Изобретение относится к химии органических гетероциклических соединений, а именно к новым азосоединениям на основе пиридоксина и аминофенолов общей формулы (I), ингибирующим образование конечных продуктов гликирования (обладающим антигликирующей активностью):
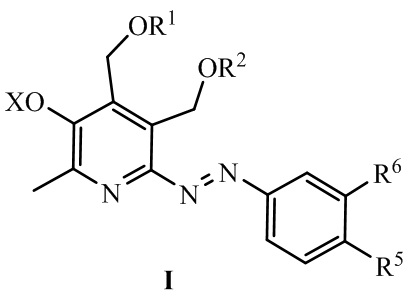
где:
R1 = R2 = H; или R1 и R2 вместе образуют заместитель вида СR3R4, где R3 = H, CH3, R4 = H, CH3, C2H5, C3H7, C5H11, C8H17;
R5=H, OS(O)2CH3, C(O)OAllyl, OH, C(O)ONa;
R6=H, OS(O)2CH3, OAllyl, OH;
X = H, Na и другие фармацевтически приемлемые катионы органической и неорганической природы.
Соединения формулы (I) являются эффективными ингибиторами образования конечных продуктов гликирования (КПГ) и могут найти широкое применение в медицине в области лечения социально-значимых заболеваний, таких как осложнения сахарного диабета, а также атеросклероз, ревматоидный артрит, остеоартрит, нейродегенеративные заболевания, включая болезни Альцгеймера и Паркинсона.
Гликирование (неферментативное гликозилирование, реакция Майяра) – это химическая реакция, при которой карбонильные группы восстанавливающих сахаров связываются с аминогруппами долгоживущих белков, липидов или пептидов, а последующий каскад реакций приводит к образованию КПГ. Процесс гликирования протекает медленно и зависит от уровня и длительности экспозиции глюкозы в организме [Severin F.F. The possible role of protein glycation in the “device of a large biological clock” / F.F. Severin, B.F. Fenyuk, V.N. Skulachev // Biokhimiya. – 2013. – Т.78. – №.9. – С.1331-1336].
Образование и накопление в организме КПГ считается основной причиной развития сосудистых осложнений при сахарном диабете. Все больше данных свидетельствует о том, что с образованием КПГ связаны также нейродегенеративные заболевания (болезнь Альцгеймера и Паркинсона), атеросклероз, ревматоидный артрит и ряд других заболеваний [Nolan C.J. Type 2 diabetes across generations: from pathophysiology to prevention and management / C.J. Nolan, P. Damm, M. Prentki // The Lancet. – 2011. – Т.378. – №.9786. – С.169-181].
Эффекты КПГ можно разделить на рецептор-независимые и рецептор-зависимые, КПГ могут действовать непосредственно на внутриклеточные белки, нарушая их конфигурацию, или взаимодействовать на поверхности клеток с особыми рецепторами для КПГ (рКПГ). Гликирование происходит в течение длительного периода и поэтому затрагивает в основном долгоживущие белки, например, белки базальной мембраны (коллаген, эластин, миелин), белки хрусталика глаза (кристаллины). Нарушение конфигурации структурных белков приводит к функциональным изменениям в стенках сосудов. Гликирование белков базальной мембраны почечных клубочков (коллаген, ламинин, гепаринсульфат) является ключевым фактором развития нефропатии у больных сахарным диабетом [Gavrilova A.O. The role of advanced glycation end-products in patogenesis of diabetic nephropathy / A.O. Gavrilova, A.S. Severina, M.S. Shamhalova, M.V. Shestakova // Diabetes mellitus. – 2022. – Т.24. – №.5. – С.461-469].
Взаимодействие КПГ с мембранными рКПГ запускает несколько внутриклеточных сигнальных путей – активацию фермента NADPH-оксидазы, митоген-активируемой протеинкиназы (МАПК), внеклеточной сигнал-регулируемой киназы, ГТФ-азы. Это приводит к развитию в организме окислительного стресса и увеличению продукции провоспалительных цитокинов [Ramasamy R. Advanced glycation endproducts: from precursors to RAGE: round and round we go / R. Ramasamy, S.F. Yan, A.M. Schmidt // Amino acids. – 2012. – Т.42. – С.1151-1161].
Применение соединений с высокой антигликирующей активностью, ингибирующих образование КПГ в организме, позволит улучшить качество жизни пациентов и снизить риск развития осложнений сахарного диабета (диабетический атеросклероз, нефропатия, нейропатия, ретинопатия, кардиопатия или ангиопатия), а также таких заболеваний, как атеросклероз, ревматоидный артрит, остеоартрит, нейродегенеративные заболевания (включая болезнь Альцгеймера и Паркинсона).
Первым и наиболее изученным веществом, ингибирующим гликирование белков, является аминогуанидин (АГ) [A. Cerami, P.C. Ulrich, M. Brownlee, Pat US 4758583 A, опубл. 19.07.1988]. Это соединение предотвращает формирование КПГ и глюкозо-производных поперечно-сшитых молекул коллагена. Механизм антигликирующего действия аминогуанидина основан на его способности захватывать реакционноспособные дикарбонильные интермедиаты. Однако клинические испытания данного лекарственного средства были остановлены из-за серьезных побочных эффектов.
Также проводились клинические испытания пиридоксамина [R. Khalifah, B.G. Hudson, Pat US 6716858 B1, опубл. 06.04.2004], обладающего антигликирующими свойствами. Результаты второй фазы клинических исследований этого препарата показали недостаточную эффективность и наличие побочных эффектов, из-за чего клинические исследования пиридоксамина были завершены.
Известны ингибиторы образования КПГ на основе азопроизводных фенилсульфокислот, содержащих пиридоксиновый фрагмент [А.А. Спасов, Ю.Г. Штырлин, К.В. Балакин, В.А. Кузнецова, В.И. Петров, А.Д. Стрельник, Патент РФ №2634594 от 01.11.2017; А.А. Спасов, Ю.Г. Штырлин, К.В. Балакин, А.У. Зиганшин, В.А. Кузнецова, В.И. Петров, А.Д. Стрельник, Патент РФ №2628605 от 21.08.2017].
При этом, следует отметить, что описанные выше лекарственные средства, по мнению заявителя, не могут рассматриваться в качестве аналогов к заявленному изобретению вследствие того, что они не совпадают с заявленным соединением по химической структуре, хотя и обладают сходной в целом антигликирующей активностью (совпадают по назначению), сопоставимой с заявленным изобретением в большей или меньшей степени.
Технической проблемой, решаемой заявленным изобретением, является разработка ингибиторов образования КПГ для профилактики и лечения заболеваний, вызванных образованием и накоплением в организме конечных продуктов гликирования, обладающих значительно более высокой антигликирующей активностью по сравнению с лекарственными средствами, вышедшими на стадию клинических исследований
Техническим результатом заявленного технического решения разработка новых соединений общей формулы I, обладающих способностью ингибировать образование конечных продуктов гликирования.
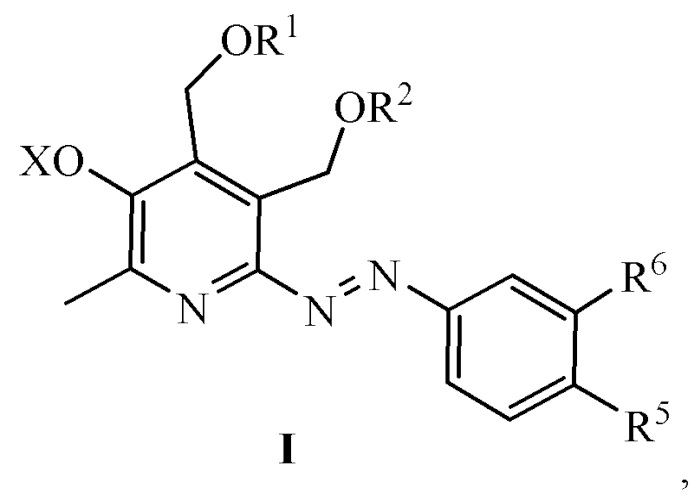
Сущностью заявленного технического решения являются азопроизводные аминофенолов общей формулы (I):
где:
R1 = R2 = H; или R1 и R2 вместе образуют заместитель вида СR3R4, где R3 = H, CH3, R4 = H, CH3, C2H5, C3H7, C5H11, C8H17;
R5 = H, R6 = OS(O)2CH3, OH, или R5 = OS(O)2CH3, OH, R6 = H, или R5 = C(O)OAllyl, C(O)ONa, R6 = OAllyl, OH;
X = H, Na. Азопроизводные аминофенолов формулы (I), обладающие способностью ингибировать образование конечных продуктов гликирования.
Заявленное техническое решение иллюстрируется Фиг.1 и Фиг.2.
На Фиг.1 приведена Таблица 1, в которой представлены значения полумаксимальных концентраций ингибирования (IC50 мкМ/л) соединений формулы I и препарата сравнения (аминогуанидина) в реакции образования КПГ на модельной системе in vitro, основанной на взаимодействии глюкозы и бычьего сывороточного альбумина при температуре 60°С.
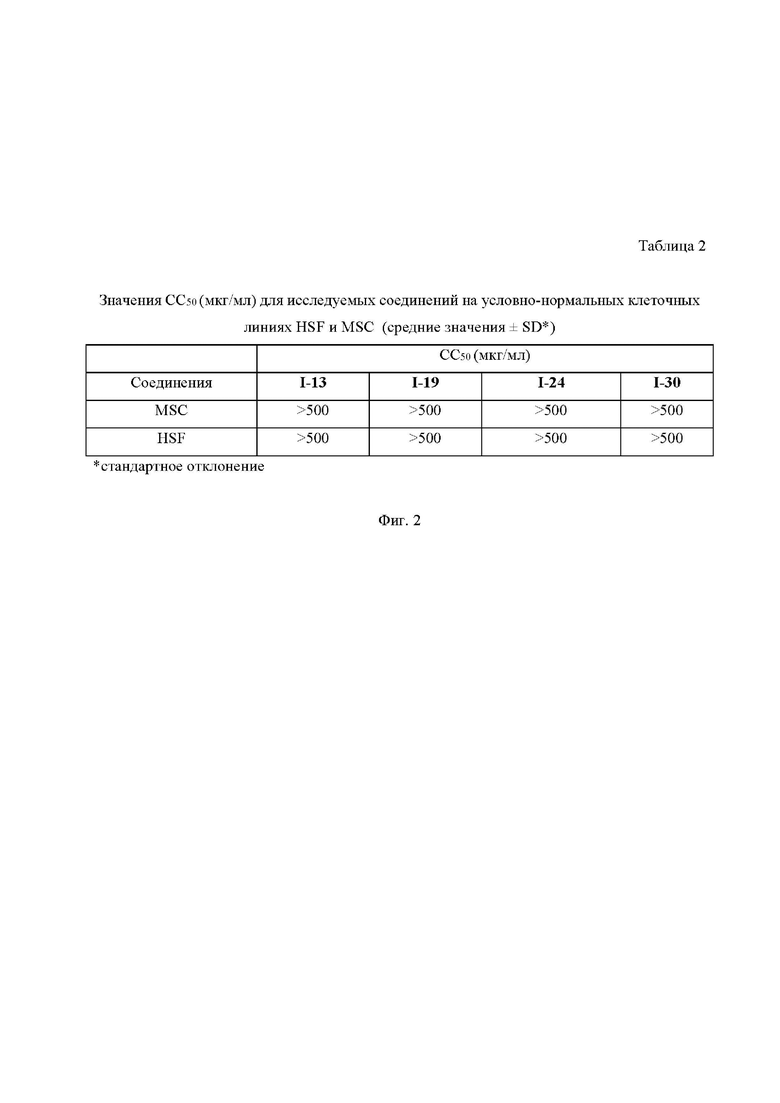
На Фиг.2 приведена Таблица 2, в которой представлены значения полумаксимальной концентрации ингибирования роста (СC50 мкМ/л) условно-нормальных клеток (MSC - мультипотентные стволовые клетки из жировой ткани и HSF – первичные фибробласты кожи человека) некоторыми из исследуемых соединений.
Представленные результаты свидетельствуют о том, что заявляемые соединения не только значительно превосходят препарат сравнения аминогуанидин по эффективности (ингибируют образование КПГ в гораздо более низких концентрациях), но и демонстрируют благоприятный профиль безопасности in vitro.
Далее заявителем приведено осуществление заявленного технического решения.
Заявленные соединения синтезируют согласно нижеприведенным схемам 1-4.
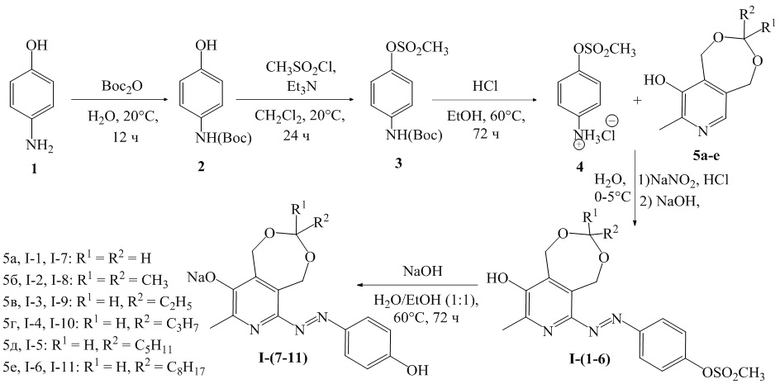
Схема 1
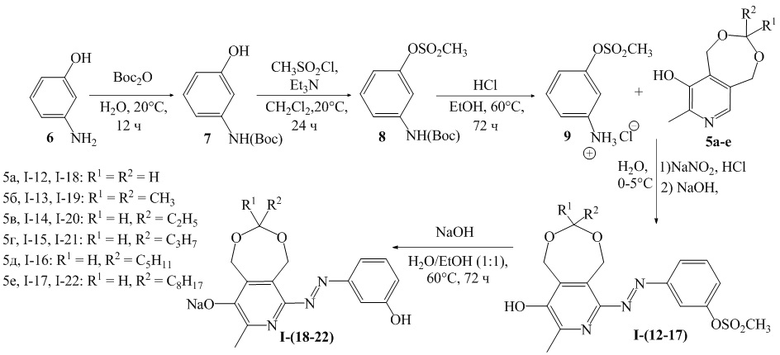
Схема 2
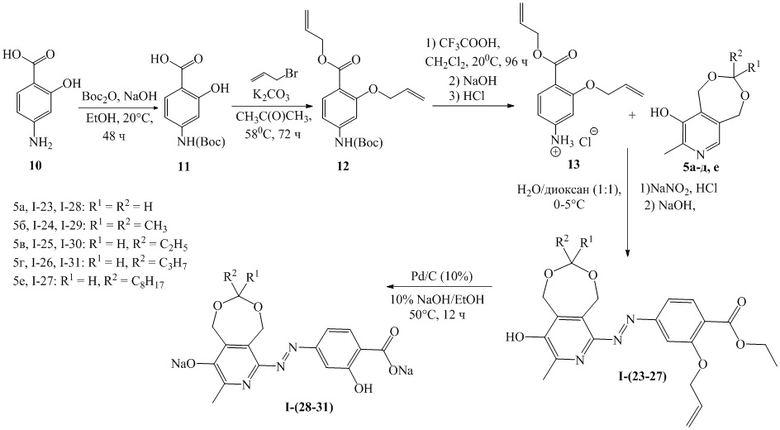
Схема 3
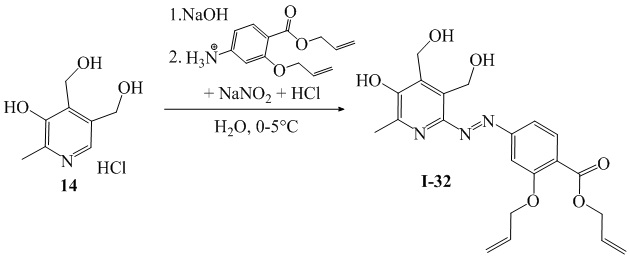
Схема 4
Характеристики новых соединений приведены далее в примерах конкретного выполнения. Структуры полученных соединений подтверждены методами масс-спектрометрии, ЯМР-спектроскопии. Спектры ЯМР регистрировали на приборе Bruker AVANCE-400. Химический сдвиг определялся относительно сигналов остаточных протонов дейтерированных растворителей. Температуры плавления определялись с помощью прибора Stanford Research Systems MPA-100 OptiMelt. Контроль за ходом реакций и чистотой соединений проводили методом ТСХ на пластинах Sorbfil Plates.
Масс-спектры MS были получены на тройном квадрупольном масс-спектрометре c линейной ионной ловушкой AB Sciex QTrap 6500 (AB SCIEX PTE. Ltd., Сингапур) с использованием детектора высокой энергии IonDriveTM и источником ионизации IonDrive Turbo V (газообразный азот распылителя, положительная полярность ионизации, напряжение иглы 5200 В). Запись спектров проводилась в режиме “Q1” с энергией столкновения 10 эВ, потенциалом декластеризации 90 эВ. Образцы с концентрацией анализируемого вещества 1 мкмоль/л готовили путем растворения исследуемых соединений в метаноле (LC-MS, Merck).
Примеры конкретного выполнения заявленного технического решения
Соединения 5а-е были получены по известным литературным методикам из пиридоксина гидрохлорида [Korytnyk W.A. Seven-Membered Cyclic Ketal of Pyridoxol // The Journal of Organic Chemistry. – 1962. – Т.27. – №.10. – С.3724-3726, 82; Sapozhnikov S. New quaternary ammonium pyridoxine derivatives: synthesis and antibacterial activity / N. Shtyrlin, A. Kayumov, A. Zamaldinova, A. Iksanova, Е. Nikitina, Е. Krylova, D. Grishaev, K. Balakin, Y. Shtyrlin // Medicinal Chemistry Research. – 2017. – Т.26. – С.3188-3202].
Пример 1. Получение 4-((9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино[5,6-c] пиридин-6-ил)диазенил)фенил метансульфоната (I-1)
Синтез 4-((трет-бутоксикарбонил)аминофенола ( 2 )
К раствору пара-аминофенола (1) (2.0 г, 18.3 ммоль, 1 экв) в 20 мл воды добавили ди-трет-бутилдикарбонат (4.2 мл, 18.3 ммоль, 1 экв) и перемешивали при комнатной температуре 12 часов. Контроль за ходом реакции осуществлялся с помощью метода тонкослойной хроматографии (элюент – CHCl3/EtOH 9:1). Завершение реакции сопровождалось появлением белого осадка, который отфильтровали и промыли водой (3 × 10 мл), а затем высушили в вакууме. Выход: 3.7 г, 97%, белый порошок. ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.44 (c, 9H, -C(CH3)3); 6.61-6.65 (м, 2H, H-Ar); 7.20 (уш.д, 2H, 3JHH = 7.9 Гц, H-Ar), 9.01 (уш.с, 1H, OH); 9.05 (с, 1H, NH).
Синтез 4-((трет-бутоксикарбонил)амино)фенил метансульфоната ( 3 )
К раствору 4-((трет-бутоксикарбонил)аминофенола (2) (2.9 г, 13.9 ммоль, 1 экв) и триэтиламина (2.9 мл, 20.8 ммоль, 1.5 экв) в 50 мл хлористого метилена по каплям добавили метансульфонилхлорид (1.3 мл, 16.7 ммоль, 1.2 экв) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Контроль за ходом реакции осуществлялся с помощью метода тонкослойной хроматографии (элюент – CHCl3/EtOH 9:1). Полученную реакционную массу промыли водой в делительной воронке (3 × 15 мл), органический слой собрали и отогнали растворитель в вакууме. Выход: 4.8 г, 92%, белый порошок. ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 1.51 (c, 9H, -C(CH3)3); 3.10 (с, 3Н, CH3SO2); 6.60 (уш.с, 1H, NH); 7.19-7.21 (м, 2H, H-Ar); 7.40-7.42 (м, 2H, H-Ar).
Синтез 4-(метилсульфонил)фениламмония хлорида ( 4 )
4-((трет-бутоксикарбонил)амино)фенил метансульфонат (3) (3.6 г, 12.5 ммоль, 1 экв) растворили в 40 мл этилового спирта, к полученному раствору добавили концентрированную (33%) соляную кислоту (3.3 г, 30.0 ммоль, 1 экв. + 2 г изб), реакционную массу перемешивали при температуре 60°С в течение 72 часов. Контроль за ходом реакции осуществлялся с помощью метода тонкослойной хроматографии (элюент – CHCl3/EtOH 9:1). По окончании реакции растворитель отогнали в вакууме. Выход: 3.2 г, 78%, желтый кристаллический порошок. Т.пл. 182°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 3.41 (с, 3H, СH3SO2); 7.42, 7.42 (АВ, 4H, 3JHH = 9.3 Гц, 4H-Ar), 10.00 (уш.с, 3H, NH3+).
Синтез 4-((9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино[5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-1)
К раствору 4-(метилсульфонил)фениламмония хлорида (4) (0.66 г, 2.8 ммоль, 1 экв) в 20 мл воды добавили концентрированную (33%) соляную кислоту (0.78 мл, 8.4 ммоль, 3 экв), раствор охладили до 0-5°C и добавили к нему предварительно охлажденный раствор нитрита натрия (0.21 г, 3.1 ммоль, 1.1 экв) в 5 мл воды. Реакционную массу оставили перемешиваться в течение 5 минут при охлаждении на бане со льдом, затем прилили ее к охлажденному раствору 9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино[5,6-c]пиридина (0.50 г, 2.8 ммоль, 1 экв), и гидроксида натрия (0.39 г, 9.8 ммоль, 3.5 экв) в 30 мл воды. Реакционную массу перемешивали при комнатной температуре в течение 15 минут, после чего нейтрализовали 15% соляной кислотой до рН 6. Осадок отфильтровали, промыли водой (3 × 5 мл) и высушили в вакууме. Выход: 65%, 0.69 г, красный кристаллический порошок. Т.пл. 170°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 2.42 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.98 (c, 4Н, 2CH2); 5.32 (с, 2Н, CH2); 7.53 (уш.с, 2H, 2H-Ar); 7.99 (уш.с, 2H, 2H-Ar); 9.84 (уш.с, 1H, OH). MS-ESI: найдено [M+H]+ 380.0, вычислено для C16H18N3O6S 380.0.
Пример 2. Получение 4-((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-2)
Получали аналогично соединению I-1 с использованием соединения 5б (0.50 г, 2.4 ммоль, 1 экв) вместо 5а. Нейтрализация реакционной массы по окончании реакции проводилась при 0°С. Выход: 80%, 0.78 г, красный кристаллический порошок. Т.пл. 153°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.46 (с, 6H, 2СH3); 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.91 (с, 2Н, CH2); 5.30 (с, 2Н, CH2); 7.55 (уш.с, 2H, 2H-Ar); 8.01 (уш.с, 2H, 2H-Ar); 9.74 (уш.с, 1H, OH). MS-ESI: найдено [M+H]+ 408.3, вычислено для C18H22N3O6S 408.1.
Пример 3. Получение 4-((3-этил-9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-3)
Получали аналогично соединению I-1 с использованием соединения 5в (0.50 г, 2.4 ммоль, 1 экв) вместо 5а. Выход: 83%, 0.81 г, красный кристаллический порошок. Т.пл. 85°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.91 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2); 1.63-1.70 (м, 2H, CH3CH2); 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.77, 5.10 (AB, 2Н, 2JHH = 15.2 Гц, CH2); 4.92 (т, 1Н, 3JHH=5.6 Гц, CHC2H5); 5.12, 5.50 (AB, 2Н, 2JHH = 15.8 Гц, CH2); (д, 2H, 3JHH = 8.3 Гц, 2H-Ar); 7.97 (д, 2H, 3JHH = 7.8 Гц, 2H-Ar); 9.85 (уш.с, 1H, OH). MS-ESI: найдено [M+H]+ 408.4, вычислено для C18H22N3O6S 408.1.
Пример 4. Получение 4-((9-гидрокси-8-метил-3-пропил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-4)
Получали аналогично соединению I-1 с использованием соединения 5г (0.50 г, 2.2 ммоль, 1 экв) вместо 5а. Выход: 85%, 0.84 г, красный кристаллический порошок. Т.пл. 156°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.85 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2); 1.34-1.43 (м, 2H, CH3CH2); 1.60-1.68 (м, 2H, CH3CH2CH2); 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.77, 5.10 (AB, 2Н, 2JHH = 15.6 Гц, CH2); 5.1 (т, 1Н, 3JHH=5.6 Гц, CHC3H7); 5.12, 5.50 (AB, 2Н, 2JHH = 15.4 Гц, CH2); 7.52 (д, 2H, 3JHH = 8.3 Гц, 2H-Ar); 7.97 (уш.с, 2H, 2H-Ar); 9.90 (с, 1H, OH). MS-ESI: найдено [M+H]+ 422.3, вычислено для C19H24N3O6S 422.3.
Пример 5. Получение 4-((9-гидрокси-8-метил-3-пентил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-5)
Получали аналогично соединению I-1 с использованием соединения 5д (0.50 г, 2.0 ммоль, 1 экв) вместо 5а. Продукт промыли 10 мл петролейного эфира при температуре 70°С, осадок отфильтровали и высушили в вакууме. Выход: 75%, 0.70 г, красный кристаллический порошок. Т.пл. 121°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.88 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2); 1.26-1.41 (м, 6H, 3CH2); 1.61-1.69 (м, 2H, CH2); 2.41 (с, 3H, СH3-Pyr); 3.47 (с, 3H, СH3SO2); 4.80, 5.14 (AB, 2Н, 2JHH = 15.6 Гц, CH2); 5.01 (уш.с, 1Н, CHC5H11); 5.15, 5.55 (AB, 2Н, 2 JHH = 15.1 Гц, CH2); 7.55 (уш.с, 2H, H-Ar); 8.02 (уш.с, 2H, H-Ar); 9.82 (уш.с, 1H, OH). MS-ESI: найдено [M+H]+ 450.2, вычислено для C21H28N3O6S 450.2.
Пример 6. Получение 4-((9-гидрокси-8-метил-3-октил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-6)
Получали аналогично соединению I-1 с использованием соединения 5е (0.50 г, 1.7 ммоль, 1 экв) вместо 5а. Продукт очищали перекристаллизацией из метанола. Выход: 58%, 0.49 г, красный кристаллический порошок. Т.пл. 110°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.85 (т, 3Н, 3JHH = 6.5 Гц, CH3CH2); 1.18-1.39 (м, 12H, 6CH2); 1.60-1.69 (м, 2H, CH2); 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.76, 5.07 (AB, 2Н, 2JHH = 15.7 Гц, CH2); 5.01 (т, 1Н, 3JHH=5.6 Гц, CHC8H17); 5.11, 5.49 (AB, 2Н, 2 JHH = 15.4 Гц, CH2); 7.52 (д, 2H, 3JHH = 8.8 Гц, 2H-Ar); 7.96 (д, 2H, 3JHH = 8.6 Гц, 2H-Ar); 10.18 (уш.с, 1H, OH). MS-ESI: найдено [M+H]+ 492.2, вычислено для C24H34N3O6S 492.2.
Пример 7. Получение 3-((9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино[5,6-c] пиридин-6-ил)диазенил)фенил метансульфоната (I-12)
Синтез 3-((трет-бутоксикарбонил)аминофенола (7)
Получали аналогично соединению 2 с использованием мета-аминофенола (6), реакция проходила 48 часов. Выход: 3.4 г, 90%, белый порошок. ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.46 (c, 9H, -C(CH3)3); 6.34 (дд, 1H, 3JHH = 8.0 Гц, 4JHH = 1.9 Гц, H-Ar); 6.82 (д, 1H, 3JHH = 8.0 Гц, H-Ar); 6.99 (т, 1H, 3JHH = 8.0 Гц, H-Ar); 9.01 (уш.с, OH, 1H); 9.27 (с, NH, 1H).
Синтез 3-((трет-бутоксикарбонил)амино)фенил метансульфоната (8)
Получали аналогично соединению 3 с использованием 3-((трет-бутоксикарбонил)аминофенола (7). Выход: 4.6 г, 87%, желтое масло. ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 1.51 (c, 9H, -C(CH3)3); 3.14 (с, 3Н, CH3SO2); 6.67 (уш.с, 1H, NH); 6.95 (ддд, 1H, 3JHH = 8.1 Гц, 4JHH = 2.0 Гц, 4JHH = 1.0 Гц, H-Ar); 7.20 (уш.д, 1H, 3JHH = 8.1 Гц, H-Ar); 7.30 (дд, 1H, 3JHH = 8.1 Гц, 3JHH = 8.1 Гц, H-Ar); 7.48 (уш.дд, 1H, 4JHH = 2.0 Гц, 4JHH = 2.0 Гц, H-Ar).
Синтез 3-(метилсульфонил)фениламмония хлорида (9)
Получали аналогично соединению 4 с использованием 3-((трет-бутоксикарбонил)амино)фенилметан сульфоната (8). Выход: 3.1 г, 75%, желтый кристаллический порошок. ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 3.42 (с, 3H, СH3SO2); 7.16-7.22 (м, 3H, 3H-Ar); 7.46-7.50 (м, 1H, H-Ar), 8.64 (уш.с, 3H, NH3+). Т.пл. 150°С (с разложением).
Синтез 3-((9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино[5,6-c]пиридин -6-ил)диазенил)фенил метансульфоната (I-12)
Получали аналогично соединению I-1 с использованием соединения 9 вместо соединения 4. Выход: 76%, 0.83 г, кристаллический порошок красного цвета. Т.пл. 166°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.90 (с, 2Н, CH2); 4.97 (с, 2Н, CH2); 5.30 (с, 2Н, CH2); 7.42 (д, 1H, 3JHH = 7.3 Гц, H-Ar); 7.66 (т, 1H, 3JHH = 8.1 Гц, H-Ar); 7.77 (с, 1H, H-Ar); 7.87 (д, 1H, 3JHH = 7.9 Гц, H-Ar). MS-ESI: найдено [M+H]+ 380.0, вычислено для C16H17N3O6S 380.0.
Пример 8. Получение 3-((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-13)
Получали аналогично соединению I-1 с использованием соединения 9 вместо соединения 4 и соединения 5б (0.50 г, 2.4 ммоль, 1 экв) вместо соединения 5а. Выход: 77%, 0.78 г, красный кристаллический порошок. Т.пл. 149°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.46 (с, 6H, 2СH3); 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.92 (с, 2Н, CH2); 5.31 (с, 2Н, CH2); 7.45-8.00 (м, 4H, 4H-Ar); 9.78 (с, 1H, OH). MS-ESI: найдено [M+H]+ 408.3, вычислено для C18H22N3O6S 408.3.
Пример 9. Получение 3-((3-этил-9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-14)
Получали аналогично соединению I-1 с использованием соединения 9 вместо соединения 4 и соединения 5в (0.50 г, 2.4 ммоль, 1 экв) вместо соединения 5а. Выход: 81%, 0.82 г, красный кристаллический порошок. Т.пл. 157°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.91 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2); 1.63-1.70 (м, 2H, CH3CH2); 2.41 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.75, 5.10 (AB, 2Н, 2JHH = 15.5 Гц, CH2); 4.92 (т, 1Н, 3JHH = 5.5 Гц, CHC2H5); 5.06, 5.49 (AB, 2Н, 2JHH = 15.3 Гц, CH2); 7.43 (уш.с, 1H, H-Ar); 7.65 (т, 1H, 3JHH = 7.8 Гц, H-Ar); 7.87 (с, 1H, H-Ar); 7.86-7.90 (м, 1H, H-Ar); 9.90 (уш.с, 1H, ОH). MS-ESI: найдено [M+H]+ 408.4, вычислено для C18H22N3O6S 408.3.
Пример 10. Получение 3-((9-гидрокси-8-метил-3-пропил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-15)
Получали аналогично соединению I-1 с использованием соединения 9 вместо соединения 4 и соединения 5г (0.50 г, 2.2 ммоль, 1 экв) вместо 5а. Выход: 64%, 0.62 г, красный кристаллический порошок. Т.пл. 110°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.92 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2CH2); 1.34-1.43 (м, 2H, CH3CH2CH2); 1.60-1.68 (м, 2H, CH3CH2CH2); 2.42 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.79 (с, 1Н, CH2); 5.01 (с, 1Н, CHC3H7); 5.12 (с, 2Н, CH2); 5.54 (с, 1Н, CH2); 7.46-8.02 (м, 4H, 4H-Ar); 9.89 (уш.с, 1H, ОH). MS-ESI: найдено [M+H]+ 422.3, вычислено для C19H24N3O6S 422.2.
Пример 11. Получение 3-((9-гидрокси-8-метил-3-пентил-1,5-дигидро-[1,3] диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-16)
Получали аналогично соединению I-1 с использованием соединения 9 вместо соединения 4 и соединения 5д (0.50 г, 2.0 ммоль, 1 экв) вместо соединения 5а. Продукт промыли 10 мл петролейного эфира при температуре 70°С, осадок отфильтровали и высушили в вакууме. Выход: 52%, 0.47 г, красный кристаллический порошок. Т.пл. 112°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.88 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2CH2); 1.26-1.41 (м, 6H, 3CH2); 1.62-1.69 (м, 2H, CH2); 2.42 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.79 (уш.с, 1Н, CH2); 5.01 (уш.с, 1Н, CHC5H11); 5.12 (уш.с, 2Н, CH2); 5.54 (уш.с, 1Н, CH2); 7.48-8.02 (м, 4H, 4H-Ar); 9.89 (уш.с, 1H, ОH). MS-ESI: найдено [M+H]+ 450.2, вычислено для C21H28N3O6S 450.2.
Пример 12. Получение 3-((9-гидрокси-8-метил-3-октил-1,5-дигидро-[1,3] диоксепино [5,6-c]пиридин-6-ил)диазенил)фенил метансульфоната (I-17)
Получали аналогично соединению I-1 с использованием соединения 9 вместо соединения 4 и соединения 5е (0.50 г, 1.7 ммоль, 1 экв) вместо 5а. Продукт очищали перекристаллизацией из метанола. Выход: 57%, 0.48 г, красный кристаллический порошок. Т.пл. 103°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.88 (т, 3Н, 3JHH = 7.5 Гц, CH3CH2CH2); 1.21-1.39 (м, 12H, 6CH2); 1.60-1.70 (м, 2H, CH2); 2.42 (с, 3H, СH3-Pyr); 3.46 (с, 3H, СH3SO2); 4.76 (уш.с, 1Н, CH2); 4.99 (с, 1Н, CHC8H17); 5.11 (уш.с, 2Н, CH2); 5.50 (уш.с, 2Н, CH2); 7.40-8.01 (м, 4H, 4H-Ar); 9.91 (уш.с, 1H, ОH). MS-ESI: найдено [M+H]+ 492.3, вычислено для C24H33N3O6S 492.2.
Пример 13. Получение 6-((4-гидроксифенил)диазенил)-8-метил-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-9-олата натрия (I-7)
Азопроизводное I-1 (0.36 г, 0.9 ммоль, 1 экв) растворили в 20 мл смеси вода/этанол (1:1), к раствору добавили гидроксид натрия (0.11 г, 2.7 ммоль, 3 экв) и перемешивали реакционную массу при температуре 60°С в течение 72 часов. Для выделения продукта реакционную смесь упарили в 2 раза, затем нейтрализовали 15% соляной кислотой до рН = 6-7. Выпавший осадок отфильтровали, промыли водой (3 × 5 мл) и высушили в вакууме. К полученному в протонированной форме продукту добавили 15 мл воды и раствор 1 экв. гидроксида натрия в 5 мл воды. Полученный раствор натриевой соли I-7 высушили в вакууме. Продукт очищали перекристаллизацией из изопропанола. Выход: 77%, 0.24 г, красный кристаллический порошок. Т.пл. 111°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 2.41 (с, 3H, СH3-Pyr); 4.99 (с, 2Н, CH2); 5.00 (с, 2Н, CH2); 5.29 (с, 2Н, CH2); 6.97 (д, 2H, 3JHH = 8.7 Гц, 2H-Ar); 7.86 (д, 2H, 3JHH = 8.7 Гц, 2H-Ar); 10.59 (уш.с, 1H, OH). MS-ESI: найдено [M-Na+H]+ 302.3, вычислено для C15H16N3O4 302.3.
Пример 14. Получение 6-((4-гидроксифенил)диазенил)-3,3,8-триметил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-8)
Получали аналогично соединению I-7 с использованием соединения I-2 (0.65 г, 1.5 ммоль, 1 экв) вместо I-1. Вместо нейтрализации соляной кислотой реакционную массу высушили в вакууме. К получившейся массе добавили 10 мл системы этанол/ацетон (7:3), нерастворившийся осадок отфильтровали, фильтрат высушили в вакууме. Полученный продукт очистили перекристаллизацией из смеси изопропанол/H2O (9:1). Выход: 65%, 0.30 г, красный кристаллический порошок. Т.пл. 188°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.40 (с, 6H, 2СH3); 2.13 (с, 3H, СH3-Pyr); 4.70 (с, 2Н, CH2); 5.19 (с, 2Н, CH2); 6.77 (д, 2H, 3JHH = 8.7 Гц, 2H-Ar); 7.53 (д, 2H, 3JHH = 8.7 Гц, 2H-Аr); 9.74 (уш.с, 1H, OH). ЯМР 13С {H} (100 МГц, D2О), δ, м.д: 23.91 (с, СH3.); 24.69 (с, СH3.); 59.87 (с, СH2); 61.62 (с, СH2); 104.24 (с, С); 110.36 (с, Car.); 113.80 (с, Car.); 119.56 (с, Car.); 130.93 (с, Car.); 133.78 (с, Car.); 135.03 (с, Car.); 144.52 (с, Car.); 149.51 (с, Car.); 154.83 (с, Car.); 161.02 (с, Car.); 164.76 (с, Car.). MS-ESI: найдено [M-Na+H]+ 330.3, вычислено для C17H20N3O4 330.3.
Пример 15. Получение 3-этил-6-((4-гидроксифенил)диазенил)-8-метил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-9)
Получали аналогично соединению I-7 с использованием соединения I-3 (0.71 г, 1.7 ммоль, 1 экв) вместо I-1. Продукт очищали методом колоночной хроматографии на силикагеле (элюент – изопропанол). Выход: 52%, 0.29 г, красный кристаллический порошок. Т.пл. 138°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.91 (т, 3Н, 3JHH = 7.5 Гц, CH3CH2); 1.63-1.70 (м, 2H, CH3CH2); 2.41 (с, 3H, СH3-Pyr); 4.78, 5.13 (AB, 2Н, 2JHH = 15.7 Гц, CH2); 5.00 (т, 1Н, 3JHH=5.5 Гц, CHC2H5); 5.07, 5.47 (AB, 2Н, 2JHH = 15.6 Гц, CH2); 6.92 (д, 2H, 3JHH = 8.4 Гц, 2H-Ar); 7.80 (д, 2H, 3JHH = 8.4 Гц, 2H-Ar); 10.27 (с, 1H, OH). MS-ESI: найдено [M-Na+H]+ 330.3, вычислено для C17H20N3O4 330.3.
Пример 16. Получение 6-((4-гидроксифенил)диазенил)-8-метил-3-пропил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-10)
Получали аналогично соединению I-7 с использованием соединения I-4 (0.72 г, 1.6 ммоль, 1 экв) вместо I-1. Продукт очищали методом колоночной хроматографии на силикагеле (элюент – изопропанол). Выход: 50%, 0.28 г, красный кристаллический порошок. Т.пл. 91°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.92 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2CH2); 1.34-1.43 (м, 6H, СН3CH2CH2); 1.60-1.67 (м, 2H, CH3CH2CH2); 2.41 (с, 3H, СH3-Pyr); 4.80, 5.14 (AB, 2Н, 2JHH = 15.5 Гц, CH2); 5.01 (т, 1Н, 3JHH=5.6 Гц, CHC3H7); 5.15, 5.55 (AB, 2Н, 2JHH = 15.5 Гц, CH2); 7.55 (д, 2H, 3JHH = 8.8 Гц, 2H-Ar); 8.02 (д, 3JHH = 8.8 Гц, 2H, 2H-Ar); 9.82 (с, 1H, OH). ЯМР 13С {H} (100 МГц, D2О), δ, м.д: 13.83 (с, СH3); 16.89 (с, СH2); 17.59 (с, СH2); 34.19 (с, СH3); 60.98 (с, СH2); 62.54 (с, СH2); 104.63 (с, С); 115.24 (с, Car.); 116.41 (с, Car.); 125.56 (с, Car.); 126.20 (с, Car.); 133.42 (с, Car.); 141.78 (с, Car.); 143.84 (с, Car.); 145.46 (с, Car.); 145.85 (с, Car.); 150.63 (с, Car.). MS-ESI: найдено [M-Na+H]+ 344.3, вычислено для C18H22N3O4 344.3.
Пример 17. Получение 6-((4-гидроксифенил)диазенил)-8-метил-3-октил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-11)
Получали аналогично соединению I-7 с использованием соединения I-6 (0.37 г, 0.74 ммоль, 1 экв) вместо I-1. Выход: 24%, 0.08 г, красный кристаллический порошок. ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.86 (т, 3Н, 3JHH = 6.7 Гц, CH3CH2-); 1.14-1.41 (м, 12H, 6СH2); 1.53-1.73 (м, 2H, CH2); 2.50 (с, 3H, СH3-Pyr); 4.83, 5.13 (AB, 2Н, 2JHH = 16.0 Гц, CH2); 5.02 (т, 1Н, 3JHH=5.3 Гц, CHC8H17); 5.13, 5.46 (AB, 2Н, 2JHH = 15.4 Гц, CH2); 6.97 (д, 2H, 3JHH = 8.6 Гц, 2H-Ar); 7.86 (д, 3JHH = 8.6 Гц, 2H, 2H-Ar); 10.59 (с, 1H, OH). MS-ESI: найдено [M-Na+2H]+ 414.3, вычислено для C23H30N3NaO4 414.2.
Пример 18. Получение 6-((3-гидроксифенил)диазенил)-8-метил-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-9-олата натрия (I-18)
Получали аналогично соединению I-7 с использованием соединения I-12 (0.73 г, 1.8 ммоль, 1 экв) вместо I-1. Выход: 29%, 0.18 г, красный кристаллический порошок. Т.пл. 100°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 2.42 (с, 3H, СH3-Pyr); 4.98 (с, 4Н, 2CH2); 5.30 (с, 2Н, CH2); 7.11-7.48 (м, 3H, 3H-Ar); 9.73 (с, 1H, OH).
Пример 19. Получение 6-((3-гидроксифенил)диазенил)-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-19)
Получали аналогично соединению I-7 с использованием соединения I-13 (0.66 г, 1.6 ммоль, 1 экв) вместо I-1. Вместо нейтрализации соляной кислотой реакционную массу высушили в вакууме. К получившейся массе добавили 10 мл системы этанол/ацетон (7:3), нерастворившийся осадок отфильтровали, фильтрат высушили в вакууме. Полученный продукт очистили перекристаллизацией из этанола. Выход: 65%, 0.69 г, красный кристаллический порошок. Т.пл. 91°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.40 (с, 6H, 2СH3); 2.14 (с, 3H, СH3-Pyr); 4.70 (с, 2Н, CH2); 5.21 (с, 2Н, CH2); 6.63-6.68 (м, 1H, H-Ar); 7.08-7.20 (м, 3H, 3H-Ar); 8.34 (с, 1H, OH). ЯМР 13С {H} (100 МГц, D2О), δ, м.д: 19.54 (с, СH3.); 23.91 (с, СH3.); 59.97 (с, СH2); 61.50 (с, СH2); 104.24 (с, С); 110.36 (с, Car.); 113.80 (с, Car.); 119.56 (с, Car.); 130.93 (с, Car.); 133.78 (с, Car.); 135.03 (с, Car.); 144.52 (с, Car.); 149.51 (с, Car.); 154.83 (с, Car.); 161.02 (с, Car.); 164.76 (с, Car.). MS-ESI: найдено [M-Na+H]+ 330.3, вычислено для C17H20N3O4 330.3.
Пример 20. Получение 3-этил-6-((3-гидроксифенил)диазенил)-8-метил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-20)
Получали аналогично соединению I-7 с использованием соединения I-14 (0.75 г, 1.8 ммоль, 1 экв) вместо I-1. Продукт промыли 10 мл петролейного эфира при температуре 70°С, осадок отфильтровали и высушили в вакууме. Выход: 72%, 0.44 г, красный кристаллический порошок. Т.пл. 105°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.91 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2); 1.61-1.72 (м, 2H, CH3CH2); 2.43 (с, 3H, СH3-Pyr); 4.77, 5.10 (AB, 2Н, 2JHH = 15.7 Гц, CH2); 4.93 (т, 1Н, 3JHH=5.6 Гц, CHC2H5); 5.10, 5.47 (AB, 2Н, 2JHH = 15.1 Гц, CH2); 6.90 (с, 1H, H-Ar); 7.24 (с, 1H, H-Ar); 7.32-7.39 (м, 2H, 2H-Ar); 9.83 (уш.с, 1H, OH). MS-ESI: найдено [M-Na+H]+ 330.3, вычислено для C17H20N3O4 330.3.
Пример 21. Получение 6-((3-гидроксифенил)диазенил)-8-метил-3-пропил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-21)
Получали аналогично соединению I-7 с использованием соединения I-15 (0.55 г, 1.3 ммоль, 1 экв) вместо I-1. Выход: 71%, 0.31 г, красный кристаллический порошок. Т.пл. 88°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.92 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2); 1.32-1.43 (м, 6H, 3CH2); 1.59-1.69 (м, 2H, CH2); 2.46 (с, 3H, СH3-Pyr); 4.79, 5.12 (AB, 2Н, 2JHH = 15.9 Гц, CH2); 5.01 (т, 1Н, 3JHH=5.4 Гц, CHC3H7); 5.12, 5.47 (AB, 2Н, 2 JHH = 15.5 Гц, CH2); 6.90-6.97 (м, 1H, H-Ar); 7.26 (с, 1H, H-Ar); 7.35-7.40 (м, 2H, 2H-Ar); 9.87 (уш.с, 1H, OH). MS-ESI: найдено [M-Na+H]+ 344.3, вычислено для C18H22N3O4 344.3.
Пример 22. Получение 6-((3-гидроксифенил)диазенил)-8-метил-3-октил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-9-олата натрия (I-22)
Получали аналогично соединению I-7 с использованием соединения I-17 (0.36 г, 0.7 ммоль, 1 экв) вместо I-1. Выход: 43%, 0.13 г, красный кристаллический порошок. ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.86 (т, 3Н, 3JHH = 7.4 Гц, CH3CH2CH2); 1.20-1.40 (м, 12H, 6CH2); 1.59-1.69 (м, 2H, CH2); 2.42 (с, 3H, СH3-Pyr); 4.76, 5.10 (AB, 2Н, 2JHH = 15.6 Гц, CH2); 4.99 (т, 1Н, 3JHH=5.4 Гц, CHC8H17); 5.10, 5.46 (AB, 2Н, 2 JHH = 15.3 Гц, CH2); 6.87-6.94 (м, 1H, H-Ar); 7.23 (с, 1H, H-Ar); 7.31-7.40 (м, 2H, 2H-Ar); 9.82 (уш.с, 1H, OH). MS-ESI: найдено [M-Na+H]+ 412.2, вычислено для C23H32N3O4 412.2.
Пример 23. Получение 2-(аллилокси)-4-((9-гидрокси-8-метил-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-6-ил)диазенил)аллилбензоата (I-23)
Синтез 4-((трет-бутоксикарбонил)амино)-2-гидроксибензойной кислоты (11)
Раствор гидроксида натрия (1.52 г, 38.0 ммоль, 2 экв) в 15 мл воды и раствор ди-трет-бутилдикарбоната (8.28 г, 38.0 ммоль, 2 экв) в 30 мл этанола добавили к раствору 4-аминосалициловой кислоты (10) (3.00 г, 19.0 ммоль, 1 экв) в 15 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Далее раствор упарили до 7 мл в вакууме и нейтрализовали 1.0 М соляной кислотой (9-15 мл) до рН = 3. Осадок отфильтровали, промыли водой и высушили в вакууме. Выход: 84%, 4.2 г, белый порошок. ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 1.47 (с, 9Н, С(СН3)3); 6.98 (д, 1H, 3JHH = 8.6 Гц, H-Ar); 7.12 (с, 1Н, H-Ar); 7.64 (д, 1H, 3JHH = 8.8 Гц, H-Ar); 9.73 (с, 1Н, NH); 11.46 (уш.с, 1Н, ОН); 13.62 (уш.с, 1Н, ОН).
Синтез 2-(аллилокси)-4-((трет-бутоксикарбонил)амино)аллилбензоата (12)
К раствору соединения 11 (4.2 г, 16.6 ммоль, 1 экв) в 20 мл ацетона добавили аллилбромид (6.0 г, 49.8 ммоль, 3 экв) и K2CO3 (6.9 г, 49.8 ммоль, 3 экв). Реакционную массу перемешивали при кипячении (58°С) с обратным холодильником в течение 72 часов. Затем раствор отфильтровали, осадок промыли 10 мл ацетона, фильтрат собрали и высушили в вакууме. Выход: 98%, 5.5 г, желтое масло. ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 1.51 (с, 9Н, С(СН3)3); 4.60-4.64 (м, 2Н, СН2); 4.76-4.79 (м, 2Н, СН2); 5.22-5.27 (м, 1Н, СН2); 5.27-5.31 (м, 1Н, СН2); 5.37-5.43 (м, 1Н, СН2); 5.50-5.57 (м, 1Н, СН2); 5.97-6.11 (м, 2Н, 2СН); 6.72 (дд, 1H, 3JHH = 8.5 Гц, 4JHH = 2.0 Гц, H-Ar); 6.76 (с, 1Н, H-Ar); 7.37 (с, 1Н, NH); 7.8 (д, 1H, 3JHH = 8.5 Гц, H-Ar).
Синтез 3-(аллилокси)-4-((аллилокси)карбонил)бензиламмоний хлорида (13)
К раствору соединения 12 (4.7 г, 14.1 ммоль, 1 экв) в 40 мл хлористого метилена добавили трифторуксусную кислоту (5.8 мл, 76.0 ммоль, 5.4 экв). Реакционную массу перемешивали 96 часов при комнатной температуре, затем перенесли на баню со льдом и нейтрализовали 10% раствором гидроксида натрия (2.8 г, 70.5 ммоль, 5 экв) при охлаждении и интенсивном перемешивании до рН = 7. Полученный раствор промыли водой в делительной воронке, органический слой собрали и отогнали растворитель в вакууме. Полученный продукт растворили в 30 мл этанола, добавили 33% соляную кислоту (2 мл, 21.0 ммоль, 1.5 экв) и отогнали растворитель. Сухой остаток растворили в 50 мл воды при кипячении, раствор охладили. Выпавший осадок отфильтровали и высушили в вакууме. Выход: 69%, 2.3 г, желтый кристаллический порошок. Т.пл. 84°С. ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 4.54-4.58 (м, 2Н, СН2); 4.74-4.77 (м, 2Н, СН2); 5.21-5.26 (м, 1Н, СН2); 5.26-5.32 (м, 1Н, СН2); 5.36-5.43 (м, 1Н, СН2); 5.50-5.57 (м, 1Н, СН2); 5.97-6.11 (м, 2Н, 2СН); 6.18-6.20 (м, 1H, H-Ar); 6.24 (дд, 1H, 3JHH = 8.6 Гц, 4JHH = 2.1 Гц, H-Ar).
Синтез 2-(аллилокси)-4-((9-гидрокси-8-метил-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)аллилбензоата (I-23)
Раствор соединения 13 (0.74 г, 2.8 ммоль, 1 экв) и концентрированной (33%) соляной кислоты (0.79 мл, 8.4 ммоль, 3 экв) в 100 мл смеси диоксан/вода (1:1) охладили до 0-5°C и добавили к нему предварительно охлажденный раствор нитрита натрия (0.21 г, 3.1 ммоль, 1.1 экв) в 5 мл воды. Реакционную массу перемешивали в течение 5 минут, затем прилили ее к охлажденному раствору соединения 5а (0.50 г, 2.8 ммоль, 1 экв) и гидроксида натрия (0.39 г, 9.8 ммоль, 3.5 экв) в 30 мл воды. Реакционную массу перемешивали при комнатной температуре в течение 15 минут, после чего нейтрализовали 15% соляной кислотой до рН 6-7. Осадок отфильтровали, промыли водой (3 × 5 мл) и высушили в вакууме. Выход: 76%, 0.89 г, красный кристаллический порошок. Т.пл. 125°С (с разложением). ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 1.51 (с, 6Н, 2CH3); 2.39 (с, 3H, СH3-Pyr); 4.68 (д, 2H, 3JHH=4.2 Гц, СH2); 4.77 (c, 2Н, CH2); 4.80 (с, 2H, СH2); 5.09 (c, 2Н, CH2); 5.27 (д, 1H, 3JHH=10.4 Гц, СH2); 5.35 (д, 1H, 3JHH=10.6 Гц, СH2); 5.42 (д, 1H, 3JHH=17.1 Гц, СH2); 5.58 (д, 1H, 3JHH=17.2 Гц, СH2); 5.98-6.16 (м, 2Н, 2СН); 6.87 (д, 1H, 3JHH=8.5 Гц, H-Ar); 6.96 (с, 1H, H-Ar); 7.93 (д, 1H, 3JHH=8.5 Гц, H-Ar); 10.77 (c, 1H, OH). MS-ESI: найдено [M+H]+ 426.2, вычислено для C22H24N3O6 426.2.
Пример 24. Получение 2-(аллилокси)-4-((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-6-ил)диазенил)аллилбензоата (I-24)
Получали аналогично соединению I-23 с использованием соединения 5б (0.50 г, 2.4 ммоль, 1 экв) вместо 5а. Нейтрализация реакционной массы по окончании реакции проводилась при 0°С. Выход: 75%, 0.81 г, красный кристаллический порошок. Т.пл. 120°С (с разложением). ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 1.51 (с, 6Н, 2CH3); 2.39 (с, 3H, СH3-Pyr); 4.66-4.70 (м, 2H, СH2); 4.77 (c, 2Н, CH2); 4.78-4.82 (м, 2H, СH2); 5.09 (c, 2Н, CH2); 5.25-5.30 (м, 1H, СH2); 5.32-5.37 (м, 1H, СH2); 5.39-5.45 (м, 1H, СH2); 5.54-5.61 (м, 1H, СH2); 5.98-6.15 (м, 2Н, 2СН); 6.87 (дд, 1H, 3JHH = 8.5 Гц, 4JHH = 1.6 Гц, H-Ar); 6.95-6.97 (м, 1H, H-Ar); 7.93 (д, 1H, 3JHH=8.5 Гц, H-Ar); 10.77 (c, 1H, OH). MS-ESI: найдено [M+H]+ 454.2, вычислено для C24H28N3O6 454.2.
Пример 25. Получение 2-(аллилокси)-4-((3-этил-9-гидрокси-8-метил-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-6-ил)диазенил)аллилбензоата (I-25)
Получали аналогично соединению I-23 с использованием соединения 5в (0.50 г, 2.4 ммоль, 1 экв) вместо 5а. Выход: 96%, 1.04 г, красный кристаллический порошок. Т.пл. 80°С (с разложением). ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 0.99 (т, 3Н, 3JHH = 7.5 Гц, СН3СН2); 1.70-1.79 (м, 2Н, СН3СН2); 2.39 (с, 3H, СH3-Pyr); 4.63, 4.91 (AB, 2Н, 2JHH = 16.4 Гц, CH2); 4.67-4.71 (м, 2H, СH2); 4.78-4.82 (м, 2H, СH2); 4.85 (т, 1Н, 3JHH=5.9 Гц, CHC2H5); 4.95, 5.23 (AB, 2Н, 2JHH = 16.4 Гц, CH2); 5.26-5.30 (м, 1H, СH2); 5.32-5.37 (м, 1H, СH2); 5.39-5.46 (м, 1H, СH2); 5.55-5.61 (м, 1H, СH2); 5.98-6.17 (м, 2Н, 2СН); 6.86-6.90 (м, 1H, H-Ar); 6.95-6.98 (м, 1H, H-Ar); 7.93 (д, 1H, 3JHH=8.5 Гц, H-Ar); 10.79 (c, 1H, OH). MS-ESI: найдено [M+H]+ 454.2, вычислено для C24H28N3O6 454.2.
Пример 26. Получение 2-(аллилокси)-4-((9-гидрокси-8-метил-3-пропил-1,5-дигидро-[1,3]диоксепино[5,6-c]пиридин-6-ил)диазенил)аллилбензоата (I-26)
Получали аналогично соединению I-23 с использованием соединения 5г (0.50 г, 2.2 ммоль, 1 экв) вместо 5а. Выход: 84%, 0.88 г, красный кристаллический порошок. Т.пл. 86°С (с разложением). ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 0.98 (т, 3Н, 3JHH = 7.4 Гц, СН3СН2СН2); 1.41-1.52 (м, 2Н, СН3СН2СН2); 1.66-1.74 (м, 2Н, СН3СН2СН2); 2.40 (с, 3H, СH3-Pyr); 4.63, 4.91 (AB, 2Н, 2JHH = 16.4 Гц, CH2); 4.67-4.71 (м, 2H, СH2); 4.78-4.82 (м, 2H, СH2); 4.85 (т, 1Н, 3JHH=5.7 Гц, CHC3H7); 4.95, 5.23 (AB, 2Н, 2JHH = 16.7 Гц, CH2); 5.26-5.30 (м, 1H, СH2); 5.32-5.37 (м, 1H, СH2); 5.39-5.46 (м, 1H, СH2); 5.54-5.61 (м, 1H, СH2); 5.99-6.16 (м, 2Н, 2СН); 6.85-6.90 (дд, 1Н, 3JHH=8.5 Гц, 4JHH=1.8 Гц, H-Ar); 6.95-6.98 (д, 4JHH=1.8 Гц, 1H, H-Ar); 7.93 (д, 1H, 3JHH=8.5 Гц, H-Ar); 10.78 (c, 1H, OH). MS-ESI: найдено [M+H]+ 468.3, вычислено для C25H30N3O6 468.2.
Пример 27. Получение 2-(аллилокси)-4-((9-гидрокси-8-метил-3-октил-1,5-дигидро [1,3]диоксепино[5,6-c]пиридин-6-ил)диазенил)аллилбензоата (I-27)
Получали аналогично соединению I-23 с использованием соединения 5е (0.50 г, 2.2 ммоль, 1 экв) вместо 5а. Выход: 87%, 0.80 г, темно-красное маслообразное вещество. ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 0.88 (т, 3Н, 3JHH = 7.0л Гц, CH3CH2-); 1.21-1.42 (м, 12H, 6СH2); 1.68-1.73 (м, 2H, CH2); 2.40 (с, 3H, СH3-Pyr); 4.63, 4.91 (AB, 2Н, 2JHH = 16.2 Гц, CH2); 4.67-4.70 (м, 2H, СH2); 4.78-4.82 (м, 2H, СH2); 4.92 (т, 1Н, 3JHH=6.2 Гц, CHC8H17); 5.20, 5.26 (AB, 2Н, 2JHH = 16.2 Гц, CH2); 5.26-5.30 (м, 1H, СH2); 5.32-5.37 (м, 1H, СH2); 5.39-5.45 (м, 1H, СH2); 5.54-5.61 (м, 1H, СH2); 5.98-6.16 (м, 2Н, 2СН); 6.86-6.92 (м, 1Н, H-Ar); 6.97 (с, 1H, H-Ar); 7.93 (д, 1H, 3JHH=8.8 Гц, H-Ar); 10.76 (c, 1H, OH). MS-ESI: найдено [M+H]+ 538.3, вычислено для C30H39N3O6 538.3.
Пример 28. Получение 2-гидрокси-4-((8-метил-9-оксидо-1,5-дигидро-[1,3]диоксепино [5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-28)
Соединение I-23 растворили (0.77 г, 1.8 ммоль, 1 экв) в 6.93 г 10% раствора гидроксида натрия в этаноле, добавили 10% Pd/C (0.08 г, 0.1 экв) и перемешивали при 50°С в течение 48 часов. К реакционной массе добавили 5 мл воды и отфильтровали. Фильтрат собрали и высушили в вакууме. К сухому остатку добавили 30 мл системы ацетон/этанол (3:7), суспензию нагрели до 50°С и отфильтровали нерастворившийся осадок, фильтрат высушили в вакууме. Продукт очищали перекристаллизацией из изопропанола. Выход: 85%, 0.60 г, красный кристаллический порошок. Т.пл. 144°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 2.15 (с, 3H, СH3-Pyr); 4.78 (с, 2Н, СН2); 4.92 (с, 2Н, СН2); 5.27 (с, 2Н, СН2); 6.93 (д, 4JHH=1.7 Гц, 1Н, Н-Ar); 6.98 (дд, 3JHH=8.2 Гц, 4JHH=1.8 Гц, H-Ar); 7.69 (д, 3JHH=8.2 Гц, 1H, H-Ar); 16.25 (c, 1H, OH). ЯМР 13С {H} (100 МГц, D2О), δ, м.д: 19.96 (с, СH3.); 66.30 (с, СH2); 66.83 (с, СH2); 98.59 (с, С); 109.87 (с, Car.); 114.40 (с, Car.); 120.01 (с, Car.); 131.99 (с, Car.); 133.99 (с, Car.); 137.35 (с, Car.); 144.59 (с, Car.); 151.52 (с, Car.); 156.76 (с, Car.); 161.31 (с, Car.); 165.83 (с, Car.); 168.92 (с, C=O); MS-ESI: найдено [M-2Na+2H]+ 346.3, вычислено для C16H18N3O6 346.3.
Пример 29. Получение 2-гидрокси-4-((3,3,8-триметил-9-оксидо-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-29)
Получали аналогично соединению I-28 с использованием соединения I-24 (0.63 г, 1.4 ммоль, 1 экв) вместо I-23. Продукт очищали перекристаллизацией из смеси растворителей изопропанол/H2O (9:1). Выход: 36%, 0.21 г, красный кристаллический порошок. Т.пл. 131°С (с разложением). ЯМР 1H (400 МГц, D2O): δ, м.д.: 1.45 (с, 6H, 2СH3); 2.32 (с, 3H, СH3-Pyr); 4.90 (с, 2Н, СН2); 5.37 (с, 2Н, СН2); 7.19 (с, 1H, H-Ar); 7.25 (дд, 3JHH=8.4 Гц, 4JHH=1.7 Гц, H-Ar); 7.81 (д, 3JHH=8.4 Гц, 1H, H-Ar). MS-ESI: найдено [M-2Na+2H]+ 374.1, вычислено для C18H20N3O6 374.1.
Пример 30. Получение 4-((3-этил-8-метил-9-оксидо-1,5-дигидро[1,3]диоксепино [5,6-c] пиридин-6-ил)диазенил)-2-гидроксибензоата натрия (I-30)
Получали аналогично соединению I-28 с использованием соединения I-25 (1.04 г, 2.3 ммоль, 1 экв) вместо I-23. Продукт очищали перекристаллизацией из смеси растворителей изопропанол/H2O (9:1). Выход: 40%, 0.36 г, красный кристаллический порошок. Т.пл. 102°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.99 (т, 3H, 3JHH = 7.4 Гц, СH3СН2); 1.58-1.64 (м, 2Н, СH3СН2); 2.15 (с, 3H, СH3-Pyr); 4.50-5.06 (AB, 2Н, 2JHH = 14.7 Гц, CH2); 4.79 (т, 1Н, 3JHH = 5.4 Гц, СНС2Н5); 4.97-5.55 (AB, 2Н, 2JHH = 14.6 Гц, CH2); 6.88-7.08 (м, 2H, H-Ar); 7.63-7.79 (м, 1H, H-Ar); 16.31 (c, 1H, OH). MS-ESI: найдено [M-2Na+2H]+ 374.1, вычислено для C18H20N3O6 374.1.
Пример 31. Получение 2-гидрокси-4-((8-метил-9-оксидо-3-пропил-1,5-дигидро-[1,3] диоксепино[5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-31)
Получали аналогично соединению I-28 с использованием соединения I-26 (0.66 г, 2.3 ммоль, 1 экв) вместо I-23. Продукт очищали перекристаллизацией из смеси растворителей изопропанол/H2O (9:1). Выход: 53%, 0.33 г, красный кристаллический порошок. Т.пл. 101°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6): δ, м.д.: 0.99 (т, 3H, 3JHH = 7.4 Гц, СH3СН2СН2); 1.30-1.40 (м, 2Н, СH3СН2СН2); 1.53-1.61 (м, 2Н, СH3СН2СН2); 2.17 (с, 3H, СH3-Pyr); 4.53-5.05 (AB, 2Н, 2JHH = 15.0 Гц, CH2); 4.88 (т, 1Н, 3JHH = 5.5 Гц, СНС3Н7); 5.00-5.53 (AB, 2Н, 2JHH = 14.8 Гц, CH2); 7.00 (д, 4JHH=1.7 Гц, 1H, H-Ar); 7.09 (дд, 3JHH=8.2 Гц, 4JHH=1.7 Гц, H-Ar); 7.74 (д, 3JHH=8.2 Гц, 1H, H-Ar). MS-ESI: найдено [M-2Na+2H]+ 388.3, вычислено для C19H22N3O6 388.2.
Пример 32. Получение 2-(аллилокси)-4-((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)диазенил)аллилбензоата (I-32)
Получали аналогично соединению I-23 с использованием соединения 14 (0.50 г, 2.4 ммоль, 1 экв) вместо 5а. Нейтрализация реакционной массы по окончании реакции проводилась при 0°С. Выход: 77%, 0.77 г, красный кристаллический порошок. Т.пл. 237°С (с разложением). ЯМР 1H (400 МГц, CDCl3): δ, м.д.: 2.41 (с, 3H, СH3-Pyr); 4.66-4.70 (м, 2H, СH2); 4.76 (c, 2Н, CH2); 4.75-4.84 (м, 4H, 2СH2); 4.99 (c, 2Н, CH2); 5.25-5.28 (м, 1H, СH2); 5.28-5.32 (м, 1H, СH2); 5.39-5.46 (м, 1H, СH2); 5.50-5.57 (м, 1H, СH2); 5.98-6.13 (м, 2Н, 2СН); 6.87 (дд, 1H, 3JHH = 8.5 Гц, 4JHH = 1.6 Гц, H-Ar); 6.95-6.97 (м, 1H, H-Ar); 7.46 (д, 1H, 3JHH=7.8 Гц, H-Ar); 7.59 (с, 1H, H-Ar); 7.86 (д, 1H, 3JHH=7.8 Гц, H-Ar).
Пример 33. Определение антигликирующей активности – способности ингибировать образование конечных продуктов гликирования
Реакцию гликирования проводили в фосфатном буферном растворе 0.05 М рН 7.4. Состав реакционной среды: 0.5 М раствор глюкозы, 1 мг/мл БСА, 0.02% натрия азид. Для предупреждения бактериального роста все манипуляции проводили в асептических условиях (с использованием ламинарного шкафа, предварительно проверенного на микробиологическую чистоту), с этой же целью в буферный раствор вносили азид натрия. Исследования выполняли в дубликатах.
В качестве препаратов сравнения использовали известные ингибиторы гликирования – аминогуанидин (АГ) и пиридоксамин (ПА). Все вещества растворяли в диметилсульфоксиде (ДМСО). В экспериментальные образцы добавляли 30 мкл раствора изучаемых веществ в различных концентрациях, в контрольные образцы добавляли ДМСО в аналогичном объеме. Образцы инкубировали в течение 24 часов при температуре 60°С. Через 24 часа отбирали по 150 мкл и проводили определение специфической флуоресценции гликированного бычьего сывороточного альбумина на спектрофотометре Tecan Infinite 200 PRO (TECAN, Швейцария) при длине волны возбуждения 370 нм и испускания 440.
Статистическую обработку результатов проводили с использованием редактора Microsoft Excel 2007, для расчета показателя IС50 использовали программное обеспечение OriginPro 8, в котором строили графики доза-ответ (х – концентрация, мкМ; у – иобразование КПГ, в процентах, вычисленное исходя из значений флуоресценции контроля и исследуемых образцов).
Результаты исследования антигликирующей активности представлены на Фиг. 1. Полученные данные свидетельствуют о том, что все полученные соединения обладают высокой антигликирующей активностью – способностью ингибировать образование конечных продуктов гликирования, при этом антигликирующая активность значительно превышает активность препаратов сравнения.
Пример 34. Исследование цитотоксичности некоторых соединений формулы I in vitro
Для оценки возможной токсичности in vitro для ряда представителей из числа заявленных соединений (I-13, I-19, I-24, I-30) была исследована их цитотоксичность в отношении условно-нормальных клеток (клетки HSF и MSC). Исследование проведено с помощью пролиферативного МТТ-теста с последующим построением кривой доза-эффект и определения концентраций полумаксимального ингибирования роста условно-нормальных клеток (СС50).
Клетки культивировали в среде α-MEM (содержащей глутамин, 10%-ную эмбриональную телячью сыворотку, 1% пенициллин-стрептомицин) в стандартных условиях (в атмосфере 5%-го СО2 при 37°С до образования монослоя). Для получения клеточной суспензии монослой клеток трипсинизировали с последующей инактивацией трипсина добавлением среды α-MEM с сывороткой. Подсчёт клеток производили в камере Нэйбауэра методом исключения трипанового синего. Клетки пересевали 2 раза в неделю в отношении 1:6. Для осуществления трехдневного теста в лунки 96-луночного планшета вносили 40000 клеток в 200 микролитрах культуральной среды. Далее планшеты с клетками инкубировали 24 часа в стандартных условиях для адгезии клеток к субстрату.
Далее в стерильном 96-луночном планшете готовили серийные разведения (8 концентраций) анализируемых соединений в стерильной очищенной воде. В лунки планшета с клетками вносили аликвоты приготовленных растворов исследуемых соединений в объеме 22 микролитра в каждую лунку. После внесения исследуемых веществ клетки культивировали в стандартных условиях в течение 72 часов. По истечении времени инкубации культуральную среду с исследуемыми веществами удаляли из планшета с помощью вакуумного аспиратора и добавляли MTT-реагента (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид). Приготовленную реакционную смесь вносили в объеме 100 мкл в каждую лунку планшета и инкубировали в СО2 инкубаторе 3 часа. Культуральную среду с исследуемыми соединениями удаляли вакуумным аспиратором. В каждую лунку планшета вносили 100 мкл ДМСО и инкубировали 5-10 минут. Появившееся фиолетовое окрашивание детектировали на планшетном ридере Tecan при 550 нм (референтная длина волны – 700 нм).
Далее рассчитывали концентрацию полумаксимального ингибирования роста клеток (CС50), принимая жизнеспособность клеток в контрольных лунках за 100%. Результаты представляли в процентном отношении к контролю, который не обрабатывали исследуемыми препаратами. Для препаратов строили кривую «доза-эффект» и определяли величины СC50 с помощью программного обеспечения OriginPro8. Результаты исследования представлены в таблице 2 на Фиг. 2.
По результатам исследования цитотоксической активности можно сделать вывод, что все изученные соединения не проявляют токсических свойств в изученном диапазоне концентраций и значение CС50 превышает 500 мкМ. Следовательно, для данных соединений можно прогнозировать благоприятный профиль безопасности.
Таким образом, из описанного выше можно сделать вывод, что заявителем решена техническая проблема и достигнут заявленный технический результат, а именно, получен ряд соединений общей формулы (I), обладающих способностью ингибировать образование конечных продуктов гликирования, превосходящей активность известных лекарственных средств, вышедших на стадию клинических исследований (например, антигликирующих препаратов аминогуанидин и пиридоксамин), в 2-53 раза.
Заявленное техническое решение соответствует критерию «новизна», предъявляемому к изобретениям, так как из исследованного уровня техники не выявлены технические решения, обладающие заявленной совокупностью отличительных признаков, обеспечивающих достижение заявленных результатов.
Заявленное техническое решение соответствует критерию «изобретательский уровень», предъявляемому к изобретениям, так как не является очевидным для специалиста в данной области науки и техники.
Заявленное техническое решение соответствует критерию «промышленная применимость», так как может быть реализовано на любом специализированном предприятии с использованием стандартного оборудования, известных отечественных материалов и технологий.
| название | год | авторы | номер документа |
|---|---|---|---|
| Азопроизводные 5-аминосалициловой кислоты, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2837878C1 |
| Оксазолидиноны на основе производных пиридоксина, обладающие антибактериальной активностью | 2024 |
|
RU2836570C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| Четвертичные аммониевые соединения на основе производных пиридоксина и жирных карбоновых кислот, обладающие антибактериальной активностью | 2022 |
|
RU2795265C1 |
| ПРОИЗВОДНЫЕ ПИРИДОКСИНА С НЕЛИНЕЙНЫМИ ОПТИЧЕСКИМИ СВОЙСТВАМИ | 2012 |
|
RU2501801C1 |
| Четвертичные аммониевые соединения на основе 3-гидроксипиридина, обладающие антибактериальной активностью | 2021 |
|
RU2778507C1 |
| ФОСФОНИЕВЫЕ СОЛИ НА ОСНОВЕ ПРОИЗВОДНЫХ ПИРИДОКСИНА | 2011 |
|
RU2466728C1 |
| Четвертичные аммониевые соединения на основе производных пентаэритрита и пиридоксина, обладающие антибактериальной активностью | 2023 |
|
RU2811203C1 |
| ПРОИЗВОДНЫЕ ПИРИДОКСИНА С АНТИХОЛИНЭСТЕРАЗНОЙ АКТИВНОСТЬЮ | 2014 |
|
RU2550080C1 |
| Ингибиторы образования конечных продуктов гликирования на основе азопроизводных фенилсульфокислот | 2016 |
|
RU2634594C1 |

Изобретение относится к химии органических гетероциклических соединений, а именно к азопроизводным аминофенолов формулы (I), где R1 = R2 = H; или R1 и R2 вместе образуют заместитель вида СR3R4, где R3 = H, CH3, R4 = H, CH3, C2H5, C3H7, C5H11, C8H17; R5 = H, R6 = OS(O)2CH3, OH, или R5 = OS(O)2CH3, OH, R6 = H, или R5 = C(O)OAllyl, C(O)ONa, R6 = OAllyl, OH; X = H, Na. Соединения формулы (I) являются эффективными ингибиторами образования конечных продуктов гликирования (КПГ). 1 з.п. ф-лы, 2 ил., 34 пр. 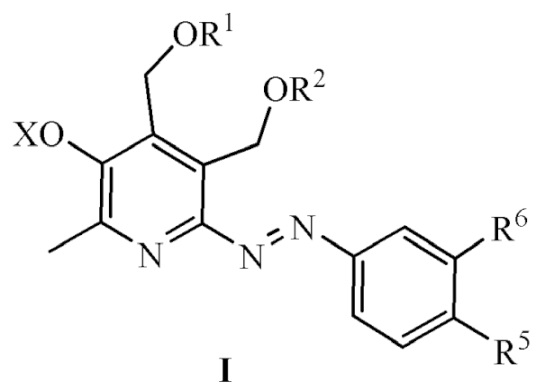
1. Азопроизводные аминофенолов общей формулы (I)
где R1 = R2 = H; или R1 и R2 вместе образуют заместитель вида СR3R4, где R3 = H, CH3, R4 = H, CH3, C2H5, C3H7, C5H11, C8H17;
R5 = H, R6 = OS(O)2CH3, OH, или R5 = OS(O)2CH3, OH, R6 = H, или R5 = C(O)OAllyl, C(O)ONa, R6 = OAllyl, OH;
X = H, Na.
2. Азопроизводные аминофенолов по п. 1, обладающие способностью ингибировать образование конечных продуктов гликирования.
| A.D | |||
| Strelnik et al | |||
| Inhibitors of the formation of advanced glycation end products based on pyridoxine azo derivatives | |||
| Russian Journal of General Chemistry, 2023, v | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Передвижной дровокольный станок | 1913 |
|
SU522A1 |
| Aliyev Hafiz M | |||
| et al | |||
| Synthesis of mercurions and their standardization | |||
| Azerbaycan Eczaciliq Jurnali, 2005, 5 (1), pp | |||
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Применение азопроизводных фенилсульфокислот в качестве ингибиторов образования конечных продуктов гликирования | 2016 |
|
RU2628605C1 |

