Область техники
Данное изобретение относится к химии органических соединений, фармакологии и медицине и касается новых химических соединений, обладающих высокой эффективностью и селективностью в ингибировании фермента СОХ-2. Данные соединения могут быть использованы в терапии заболеваний, ассоциированных с развитием воспаления, а также заболеваний, опосредованных избыточной секрецией СОХ-2.
Уровень техники
Простагландины (PG), продуцируемые из арахидоновой кислоты посредством фермента циклооксигеназы (СОХ), играют важную роль в развитии воспалительных реакций у человека и животных. Ингибирование фермента циклооксигеназы позволяет остановить биосинтез таких простагландинов, как PGE2, PGG2 и PGH2 и нивелировать симптомы воспаления (отек, боль, гиперемию), связанные с их действием. Известны как минимум две разновидности фермента циклооксигеназы: СОХ-1 и СОХ-2, из которых за развитие воспалительного процесса отвечает только СОХ-2, тогда как СОХ-1 участвует в жизненно важной регуляции секреции в желудочно-кишечном тракте и почках (ЖКТ). Структурно эти ферменты чрезвычайно схожи, и их избирательное ингибирование затруднительно [1]. Длительное ингибирование функций фермента СОХ-1, например, при применении неселективных нестероидных противовоспалительных препаратов (НПВС) таких как аспирин, ибупрофен, диклофенак, индометацин, напроксен и других, часто приводит к развитию желудочно-кишечных язв, кровотечений, деструкции клеточной стенки ЖКТ, может спровоцировать перфорацию органов пищеварения и развитие раковых опухолей [2, 3].
В начале 2000-х годов на фармацевтический рынок были выведены НПВС второго поколения, относящихся к классу селективных ингибиторов фермента СОХ-2 (коксибы). Типичные примеры таких НПВС человека: рофекоксиб ("Vioxx", Merck), целекоксиб ("Celebrex", Pfizer), вальдекоксиб ("Bextra", G.D. Searle & Company), эторикоксиб ("Arcoxia", Merck), и животных: фирококсиб ("Previcox"), деракоксиб ("Deramaxx", Novartis), капрофен ("Rimadyl"). Данный класс препаратов объединяет строение молекулы действующего вещества - все они 1,2-диарилзамещенные гетероциклические соединения, содержащие в своем составе группы SO2NH2 или SO2Me, отвечающие за селективность ингибирования СОХ-2 [4]. Однако большинство из известных селективных ингибиторов СОХ-2 при длительном применении вызывают повышенный риск сердечнососудистых осложнений (испытания VIGOR [5], APPROVe [6]), инфаркта миокарда и инсульта, в результате чего, в 2004-2006 годах большая часть селективных ингибиторов СОХ-2 была отозвана с рынков и развитие данной области фармакологии было несколько затруднено. Таким образом, на сегодняшний день нет удовлетворительного решения проблемы длительной и безопасной противовоспалительной терапии различных заболеваний, в частности, терапии различных форм хронического артрита, остеоартрита, ревматоидного артрита и других заболеваний суставов.
Тем не менее, причина и степень выраженности осложнений со стороны сердечнососудистой системы достаточно широко варьируется в зависимости от строения ингибитора, так как влияние коксибов в каскаде арахидоновой кислоты распространяется не только на СОХ-пути, но и косвенно также на LOX и CYT-P450 траектории [7]. Широкомасштабные исследования сравнительной кардиоваскулярной безопасности НПВС, выполненные после запрещения рофекоксиба (2006-2012 г.), показали [7], что такие классические НПВС как ибупрофен, диклофенак, индометацин - часто превосходят селективные ингибиторы СОХ-2 по выраженности побочных эффектов. При этом анальгетическое действие более выражено у последних, степень опасности которых, в целом, является переоцененной [8-9]. В настоящее время, особое внимание уделяется также новым областям применения ингибиторов СОХ-2 - это терапия некоторых форм рака (2,5-диметил целекоксиб, априкоксиб, тилмакоксиб (JTE-522)) [10], а также психических [11] и нейродегенеративных заболеваний (болезнь Альцгеймера) [12].
Необходимо также отметить, что большинство (90%) известных гетероциклических cis-диарил коксибов имеют совершенно определенный порядок арильных заместителей -идрофобный арил соседствует с гидрофильной (и/или стерически объемной) группой или гетероатомом в гетероцикле. Перестановка положения SO2X-замещенного арила (т.е. гидрофильного арила) и гидрофильного арила (например, галоген-замещенного фенила) в ряде случаев приводит к полной утрате активности соединения по отношению к ферменту СОХ-2 [13]. Тем не менее, имеется ряд обратных примеров, когда перестановка арильных групп не ведет к утрате ингибирующей способности [14]. В этом случае, можно ожидать проявление каких-то новых свойств ингибитора, например, данная структурная особенность может повлиять на степень выраженности кардиоваскулярных побочных эффектов применения таких соединений.
В этой связи поиск новых высокоэффективных лекарственных препаратов, селективных ингибиторов циклооксигеназы-2, не вызывающих развития побочных эффектов, является одним из основных направлений создания новых фармакологических средств для лечения воспалительных заболеваний и острой боли, в том числе: суставного синдрома при ревматизме и обострении подагры, ревматоидного артрита, псориатического артрита, остеоартроза, радикулита, воспалительных процессов связок и сухожилий; воспалений, вызванных укусами ядовитых змей, скорпионов и других животных; воспалений, вызванных солнечными ожогами, повреждениями кожного покрова под действием ультрафиолетового света; воспалений, вызванных различными формами онкологических заболеваний, такими как рак кожных покровов, колоректальный рак, рак молочной железы, рак предстательной железы, и другими формами рака.
Раскрытие изобретения
Задачей настоящего изобретения является разработка и получение новых эффективных ингибиторов циклооксигеназы-2 (СОХ-2), являющихся перспективными для лечения заболеваний воспалительного генеза или заболеваний, ассоциированных с избыточной секрецией циклооксигеназы-2.
Техническим результатом данного изобретения является разработка и получение новых соединений, обладающих высокой ингибирующей активностью и селективностью по отношению к ферменту циклооксигеназе-2. Указанные соединения предпочтительно ингибируют циклооксигеназу-2 по сравнению с циклооксигеназой-1, что, в свою очередь, снижает вероятность возникновения побочных эффектов, связанных с деструктивными поражениями желудочно-кишечного тракта, почек и печени [2, 3]. Данные соединения являются перспективными для применения в терапии заболеваний воспалительного генеза и/или заболеваний, ассоциированных с избыточной секрецией циклооксигеназы-2, в частности, для лечения боли (как хронической, так и острой), лихорадки и воспаления при различных состояниях и заболеваниях.
Указанный технический результат достигается путем получения соединений общей формулы (I):
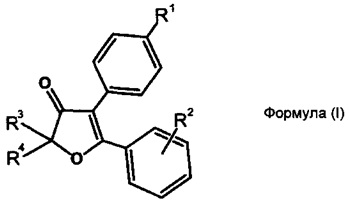
или их стереоизомеров, энантиомеров, фармацевтически приемлемых солей, сольватов или гидратов, где:
R1 представляет собой сульфонамидную группу -SO2NHAlk, где Alk выбирают из метила, этила, изопропила или н-пропила или незамещенную сульфонамидную (-SO2NH2) или сульфо (-SO2OH) группу;
R2 выбирается независимо и представляет собой галоген или водород, причем указанные заместители находятся в мета- или параположении по отношению к точке присоединения фенильной группы;
R3 и R4 выбираются независимо и представляют собой -С1-5-алкил или -С3-7-циклоалкилалкил, причем R3 и R4 совместно с атомом углерода, к которому они присоединены, могут образовывать насыщенный цикл, содержащий от 3 до 7 атомов.
В некоторых предпочтительных вариантах воплощения изобретения R2 выбирается независимо и представляет собой фтор или хлор.
Отдельный подкласс соединений, представляющих интерес, включает соединения формулы (I), выбранных из группы:
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-фенилдигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(4'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(3'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(4'-хлорфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2H)-он;
4-(5,5-диметил-4-оксо-2-фенил-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(4'-фторфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(4'-хлорфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(3'-фторфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(3'-хлорфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
Указанный технический результат также достигается путем получения соединений общий формулы (II):
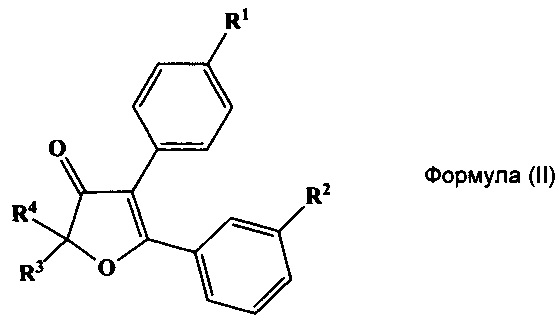
или их стереоизомеров, энантиомеров, фармацевтически приемлемых солей, сольватов или гидратов, где:
R1 выбирается независимо и представляет собой C1-3-алкилсульфонил или C1-3-алкилсульфинил группу;
R2 выбирается независимо и представляет собой галоген;
R3 и R4 выбираются независимо и представляют собой -C1-5-алкил или -С3-7-циклоалкилалкил, причем R3 и R4 совместно с атомом углерода, к которому они присоединены, могут образовывать насыщенный цикл, содержащий от 3 до 7 атомов.
В некоторых предпочтительных вариантах воплощения изобретения R2 выбирается независимо и представляет собой фтор или хлор.
Еще один отдельный подкласс соединений, представляющих интерес, включает соединения формулы (II), выбранных из группы:
2,2-Диметил-4-[4'-(метилсульфинил)фенил]-5-(3'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(метилсульфонил)фенил]-5-(3'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(метилсульфинил)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(метилсульфонил)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2H)-он.
Кроме того, указанный технический результат также достигается путем получения соединений общий формулы (III):
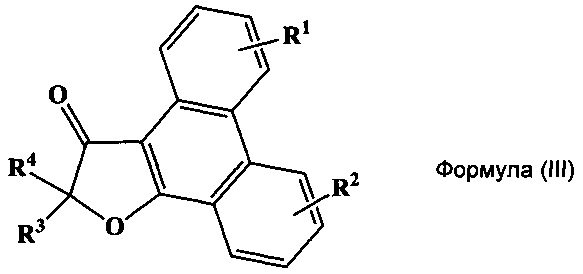
или их стереоизомеров, энантиомеров, фармацевтически приемлемых солей, сольватов или гидратов, где:
R1 выбирается независимо и представляет собой C1-3-алкилсульфонил, C1-3-алкилсульфинил, замещенный сульфонамид (-SO2NHAlk, где Alk выбирают из метила, этила, изопропила или н-пропила) или незамещенный сульфонамид (-SO2NH2), сульфо (-SO2OH) группу, C1-3-алкилтио группу, водород или галоген, причем указанные заместители находятся в третьем или четвертом положении фенантренового фрагмента;
R2 выбирается независимо и представляет собой C1-3-алкилсульфонил, С1-3-алкилсульфинил, замещенный сульфонамид (-SO2NHAlk, где Alk выбирают из метила, этила, изопропила или н-пропила) или незамещенный сульфонамид (-SO2NH2), сульфо (-SO2OH) группу, C1-3-алкилтио группу, водород или галоген, причем указанные заместители находятся в пятом или шестом положении фенантренового фрагмента;
причем если R1 представляет собой водород, то R2 не может быть водородом и наоборот;
R3 и R4 выбираются независимо и представляют собой -С1-5-алкил или -С3-7-циклоалкилалкил, причем R3 и R4 совместно с атомом углерода, к которому они присоединены, могут образовывать насыщенный цикл, содержащий от 3 до 7 атомов.
В некоторых предпочтительных вариантах воплощения изобретения R1 и R2 выбираются независимо и представляют собой фтор или хлор.
Другой отдельный подкласс соединений, представляющих интерес, включает соединения формулы (III), выбранных из группы:
2',2'-Диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'H)-он;
2',2'-Диметил-3-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
2',2'-Диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
2',2'-Диметил-3-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он
2',2'-Диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
2',2'-Диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
2',2'-Диметил-6-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
6-фтор-2',2'-Диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
6-фтор-2',2'-Диметил-3-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'H)-он;
6-фтор-2',2'-Диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
6-фтор-2',2'-Диметил-3-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-фтор-2',2'-Диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2H)-он;
3-фтор-2',2'-Диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'H)-он;
3-фтор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-фтор-2',2'-Диметил-6-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
6-хлор-2',2'-Диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
6-хлор-2',2'-Диметил-3-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
6-хлор-2',2'-Диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
6-хлор-2',2'-Диметил-3-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
3-хлор-2',2'-Диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-хлор-2',2'-Диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'H)-он;
3-хлор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
3-хлор-2',2'-Диметил-6-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
5-хлор-2',2'-Диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
5-хлор-2',2'-Диметил-3-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
5-хлор-2',2'-Диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
5-хлор-2',2'-Диметил-3-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
4-хлор-2',2'-Диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
4-хлор-2',2'-Диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
4-хлор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
4-хлор-2',2'-Диметил-6-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
5-фтор-2',2'-Диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
5-фтор-2',2'-Диметил-3-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'H)-он;
5-фтор-2',2'-Диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
5-фтор-2',2'-Диметил-3-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
4-фтор-2',2'-Диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2H)-он;
4-фтор-2',2'-Диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
4-фтор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
4-фтор-2',2'-Диметил-6-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он.
Данное изобретение также относится к применению соединений, являющихся предметом изобретения, в качестве ингибиторов фермента циклооксигеназы-2.
В некоторых вариантах воплощения изобретения соединения, являющиеся предметом настоящего изобретения и проявляющие свойства ингибиторов циклооксигеназы-2, могут быть использованы в качестве противовоспалительных, анальгетических, жаропонижающих, противоартритных, противоопухолевых средств [15, 16], средств, замедляющих ангиогенез новых сосудов раковых опухолей [17], средств, повышающих иммунную реакцию организма по отношению к раковым клеткам [18], средств, замедляющих пролиферацию раковых клеток [19, 20], а также для лечения болезни Альцгеймера.
Соединения по настоящему изобретению могут применяться для лечения боли (как хронической, так и острой), лихорадки и воспаления при различных состояниях и заболеваниях, в частности, таких заболеваниях как: артрит, суставной синдром при ревматизме и обострении подагры, ревматоидный артрит, псориатический артрит, остеоартроз, радикулит, болезни Альцгеймера, головной, зубной или менструальной боли воспалительные процессы связок, сухожилий, ишиас, люмбаго, воспаление, связанное с солнечными ожогами, повреждениями кожного покрова под действием ультрафиолетового света, различными формами онкологических заболеваний, в частности онкологических заболеваний кожного покрова.
Настоящее изобретение также относится к применению соединений, являющихся предметом изобретения, для получения фармацевтической композиции для лечения заболеваний воспалительного генеза или заболеваний, ассоциированных с избыточной секрецией фермента циклооксигеназы-2.
Кроме того, изобретение предусматривает фармацевтические композиции для лечения заболеваний воспалительного генеза или заболеваний, ассоциированных с избыточной секрецией фермента циклооксигеназы-2, содержащие эффективное количество, по меньшей мере, одного соединения, являющегося предметом настоящего изобретения, и, по меньшей мере, один фармацевтически приемлемый носитель и/или разбавитель. В некоторых частных вариантах воплощения изобретения, фармацевтическая композиция характеризуется тем, что содержит соединения по изобретению и его региоизомер относительно перестановки местами заместителей R1 и R2.
В некоторых вариантах воплощения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, могут быть использованы в качестве противовоспалительных, анальгетических, жаропонижающих средств.
В некоторых других вариантах воплощения изобретения фармацевтические композиции по изобретению могут применяться для лечения заболеваний, представляющих собой артрит, кожное воспалительное заболевание, воспаление при сосудистых заболеваниях, легочное воспаление, тиреоидит, воспаление, вязанное с общим переохлаждением, гриппом или вирусной инфекцией. Кроме того, более конкретно артрит представляет собой ревматоидный артрит, остеоартрит, подагрический артрит, ювенильный артрит, спондилолистез, а кожное воспалительное заболевание представляет собой экзему, дерматит, псориаз.
Соединения по изобретению могут быть пригодны для лечения млекопитающих, предпочтительно человека, а также животных, включая, но не ограничиваясь, овец, собак, кошек, лошадей, свиней, крыс, мышей, кроликов и других.
Соединения по изобретению могут быть использованы в комбинации с, по меньшей мере, одним другим терапевтическим агентом.
Изобретение также включает получение соединений общих формул (I), (II) и (III).
Изобретение также относится к способу лечения заболеваний воспалительного генеза или заболеваний, ассоциированных с избыточной секрецией фермента циклооксигеназы-2 путем введения пациенту фармацевтически эффективного количества соединения по изобретению или фармацевтической композиции по изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В результате проведенных исследований было обнаружено, что соединения по изобретению избирательно и эффективно ингибируют СОХ-2. Указанные соединения полезны при лечении очень широкого спектра нарушений, опосредованных СОХ-2, включая, но не ограничиваясь, нарушениями, характеризующимися воспалением, болью и/или лихорадкой. Такие композиции особенно полезны в качестве противовоспалительных агентов, в частности, при лечении артрита, имея дополнительное преимущество, состоящее в проявлении значительно менее опасных побочных эффектов, чем известные из уровня техники нестероидные противовоспалительные препараты, которые обладают невысокой селективностью к СОХ-2 относительно СОХ-1. В частности, соединения по изобретению имеют сниженный потенциал желудочно-кишечной токсичности и раздражения желудочно-кишечного тракта, включая образование язвы в верхней части желудочно-кишечного тракта и кровотечение, сниженный потенциал почечных побочных эффектов. Поэтому соединения по изобретению, селективно ингибирующие СОХ-2, особенно полезны в качестве альтернативы общеизвестным нестероидным противовоспалительным препаратам, где такие препараты противопоказаны, например, у пациентов с язвами желудка, гастритом, регионарным энтеритом, язвенным колитом, дивертикулитом или с рецидивирующими желудочно-кишечными нарушениями; при желудочно-кишечном кровотечении, нарушениях свертываемости крови, включая анемию, такую как гипопротромбинемия, гемофилию или других проблемах, связанных с кровотечением; при болезни почек; или у пациентов перед хирургическим вмешательством, или у пациентов, принимающих антикоагулянты.
Такие соединения полезны при лечении боли, включая, но не ограничиваясь, болью после операции, зубной болью, мышечной болью и болью, возникающей при онкологическом заболевании. Такие композиции полезны, например, для ослабления боли, лихорадки и воспаления при различных состояниях, включая поясничную и шейную боль, дисменорею, головную боль, зубную боль, растяжения и деформации, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, дегенеративные суставные заболевания (остеоартрит), подагру, бурсит, ожоги и травмы после хирургических и стоматологических вмешательств.
Определения и термины
Термин «алкил» в настоящем документе означает как неразветвленные, так и разветвленные алкилы. Кроме того, термин «алкил» в настоящем документе относится к группам, обычно имеющим от одного до трех атомов углерода. Например, термин -С1-3-алкил означает метил, этил, изопропил, н-пропил.
Термин «циклоалкил» в настоящем документе относится к группам, имеющим от трех до семи атомов углерода в моноциклической структуре. В качестве иллюстрации, циклоалкилы включают, но не ограничиваются, следующими радикалами: циклопропил, циклобутил, циклопентил, циклогексил.
Термин «арил» в настоящем документе означает группы, содержащие ароматический цикл, имеющий шесть атомов углерода. Примером арильных циклических групп является фенил.
Термин «галоген» сам по себе или в части другого термина относится к атому фтора, хлора, брома или йода.
Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40°C в отсутствие химически активных условий, в течение, по крайней мере, одной недели.
Термин «заместитель» означает химический радикал, который присоединяется к молекулярному остову (фрагменту), например «заместитель алкильный», «заместитель циклической системы», значения которых определены в данном документе.
Если не указано иначе, изображенные здесь структуры также подразумевают и все стереоизомеры, то есть R- и S- изомеры для каждого ассиметричного центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси настоящих соединений, также являются предметом данного изобретения. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%, а предпочтительно >95% мольной чистоты), так же как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например путем получения диастереоизомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания, эфира или соли.
Термин «региоизомер» охватывает в настоящем документе соединения, получающиеся перестановкой заместителей R1 и R2 в общих формулах (I), (II), или (III) вместе с их положениями в бензольном кольце, между собой. Так, например, региоизомерами, в данном документе, для соединений формул (I), (II), (III) будут соединения формулы (I*), (II*), (III*):
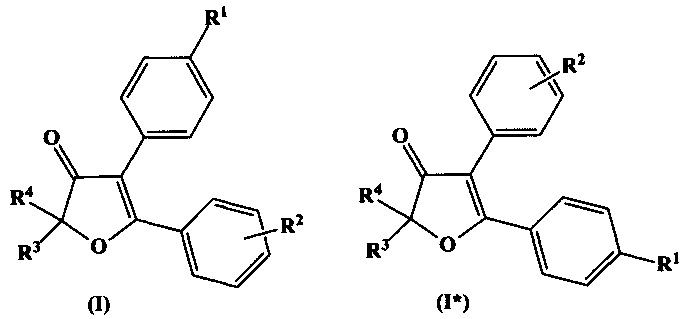
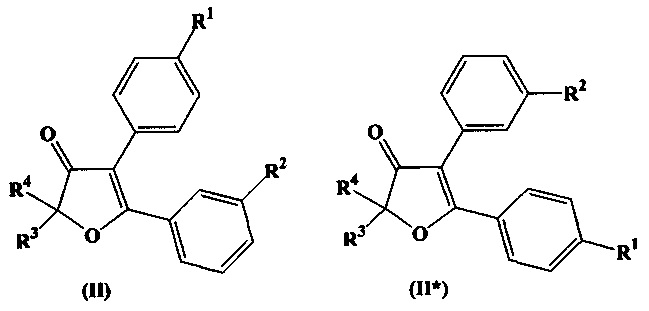
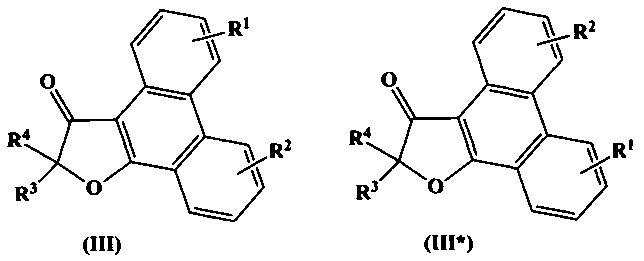
При этом, например, в формулах (II) и (II*) заместитель R1 сохраняет параположение, а заместитель R2 - сохраняет метаположение, в соответствии с данным определением.
Нумерация в фенантреновом фрагменте соединений общей формулы (III) в данном документе полагается жесткозакрепленной, постоянной и независящей от заместителей R1 и R2 так, как это показано на следующем рисунке:
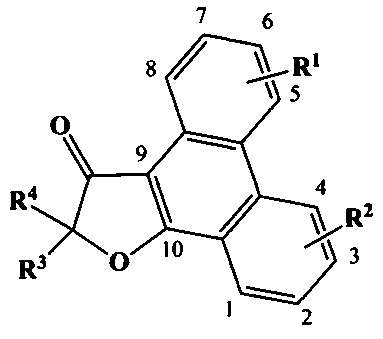
Термин «пациент» охватывает все виды млекопитающих, предпочтительно человека.
Термины «лечение», «терапия» охватывают лечение патологических состояний у млекопитающих, предпочтительно у человека, и включают: а) блокирование (приостановку) течения заболевания, б) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания.
Термин «воспаление» - (лат. inflammatio) - это комплексный, местный и общий патологический процесс, возникающий в ответ на повреждение (alteratio) клеточных структур организма или действие патогенного раздражителя и проявляющийся в реакциях (exudatio и др.), направленных на устранение продуктов повреждения, а если возможно, то и агентов (раздражителей), а также приводящий к максимальному для данных условий восстановлению (proliferatio и др.) в зоне повреждения.
Термин «терапевтически эффективное количество» подразумевает такое количество соединения, которое при введении в качестве моно- или комбинированной терапии вызывает ингибирование фермента СОХ-2, достаточное для лечения заболевания млекопитающего, ассоциированного с развитием воспаления, а также заболевания, опосредованного избыточной секрецией СОХ-2. При применении соединений по изобретению в комбинированной терапии термин «терапевтически эффективное количество» относится к комбинации количеств активных ингредиентов, прием которых ведет к превентивному или терапевтическому эффекту при последовательном или одновременном приеме. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от вида млекопитающего, возраста и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Термин «сольват» относится к ассоциации или комплексу из одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин «гидрат» относится к комплексу, где молекулами растворителя является вода.
Краткое описание чертежей
Рисунок 1. Значения нормализованных потенциальных энергий (ккал/моль) для аминокислот в составе фермента hCOX-2 после процедуры «отжига».
Рисунок 2. Значения нормализованных потенциальных энергий (ккал/моль) для аминокислот в составе фермента hCOX-1 после процедуры «отжига».
Рисунок 3. Аминокислоты в hCOX-1 с высокими значениями стресса (серые сферы), сайт связывания показан желтыми сферами.
Рисунок 4. Референсный набор известных ингибиторов hCOX-1 и hCOX-2. Соединения Е-8, Е-9, Е-13, Е-14 относятся к СОХ-ингибиторам с измененным порядком следования арильных групп относительно общепринятого.
Рисунок 5. Внутренняя валидация модели с использованием молекулы целекоксиба, активная конформация: оранжевый - данные рентгеноструктурного анализа, серый - докинг: а) hCOX-2 (скоринг: - 14.27 ккал/моль), RMSD=0.13 б) hCOX-1 (скоринг: - 13.39 ккал/моль); RMSD=0.21.
Рисунок 6. Общий вид трехцентрового фармакофора с молекулой целекоксиба. Фармакофор интегрирован в in silico модель (Acc - потенциальный акцептор водородной связи, Aro - ароматическая область).
Рисунок 7. QSAR модель для СОХ-2 в форме линейной аппроксимации.
Рисунок 8. QSAR модель для СОХ-1 в форме линейной аппроксимации.
Рисунок 9. Исследованные структуры ингибиторов СОХ-1 и СОХ-2 по изобретению.
Рисунок 10. Структуры некоторых известные ингибиторы циклооксигеназы:
а) Рофекоксиб: IC50 (СОХ-2)=0.06 мкг/мл, IC50 (СОХ-1)=100 мкг/мл, селективность 1700;
б) Целекоксиб: IC50 (СОХ-2)=0.02 мкг/мл, IC50 (СОХ-1)=1.9 мкг/мл, селективность 100;
в) Кетопрофен: IC50 (СОХ-2)=0.13 мкг/мл, IC50 (СОХ-1)=0.6 мкг/мл, селективность 4.6.
Рисунок 11. Докинг 2,2-диметил-4-[4'-(аминосульфонил)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (1u) в сайте связывания СОХ-2. Сравнение с целекоксибом (оранжевая структура). Скоринг: - 8.631 ккал/моль.
Рисунок 12. Докинг 2,2-диметил-4-[4'-(аминосульфонил)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (1v) в сайте связывания СОХ-2. Сравнение с целекоксибом (оранжевая структура). Скоринг: - 8.495 ккал/моль.
Рисунок 13. Докинг 3-хлор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-она (2е) в сайте связывания СОХ-2. Сравнение с целекоксибом (оранжевая структура). Скоринг: - 7.499 ккал/моль.
Рисунок 14. Докинг 4-фтор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-она (2h) в сайте связывания СОХ-2. Сравнение с целекоксибом (оранжевая структура). Скоринг: - 7.138 ккал/моль.
Осуществление изобретения
Обзор методов получения соединений изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Следует понимать, что эти и все приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Спектры ЯМР 1Н (400 МГц) и 13С (100 МГц) регистрировали на спектрометре «Bruker 400 MHz Avance» в дейтерохлороформе, CHCl3 (реперный сигнал 7.26 м.д.). ИК спектры получены на спектрофотометре «Perkin-Elmer ВХII» в диапазоне 4000-400 см-1 в растворителе тетрахлоруглероде или в таблетке KBr. Для снятия масс-спектров использовали хромато-масс спектрометр «Bruker microTOF». Температуру плавления измеряли на приборе «Buchi В-540».
Реакции проводили в безводных растворителях, очищенных по стандартным методикам. Для препаративного разделения реакционных смесей использовали колоночную и флеш-хроматографию на силикагеле Silicagel L 40/100 мкм в градиентном режиме, а также препаративную ТСХ на пластинах Silufol UV-254 (Kavalier,  ). Аналитическую ТСХ проводили на пластинах Silufol UV-254, элюенты - гексан, петролейный эфир, хлористый метилен, ацетон, этилацетат и смесь растворителей в различном соотношении.
). Аналитическую ТСХ проводили на пластинах Silufol UV-254, элюенты - гексан, петролейный эфир, хлористый метилен, ацетон, этилацетат и смесь растворителей в различном соотношении.
Соединения, приведенные в описании без ссылок, приготовлены по стандартным лабораторным методикам либо являются коммерчески доступными веществами.
Промежуточные продукты для получения некоторых соединений по изобретению могут быть получены по нижеописанным методикам.
Получение интермедиата 2,2-диметил-4-[4'-(метилтио)фенил]-5-фенилфуран-3(2H)-она (Схема 1:1а)
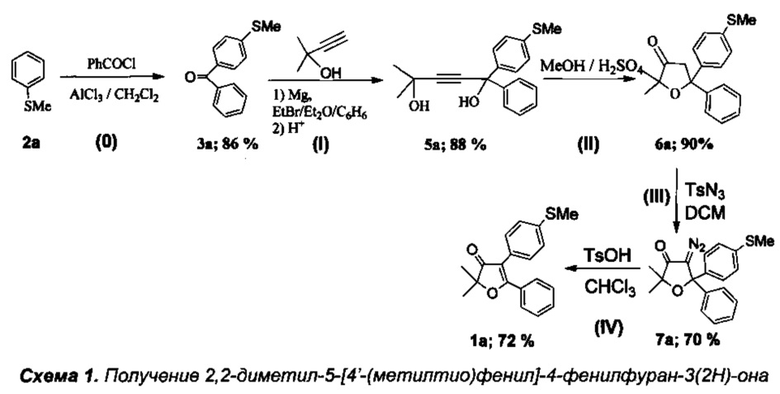
Стадия 0. Получение 4'-(метилтио)фенил(фенил)метанона (3а).
К охлажденному до 5°C раствору 25 мл (25.5 г, 0.2 моль) тиоанизола (2а) в 200 мл хлористого метилена при перемешивании добавили 30 г безводного AlCl3. Затем в течение 1.5 ч при интенсивном охлаждении до 0-5°C по каплям добавили 23 мл (28 г, 0.2 моль) хлористого бензоила, при этом смесь приобрела оранжевый цвет и большую вязкость. После добавления всего хлористого бензоила смесь кипятили с обратным холодильником в течение 3.5 ч. По завершению реакции оранжевый осадок комплекса с хлоридом алюминия отфильтровали на фильтре Шотта, промыли дихлорметаном и гидролизовали: в начале холодной 7% HCl, а затем довели гидролиз до полного обесцвечивания продукта путем кипячения. Затвердевший при охлаждении кетон 3а был растворен в дихлорметане, промыт H2O и высушен над Na2SO4 в течение 12 часов. После отгонки растворителя получили 33.3 г 4'-(метилтио)фенил(фенил)метанона (3а). Темно-оранжевый маточный раствор комплекса в дихлорметане гидролизовали кипячением с 7% водным раствором HCl до полного обесцвечивания, затем органический слой отделили от водного, водный слой экстрагировали дихлорметаном (3×15 мл). Объединенные органические фракции были промыты водой и высушены над Na2SO4. После отгонки растворителя продукт был перекристаллизован из гексана и высушен на воздухе (1 сутки). Получили 5.95 г соединения 3a. Общий выход 39.2 г (86%), бесцветные кристаллы, т.пл. 79-80°C.
Спектр ЯМР 1Н (400 МГц, 32 мг в 0.8 мл CDCl3), δ, м.д.: 2.53 с (3Н, SCH3), 7.29 д (2H), 7.46 т (2H), 7.52-7.60 м (1Н), 7.73 д (2H), 7.75 д (2H).
Спектр ЯМР 13С (100 МГц, 32 мг в 0.8 мл CDCl3), δ, м.д.: 14.8 (SCH3), 124.8, 128.2, 129.7, 130.5, 132.1, 133.5, 137.7, 145.2 (все CArAr'), 195.8 (С=O).
ИК спектр (в CCl4), см-1: 836 ср., 920 ср., 936 ср., 1090 с., 1175 ср., 1282 с., 1315 с., 1400 ср., 1445 ср., 1589 с., 1658 с. (С=O), 2923 сл., 2990 сл., 3025 сл., 3062 сл., 3083 сл.
Масс-спектр HRMS, m/z найдено: 229.0689 [МН]+, вычислено: 229.0682.
Стадия I. Получение 2-метил-5-фенил-5-(4'-метилтиофенил)пент-3-ин-2,5-диола (5а).
К раствору EtMgBr, приготовленному из 5 г (0.21 моль) магния и 22.7 г (0.21 моль) этилбромида, в 60 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 7.85 мл (0.081 моль) 2-метилбут-3-ин-2-ола в 50 мл абсолютного бензола. Смесь кипятили в течение 2 ч, затем при охлаждении добавили по каплям раствор 13 г (0.0575 моль) соединения 3а в 50 мл бензола и кипятили в течение 5 ч. Затем смесь охладили в водяной бане и осторожно гидролизовали при помощи 150 мл 5% холодного раствора соляной кислоты. Органический слой отделили от водного. Водный слой экстрагировали бензолом (3×20 мл). Объединенные органические фракции промыли раствором NaHCO3, водой и высушили над Na2SO4. После отгонки растворителя полученное желтое маслообразное вещество высушили в вакууме при давлении 0.01÷0.05 торр в течение 5 ч. Выход 5а 16.2 г (88%), бесцветное маслообразное вещество. Без дополнительной очистки полученное соединение использовали на следующей стадии.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 1.57 с (6Н, 2СН3), 2.53 с (3Н, SCH3), 7.17 д (2H), 7.22-7.27 м (1Н), 7.31 т (2H), 7.48 д (2H), 7.52-7.58 м (2H).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 15.5 (SCH3), 31.3 (2СН3), 65.2 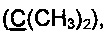 73.9, 84.7
73.9, 84.7 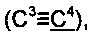 91.8
91.8 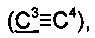 125.8, 126.1, 126.4, 127.8, 128.4, 138.1, 141.8, 144.8.
125.8, 126.1, 126.4, 127.8, 128.4, 138.1, 141.8, 144.8.
ИК спектр (в CCl4), см-1: 700.9 сл., 908.5 сл., 951.7 сл., 999.6 сл., 1167.6 сл., 1325.6 сл., 1449.0 сл., 1490.0 сл., 2854.7 ср., 2926.6 с., 3606.5 сл.
Масс-спектр HRMS, m/z, рассчитано: 335.1077, найдено: 335.1087 [MNa]+.
Стадия II. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-5-фенил-дигидрофуран-3(2H)-она (6а).
15.8 г (0.051 моль) соединения 5а растворили в 50 мл кипящего метанола и при перемешивании по каплям добавили 2.5 мл (0.045 моль) 98% серной кислоты, растворенной в 7.5 мл метанола. Смесь кипятили в течение 2 ч до окончания реакции (контроль по ТСХ). Затем смесь вылили в 100 мл воды и тщательно экстрагировали хлористым метиленом (3×50 мл). Органический слой высушили над Na2SO4, растворитель отогнали на роторе, а полученное вещество высушили от метанола и дихлорметана в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Полученное масло закристаллизовали с использованием затравки и повторно высушили в вакууме. Получено 14.2 г вещества (выход 90%) 6а, бесцветные кристаллы, т.пл. 64-65°C. Без дополнительной очистки полученное соединение использовали на следующей стадии.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.7 мл CDCl3), δ, м.д.: 1.21 д (6Н, 2СН3), 2.45 с (3Н, SCH3), 3.30 с (2Н, СН2), 7.17 д (2H), 7.21-7.26 м (1Н), 7.26-7.36 м (4Н), 7.40 д (2H).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.7 мл CDCl3), δ, м.д.: 15.6 (SCH3), 25.3 (2СН3), 47.9 (СН2), 81.4 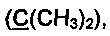 82.4, 125.9, 126.1, 126.6, 127.3, 128.3, 137.7, 142.8, 146.0, 216.9 (С=O).
82.4, 125.9, 126.1, 126.6, 127.3, 128.3, 137.7, 142.8, 146.0, 216.9 (С=O).
ИК спектр (в CCl4), см-1: 961 сл., 994 сл., 1095 сл., 1133 сл., 1173 сл., 1281 сл., 1376 сл., 1447 сл., 1493 сл., 1599 сл., 1760 с., 2925 сл., 2984 сл., 3028 сл., 3064 сл.
Масс-спектр HRMS, m/z, рассчитано: 313.1257, найдено: 313.1259 [МН]+.
Стадия III. Получение 4-диазо-2,2-диметил-5-[4'-(метилтио)фенил]-5-фенил-дигидрофуран-3(2H)-она (7а).
К смеси 250 мг (0.8 ммоль) фуранона 6а и 195 мг (0.99 ммоль) паратолуолсульфонилазида в 15 мл дихлорметана добавили по каплям раствор 50 мг (0.33 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 1 мл дихлорметана, при этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 35°C в течение 6 ч до завершения реакции (контроль с помощью ТСХ), затем охладили, удалили в вакууме избыток растворителя, перенесли остаток на колонку с силикагелем, и хроматографировали в градиентном режиме смесью н-гексан-хлористый метилен. После удаления растворителя кристаллический остаток высушили в вакууме и получили 191 мг (70%) диазокетона 7а, желтое кристаллическое вещество, т.пл. 134-135°C.
Спектр ЯМР 1Н (400 МГц, 30 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.35 с (6Н, 2СН3), 2.48 с (3Н, SCH3), 7.23 с (4Н), 7.29-7.41 м (5Н).
Спектр ЯМР 13С (100 МГц, 30 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.4 (SCH3), 25.2 с (2СН3), 83.9 (С5), 85.5 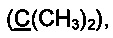 126.1, 126.3, 126.9, 128.4, 128.5, 139.1, 140.4, 143.5 (все CArAr'), 196.9 (С=O).
126.1, 126.3, 126.9, 128.4, 128.5, 139.1, 140.4, 143.5 (все CArAr'), 196.9 (С=O).
ИК спектр (в CCl4), см-1: 1025 сл., 1094 сл., 1165 сл., 1226 сл., 1346 ср., 1378 сл., 1493 сл., 1696 ср. (С=O), 2091 с. (C=N2), 2926 сл., 2982 сл.
Масс-спектр, m/z, рассчитано: 361.0982, найдено: 361.0987 [MNa]+.
Стадия IV. Получение 2,2-диметил-4-[4'-(метилтио)фенил]-5-фенилфуран-3(2H)-она (1а).
Реакция проводится в атмосфере азота. К раствору 200 мг (0.59 ммоль) диазокетона 7а в 10 мл дихлорметана добавили по каплям 0.1 мл трифторметансульфоновой кислоты (TfOH), растворенной в 5 мл хлористого метилена. Смесь кипятили 3 часа до полного исчезновения исходного диазокетона (контроль с помощью ТСХ), а по завершении реакции смесь промыли водным раствором NaHCO3, водой (2×5 мл) и высушили над K2CO3. После удаления растворителя получили 180 мг (98%) смеси двух региоизомерных 2,2-диалкил-4,5-диарил-дигидрофуран-3(2H)-онов в примерном соотношении 3:1 в виде светло-желтого маслообразного продукта. Реакционную смесь (180 мг) разделили на колонке с силикагелем и получили фуранон 1а с выходом 130 мг (72%), светло-желтые кристаллы, т.пл. 120-121°C.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.55 с (6Н, 2СН3), 2.49 с (3Н, SCH3), 7.23-7.26 м (4Н), 7.37 т (2Н, 3JHH=7.6 Гц), 7.44-7.49 м (1Н), 7.67 д (2Н, 3JHH=7.4 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.7 (SCH3), 23.4 (2СН3), 87.0 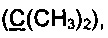 113.0 (С4), 126.6, 128.5, 128.4, 129.7, 131.8, 137.7 (все CArAr'), 178.1 (С5), 205.4 (С=O).
113.0 (С4), 126.6, 128.5, 128.4, 129.7, 131.8, 137.7 (все CArAr'), 178.1 (С5), 205.4 (С=O).
Спектр ИК (в CCl4), см-1: 1051 сл., 1094 сл., 1167 сл., 1242 сл., 1384 ср., 1502 сл., 1574 сл., 1591 сл., 1618 сл., 1699 с., 2926 сл., 2982 сл.
Масс-спектр HRMS, m/z, рассчитано: 311.1101, найдено: 311.1107 [МН]+.
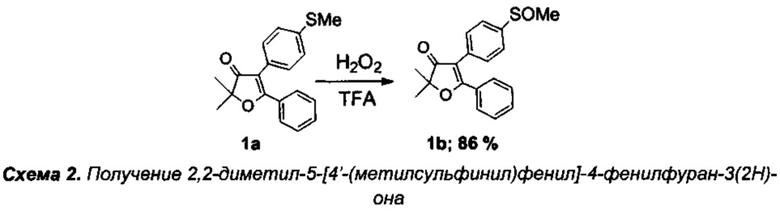
Получение интермедиата 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-фенилфуран-3(2H)-она (Схема 2:1b)
Получение соединения 1а описано выше.
Получение 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-фенилфуран-3(2H)-она (1b).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты, растворили в ней при +50°C 300 мг (0.96 ммоль) соединения 1а, и по каплям при интенсивном перемешивании в течение 1 ч осторожно добавили смесь 100 мг 35% (0.96 ммоль) водного раствора пероксида водорода в 3 мл трифторуксусной кислоты и 0.05 мл (1 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное эквимолярное соотношение пероксида водорода и 1а. Реакционную смесь перемешивали еще в течение 3 ч при +50-55°C (контроль с помощью ТСХ), по завершении реакции смесь вылили в воду, экстрагировали этилацетатом (4×20 мл), органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и вакуумной сушки, получили 270 мг кристаллического вещества. Выход целевого соединения 1b составил 270 мг (86%), бесцветное маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.58 с (6Н, 2СН3), 2.74 с (3Н, SOCH3), 7.38 т (2Н, 3JHH=7.8 Гц), 7.49-7.51 м (3H), 7.63 т (4Н, 3JHH=7.3 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.3 (СН3), 23.4 (СН3), 43.9 (SOCH3), 87.6  112.4 (С4), 123.9, 127.9, 128.5, 128.7, 129.5, 130.3, 132.2, 144.6 (все CArAr'), 179.2 (С5), 204.9 (С=O).
112.4 (С4), 123.9, 127.9, 128.5, 128.7, 129.5, 130.3, 132.2, 144.6 (все CArAr'), 179.2 (С5), 204.9 (С=O).
ИК спектр (в CCl4), см-1: 955 сл., 1051 ср., 1090 сл., 1383 с., 1616 ср., 1701 с. (С=O), 2931 сл., 2982 сл.
Масс-спектр HRMS, m/z, рассчитано 349.0869, найдено: 349.0876 [MNa]+.
Получение интермедиата 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-фенилфуран-3(2H)-она (Схема 3:1с)
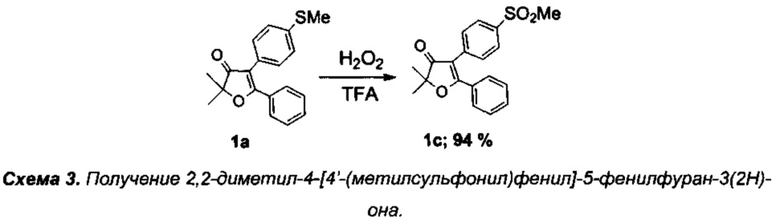
Получение соединения 1а описано выше.
Получение 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-фенилфуран-3(2H)-она (1с).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты, растворили в ней при +40°C 300 мг (0.96 ммоль) соединения 1а и добавили по каплям смесь 0.21 г (1.96 ммоль) 35% водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.05 мл (1 ммоль) 98% H2SO4. Реакционную смесь перемешивали в течение 45 мин при +65-70°C (контроль с помощью ТСХ), по завершении реакции смесь вылили в воду, экстрагировали CH2Cl2 (4×20 мл), органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над K2CO3. После удаления растворителя получили 310 мг (94%) соединения 1с, бесцветное кристаллическое вещество, т.пл. 181-182°C.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.58 с (6Н, 2СН3), 3.04 с (3Н, SO2CH3), 7.38 т (2Н, 3JHH=7.7 Гц), 7.50-7.54 м (3H), 7.61 д (2Н, 3JHH=8.5 Гц), 7.91 д (2Н, 3JHH=8.4 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.3 (2СН3), 43.5 (SO2CH3), 87.9  111.6 (С4), 127.5, 128.4, 128.6, 129.2, 130.1, 132.4, 136.1, 139.0 (все CArAr'), 179.8 (С5), 204.4 (С=O).
111.6 (С4), 127.5, 128.4, 128.6, 129.2, 130.1, 132.4, 136.1, 139.0 (все CArAr'), 179.8 (С5), 204.4 (С=O).
ИК спектр (в CCl4), см-1: 955 ср., 1051 ср., 1157 с., 1327 ср., 1383 ср., 1614 ср., 1701 с. (С=O), 2932 сл., 2982 сл.
Масс-спектр HRMS, m/z, рассчитано: 365.0818, найдено 365.0823 [MNa]+.
Пример 1.
Получение 2',2'-диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-она (Схема 4, 2а).
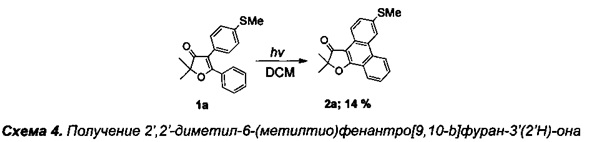
Получение соединения 1а описано выше.
Получение 2',2'-диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-она (2а).
В кварцевый фотохимический реактор с водяным охлаждением и объемом 50 мл, поместили раствор 500 мг (1.61 ммоль) 4,5-диарил-дигидрофуран-3(2H)-она 1а в 50 мл дихлорметана. Облучение проводили ртутной лампой мощностью 150 Вт в течение 4 ч в присутствии кислорода воздуха. По завершении реакции растворитель отогнали на роторном испарителе, смесь разделили при помощи хроматографии на фракции, при этом исходное вещество может быть использовано для повторного фотолиза (элюэнт : гексан-этилацетат-дихлорметан). Выход продукта 2а: 70 мг (14%). Степень конверсии исходного фуранона 6а составила 14%.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.62 с (6Н, 2СН3), 2.64 с (3Н, SCH3), 7.60 дд (1Н, 3JHH=8.5 Гц, 4JHH=1 8 Гц), 7.70 т (1Н, 3JHH=7.3 Гц), 7.90-7.84 м (1Н), 8.33 дд (1Н, 3JHH=8.0 Гц, 4JHH=1.0 Гц), 8.45 д (1Н, 4JHH=1.5 Гц), 8.63 д (1Н, 3JHH=8.4 Гц), 8.76 д (1Н, 3JHH=8.4 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 16.6 (SCH3), 23.3 (2СН3), 89.6 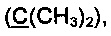 96.1 (С4), 108.2, 121.0, 122.3, 123.4, 123.4, 124.2, 125.2, 127.3, 127.9, 131.4, 135.0, 135.6, 172.4, 203.4 (С=O).
96.1 (С4), 108.2, 121.0, 122.3, 123.4, 123.4, 124.2, 125.2, 127.3, 127.9, 131.4, 135.0, 135.6, 172.4, 203.4 (С=O).
ИК спектр: 2982 сл., 2927 сл., 2855 сл., 1701 с. (С=O), 1626 ср., 1591 ср., 1521 с., 1431 ср., 1181 ср., 1126 ср., 1095 сл., 891 ср.
Масс-спектр HRMS, m/z, рассчитано 331.0764, найдено: 331.0769 [MNa]+.
Пример 2.
Получение 2',2'-диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-она (Схема 5, 2b).
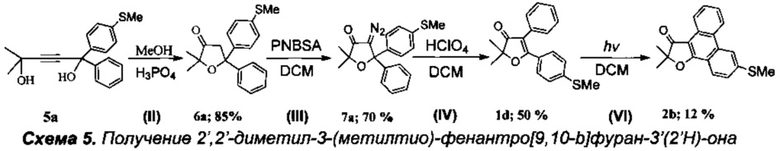
Получение соединения 5а описано выше.
Стадия II. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-5-фенил-дигидрофуран-3(2H)-она (6а).
15.8 г (0.051 моль) соединения 5а растворили в 40 мл кипящего метанола и при перемешивании по каплям добавили смесь 15 мл 98% ортофосфорной кислоты и 10 мл метанола. Смесь кипятили в течение 3 ч до окончания реакции (контроль по ТСХ). Затем смесь вылили в 100 мл воды и тщательно экстрагировали хлористым метиленом (3×50 мл). Органический слой высушили Na2SO4, растворитель отогнали на роторе, а полученное вещество высушили от метанола и дихлорметана в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Полученное масло закристаллизовали с использованием затравки и повторно высушили в вакууме. Получено 12.6 г вещества (выход 80%) 6а. Без дополнительной очистки полученное соединение использовали на следующей стадии.
Стадия III. Получение 4-диазо-2,2-диметил-5-[4'-(метилтио)фенил]-5-фенил-дигидрофуран-3(2H)-она (7а).
К смеси 250 мг (0.8 ммоль) фуранона 6а и 228 мг (0.99 ммоль) паранитробензолсульфонилазида в 15 мл дихлорметана добавили по каплям раствор 50 мг (0.33 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 1 мл дихлорметана, при этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 35°C в течение 6 ч до завершения реакции (контроль с помощью ТСХ), затем охладили и перенесли на колонку с силикагелем, и хроматографировали в градиентном режиме смесью н-гексан - хлористый метилен. После удаления растворителя (азеотропной отгонкой с этанолом) кристаллический остаток высушили в вакууме и получили 189 мг (70%) диазокетона 7а.
Стадия IV. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-4-фенилфуран-3(2H)-она (1d).
Реакция проводится в атмосфере азота. К раствору 200 мг (0.59 ммоль) диазокетона 7а в 10 мл дихлорметана добавили по каплям 5 мл 70% водной хлорной кислоты (HClO4). Смесь кипятили с обратным холодильником при интенсивном перемешивании 3 часа до полного исчезновения исходного диазокетона (контроль с помощью ТСХ), а по завершении реакции органический слой отделили, водный - экстрагировали 10 мл дихлорметана, затем объединенный органический слой промыли водным раствором NaHCO3, водой (2×5 мл) и высушили Na2SO4 и K2CO3. После удаления растворителя получили 180 мг (98%) смеси двух региоизомерных 2,2-диалкил-4,5-диарил-дигидрофуран-3(2H)-онов (в соотношении 1:1) в виде светло-желтого маслообразного продукта. Реакционную смесь (180 мг) разделили на колонке с силикагелем и получили фуранон 1d с выходом 91 мг (50%).
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.55 с (6Н, 2СН3), 2.47 с (3Н, SCH3), 7.15-7.17 м (1Н), 7.25 д (2Н, 3JHH=7.6 Гц), 7.30-7.41 м (4Н), 7.57 д (2Н, 3JHH=7.4 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.7 (SCH3), 23.4 (2СН3), 86.9 (С(СН3)2), 113.1 (С4), 125.0, 128.6, 128.6, 129.5, 131.7, 144.2 (все CArAr'), 177.5 (С5), 205.2 (С=O).
Спектр ИК (в CCl4), см-1: 676 сл., 1052 сл., 1095 сл., 1147 сл., 1168 сл., 1239 сл., 1264 сл., 1386 ср., 1485 сл., 1598 ср., 1615 ср., 1698 с., 2858 сл., 2927 ср., 2960 ср.
Масс-спектр HRMS, m/z, рассчитано: 311.1101, найдено: 311.1113 [МН]+.
Стадия VI. Получение 2',2'-диметил-3-(метилтио)-фенантро[9,10-b]фуран-3'(2'Н)-она (2b)
В кварцевый фотохимический реактор с водяным охлаждением и объемом 50 мл поместили раствор 500 мг (1.61 ммоль) 4,5-диарил-дигидрофуран-3(2H)-она 1d в 40 мл дихлорметана. Облучение проводили ртутной лампой мощностью 150 Вт в течение 4 ч в присутствии кислорода воздуха. По завершении реакции растворитель отогнали на роторном испарителе, смесь нанесли на пластину для препаративной хроматографии и разделили на фракции (элюэнт: гексан-этилацетат-дихлорметан). Выход продукта 2b. 60 мг (12%). Степень конверсии исходного фуранона 1d составила 60%.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.62 с (6Н, 2СН3), 2.71 с (3Н, SCH3), 7.53-7.58 м (2H), 7.66 т (1Н, 3JHH=7.0 Гц), 8.23 д (1Н, 3JHH=8.5 Гц), 8.42 д (1Н, 4JHH=1.5 Гц), 8.51 д (1Н, 3JHH=8.3 Гц), 8.83 дд (1Н, 3JHH=8 Гц, 4JHH=1 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.0 м.д.), δ, м.д.: 15.3 (SCH3), 23.3 (2СН3), 89.5 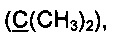 96.1 (С4), 107.5, 119.3, 122.7, 123.6, 123.9, 125.1, 125.4, 125.9, 128.2, 128.8, 136.1, 144.1, 172.6, 203.1 (С=O).
96.1 (С4), 107.5, 119.3, 122.7, 123.6, 123.9, 125.1, 125.4, 125.9, 128.2, 128.8, 136.1, 144.1, 172.6, 203.1 (С=O).
ИК спектр (в CCl4), см-1: 2982 сл., 2929 сл., 2857 сл., 1700 с. (С=O), 1624 с., 1614 с., 1592 ср., 1507 ср., 1458 ср., 1430 с., 1324 ср., 1187 ср., 1145 ср., 1114 ср., 892 ср.
Масс-спектр HRMS, m/z, вычислено: 347.0503, найдено 347.0518 [MK]+.
Пример 3.
Получение 2',2'-диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-она (Схема 6, 2с).
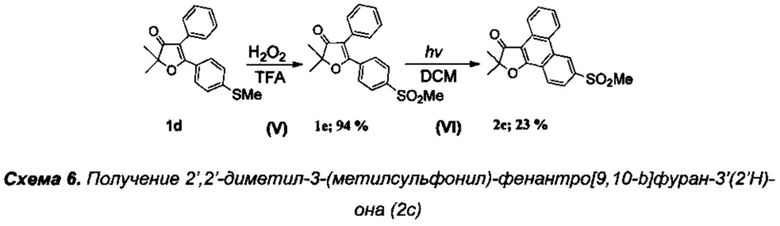
Проведение соединения 1d описано в примере 2 выше.
Стадия V. Получение 2,2-диметил-5-[4'-(метилсульфонил)фенил]-4-фенилфуран-3(2H)-она (1е).
В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты, растворили в ней при +40°C 300 мг (0.96 ммоль) соединения 1d и добавили по каплям смесь 0.21 г (1.96 ммоль) 35% водного раствора пероксида водорода в смеси 5 мл трифторуксусной кислоты и 0.05 мл (1 ммоль) 98% H2SO4. Реакционную смесь перемешивали в течение 1 ч при +65-70°C (контроль с помощью ТСХ), по завершении реакции смесь вылили в воду, экстрагировали CH2Cl2 (4×20 мл), органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над K2CO3. После удаления растворителя получили 310 мг (94%) соединения 1е, бесцветные кристаллы, т.пл. 201-202°C.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.58 с (6Н, 2СН3), 3.06 с (3Н, SO2CH3), 7.26 - 7.40 м (5Н), 7.73 д (2Н, 3JHH=8.5 Гц), 8.03 д (2Н, 3JHH=8.4 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 23.3 (2СН3), 43.3 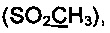 87.5 (С2), 115.6 (С4), 127.4, 128.2, 128.9, 129.2, 129.4, 135.1, 142.8, 175.2 (С5), 205.2 (С=O).
87.5 (С2), 115.6 (С4), 127.4, 128.2, 128.9, 129.2, 129.4, 135.1, 142.8, 175.2 (С5), 205.2 (С=O).
ИК спектр (в CCl4), см-1: 668 ср., 955 ср., 1152 с., 1332 ср., 1704 ср (С=O).
Масс-спектр HRMS: m/z=343.0996 [МН+] (расчет: 343.0999).
Стадия VI. Получение 2',2'-диметил-3-(метилсульфонил)-фенантро[9,10-b]фуран-3'(2'Н)-она (2с).
В кварцевый фотохимический реактор с водяным охлаждением и объемом 50 мл поместили раствор 50 мг (0.2 ммоль) йода и 545 мг (1.6 ммоль) 4,5-диарил-дигидрофуран-3(2H)-она 1е в 50 мл тетрагидрофурана. Облучение проводили ртутной лампой мощностью 150 Вт в течение 4 ч. По завершении реакции растворитель отогнали на роторном испарителе, смесь нанесли на пластину для препаративной хроматографии и разделили на фракции (элюэнт: гексан-этилацетат-дихлорметан). Выход продукта 2с: 125 мг (23%), бесцветное кристаллическое вещество, фиолетовая флуоресценция (при 254 нм). Степень конверсии исходного фуранона 1е составила 90%.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.67 с (6Н, 2СН3), 3.21 с (3Н, SO2CH3), 7.71 т (1Н, 3JHH=8.4 Гц), 7.79 т (1Н, 3JHH=8.1 Гц), 8.22 дд (1Н, 3JHH=8.4 Гц, 4JHH=1.5 Гц), 8.57 д (1Н, 3JHH=8.4 Гц), 8.68 д (1Н, 3JHH=8.2 Гц), 8.91 дд (1Н, 3JHH=8 Гц, 4JHH=1 Гц), 9.31 д (1Н, 4JHH=1-5 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.2 (2СН3), 44.7 (SO2CH3), 90.2, 110.5, 123.2, 123.5, 124.0, 124.3, 124.8, 125.1, 126.5, 128.0, 129.8, 135.8, 142.5, 171.0, 203.5 (С=O).
ИК спектр (в CCl4), см-1: 3019 сл., 2981 сл., 2930 сл., 1705 с., 1627 сл., 1517 ср., 1427 ср., 1326 е., 1215 с., 1154 с., 955 ср., 890 сл., 669 ср., 555 ср.
Масс-спектр HRMS, m/z, рассчитано 363.0662, найдено: 363.0675 [MNa]+.
Получение интермедиата 2,2-диметил-4-[4'-(метилтио)фенил]-5-(4'-фторфенил)фуран-3(2H)-она (Схема 7,1f).
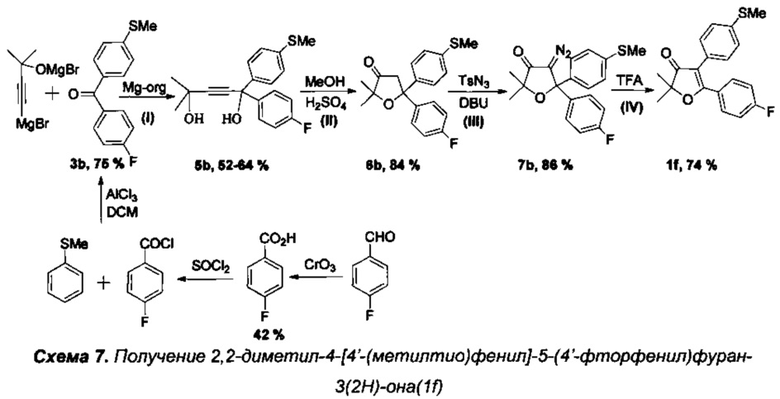
Стадия 0. Получение 4'-(метилтио)фенил(4'-фторфенил)метанона (3b).
10.7 г парафторбензойного альдегида растворили в 60 мл уксусной кислоты при 70°C и перемешивании на магнитной мешалке. Затем, по каплям в течение 1.5 часов добавили раствор 5.5 г CrO3 в 60 мл водной уксусной кислоты. Смесь перемешивали на водяной бане 3 часа. После окончания реакции в раствор добавили 100 мл ледяной HCl (35%), выпавший осадок кислоты отфильтровали на фильтре Шотта, дважды промыли 10% соляной кислотой. Для дополнительной очистки от остатков альдегида и солей хрома, полученный продукт был перекристаллизован из смеси хлористый метилен-метанол, а маточный раствор был пропущен через колонку с силикагелем. Выход продукта после вакуумной сушки составил 4.96 г (42%). В круглодонную колбу с магнитной мешалкой добавили 4.96 г парафторбензойной кислоты, 25 мл тетрахлоруглерода и затем осторожно добавили смесь 4 мл перегнанного тионилхлорида SOCl2 с 0.05 мл диметилформамида в 25 мл тетрахлоруглерода. Смесь кипятили с обратным холодильником 5 часов до окончания выделения газообразных продуктов. Затем растворитель и остатки тионилхлорида были удалены в вакууме роторного испарителя. Полученное масло, массой 5.6 г (97% теоретического выхода), после короткой вакуумной сушки, без дополнительной очистки, было растворено в 20 мл хлористого метилена и при охлаждении по каплям добавлено к суспензии 5 г безводного хлорида алюминия, 4.38 г (35 ммоль) тиоанизола и 30 мл хлористого метилена. После добавления всего количества хлорангидрида, смесь кипятили с обратным холодильником в течение 4 часов до полного прекращения выделения HCl. Полученный продукт осторожно гидролизовали 5% соляной кислотой, экстрагировали хлористым метиленом (3×30 мл), высушили над Na2SO4, а после отгонки растворителя - перекристаллизовали из гексана для очистки от избытка тиоанизола. Выход кристаллического 4'-(метилтио)фенил(4'-фторфенил)метанона (3b) составил 6.48 г (75%), бесцветные кристаллы, т.пл. 107-108°C.
Спектр ЯМР 1Н (400 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: 2.535 с (3Н, SCH3), 7.15 т (2Н, 3JHH=8.6 Гц), 7.29 д (2Н, 3JHH=8.4 Гц), 7.71 д (2Н, 3JHH=8.5 Гц), 7.81 дд (2Н, 3JHH=8.7 Гц, 4JHF=5.5 Гц).
Спектр ЯМР 13С (100 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: 14.8 (SCH3), 115.4 д (2С), 124.9 (2С), 130.4 (2С), 132.4 д (2С), 133.4, 133.8, 145.4, 137.7, 145.2, 165.2 д (1JCF=254 Гц), 194.3 (С=O).
Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -106.3.
Масс-спектр HRMS, m/z, рассчитано: 247.0588, найдено: 247.0598 [МН]+.
Парафторбензойная кислота, спектр ЯМР 1Н (400 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: 7.154 т (2Н, 3JHH=8.6 Гц), 8.135 дд (2Н, 3JHH=8.7 Гц, 4JHF=5.4 Гц).
Стадия I. Получение 2-метил-5-(4'-фторфенил)-5-(4'-метилтиофенил)пент-3-ин-2,5-диола (5b).
К раствору EtMgBr, приготовленному из 2.3 г (95 ммоль) магния и 10.3 г (95 ммоль) этилбромида, в 40 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 3.1 г (37 ммоль) 2-метилбут-3-ин-2-ола в 25 мл абсолютного бензола. Смесь кипятили в течение 3 ч, затем, не прекращая кипячения, в течение 2 часов добавили по каплям раствор 6.4 г (26 ммоль) соединения 3b в 25 мл бензола и кипятили далее в течение 5 ч. Затем смесь охладили и осторожно гидролизовали при помощи 100 мл 5% холодного раствора соляной кислоты. Органический слой отделили от водного. Водный слой экстрагировали бензолом (3×20 мл) и хлористым метиленом (1×20 мл). Объединенные органические фракции промыли раствором NaHCO3, водой и высушили над Na2SO4. После отгонки растворителя полученный желтый маслообразный продукт был разделен хроматографически на 3 фракции: целевой 1,4-диол, исходный непрореагировавший замещенный бензофенон и побочный продукт димеризации бензофенона до 1,2-диола. С целью уменьшения количества побочных продуктов и увеличения процента конверсии необходимо использовать недостаток магния и очень медленное добавление бензофенона в процессе реакции, так чтобы его концентрация оставалась незначительной. Очищенный γ-гликоль высушили в вакууме при давлении 0.01÷0.05 торр в течение 5 ч. Выход 1,4-диола 5b 4.5 г (52%), желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 1.54 с (6Н, 2СН3), 2.45 с (3Н, SCH3), 3.32 с (1Н, -ОН), 6.97 т (2Н, 3JHH=8.7 Гц), 7.18 д (2Н, 3JHH=8.6 Гц), 7.44 д (2Н, 3JHH=8.6 Гц), 7.51 дд (2Н, 3JHH=8.9 Гц, 4JHF=5.3 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 15.6 (SCH3), 31.2 (2СН3), 65.3 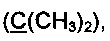 73.4, 84.5
73.4, 84.5 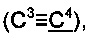 92.0
92.0 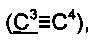 115.0 д (2С, 2JHF=21.6 Гц), 126.3 д (2С), 127.8 д (2С), 138.2, 140.7, 141.7, 162.1 д (1С, 1JHF=246.6 Гц). Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -114.8.
115.0 д (2С, 2JHF=21.6 Гц), 126.3 д (2С), 127.8 д (2С), 138.2, 140.7, 141.7, 162.1 д (1С, 1JHF=246.6 Гц). Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -114.8.
Спектр ИК (в CCl4), см-1: 836 с., 957 ср., 1081 ср., 1158 с., 1232 с., 1365 сл., 1506 с., 1601 ср., 1682 сл., 2930 сл., 2984 ср. (С-Н), 3469 сл. (шир., ОН), 3605 сл. (ОН).
Масс-спектр HRMS, m/z, рассчитано: 353.0982, найдено: 353.1064 [MNa]+.
Стадия II. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-5-(4'-фторфенил)-дигидрофуран-3(2H)-она (6b).
3.55 г соединения 5b растворили в 30 мл кипящего метанола и при перемешивании по каплям добавили раствор 2.5 мл 98% серной кислоты, растворенной в 30 мл метанола. Смесь кипятили в течение 3 ч до окончания реакции (контроль по ТСХ). Затем смесь вылили в 100 мл воды и тщательно экстрагировали хлористым метиленом (3×50 мл). Органический слой высушили над Na2SO4, растворитель отогнали на роторе, а полученное вещество высушили от метанола и дихлорметана в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Получено 3.30 г (84%) маслообразного продукта 6b, бесцветное маслообразное вещество. Без дополнительной очистки полученное соединение использовали на следующей стадии.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.7 мл CDCl3), δ, м.д.: 1.20 с (3Н, СН3), 1.22 с (3Н, СН3), 2.45 с (3Н, SCH3), 3.30 дд (2Н, СН2), 7.00 т (2Н, 3JHH=8.7 Гц), 7.19 д (2Н, 3JHH=8.5 Гц), 7.30 д (2Н, 3JHH=8.5 Гц), 7.36 дд (2Н, 3JHH=8.8 Гц, 4JHF=5.3 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.7 мл CDCl3), δ, м.д.: 15.3 (SCH3), 25.2 д (2СН3), 48.0 (СН2), 81.4 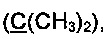 82.0, 115.1 д (2С), 126.2 (2С), 126.4 (2С), 127.7 д (2С), 138.0, 141.9 д (1С), 142.6, 162.0 d (1JCF=254 Гц), 216.4 (С=O).
82.0, 115.1 д (2С), 126.2 (2С), 126.4 (2С), 127.7 д (2С), 138.0, 141.9 д (1С), 142.6, 162.0 d (1JCF=254 Гц), 216.4 (С=O).
Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -115.1. Спектр ИК (в CCl4), см-1: 837 ср., 993 ср., 1097 ср., 1159 ср., 1236 с., 1508 с., 1604 сл., 1761 с. (С=O), 2983 сл. (С-Н).
Масс-спектр HRMS, m/z, рассчитано: 353.0982, найдено: 353.1026 [MNa]+.
Стадия III. Получение 4-диазо-2,2-диметил-5-[4'-(метилтио)фенил]-5-(4'-фторфенил-дигидрофуран-3(2H)-она (7b).
К смеси 3 г фуранона 6b и 1.8 г паратолуолсульфонилазида в 50 мл дихлорметана добавили по каплям раствор 350 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 25 мл дихлорметана, при этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 35°C в течение 7 ч до завершения реакции (контроль с помощью ТСХ), затем охладили, отогнали избыток растворителя, перенесли на колонку с силикагелем, и хроматографировали в градиентном режиме смесью н-гексан - хлористый метилен. После удаления растворителя кристаллический остаток высушили в вакууме и получили 2.80 г (84%) диазокетона 7b, желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.33 с (3Н, СН3), 1.34 с (3Н, СН3), 2.48 с (3Н, SCH3), 7.05 т (2Н, 3JHH=8.6 Гц), 7.25-7.18 м (4Н, PhSCH3), 7.29 дд (2Н, 3JHH=8.8 Гц, 4JHF=5.2 Гц). Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 15.3 (SCH3), 25.2 (2СН3), 64.3 (C=N2), 83.6 (С5), 85.5 (С2), 115.4 д (2С, 2JCF=21.7 Гц), 126.1 (2С), 126.8 (2С), 128.3 д (2С, 3JCF=8.3 Гц), 139.3, 139.4 д (1С, 4JCF=3.2 Гц), 140.1, 162.5 д (1С, 1JCF=248.2 Гц), 196.7 (С=O). Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -113.40.
ИК спектр (в CCl4), см-1: 909 сл., 1025 сл., 1093 сл., 1157 ср., 1231 ср., 1344 с., 1507 ср., 1695 с. (С=O), 2089 с. (CN2), 2928 сл., 2986 сл.
Масс-спектр HRMS, m/z, рассчитано для C19H17FN2O2SNa+: 379.0887; найдено: 379.0922 [M+Na+].
Стадия IV. Получение 2,2-диметил-4-(4'-(метилтио)фенил)-5-(4'-фторфенил)фуран-3(2H)-она (1f).
Реакция проводится в атмосфере азота. Раствор 2.0 г диазокетона 7а в 30 мл трифторуксусной кислоты кипятили 3 часа до полного исчезновения исходного диазокетона (контроль с помощью ТСХ). По завершении реакции трифторуксусная кислота была удалена в вакууме, осадок растворили в 25 мл хлористого метилена, тщательно промыли водным раствором NaHCO3, водой (2×5 мл) и высушили над K2CO3. После удаления растворителя получили 1.80 г (98%) смеси двух региоизомерных 2,2-диалкил-4,5-диарил-дигидрофуран-3(2H)-онов в примерном соотношении 3:1 в виде светло-желтого маслообразного продукта. Реакционную смесь (1.8 г) разделили на колонке с силикагелем и получили фуранон 1f с выходом 1.33 г (74%), светло-желтое маслообразное вещество или кристаллы.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.54 с (6Н, 2СН3), 2.49 с (3Н, SCH3), 7.05 т (2Н, 3JHH=8.7 Гц), 7.20-7.25 м (4Н, PhSCH3), 7.68 дд (2Н, 3JHH=8.9 Гц, 4JHF=5.4 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.6 (SCH3), 23.4 (2СН3), 87.1 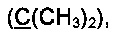 112.8 (С4), 115.7 д (2С, 2JCF=21.9 Гц), 126.4, 126.6 (2С), 128.5, 129.8 (2С), 130.7 д (2С, 3JCF=8.9 Гц), 138.0, 164.7 д (1С, 1JCF=254.0 Гц), 176.8 (С5), 205.2 (С=O).
112.8 (С4), 115.7 д (2С, 2JCF=21.9 Гц), 126.4, 126.6 (2С), 128.5, 129.8 (2С), 130.7 д (2С, 3JCF=8.9 Гц), 138.0, 164.7 д (1С, 1JCF=254.0 Гц), 176.8 (С5), 205.2 (С=O).
Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -106.3.
Масс-спектр HRMS, m/z, рассчитано 329.1007, найдено 329.1021 [МН]+.
Получение интермедиата 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-(4'-фторфенил)фуран-3(2H)-она (Схема 8, 1g).
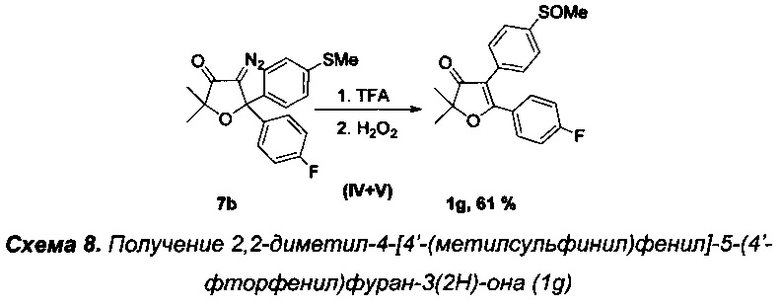
Получение соединения 7b описано выше.
Стадии IV-V. Получение 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-(4'-фторфенил)фуран-3(2H)-она (1g).
Реакцию проводили в атмосфере азота. В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты и растворили в ней 342 мг (0.96 ммоль) соединения 7b и кипятили данный раствор в течение 3-х часов. Затем охладили до 50°C и по каплям при интенсивном перемешивании в течение 1 ч осторожно добавили смесь 100 мг 35% (0.96 ммоль) свежего водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.025 мл (0.5 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное эквимолярное соотношение пероксида водорода к 7b. Реакционную смесь перемешивали еще в течение 1 ч при +50-55°C (контроль с помощью ТСХ). По завершении реакции смесь вылили в воду, нейтрализовали карбонатом натрия, экстрагировали этилацетатом (4×20 мл). Органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и хроматографического разделения региоизомерных сульфоксидов, получили 190 мг (61%) соединения 1д, светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.54 с (6Н, 2СН3), 2.72 с (3Н, SOCH3), 7.04 т (2Н, 3JHH=8.7 Гц), 7.45-7.47 м (2H), 7.60-7.65 дд (4Н).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.4 (2СН3), 43.6 (SOCH3), 87.6  112.0, 115.9 д (2С, 2JCF=21.9 Гц), 123.9 (2С), 130.3 (2С), 130.7 д (2С, 3JCF=8.9 Гц), 133.1, 144.5, 164.8 д (1С, 1JCF=254.0 Гц), 177.7 (С5), 204.5 (С=O).
112.0, 115.9 д (2С, 2JCF=21.9 Гц), 123.9 (2С), 130.3 (2С), 130.7 д (2С, 3JCF=8.9 Гц), 133.1, 144.5, 164.8 д (1С, 1JCF=254.0 Гц), 177.7 (С5), 204.5 (С=O).
Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -105.4. ИК спектр (в CCl4), см-1: 842 ср., 902 сл., 1049 ср. (S=O), 1161 ср., 1242 с., 1381 с., 1515 ср., 1616 с., 1703 с. (С=O), 2980 сл. (С-Н).
Масс-спектр LCMS, m/z, рассчитано 345.0956, найдено 353.1015 [МН]+.
Получение интермедиата 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-(4'-фторфенил)фуран-3(2H)-она (Схема 9, 1h).
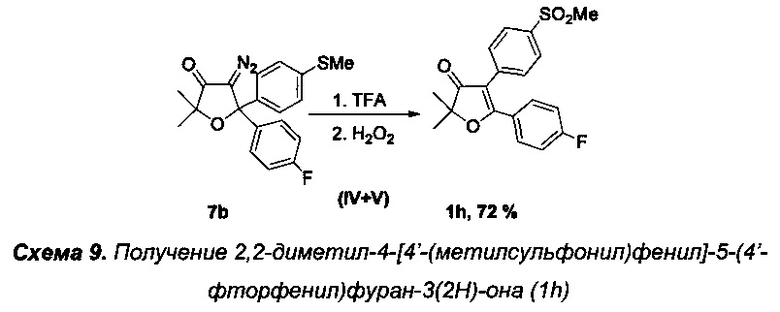
Получение соединения 7b описано выше.
Стадии IV-V. Получение 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-(4'-фторфенил)фуран-3(2H)-она (1h).
Реакцию проводили в атмосфере азота. В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты и растворили в ней 342 мг (0.96 ммоль) соединения 7b и кипятили данный раствор в течение 3-х часов. Затем, не прекращая кипячения, по каплям в течение 10 мин добавили смесь 200 мг 34% (1.98 ммоль) водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.025 мл (0.5 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное 2х кратное молярное соотношение пероксида водорода к 7b для предотвращения окисления двойной связи в соединении 1h. Реакционную смесь перемешивали еще в течение 2 ч при +65-70°C (контроль с помощью ТСХ). По завершении реакции смесь вылили в воду, кислоту нейтрализовали карбонатом натрия, органические вещества экстрагировали этилацетатом (4×20 мл). Органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и хроматографического разделения региоизомерных сульфонов, получили 225 мг (72%) соединения 1h, светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.57 с (6Н, 2СН3), 3.06 с (3Н, SO2CH3), 7.09 т (2Н, 3JHH=8.5 Гц), 7.52 д (2Н, 3JHH=8.0 Гц), 7.63 дд (2Н, 3JHH=8.8 Гц, 4JHF=5.4 Гц), 7.92 д (2Н, 3JHH=8.0 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.3 (2СН3), 44.5 (SO2CH3), 88.0 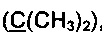 116.2 д (2С, 2JCF=22.0 Гц), 126.3 (1С), 127.8 (2С), 130.2 (2С), 130.9 д (2С, 3JCF=9.0 Гц), 136.1, 139.3, 165.1 д (1С, 1JCF=255.4 Гц), 178.4 (С5), 204.2 (С=O). Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -104.8.
116.2 д (2С, 2JCF=22.0 Гц), 126.3 (1С), 127.8 (2С), 130.2 (2С), 130.9 д (2С, 3JCF=9.0 Гц), 136.1, 139.3, 165.1 д (1С, 1JCF=255.4 Гц), 178.4 (С5), 204.2 (С=O). Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -104.8.
ИК спектр (в CCl4), см-1: 955 ср., 1157 с. (SO2) 1243 ср., 1327 с. (SO2), 1383 ср., 1613 ср., 1704 с. (С=O), 2933 сл. (С-Н).
Масс-спектр LCMS, m/z, рассчитано 361.0905, найдено 361.0953 [МН]+.
Пример 4.
Получение 2',2'-диметил-6-(метилсульфонил)-3-фторфенантро[9,10-b]фуран-3'(2'Н)-она (Схема 10, 2d).
Получение соединения 1f дописано выше.
Стадия VI. Получение 2',2'-диметил-6-(метилсульфонил)-3-фторфенантро[9,10-b]фуран-3'(2'Н)-она (2d).
В кварцевый фотохимический реактор с водяным охлаждением и объемом 50 мл поместили раствор 50 мг (0.2 ммоль) йода и 525 мг (1.46 ммоль) 4,5-диарил-дигидрофуран-3(2H)-она 1f в 50 мл тетрагидрофурана. Облучение проводили ртутной лампой мощностью 150 Вт в течение 4 ч. По завершении реакции растворитель отогнали на роторном испарителе, смесь нанесли на пластину для препаративной хроматографии и разделили на фракции (элюэнт: гексан-этилацетат-дихлорметан).
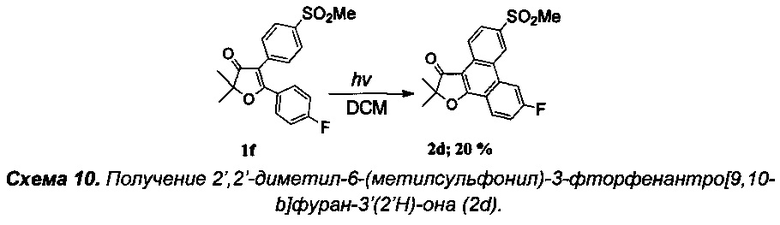
Получение интермедиата 2,2-диметил-4-[4'-(метилтио)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (Схема 11, 1i).
Стадия 0. Получение 4'-(метилтио)фенил(4'-хлорфенил)метанона (3с).
5 г (36 ммоль) парахлорбензойного альдегида растворили в 30 мл уксусной кислоты при 70°C и перемешивании на магнитной мешалке. Затем, по каплям в течение 1.5 часов добавили раствор 2.4 г (24 ммоль) CrO3 в 30 мл водной уксусной кислоты. Смесь перемешивали на водяной бане 3 часа (100°C). После окончания реакции в раствор добавили 50 мл ледяной HCl (35%), выпавший осадок кислоты отфильтровали на фильтре Шотта и дважды промыли 10% соляной кислотой. Для дополнительной очистки от остатков альдегида и солей хрома, полученный продукт был перекристаллизован из смеси хлористый метилен-метанол, а маточный раствор был пропущен через колонку с силикагелем. Выход парахлорбензойной кислоты после вакуумной сушки составил 3.71 г (66%). В круглодонную колбу с магнитной мешалкой добавили 3.71 г парахлорбензойной кислоты, 20 мл тетрахлоруглерода и затем осторожно добавили смесь 2.6 мл перегнанного тионилхлорида SOCl2 с 0.025 мл диметилформамида в 20 мл тетрахлоруглерода. Смесь кипятили с обратным холодильником 5 часов до окончания выделения газообразных продуктов. Затем растворитель и остатки тионилхлорида были удалены в вакууме роторного испарителя. Полученное масло, массой 4.1 г (99% теоретического выхода), после короткой вакуумной сушки, без дополнительной очистки, было растворено в 15 мл хлористого метилена и при охлаждении по каплям добавлено к суспензии 3.4 г безводного хлорида алюминия, 2.9 г (23 ммоль) тиоанизола и 15 мл хлористого метилена. После добавления всего количества хлорангидрида, смесь кипятили с обратным холодильником в течение 4 часов до полного прекращения выделения HCl. Полученный продукт осторожно гидролизовали 5% соляной кислотой, экстрагировали хлористым метиленом (3×30 мл), высушили над Na2SO4, а после отгонки растворителя - перекристаллизовали из гексана для очистки от избытка тиоанизола. Выход кристаллического 4'-(метилтио)фенил(4'-хлорфенил)метанона (3c) составил 4.3 г (70%), бесцветные кристаллы, т.пл. 132-133°C.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 2.524 с (3Н, SCH3), 7.28 д (2Н, 3JHH=8.5 Гц), 7.44 д (2Н, 3JHH=8.5 Гц), 7.70 д (2Н, 3JHH=8.7 Гц), 7.71 д (2Н, 3JHH=8.8 ГЦ).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 14.7 (SCH3), 124.8 (2С), 128.5 (2С), 130.4 (2С), 131.2 (2С), 133.1, 136.1, 138.5, 145.6, 194.4 (С=O).
Парахлорбензойная кислота, спектр ЯМР 1Н (400 МГц, 10 мг в 0.8 мл (CD3)2CO), δ, м.д.: 7.57 д (2Н, 3JHH=8.8 Гц), 8.05 д (2Н, 3JHH=8.7 Гц).
Стадия I. Получение 2-метил-5-(4'-хлорфенил)-5-(4'-метилтиофенил)пент-3-ин-2,5-диола (5b).
К раствору EtMgBr, приготовленному из 1.65 г (69 ммоль) магния и 7.5 г (69 ммоль) этилбромида, в 30 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 2.25 г (27 ммоль) 2-метилбут-3-ин-2-ола в 20 мл абсолютного бензола. Смесь кипятили в течение 3 ч, затем, не прекращая кипячения, в течение 2 часов добавили по каплям раствор 4.9 г (19 ммоль) соединения 3с в 20 мл бензола и кипятили далее в течение 4 ч. Затем смесь охладили и осторожно гидролизовали при помощи 100 мл 5% холодного раствора соляной кислоты. Органический слой отделили от водного. Водный слой экстрагировали бензолом (3×20 мл) и хлористым метиленом (1×20 мл). Объединенные органические фракции промыли раствором NaHCO3, водой и высушили над Na2SO4. После отгонки растворителя полученный желтый маслообразный продукт был разделен хроматографически на 3 фракции: целевой 1,4-диол, исходный непрореагировавший замещенный бензофенон и побочный продукт димеризации бензофенона до 1,2-диола. С целью уменьшения количества побочных продуктов и увеличения процента конверсии необходимо использовать недостаток магния и очень медленное добавление бензофенона в процессе реакции, так чтобы его концентрация оставалась незначительной. Очищенный γ-гликоль высушили в вакууме при давлении 0.01÷0.05 торр в течение 5 ч. Выход 1,4-диола 5с 1.56 г (23%), желтое кристаллическое вещество, т.пл. 112-113°C. Основным продуктом реакции является 1,2-бис(4-хлорфенил)-1,2-бис(4-(метилтио)фенил)этан-1,2-диол.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 1.54 с (6Н, 2СН3), 2.44 с (3Н, SCH3), 7.17 д (2Н, 3JHH=8.7 Гц), 7.25 д (2Н, 3JHH=8.8 Гц), 7.43 д (2Н, 3JHH=8.8 Гц), 7.46 д (2Н, 3JHH=8.9 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 15.5 (SCH3), 31.2 (2СН3), 65.3 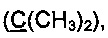 73.4, 84.3
73.4, 84.3 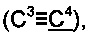 92.0
92.0 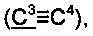 126.2 (2С), 126.3 (2С), 127.3 (2С), 128.3 (2С), 133.5, 138.3, 141.4, 143.4.
126.2 (2С), 126.3 (2С), 127.3 (2С), 128.3 (2С), 133.5, 138.3, 141.4, 143.4.
ИК спектр (в KBr), см-1: 829 с., 993 с., 1015 с., 1093 с., 1135 ср., 1161 ср., 1239 ср., 1399 ср., 1491 с., 1592 ср., 1707 сл., 2923 сл., 2979 ср., 3309 оч.с, 3402 оч.с.
Масс-спектр LCMS, m/z, рассчитано 369.0687, найдено 369.0742 [MNa]+.
Стадия II. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-5-(4'-хлорфенил)-дигидрофуран-3(2H)-она (6с).
1.15 г соединения 5с растворили в 20 мл кипящего метанола и при перемешивании по каплям добавили раствор 1.9 мл 98% серной кислоты, растворенной в 20 мл метанола. Смесь кипятили в течение 4 ч до окончания реакции (контроль по ТСХ). Затем смесь вылили в 50 мл воды и тщательно экстрагировали хлористым метиленом (3×25 мл). Органический слой высушили над Na2SO4, растворитель отогнали на роторе, а полученное вещество высушили от метанола и дихлорметана в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Получено 1.19 г (ЯМР с внутренним стандартом показывает выход 91%) маслообразного продукта 6с. Без дополнительной очистки полученное соединение использовали на следующей стадии.
Спектр ЯМР 1Н (400 МГц, 15 мг в 0.7 мл CDCl3), δ, м.д.: 1.19 с (3Н, СН3), 1.21 с (3Н, СН3), 2.44 с (3Н, SCH3), 3.31 дд (2Н, СН2), 7.21-7.38 м (8Н).
Масс-спектр LCMS, m/z, рассчитано 369.0687, найдено 369.0720 [MNa]+.
Стадия III. Получение 4-диазо-2,2-диметил-5-[4'-(метилтио)фенил]-5-(4'-хлорфенил)дигидрофуран-3(2H)-она (7с).
К смеси 1.1 г фуранона 6с и 0.63 г паратолуолсульфонилазида в 25 мл дихлорметана добавили по каплям раствор 120 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 25 мл дихлорметана, при этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 35°C в течение 8 ч до завершения реакции (контроль с помощью ТСХ), затем отогнали избыток растворителя, перенесли на колонку с силикагелем и хроматографировали в градиентном режиме смесью н-гексан - хлористый метилен. После удаления растворителя кристаллический остаток высушили в вакууме и получили 0.9 г (75%) диазокетона 7с, желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.33 s (6Н, 2СН3), 2.48 s (3Н, SCH3), 7.21-7.26 m (6Н), 7.34 d (2Н, 3JHH=8.4 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 15.3 (SCH3), 25.2 (2СН3), 64.0 (C=N2), 83.5 (С5), 85.6 (С2), 126.1 (2С), 126.8 (2С), 127.8 (2С), 128.7 (2С), 134.4, 139.4, 139.8, 142.2, 196.6 (С=O).
ИК спектр (в CCl4), см-1: 1093 ср., 1165 сл., 1227 сл., 1344 с., 1491 ср., 1697 с. (С=O), 2091 оч. с. (CN2), 2924 сл., 2982 сл.
Масс-спектр HRMS, m/z рассчитано для C19H17ClN2O2SNa+: 395.0592, найдено 395.0612 [MNa+].
Стадия IV. Получение 2,2-диметил-4-[4'-(метилтио)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (1i).
Реакция проводится в атмосфере азота. К раствору 300 мг диазокетона 7 с в 15 мл дихлорметана добавили по каплям 0.2 мл трифторметансульфоновой кислоты (TfOH), растворенной в 5 мл хлористого метилена. Смесь кипятили 3 часа до полного исчезновения исходного диазокетона (контроль с помощью ТСХ), а по завершении реакции смесь промыли водным раствором NaHCO3, водой (2×5 мл) и высушили над K2CO3. После удаления растворителя получили 273 мг (98%) смеси двух региоизомерных 2,2-диалкил-4,5-диарил-дигидрофуран-3(2H)-онов в примерном соотношении 3:1 в виде светло-желтого маслообразного продукта. Реакционную смесь (273 мг) разделили на колонке с силикагелем и получили 205 мг (75%) фуранона 1i в виде светло-желтого маслообразного вещества.
Спектр ЯМР 1Н (400 МГц, 5 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.54 s (6Н, 2СН3), 2.49 s (3Н, SCH3), 7.20-7.24 m (4Н, PhSCH3), 7.33 d (2Н, 3JHH=8.6 Гц), 7.60 d (2Н, 3JHH=8.6 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 15.6 (SCH3), 23.3 (2СН3), 87.1 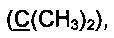 113.3 (С4), 126.6 (2С), 128.8 (2С), 128.9, 129.7 (2С), 129.8 (2С), 130.8, 138.0, 138.2, 176.6 (С5), 205.2 (С=O).
113.3 (С4), 126.6 (2С), 128.8 (2С), 128.9, 129.7 (2С), 129.8 (2С), 130.8, 138.0, 138.2, 176.6 (С5), 205.2 (С=O).
Масс-спектр HRMS, m/z рассчитано 345.0711, найдено 345.0728 [МН]+.
Получение интермедиата 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (Схема 12, 1j).
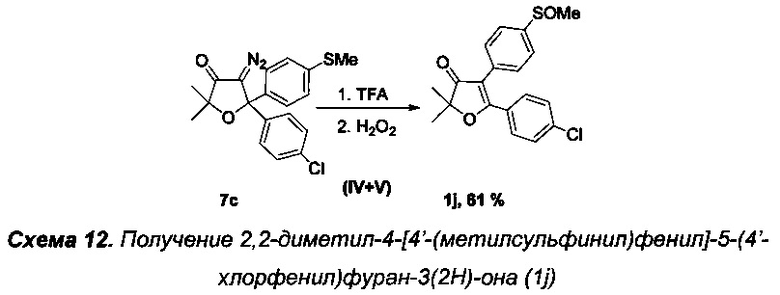
Получение соединения 7 с описано выше.
Стадии IV-V. Получение 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (1j).
Реакцию проводили в атмосфере азота. В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты и растворили в ней 360 мг (0.96 ммоль) соединения 7 с и кипятили данный раствор в течение 3-х часов. Затем охладили до 40°C и по каплям при интенсивном перемешивании в течение 1 ч осторожно добавили смесь 100 мг 35% (0.96 ммоль) свежего водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.025 мл (0.5 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное эквимолярное соотношение пероксида водорода к 7 с. Реакционную смесь перемешивали еще в течение 1 ч при +50-55°C (контроль реакции с помощью ТСХ). По завершении реакции смесь вылили в воду, нейтрализовали карбонатом натрия, тщательно экстрагировали этилацетатом (4×20 мл). Органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и хроматографического разделения региоизомерных сульфоксидов, получили 217 мг (63%) соединения 1j, светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.54 с (6Н, 2СН3), 2.74 с (3Н, SOCH3), 7.32 д (2Н, 3JHH=7.4 Гц), 7.45 (2Н, 3JHH=7.5 Гц), 7.58 д (2Н, 3JHH=7.4 Гц), 7.73 д (2Н, 3JHH=7.5 Гц).
Масс-спектр LCMS, m/z рассчитано 361.0660, найдено 361.0740 [МН]+.
Получение интермедиата 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (Схема 13, 1k).
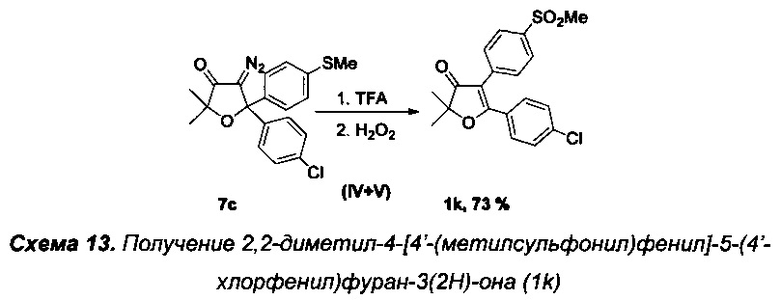
Получение соединения 7с описано выше.
Стадии IV-V. Получение 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-(4'-хлорфенил)фуран-3(2H)-она (1k).
Реакцию проводили в атмосфере азота. В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты и растворили в ней 360 мг (0.96 ммоль) соединения 7с и кипятили данный раствор в течение 3-х часов. Затем, не прекращая кипячения, по каплям в течение 10 мин добавили смесь 200 мг 34% (1.98 ммоль) водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.025 мл (0.5 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное 2х кратное молярное соотношение пероксида водорода к 7с для предотвращения окисления двойной связи в соединении 1k. Реакционную смесь перемешивали еще в течение 2 ч при +65-70°C (контроль с помощью ТСХ). По завершении реакции смесь вылили в воду, кислоту нейтрализовали карбонатом натрия, органические вещества экстрагировали этилацетатом (4×20 мл). Органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и хроматографического разделения региоизомерных сульфонов, получили 260 мг (73%) соединения 1k, светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.57 с (6Н, 2СН3), 3.06 с (3Н, SO2CH3), 7.37 д (2Н, 3JHH=8.8 Гц), 7.53 дд (4Н, 3JHH=8.8 Гц, 4JHF=5.4 Гц), 7.92 д (2Н, 3JHH=8.8 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.3 (2СН3), 44.5 (SO2CH3), 88.1 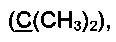 112.0, 127.7 (2С), 129.2 (2С), 129.7 (2С), 130.2 (2С), 130.6, 135.9, 138.8, 139.3, 178.3 (С5), 204.2 (С=O).
112.0, 127.7 (2С), 129.2 (2С), 129.7 (2С), 130.2 (2С), 130.6, 135.9, 138.8, 139.3, 178.3 (С5), 204.2 (С=O).
ИК спектр (в CCl4), см-1: 956 ср., 1095 ср., 1158 с. (SO2), 1329 с. (SO2), 1384 с., 1616 ср., 1703 с. (С=O), 2931 сл., 2982 сл. (С-Н).
Масс-спектр LCMS, m/z рассчитано 377.0609, найдено 377.0679 [МН]+.
Пример 5.
Получение 2',2'-диметил-6-(метилсульфонил)-3-хлорфенантро[9,10-b]фуран-3'(2'Н)-она (Схема 14, 2е).
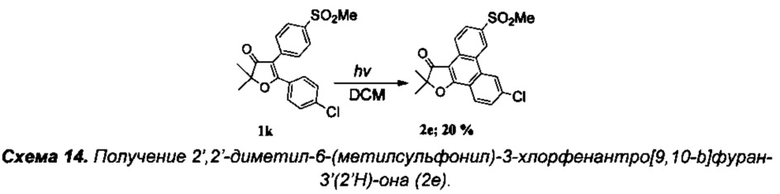
Получение соединения 1k описано выше.
Стадия VI. Получение 2',2'-диметил-6-(метилсульфонил)-3-хлорфенантро[9,10-b]фуран-3'(2'Н)-она (2е).
В кварцевый фотохимический реактор с водяным охлаждением и объемом 50 мл поместили раствор 50 мг (0.2 ммоль) йода и 550 мг (1.46 ммоль) 4,5-диарил-дигидрофуран-3(2H)-она 1k в 50 мл тетрагидрофурана. Облучение проводили ртутной лампой мощностью 150 Вт в течение 4 ч. По завершении реакции растворитель отогнали на роторном испарителе, смесь нанесли на пластину для препаративной хроматографии и разделили на фракции (элюэнт: гексан-этилацетат-дихлорметан).
Получение интермедиата 2,2-диметил-4-[4'-(метилтио)фенил]-5-(3'-фторфенил)фуран-3(2H)-она (Схема 15, 1l).
Стадия 0. Получение 4'-(метилтио)фенил(3'-фторфенил)метанона (3d).
В пластиковой посуде смешали 6.2 г борной кислоты (H3BO3) и 23 г водного 35% раствора плавиковой кислоты (HF) и оставили на 20 минут охлаждаться до 0°C. Затем в 7 граммах полученного раствора растворили при перемешивании 3.24 г метааминобензойной кислоты, затем при охлаждении по каплям добавили раствор 1.7 г нитрита натрия (NaNO2) в 5-7 мл воды. Выпавший осадок тетрафторбората отфильтровали на бумажном фильтре с использованием вакуума, промыли водой и высушили, пропуская через слой соли абсолютный бензол, а затем абсолютный толуол (тщательное удаление воды необходимо для недопущения образования фенолов). Получили 3.48 г (62%) тетрафторбората диазония. Полученная соль была порциями по 100-200 мг осторожно добавлена к 100 мл кипящего толуола. Процесс сопровождается термическим разложением соли с бурным выделением BF3 (в виде дыма) и протекает в интервале 100-110°C в течение 2.5 часов. По завершении реакции толуол был удален азеотропной отгонкой в вакууме, а остаток переведен в водорастворимую соль путем обработки раствором гидроксида натрия (или содой). К полученному раствору добавили 30-50 мл концентрированной соляной кислоты, выделившиеся белые хлопья кислоты были отфильтрованы на бумажном фильтре, промыты ледяной соляной кислотой, гексаном и бензолом. Масса метафторбензойной кислоты после вакуумной сушки составила 1.18 г (35%).
В круглодонную колбу с магнитной мешалкой добавили 1.18 г парафторбензойной кислоты, 10 мл тетрахлоруглерода и затем осторожно добавили смесь 1 мл перегнанного тионилхлорида SOCl2 с 0.01 мл диметилформамида в 5 мл тетрахлоруглерода. Смесь кипятили с обратным холодильником 4 часа до окончания выделения газообразных продуктов. Затем растворитель и остатки тионилхлорида были удалены в вакууме роторного испарителя. Полученное масло, после короткой вакуумной сушки и без дополнительной очистки, было растворено в 10 мл хлористого метилена и при охлаждении по каплям добавлено к суспензии 1.2 г безводного хлорида алюминия, 0.91 г (7.3 ммоль) тиоанизола и 8 мл хлористого метилена. После добавления всего количества хлорангидрида, смесь кипятили с обратным холодильником в течение 5 часов до полного прекращения выделения HCl. Полученный продукт осторожно гидролизовали 5% соляной кислотой, экстрагировали хлористым метиленом (3×20 мл), высушили Na2SO4, а после отгонки растворителя - для дополнительной очистки пропустили через колонку с силикагелем. Выход кристаллического 4'-(метилтио)фенил(3'-фторфенил)метанона (3d) составил 0.71 г (39%), бесцветные кристаллы, т.пл. 67-68°C.
Спектр ЯМР 1Н (400 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: 2.54 с (3Н, SCH3), 7.30 д (2Н, 3JHH=8.4 Гц), 7.46-7.48 м (2H), 7.53 д (1Н, 3JHH=7.6 Гц), 7.74 д (2Н, 3JHH=8.5 Гц).
Спектр ЯМР 13С (100 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: 14.8 (SCH3), 116.5 д (2С), 119.1 д, 124.9 (2С), 125.5 д (1С), 129.9 д (1С), 130.5 (2С), 133.0, 140.0 д, 145.9, 162.4 д (1JCF=247 Гц), 194.3 (С=O).
Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -112.0.
Метафторбензойная кислота, спектр ЯМР 1Н (400 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: 7.32 т (1Н, 3JHH=8.6 Гц), 7.40-7.50 м (1Н), 7.80 д (1Н, 3JHH=8.8 Гц), 7.92 д (1Н, 3JHH=7.6 Гц). Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -112.0.
Стадия I. Получение 2-метил-5-(3'-фторфенил)-5-(4'-метилтиофенил)пент-3-ин-2,5-диола (5d).
К раствору EtMgBr, приготовленному из 250 мг магния и 1.13 г этилбромида, в 10 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 340 мг 2-метилбут-3-ин-2-ола в 10 мл абсолютного бензола. Смесь кипятили в течение 4 ч, затем, не прекращая кипячения, в течение 2 часов добавили по каплям раствор 700 мг соединения 3d в 5 мл бензола и кипятили далее в течение 5 ч. Затем смесь охладили и осторожно гидролизовали при помощи 100 мл 5% холодного раствора соляной кислоты. Органический слой отделили от водного. Водный слой экстрагировали бензолом (3×20 мл) и хлористым метиленом (1×20 мл). Объединенные органические фракции промыли раствором NaHCO3, водой и высушили над Na2SO4. После отгонки растворителя полученный желтый маслообразный продукт (813 мг) был разделен хроматографически на 3 фракции: целевой 1,4-диол, исходный непрореагировавший замещенный бензофенон и побочный продукт димеризации бензофенона до 1,2-диола. С целью уменьшения количества побочных продуктов и увеличения процента конверсии необходимо использовать недостаток магния и очень медленное добавление бензофенона в процессе реакции, так чтобы его концентрация оставалась незначительной. Очищенный γ-гликоль 5d высушили в вакууме 0.01÷0.05 торр в течение 5 ч. Выход 1,4-диола 5d - 235 мг (25%), светло-желтое маслообразное вещество. Степень конверсии исходного кетона 3d - 75%.
Спектр ЯМР 1Н (400 МГц, 15 мг в 0.8 мл CDCl3), δ, м.д.: 1.52 с (6Н, 2СН3), 2.43 с (3Н, SCH3), 3.17 с (1Н, -ОН), 4.05 с (1Н, -ОН), 7.16 д (2Н, 3JHH=8.4 Гц), 7.22-7.32 м (4Н), 7.45 д (2Н, 3JHH=8.4 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 15.5 (SCH3), 31.0 (2СН3), 65.2  73.3, 84.1
73.3, 84.1  91.9
91.9 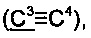 113.0 д (2С, 2JHF=21.6 Гц), 114.4 д, 121.5 д, 126.2 д (2С), 129.6 д, 138.1, 141.4, 162.5 д (1С, 1JHF=246.6 Гц).
113.0 д (2С, 2JHF=21.6 Гц), 114.4 д, 121.5 д, 126.2 д (2С), 129.6 д, 138.1, 141.4, 162.5 д (1С, 1JHF=246.6 Гц).
Спектр ЯМР 19F (376 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: -112.5.
Масс-спектр LCMS, m/z рассчитано 331.1163, найдено 331.1173 [МН]+.
Стадия II. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-5-(3'-фторфенил)-дигидрофуран-3(2H)-она (6d).
230 мг соединения 5d растворили в 10 мл кипящего метанола и при перемешивании по каплям добавили раствор 0.7 мл 98% серной кислоты, растворенной в 5 мл метанола. Смесь кипятили в течение 5 ч до окончания реакции (контроль по ТСХ). Затем смесь вылили в 100 мл воды и тщательно экстрагировали хлористым метиленом (3×10 мл). Органический слой высушили над Na2SO4, растворитель отогнали на роторе, а полученную смесь разделили на компоненты хроматографическим методом. Основным продуктом реакции является смесь изомерных α-оксиенонов. Выделенный целевой фуранон высушили от метанола и дихлорметана в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Получено 70 мг (31%) маслообразного продукта 6d, бесцветное маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.7 мл CDCl3), δ, м.д.: 1.19 с (3Н, СН3), 1.26 с (3Н, СН3), 2.46 с (3Н, SCH3), 3.24 д (1Н, СН2, 2JHH=17.6 Гц), 3.33 д (1Н, СН2, 2JHH=17.7 Гц), 7.13-7.33 м (8Н).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.7 мл CDCl3), δ, м.д.: 15.5 (SCH3), 25.2 д (2СН3), 47.7 (СН2), 81.5 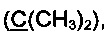 81.9 д, 113.1 д, 114.3 д, 121.3, 126.3 д (2С), 129.8 д, 138.1, 142.1, 149.0, 162.7 д (1JCF=254 Гц), 216.2 (С=O).
81.9 д, 113.1 д, 114.3 д, 121.3, 126.3 д (2С), 129.8 д, 138.1, 142.1, 149.0, 162.7 д (1JCF=254 Гц), 216.2 (С=O).
Спектр ЯМР 19F (376 МГц, 30 мг в 0.8 мл CDCl3), δ, м.д.: -112.2.
Масс-спектр LCMS, m/z рассчитано 331.1163, найдено 331.1187 [МН]+.
Стадия III. Получение 4-диазо-2,2-диметил-5-[4'-(метилтио)фенил]-5-(3'-фторфенил)дигидрофуран-3(2H)-она (7d).
К смеси 70 мг (0.21 ммоль) фуранона 6d и 42 мг паратолуолсульфонилазида в 5 мл дихлорметана добавили по каплям раствор 10 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 5 мл дихлорметана, при этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 35°C в течение 24 ч до завершения реакции (контроль с помощью ТСХ), затем охладили, отогнали избыток растворителя, перенесли на колонку с силикагелем, и хроматографировали в градиентном режиме смесью н-гексан - хлористый метилен. После удаления растворителя кристаллический остаток высушили в вакууме и получили 54 мг (71%) диазокетона 7d, желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.34 с (3Н, СН3), 1.36 с (3Н, СН3), 2.49 с (3Н, SCH3), 7.03-7.10 м (2H), 7.20-7.25 м (4Н), 7.30-7.35 м (2H).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 15.4 (SCH3), 25.2 (2СН3), 64.1 (C=N2), 85.6 (С5), 113.7 д (J=23 Гц), 115.4 д (J=21.3 Гц), 121.9 д (1С, 4JCF=2.9 Гц), 126.2 (2С), 126.8 (2С), 130.2 д, 139.4 д (J=3.2 Гц), 139.6 д (J=26.2 Гц) 146.4 д, 162.8 д (1С, 1JCF=247.6 Гц), 196.6 (С=O).
Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -111.5.
Масс-спектр HRMS, m/z рассчитано 357.1068, найдено 357.1075 [МН]+.
Стадия IV. Получение 2,2-диметил-4-(4'-(метилтио)фенил)-5-(3'-фторфенил)фуран-3(2H)-она  .
.
Реакция проводится в атмосфере азота. Раствор 54 мг диазокетона 7d в 5 мл трифторуксусной кислоты кипятили 3 часа до полного исчезновения исходного диазокетона (контроль с помощью ТСХ). По завершении реакции трифторуксусная кислота была удалена в вакууме, осадок растворили в 5 мл хлористого метилена, тщательно промыли водным раствором NaHCO3, водой (2×1 мл) и высушили над K2CO3. После удаления растворителя получили 37 мг (75%) смеси двух региоизомерных 2,2-диалкил-4,5-диарил-дигидрофуран-3(2H)-онов в примерном соотношении 7:1 в виде светло-желтого маслообразного продукта. Реакционную смесь (37 мг) разделили на колонке с силикагелем и получили фуранон  с выходом 32 мг (66%), светло-желтое маслообразное вещество.
с выходом 32 мг (66%), светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.56 с (6Н, 2СН3), 2.50 с (3Н, SCH3), 7.10-7.45 м (8Н).
Спектр 19F NMR (376 МГц, CDCl3) δ, м.д.: -111.4.
Получение интермедиата 2,2-диметил-4-[4'-(метилтио)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (Схема 16, 1m).
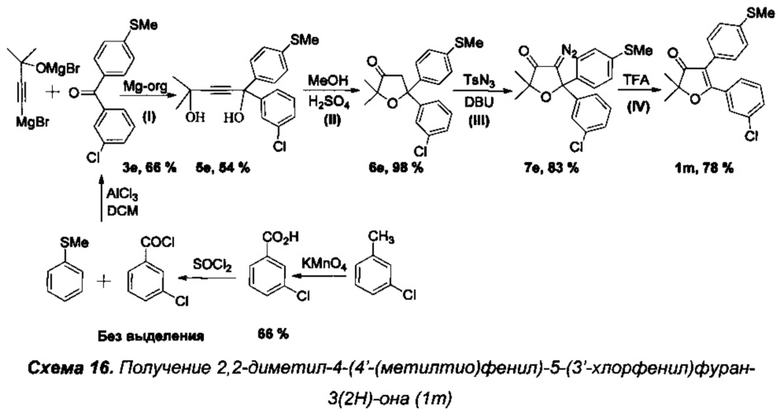
Стадия 0. Получение 4'-(метилтио)фенил(3'-хлорфенил)метанона (3е).
10 г метахлортолуола смешали с горячим раствором 30 г перманганата калия в 300 мл воды. Реакционную смесь кипятили с обратным холодильником в течение 4 часов, а затем в течение 1 часа с водяным паром отогоняли остатки непрореагировавшего метахлортолуола. Затем, реакционная смесь была профильтрована от диоксида марганца, твердая фаза дополнительно была промыта горячей водой, а к объединенным охлажденным фильтратам добавили 50 мл ледяной концентрированной соляной кислоты. Выпавший белый осадок метахлорбензойной кислоты был отфильтрован, промыт ледяной водой и высушен путем тройной отгонки азетропа вода-изопропанол (этанол) в вакууме роторного испарителя. Выход метахлорбензойной кислоты составил 4.88 г (45% на вошедший в реакцию хлортолуол).
В круглодонную колбу с магнитной мешалкой поместили 4.86 г метахлорбензойной кислоты, 40 мл тетрахлоруглерода и затем осторожно добавили смесь 5 мл перегнанного тионилхлорида SOCl2 с 0.05 мл диметилформамида в 20 мл тетрахлоруглерода. Смесь кипятили с обратным холодильником 5 часов до окончания выделения газообразных продуктов. Затем растворитель и остатки тионилхлорида были удалены в вакууме роторного испарителя. Полученное масло, после короткой вакуумной сушки, без дополнительной очистки, было растворено в 40 мл хлористого метилена и при охлаждении по каплям добавлено к суспензии 4.5 г безводного хлорида алюминия, 3.7 г тиоанизола и 40 мл хлористого метилена. После добавления всего количества хлорангидрида, смесь кипятили с обратным холодильником в течение 5 часов до полного прекращения выделения HCl. Полученный продукт гидролизовали 5-10% соляной кислотой, экстрагировали хлористым метиленом (3×40 мл), высушили над Na2SO4, а после отгонки растворителя - перекристаллизовали из гексана для очистки от избытка тиоанизола и комплексов алюминия (зеленого цвета). Выход кристаллического 4'-(метилтио)фенил(3'-хлорфенил)метанона (3е) составил 5.15 г (66%), бесцветные кристаллы, т.пл. 73-74°C.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 2.54 с (3Н, SCH3), 7.30 д (2Н, 3JHH=8.5 Гц), 7.41 т (1Н, 3JHH=7.8 Гц), 7.54 дм (1Н, JHH=8.0 Гц), 7.63 д (1Н, 3JHH=7.7 Гц), 7.73 д (3Н, 3JHH=8.5 Гц).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 14.8 (SCH3), 124.9 (2С), 127.8, 129.6, 129.7, 130.5 (2С), 132.1, 132.9, 134.5, 139.5, 146.0, 194.2 (С=O).
Стадия I. Получение 2-метил-5-(4'-хлорфенил)-5-(4'-метилтиофенил)пент-3-ин-2,5-диола (5е).
К раствору EtMgBr, приготовленному из 1.65 г (69 ммоль) магния и 7.5 г (69 ммоль) этилбромида, в 30 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 2.25 г (27 ммоль) 2-метилбут-3-ин-2-ола в 20 мл абсолютного бензола. Смесь кипятили в течение 3 ч, затем, не прекращая кипячения, в течение 2 часов добавили по каплям раствор (суспензию) 5.0 г (19 ммоль) соединения 3е в 40 мл бензола и кипятили далее в течение 4-5 ч. Затем смесь охладили и осторожно гидролизовали при помощи 100 мл холодного 5% раствора соляной кислоты. Органический слой отделили от водного. Водный слой экстрагировали бензолом (3×30 мл) и хлористым метиленом (2×20 мл). Объединенные органические фракции промыли раствором NaHCO3, водой и высушили над Na2SO4. После отгонки растворителя полученный желтый маслообразный продукт был разделен хроматографически на 3 фракции: целевой 1,4-диол, исходный непрореагировавший замещенный бензофенон и побочный продукт димеризации бензофенона до 1,2-диола. С целью уменьшения количества побочных продуктов и увеличения процента конверсии необходимо использовать недостаток магния и очень медленное добавление бензофенона в процессе реакции, так чтобы его концентрация оставалась незначительной. Очищенный γ-гликоль высушили в вакууме при давлении 0.01÷0.05 торр в течение 5 ч. Выход 1,4-диола 5е 3.56 г (54%), желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 1.57 с (6Н, 2СН3), 2.45 с (3Н, SCH3), 3.14 (1Н, -ОН), 7.18-7.25 м (4Н), 7.40-7.48 м (3H), 7.56-7.57 м (1Н).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3), δ, м.д.: 15.6 (SCH3), 31.2 (2СН3), 65.3 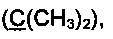 73.4, 84.1
73.4, 84.1 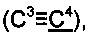 92.3
92.3 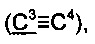 124.1, 126.1, 126.2 (2С), 126.4 (2С), 127.8, 129.5, 134.2, 138.4, 141.2, 146.9.
124.1, 126.1, 126.2 (2С), 126.4 (2С), 127.8, 129.5, 134.2, 138.4, 141.2, 146.9.
ИК спектр (в CCl4), см-1: 3603 ср., 3381 с. шир., 2984 ср., 2924 сл., 1595 ср., 1491 ср., 1423 ср., 1323 с., 1167 с., 1095 ср., 1006 ср.
Масс-спектр LCMS, m/z, рассчитано: 369.0687, найдено 369.0729 [MNa+].
Стадия II. Получение 2,2-диметил-5-[4'-(метилтио)фенил]-5-(3'-хлорфенил)-дигидрофуран-3(2H)-она (6е).
3.56 г соединения 5е растворили в 30 мл кипящего метанола и при перемешивании по каплям добавили раствор 2.9 мл 98% серной кислоты, растворенной в 30 мл метанола. Смесь кипятили в течение 5 ч до окончания реакции (контроль по ТСХ). Затем смесь вылили в 70 мл воды и тщательно экстрагировали хлористым метиленом (4×25 мл). Органический слой высушили Na2SO4, растворитель отогнали на роторе, а полученное масло высушили от метанола и дихлорметана в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Получено 3.49 г (выход 98%) маслообразного продукта 6е. Без дополнительной очистки полученное соединение использовали на следующей стадии.
Спектр ЯМР 1Н (400 МГц, 15 мг в 0.7 мл CDCl3), δ, м.д.: 1.18 с (3Н, СН3), 1.26 с (3Н, СН3), 2.46 с (3Н, SCH3), 3.21 д (1Н, СН2, 2JHH=18 Гц), 3.33 д (1Н, СН2, 2JHH=18 Гц), 7.16-7.27 м (5Н), 7.31 д (2Н, 3JHH=8.8 Гц), 7.41-7.44 м (1Н).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.7 мл CDCl3), δ, м.д.: 15.5 (SCH3), 25.1 (СН3), 25.4 (СН3), 47.7 (СН2), 81.6 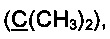 81.9, 123.9, 126.0, 126.3 (2С), 126.5 (2С), 127.5, 129.6, 134.4, 138.2, 142.1, 148.4, 216.2 (С=O).
81.9, 123.9, 126.0, 126.3 (2С), 126.5 (2С), 127.5, 129.6, 134.4, 138.2, 142.1, 148.4, 216.2 (С=O).
ИК спектр (в CCl4), см-1: 880 сл., 997 ср., 1135 ср., 1172 ср., 1422 ср., 1493 ср., 1596 ср., 1762 с. (С=O), 2982 ср. (С-Н).
Масс-спектр HRMS, m/z, рассчитано: 369.0687, найдено 369.0696 [MNa+].
Стадия III. Получение 4-диазо-2,2-диметил-5-[4'-(метилтио)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2H)-она (7е).
К смеси 3.49 г фуранона 6е и 2.0 г паратолуолсульфонилазида в 50 мл дихлорметана добавили по каплям раствор 400 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 25 мл дихлорметана, при этом смесь сразу же окрасилась в оранжево-желтый цвет. Реакционную смесь перемешивали при температуре 35°C в течение 8 ч до завершения реакции (контроль с помощью ТСХ), затем отогнали избыток растворителя, перенесли на колонку с силикагелем и хроматографировали в градиентном режиме смесью н-гексан - хлористый метилен. После удаления растворителя продукт высушили в вакууме и получили 3.10 г (83%) диазокетона 7е, желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.) δ, м.д.: 1.34 с (3Н, СН3), 1.35 с (3Н, СН3), 2.49 с (3Н, SCH3), 7.15-7.35 м (8Н).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.) δ, м.д.: 15.3 (SCH3), 25.2 (2СН3), 64.0 (C=N2), 83.5 (С5), 85.7 (С2), 124.5, 126.2 (2С), 126.6, 126.8 (2С), 128.6, 129.8, 134.7, 139.5, 139.6, 145.8, 196.5 (С=O).
ИК спектр (в CCl4), см-1: 1026 сл., 1094 сл., 1163 сл., 1225 сл., 1344 с., 1377 сл., 1595 сл., 1697 с (С=O), 2093 оч.с. (CN2), 2930 сл., 2982 сл.
Масс-спектр HRMS, m/z, рассчитано для C19H17ClN2O2SNa+: 395.0592, найдено 395.0598 [MNa+].
Стадия IV. Получение 2,2-диметил-4-[4'-(метилтио)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (1m).
Реакция проводится в атмосфере азота. К раствору 1.0 г диазокетона 7е в 35 мл дихлорметана добавили по каплям 0.3 мл трифторметансульфоновой кислоты (TfOH), растворенной в 10 мл хлористого метилена. Смесь кипятили 3 часа до полного исчезновения исходного диазокетона (контроль с помощью ТСХ), а по завершении реакции смесь промыли водным раствором NaHCO3, водой (2×10 мл) и высушили K2CO3. После удаления растворителя получили 870 мг (94%) смеси двух региоизомерных 2,2-диалкил-4,5-диарил-дигидрофуран-3(2H)-онов в примерном соотношении 5:1 в виде светло-желтого маслообразного продукта. Реакционную смесь (273 мг) разделили на колонке с силикагелем и получили фуранон 1m с выходом 720 мг (78%), светло-желтое маслообразное вещество или кристаллы.
Спектр ЯМР 1Н (400 МГц, 5 мг в 0.8 мл CDCl3, репер: CHCl3=7.260 м.д.), δ, м.д.: 1.55 с (6Н, 2СН3), 2.49 с (3Н, SCH3), 7.21-7.26 м (4Н, PhSCH3), 7.36 с (1Н), 7.43 д (1Н, 3JHH=9.1 Гц), 7.48 д (1Н, 3JHH=7.8 Гц), 7.72 с (1Н).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.0 м.д.), δ, м.д.: 15.6 (SCH3), 23.3 (2СН3), 87.2 (С(СН3)2), 113.7 (С4), 126.6 (2С), 126.7, 128.0, 128.3, 129.7, 129.8 (2С), 131.7, 131.8, 134.7, 138.2, 176.1 (С5), 205.3 (С=O).
Масс-спектр HRMS, m/z, рассчитано 345.0711, найдено 345.0718 [МН]+.
Пример 6.
Получение 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (Схема 17, 1n).
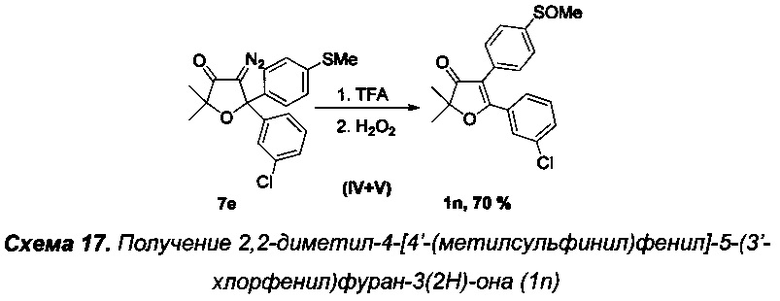
Получение соединения 7е описано выше.
Стадии IV-V. Получение 2,2-диметил-4-[4'-(метилсульфинил)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (1n).
Реакцию проводили в атмосфере азота. В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 15 мл трифторуксусной кислоты и растворили в ней 360 мг (0.96 ммоль) соединения 7е и кипятили данный раствор в течение 3-х часов. Затем охладили до 40°C и по каплям при интенсивном перемешивании в течение 1 ч осторожно добавили смесь 100 мг 35% (0.99 ммоль) свежего водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.025 мл (0.5 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное эквимолярное соотношение пероксида водорода к 7е. Реакционную смесь перемешивали еще в течение 1.5 ч при +50-55°C (контроль реакции с помощью ТСХ). По завершении реакции смесь вылили в воду, нейтрализовали карбонатом натрия, тщательно экстрагировали этилацетатом (4×20 мл). Органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и хроматографического разделения региоизомерных сульфоксидов, получили 240 мг (70%) соединения 1n, светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.53 с (6Н, 2СН3), 2.79 с (3Н, SOCH3), 7.23-7.27 м (1Н), 7.37-7.50 м (4Н), 7.60-7.70 м (3H).
Спектр ЯМР 13С (100 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.3 (2СН3), 43.6 (SOCH3), 87.9 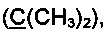 113.0 (С4), 124.2, 126.8, 128.1, 130.0, 130.3 (2С), 131.2, 132.2, 132.9, 134.9, 144.0, 177.4 (С5), 204.7 (С=O).
113.0 (С4), 124.2, 126.8, 128.1, 130.0, 130.3 (2С), 131.2, 132.2, 132.9, 134.9, 144.0, 177.4 (С5), 204.7 (С=O).
ИК спектр (в CCl4), см-1: 1051 ср. (S=O), 1145 ср., 1238 сл., 1384 с., 1618 ср., 1706 с. (С=O), 2983 сл. (С-Н).
Масс-спектр HRMS, m/z, рассчитано 361.0660, найдено 361.0740 [МН]+.
Пример 7.
Получение 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (Схема 18, 1о).
Получение соединения 7е описано выше.
Стадии IV-V. Получение 2,2-диметил-4-[4'-(метилсульфонил)фенил]-5-(3'-хлорфенил)фуран-3(2H)-она (1о).
Реакцию проводили в атмосфере азота. В круглодонную колбу с магнитной мешалкой и воздушным холодильником поместили 10 мл трифторуксусной кислоты и растворили в ней 360 мг (0.96 ммоль) соединения 7е и кипятили данный раствор в течение 3-х часов. Затем, не прекращая кипячения, по каплям в течение 10 мин добавили смесь 200 мг 34% (2 ммоль) водного раствора пероксида водорода в 5 мл трифторуксусной кислоты и 0.025 мл (0.5 ммоль) 98% H2SO4. В данной реакции очень важно соблюдать точное 2х кратное молярное соотношение пероксида водорода к 7е для предотвращения окисления двойной связи в соединении 1о. Реакционную смесь перемешивали еще в течение 2 ч при +65-70°C (контроль с помощью ТСХ). По завершении реакции смесь вылили в воду, кислоту нейтрализовали карбонатом натрия, органические вещества экстрагировали этилацетатом (4×20 мл). Органические вытяжки промыли раствором NaHCO3, водой (3×5 мл), высушили над Na2SO4 и K2CO3, и, после удаления растворителя и хроматографического разделения региоизомерных сульфонов, получили 278 мг (77%) соединения 1о, бесцветное или светло-желтое маслообразное вещество.
Спектр ЯМР 1Н (400 МГц, 20 мг в 0.8 мл CDCl3, репер: CHCl3=7.26 м.д.), δ, м.д.: 1.56 с (6Н, 2СН3), 3.04 с (3Н, SO2CH3), 7.29 д (1Н, 3JHH=7.8 Гц), 7.38 д (1Н, 3JHH=7.9 Гц), 7.51 д (2Н, 3JHH=8.4 Гц), 7.67 с (1Н), 7.91 д (2Н, 3JHH=8.4 Гц).
Спектр ЯМР 13С (100 МГц, 25 мг в 0.8 мл CDCl3, репер: CHCl3=77.00 м.д.), δ, м.д.: 23.2 (2СН3), 44.4 (SO2CH3), 88.1  112.4, 126.7, 127.6 (2С), 128.0, 130.0, 130.1 (2С), 131.0, 132.3, 135.0, 135.6, 139.3, 177.8 (С5), 204.2 (С=O).
112.4, 126.7, 127.6 (2С), 128.0, 130.0, 130.1 (2С), 131.0, 132.3, 135.0, 135.6, 139.3, 177.8 (С5), 204.2 (С=O).
ИК спектр (в CCl4), см-1: 957 ср., 1157 с. (SO2), 1330 с. (SO2), 1384 ср., 1616 ср., 1705 с. (С=O), 2931 сл., 2982 сл. (С-Н).
Масс-спектр LCMS, m/z, рассчитано 377.0609, найдено 377.0667 [МН]+.
Пример 8.
Получение 2,2-диметил-4-(4'-(аминосульфонил)фенил)-5-фенилфуран-3(2H)-она (Схема 19, 1q).
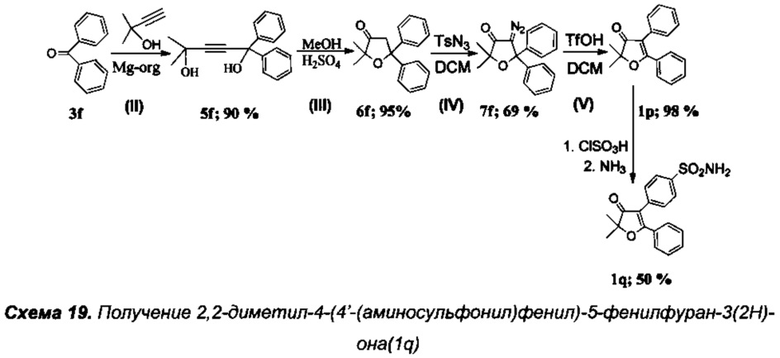
Стадия II. Получение 2-метил-5,5-дифенилпент-3-ин-2,5-диола (5f).
К раствору EtMgBr, приготовленного из 10 г (0.41 моль) магния и 46 г (0.42 моль) этилбромида, в 160 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 15.7 мл (0.16 моль) 2-метилбут-3-ин-2-ола в 90 мл абсолютного бензола. Далее смесь кипятили в течение 3 ч, после чего добавили 21 г (0.12 моль) бензофенона (3f) и кипятили в течение 8 ч. После охлаждения реакционной смеси в водяной бане через капельную воронку прилили 250 мл раствора соляной кислоты, приготовленного из 200 мл воды и 50 мл 36% соляной кислоты. Органический слой отделили от водного, последний подвергли экстракции абсолютным хлористым метиленом (3×30 мл). Объединенные органические вытяжки промыли NaHCO3, затем водой и высушили над K2CO3. Растворитель отогнали в вакууме, полученный остаток перекристаллизовали из смеси гексана и хлороформа. Выход 28.8 г (90%) соединения 5f, т.пл. 119-120°C (литературные данные 118-119°C [21]).
Стадия III. Получение 2,2-диметил-5,5-дифенилдигидрофуран-3(2H)-она (6f).
К раствору 19.13 г (72 ммоль) соединения 5f в 140 мл СН3ОН осторожно прибавили 55 мл раствора, представляющего собой 15 мл концентрированной серной кислоты в 40 мл метанола. Смесь кипятили с обратным холодильником в течение 1-2 часов. По завершению реакции смесь вылили в 400 мл воды и подвергали экстракции дихлорметаном (4×40 мл). Органические фазы промыли 50 мл раствора Na2CO3(нас.), затем насыщенным раствором NaCl (2×20 мл) и высушили над Na2SO4. Отогнав растворитель в вакууме роторного испарителя, выпавший осадок перекристаллизовали из гексана. Выход 18 г (95%) соединения 6f, бесцветные кристаллы т.пл. 64-65°C.
Спектр 1Н ЯМР (400 МГц, 5 мг в 0.7 мл CDCl3,TMS=0 м.д.), δ, м.д.: 1.210 с (6Н, 2СН3), 3.330 с (2Н, СН2), 7.240 д (2H, 3JHH=6.7 Гц), 7.307 т (4Н, 3JHH=7.6 Гц), 7.410 д (4Н, 3JHH=8.2 Гц).
Спектр 13С ЯМР (100 МГц, 30 мг в 0.7 мл CDCl3), δ, м.д.: 25.31 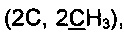 48.04
48.04 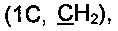 81.35 (1С, С2), 82.64 (1С, С5), 126.0 (4С), 127.34 (2С), 128.28 (4С), 146.09 (2С), 217.0
81.35 (1С, С2), 82.64 (1С, С5), 126.0 (4С), 127.34 (2С), 128.28 (4С), 146.09 (2С), 217.0 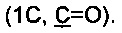
Спектр ИК (в CCl4), см-1: 699.4 м., 740.2 сл., 994.5 сл., 1131.1 сл., 1173.0 сл., 1376.1 сл., 1448.4 сл., 1492.6 сл., 1760.1 с., 2931.6 сл., 2983.2 сл., 3027.8 сл., 3064.9 сл. HRMS, m/z, вычислено: 267.1380, найдено: 267.1381 [МН]+.
Стадия IV. Получение 4-диазо-2,2-диметил-5,5-дифенилдигидрофуран-3(2H)-она (7f).
К смеси 1 грамма (3.76 ммоль) соединения фуранона 6f, 770 мг (3.9 ммоль) паратолуолсульфанилазида в 70 мл дихлорметана при интенсивном перемешивании добавили по каплям 200 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 10 мл дихлорметана. Смесь перемешивали при 35°C в течение 4 ч. После завершения реакции органический слой промыли насыщенным раствором соли (2×20 мл), водные вытяжки подвергли экстракции хлористым метиленом (2×20 мл). Объединенные органические фазы промыли водой (5×10 мл) до нейтральной среды и сушили над сульфатом магния. Растворитель отогнали, остаток пропустили через колонку с силикагелем (элюент: гексан/хлористый метилен в градиентном режиме). Выход: 760 мг (69%) соединения 7f, желтое кристаллическое вещество, т.пл. 118.7-118.9°C.
Спектр 1Н ЯМР (400 МГц, 5 мг в 0.7 мл CDCl3,TMS=0 м.д.), δ, м.д.: 1.35 с (6Н, 2СН3); 7.30-7.38 м (10Н, 2Ph).
Спектр 13С ЯМР (100 МГц, 30 мг в 0.7 мл CDCl3), δ, м.д.: 25.3 (2СН3), 64.4 (C=N2), 84.2, 85.5, 128.5, 128.3, 126.4, 143.8, 197.0 (С=O).
Спектр ИК (в KBr), см-1: 3065 сл., 3045 сл., 2981 сл., 2932 сл., 2866 сл., 2093 оч. с. (C=N2), 1832 сл., 1806 сл., 1692 оч. с. (С=O), 1599 сл., 1490 ср., 1446 с., 1377 с., 1347 оч. с., 1226 с., 1185 ср., 1165 ср., 1153 ср., 1083 сл., 1022 с., 1005 с., 931 сл., 921 сл., 910 ср., 894 ср., 761 с., 701 с.
CHN анализ, вычислено для C18H16N2O2: С - 73.96, Н - 5.52, N - 9.58, найдено: С - 73.93, Н - 5.52, N - 9.57.
Стадия V. Получение 2,2-диметил-4,5-дифенилфуран-3(2H)-она (1р).
К раствору 290 мг (1 ммоль) соединения 7f в 10 мл дихлорметана по каплям добавляли раствор 0.1 мл трифторметансульфоновой кислоты (TfOH) в 5 мл дихлорметана. Смесь кипятили 2 часа до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×10 мл) и высушили над K2CO3. После отгонки растворителя получено 260 мг (98%) соединения 1р, бесцветное кристаллическое вещество.
Спектр 1Н ЯМР (400 МГц, 5 мг в 0.7 мл CDCl3, TMS=0 м.д.), δ, м.д.: 1.58 с (6Н, 2СН3), 7.28-7.68 м (10Н, 2Ph).
Спектр 13С ЯМР (100 МГц, 30 мг в 0.7 мл CDCl3), δ, м.д.: 23.8, 87.4, 114.0, 127.9, 128.8, 129.0, 129.9, 130.4, 132.2, 178.4, 205.8. HRMS, m/z, вычислено: 265.1224, найдено: 265.1231 [МН]+.
Стадия VI. Получение 2,2-диметил-4-(4'-(аминосульфонил)фенил)-5-фенилфуран-3(2H)-она (1q).
К 1.5 г (10-кратный мольный избыток) хлорсульфоновой кислоты осторожно добавили 325 мг фуранона 1р, затем добавили 10 мл тетрахлоруглерода и 100 мг хлорида натрия. Смесь перемешивали при 60°C в течение 3 часов. После завершения реакции сульфохлорирования смесь поставили на ледяную баню и при интенсивном перемешивании гидролизовали непрореагировавшую хлорсульфоновую кислоту медленно прикапывая 50% серную кислоту (10 мл) к реакционной смеси. После чего через реакционный сосуд при 0°C пропускали сухой аммиак до нейтрализации всех кислот, затем добавили 10 мл 25% раствора аммиака в воде и перемешивали при 50-60°C в течение 3 часов. По завершению реакции амидирования, воду и тетрахлорид углерода удалили путем азеотропной отгонки на роторном испарителе. Полученную кристаллическую смесь нанесли на колонку с силикагелем и отделили непрореагировавший исходный фуранон (элюэнт: дихлорметан) от целевого амида 1q (элюэнт: метиловый спирт). Неорганические соли и соли сульфокислот остаются на старте колонки. Отделенный таким образом амид дополнительно перекристаллизовали из этанола и высушили в вакууме при давлении 0.02÷0.05 торр в течение 5 ч. Выход 1q: 330 мг (79%), бесцветное кристаллическое вещество..
Примечание 1. В предложенном методе тетрахлорид углерода служит инертным разбавителем реакционной смеси, уменьшающим ее вязкость. Разбавление 50% серной кислотой проводится, так как прямой гидролиз водой или гидролиз с амидированием раствором аммиака очень часто протекает неуправляемо и со взрывом, несмотря на интенсивное охлаждение. Нейтрализация кислот газообразным аммиаком проводится с той же целью уменьшения интенсивности тепловых эффектов. В данном методе мы не используем экстракцию полученного вещества из водного раствора по причине возможных больших потерь амида 1q за счет его хорошей растворимости в воде. Следует обратить внимание, также на способность 1q образовывать сольваты со спиртами. Амиды малорастворимы в хлороформе, поэтому снятие ЯМР спектров осуществлялось в ацетоне. По спектру имеется только один набор сигналов, соответствующих одному региоизомеру.
Спектр 1Н ЯМР (400 МГц, 5 мг в 0.7 мл ацетон-d6, реперный мультиплет ацетона = 2.05 м.д.), δ, м.д.: 1.53 с (6Н, 2СН3), 6.60 с (2Н, -NH2 ассоц.), 7.44-7.49 м (4Н), 7.58 т (1Н, 3JHH=7.4 Гц), 7.65 д (2Н, 3JHH=7.2 Гц), 7.87 д (2Н, 3JHH=8.5 Гц).
Спектр 1Н ЯМР (400 МГц, 3 мг в 0.6 мл CDCl3), δ, м.д.: 1.58 с (6Н, 2СН3), 4.78 с (2Н, NH2), 7.40 т (2Н, 3JHH=7.7 Гц), 7.50 т (3Н, 3JHH=8.2 Гц), 7.62 д (2Н, 3JHH=7.2 Гц), 7.90 д (2Н, 3JHH=8.5 Гц).
Спектр 13С ЯМР (100 МГц, 15 мг в 0.7 мл ацетон-d6, реперный мультиплет ацетона = 29.84 м.д.), δ, м.д.: 23.4 (2С), 88.0 (С2), 113.0 (С4), 127.0 (2С), 129.2 (2С), 129.6 (2С), 130.6 (2С), 130.7, 133.0, 135.3, 143.7, 179.7 (С5), 204.5 (С=O).
Спектр ИК (в KBr), см-1: 3346 шир. (-SO2NH2), 3234 шир. (-SO2NH2), 2988 сл. (СН3, Ar), 2938 сл. (СН3, Ar), 1662 оч. с., 1607 с., 1568 с., 1485 ср., 1386 с., 1342 с., 1238 ср., 1170 с., 1058 ср., 904 ср., 746 ср., 658 ср., 597 ср., 553 ср.
HRMS, m/z, вычислено: 344.0952, найдено: 344.1006 [МН]+.
Пример 9.
Получение 2,2-диметил-4-(4'-(аминосульфонил)фенил)-5-(4'-хлорфенил)фуран-3(2H)-она и 4-(2'-(4''-хлорфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислоты (Схема 20, 1v и 1vv).
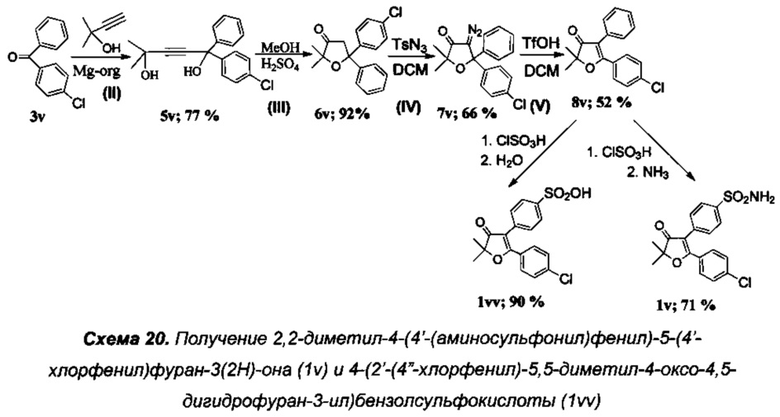
Стадия II. Получение 1-(4-хлорфенил)-4-метил-1-фенилпент-2-ин-1,4-диола (5v).
К раствору EtMgBr, приготовленного из 10 г (0.41 моль) магния и 46 г (0.42 моль) этилбромида, в 160 мл абсолютного диэтилового эфира при охлаждении на ледяной бане медленно прибавили по каплям раствор 15.7 мл (0.16 моль) 2-метилбут-3-ин-2-ола в 90 мл абсолютного бензола. Далее смесь кипятили в течение 3 ч, после чего добавили 25.9 г (0.12 моль) (4-хлорфенил)(фенил)метанона (3v) и кипятили в течение 7 ч. После охлаждения реакционной смеси в водяной бане через капельную воронку прилили 230 мл раствора соляной кислоты, приготовленного из 180 мл воды и 50 мл 36% соляной кислоты. Органический слой отделили от водного, последний подвергли экстракции абсолютным хлористым метиленом (3×30 мл). Объединенные органические вытяжки промыли NaHCO3, затем водой и высушили над K2CO3. Растворитель отогнали в вакууме, полученный остаток перекристаллизовали из смеси гексана и хлороформа. Выход 27.7 г (77%) соединения 5v, т.пл. 91-92°C (литературные данные 89-95°C, [22]).
Стадия III. Получение 2,2-диметил-5-фенил-5-(4'-хлорфенил)дигидрофуран-3(2H)-она (6v).
К раствору 21.6 г (72 ммоль) соединения 5v в 140 мл СН3ОН осторожно прибавили 55 мл раствора, представляющего собой смесь 15 мл концентрированной серной кислоты и 40 мл метанола. Смесь кипятили с обратным холодильником в течение 1-2 часов. По завершению реакции смесь вылили в 400 мл воды и подвергали экстракции дихлорметаном (4×40 мл). Органические фазы промыли 50 мл раствора Na2CO3(нас.), затем насыщенным раствором NaCl (2×20 мл) и высушили над Na2SO4. Отогнав растворитель в вакууме роторного испарителя, выпавший осадок перекристаллизовали из гексана. Выход 18 г (92%) соединения 6v, бесцветные кристаллы т.пл. 68°C. (литературные данные 67.5-69°C, [22]).
Стадия IV. Получение 4-диазо-2,2-диметил-5-фенил-5-(4'-хлорфенил)дигидрофуран-3(2H)-она (7v).
К смеси 1.13 грамма (3.76 ммоль) фуранона 6v, 770 мг (3.9 ммоль) паратолуолсульфанилазида в 60 мл дихлорметана при интенсивном перемешивании добавили по каплям 190 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 10 мл дихлорметана. Смесь перемешивали при 30°C в течение 6 ч. После завершения реакции органический слой промыли насыщенным раствором соли (2×20 мл), водные вытяжки подвергли экстракции хлористым метиленом (2×20 мл). Объединенные органические фазы промыли водой (5×10 мл) до нейтральной среды и сушили над сульфатом магния. Растворитель отогнали, остаток пропустили через колонку с силикагелем (элюент: гексан/хлористый метилен в градиентном режиме). Выход: 749 мг (66%) соединения 7v, желтое кристаллическое вещество, т.пл. 131-132°C.
Спектр 1Н ЯМР (400 МГц, 6 мг в 0.7 мл CDCl3, TMS=0 м.д.), δ, м.д.: 1.35 с (6Н, 2СН3); 7.30-7.36 м (9Н).
CHN анализ, вычислено для C18H15N2O2Cl: С - 66.16, Н - 4.63, N - 8.57, найдено: С - 66.0, Н - 4.65, N - 9.59.
Стадия V. Получение 2,2-диметил-4-фенил-5-(4'-хлорфенил)фуран-3(2H)-она (8v).
К раствору 326 мг (1 ммоль) соединения 7v в 15 мл дихлорметана по каплям добавляли раствор 0.1 мл трифторметансульфоновой кислоты (TfOH) в 5 мл дихлорметана. Смесь кипятили 2 часа до полного исчезновения исходного диазокетона (контроль реакции по ТСХ). По завершении реакции смесь промыли раствором NaHCO3, водой (2×10 мл) и высушили над K2CO3. Растворитель отогнали, смесь 2х региоизомеров разделили на колонке с силикагелем (элюент: гексан/хлористый метилен в градиентном режиме). Получено 155 мг (52%) соединения 8v, бесцветное кристаллическое вещество.
Спектр 1Н ЯМР (400 МГц, 7 мг в 0.7 мл CDCl3, TMS=0 м.д.), δ, м.д.: 1.55 с (6Н, 2СН3), 7.30-7.61 м (9Н).
HRMS, m/z, вычислено: 299.0834, найдено: 299.0885 [МН]+.
Получение 4-(2'-(4''-хлорфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислоты (1vv).
К 1.5 г (10-кратный мольный избыток) хлорсульфоновой кислоты осторожно добавили 298 мг (1 ммоль) фуранона 8v, затем добавили 10 мл тетрахлоруглерода и 10 мг хлорида натрия. Смесь перемешивали при 60°C в течение 3 часов. После завершения реакции сульфохлорирования смесь поставили на ледяную баню и при интенсивном перемешивании гидролизовали непрореагировавшую хлорсульфоновую кислоту медленно прикапывая вначале ледяную воду (10 мл), а затем 30% водный раствор KOH до полной нейтрализации. По завершению реакции, воду и тетрахлорид углерода удалили путем азеотропной отгонки на роторном испарителе. Полученную кристаллическую смесь солей растворили в минимальном количестве кипящей воды (1-3 мл) и добавили 4-5 мл ледяной 30% HCl. Выпавший хлопьевидный осадок сульфокислоты 1vv отфильтровали на фильтре Шотта, промыли ледяной 25% HCl и высушили в вакууме 0.1 торр в течение 24 часов до постоянной массы. Альтернативно, сушка сульфокислоты может быть осуществлена путем ее растворения в достаточном количестве дихлорметана и выдерживании в течение 12 часов над безводным Na2SO4. Выход сульфокислоты 1vv: 340 мг (90%), бесцветное кристаллическое вещество.
Спектр 1Н ЯМР (400 МГц, 5 мг в 0.7 мл ацетон-d6, реперный мультиплет ацетона = 2.05 м.д.), δ, м.д.: 1.53 с (6Н, 2СН3), 7.44-7.51 м (4Н), 7.55-7.90 м (4Н).
HRMS, m/z, вычислено: 401.0221, найдено: 401.0210 [MNa]+.
Получение 2,2-диметил-4-(4'-(аминосульфонил)фенил)-5-(4'-хлорфенил)фуран-3(2H)-она (1v).
К 1.5 г (10-кратный мольный избыток) хлорсульфоновой кислоты осторожно добавили 298 мг фуранона 1v, затем добавили 10 мл тетрахлоруглерода и 100 мг хлорида натрия. Смесь перемешивали при 60°C в течение 3 часов. После завершения реакции сульфохлорирования смесь поставили на ледяную баню и при интенсивном перемешивании гидролизовали непрореагировавшую хлорсульфоновую кислоту медленно прикапывая 50% серную кислоту (10 мл) к реакционной смеси. После чего через реакционный сосуд при 0°C при интенсивном перемешивании пропускали сухой аммиак до нейтрализации всех кислот, затем добавили 10 мл 25% раствора аммиака в воде и перемешивали при 50-60°C в течение 3 часов. По завершению реакции амидирования, воду и тетрахлорид углерода удалили путем азеотропной отгонки с изопропиловым спиртом на роторном испарителе. Полученную кристаллическую смесь нанесли на колонку с силикагелем и отделили непрореагировавший исходный фуранон (элюэнт: дихлорметан) от целевого амида 1v (элюэнт: метиловый спирт). Неорганические соли и соли сульфокислот остаются на старте колонки. Отделенный таким образом амид дополнительно перекристаллизовали из этанола и высушили в вакууме при давлении 0.02÷0.05 торр в течение 6 ч. Выход 1v: 268 мг (71%), бесцветное кристаллическое вещество.
Спектр 1Н ЯМР (400 МГц, 5 мг в 0.7 мл ацетон-d6, реперный мультиплет ацетона = 2.05 м.д.), δ, м.д.: 1.53 с (6Н, 2СН3), 4.82 с (2Н, -NH2), 7.44-7.49 м (4Н), 7.64 д (2Н, 3JHH=7.2 Гц), 7.86 д (2Н, 3JHH=8.5 Гц).
HRMS, m/z, вычислено: 378.0562, найдено: 378.0578 [МН]+.
Примечание 2. Получение остальных парафтор, метафтор и метахлор замещенных бензолсульфокислот и сульфамидов осуществляется по вышеописанным методикам исходя из замещенных бензофенонов и не представляет трудностей. Замещенные сульфамиды получаются при использовании вместо аммиака первичных аминов, таких как метиламин и этиламин, в виде растворов в подходящих растворителях, например, в тетрахлоруглероде.
Фармацевтически приемлемые производные соединений
Соединения данного изобретения могут существовать в свободной форме в процессе обработки или, если требуется, в виде фармацевтически приемлемой соли или другого производного.
Используемый здесь термин «фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в [23]). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные.
Фармацевтические композиции
Изобретение также относится с фармацевтическим композициям, которые содержат, по меньшей мере, одно из описанных здесь соединений (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, растворителей и/или наполнителей. Данные композиции также могут содержать один или несколько дополнительных терапевтических агентов. С другой стороны, соединение данного изобретения может быть введено пациенту, нуждающемуся в соответствующей терапии, в комбинации с одним или более других терапевтических агентов, например в сочетании с антиагрегантами, препаратами для лечения сопутствующих заболеваний (в том числе противоопухолевыми препаратами) и т.д.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. [24] раскрывает различные носители, использованные при разработке фармацевтических композиций и известные методы их приготовления. За исключением таких случаев, когда среда обычных носителей несовместима с соединением изобретения, например, при появлении любых нежелательных биологических эффектов и иных нежелательных взаимодействий с любым другим компонентом (компонентами) фармацевтической композиции, использование таких композиций находится в рамках данного изобретения. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахаридами, а также их производными; солодом, желатином; тальком; эксципиентами, такими как: какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Применение соединений в комбинированной терапии
Несмотря на то, что соединения данного изобретения могут вводиться в качестве индивидуального активного фармацевтического средства, их также можно использовать в сочетании с одним или несколькими соединениями изобретения, и/или одним или несколькими другими агентами. При совместном приеме внутрь терапевтические агенты могут представлять собой разные лекарственные формы, которые вводятся одновременно или последовательно в разное время, либо терапевтические агенты могут быть объединены в одну лекарственную форму.
Фраза «комбинированная терапия» в отношении соединений данного изобретения в сочетании с другими фармацевтическими агентами, означает одновременный или последовательный прием всех агентов, который, так или иначе, обеспечит благоприятное воздействие сочетания лекарств. Совместное введение подразумевает, в частности, совместную доставку, например, в одной таблетке, капсуле, инъекции или в другой форме, имеющий фиксированное соотношение активных веществ, также как и одновременную доставку в нескольких, отдельных лекарственных формах для каждого соединения соответственно.
Таким образом, введение соединений данного изобретения может быть осуществлено в сочетании с дополнительными методами лечения, известными специалистам в области лечения заболеваний, ассоциированных с развитием воспаления, а также заболеваний, опосредованных избыточной секрецией СОХ-2.
Характеристика биологической активности соединений
Компьютерное моделирование процесса ингибирования ферментов циклооксигеназы-1 и -2 соединениями по изобретению.
Поскольку на сегодняшний день проведено достаточно большое число биологических тестов различных классов ингибиторов циклооксигеназы человека и животных [14], и накоплен значительный массив экспериментальных данных по оценке активности тех или иных молекул в отношении СОХ-1 и -2, такие параметры, как IC50 (концентрация вещества, приводящая к 50% ингибирования циклооксигеназы стандартной концентрации) и SI (индекс селективности, IC50 (СОХ-1)/IC50 (СОХ-2)), могут быть с достаточной точностью рассчитаны методами компьютерного моделирования с помощью построения QSAR модели по ряду наиболее близких исследованных структур [25]. Ниже проведено компьютерное моделирование ингибирующей активности на примере соединений по изобретению.
Список общепринятых сокращений и терминов в области виртуального скрининга.
- Докинг (Docking) - метод, позволяющий на основе трехмерной структуры мишени (фермента), прогнозировать возможные супрамолекулярные взаимодействия с лигандом (молекулой СОХ-ингибитора).
- Преподготовка - процедура минимизации потенциальной энергии и геометрической оптимизации трехмерной структуры для малых и биомолекул, с применением стандартных подходов молекулярной механики и динамики.
- Отжиг (Annealing) - метод получения трехмерной конформации мишени (белковая молекула) с минимальными расчетными значениями потенциальной энергии с использованием подходов молекулярной динамики.
- Стресс (Stress) аминокислот - конформационное напряжение (энергетический терм) аминокислот в третичной структуре белка-мишени.
- Скоринг (Score) - энергетическая (ккал/моль) функция, характеризующая аффинность лиганда по отношению к сайту связывания.
- RMSD - среднеквадратичное отклонение трехмерных атомных координат.
- Фармакофор - это набор пространственных и электронных признаков молекулы, необходимых для обеспечения ее оптимальных супрамолекулярных взаимодействий с определенной биологической мишенью, которые могут вызывать (или блокировать) биологический ответ.
Преподготовку и построение гомологичных моделей белков-мишеней проводили в программе ICM Pro v. 1.8а (MolSoft, официальный сайт: http://www.molsoft.com), докинг осуществляли в программе МОЕ v. 2014.09.01 (Molecular Operating Environment, CCG, официальный сайт: http://www.chemcomp.com). Для построения трехмерных моделей изоформ hCOX-1 и hCOX-2 в качестве исходных использовали кристаллографические данные, полученные ранее для лекарственного вещества целекоксиба (Celebrex®, Pfizer), который является наиболее известным селективным ингибитором циклооксигеназы-2. Номера записей в базе данных PDB (Protein Data Bank, http://www.rcsb.org) 3KK6 (СОХ-1 овечья изоформа, DOI: 10.2210/pdb3kk6/pdb) и 3LN1 (СОХ-2 мышиная изоформа, DOI: 10.2210/pdb3ln1/pdb). Указанные белки были получены рекомбинантным путем в клеточной линии насекомых Spodoptera frugiperda.
Гомологичные модели строили с использованием функции Homology Model. Использовали стандартный FASTA код первичной последовательности аминокислот hCOX-1 (номер sp_P23219_PGH1_HUMAN, источник http://www.uniprot.org/uniprot/P23219) и hCOX-2 (номер sp_P35354_PGH2_HUMAN, источник http://www.uniprot.org/uniprot/P35354). Молекулы воды были исключены из рассмотрения с целью упрощения процедуры моделирования и построения QSAR модели. На первом этапе получали трехмерные конформации целевых аминокислотных последовательностей путем «грубого» наложения на соответствующий темплейт (т.е. вторичная и третичная структуры ферментов 3KK6 и 3LN1), затем «отжигали» белки при различных начальных температурах до достижения минимальных значений стресса аминокислот. Диаграммы стресса для финальных трехмерных конформаций представлены на рисунках 1, 2.
Как следует из рисунков 1 и 2, две изоформы hCOX-1 и hCOX-2 характеризуются относительно низким значением стресса, что говорит о достоверности построенной модели. Наиболее напряженные аминокислоты находятся сравнительно далеко от сайта связывания (рисунок 3). Особое внимание было уделено минимизации потенциальной энергии радикалов наиболее близких аминокислот (HOOC(R)CHNH2) и атомов водорода в сайте связывания с ингибитором (целекоксиб), а также в его ближайшей окрестности (ячейка размером  ).
).
Таким образом, в результате процедуры минимизации стресса были получены две трехмерные конформации ферментов hCOX-1 и hCOX-2 с потенциальной энергией, лежащей в окрестности расчетного минимума. Отметим, что сайты связывания целекоксиба в темплейтных структурах характеризуются высокой гомологией по отношению к их человеческим аналогам.
Построенные модели протестировали с использованием серии известных селективных ингибиторов СОХ-1 и -2 (Таблица 1). Для получения реперного значения энергетического скоринга осуществили докинг целекоксиба в сайт связывания hCOX-1 и -2 (Рисунок 5). В ходе эксперимента генерировалось 50 различных активных конформаций, соответствующих оптимальным позициям лиганда в указанном сайте связывания. Докинг осуществляли в динамическом режиме (активный сайт белка - подвижная система). Как видно из рисунка 5, в обоих случаях результаты молекулярного докинга хорошо коррелируют с данными рентгеноструктурного анализа (по RMSD).
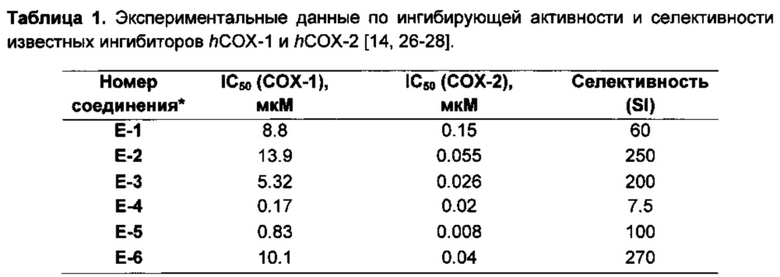
Далее с использованием построенной модели спрогнозировали активность для ряда реперных молекул (Таблица 2). Докинг осуществляли в динамическом режиме с интегрированным трехцентровым фармакофором (Рисунок 6), основанным на особенностях связывания целекоксиба, который необходим для ограничения степени свободы тестируемого лиганда в сайте связывания. Для каждой структуры генерировались 50 различных возможных конформаций с соответствующими значениями энергетического in silico скоринга. Средние арифметические значения скоринга по всем спрогнозированным конформациям были приняты за истинные. Они использовались для построения соответствующих QSAR моделей (Рисунки 7, 8). Как видно из Таблицы 2 и рисунков 7 и 8, модели продемонстрировали высокие значения корреляции (RMSD): R2=0.82 (СОХ-2) и 0.87 (СОХ-1).
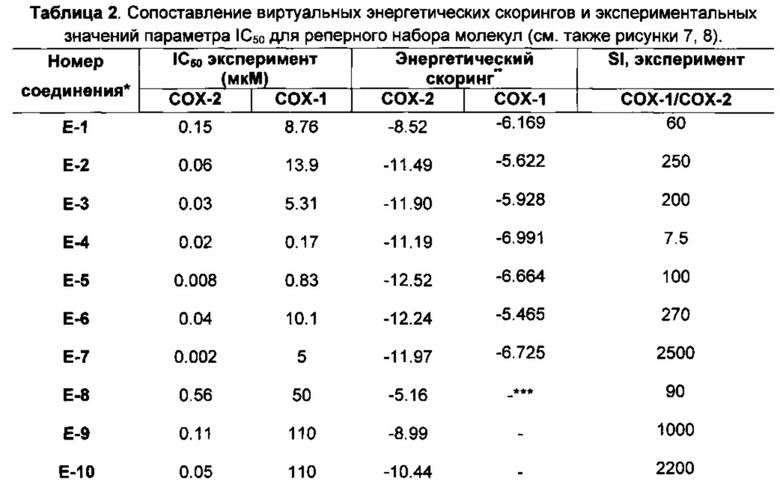

Разработанные модели были использованы для оценки активности новых молекул in silico. Так, для соединений по изобретению (Рисунок 9) был осуществлен молекулярный докинг по аналогии с описанными ранее процедурами. Полученные значения энергетического скоринга усредняли и с использованием двух QSAR моделей, указанных на рисунках 7 и 8, рассчитывали значения параметра IC50 для исследуемых соединений (Таблица 3).
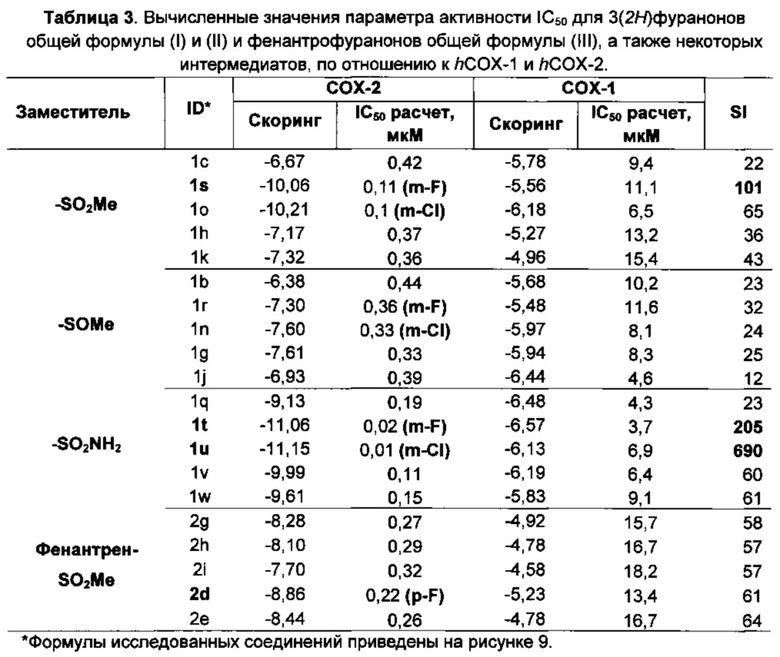
Основываясь на значениях ошибки линейной аппроксимации (1-R2), можно утверждать, что в рамках рассматриваемого класса ингибиторов, построенные QSAR модели обладают высокой прогностической способностью.
Как видно из таблицы 3, структуры 1q, 1t, 1u, 1v, 1w общей формулы (I), содержащие NH2SO2-группу, обладают особенно высокими значениями скоринга, индекса селективности (до 690 для 1u) и, как следствие, наиболее более низкими значениями IC50 (СОХ-2), по сравнению с аналогичными молекулами с сульфонилметильным фрагментом. Таким образом, применение сульфамидов является предпочтительным с точки зрения введения минимального количества действующего вещества в готовую лекарственную форму. Применение соединений (I) в качестве действующего вещества перспективно еще и по той причине, что, как известно из уровня техники, сульфамиды и соли сульфоновых кислот общей формулы (I) обладают существенно большей растворимостью в воде и физиологическом растворе, чем алкилсульфоны и алкилсульфоксиды.
Соединения общей формулы (I), предложенные в данном изобретении, являются региоизомерными к соединениям патента US 6492416 В1 [28], с которыми они наиболее близки структурно (примеры 150-165). Однако, соединения по изобретению обладают существенно более низкими (в 3-5 раз) концентрациями in vitro ингибирования СОХ-2 (1t, 1u: 0.004-0.007 мкг/мл) по сравнению с вышеуказанными примерами патента US 6492416 В1 [28] (№151 - 0.02 мкг/мл, №155 - 0.016 мкг/мл, №157 - 0.038 мкг/мл, №164 - 0.03 мкг/мл), что дает преимущество соединениям по данному изобретению, так как позволяет обойтись меньшими терапевтическими дозами в случае применения соединений настоящего патента в фармацевтической композиции.
Также из таблицы 3 следует, что структуры общей формулы (II) и (III) обладают средней ингибирующей активностью и селективностью, как по отношению к известному соединению целекоксибу, так и по отношению соединений общей формулы (I). Однако, из уровня техники [5-7] известно, что высокая активность сульфоамидов может приводить и к более выраженным побочным эффектам со стороны сердечно-сосудистой системы, а именно, к избыточной агрегации тромбоцитов в кровеносных сосудах и образованию тромбов с развитием ишемии и инсультов. Примером такого «избыточного» действия является рофекоксиб [5, 6], селективная ингибиторная активность которого крайне высока (SI>1700), однако, он также занимает одно из первых мест по выраженности побочных явлений [7] и запрещен к применению. Таким образом, вещества с более низкими значениями индекса селективности SI, характерные для алкилсульфонов (например, -SO2Me), должны быть также исследованы с точки зрения создания наиболее безопасного лекарственного препарата. На настоящий момент нет общепризнанной точки зрения относительно механизма развития побочных явлений при применении селективных ингибиторов циклооксигеназы-2, и возможностей их нивелирования путем модификации структуры ингибитора. Этот вопрос должен быть решен экспериментальным путем.
Сульфоксиды общей формулы (II) представляют интерес как пролекарственная форма ("prodrugs") алкилсульфонов, которая, несмотря на свою меньшую активность, в организме легко превращается в активную сульфоновую форму в результате нормального окислительного метаболизма [29]. Несмотря на меньшую активность, сульфоксид имеет существенно большую биодоступность, что приводит к снижению риска осложнений в связи с передозировкой, а также улучшению кинетических параметров соединения (пролонгированное действие). Таким образом, сульфоксиды также представляют интерес в данном изобретении и вопрос о возможности их применения при лечении воспалительных заболеваний должен решаться экспериментально.
Наиболее близкими соединениями к алкилсульфонам общей формулы (II) являются соединения патента RU 2563876 [30], в которых галогенирование гидрофобного арильного заместителя касается параположения. Однако, проведенное нами компьютерное моделирование, показывает, что паразамещенные алкилсульфоны проявляют в 2-3 раза меньшую ингибирующую активность, нежели метазамещенные. Так, например, для паразамещенных соединений 1с, 1h, 1k из патента RU 2563 876 [30] мы видим IC50 на уровне 0.36-0.42 мкМ, тогда как метазамещение (1s, 1о) существенно улучшает IC50 (в 3-4 раза) до 0.11 мкМ. Кроме того, индекс селективности у паразамещенных соединений также ниже (в 2-3 раза, на уровне 22-43), нежели в случае метазамещенных (65-101) (см. табл. 4). Таким образом, имеется несколько существенных причин предпочтения введения галогена в метаположение в соединениях по настоящему изобретению, по сравнению с его введением в параположение.
Методика выполнения in vitro эксперимента по определению параметров ингибирования IC50 для ферментов циклооксигеназы-1 (СОХ-1) и циклооксигеназы-2 (СОХ-2) для исследуемых соединений.
Активность ингибирования СОХ-1 и СОХ-2 для заявленных соединений будет измерена с использованием овечьей изоформы фермента СОХ-1 и человеческой рекомбинантной изоформы энзима СОХ-2, которые включены в набор (kit) "СОХ Inhibitor Screening Assay" поставляемый компанией «Кайман» (Cayman Chemical Co., Ann Arbor, Ml), по стандартной, для данного экспериментального набора, методике. В данном эксперименте напрямую флюорометрически измеряется концентрация простагландина PGF2a продуцируемого при восстановлении дихлоридом олова (SnCl2) простогландина PGH2, образование которого контролируется СОХ-ферментами. Продуцирование простагландинов в системе оценивается посредством их реакции с широко специфичными антителами, которые взаимодействуют со всеми основными простагландинами [31]. Эксперимент по определению ингибиторной активности проводится при различных концентрациях арахидоновой кислоты (от 0.1 до 100 мкМ). Определение целевого % IC50, по отношению к плацебо, производится при существенно меньшей концентрации изучаемого ингибитора, чем его концентрация в насыщенном растворе. Для лучшей визуализации и повышения точности результатов эксперимента, ингибиторная активность по отношению к ферменту СОХ-1 измеряется при концентрации арахидоновой кислоты около 1 мкМ (микромоль на литр), тогда как для случая фермента СОХ-2 используется концентрация 0.1 мкМ. Используемая концентрация исследуемых ингибиторов в растворе составляет 200 мкМ. В качестве реперных величин используются параметры IC50 для напроксена и индометацина (используемые в той же концентрации 200 мкМ).
Методика выполнения in vivo эксперимента по определению степени ингибирования карраген-индуцированного отека лап мышей (СРЕ %) при инъекции исследуемых соединений.
Отек индуцируется путем подкожного введения в правую заднюю лапку мыши 0.05 мл 2% раствора каррагена в воде. Используются мыши обоего пола за исключением беременных самок. Каждое соединение испытывается в группе из 6-10 животных, которые перед экспериментом содержатся при стандартных условиях и получают стандартный коммерческий корм для лабораторных животных. После начала эксперимента животные корм не получают. Исследуемые соединения, в концентрации 0.01 ммоль на 1 кг веса животного), предварительно суспензируются в воде с несколькими каплями поверхностно-активного вещества ТВИН-80 и, после фильтрования, вводятся в внутрибрюшинно, одновременно с инъекцией каррагена. Через 3.5 часа после инъекций мыши усыпляются. Различия в весе отечной лапки и здоровой определяются для всех животных, усредняются (стандартное отклонение должно быть менее 10%) и сравниваются с тем же параметром в контрольной группе (внутрибрюшинная инъекция физиологического раствора вместо раствора ингибитора). Так определяется СРЕ % - процент ингибирования отека лап мышей.
Общая методика выполнения in vitro эксперимента по определению степени ингибирования скорости роста раковых клеток исследуемыми соединениями.
Активность в отношении клеточных линий, полученных из твердых опухолей (НТ-29, NCl-Н460, MCF-7, и др.) оценивается в экспоненциально растущих культурах, посеянных в концентрации 5*104 клеток/мл, которым дают возможность прилипать к донышку планшета в течение 18 ч перед добавлением препаратов. Жизнеспособность клеток определяется через 72 ч при 37°C методом МТТ [32]. Линию клеток рака толстого кишечника (альтернативно: рака желудка, рака легкого, рака молочной железы, рака поджелудочной железы), стойкую к действию доксорубицина - LoVo/Doxo, культивируют в среде Хама F12; при этом доксорубицин (100 нг/мл) добавляют к стойким сублиниям рака при каждом проходе. Скорость роста клеток при каждой концентрации лекарственного средства выражают в процентах от скорости роста контрольного посева; концентрацию, обеспечивающую 50% ингибирование роста (IC50) определяют с помощью анализа линейной регрессии.
Список литературы:
[1] P.N. Praveen Rao, Е.Е. Knaus, J. Pharm. Pharmaceut. Sci. 2008, 11, 81.
[2] Е.Л. Насонов, Consilium Medicum, 1999, том 1, №5.
[3] Ю. Муравьев, В. Лебедева, Российский гастроэнтерологический журнал, 2000, №4.
[4] J.J. Talley, Prog. Med. Chem. Res. 1999, 36, 201.
[5] Bombardier, Laine, Reicin et al., N. Engl. J. Med. 2000, 343, 1520.
[6] R.S. Bresalier, R.S. Sandler, H. Quan et al., N. Engl. J. Med. 2005, 352, 1092.
[7] Sven Trelle et al., BMJ 2011, 342, 7086.
[8] Svetla Gadzhanova, et al., Drugs Aging 2013, 30, 23.
[9] Meheroz, Rabadi et al., Ann Indian Acad Neurol. 2013, 16 (1), 47.
[10] H. Sheng et al., J. Clin. Invest. 1997, 99, 2254.
[11] N.  , Curr Opin Investig Drugs, 2010, W(1), 31-42.
, Curr Opin Investig Drugs, 2010, W(1), 31-42.
[12] W.F. Stewart, Neurology, 1997, 48, 626.
[13] P. Prasit et al., Bioorg. Med. Chem. Lett., 1999, 9, 1773-1778.
[14] A.K. Chakraborti et al., Current Medicinal Chemistry, 2010, 17, 1563-1593.
[15] B. Rigas, I. Goldman, L. Levine, "Altered eicosanoid levels in human colon cancer", J. Lab. Clin. Med, 122, 1993, p. 518-523.
[16] С. Koehne, R. Dubois "COX-2 inhibition and colorectal cancer", Semin. Oncol., 2004, p. 12-21.
[17] K. Leahy, A. Koki, J. Masferrer, "Role of cyclooxygenases in angiogenesis", Curr. Med. Chem., 7, 2000, p. 1163-1170.
[18] S. Zelenay et al., "Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity", Cell, 162, 6, 2015, p. 1257-1270.
[19] M. Dore, Vet. Pathol., 48, 2011, p. 254.
[20] Brunelle, Vet. Pathol., 43, 2006, p. 656-666.
[21] S.A. Malashikhin et. al., Helv. Chim. Acta, 91, 2008, p. 1662.
[22] G. Bennett et al., Journal of Medicinal Chemistry, 19(5), 1976, p. 709.
[23] S.M. Berge et al., "Pharmaceutical Salts", J. Pharm. Sci., 66, 1977, p. 1-19.
[24] E.W. Martin "Remington's pharmaceutical sciences", 15th edition, Mack Publishing Co.,
Easton, Pa., 1975.
[25] Philippe Chavatte et al., J. Med. Chem., 2001, 44, 3223-3230.
[26] Song Seok Shin and coworkers, Bioorganic & Medicinal Chemistry Letters 2001, 11, 165.
[27] Song Seok Shin and coworkers, J. Med. Chem, 2004, 47, 792.
[28] Патент S. Shin et al., US 6492416 B1 (2002).
[29] J.H. Moh et al., Bioorg. Med. Chem. Lett., 14, 2004, p. 1757.
[30] Ю. Медведев, Д. Семенок, В. Николаев, Л. Родина, RU 2563876 (приоритет 11.11.2014).
[31] G. Ziakas et al., Bioorg. Med. Chem., 14 (16), 2006, p. 5616-5624.
[32] T. Mosmann, J. Immunol. Methods, 1983, 65, p. 55-63.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,2-ДИАЛКИЛ-4,5-ДИАРИЛФУРАН-3(2Н)-ОНОВ | 2014 |
|
RU2563876C1 |
| Четвертичные аммониевые соединения на основе 3-гидроксипиридина, обладающие антибактериальной активностью | 2021 |
|
RU2778507C1 |
| Четвертичные аммониевые соли на основе производных витамина В6 | 2015 |
|
RU2607522C1 |
| ФОСФОНИЕВЫЕ СОЛИ НА ОСНОВЕ ПРОИЗВОДНЫХ ПИРИДОКСИНА | 2011 |
|
RU2466728C1 |
| Производные пиридоксина и ацетона с противоопухолевой активностью | 2017 |
|
RU2639879C1 |
| АНТИБАКТЕРИАЛЬНЫЕ СРЕДСТВА НА ОСНОВЕ ЧЕТВЕРТИЧНЫХ АММОНИЕВЫХ СОЛЕЙ | 2014 |
|
RU2561281C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| Оксазолидиноны на основе производных пиридоксина, обладающие антибактериальной активностью | 2024 |
|
RU2836570C1 |
| N,N-бис(2-(диалкиламино)этил)карбоксамиды и их дигидрохлориды, проявляющие антиаритмическую активность, и фармацевтические композиции на их основе | 2017 |
|
RU2645080C1 |
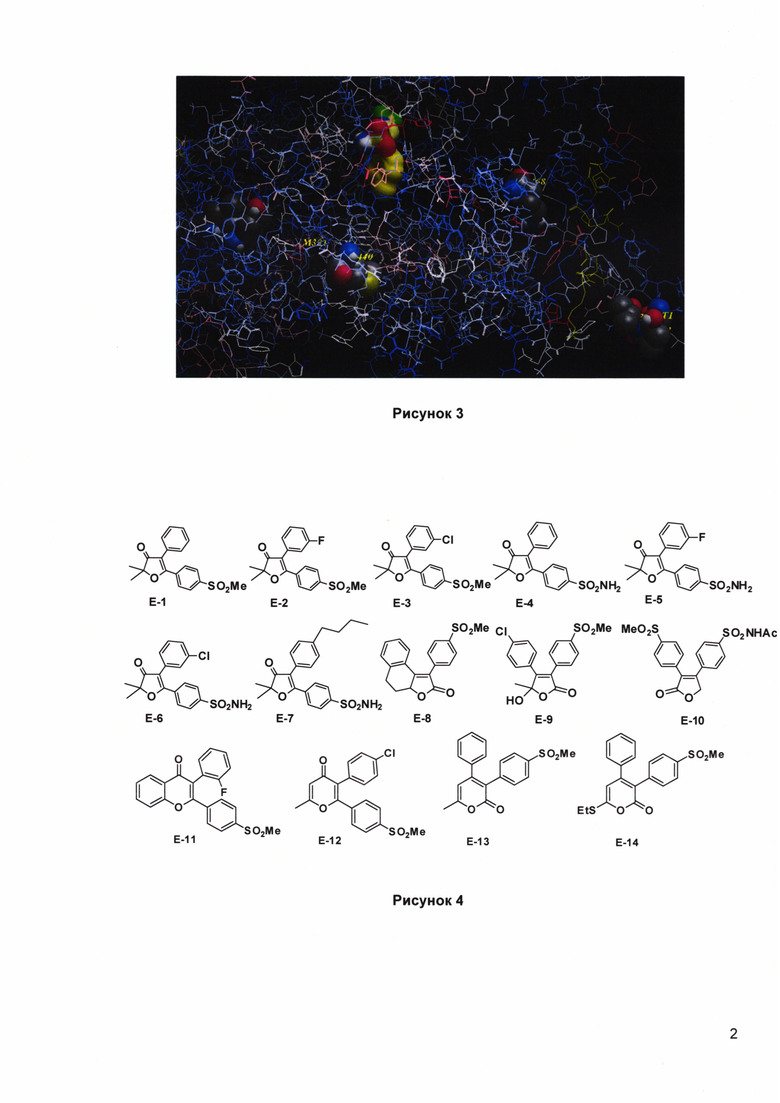
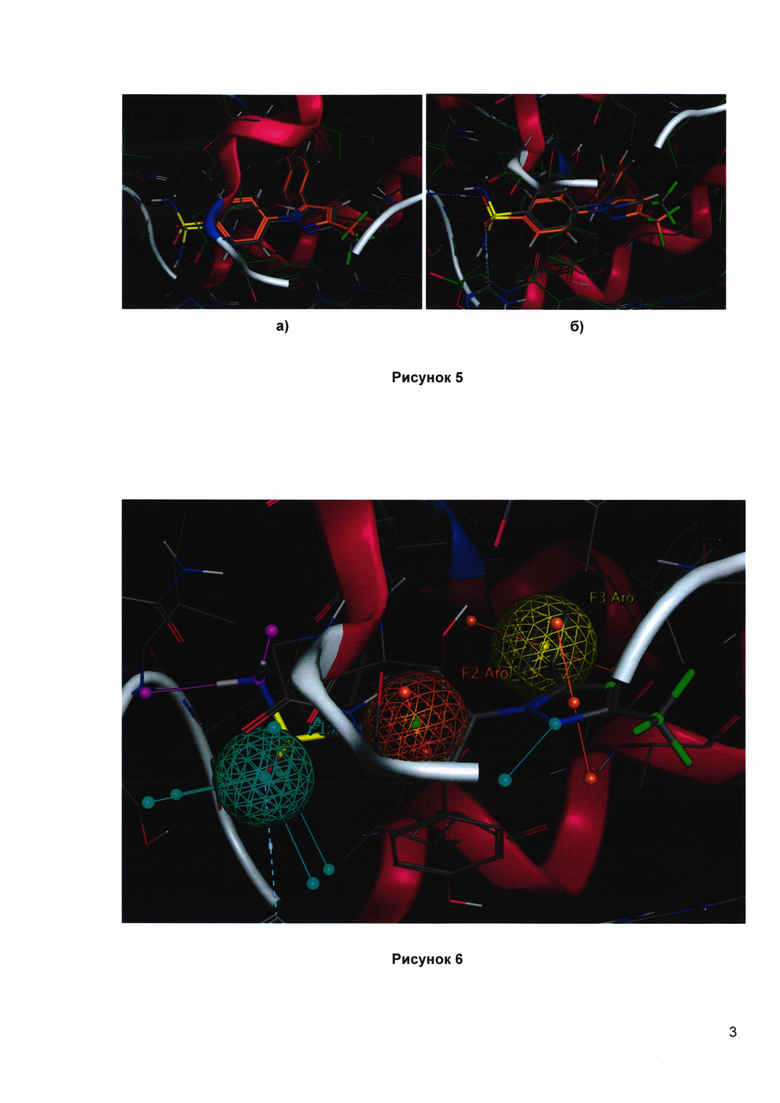
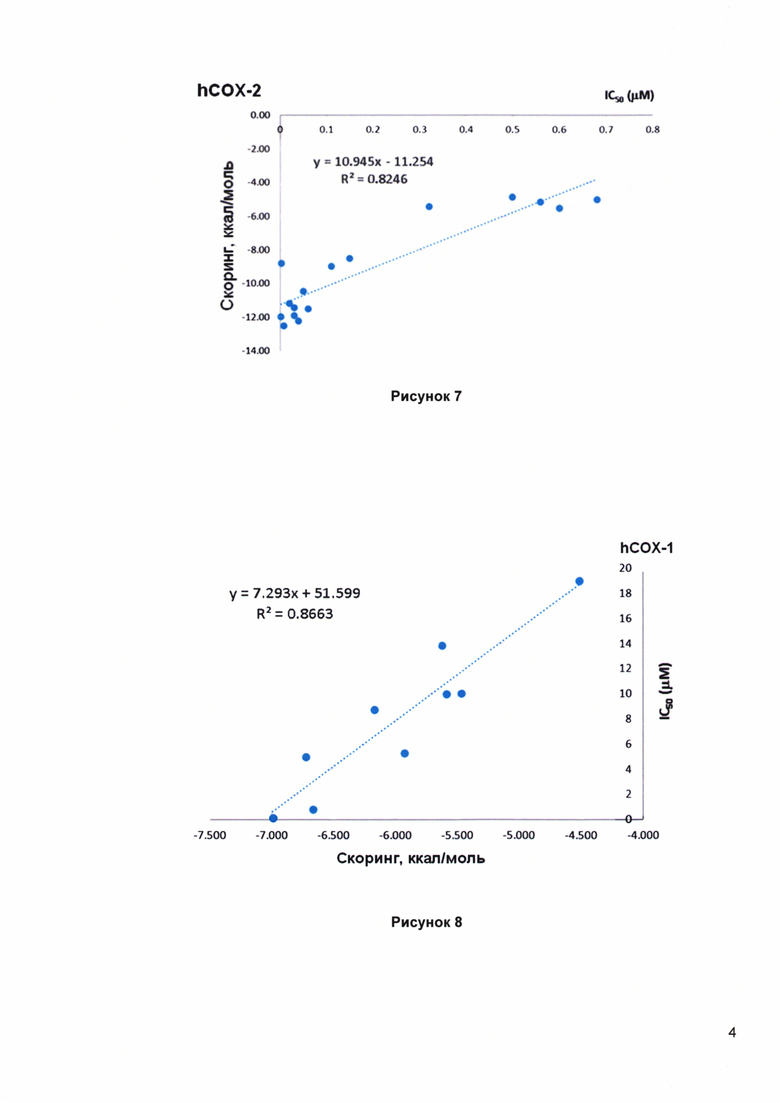
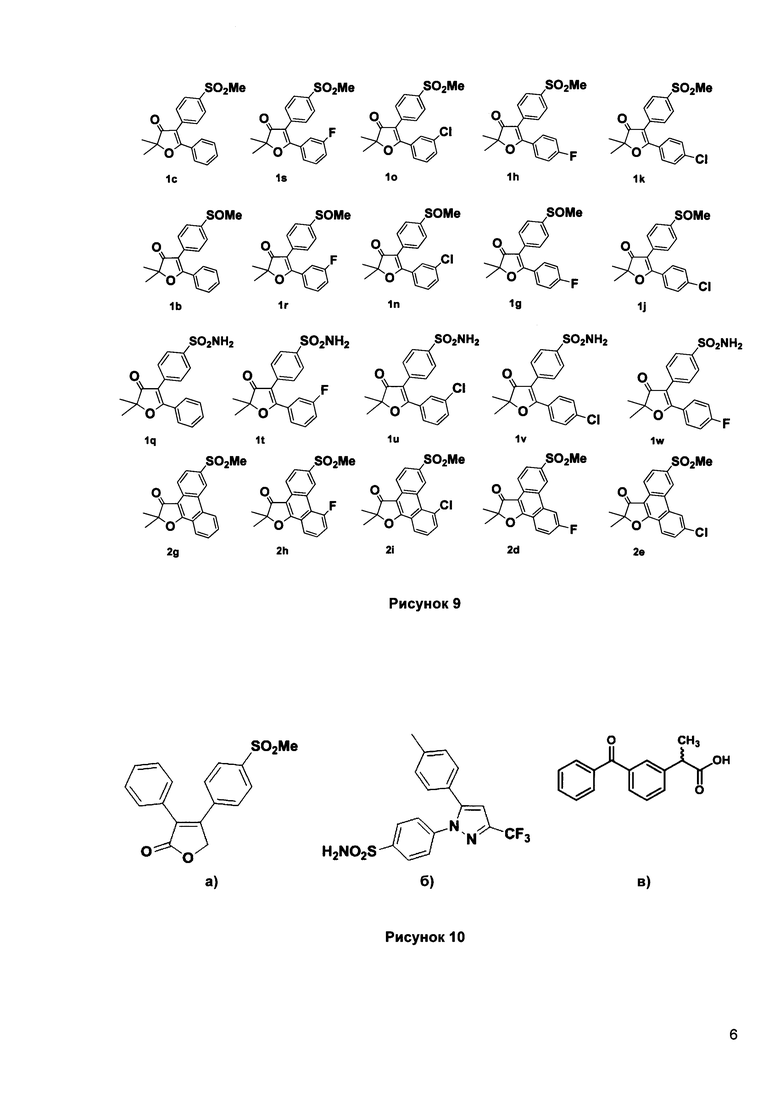
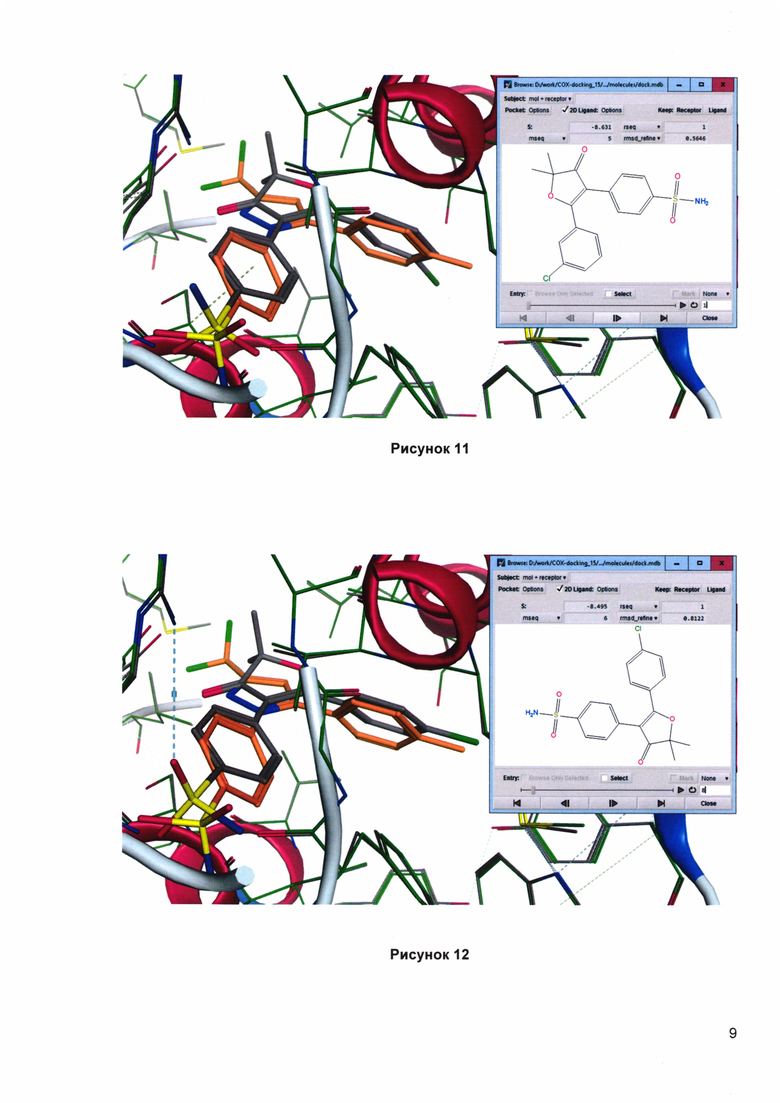
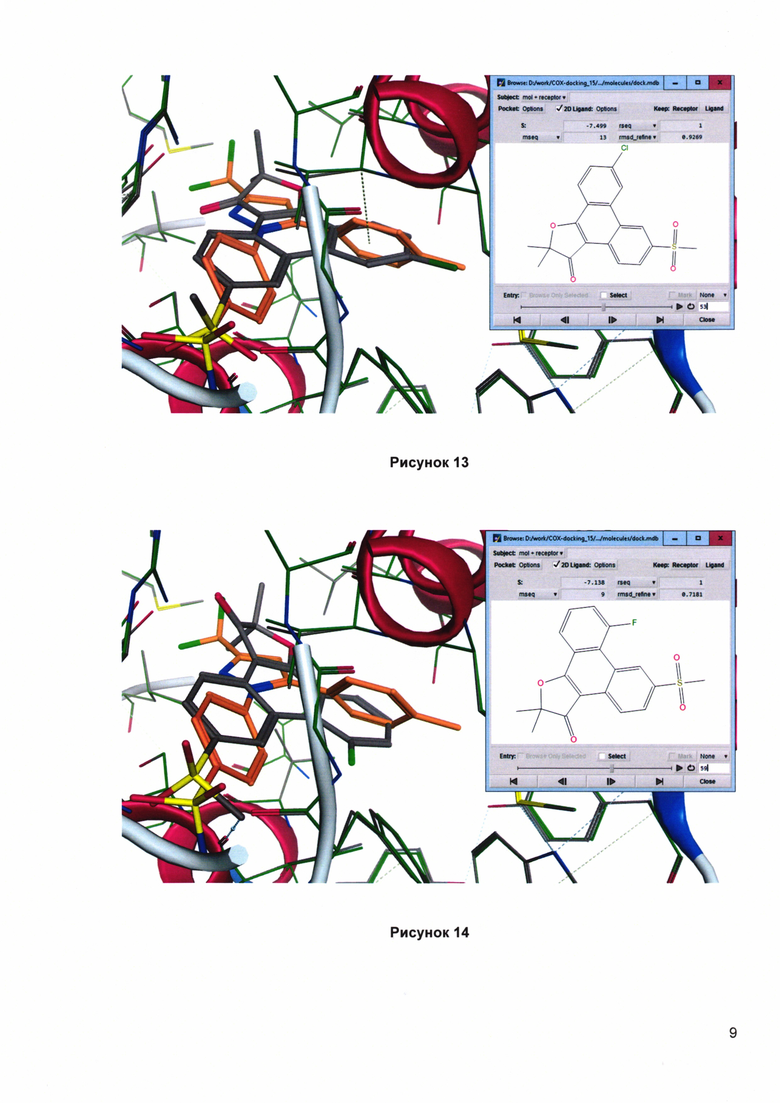
Изобретение относится к селективным ингибиторам фермента циклооксигеназы-2 (СОХ-2) класса замещенных 3(2Н)-фуранонов общей формулы (I) и (II) и замещенных фенантро[9,10-b]фуран-3(2Н)-онов общей формулы (III), а также фармацевтической композиции на их основе. Данные соединения являются эффективными селективными ингибиторами циклооксигеназы-2 (IC50 до 0.004 мкг/мл, SI до 690) и могут быть использованы в лечении заболеваний, ассоциированных с развитием воспаления и гиперэкспрессией фермента СОХ-2, таких как хронические формы артрита, мигрени, травмы, заболевания кожи, а также для ингибирования ангиогенеза и замедления роста клеток некоторых видов рака, таких как нейробластома, рак молочной железы, желудка, легких и толстого кишечника. 4 н. и 11 з.п. ф-лы, 4 табл., 9 пр., 14 ил.
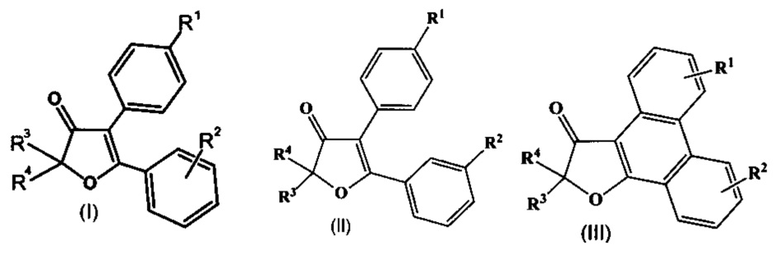
1. Соединение общей формулы (I)
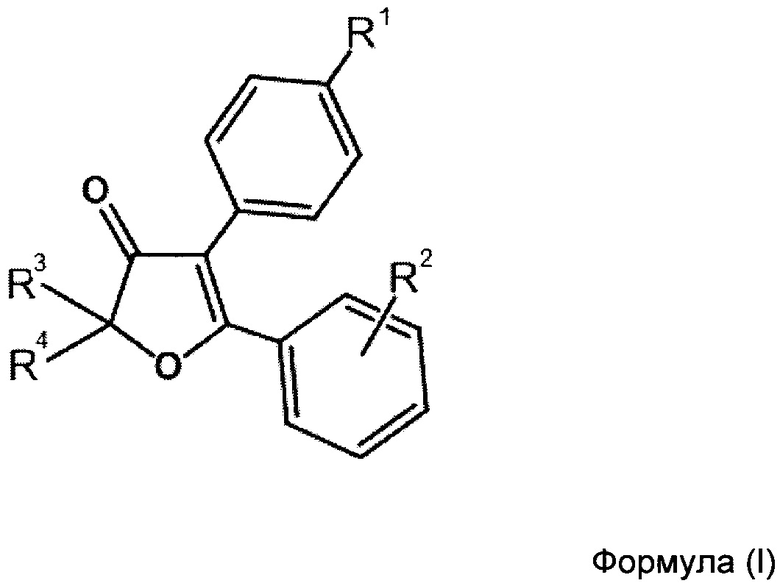
или его фармацевтически приемлемая соль, где
R1 представляет собой замещенную сульфонамидную группу -SO2NHAlk, где Alk выбирают из метила, этила, изопропила или н-пропила, или незамещенную сульфонамидную (-SO2NH2) или сульфо (-SO2OH) группу;
R2 выбирается независимо и представляет собой галоген или водород, причем указанные заместители находятся в мета- или параположении по отношению к точке присоединения фенильной группы;
R3 и R4 выбираются независимо и представляют собой C1-5-алкил.
2. Соединение по п. 1, выбранное из группы:
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-фенилдигидрофуран-3(2Н)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(4'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(3'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(4'-хлорфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(аминосульфонил)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2H)-он;
4-(5,5-диметил-4-оксо-2-фенил-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(4'-фторфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(4'-хлорфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(3'-фторфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота;
4-(2-(3'-хлорфенил)-5,5-диметил-4-оксо-4,5-дигидрофуран-3-ил)бензолсульфокислота.
3. Соединение общей формулы (II)
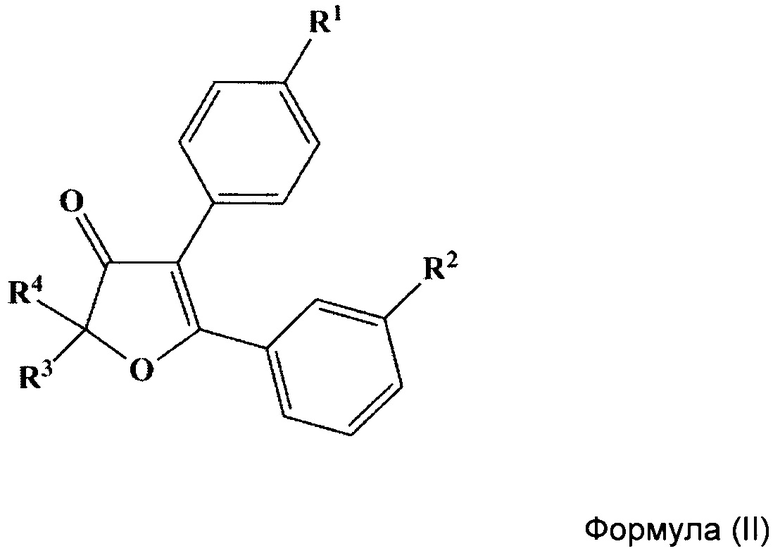
или его фармацевтически приемлемая соль, где
R1 выбирается независимо и представляет собой C1-3-алкилсульфонил или С1-3-алкилсульфинил группу;
R2 выбирается независимо и представляет собой галоген;
R3 и R4 выбираются независимо и представляют собой C1-5-алкил.
4. Соединение по п. 2, в котором
R2 выбирается независимо и представляет собой фтор или хлор.
5. Соединение по п. 4, выбранное из группы:
2,2-Диметил-4-[4'-(метилсульфинил)фенил]-5-(3'-фторфенил)дигидрофуран-3(2Н)-он;
2,2-Диметил-4-[4'-(метилсульфонил)фенил]-5-(3'-фторфенил)дигидрофуран-3(2H)-он;
2,2-Диметил-4-[4'-(метилсульфинил)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2Н)-он;
2,2-Диметил-4-[4'-(метилсульфонил)фенил]-5-(3'-хлорфенил)дигидрофуран-3(2Н)-он.
6. Соединение общей формулы (III)
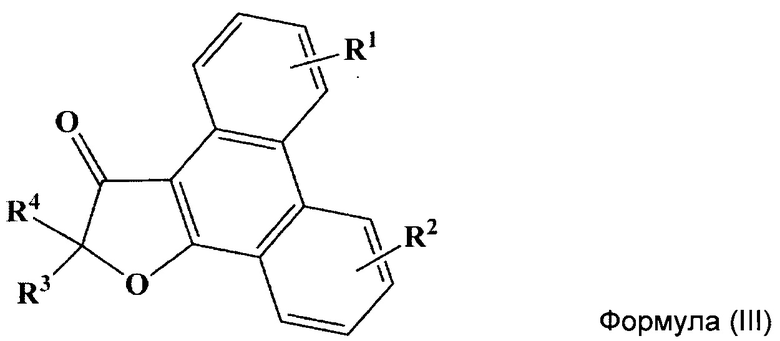
или его фармацевтически приемлемая соль, где
R1 выбирается независимо и представляет собой C1-3-алкилсульфонил, С1-3-алкилсульфинил, незамещенный сульфонамид (-SO2NH2), C1-3-алкилтио группу, водород или галоген, причем указанные заместители находятся в третьем или четвертом положении фенантренового фрагмента;
R2 выбирается независимо и представляет собой C1-3-алкилсульфонил, С1-3-алкилсульфинил, незамещенный сульфонамид (-SO2NH2), C1-3-алкилтио группу, водород или галоген, причем указанные заместители находятся в пятом или шестом положении фенантренового фрагмента;
причем если R1 представляет собой водород, то R2 не может быть водородом и наоборот;
R3 и R4 выбираются независимо и представляют собой C1-5-алкил.
7. Соединение по п. 6, в котором
R1 и R2 выбираются независимо и представляют собой фтор или хлор.
8. Соединение по п. 6, выбранное из группы:
2',2'-диметил-3-(метилтио)фенантро[9,10-b]фуран-3'(2'H)-он;
2',2'-диметил-3-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
2',2'-диметил-3-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
2',2'-диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
2',2'-диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'H)-он;
2',2'-диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-фтор-2',2'-диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2H)-он;
3-фтор-2',2'-диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-фтор-2',2'-диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
3-хлор-2',2'-Диметил-6-(метилтио)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-хлор-2',2'-Диметил-6-(метилсульфинил)фенантро[9,10-b]фуран-3'(2'H)-он;
3-хлор-2',2'-Диметил-6-(метилсульфонил)фенантро[9,10-b]фуран-3'(2'Н)-он;
3-хлор-2',2'-Диметил-6-(аминосульфонил)фенантро[9,10-b]фуран-3'(2'H)-он;
или их фармацевтически приемлемые соли.
9. Фармацевтическая композиция для лечения заболеваний воспалительного генеза или заболеваний, ассоциированных с избыточной секрецией ферментов циклооксигеназы-1 и циклооксигеназы-2, обладающая свойством ингибировать предпочтительно СОХ-2 по сравнению с СОХ-1 и содержащая эффективное количество по меньшей мере одного соединения по любому из пп. 1-8 и по меньшей мере один фармацевтически приемлемый носитель и/или разбавитель.
10. Фармацевтическая композиция по п. 9, характеризующаяся тем, что содержит соединение по любому из пп. 1-8 и дополнительно содержит его региоизомер.
11. Фармацевтическая композиция по п. 9, характеризующаяся тем, что обладает противовоспалительным, анальгетическим и/или жаропонижающим действием.
12. Фармацевтическая композиция по п. 9, характеризующаяся тем, что заболевание представляет собой артрит, суставной синдром при ревматизме и обострении подагры, кожное воспалительное заболевание, воспаление при сосудистых заболеваниях, легочное воспаление, тиреоидит, воспаление, связанное с общим переохлаждением, гриппом или другой острой респираторной вирусной инфекцией, воспалительные процессы связок, сухожилий, ишиас, люмбаго.
13. Фармацевтическая композиция по п. 12, характеризующаяся тем, что артрит представляет собой ревматоидный артрит, остеоартрит, подагрический артрит, ювенильный артрит, псориатический артрит, спондилолистез.
14. Фармацевтическая композиция по п. 12, характеризующаяся тем, что кожное воспалительное заболевание представляет собой экзему, дерматит, псориаз.
15. Фармацевтическая композиция по п. 9, характеризующаяся тем, что обладает противоопухолевой активностью.
| WO 0061571 A1 19.10.2000 | |||
| СПОСОБ ПОЛУЧЕНИЯ 2,2-ДИАЛКИЛ-4,5-ДИАРИЛФУРАН-3(2Н)-ОНОВ | 2014 |
|
RU2563876C1 |
| Hongmiao Sheng et al, "Inhibition of Human Colon Cancer Cell Growth by Selective Inhibition of Cyclooxygenase-2", J.of Clinical Invetigation, 1997,v.99, p.2254-2259. | |||

