-5-[4′-(метилтио)фенил]-4-фенилфуран-3(2Н)-она (1с).
К раствору 200 мг (0.59 ммоль) соединения 6с в 10 мл муравьиной кислоты (99.8%) по каплям добавили 0.3 мл (3.83 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты (TFA). Смесь кипятили 1 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь нейтрализовали раствором NaHCO3, экстрагировали хлористым метиленом (3×15 мл) и высушили K2CO3. После отгонки растворителя получено 183 мг (99%) смеси двух региоизомерных 2,2-диалкил-4,5-диарилфуран-3(2H)-онов в виде светло-желтого масла. Реакционная смесь (183 мг) была разделена хроматографически на колонке с силикагелем. Анализ с помощью 1H ЯМР с внутренним стандартом (1,2-тетрахлорэтан) показал образование двух основных продуктов в соотношении 2.22 (69%): 1 (31%). Выход 55 мг (30%) соединения 1с.
2,2-Диметил-5-[4′-(метилтио)фенил]-4-фенилфуран-3(2H)-он (1с), светло-желтое масло или кристаллы. Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.55 с (6Н, 2CH3), 2.48 с (3Н, SCH3), 7.15-7.17 м (1H), 7.25 д (2Н), 7.30-7.40 м (4Н), 7.56 д (2Н). Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.7 (SCH3), 23.4 (2CH3), 86.9 (C(CH3)2), 113.1 (C4), 125.0, 128.6, 128.6, 129.6, 131.8, 144.3 (все CArAr′), 177.6 (C5), 205.2 (C=O). Спектр ИК (в CCl4), см-1: 676.1 сл., 1051.8 сл., 1094.6 сл., 1147.1 сл., 1168.1 сл., 1238.6 сл., 1264.5 сл., 1385.9 ср., 1484.6 сл., 1598.0 ср., 1614.6 ср., 1697.9 с, 2858.3 сл., 2927.6 ср., 2959.7 ср. Масс-спектр HRMS, m/z: 311.1113 [MH]+, 333.0935 [MNa]+.
Пример 4.
Получение 2,2-диметил-4-[4′-(метилсульфонил)фенил]-5-фенилфуран-3(2Н)-она (Схема 13, 1d).
Проведение реакций на стадиях I-IV аналогично примеру 3.
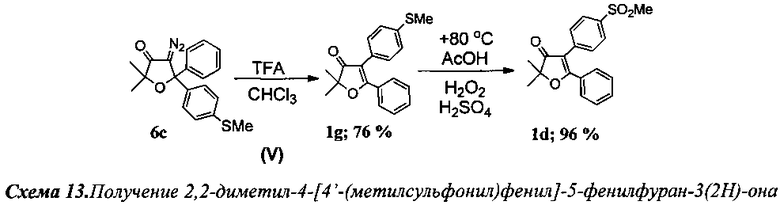
Стадия V. Получение 2,2-диметил-4-[4′-(метилтио)фенил]-5-фенилфуран-3(2Н)-она (1g).
К раствору 200 мг (0.59 ммоль) соединения 6с в 10 мл хлороформа по каплям добавили 0.3 мл (3.83 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты (TFA). Смесь кипятили 2 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×3 мл) и высушили K2CO3. После отгонки растворителя получено 183 мг (99%) смеси двух региоизомерных 2,2-диалкил-4,5-диарилфуран-3(2H)-онов в виде светло-желтого маслообразного продукта. Реакционная смесь (183 мг) была разделена на колонке с силикагелем. Анализ с помощью 1H ЯМР с внутренним стандартом (1,2-тетрахлорэтан) показал образование двух основных продуктов в соотношении 3.17 (76%): 1 (24%). Выход 139 мг (76%) соединения (1g).
2,2-Диметил-4-[4′-(метилтио)фенил]-5-фенилфуран-3(2H)-он (1g), светло-желтые кристаллы, т.пл. 120-121°C. Спектр ЯМР 1H (400 МГц, 24 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.55 с (6Н, 2CH3), 2.49 с (3Н, SCH3), 7.24-7.25 м (4Н), 7.36 т (2Н), 7.45-7.49 м (1H), 7.66 д (2Н). Спектр ЯМР 13С (100 МГц, 24 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.7 (SCH3), 23.37 (2CH3), 86.99 (C(CH3)2), 113.0 (C4), 126.6, 128.4, 128.4, 129.8, 131.8, 137.8 (все CArAr′) 178.1 (C5), 205.4 (C=O). Спектр ИК (в CCl4), см-1: 639.1 сл., 1050.7 сл., 1093.6 сл., 1166.6 сл., 1242.4 сл., 1383.9 ср., 1502.1 сл., 1573.6 сл., 1591.1 сл., 1618.5 сл., 1698.9 с, 2925.8 сл., 2982.3 сл. Масс-спектр HRMS, m/z: 311.1107 [МН]+, 333.0935 [MNa]+.
Стадия окисления. Получение 2,2-диметил-4-[4′-(метилсульфонил)фенил]-5-фенилфуран-3(2Н)-она (1d).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл уксусной кислоты и растворили в ней при +50°C 300 мг (0.96 ммоль) соединения 1g. Затем по каплям добавили смесь из 1 г 35% водного раствора пероксида водорода (9.6 ммоль) в 5 мл уксусной кислоты и 0.05 мл 98% H2SO4. Реакционную смесь перемешивали в течение 20 мин при +80-85°C (контроль реакции с помощью ТСХ). После завершения реакции смесь вылили в воду, экстрагировали CH2Cl2 (4×15 мл), органическую фракцию промыли раствором NaHCO3 и водой (3×5 мл), а затем высушили K2CO3. После отгонки растворителя получено 318 мг кристаллического вещества. Выход: 318 мг (96%) соединения 1d.
2,2-Диметил-4-[4′-(метилсульфонил)фенил]-5-фенилфуран-3(2H)-он (1d), кристаллическое бесцветное вещество. Спектр ЯМР 1H (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.58 с (6Н, 2CH3), 3.05 с (3Н, SO2CH3), 7.39 т (2Н), 7.50-7.55 м (3Н), 7.60 д (2Н), 7.90 д (2Н). Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.3 (2CH3), 43.5 (SO2CH3), 87.9 (C(CH3)2), 111.7 (C4), 127.6, 128.4, 128.7, 129.2, 130.1, 132.4, 136.2, 139.0 (все CArAr′), 179.8 (C5), 204.4 (C=O). ИК спектр (в CCl4), см-1: 954.6 ср., 1050.87 ср., 1157.1 с, 1326.6 ср., 1382.9 ср., 1613.8 ср., 1700.7 с. (C=O), 2931.9 сл., 2982.3 сл. Масс-спектр HRMS, m/z=365.0823 [MNa]+.
Пример 5.
Получение 2,2-диметил-4-[4′-(метилсульфинил)фенил]-5-фенилфуран-3(2Н)-она (Схема 14, 1f).
Проведение реакций на стадиях I-IV аналогично примеру 3.
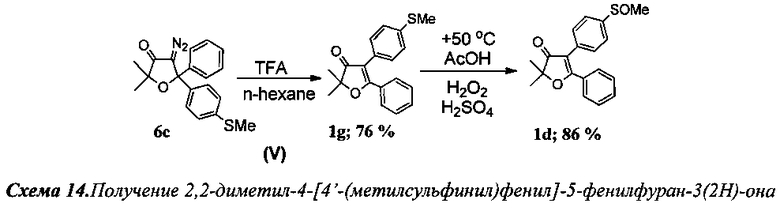
Стадия V. Получение 2,2-диметил-4-[4′-(метилтио)фенил]-5-фенилфуран-3(2Н)-она (1g).
К раствору 200 мг (0.59 ммоль) соединения 6с в 20 мл н-гексана по каплям добавили 0.3 мл (3.83 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты (TFA). Смесь кипятили 4 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×5 мл) и высушили K2CO3. После отгонки растворителя получено 183 мг (99%) смеси двух региоизомерных 2,2-диалкил-4,5-диарилфуран-3(2H)-онов в виде светло-желтого масла. Реакционная смесь (183 мг) была разделена хроматографически на колонке с силикагелем. Анализ с помощью 1H ЯМР с внутренним стандартом (1,2-тетрахлорэтан) показал образование двух основных продуктов в соотношении 3.17 (76%): 1 (24%). Выход: 139 мг (76%) соединения 1g.
Стадия окисления. Получение 2,2-диметил-4-[4′-(метилсульфинил)фенил]-5-фенилфуран-3(2Н)-она (1f).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл уксусной кислоты и растворили в ней при +50°C 300 мг (0.96 ммоль) соединения 1g. Затем по каплям при интенсивном перемешивании в течение 1 ч осторожно добавили смесь из 100 мг 35% водного раствора пероксида водорода (0.96 ммоль) в 3 мл уксусной кислоты и 0.05 мл 98% H2SO4. Реакционную смесь перемешивали в течение 3 часов при +50-55°C (контроль реакции по ТСХ). После завершения реакции смесь вылили в воду, экстрагировали CH2Cl2 (4×15 мл), органическую фракцию промыли раствором NaHCO3 и водой (3×5 мл), а затем высушили K2CO3. После отгонки растворителя получено 272 мг кристаллического вещества. Выход: 272 мг (86%) соединения 1f.
2,2-Диметил-4-[4′-(метилсульфинил)фенил]-5-фенил-дигидрофурано-3(2H)-он (1f), кристаллическое бесцветное вещество. Спектр ЯМР Ή (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.58 с (6Н, 2CH3), 2.74 с (3Н, SOCH3), 7.38 т (2Н), 7.49-7.51 м (3Н), 7.63 т (4Н). ИК спектр (в CCl4), см-1: 954.8 сл., 1050.6 ср., 1090.2 сл., 1383.4 с, 1616.1 ср., 1700.9 с. (C=O), 2931.4 сл., 2981.6 сл. Масс-спектр HRMS, m/z=349.0876 [MNa]+.
Пример 6.
Получение 2,2-диметил-4-[4′-(метилтио)фенил]-5-фенилфуран-3(2Н)-она (Схема 15, 1g).
Проведение реакций на стадиях I-III аналогично примеру 1.
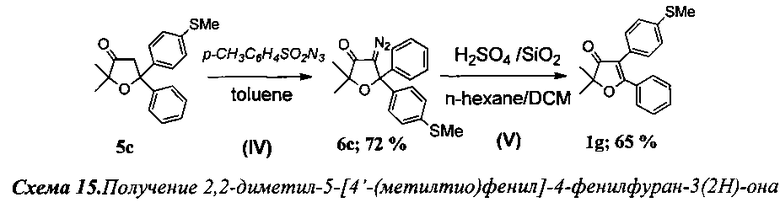
Стадия IV. Получение 4-диазо-2,2-диметил-5-[4′-(метилтио)фенил]-5-фенил-дигидрофуран-3(2Н)-она (6с)
К смеси 3.62 г (11.6 ммоль) соединения 5с, 1.58 г (4.9 ммоль) (n-Bu)4NBr, 2.3 г (11.7 ммоль) пара-толуолсульфонилазида и 253 мг (14.5 ммоль) 18-краун-6 в 75 мл абсолютного толуола при охлаждении по каплям добавили 50 мл холодного 50% раствора КОН. При этом смесь сразу же окрашивалась в темно-красный цвет. Реакционную смесь перемешивали при температуре 40°C в течение 4 ч до завершения реакции (контроль по ТСХ). Органический слой отделили, водный слой подвергли экстракции толуолом (4×30 мл). Объединенные органические фракции промыли водой (3×20 мл) и высушили CaCl2. После азеотропной отгонки растворителя (с этанолом) продукт очистили на колонке с силикагелем. Выход 2.82 г (72%) соединения 6с.
Стадия 5. Получение 2,2-диметил-4-[4′-(метилтио)фенил]-5-фенилфуран-3(2Н)-она (1g).
200 мг (0.59 ммоль) соединения 6 с в 3 мл хлористого метилена нанесли на колонку (30×8 см2) с силикагелем, содержавшую слой силикагеля с осажденной на нем 98% серной кислотой. Затем, при постоянном токе элюэнта н-гексан-хлористый метилен (1:2), диазокетон был пропущен через кислотный слой. После отгонки растворителя получено 167 мг (91%) смеси двух региоизомерных 2,2-диалкил-4,5-диарилфуран-3(2H)-онов в виде светло-желтого масла. Анализ с помощью 1Н ЯМР с внутренним стандартом (1,2-тетрахлорэтан) показал образование двух основных продуктов в соотношении 2.45 (71%): 1 (29%). Продукты реакции были разделены хроматографически. Выход 119 мг (65%) соединения 1g.
Альтернативно, 2,2-диметил-4-[4′-(метилтио)фенил]-5-фенилфуран-3(2Н)-он (1g) был получен на стадии V примера 4.
Пример 7.
Иллюстрация возможности проведения окисления на стадии диазосоединений. Получение 4-диазо-2,2-диметил-5-[4′-(метилсульфонил)фенил]-5-фенил-дигидрофуран-3(2Н)-она (6а).
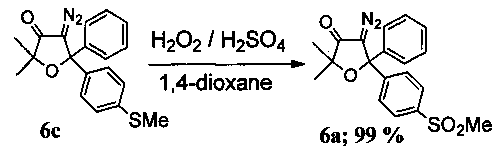
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 5 мл 1,4-диоксана и растворили в нем при +50°C 100 мг (0.29 ммоль) соединения 6с. Затем по каплям добавили смесь из 0.25 мл 35% водного раствора пероксида водорода (2.6 ммоль) и 0.05 мл 98% H2SO4. Реакционную смесь перемешивали в течение 1 ч при +90-95°C (контроль реакции по ТСХ). После завершения реакции смесь вылили в воду, экстрагировали CH2Cl2 (4×15 мл), органическую фракцию промыли раствором NaHCO3 и водой (3×5 мл), а затем высушили K2CO3. После отгонки растворителя под вакуумом получено 110 мг желтого масла. Продукт чист по ЯМР 1H. Выход: 110 мг (99%) соединения 6а.
Альтернативно, 4-диазо-2,2-диметил-5-[4′-(метилсульфонил)фенил]-5-фенил-дигидрофуран-3(2Н)-он (6а) был получен на стадии TV примера 1.
Пример 8.
Получение 2,2-диметил-4,5-дифенилфуран-3(2Н)-она (Схема 16, 1h).
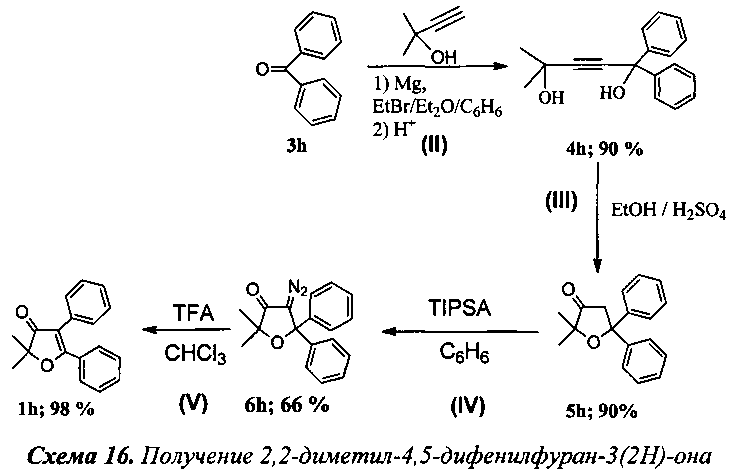
Стадия II. Получение 2-метил-5,5-дифенилпент-3-ин-2,5-диола (4h).
К раствору EtMgBr, приготовленного из 10 г (0.41 моль) магния и 46 г (0.42 моль) этилбромида, в 170 мл абсолютного диэтилового эфира при охлаждении ледяной бане медленно прибавили по каплям раствор 15.7 мл (0.16 моль) 2-метилбут-3-ин-2-ола в 100 мл абсолютного бензола. Далее смесь кипятили в течение 2 ч, после чего добавили 21 г (0.12 моль) бензофенона (3h) и кипятили в течение 7 ч. После охлаждения реакционной смеси в водяной бане через капельную воронку прилили 260 мл раствора соляной кислоты, приготовленного из 200 мл воды и 60 мл 36% соляной кислоты. Органический слой отделили от водного, последний подвергли экстракции абсолютным хлористым метиленом (1×50 мл, 1×40 мл, 2×30 мл). Объединенные органические вытяжки промыли NaHCO3, затем водой и высушили K2CO3. Растворитель отогнали в вакууме, полученный остаток перекристаллизовали из смеси петролейного эфира и хлористого метилена (2:1). Выход 28.8 г (90%) соединения 4h, т.пл. 119-120°C (литературные данные 118-119°C, [10]).
Стадия III. Получение 2,2-диметил-5,5-дифеншдигидрофуран-3(2Н)-она (5h).
К раствору 19.13 г (72 ммоль) соединения 4h в 140 мл EtOH (95-96%) по каплям прибавили 13 мл (0.24 моль) концентрированной серной кислоты. Смесь перемешивали при температуре 63-66°C в течение 8 ч. После чего добавили 400 мл воды и подвергали реакционную смесь экстракции абсолютным хлороформом (2×40 мл, 1×30 мл). Органические фазы промыли 50 мл раствора Na2CO3(нас.), затем насыщенным раствором соли (2×20 мл) и высушили Na2SO4. Отогнав растворитель в вакууме водоструйного насоса, выпавший осадок перекристаллизовали из петролейного эфира. Выход 17 г (90%) соединения 5h (литературные данные [10]).
Стадия TV. Получение 4-диазо-2,2-диметил-5,5-дифенилдигидрофуран-3(2Н)-она (6h).
К смеси 1 грамма (3.76 ммоль) соединения 5h, 0.435 г (1.35 ммоль) Bu4NBr, 1.2 г (3.9 ммоль) 2,4,6-трис-изопропилбензолсульфанилазида и 0.035 г (0.133 ммоль) 18-краун-6 в 70 мл бензола при интенсивном перемешивании добавили по каплям 50 мл 50% раствора КОН. Смесь поставили перемешиваться при 45°C в течение 4 ч. После завершения реакции органический слой промыли насыщенным раствором соли (2×50 мл), водные вытяжки подвергли экстракции хлористым метиленом (2×50 мл). Объединенные органические фазы промыли водой (5×50 мл) до нейтральной среды и сушили над сульфатом магния. Растворитель отогнали, остаток пропустили через колонку с силикагелем (элюент: петролеиновый эфир/ хлористый метилен в градиентном режиме). Выход: 720 мг (66%) соединения 6h (литературные данные [10]).
Стадия V. Получение 2,2-диметил-4,5-дифенилфуран-3(2Н)-она (1h).
К раствору 58 мг (0.2 ммоль) соединения 6h в 5 мл хлороформа по каплям было добавлено 0.1 мл (1.3 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты. Смесь кипятили 1 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×5 мл) и высушили над K2CO3. После отгонки растворителя получено 52 мг (98%) соединения 1h [11].
Пример 9.
Получение 2,2-диметил-4,5-ди(4-фторфенил)фуран-3(2Н)-она (Схема 17, 1i).
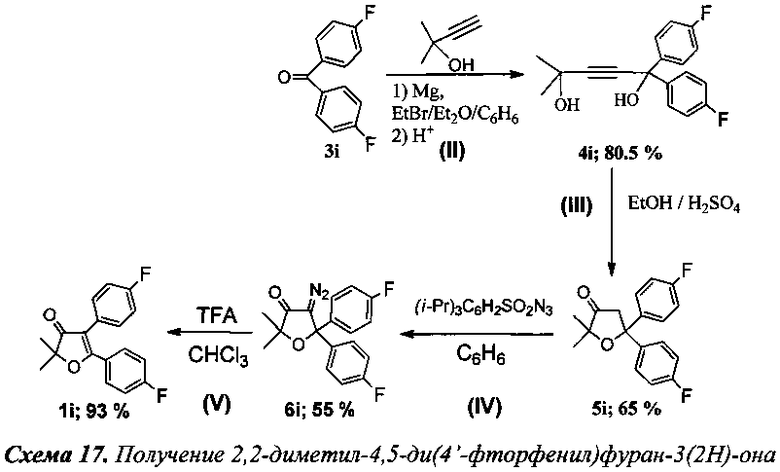
Стадия II. Получение 2-метил-5,5-ди(4-фторфенил)пент-3-ин-2,5-диола (4i).
К раствору EtMgBr, приготовленного из 4.3 г (180 ммоль) магния и 19.6 г (180 ммоль) этилбромида, в 75 мл абсолютного диэтилового эфира при охлаждении ледяной бане медленно прибавили по каплям раствор 6.8 мл (70 ммоль) 2-метилбут-3-ин-2-ола в 70 мл абсолютного бензола. Далее смесь кипятили в течение 2 ч, после чего добавили 10.9 г (50 ммоль) 4,4′-дифторбензофенона (3i) и кипятили в течение 7 ч. После охлаждения реакционной смеси в водяной бане через капельную воронку прилили 130 мл раствора соляной кислоты, приготовленного из 100 мл фильтрованной воды и 30 мл 36% соляной кислоты. Органический слой отделили от водного, последний подвергли экстракции абсолютным хлористым метиленом (1×30 мл, 1×20 мл, 2×10 мл). Объединенные органические вытяжки промыли NaHCO3, затем водой и высушили K2CO3. Растворитель отогнали в вакууме, полученный остаток перекристаллизовали из смеси петролейного эфира и хлористого метилена (1.5:1). Выход 12.2 г (80.5%) соединения 4i, т.пл. 95-96°C (литературные данные 95-96°C, [10-11]).
Стадия III. Получение 2,2-диметил-5,5-ди(4′-фторфенил)дигидрофуран-3(2Н)-она (5i).
К раствору 6.05 г (20 ммоль) соединения 4i в 40 мл EtOH (95-96%) по каплям прибавили 3.5 мл (64 ммоль) концентрированной серной кислоты. Смесь перемешивали при температуре 63-66°C в течение 8 ч. После чего добавляли 100 мл фильтрованной воды и подвергали реакционную смесь экстракции абсолютным хлороформом (2×30 мл, 1×20 мл). Органические фазы промыли 25 мл раствора Na2CO3(нас.), затем насыщенным раствором соли (2×20 мл) и сушили Na2SO4. Отогнав растворитель в вакууме водоструйного насоса, выпавший осадок перекристаллизовали из петролейного эфира. Выход 3.95 г (65%) соединения 5i, т.пл. 59-60°C (литературные данные 59-60°C [10-11]).
Стадия IV. Получение 4-диазо-2,2-диметил-5,5-ди(4′-фторфенил)дигидрофуран-3(2Н)-она (6i).
К смеси 1 грамма соединения 5i, 0.435 г (1.35 ммоль) Bu4NBr, 1.301 г (4.21 ммоль) 2,4,6-трис(изопропил)бензолсульфанилазида и 0.035 г (0.133 ммоль) 18-краун-6 в 70 мл бензола при интенсивном перемешивании добавили по каплям 70 мл 50% раствора KOH. Смесь поставили перемешиваться при 37°C в течение 4 ч. После завершения реакции органический слой промыли насыщенным раствором соли (2×50 мл), водные вытяжки подвергли экстракции хлористым метиленом (2×50 мл). Объединенные органические фазы промыли водой (5×50 мл) до нейтральной среды и сушили сульфатом магния. Растворитель отогнали, остаток пропустили через колонку с силикагелем (элюент: петролеиновый эфир/ хлористый метилен в градиентном режиме). Выход: 597 мг (55%) соединения 6i, т.пл 113-114°C (литературные данные 113-114°C [10-11]).
Стадия V. Получение 2,2-диметил-4,5-ди(4′-фторфенил)фуран-3(2Н)-она (1i).
К раствору 70 мг (0.2 ммоль) соединения 6i в 5 мл хлороформа по каплям было добавлено 0.1 мл (1.3 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты. Смесь кипятили 6 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×5 мл) и высушили K2CO3. После отгонки растворителя получено 56 мг (93%) соединения 1i [11].
Пример 10.
Получение 2,2-диметил-4,5-ди(4′-метоксифенил)фуран-3(2Н)-она.
(Схема 18, 1j).
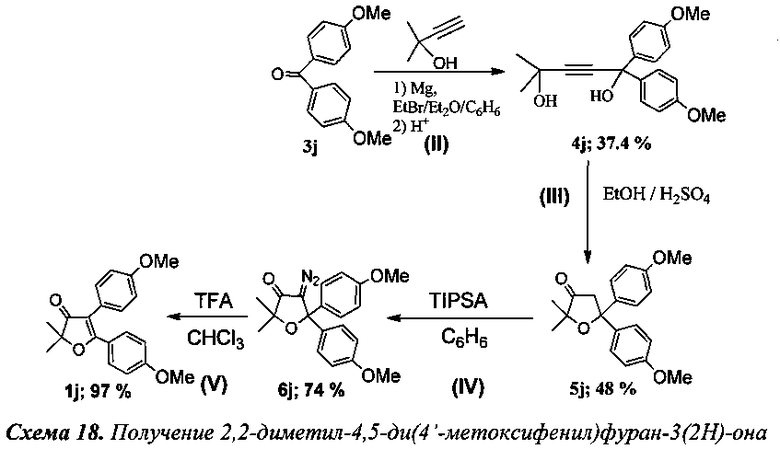
Стадия II. Получение 2-метил-5,5-ди(4′-метоксифенил)пент-3-ин-2,5-диола (4J).
К раствору EtMgBr, приготовленного из 4.3 г (180 ммоль) магния и 19.6 г (180 ммоль) этилбромида в 50 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 6.8 мл (70 ммоль) 2-метилбут-3-ин-2-ола в 70 мл абсолютного бензола. Далее смесь кипятили в течение 2 ч, после чего добавили 12.1 г (50 ммоль) 4,4′-диметоксибензофенона (3j) и кипятили в течение 5 ч. После чего при охлаждении в водяной бане через капельную воронку прилили 130 мл раствора соляной кислоты, приготовленного из 100 мл фильтрованной воды и 30 мл 36% соляной кислоты. Органический слой отделили от водного, последний подвергли экстракции абсолютным хлористым метиленом (1×20 мл, 1×15 мл, 1×10 мл, 1×5 мл). Объединенные органические вытяжки промыли NaClнас (30 мл), Na2CO3(нас) (20 мл), затем еще раз NaCl (20 мл) и оставили сушиться над K2CO3, далее растворитель отогнали в вакууме, полученный остаток перекристаллизовали из смеси петролейного эфира и хлористого метилена (1:1). Выход 6.1 г (37.4%) соединения 4j, т.пл. 111-112°C (литературные данные 112-113°C [10-11]).
Стадия III. Получение 2,2-диметил-5,5-ди(4′-метоксифенил)дигидрофуран-3(2Н)-она (5j).
К раствору 6 г (18 ммоль) соединения 4j в 40 мл ЕtOН (95-96%) по каплям прибавили 3.7 мл (68 ммоль) концентрированной серной кислоты. Раствор быстро приобрел темную окраску, через 20 мин на ТСХ исчезло пятно исходного диола. После чего добавили 70 мл фильтрованной воды. Из смеси экстрагировали абсолютным хлороформом (1×50 мл, 2×20 мл). Органические фазы промыли 25 мл раствора Na2CO3(нас.), затем насыщенным раствором соли (25 мл) и оставили сушиться MgSO4. Отогнали растворитель в вакууме водоструйного насоса, выпавший осадок перекристаллизовали из смеси петролейного эфира и хлористого метилена. Выход 2.9 г (48%) соединения 5j, т.пл. 127-128°C (литературные данные 128-129°C [10-11]).
Стадия IV. Получение 4-диазо-2,2-диметил-5,5-ди(4′-метоксифенил)дигидрофуран-3(2Н)-она (6j).
К смеси 1 грамма (3.1 ммоль) соединения 5j, 0.435 г (1.35 ммоль) Bu4NBr, 1 г (3.24 ммоль) 2,4,6-трис-изопропилбензолсульфанилазида и 0.035 г (0.133 ммоль) 18-краун-6 в 70 мл бензола при интенсивном перемешивании добавили по каплям 50 мл 50% раствора КОН. Смесь поставили перемешиваться при 45°C в течение 4 ч. После завершения реакции органический слой промыли насыщенным раствором соли (2×50 мл), водные вытяжки подвергли экстракции хлористым метиленом (2×50 мл). Объединенные органические фазы промыли водой (5×50 мл) до нейтральной среды и сушили сульфатом магния. Растворитель отогнали, остаток пропустили через колонку с силикагелем (элюент: петролеиновый эфир/ хлористый метилен в градиентном режиме). Выход 808 мг (74%) соединения 6j (литературные данные [10-11]).
Стадия V. Получение 2,2-диметил-4,5-ди(4′-метоксифенил)фуран-3(2Н)-она (1j).
К раствору 70 мг (0.2 ммоль) соединения 6j в 5 мл хлороформа по каплям было добавлено 0.1 мл (1.3 ммоль, 6.5 кратный мольный избыток) трифторуксусной кислоты. Смесь кипятили 6 ч до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×5 мл) и высушили над K2CO3. После отгонки растворителя получено 63 мг (97%) соединения 1j [11].
Таким образом, приведенные результаты апробации заявленного способа получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов общей формулы (1) на примерах 1-10 показывают, что этот способ является универсальным и эффективным 5-6 стадийным методом синтеза соединений этого класса, для выполнения которого в качестве исходных соединений используются коммерчески доступные 1,1-диалкилпроп-2-ин-1-олы и замещенные бензофеноны. Способ не предполагает использования металл-катализируемых реакций, а в качестве ключевой стадии процесса применяется 1,2-нуклеофильная перегруппировка диазокетонов ряда тетрагидрофуранонов, протекающая под действием сильных минеральных или органических кислот с высокими выходами (91-99%), что обеспечивает более высокую эффективность и более низкую себестоимость целевого продукта реакции, чем известные способы получения аналогичных соединений.
Список литературы
1. Song Seok Shin and coworkers, J. Med. Chem., 2004, 47, p. 792.
2. Е.Л. Насонов, Consilium Medicum, 1999, том 1, №5.
3. Ю. Муравьев, В. Лебедева, Российский гастроэнтерологический журнал, 2000, №4.
4. Carol A. Rouzer and Lawrence J. Marnett, Chem. Rev., 2003, 103, p. 2239.
5. Sven Trelle et al., BMJ, 2011, 342, p. 7086.
6. Song Seok Shin and coworkers, Bioorganic & Medicinal Chemistry Letters, 2001, 11, p. 165.
7. Патент KR 20010094519.
8. Патенты US 6,492,416 B1 (прототип), KR 20010111584, KR 20010027342, KR 20000066223, KR 20010094161, KR 20010010728.
9. J. Bunger, J. Stork, K. Stalder, Int. Arch. Occup. Environ. Health, 1996, vol. 69, pp. 33-38.
10. S.A. Malashikhin et. al, Helv. Chim. Acta, 2008, 91, p. 1662.
11. L.L. Rodina et. al., EuroJOC, 2014, 14, pp. 2993-3000.
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ЦИКЛООКСИГЕНАЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2631317C1 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАЗАМЕЩЕННЫХ 4-ДИАЗО-5,5-ДИАЛКИЛ-2,2-ДИАРИЛДИГИДРОФУРАН-3(2Н)-ОНОВ | 2014 |
|
RU2562247C1 |
| СПОСОБ ПОЛУЧЕНИЯ [1,2,3,4]ТЕТРАЗИНО[5,6-е][1,2,3,4]ТЕТРАЗИН-1,3,6,8-ТЕТРАОКСИДА | 2015 |
|
RU2593993C1 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 | 2009 |
|
RU2539979C2 |
| ДИСПИРО 1,2,4-ТРИОКСОЛАНЫ КАК ПРОТИВОМАЛЯРИЙНЫЕ СРЕДСТВА | 2008 |
|
RU2493159C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 | 2009 |
|
RU2531272C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ МОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2664541C2 |
| НОВЫЕ 2',5'-ДИАРИЛСПИРО[ИНДОЛ-3,3'-ПИРРОЛИДИН]-2(1Н)-ОНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2730287C1 |
| ГЕТЕРОАРИЛАМИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2100357C1 |
Изобретение относится к новому способу получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов общей формулы 1, где Alk и Alk1=CH3, CH3CH2 или другие алкильные или циклоалкильные группы, а Ar и Ar1=Ph, p-X-C6H4 (X=H, MeO, MeS, MeSO, MeSO2, Hal) и другие замещенные или незамещенные арильные группы, заключающемуся в шестистадийном синтезе на основе реакций 1,1-диалкилпроп-2-ин-1-олов и замещенных бензофенонов, приводящему к целевым 2,2-диалкил-4,5-диарилфуран-3(2H)-онам, и содержащему в качестве ключевой стадии 1,2-нуклеофильную перегруппировку 4-диазо-2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов, катализируемую органической или минеральной кислотой. Полученные соединения проявляют свойства селективных ингибиторов фермента циклооксигеназы-2 и могут быть использованы в качестве нестероидных противовоспалительных лекарственных препаратов для длительной терапии заболеваний суставов. Технический результат - упрощение процесса и повышение выхода конечного продукта. 10 пр.
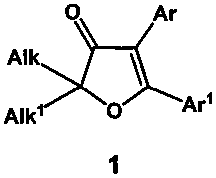
Способ получения 2,2-диалкил-4,5-диарилфуран-3(2H)-онов общей формулы (1)
где Alk и Alk1=CH3, CH3CH2 или другие алкильные или циклоалкильные группы, а Ar и Ar1=Ph, p-X-C6H4 (X=H, MeO, MeS, MeSO, MeSO2, Hal) и другие замещенные или незамещенные арильные группы, многостадийным процессом из 1,1-диалкилпроп-2-ин-1-олов и бензофенонов, отличающийся тем, что введение арильной группы в 4-е положение фуранового цикла осуществляют путем каталитической 1,2-нуклеофильной перегруппировки соответствующих 4-диазо-2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов под действием органической или минеральной кислоты с выходами 91-99%.
| KR 20010111584 A 19.12.2001 | |||
| KR 20010094518 A 01.11.2001 | |||
| L.L.Rodina er al, "Thermolis of 4-Diazotetrahydrofuran-3-ones",Eur.L.Org.Chem.,2014,p.2993-3000 | |||
| ДИЗАМЕЩЕННЫЕ ФУРАНОНЫ, ТИАЗОЛЫ И ПЕНТЕНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЦИКЛООКСИГЕНАЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2131423C1 |

