Изобретение относится к аналитической химии, гидрохимии, биохимии, экологии, криомедицине, фармакологии, судебной медицине, криминалистике и может быть использовано для выделения как природных, так и синтетических, техногенных органических веществ из водных сред, водосодержащих биологических жидкостей (моча, кровь и др.) и водных экстрактов-вытяжек различных объектов.
Известен способ выделения органических веществ, сочетающий экстракцию и вымораживание [1]. Растворенные органические соединения целевые органические вещества извлекают из воды, водосодержащих биологических жидкостей, водных вытяжек различных объектов путем предварительного добавления в объем пробы растворимого или ограниченно растворимого органического экстрагента (ацетонитрил, ацетон, диэтиловый эфир и т.п.) и последующего охлаждения приготовленного раствора до кристаллизации водной части. В результате в выделяющийся в отдельную жидкую фазу добавленный органический растворитель переходят извлекаемые органические вещества. Полученный жидкий органический экстракт с целевыми компонентами отделяют от замороженной части образца. Данный способ впоследствии получил название метод экстракционного вымораживания (Extractive freezing-out) [2-3]. В зависимости от условий проведения процесса целевые компоненты можно концентрировать и в твердой фазе льда [4].
Однако указанный способ [1] имеет ряд серьезных недостатков:
- несмотря на идентичность условий масса получаемых жидких экстрактов сильно варьирует (изменяется) даже в параллельных определениях-опытах [2-3];
- снижение температуры экстракционного вымораживания сопровождается резким снижением количества экстракта в результате втягивания его значительной части в образующиеся при кристаллизации трещины льда за счет капиллярных сил вплоть до полного отсутствия [4, 5];
- степень концентрирования извлекаемых компонентов обычно не превышает 3÷4 крат, поскольку не удается снизить долю добавляемого экстрагента менее 25% [3] по причине существенных потерь экстракта в виде жидких микровключений в кристаллической фазе, а также вследствие втягивания его значительной части в образующиеся при кристаллизации трещины льда за счет капиллярных сил [4].
Задачей предлагаемого способа было улучшение воспроизводимости результатов экстракции в отношении количества получаемого экстракта, снижение количества экстрагента в экстракционной системе и содержания воды в получаемом экстракте, улучшение экономических показателей.
Это достигается тем, что целевые органические вещества извлекают из водосодержащей среды с помощью экстракционного вымораживания (ЭВ) в условиях действия поля центробежных сил. А именно после предварительного добавления в пробу водного раствора, водосодержащей биологической жидкости, водной вытяжки различных объектов органического экстрагента, в т.ч. растворимого или ограниченно растворимого (ацетонитрил, ацетон, диэтиловый эфир, этилацетат и т.п.) кристаллизацию водной части осуществляют охлаждением смеси в условиях центрифугирования, т.е. под воздействием поля центробежных сил. За счет действия центробежных сил, в условиях сжатия, разности плотностей контактирующих фаз, в т.ч. воды, экстрагента, растворенных газов, твердой фазы (ее гранулометрического состава), гидродинамических факторов, вязкости жидких, газообразных составляющих и т.д., а также вибрации, при замораживании удается достичь более гомогенной структуры кристаллической водной фазы. Следствием этого является значительное возрастание массы (объема) экстракта, получаемого в результате предлагаемого нового способа экстракции, по сравнению с процедурой ЭВ в отсутствии центрифугирования, а также резкое повышение воспроизводимости результатов извлечения (масса получаемого экстракт, содержание в нем целевого компонента). Снижение температуры проведения ЭВ выгодно с точки зрения уменьшения содержания воды в получаемом экстракте за счет снижении ее растворимости органическом растворителе, используемом в качестве экстрагента. Во-первых, это уменьшает объем получаемого экстракта и повышает коэффициент (степень) концентрирования в нем целевых компонентов, во-вторых, является важным условием сохранения работоспособности и физико-химических характеристик разделительной колонки при газохроматографическом исследовании пробы. Кроме того, в химическом анализе также часто встречаются ситуации, когда присутствие воды в экстрактах нежелательно. Вместе с тем, из-за неоднородности кристаллической фазы льда может наступить момент, когда практически весь экстракт втягивается в ее трещины. Предлагаемый способ решает эти проблемы, что подтверждается приведенными ниже экспериментами.
Приведенные ниже примеры демонстрируют возможность использования различных растворителей для извлечения органических веществ разных классов, а также варьирования условий экстракционного вымораживания в поле центробежных сил, в т.ч. температуры, силы поля центробежных сил через изменение скорости вращения ротора центрифуги. Указанные параметры важны для оптимизации процесса экстракции, повышения ее эффективности и селективности в отношении извлечения целевых компонентов из сложных биологических матриц.
Пример 1.
Во флаконы (8 шт. ) с завинчивающимися пробками емкостью 11 мл поместили по 3,0 мл водного раствора органических кислот. Индивидуальное содержание в воде Свод. каждой из них указано в табл. 1. Добавили по 1,5 мл ацетонитрила марки «Сорт 2». Тщательно перемешав, четыре пробы поместили в ротор охлаждаемой центрифуги, температура - 25±1°С. Произвели центрифугирование при 8000 обор./мин в течение 90 мин. Оставшиеся четыре подвергли процедуре ЭВ по способу [1] в тех же самых температурных условиях. После того как водная часть пробы замерзла, декантацией отделили органический прозрачный слой жидкого экстракта ацетонитрила. Его масса в среднем составила 0,12±0,005 г. В пробах, параллельно подвергнутых ЭВ по способу [1], экстракта в виде отдельной жидкой фазы образовалось в количестве менее 5-10 мкл, что не позволило его отделить от твердой фазы льда и проанализировать. Методом газовой хроматографии с пламенно-ионизационным детектированием определили содержание кислот в экстрактах, полученных предлагаемым способом экстракционного вымораживания с одновременным центрифугированием. Результаты опыта представлены в табл. 1.

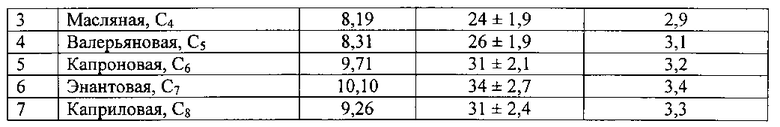
Пример 2.
Образцы воды массой 8,5 мл, подкисленные фосфорной кислотой до рН3 и содержащие сорбиновую кислоту в концентрации 1,9 мкг/мл, поместили в стеклянные флаконы с завинчивающимися пробками емкостью 11 мл. Добавили 1,5 мл ацетонитрила марки «Сорт 2». Герметично закрыв завинчивающимися пробками флаконы (8 шт. ), содержимое перемешали до растворения. Часть флаконов в количестве 4 шт. поместили в морозильную камеру при температуре - 25±1°С, остальные (4 шт. ) - в ротор охлаждаемой центрифуги, термостатируемой при - 25±1°С, и подвергли центрифугированию до кристаллизации водной части образца. Во флаконах, охлаждаемых без центрифугирования, т.е. по способу [1], после кристаллизации водной части образца с поверхности льда собрать экстракт не удалось. Он полностью впитался в твердую фазу. В то же время во флаконах, находившихся в охлаждаемой центрифуге, на поверхности льда образовался слой жидкой фазы органического растворителя (экстракт), который отделили декантацией в отдельную пробирку. Результаты опыта по определению массы экстракта и содержания в нем органической кислоты представлены в табл. 2.
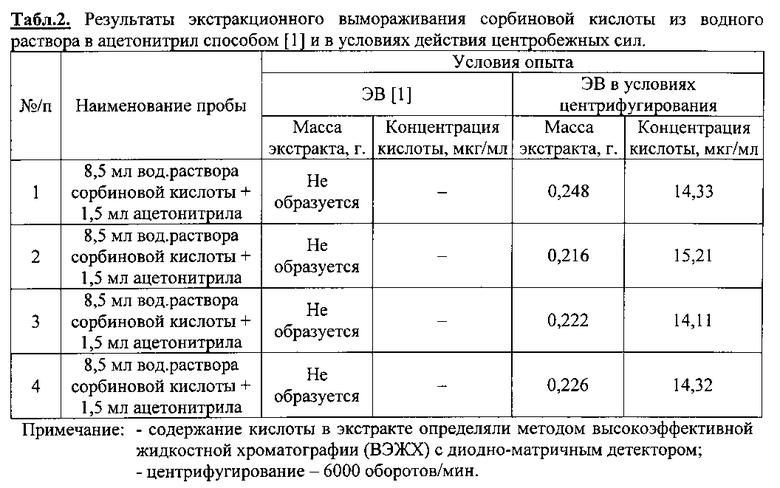
Представленные в табл. 2 данные свидетельствуют о высокой воспроизводимости результатов опыта в отношении величины массы получаемого экстракта предлагаемым способом: доверительный интервал для среднего значения равен 0,23±0,014 (n=4, Р=0,95). Это значительно превосходит показатели известного метода ЭВ [1, 5] при осуществлении процесса в аналогичном температурном интервале.
Пример 3.
Во флакон №1 с завинчивающейся пробкой емкостью 11 мл поместили 8,5 мл утренней мочи пациента Б. и 1,5 мл ацетонитрила марки «Сорт 2». Во флакон №2 с завинчивающейся пробкой емкостью 11 мл поместили 8,5 мл утренней мочи пациента Б., добавили 50 мкл раствора салициловой кислоты в воде концентрации 1350 мкг/мл и 1,5 мл ацетонитрила марки «Сорт 2». Пробы, тщательно перемешав, поместили в ротор охлаждаемой центрифуги, температура - 25±1°С. Произвели центрифугирование при 8000 об/мин в течение 120 мин. После кристаллизации водной части исследуемых образцов с поверхности льда декантацией собрали жидкую органическую фазу (экстракт). Масса экстракта составила: из флакона №1 - 0,218 г, флакона №2 - 0,224 г. В результате исследования методом высокоэффективной жидкостной хроматографии с диодно-матричным детектированием установлено содержание салициловой кислоты в полученном экстракте из флакона №2 - 25,2 мкг/мл. В экстракте из флакона №1 салициловая кислота не обнаружена.
Пример 4.
Во флаконы №1-4 (4 шт. ) с завинчивающимися пробками емкостью 11 мл поместили по 9 мл морской воды плавательного бассейна. Добавили 10 мкл серной кислоты (50%) и 1 мл ацетонитрила марки «Сорт 2». В аналогичные флаконы №5-8 (4 шт.) с завинчивающимися пробками емкостью 11 мл поместили по 9 мл морской воды бассейна с добавкой органических кислот С2-С8 в каждый по 4 мкл из стандартного раствора кислот в ацетонитриле с содержанием С2 - 656, С3 - 660, С4 - 844, С5 - 856, С6 - 1000, С7 - 1040 и С8 - 954 мкг/мл. Добавили 10 мкл серной кислоты (50%) и 1 мл ацетонитрила марки «Сорт 2». Тщательно перемешав, все пробы поместили в ротор охлаждаемой центрифуги, температура - 25±1°С. Произвели центрифугирование при 8000 об/мин в течение 120 мин. После того как водная часть пробы замерзла, декантацией отделили органический прозрачный слой жидкого экстракта ацетонитрила. Его масса в среднем составила 0,06±0,003 г. Методом газовой хроматографии с пламенно-ионизационным детектированием определили содержание кислот в экстрактах. Результаты опыта представлены в табл. 3.
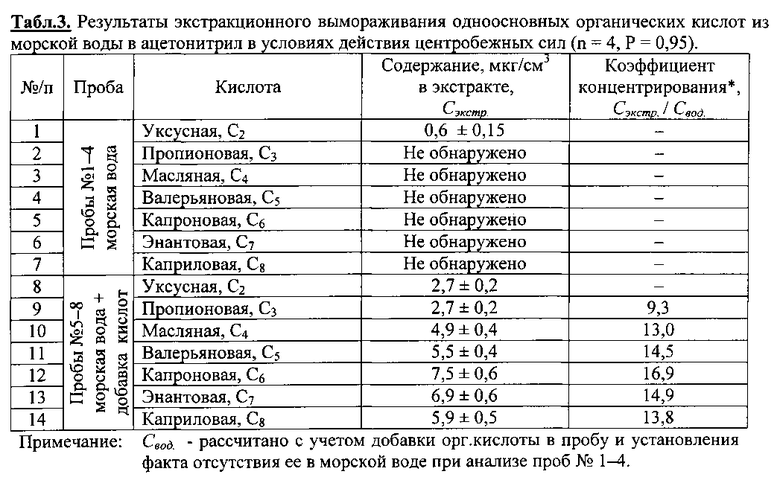
Как следует из полученных результатов (табл. 3), коэффициент концентрирования целевого компонента предлагаемым способом может достигать практически 17 крат (например, в случае капроновой кислоты), что почти в четыре раза превосходит возможности способа [1] в подобном температурном интервале экстракционного вымораживания [5].
Пример 5.
В опыте изучено влияние скорости вращения ротора центрифуги или величины центробежного ускорения, поскольку это взаимосвязанные величины, на массу получаемого экстракта. Во флаконы с завинчивающимися пробками емкостью 11 мл наливали по 9 мл дистиллированной воды и 1 мл ацетонитрила. После этого флаконы с содержимым помещали в предварительно охлажденный до температуры - 32±2°С ротор центрифуги. Контроль температуры вели с помощью цифрового термометра «Testo 174Т» (фирма Testo AG, Germany), находящегося внутри морозильной камеры. Затем проводили центрифугирование в течение 45 мин. Заданную скорость вращения ротора контролировали с помощью тахометра «UT372» (фирма UNIT, Hong Kong). Полученная экспериментальная зависимость представлена рис. 1.
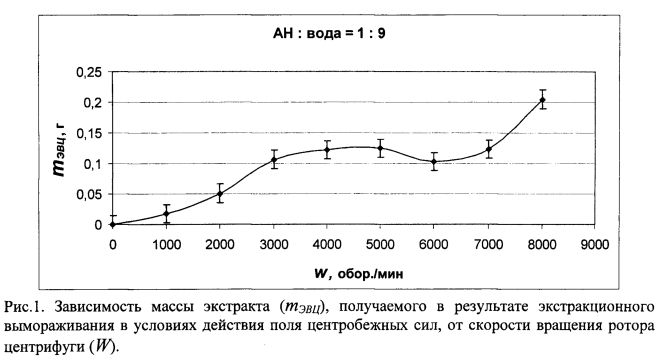
При ЭВ без центрифугирования, т.е. в условиях W=0, экстракт (ацетонирил) полностью поглощался образующимся льдом, имеющим в этом случае поликристаллическую (с трещинами) структуру. Начиная со скорости вращения ротора 3000-4000 об/мин и выше большая часть замерзшей водной фазы образца имеет вид прозрачного монокристаллического льда. Лишь в центральной его части остается небольшая поликристаллическая область с некоторым количеством вмерзших шарообразных пузырьков воздуха и, возможно, ацетонитрильного экстракта. Представленная на рис. 1 зависимость показывает, что при прочих равных условиях с увеличением скорости вращения ротора, т.е. с ростом центробежной силы, количество получаемого ацетонитрильного экстракта увеличивается на участках от 0 до 3000 и от 7000 до 8000 об/мин, в диапазоне от 3000 до 7000 об/мин его масса стабильна (0,1-0,12 г).
Пример 6.
Во флаконы (4 шт.) с завинчивающейся пробкой емкостью 11 мл поместили по 5 мл раствора фенола в дистиллированной воде с концентрацией 2,44 мкг/мл и 1 мл 95% этилового спирта. Приготовленные образцы, тщательно перемешав, поместили в ротор охлаждаемой центрифуги, температура - 27°С. Произвели центрифугирование при 4000 обор./мин в течение 45 мин. После кристаллизации водной части исследуемых образцов с поверхности льда декантацией собрали жидкую органическую фазу (экстракт). Масса экстракта составила: из флакона №1 - 0,398 г, флакона №2 - 0,249 г, флакона №3 - 0,262 г и флакона №4 - 0,288 г. В результате исследования методом газовой хроматографии с пламенно-ионизационным детектированием установлено содержание фенола в полученных экстрактах из флакона №1 - 5,07 мкг/мл, флакона №2 - 5,48 мкг/мл, флакона №3 - 5,18 мкг/мл и из флакона №4 - 5,30 мкг/мл.
В аналогичных температурных условиях указанные выше смеси этанола и водного раствора фенола без центрифугирования замерзали без образования отдельной жидкой фазы спиртового экстракта.
Пример 7.
Во флаконы (4 шт.) с завинчивающейся пробкой емкостью 11 мл поместили 4 мл раствора фенола в дистиллированной воде с концентрацией 2,29 мкг/мл и 1,75 мл свежеперегнанного ацетилацетона. Приготовленные образцы, тщательно перемешав, поместили в ротор охлажденной до температуры - 17°С центрифуги. Произвели центрифугирование при 4000 об/мин в течение 40 мин. После кристаллизации водной части исследуемых образцов с поверхности льда декантацией собрали жидкую органическую фазу (экстракт). Масса экстракта составила: из флакона №1 - 0,852 г, флакона №2 - 0,784 г, флакона №3 - 0,755 г и флакона №4 - 0,796 г. В результате исследования методом газовой хроматографии с пламенно-ионизационным детектированием установлено содержание фенола в полученных экстрактах из флакона №1 - 0,20 мкг/мл, флакона №2 - 0,21 мкг/мл, флакона №3 - 0,20 мкг/мл и из флакона №4 - 0,18 мкг/мл.
Пример 8.
Образцы молока объемом по 8 мл, приготовленного из порошка сухого молока «Test Material 0590 Pesticide Residues Milk Powder» фирмы FAPAS с аттестованным значением содержания пестицидов путем растворения 0,80±0,002 г в 8 мл воды, поместили во флаконы (4 шт.) с завинчивающейся пробкой емкостью 11 мл. В два из них (№1-2) добавили по 1 мл ацетонитрила в каждый, в оставшиеся два (№3-4) по 1 мл раствора α-гексахлорциклогексана (изомер пестицида линдана) в ацетонитриле с концентрацией 0,0678 мкг/мл. Приготовленные образцы, тщательно перемешав, поместили в ротор охлаждаемой центрифуги, температура - 25°С. Произвели центрифугирование при 4000 об/мин в течение 45 мин. После кристаллизации водной части исследуемых образцов с поверхности льда, в объеме которого остались все дисперсные частицы молока, декантацией собрали прозрачную жидкую органическую фазу ацетонитрильного экстракта. Масса экстракта составила: из флакона №1 - 0,046 г, флакона №2 - 0,044 г, флакона №3 - 0,059 г и флакона №4 - 0,047 г. В результате газохроматографического исследования с электронозахватным детектированием расчетом по методу добавки с усреднением по двум параллельным пробам установлено содержание α-гексахлорциклогексана в сухом молоке 63 мкг/кг. Аттестованное значение содержания этого химического соединения в данном образце сухого молока согласно паспорту - 89,9 мкг/кг.
Пример 9.
Во флаконы (4 шт.) с завинчивающейся пробкой емкостью 11 мл поместили навески мелко нарезанной массы листьев персика массой 1,0±0,02 г, обработанного накануне за 1 сутки водным раствором салициловой кислоты. В два из них (№1-2) добавили по 2 мл дистиллированной воды и 2 мл ацетонитрила в каждый, в оставшиеся два (№3-4) по 2 мл дистиллированной воды и 2 мл раствора салициловой кислоты в ацетонитриле с концентрацией 6,215 мкг/мл. Флаконы поместили на вибро-встряхиватель «Heidolph» для экстракции в течение 30 мин (скоростной режим 1). Затем флаконы с содержимым поместили в ротор охлажденной до температуры - 27°С центрифуги. Произвели центрифугирование при 4000 об/мин в течение 45 мин. После кристаллизации водной части исследуемых образцов с поверхности льда, в объеме которого содержалась вся масса дисперсных растительных частиц, декантацией собрали прозрачную жидкую органическую фазу ацетонитрильного экстракта. Массы экстрактов составили: из флакона №1 - 0,556 г, флакона №2 - 0,548 г, флакона №3 - 0,559 г флакона №4 - 0,529 г. В результате исследования с помощью высоко эффективной жидкостной хроматографии с диодно-матричным детектированием расчетом по методу добавки с усреднением по двум параллельным пробам установлено содержание салициловой кислоты в листьях персика 19,34 мкг/г.
Пример 10.
Во флаконы (4 шт.) с завинчивающейся пробкой емкостью 11 мл поместили по 9 мл дистиллированной воды, в каждый из которых затем внесли микрошприцем МШ-10 по 2,5 мкл раствора нафталина в ацетонитриле в концентрации 766 мкг/мл и тщательно перемешали. В каждый из них затем также пипеткой-автодозатором BIOHIT добавили 1 мл этилацетата. Приготовленные таким образом смеси, тщательно перемешав, поместили в ротор охлажденной до температуры - 26°С центрифуги. Произвели центрифугирование при 4000 об/мин в течение 45 мин. После кристаллизации водной части исследуемых образцов с поверхности льда декантацией собрали жидкую органическую фазу этилацетата (экстракт). Его масса экстракта составила: из флакона №1 - 0,093 г, флакона №2 - 0,078 г, флакона №3 - 0,082 г и флакона №4 - 0,087 г. В результате исследования методом газовой хроматографии с пламенно-ионизационным детектированием установлено содержание нафталина в полученных экстрактах из флакона №1 - 12,5 мкг/мл, флакона №2 - 15,0 мкг/мл, флакона №3 - 14,4 мкг/мл и из флакона №4 - 12,9 мкг/мл.
Предлагаемый способ позволяет:
- существенно повысить степень концентрирования целевых компонентов в получаемом экстракте в сравнении с ранее известным способом [1];
- значительно снизить количество используемого экстрагента в экстракционной системе во время процедуры экстракционного вымораживания;
- добиться высокой стабильности массы (количества) получаемого экстракта в параллельных опытах;
- проводить одностадийную экстракцию целевых компонентов из биологических объектов;
- уменьшить материальные (необходимо меньшее количество экстрагента) и трудовые затраты, поскольку с ростом степени концентрирования целевых компонентов в получаемом экстракте нет необходимости проводить повторное ЭВ.
Библиография
1. Патент РФ на изобретение №2303476 // Способ извлечения органических веществ из водных сред экстракцией в сочетании с вымораживанием // Бехтерев В.Н. // Б.И. №21, 2007.
2. Бехтерев В.Н. Выделение фенолов из воды экстракционным вымораживанием // Журнал аналитической химии. 2008. Т. 63. №10. С. 1045-1049.
3. Bekhterev V.N. Extractive freezing-out in the analysis of organic compounds in the aqueous mediums // Mendeleev Communications. 2007. V. 17. P. 241-243.
4. Бехтерев В.Н. Закономерности поведения растворенных органических веществ в условиях экстракционного вымораживания // Журнал аналитической химии. 2011. Т. 66. №6. С. 608-613.
5. Бехтерев В.Н. // Автореф. дисс … докт. хим. наук. - М., 2011. - 41 с.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИЗВЛЕЧЕНИЯ ОРГАНИЧЕСКИХ ВЕЩЕСТВ ИЗ ВОДНЫХ СРЕД ЭКСТРАКЦИЕЙ В СОЧЕТАНИИ С ВЫМОРАЖИВАНИЕМ | 2005 |
|
RU2303476C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОДНООСНОВНЫХ КАРБОНОВЫХ КИСЛОТ C-С В ВОДЕ | 2008 |
|
RU2364864C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ОРГАНИЧЕСКИХ ВЕЩЕСТВ ИЗ ВОДНОЙ СРЕДЫ | 2005 |
|
RU2296716C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МИКРОКОЛИЧЕСТВ КЛОЗАНТЕЛА В ПЛАЗМЕ, МОЛОКЕ, ТКАНЯХ | 2007 |
|
RU2366950C2 |
| ЭКСТРАКЦИОННЫЙ ГЕНЕРАТОР ТЕХНЕЦИЯ - 99 М | 2000 |
|
RU2161132C1 |
| Способ качественного и количественного колориметрического определения формальдегида в молоке | 2022 |
|
RU2795470C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПОЛИЦИКЛИЧЕСКИХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ В ПОЧВАХ И ДОННЫХ ОТЛОЖЕНИЯХ | 2019 |
|
RU2719578C1 |
| Легкоплавкий экстрагент и способ извлечения цинка (II) из кислых водных растворов | 2020 |
|
RU2767313C1 |
| ЭКСТРАГЕНТ ДЛЯ ВЫДЕЛЕНИЯ ИОНОВ МЕТАЛЛОВ ИЗ ВОДНЫХ РАСТВОРОВ | 2007 |
|
RU2333028C1 |
| СПОСОБ ЭКСТРАКЦИОННОГО АФФИНАЖА УРАНА | 2014 |
|
RU2562604C1 |
Изобретение может быть использовано для выделения органических веществ из водных сред, водосодержащих биологических жидкостей и водных экстрактов-вытяжек. Для осуществления способа проводят экстракцию органических веществ из водной среды в органический растворитель в сочетании с вымораживанием в условиях действия поля центробежных сил. Для получения экстракта целевых органических веществ в пробу предварительно добавляют подходящий органический растворитель, в том числе растворимый или ограниченно растворимый в воде, который выделяется в отдельную жидкую фазу в процессе замораживания водной части в условиях центрифугирования. Способ обеспечивает улучшение воспроизводимости результатов экстракции в отношении количества получаемого экстракта, снижение количества экстрагента в экстракционной системе и снижение содержания воды в получаемом экстракте. 10 пр.
Способ извлечения органических веществ из водных сред, включающий вымораживание водной части и экстракцию растворенных соединений в добавленный растворимый в воде органический растворитель, выделяющийся в отдельную жидкую фазу в процессе замораживания, когда водная часть заморожена, отличающийся тем, что процесс осуществляют в поле центробежных сил.
| СПОСОБ ИЗВЛЕЧЕНИЯ ОРГАНИЧЕСКИХ ВЕЩЕСТВ ИЗ ВОДНЫХ СРЕД ЭКСТРАКЦИЕЙ В СОЧЕТАНИИ С ВЫМОРАЖИВАНИЕМ | 2005 |
|
RU2303476C2 |
| ЛЕЙТЕ В., Определение органических загрязнений питьевых, природных и сточных вод, Москва, Химия, 1975, с.37 | |||
| СПОСОБ УЛУЧШЕНИЯ КАЧЕСТВА ПИТЬЕВОЙ ВОДЫ ЗАМОРАЖИВАНИЕМ И ОТТАИВАНИЕМ | 2001 |
|
RU2186033C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОДНООСНОВНЫХ КАРБОНОВЫХ КИСЛОТ C-С В ВОДЕ | 2008 |
|
RU2364864C1 |
| СПОСОБ ПРОМЫШЛЕННОЙ ПЕРЕРАБОТКИ БЕЛОКСОДЕРЖАЩИХ ОРГАНИЧЕСКИХ ОТХОДОВ | 2012 |
|
RU2516759C2 |
| US 6367285 A, 09.04.2002 | |||
| US 5394706 A, 07.03.1995 | |||
| JPS62135437 A, 18.06.1987 | |||

