Изобретение относится к области органического синтеза, в частности, к способу получения 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов посредством пятистадийного синтеза из 1,1-диалкилпроп-2-ин-1-олов и паразамещенных бензофенонов. Эти диазокарбонильные соединения представляют значительный практический интерес, поскольку их можно использовать в реакциях сужения циклов и получать труднодоступные другими путями производные оксетана [1-4], входящие в состав природных соединений и многих фармакологически активных веществ.
Так, ключевой стадией синтеза оксетаноцина является перегруппировка Вольфа замещенного диазотетрагидрофуранона в производное оксетана [3]. Оксетаноцин - оптически активный четырехчленный нуклеозид-антибиотик, in vitro ингибирующий репликацию ВИЧ и обладающий очень сильным антивирусным действием. Его производные входят в состав фармацевтических препаратов против СПИДа, гепатита-B, герпеса и других вирусов [5-7].
Другим примером перспективного использования диазокетонов ряда ТГФ является получение на их основе 3(2H)-фуранонов - потенциальных высокоактивных нестероидных противовоспалительных препаратов [8-9]. 5-Арил-2,2-диалкил-4-фенил-3(2H)-фураноны с SO2NH2 и SO2Me группами в параположениях арильного кольца являются селективными ингибиторами фермента COX-2, что обуславливает их противовоспалительные свойства [10].
Таким образом, высокая биологическая активность соединений, получаемых из диазотетрагидрофуранонов, стимулирует разработку эффективных методов синтеза последних.
Известен способ получения диазосоединений окислением гидразонов надуксусной кислотой в двухфазной системе в присутствии катализаторов окисления и межфазного переноса - йода и четвертичных аммониевых солей [11]. Недостатком этого способа является пригодность метода для синтеза исключительно диазоалканов, а при его реализации используют токсичные реагенты. Так, йод в чистом виде может вызывать раздражение кожи [12], а надуксусная кислота и четвертичные аммониевые соли воздействуют на дыхательные пути [13], в отдельных случаях приводя к развитию астмы [14-15].
Другой способ получения диазосоединений, в частности диазокетонов, заключается во взаимодействии карбонилсодержащих третичных фосфинов с азидами и последующий термолиз или обработку основанием промежуточно образующихся ацилтриазенов в целевые диазокарбонильные соединения [16]. Недостатком этого аналога является сравнительно высокая токсичность третичных фосфинов [17] и потенциальная взрывоопасность азидов [18].
Наиболее близким аналогом заявляемого изобретения является способ получения диазокетонов, основанный на обработке α-дикетонов гидразин-гидратом и последующем окислении образующихся гидразонов окисью ртути(II) [19], который принят в качестве прототипа. При этом исходные дикетоны получают окислением α-оксикетонов ацетатом меди(II) в водно/спиртовом растворе.
К недостаткам прототипа следует отнести, во-первых, необходимость проведения процесса окисления гидразонов окисью ртути в течение весьма длительного времени (до 24 часов), а во-вторых, использование в качестве реагентов высокотоксичных оксида ртути(II) [20] и гидразин-гидрата [12], причисленного к вредным веществам первого класса опасности [21]. В-третьих, введение в реакцию оксида ртути(II) и ацетата меди(II) в качестве окислителей сопровождается образованием свободной ртути и оксида меди(II), которые загрязняют реакционную смесь и с трудом удаляются из продуктов реакции.
Техническая задача, решаемая заявляемым изобретением, направлена на устранение этих недостатков и заключается в использовании более эффективного, а также экологически более чистого и нетоксичного способа получения тетразамещенных 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов общей формулы I
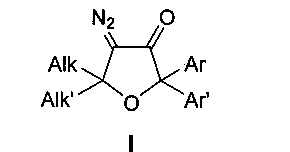
где Alk, Alk′ = Me, Et, cycloalkyl или другие алкильные группы, а Ar, Ar′ = Ph, p-MeO-C6H4, p-F-C6H4, p-Cl-C6H4 и другие замещенные или незамещенные арильные группы.
Технический результат, получаемый при реализации заявленного изобретения, заключается в более эффективном методе синтеза диазокетонов, пригодных в качестве прекурсоров при получении биологически активных и лекарственных препаратов, а также использовании нетоксичных реагентов и растворителей на каждой стадии процесса.
Указанный технический результат достигается тем, что:
во-первых, для получения 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов используется 5-стадийная схема синтеза из (3,3-диалкил-3-гидроксипроп-1-ин-1-ил)магний бромидов (II) и паразамещенных бензофенонов, взаимодействие которых по методу Йоцича (Стадия 1) приводит к образованию 1,1-диалкил-4,4-диарилбут-2-ин-1,4-диолов (III):

во-вторых, 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-оны (IV) получают дегидратацией с одновременной гидратацией ацетиленовых γ-гликолей (III) в присутствии катализатора - серной кислоты (Стадия 2);
в-третьих, при получении 2,2-диарил-5,5-диалкилтетрагидрофуран-3,4-дионов (V) используют нитрозирование 2,2-диарил-5,5-диалкил-тетрагидрофуран-3-онов (IV) нитритом натрия в водно-органическом растворе соляной кислоты и их последующий гидролиз в α-дикетоны (V) при 63°C (Стадия 3);
в-четвертых, для синтеза моногидразонов α-дикетонов (VI) вместо гидразин-гидрата применяют p-толуолсульфонилгидразид, который прибавляют непосредственно в реакционную смесь с α-дикетоном (V) в том же растворителе (ТГФ) и при той же температуре (63°C) (Стадия 4);
в-пятых, превращение тозилгидразонов (VI) в целевые 4-диазо-5,5-диарил-2,2-диалкилдигидрофуран-3(2H)-оны (I) на заключительной стадии синтеза (Стадия 5) проводят в водно-щелочном растворе при комнатной температуре вместо окисления гидразонов окисью ртути, используемого в известном прототипе.
Заявляемое изобретение имеет следующие отличительные признаки и преимущества:
1. значительное увеличение выходов диазокетонов (I) на последней стадии синтеза (до 97%);
2. многократное уменьшение длительности превращения гидразонов (VI) в диазокетоны (I) (с 8-24 часов до 1 часа);
3. заметное упрощение методики обработки реакционных смесей, выделение целевых продуктов (I) лишь путем перекристаллизации;
4. последовательное превращение монокетонов (IV) в тозилгидразоны (VI) без выделения промежуточных α-дикетонов (V) и отсутствие необходимости удаления твердых осадков из реакционной смеси, образующихся в известном способе;
5. отказ от использования токсичных и ядовитых реагентов на всех стадиях синтеза;
6. проведение реакций только в нетоксичных растворителях - диэтиловом эфире, этаноле и тетрагидрофуране.
Апробация заявленного изобретения была выполнена в Санкт-Петербургском государственном университете с использованием оборудования ресурсных центров СПбГУ «Магнитно-резонансные методы исследования» и «Методы анализа состава вещества». Результаты исследования представлены в виде конкретных примеров реализации.
Примеры выполнения способа
Спектры ЯМР 1H (400.13 МГц) и 13C (100.61 МГц) регистрировали на приборе "Bruker 400 МГц Avance" в растворе CDCl3, внутренний стандарт - (CH3)4Si. ИК-спектры получены на спектрофотометре "Perkin-Elmer BXII" в диапазоне 4000-400 см-1 в таблетках KBr. Для снятия масс-спектров использовали хромато-масс-спектрометры Brucker micrOTOF и «MaXis», Bruker Daltonik GmbH. Элементный анализ проводили на приборе Euro EA3028-HT. Температуру плавления измеряли на приборе Büchi B-540.
Реакции проводили в безводных растворителях, очищенных по стандартным методикам. Для препаративного разделения реакционных смесей использовали колоночную и флеш-хроматографию на силикагеле (Silicagel L, 40-100 µm; Chemapol и Alugram Sil G/UV254) в градиентном режиме. Аналитическую TCX проводили на пластинах Silica gel Merck 60 F254 (Германия) и Alugram Xtra Sil G/UV254 (Германия), элюенты - петролейный эфир, гексан и ацетон.
Соединения, приведенные без ссылок, приготовлены по стандартным лабораторным методикам либо являются коммерчески доступными веществами.
Пример 1.
Получение 4-диазо-5,5-диметил-2,2-ди(p-метоксифенил)тетрагидрофуран-3-она.
Стадия 1. К раствору этилмагний-бромида, полученному из 2.16 г (90 ммоль) магния и 9.81 г (90 ммоль) этилбромида в 35 мл диэтилового эфира, при 0°C прибавили по каплям раствор 2.95 г (35 ммоль) 2-метилбут-3-ин-2-ола в 35 мл эфира. Реакционную смесь кипятили в течение 3 часов, охладили и при 0°C прибавили в виде сухого порошка 6.05 г (25 ммоль) 4,4′-диметоксибензофенона. После этого смесь кипятили с обратным холодильником 4 часа (контроль по ТСХ), охладили, прибавили 40 мл соляной кислоты (3:1) при охлаждении, водный слой экстрагировали хлористым метиленом (3×35 мл), объединенные органические вытяжки сушили безводным K2CO3, растворитель отогнали в вакууме, остаток перекристаллизовали из смеси петролейный эфир/хлороформ. Выход 2-метил-5,5-ди(p-метоксифенил)пент-3-ин-2,5-диола 7.7 г (94%).
Бесцветное твердое вещество.
Т.пл. 112-113°C (петролейный эфир/хлороформ). Rf (петролейный эфир/ацетон, 5:3) 0.57.
FT-IR (KBr) 
1H-NMR (400 MHz, CDCl3; 25°C): 1.54 (s, 6H, 2 Me); 2.52 (s, 1H, OH); 3.15 (s, 1H, OH); 3.76 (s, 6H, 2 OMe) 6.79-6.83 (m, 4 HAr), 7.42-7.46 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 31.3 (2 Me); 55.3 (2 OMe); 65.3, 73.5 (CMe2, CAr2); 85.2 (Me2C-C≡), 91.4 (≡C-CAr2,); 113.5, 127.3, 137.6, 158.9 (все CAr).
HRMS (ESI) для C20H22O4 (326.1518): вычисл. для [M+H]+ 327.1591, найдено 327.1591.
Аналит. вычисл. для C20H22O4 (326.1518): C 73.62, H 6.75; найдено: C 73.64, H 6.93.
Стадия 2. К раствору 6.52 г (20 ммоль) 2-метил-5,5-ди(p-метоксифенил)пент-3-ин-2,5-диола в 40 мл абсолютного этанола по каплям прибавили 4 мл серной кислоты (d 1.83 г/мл). Смесь перемешивали при 65-68°C в течение 7 часов, после чего охладили и поставили на ночь в холодильник. Образовавшиеся бесцветные кристаллы отфильтровали и промыли ледяной водой. К маточному раствору прибавили 50 мл воды, водный слой экстрагировали хлористым метиленом (3×25 мл), объединенные органические вытяжки промыли водой (2×30 мл), затем насыщенным раствором поваренной соли (30 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме, остаток перекристаллизовали из петролейного эфира. Общий выход 2,2-диметил-5,5-ди(p-метоксифенил)дигидрофуран-3(2H)-она 4.2 г (65%).
Бесцветное твердое вещество.
Т.пл. 76-77°C (петролейный эфир). Rf (петролейный эфир/ацетон, 5:1) 0.37.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 25°C): 1.20 (s, 6H, 2 Me); 3.25 (s, 2H, CH2); 3.77 (s, 3H, OMe); 6.81-6.85 (m, 4 HAr); 7.27-7.31 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 25.4 (2 CH3); 48.5 (CH2); 55.2 (OCH3); 81.2, 82.3 (CAr2, CMe2); 113.5, 127.4, 138.4, 158.8 (все CAr); 217.4 (C=O).
HRMS (ESI) для C20H22O4 (326.1518): вычисл. для [M+Na]+ 349.1410, найдено 349.1410.
Аналит. вычисл. для C20H22O4 (326.1518): C 73.60, H 6.79; найдено: C 73.62, H 6.75.
Стадии 3 и 4. К смеси 0.8 г (2.45 ммоль) 2,2-диметил-5,5-ди(p-метоксифенил)дигидрофуран-3(2H)-она и 0.5 г (7.35 ммоль) нитрита натрия в 15 мл ТГФ при перемешивании по каплям прибавили 3.7 мл (122 ммоль) концентрированной соляной кислоты. Нагревание до температуры 53-56°C привело к интенсивному выделению бурого газа и кипению реакционной смеси. После окончания выделения газа смесь перемешивали при 65°C в течение 1 часа (контроль по TCX), при этом она приобрела темно-малиновую окраску. Далее прибавили 0.46 г (2.45 ммоль) p-толуолсульфонилгидразида в 4 мл ТГФ, реакционная смесь изменила цвет на темно-желтый. Смесь кипятили еще 40 минут (контроль по TCX), охладили, прибавили 50 мл хлористого метилена. Реакционную смесь перенесли в делительную воронку, промыли водой (2×50 мл), затем насыщенным раствором поваренной соли (50 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме. Остаток хроматографировали на колонке с SiO2 (элюент : гексан/ацетон 10:1→4:1). Выход N′-(2,2-диметил-5,5-ди(p-метоксифенил)-4-оксодигидрофуран-3(2H)-илиден)-4-метилбензолсульфоногидразида на двух стадиях составил 0.54 г (43%).
Желтое твердое вещество. Т.пл. 162-164° (гексан). Rf (гексан/ацетон, 3:1,2 раза) 0.24.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 26°C): 1.40 (s, 6H, 2 Me); 2.42 (s, 3H, Me); 3.77 (s, 6H, 2 OMe); 6.82 (d, J=8.9 Hz, 4 CHAr); 7.28 (d, J=8.0 Hz, 4 CHAr); 7.31 (d, J=8.9 Hz, 2 CHTs); 7.80 (d, J=8.0 Hz, 2 CHTs); 12.22 (5, 1H, NH).
13C-NMR (100 MHz, CDCl3; 26°C): 21.6 (Me); 28.5 (2 Me); 55.3 (2 OMe); 78.7, 85.5 (CMe2, CAr2); 113.8, 127.8, 133.0, 159.4 (все CAr); 127.8, 129.8, 144.8, 146.8 (все CTs); 135.1 (C=N-NH2); 199.0 (C=O).
HRMS (ESI) для C27H28N2O6S (508.1668): вычисл. для [M+H]+ 509.1741, найдено 509.1741; вычисл. для [M+Na]+ 531.1560, найдено 531.1560.
Стадия 5. К раствору 508 мг (1 ммоль) N′-(2,2-диметил-5,5-ди(p-метоксифенил)-4-оксодигидрофуран-3(2H)-илиден)-4-метилбензолсульфоногидразида в 15 мл диэтилового эфира прибавили раствор 60 мг NaOH в 10 мл H2O. Реакционную смесь интенсивно перемешивали в течение часа (контроль по TCX), перенесли в делительную воронку и прибавили еще 30 мл воды. Водный слой отделили от органического, экстрагировали Et2O (2×10 мл). Объединенные органические вытяжки промыли водой (2×20 мл), затем насыщенным раствором поваренной соли (20 мл), высушили безводным MgSO4. Растворитель отогнали в вакууме. Остаток перекристаллизовали из петролейного эфира. Выход 4-диазо-5,5-диметил-2,2-ди(p-метоксифенил)дигидрофуран-3(2H)-она 317 мг (90%).
Желтое твердое вещество. Т.пл. 131-132°C (петролейный эфир). Rf (циклогексан/ацетон, 10:1) 0.28.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 26°C): 1.61 (s, 6H, 2 Me); 3.78 (s, 6H, 2 OMe); 6.84 (d, J=8.7 Hz, 4 HAr), 7.37 (d, J=8.5 Hz, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 28.9 (2 Me); 55.2 (2 OMe); 65.1 (C=N2); 78.1, 90.2 (CMe2, CAr2); 113.5,128.0,134.0,159.2 (все CAr); 193.3 (C=O).
HRMS (ESI) для C20H20N2O4 (352.1423): вычисл. для [M+H]+ 353.1496, найдено 353.1496.
Аналит. вычисл. для C20H20N2O4 (352.1423): C 68.17, H 5.72, N 7.95; найдено: C 68.11, H 5.92, N 7.72.
Пример 2.
Получение 4-диазо-5,5-диметил-2,2-ди(p-фторфенил)дигидрофуран-3(2H)-она.
Стадия 1. К раствору этилмагний-бромида, полученному из 4.32 г (180 ммоль) магния и 19.62 г (180 ммоль) этилбромида в 75 мл диэтилового эфира, при 0°C прибавили по каплям раствор 5.88 г (70 ммоль) 2-метилбут-3-ин-2-ола в 70 мл эфира. Реакционную смесь кипятили в течение 3 часов, охладили и при 0°C прибавили в виде сухого порошка 10.9 г (50 ммоль) 4,4′-дифторбензофенона. Полученную смесь кипятили с обратным холодильником 4 часа (контроль по TCX), охладили, прибавили 55 мл соляной кислоты (3:1) при охлаждении, водный слой экстрагировали хлористым метиленом (3×70 мл). Объединенные органические вытяжки сушили безводным K2CO3, растворитель отогнали в вакууме, полученный остаток перекристаллизовали из петролейного эфира. Выход 2-метил-5,5-ди(p-фторфенил)пент-3-ин-2,5-диола 14.7 г (97%).
Бесцветное твердое вещество. Т.пл. 95-96°C (петролейный эфир). Rf (петролейный эфир/ацетон, 5:3) 0.66.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 25°C): 1.56 (s, 6H, 2 Me); 2.27 (s, 1H, OH); 3.11 (s, 1H, OH); 6.96-7.02 (m, 4 HAr), 7.48-7.53 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 31.3 (s, 2 Me); 65.4, 73.3 (CMe2, CAr2); 84.5 (Me2C-C≡), 92.2 (≡C-CAr2,); 115.1 (d, 2JC,F=21.6 Hz), 127.7 (d, 3JC,F=8.2 Hz); 140.7 (d, 4JC,F=2.7 Hz); 162.2 (d, 1JC,F=246.9 Hz).
HRMS (ESI) для C18H16F2O2 (302.1118): вычисл. для [M+H]+ 303.1191, найдено 303.1191.
Аналит. вычисл. для C18H16F2O2 (302.1118): C 71.52, H 5.29; найдено: C 71.26, H 5.38.
Стадия 2. К раствору 12.08 г (40 ммоль) 2-метил-5,5-ди(p-фторфенил)пент-3-ин-2,5-диола в 80 мл абсолютного этанола по каплям прибавили 7.5 мл серной кислоты (d 1.83 г/мл). Смесь перемешивали при 65-68°C в течение 6 часов, после чего охладили, поставили на ночь в холодильник. Образовавшиеся бесцветные кристаллы отфильтровали и промыли ледяной водой. К маточному раствору прибавили 100 мл воды, водный слой экстрагировали хлористым метиленом (3×30 мл), объединенные органические вытяжки промыли водой (2×60 мл), затем насыщенным раствором поваренной соли (60 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме, остаток перекристаллизовали из петролейного эфира. Общий выход 2,2-диметил-5,5-ди(p-фторфенил)дигидрофуран-3(2H)-она 10.8 г (89%).
Бесцветное твердое вещество. Т.пл. 57-58°C (петролейный эфир). Rf (петролейный эфир/ацетон, 5:1) 0.55.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 28°C): 1.20 (s, 6H, 2 Me); 3.27 (s, 2H, CH2); 6.97-7.03 (m, 4 HAr); 7.33-7.38 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 28°C): 25.3 (2 Me); 48.2 (CH2); 81.5, 81.9 (CMe2, CAr2); 115.2 (d, 2JC,F=21.5 Hz), 127.7 (d, 3JC,F=8.1 Hz); 141.8 (d, 4JC,F=3.2 Hz); 162.0 (d, 1JC,F=247.0 Hz); 216.3 (C=O).
HRMS (ESI) для C18H16F2O2 (302.1118): вычисл. для [M+H]+ 303.1191, найдено 303.1191; вычисл. для [M+Na]+ 325.1011, найдено 325.1011.
Аналит. вычисл. для C18H16F2O2 (302.1118): C 71.51, H 5.33; найдено: C 71.66, H 5.24.
Стадии 3 и 4. К смеси 0.6 г (2.0 ммоль) 2,2-диметил-5,5-ди(p-фторфенил)дигидрофуран-3(2H)-она и 0.41 г (6.0 ммоль) нитрита натрия в 15 мл ТГФ при перемешивании по каплям прибавили 3 мл (100 ммоль) концентрированной соляной кислоты. Нагревание до температуры 53-56°C привело к интенсивному выделению бурого газа и кипению реакционной смеси. После окончания выделения газа смесь перемешивали при 65°C в течение 1 часа (контроль по TCX), при этом она приобрела темно-малиновую окраску. Далее прибавили 0.37 г (2 ммоль) p-толуолсульфонилгидразида в 4 мл ТГФ, реакционная смесь изменила цвет на темно-желтый. Смесь кипятили еще 30 минут (контроль по TCX), охладили, прибавили 40 мл хлористого метилена. Реакционную смесь перенесли в делительную воронку, промыли водой (2×40 мл), затем насыщенным раствором поваренной соли (40 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме. Остаток хроматографировали на колонке с SiO2 (элюент : гексан/ацетон 15:1→10:1). Выход Ν′-(2,2-диметил-5,5-ди(p-фторфенил)-4-оксодигидрофуран-3(2Н)-илиден)-4-метилбензолсульфоногидразида на двух стадиях составил 0.39 г (40%).
Желтое твердое вещество. Т.пл. 171-173° (гексан). Rf (гексан/ацетон, 3:1) 0.22.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 26°C): 1.40 (s, 6H, 2 Me); 2.42 (s, 3H, Me); 6.98-7.02 (m, 4 HAr), 7.31 (d, J=8.0 Hz, 2 CHTs); 7.36-7.39 (m, 4 HAr); 7.81 (d, J=8.0 Hz, 2 CHTs); 12.18 (s, 1Η, ΝΗ).
13C-NMR (100 MHz, CDCl3; 27°C): 21.6 (Me); 28.5 (2 Me); 79.2, 84.8 (CMe2, CAr2); 115.5 (d, 2JC,F=21.6 Hz); 127.9, 129.8, 144.9, 145.9 (все CTs); 128.1 (d, 3JC,F=8.1 Hz); 134.9 (C=N); 136.5 (d, 4JC,F=2.9 Hz); 162.5 (d, 1JC,F=248.3 Hz); 197.9 (C=O).
HRMS (ESI) для C25H22F2N2O4S (484.1268): вычисл. для [M+H]+ 485.1341, найдено 485.1341; вычисл. для [M+Na]+ 507.1161, найдено 507.1161.
Стадия 5. К раствору 484 мг (1 ммоль) N′-(2,2-диметил-5,5-ди(p-фторфенил)-4-оксодигидрофуран-3(2H)-илиден)-4-метилбензолсульфоногидразида в 15 мл диэтилового эфира прибавили раствор 60 мг NaOH в 10 мл H2O. Реакционную смесь интенсивно перемешивали в течение 40 минут (контроль по TCX), перенесли в делительную воронку и прибавили еще 30 мл воды. Водный слой отделили от органического, экстрагировали Et2O (2×10 мл). Объединенные органические вытяжки промыли водой (2×20 мл), затем насыщенным раствором поваренной соли (20 мл), высушили безводным MgSO4. Растворитель отогнали в вакууме. Остаток перекристаллизовали из петролейного эфира. Выход 4-диазо-5,5-диметил-2,2-ди(p-фторфенил)дигидрофуран-3(2H)-она 308 мг (94%).
Желтое твердое вещество. Т.пл. 115-116°C (петролейный эфир). Rf (петролейный эфир/ацетон, 3:1) 0.49.
FT-IR (KBr)
1H-NMR (400 MHz, CDCl3; 25°C): 1.61 (s, 6H, 2 Me); 6.98-7.04 (m, 4 HAr); 7.43-7.48 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 28.9 (2 CH3); 65.5 (C=N2); 78.5, 89.3 (CAr2, CMe2); 115.1 (d, 2JC,F=21.5 Hz), 128.3 (d, 3JC,F=8.2 Hz); 137.4 (d, 4JC,F=3.1 Hz); 162.4 (d, 1JC,F=247.3 Hz); 196.5 (C=O).
HRMS (ESI) для C18H14F2N2O2 (328.1023): вычисл. для [M+H]+ 329.1096, найдено 329.1096.
Аналит. вычисл. для C18H14F2N2O2 (328.1023): C 65.85, H 4.30, N 8.53; найдено: C 65.63, H 4.31. N 8.18.
Таким образом, как показывают результаты апробации, заявленный способ получения тетразамещенных 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов общей формулы I является более эффективным и исключает применение на всех стадиях процесса вредных для окружающей среды веществ, т.е. является экологически более чистым, чем известные аналоги, и может быть с уверенностью отнесен к Зеленой химии.
Использованные источники информации
1. W. Kirmse, 100 years of the Wolff rearrangement // Eur. J. Org. Chem., 2002, V. No. 14, pp. 2193-2256.
2. H.D. Stachel, H. Poschenrieder, J. Redlin, Thermolysis and photolysis of cyclic diazo compounds // Z. Naturforsch, Teil B, 1996, V. 51, No. 9, pp. 1325-1333.
3. D.W. Norbeck, J.B. Kramer, Synthesis of (-)-Oxetanocin // J. Am. Chem. Soc., 1988, V. 110, No. 21, pp. 7217-7218.
4. J. Beneke, R. Schobert, α-Carboxy-β-Lactones from Photoinduced Ring Contraction of 3-Diazodihydrofuran-2,4-diones // Synthesis, 2013, V. 45, No. 6, pp. 773-776.
5. Y. Nishiyama, N. Yamamoto, K. Takahash, N. Shimada, Selective inhibition of human cytomegalovirus replication by a novel nucleoside, oxetanocin G // Antimicrob. Agents Chemother., 1988, V. 32, No. 7, pp. 1053-1056.
6. S. Hayashi, D.W. Norbeck, W. Rosenbrook, R.L. Fine, M. Matsukura, J.J. Planner, S. Broder, H. Mitsuya, Cyclobut-A and cyclobut-G, carbocyclic oxetanocin analogs that inhibit the replication of human immunodeficiency virus in Τ cells and monocytes and macrophages in vitro // Antimicrob. Agents Chemother., 1990, V. 34, No. 2, pp. 287-294.
7. Y. Wang, G.W.J. Fleet, R. Storer, P.L. Myers, C.J. Wвсеis, O. Doherty, D.J. Watkin, K. Vogt, D.R. Witty, F.X. Wilson, J.M. Peach, Synthesis of the potent antiviral oxetane nucleoside epinoroxetanocin from D-lyxonolactone // Tetrahedron-Asymmetry, 1990, V. 1, No. 8, pp. 527-530.
8. L.L. Rodina, J.J. Medvedev, O.S. Galkina, V.A. Nikolaev, Thermolysis of 4-Diazotetrahydrofuran-3-ones: Total Change of Reaction Course Compared to Photolysis // Eur. J. Org. Chem., 2014, V. No. 14, pp. 2993-3000.
9. J.J. Medvedev, D.V. Semenok, L.L. Rodina, Effective approach to 4,5-Diaryl-3(2H)-furanones - a promising inhibitors for COX-2 // Journal of Medical & Biological Sciences, 2014, V. 1, Νο. pp. 84-88.
10. Патент WO 2000/061571. 4,5-Diaryl-3(2H)-furanone derivatives as cyclooxygenase-2 inhibitors. / Y.J. Byun, J.K. Choi, Y.H. Choi, S. Chung, J.Y. Ha, Y.S. Jeong, Y.H. Joo, S.H. Kang, J.Y. Kim, J.K. Kim. - Заявлено 12.04.2000. - Опубликовано 19.10.2000.
11. Патент US 4092306. Oxidation of hydrazones to the corresponding diazo compounds in the presence of a phase transfer and an oxidation catalyst which is iodine, an iodide or an iodonium salt. / D.T. Eastlick. - Заявлено 21.09.1976. - Опубликовано 30.05.1978.
12. Ν.Η. Proctor, J.P. Hughes, G.J. Hathaway, Proctor and Hughes′ Chemical hazards of the workplace. 5th ed. / [edited by] Gloria J. Hathaway and Nick H. Proctor, ed.; Wiley-Interscience: Hoboken; Chichester, 2004.
13. A. Bello, M.M. Quinn, M.J. Perry, D.K. Milton, Characterization of occupational exposures to cleaning products used for common cleaning tasks - a pilot study of hospital cleaners // Environ. Health, 2009, V. 8, No.
14. E. Cristofari-Marquand, M. Kacel, F. Milhe, A. Magnan, M. P. Lehucher-Michel, Asthma caused by peracetic acid-hydrogen peroxide mixture // J. Occup. Health, 2007, V. 49, No. 2, pp. 155-158.
15. A. Purohit, M.C. Kopferschmitt-Kubler, C. Moreau, E. Popin, M. Blaumeiser, G. Pauli, Quaternary ammonium compounds and occupational asthma // Int. Arch. Occup. Environ. Health, 2000, V. 73, No. 6, pp. 423-427.
16. Патент US 8350014. Preparation of diazo and diazonium compounds. / R.T. Raines, E.L. Myers. - Заявлено 17.11.2009. - Опубликовано 08.01.2013.
17. D.Ε.С. Corbridge, Phosphorus: chemistry, biochemistry and technology. 6th ed.; Taylor & Francis: Boca Raton, 2013.
18. S. Brase, C. Gil, K. Knepper, V. Zimmermann, Organic azides: An exploding diversity of a unique class of compounds // Angew. Chem., Int. Ed., 2005, V. 44, No. 33, pp. 5188-5240.
19 Патент US 2933494. Diazo pregnane compounds and method of preparing same. / B.G. Christensen, R.F. Hirschmann. - Заявлено 27.01.1958. - Опубликовано 19.04.1960 (Прототип).
20. A. Picot, N. Proust, Mercury and its compounds: from speciation to toxicity // Actual. Chim., 1998, V. No. 4, pp. 16-24.
21. ГОСТ 19503-88. Гидразин-гидрат технический. Технические условия. / - Введен в действие 01.07.1989.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ ВИТАМИНА D, СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2153491C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,2-ДИАЛКИЛ-4,5-ДИАРИЛФУРАН-3(2Н)-ОНОВ | 2014 |
|
RU2563876C1 |
| МОЛЕКУЛЫ С ПЕСТИЦИДНОЙ ФУНКЦИЕЙ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2016 |
|
RU2735602C2 |
| 1,3-Диметоксипропаны, содержащие 6,6-диметилбицикло[3.1.1]гептановый фрагмент природного происхождения и н-алкильный заместитель, способ их получения и титан-магниевый катализатор полимеризации пропилена, содержащий эти соединения в своем составе | 2024 |
|
RU2839765C1 |
| Способ получения N-замещенных мостиковых 1,2,4-диоксазолидинов | 2023 |
|
RU2804396C1 |
| Способ синтеза 3-арилиндан-1-онов реакцией суперэлектрофильного ароматического замещения коричных нитрилов | 2020 |
|
RU2768714C2 |
| СПОСОБ ПОЛУЧЕНИЯ α,ω-ДИ-(СПИРО[АДАМАНТАН-2,3'-[1,2,4,5,7]ТЕТРАОКСАЗОКАН]-7'-ИЛ)АЛКАНОВ | 2019 |
|
RU2727139C1 |
| СПОСОБЫ ФОРМИРОВАНИЯ ГЛИКОЗИДНЫХ СВЯЗЕЙ, ХИМИЧЕСКАЯ КОМПОЗИЦИЯ, ГЛИКОЗИД И ГЛИКОЗИДНАЯ БИБЛИОТЕКА | 1994 |
|
RU2134693C1 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГРИППА | 1997 |
|
RU2169145C2 |
| АРИЛСУЛЬФОНИЛМЕТИЛЬНЫЕ ИЛИ АРИЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ, ВОСПРИИМЧИВЫХ К ЛЕЧЕНИЮ ЛИГАНДАМИ ДОФАМИНОВЫХ D РЕЦЕПТОРОВ, С ИХ ПОМОЩЬЮ | 2005 |
|
RU2442781C2 |
Изобретение относится к эффективному и экологически чистому способу получения 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов формулы 1, где Alk, Alk′ = Me, Et, циклоалкил или другие алкильные группы, а Ar, Ar′ = Ph, p-MeO-C6H4, p-F-C6H4, p-Cl-C6H4 и другие замещенные или незамещенные арильные группы, мультистадийным процессом из 1,1-диалкилпроп-2-ин-1-олов и бензофенонов, заключающийся в том, что введение диазофункции в структуру молекулы осуществляют нитрозированием 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов нитритом натрия в водно-органическом растворе соляной кислоты, конденсацией образующихся 2,2-диалкил-5,5-диарилфуран-3,4(2H,5H)-дионов с p-толуолсульфонилгидразидом и превращением тозилгидразонов в целевые 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-оны в водно-щелочном растворе с выходами до 97% на заключительной стадии этого экологически чистого процесса и общим выходом до 37%. Способ позволяет исключить применение вредных для окружающей среды веществ на всех его этапах. Полученные соединения находят применение в качестве прекурсоров в синтезе биологически активных и лекарственных препаратов. 2 пр.
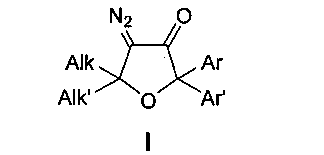
Способ получения тетразамещенных 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов общей формулы I
где Alk, Alk′ = Me, Et, циклоалкил или другие алкильные группы, а Ar, Ar′ = Ph, p-MeO-C6H4, p-F-C6H4, p-Cl-C6H4 и другие замещенные или незамещенные арильные группы, мультистадийным процессом из 1,1-диалкилпроп-2-ин-1-олов и бензофенонов, отличающийся тем, что введение диазофункции в структуру молекулы осуществляют нитрозированием 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов нитритом натрия в водно-органическом растворе соляной кислоты, конденсацией образующихся 2,2-диалкил-5,5-диарилфуран-3,4(2H,5H)-дионов с p-толуолсульфонилгидразидом и превращением тозилгидразонов в целевые 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-оны в водно-щелочном растворе с выходами до 97% на заключительной стадии этого экологически чистого процесса.
| S.A.Malashikhin et al,Synthesis and Structures of Two Isomeric 4-Diazo-2,3,4,5-tetrahydrofuran-3-ones,Helvetica Acta,2008,v.91,no | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| V.A.Nicolaev et al,Surprising secondary photochemical reaction observed on conventional photolysis of diazotetrahydrofuranones,Tetrahedron Letters, 2010,v.51,p | |||
| Воздушная турбина | 1924 |
|
SU2713A1 |
| L.L.Rodina et al, Photochical | |||

