Изобретение относится к области фармации и касается способа получения средства из растительного сырья, обладающего иммуностимулирующим действием.
Известен способ получения фракции флавоноидов из корней софоры лисохвостной, обладающей иммуностимулирующим, гепатозащитным, противовоспалительным, детоксицирующим и антиоксидантым действием [1], заключающийся в последовательной экстракции сырья водой и этанолом, с последующим концентрированием, нанесением на колонку с активированным углем водного остатка, элюированием колонки смесью хлороформ-изопропанол (1:1), концентрированием элюата, растворением последнего в растворе гидроксида натрия и осаждением флавоноидов раствором соляной кислоты. Недостатками известного метода является длительность технологического процесса (один цикл - около 4 дней), использование в ходе выделения токсичных реагентов (хлороформ, изопропанол), применение процедуры растворения суммы флавоноидов в щелочном растворе, что приводит к частичной деструкции фенольных соединений и снижает выход и качество конечного продукта. Более того, для производства препарата используется сырье софоры лисохвостной, имеющей ограниченные сырьевые запасы на территории России. В качестве альтернативного источника получения подобного средства предлагаются корни софоры желтоватой, которая, несмотря на узкий ареал распространения на территории Сибири и Дальнего Востока, прекрасно воспроизводится в условиях культуры и разводится в настоящее время в Забайкалье. Следует также отметить, что для софоры лисохвостной характерна более низкая концентрация флавоноидов, чем для софоры желтоватой, что, в свою очередь, также оказывает влияние на активность конечных препаратов.
Задачей изобретений является упрощение способа получения суммарного флавоноидсодержащего препарата, повышение иммуностимулирующего действия средства за счет более высокого содержания флавоноидов.
Технический результат изобретения (получение субстанции флавоноидов из корней софоры желтоватой - Sophora flavescens Soland., семейство Бобовые) достигается за счет использования в ходе технологического процесса ультразвуковой экстракции растительного сырья и дополнительной процедуры экстракции полупродукта.
Для достижения указанного технического результата измельченный растительный материал (корни софоры желтоватой) заливают водой в соотношении сырье:экстрагент 1:(10-12) и подвергают смесь ультразвуковой обработке частотой 50 кГц в течение 20-30 мин. Далее смесь нагревают до температуры 30-40°C и оставляют при постоянном перемешивании в течение 1-1.5 ч. Извлечение фильтруют, маточный раствор отбрасывают. Шрот заливают водой в соотношении сырье:экстрагент 1:(18-20) и экстрагируют при температуре 50-60°C в течение 1.5-2 ч. Извлечение фильтруют, маточный раствор отбрасывают. Далее шрот заливают 90-95% этанолом в соотношении сырье:экстрагент 1:(15-17), подвергают смесь ультразвуковой обработке частотой 50 кГц в течение 20-30 мин, после чего смесь экстрагируют при температуре 80-85°C в течение 1-1.5 ч. Извлечение фильтруют в сборник. Экстракцию повторяют в тех же условиях еще один раз. Спиртовые извлечения объединяют и концентрируют до 1/50 от первоначального объема. Сгущенный экстракт высушивают в вакуум-сушильном шкафу и измельчают на мельнице пропеллерного типа. Выход полупродукта составляет 9-11% от массы растительного сырья. Измельченный полупродукт смешивают с 10% этанолом в соотношение сырье:экстрагент 1:(6-8) и оставляют при 20-25°C и постоянном перемешивании в течение 0.5-1 ч. Нерастворившийся остаток отфильтровывают и высушивают в вакуум-сушильном шкафу. Выход готового продукта (субстанции флавоноидов) составляет 6-7% от массы растительного сырья.
Выявленные отличительные признаки позволяют сделать вывод о соответствии предлагаемого технологического решения критерию "новизна".
Предложенный способ позволяет получить субстанцию флавоноидов (далее - субстанция) в виде рассыпчатого негигроскопичного порошка желтого цвета, без вкуса и запаха. Потеря массы при высушивании - 3-5%.
Способ иллюстрируется нижеследующими примерами.
Пример 1. 1.1 кг корней софоры желтоватой измельчают на мельнице до размера частиц диаметром 2-3 мм. 1 кг измельченного сырья загружают в экстракционный аппарат c мешалкой и внешним паровым обогревателем и заливают 11 л воды. Полученную смесь перемешивают, вносят стержневой ультразвуковой процессор и обрабатывают смесь ультразвуком мощностью 50 кГц в импульсном режиме в течение 25 мин. После этого ультразвуковой процессор удаляют, смесь нагревают до температуры 35°C и проводят экстракцию при постоянном перемешивании в течение 1.5 ч. Извлечение фильтруют, маточный раствор отбрасывают. Шрот заливают 19 л воды и экстрагируют при температуре 55°C в течение 1.5 ч. Извлечение фильтруют, маточный раствор отбрасывают. Далее шрот заливают 16 л 90% этанола, подвергают смесь ультразвуковой обработке частотой 50 кГц в течение 25 мин, после чего смесь экстрагируют при температуре 80-85°C в течение 1.5 ч. Извлечение фильтруют в сборник. Проводят еще одну экстракцию в тех же условиях, подавая в экстрактор 90% этанол в количестве, равном объему слитого (1 слив - 13.9 л, 2 слив - 13.8 л). Объединенные спиртовые извлечения упаривают примерно до 1/50 от первоначального объема, сушат в вакуум-сушильном шкафу при температуре 55°C и давлении 0.1 атм в течение 5 часов и измельчают на мельнице пропеллерного типа. Получают 0.114 кг полупродукта, что составляет 10.36% от массы растительного сырья. Измельченный полупродукт смешивают с 7 л 10% этанола и оставляют при 20-25°C и постоянном перемешивании в течение 0.75 ч. Нерастворившийся остаток отфильтровывают и высушивают в вакуум-сушильном шкафу. Получают 0.072 кг готового продукта, что составляет 6.55% от массы исходного сырья. Субстанция представляет собой рассыпчатый негигроскопичный порошок желтого цвета, без вкуса и запаха. Потеря массы при высушивании составляет 3.22%.
Пример 2. 1.1 кг корней софоры желтоватой измельчают на мельнице до размера частиц диаметром 2-3 мм. 1 кг измельченного сырья загружают в экстракционный аппарат c мешалкой и внешним паровым обогревателем и заливают 12 л воды. Полученную смесь перемешивают, вносят стержневой ультразвуковой процессор и обрабатывают смесь ультразвуком мощностью 50 кГц в импульсном режиме в течение 30 мин. После этого ультразвуковой процессор удаляют, смесь нагревают до температуры 40°C и проводят экстракцию при постоянном перемешивании в течение 2 ч. Извлечение фильтруют, маточный раствор отбрасывают. Шрот заливают 18 л воды и экстрагируют при температуре 60°C в течение 2 ч. Извлечение фильтруют, маточный раствор отбрасывают. Далее шрот заливают 17 л 95% этанола, подвергают смесь ультразвуковой обработке частотой 50 кГц в течение 30 мин, после чего смесь экстрагируют при температуре 80-85°C в течение 1.2 ч. Извлечение фильтруют в сборник. Проводят еще одну экстракцию в тех же условиях, подавая в экстрактор 95% этанол в количестве, равном объему слитого (1 слив - 15.0 л, 2 слив - 14.8 л). Объединенные спиртовые извлечения упаривают примерно до 1/50 от первоначального объема, сушат в вакуум-сушильном шкафу при температуре 55°C и давлении 0.1 атм в течение 5 часов и измельчают на мельнице пропеллерного типа. Получают 0.120 кг полупродукта, что составляет 10.91% от массы растительного сырья. Измельченный полупродукт смешивают с 8 л 10% этанола и оставляют при 20-25°C и постоянном перемешивании в течение 1 ч. Нерастворившийся остаток отфильтровывают и высушивают в вакуум-сушильном шкафу. Получают 0.086 кг готового продукта, что составляет 7.82% от массы исходного сырья. Субстанция представляет собой рассыпчатый негигроскопичный порошок коричневато-желтого цвета, без вкуса и запаха. Потеря массы при высушивании составляет 3.59%.
Пример 3. 1.1 кг корней софоры желтоватой измельчают на мельнице до размера частиц диаметром 2-3 мм. 1 кг измельченного сырья загружают в экстракционный аппарат с мешалкой и внешним паровым обогревателем и заливают 10 л воды. Полученную смесь перемешивают, вносят стержневой ультразвуковой процессор и обрабатывают смесь ультразвуком мощностью 50 кГц в импульсном режиме в течение 20 мин. После этого ультразвуковой процессор удаляют, смесь нагревают до температуры 30°C и проводят экстракцию при постоянном перемешивании в течение 1.75 ч. Извлечение фильтруют, маточный раствор отбрасывают. Шрот заливают 20 л воды и экстрагируют при температуре 50°C в течение 1.5 ч. Извлечение фильтруют, маточный раствор отбрасывают. Далее шрот заливают 15 л 95% этанола, подвергают смесь ультразвуковой обработке частотой 50 кГц в течение 20 мин, после чего смесь экстрагируют при температуре 80-85°C в течение 1 ч. Извлечение фильтруют в сборник. Проводят еще одну экстракцию в тех же условиях, подавая в экстрактор 95% этанол в количестве, равном объему слитого (1 слив - 13.0 л, 2 слив - 12.9 л). Объединенные спиртовые извлечения упаривают примерно до 1/50 от первоначального объема, сушат в вакуум-сушильном шкафу при температуре 55°C и давлении 0.1 атм в течение 5 часов и измельчают на мельнице пропеллерного типа. Получают 0.102 кг полупродукта, что составляет 9.27% от массы растительного сырья. Измельченный полупродукт смешивают с 6 л 10% этанола и оставляют при 20-25°C и постоянном перемешивании в течение 0.5 ч. Нерастворившийся остаток отфильтровывают и высушивают в вакуум-сушильном шкафу. Получают 0.068 кг готового продукта, что составляет 6.18% от массы исходного сырья. Субстанция представляет собой рассыпчатый негигроскопичный порошок желтого цвета, без вкуса и запаха. Потеря массы при высушивании составляет 4.25%.
Химический состав субстанции
Для проведения качественного и количественного анализа средства было проведено хроматографическое разделение флавоноидов с использованием колоночной хроматографии на прямом и обращено-фазовом силикагеле, полиамиде, сефадексе LH-20, а также применен комплекс спектральных (спектрофотометрия) и хроматографических методов анализа (ВЭЖХ).
В результате хроматографического разделения было выделено 19 соединений, в т.ч. халконы - кураридин, кураридинол, ксантогумол, кушенол D; флаваноны - кушенол A, кушенол B, норкураринон, кураринол, неокураринол, кураринон, изокураринон, 2′-метоксикураринон, софорафлаванон B, софорафлаванон G, изоксантогумол; флаванонолы - кушенол I, кушенол N; изофлавон - формононетин; птерокарпан - маакиаин.
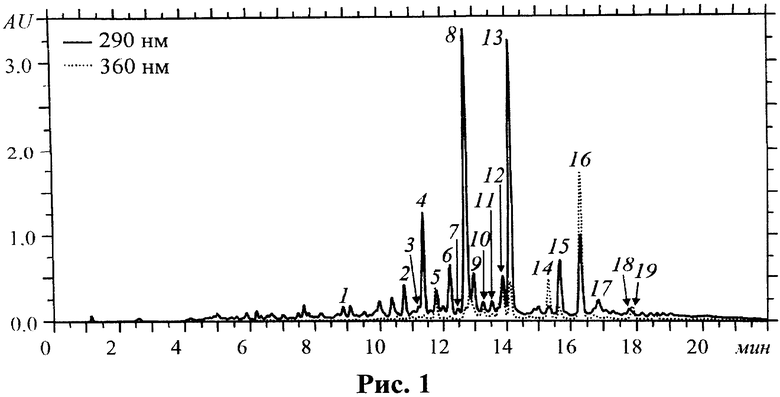
ВЭЖХ. 10 мг средства переносили в пробирку Эппендорфа, приливали 1 мл метанола и встряхивали на вортексе до полного растворения (1-2 мин). Полученный раствор фильтровали через кварцевый фильтр Millipore (⌀ 0.45 мкм) и использовали для анализа методом микроколоночной ВЭЖХ-УФ. Условия ВЭЖХ-УФ: жидкостный хроматограф Милихром A-02 (Эконова); колонка ProntoSIL-120-5-C18 AQ (2×75 мм, ⌀ 5 мкм; Metrohm AG); подвижная фаза: (4.1 М LiClO4 в 0.1 М HClO4):H2O 5:95 (A), MeCN (B); градиентный режим B в A: 5-100% (0-20 мин), 100% (20-22 мин); ν 150 мкл/мин; Тколонки 35°C; Vпробы 2 мкл; детектор при 290 и 360 нм. В качестве внешних стандартов использовали растворы индивидуальных соединений с известной концентрацией. Чистота соединений по данным ВЭЖХ составляла не менее 95%. Суммарная погрешность измерений не превышала 5%. Хроматограмма метанольного раствора субстанции приведена на рис.1.
Согласно данным ВЭЖХ, выделенные соединения находятся в субстанции в детектируемых количествах, что позволяет установить их количественный состав. Установлено, что доминирующими соединениями субстанции являются флаваноны (27.35%) и халконы (12.30%) (табл.1). Суммарное содержание идентифицированных компонентов субстанции составило около 42%.
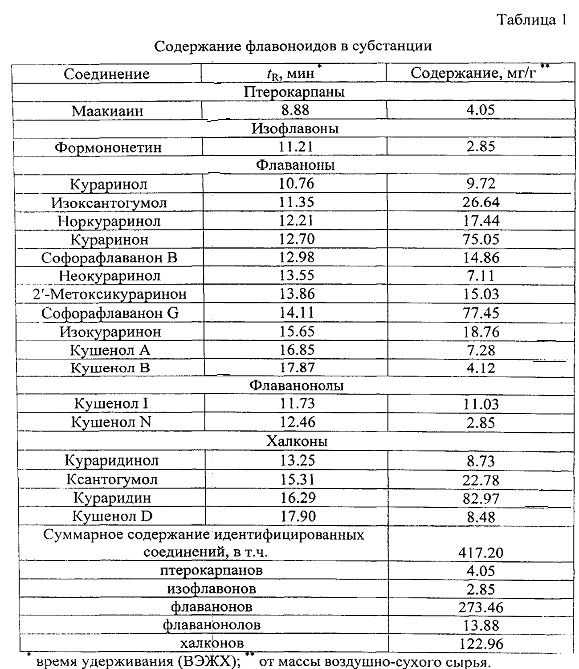
К числу основных флавоноидов субстанции относятся кураридин (8.30%), софорафлаванон G (7.75%), кураринон (7.51%). Следует отметить относительно высокое содержание таких биологически активных компонентов, как изоксантогумол (2.66%), ксантогумол (2.28%), изокураринон (1.88%) и норкураринон (1.74%).
Количественный анализ и показатели качества субстанции
Для проведения количественного анализа субстанции разработана методика определения суммарного содержания флавоноидов, а также раздельного определения доминирующих классов флавоноидов - флаванонов и халконов, с применением спектрофотометрического метода.
Методика основана на регистрации оптической плотности спиртового раствора субстанции при 290 нм (флаваноны) и 380 нм (халконы). В качестве рабочих стандартных образцов (РСО) выбраны кураринон и кураридин - доминирующие компоненты в составе групп флаванонов и халконов.
Методика количественного анализа флавоноидов. Около 0.1 г (точная навеска) субстанции помещают в колбу со шлифом вместимостью 250 мл, приливают 100 мл 95% этанола, присоединяют к обратному холодильнику и нагревают на кипящей водяной бане в течение 15 мин. После охлаждения содержимое колбы фильтруют в мерную колбу вместимостью 100 мл и доводят объем раствора до метки 95% этанолом (раствор А). 1 мл раствора А переносят в мерную колбу вместимостью 25 мл и доводят объем раствора до метки 95% этанолом (раствор Б). Оптическую плотность раствора Б измеряют при длинах волн 290 и 380 нм. В качестве раствора сравнения используют 95% этанол.
Суммарное содержание флаванонов в пересчете на кураринон в субстанции в процентах (X1) вычисляют по формуле
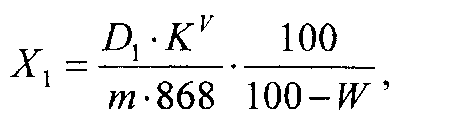
где D1 - оптическая плотность раствора Б при 290 нм; KV - коэффициент разбавления исследуемого раствора (2500); m - масса навески субстанции, г; 868 - удельный коэффициент погашения кураринона при 290 нм в 95% этаноле; W - потеря в массе при высушивании субстанции, %.
Суммарное содержание халконов в пересчете на кураридин в субстанции в процентах (Х2) вычисляют по формуле
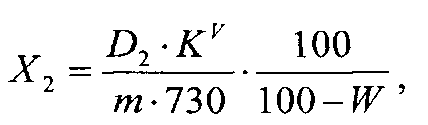
где D2 - оптическая плотность раствора Б при 380 нм; KV - коэффициент разбавления исследуемого раствора (2500); m - масса навески субстанции, г; 730 - удельный коэффициент погашения кураридина при 380 нм в 95% этаноле; W - потеря в массе при высушивании субстанции, %.
Суммарное содержание флавоноидов (X) вычисляют по формуле
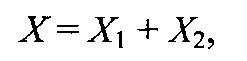
где X1 - суммарное содержание флаванонов в субстанции, %; X2 - суммарное содержание халконов в субстанции, %.
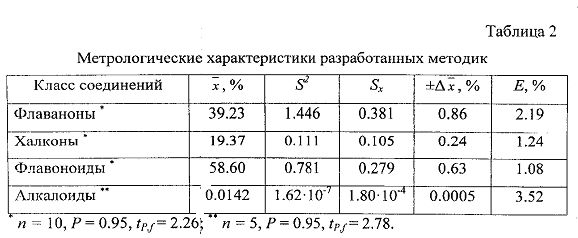
В табл.2 приведены результаты метрологического анализа разработанной методики. Исследования 8 серий субстанции показали, что содержание флаванонов может составлять 36.01-43.89%, халконов - 17.44-27.54%, суммарное содержание флавоноидов - 53.45-71.43%.
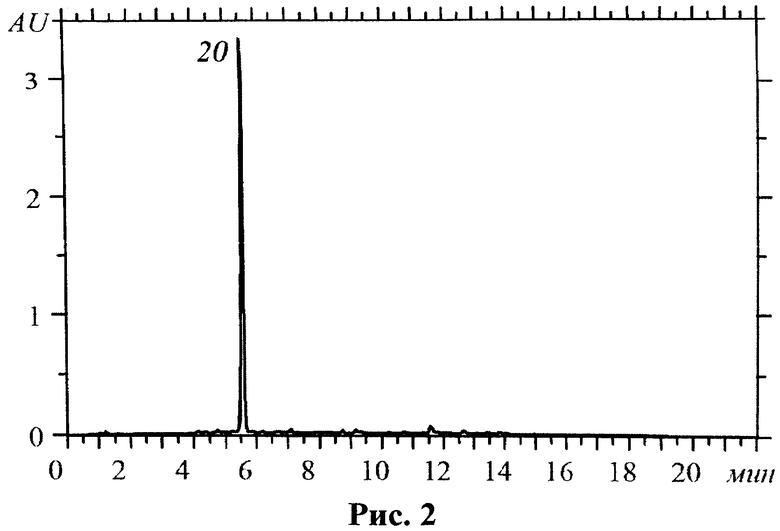
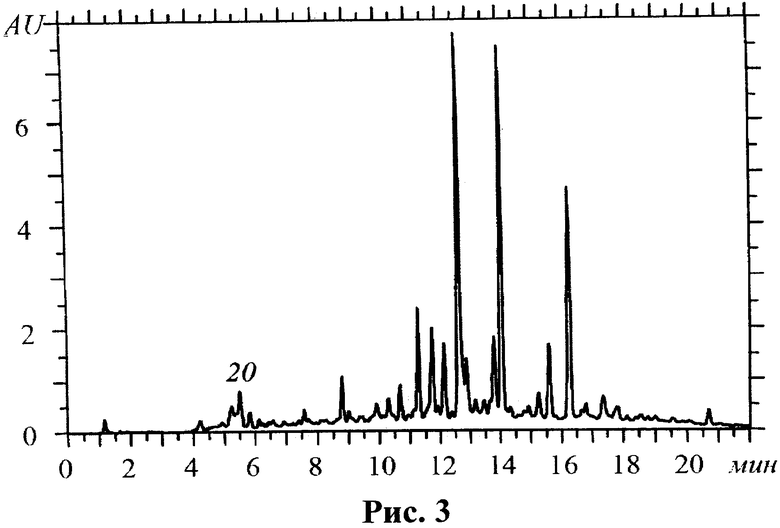
Учитывая тот факт, что субстанция представляет собой суммарный препарат флавоноидов, в ходе контроля качества необходимо определять содержание примесных соединений, каковыми в данном случае являются алкалоиды, присутствующие в растительном сырье и извлекаемые в результате водной экстракции в ходе технологического процесса. Как показали исследования, невозможно добиться полной экстракции данной группы соединений, но выбранные условия экстракции позволяют удалить до 95% алкалоидов (от исходного содержания в растительном сырье). Контроль наличия алкалоидов в субстанции предлагается проводить с применением метода ВЭЖХ по содержанию доминирующего соединения - матрина. На рис.2 представлена хроматограмма стандартного образца матрина; на рис.3 - хроматограмма субстанции флавоноидов.
Исследования показали, что содержание матрина в субстанции может составлять 0.01-0.03% и не превышает значения 0.1%, которое выбрано в качестве верхней границы содержания данного соединения. Метрологические характеристики методики определения матрина с применением ВЭЖХ приведены в табл.2.
Методика количественного анализа матрина. Около 0.1 г (точная навеска) субстанции помещают в колбу со шлифом вместимостью 250 мл, приливают 100 мл 95% этанола, присоединяют к обратному холодильнику и нагревают на кипящей водяной бане в течение 15 мин. После охлаждения содержимое колбы фильтруют в мерную колбу вместимостью 100 мл и доводят объем раствора до метки 95% этанолом. 1 мл полученного раствора фильтруют через кварцевый фильтр Millipore (⌀ 0.45 мкм) в хромаографическую виалу и используют для анализа методом микроколоночной ВЭЖХ-УФ. Условия ВЭЖХ-УФ: жидкостный хроматограф Милихром А-02 (Эконова); колонка ProntoSIL-120-5-C18 AQ (2×75 мм, ⌀ 5 мкм; Metrohm AG); подвижная фаза: (4.1 M LiClO4 в 0.1 M HClO4):H2O 5:95 (A), MeCN (В); градиентный режим B в A: 5-100% (0-20 мин), 100% (20-22 мин); ν 150 мкл/мин; Тколонки 35°C; Vпробы 2 мкл; детектор при 210 нм.
Содержание матрина в субстанции в процентах (X1) вычисляют по формуле
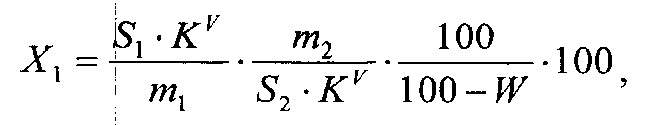
где S1 - площадь пика матрина в исследуемом растворе; KV - коэффициент разбавления исследуемого раствора (100); m1 - масса навески субстанции, г; S2 - площадь пика матрина в растворе РСО; KV - коэффициент разбавления раствора РСО матрина (1); m2 - масса навески матрина, г; W - потеря в массе при высушивании субстанции, %.
Приготовление раствора PCO матрина. Около 1 мг (точная навеска) матрина переносят в пробирку Эппендорфа, приливают 1 мл метанола и перемешивают.
Метрологический анализ разработанных методик показал, что относительная ошибка определения флавоноидов спектрофотометрическим методом не превышает 2.5%, матрина методом ВЭЖХ - не превышает 4%. Полученные результаты свидетельствуют об удовлетворительных валидационных параметрах методик, что указывает на возможность их использования в практике фармакопейного анализа для определения показателей качества субстанции флавоноидов из корней Sophora flavescens.
По данным проведенных исследований определены общие показатели качества субстанции, обобщенные в табл.3. Для стандартизации субстанции было предложено определение внешнего вида, подлинности (присутствие трех флавоноидов методом ВЭТСХ), потери в массе при высушивании, золы сульфатной, тяжелых металлов, количественное определение суммарного содержания флаванонов, халконов и флавоноидов (спектрофотометрия), матрина (ВЭЖХ) и микробиологической чистоты.
Предлагаемый способ, по сравнению с известным, позволяет получить препарат постоянного состава с более выраженной активностью за счет более высокого содержания действующих веществ (флавоноидов).
Рассчитать экономическую целесообразность предлагаемого способа в настоящее время не представляется возможным, однако вышеуказанные преимущества в сочетании с простой схемой получения способствуют рациональному использованию лекарственного растительного сырья и определяют перспективность внедрения данного способа в фармацевтическую промышленность.
Фармакотерапевтические свойства субстанции
Острая токсичность средства. Опыты проведены на крысах-самцах линии Wistar массой 180-190 г. Острую токсичность средства оценивали с использованием метода Кербера [2] при однократном внутрибрюшинном введении в дозах 500-5000 г/кг и внутрижелудочном введении в дозах 1000-6000 мг/кг. Все испытанные дозы растворяли в ДМСО и доводили дистиллированной водой до конечного объема, составляющего 1.0 мл/100 г массы животного. Наблюдение за общим состоянием подопытных крыс и их поведением осуществляли в течение 14 дней. При этом в первый день после введения указанных средств животные находились под постоянным наблюдением. Регистрировали наблюдаемые признаки интоксикации: общее состояние животных, поведение, двигательную активность, характер дыхательных движений, состояние волосяного и кожного покрова, окраску слизистых оболочек, потребление корма и воды, количество и консистенцию фекальных масс, частоту мочеиспускания и окраску мочи. Также регистрировали сроки развития интоксикации и гибели животных. На 14 сутки эксперимента осуществляли эвтаназию под эфирным наркозом и проводили макроскопический осмотр внутренних органов с помощью бинокулярной лупы, а также осуществляли патоморфологическое исследование жизненно важных органов.
Установлено, что при внутрижелудочном и внутрибрюшинном введении средства во всех исследованных дозах гибели животных в течение всего периода наблюдения не отмечалось. При внутрибрюшинном введении наиболее высоких доз испытуемого средства (2000-5000 мкг/кг) в течение 1-2 суток видимые признаки интоксикации животных не наблюдались. Вскрытие животных, осуществленное через 14 суток, показало, что визуально внутренние органы не отличались от таковых у интактных крыс; патоморфологические изменения у животных, получавших средство в высоких дозах, также не были отмечены. Таким образом, полученные данные позволяют отнести испытуемое средство к группе малотоксичных веществ по классификации К.К. Сидорова [3].
Влияние средства на иммунную систему. Эксперименты проведены на мышах самцах линии CBA массой 18-20 г. Действие исследуемых фракций и индивидуальных соединений было изучено на животных, находящихся в состоянии иммунодепрессии, вызванной цитостатиком азатиоприном, который вводили контрольной группе животных в дозе 50 мг/кг перорально 1 раз в сутки в течение 5 дней [4]. Исследуемые фракции и соединения вводили на фоне азатиоприна перорально ежедневно в течение 14 дней. Интактная группа животных получала изотонический раствор хлорида натрия по аналогичной схеме. Состояние гуморального иммунитета оценивали по количеству антителообразующих клеток (АОК), определяемых методом локального гемолиза по методу Cunningham [5]. Мышей иммунизировали внутрибрюшинно эритроцитами барана в дозе 2·108 клеток/мышь. Величину иммунного ответа оценивали по числу АОК на селезенку и на 106 клеток с ядрами на 5 сутки после иммунизации. Действие испытуемых средств на состояние клеточного звена иммунного ответа оценивали в реакции гиперчувствительности замедленного типа (ГЗТ) [6]. При статистической обработке экспериментальных данных вычислялась средняя арифметическая, ошибка средней арифметической, критерий Стьюдента и достоверность различий (P). Различие считали достоверным при вероятности 95% (Р≤0.05) [7].
В число объектов исследования, кроме субстанции флавоноидов, были включены суммарный спиртовой экстракт корней S. flavescens (сухой экстракт), различные фракции экстракта (хлороформная, этилацетатная, бутанольная, водная) и три индивидуальных соединения (кураринон, софорафлаванон G, кураридин). В качестве препарата сравнения использовали высушенный до воздушно-сухого состояния аптечный экстракт элеутерококка колючего.
Получение сухого экстракта. 200 г измельченных корней S. flavescens экстрагировали 70% этанолом в соотношении сырье:экстрагент 1:15 трехкратно при 90°С. Спиртовые извлечения отфильтровывали, объединяли, концентрировали в вакууме до 1/60 начального объема и высушивали в вакуум-сушильном шкафу до воздушно-сухого состояния. Выход сухого экстракта составил 53.4 г (26.7% от массы сырья).
Получение фракций сухого экстракта. 30 г сухого экстракта последовательно экстрагировали в аппарате Сокслета хлороформом и этилацетатом. К остатку экстракта после обработки органическими растворителями приливали 200 мл смеси бутанол: вода и встряхивали до полного распределения между фазами (40-50 мин), после чего смесь оставляли до полного расслоения. Полученные органические и водное извлечения концентрировали в вакууме до воздушно-сухого состояния. В результате было получено 2.25 г хлороформной фракции (выход 7.5% от массы сухого экстракта), 2.25 г этилацетатной фракции (выход 3.5%), 11.02 г бутанольной фракции (выход 36.7%) и 15.31 г водной фракции (выход 51.0%).
При исследовании влияния сухого экстракта и фракций S. flavescens на процессы антителообразования установлено, что данные средства восстанавливают показатели гуморального иммунного ответа в условиях азатиоприновой иммуносупрессии. Введение азатиоприна приводило к снижению как абсолютного числа АОК, так и числа АОК на 106 спленоцитов на 47% и 39% соответственно по сравнению с теми же показателями в контрольной группе (табл.4).
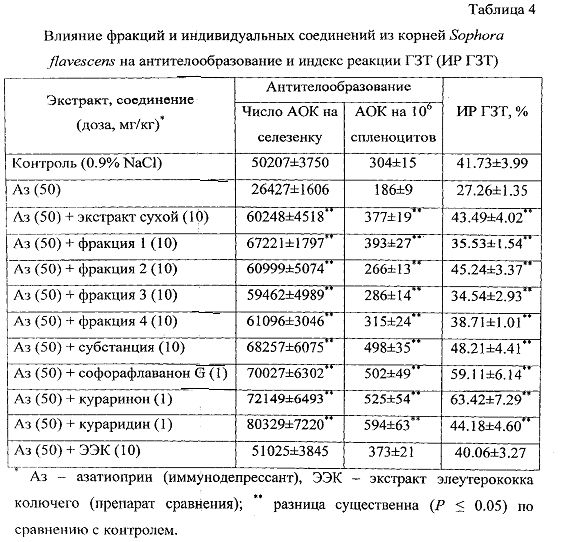
При введении сухого экстракта и фракций из него на фоне иммуносупрессии наблюдали достоверное увеличение количества АОК как в абсолютных значениях, так и при расчете на 106 спленоцитов; при этом первый показатель превышал уровень азатиоприновой супрессии в 2.3-2.5 раза, а второй - в 1.4-2.1 раза соответственно. Наиболее активными являются хлороформная и этилацетатная фракции, причем первая оказывала наиболее выраженное стимулирующее действие на показатели гуморального иммунного ответа.
При исследовании влияния средств на клеточно-опосредованную реакцию ГЗТ установлено, что они восстанавливают индекс данной реакции (ИР ГЗТ) в условиях азатиоприновой иммуносупрессии. Введение азатиоприна приводило к снижению ИР ГЗТ на 35% по сравнению с тем же показателем в контрольной группе. При введении средств наблюдали увеличение ИР ГЗТ в 1.3-1.7 раза по сравнению с контролем. Наибольшая выраженность действия была выявлена для группы животных, получавших этилацетатную фракцию (45.24%).
Согласно данным химического анализа наиболее активные хлороформная и этилацетатная фракции сухого экстракта содержат в качестве основных соединений флавоноиды, причем в хлороформной доминировали халконы, а в этилацетатной - флаваноны. В субстанции флавоноидов корней S. flavescens присутствуют обе группы соединений, что позволяет предположить наличие у данной фракции способности к одновременному стимулированию как гуморального, так и клеточного звеньев иммунитета, что было подтверждено экспериментальными данными. Так, было установлено, что применение субстанции в условиях иммуносупрессии повышает число АОК на 158% по сравнению с показателем группы, принимавшей азатиоприн, и на 168% увеличивает количество АОК на 106 спленоцитов. Прием субстанции увеличивает значение ИР ГЗТ до 48.21%, что в 1.8 раз больше, чем у азатиоприновой группы животных.
Флавоноиды S. flavescens являются основной группой веществ, обуславливающих наличие иммуностимулирующего эффекта, что подтверждается сведениями об активности индивидуальных соединений. Следует отметить, что флаваноны софорафлаванон G и кураринон оказывают наиболее выраженное действие на клеточное звено иммунитета, а более липофильный халкон кураридин - на гуморальное звено. Активность субстанции флавоноидов превышает таковую препарата сравнения (экстракта элеутерококка колючего) на 20 и 34% соответственно для ИР ГЗТ и АОК.
Таким образом, полученные данные позволяют заключить, что субстанция флавоноидов софоры желтоватой является эффективным иммунокорригирующим средством, способным ослаблять супрессивное действие азатиоприна на состояние клеточного и гуморального звеньев иммунного ответа. Исследуемое средство не изменяет показатели иммунитета интактных животных, что присуще лишь истинным иммуномодуляторам, обладающим активностью только в условиях повреждения иммунитета.
Источники информации
1. Халилов P.M., Маматханов А.У., Сотимов Г.Б., Маматханова М.А. Разработка технологии получения фланорина из Pseudosophora alopecuroides // Химия растительного сырья. 2009. №2. С.93-96 - прототип.
2. Требования по доклиническому изучению общетоксического действия новых фармакологических веществ. М., 1984. 49 с.
3. Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ. МЗ РФ. Департамент контроля качества, эффективности и безопасности фармакологических средств. Фармакологический комитет МЗ РФ. 2 изд., перераб. и доп. М., 2005. С.138-145.
4. Лазарева Д.Н., Алехин E.K. Стимуляторы иммунитета. М., 1985. 256 с.
5. Cunningham A.J. A method of increased sensitivity for detecting single antibodyforming cells // Nature. 1965. V. 207. №5001. P. 1106-1107.
6. Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ. М., 2000. 678 с.
7. Лакин Г.Ф. Биометрия. М., 1990. 352 с.
Краткое описание чертежей
На рис.1 изображена хроматограмма субстанции флавоноидов корней Sophora flavescens, на которой числами указано положение соединений: 1 - маакиаин, 2 - кураринол, 3 - формононетин, 4 - изоксантогумол, 5 - кушенол I, 6 - норкураринол, 7 - кушенол N, 8 - кураринон, 9 - софорафлаванон B, 10 - кураридинол, 11 - неркураринол, 12 - 2′-метоксикураринон, 13 - софорафлаванон G, 14 - ксантогумол, 15 - изокураринон, 16 - кураридин, 17 - кушенол A, 18 - кушенол B, 19 - кушенол D.
На рис.2. изображена хроматограмма стандартного образца матрина, на которой обозначено положение матрина (20).
На рис.3. изображена хроматограмма субстанции флавоноидов из корней Sophora flavescens при 210 нм, на которой обозначено положение матрина (20).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения иммуностимулирующего средства из шрота ягод брусники обыкновенной | 2021 |
|
RU2775065C1 |
| СПОСОБ ПОЛУЧЕНИЯ РАСТИТЕЛЬНОГО СРЕДСТВА, ОБЛАДАЮЩЕГО ИММУНОСТИМУЛИРУЮЩЕЙ АКТИВНОСТЬЮ | 2016 |
|
RU2639132C1 |
| СПОСОБ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО АДАПТОГЕННЫМ ДЕЙСТВИЕМ | 2012 |
|
RU2582952C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-ГИДРОКСИАЦЕТОФЕНОН-4-О-БЕТА-D-ГЛЮКОПИРАНОЗИДА (ПИЦЕИНА) ИЗ РАСТИТЕЛЬНОГО СЫРЬЯ | 2012 |
|
RU2532167C2 |
| СПОСОБ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО ГЕПАТОЗАЩИТНОЙ АКТИВНОСТЬЮ | 2008 |
|
RU2366445C1 |
| СПОСОБ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО ИММУНОМОДУЛИРУЮЩЕЙ АКТИВНОСТЬЮ | 2012 |
|
RU2496510C2 |
| Средство, обладающее капилляроукрепляющей и антиагрегационной активностью, и способ его получения | 2023 |
|
RU2814315C1 |
| СПОСОБ ПОЛУЧЕНИЯ РАНОЗАЖИВЛЯЮЩЕГО И АНТИМИКРОБНОГО СРЕДСТВА | 1999 |
|
RU2152797C1 |
| СПОСОБ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО ГИПОЛИПИДЕМИЧЕСКОЙ АКТИВНОСТЬЮ | 2000 |
|
RU2173161C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПАТУЛЕТИНА И ПАТУЛЕТИН-7-О-β-D-ГЛЮКОПИРАНОЗИДА (ПАТУЛИТРИНА) ИЗ РАСТИТЕЛЬНОГО СЫРЬЯ | 2013 |
|
RU2546298C2 |
Изобретение относится к фармацевтической промышленности и касается способа получения средства, обладающего иммуностимулирующем действием. Способ получения средства, обладающего иммуностимулирующим действием, из корней софоры желтоватой заключается в том, что измельченные корни софоры желтоватой заливают водой, подвергают ультразвуковой обработке, далее смесь нагревают и экстрагируют при постоянном перемешивании, извлечение фильтруют, маточный раствор отбрасывают, шрот заливают водой и экстрагируют, извлечение фильтруют, маточный раствор отбрасывают, далее шрот заливают 90-95% этанолом, подвергают смесь ультразвуковой обработке, после чего смесь экстрагируют дважды, извлечения фильтруют, спиртовые извлечения объединяют и концентрируют, сгущенный экстракт высушивают и измельчают, смешивают с 10% этанолом и оставляют при постоянном перемешивании, нерастворившийся остаток отфильтровывают и высушивают, при определенных условиях. Средство, полученное вышеописанным способом, обладает выраженным иммуностимулирующим действием. 3 ил., 4 табл.
Способ получения средства, обладающего иммуностимулирующим действием, из корней софоры желтоватой, заключающийся в том, что измельченные корни софоры желтоватой заливают водой в соотношении сырье:экстрагент 1:(10-12), подвергают ультразвуковой обработке частотой 50 кГц в течение 20-30 мин, далее смесь нагревают до температуры 30-40°C и экстрагируют при постоянном перемешивании в течение 1-1,5 ч, извлечение фильтруют, маточный раствор отбрасывают, шрот заливают водой в соотношении сырье:экстрагент 1:(18-20) и экстрагируют при температуре 50-60°C в течение 1,5-2 ч, извлечение фильтруют, маточный раствор отбрасывают, далее шрот заливают 90-95% этанолом в соотношении сырье:экстрагент 1:(15-17), подвергают смесь ультразвуковой обработке частотой 50 кГц в течение 20-30 мин, после чего смесь экстрагируют дважды при температуре 80-85°C в течение 1-1,5 ч, извлечения фильтруют, спиртовые извлечения объединяют и концентрируют до 1/50 от первоначального объема, сгущенный экстракт высушивают и измельчают, смешивают с 10% этанолом в соотношении 1:(6-8) и оставляют при 20-25°C и постоянном перемешивании в течение 0,5-1 ч, нерастворившийся остаток отфильтровывают и высушивают.

